 Jianan Zhao
Jianan Zhao Shicheng Guo
Shicheng Guo Steven J. Schrodi
Steven J. Schrodi Dongyi He
Dongyi He
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Immunol., 04 August 2022
Sec. Autoimmune and Autoinflammatory Disorders
Volume 13 - 2022 | https://doi.org/10.3389/fimmu.2022.930278
This article is part of the Research TopicThe Potential Drug Regulation in Arthritic DisordersView all 11 articles
Rheumatoid arthritis (RA) is an autoimmune disease that severely affects patients’ physical and mental health, leading to chronic synovitis and destruction of bone joints. Although various available clinical treatment options exist, patients respond with varying efficacies due to multiple factors, and there is an urgent need to discover new treatment options to improve clinical outcomes. Cuproptosis is a newly characterized form of cell death. Copper causes cuproptosis by binding to lipid-acylated components of the tricarboxylic acid cycle, leading to protein aggregation, loss of iron-sulfur cluster proteins, and eventually proteotoxic stress. Targeting copper cytotoxicity and cuproptosis are considered potential options for treating oncological diseases. The synovial hypoxic environment and the presence of excessive glycolysis in multiple cells appear to act as inhibitors of cuproptosis, which can lead to excessive survival and proliferation of multiple immune cells, such as fibroblast-like synoviocytes, effector T cells, and macrophages, further mediating inflammation and bone destruction in RA. Therefore, in this study, we attempted to elaborate and summarize the linkage of cuproptosis and key genes regulating cuproptosis to the pathological mechanisms of RA and their effects on a variety of immune cells. This study aimed to provide a theoretical basis and support for translating preclinical and experimental results of RA to clinical protocols.
Rheumatoid arthritis (RA) is an autoimmune disease characterized by chronic synovitis, presence of multiple autoantibodies, and bone and joint destruction (1). Genetic factors (common risk variants), environmental factors (smoking), genetic and environmental interactions (epigenetic mechanisms), and metabolic abnormalities are risk factors for RA (2). RA affects 1% of the global population and is more prevalent in women than in men (3). Current clinical treatment options for RA include disease-modifying anti-rheumatic drugs, non-steroidal anti-inflammatory drugs (NSAIDs), and biological and non-biological agents. Painkillers and NSAIDs reduce pain and stiffness, but NSAIDs have limited effectiveness and may cause stomach irritation, heart problems, and kidney damage (1). Disease-modifying antirheumatic drugs (DMARDs) are the primary treatment, and when used in combination, these drugs can slow the progression of RA and protect joints and other tissues from permanent damage. However, some DMARDs have multiple adverse effects, such as nausea, liver damage, bone marrow suppression, and development of lung infections (1). Biological agents, including anti-TNF-α antibodies, are also effective, but there are still adverse events, such as infection at the injection site and variation in the efficacy (1). In addition, proper lifestyle management, exercise, and food supplementation are prescribed as complementary therapies. However, due to multiple heterogeneous factors and a complex network of immune-inflammatory pathological mechanisms in RA, available therapies have shown limited clinical efficacy in some patients (2). Therefore, innovative discovery of new drug targets and elucidation of new mechanisms are of great importance for the clinical management of RA.
Cell death is closely associated with RA. Reduced apoptosis in fibroblast-like synoviocytes (FLS) leads to harmful and excessive proliferation, and other pro-inflammatory cell death mechanisms (e.g., pyroptosis and necroptosis) promote inflammation in RA (4). Tsvetkov et al. have characterized a novel form of cell death called “cuproptosis” (5). Cuproptosis in human cells occurs when mitochondrial respiration is disrupted, primarily by the direct binding of excess copper to the lipid-acylated components of the tricarboxylic acid (TCA) cycle. This leads to aggregation of lipid-acylated-related proteins, loss of iron-sulfur cluster proteins, and ultimately, cell death due to intracellular proteotoxic stress (5). In addition, Tsvetkov et al. identified 10 key genes for cuproptosis, including positive regulation factors (ferredoxin 1(FDX1), lipoic acid synthetase (LIAS), lipoyltransferase 1 (LIPT1), dihydrolipoamide dehydrogenase (DLD), drolipoamide S-acetyltransferase (DLAT), pyruvate dehydrogenase E1 subunit alpha 1(PDHA1), and pyruvate dehydrogenase E1 subunit beta (PDHB)) and negative regulatory factors (metal-regulatory transcription factor-1 (MTF1), glutaminase (GLS), and cyclin-dependent kinase inhibitor 2A (CDKN2A)) (5). A meta-analysis of 1444 patients with RA showed that their serum copper levels were significantly higher when compared to that of healthy controls (6). Similarly, Ma et al. found elevated serum copper and decreased zinc and selenium levels in RA patients by systematic evaluation and meta-analysis of common trace metals in them, with possible geographical differences in all three, and that serum selenium levels positively correlated with steroid treatment (7). In addition, serum copper levels were higher in patients with active RA, positively correlated with erythrocyte sedimentation rate (ESR) and morning stiffness, and negatively correlated with hemoglobin levels, which are auxiliary markers for disease assessment (8). Therefore, given the excess copper levels in RA, we sought to elucidate its potential association with RA by searching for cuproptosis and cuproptosis–related genes in PubMed to provide theoretical references and guidance for the discovery and innovative development of clinical treatment options for RA.
Tsvetkov et al. characterized an extensive and detailed characterization of cuproptosis (5). First, the factors necessary for cuproptosis include the presence of glutathione, and the mitochondrial metabolism of galactose and pyruvate (5). Second, cuproptosis appears to be more dependent on mitochondrial respiration, which is inhibited under various conditions such as hypoxia, and presence of mitochondrial antioxidants, inhibitors of mitochondrial function and fatty acids (5). Finally, the knockdown of seven genes that positively regulate cuproptosis may inhibit cuproptosis. For example, knockdown of FDX1 results in the loss of protein-lipid acylation, decreased mitochondrial respiration, accumulation of pyruvate and α-glutarate, and loss of iron-sulfur cluster proteins (5). In addition, accumulation of regulatory gene oligomers is important for the occurrence of cuproptosis (5). Synovial tissue of patients with RA presents a hypoxic environment due to chronic inflammation, vascular proliferation, and excessive cell proliferation (9). Under hypoxic conditions, multiple mediators of bone destruction (matrix metallopeptidases (MMPs)), pro-inflammatory factors (interleukin 8 (IL-8) and IL-6), and chemokines (chemokine (C-C motif) ligand 20 (CCL20)) are involved in bone destruction and inflammatory processes in RA (10, 11). Multiple cells in RA are characterized by an imbalance between cell survival and cell death. The metabolic mechanisms associated with cuproptosis may be linked to these cells. For example, the overall glucose and glutamine levels were reduced in RA FLS, showing enhanced depletion, and indicating that glutamine plays an essential role in FLS proliferation. Glutamine is a critical factor in cuproptosis and its reduced levels, which lead to significant inhibition of cuproptosis, may contribute to the abnormal proliferation of FLS. RA FLS have multiple tumor-like features and survive and over proliferate in a tumor-like microenvironment. The aberrant proliferation of RA FLS is partially attributed to the inhibition of apoptosis (12, 13). The hypoxic environment may also inhibit cuproptosis and thus may contribute to abnormal cell survival and proliferation. The link between copper and hypoxia is complex. Hypoxic conditions promote copper cytotoxicity by inhibiting antioxidant defense mechanisms by increasing reactive oxygen species (ROS), copper transport, and mitotic phagocytosis, with specific molecular mechanisms possibly involving MTF1 and the forkhead box O-3 (FoxO3) signaling pathway (14). Additionally, similar to the inhibition of cuproptosis by the glycolytic effect of FLS, effector T cells exert their effect through the mTOR-dependent pathway, using glycolysis to take in large amounts of glutamine and glucose to provide energy, which may also inhibit cuproptosis, thereby exerting a pro-inflammatory effect. Overactivation of the glycolytic pathway may also inhibit Treg cell function (15). Activated M1 pro-inflammatory macrophages are glycolytic and release pro-inflammatory mediators through multiple mechanisms to destroy tissues (15). These factors may promote inflammatory effects by inhibiting the cuproptosis process in pro-inflammatory cell populations (Table 1). Next, we describe the potential association between critical genes associated with cuproptosis and RA development.

Table 1 The potential function of cuproptosis-related genes in RA.
Lactate levels are significantly increased and glucose concentrations are significantly decreased in RA synovial membranes, suggesting excessive activation of glycolytic pathways (16). Glycolysis converts glucose to pyruvate, and the downstream pathways of glycolysis include lactate fermentation and oxidation of pyruvate (17). PDHA has been extensively studied in tumor cells. Tumor cells promote their growth primarily by enhancing the glycolytic pathway and attenuating oxidative phosphorylation, which appears to also like the excessive glycolysis in RA FLS. During oxidative phosphorylation, the pyruvate dehydrogenase complex (PDHC) converts pyruvate to acetyl coenzyme A. PDHA1, a subunit of PDHC, is a key component linking glycolysis and the TCA cycle (18). PDHA1 inhibition affects PDHC activity, leading to tumor cell glycolysis, enhanced consumption of glucose and glutamine, and inhibition of oxidative phosphorylation (19). Gut microbial-derived butyrate inhibits sirtuin 3 and mitochondrial complex I in tumor cells to prevent the conversion of TCA cycle intermediates to adenosine triphosphate (ATP). Butyrate induces hyperacetylation of PDHA1 to relieve the inhibition of PDHA1 phosphorylation at serine 293 to promote tumor cell apoptosis (20). The transcription factor RUNX family transcription factor 2 (RUNX2) promotes the expression of several glycolytic proteins (phosphorylated protein kinase B (PKB), hexokinase 2 (HK2), and PDH kinase 1(PDHK1)), inhibits the expression of PDHA1 and sirtuin 6 (SIRT6), and suppresses the rate of mitochondrial oxygen consumption (a marker of mitochondrial oxidative phosphorylation), thereby promoting tumor cell proliferation (21). Therefore, it can be speculated that PDHA1 may be involved in RA FLS by regulating the glycolytic process. PDHA1 in RA FLS may be in an inhibited state, thus contributing to the excessive glycolytic and hyperproliferative state of FLS.
In addition to what has been described above, PDHA1 can also be potentially linked to RA through the regulation of inflammation. The release of the NLRP3 inflammasome and related pro-inflammatory mediators plays an important role in the inflammation in RA (4). Activation of the nucleotide-binding oligomerization domain (NOD)-like receptor pyrin domain containing 3 (NLRP3) inflammasome requires lactate fermentation and inhibition of PDHA1 leads to impaired pyruvate oxidation. NLRP3 inflammasome activation leads to release of IL-1β pro-inflammatory mediators (17). Macrophages are important effector cells that are involved in the inflammatory response to RA. Macrophage SIRT-3 is deacetylated at lysine 83, which activates PDHA1, and inhibits NLRP3 inflammasome activation and IL-1β release (22). In addition, the LPS-induced in vitro cell model is an important model for RA inflammation (23). Melatonin receptor 1 (MT1) inhibits LPS-induced aerobic glycolysis and impairs oxidative phosphorylation by promoting PDHA1 expression to suppress inflammation (24).The role of MT1 has been extensively studied in RA. MT1 plays critical roles such as altering the Th1/Th17 balance to suppress inflammation (25) and reducing inflammation and cartilage degradation through the phosphatidylinositol 3−kinase (PI3K)/protein kinase B (AKT), extracellular signal-regulated kinase (ERK), and nuclear factor-κB (NF-κB) signaling pathways, as well as tumor necrosis factor α (TNFα) and IL-1β (26). In summary, PDHA1 appears to be a potent regulator of excessive glycolysis and inflammation and is regulated by different transcriptional mechanisms. Further studies specific to RA are still needed.
PDHB is a subunit of pyruvate dehydrogenase, which is similar in function to PDHA1 in that they both catalyze pyruvate to acetyl coenzyme A (27). PDHB has been identified as a susceptibility gene for RA and its expression is downregulated in various tissues and cells (28). Deglycase DJ-1 was found to bind PDHB in Tregs, inhibit PDHA phosphorylation, and promote PDH activity and oxidative phosphorylation to maintain Treg cell differentiation and the functional integrity of T cells (29). In addition, PDHB has also been studied in various tumor cells. As previously mentioned, it may be linked to abnormalities in RA FLS. Maternally expressed gene 3 (MEG3) inhibits miRNA (miR)-103a-3p, upregulates PDHB-induced endoplasmic reticulum stress proteins’ expressions (glucose-regulated protein 78 (GRP78), activating transcription factor 6 (ATF6), C/EBP homologous protein (CHOP), caspase-3, and caspase-9), inhibits cell viability, colony formation ability and invasion, blocks the cell cycle, and induces apoptosis in tumor cells (30). MiR-203, miR-146b-5p, and miR-363-3p promote pro-tumor cell growth, invasion, inhibition of apoptosis, and enhancement of glycolysis by targeting PDHB (31–33). PDHB also inhibits RasV12-driven ERK signaling and tumor cell proliferation (34). The interaction between PDHB and NIMA-related kinase 10 (NEK10) may be necessary for maintaining mitochondrial homeostasis, and NEK10 knockdown leads to increased mitochondrial damage and dysfunction (35). Thus, PDHB appears to be regulated by multiple miRNAs, while abnormalities in multiple miRNAs contribute to the pathological progression of RA, and the interconnection between the two deserves further exploration (36). In conclusion, downregulation of PDHB may contribute to the abnormal proliferative state of FLS in RA and may lead to defective Treg function through reduced binding to DJ-1.
GLS primarily includes two isoforms, GLS1 and GLS2, which are the key enzymes for glutamine metabolism. GLS1 exists in two splice variants: KGA and GAC (37). GLS1 may promote abnormal proliferative processes in RA FLS. In response to the inflammatory factor IL-17, the mRNA expression of GLS1 was upregulated, whereas the expression of GLS2 was extremely low, implying that GLS1 is primarily responsible for glutamine metabolism. Furthermore, the inhibition of GLS1 suppresses the proliferation of RA FLS and improves joint inflammation in arthritic mice (38).
GLS1 inhibition has multiple effects on CD4+ T cells and their subpopulations. First, it leads to α-CD3/CD28-induced suppression of CD4+ T cell proliferation and decreased expression of T cell activation markers CD25 and CD226 (39). Second, it inhibits cytokine secretion from multiple CD4+ T cell-differentiated T cell subsets, e.g., IL-2 and interferon gamma (IFN-γ) (Th1 cytokines), TNF-α, IL-6, IL-4 (Th2 cytokines), and IL-17a (Th17 cytokines) (39). Finally, the percentage of CD4+ T cells expressing chemokine (C-C motif) receptor 6 (CCR6) and C-X-C chemokine receptor 3 (CXCR3) is reduced (39), both of which have essential roles in inflammatory chemotaxis in RA (40, 41). Th17 is a critical pro-inflammatory mediator in RA that releases IL-17 pro-inflammatory factors to promote inflammation, which preferentially uses glycolysis and glutamine catabolism to provide energy (42). Peroxisome proliferator-activated receptor gamma (PPAR-γ) expression is significantly reduced in the synovial membranes of RA patients (43). PPAR-γ activation inhibits Th17 differentiation by suppressing glutamine catabolism. On one hand, the specific mechanism may involve PPAR-γ inhibiting GLS1 and decreasing 2-hydroxyglutarate (2-HG) levels, thereby regulating lysine demethylase 5 (KDM5)-specific trimethylation of Histone H3 at Lysine 4 (H3K4me3) modifications in the promoter and CNS2 binding regions of the IL-17 locus. In contrast, PPAR-γ inhibits GLS1 and reduces GSH levels, increases ROS levels, and downregulates retinoic acid-related orphan receptor gamma (RORγt) expression (44). In conclusion, GLS1 may primarily affect FLS, B cell and CD4+ T cell subsets in RA by promoting FLS cell proliferation, inflammatory cell differentiation, and pro-inflammatory cytokine release.
LIAS is an iron-sulfur cluster mitochondrial enzyme that replicates the final step of the ab initio pathway that catalyzes lipoic acid biosynthesis, in which lipoic acid is a powerful antioxidant (45). Lipoic acid can be synthesized in the mitochondria by an enzymatic reaction involving octanoic acid. Lipoic acid is essential for mitochondrial α-keto acid dehydrogenase activity and plays an important role in mitochondrial energy metabolism (46). Mitochondria are important organelles in organisms and play several roles, including providing energy to the cell through oxidative phosphorylation and ATP synthesis. When mitochondria produce energy, they store the electrochemical potential energy in the inner mitochondrial membrane. On both sides of the inner membrane, an asymmetric distribution of protons and other ion concentrations results in the mitochondrial membrane potential. Glycolysis oxidizes pyruvate and combines it with coenzyme A, a reaction coupled with the reduction of NAD+, to produce CO2 and acetyl coenzyme A. Acetyl coenzyme A can enter the tricarboxylic acid cycle, which produces ATP (or GTP), more CO2, FADH2, and NADH (47). NADH is then involved in the electron transport chain and oxidative phosphorylation. Oxidative stress is an important factor in mitochondrial dysfunction that leads to RA injury and RA-related atherosclerosis (48). LIAS is primarily associated with oxidative stress, inflammation, and RA. Significantly lower LIAS expression in mice after LPS induction is accompanied by enhanced inflammatory response and tissue damage (49). LIAS overexpression in experimental atherosclerotic mice significantly increases the number of Tregs and reduces T-cell infiltration (50). Similarly, reduced liver LIAS in mice with hepatic fibrosis is accompanied by mitochondrial dysfunction and morphological abnormalities, including mitochondrial edema, reduced density or vacuolization of mitochondrial cristae and matrix, reduced activity of mitochondrial complexes I, II, IV, and V, increased mitochondrial fission activity, and reduced mitochondrial fusion activity (51). Overexpression of LIAS reduced hepatic oxidative stress in non-alcoholic fatty liver disease in mice and protected mitochondrial function by upregulating the nuclear factor erythroid 2–related factor 2 to reduce ROS production (52), attenuated the chronic inflammatory response, inhibited NF-kB activity in lung fibrosis in mice (53), significantly increased Treg cell numbers, and reduced T cell infiltration (50). Mutations in LIAS stabilize HIF-1a in its non-hydroxylated form and promote HIF-1 activation by inhibiting the activity of prolyl hydroxylases (PHDs), which potentially leads to enhanced glycolytic effects in cells (54). Therefore, LIAS and HIF-1 may be involved in RA progression.
E4 transcription factor 1 (E4F1) is a crucial gene involved in controlling mitochondrial function and cell cycle checkpoints that can interact with RA via P53 (55, 56). E4F1 regulates DLAT. These two factors may synergistically regulate the pathogenesis of RA (57). Mitochondrial PDHC is primarily involved in pyruvate oxidation and the TCA cycle, and provides energy to the body (57). Sirtuin 4 (SIRT4) has enzymatic hydrolytic activity and it was significantly downregulated and markedly correlated positively with anti-cyclic citrullinated peptide (anti-CCP) antibody, ESR, and C-reactive protein (CRP) levels in patients with RA (58, 59). SIRT4 can hydrolyze the lipoamide cofactors of DLAT, thereby inhibiting PDH activity (59). In addition, component 1 Q subcomponent-binding protein (C1QBP) in the mitochondria is associated with histological inflammation scores in RA. It can regulate mitochondrial metabolism by affecting PDGH activity through binding to DLAT (60, 61). Therefore, DLAT may influence the development of RA primarily by affecting pyruvate oxidation in PDHC, the TCA cycle, and mitochondrial function.
FDX1 is a member of the ferredoxin family, which comprises iron-sulfur (Fe/S) proteins (62). The transcription factors c-Jun and SF1 can synergistically promote the transcription and expression of FDX1 (63). FDX1 influences immune cells (dendritic cells, monocytes, macrophages, and iTreg cells) (64). Monocytes in RA prefer to use fatty acid oxidation to provide energy and drive receptor activator of nuclear factor kappa-B ligand (RANKL)-induced osteoclast survival and the associated bone destruction (65).FDX1 was found to significantly promote ATP production in these cells. FDX1 knockdown significantly promotes production of fructose 6-phosphate, thus affecting downstream glycolysis, and decreases the levels of many long-chain fatty acids, indicating that it promotes fatty acid oxidation (64).
Abnormalities in and regulation of steroid production play an important role in RA. For example, there are multiple abnormal steroid-related metabolites in patients with RA (66). Increased pro-inflammatory factors in RA may be associated with the reduced renal clearance of steroids (67). FDX1 may be involved in RA development, by potentially influencing this process. Ferredoxin reductase transfers electrons from nicotinamide adenine dinucleotide phosphate (NADPH) to FDX1, reducing members of the mitochondrial cytochrome P450 protein family such as cytochrome P450 11A1 (CYP11A1) and CYP11B (62). CYP11A1 catalyzes the conversion of cholesterol to pregnenolone via side-chain cleavage in the mitochondria, which is the rate-limiting step in adrenal steroid biosynthesis (63). CYP11B promotes the conversion of cortisol to corticosterone, or aldosterone (62). CYP11A1 also converts vitamin D3 to the non-calcemic analog 20S-hydroxyvitamin, which significantly reduces the release of pro-inflammatory T cell subsets and pro-inflammatory cytokines, increases the proportion of Treg cells, and improves symptoms in a mouse model of arthritis (68).
MTF1 is a classical metal-binding transcription factor closely associated with copper homeostasis in eukaryotic organisms (69). Copper loading induces transcriptional activation of metallothionein (MT) through MTF1 and metal responsive element (MRE)-dependent pathways and promotes the nuclear expression of MTF1, which promotes metallothionein expression (70). When copper is depleted, MTF1 also binds to the MRE of CTR1B to promote its transcription and expression of CTR1B, facilitating the introduction of copper to maintain copper homeostasis (71). In addition to MTF1 to maintain copper homeostasis, mammalian cells express a variety of copper transporter proteins or enzymes, such as copper transporter 1 (CTR1), cytochrome c-oxidase 1 (Cox1), Cox2, Cox11, Cox17, synthesis of cytochrome c oxidase 1 (Sco1), Sco2, superoxide dismutase 1 (SOD1), antioxidant-1 (Atox1), ATPase copper transporting alpha (ATP7A), ATPase copper transporting beta (ATP7B), extracellular superoxide dismutase (ecSOD, SOD3), and lysyl oxidase (LOX). Copper homeostasis can be divided into several stages. Firstly, CTR1 uptake of copper, where the copper is transported via protein interactions to three different sites for further processing. For example, ligand-bound copper ions and copper transport proteins, such as Cox1, Cox2, Cox11, and Cox17, are subsequently transported to Sco1 and Sco2 in mitochondria (72) whereas in the cytoplasmic lysates and mitochondrial gap copper is transported to SOD1 (72). Copper is transported via ATP7A or ATP7B to the secreted enzymes EcSOD, SOD3, and LOX (72). Other copper transporter proteins and their specific roles have been clearly described, and here we focus only on cuproptosis-related genes (72).
MTF-1 can directly or indirectly regulate a variety of cellular functions, and is mainly associated with hypoxic conditions in patients with RA. The RA risk SNP (rs28411362) forms a 3D contact with the MTF1 promoter during inflammatory factor-stimulated chromatin remodeling of RA FLS, whose binding motif stimulates FLS recruitment, and MTF1 inhibition significantly suppresses FLS cytokine and chemokine production and improves the mouse arthritis model (73). Under hypoxic conditions, MTF-1 expression promotes the transcriptional activation of phosphatidylinositol glycan anchor biosynthesis class F(PIGF) to promote angiogenesis and enhance endothelial growth and permeability via the vascular endothelial growth factor (VEGF) (74). MTF1 also promotes the activity of hypoxia-inducible factor-1 (HIF-1) (75). HIF-1a is a major regulator of cells under hypoxic conditions and is highly expressed in the RA synovium (10, 76, 77). HIF-1a can also induce MMP-3 production to promote bone destruction (10). HIF-1a promotes pro-inflammatory T cell arrest in joints and Th17 differentiation through transcriptional activation of RORγT and tertiary complex formation with RORγt and p300 recruitment to the IL-17 promoter. HIF-1 inhibits Treg development by targeting forkhead box P3 (FOXP3) for proteasomal degradation (77). HIF-1a promotes the conversion of pyruvate to lactate by increasing LDHA activity. High concentrations of lactate promote cell proliferation of FLS (10, 76, 77). Furthermore, in addition to its effects on RA FLS, high lactate concentrations can promote pro-inflammatory T-cell arrest in the joints. It is worth noting that MTF1 responds to copper stimulation through different binding genes (78), and phosphorylation of MTF1 is essential for the functional activation of MTF (79). For example, MTF1 promotes ATP7B expression by binding to the MRE in the promoter region of ATP7B to promote Wilson’s disease caused by copper overload (80). Phosphorylation of the kinase LATS of the Hippo pathway and inhibition of MTF1 protects cells from heavy metal-induced cytotoxicity (81). Thus, MTF1 primarily responds to excess copper levels in RA, and the hypoxic environment affects multiple pathological aspects of RA.
The fraction of cells expressing p16 (CDKN2A) is a typical marker of cellular senescence (82). Cellular senescence has been associated with RA in various cell types. For example, senescent T cells are highly inflammatory, secrete cytotoxic mediators, and express natural killer receptors (NKR), bypassing their antigenic specificity (83, 84). Histone deacetylase1 (HDAC1) is overexpressed in RA FLS and promotes cell proliferation in FLS (85). The deacetylase (HDA) inhibitor FK228 inhibits joint swelling, synovial inflammation, and bone destruction in mice with experimentally induced arthritis. It also inhibits the proliferation of RA FLS in vitro by a mechanism that involves FK228, thereby inducing high histone acetylation and DKN2A expression in synovial cells, upregulating p21, and decreasing the release of TNF and IL-1β (86). However, it is noteworthy that the senescent phenotype of RA FLS highly expresses CDKN2A and releases more pro-inflammatory mediators in response to TNF or oxidative stress stimuli to promote inflammation (87). The histone methyltransferase EZH2 is strongly induced in chronic inflammation of RA FLS, which may suppress CDKN2A expression and thus contribute to the abnormal response to FLS (88). In addition to its potential effects on RA FLS, CDKN2A may affect RA by influencing the function of macrophages, T cells, and leukocytes. Oxidized low-density lipoprotein (ox-LDL) activates multiple immune cells in RA to promote the secretion of pro-inflammatory mediators and assemble Abs to promote the production of immune complexes to mediate RA pathological progression (89). Ox-LDL promotes the secretion of TNF-α and IL-1β by macrophages and functions via the MEG3/miR-204/CDKN2A axis (90). CDKN2A expression in macrophages inhibits LPS-induced IL-6 production by a specific mechanism involving CDKN2A, promoting ubiquitin-dependent degradation of IRAK1 and impairing the activation of AP-1 (91). Reduced expression of CDKN2A in leukocytes appears to be associated with increased CD14++CD16++ monocyte subsets, increased immune complex responses, and overproduction of pro-inflammatory factors in RA (2, 92). EZH2 is also thought to be essential for B and T cell development, and IL-17 in RA patients with RA synovial fluid may inhibit EZH2 expression downregulation in CD4+ T cells and suppress Treg differentiation (93). EZH2 also suppresses CDKN2A expression in naive CD8+ T cells by reducing H3K27me3 levels at two loci (50) and by controlling B-cell maturation (94). Therefore, EZH2 may work in combination with CDKN2A to regulate abnormal T and B cell responses in RA.
LIPT1 primarily encodes LIPT1, which catalyzes the transfer of lipoic acid from the H protein of the glycine cleavage system to the E2 subunit of 2-ketoacid dehydrogenase, an essential step in lipid acylation (95, 96). LIPT1 is primarily responsible for regulating glutamine metabolism to support mitochondrial respiration, the TGA cycle, and fatty acid production (95). Mutations in LIPT1 impair mitochondrial proteolipid acylation and TGA cycling, and promote the accumulation of lactate and pyruvate (95). Among them, lactate and pyruvate can stimulate synovial cell proliferation, angiogenesis, and vascular opacification in patients with RA (97). Little research has been conducted on LIPT1 in diseases, and further studies are still needed.
A specific concentration of copper in an organism contributes to organismal homeostasis. However, the imbalance in copper homeostasis may affect the organism by triggering cuproptosis, leading to disease development. Cuproptosis is considered a potential therapeutic option for oncological diseases, and its possible association with RA is multifaceted (Figure 1). First, cuproptosis in multiple immune cells may be suppressed, and this suppression contributes to their over-proliferation in RA. Secondly, several essential regulatory genes of cuproptosis have been identified to be associated with multiple RA processes, such as aberrant FLS proliferation and inflammatory processes in various immune cells. PDHA1 regulates glycolysis and inflammation; miRNAs primarily regulate PDHB, GLS1, and LIPT1 regulate glutamine metabolism; DLAT regulates mitochondrial function and the TCA cycle metabolism; and FDX1 regulates fatty acid oxidation and steroidogenesis; MTF1 and LIAS regulate copper homeostasis; and HIF-1 and CDKN2A regulate cellular senescence. Finally, it is worth noting that cuproptosis is a newly characterized form of cell death, and its specific mechanisms and effects on disease are not as well studied as other forms of cell death, such as apoptosis and ferroptosis. Well-designed preclinical experiments and clinical trials are still required for in-depth studies of cuproptosis and its associated genes in the context of RA, which still present a significant challenge. However, it is undeniably a research direction with great potential.
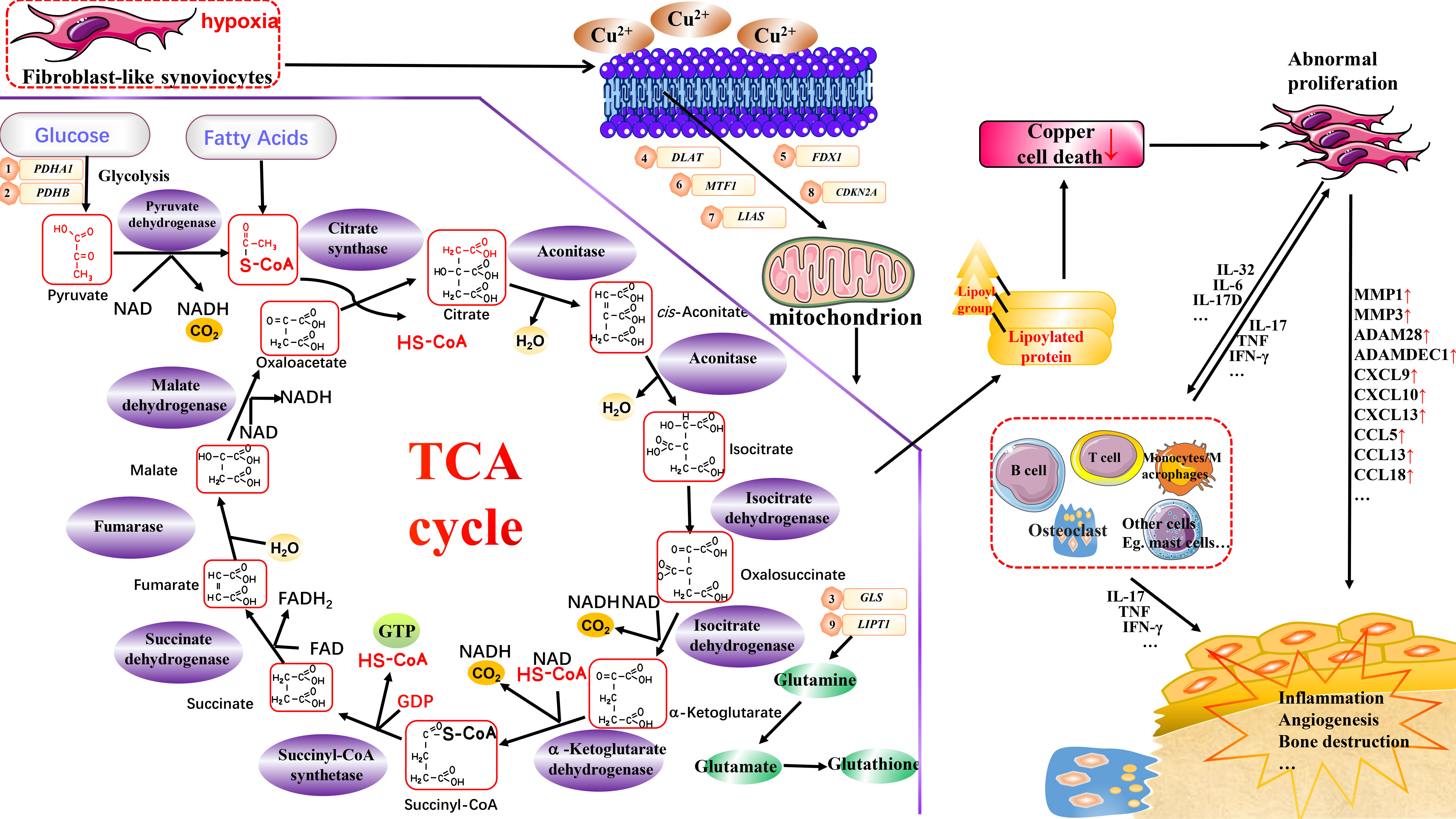
Figure 1 Potential association of cuproptosis and cuproptosis–related genes with RA. Copper can induce cuproptosis by binding to lipid-acylated TCA cycle components, promoting lipid-acylated protein aggregation, and inducing protein stress. This may affect various cells in RA, such as FLS and monocytes/macrophages, thereby aiding in inflammation, angiogenesis, and the bone destruction processes. Vital regulatory genes for cuproptosis are potentially linked to RA through distinct biological functions. However, the specific mechanisms require further investigation.
JZ is responsible for the collection, collation, and writing of the original manuscript. SG, SS, and DH are responsible for the concept development, revision, and manuscript review. All authors reviewed and accepted the final version.
This work was funded by the National Natural Science Funds of China (82074234 and 82071756), National Key Research and Development Project (2018YFC1705200 and 2018YFC1705203), Shanghai Chinese Medicine Development Office, National Administration of Traditional Chinese Medicine, Regional Chinese Medicine (Specialist) Diagnosis and Treatment Center Construction Project-Rheumatology, State Administration of Traditional Chinese Medicine, National TCM Evidence-Based Medicine Research and Construction Project, Basic TCM Evidence-Based Capacity Development Program, Shanghai Municipal Health Commission, and East China Region-based Chinese and Western Medicine Joint Disease Specialist Alliance.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
RA, rheumatoid arthritis; TCA cycle, tricarboxylic acid cycle; NSAIDs, non-steroidal anti-inflammatory drugs; FLS, fibroblast-like synoviocytes; FDX1, ferredoxin 1; LIAS, lipoic acid synthetase; LIPT1, lipoyltransferase 1; DLD, dihydrolipoamide dehydrogenase; DLAT, drolipoamide S-acetyltransferase; PDHA1, pyruvate dehydrogenase E1 subunit alpha 1; PDHB, pyruvate dehydrogenase E1 subunit beta; MTF1, metal-regulatory transcription factor-1; GLS, glutaminase; CDKN2A, cyclin-dependent kinase inhibitor 2A; MMPs, matrix metallopeptidases; IL, interleukin; CCL20, chemokine (C-C motif) ligand 20; FoxO3, forkhead box O-3; PDHC, the pyruvate dehydrogenase (PDH) complex; ATP, adenosine triphosphate; RUNX2, RUNX family transcription factor 2; PKB, phosphorylated protein kinase B; HK2, hexokinase 2; PDHK1, PDH kinases 1; SIRT6, sirtuin 6; PI3K, phosphatidylinositol 3−kinase; AKT, protein kinase B; ERK, the extracellular signal-regulated kinase; NF-κB, nuclear factor-κB; TNFα, tumor necrosis factor α; NLRP3, the nucleotide-binding oligomerization domain (NOD)-like receptor pyrin domain containing 3; MEG3, maternally expressed gene 3; miR, miRNA; GRP78, glucose-regulated protein 78; ATF6, activating transcription factor 6; CHOP, C/EBP homologous protein; NEK10, NIMA-related kinase 10; IFN-γ, interferon gamma; CCR6, chemokine (C-C motif) receptor 6; CXCR3, C-X-C chemokine receptor 3; PPAR-γ, peroxisome proliferator- activated receptor gamma; 2-HG, 2-hydroxyglutarate; KDM5, lysine demethylase 5; H3K4me3, trimethylation of Histone H3 at Lysine 4; ROS, reactive oxygen species; RORγt, retinoic acid-related orphan receptor gamma t; E4F1, E4 transcription factor 1; SIRT4, sirtuin 4; anti-CCP, the antibodies cyclic citrullinated peptides; ESR, erythrocyte sedimentation rate; CRP, C-reactive protein; C1QBP, component 1, q subcomponent-binding protein; Fe/S, iron-sulfur; RANKL, receptor activator of nuclear factor kappa-B ligand; NADPH, nicotinamide adenine dinucleotide phosphate: CYP11A1, cytochrome P450 11A1; MRE, metal responsive element; PIGF, phosphatidylinositol glycan anchor biosynthesis class F; HIF-1, hypoxia-inducible factor-1; FOXP3, forkhead box P3; PHDs, prolyl hydroxylases; ox-LDL, oxidized low-density lipoprotein; NKR, natural killer receptors.

1. Scott DL, Wolfe F, Huizinga TW. Rheumatoid arthritis. Lancet (2010) 376(9746):1094–108. doi: 10.1016/s0140-6736(10)60826-4
2. Zhao J, Guo S, Schrodi SJ, He D. Molecular and cellular heterogeneity in rheumatoid arthritis: Mechanisms and clinical implications. Front Immunol (2021) 12:790122. doi: 10.3389/fimmu.2021.790122
3. McInnes IB, Schett G. Pathogenetic insights from the treatment of rheumatoid arthritis. Lancet (2017) 389(10086):2328–37. doi: 10.1016/s0140-6736(17)31472-1
4. Zhao J, Jiang P, Guo S, Schrodi SJ, He D. Apoptosis, autophagy, NETosis, necroptosis, and pyroptosis mediated programmed cell death as targets for innovative therapy in rheumatoid arthritis. Front Immunol (2021) 12:809806. doi: 10.3389/fimmu.2021.809806
5. Tsvetkov P, Coy S, Petrova B, Dreishpoon M, Verma A, Abdusamad M, et al. Copper induces cell death by targeting lipoylated TCA cycle proteins. Science (2022) 375(6586):1254–61. doi: 10.1126/science.abf0529
6. Xin L, Yang X, Cai G, Fan D, Xia Q, Liu L, et al. Serum levels of copper and zinc in patients with rheumatoid arthritis: a meta-analysis. Biol Trace Elem Res (2015) 168(1):1–10. doi: 10.1007/s12011-015-0325-4
7. Ma Y, Zhang X, Fan D, Xia Q, Wang M, Pan F. Common trace metals in rheumatoid arthritis: A systematic review and meta-analysis. J Trace Elem Med Biol (2019) 56:81–9. doi: 10.1016/j.jtemb.2019.07.007
8. Chakraborty M, Chutia H, Changkakati R. Serum copper as a marker of disease activity in rheumatoid arthritis. J Clin Diagn Res (2015) 9(12):Bc09–11. doi: 10.7860/jcdr/2015/14851.7001
9. Hitchon CA, El-Gabalawy HS, Bezabeh T. Characterization of synovial tissue from arthritis patients: a proton magnetic resonance spectroscopic investigation. Rheumatol Int (2009) 29(10):1205–11. doi: 10.1007/s00296-009-0865-z
10. Ahn JK, Koh EM, Cha HS, Lee YS, Kim J, Bae EK, et al. Role of hypoxia-inducible factor-1alpha in hypoxia-induced expressions of IL-8, MMP-1 and MMP-3 in rheumatoid fibroblast-like synoviocytes. Rheumatol (Oxford) (2008) 47(6):834–9. doi: 10.1093/rheumatology/ken086
11. Bosco MC, Delfino S, Ferlito F, Battaglia F, Puppo M, Gregorio A, et al. Hypoxic synovial environment and expression of macrophage inflammatory protein 3gamma/CCL20 in juvenile idiopathic arthritis. Arthritis Rheumatol (2008) 58(6):1833–8. doi: 10.1002/art.23516
12. Bartok B, Firestein GS. Fibroblast-like synoviocytes: key effector cells in rheumatoid arthritis. Immunol Rev (2010) 233(1):233–55. doi: 10.1111/j.0105-2896.2009.00859.x
13. Bottini N, Firestein GS. Duality of fibroblast-like synoviocytes in RA: passive responders and imprinted aggressors. Nat Rev Rheumatol (2013) 9(1):24–33. doi: 10.1038/nrrheum.2012.190
14. Pan Y, Ai CX, Zeng L, Liu C, Li WC. Modulation of copper-induced antioxidant defense, Cu transport, and mitophagy by hypoxia in the large yellow croaker (Larimichthys crocea). Fish Physiol Biochem (2020) 46(3):997–1010. doi: 10.1007/s10695-020-00765-0
15. Murray PJ, Rathmell J, Pearce E. SnapShot: Immunometabolism. Cell Metab (2015) 22(1):190–.e1. doi: 10.1016/j.cmet.2015.06.014
16. Ciurtin C, Cojocaru VM, Miron IM, Preda F, Milicescu M, Bojincă M, et al. Correlation between different components of synovial fluid and pathogenesis of rheumatic diseases. Rom J Intern Med (2006) 44(2):171–81.
17. Lin HC, Chen YJ, Wei YH, Lin HA, Chen CC, Liu TF, et al. Lactic acid fermentation is required for NLRP3 inflammasome activation. Front Immunol (2021) 12:630380. doi: 10.3389/fimmu.2021.630380
18. Liu Z, Yu M, Fei B, Fang X, Ma T, Wang D. miR−21−5p targets PDHA1 to regulate glycolysis and cancer progression in gastric cancer. Oncol Rep (2018) 40(5):2955–63. doi: 10.3892/or.2018.6695
19. Liu L, Cao J, Zhao J, Li X, Suo Z, Li H. PDHA1 gene knockout in human esophageal squamous cancer cells resulted in greater warburg effect and aggressive features In vitro and in vivo. Onco Targets Ther (2019) 12:9899–913. doi: 10.2147/ott.S226851
20. Xu S, Liu CX, Xu W, Huang L, Zhao JY, Zhao SM. Butyrate induces apoptosis by activating PDC and inhibiting complex I through SIRT3 inactivation. Signal Transduct Target Ther (2017) 2:16035. doi: 10.1038/sigtrans.2016.35
21. Choe M, Brusgard JL, Chumsri S, Bhandary L, Zhao XF, Lu S, et al. The RUNX2 transcription factor negatively regulates SIRT6 expression to alter glucose metabolism in breast cancer cells. J Cell Biochem (2015) 116(10):2210–26. doi: 10.1002/jcb.25171
22. Wei T, Gao J, Huang C, Song B, Sun M, Shen W. SIRT3 (Sirtuin-3) prevents ang II (Angiotensin II)-induced macrophage metabolic switch improving perivascular adipose tissue function. Arterioscler Thromb Vasc Biol (2021) 41(2):714–30. doi: 10.1161/atvbaha.120.315337
23. Kitamura K, Sasaki M, Matsumoto M, Shionoya H, Iida K. Protective effect of bacteroides fragilis LPS on escherichia coli LPS-induced inflammatory changes in human monocytic cells and in a rheumatoid arthritis mouse model. Immunol Lett (2021) 233:48–56. doi: 10.1016/j.imlet.2021.03.008
24. Gu C, Wang F, Zhang YT, Wei SZ, Liu JY, Sun HY, et al. Microglial MT1 activation inhibits LPS-induced neuroinflammation via regulation of metabolic reprogramming. Aging Cell (2021) 20(6):e13375. doi: 10.1111/acel.13375
25. Sun J, Li L, Li L, Ding L, Liu X, Chen X, et al. Metallothionein-1 suppresses rheumatoid arthritis pathogenesis by shifting the Th17/Treg balance. Eur J Immunol (2018) 48(9):1550–62. doi: 10.1002/eji.201747151
26. Huang CC, Chiou CH, Liu SC, Hu SL, Su CM, Tsai CH, et al. Melatonin attenuates TNF-α and IL-1β expression in synovial fibroblasts and diminishes cartilage degradation: Implications for the treatment of rheumatoid arthritis. J Pineal Res (2019) 66(3):e12560. doi: 10.1111/jpi.12560
27. Li A, Zhang Y, Zhao Z, Wang M, Zan L. Molecular characterization and transcriptional regulation analysis of the bovine PDHB gene. PloS One (2016) 11(7):e0157445. doi: 10.1371/journal.pone.0157445
28. Wu C, Tan S, Liu L, Cheng S, Li P, Li W, et al. Transcriptome-wide association study identifies susceptibility genes for rheumatoid arthritis. Arthritis Res Ther (2021) 23(1):38. doi: 10.1186/s13075-021-02419-9
29. Danileviciute E, Zeng N, Capelle CM, Paczia N, Gillespie MA, Kurniawan H, et al. PARK7/DJ-1 promotes pyruvate dehydrogenase activity and maintains t(reg) homeostasis during ageing. Nat Metab (2022) 4(5):589–607. doi: 10.1038/s42255-022-00576-y
30. Wang G, Ye Q, Ning S, Yang Z, Chen Y, Zhang L, et al. LncRNA MEG3 promotes endoplasmic reticulum stress and suppresses proliferation and invasion of colorectal carcinoma cells through the MEG3/miR-103a-3p/PDHB ceRNA pathway. Neoplasma (2021) 68(2):362–74. doi: 10.4149/neo_2020_200813N858
31. Zhu Y, Wu G, Yan W, Zhan H, Sun P. miR-146b-5p regulates cell growth, invasion, and metabolism by targeting PDHB in colorectal cancer. Am J Cancer Res (2017) 7(5):1136–50.
32. Xu DX, Guo JJ, Zhu GY, Wu HJ, Zhang QS, Cui T. MiR-363-3p modulates cell growth and invasion in glioma by directly targeting pyruvate dehydrogenase b. Eur Rev Med Pharmacol Sci (2018) 22(16):5230–9. doi: 10.26355/eurrev_201808_15721
33. Xiaohong Z, Lichun F, Na X, Kejian Z, Xiaolan X, Shaosheng W. MiR-203 promotes the growth and migration of ovarian cancer cells by enhancing glycolytic pathway. Tumour Biol (2016) 37(11):14989–97. doi: 10.1007/s13277-016-5415-1
34. Tang H, Luo X, Li J, Zhou Y, Li Y, Song L, et al. Pyruvate dehydrogenase b promoted the growth and migration of the nasopharyngeal carcinoma cells. Tumour Biol (2016) 37(8):10563–9. doi: 10.1007/s13277-016-4922-4
35. Peres de Oliveira A, Basei FL, Slepicka PF, de Castro Ferezin C, Melo-Hanchuk TD, de Souza EE, et al. NEK10 interactome and depletion reveal new roles in mitochondria. Proteome Sci (2020) 18:4. doi: 10.1186/s12953-020-00160-w
36. Chang C, Xu L, Zhang R, Jin Y, Jiang P, Wei K, et al. MicroRNA-mediated epigenetic regulation of rheumatoid arthritis susceptibility and pathogenesis. Front Immunol (2022) 13:838884. doi: 10.3389/fimmu.2022.838884
37. Taylor L, Curthoys NP. Glutamine metabolism: Role in acid-base balance*. Biochem Mol Biol Educ (2004) 32(5):291–304. doi: 10.1002/bmb.2004.494032050388
38. Takahashi S, Saegusa J, Sendo S, Okano T, Akashi K, Irino Y, et al. Glutaminase 1 plays a key role in the cell growth of fibroblast-like synoviocytes in rheumatoid arthritis. Arthritis Res Ther (2017) 19(1):76. doi: 10.1186/s13075-017-1283-3
39. Sener Z, Cederkvist FH, Volchenkov R, Holen HL, Skålhegg BS. T Helper cell activation and expansion is sensitive to glutaminase inhibition under both hypoxic and normoxic conditions. PloS One (2016) 11(7):e0160291. doi: 10.1371/journal.pone.0160291
40. Meitei HT, Jadhav N, Lal G. CCR6-CCL20 axis as a therapeutic target for autoimmune diseases. Autoimmun Rev (2021) 20(7):102846. doi: 10.1016/j.autrev.2021.102846
41. Lacotte S, Brun S, Muller S, Dumortier H. CXCR3, inflammation, and autoimmune diseases. Ann N Y Acad Sci (2009) 1173:310–7. doi: 10.1111/j.1749-6632.2009.04813.x
42. Sun L, Fu J, Zhou Y. Metabolism controls the balance of Th17/T-regulatory cells. Front Immunol (2017) 8:1632. doi: 10.3389/fimmu.2017.01632
43. Li XF, Sun YY, Bao J, Chen X, Li YH, Yang Y, et al. Functional role of PPAR-γ on the proliferation and migration of fibroblast-like synoviocytes in rheumatoid arthritis. Sci Rep (2017) 7(1):12671. doi: 10.1038/s41598-017-12570-6
44. Miao Y, Zheng Y, Geng Y, Yang L, Cao N, Dai Y, et al. The role of GLS1-mediated glutaminolysis/2-HG/H3K4me3 and GSH/ROS signals in Th17 responses counteracted by PPARγ agonists. Theranostics (2021) 11(9):4531–48. doi: 10.7150/thno.54803
45. Krishnamoorthy E, Hassan S, Hanna LE, Padmalayam I, Rajaram R, Viswanathan V. Homology modeling of homo sapiens lipoic acid synthase: Substrate docking and insights on its binding mode. J Theor Biol (2017) 420:259–66. doi: 10.1016/j.jtbi.2016.09.005
46. Shay KP, Moreau RF, Smith EJ, Smith AR, Hagen TM. Alpha-lipoic acid as a dietary supplement: molecular mechanisms and therapeutic potential. Biochim Biophys Acta (2009) 1790(10):1149–60. doi: 10.1016/j.bbagen.2009.07.026
47. King A, Selak MA, Gottlieb E. Succinate dehydrogenase and fumarate hydratase: linking mitochondrial dysfunction and cancer. Oncogene (2006) 25(34):4675–82. doi: 10.1038/sj.onc.1209594
48. Wójcik P, Gęgotek A, Žarković N, Skrzydlewska E. Oxidative stress and lipid mediators modulate immune cell functions in autoimmune diseases. Int J Mol Sci (2021) 22(2):723. doi: 10.3390/ijms22020723
49. Yi X, Kim K, Yuan W, Xu L, Kim HS, Homeister JW, et al. Mice with heterozygous deficiency of lipoic acid synthase have an increased sensitivity to lipopolysaccharide-induced tissue injury. J Leukoc Biol (2009) 85(1):146–53. doi: 10.1189/jlb.0308161
50. Tian S, Nakamura J, Hiller S, Simington S, Holley DW, Mota R, et al. New insights into immunomodulation via overexpressing lipoic acid synthase as a therapeutic potential to reduce atherosclerosis. Vascul Pharmacol (2020) 133-134:106777. doi: 10.1016/j.vph.2020.106777
51. Luo J, Shen S. Lipoic acid alleviates schistosomiasis-induced liver fibrosis by upregulating Drp1 phosphorylation. Acta Trop (2020) 206:105449. doi: 10.1016/j.actatropica.2020.105449
52. Xu G, Yan T, Peng Q, Li H, Wu W, Yi X, et al. Overexpression of the lias gene attenuates hepatic steatosis in leprdb/db mice. J Endocrinol (2021) 248(2):119–31. doi: 10.1530/joe-19-0606
53. Zhao Y, Xu G, Li H, Chang M, Guan Y, Li Y, et al. Overexpression of endogenous lipoic acid synthase attenuates pulmonary fibrosis induced by crystalline silica in mice. Toxicol Lett (2020) 323:57–66. doi: 10.1016/j.toxlet.2020.01.023
54. Burr SP, Costa AS, Grice GL, Timms RT, Lobb IT, Freisinger P, et al. Mitochondrial protein lipoylation and the 2-oxoglutarate dehydrogenase complex controls HIF1α stability in aerobic conditions. Cell Metab (2016) 24(5):740–52. doi: 10.1016/j.cmet.2016.09.015
55. Rodier G, Kirsh O, Baraibar M, Houlès T, Lacroix M, Delpech H, et al. The transcription factor E4F1 coordinates CHK1-dependent checkpoint and mitochondrial functions. Cell Rep (2015) 11(2):220–33. doi: 10.1016/j.celrep.2015.03.024
56. Taghadosi M, Adib M, Jamshidi A, Mahmoudi M, Farhadi E. The p53 status in rheumatoid arthritis with focus on fibroblast-like synoviocytes. Immunol Res (2021) 69(3):225–38. doi: 10.1007/s12026-021-09202-7
57. Lacroix M, Rodier G, Kirsh O, Houles T, Delpech H, Seyran B, et al. E4F1 controls a transcriptional program essential for pyruvate dehydrogenase activity. Proc Natl Acad Sci USA (2016) 113(39):10998–1003. doi: 10.1073/pnas.1602754113
58. Hussain MZ, Haris MS, Khan MS, Mahjabeen I. Role of mitochondrial sirtuins in rheumatoid arthritis. Biochem Biophys Res Commun (2021) 584:60–5. doi: 10.1016/j.bbrc.2021.11.016
59. Mathias RA, Greco TM, Oberstein A, Budayeva HG, Chakrabarti R, Rowland EA, et al. Sirtuin 4 is a lipoamidase regulating pyruvate dehydrogenase complex activity. Cell (2014) 159(7):1615–25. doi: 10.1016/j.cell.2014.11.046
60. Chen R, Xiao M, Gao H, Chen Y, Li Y, Liu Y, et al. Identification of a novel mitochondrial interacting protein of C1QBP using subcellular fractionation coupled with CoIP-MS. Anal Bioanal Chem (2016) 408(6):1557–64. doi: 10.1007/s00216-015-9228-7
61. de Seny D, Bianchi E, Baiwir D, Cobraiville G, Collin C, Deliège M, et al. Proteins involved in the endoplasmic reticulum stress are modulated in synovitis of osteoarthritis, chronic pyrophosphate arthropathy and rheumatoid arthritis, and correlate with the histological inflammatory score. Sci Rep (2020) 10(1):14159. doi: 10.1038/s41598-020-70803-7
62. Sheftel AD, Stehling O, Pierik AJ, Elsässer HP, Mühlenhoff U, Webert H, et al. Humans possess two mitochondrial ferredoxins, Fdx1 and Fdx2, with distinct roles in steroidogenesis, heme, and Fe/S cluster biosynthesis. Proc Natl Acad Sci U S A (2010) 107(26):11775–80. doi: 10.1073/pnas.1004250107
63. Roumaud P, Rwigemera A, Martin LJ. Transcription factors SF1 and cJUN cooperate to activate the Fdx1 promoter in MA-10 leydig cells. J Steroid Biochem Mol Biol (2017) 171:121–32. doi: 10.1016/j.jsbmb.2017.03.003
64. Zhang Z, Ma Y, Guo X, Du Y, Zhu Q, Wang X, et al. FDX1 can impact the prognosis and mediate the metabolism of lung adenocarcinoma. Front Pharmacol (2021) 12:749134. doi: 10.3389/fphar.2021.749134
65. Yoon N, Jang AK, Seo Y, Jung BH. Metabolomics in autoimmune diseases: Focus on rheumatoid arthritis, systemic lupus erythematous, and multiple sclerosis. Metabolites (2021) 11(12):812. doi: 10.3390/metabo11120812
66. Yousri NA, Bayoumy K, Elhaq WG, Mohney RP, Emadi SA, Hammoudeh M, et al. Large Scale metabolic profiling identifies novel steroids linked to rheumatoid arthritis. Sci Rep (2017) 7(1):9137. doi: 10.1038/s41598-017-05439-1
67. Straub RH, Weidler C, Demmel B, Herrmann M, Kees F, Schmidt M, et al. Renal clearance and daily excretion of cortisol and adrenal androgens in patients with rheumatoid arthritis and systemic lupus erythematosus. Ann Rheum Dis (2004) 63(8):961–8. doi: 10.1136/ard.2003.014274
68. Postlethwaite AE, Tuckey RC, Kim TK, Li W, Bhattacharya SK, Myers LK, et al. 20S-hydroxyvitamin D3, a secosteroid produced in humans, is anti-inflammatory and inhibits murine autoimmune arthritis. Front Immunol (2021) 12:678487. doi: 10.3389/fimmu.2021.678487
69. Balamurugan K, Schaffner W. Copper homeostasis in eukaryotes: teetering on a tightrope. Biochim Biophys Acta (2006) 1763(7):737–46. doi: 10.1016/j.bbamcr.2006.05.001
70. Chen GH, Lv W, Xu YH, Wei XL, Xu YC, Luo Z. Functional analysis of MTF-1 and MT promoters and their transcriptional response to zinc (Zn) and copper (Cu) in yellow catfish pelteobagrus fulvidraco. Chemosphere (2020) 246:125792. doi: 10.1016/j.chemosphere.2019.125792
71. Selvaraj A, Balamurugan K, Yepiskoposyan H, Zhou H, Egli D, Georgiev O, et al. Metal-responsive transcription factor (MTF-1) handles both extremes, copper load and copper starvation, by activating different genes. Genes Dev (2005) 19(8):891–6. doi: 10.1101/gad.1301805
72. Fukai T, Ushio-Fukai M, Kaplan JH. Copper transporters and copper chaperones: roles in cardiovascular physiology and disease. Am J Physiol Cell Physiol (2018) 315(2):C186–c201. doi: 10.1152/ajpcell.00132.2018
73. Tsuchiya H, Ota M, Sumitomo S, Ishigaki K, Suzuki A, Sakata T, et al. Parsing multiomics landscape of activated synovial fibroblasts highlights drug targets linked to genetic risk of rheumatoid arthritis. Ann Rheum Dis (2020) 80(4):440–50. doi: 10.1136/annrheumdis-2020-218189
74. Green CJ, Lichtlen P, Huynh NT, Yanovsky M, Laderoute KR, Schaffner W, et al. Placenta growth factor gene expression is induced by hypoxia in fibroblasts: a central role for metal transcription factor-1. Cancer Res (2001) 61(6):2696–703.
75. Murphy BJ, Sato BG, Dalton TP, Laderoute KR. The metal-responsive transcription factor-1 contributes to HIF-1 activation during hypoxic stress. Biochem Biophys Res Commun (2005) 337(3):860–7. doi: 10.1016/j.bbrc.2005.09.124
76. Gaber T, Häupl T, Sandig G, Tykwinska K, Fangradt M, Tschirschmann M, et al. Adaptation of human CD4+ T cells to pathophysiological hypoxia: a transcriptome analysis. J Rheumatol (2009) 36(12):2655–69. doi: 10.3899/jrheum.090255
77. Dang EV, Barbi J, Yang HY, Jinasena D, Yu H, Zheng Y, et al. Control of T(H)17/T(reg) balance by hypoxia-inducible factor 1. Cell (2011) 146(5):772–84. doi: 10.1016/j.cell.2011.07.033
78. Tavera-Montañez C, Hainer SJ, Cangussu D, Gordon SJV, Xiao Y, Reyes-Gutierrez P, et al. The classic metal-sensing transcription factor MTF1 promotes myogenesis in response to copper. FASEB J (2019) 33(12):14556–74. doi: 10.1096/fj.201901606R
79. LaRochelle O, Gagné V, Charron J, Soh JW, Séguin C. Phosphorylation is involved in the activation of metal-regulatory transcription factor 1 in response to metal ions. J Biol Chem (2001) 276(45):41879–88. doi: 10.1074/jbc.M108313200
80. Stalke A, Pfister ED, Baumann U, Illig T, Reischl E, Sandbothe M, et al. MTF1 binds to metal-responsive element e within the ATP7B promoter and is a strong candidate in regulating the ATP7B expression. Ann Hum Genet (2020) 84(2):195–200. doi: 10.1111/ahg.12355
81. Han H, Nakaoka HJ, Hofmann L, Zhou JJ, Yu C, Zeng L, et al. The hippo pathway kinases LATS1 and LATS2 attenuate cellular responses to heavy metals through phosphorylating MTF1. Nat Cell Biol (2022) 24(1):74–87. doi: 10.1038/s41556-021-00813-8
82. Uyar B, Palmer D, Kowald A, Murua Escobar H, Barrantes I, Möller S, et al. Single-cell analyses of aging, inflammation and senescence. Ageing Res Rev (2020) 64:101156. doi: 10.1016/j.arr.2020.101156
83. Goronzy JJ, Henel G, Sawai H, Singh K, Lee EB, Pryshchep S, et al. Costimulatory pathways in rheumatoid synovitis and T-cell senescence. Ann N Y Acad Sci (2005) 1062:182–94. doi: 10.1196/annals.1358.022
84. Covre LP, De Maeyer RPH, Gomes DCO, Akbar AN. The role of senescent T cells in immunopathology. Aging Cell (2020) 19(12):e13272. doi: 10.1111/acel.13272
85. Horiuchi M, Morinobu A, Chin T, Sakai Y, Kurosaka M, Kumagai S. Expression and function of histone deacetylases in rheumatoid arthritis synovial fibroblasts. J Rheumatol (2009) 36(8):1580–9. doi: 10.3899/jrheum.081115
86. Nishida K, Komiyama T, Miyazawa S, Shen ZN, Furumatsu T, Doi H, et al. Histone deacetylase inhibitor suppression of autoantibody-mediated arthritis in mice via regulation of p16INK4a and p21(WAF1/Cip1) expression. Arthritis Rheumatol (2004) 50(10):3365–76. doi: 10.1002/art.20709
87. Del Rey MJ, Valín Á, Usategui A, Ergueta S, Martín E, Municio C, et al. Senescent synovial fibroblasts accumulate prematurely in rheumatoid arthritis tissues and display an enhanced inflammatory phenotype. Immun Ageing (2019) 16:29. doi: 10.1186/s12979-019-0169-4
88. Trenkmann M, Brock M, Gay RE, Kolling C, Speich R, Michel BA, et al. Expression and function of EZH2 in synovial fibroblasts: epigenetic repression of the wnt inhibitor SFRP1 in rheumatoid arthritis. Ann Rheum Dis (2011) 70(8):1482–8. doi: 10.1136/ard.2010.143040
89. Rhoads JP, Lukens JR, Wilhelm AJ, Moore JL, Mendez-Fernandez Y, Kanneganti TD, et al. Oxidized low-density lipoprotein immune complex priming of the Nlrp3 inflammasome involves TLR and FcγR cooperation and is dependent on CARD9. J Immunol (2017) 198(5):2105–14. doi: 10.4049/jimmunol.1601563
90. Yan L, Liu Z, Yin H, Guo Z, Luo Q. Silencing of MEG3 inhibited ox-LDL-induced inflammation and apoptosis in macrophages via modulation of the MEG3/miR-204/CDKN2A regulatory axis. Cell Biol Int (2019) 43(4):409–20. doi: 10.1002/cbin.11105
91. Murakami Y, Mizoguchi F, Saito T, Miyasaka N, Kohsaka H. p16(INK4a) exerts an anti-inflammatory effect through accelerated IRAK1 degradation in macrophages. J Immunol (2012) 189(10):5066–72. doi: 10.4049/jimmunol.1103156
92. Martínez-Hervás S, Sánchez-García V, Herrero-Cervera A, Vinué Á, Real JT, Ascaso JF, et al. Type 1 diabetic mellitus patients with increased atherosclerosis risk display decreased CDKN2A/2B/2BAS gene expression in leukocytes. J Transl Med (2019) 17(1):222. doi: 10.1186/s12967-019-1977-1
93. Xiao XY, Li YT, Jiang X, Ji X, Lu X, Yang B, et al. EZH2 deficiency attenuates treg differentiation in rheumatoid arthritis. J Autoimmun (2020) 108:102404. doi: 10.1016/j.jaut.2020.102404
94. Jacobsen JA, Woodard J, Mandal M, Clark MR, Bartom ET, Sigvardsson M, et al. EZH2 regulates the developmental timing of effectors of the pre-antigen receptor checkpoints. J Immunol (2017) 198(12):4682–91. doi: 10.4049/jimmunol.1700319
95. Ni M, Solmonson A, Pan C, Yang C, Li D, Notzon A, et al. Functional assessment of lipoyltransferase-1 deficiency in cells, mice, and humans. Cell Rep (2019) 27(5):1376–86.e6. doi: 10.1016/j.celrep.2019.04.005
96. Mayr JA, Feichtinger RG, Tort F, Ribes A, Sperl W. Lipoic acid biosynthesis defects. J Inherit Metab Dis (2014) 37(4):553–63. doi: 10.1007/s10545-014-9705-8
Keywords: rheumatoid arthritis, autoimmune disease, inflammation, cuproptosis, cuproptosis-related genes
Citation: Zhao J, Guo S, Schrodi SJ and He D (2022) Cuproptosis and cuproptosis–related genes in rheumatoid arthritis: Implication, prospects, and perspectives. Front. Immunol. 13:930278. doi: 10.3389/fimmu.2022.930278
Received: 27 April 2022; Accepted: 18 July 2022;
Published: 04 August 2022.
Edited by:
Shanshan Hu, Anhui Medical University, ChinaReviewed by:
Weinan Zhou, University of Illinois at Urbana-Champaign, United StatesCopyright © 2022 Zhao, Guo, Schrodi and He. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Shicheng Guo, U2hpY2hlbmcuR3VvQHdpc2MuZWR1; Steven J. Schrodi, U2Nocm9kaUB3aXNjLmVkdQ==; Dongyi He, ZG9uZ3lpaGVAbWVkbWFpbC5jb20uY24=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.