
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 05 August 2022
Sec. T Cell Biology
Volume 13 - 2022 | https://doi.org/10.3389/fimmu.2022.934624
This article is part of the Research Topic Molecular Switches of the Immune System: The E Protein/Id Axis in Hematopoietic Development and Function View all 11 articles
 Shweta Singh1†
Shweta Singh1† Tanmoy Sarkar1†
Tanmoy Sarkar1† Brad Jakubison1,2
Brad Jakubison1,2 Stephen Gadomski1Andrew Spradlin1
Stephen Gadomski1Andrew Spradlin1 Kristbjorn O. Gudmundsson1,2
Kristbjorn O. Gudmundsson1,2 Jonathan R. Keller1,2*
Jonathan R. Keller1,2*Adult mammalian hematopoiesis is a dynamic cellular process that provides a continuous supply of myeloid, lymphoid, erythroid/megakaryocyte cells for host survival. This process is sustained by regulating hematopoietic stem cells (HSCs) quiescence, proliferation and activation under homeostasis and stress, and regulating the proliferation and differentiation of downstream multipotent progenitor (MPP) and more committed progenitor cells. Inhibitor of DNA binding (ID) proteins are small helix-loop-helix (HLH) proteins that lack a basic (b) DNA binding domain present in other family members, and function as dominant-negative regulators of other bHLH proteins (E proteins) by inhibiting their transcriptional activity. ID proteins are required for normal T cell, B cell, NK and innate lymphoid cells, dendritic cell, and myeloid cell differentiation and development. However, recent evidence suggests that ID proteins are important regulators of normal and leukemic hematopoietic stem and progenitor cells (HSPCs). This chapter will review our current understanding of the function of ID proteins in HSPC development and highlight future areas of scientific investigation.
Hematopoiesis is sustained by a limited number of hematopoietic stem cells (HSCs) that reside in a quiescent state protected from proliferation-induced damage and exhaustion (1, 2). HSCs proliferate and differentiate to give rise to multipotent progenitor (MPP) cells with limited self-renewal potential that give rise to more restricted progenitor cells to maintain normal numbers of differentiated blood cells (3, 4). Recent evidence suggests that HSCs have a limited number of self-renewing divisions indicating that HSC quiescence and proliferation is a tightly regulated process that protects the host from HSC exhaustion and hematopoietic failure (5, 6). While HSCs are one of the best characterized vertebrate stem cells, a significant effort is still focused on understanding the molecular and cellular mechanisms that regulate HSC quiescence, self-renewal and differentiation to 1) improve methods to expand adult HSCs and their differentiated progeny for bone marrow transplantation (BMT) and cell therapies, 2) identify molecular pathways that direct adult HSC development and expansion from pluripotential stem cells, 3) improve gene editing technology in hematopoietic stem and progenitor cells (HSPCs), and 4) develop new methods to detect, prevent and treat hematologic malignancies.
Inhibitor of DNA binding proteins (ID1-4) are members of the helix-loop-helix (HLH) family of proteins that regulate cell proliferation and differentiation (7–10). ID proteins function as dominant negative regulators of other HLH proteins (E proteins) by inhibiting their DNA binding and transcriptional activity, which is essential for the differentiation and proliferation of normal tissues including muscle, nerve, lymphoid, and embryonic stem cells (11–14). ID proteins also inhibit the transcriptional activity of erythroblast transformation specific (ETS), retinoblastoma (RB), and paired box proteins (PAX) proteins, which affect cell growth and differentiation (15–17). ID proteins are critical transcriptional regulators of hematopoietic cell differentiation, and are required for the proper development of T cells, B cells, dendritic and NK and innate lymphoid cells (13, 14, 18–23). ID proteins have emerged as critical regulators of HSPC quiescence, proliferation and differentiation under homeostasis, inflammatory and genotoxic stress, and aging. These findings suggest that ID proteins could have therapeutic potential to treat myeloid proliferative neoplasia’s (MPN), myelodysplastic syndromes (MDS), and clonal hematopoiesis. Therefore, this prospective will review data related to the function of Id genes in regulating normal HSPC quiescence and differentiation, and their roles in hematopoietic stress and hematologic malignancies.
Id gene expression in hematopoietic cells was first reported in the murine erythroleukemia cell line, MEL cells, and its expression was shown to decrease during erythroid differentiation (11, 24–26). Subsequently, Id1 was detected in an interleukin-3 (IL-3) dependent myeloid progenitor cell line, 32Dcl3 cells, which can be induced to differentiate into neutrophils when cultured in media containing granulocyte-colony stimulating factor (G-CSF) (27). The expression of Id1 decreased during G-CSF-induced differentiation of 32Dcl3 cells, while the binding of bHLH proteins to a canonical E-box motif increased. Furthermore, enforced expression of Id1 blocked G-CSF-induced differentiation of 32Dcl3 cells. Taken together, these results suggested that ID1 may function during myeloid cell differentiation by disrupting bHLH protein function, the canonical target of ID proteins. Subsequent studies demonstrated that IL-3 and other myeloid growth factors (G-CSF and granulocyte/macrophage CSF, GM-CSF) induce Id1 expression and cell proliferation in other IL-3-dependent progenitor cell lines including NFS-60 and FDC-P1 cells, while Id2 expression increases upon withdrawal of IL-3 and cell cycle arrest or differentiation (28–30). The HSC/MPP-like stem cell factor (SCF) -dependent progenitor cell line, EML cells, express ID2 but not ID1, while more committed IL-3-dependent progenitors cell lines derived from EML cells express ID1 but not ID2, suggesting ID2 may function in more primitive hematopoietic progenitor cells. Taken together, these studies demonstrated that ID1 is correlated with increased proliferation/growth and decreased differentiation of hematopoietic progenitor cell (HPC) lines, which is consistent with results from previous studies in other tissues suggesting that ID1 promotes cell proliferation and inhibits cellular differentiation (7, 9, 10, 31, 32). In addition, these studies suggest that ID2 may have functions distinct from ID1 in HSPCs.
Id1 gene expression was increased in normal murine progenitor cells during the early proliferative phase of colony formation in soft agar assays stimulated by myeloid growth factors (IL-3/SCF/IL-1/erythropoietin, EPO), while Id2 was induced to a lesser extent and no effect on Id3 or Id4 expression was observed in these assays (29). In comparison, Id2 levels were increased and Id1 levels decreased in cells from colonies that contained differentiated progeny after 7-10 days. Thus, Id1 is expressed in normal proliferating HPCs, while Id2 expression increases and Id1 levels decrease as cells exit the cell cycle and differentiate into myeloid cells. Subsequent studies confirmed that myeloid hematopoietic growth factors (HGFs) (SCF, IL-3 and GM-CSF) but not lymphoid HGFs (IL-7) and erythroid/megakaryocyte HGFs (EPO/thrombopoietin, TPO) induce Id1 expression in purified lineage-negative, Lin-/Sca-1+/c-Kit+ (LSK) BMCs that are enriched for HSCs/MPPs, suggesting that Id1 may be required for myeloid development, but not lymphoid or erythroid development (33, 34). Analysis of Id gene expression in purified HSPC populations showed low levels of Id1 expression in LSK cells and clonogenic lymphoid progenitors (CLPs), while Id1 expression was increased in common myeloid progenitors (CMP), and further increased in more differentiated granulocyte/macrophage progenitors (GMPs), but not in megakaryocyte/erythroid progenitors (MEP), indicating a potential role for Id1 in myeloid cell development (Figure 1) (33–35). Subsequent analysis of purified HSCs demonstrated that roughly 5-10% of normal HSCs defined as LSK/Flk2-/CD150+/CD48- express Id1 during steady state hematopoiesis, and that myeloid HGFs induce Id1 in HSCs (36, 37). While the expression of Id1 increases in committed myeloid progenitor cells (CMP and GMP), the expression of Id1 decreases during the final stages of neutrophil maturation. Specifically, FACS purified normal neutrophils express low levels of ID1 protein compared to Lin- cells that are comprised of LSK cells and more committed Lin-/cKit+/Sca-1- (LK) progenitor cells that include CMPs/GMPs (33). ID1 expression decreases during G-CSF- and GM-CSF-induced neutrophil differentiation of myeloid progenitor cell lines, 32Dcl3 and MPRO, respectively (27, 33). In contrast, mature neutrophils in Id1EGFP reporter mice show high levels of ID1/EGFP expression compared to CMP’s and GMP’s, suggesting that neutrophils express high levels of ID1 (34, 36). However, EGFP might not accurately reflect ID1 protein levels in neutrophils since ID proteins are rapidly degraded and the stability of EGFP and Id1 may differ significantly in these terminally differentiated cells. Future studies are needed to define the precise expression and function of ID1 and other ID genes in maturing neutrophils and macrophages.
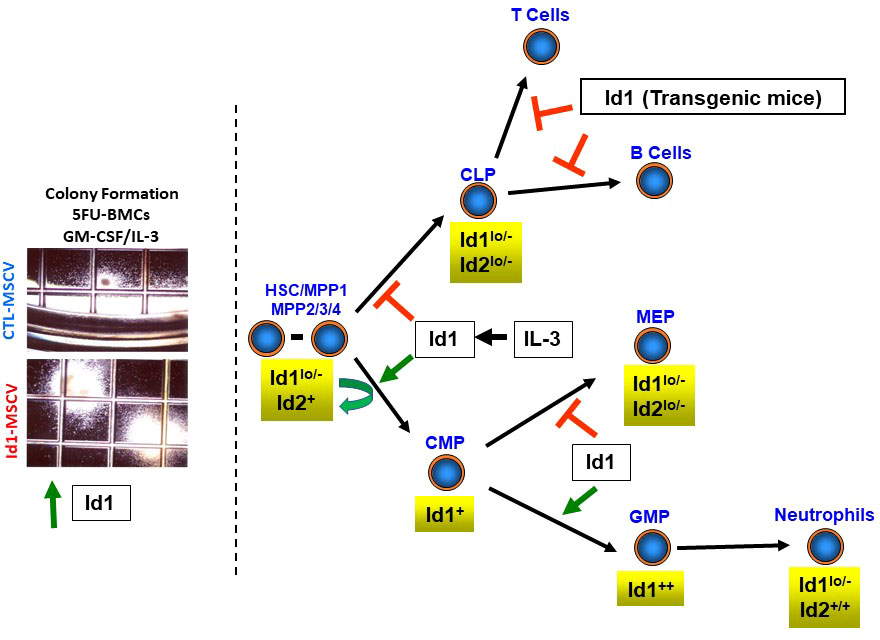
Figure 1 Summary of Id gene expression in hematopoietic stem and progenitor cells. Levels of Id gene expression are shown for hematopoietic stem cells (HSCs), multipotent progenitor (MPP) cells, clonogenic lymphoid progenitors (CLPs), common myeloid progenitors (CMPs), granulocyte/macrophage progenitors (GMPs) and megakaryocyte/erythroid progenitor cells. Photomicrographs of BMCs harvested from mice three days after the administration of 5-FU, transduced with control and Id1 expressing retroviral vectors and cultured in methycellulose for 7-10 days (colony formation assay).
ID2 expression is 3-fold higher than ID1 and 6.5-fold higher than ID3 in purified human cord blood (CB) HSCs suggesting that human HSCs express high levels of ID2 (38). Little or no expression of ID1 was detected in purified CD34+38- human bone marrow HSPCs, but was induced by myeloid HGFs including GM-CSF/IL-3, but not SCF/EPO (erythroid) and SCF/TPO (megakaryocytes) HGFs (39). Thus, ID1 is expressed at low levels in human HSPCs and is induced by myeloid HGFs, and is consistent with results obtained with murine bone marrow cells (BMCs). Similarly, ID1 protein expression is induced during the early proliferative phase of normal human neutrophil/eosinophil differentiation in culture; after which, ID1 expression rapidly declines, while ID2 steadily increases (40). Furthermore, gain and loss of function studies showed that ID2 expression promotes eosinophil/neutrophil differentiation, and ID1 expression promotes neutrophil differentiation of human CD34+ progenitor cells in vitro and in vivo (40). Collectively, these results demonstrate that ID1 functions during the early proliferative stages of myeloid development, while ID2 may be required for the final stages of myeloid cell development. Further studies are needed to more precisely define, 1) the cell-specific and temporal expression of Id1 and Id2, 2) how Id1 and Id2 gene expression is regulated, and 3) their molecular mechanism(s) of action during myeloid cell differentiation in normal human and murine cells.
Initial studies to define the physiological function of Id genes in hematopoietic cells in vivo demonstrated that transgenic mice overexpressing Id1 or Id2 during T cell differentiation manifest a severe block in T cell development (Figure 1) (41, 42). Similarly, transgenic mice that overexpress Id1 during B cell differentiation show impaired B cell development (43). These studies are consistent with the strict requirement for E2A homodimers in B cell development and E2A homo and heterodimers in T cell development, and the dominant negative regulation of E protein function by ID proteins (20, 44).
To study Id gene function in HSPCs, Id genes were overexpressed in normal murine HSPCs using retroviral vectors and analyzed for growth and differentiation in vitro (33). Enforced expression of Id1 in HSPCs resulted in a significant increase in the size and number of colonies in soft agar in the presence of SCF, IL-3 and GM-CSF (Figure 1) (39). Furthermore, these colonies contained increased numbers of primitive and differentiating myeloid cells, suggesting that Id1 promotes myeloid cell hyperplasia without blocking differentiation in vitro. However, immortalized hematopoietic progenitor cell (HPC) lines that escape normal senescence and differentiation could be readily derived from BMCs overexpressing Id1 in liquid culture containing SCF (39). The HPC lines resembled CMP-like progenitors by immunophenotype and lineage specific gene expression, and continued to divide in SCF; however, these cells were not arrested in differentiation and retained the ability to differentiate in response to G-CSF and GM-CSF in vitro. In other studies, overexpression of Id1 in purified LSK cells inhibited B cell development and promoted myeloid/dendritic cell development in cultures supplemented with SCF/FLT3/IL-7 (34). Furthermore, LSK/Flk2- and lymphoid-primed multipotent progenitor (LMPP) cells isolated from mice that overexpress a dominant form of E protein (ET2) that prevents ID protein binding, showed increased B cell, and reduced myeloid cell development when cultured in vitro. Collectively these studies demonstrate that Id1 promotes the proliferation of murine HSPCs and myeloid development, while inhibiting lymphoid development in vitro.
Id1-overexpressing HSPCs showed increased myeloid and decreased B cell repopulation potential when transplanted into γ-irradiated (IR) recipients in vivo (Figure 1) (33, 39). In addition, thymocyte repopulation was inhibited during the early stages of T cell development after transplantation; however, no effect on T cell repopulation was observed in PBCs 4-6 months after BMT. A comparison of Id1/EGFP expression in repopulating T cells and myeloid cells in the same mouse by flow cytometry showed a significant reduction in Id1/EGFP expression in T cells compared to myeloid cells. Thus, it was reasoned that T cells expressing high levels of Id1 do not repopulate, while low levels of Id1 expression are permissive for T cell development in vivo, and account for the recovered T cell development over time. Historically, murine BMCs cultured in HGFs cocktails that included IL-3 to promote HSC cycling and increase retroviral vector transduction, show significantly reduced lymphoid repopulation when transplanted in vivo. It is tempting to speculate that inhibition of lymphoid cell repopulation in these BMT experiments was due to the induction of Id1 by IL-3. In this regard, HGFs that promote Id1 expression and inhibit lymphoid development of HSCs should be avoided in HSC expansion media. Finally, mice transplanted with Id1-overexpressing HSPCs become moribund within a year after transplantation and develop a myeloid proliferative neoplasia (MPN) including 1) increased myeloid blasts in BM, 2) splenomegaly and extramedullary hematopoiesis (myeloid and erythroid), and 3) myeloid cell infiltration in liver, which does not progress to acute myeloid leukemia (AML) (39). Collectively, enforced expression of Id1 regulates HSPC fate by promoting myeloid and inhibiting lymphoid development in vitro and in vivo, and Id1 can promote HSPC proliferation and immortalize HSPCs in vitro, and promote a myeloproliferative disease in vivo.
HPC lines were also derived from murine BMCs transduced with lentiviral vectors expressing inducible Id2 (Id2-HPCs), which were established in conditions that support primitive B cell growth including SCF, FLT-3 and IL-7 on S17 stromal cells (45). The Id2-HPC lines express c-Kit, CD43 and low levels of B220, but lack the expression of other cell surface markers expressed on more mature myeloid and lymphoid lineage cells. Id2-HPC lines are multipotential and differentiate into myeloid cells in the presence of IL-3, GM-CSF, and M-CSF in vitro and in vivo, under conditions that maintain Id2 expression. Id2-HPCs differentiate into lymphoid cells (B cell and T cell) upon withdrawal of ID2 in vitro and in vivo consistent with the ability of ID2 to inhibit E protein function. The lymphoid potential of the Id2-HPC lines was lost with cell passaging suggesting that the Id2-HPC lines may undergo further myeloid differentiation and commitment in vitro. It is possible that the Id2-HPC lines that have lost lymphoid potential may resemble the SCF-dependent CMP-like HPC lines that overexpress Id1 (39). The Id2-HPC line transcriptome resembles the transcriptome of the previously described E2a-/- multipotential cell lines (pre-pro-B cells) that were established in SCF, FLT-3, IL-7 and stromal cells (45), which is consistent with the ability of ID2 to inhibit the function of E2A and prevent lymphoid development. In comparison, over expression of Id2 in human cord blood CD34+ HSPCs resulted in expansion of CD34+/CD38-/CD90+/CD45RA- HSCs, and a skewing toward myeloid (CMP/MEP) versus lymphoid (B/NK/GMP) by immunophenotype analysis in vitro, and reduced lymphoid development when transplanted into NSG mice (38). HSPCs isolated from NSG mice transplanted with ID2-overexpressing CB-HSPCs show increased expression of stem cell genes and genes expressed in primitive myeloid differentiation programs, and decreased expression of B cell factors, EBF1 and FOXO1, which are E2A target genes (46). ID2-mediated expansion of HSCs was rescued by overexpressing E2A in vitro, and overexpression of E2A promoted lymphoid development in vivo. Knock down of the E2A target gene, EBF1, which is required for lymphoid development, resulted in an increase in HSC numbers. These results suggested that ID2 expands or maintains HSC numbers by inhibiting lymphoid lineage priming, which leads to a reduction in HSC numbers (38). Taken together, overexpression of ID2 expands HSCs and restrains B cell development in human HSPCs. These studies suggest that ID2 may function to regulate human and mouse HSCs function in vivo.
Hematopoietic progenitor cell lines were also established by overexpressing Id3 in fetal liver cells and BMC cultures containing SCF, FLT-3, IL-7, and stromal cells, and resemble pre-pro B cells (Id3-HPC lines) (47). Id3-HPC lines are multipotential and retain myeloid, B cell and T cell potential in vitro and in vivo. The lymphoid potential of Id3-HPC lines was induced by down regulation of Id3 expression in developing B cells in vivo, supporting previous studies that high levels of Id3 impair B and T cell development (48–51). Secondary transplantation of BMCs from primary BMT recipients that received Id3-HPCs showed residual myeloid reconstitution, but no lymphoid reconstitution suggesting that Id3-HPCs are not HSC/MPP-like cells (47). Id3-HPC lines could not be established from human CB HSPCs; however, Id3 overexpressing CB HSPCs cells showed enhanced proliferation in vitro and limited repopulation potential in NSG mice. Additional experiments are required to further understand the mechanism(s) of Id3-mediated immortalization in murine and human HSPCs compared to Id1 and Id2 immortalization.
The first Id1- deficient (Id1-/-) mouse model was generated by gene targeting, which replaced the first exon and part of the promoter of Id1 with a neomycin resistance gene cassette. Conventional Id1-/- mice were born at normal mendelian frequencies, were fertile and showed normal lifespan with no overt abnormalities (52). No significant difference in the number of mature hematopoietic cells was reported in PBCs, BMCs and spleen cells of Id1-/- mice, suggesting that Id1 is not required for normal hematopoietic development. However, a more detailed analysis of these mice revealed that Id1-/- mice have impaired hematopoietic development including 1) increased myeloid cells and decreased lymphoid cells in PBCs, 2) decreased BM cellularity, 3) decreased numbers of LSK cells, 4) decreased numbers of LSK/CD34-/Flk2- cells enriched for HSCs, 5) increased cycling of LSK cells, and 6) increased proliferation and differentiation of LSK cells in vitro (35). No difference in the repopulation potential of Id1-/- and Id1+/+ BMCs was observed in primary BMT recipients; however, Id1-/- BMCs showed impaired secondary repopulation ability albeit with low statistical significance, suggesting that Id1-/- HSCs have a defect in self-renewal. Collectively, these investigators concluded that Id1-/- HSCs show increased cycling and increased myeloid commitment, which resulted in decreased HSC self-renewal in secondary BMT recipients, suggesting that Id1 is required to maintain HSCs.
A second Id1-/- mouse model was generated by inserting the EGFP coding sequence downstream of the Id1 transcriptional start site, which results in an Id1 null allele (Id1EGFP/EGFP) (37). In comparison to the Jankovic et al. Id1-/- mouse model described above, the hematopoietic phenotypes observed in Id1EGFP/EGFP mice included decreased numbers of HSCs (LSK/CD150+/CD48-); however, this report showed no effect on the cycling of HSC enriched populations, no difference in the development of mature lymphoid and myeloid cell populations in BMCs or PBCs. Id1EGFP/EGFP mice showed no difference in repopulation in primary BMT recipients, but reduced secondary repopulation potential suggesting that Id1 was required for HSC development and maintenance. However, it should be noted that the secondary BMT was performed 16 days after primary BMT, which is a significant deviation from the typical secondary BMT protocol, which is usually performed 10-16 weeks after primary BMT, when hematopoiesis resembles more steady-state conditions (37). Therefore, it is difficult to conclude from these studies if Id1 is required for HSC self-renewal.
A third study also analyzed hematopoietic development in the Id1-/- mouse model used in the Jankovic et al. study discussed above (35) and confirmed that 1) myeloid cells were increased and lymphoid cells decreased in BMCs and PBCs, 2) BM cellularity was decreased, and 3) LSK cycling was increased (53). This study showed no difference in the number of HSC-enriched cells (LSK/CD34- cells) and HSC function in vivo; however, the two studies used different cell surface antigens to immunophenotype the HSCs, which could explain the differences in HSC numbers. Finally, the two studies agreed and showed no difference in HSC repopulation potential in primary BMT recipient mice and differed in serial repopulation potential leaving open the question whether Id1 functions in HSC self-renewal.
Based on the known function of Id1 in other cellular contexts, it might be predicted that loss of Id1 would lead to reduced cell cycling and increased B cell development in the hematopoietic compartment of Id1-/- mice. However, Id1-/- mice show the opposite hematopoietic phenotypes including increased LSK cycling and proliferation, and decreased B cell development (35, 53). Since Id1 expression is ablated in all cells in conventional Id1-/- mice, and Id genes are widely expressed in other tissues including endothelial cells (ECs) and skeletal stem cells (SSCs) and their progeny, which are cellular constituents of the hematopoietic microenvironment (HME) (8, 54–57), it is possible that loss of Id1 function in the HME could contribute to the hematopoietic phenotypes observed in conventional Id1-/-. Therefore, in the third study, Id1+/+ and Id1-/- BMCs were transplanted into γ-IR Id1-/- or Id1+/+ recipient mice and monitored for hematopoietic development (53). Id1+/+ BMCs transplanted into Id1-/- recipients showed impaired hematopoietic development similar to the hematopoietic phenotypes observed in the conventional Id1-/- mice including decreased BM cellularity, increased myeloid and decreased erythroid development. Importantly, Id1-/- BMCs showed normal hematopoietic development when transplanted into Id1+/+ recipient mice. These results were confirmed in a recent study using a mouse model that lacked Id1 and Id3 expression in the HME, since HME phenotype was less severe in mice on a pure C57BL/6 background and Id3 can compensate for loss of Id1 in some models (58). Transplantation of normal BMCs into γ-IR Id1-/-;Id3-/- recipient mice showed a significant decrease in BM cellularity, decreased B cell development, increased HSC cycling and decreased HSC numbers (56). Finally, Id1-/- stromal cells show altered cytokine production in vitro, and cytokine levels were deregulated in conventional Id1-/- mice in vivo. Collectively, these results demonstrated that Id1 is required for the proper function of the HME, and that the hematopoietic phenotypes observed in conventional Id1-/- mice could, in part, be explained by the loss of Id1 function in the HME.
Endothelial cells are critical cellular components of the HME and are required to maintain steady state hematopoiesis (59–63). SECs are critical cellular components of the HME and are required to maintain steady state hematopoiesis, in part, through angiocrine signaling (64, 65). Lineage tracing studies using vascular endothelial cadherin (VE-Cad) transgenic mice Cdh5(PAC)-CreERT2 bred to Rosa26-mT/mG mice showed that transition endothelial vessels (H vessels), proliferate to regenerate diaphyseal sinusoidal ECs (SECs) within forty days under homeostasis (66). Recent evidence suggest that Id genes promote EC proliferation and vessel regeneration under stress (67–69); thus, it was hypothesized that Id genes may be required for proper HME function by maintaining ECs under steady state conditions and stress (56). Therefore, Cdh5(PAC)-CreERT2 mice were bred to Id1F/F;Id3-/- mice to specifically ablate Id1 and Id3 expression in ECs, since Id1 and Id3 are required for proper HME in mice on a pure C57/BL6 background. Loss of sinusoidal integrity was observed in Id1-/-; Id3-/- mice characterized by dilated, leaky, and apoptotic BM SECs that increased in severity over time (56). The proliferation of Id1-/-; Id3-/- SECs, and transition endothelial vessels was significantly reduced in vitro and in vivo, leading to impaired vascular integrity under steady state conditions, which was more severe following acute stress. The disruption in sinusoidal integrity and neovascularization in Id1-/-;Id3-/- mice led to a progressive decline in hematopoiesis, marked by increased HSC activation, proliferation, differentiation, migration, and exhaustion. Thus, Id1 and Id3 are required for the survival and steady state regeneration of BM SECs, which provide a supportive niche for HSC quiescence and survival. Future studies are needed to examine if Id genes regulate other cells in the HME including subtypes of SSCs (Leptin-cre+, Nestin-ER-cre+, and NG2-cre+), which functionally support hematopoiesis, and their downstream progeny including osteoblasts and chondrocytes (60, 63, 70). Future studies are needed to examine Id gene function in the neural niche, since nerve fibers such as adrenergic and cholinergic nerves are instructive for hematopoietic mobilization and quiescence respectively.
Id3-/- mice are born at normal mendelian frequencies, are fertile, and young mice show no overt phenotypes. Id3-/- mice have normal numbers of developing B cells, but show impaired humoral immunity, B-cell proliferation, and develop a unique autoimmune disease, Sjogren’s syndrome, and γδ-T cell hyperplasia with age (49, 50, 71, 72). In addition, Id3-/- mice show severely impaired positive and negative thymocyte selection (50). Transplantation of Id3-/- BMCs into γ-IR mice show normal myeloid and B cell repopulation, but impaired T cell repopulation in secondary recipient mice, suggesting that Id3-/- is not required for HSC maintenance (37). However, further studies are needed to examine if Id3 is required for HSC self-renewal in serial BMT assays separated by 10-12 weeks (37). In other studies, no difference in donor B cells, T cells, neutrophils, HSCs and MPP repopulation were observed in mice 12 weeks after competitive BMT of Id1-/-Id3-/- BMCs compared to controls, confirming that Id3 is not required for HSC repopulation of primary BMT recipient mice; however, HSC self-renewal was not evaluated in these assays (56). Therefore, additional studies are needed to examine the requirement of Id3 in HSC development.
The majority of conventional Id2-/- mice show perinatal lethality and are born at less than ten percent of the normal mendelian frequencies. The surviving mice lack Langerhans cells, splenic dendritic cells, and NK cells, and show absence of lymph nodes and Peyer’s patches, which demonstrates the requirement for ID2 in the development of these cells (18, 22). Surviving Id2-/- mice also show increased B cell development, decreased erythroid cells and no effect on myeloid cell development (73). Gain and loss of Id2 function studies in normal HSPCs confirmed that Id2 intrinsically inhibits B cell development in vivo by negatively regulating E2A. In addition, these studies showed that ID2 binds to PU.1 and interferes with PU.1’s ability to inhibit GATA-1 transcriptional activity, suggesting a potential mechanism of action for how ID2 promotes erythroid development (73). Additional experiments are needed to further explore this mechanism of action, and more precisely define the progenitors that express Id2, and where cell fate is determined and how the expression of Id2 is regulated. In this regard, Gfi-1 has been identified as a direct transcriptional repressor of Id2, and high levels of Id2 are expressed in Gfi-1-/- BMCs, and these mice show defects in B cell, T cell, and neutrophil development (74, 75). In addition, these mice have impaired short term reconstituting cell (STRC) activity or ability to radio-protect lethally irradiated recipient mice, and have significantly reduced numbers of HSCs (74–76). Reducing Id2 levels in Gfi-1-/- mice (Gfi-1-/-;Id2+/- mice) partially restores B cell development by overcoming the block in B cell development at the pro-B cell stage, and reduces myeloid hyperplasia, but does not rescue T cell development. These results provide evidence for a direct link between Gfi-1 and the B cell transcriptional network via Id2, which inhibits E2A function required for B cell development (76). While reducing the levels of Id2 in Gfi-1-/- mice rescued the myeloid hyperplasia in the spleen, it did not rescue neutrophil differentiation. Additional studies are needed to uncover how Id2 promotes myeloid expansion in Gfi-1-/- mice and why normal myeloid development is not restored. Finally, reducing Id2 levels in Gfi-1-/- mice partially restores the number of STRCs, CMP and MEP progenitors and differentiating erythroid cells in the BM bone marrow, which is sufficient to radio-protect lethally irradiated BMT recipient mice (77). Increased red cell production in Gfi-1-/-;Id2+/- mice was correlated with increased expression of Gata1, Eklf and EpoR, which are required for erythroid development. It was proposed that ID2 inhibits E2A/Scl complexes that regulate erythroid gene expression via a multiprotein transcription factor complex that binds paired E-box/GATA sites in the promoters of Gata1, Eklf and EpoR. However; the precise molecular mechanism(s) of Id2 action that rescue the erythroid lineage in Gfi-1-/- mice remain to be defined. Finally, reducing Id2 levels in Gfi-1-/- mice does not increase HSC numbers and rescue the defect in HSC function in competitive repopulation assays indicating that other genes mediate HSC loss in Gfi-1-/- mice. Collectively, these observations suggest that Id2 regulates HSPC fate at multiple cellular levels, but leaves open the question whether Id2 regulates HSC development.
Few conventional Id2-/- mice survive beyond birth suggesting that the surviving Id2-/- mice may have compensated in some way to promote the survival of Id2-/- mice. Therefore, the intrinsic requirement of Id2 in HSCs was evaluated in conditional Id2F/F mice. Specifically, BMCs from Mx1-cre;Id2F/F mice (78) were transplanted in γ-IR recipient mice (chimeric mice) to reconstitute the host hematopoietic system and eliminate any contribution that loss of Id2 function might have in the HME. Chimeric mice were treated with pIpC to induce interferon production and ablate Id2 expression in hematopoietic cells six weeks after BMT, and then examined for hematopoietic development after ten weeks. Chimeric mice showed a significant reduction in the total number immunophenotypic HSCs, and decreased donor reconstitution of competitively transplanted primary recipient mice confirming that Id2 is required to maintain HSCs in chimeric mice (Figure 2). Furthermore, Id2-/- chimeric mice showed reduced overall survival due to anemia and BM failure compared to Id2+/+ chimeric mice indicating that ID2 is intrinsically required for HSC maintenance. Mechanistically, Id2-/- HSCs showed increased proliferation and cycling, mitochondrial activation, reactive oxygen species (ROS) production and differentiation in vitro and in vivo. Pathway analysis of differentially expressed genes in Id2+/+ and Id2-/- HSCs revealed increased expression of genes that regulate cellular proliferation and genes that regulate oxidative phosphorylation in Id2-/- HSCs compared to Id2+/+ HSCs. In addition, gene set expression analysis (GSEA) and pathway analysis revealed that HIF-1α target genes were decreased Id2-/- HSCs suggesting that ID2 might affect the levels or function of HIF-1α. In this regard, HIF-1α-/- mice show similar hematopoietic phenotypes with Id2-/- mice including increased cycling, decreased quiescence, and increased susceptibility to 5-FU treatment (79). HIF-1α protein levels were reduced in purified Id2-/- HSCs, and loss of HSC function in Id2-/- mice could be restored by chemically stabilizing HIF-1α and overexpression of stabilized HIF-1α in vitro and in vivo. Mechanistically, ID2 stabilizes HIF-1α by binding to VHL and interfering with HIF-1α ubiquitination and proteasomal degradation (Figure 2). Collectively, ID2 is required to maintain HSC quiescence, a function that is distinct from ID1, which promotes HSC proliferation. Id2 and Id1 expression are inversely correlated during hematopoietic stress, where Id2 expression decreases in HSCs as they exit quiescence and Id1 expression increases; after which, Id1 decreases and Id2 increases when HSCs resume quiescence (Figure 3). Future experiments are needed to determine if Id2 regulates quiescence of leukemic stem cells (LSCs), and if there are additional mechanism(s) of Id2 action in normal and LSCs.
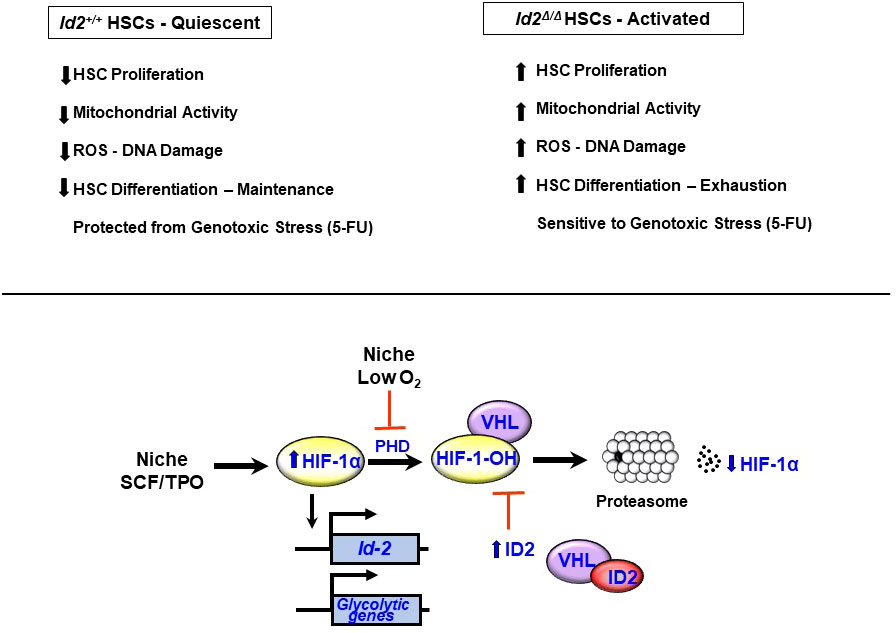
Figure 2 Id2 is required for HSC quiescence and maintenance. Summary of hematopoietic phenotypes in Id2 ablated mice. Hif-1α expression is induced in HSCs by stem cell factor (SCF) and thombopoietin (TPO). HIF-1α protein levels are maintained at low levels under normoxic conditions via the action of proline hydroxylases (PHDs), which hydroxylate HIF-1α and promotes its association with the VHL complex, ubiquitination and proteasomal degradation. Under low O2 conditions, PHD is inhibited resulting in reduced levels of hydroxylated HIF-1α, and reduced ubiquitination and stabilization of HIF-1α. ID2 also acts to stabilize HIF-1α by binding to VHL, which prevents ubiquination and proteosomal degradation of HIF-1α and promotes HSC quiescence.
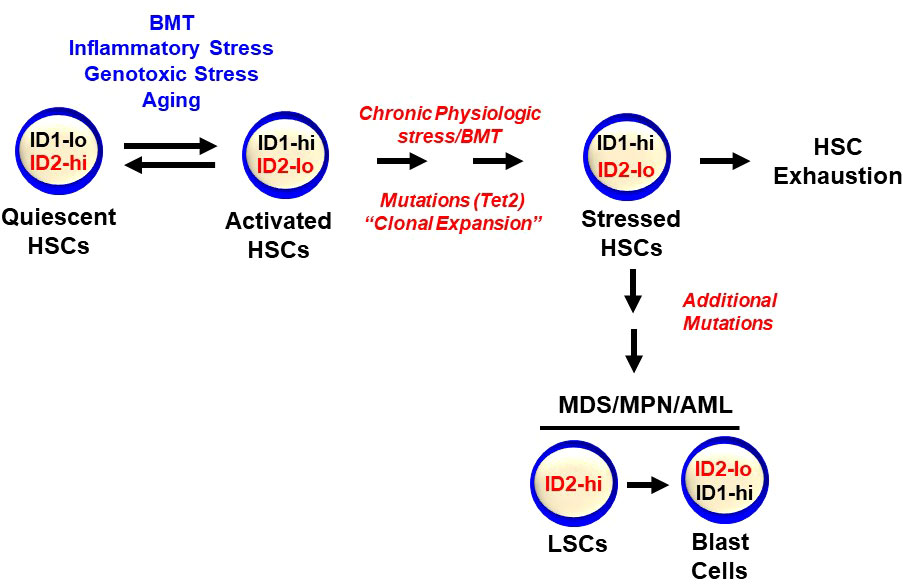
Figure 3 Id1 and Id2 are differentially expressed during HSC activation and quiescence. HSCs express low levels of Id1 that are induced during stress including BMT, genotoxic and inflammatory stress, and aging. Id2 is required to maintain HSC quiescence and Id2 decreases during HSC proliferation and activation, after which, Id1 levels decrease and Id2 levels increase as HSCs return to quiescence. Chronic stress can lead to HSC exhaustion and clonal hematopoiesis and hematopoietic malignancies.
Id genes are early response genes that are induced in NIH-3T3 fibroblasts by serum and growth factors, and are required for re-entry of serum-starved cells into cell cycle, demonstrating the requirement for Id genes in HGF-induced proliferation (stress) (80). Id genes are expressed at low levels in most adult tissues, and are induced by a wide array of extracellular signals in response to stress or injury to promote tissue repair and regeneration (10, 67, 68). HSCs express low levels of Id1 under steady state conditions, and Id1 is induced by HGFs that promote myeloid proliferation and differentiation including IL-3 (33, 34). Furthermore, enforced expression of Id1 in HSPCs promotes myeloid cell proliferation at the expense of lymphoid development, suggesting that ID1 may function during hematopoietic stress (33, 39). Therefore, to evaluate the role of Id1 in hematopoietic stress, Id1-/- BMCs were serially transplanted in γ-IR recipient mice. No significant difference in the repopulation potential of Id1-/- BMCs compared to Id1-/- BMCs was observed in primary BMT recipients. However, Id1-/- BMCs showed enhanced self-renewal potential and promoted the survival of serially transplanted mice significantly beyond the potential of Id1+/+ BMCs (Figure 4) (36). Increased numbers of HSCs were detected in mice serially transplanted with Id1-/- BMCs compared to Id1+/+ BMCs, demonstrating that Id1-/- HSCs are maintained and protected from exhaustion during chronic stress. Furthermore, Id1-/- HSCs in serially transplanted mice showed reduced cycling, DNA damage (γ-H2AX phosphorylation), mitochondrial biogenesis and activation, and ROS levels compared to Id1+/+ HSCs, indicating that Id1-/- HSCs show increased quiescence during stress (36). Comparative transcriptome analysis of purified donor Id1-/- and Id1+/+ HSCs confirmed that Id1-/- HSCs have an increased quiescent molecular signature including reduced expression of genes involved in cell cycle, oxidative phosphorylation, ribosomal biogenesis, and protein synthesis. Taken together, Id1-/- HSCs show increased quiescence and reduced proliferation and activation during hematopoietic stress compared to Id1+/+ HSCs.
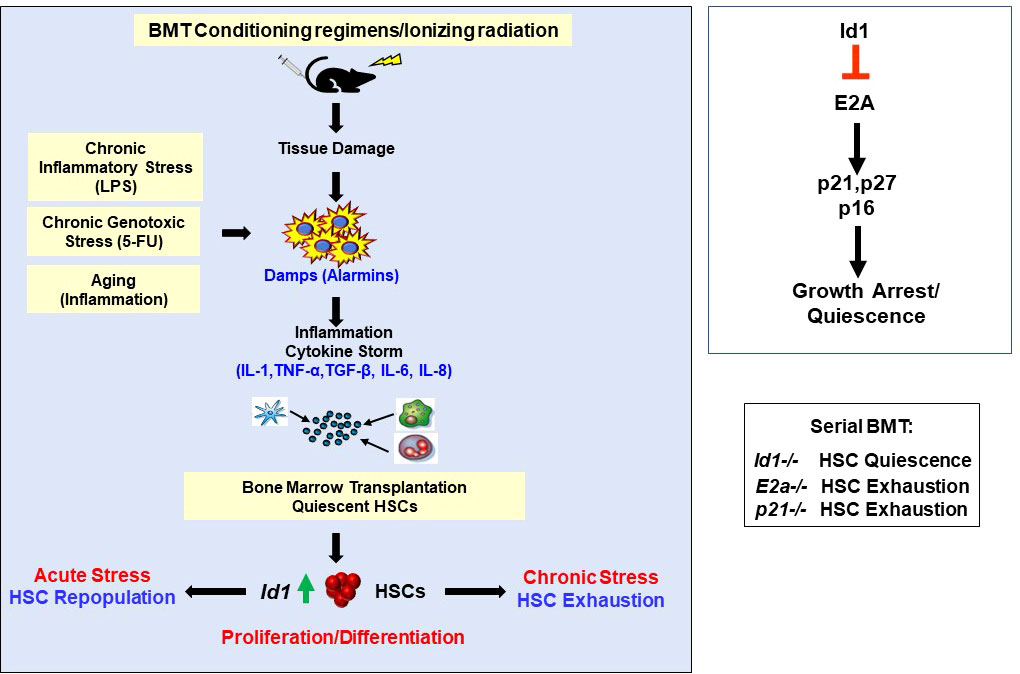
Figure 4 Ablation of Id1 Protects HSCs from chronic proliferative stress including BMT, chronic genotoxic and inflammatory stress, and aging. Summary of mechanism of ID1 action in HSCs, and gene knockout mouse models in the molecular pathway.
BMT conditioning regimens including γ-IR damages the HME resulting in acute and chronic inflammation due to the production of alarmins, recruitment of inflammatory cells and the production of pro-inflammatory cytokines (cytokine storm) (81–85). Initial reports demonstrated that myeloid HGFs induce Id1 expression and proliferation of HSPCs in vitro (33, 34). Subsequent studies showed that Id1 can be induced in HSCs by a variety of proinflammatory cytokines in vitro, and HSCs that express Id1 are actively proliferating (36). In addition, these studies found that Id1 is induced in HSCs by proinflammatory cytokines in vivo, and in HSCs after 6 Gy γ-IR. Thus, during chronic stress Id1-/- HSCs fail to properly respond to cytokine-induced proliferation and differentiation and are protected from exhaustion in vitro and vivo during serial BMT.
It is difficult to predict which cytokines in the cytokine storm induce Id1 in HSCs after BMT in vivo; however, many cytokines signal via the Jak/Stat pathway suggesting that JAK inhibitors could protect HSCs from chronic proliferative stress. In this regard, JAK1 is an intracellular tyrosine kinase signaling molecule required for HSCs to respond to stress cytokines including IFN-α/β/γ and IL-3 (86). Jak1 deficient HSCs exhibited increased quiescence, inability to enter cell cycle, reduced response to type I interferons and IL-3, and impaired ability to reconstitute hematopoiesis during BMT and stress (86). Since ID1 proteins are induced by IL-3 in normal HSPCs and cell lines (33), the increased quiescence of Jak1-/- HSCs during stress may be mediated, in part, by reduced expression of Id1. Therefore, mice were treated with small molecule inhibitors of the JAK/STAT pathway after BMT to inhibit Id1 induction in HSCs. JAK/STAT inhibitors partially inhibited the induction of Id1 in HSCs after BMT in γ-IR recipient mice demonstrating that pro-inflammatory cytokines produced after γ-IR induce Id1 in HSCs in vivo (36). Future studies could target other relevant ligands (cytokines), receptors, and downstream signaling pathways involved in proinflammatory cytokine signaling during BMT including IL-6, IL-1, tumor necrosis factor (TNF), IL-8 and others to prevent HSC exhaustion and improve BMT.
It was hypothesized that since Id1-/- HSCs are protected from chronic proliferative stress and exhaustion during serial BMT, ablation of Id1 expression in HSPCs during other conditions of chronic stress including inflammatory and genotoxic stress, and aging could prevent HSC exhaustion (Figure 4). Indeed, studies found that Id1-/- HSCs were also preserved in models of chronic proliferative stress including chronic inflammatory stress (LPS) and genotoxic stress (5-FU) (36, 87). In addition, HSCs from aged (2 years old) Id1-/- mice resemble more youthful HSCs including, 1) an immunophenotype of young HSCs, 2) increased HSC function in serial BMT assays, 3) increased HSCs in G0, 4) decreased HSC DNA damage, 4) reduced HSC ROS levels, and 5) reduced HSC mitochondrial stress compared to Id1+/+ HSCs. Future experiments are needed to further define the molecular mechanism(s) that protect Id1-/- HSCs from aging. Collectively, these results demonstrate that Id1-/- HSCs are more quiescent and resistant to chronic proliferative stress including inflammatory and genotoxic stress and aging. Clonal hematopoiesis, MPN and MDS are associated with increased inflammation (84, 88–90), which could induce Id1, promote HSPC proliferation during clonal hematopoiesis, and increase mutational load and genomic instability over time.
Id1-/- HSCs fail to properly respond to cytokine-induced proliferation and differentiation and show decreased BrdU incorporation and cell cycling after stress in vitro and in vivo (36). Since E proteins inhibit cell proliferation, in part, by promoting the expression of cyclin dependent kinase inhibitors (Cdki’s) (91–94), and ID proteins inhibit E-proteins resulting in increased proliferation, it is reasonable to hypothesize that the reduced cycling of Id1-/- HSCs is due to unrestrained E protein transcriptional activity and increased Cdki expression (Id1-E2a-Cdki pathway) (Figure 4). Increased p21 expression was observed in Id1-/- LSK cells compared to Id1+/+ HSCs (35). Subsequent reports demonstrated that Id1-/- HSCs show increased p27 and p16 expression and reduced proliferation in expansion cultures compared to Id1+/+ HSCs, and Id1-/- HSCs show increased expression of p21 and p27 after 14 weeks in competitive BMT assays compared to Id1+/+ HSCs (36). Furthermore, knock down of E2a and p16 expression in Id1-/- HSPC expansion cultures increased HSC proliferation suggesting that Id1 increases HSC proliferation, in part, by restraining E protein function and reducing p16 expression (36). Importantly, E2a-/- HSCs show increased cycling, decreased serial repopulation potential, and decreased expression of p21 and p27 expression (95–97), and p21-/- HSCs have decreased serial repopulation populations potential (98). Thus, Id1-/- HSCs demonstrate opposite hematopoietic phenotypes to E2a-/- and p21-/- HSCs, whereby Id1-/- HSCs show increased quiescence and are protected from hematopoietic stress and exhaustion, and E2a-/- and p21-/- HSCs show increased proliferation and activation and are sensitive to hematopoietic stress and premature exhaustion. Thus, the Id-E-Cdki pathway critically regulates HSC cycling during stress. In other studies, the E protein(E47) – Cdki (p21) pathway was shown to be important in preventing HSC exhaustion during BMT and 5-FU mediated stress (99). Specifically, genetic experiments examining the requirement for E47- p21 pathway in maintaining HSCs under stress showed that E47hetp21het HSCs exhibit decreased serial BMT repopulation when compared to E47hetp21WT, which have increased p21 levels. The E-Cdki pathway is conserved in endothelial cells (ECs), where p21 and p27 RNA expression is increased in ECs that lack Id gene expression, and growth inhibition of Id1-/-Id3-/- ECs in vitro was rescued by knocking down E2-2 expression (56). In comparison, E2a and p16 shRNAs partially restore the proliferation of Id1-/- HSCs in vitro, suggesting that other E and Cdki proteins may regulate quiescence in Id1 ablated HSCs, or Id1 regulates HSC functions through other target genes and pathways. Therefore, it will be important to evaluate if knock down of other E proteins, E2-2 and Heb, and other Cdki’s rescue Id1-/- HSC proliferation in vitro. In this regard, the role of E2-2 and Heb in HSC development in conditional mouse models has not been evaluated. Furthermore, it would be important to know if reducing the expression of p21 and other Cdki’s via genetic experiments can rescue the hematopoietic phenotypes in Id1-/- mice in vivo. Finally, additional transcriptomic, proteomic, and single cell analysis of normal, stressed, or aged Id1-/- HSCs might uncover additional molecular pathways that regulate HSC quiescence.
Id gene expression has been correlated with the initiation, progression, and metastasis of cancer in many tissues (7, 10, 100). Id genes are frequently overexpressed in advanced stage chronic myeloid leukemia (CML), AML and MDS patient samples while low or no levels of ID1 were detected in normal CD34+ HSPCs (39, 101–110). Knock down of ID1 expression in the AML cell line, MO-7e, resulted in decreased growth and suggested that some AML cells may require ID1 for growth. Furthermore, analysis of ID gene expression in an AML patient cohort showed that high levels of ID gene expression were correlated with FLT3-ITD, RAS, EVI-1 and C/EBPA mutations, suggesting ID gene expression may be induced downstream of oncogene activated signal transduction pathways (111). ID1 was identified as a common downstream target of oncogenic tyrosine kinases including FLT3-ITD and BCR-ABL (112). Stable KD of ID1 in K562 (BCR-ABL) and Molm14 (FLT3-ITD) AML cell lines inhibited their growth in vitro confirming that ID1 is required for the growth of some AML cell lines. Analysis of Id1 expression in a 237 AML patient cohort demonstrated that AML patients with high levels of ID1 gene expression were less likely to achieve complete remission and were correlated with shorter disease-free survival and overall survival (113). Thus, it was suggested that ID1 expression levels may provide a molecular tool for refining risk classification of AML. In addition, these studies suggest that Id1 may represent a candidate for targeted therapy to treat AMLs (106).
To explore the intrinsic role of Id genes in hematopoietic malignancies, Id1-overexpressing HSPCs were transplanted into lethally irradiated recipient mice and monitored for survival. Id1-overexpressing mice died roughly one year after BMT compared to control mice, and sick mice showed 1) myeloid/erythroid cell hyperplasia and increased numbers of immature cells in the BM, 2) splenomegaly and extramedullary hematopoiesis, and 3) peripheral blood monocytosis, indicating that these mice succumbed to a MPN that did not progress to AML (39). ID1 expression is increased in leukemic cells from AML patients with t (8, 21) translocations, and AML1-ETO regulates the ID1 promoter, suggesting a role for ID1 in AML1-ETO leukemia (114). Ablation of Id1 in AML1-ETO transduced murine fetal liver cells delays the onset and development of leukemia by roughly 130 days after transplantation in vivo (115). Furthermore, conditional loss of Id1 in established AML-ETO-leukemia’s slows the development of leukemia and promotes animal survival, suggesting that Id1 is also required for the maintenance of leukemia. Mechanistically, these studies provided evidence that Id1 promotes AML-ETO leukemic cell growth by interacting with Akt and increasing its activity. In other studies, the progression of leukemia was significantly delayed after transplantation of MLL-AF9-transduced Id1-/- FL cells compared to MLL-AF9-transduced Id1+/+ fetal liver cells (116). The delay in Id1-/- MLL-AF9 leukemia was reversed in FL cells that lack p21, which is consistent with previous studies demonstrating that ID proteins promote HSPC proliferation by inhibiting the E-Cdki pathway. Interestingly, loss of Id1 accelerated the progression of MLL-AF9-induced leukemia of BMCs, suggesting that Id1 is not required for MLL-AF9-induced leukemia of BMCs, and that leukemogenesis may differ significantly for FL and BM HSPCs. The cellular and molecular mechanism(s) that account for the differences in the progression of leukemia in these models is not currently known. Taken together, these studies provide evidence that ID1 is required for the initiation and progression of oncogene driven leukemias. Future studies are needed to determine if ID1 is required for AML cell line growth in xenotransplantation and human AML PDX models, and if the recently identified inhibitors of ID1 including cannabidiol and AGX51 show any therapeutic benefit in these models (117–119). Since MPN and MDS and myeloid malignancies that develop with age are strongly correlated with inflammation and Id1 is induced in HSPCs downstream of pro-inflammatory signals, future studies are needed to better understand the role of ID1 in these diseases and the molecular mechanism(s) of ID1 action (84, 89, 120).
Ablation of Id2 in HSCs results in increased proliferation and HSC activation suggesting that ID2 may function as a tumor suppressor. Interestingly, Ko et al. showed that mice transplanted with Id2-/- fetal liver cells develop leukocytosis after 6 months that resembles a myeloproliferative disorder, and that over expression of ID2 delays the onset BCR-ABL-induced CML in vivo (121). In addition, loss of Id2 expression is associated with increased MLL-AF9-induced leukemia in mice, and over expression of Id2 inhibits the growth of MV4-11 and MOLM-13 AML cell lines that express MLL-AF9, and Kasumi AML cells that express AML-ETO (122). Together, these results suggest that ID2 may function as a tumor suppressor in hematopoietic malignancies. In addition, mice that lack Id2 develop intestinal adenomas, and show a hyperproliferation of colon stem cells during embryonic development due to increased Wnt/B-catenin signaling, suggesting that ID2 may function as a tumor suppressor in other cell types (123, 124). Analysis of ID2 expression in 145 AML patient BMCs showed that AML patient cells with high levels of ID2 expression were correlated with lower complete remission and shorter overall survival, and was a predictor of poor chemotherapy response (103). Analysis of ID2 expression in a subset of MLL-rearranged AML patient cells indicated that MLL patients (35 patients) with high levels of ID2 expression had a significantly better overall and event free survival than patients with low levels of ID2 (122). Further studies are needed to examine if levels of ID2 expression are prognostic for AML patient subsets, and to determine if ID2 is expressed in LSCs and functions to regulate their quiescence and survival (Figure 3).
ID proteins have emerged as important regulators of HSPC quiescence (ID2) and proliferation (ID1). Specifically, low levels of ID1 are expressed in primitive HSPCs, but are induced in HSPC after acute stress including BMT, inflammatory and genotoxic stress, and promote HSPC proliferation and myeloid development, while inhibiting lymphoid development (Figure 3). Upon resolution of an acute stress, HSCs return to quiescence with low levels of ID1 and hematopoiesis resumes under steady state conditions. However, under chronic proliferative stress ID1 levels remain high and HSCs undergo excessive proliferation, exhaustion and bone marrow failure. Reducing ID1 levels during serial BMT, chronic inflammatory and genotoxic stress and aging may be therapeutic to protect HSCs from exhaustion. In addition, since hematopoietic malignancies and bone marrow failure syndromes are often accompanied by inflammation and increased ID1 expression, reducing ID1 levels could be therapeutic by reducing preleukemic proliferation and clonal expansion, which could delay the onset and reduce the incidence of hematopoietic malignancies and bone marrow failure syndromes. ID2 has emerged as a critical regulator of normal HSC quiescence and shows opposite expression to ID1, where ID2 levels decrease as HSCs exit quiescence and ID1 levels increase during cell proliferation. Maintaining high levels of ID2 in vitro and in vivo could be exploited to expand HSCs for gene and cell therapies including BMT. Opposing expression of ID1 and ID2 is also observed during the final stages of myeloid development, where ID1 expression is increased in myeloid progenitors (CMP/GMP), and then decreases as cells exit the cell cycle and differentiate, while ID2 expression is increased in mature neutrophils. Furthermore, current evidence suggests that ID1 and ID2 may function during neutrophil and eosinophil development, however, gain and loss of function studies are needed to reveal if these genes are required for the differentiation and function of these cells. Since ID2 regulates normal HSC quiescence, the potential role of ID2 in LSC quiescence and resistance to current therapies remains to be explored.
SS, TS, BJ, SG, AS, KG, and JK contributed to writing and editing the review and figures. All authors contributed to the article and approved the submitted version.
This project has been funded in part with federal funds from the Frederick National Laboratory for Cancer Research, NIH (HHSN261200800001E). The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does the mention of trade names, commercial products, or organizations imply endorsements by the US Government.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Trumpp A, Essers M, Wilson A. Awakening dormant haematopoietic stem cells. Nat Rev Immunol (2010) 10(3):201–9. doi: 10.1038/nri2726
2. Pinho S, Frenette PS. Haematopoietic stem cell activity and interactions with the niche. Nat Rev Mol Cell Biol (2019) 20(5):303–20. doi: 10.1038/s41580-019-0103-9
3. Seita J, Weissman IL. Hematopoietic stem cell: self-renewal versus differentiation. Wiley Interdiscip Rev Syst Biol Med (2010) 2(6):640–53. doi: 10.1002/wsbm.86
4. Ito K, Frenette PS. HSC contribution in making steady-state blood. Immunity (2016) 45(3):464–6. doi: 10.1016/j.immuni.2016.09.002
5. Bernitz JM, Kim HS, MacArthur B, Sieburg H, Moore K. Hematopoietic stem cells count and remember self-renewal divisions. Cell (2016) 167(5):1296–309.e10. doi: 10.1016/j.cell.2016.10.022
6. Qiu J, Papatsenko D, Niu X, Schaniel C, Moore K. Divisional history and hematopoietic stem cell function during homeostasis. Stem Cell Rep (2014) 2(4):473–90. doi: 10.1016/j.stemcr.2014.01.016
7. Roschger C, Cabrele C. The id-protein family in developmental and cancer-associated pathways. Cell Commun Signal (2017) 15(1):7. doi: 10.1186/s12964-016-0161-y
8. Wang LH, Baker NE. E proteins and id proteins: helix-loop-helix partners in development and disease. Dev Cell (2015) 35(3):269–80. doi: 10.1016/j.devcel.2015.10.019
9. Norton JD. ID Helix-loop-helix proteins in cell growth, differentiation and tumorigenesis. J Cell Sci (2000) 113(Pt 22):3897–905. doi: 10.1242/jcs.113.22.3897
10. Lasorella A, Benezra R, Iavarone A. The ID proteins: master regulators of cancer stem cells and tumour aggressiveness. Nat Rev Cancer (2014) 14(2):77–91. doi: 10.1038/nrc3638
11. Benezra R, Davis RL, Lockshon D, Turner DL, Weintraub H. The protein id: a negative regulator of helix-loop-helix DNA binding proteins. Cell (1990) 61(1):49–59. doi: 10.1016/0092-8674(90)90214-Y
12. Murre C. Helix-loop-helix proteins and lymphocyte development. Nat Immunol (2005) 6(11):1079–86. doi: 10.1038/ni1260
13. de Pooter RF, Kee BL. E proteins and the regulation of early lymphocyte development. Immunol Rev (2010) 238(1):93–109. doi: 10.1111/j.1600-065X.2010.00957.x
14. Engel I, Murre C. The function of e- and id proteins in lymphocyte development. Nat Rev Immunol (2001) 1(3):193–9. doi: 10.1038/35105060
15. Ohtani N, Zebedee Z, Huot TJ, Stinson JA, Sugimoto M, Ohashi Y, et al. Opposing effects of ets and id proteins on p16INK4a expression during cellular senescence. Nature (2001) 409(6823):1067–70. doi: 10.1038/35059131
16. Lasorella A, Noseda M, Beyna M, Yokota Y, Iavarone A. Id2 is a retinoblastoma protein target and mediates signalling by myc oncoproteins. Nature (2000) 407(6804):592–8. doi: 10.1038/35036504
17. Roberts EC, Deed RW, Inoue T, Norton JD, Sharrocks AD. Id helix-loop-helix proteins antagonize pax transcription factor activity by inhibiting DNA binding. Mol Cell Biol (2001) 21(2):524–33. doi: 10.1128/MCB.21.2.524-533.2001
18. Hacker C, Kirsch RD, Ju XS, Hieronymus T, Gust TC, Kuhl C, et al. Transcriptional profiling identifies Id2 function in dendritic cell development. Nat Immunol (2003) 4(4):380–6. doi: 10.1038/ni903
19. Jackson JT, Hu Y, Liu R, Masson F, D’Amico A, Carotta S, et al. Id2 expression delineates differential checkpoints in the genetic program of CD8alpha+ and CD103+ dendritic cell lineages. EMBO J (2011) 30(13):2690–704. doi: 10.1038/emboj.2011.163
21. Kee BL, Quong MW, Murre C. E2A proteins: essential regulators at multiple stages of b-cell development. Immunol Rev (2000) 175:138–49. doi: 10.1111/j.1600-065X.2000.imr017514.x
22. Yokota Y, Mansouri A, Mori S, Sugawara S, Adachi S, Nishikawa S, et al. Development of peripheral lymphoid organs and natural killer cells depends on the helix-loop-helix inhibitor Id2. Nature (1999) 397(6721):702–6. doi: 10.1038/17812
23. Boos MD, Yokota Y, Eberl G, Kee BL. Mature natural killer cell and lymphoid tissue-inducing cell development requires Id2-mediated suppression of e protein activity. J Exp Med (2007) 204(5):1119–30. doi: 10.1084/jem.20061959
24. Sun XH, Copeland NG, Jenkins NA, Baltimore D. Id proteins Id1 and Id2 selectively inhibit DNA binding by one class of helix-loop-helix proteins. Mol Cell Biol (1991) 11(11):5603–11.
25. Lister J, Forrester WC, Baron MH. Inhibition of an erythroid differentiation switch by the helix-loop-helix protein Id1. J Biol Chem (1995) 270(30):17939–46. doi: 10.1074/jbc.270.30.17939
26. Shoji W, Yamamoto T, Obinata M. The helix-loop-helix protein id inhibits differentiation of murine erythroleukemia cells. J Biol Chem (1994) 269(7):5078–84. doi: 10.1016/S0021-9258(17)37657-3
27. Kreider BL, Benezra R, Rovera G, Kadesch T. Inhibition of myeloid differentiation by the helix-loop-helix protein id. Science (1992) 255(5052):1700–2. doi: 10.1126/science.1372755
28. Quesenberry PJ, Iscove NN, Cooper C, Brady G, Newburger PE, Stein GS, et al. Expression of basic helix-loop-helix transcription factors in explant hematopoietic progenitors. J Cell Biochem (1996) 61(3):478–88. doi: 10.1002/(SICI)1097-4644(19960601)61:3<478::AID-JCB15>3.0.CO;2-F
29. Cooper CL, Brady G, Bilia F, Iscove NN, Quesenberry PJ. Expression of the id family helix-loop-helix regulators during growth and development in the hematopoietic system. Blood (1997) 89(9):3155–65. doi: 10.1182/blood.V89.9.3155
30. Cooper CL, Newburger PE. Differential expression of id genes in multipotent myeloid progenitor cells: Id-1 is induced by early-and late-acting cytokines while id-2 is selectively induced by cytokines that drive terminal granulocytic differentiation. J Cell Biochem (1998) 71(2):277–85. doi: 10.1002/(SICI)1097-4644(19981101)71:2<277::AID-JCB12>3.0.CO;2-I
31. Jen Y, Weintraub H, Benezra R. Overexpression of id protein inhibits the muscle differentiation program: in vivo association of id with E2A proteins. Genes Dev (1992) 6(8):1466–79. doi: 10.1101/gad.6.8.1466
32. Desprez PY, Hara E, Bissell MJ, Campisi J. Suppression of mammary epithelial cell differentiation by the helix-loop-helix protein id-1. Mol Cell Biol (1995) 15(6):3398–404. doi: 10.1128/MCB.15.6.3398
33. Leeanansaksiri W, Wang H, Gooya JM, Renn K, Abshari M, Tsai S, et al. IL-3 induces inhibitor of DNA-binding protein-1 in hemopoietic progenitor cells and promotes myeloid cell development. J Immunol (2005) 174(11):7014–21. doi: 10.4049/jimmunol.174.11.7014
34. Cochrane SW, Zhao Y, Welner RS, Sun XH. Balance between id and e proteins regulates myeloid-versus-lymphoid lineage decisions. Blood (2009) 113(5):1016–26. doi: 10.1182/blood-2008-06-164996
35. Jankovic V, Ciarrocchi A, Boccuni P, DeBlasio T, Benezra R, Nimer SD. Id1 restrains myeloid commitment, maintaining the self-renewal capacity of hematopoietic stem cells. Proc Natl Acad Sci USA (2007) 104(4):1260–5. doi: 10.1073/pnas.0607894104
36. Singh SK, Singh S, Gadomski S, Sun L, Pfannenstein A, Magidson V, et al. Id1 ablation protects hematopoietic stem cells from stress-induced exhaustion and aging. Cell Stem Cell (2018) 23(2):252–65.e8. doi: 10.1016/j.stem.2018.06.001
37. Perry SS, Zhao Y, Nie L, Cochrane SW, Huang Z, Sun XH. Id1, but not Id3, directs long-term repopulating hematopoietic stem-cell maintenance. Blood (2007) 110(7):2351–60. doi: 10.1182/blood-2007-01-069914
38. van Galen P, Kreso A, Wienholds E, Laurenti E, Eppert K, Lechman ER, et al. Reduced lymphoid lineage priming promotes human hematopoietic stem cell expansion. Cell Stem Cell (2014) 14(1):94–106. doi: 10.1016/j.stem.2013.11.021
39. Suh HC, Leeanansaksiri W, Ji M, Klarmann KD, Renn K, Gooya J, et al. Id1 immortalizes hematopoietic progenitors in vitro and promotes a myeloproliferative disease in vivo. Oncogene (2008) 27(42):5612–23. doi: 10.1038/onc.2008.175
40. Buitenhuis M, van Deutekom HW, Verhagen LP, Castor A, Jacobsen SE, Lammers JW, et al. Differential regulation of granulopoiesis by the basic helix-loop-helix transcriptional inhibitors Id1 and Id2. Blood (2005) 105(11):4272–81. doi: 10.1182/blood-2004-12-4883
41. Morrow MA, Mayer EW, Perez CA, Adlam M, Siu G. Overexpression of the helix-Loop-Helix protein Id2 blocks T cell development at multiple stages. Mol Immunol (1999) 36(8):491–503. doi: 10.1016/S0161-5890(99)00071-1
42. Kim D, Peng XC, Sun XH. Massive apoptosis of thymocytes in T-cell-deficient Id1 transgenic mice. Mol Cell Biol (1999) 19(12):8240–53. doi: 10.1128/MCB.19.12.8240
43. Sun XH. Constitutive expression of the Id1 gene impairs mouse b cell development. Cell (1994) 79(5):893–900. doi: 10.1016/0092-8674(94)90078-7
44. Belle I, Zhuang Y. E proteins in lymphocyte development and lymphoid diseases. Curr topics Dev Biol (2014) 110:153–87. doi: 10.1016/B978-0-12-405943-6.00004-X
45. Mercer EM, Lin YC, Benner C, Jhunjhunwala S, Dutkowski J, Flores M, et al. Multilineage priming of enhancer repertoires precedes commitment to the b and myeloid cell lineages in hematopoietic progenitors. Immunity (2011) 35(3):413–25. doi: 10.1016/j.immuni.2011.06.013
46. Mercer EM, Lin YC, Murre C. Factors and networks that underpin early hematopoiesis. Semin Immunol (2011) 23(5):317–25. doi: 10.1016/j.smim.2011.08.004
47. Ikawa T, Masuda K, Huijskens M, Satoh R, Kakugawa K, Agata Y, et al. Induced developmental arrest of early hematopoietic progenitors leads to the generation of leukocyte stem cells. Stem Cell Rep (2015) 5(5):716–27. doi: 10.1016/j.stemcr.2015.09.012
48. Kee BL, Rivera RR, Murre C. Id3 inhibits b lymphocyte progenitor growth and survival in response to TGF-beta. Nat Immunol (2001) 2(3):242–7. doi: 10.1038/85303
49. Pan L, Sato S, Frederick JP, Sun XH, Zhuang Y. Impaired immune responses and b-cell proliferation in mice lacking the Id3 gene. Mol Cell Biol (1999) 19(9):5969–80. doi: 10.1128/MCB.19.9.5969
50. Rivera RR, Johns CP, Quan J, Johnson RS, Murre C. Thymocyte selection is regulated by the helix-loop-helix inhibitor protein, Id3. Immunity (2000) 12(1):17–26. doi: 10.1016/S1074-7613(00)80155-7
51. Thal MA, Carvalho TL, He T, Kim HG, Gao H, Hagman J, et al. Ebf1-mediated down-regulation of Id2 and Id3 is essential for specification of the b cell lineage. Proc Natl Acad Sci USA (2009) 106(2):552–7. doi: 10.1073/pnas.0802550106
52. Yan W, Young AZ, Soares VC, Kelley R, Benezra R, Zhuang Y. High incidence of T-cell tumors in E2A-null mice and E2A/Id1 double-knockout mice. Mol Cell Biol (1997) 17(12):7317–27. doi: 10.1128/MCB.17.12.7317
53. Suh HC, Ji M, Gooya J, Lee M, Klarmann KD, Keller JR. Cell-nonautonomous function of Id1 in the hematopoietic progenitor cell niche. Blood (2009) 114(6):1186–95. doi: 10.1182/blood-2008-09-179788
54. Yang J, Li X, Morrell NW. Id proteins in the vasculature: from molecular biology to cardiopulmonary medicine. Cardiovasc Res (2014) 104(3):388–98. doi: 10.1093/cvr/cvu215
55. Lyden D, Young AZ, Zagzag D, Yan W, Gerald W, O’Reilly R, et al. Id1 and Id3 are required for neurogenesis, angiogenesis and vascularization of tumour xenografts. Nature (1999) 401(6754):670–7. doi: 10.1038/44334
56. Gadomski S, Singh SK, Singh S, Sarkar T, Klarmann KD, Berenschot M, et al. Id1 and Id3 maintain steady-state hematopoiesis by promoting sinusoidal endothelial cell survival and regeneration. Cell Rep (2020) 31(4):107572. doi: 10.1016/j.celrep.2020.107572
57. Peng Y, Kang Q, Luo Q, Jiang W, Si W, Liu BA, et al. Inhibitor of DNA binding/differentiation helix-loop-helix proteins mediate bone morphogenetic protein-induced osteoblast differentiation of mesenchymal stem cells. J Biol Chem (2004) 279(31):32941–9. doi: 10.1074/jbc.M403344200
58. Fraidenraich D, Stillwell E, Romero E, Wilkes D, Manova K, Basson CT, et al. Rescue of cardiac defects in id knockout embryos by injection of embryonic stem cells. Science (2004) 306(5694):247–52. doi: 10.1126/science.1102612
59. Morrison SJ, Scadden DT. The bone marrow niche for haematopoietic stem cells. Nature (2014) 505(7483):327–34. doi: 10.1038/nature12984
60. Wei Q, Frenette PS. Niches for hematopoietic stem cells and their progeny. Immunity (2018) 48(4):632–48. doi: 10.1016/j.immuni.2018.03.024
61. Kusumbe AP, Ramasamy SK, Itkin T, Mae MA, Langen UH, Betsholtz C, et al. Age-dependent modulation of vascular niches for haematopoietic stem cells. Nature (2016) 532(7599):380–4. doi: 10.1038/nature17638
62. Itkin T, Gur-Cohen S, Spencer JA, Schajnovitz A, Ramasamy SK, Kusumbe AP, et al. Distinct bone marrow blood vessels differentially regulate haematopoiesis. Nature (2016) 532(7599):323–8. doi: 10.1038/nature17624
63. Comazzetto S, Shen B, Morrison SJ. Niches that regulate stem cells and hematopoiesis in adult bone marrow. Dev Cell (2021) 56(13):1848–60. doi: 10.1016/j.devcel.2021.05.018
64. Rafii S, Butler JM, Ding BS. Angiocrine functions of organ-specific endothelial cells. Nature (2016) 529(7586):316–25. doi: 10.1038/nature17040
65. Kobayashi H, Butler JM, O’Donnell R, Kobayashi M, Ding BS, Bonner B, et al. Angiocrine factors from akt-activated endothelial cells balance self-renewal and differentiation of haematopoietic stem cells. Nat Cell Biol (2010) 12(11):1046–56. doi: 10.1038/ncb2108
66. Kusumbe AP, Ramasamy SK, Adams RH. Coupling of angiogenesis and osteogenesis by a specific vessel subtype in bone. Nature (2014) 507(7492):323–8. doi: 10.1038/nature13145
67. Ding BS, Cao Z, Lis R, Nolan DJ, Guo P, Simons M, et al. Divergent angiocrine signals from vascular niche balance liver regeneration and fibrosis. Nature (2014) 505(7481):97–102. doi: 10.1038/nature12681
68. Ding BS, Nolan DJ, Butler JM, James D, Babazadeh AO, Rosenwaks Z, et al. Inductive angiocrine signals from sinusoidal endothelium are required for liver regeneration. Nature (2010) 468(7321):310–5. doi: 10.1038/nature09493
69. Poulos MG, Guo P, Kofler NM, Pinho S, Gutkin MC, Tikhonova A, et al. Endothelial jagged-1 is necessary for homeostatic and regenerative hematopoiesis. Cell Rep (2013) 4(5):1022–34. doi: 10.1016/j.celrep.2013.07.048
70. Kfoury Y, Scadden DT. Mesenchymal cell contributions to the stem cell niche. Cell Stem Cell (2015) 16(3):239–53. doi: 10.1016/j.stem.2015.02.019
71. Li H, Dai M, Zhuang Y. A T cell intrinsic role of Id3 in a mouse model for primary sjogren’s syndrome. Immunity (2004) 21(4):551–60. doi: 10.1016/j.immuni.2004.08.013
72. Li J, Maruyama T, Zhang P, Konkel JE, Hoffman V, Zamarron B, et al. Mutation of inhibitory helix-loop-helix protein Id3 causes gammadelta T-cell lymphoma in mice. Blood (2010) 116(25):5615–21. doi: 10.1182/blood-2010-03-274506
73. Ji M, Li H, Suh HC, Klarmann KD, Yokota Y, Keller JR. Id2 intrinsically regulates lymphoid and erythroid development via interaction with different target proteins. Blood (2008) 112(4):1068–77. doi: 10.1182/blood-2008-01-133504
74. Hock H, Hamblen MJ, Rooke HM, Schindler JW, Saleque S, Fujiwara Y, et al. Gfi-1 restricts proliferation and preserves functional integrity of haematopoietic stem cells. Nature (2004) 431(7011):1002–7. doi: 10.1038/nature02994
75. Zeng H, Yucel R, Kosan C, Klein-Hitpass L, Moroy T. Transcription factor Gfi1 regulates self-renewal and engraftment of hematopoietic stem cells. EMBO J (2004) 23(20):4116–25. doi: 10.1038/sj.emboj.7600419
76. Li H, Ji M, Klarmann KD, Keller JR. Repression of Id2 expression by gfi-1 is required for b-cell and myeloid development. Blood (2010) 116(7):1060–9. doi: 10.1182/blood-2009-11-255075
77. Kim W, Klarmann KD, Keller JR. Gfi-1 regulates the erythroid transcription factor network through Id2 repression in murine hematopoietic progenitor cells. Blood (2014) 124(10):1586–96. doi: 10.1182/blood-2014-02-556522
78. Jakubison BL, Sarkar T, Gudmundsson KO, Singh S, Sun L, Morris HM, et al. ID2 and HIF-1α collaborate to protect quiescent hematopoietic stem cells from activation, differentiation, and exhaustion. J Clin Invest (2022) 132(13). doi: 10.1172/JCI152599
79. Takubo K, Goda N, Yamada W, Iriuchishima H, Ikeda E, Kubota Y, et al. Regulation of the HIF-1alpha level is essential for hematopoietic stem cells. Cell Stem Cell (2010) 7(3):391–402. doi: 10.1016/j.stem.2010.06.020
80. Barone MV, Pepperkok R, Peverali FA, Philipson L. Id proteins control growth induction in mammalian cells. Proc Natl Acad Sci USA (1994) 91(11):4985–8. doi: 10.1073/pnas.91.11.4985
81. Ganuza M, McKinney-Freeman S. Hematopoietic stem cells under pressure. Curr Opin Hematol (2017) 24(4):314–21. doi: 10.1097/MOH.0000000000000347
82. Termini CM, Chute JP. Hematopoietic stem cell stress and regeneration. Curr Stem Cell Rep (2020) 6(4):134–43. doi: 10.1007/s40778-020-00181-3
83. Boettcher S, Manz MG. Regulation of inflammation- and infection-driven hematopoiesis. Trends Immunol (2017) 38(5):345–57. doi: 10.1016/j.it.2017.01.004
84. Caiado F, Pietras EM, Manz MG. Inflammation as a regulator of hematopoietic stem cell function in disease, aging, and clonal selection. J Exp Med (2021) 218(7). doi: 10.1084/jem.20201541
85. Singh S, Jakubison B, Keller JR. Protection of hematopoietic stem cells from stress-induced exhaustion and aging. Curr Opin Hematol (2020) 27(4):225–31. doi: 10.1097/MOH.0000000000000586
86. Kleppe M, Spitzer MH, Li S, Hill CE, Dong L, Papalexi E, et al. Jak1 integrates cytokine sensing to regulate hematopoietic stem cell function and stress hematopoiesis. Cell Stem Cell (2018) 22(2):277. doi: 10.1016/j.stem.2017.12.018
87. Zhao Y, Ling F, Wang HC, Sun XH. Chronic TLR signaling impairs the long-term repopulating potential of hematopoietic stem cells of wild type but not Id1 deficient mice. PloS One (2013) 8(2):e55552. doi: 10.1371/journal.pone.0055552
88. Barreyro L, Chlon TM, Starczynowski DT. Chronic immune response dysregulation in MDS pathogenesis. Blood (2018) 132(15):1553–60. doi: 10.1182/blood-2018-03-784116
89. Trowbridge JJ, Starczynowski DT. Innate immune pathways and inflammation in hematopoietic aging, clonal hematopoiesis, and MDS. J Exp Med (2021) 218(7). doi: 10.1084/jem.20201544
90. Craver BM, El Alaoui K, Scherber RM, Fleischman AG. The critical role of inflammation in the pathogenesis and progression of myeloid malignancies. Cancers (Basel) (2018) 10(4):104–20.
91. Prabhu S, Ignatova A, Park ST, Sun XH. Regulation of the expression of cyclin-dependent kinase inhibitor p21 by E2A and id proteins. Mol Cell Biol (1997) 17(10):5888–96.
92. Rothschild G, Zhao X, Iavarone A, Lasorella A. E proteins and Id2 converge on p57Kip2 to regulate cell cycle in neural cells. Mol Cell Biol (2006) 26(11):4351–61. doi: 10.1128/MCB.01743-05
93. Mern DS, Hoppe-Seyler K, Hoppe-Seyler F, Hasskarl J, Burwinkel B. Targeting Id1 and Id3 by a specific peptide aptamer induces e-box promoter activity, cell cycle arrest, and apoptosis in breast cancer cells. Breast Cancer Res Treat (2010) 124(3):623–33. doi: 10.1007/s10549-010-0810-6
94. Zheng W, Wang H, Xue L, Zhang Z, Tong T. Regulation of cellular senescence and p16(INK4a) expression by Id1 and E47 proteins in human diploid fibroblast. J Biol Chem (2004) 279(30):31524–32. doi: 10.1074/jbc.M400365200
95. Semerad CL, Mercer EM, Inlay MA, Weissman IL, Murre C. E2A proteins maintain the hematopoietic stem cell pool and promote the maturation of myelolymphoid and myeloerythroid progenitors. Proc Natl Acad Sci USA (2009) 106(6):1930–5. doi: 10.1073/pnas.0808866106
96. Yang Q, Esplin B, Borghesi L. E47 regulates hematopoietic stem cell proliferation and energetics but not myeloid lineage restriction. Blood (2011) 117(13):3529–38. doi: 10.1182/blood-2010-07-297689
97. Dias S, Mansson R, Gurbuxani S, Sigvardsson M, Kee BL. E2A proteins promote development of lymphoid-primed multipotent progenitors. Immunity (2008) 29(2):217–27. doi: 10.1016/j.immuni.2008.05.015
98. Cheng T, Rodrigues N, Shen H, Yang Y, Dombkowski D, Sykes M, et al. Hematopoietic stem cell quiescence maintained by p21cip1/waf1. Science (2000) 287(5459):1804–8. doi: 10.1126/science.287.5459.1804
99. Santos PM, Ding Y, Borghesi L. Cell-intrinsic in vivo requirement for the E47-p21 pathway in long-term hematopoietic stem cells. J Immunol (2014) 192(1):160–8. doi: 10.4049/jimmunol.1302502
100. Nair R, Teo WS, Mittal V, Swarbrick A. ID Proteins regulate diverse aspects of cancer progression and provide novel therapeutic opportunities. Mol Ther: J Am Soc Gene Ther (2014) 22(8):1407–15. doi: 10.1038/mt.2014.83
101. Radich JP, Dai H, Mao M, Oehler V, Schelter J, Druker B, et al. Gene expression changes associated with progression and response in chronic myeloid leukemia. Proc Natl Acad Sci USA (2006) 103(8):2794–9. doi: 10.1073/pnas.0510423103
102. Rucker FG, Bullinger L, Schwaenen C, Lipka DB, Wessendorf S, Frohling S, et al. Disclosure of candidate genes in acute myeloid leukemia with complex karyotypes using microarray-based molecular characterization. J Clin Oncol (2006) 24(24):3887–94. doi: 10.1200/JCO.2005.04.5450
103. Zhou JD, Ma JC, Zhang TJ, Li XX, Zhang W, Wu DH, et al. High bone marrow ID2 expression predicts poor chemotherapy response and prognosis in acute myeloid leukemia. Oncotarget (2017) 8(54):91979–89. doi: 10.18632/oncotarget.20559
104. Zhou JD, Yang L, Zhu XW, Wen XM, Yang J, Guo H, et al. Clinical significance of up-regulated ID1 expression in Chinese de novo acute myeloid leukemia. Int J Clin Exp Pathol (2015) 8(5):5336–44.
105. Tochareontanaphol C, Sinthuwiwat T, Buathong B, Thita T, Promso S, Paca-Uccaralertkun S. New mutations of the id1 gene in acute myeloid leukemia patients. Pathobiology (2015) 82(1):43–7. doi: 10.1159/000370243
106. Klarmann K, Ji M, Li H, Satyanarayana A, Kim W, Bowers E, et al Novel targets in myelogenous leukemia: the id family of proteins. myeloid leukemia – basic mechanisms of leukemogenesis. Koschmieder S, Krug U editors. Rijeka, Croatia: InTech (2011). 215–38 p.
107. Bera R, Chiu MC, Huang YJ, Lin TH, Kuo MC, Shih LY. RUNX1 mutations promote leukemogenesis of myeloid malignancies in ASXL1-mutated leukemia. J Hematol Oncol (2019) 12(1):104. doi: 10.1186/s13045-019-0789-3
108. Liquori A, Ibanez M, Sargas C, Sanz MA, Barragan E, Cervera J. Acute promyelocytic leukemia: A constellation of molecular events around a single pml-rara fusion gene. Cancers (Basel) (2020) 12(3):624–6. doi: 10.3390/cancers12030624
109. Sayar H, Liu Y, Gao R, Zaid MA, Cripe LD, Weisenbach J, et al. Consecutive epigenetically-active agent combinations act in ID1-RUNX3-TET2 and HOXA pathways for Flt3ITD+ve AML. Oncotarget (2018) 9(5):5703–15. doi: 10.18632/oncotarget.23655
110. Eisfeld AK, Kohlschmidt J, Mrozek K, Volinia S, Blachly JS, Nicolet D, et al. Mutational landscape and gene expression patterns in adult acute myeloid leukemias with monosomy 7 as a sole abnormality. Cancer Res (2017) 77(1):207–18. doi: 10.1158/0008-5472.CAN-16-1386
111. Valk PJ, Verhaak RG, Beijen MA, Erpelinck CA, Barjesteh van Waalwijk van Doorn-Khosrovani S, Boer JM, et al. Prognostically useful gene-expression profiles in acute myeloid leukemia. N Engl J Med (2004) 350(16):1617–28. doi: 10.1056/NEJMoa040465
112. Tam WF, Gu TL, Chen J, Lee BH, Bullinger L, Frohling S, et al. Id1 is a common downstream target of oncogenic tyrosine kinases in leukemic cells. Blood (2008) 112(5):1981–92. doi: 10.1182/blood-2007-07-103010
113. Tang R, Hirsch P, Fava F, Lapusan S, Marzac C, Teyssandier I, et al. High Id1 expression is associated with poor prognosis in 237 patients with acute myeloid leukemia. Blood (2009) 114(14):2993–3000. doi: 10.1182/blood-2009-05-223115
114. Wang L, Gural A, Sun XJ, Zhao X, Perna F, Huang G, et al. The leukemogenicity of AML1-ETO is dependent on site-specific lysine acetylation. Science (2011) 333(6043):765–9. doi: 10.1126/science.1201662
115. Wang L, Man N, Sun XJ, Tan Y, Garcia-Cao M, Liu F, et al. Regulation of AKT signaling by Id1 controls t (8,21)Leukemia initiation and progression. Blood (2015) 126(5):640–50. doi: 10.1182/blood-2015-03-635532
116. Man N, Sun XJ, Tan Y, Garcia-Cao M, Liu F, Cheng G, et al. Differential role of Id1 in MLL-AF9-driven leukemia based on cell of origin. Blood (2016) 127(19):2322–6. doi: 10.1182/blood-2015-11-677708
117. Wojnarowicz PM, Escolano MG, Huang YH, Desai B, Chin Y, Shah R, et al. Anti-tumor effects of an ID antagonist with no observed acquired resistance. NPJ Breast Cancer (2021) 7(1):58. doi: 10.1038/s41523-021-00266-0
118. Soroceanu L, Murase R, Limbad C, Singer E, Allison J, Adrados I, et al. Id-1 is a key transcriptional regulator of glioblastoma aggressiveness and a novel therapeutic target. Cancer Res (2013) 73(5):1559–69. doi: 10.1158/0008-5472.CAN-12-1943
119. McAllister SD, Christian RT, Horowitz MP, Garcia A, Desprez PY. Cannabidiol as a novel inhibitor of id-1 gene expression in aggressive breast cancer cells. Mol Cancer Ther (2007) 6(11):2921–7. doi: 10.1158/1535-7163.MCT-07-0371
120. Kovtonyuk LV, Fritsch K, Feng X, Manz MG, Takizawa H. Inflamm-aging of hematopoiesis, hematopoietic stem cells, and the bone marrow microenvironment. Front Immunol (2016) 7:502. doi: 10.3389/fimmu.2016.00502
121. Ko J, Patel N, Ikawa T, Kawamoto H, Frank O, Rivera RR, et al. Suppression of e-protein activity interferes with the development of BCR-ABL-mediated myeloproliferative disease. Proc Natl Acad Sci U S A (2008) 105(35):12967–72. doi: 10.1073/pnas.0805073105
122. Ghisi M, Kats L, Masson F, Li J, Kratina T, Vidacs E, et al. Id2 and e proteins orchestrate the initiation and maintenance of MLL-rearranged acute myeloid leukemia. Cancer Cell (2016) 30(1):59–74. doi: 10.1016/j.ccell.2016.05.019
123. Nigmatullina L, Norkin M, Dzama MM, Messner B, Sayols S, Soshnikova N. Id2 controls specification of Lgr5(+) intestinal stem cell progenitors during gut development. EMBO J (2017) 36(7):869–85. doi: 10.15252/embj.201694959
Keywords: ID proteins, hematopoiesis, stem cells, quiescence, stress
Citation: Singh S, Sarkar T, Jakubison B, Gadomski S, Spradlin A, Gudmundsson KO and Keller JR (2022) Inhibitor of DNA binding proteins revealed as orchestrators of steady state, stress and malignant hematopoiesis. Front. Immunol. 13:934624. doi: 10.3389/fimmu.2022.934624
Received: 02 May 2022; Accepted: 12 July 2022;
Published: 05 August 2022.
Edited by:
Barbara L. Kee, The University of Chicago, United StatesReviewed by:
Ivan Maillard, University of Pennsylvania, United StatesCopyright © 2022 Singh, Sarkar, Jakubison, Gadomski, Spradlin, Gudmundsson and Keller. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jonathan R. Keller, S2VsbGVyam9AbWFpbC5uaWguZ292
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.