Abstract
NF-κB-inducing kinase (NIK) is a key player in non-canonical NF-κB signaling, involved in several fundamental cellular processes, and is crucial for B cell function and development. In response to certain signals and ligands, such as CD40, BAFF and lymphotoxin-β activation, NIK protein stabilization and subsequent NF-κB activation is achieved. Overexpression or overactivation of NIK is associated with several malignancies, including activating mutations in multiple myeloma (MM) and gain-of-function in MALT lymphoma as a result of post-translational modifications. Consequently, drug discovery studies are devoted to pharmacologic modulation of NIK and development of specific novel small molecule inhibitors. However, disease-specific in vitro and in vivo studies investigating NIK inhibition are as of yet lacking, and clinical trials with NIK inhibitors remain to be initiated. In order to bridge the gap between bench and bedside, this review first briefly summarizes our current knowledge on NIK activation, functional activity and stability. Secondly, we compare current inhibitors targeting NIK based on efficacy and specificity, and provide a future perspective on the therapeutic potential of NIK inhibition in B cell malignancies.
Introduction
Nuclear factor kappa B (NF-κB) comprises a family of transcription factors that regulate a wide array of genes involved in inflammation, immunity, differentiation, proliferation and cell survival. As NF-κB is ubiquitously expressed, dysregulation of NF-κB is an important contributor to both the development of various cancers and their progression (1). NF-κB signaling can be distinguished into two distinct pathways, the canonical and non-canonical pathway. NF-κB inducing kinase (NIK), which is encoded by the gene MAP3K14, has been identified as the central kinase controlling non-canonical NF-κB activation (Figure 1). The non-canonical NF-κB pathway can be activated upon stimulation of a subset of upstream activators, including activation of the BAFF receptor (BAFF-R), CD40, receptor activator of NF-κB (RANK) or the lymphotoxin beta receptor (LT-βR) (2, 3). Stimulation of these receptors will recruit TRAF2/3 to the receptor, resulting in the accumulation of NIK protein levels. NIK is able to directly phosphorylate the precursor p100 at Ser866/870 and induce phosphorylation of IKKα homodimers at Ser176/180 (4–6). In turn, IKKα complexes will phosphorylate p100 at Ser872, which together with Ser866/870 phosphorylation by NIK is required to result in the proteasomal processing of p100 into p52 (7, 8). Whereas p100/RelB dimers are sequestered in the cytoplasm, p52/RelB dimers are free to translocate to the nucleus in order to activate target genes. Because of this role of NIK in non-canonical NF-κB signaling, NIK is crucial in regulating immunity and inflammation, where loss of NIK is associated with severe immune defects whereas NIK overexpression is observed in inflammatory diseases and malignancies (9). For this reason, targeting of NIK and the non-canonical NF-κB pathway may have therapeutic potential in various diseases.
Figure 1
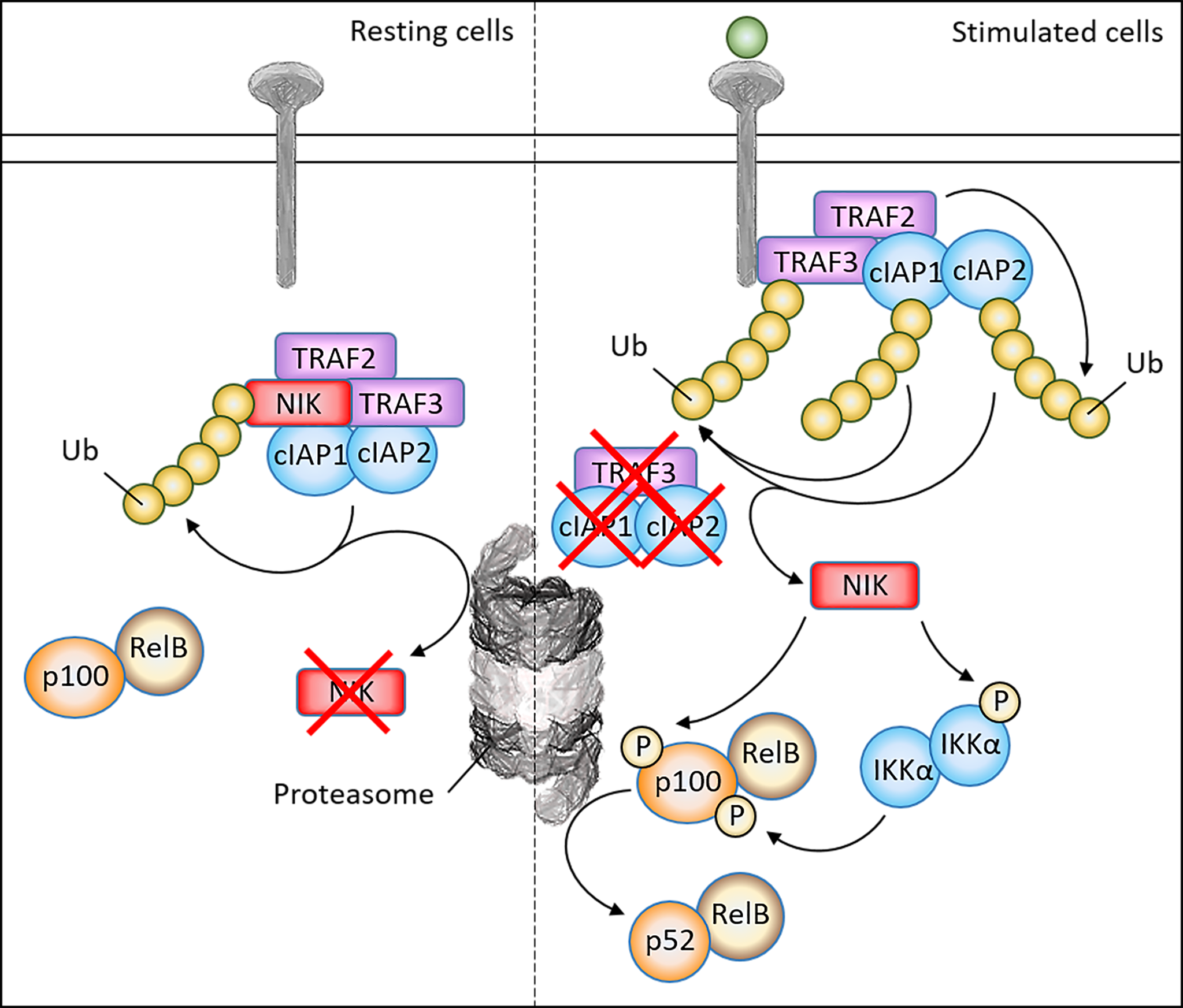
Schematic overview of non-canonical signaling pathway and NIK regulatory mechanisms. In resting cells, NIK forms a complex with TRAF2/3 and cIAP1/2, which leads to the continuous ubiquitination and proteasomal degradation of NIK. Upon ligand binding, TRAF3 is recruited to the receptor, where TRAF2 induces ubiquitination of cIAP1/2 which in turn induces ubiquitination of TRAF3. As a result, TRAF3 is degraded and NIK is stabilized. After NIK stabilization, NIK is able to phosphorylate the IKKα complex to induce activation. After direct phosphorylation of the precursor p100 by both NIK and the IKKα complex, p100 will undergo proteasomal processing into the active subunit p52, which together with RelB may translocate to the nucleus to activate target genes.
NIK Activity
NIK is a kinase that does not require phosphorylation in order to become activated (10). It was previously thought that phosphorylation of Thr559 would result in NIK activation (11). However, subsequent studies could not identify phosphorylation of Thr559 and in addition showed that mutation of this site results in similar kinase activity (12). Structural studies revealed a constitutively active conformation of NIK (13). This suggests that NIK is predominantly regulated at the protein level by inducing its degradation as previously described. Several proteins have been suggested to regulate the activity of NIK rather than its stability, such as TRAFs and NIK-associated protein encoded by the TNAP gene, and Phosphatidylethanolamine binding protein 1 encoded by the PEBP1 gene, though their mechanism of NIK inactivation has not been well elucidated (14, 15).
NIK Regulation
The basal level of NIK protein is very low in resting cells yet NIK is stabilized after stimulation of upstream receptors such as CD40 or BAFF-R after which the half-life of NIK increases to about 3 hours (16). The accumulation of NIK protein levels is the key event which permits the activation of the alternative NF-κB pathway. As a consequence, activation of non-canonical NF-κB dimers takes more time compared to canonical NF-κB dimers, due to the requirement for the accumulation of NIK by de novo protein synthesis (17).
There has been a lot of investigation into factors that regulate NIK degradation and stability, which is important in order to design potential therapies targeting this key kinase. Arguably the most important proteins involved in the regulation of NIK are the TNF receptor associated factor (TRAF) proteins and cellular inhibitor of apoptosis (cIAP) proteins. In resting cells, NIK forms a complex with TRAF2/3 and cIAP1/2, which leads to the continuous ubiquitination and proteasomal degradation of NIK. Upon ligand binding, TRAF3 is recruited to the respective TNFR, where TRAF2 induces ubiquitination of cIAP1/2 which in turn induces ubiquitination of TRAF3. As a result, TRAF3 is degraded and NIK is stabilized (18).
In addition to constitutive regulation by the TRAF-cIAP complex, it has been demonstrated that NIK is also subject to negative feedback regulation (19). IKKα phosphorylates NIK at Ser809/812/815 to induce NIK proteasomal degradation thereby limiting its own activation. Phosphorylation of NIK by TBK1 at Ser862 has also been identified to destabilize and induce degradation of NIK, in a TRAF3-independent manner (20, 21). Additionally, expression of NEMO was found to suppress NIK protein levels, suggesting a mechanism for the canonical NF-κB signaling pathway to limit activity of non-canonical NF-κB activation (22).
In addition to these well-described mechanisms, there are several additional factors that are thought to be involved in the regulation of NIK. One such example is Hsp90, which has been proposed to control NIK protein folding and stabilization (23). Inhibition of Hsp90 disrupts its interaction with NIK, resulting in NIK degradation independent of ubiquitination and which is mediated by autophagy. Moreover, the protein CHIP, encoded by the gene STUB1, is shown to be able to directly bind NIK and thereby induce NIK degradation and inhibit non-canonical NF-κB activity (24). Although CHIP contains ubiquitin E3 ligase activity, this is not actually required for the ubiquitination of NIK. This suggests that many more NIK-interacting proteins could serve as adaptor proteins coordinating the regulation of NIK, possibly by functioning in concordance with the TRAF/cIAP machinery. However, a few proteins have also been reported to directly induce ubiquitination of NIK independent of TRAF/cIAP, like the E3 ligase CRL4 (25).
NIK is encoded by the MAP3K14 gene which requires active transcription before NIK can be regulated at the protein level. Importantly, NIK seems to positively regulate its own transcription (26), creating a positive feedback loop when TRAF3 is degraded. At the same time, NIK may shape a transcriptional negative feedback loop, as cells with constitutively active NIK mutants show increased TRAF2 and TRAF3 mRNA expression (27). Although not much is known about the transcriptional regulation of MAP3K14, an increasing number of observations point towards a complex transcriptional regulation. It was shown that MAP3K14 mRNA may be targeted by nonsense-mediated decay to prevent NIK overactivity which is disrupted in myofibroblastic tumors (28). Additionally, there is evidence of regulation of MAP3K14 via various microRNAs (29–33).
Role of NIK in B Cell Malignancies
A tumor can establish elevated NF-κB activity by either extrinsic or intrinsic factors. BAFF-R, CD40, RANK and LT-βR signaling play a role in several B cell malignancies associated with increased non-canonical NF-κB activation (34–40). Whereas in some B cell malignancies this is established by the microenvironment, others may acquire mutations that promote non-canonical NF-κB signaling. For example, up to 20% of MM cases display constitutive non-canonical NF-κB signaling as a result from genetic aberrations of NF-κB regulatory genes. Bi-allelic deletions observed in MM include TRAF2, TRAF3, BIRC2 (encoding cIAP1) and BIRC3 (encoding cIAP2), which lead to their inactivation and subsequent activation of NIK (41). Gene expression profiling cohorts also indicate overexpression of MAP3K14 itself, indicating increased transcription of NIK. TRAF3 deletions and MAP3K14 gains are also frequently observed in Hodgkin lymphoma (42). Additionally, MAP3K14 translocation to the IGH or IGL locus has been found in MM patients and leads to a significant increase of NIK expression. One specific MAP3K14 translocation to the EFTUD2 gene results in an EFTUD2-NIK fusion protein which results in a stable form of NIK that lacks the binding region to TRAF3 (43). Other overexpressed genes include LTBR, CD40 and TNFRSF13B, which all encode for upstream activators of non-canonical signaling and thus prime cells for NIK activation, respectively. Additionally, TRAF3 mutations are observed resulting in truncated TRAF3 protein that is no longer able to bind and regulate NIK. The most common cytogenetic abnormality in MALT lymphoma is the chromosomal translocation t(11;18)(q21;q21) which creates the API2-MALT1 fusion protein (44). This fusion protein cleaves several substrates, including NIK at Arg325, which generates a stable NIK fragment that is catalytically active, resulting in constitutive non-canonical NF-κB signaling. 5-10% of acute myeloid leukemias harbor genomic rearrangements resulting in the MLL-AF9 fusion gene, associated with stabilized NIK and constitutive non-canonical NF-κB activation, thereby exerting anti-apoptotic effects (45). Furthermore, NIK is overexpressed in anaplastic large cell lymphoma, adult T cell leukemia and peripheral T cell lymphoma (46–48). In mantle cell lymphoma, ibrutinib-resistant patients show recurrent mutations in TRAF2 or BIRC3 associated with constitutive non-canonical NF-κB signaling and a complete dependence on NIK (49). Therefore, genomic profiling reveals NIK to be a valuable therapeutic target in several B cell malignancies and NIK may become even more important in the case of resistance or non-responsiveness to B cell receptor inhibitors.
Genetic Targeting of NIK
As most cells under physiological conditions do not have nor require abundant NIK expression and NIK becomes stabilized in pathological contexts, NIK is a potential clinical target. Notably, NIK is not essential for the activation of the canonical NF-κB pathway which may translate to limited side effects upon inhibition in vivo. The fact that NIK is overexpressed and/or hyperactivated in many B cell malignancies including other cancers, suggests that inhibition of NIK has clear clinical potential. Yet, most preclinical studies have relied on genetic targeting of NIK. NIK knockout mice and alymphoplasia (aly/aly) mice deficient for NIK suffer from a lack of lymph nodes, Peyer’s patches and abnormal spleen architecture (50–52). NIK-deficient mice have impaired germinal center formation, somatic hypermutation and antigen-specific antibody production (51, 53, 54), which is consistent with biallelic mutation of NIK in B cell lymphopenia patients who have decreased numbers of class-switched B cells and hypogammaglobulinemia (55). On the other hand, NIK deficiency has little effect on thymocyte development (56), though NIK is necessary for Th17 differentiation (57). Moreover, NIK is required for antigen-specific signaling in effector and memory T cells (58). Additionally, NIK knockout and aly/aly mice have fewer regulatory T cells (59). NIK knockout results in defective LT-βR signaling and conditional deletion of NIK in adult mice results in defects in B cell activation and survival as well as defective signaling upon BAFF, anti-CD40 or anti-IgM stimulation (53, 60), which may indicate great promise of NIK inhibition in B cell malignancies that are largely dependent on microenvironmental signals, such as chronic lymphocytic leukemia (CLL).
Role of NIK in Crosstalk Between Canonical and Non-Canonical NF-κB Signaling
Ideally, a specific NF-κB inhibitor should only target the corresponding canonical or non-canonical pathway without effects on other signaling pathways. Despite the seemingly clear delineation of canonical versus non-canonical NF-κB signaling, there appears to be crosstalk between these two pathways. For example, it has been demonstrated that sustained high NIK protein levels resulted in enhanced canonical NF-κB activity in response to a variety of stimuli (61). Similar findings were shown in MM where NIK overexpression triggered constitutive non-canonical signaling and often activation of the canonical NF-κB pathway as well (41, 43, 62). It may be the case that NIK overexpression enables binding of NIK to additional NF-κB regulatory proteins that have a lower binding affinity, such as IKKβ in the canonical pathway. In support, NIK-dependent activation of canonical NF-κB signaling has been shown via the interaction of NIK with the IKKα/IKKβ complex in response to LT-βR ligation (63). Importantly, p100 contains inhibitory functions by sequestering both canonical and non-canonical NF-κB dimers in the cytoplasm (64). In this way, NIK may promote both canonical and non-canonical NF-κB activation as a result of the processing of p100 into p52 and therefore targeting of NIK may inhibit both NF-κB pathways. The ability of NIK to contribute to both NF-κB pathways is a probable cause why its dysregulation is associated with malignancies, especially in B cells which are reliant on continuous signaling through both pathways for cell proliferation and survival. On one hand, this makes NIK an excellent therapeutic target as it can modulate both NF-κB signaling pathways, but on the other hand, targeting of NIK may result in unwanted toxicities or side effects when selective inhibition of non-canonical NF-κB is preferable. The exact conditions and mechanisms on when and how NIK contributes to canonical NF-κB signaling are important to understand. If the contribution of NIK to canonical NF-κB is restricted to its overexpression, then NIK will indeed be a highly interesting target for therapy as its inhibition should have limited side effects on healthy cells yet be detrimental to cancer cells (65). If NIK is implicated in conventional canonical NF-κB activation, targeting of NIK may cause severe toxicities. Although NIK-deficient mouse models display clear impairments in adaptive immunity, de novo targeting of NIK may have a different impact compared to NIK deficiency during development. Despite the fact that no NIK inhibitor has been clinically approved yet, a number of small molecule inhibitors targeting NIK have been developed. In the next section, we provide an overview and comparison of NIK inhibitors published in literature based on efficacy and specificity.
Overview of In Vitro NIK Inhibitors
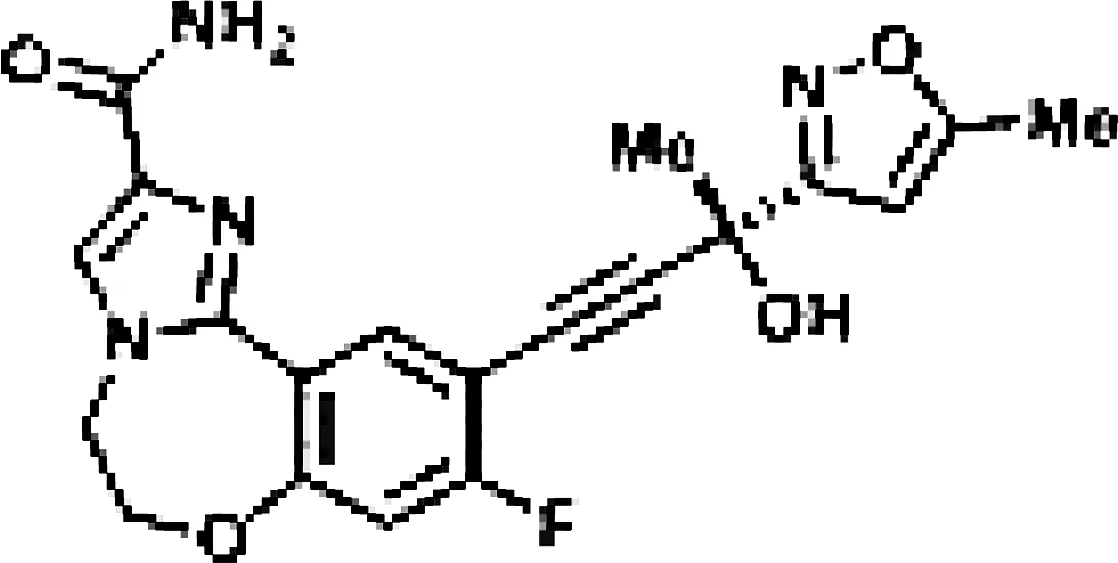
Though NIK seems a promising potential therapeutic target, studies on NIK inhibitors were limited due to missing structural data. Some in vitro studies have shown promising results with NIK inhibition in Hodgkin lymphoma (66) and mantle cell lymphoma (MCL) (49), yet these studies did apply inhibitors designed before publication of the crystal structure of NIK in 2012 (12, 13). Furthermore, some studies report NIK inhibition after application of pan-kinase inhibitors or naturally occurring compounds that inhibit a wide spectrum of kinases (67–69). Virtually all inhibitors designed to specifically target NIK are small molecule inhibitors aimed at binding the catalytic ATP-binding site, and this type we will discuss here. The structure-based study by Li et al. was one of the pioneering studies in the development of NIK-inhibiting compounds (70). By using a small-molecule compound library based on the inhibition of a truncated form of NIK containing only the catalytic domain, they identified imidazopyridinyl pyrimidinamine 1, which laid the foundation of the development of more potent and selective NIK inhibitors (Table 1). Next, Demchenko et al. published the novel Amgen compound AM-0216 which is now commercially available as Amgen16 (71). The authors demonstrated inhibition of non-canonical NF-κB signaling in NIK-dependent MM cell lines whereas MM cell lines containing mutations that activate non-canonical NF-κB signaling independent of NIK, were not affected. Inhibition of p52 nuclear translocation upon in vitro Amgen16 treatment was later also demonstrated in MCL and another study in the context of inflammation (72, 73). However, the poor pharmacokinetic properties of this inhibitor prevented further in vivo studies. In support, other studies also report poor bioavailability in vivo and a short metabolic half-life of similar NIK-inhibiting compounds (78, 79). Another structure-based study identified a new NIK-inhibiting compound that was able to specifically inhibit nuclear translocation of non-canonical NF-κB while maintaining canonical NF-κB translocation (74). A novel compound screening study by Pippione et al. identified the NIK inhibitor aminopyrazole 3a (75). Though its specificity remains partly unclear, the authors showed efficacy in a NIK-dependent MM cell line using a general NF-κB gene reporter assay readout, with no significant inhibition in two NIK-independent MM cell lines. One year later, a new NIK-targeting compound was identified by Cheng et al. with a relatively high IC50 value (76). It would be interesting to test this inhibitor in a B cell malignancy model for more accurate comparison. Finally, we have recently published the novel NIK inhibitor CW15337 and applied it to primary CLL cells (77). We showed that lower concentrations of up to 0.25µM CW15337 resulted in specific non-canonical NF-κB inhibition without an effect on cell viability. Higher concentrations of the inhibitor resulted in a full blockade of non-canonical NF-κB signaling along with partial inhibition of canonical NF-κB signaling. Furthermore, we demonstrated that NIK inhibition in CLL cells abrogated CD40-mediated drug resistance and may sensitize CLL cells to venetoclax.
Table 1
| Inhibitor | Structure | Model | Cells | IC50 | Reference |
|---|---|---|---|---|---|
| Imidazopyridinyl pyrimidinamine 1 |
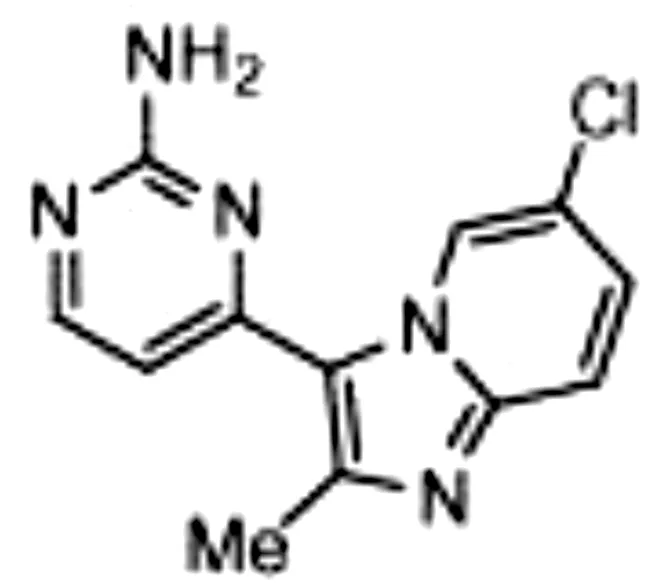
|
Colorectal adenocarcinoma | HT-29 cell line | 16 µM | 70 |
| Amgen16 |
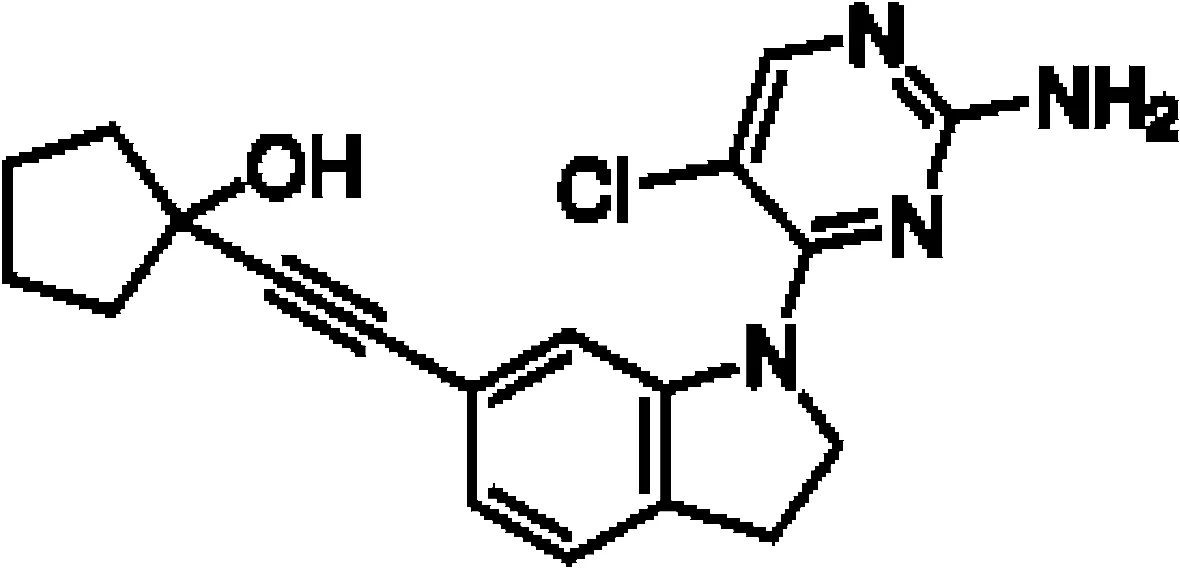
|
Multiple myeloma Mantle cell lymphoma Osteosarcoma |
L363, KMS11 and JMW1 cell lines Z138 and MAVER-1 cell lines U-2 OS cell line |
0.417-2.517 µM |
71
72 73 |
| Compound 10 |
|
Cervical cancer | HEK293 cell line | 123-189 nM | 74 |
| Aminopyrazole 3a |
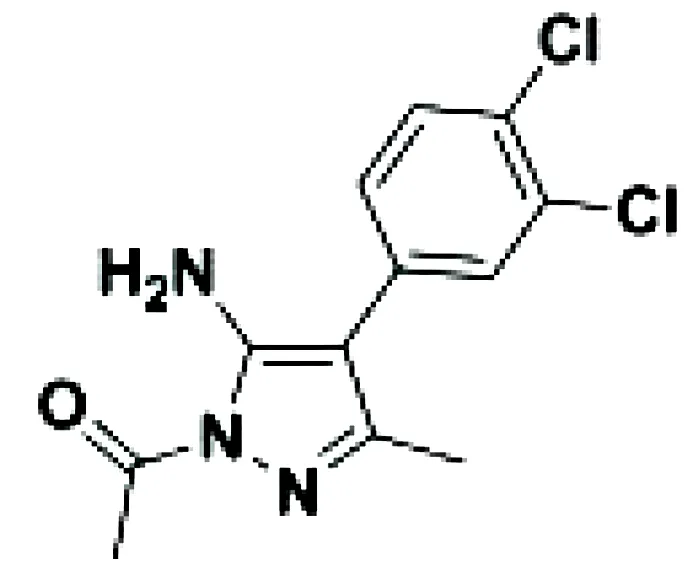
|
Multiple myeloma | EJM cell line | 7.1-9.7 µM | 75 |
| N-(3-(6-benzamido-3a,7a-dihydrobenzo[d]oxazol-2-yl)-phenyl)benzamide |
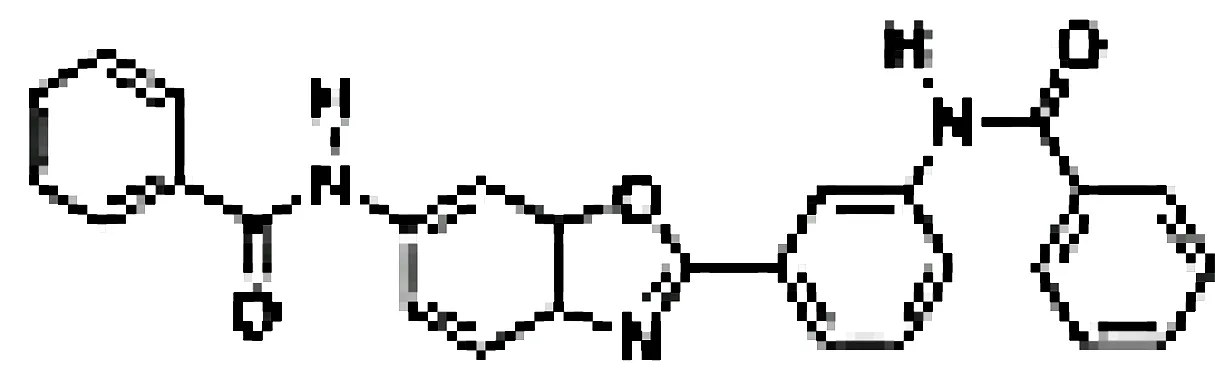
|
Pancreatic cancer | SW1990 cell line | 42-55.8 µM | 76 |
| CW15337 |
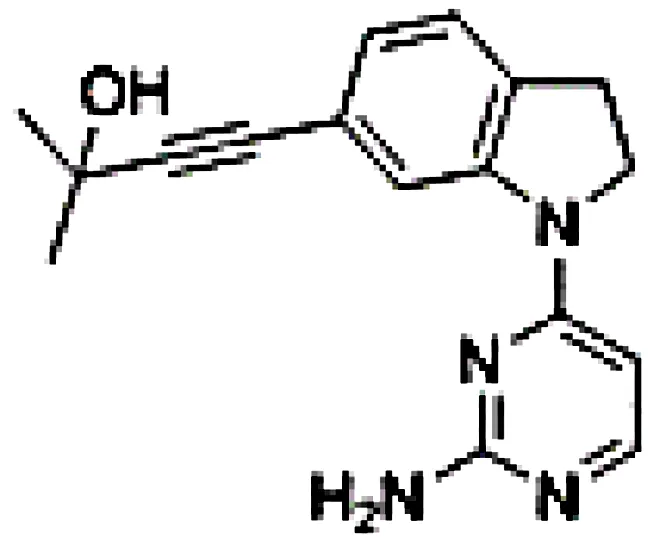
|
Chronic lymphocytic leukemia | Primary cells | 0.5-1 µM | 77 |
Overview of NIK inhibitors applied in vitro.
Overview of In Vivo NIK Inhibitors
Next, we will discuss some NIK inhibitors that have also been applied in in vivo models. Due to the lack of in vivo NIK inhibition in the context of B cell malignancies, we will discuss some alternative mouse models, which allow us to learn about the efficacy and toxicities of these NIK inhibitors (Table 2).
Table 2
| Inhibitor | Structure | Model | Mice | IC50 | Reference |
|---|---|---|---|---|---|
| B022 |
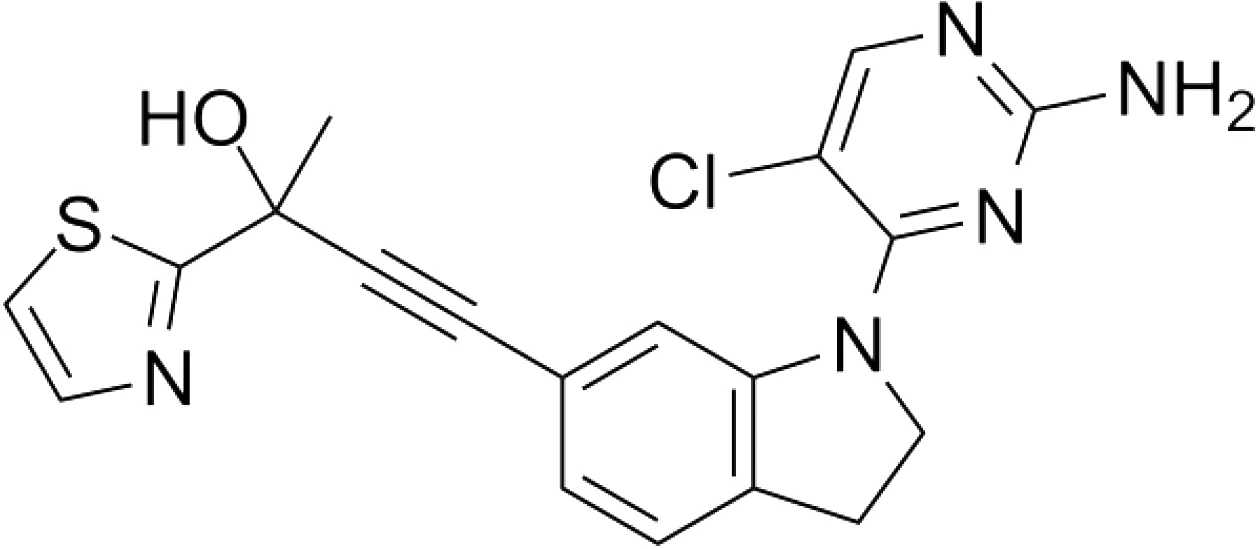
|
Liver injury & fibrosis Diabetes Alcoholic liver disease |
Liver-restricted NIK overexpression β cell-restricted NIK overexpression WT mice on ethanol diet |
Unknown |
80
81 82 |
| NIK SMI1 |
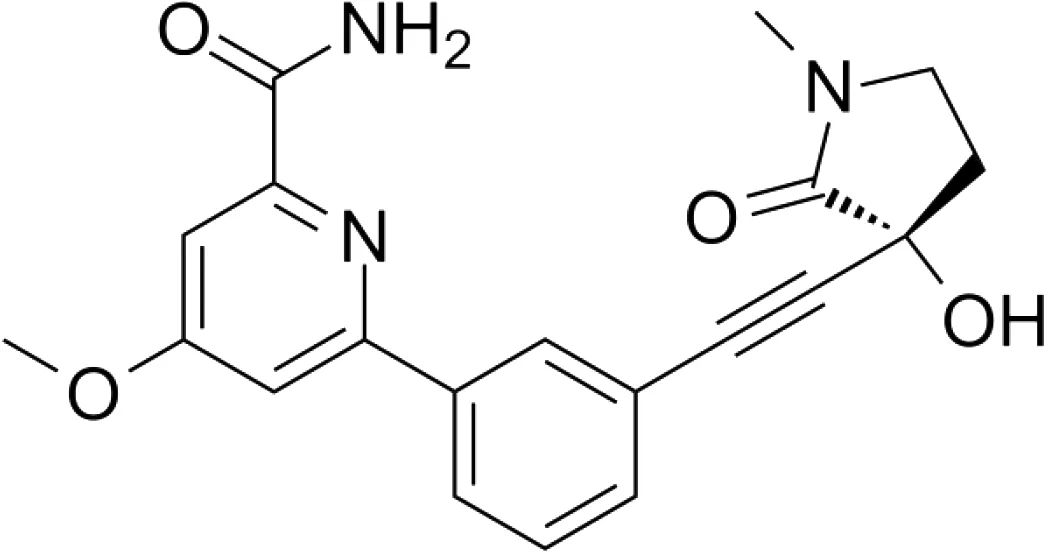
|
Systemic lupus erythematosus | WT mice, NZB/W F1 lupus mice Intrahepatic cholangiocarcinoma xenografts |
108.6-269.4 nM |
83
84 |
| Compound 4f |
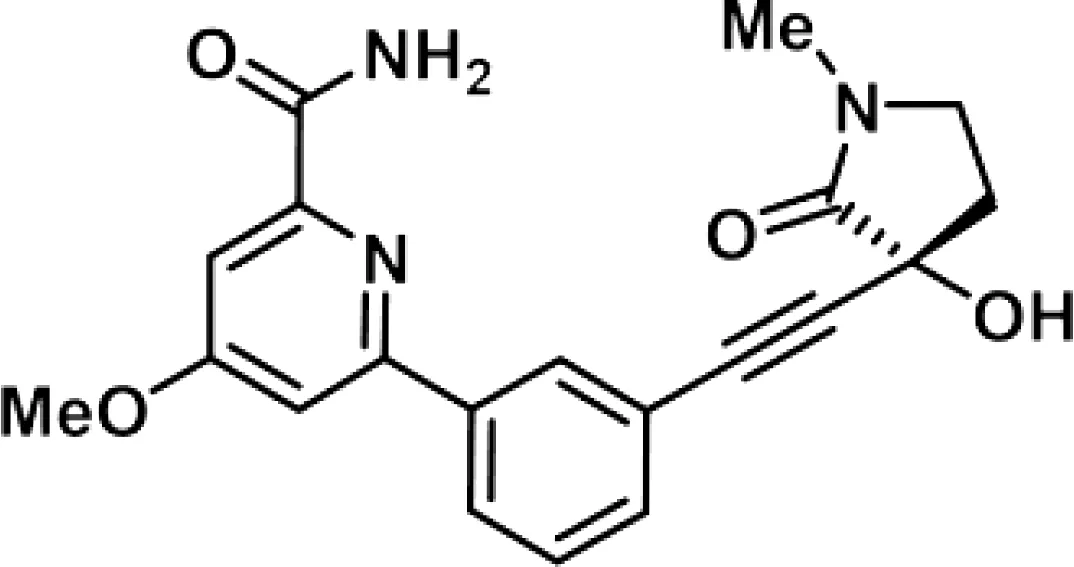
|
WT mice | 309-437 nM | 85 |
Overview of NIK inhibitors applied in vivo.
The commercially available NIK inhibitor B022, which is structurally very similar to Amgen16, was the first small molecule inhibitor targeting NIK to be applied in in vivo models (80). Using this inhibitor, Ren et al. showed inhibition of p100-to-p52 processing and utilized a liver-specific NIK overexpression mouse model. Intravenous B022 administration completely abrogated death as a result of liver inflammation and injury, though general toxicities remain unclear. The authors do note that, just like Amgen16 (71) and in line with another study in diabetic mice (81), due to the bad in vivo pharmacokinetic properties of B022, further modifications are needed to achieve efficient long-term in vivo NIK inhibition. B022 has also been used to treat mice in the context of alcoholic liver disease, where it alleviated alcoholic steatosis, yet toxicities remain unclear (82). Finally, in vitro B022 treatment of mouse stem cells prevented adipogenesis and inhibited non-canonical NF-κB signaling (86). Although these cells do not show nuclear canonical NF-κB, B022 treatment did reduce cytoplasmic RelA phosphorylation levels.
NIK SMI1 is another commercially available small molecule inhibitor of NIK that was generated and published by Brightbill et al. (83). The authors demonstrate an in vitro IC50 value in the nanomolar range in primary B cells with a high selectivity for non-canonical NF-κB signaling as RelA activity was not affected. NKI SMI1 administration to WT mice also indicated a dose-dependent effect on B cell survival with a reduction of marginal zone and follicular B cells as well as reduced serum IgA levels, consistent with the phenotype of NIK-deficient mice (53). Consistently, NIK inhibition suppressed in vivo immune responses based on reduced germinal center development and IgG1 production, as well as effector/memory T cell generation. NIK SMI1 treatment of a systemic lupus erythematosus mouse model with an autoantibody response inhibited germinal center B cell and plasma cell differentiation, while maintaining normal splenocyte counts. The same was true for the differentiation of CD4 effector memory and follicular helper T cells, without changes in total CD4 T cell numbers. Finally, NIK SMI1 treatment significantly reduced mortality, demonstrating a clear therapeutic effect in hyperinflammatory diseases. In the same year of 2018, Blaquiere et al. published a compound screen where they selected a compound with a favorable in vivo pharmacokinetic profile and a molecular structure that is almost identical to NIK SMI1 (85). Consistent with the study of Brightbill et al., the authors found a dose-dependent inhibition of B cell survival in vitro and in vivo. In mice, NIK inhibition significantly reduced the number of marginal zone B cells, similar to BAFF blockade. A recent study using mouse intrahepatic cholangiocarcinoma xenografts reported good tolerance of NIK SMI1 without weight loss or changes in blood test results (84). They reported an inhibition of tumor cell proliferation without apoptosis both in vivo and in vitro, pointing towards clinical relevance, potentially in combination with cytotoxic agents.
Discussion
We have highlighted the importance and advances in the development of NIK inhibitors. The increasing number of studies focusing on the development of compounds targeting NIK may bring NIK closer to the clinic as a therapeutic candidate. However, there are relatively little studies investigating NIK inhibition in vitro and in vivo in an applied setting. In vitro studies and compound screens are mostly performed in standard cell lines whereas in vivo studies are mostly outside the context of malignancy. Notably, most of the published studies applying NIK inhibitors lack data on the specificity of the applied NIK inhibitors, and should show the effects of NIK inhibition on both canonical and non-canonical NF-κB pathways, including off-target effects. Importantly, we still lack clinical information on the off-target effects, safety and efficacy of NIK-targeting drugs in vivo. Altogether, the investigation of NIK inhibitors has been relatively underexplored, and to date, no specific NIK nor other NF-κB inhibitor has been clinically approved. Existing concerns about toxicities as a result of the broad spectrum of physiological roles of NF-κB need to be further resolved and likely contribute to the fact that none of the NIK inhibitors developed by various pharmaceutical companies has entered clinical trials. Regarding potential adverse effects of NIK inhibition in patients, the best guide is provided by human genetic NIK deficiency. Specifically in the context of B cell malignancies, the requirement for NIK in hematopoiesis may pose potential risks for patients (87). Although the impact of de novo NIK targeting may differ from NIK deficiency during development, kinase inhibitors will likely have off-target effects as well. In addition to inhibition of key components of the non-canonical NF-κB pathway, an alternative clinically relevant approach is to block downstream targets or upstream stimulators, or to target crosstalk mechanisms with parallel signaling networks in order to limit toxicities. Due to the many roles of NIK and non-canonical NF-κB signaling in immunity, long-term application of a NIK inhibitor could result in immunodeficiency. Therefore, NIK inhibition in therapy should preferably be used in shorter periods of time (88). While currently only hypothetical, time-limited application of NIK inhibitors may complement existing therapies in the future (77). In conclusion, despite NIK being a promising therapeutic target in many B cell malignancies, current studies on NIK inhibition lack information on specificity, toxicities as well as data on efficacy in the context of malignancy.
Funding
This work was funded by the Blaauboer Fund via the AMC foundation.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Statements
Author contributions
MH performed the literature study, designed the article, and drafted the manuscript. EE reviewed and edited the manuscript. All authors contributed to the article and approved the submitted version.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1
Staudt LM . Oncogenic Activation of NF-kappaB. Cold Spring Harb Perspect Biol (2010) 2(6):a000109. doi: 10.1101/cshperspect.a000109
2
Coope HJ Atkinson PG Huhse B Belich M Janzen J Holman MJ et al . CD40 Regulates the Processing of NF-Kappab2 P100 to P52. EMBO J (2002) 21(20):5375–85. doi: 10.1093/emboj/cdf542
3
Kayagaki N Yan M Seshasayee D Wang H Lee W French DM et al . BAFF/BLyS Receptor 3 Binds the B Cell Survival Factor BAFF Ligand Through a Discrete Surface Loop and Promotes Processing of NF-Kappab2. Immunity (2002) 17(4):515–24. doi: 10.1016/S1074-7613(02)00425-9
4
Xiao G Harhaj EW Sun SC . NF-kappaB-Inducing Kinase Regulates the Processing of NF-Kappab2 P100. Mol Cell (2001) 7(2):401–9. doi: 10.1016/S1097-2765(01)00187-3
5
Senftleben U Cao Y Xiao G Greten FR Krahn G Bonizzi G et al . Activation by IKKalpha of a Second, Evolutionary Conserved, NF-Kappa B Signaling Pathway. Science (2001) 293(5534):1495–9. doi: 10.1126/science.1062677
6
Ling L Cao Z Goeddel DV . NF-kappaB-Inducing Kinase Activates IKK-Alpha by Phosphorylation of Ser-176. Proc Natl Acad Sci USA (1998) 95(7):3792–7. doi: 10.1073/pnas.95.7.3792
7
Xiao G Fong A Sun SC . Induction of P100 Processing by NF-kappaB-Inducing Kinase Involves Docking IkappaB Kinase Alpha (IKKalpha) to P100 and IKKalpha-Mediated Phosphorylation. J Biol Chem (2004) 279(29):30099–105. doi: 10.1074/jbc.M401428200
8
Liang C Zhang M Sun SC . Beta-TrCP Binding and Processing of NF-Kappab2/P100 Involve its Phosphorylation at Serines 866 and 870. Cell Signal (2006) 18(8):1309–17. doi: 10.1016/j.cellsig.2005.10.011
9
Pflug KM Sitcheran R . Targeting NF-kappaB-Inducing Kinase (NIK) in Immunity, Inflammation, and Cancer. Int J Mol Sci (2020) 21(22). doi: 10.3390/ijms21228470
10
Tao Z Ghosh G . Understanding NIK Regulation From its Structure. Structure (2012) 20(10):1615–7. doi: 10.1016/j.str.2012.09.012
11
Lin X Mu Y Cunningham ET Jr. Marcu KB Geleziunas R Greene WC . Molecular Determinants of NF-kappaB-Inducing Kinase Action. Mol Cell Biol (1998) 18(10):5899–907. doi: 10.1128/MCB.18.10.5899
12
de Leon-Boenig G Bowman KK Feng JA Crawford T Everett C Franke Y et al . The Crystal Structure of the Catalytic Domain of the NF-kappaB Inducing Kinase Reveals a Narrow But Flexible Active Site. Structure (2012) 20(10):1704–14. doi: 10.1016/j.str.2012.07.013
13
Liu J Sudom A Min X Cao Z Gao X Ayres M et al . Structure of the Nuclear Factor kappaB-Inducing Kinase (NIK) Kinase Domain Reveals a Constitutively Active Conformation. J Biol Chem (2012) 287(33):27326–34. doi: 10.1074/jbc.M112.366658
14
Hu WH Mo XM Walters WM Brambilla R Bethea JR . TNAP, a Novel Repressor of NF-kappaB-Inducing Kinase, Suppresses NF-kappaB Activation. J Biol Chem (2004) 279(34):35975–83. doi: 10.1074/jbc.M405699200
15
Yeung KC Rose DW Dhillon AS Yaros D Gustafsson M Chatterjee D et al . Raf Kinase Inhibitor Protein Interacts With NF-kappaB-Inducing Kinase and TAK1 and Inhibits NF-kappaB Activation. Mol Cell Biol (2001) 21(21):7207–17. doi: 10.1128/MCB.21.21.7207-7217.2001
16
Qing G Qu Z Xiao G . Stabilization of Basally Translated NF-kappaB-Inducing Kinase (NIK) Protein Functions as a Molecular Switch of Processing of NF-Kappab2 P100. J Biol Chem (2005) 280(49):40578–82. doi: 10.1074/jbc.M508776200
17
Saccani S Pantano S Natoli G . Modulation of NF-kappaB Activity by Exchange of Dimers. Mol Cell (2003) 11(6):1563–74. doi: 10.1016/S1097-2765(03)00227-2
18
Vallabhapurapu S Matsuzawa A Zhang W Tseng PH Keats JJ Wang H et al . Nonredundant and Complementary Functions of TRAF2 and TRAF3 in a Ubiquitination Cascade That Activates NIK-Dependent Alternative NF-kappaB Signaling. Nat Immunol (2008) 9(12):1364–70. doi: 10.1038/ni.1678
19
Razani B Zarnegar B Ytterberg AJ Shiba T Dempsey PW Ware CF et al . Negative Feedback in Noncanonical NF-kappaB Signaling Modulates NIK Stability Through IKKalpha-Mediated Phosphorylation. Sci Signal (2010) 3(123):ra41. doi: 10.1126/scisignal.2000778
20
Jin J Xiao Y Chang JH Yu J Hu H Starr R et al . The Kinase TBK1 Controls IgA Class Switching by Negatively Regulating Noncanonical NF-kappaB Signaling. Nat Immunol (2012) 13(11):1101–9. doi: 10.1038/ni.2423
21
Bram RJ . TBK1 Suppression of IgA in the NIK of Time. Nat Immunol (2012) 13(11):1027–9. doi: 10.1038/ni.2451
22
Gray CM Remouchamps C McCorkell KA Solt LA Dejardin E Orange JS et al . Noncanonical NF-kappaB Signaling is Limited by Classical NF-kappaB Activity. Sci Signal (2014) 7(311):ra13. doi: 10.1126/scisignal.2004557
23
Qing G Yan P Qu Z Liu H Xiao G . Hsp90 Regulates Processing of NF-Kappa B2 P100 Involving Protection of NF-Kappa B-Inducing Kinase (NIK) From Autophagy-Mediated Degradation. Cell Res (2007) 17(6):520–30. doi: 10.1038/cr.2007.47
24
Jiang B Shen H Chen Z Yin L Zan L Rui L . Carboxyl Terminus of HSC70-Interacting Protein (CHIP) Down-Regulates NF-kappaB-Inducing Kinase (NIK) and Suppresses NIK-Induced Liver Injury. J Biol Chem (2015) 290(18):11704–14. doi: 10.1074/jbc.M114.635086
25
Huang T Gao Z Zhang Y Fan K Wang F Li Y et al . CRL4(DCAF2) Negatively Regulates IL-23 Production in Dendritic Cells and Limits the Development of Psoriasis. J Exp Med (2018) 215(8):1999–2017. doi: 10.1084/jem.20180210
26
Bista P Zeng W Ryan S Bailly V Browning JL Lukashev ME . TRAF3 Controls Activation of the Canonical and Alternative NFkappaB by the Lymphotoxin Beta Receptor. J Biol Chem (2010) 285(17):12971–8. doi: 10.1074/jbc.M109.076091
27
Sasaki Y Calado DP Derudder E Zhang B Shimizu Y Mackay F et al . NIK Overexpression Amplifies, Whereas Ablation of its TRAF3-Binding Domain Replaces BAFF:BAFF-R-Mediated Survival Signals in B Cells. Proc Natl Acad Sci USA (2008) 105(31):10883–8. doi: 10.1073/pnas.0805186105
28
Lu J Plank TD Su F Shi X Liu C Ji Y et al . The Nonsense-Mediated RNA Decay Pathway is Disrupted in Inflammatory Myofibroblastic Tumors. J Clin Invest (2016) 126(8):3058–62. doi: 10.1172/JCI86508
29
Yamagishi M Nakano K Miyake A Yamochi T Kagami Y Tsutsumi A et al . Polycomb-Mediated Loss of miR-31 Activates NIK-Dependent NF-kappaB Pathway in Adult T Cell Leukemia and Other Cancers. Cancer Cell (2012) 21(1):121–35. doi: 10.1016/j.ccr.2011.12.015
30
Fei X Zhang P Pan Y Liu Y . MicroRNA-98-5p Inhibits Tumorigenesis of Hepatitis B Virus-Related Hepatocellular Carcinoma by Targeting NF-kappaB-Inducing Kinase. Yonsei Med J (2020) 61(6):460–70. doi: 10.3349/ymj.2020.61.6.460
31
Zhang S Shan C Kong G Du Y Ye L Zhang X . MicroRNA-520e Suppresses Growth of Hepatoma Cells by Targeting the NF-kappaB-Inducing Kinase (NIK). Oncogene (2012) 31(31):3607–20. doi: 10.1038/onc.2011.523
32
Qu LL He L Zhao X Xu W . Downregulation of miR-518a-3p Activates the NIK-Dependent NF-kappaB Pathway in Colorectal Cancer. Int J Mol Med (2015) 35(5):1266–72. doi: 10.3892/ijmm.2015.2145
33
Jang H Park S Kim J Kim JH Kim SY Cho S et al . The Tumor Suppressor, P53, Negatively Regulates Non-Canonical NF-kappaB Signaling Through Mirnainduced Silencing of NF-kappaB-Inducing Kinase. Mol Cells (2020) 43(1):23–33. doi: 10.14348/molcells.2019.0239
34
Sevdali E Katsantoni E Smulski CR Moschovi M Palassopoulou M Kolokotsa EN et al . BAFF/APRIL System Is Functional in B-Cell Acute Lymphoblastic Leukemia in a Disease Subtype Manner. Front Oncol (2019) 9:594. doi: 10.3389/fonc.2019.00594
35
Troeger A Glouchkova L Ackermann B Escherich G Meisel R Hanenberg H et al . High Expression of CD40 on B-Cell Precursor Acute Lymphoblastic Leukemia Blasts is an Independent Risk Factor Associated With Improved Survival and Enhanced Capacity to Up-Regulate the Death Receptor CD95. Blood (2008) 112(4):1028–34. doi: 10.1182/blood-2007-11-123315
36
Haselager MV Kater AP Eldering E . Proliferative Signals in Chronic Lymphocytic Leukemia; What Are We Missing? Front Oncol (2020) 10:592205. doi: 10.3389/fonc.2020.592205
37
Rajakumar SA Papp E Lee KK Grandal I Merico D Liu CC et al . B Cell Acute Lymphoblastic Leukemia Cells Mediate RANK-RANKL-Dependent Bone Destruction. Sci Transl Med (2020) 12(561). doi: 10.1126/scitranslmed.aba5942
38
Alankus B Ecker V Vahl N Braun M Weichert W Macher-Goppinger S et al . Pathological RANK Signaling in B Cells Drives Autoimmunity and Chronic Lymphocytic Leukemia. J Exp Med (2021) 218(2). doi: 10.1084/jem.20200517
39
Fernandes MT Ghezzo MN Silveira AB Kalathur RK Povoa V Ribeiro AR et al . Lymphotoxin-Beta Receptor in Microenvironmental Cells Promotes the Development of T-Cell Acute Lymphoblastic Leukaemia With Cortical/Mature Immunophenotype. Br J Haematol (2015) 171(5):736–51. doi: 10.1111/bjh.13760
40
Nagy B Ferrer A Larramendy ML Galimberti S Aalto Y Casas S et al . Lymphotoxin Beta Expression is High in Chronic Lymphocytic Leukemia But Low in Small Lymphocytic Lymphoma: A Quantitative Real-Time Reverse Transcriptase Polymerase Chain Reaction Analysis. Haematologica (2003) 88(6):654–8.
41
Keats JJ Fonseca R Chesi M Schop R Baker A Chng WJ et al . Promiscuous Mutations Activate the Noncanonical NF-kappaB Pathway in Multiple Myeloma. Cancer Cell (2007) 12(2):131–44. doi: 10.1016/j.ccr.2007.07.003
42
Otto C Giefing M Massow A Vater I Gesk S Schlesner M et al . Genetic Lesions of the TRAF3 and MAP3K14 Genes in Classical Hodgkin Lymphoma. Br J Haematol (2012) 157(6):702–8. doi: 10.1111/j.1365-2141.2012.09113.x
43
Annunziata CM Davis RE Demchenko Y Bellamy W Gabrea A Zhan F et al . Frequent Engagement of the Classical and Alternative NF-kappaB Pathways by Diverse Genetic Abnormalities in Multiple Myeloma. Cancer Cell (2007) 12(2):115–30. doi: 10.1016/j.ccr.2007.07.004
44
Rosebeck S Madden L Jin X Gu S Apel IJ Appert A et al . Cleavage of NIK by the API2-MALT1 Fusion Oncoprotein Leads to Noncanonical NF-kappaB Activation. Science (2011) 331(6016):468–72. doi: 10.1126/science.1198946
45
Studencka-Turski M Maubach G Feige MH Naumann M . Constitutive Activation of Nuclear Factor Kappa B-Inducing Kinase Counteracts Apoptosis in Cells With Rearranged Mixed Lineage Leukemia Gene. Leukemia (2018) 32(11):2498–501. doi: 10.1038/s41375-018-0128-7
46
Wang H Wei W Zhang JP Song Z Li Y Xiao W et al . A Novel Model of Alternative NF-kappaB Pathway Activation in Anaplastic Large Cell Lymphoma. Leukemia (2021) 35(7):1976–89. doi: 10.1038/s41375-020-01088-y
47
Saitoh Y Yamamoto N Dewan MZ Sugimoto H Martinez Bruyn VJ Iwasaki Y et al . Overexpressed NF-kappaB-Inducing Kinase Contributes to the Tumorigenesis of Adult T-Cell Leukemia and Hodgkin Reed-Sternberg Cells. Blood (2008) 111(10):5118–29. doi: 10.1182/blood-2007-09-110635
48
Odqvist L Sanchez-Beato M Montes-Moreno S Martin-Sanchez E Pajares R Sanchez-Verde L et al . NIK Controls Classical and Alternative NF-kappaB Activation and is Necessary for the Survival of Human T-Cell Lymphoma Cells. Clin Cancer Res (2013) 19(9):2319–30. doi: 10.1158/1078-0432.CCR-12-3151
49
Rahal R Frick M Romero R Korn JM Kridel R Chan FC et al . Pharmacological and Genomic Profiling Identifies NF-kappaB-Targeted Treatment Strategies for Mantle Cell Lymphoma. Nat Med (2014) 20(1):87–92. doi: 10.1038/nm.3435
50
Miyawaki S Nakamura Y Suzuka H Koba M Yasumizu R Ikehara S et al . A New Mutation, Aly, That Induces a Generalized Lack of Lymph Nodes Accompanied by Immunodeficiency in Mice. Eur J Immunol (1994) 24(2):429–34. doi: 10.1002/eji.1830240224
51
Yamada T Mitani T Yorita K Uchida D Matsushima A Iwamasa K et al . Abnormal Immune Function of Hemopoietic Cells From Alymphoplasia (Aly) Mice, a Natural Strain With Mutant NF-Kappa B-Inducing Kinase. J Immunol (2000) 165(2):804–12. doi: 10.4049/jimmunol.165.2.804
52
Koike R Nishimura T Yasumizu R Tanaka H Hataba Y Hataba Y et al . The Splenic Marginal Zone is Absent in Alymphoplastic Aly Mutant Mice. Eur J Immunol (1996) 26(3):669–75. doi: 10.1002/eji.1830260324
53
Brightbill HD Jackman JK Suto E Kennedy H Jones C 3rd Chalasani S et al . Conditional Deletion of NF-kappaB-Inducing Kinase (NIK) in Adult Mice Disrupts Mature B Cell Survival and Activation. J Immunol (2015) 195(3):953–64. doi: 10.4049/jimmunol.1401514
54
Hahn M Macht A Waisman A Hovelmeyer N . NF-kappaB-Inducing Kinase is Essential for B-Cell Maintenance in Mice. Eur J Immunol (2016) 46(3):732–41. doi: 10.1002/eji.201546081
55
Willmann KL Klaver S Dogu F Santos-Valente E Garncarz W Bilic I et al . Biallelic Loss-of-Function Mutation in NIK Causes a Primary Immunodeficiency With Multifaceted Aberrant Lymphoid Immunity. Nat Commun (2014) 5:5360. doi: 10.1038/ncomms6360
56
Sun L Luo H Li H Zhao Y . Thymic Epithelial Cell Development and Differentiation: Cellular and Molecular Regulation. Protein Cell (2013) 4(5):342–55. doi: 10.1007/s13238-013-3014-0
57
Li Y Wang H Zhou X Xie X Chen X Jie Z et al . Cell Intrinsic Role of NF-kappaB-Inducing Kinase in Regulating T Cell-Mediated Immune and Autoimmune Responses. Sci Rep (2016) 6:22115. doi: 10.1038/srep22115
58
Rowe AM Murray SE Raue HP Koguchi Y Slifka MK Parker DC . A Cell-Intrinsic Requirement for NF-kappaB-Inducing Kinase in CD4 and CD8 T Cell Memory. J Immunol (2013) 191(7):3663–72. doi: 10.4049/jimmunol.1301328
59
Murray SE . Cell-Intrinsic Role for NF-Kappa B-Inducing Kinase in Peripheral Maintenance But Not Thymic Development of Foxp3+ Regulatory T Cells in Mice. PloS One (2013) 8(9):e76216. doi: 10.1371/journal.pone.0076216
60
Yin L Wu L Wesche H Arthur CD White JM Goeddel DV et al . Defective Lymphotoxin-Beta Receptor-Induced NF-kappaB Transcriptional Activity in NIK-Deficient Mice. Science (2001) 291(5511):2162–5. doi: 10.1126/science.1058453
61
Zarnegar B Yamazaki S He JQ Cheng G . Control of Canonical NF-kappaB Activation Through the NIK-IKK Complex Pathway. Proc Natl Acad Sci USA (2008) 105(9):3503–8. doi: 10.1073/pnas.0707959105
62
Demchenko YN Glebov OK Zingone A Keats JJ Bergsagel PL Kuehl WM . Classical and/or Alternative NF-kappaB Pathway Activation in Multiple Myeloma. Blood (2010) 115(17):3541–52. doi: 10.1182/blood-2009-09-243535
63
Kucharzewska P Maracle CX Jeucken KCM van Hamburg JP Israelsson E Furber M et al . NIK-IKK Complex Interaction Controls NF-kappaB-Dependent Inflammatory Activation of Endothelium in Response to LTbetaR Ligation. J Cell Sci (2019) 132(7). doi: 10.1242/jcs.225615
64
Novack DV Yin L Hagen-Stapleton A Schreiber RD Goeddel DV Ross FP et al . The IkappaB Function of NF-Kappab2 P100 Controls Stimulated Osteoclastogenesis. J Exp Med (2003) 198(5):771–81. doi: 10.1084/jem.20030116
65
Gardam S Beyaert R . The Kinase NIK as a Therapeutic Target in Multiple Myeloma. Expert Opin Ther Targets (2011) 15(2):207–18. doi: 10.1517/14728222.2011.548861
66
Ranuncolo SM Pittaluga S Evbuomwan MO Jaffe ES Lewis BA . Hodgkin Lymphoma Requires Stabilized NIK and Constitutive RelB Expression for Survival. Blood (2012) 120(18):3756–63. doi: 10.1182/blood-2012-01-405951
67
Mortier J Masereel B Remouchamps C Ganeff C Piette J Frederick R . NF-kappaB Inducing Kinase (NIK) Inhibitors: Identification of New Scaffolds Using Virtual Screening. Bioorg Med Chem Lett (2010) 20(15):4515–20. doi: 10.1016/j.bmcl.2010.06.027
68
Takeda T Tsubaki M Sakamoto K Ichimura E Enomoto A Suzuki Y et al . Mangiferin, a Novel Nuclear Factor Kappa B-Inducing Kinase Inhibitor, Suppresses Metastasis and Tumor Growth in a Mouse Metastatic Melanoma Model. Toxicol Appl Pharmacol (2016) 306:105–12. doi: 10.1016/j.taap.2016.07.005
69
Shen G Liu X Lei W Duan R Yao Z . Plumbagin is a NF-kappaB-Inducing Kinase Inhibitor With Dual Anabolic and Antiresorptive Effects That Prevents Menopausal-Related Osteoporosis in Mice. J Biol Chem (2022) 298(4):101767. doi: 10.1016/j.jbc.2022.101767
70
Li K McGee LR Fisher B Sudom A Liu J Rubenstein SM et al . Inhibiting NF-kappaB-Inducing Kinase (NIK): Discovery, Structure-Based Design, Synthesis, Structure-Activity Relationship, and Co-Crystal Structures. Bioorg Med Chem Lett (2013) 23(5):1238–44. doi: 10.1016/j.bmcl.2013.01.012
71
Demchenko YN Brents LA Li Z Bergsagel LP McGee LR Kuehl MW . Novel Inhibitors are Cytotoxic for Myeloma Cells With NFkB Inducing Kinase-Dependent Activation of NFkB. Oncotarget (2014) 5(12):4554–66. doi: 10.18632/oncotarget.2128
72
Vidal-Crespo A Rodriguez V Matas-Cespedes A Lee E Rivas-Delgado A Gine E et al . The Bruton Tyrosine Kinase Inhibitor CC-292 Shows Activity in Mantle Cell Lymphoma and Synergizes With Lenalidomide and NIK Inhibitors Depending on Nuclear factor-kappaB Mutational Status. Haematologica (2017) 102(11):e447–51. doi: 10.3324/haematol.2017.168930
73
Henry KL Kellner D Bajrami B Anderson JE Beyna M Bhisetti G et al . CDK12-Mediated Transcriptional Regulation of Noncanonical NF-kappaB Components is Essential for Signaling. Sci Signal (2018) 11(541). doi: 10.1126/scisignal.aam8216
74
Castanedo GM Blaquiere N Beresini M Bravo B Brightbill H Chen J et al . Structure-Based Design of Tricyclic NF-kappaB Inducing Kinase (NIK) Inhibitors That Have High Selectivity Over Phosphoinositide-3-Kinase (PI3K). J Med Chem (2017) 60(2):627–40. doi: 10.1021/acs.jmedchem.6b01363
75
Pippione AC Sainas S Federico A Lupino E Piccinini M Kubbutat M et al . N-Acetyl-3-Aminopyrazoles Block the non-Canonical NF-kB Cascade by Selectively Inhibiting NIK. Medchemcomm (2018) 9(6):963–8. doi: 10.1039/C8MD00068A
76
Cheng G Mei XB Yan YY Chen J Zhang B Li J et al . Identification of New NIK Inhibitors by Discriminatory Analysis-Based Molecular Docking and Biological Evaluation. Arch Pharm (Weinheim) (2019) 352(7):e1800374. doi: 10.1002/ardp.201800374
77
Haselager M Thijssen R West C Young L Van Kampen R Willmore E et al . Regulation of Bcl-XL by non-Canonical NF-kappaB in the Context of CD40-Induced Drug Resistance in CLL. Cell Death Differ (2021) 28(5):1658–68. doi: 10.1038/s41418-020-00692-w
78
Li Z Li X Su MB Gao LX Zhou YB Yuan B et al . Discovery of a Potent and Selective NF-kappaB-Inducing Kinase (NIK) Inhibitor That Has Anti-Inflammatory Effects in Vitro and in Vivo. J Med Chem (2020) 63(8):4388–407. doi: 10.1021/acs.jmedchem.0c00396
79
Song J Zhu Y Zu W Duan C Xu J Jiang F et al . The Discovery of Quinoline Derivatives, as NF-kappaB Inducing Kinase (NIK) Inhibitors With Anti-Inflammatory Effects In Vitro, Low Toxicities Against T Cell Growth. Bioorg Med Chem (2021) 29:115856. doi: 10.1016/j.bmc.2020.115856
80
Ren X Li X Jia L Chen D Hou H Rui L et al . A Small-Molecule Inhibitor of NF-kappaB-Inducing Kinase (NIK) Protects Liver From Toxin-Induced Inflammation, Oxidative Stress, and Injury. FASEB J (2017) 31(2):711–8. doi: 10.1096/fj.201600840R
81
Li X Wu Y Song Y Ding N Lu M Jia L et al . Activation of NF-kappaB-Inducing Kinase in Islet Beta Cells Causes Beta Cell Failure and Diabetes. Mol Ther (2020) 28(11):2430–41. doi: 10.1016/j.ymthe.2020.07.016
82
Li Y Chen M Zhou Y Tang C Zhang W Zhong Y et al . NIK Links Inflammation to Hepatic Steatosis by Suppressing PPARalpha in Alcoholic Liver Disease. Theranostics (2020) 10(8):3579–93. doi: 10.7150/thno.40149
83
Brightbill HD Suto E Blaquiere N Ramamoorthi N Sujatha-Bhaskar S Gogol EB et al . NF-kappaB Inducing Kinase is a Therapeutic Target for Systemic Lupus Erythematosus. Nat Commun (2018) 9(1):179. doi: 10.1038/s41467-017-02672-0
84
Shiode Y Kodama T Shigeno S Murai K Tanaka S Newberg JY et al . TNF Receptor-Related Factor 3 Inactivation Promotes the Development of Intrahepatic Cholangiocarcinoma Through NF-kappaB-Inducing Kinase-Mediated Hepatocyte Transdifferentiation. Hepatology (2022). doi: 10.1002/hep.32317
85
Blaquiere N Castanedo GM Burch JD Berezhkovskiy LM Brightbill H Brown S et al . Scaffold-Hopping Approach To Discover Potent, Selective, and Efficacious Inhibitors of NF-kappaB Inducing Kinase. J Med Chem (2018) 61(15):6801–13. doi: 10.1021/acs.jmedchem.8b00678
86
Pflug KM Lee DW Sitcheran R . NF-κb-Inducing Kinase Rewires Metabolic Homeostasis and Promotes Diet-Induced Obesity. bioRxiv (2021). doi: 10.1101/2021.08.26.457753
87
Valino-Rivas L Vaquero JJ Sucunza D Gutierrez S Sanz AB Fresno M et al . NIK as a Druggable Mediator of Tissue Injury. Trends Mol Med (2019) 25(4):341–60. doi: 10.1016/j.molmed.2019.02.005
88
Yu H Lin L Zhang Z Zhang H Hu H . Targeting NF-kappaB Pathway for the Therapy of Diseases: Mechanism and Clinical Study. Signal Transduct Target Ther (2020) 5(1):209. doi: 10.1038/s41392-020-00312-6
Summary
Keywords
NIK, therapeutic targets, B cell malignancies, small molecule inhibitors, in vitro , in vivo
Citation
Haselager MV and Eldering E (2022) The Therapeutic Potential of Targeting NIK in B Cell Malignancies. Front. Immunol. 13:930986. doi: 10.3389/fimmu.2022.930986
Received
28 April 2022
Accepted
08 June 2022
Published
12 July 2022
Volume
13 - 2022
Edited by
Matthew Cook, Australian National University, Australia
Reviewed by
Laura Patrussi, University of Siena, Italy; Ursula Zimber-Strobl, Helmholtz Association of German Research Centres (HZ), Germany
Updates
Copyright
© 2022 Haselager and Eldering.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Eric Eldering, e.eldering@amsterdamumc.nl
This article was submitted to Molecular Innate Immunity, a section of the journal Frontiers in Immunology
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.