
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
PERSPECTIVE article
Front. Immunol. , 11 July 2022
Sec. Parasite Immunology
Volume 13 - 2022 | https://doi.org/10.3389/fimmu.2022.931034
This article is part of the Research Topic New advances in knowledge of the parasite-host relationship during malarial infection View all 7 articles
 André Barateiro1
André Barateiro1 Alexsander Rodrigues Carvalho Junior1
Alexsander Rodrigues Carvalho Junior1 Sabrina Epiphanio2
Sabrina Epiphanio2 Claudio Romero Farias Marinho1*
Claudio Romero Farias Marinho1*Malaria represents a significant public health burden to populations living in developing countries. The disease takes a relevant toll on pregnant women, who are more prone to developing severe clinical manifestations. Inflammation triggered in response to P. falciparum sequestration inside the placenta leads to physiological and structural changes in the organ, reflecting locally disrupted homeostasis. Altogether, these events have been associated with poor gestational outcomes, such as intrauterine growth restriction and premature delivery, contributing to the parturition of thousands of African children with low birth weight. Despite significant advances in the field, the molecular mechanisms that govern these outcomes are still poorly understood. Herein, we discuss the idea of how some housekeeping molecular mechanisms, such as those related to autophagy, might be intertwined with the outcomes of malaria in pregnancy. We contextualize previous findings suggesting that placental autophagy is dysregulated in P. falciparum-infected pregnant women with complementary research describing the importance of autophagy in healthy pregnancies. Since the functional role of autophagy in pregnancy outcomes is still unclear, we hypothesize that autophagy might be essential for circumventing inflammation-induced stress in the placenta, acting as a cytoprotective mechanism that attempts to ensure local homeostasis and better gestational prognosis in women with malaria in pregnancy.
Malaria represents a burden for multiple communities worldwide. Despite continuous eradication efforts, 241 million cases were recorded in 2020, resulting in approximately 627 thousand deaths. The disease affects mostly African children and pregnant women, who are highly susceptible to developing severe disease resulting from P. falciparum infections (1). Malaria in pregnancy (MiP) is often neglected, yet in 2019, the World Health Organization raised awareness of this issue, describing MiP as “a significant public health problem”. In 2020, the disease affected nearly 12 million African women who gave birth to more than 800 thousand babies with low birth weight, contributing to high infant mortality (1, 2).
P. falciparum infections during pregnancy often lead to poor outcomes, such as maternal and fetal mortality, anemia, fetal growth restriction, preterm delivery, and low birth weight (3, 4). Disease susceptibility varies according to P. falciparum endemicity spectrum, as pregnant women living in high transmission settings tend to have symptomless infections due to recurrent exposure during pregnancy. Nevertheless, women in their first pregnancy may experience more severe symptoms due to low immunity (5). Immunity is governed by protective antibodies to VAR2CSA (6, 7), which mediates the binding of P. falciparum-infected erythrocytes to chondroitin sulfate A, abundantly expressed by trophoblasts in the placenta (8, 9). This event promotes parasite sequestration inside this organ, which characterizes placental malaria (PM) (10). Trophoblasts (and eventually maternal monocytes) in the placenta recognize P. falciparum antigens, activating innate immune responses, which culminate with the production of chemokines such as MIP-1α, MCP-1, I-309, and IL-8 (responsible for monocyte recruitment) and cytokines, such as IFN-γ, TNF-α, IL-2, IL-6, IL-10, and IL-1β, which are frequently found in placentas from P. falciparum-infected women (11–15). Inflammation is known to control infection, yet it is etiologically linked to poor outcomes (16, 17) since both placental monocytes and cytokines have been associated with maternal anemia, pregnancy loss, preterm delivery, and low birth weight in children born to P. falciparum-infected women (13, 14, 18–20).
The placenta ensures immunological tolerance to the maternal immune system (21), promotes nutrient and gas exchanges between the mother and the fetus (22), and works as a physical and immunological barrier against pathogens (23). These properties are lost during infections due to significant tissue disarrangement. P. falciparum-infected placentas often suffer from histological modifications, such as the presence of malarial pigment hemozoin, leukocyte infiltrate, syncytial nuclear aggregates, fibrinoid necrosis, and trophoblast barrier thickening (24–26). Therefore, it is possible that this reflects local homeostasis dysregulation, which would predict fetal growth restrictions due to placental insufficiency resulting from impaired placental blood flow and vasculogenesis/angiogenesis, hypoxia, oxidative stress, and reduced transplacental nutrient transportation (3, 27).
Although several factors are known to be associated with MiP outcomes, the molecular mechanisms involved in disease pathogenesis are still unclear. Therefore, it is possible that other mechanisms less studied to date, such as those involving autophagy, might be linked to MiP pathogenesis.
Autophagy is conventionally known as a conserved mechanism of lysosomal degradation of cytoplasmic components and is involved in cellular differentiation and development (28), homeostasis and survival (29). It can be activated in response to a plethora of stress-inducing agents that range from starvation, hypoxia, oxidative stress and damaged organelles to immune activation and pathogens, promoting degradation of undesired microorganisms, nutrient recycling, and organelle turnover (29–31) (Figure 1). In mammalian cells, the most well studied type of autophagy is macroautophagy (hereafter referred to simply as autophagy), which relies on the de novo formation of a vesicular structures capable of selecting and transporting cargo to lysosomes for degradation (32). Proteins transcribed by autophagy-related genes (ATGs) tightly regulate this process, which is primarily characterized by the formation of an intermediate double-membrane structure (phagophore) that surrounds specific cargo (forming the autophagosome), ultimately fusing with lysosomes (autolysosomes), which promote content digestion (29). The system seems to be redundant, as different triggers converge to activate almost identical transduction pathways inside cells (31). At its core, autophagy is regulated by at least some primary core complexes (Figure 1A): 1) the ULK complex (2), the Beclin1 interactome, 3) ATG9 and VMP1 transmembrane proteins, 4) two ubiquitin-like conjugation systems associated with ATG12 and LC3, and 5) autophagosome-lysosome fusion mediators (i.e., Rab7). Some of these components can be directly activated by distinct stress-inducing signals, likely ending with autophagosome-lysosome fusion and digestion of inner-vesicle cargo (29).
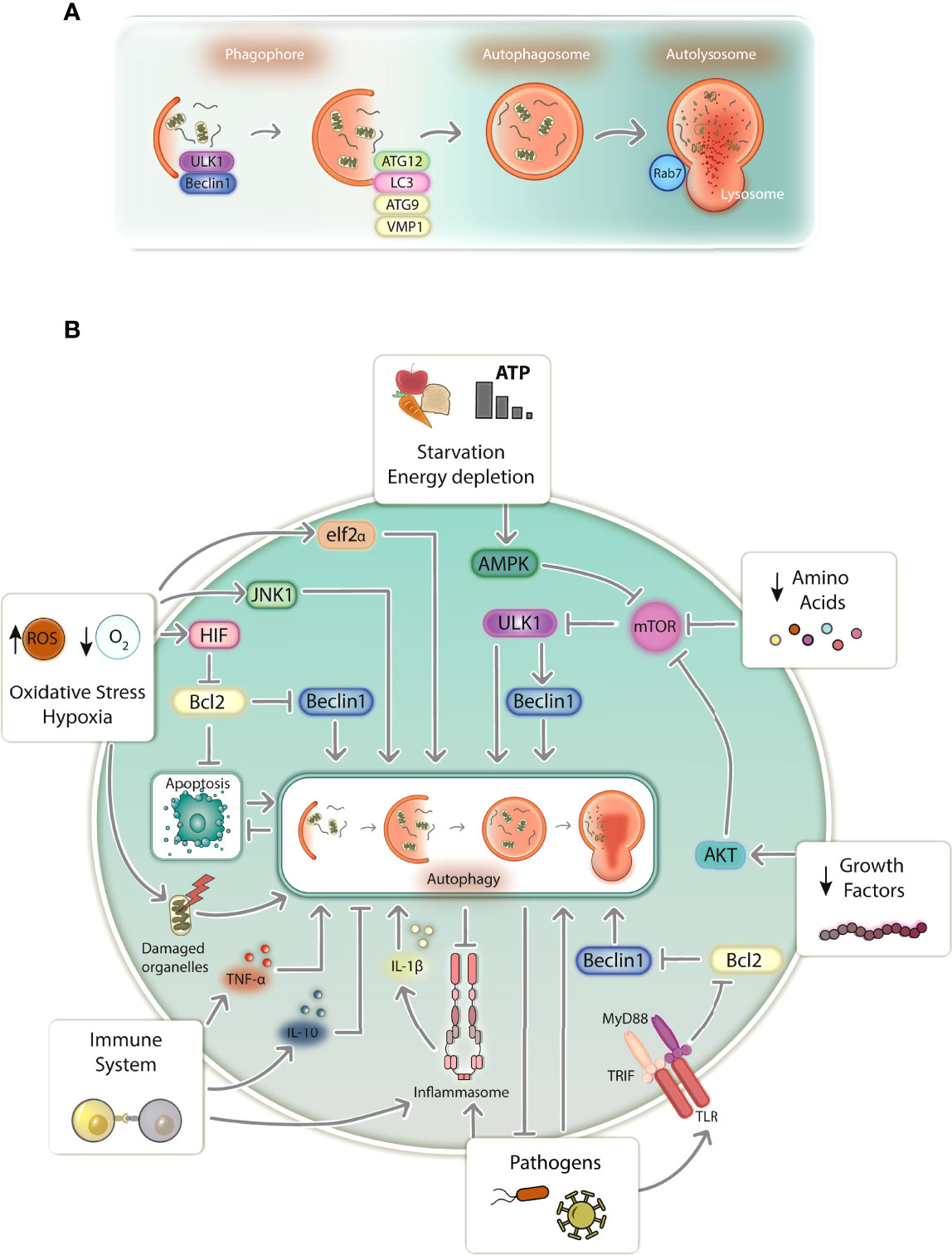
Figure 1 Schematic representation of autophagy in mammalian cells. (A) Autophagy proceeds through a series of events that begin with phagophore formation, elongation, and maturation into an autophagosome. The autophagosome fuses with a lysosome to form the autolysosome, followed by the degradation of selected cargo. Autophagy is tightly regulated by protein complexes produced from autophagy-related genes (ATGs) and its execution relies mostly on ULK1 and Beclin1 complexes (phagophore isolation), ATG9 and VMP1 (phagophore elongation), the ATG12 complex and LC3 (phagophore maturation into autophagosomes), and Rab7 GTPase (autophagosome-lysosome fusion for autophagy completion). (B) Cells transduce distinct signals that equally induce autophagy. Starvation, growth factor and amino acid depletion induces autophagy via AMPK and AKT, which blocks the inhibitory action of mTOR over the autophagy inducers ULK1 and Beclin1. Oxidative stress and hypoxia induce autophagy via elf2α, JNK1 and HIF, being the latter responsible for disrupting the inhibitory action of Bcl2 over Beclin1. Damaged organelles such as mitochondria induce autophagy, which digests damaged organelles and prevents cells death resulting from apoptosis. Immune system activation induces autophagy via TLR signaling upon pathogen sensing by destabilizing Bcl2-Beclin1 interaction. Autophagy is also activated or inhibited by TNF-α and IL-10, respectively. Inflammasome activity also induces autophagy, which works to counteract its overactivation resulting from excessive inflammatory signals. Pathogens directly induce autophagy, besides activating the immune system, which suppresses pathogen development by directly targeting them for degradation. ROS, reactive oxygen species; TLR, Toll-like receptor.
Several molecules can induce autophagy in response to starvation (Figure 1B); however, the best studied are those associated with mTOR, which constitutively inhibits autophagy in homeostasis. Signals such as starvation, energy depletion, absence of growth factors and amino acids, are transduced by AMPK or AKT kinases, leading to mTOR inhibition (29, 33). Consequently, ULK1 complex activation occurs, promoting Beclin1 interactome phosphorylation, which enables PI3K Vps34 to produce PI3P, relevant to phagophore isolation from intracellular membrane structures such as the endoplasmic reticulum (ER) (34, 35). In parallel, ATG9 and VMP1 are responsible for lipid recruitment to the isolated membrane, participating in its biogenesis (29). Afterward, phagophore elongation and maturation occurs, which depends on two ubiquitin-like conjugation systems: 1) the oligomeric ATG12-ATG5-ATG16 complex and 2) LC3 (LC3I), after being conjugated with a phosphatidylethanolimine (LC3II), which together promote phagophore elongation and membrane closure to form the autophagosome (29, 35). Selective autophagy occurs in response to misfolded proteins and damaged organelles, which are targeted for degradation by sequestosome-like proteins such as SQSTM1/p62 and BNIP3. These proteins, which have LC3-interacting regions, recognize ubiquitinated proteins and damaged mitochondria, for instance, taking them to the emerging autophagosome to be engulfed (29, 36).
Other kinases that also regulate starvation-induced autophagy, such as JNK1 and eIF2α, initiate autophagy in response to oxidative and ER stress, and hypoxia (Figure 1B). eIF2α transduces stress signals imposed by ER-misfolded proteins, reduced oxygen levels and increased levels of reactive oxygen species (ROS). Hypoxia and ROS can also induce autophagy via JNK1 (29). Additionally, hypoxia induces autophagy via HIF by weakening the Bcl2 inhibitory interaction with Beclin1 or by inducing BNIP3 to deliver mitochondria for autophagic degradation (29, 37). Abnormal membrane potential in damaged mitochondria (Figure 1B), which are potent autophagy inducers, can also be sensed by voltage-dependent kinases, inducing ubiquitination of mitochondrial proteins, which are subsequently targeted by SQSTM1/p62 and BNIP3, directing mitochondria to be degraded by autophagy (29, 38). Apoptosis (Figure 1B), which is often a consequence of mitochondrial damage, can be counteracted by autophagy, both occurring almost in a mutually exclusive manner. Upon dissociation from Bcl2, Beclin1 induces autophagy via PI3K Vps34 (34), while Bcl2 inhibits apoptosis by counter regulating mitochondrial membrane permeabilization (39, 40). This supports autophagy’s role as a cytoprotective mechanism that can circumvent cell death by abrogating manageable levels of cellular stress (41, 42).
Innate immunity (Figure 1B) also modulates autophagy, especially in the context of infections. Since infections have been linked to nutritional stress, it is plausible to hypothesize that evolution led to the development of cellular mechanisms shared between the immune system and autophagy (31). TLRs trigger autophagy via TRIF/MyD88 signaling, weakening the Bcl2-Beclin1 interaction, which normally represses autophagy (43). Likewise, NLRs were also shown to induce autophagy by activating ATG16L1 in response to bacterial infections (44). Immune system activation by pathogens frequently culminates in cytokine production and inflammation. As such, autophagy can be activated to 1) attenuate inflammation by controlling immune system activation or 2) directly destroying pathogens (30, 31, 45). Th1 proinflammatory cytokines, such as TNF-α, are produced during infections and contribute to autophagy induction by mechanisms that are still poorly understood (46). TNF-α was shown to induce LC3 transcription and conversion into LC3II, Beclin1 synthesis and autophagosome accumulation in human muscle cells (47, 48). In addition, elimination of intracellular pathogens such as T. gondii, Shigella spp., and Listeria spp. by autophagy can also be tuned by TNF-α, supporting the role of proinflammatory cytokines in autophagy-dependent pathogen clearance (49, 50). On the other hand, Th2 cytokines, such as IL-10, are known to inhibit autophagy (46). IL-10 activation of its specific receptor was shown to inhibit LPS- (51) and starvation-induced autophagy in a PI3K- and AKT-dependent process (52).
Inflammation often promotes tissue damage while attempting to clear infections. Therefore, autophagy can be cytoprotective by degrading deleterious stimuli and controlling inflammation (Figure 1B). Autophagy-inflammasome interplay might represent the best example of how autophagy modulates inflammation (53, 54). Autophagy is known to negatively regulate inflammasomes and IL-1β production (54). NLRP3 and IL-1β levels were shown to be increased in ATG16L1 knockout mice challenged with bacterial toxins, suggesting a negative regulation of the NLRP3 inflammasome by autophagy (55). This probably occurs due to direct targeting of inflammasome-associated proteins for degradation. AIM2, ASC, Caspase-1, and IL-1β were shown to increase during autophagy inhibition, which was shown to be dependent on ubiquitination and SQSTM1/p62 selection (56, 57). Alternatively, autophagy can target pathogens for degradation (Figure 1B). It was shown that Sindbis and Chikungunya viral proteins (58, 59) and bacteria such as L. monocytogenes and S. typhimurium (60, 61) are selected by SQSTM1/p62 for autophagic degradation. To a similar extent, intracellular parasites such as Plasmodium spp. and T. gondii are also targeted by autophagy (62). However, unlike conventional autophagy, proteins such as LC3 may directly coat the parasitophorous vacuole instead of directing the parasite to be enwrapped by autophagosomes in a process known as LC3-associated phagocytosis (62).
Autophagy has an important role in maintaining tissue homeostasis in response to nutritional, oxidative and immunological stress. Likewise, gestation demands significant levels of placental plasticity and adaptation to ensure fetal development (63). Therefore, placental autophagy is likely to be fundamental to a healthy pregnancy, maintaining local homeostasis in response to metabolic and immunological stressors with the potential to jeopardize healthy gestational outcomes.
The role of placental autophagy during gestation is unclear. However, it is believed that placental autophagy functions as a survival mechanism while maintaining local homeostasis (64), opposing the misleading definition that classifies autophagy as a cell death mechanism (41). Embryonic development was reported to be autophagy dependent, as genetic ablation of ATG5 (65), Ambra1 (66), and Beclin1 (67) promotes embryonic lethality by impairing germinal and embryonic development. Knocking-out genes, such as Atg3, Atg9, Atg16l1 and Atg7, failed to induce embryonic lethality but promoted neonatal death after birth (28). Despite current knowledge on this topic, the mechanisms leading to poor outcomes in autophagy-deficient pregnant mice are still unclear.
Logically, one could expect autophagic abnormalities in complicated pregnancies. In uncomplicated pregnancies, placental levels of Beclin1 and LC3 should not vary significantly throughout gestation (68). However, the same is not true during pregnancy-related disorders. Preeclamptic women, for instance, who often experience hypertension, placental insufficiency, fetal growth restrictions and preterm delivery (69), frequently exhibit placental autophagy dysregulation. Increased LC3II and Beclin1 levels and diminished SQSTM1/p62 levels were found in placentas from preeclamptic women with fetal growth restrictions, suggesting overinduction of placental autophagy (70–73). Accordingly, autophagy was hypothesized to be etiologically linked to these outcomes during preeclampsia (71, 73, 74). However, pregnant mice knockout for Atg7 and Atg9a present preeclamptic-like symptoms and evidence of fetal growth restrictions, suggesting that functional autophagy is necessary for healthy gestations (75–77). The reason that placental autophagy is induced above basal levels during complicated pregnancies remains a mystery. Evidence indicates that autophagy is activated in response to hypoxia, protein aggregation, and oxidative stress or to maintain local homeostasis in response to inflammation (74, 78).
Other studies have shown an apparent link between impaired autophagy and inflammation-induced preterm labor. Women experiencing preterm delivery due to infection-induced inflammation have impaired placental autophagy, characterized by reduced levels of ATG16L1 and LC3II and increased levels of SQSTM1/p62 (79). Likewise, downregulation of placental autophagy genes such as Atg4c, Atg7, and LC3B was also reported in a murine model of inflammation-induced preterm delivery (80). Since delivery itself (either preterm or at term) relies on mechanisms of inflammation, such as the secretion of proinflammatory cytokines such as IL-1β (81, 82), its exacerbation frequently results in labor anticipation; therefore, counterregulatory mechanisms are necessary to ensure placental homeostasis. Since autophagy was shown to downregulate inflammasomes, for instance (54–57), it is plausible to hypothesize that it might participate in the regulation of placental inflammation. In fact, one study reported diminished levels of Beclin1, ATG3, ATG5, ATG12 and ATG16L1 in placental tissue after spontaneous delivery. Ex vivo stimulation of this tissue with LPS led to increased liberation of IL-1β that was augmented by autophagy inhibition and attenuated during autophagy induction, suggesting that this homeostatic process downmodulates placental inflammation in preterm deliveries (83).
Therefore, placental autophagy might have a cytoprotective role against stress induced during pregnancy (64). In addition, studies have provided evidence of a link between infection-induced inflammation during pregnancy and placental autophagy (79, 80), which might be critical for dysregulating placental homeostasis, contributing to poor outcomes during pregnancy-related diseases, such as MiP.
Low birth weight in MiP is known to have a multifactorial etiology. A combination of impaired placental perfusion, dysfunctional endocrine signaling and transplacental nutrient transportation seems to be the most significant cause (3). However, a link between placental autophagy and MiP pathogenesis was established, especially in the context of local inflammation. Placental intervillositis (monocyte infiltrate in the tissue) during MiP was shown to be associated with low birth weight and reduced IGF levels, a hormone with significant importance to fetal growth and transplacental nutrient transportation (3, 84). Accordingly, it was observed that placental inflammation characterized by the presence of cytokines such as IL-1β and intervillositis was also associated with decreased expression of amino acid and glucose transporters, and low birth weight (85, 86). It was hypothesized that reduced transplacental nutrient uptake due to decreased expression of nutrient transporters was controlled by mTOR as a consequence of diminished levels of circulating IGF and local inflammation (63, 87). Kinases rpS6, 4E-BP1, and AKT (mTOR targets) phosphorylation levels were diminished in placentas from women with MiP-associated intervillositis and constitutive mTOR activation in trophoblasts was shown to upregulate amino acid transporter activity even when incubated with conditioned culture medium from monocytes co-cultured with P. falciparum-infected erythrocytes, proving the existing link between mTOR activity, downregulation of transplacental amino acid transport and MiP-associated intervillositis (88). Although autophagy activation is directly associated with mTOR inhibition in the absence of nutrients and growth factors (29), no hypotheses were raised at that time. Afterward, evidence of placental autophagy dysregulation in P. falciparum-infected women was presented. Placental parasitemia and intervillositis were associated with increased LC3II protein levels and autophagosomes/lysosomes in the placenta (induced autophagy); however, reduced Rab7 and unaltered SQSTM1/p62 levels supported autophagy blockage with consequent impaired degradation of autophagosome content (89). Likewise, our findings suggested abnormal autophagy regulation in placentas from P. falciparum-infected women, characterized by reduced ULK1, BECN1 and MAP1LC3B mRNA levels and reduced ULK1, Beclin1 and LC3II protein levels. These differences were more prominent in placentas diagnosed with PM, which experience higher levels of local inflammation (90).
Despite evidence supporting the existence of impaired placental autophagy during MiP, its role in disease pathogenesis remains unclear. Dissecting placental autophagy’s role during MiP is extremely important to understand whether this mechanism has a pathogenic or a cytoprotective function that can be exploited to improve MiP prognosis.
One can consider that parasite virulence factors such as VAR2CSA and microvesicles might have a role on placental autophagy modulation. Pregnant women diagnosed with PM (VAR2CSA-dependent sequestration of infected erythrocytes) experience a significant modulation of autophagy when compared to women without PM (90). Additionally, infected erythrocytes selected for binding trophoblasts activate JNK1 in these cells (12), which is a known autophagy inducer (29). However, experiments with null-VAR2CSA parasites were missing so we cannot rule out the hypothesis that observed effects might still occur in a binding independent manner and simply due to parasite antigen recognition. Plasmodium-derived microvesicles, which carry parasite-derived antigens and activate the immune system, are transferred to astrocytes by LC3-associated phagocytosis, with implications to neuroinflammation and cerebral malaria pathogenesis (91). Besides, microvesicles can also trigger inflammation by activating TLR4-MyD88 axis (92), which is linked to autophagy modulation as previously discussed. Although we cannot discard the hypothesis that some of these virulence factors regulate autophagy, it seems that placental autophagy during MiP is mostly modulated by immune signals and inflammation with possible implications to disease pathogenesis (Figure 2).
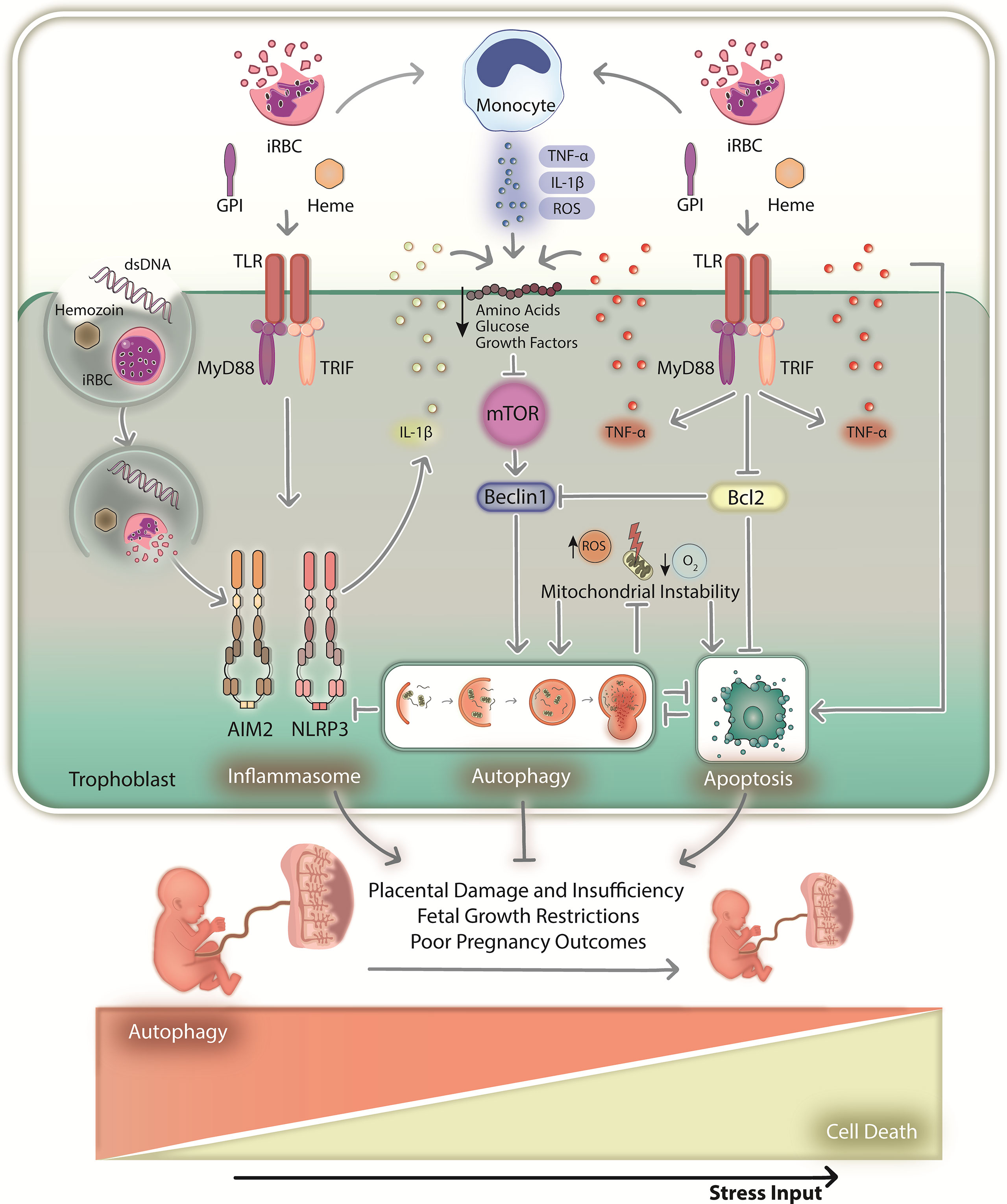
Figure 2 The hypothetical role of placental autophagy during malaria in pregnancy. During MiP, immune system activation in response to local infection results in inflammation and placental homeostasis imbalance. Monocytes and trophoblasts recognize infected red blood cells and parasite-derived antigens (dsDNA, GPI and hemozoin) via TLRs, which promote the downstream production of cytokines such as TNF-α and IL-1β (upon being processed by the inflammasome). TLR-MyD88-TRIF signaling might trigger autophagy by disrupting the Bcl2-Beclin1 interaction or through the paracrine action of the produced cytokines. In addition, inflammation leads to the downregulation of glucose/amino acid transporters, consequently activating autophagy upon mTOR inhibition. The hypoxic environment and ROS produced by monocytes might also induce autophagy by promoting oxidative stress and mitochondrial instability. Consequent activation of apoptotic machinery might also activate autophagy. As such, a multitude of stimuli might induce placental autophagy above basal levels, which might function as a cytoprotective mechanism that maintains local homeostasis and proper gestational development when stress intensity is low. In these circumstances, autophagy might circumvent lethal outcomes that may arise from activation of cell death mechanisms. However, persisting infection and sustained inflammation might increase cellular stress by activating programmed cell death mechanisms that regulate pyroptosis and apoptosis, for instance. First, autophagy might act as a survival mechanism by promoting nutrient recycling and destroying death signals such as inflammasome machinery and damaged mitochondria. However, if stress input increases above a given threshold, cell death executors such as apoptotic caspases might degrade autophagy-related proteins, consequently impairing their cytoprotective functions. Therefore, tissue homeostasis will not be maintained, resulting in the dysregulation of placental physiology, eventually leading to MiP poor pregnancy outcomes. dsDNA, parasite double-stranded DNA; iRBC, infected red blood cells; GPI, glycosylphosphatidylinositol; ROS, reactive oxygen species; TLR – Toll-like receptor.
During MiP, trophoblasts produce chemokines upon parasite recognition, recruiting monocyte-producing cytokines and ROS to clear local infection. However, excessive inflammation downregulates IGF signaling and glucose/amino acid transportation, activating autophagy upon mTOR inhibition (85, 86, 88, 89). Autophagy might also be triggered upon parasite sensing by TLRs (92), which induce autophagy through the MyD88-TRIF interaction with the Bcl2-Beclin1 complex (43). In fact, TLR-MyD88 signaling, which initiates inflammation by promoting the production of cytokines such as TNF-α, and IL-1β (by inducing its transcription and posttranslational maturation through the inflammasomes), is implicated in the outcomes of MiP, since genetic depletion of TLR4 and MyD88 improves pregnancy outcomes in an experimental murine model of MiP (93–96). In parallel, MiP-associated hypoxia and ROS (3, 27) can promote mitochondrial instability, increasing oxidative stress and cell death via apoptotic pathways. Placental apoptosis occurs in pregnant mice and women with MiP, characterized by increased trophoblast apoptosis, active Caspase-3, and reduced Bcl2 levels (97, 98). Accordingly, reduced Bcl2 levels and damaged mitochondria might also activate autophagy via a canonical pathway dependent on Beclin1. Inflammasomes, which potentiate inflammation and cell death via pyroptosis in response to parasite antigens and danger signals, can also induce autophagy. Placental NLRP3- and AIM2-dependent IL-1 axis activation is associated with MiP outcomes (99), which in principle can be downregulated by autophagy if overactivated (54). In addition, autophagy can be induced by proinflammatory cytokines such as TNF-α, which induce apoptosis via death receptors and are associated with MiP poor outcomes (14, 19, 20).
If autophagy promotes survival in response to stress, if it is essential for gestational development, and is downregulated in pregnancies complicated by infection-induced inflammation such as MiP, one can hypothesize that poor outcomes will result from placental homeostasis imbalance, which cannot be maintained by autophagy due to nonmanageable stress levels caused by infection. A cytoprotective role for placental autophagy during MiP can be hypothesized when stress levels are not lethal and cell death mediated by apoptotic mechanisms, for instance, is not triggered in response to stress (Figure 2). Autophagy can counteract cell death via apoptosis, for instance, by degrading damaged mitochondria and proapoptotic cytosolic signals; however, stress levels will eventually exceed a given threshold, and autophagy will not be able to circumvent the lethal outcome. The interplay between autophagy and apoptosis is mostly of an inhibitory nature (42). Therefore, proapoptotic caspases may eventually cleave regulatory proteins, downmodulating autophagy and possibly aborting its cytoprotective function, resulting in total dysregulation of cellular homeostasis and ultimately, cell death. This sequence of events may promote placental damage and dysregulation of local homeostasis, ultimately contributing to MiP poor outcomes.
In this context, we hypothesize that placental autophagy plays a cytoprotective role during MiP by promoting nutrient recycling and controlling inflammation and cellular damage. However, this activity cannot be properly exerted due to the nonmanageable levels of inflammation, cell death and tissue damage observed in placentas from women infected with P. falciparum, contributing to impaired gestational development and poor pregnancy outcomes.
The original contributions presented in the study are included in the article. Further inquiries can be directed to the corresponding author.
AB drafted the manuscript, collected information from the literature and drafted the figures. AJ drafted the manuscript. CM and SE supervised and reviewed the manuscript. All authors contributed to the article and approved the submitted version.
This manuscript was primarily funded by the São Paulo Research Foundation – FAPESP (2018/20468-0 and 2020/06747-4 to CM, 2020/03163-1 to SE) and National Council for Scientific and Technological Development – CNPq (408636/2018-1 and 302917/2019-5 to CM, 304033/2021-9 to SE). AB and AJ were supported by FAPESP fellowships (2017/03939-7 and 2019/12078-0, respectively).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We acknowledge Dra. Ana Carolina Kayano from BioPic3D Biomedical Art Studio (https://biopic3d.myportfolio.com) for creating the scientific illustrations presented in this manuscript.
1. World Health Organization. World Malaria Report 2021 Geneva: World Health Organization (2021). 322 p. Licence: CC BY-NC-SA 3.0 IGO.
2. World Health Organization. World Malaria Report 2019. Geneva: World Health Organization (2019). 232 p. Licence: CC BY-NC-SA 3.0 IGO.
3. Umbers AJ, Aitken EH, Rogerson SJ. Malaria in Pregnancy: Small Babies, Big Problem. Trends Parasitol (2011) 27:168–75. doi: 10.1016/j.pt.2011.01.007
4. Rogerson SJ, Desai M, Mayor A, Sicuri E, Taylor SM, van Eijk AM. Burden, Pathology, and Costs of Malaria in Pregnancy: New Developments for an Old Problem. Lancet Infect Diseases (2018) 18(4):e107–18. doi: 10.1016/S1473-3099(18)30066-5
5. Nosten F, Rogerson SJ, Beeson JG, McGready R, Mutabingwa TK, Brabin B. Malaria in Pregnancy and the Endemicity Spectrum: What can We Learn? Trends Parasitol (2004) 20(9):425–32. doi: 10.1016/j.pt.2004.06.007
6. Salanti A, Staalsoe T, Lavstsen T, Jensen TR, Sowa MPK, Arnot DE, et al. Selective Upregulation of a Single Distinctly Structured Var Gene in Chondroitin Sulphate A-Adhering Plasmodium Falciparum Involved in Pregnancy-Associated Malaria. Mol Microbiol (2003) 49(1):179–91. doi: 10.1046/j.1365-2958.2003.03570.x
7. Salanti A, Dahlbäck M, Turner L, Nielsen MA, Barfod L, Magistrado P, et al. Evidence for the Involvement of VAR2CSA in Pregnancy-Associated Malaria. J Exp Med (2004) 200(9):1197–203. doi: 10.1084/jem.20041579
8. Rogerson SJ, Chaiyaroj SC, Ng K, Reeder JC, Brown GV. Chondroitin Sulfate A is a Cell Surface Receptor for Plasmodium Falciparum-Infected Erythrocytes. J Exp Med (1995) 182(1):15–20. doi: 10.1084/jem.182.1.15
9. Fried M, Duffy PE. Adherence of Plasmodium Falciparum to Chondroitin Sulfate A in the Human Placenta. Science (1996) 272(5267):1502–4. doi: 10.1126/science.272.5267.1502
10. Beeson JG, Amin N, Kanjala M, Rogerson SJ. Selective Accumulation of Mature Asexual Stages of Plasmodium Falciparum -Infected Erythrocytes in the Placenta. Infect Immunity (2002) 70(10):5412–5. doi: 10.1128/IAI.70.10.5412-5415.2002
11. Abrams ET, Brown H, Chensue SW, Turner GDH, Molyneux ME, Meshnick SR, et al. Host Response to Malaria During Pregnancy: Placental Monocyte Recruitment Is Associated With Elevated β Chemokine Expression. J Immunol (2003) 170:2759–64. doi: 10.4049/jimmunol.170.5.2759
12. Lucchi NW, Peterson DS, Moore JM. Immunologic Activation of Human Syncytiotrophoblast by Plasmodium Falciparum. Malaria J (2008) 7:42. doi: 10.1186/1475-2875-7-42
13. Fried M, Muga RO, Misore AO, Duffy PE. Malaria Elicits Type 1 Cytokines in the Human Placenta: IFN-Gamma and TNF-Alpha Associated With Pregnancy Outcomes. J Immunol (1998) 160(5):2523–30. http://www.jimmunol.org/content/160/5/2523
14. Moormann AM, Sullivan AD, Rochford RA, Chensue SW, Bock PJ, Nyirenda T, et al. Malaria and Pregnancy: Placental Cytokine Expression and its Relationship to Intrauterine Growth Retardation. J Infect Diseases (1999) 180(6):1987–93. doi: 10.1086/315135
15. Fievet N, Moussa M, Tami G, Maubert B, Cot M, Deloron P, et al. Plasmodium Falciparum Induces a Th1/Th2 Disequilibrium, Favoring the Th1-Type Pathway, in the Human Placenta. J Infect Diseases (2001) 183(10):1530–4. doi: 10.1086/320201
16. Schofield L, Grau GE. Immunological Processes in Malaria Pathogenesis. Nat Rev: Immunol (2005) 5(9):722–35. doi: 10.1038/nri1686
17. Rogerson SJ, Mwapasa V, Meshnick SR. Malaria in Pregnancy: Linking Immunity and Pathogenesis to Prevention. Am J Trop Med Hyg (2007) 77(SUPPL. 6):14–22. doi: 10.4269/ajtmh.77.6.suppl.14
18. Rogerson SJ, Pollina E, Getachew A, Tadesse E, Lema VM, Molyneux ME. Placental Monocyte Infiltrates in Response to Plasmodium Falciparum Malaria Infection and Their Association With Adverse Pregnancy Outcomes. Am J Trop Med Hyg (2003) 68(1):115–9. doi: 10.4269/ajtmh.2003.68.1.0680115
19. Rogerson SJ, Brown HC, Pollina E, Abrams ET, Tadesse E, Lema VM, et al. Placental Tumor Necrosis Factor Alpha But Not Gamma Interferon is Associated With Placental Malaria and Low Birth Weight in Malawian Women. Infect Immunity (2003) 71(1):267–70. doi: 10.1128/IAI.71.1.267-270.2003
20. Fried M, Kurtis JD, Swihart B, Pond-Tor S, Barry A, Sidibe Y, et al. Systemic Inflammatory Response to Malaria During Pregnancy Is Associated With Pregnancy Loss and Preterm Delivery. Clin Infect Diseases (2017) 65(10):1729–35. doi: 10.1093/cid/cix623
21. Kanellopoulos-Langevin C, Caucheteux SM, Verbeke P, Ojcius DM. Tolerance of the Fetus by the Maternal Immune System: Role of Inflammatory Mediators at the Feto-Maternal Interface. Reprod Biol Endocrinol (2003) 1:121. doi: 10.1186/1477-7827-1-121
22. Lager S, Powell TL. Regulation of Nutrient Transport Across the Placenta. J Pregnancy (2012) 2012:1–14. doi: 10.1155/2012/179827
23. Robbins JR, Bakardjiev AI. Pathogens and Placental Fortress. Curr Opin Microbiol (2012) 15(1):36–43. doi: 10.1016/j.mib.2011.11.006
24. Walter P, Garin Y, Blot P. Placental Pathologic Changes in Malaria: A Histologic and Ultrastructural Study. Am J Pathol (1982) 109(3):330–42.
25. Ismail MR, Ordi J, Menendez C, Ventura PJ, Aponte JJ, Kahigwa E, et al. Placental Pathology in Malaria: A Histological, Immunohistochemical, and Quantitative Study. Hum Pathol (2000) 31(1):85–93. doi: 10.1016/S0046-8177(00)80203-8
26. Souza RM, Ataíde R, Dombrowski JG, Ippólito V, Aitken EH, Valle SN, et al. Placental Histopathological Changes Associated With Plasmodium Vivax Infection During Pregnancy. PloS Negl Trop Diseases (2013) 7(2):1–11. doi: 10.1371/journal.pntd.0002071
27. Sharma L, Shukla G. Placental Malaria: A New Insight Into the Pathophysiology. Front Med (2017) 4:1–6. doi: 10.3389/fmed.2017.00117
28. Mizushima N, Levine B. Autophagy in Mammalian Development and Differentiation. Nat Cell Biol (2010) 12(9):823–30. doi: 10.1038/ncb0910-823
29. Kroemer G, Mariño G, Levine B. Autophagy and the Integrated Stress Response. Mol Cell (2010) 40(2):280–93. doi: 10.1016/j.molcel.2010.09.023
30. Levine B, Mizushima N, Virgin HW. Autophagy in Immunity and Inflammation. Nature (2011) 469(7330):323–35. doi: 10.1038/nature09782
31. Deretic V, Saitoh T, Akira S. Autophagy in Infection, Inflammation and Immunity. Nat Rev Immunol (2013) 13(10):722–37. doi: 10.1038/nri3532
32. Parzych KR, Klionsky DJ. An Overview of Autophagy: Morphology, Mechanism, and Regulation. Antioxid Redox Signaling (2014) 20(3):460–73. doi: 10.1089/ars.2013.5371
33. Sengupta S, Peterson TR, Sabatini DM. Regulation of the mTOR Complex 1 Pathway by Nutrients, Growth Factors, and Stress. Mol Cell (2010) 40(2):310–22. doi: 10.1016/j.molcel.2010.09.026
34. He C, Levine B. The Beclin 1 Interactome. Curr Opin Cell Biol (2010) 22(2):140–9. doi: 10.1016/j.ceb.2010.01.001
35. Mizushima N, Yoshimori T, Ohsumi Y. The Role of Atg Proteins in Autophagosome Formation. Annu Rev Cell Dev Biol (2011) 27(1):107–32. doi: 10.1146/annurev-cellbio-092910-154005
36. Sánchez-Martın P, Komatsu M. P62/SQSTM1 – Steering the Cell Through Health and Disease. J Cell Sci (2018) 131(21):1–13. doi: 10.1242/jcs.222836
37. Daskalaki I, Gkikas I, Tavernarakis N. Hypoxia and Selective Autophagy in Cancer Development and Therapy. Front Cell Dev Biol (2018) 6. doi: 10.3389/fcell.2018.00104
38. Palikaras K, Lionaki E, Tavernarakis N. Mechanisms of Mitophagy in Cellular Homeostasis, Physiology and Pathology. Nat Cell Biol (2018) 20(9):1013–22. doi: 10.1038/s41556-018-0176-2
39. Green DR, Llambi F. Cell Death Signaling. Cold Spring Harbor Perspect Biol (2015) 7(12):a006080. doi: 10.1101/cshperspect.a006080
40. Llambi F, Moldoveanu T, Tait SWG, Bouchier-Hayes L, Temirov J, McCormick LL, et al. A Unified Model of Mammalian BCL2 Protein Family Interactions at the Mitochondria. Mol Cell (2011) 44(4):517–31. doi: 10.1016/j.molcel.2011.10.001
41. Kroemer G, Levine B. Autophagic Cell Death: The Story of a Misnomer. Nat Rev Mol Cell Biol (2008) 9(12):1004–10. doi: 10.1038/nrm2529
42. Mariño G, Niso-Santano M, Baehrecke EH, Kroemer G. Self-Consumption: The Interplay of Autophagy and Apoptosis. Nat Rev Mol Cell Biol (2014) 15(2):81–94. doi: 10.1038/nrm3735
43. Shi CS, Kehrl JH. MyD88 and Trif Target Beclin 1 to Trigger Autophagy in Macrophages. J Biol Chem (2008) 283(48):33175–82. doi: 10.1074/jbc.M804478200
44. Travassos LH, Carneiro LAM, Ramjeet M, Hussey S, Kim YG, Magalhes JG, et al. Nod1 and Nod2 Direct Autophagy by Recruiting ATG16L1 to the Plasma Membrane at the Site of Bacterial Entry. Nat Immunol (2010) 11(1):55–62. doi: 10.1038/ni.1823
45. Sumpter R, Levine B. Autophagy and Innate Immunity: Triggering, Targeting and Tuning. Semin Cell Dev Biol (2010) 21(7):699–711. doi: 10.1016/j.semcdb.2010.04.003
47. Jia G, Cheng G, Gangahar DM, Agrawal DK. Insulin-Like Growth Factor-1 and TNF-α Regulate Autophagy Through C-Jun N-Terminal Kinase and Akt Pathways in Human Atherosclerotic Vascular Smooth Cells. Immunol Cell Biol (2006) 84(5):448–54. doi: 10.1111/j.1440-1711.2006.01454.x
48. Keller CW, Fokken C, Turville SG, Lünemann A, Schmidt J, Münz C, et al. TNF-α Induces Macroautophagy and Regulates MHC Class II Expression in Human Skeletal Muscle Cells. J Biol Chem (2011) 286(5):3970–80. doi: 10.1074/jbc.M110.159392
49. Andrade RM, Wessendarp M, Gubbels MJ, Striepen B, Subauste CS. CD40 Induces Macrophage Anti-Toxoplasma Gondii Activity by Triggering Autophagy-Dependent Fusion of Pathogen-Containing Vacuoles and Lysosomes. J Clin Invest (2006) 116(9):2366–77. doi: 10.1172/JCI28796
50. Mostowy S, Sancho-Shimizu V, Hamon MA, Simeone R, Brosch R, Johansen T, et al. P62 and NDP52 Proteins Target Intracytosolic Shigella and Listeria to Different Autophagy Pathways. J Biol Chem (2011) 286(30):26987–95. doi: 10.1074/jbc.M111.223610
51. Ní Cheallaigh C, Keane J, Lavelle EC, Hope JC, Harris J. Autophagy in the Immune Response to Tuberculosis: Clinical Perspectives. Clin Exp Immunol (2011) 164(3):291–300. doi: 10.1111/j.1365-2249.2011.04381.x
52. Park HJ, Lee SJ, Kim SH, Han J, Bae J, Kim SJ, et al. IL-10 Inhibits the Starvation Induced Autophagy in Macrophages via Class I Phosphatidylinositol 3-Kinase (PI3K) Pathway. Mol Immunol (2011) 48(4):720–7. doi: 10.1016/j.molimm.2010.10.020
53. Deretic V, Levine B. Autophagy Balances Inflammation in Innate Immunity. Autophagy (2018) 14(2):243–51. doi: 10.1080/15548627.2017.1402992
54. Harris J, Lang T, Thomas JPW, Sukkar MB, Nabar NR, Kehrl JH. Autophagy and Inflammasomes. Mol Immunol (2017) 86:10–5. doi: 10.1016/j.molimm.2017.02.013
55. Saitoh T, Fujita N, Jang MH, Uematsu S, Yang BG, Satoh T, et al. Loss of the Autophagy Protein Atg16L1 Enhances Endotoxin-Induced IL-1β Production. Nature (2008) 456(7219):264–8. doi: 10.1038/nature07383
56. Shi CS, Shenderov K, Huang NN, Kabat J, Abu-Asab M, Fitzgerald KA, et al. Activation of Autophagy by Inflammatory Signals Limits IL-1β Production by Targeting Ubiquitinated Inflammasomes for Destruction. Nat Immunol (2012) 13(3):255–63. doi: 10.1038/ni.2215
57. Harris J, Hartman M, Roche C, Zeng SG, O’Shea A, Sharp FA, et al. Autophagy Controls IL-1β Secretion by Targeting Pro-IL-1β for Degradation. J Biol Chem (2011) 286(11):9587–97. doi: 10.1074/jbc.M110.202911
58. Orvedahl A, MacPherson S, Sumpter R, Tallóczy Z, Zou Z, Levine B. Autophagy Protects Against Sindbis Virus Infection of the Central Nervous System. Cell Host Microbe (2010) 7(2):115–27. doi: 10.1016/j.chom.2010.01.007
59. Judith D, Mostowy S, Bourai M, Gangneux N, Lelek M, Lucas-Hourani M, et al. Species-Specific Impact of the Autophagy Machinery on Chikungunya Virus Infection. EMBO Rep (2013) 14(6):534–44. doi: 10.1038/embor.2013.51
60. Yoshikawa Y, Ogawa M, Hain T, Yoshida M, Fukumatsu M, Kim M, et al. Listeria Monocytogenes ActA-Mediated Escape From Autophagic Recognition. Nat Cell Biol (2009) 11(10):1233–40. doi: 10.1038/ncb1967
61. Zheng YT, Shahnazari S, Brech A, Lamark T, Johansen T, Brumell JH. The Adaptor Protein P62/SQSTM1 Targets Invading Bacteria to the Autophagy Pathway. J Immunol (2009) 183(9):5909–16. doi: 10.4049/jimmunol.0900441
62. Coppens I. How Toxoplasma and Malaria Parasites Defy First, Then Exploit Host Autophagic and Endocytic Pathways for Growth. Curr Opin Microbiol (2017) 40:32–9. doi: 10.1016/j.mib.2017.10.009
63. Dimasuay KG, Boeuf P, Powell TL, Jansson T. Placental Responses to Changes in the Maternal Environment Determine Fetal Growth. Front Physiol (2016) 7:1–9. doi: 10.3389/fphys.2016.00012
64. Bildirici I, Longtine MS, Chen B, Nelson DM. Survival by Self-Destruction: A Role for Autophagy in the Placenta? Placenta (2012) 33(8):591–8. doi: 10.1016/j.placenta.2012.04.011
65. Tsukamoto S, Kuma A, Murakami M, Kishi C, Yamamoto A, Mizushima N. Autophagy Is Essential for Preimplantation Development of Mouse Embryos. Science (2008) 321(5885):117–20. doi: 10.1126/science.1154822
66. Maria Fimia G, Stoykova A, Romagnoli A, Giunta L, di Bartolomeo S, Nardacci R, et al. Ambra1 Regulates Autophagy and Development of the Nervous System. Nature (2007) 447(7148):1121–5. doi: 10.1038/nature05925
67. Yue Z, Jin S, Yang C, Levine AJ, Heintz N. Beclin 1, an Autophagy Gene Essential for Early Embryonic Development, is a Haploinsufficient Tumor Suppressor. Proc Natl Acad Sci (2003) 100(25):15077–82. doi: 10.1073/pnas.2436255100
68. Hung T, Hsieh T, Chen S, Li Mj, Yeh Yl. Autophagy in the Human Placenta Throughout Gestation. PloS One (2013) 8(12):1–11. doi: 10.1371/journal.pone.0083475
69. Phipps EA, Thadhani R, Benzing T, Karumanchi SA. Pre-Eclampsia: Pathogenesis, Novel Diagnostics and Therapies. Nat Rev Nephrol (2019) 15(5):275–89. doi: 10.1038/s41581-019-0119-6
70. Oh SY, Choi SJ, Kim KH, Cho EY, Kim JH, Roh CR. Autophagy-Related Proteins, LC3 and Beclin-1, in Placentas From Pregnancies Complicated by Preeclampsia. Reprod Sci (2008) 15(9):912–20. doi: 10.1177/1933719108319159
71. Hung T, Chen S, Lo Lm, Li Mj, Yeh Yl, Hsieh T. Increased Autophagy in Placentas of Intrauterine Growth-Restricted Pregnancies. PloS One (2012) 7(7):e40957. doi: 10.1371/journal.pone.0040957
72. Akaishi R, Yamada T, Nakabayashi K, Nishihara H, Furuta I, Kojima T, et al. Autophagy in the Placenta of Women With Hypertensive Disorders in Pregnancy. Placenta (2014) 35(12):974–80. doi: 10.1016/j.placenta.2014.10.009
73. Gao L, Qi HB, Kc K, Zhang XM, Zhang H, Baker PN. Excessive Autophagy Induces the Failure of Trophoblast Invasion and Vasculature: Possible Relevance to the Pathogenesis of Preeclampsia. J Hypertension (2015) 33(1):106–17. doi: 10.1097/HJH.0000000000000366
74. Oh Sy, Roh Cr. Autophagy in the Placenta. Obstet Gynecol Sci (2017) 60(3):241. doi: 10.5468/ogs.2017.60.3.241
75. Muralimanoharan S, Gao X, Weintraub S, Myatt L, Maloyan A. Sexual Dimorphism in Activation of Placental Autophagy in Obese Women With Evidence for Fetal Programming From a Placenta-Specific Mouse Model. Autophagy (2016) 12(5):752–69. doi: 10.1080/15548627.2016.1156822
76. Aoki A, Nakashima A, Kusabiraki T, Ono Y, Yoshino O, Muto M, et al. Trophoblast-Specific Conditional Atg7 Knockout Mice Develop Gestational Hypertension. Am J Pathol (2018) 188(11):2474–86. doi: 10.1016/j.ajpath.2018.07.021
77. Kojima T, Yamada T, Akaishi R, Furuta I, Saitoh T, Nakabayashi K, et al. Role of the Atg9a Gene in Intrauterine Growth and Survival of Fetal Mice. Reprod Biol (2015) 15(3):131–8. doi: 10.1016/j.repbio.2015.05.001
78. Nakashima A, Tsuda S, Kusabiraki T, Aoki A, Ushijima A, Shima T, et al. Current Understanding of Autophagy in Pregnancy. Int J Mol Sci (2019) 20(9):1–13. doi: 10.3390/ijms20092342
79. Cao B, Macones C, Mysorekar IU. ATG16L1 Governs Placental Infection Risk and Preterm Birth in Mice and Women. JCI Insight (2016) 1(21):1–10. doi: 10.1172/jci.insight.86654
80. Agrawal V, Jaiswal MK, Mallers T, Katara GK, Gilman-Sachs A, Beaman KD, et al. Altered Autophagic Flux Enhances Inflammatory Responses During Inflammation-Induced Preterm Labor. Sci Rep (2015) 5:1–11. doi: 10.1038/srep09410
81. Bowen JM, Chamley L, Keelan JA, Mitchell MD. Cytokines of the Placenta and Extra-Placental Membranes: Roles and Regulation During Human Pregnancy and Parturition. Placenta (2002) 23(4):257–73. doi: 10.1053/plac.2001.0782
82. Elliott CL, Loudon JAZ, Brown N, Slater DM, Bennett PR, Sullivan MHF. IL-1β and IL-8 in Human Fetal Membranes: Changes With Gestational Age, Labor, and Culture Conditions. Am J Reprod Immunol (2001) 46(4):260–7. doi: 10.1034/j.1600-0897.2001.d01-11.x
83. Brickle A, Tran HT, Lim R, Liong S, Lappas M. Autophagy, Which is Decreased in Labouring Fetal Membranes, Regulates IL-1β Production via the Inflammasome. Placenta (2015) 36(12):1393–404. doi: 10.1016/j.placenta.2015.10.015
84. Umbers AJ, Boeuf P, Clapham C, Stanisic DI, Baiwog F, Mueller I, et al. Placental Malaria-Associated Inflammation Disturbs the Insulin-Like Growth Factor Axis of Fetal Growth Regulation. J Infect Diseases (2011) 203(4):561–9. doi: 10.1093/infdis/jiq080
85. Boeuf P, Aitken EH, Chandrasiri U, Chua CLL, McInerney B, McQuade L, et al. Plasmodium Falciparum Malaria Elicits Inflammatory Responses That Dysregulate Placental Amino Acid Transport. PloS Pathogens (2013) 9(2):1–12. doi: 10.1371/journal.ppat.1003153
86. Chandrasiri UP, Chua CLL, Umbers AJ, Chaluluka E, Glazier JD, Rogerson SJ, et al. Insight Into the Pathogenesis of Fetal Growth Restriction in Placental Malaria: Decreased Placental Glucose Transporter Isoform 1 Expression. J Infect Diseases (2014) 209(10):1663–7. doi: 10.1093/infdis/jit803
87. Kidima WB. Syncytiotrophoblast Functions and Fetal Growth Restriction During Placental Malaria: Updates and Implication for Future Interventions. BioMed Res Int (2015) 2015:1–9. doi: 10.1155/2015/451735
88. Dimasuay KG, Aitken EH, Rosario F, Njie M, Glazier J, Rogerson SJ, et al. Inhibition of Placental mTOR Signaling Provides a Link Between Placental Malaria and Reduced Birthweight. BMC Med (2017) 15(1):1–11. doi: 10.1186/s12916-016-0759-3
89. Dimasuay KG, Gong L, Rosario F, McBryde E, Spelman T, Glazier J, et al. Impaired Placental Autophagy in Placental Malaria. PloS One (2017) 12(11):1–20. doi: 10.1371/journal.pone.0187291
90. Lima FA, Barateiro A, Dombrowski JG, de Souza RM, de Sousa Costa D, Murillo O, et al. Plasmodium Falciparum Infection Dysregulates Placental Autophagy. PloS One (2019) 14(12):e0226117. doi: 10.1371/journal.pone.0226117
91. Leleu I, Alloo J, Cazenave PA, Roland J, Pied S. Autophagy Pathways in the Genesis of Plasmodium-Derived Microvesicles: A Double-Edged Sword? Life (2022) 12(3):415. doi: 10.3390/life12030415
92. Gazzinelli RT, Kalantari P, Fitzgerald KA, Golenbock DT. Innate Sensing of Malaria Parasites. Nat Rev: Immunol (2014) 14:1–14. doi: 10.1038/nri3742
93. Barboza R, Reis AS, Silva LG da, Hasenkamp L, Pereira KRB, Câmara NOS, et al. MyD88 Signaling is Directly Involved in the Development of Murine Placental Malaria. Infect Immunity (2014) 82(2):830–8. doi: 10.1128/IAI.01288-13
94. Barboza R, Lima FA, Reis AS, Murillo OJ, Peixoto EPM, Bandeira CL, et al. TLR4-Mediated Placental Pathology and Pregnancy Outcome in Experimental Malaria. Sci Rep (2017) 7(1):1–12. doi: 10.1038/s41598-017-08299-x
95. Barboza R, Hasenkamp L, Barateiro A, Murillo O, Peixoto EPM, Lima FA, et al. Fetal-Derived MyD88 Signaling Contributes to Poor Pregnancy Outcomes During Gestational Malaria. Front Microbiol (2019) 10. doi: 10.3389/fmicb.2019.00068
96. Pandya Y, Marta A, Barateiro A, Bandeira CL, Dombrowski JG, Costa J, et al. TLR4-Endothelin Axis Controls Syncytiotrophoblast Motility and Confers Fetal Protection in Placental Malaria. Infect Immun (2021) 89(8):e00809–20. doi: 10.1128/IAI.00809-20
97. Kawahara R, Rosa-Fernandes L, dos Santos AF, Bandeira CL, Dombrowski JG, Souza RM, et al. Integrated Proteomics Reveals Apoptosis-Related Mechanisms Associated With Placental Malaria. Mol Cell Proteomics (2019) 18(2):182–99. doi: 10.1074/mcp.RA118.000907
98. Sharma L, Kaur J, Shukla G. Role of Oxidative Stress and Apoptosis in the Placental Pathology of Plasmodium Berghei Infected Mice. PloS One (2012) 7(3):e32694. doi: 10.1371/journal.pone.0032694
Keywords: Malaria, Plasmodium, Pregnancy, Placenta, Inflammation, Autophagy
Citation: Barateiro A, Junior ARC, Epiphanio S and Marinho CRF (2022) Homeostasis Maintenance in Plasmodium-Infected Placentas: Is There a Role for Placental Autophagy During Malaria in Pregnancy? Front. Immunol. 13:931034. doi: 10.3389/fimmu.2022.931034
Received: 28 April 2022; Accepted: 20 June 2022;
Published: 11 July 2022.
Edited by:
Letusa Albrecht, Carlos Chagas Institute (FIOCRUZ), BrazilReviewed by:
Maria Bernabeu, European Molecular Biology Laboratory, SpainCopyright © 2022 Barateiro, Junior, Epiphanio and Marinho. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Claudio Romero Farias Marinho, bWFyaW5ob0B1c3AuYnI=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.