
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 15 June 2022
Sec. Molecular Innate Immunity
Volume 13 - 2022 | https://doi.org/10.3389/fimmu.2022.930673
This article is part of the Research Topic Identification, Function and Mechanisms of Interferon Induced Genes Associated with Viruses View all 12 articles
 Peifeng Huang1‡
Peifeng Huang1‡ Qingwei Zuo1‡
Qingwei Zuo1‡ Yue Li2‡Patrick Kwabena Oduro3‡Fengxian Tan1Yuanyuan Wang1
Yue Li2‡Patrick Kwabena Oduro3‡Fengxian Tan1Yuanyuan Wang1 Xiaohui Liu1Jing Li2Qilong Wang3
Xiaohui Liu1Jing Li2Qilong Wang3 Fei Guo4*
Fei Guo4* Yue Li3*†Long Yang1,5*
Yue Li3*†Long Yang1,5*The coronavirus disease 2019 (COVID-19), caused by the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) virus, is one of the fastest-evolving viral diseases that has instigated a worldwide pandemic. Severe inflammatory syndrome and venous thrombosis are commonly noted in COVID-19 patients with severe and critical illness, contributing to the poor prognosis. Interleukin (IL)-6, a major complex inflammatory cytokine, is an independent factor in predicting the severity of COVID-19 disease in patients. IL-6 and tumor necrosis factor (TNF)-α participate in COVID-19-induced cytokine storm, causing endothelial cell damage and upregulation of plasminogen activator inhibitor-1 (PAI-1) levels. In addition, IL-6 and PAI-1 form a vicious cycle of inflammation and thrombosis, which may contribute to the poor prognosis of patients with severe COVID-19. Targeted inhibition of IL-6 and PAI-1 signal transduction appears to improve treatment outcomes in severely and critically ill COVID-19 patients suffering from cytokine storms and venous thrombosis. Motivated by studies highlighting the relationship between inflammatory cytokines and thrombosis in viral immunology, we provide an overview of the immunothrombosis and immunoinflammation vicious loop between IL-6 and PAI-1. Our goal is that understanding this ferocious circle will benefit critically ill patients with COVID-19 worldwide.
Since late December, coronavirus disease 2019 (COVID-19) (1) has spread worldwide and instigated a pandemic. Globally, as of April 12, 2022, more than five hundred million people have been diagnosed with COVID-19 disease, including more than 6 million deaths from the disease (WHO, https://covid19.who.int/), posing a great challenge to the health system around the world. The causative agent of the disease is the SARS-CoV-2 virus. Based on the clinical presentation of the COVID-19 disease, the mild-to-moderate disease accounts for 81% of COVID-19 infections and is accompanied by symptoms such as cough, fever, fatigue, and others. Meanwhile, only about 14% of cases have severe symptoms such as dyspnea and hypoxemia, while 5% present with respiratory failure, shock failure, multiple organ failure, and other severe conditions that can result in death. In addition, 14.8% of patients are classified as severe or critically ill patients (Table 1) (2). Emerging laboratory and pathological examination data indicate that cytokine storms and thrombosis were closely related to the disease progression, accounting for the poor prognosis in COVID-19 patients (3–8).
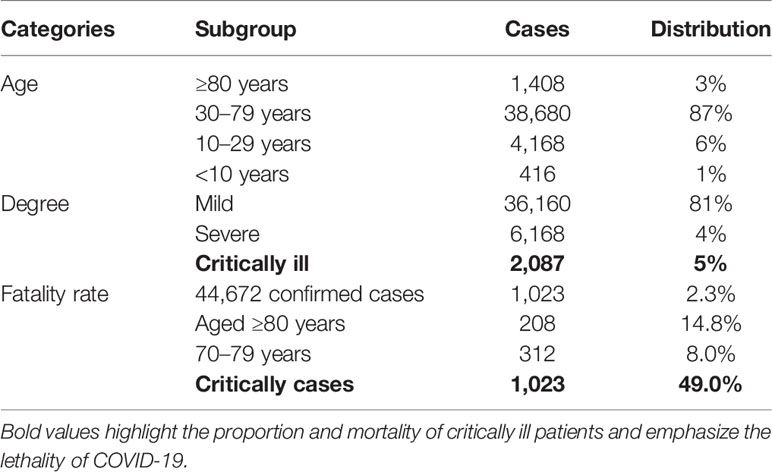
Table 1 The distribution of age, degree, and fatality rate of COVID-19 (2).
A significant reduction in spontaneous clot dissolution after activation of the external clotting pathway and increased resistance to tissue plasminogen activator (tPA) suggests a potential link between fibrinolytic disorder and thrombosis (9). Serum proteomics studies in patients with COVID-19 have found that abnormal increases in IL-6 correlate with increases in the coagulation and complement cascade components (10). PAI-1 is a serine protease inhibitor that acts as a principal inhibitor of tPA and urokinase-type plasminogen activator (uPA) to inhibit fibrinolysis. Based on PAI-1’s primary function, diseases, or disorders that increase PAI-1 levels appear to result in high coagulation states (11–13). Interestingly, in patients with mild-to-moderate disease, plasma levels of PAI-1 were normal compared to critically ill COVID-19 patients (14, 15). However, reports from studies suggested that PAI-1 levels significantly increase in critically ill (14) and hospitalized COVID-19 patients (Figure 1). In addition, previous analyses on the detection of inflammatory and prethrombotic biomarkers in the blood showed significant differences between IL-6 and PAI-1 levels. The mean concentration of IL-6 in the non-severe COVID-19 group was 430.3 pg/ml, whereas that of the control group was 419.5 pg/ml. Meanwhile, the concentration of IL-6 in severe COVID-19 and death group was 1,463 and 2,200 pg/ml, respectively (14). PAI-1 is a widely recognized biomarker of endothelial dysfunction and has been shown that increased concentration is associated with the severity of the disease (16, 17). The expression of PAI-1 may reflect the severity of SARS-CoV-2 infection to some extent (18). The plasma concentration of PAI-1 detected in patients with severe COVID-19 was 713.3 ng/ml, while in the COVID-19 death group, it was 1,223.5 ng/ml. Then again, in the non-severe COVID-19 group, the plasma concentration of PAI-1 was 465.2 ng/ml and that of healthy donors was 183.7 ng/ml (14). It is important to note that severe and critically ill patients with COVID-19 often suffer from underlying diseases (19, 20). Evidence has also suggested that most of the underlying diseases present with elevated levels of PAI-1 (21). For example, among diabetes and acute cerebral infarction patients without COVID-19, PAI-1 levels averaged 36.5 and 63.95 ng/ml (22, 23). Nonetheless, COVID-19-infected individuals have significantly higher levels of PAI-1 than those with diabetes or acute cerebral infarction, providing indirect evidence that COVID-19 could increase PAI-1 levels (Table 2).
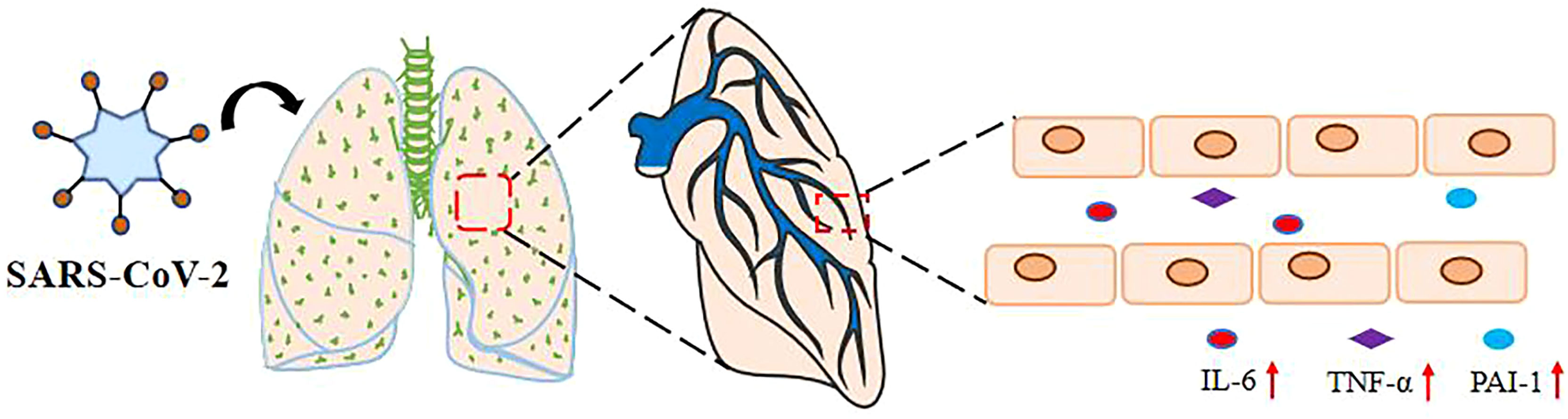
Figure 1 SARS-Co-2 upregulates plasma IL-6, TNF- α, and PAI-1 levels. The levels of IL-6, PAI-1, and TNF-α in the serum of severely and critically ill COVID-19 patients with SARS-CoV-2 pulmonary infection via the respiratory tract were significantly increased.

Table 2 The expression of IL-6 and PAI-1 in COVID-19 and underlying diseases.
Studies on coexpression-induced IL-6 and PAI-1 through the nuclear factor-kappa B (NF-κB) pathway and ligand-dependent epidermal growth factor receptor (EGFR) activation confirmed a significant correlation between IL-6 and PAI-1 (26). The same phenomenon has revealed significant differences between IL-6 and PAI-1 levels in severe and mild-to-moderate COVID-19 patients (14). Treatment with anti-TNFs can reduce the death rate and poor outcomes of COVID-19 patients (27). Below, we review the possible relationship between inflammatory levels and thrombosis in severe and critically ill COVID-19 patients.
The SARS-CoV-2 infection has a devastating effect on immune regulation, leading to a life-threatening systemic inflammatory syndrome called the cytokine storm. This systemic inflammatory syndrome involves abnormal immune-cell hyperactivation and uncontrolled release of circulatory cytokines. Elegant evidence from the COVID-19 pandemic shows that IL-6 and TNF-α are involved in the COVID-19-induced cytokine storm (28). In severe disease, IL-6 and TNF-α are major contributing factors that worsen the condition and cause poor clinical outcomes and even death (29–31). IL-6 is a multifunctional cytokine capable of transmitting cell signals. It is the main trigger of endothelial cytokine storm and an intervention target for clinical therapy (32, 33). Almost all stromal cells and immune system cells can produce IL-6, and the primary activator is IL-1β or TNF-α (34). Toll-like receptor (TLR)-stimulated monocytes and macrophages can also promote the expression of IL-6 (35).
During propagation of the SARS-CoV-2 virus, the envelope spike glycoprotein of the SARS-CoV-2 virus attaches to the angiotensin-converting enzyme (ACE)-2 on the target cell surface, resulting in ACE-2 loss (36). ACE-2 is a negative regulator that functions by activating tPA. ACE-2 deficiency disrupts the effective ACE-2/angiotensin (1–7)/Mas receptor axis, making Ang II more active and decreasing tPA activity, prompting endothelial and smooth muscle cells to synthesize and release PAI-1, leading to the balance of PAI-1/tPA to revert to its prethrombotic state (37, 38). Studies on intensive care unit (ICU) patients with critically ill COVID-19 found that low fibrinolysis was mainly associated with elevated PAI-1 levels (39). The action of recombinant SARS-CoV-2 on the ACE-2 receptor is comparable to that of live viruses, and its spiking glycoprotein induces the expression of PAI-1 in human pulmonary microvascular endothelial cells (HPMECs) (40). In individuals with severe COVID-19 illness, increased PAI-1 expression reduces tPA activity and increases thrombosis while perhaps worsening the inflammatory response (Table 3).
Table 3 The expressions of PAI-1 and IL-6 in severe COVID-19 patients.
In several studies, PAI-1 has been found at the inflammatory site after tissue damage (47, 48). PAI-1 inhibitors reduce TNF-α expression and, at the same time, decrease PAI-1 expression in diabetic mice (49). PAI-1 upregulation may be related to its capacity to activate macrophages. PAI-1 helps to regulate the lipopolysaccharide (LPS)-induced inflammatory response in NR8383 cells, possibly by influencing the TLR4-myeloid differentiation protein 2 (MD-2)/NF-κB signaling transfer pathway (50). PAI-1-induced TLR4 activation causes monocyte macrophages to release significant quantities of IL-6 and TNF-α, exacerbating the inflammatory response (51, 52). This shows that TLR4 is an essential medium for PAI-1 to activate macrophages and promote TNF-α expression. The expression spectrum of macrophages stimulated by PAI-1 occurs 2 h after the peak transcription of PAI-1 (53). PAI-1 can promote macrophage activation and may also be an initial response gene for predicting inflammation. PAI-1 promotes the recruitment of monocytes/macrophages in tumor cells. Its lipoprotein-receptor-related protein 1 interaction domain regulates macrophage migration, whereas its C-terminal uPA interaction domain auto-secretes IL-6 by activating the p38MAPK and NF-κB pathway and inducing macrophage polarization (54). There was a considerable increase in the expression of M1 macrophages in obese mice caused by a high-fat diet (HFD), but PAI-1 deficiency and PAI-039 therapy prevented the development of these markers, demonstrating that PAI-1 is required for macrophage polarization. Meanwhile, PAI-1 activates TLR4, triggering a robust inflammatory response in endothelial cells (ECs), allowing ECs to continuously secrete IL-6 (55). PAI-1 may interact with TLR4 to activate NF-κB, leading ECs to generate cytokines such as IL-6 (56, 57). This shows that PAI-1 can stimulate macrophages and endothelial cells in various ways, promoting inflammatory responses (Table 4).

Table 4 PAI-1 upregulate the expressions of IL-6 and TNF-α.
There is no clinical use of PAI-1 inhibitors in COVID-19 patients. However, it is worth noting that bortezomib upregulates KLF2 to suppress PAI-1 expression and reduce EC damage in HPME cells stimulated with rSARS-CoV-2-S1 glycoprotein (58).
Severe clotting disorder in patients with COVID-19 is closely related to the increased risk of death (59–62). Venous thromboembolism was prevalent in COVID-19 patients, with a total incidence of 31% in 184 patients with severe COVID-19 (63), and a preliminary autopsy on 11 of the COVID-19 patients revealed thrombus in the pulmonary arterioles (64). The D-dimer is a fibrin degradation product used as an alternative marker of fibrinolysis and is often elevated in thrombotic events (65). Relevant studies on COVID-19 report that D-dimer elevation is a prevalent feature (66). Low fibrinolysis is the primary cause of increased blood viscosity and is associated with elevated PAI-1 levels (39). PAI-1 circulating levels may be used as an independent predictor of severity in COVID-19 patients (41), and regulating PAI-1 expression can benefit patients with COVID-19 (42).
Alongside PAI-1, IL-6 is an independent predictor of COVID-19 severity (43–46). IL-6 levels have a substantial predictive value for mortality in COVID-19 ICUs (67). Patients with severe COVID-19 have considerable IL-6 overexpression, and IL-6 signal transduction is the most upregulated pathway in COVID-19 patients (10). IL-6 may have a significant role in the progression of severe COVID-19 disease in patients. PAI-1 expression is only found in severe COVID-19 patients and increases thrombosis (14). PAI-1 is linked to elevated levels of IL-6 in critically ill COVID-19 patients. IL-6 signals through two central pathways. The first is the classic cis signaling, and the second is the trans-signaling. In the classic cis pathway, IL-6 attaches to cells, mainly immune cells, expressing the membrane-bound interleukin-6 receptor (IL-6R) to initiate a downstream signaling response (68, 69). On the other hand, in trans-signaling, IL-6 binds to the soluble form of IL-6R, which is released from IL-6R expressing cell surfaces by proteolysis and IL-6R mRNA to form an exciting complex that associates with membrane-bound gp130 (70–72). In the presence of high circulating levels of IL-6, trans-signaling typically occurs. For instance, ECs express the membrane-bound gp130 but not the membrane-bound IL-6R (73–76), allowing for IL-6/soluble-IL-6R/gp130 downstream signaling activation.
The detection of PAI-1 expression before and after tocilizumab (TCZ) treatment demonstrates that IL-6 signaling transduction can promote PAI-1 expression in ECs (18, 42). LPS stimulates the NF-κB classical pathway to increase the PAI-1 expression and promote alveolar hypercoagulation and fibrinolysis inhibitory states. PAI-1 expression is dramatically reduced following NF-κB knockout (77, 78), indicating that the NF-κB pathway can control PAI-1 expression to some extent. At the same time, elevated plasma IL-6 levels promote NF-κB activation (79), resulting in EC-induced PAI-1 overexpression. In hepatocytes, IL-6 signals via the Janus kinase (JAK) pathway to promote C/EBPδ-induced PAI-1 expression (80). In addition, IL-6 signals and activates the IL-6R/signal transducer and activator of transcription 3 (STAT3) pathway (54), which can indirectly upregulate PAI-1 via miR-34a (81). TNF-α can also upregulate PAI-1 (82). However, it is less commonly documented in the literature, and the mechanism remains unknown (Table 5).

Table 5 IL-6 and TNF-α promote the expression of PAI-1.
According to the preceding discussion, elevated and persistent IL-6, TNF-α, and PAI-1 levels in severe COVID-19 patients potentially generate a vicious cycle of inflammatory response and thrombosis (Figure 2).
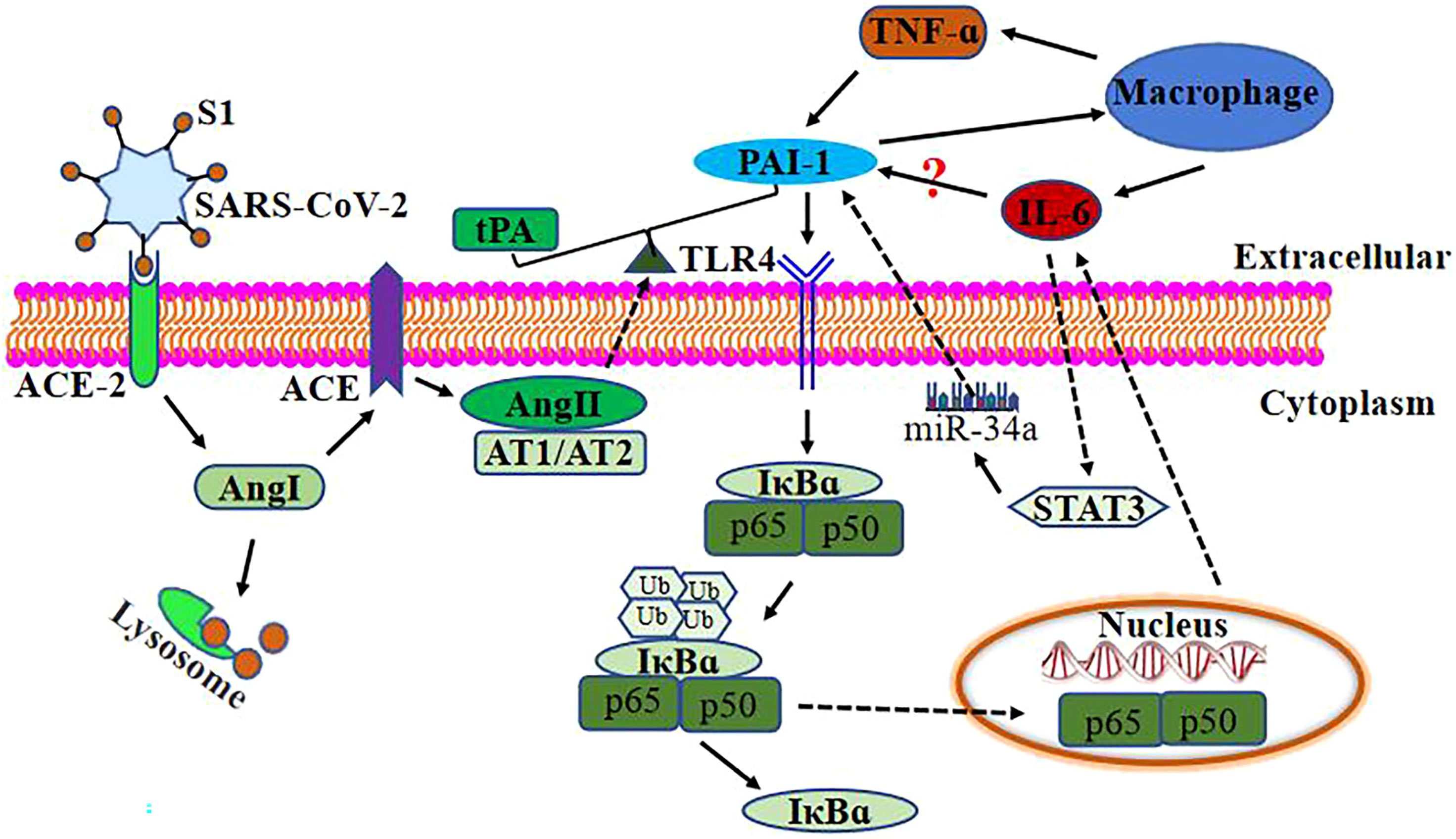
Figure 2 Relationship between PAI-1 and IL-6 after SARS-Co-2 infection. SARS-CoV-2 binds to ACE-2 on the target cell surface, resulting in the loss of ACE-2. ACE-2 is a negative regulator that works by activating tPA. ACE-2 deficiency loses the effective ACE-2/angiotensin (1–7)/Mas receptor axis and increases the level of Ang1. ACE converts Ang I to Ang II and decreases tPA activity, causing endothelial cells and smooth muscle cells to synthesize and release PAI-1. Ang II binds to AT1/AT2 to break the balance of PAI-1/tPA to its prethrombotic state. Elevated levels of PAI-1 in severely and critically ill COVID-19 patients may upregulate IL-6 expression through TLR4/NF-κB pathway and activate macrophages to upregulate IL-6 and TNF-α expression. At the same time, TNF-α can also upregulate PAI-1 expression. IL-6 upregulates the expression of PAI-1 via STAT3/miR-29a.
The probable inflammatory response and thrombus interaction mechanisms are first described in critically ill COVID-19 patients. TCZ is a recombinant human-resistant human IL-6R IgG1 monoclonal antibody (83). The use of TCZ in critically ill COVID-19 patients can decrease PAI-1 levels and improve the condition of severe COVID-19 patients (42). TCZ is authorized for the treatment of rheumatoid arthritis (84) and systemic juvenile idiopathic arthritis (85) because it selectively binds soluble and membrane-bound IL-6 receptors and inhibits IL-6-mediated classic cis and trans-signaling (86). IL-6 levels in severe COVID-19 patients are significantly higher than in other patients, prompting several researchers to recommend TCZ to inhibit IL-6 signaling in patients with severe COVID-19 to improve patient symptoms (35, 87). According to reports, TCZ can be used as an alternative therapy for COVID-19 patients who are at risk of cytokine storms (88). It is advised that in critically ill patients with elevated IL-6 levels, a repeated dose of TCZ will be necessary to reduce IL-6 levels significantly (88). However, TCZ is ineffective for patients with moderate COVID-19 (89) but can improve clinical symptoms in severely and critically ill COVID-19 patients (90). Breathing and bilateral diffuse turbidity disappear by intravenous TCZ in severe COVID-19 patients with pneumonia and acute respiratory distress syndrome (ARDS) (91). Unfortunately, thrombosis in severe COVID-19 patients was not mentioned. PAI-1 inhibition can improve the level of IL-6 and the damage to ECs. Treatment with TM5614 (PAI-1 inhibitor) eliminates the elevated circulating levels of PAI-1 and thrombin in plasma produced by particulate matter (PM) 2.5 (92). Meanwhile, TM5614 significantly reduces the elevated level of IL-6 (92). Bortezomib, a proteasomal degradation inhibitor, enhances KLF2, decreases PAI-1 expression, and reduces EC damage in HPMECs stimulated with rSARS-CoV-2-S1 glycoprotein (58). PAI-1 may have a role in prothrombotic events and inflammation in COVID-19 patients. This asserts the vicious cycle of PAI-1 and IL-6 in COVID-19.
In this review, we briefly discussed the possible link between elevated IL-6 levels and thrombosis in COVID-19 patients. From non-viral contexts, the link between PAI-1 and IL-6 forms an inflammatory–thrombus circuit (42). PAI-1 and IL-6 were not shown to be strongly connected in COVID-19 case reports, although autopsy demonstrated substantial damage to ECs (93). In COVID-19 patients, inflammation and thrombosis are two of the most significant deleterious responses (94, 95). The development of blood clots in the heart can be explained by the distribution of ECs in the heart and by the above process (96). In critically ill COVID-19 patients, EC dysfunction increases PAI-1 expression (17) and promotes macrophage recruitment and activation (54). This raises the amount of IL-6 and TNF-α in the blood, increasing the odds of a “cytokine storm” (28). TCZ can decrease IL-6 signal transduction via IL-6R and soluble IL-6R. TNF-α, on the other hand, stimulates endothelial PAI-1 production and activates macrophages, exposing ECs to prominent levels of IL-6 and TNF-α and causing sustained tissue and organ damage. In thrombosis, therapeutic use of thrombolytic treatment merely lowers fibrin production. Inability to directly suppress PAI-1 expression and break the vicious cycle between PAI-1 and IL-6 results in serum PAI-1 and IL-6 buildup, facilitating tissue damage and thrombosis development. IL-6 trans-signaling has been shown to increase PAI-1 expression. When IL-6 is coupled with soluble IL-6R and gp130, it activates the downstream JAK/STAT signal pathway and promotes the expression of IL-6 and PAI-1 (54, 97, 98) (Figure 3). STAT3-dependent transcription inhibition significantly reduces VEGF-induced vascular permeability in zebrafish, mouse, and human endothelial cells (99). Increased endothelial cell permeability can aggravate pulmonary edema and dyspnea in COVID-19 patients (100). Although the connection between PAI-1 and IL-6 has not yet been shown, the possibility of a malignant interaction between PAI-1 and IL-6 in critically ill COVID-19 patients should not be overlooked. PAI-1 and IL-6 may produce a vicious cycle in which their expression is mutually induced, but the mechanism involved remains unclear. Thrombosis and inflammatory responses in patients with severe COVID-19 are discussed from a new perspective, which provides innovative ideas for future studies.
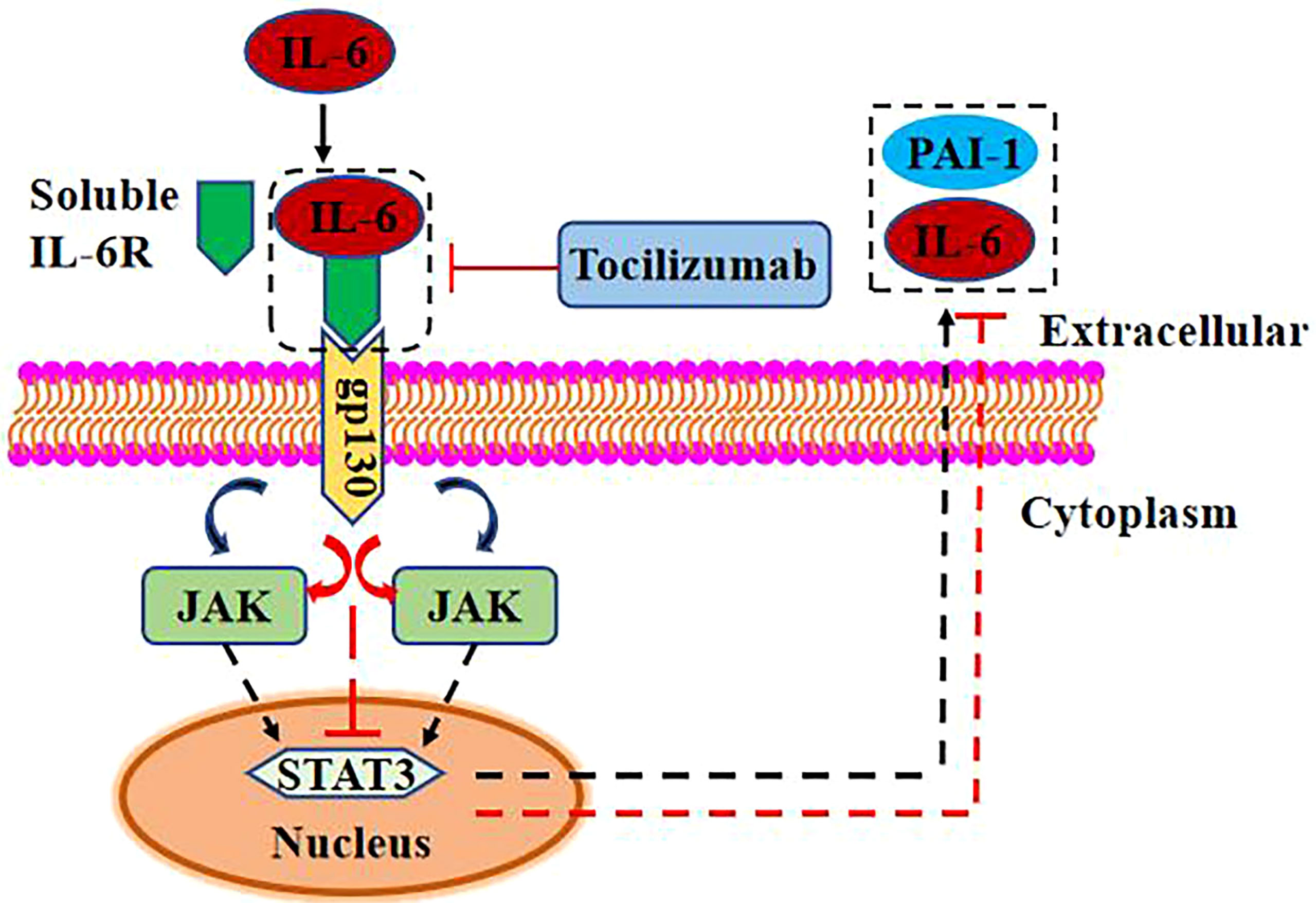
Figure 3 IL-6 promotes PAI-1 expression via trans signaling. High concentration of IL-6 combined with soluble IL-6R can activate the JAK/STAT3 signal pathway through gp130 and upregulate the expression of PAI-1 and IL-6. TCZ can reduce the expression of PAI-1 and IL-6 by inhibiting the binding of IL-6 and soluble IL-6R.
All authors have read and approved the manuscript. FG, YL (11th author), and LY supervised and edited the final manuscript with comments from co-authors. PH, QZ, YL (3rd author), and PO conceptualized and wrote the initial draft, which was further reviewed and edited by FT, YW, XL, JL, and QW for intellectual content. All authors provided crucial revisions in subsequent drafts.
This work was supported by the Tianjin Municipal Education Commission Scientific Research Project (Natural Science, Grant No. 2019ZD11 to LY), Science and Technology Program of Tianjin (21ZYJDJC00070), the National Key Research and Development Program of China (2019YFC1708803), and Innovation Team and Talents Cultivation Program of National Administration of Traditional Chinese Medicine (ZYYCXTD-C-202203).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We thank LY, YL, and FG for their assistance with conceptualization and helpful discussion. We are also grateful to the Tianjin Municipal Education Commission Scientific Research Project for the funding support.
ACE, Angiotensin-converting enzyme; COVID-19, Coronavirus disease 2019; ECs, Endothelial cells; EGFR, Epidermal growth factor receptor; HFD, High-fat diet; HPMECs, Human pulmonary microvascular endothelial cells; ICU, Intensive care unit; IL, Interleukin; IL-6R, Interleukin-6 receptor; JAK, Janus kinase; LPS, Lipopolysaccharide; MD2, Myeloid differentiation protein 2; NF-kB, Nuclear factor of kappa B; PAI-1, Plasminogen activator inhibitor 1; STAT3, Signal transducer and activator of transcription 3; TCZ, Tocilizumab; TLR, Toll-like receptors; TNF, Tumor necrosis factor; tPA, Tissue plasminogen activator; uPA, Urokinase-type plasminogen activator.
1. Li Q, Guan X, Wu P, Wang X, Zhou L, Tong Y, et al. Early Transmission Dynamics in Wuhan, China, of Novel Coronavirus-Infected Pneumonia. N Engl J Med (2020) 382(13):1199–207. doi: 10.1056/NEJMoa2001316
2. Wu ZY, McGoogan JM. Characteristics of and Important Lessons From the Coronavirus Disease 2019 (COVID-19) Outbreak in China Summary of a Report of 72 314 Cases From the Chinese Center for Disease Control and Prevention. Jama-J Am Med Assoc (2020) 323(13):1239–42. doi: 10.1001/jama.2020.2648
3. Guan WJ, Ni ZY, Hu Y, Liang WH, Ou CQ, He JX, et al. Clinical Characteristics of Coronavirus Disease 2019 in China. N Engl J Med (2020) 382(18):1708–20. doi: 10.1056/NEJMoa2002032
4. Liu K, Fang YY, Deng Y, Liu W, Wang MF, Ma JP, et al. Clinical Characteristics of Novel Coronavirus Cases in Tertiary Hospitals in Hubei Province. Chin Med J (Engl) (2020) 133(9):1025–31. doi: 10.1097/CM9.0000000000000744
5. Diao B, Wang C, Tan Y, Chen X, Liu Y, Ning L, et al. Reduction and Functional Exhaustion of T Cells in Patients With Coronavirus Disease 2019 (COVID-19). Front Immunol (2020) 11:827. doi: 10.3389/fimmu.2020.00827
6. Song F, Shi N, Shan F, Zhang Z, Shen J, Lu H, et al. Emerging 2019 Novel Coronavirus (2019-Ncov) Pneumonia. Radiology (2020) 295(1):210–7. doi: 10.1148/radiol.2020200274
7. Tian S, Xiong Y, Liu H, Niu L, Guo J, Liao M, et al. Pathological Study of the 2019 Novel Coronavirus Disease (COVID-19) Through Postmortem Core Biopsies. Mod Pathol (2020) 33(6):1007–14. doi: 10.1038/s41379-020-0536-x
8. Belen-Apak FB, Sarialioglu F. Pulmonary Intravascular Coagulation in COVID-19: Possible Pathogenesis and Recommendations on Anticoagulant/Thrombolytic Therapy. J Thromb Thrombolysis (2020) 50(2):278–80. doi: 10.1007/s11239-020-02129-0
9. Hammer S, Haberle H, Schlensak C, Bitzer M, Malek NP, Handgretinger R, et al. Severe SARS-CoV-2 Infection Inhibits Fibrinolysis Leading to Changes in Viscoelastic Properties of Blood Clot: A Descriptive Study of Fibrinolysis in COVID-19. Thromb Haemost (2021) 121(11):1417–26. doi: 10.1055/a-1400-6034
10. D'Alessandro A, Thomas T, Dzieciatkowska M, Hill RC, Francis RO, Hudson KE, et al. Serum Proteomics in COVID-19 Patients: Altered Coagulation and Complement Status as a Function of IL-6 Level. J Proteome Res (2020) 19(11):4417–27. doi: 10.1021/acs.jproteome.0c00365
11. Cesari M, Pahor M, Incalzi RA. Plasminogen Activator Inhibitor-1 (PAI-1): A Key Factor Linking Fibrinolysis and Age-Related Subclinical and Clinical Conditions. Cardiovasc Ther (2010) 28(5):e72–91. doi: 10.1111/j.1755-5922.2010.00171.x
12. Binder BR, Christ G, Gruber F, Grubic N, Hufnagl P, Krebs M, et al. Plasminogen Activator Inhibitor 1: Physiological and Pathophysiological Roles. News Physiol Sci (2002) 17:56–61. doi: 10.1152/nips.01369.2001
13. Ghosh AK, Vaughan DE. PAI-1 in Tissue Fibrosis. J Cell Physiol (2012) 227(2):493–507. doi: 10.1002/jcp.22783
14. Lopez-Castaneda S, Garcia-Larragoiti N, Cano-Mendez A, Blancas-Ayala K, Damian-Vazquez G, Perez-Medina AI, et al. Inflammatory and Prothrombotic Biomarkers Associated With the Severity of COVID-19 Infection. Clin Appl Thromb Hemost (2021) 27:1076029621999099. doi: 10.1177/1076029621999099
15. Pine AB, Meizlish ML, Goshua G, Chang CH, Zhang H, Bishai J, et al. Circulating Markers of Angiogenesis and Endotheliopathy in COVID-19. Pulm Circ (2020) 10(4):2045894020966547. doi: 10.1177/2045894020966547
16. Bernard I, Limonta D, Mahal LK, Hobman TC. Endothelium Infection and Dysregulation by SARS-CoV-2: Evidence and Caveats in COVID-19. Viruses (2020) 13(1):29. doi: 10.3390/v13010029
17. Norooznezhad AH, Mansouri K. Endothelial Cell Dysfunction, Coagulation, and Angiogenesis in Coronavirus Disease 2019 (COVID-19). Microvasc Res (2021) 137:104188. doi: 10.1016/j.mvr.2021.104188
18. Kellici TF, Pilka ES, Bodkin MJ. Therapeutic Potential of Targeting Plasminogen Activator Inhibitor-1 in COVID-19. Trends Pharmacol Sci (2021) 42(6):431–3. doi: 10.1016/j.tips.2021.03.006
19. Vogrig A, Gigli GL, Bna C, Morassi M. Stroke in Patients With COVID-19: Clinical and Neuroimaging Characteristics. Neurosci Lett (2021) 743:135564. doi: 10.1016/j.neulet.2020.135564
20. Zhou Y, Chi J, Lv W, Wang Y. Obesity and Diabetes as High-Risk Factors for Severe Coronavirus Disease 2019 (Covid-19). Diabetes Metab Res Rev (2021) 37(2):e3377. doi: 10.1002/dmrr.3377
21. Bayomy O, Rao AD, Garg R, Vaidya A, Kotin AR, Reiber B, et al. Plasminogen Activator Inhibitor-1 and Pericardial Fat in Individuals With Type 2 Diabetes Mellitus. Metab Syndr Relat Disord (2017) 15(6):269–75. doi: 10.1089/met.2017.0031
22. Sakurai S, Jojima T, Iijima T, Tomaru T, Usui I, Aso Y. Empagliflozin Decreases the Plasma Concentration of Plasminogen Activator Inhibitor-1 (PAI-1) in Patients With Type 2 Diabetes: Association With Improvement of Fibrinolysis. J Diabetes Complications (2020) 34(11):107703. doi: 10.1016/j.jdiacomp.2020.107703
23. Li L, Ren S, Hao X, Zhen Z, Ji H. Efficacy of Minimally Invasive Intervention in Patients With Acute Cerebral Infarction. J Cardiovasc Pharmacol (2019) 73(1):22–6. doi: 10.1097/FJC.0000000000000625
24. Mallard AR, Hollekim-Strand SM, Ingul CB, Coombes JS. High Day-to-Day and Diurnal Variability of Oxidative Stress and Inflammation Biomarkers in People With Type 2 Diabetes Mellitus and Healthy Individuals. Redox Rep (2020) 25(1):64–9. doi: 10.1080/13510002.2020.1795587
25. Liu Y, Qu M, Wang N, Wang L. Effects of an Evidence-Based Nursing Intervention on Neurological Function and Serum Inflammatory Cytokines in Patients With Acute Cerebral Infarction: A Randomized Controlled Trial. Restor Neurol Neurosci (2021) 39(2):129–37. doi: 10.3233/RNN-201080
26. Alberti C, Pinciroli P, Valeri B, Ferri R, Ditto A, Umezawa K, et al. Ligand-Dependent EGFR Activation Induces the Co-Expression of IL-6 and PAI-1 via the NFkB Pathway in Advanced-Stage Epithelial Ovarian Cancer. Oncogene (2012) 31(37):4139–49. doi: 10.1038/onc.2011.572
27. Robinson PC, Liew DFL, Liew JW, Monaco C, Richards D, Shivakumar S, et al. The Potential for Repurposing Anti-TNF as a Therapy for the Treatment of COVID-19. Med (NY) (2020) 1(1):90–102. doi: 10.1016/j.medj.2020.11.005
28. Azevedo RB, Botelho BG, Hollanda JVG, Ferreira LVL, Junqueira de Andrade LZ, Oei S, et al. Covid-19 and the Cardiovascular System: A Comprehensive Review. J Hum Hypertens (2021) 35(1):4–11. doi: 10.1038/s41371-020-0387-4
29. Li X, Geng M, Peng Y, Meng L, Lu S. Molecular Immune Pathogenesis and Diagnosis of COVID-19. J Pharm Anal (2020) 10(2):102–8. doi: 10.1016/j.jpha.2020.03.001
30. Godeau D, Petit A, Richard I, Roquelaure Y, Descatha A. Return-To-Work, Disabilities and Occupational Health in the Age of COVID-19. Scand J Work Environ Health (2021) 47(5):408–9. doi: 10.5271/sjweh.3960
31. Tisoncik JR, Korth MJ, Simmons CP, Farrar J, Martin TR, Katze MG. Into the Eye of the Cytokine Storm. Microbiol Mol Biol Rev (2012) 76(1):16–32. doi: 10.1128/MMBR.05015-11
32. Hantoushzadeh S, Norooznezhad AH. Possible Cause of Inflammatory Storm and Septic Shock in Patients Diagnosed With (COVID-19). Arch Med Res (2020) 51(4):347–8. doi: 10.1016/j.arcmed.2020.03.015
33. Schett G, Elewaut D, McInnes IB, Dayer JM, Neurath MF. How Cytokine Networks Fuel Inflammation: Toward a Cytokine-Based Disease Taxonomy. Nat Med (2013) 19(7):822–4. doi: 10.1038/nm.3260
34. Hunter CA, Jones SA. IL-6 as a Keystone Cytokine in Health and Disease. Nat Immunol (2015) 16(5):448–57. doi: 10.1038/ni.3153
35. Zhang C, Wu Z, Li JW, Zhao H, Wang GQ. Cytokine Release Syndrome in Severe COVID-19: Interleukin-6 Receptor Antagonist Tocilizumab may be the Key to Reduce Mortality. Int J Antimicrob Agents (2020) 55(5):105954. doi: 10.1016/j.ijantimicag.2020.105954
36. Hoffmann M, Kleine-Weber H, Schroeder S, Kruger N, Herrler T, Erichsen S, et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell (2020) 181(2):271–80.e278. doi: 10.1016/j.cell.2020.02.052
37. Nishimura H, Tsuji H, Masuda H, Nakagawa K, Nakahara Y, Kitamura H, et al. Angiotensin II Increases Plasminogen Activator Inhibitor-1 and Tissue Factor mRNA Expression Without Changing That of Tissue Type Plasminogen Activator or Tissue Factor Pathway Inhibitor in Cultured Rat Aortic Endothelial Cells. Thromb Haemost (1997) 77(6):1189–95. doi: 10.1055/s-0038-1656136
38. van Leeuwen RT, Kol A, Andreotti F, Kluft C, Maseri A, Sperti G. Angiotensin II Increases Plasminogen Activator Inhibitor Type 1 and Tissue-Type Plasminogen Activator Messenger RNA in Cultured Rat Aortic Smooth Muscle Cells. Circulation (1994) 90(1):362–8. doi: 10.1161/01.CIR.90.1.362
39. Nougier C, Benoit R, Simon M, Desmurs-Clavel H, Marcotte G, Argaud L, et al. Hypofibrinolytic State and High Thrombin Generation may Play a Major Role in SARS-COV2 Associated Thrombosis. J Thromb Haemost (2020) 18(9):2215–9. doi: 10.1111/jth.15016
40. Han M, Pandey D. ZMPSTE24 Regulates SARS-CoV-2 Spike Protein-Enhanced Expression of Endothelial Plasminogen Activator Inhibitor-1. Am J Respir Cell Mol Biol (2021) 65(3):300–8. doi: 10.1165/rcmb.2020-0544OC
41. Henry BM, Cheruiyot I, Benoit JL, Lippi G, Prohaszka Z, Favaloro EJ, et al. Circulating Levels of Tissue Plasminogen Activator and Plasminogen Activator Inhibitor-1 Are Independent Predictors of Coronavirus Disease 2019 Severity: A Prospective, Observational Study. Semin Thromb Hemost (2021) 47(4):451–5. doi: 10.1055/s-0040-1722308
42. Kang S, Tanaka T, Inoue H, Ono C, Hashimoto S, Kioi Y, et al. IL-6 Trans-Signaling Induces Plasminogen Activator Inhibitor-1 From Vascular Endothelial Cells in Cytokine Release Syndrome. Proc Natl Acad Sci USA (2020) 117(36):22351–6. doi: 10.1073/pnas.2010229117
43. Aziz M, Fatima R, Assaly R. Elevated Interleukin-6 and Severe COVID-19: A Meta-Analysis. J Med Virol (2020) 92(11):2283–5. doi: 10.1002/jmv.25948
44. Santa Cruz A, Mendes-Frias A, Oliveira AI, Dias L, Matos AR, Carvalho A, et al. Interleukin-6 Is a Biomarker for the Development of Fatal Severe Acute Respiratory Syndrome Coronavirus 2 Pneumonia. Front Immunol (2021) 12:613422. doi: 10.3389/fimmu.2021.613422
45. Liu F, Li L, Xu M, Wu J, Luo D, Zhu Y, et al. Prognostic Value of Interleukin-6, C-Reactive Protein, and Procalcitonin in Patients With COVID-19. J Clin Virol (2020) 127:104370. doi: 10.1016/j.jcv.2020.104370
46. Henry BM, de Oliveira MHS, Benoit S, Plebani M, Lippi G. Hematologic, Biochemical and Immune Biomarker Abnormalities Associated With Severe Illness and Mortality in Coronavirus Disease 2019 (COVID-19): A Meta-Analysis. Clin Chem Lab Med (2020) 58(7):1021–8. doi: 10.1515/cclm-2020-0369
47. Juhan-Vague I, Moerman B, De Cock F, Aillaud MF, Collen D. Plasma Levels of a Specific Inhibitor of Tissue-Type Plasminogen Activator (and Urokinase) in Normal and Pathological Conditions. Thromb Res (1984) 33(5):523–30. doi: 10.1016/0049-3848(84)90018-5
48. Kluft C, Verheijen JH, Jie AF, Rijken DC, Preston FE, Sue-Ling HM, et al. The Postoperative Fibrinolytic Shutdown: A Rapidly Reverting Acute Phase Pattern for the Fast-Acting Inhibitor of Tissue-Type Plasminogen Activator After Trauma. Scand J Clin Lab Invest (1985) 45(7):605–10. doi: 10.3109/00365518509155267
49. Tang S, Liu W, Pan X, Liu L, Yang Y, Wang D, et al. Specific Inhibition of Plasminogen Activator Inhibitor 1 Reduces Blood Glucose Level by Lowering TNF-A. Life Sci (2020) 246:117404. doi: 10.1016/j.lfs.2020.117404
50. Ren W, Wang Z, Hua F, Zhu L. Plasminogen Activator Inhibitor-1 Regulates LPS-Induced TLR4/MD-2 Pathway Activation and Inflammation in Alveolar Macrophages. Inflammation (2015) 38(1):384–93. doi: 10.1007/s10753-014-0042-8
51. Kawai T, Akira S. Signaling to NF-kappaB by Toll-Like Receptors. Trends Mol Med (2007) 13(11):460–9. doi: 10.1016/j.molmed.2007.09.002
52. Togbe D, Schnyder-Candrian S, Schnyder B, Doz E, Noulin N, Janot L, et al. Toll-Like Receptor and Tumour Necrosis Factor Dependent Endotoxin-Induced Acute Lung Injury. Int J Exp Pathol (2007) 88(6):387–91. doi: 10.1111/j.1365-2613.2007.00566.x
53. Gupta KK, Xu Z, Castellino FJ, Ploplis VA. Plasminogen Activator Inhibitor-1 Stimulates Macrophage Activation Through Toll-Like Receptor-4. Biochem Biophys Res Commun (2016) 477(3):503–8. doi: 10.1016/j.bbrc.2016.06.065
54. Kubala MH, Punj V, Placencio-Hickok VR, Fang H, Fernandez GE, Sposto R, et al. Plasminogen Activator Inhibitor-1 Promotes the Recruitment and Polarization of Macrophages in Cancer. Cell Rep (2018) 25(8):2177–91.e2177. doi: 10.1016/j.celrep.2018.10.082
55. Lu Z, Li Y, Jin J, Zhang X, Lopes-Virella MF, Huang Y. Toll-Like Receptor 4 Activation in Microvascular Endothelial Cells Triggers a Robust Inflammatory Response and Cross Talk With Mononuclear Cells via Interleukin-6. Arterioscler Thromb Vasc Biol (2012) 32(7):1696–706. doi: 10.1161/ATVBAHA.112.251181
56. Obi AT, Andraska E, Kanthi Y, Kessinger CW, Elfline M, Luke C, et al. Endotoxaemia-Augmented Murine Venous Thrombosis Is Dependent on TLR-4 and ICAM-1, and Potentiated by Neutropenia. Thromb Haemost (2017) 117(2):339–48. doi: 10.1160/TH16-03-0218
57. Dasu MR, Devaraj S, Du Clos TW, Jialal I. The Biological Effects of CRP Are Not Attributable to Endotoxin Contamination: Evidence From TLR4 Knockdown Human Aortic Endothelial Cells. J Lipid Res (2007) 48(3):509–12. doi: 10.1194/jlr.C600020-JLR200
58. Han M, Pandey D. ZMPSTE24 Regulates SARS-CoV-2 Spike Protein-Enhanced Expression of Endothelial PAI-1. Am J Respir Cell Mol Biol (2021) 65(3):300–8. doi: 10.1165/rcmb.2020-0544OC
59. Tang N, Li D, Wang X, Sun Z. Abnormal Coagulation Parameters are Associated With Poor Prognosis in Patients With Novel Coronavirus Pneumonia. J Thromb Haemost (2020) 18(4):844–7. doi: 10.1111/jth.14768
60. Chen N, Zhou M, Dong X, Qu J, Gong F, Han Y, et al. Epidemiological and Clinical Characteristics of 99 Cases of 2019 Novel Coronavirus Pneumonia in Wuhan, China: A Descriptive Study. Lancet (2020) 395(10223):507–13. doi: 10.1016/S0140-6736(20)30211-7
61. Huang C, Wang Y, Li X, Ren L, Zhao J, Hu Y, et al. Clinical Features of Patients Infected With 2019 Novel Coronavirus in Wuhan, China. Lancet (2020) 395(10223):497–506. doi: 10.1016/S0140-6736(20)30183-5
62. Fogarty H, Townsend L, Ni Cheallaigh C, Bergin C, Martin-Loeches I, Browne P, et al. More on COVID-19 Coagulopathy in Caucasian Patients. Br J Haematol (2020) 189(6):1060–1. doi: 10.1111/bjh.16791
63. Klok FA, Kruip M, van der Meer NJM, Arbous MS, Gommers D, Kant KM, et al. Incidence of Thrombotic Complications in Critically Ill ICU Patients With COVID-19. Thromb Res (2020) 191:145–7. doi: 10.1016/j.thromres.2020.04.013
64. Lax SF, Skok K, Zechner P, Kessler HH, Kaufmann N, Koelblinger C, et al. Pulmonary Arterial Thrombosis in COVID-19 With Fatal Outcome : Results From a Prospective, Single-Center, Clinicopathologic Case Series. Ann Intern Med (2020) 173(5):350–61. doi: 10.7326/M20-2566
65. Ordieres-Ortega L, Demelo-Rodriguez P, Galeano-Valle F, Kremers BMM, Ten Cate-Hoek AJ, Ten Cate H. Predictive Value of D-Dimer Testing for the Diagnosis of Venous Thrombosis in Unusual Locations: A Systematic Review. Thromb Res (2020) 189:5–12. doi: 10.1016/j.thromres.2020.02.009
66. Lippi G, Plebani M, Henry BM. Thrombocytopenia is Associated With Severe Coronavirus Disease 2019 (COVID-19) Infections: A Meta-Analysis. Clin Chim Acta (2020) 506:145–8. doi: 10.1016/j.cca.2020.03.022
67. Gorham J, Moreau A, Corazza F, Peluso L, Ponthieux F, Talamonti M, et al. Interleukine-6 in Critically Ill COVID-19 Patients: A Retrospective Analysis. PLos One (2020) 15(12):e0244628. doi: 10.1371/journal.pone.0244628
68. Jones SA, Scheller J, Rose-John S. Therapeutic Strategies for the Clinical Blockade of IL-6/Gp130 Signaling. J Clin Invest (2011) 121(9):3375–83. doi: 10.1172/JCI57158
69. Tanaka Y, Martin Mola E. IL-6 Targeting Compared to TNF Targeting in Rheumatoid Arthritis: Studies of Olokizumab, Sarilumab and Sirukumab. Ann Rheum Dis (2014) 73(9):1595–7. doi: 10.1136/annrheumdis-2013-205002
70. Rose-John S, Heinrich PC. Soluble Receptors for Cytokines and Growth Factors: Generation and Biological Function. Biochem J (1994) 300(Pt 2):281–90. doi: 10.1042/bj3000281
71. Peters M, Jacobs S, Ehlers M, Vollmer P, Mullberg J, Wolf E, et al. The Function of the Soluble Interleukin 6 (IL-6) Receptor In Vivo: Sensitization of Human Soluble IL-6 Receptor Transgenic Mice Towards IL-6 and Prolongation of the Plasma Half-Life of IL-6. J Exp Med (1996) 183(4):1399–406. doi: 10.1084/jem.183.4.1399
72. Schobitz B, Pezeshki G, Pohl T, Hemmann U, Heinrich PC, Holsboer F, et al. Soluble Interleukin-6 (IL-6) Receptor Augments Central Effects of IL-6 In Vivo. FASEB J (1995) 9(8):659–64. doi: 10.1096/fasebj.9.8.7768358
73. Yoshida K, Taga T, Saito M, Suematsu S, Kumanogoh A, Tanaka T, et al. Targeted Disruption of Gp130, a Common Signal Transducer for the Interleukin 6 Family of Cytokines, Leads to Myocardial and Hematological Disorders. Proc Natl Acad Sci USA (1996) 93(1):407–11. doi: 10.1073/pnas.93.1.407
74. Kopf M, Baumann H, Freer G, Freudenberg M, Lamers M, Kishimoto T, et al. Impaired Immune and Acute-Phase Responses in Interleukin-6-Deficient Mice. Nature (1994) 368(6469):339–42. doi: 10.1038/368339a0
75. Jones GW, McLoughlin RM, Hammond VJ, Parker CR, Williams JD, Malhotra R, et al. Loss of CD4+ T Cell IL-6R Expression During Inflammation Underlines a Role for IL-6 Trans Signaling in the Local Maintenance of Th17 Cells. J Immunol (2010) 184(4):2130–9. doi: 10.4049/jimmunol.0901528
76. McElvaney OJ, Curley GF, Rose-John S, McElvaney NG. Interleukin-6: Obstacles to Targeting a Complex Cytokine in Critical Illness. Lancet Respir Med (2021) 9(6):643–54. doi: 10.1016/S2213-2600(21)00103-X
77. Wu Y, Wang Y, Liu B, Cheng Y, Qian H, Yang H, et al. SN50 Attenuates Alveolar Hypercoagulation and Fibrinolysis Inhibition in Acute Respiratory Distress Syndrome Mice Through Inhibiting NF-kappaB P65 Translocation. Respir Res (2020) 21(1):130. doi: 10.1186/s12931-020-01372-6
79. Li W, Sun L, Lei J, Wu Z, Ma Q, Wang Z. Curcumin Inhibits Pancreatic Cancer Cell Invasion and EMT by Interfering With Tumorstromal Crosstalk Under Hypoxic Conditions via the IL6/ERK/NFkappaB Axis. Oncol Rep (2020) 44(1):382–92. doi: 10.3892/or.2020.7600
80. Dong J, Fujii S, Imagawa S, Matsumoto S, Matsushita M, Todo S, et al. IL-1 and IL-6 Induce Hepatocyte Plasminogen Activator Inhibitor-1 Expression Through Independent Signaling Pathways Converging on C/EBPdelta. Am J Physiol Cell Physiol (2007) 292(1):C209–215. doi: 10.1152/ajpcell.00157.2006
81. Rokavec M, Oner MG, Li H, Jackstadt R, Jiang L, Lodygin D, et al. IL-6r/STAT3/miR-34a Feedback Loop Promotes EMT-Mediated Colorectal Cancer Invasion and Metastasis. J Clin Invest (2014) 124(4):1853–67. doi: 10.1172/JCI73531
82. Cigolini M, et al. Expression of Plasminogen Activator Inhibitor-1 in Human Adipose Tissue: A Role for TNF-Alpha?. Atherosclerosis (1999) 143(1):81–90. doi: 10.1016/s0021-9150(98)00281-0
83. Alves JD, Marinho A, Serra MJ. Tocilizumab: Is There Life Beyond Anti-TNF Blockade? Int J Clin Pract (2011) 65(4):508–13. doi: 10.1111/j.1742-1241.2010.02612.x
84. Kaly L, Rosner I. Tocilizumab - A Novel Therapy for non-Organ-Specific Autoimmune Diseases. Best Pract Res Clin Rheumatol (2012) 26(1):157–65. doi: 10.1016/j.berh.2012.01.001
85. Yokota S, Miyamae T, Imagawa T, Iwata N, Katakura S, Mori M, et al. Therapeutic Efficacy of Humanized Recombinant Anti-Interleukin-6 Receptor Antibody in Children With Systemic-Onset Juvenile Idiopathic Arthritis. Arthritis Rheum (2005) 52(3):818–25. doi: 10.1002/art.20944
86. Le RQ, Li L, Yuan W, Shord SS, Nie L, Habtemariam BA, et al. FDA Approval Summary: Tocilizumab for Treatment of Chimeric Antigen Receptor T Cell-Induced Severe or Life-Threatening Cytokine Release Syndrome. Oncologist (2018) 23(8):943–7. doi: 10.1634/theoncologist.2018-0028
87. Magro G. SARS-CoV-2 and COVID-19: Is Interleukin-6 (IL-6) the 'Culprit Lesion' of ARDS Onset? What is There Besides Tocilizumab? SGP130Fc. Cytokine X (2020) 2(2):100029. doi: 10.1016/j.cytox.2020.100029
88. Luo P, Liu Y, Qiu L, Liu X, Liu D, Li J. Tocilizumab Treatment in COVID-19: A Single Center Experience. J Med Virol (2020) 92(7):814–8. doi: 10.1002/jmv.25801
89. Stone JH, Frigault MJ, Serling-Boyd NJ, Fernandes AD, Harvey L, Foulkes AS, et al. Efficacy of Tocilizumab in Patients Hospitalized With Covid-19. N Engl J Med (2020) 383(24):2333–44. doi: 10.1056/NEJMoa2028836
90. Xu X, Han M, Li T, Sun W, Wang D, Fu B, et al. Effective Treatment of Severe COVID-19 Patients With Tocilizumab. Proc Natl Acad Sci USA (2020) 117(20):10970–5. doi: 10.1073/pnas.2005615117
91. Toniati P, Piva S, Cattalini M, Garrafa E, Regola F, Castelli F, et al. Tocilizumab for the Treatment of Severe COVID-19 Pneumonia With Hyperinflammatory Syndrome and Acute Respiratory Failure: A Single Center Study of 100 Patients in Brescia, Italy. Autoimmun Rev (2020) 19(7):102568. doi: 10.1016/j.autrev.2020.102568
92. Ghosh AK, Soberanes S, Lux E, Shang M, Aillon RP, Eren M, et al. Pharmacological Inhibition of PAI-1 Alleviates Cardiopulmonary Pathologies Induced by Exposure to Air Pollutants PM2.5. Environ Pollut (2021) 287:117283. doi: 10.1016/j.envpol.2021.117283
93. Ackermann M, Verleden SE, Kuehnel M, Haverich A, Welte T, Laenger F, et al. Pulmonary Vascular Endothelialitis, Thrombosis, and Angiogenesis in Covid-19. N Engl J Med (2020) 383(2):120–8. doi: 10.1056/NEJMoa2015432
94. Gao YD, Ding M, Dong X, Zhang JJ, Kursat Azkur A, Azkur D, et al. Risk Factors for Severe and Critically Ill COVID-19 Patients: A Review. Allergy (2021) 76(2):428–55. doi: 10.1111/all.14657
95. Chan NC, Weitz JI. COVID-19 Coagulopathy, Thrombosis, and Bleeding. Blood (2020) 136(4):381–3. doi: 10.1182/blood.2020007335
96. Topol EJ. COVID-19 can Affect the Heart. Science (2020) 370(6515):408–9. doi: 10.1126/science.abe2813
97. Moore JB, June CH. Cytokine Release Syndrome in Severe COVID-19. Science (2020) 368(6490):473–4. doi: 10.1126/science.abb8925
98. Matsuyama T, Kubli SP, Yoshinaga SK, Pfeffer K, Mak TW. An Aberrant STAT Pathway Is Central to COVID-19. Cell Death Differ (2020) 27(12):3209–25. doi: 10.1038/s41418-020-00633-7
99. Wang L, Astone M, Alam SK, Zhu Z, Pei W, Frank DA, et al. Suppressing STAT3 Activity Protects the Endothelial Barrier From VEGF-Mediated Vascular Permeability. bioRxiv (2020) 14(11):dmm049029. doi: 10.1101/2020.10.27.358374
Keywords: COVID-19, PAI-1, IL-6, inflammatory reaction, venous thrombosis, tocilizumab, endothelial cells
Citation: Huang P, Zuo Q, Li Y, Oduro PK, Tan F, Wang Y, Liu X, Li J, Wang Q, Guo F, Li Y and Yang L (2022) A Vicious Cycle: In Severe and Critically Ill COVID-19 Patients. Front. Immunol. 13:930673. doi: 10.3389/fimmu.2022.930673
Received: 28 April 2022; Accepted: 12 May 2022;
Published: 15 June 2022.
Edited by:
Chang Li, Chinese Academy of Agricultural Sciences (CAAS), ChinaReviewed by:
Zhanbo Zhu, Heilongjiang Bayi Agricultural University, ChinaCopyright © 2022 Huang, Zuo, Li, Oduro, Tan, Wang, Liu, Li, Wang, Guo, Li and Yang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Fei Guo, Z3VvZmVpQGlwYi5wdW1jLmVkdS5jbg==; Yue Li, bGl5dWUyMDE4QHRqdXRjbS5lZHUuY24=; Long Yang, bG9uZy55YW5nQHRqdXRjbS5lZHUuY24=
†ORCID: Yue Li, orcid.org/0000-0001-8198-9911
‡These authors have contributed equally to this work and share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.