
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol., 28 July 2022
Sec. Alloimmunity and Transplantation
Volume 13 - 2022 | https://doi.org/10.3389/fimmu.2022.928173
This article is part of the Research TopicProgress of Allo- and Xeno-transplantation Facilitating the Initial Xeno-Kidney and Islet Clinical TrialsView all 16 articles
 Qiao Zhou1,2,3†
Qiao Zhou1,2,3† Ting Li4†
Ting Li4† Kaiwen Wang5
Kaiwen Wang5 Qi Zhang6
Qi Zhang6 Zhuowen Geng5
Zhuowen Geng5 Shaoping Deng2,3,7
Shaoping Deng2,3,7 Chunming Cheng8*
Chunming Cheng8* Yi Wang9*
Yi Wang9*Transplantation is often the last resort for end-stage organ failures, e.g., kidney, liver, heart, lung, and pancreas. The shortage of donor organs is the main limiting factor for successful transplantation in humans. Except living donations, other alternatives are needed, e.g., xenotransplantation of pig organs. However, immune rejection remains the major challenge to overcome in xenotransplantation. There are three different xenogeneic types of rejections, based on the responses and mechanisms involved. It includes hyperacute rejection (HAR), delayed xenograft rejection (DXR) and chronic rejection. DXR, sometimes involves acute humoral xenograft rejection (AHR) and cellular xenograft rejection (CXR), which cannot be strictly distinguished from each other in pathological process. In this review, we comprehensively discussed the mechanism of these immunological rejections and summarized the strategies for preventing them, such as generation of gene knock out donors by different genome editing tools and the use of immunosuppressive regimens. We also addressed organ-specific barriers and challenges needed to pave the way for clinical xenotransplantation. Taken together, this information will benefit the current immunological research in the field of xenotransplantation.
Organ failures, which are usually a consequence of diseases, trauma, or alcohol/drug abuse, represent the top causes of mortality in most population groups (1). A recent population-based cohort study covering a 3-year period and involving 9,187 adult patients admitted at the emergency department of the Odense University Hospital, Denmark, indicated that the one-year all-cause mortality of organ failure was 29.8% (2). Another study, involving a cohort of 1,023 patients sequentially admitted at ten Scottish intensive care units, revealed a one-year overall mortality of 46.5% (3). Organ transplantation is the ideal treatment for most end-stage organ failure affecting the heart, lungs, kidneys, liver, and in certain cases, the pancreas. However, the demand of human organs for transplantation purpose exceeds by far the number of organ donations, which limits this procedure in general clinical practice. The World Health Organization (Geneva) estimates that only 10% of the worldwide need for organ transplantation is being met (4). Several countries around the world have implemented different strategies to overcome human organ shortage, including financial incentives in the United States and China (5, 6), or campaigns to increase public awareness (7). France and other European countries recently declared all citizen as donors by default in case of death or brain death unless they opted out during their lifetime. Other approaches are also explored to enable the use of animals’ organs and tissues, and are referred to as xenotransplantation. In addition to addressing organ shortage issues, xenotransplants could allow for tailored transplantation and meet specific patients’ needs, or offer more flexible schedules than that imposed by organ donations, usually performed on short notice (8).
The prominent pathobiological barriers to successful clinical use of xenotransplantation include the rapid activation of innate cellular responses against the graft, and at later stage, further rejection of the organ by the adaptive immune system (9, 10). As important, coagulation dysregulation and inflammation intervene in the rejection process. The long-term survival of the grafts depends on how to reduce or even avoid the occurrence of rejection, which in turn requires a deep understanding of these different rejection mechanisms. In this review, we illustrate in detail from current literatures these different mechanisms in rejection processes and the corresponding strategies to overcome them. We hope this comprehensive overview will bring new insights into current immunological research related to xenotransplantation and future directions towards its application.
HAR generally defines as a graft destruction occurring within 24 hours and that usually lasts for few minutes to hours. It is caused by the binding of human or non-human primate (NHP) pre-existing antibodies against graft antigens (Figure 1A) (11). Among these antibodies, most frequent IgMs and IgGs recognize galactose-α 1,3-galactose (α-Gal) residues added on glycoproteins and glycolipids by the α1,3 galactosyltransferase (α1,3GT) present in the genomes of non-primates and New World monkeys (platyrrhine primates living in South and Central America, including howlers, spider monkeys, and woolly monkeys) (12, 13). Humans, Old World monkeys (catarrhine primates living in African, Asian and Europe, including baboons, colobuses and mandrills), and apes lack α-Gal epitopes because their α1,3GT gene is affected by a loss-of-function mutation (14). In addition, about 70–90% of the antibodies produced by these species target α-Gal epitopes specifically (15). Consequently, when a pig organ is transplanted into a human or a NHP, the pre-existing anti-Gal antibodies bind to α-Gal epitopes present on the graft’s vascular endothelium, and induce complement component 3b (C3b) production, complements activation (16), and formation of a membrane attacking complex (MAC). These reactions cause endothelial cells lysis, destruction of the vasculature, and ultimately, graft rejection (17, 18). The loss of endothelial vascular integrity further leads to interstitial haemorrhage, tissue ischemia, and necrosis (19, 20). Moreover, capillaries thrombotic occlusion, fibrinoid necrosis of arterial walls, and neutrophils accumulation contribute to graft failure (21). Nitric oxide species (NOS), reactive oxygen species (ROS), and other free radicals are also key components of the rejection process. The histopathological features of HAR characterize by disruption of vascular integrity, oedema, fibrin-platelet rich thrombi, and interstitial haemorrhage with widespread deposition of immunoglobulins and terminal complement products on vessel walls (21, 22).
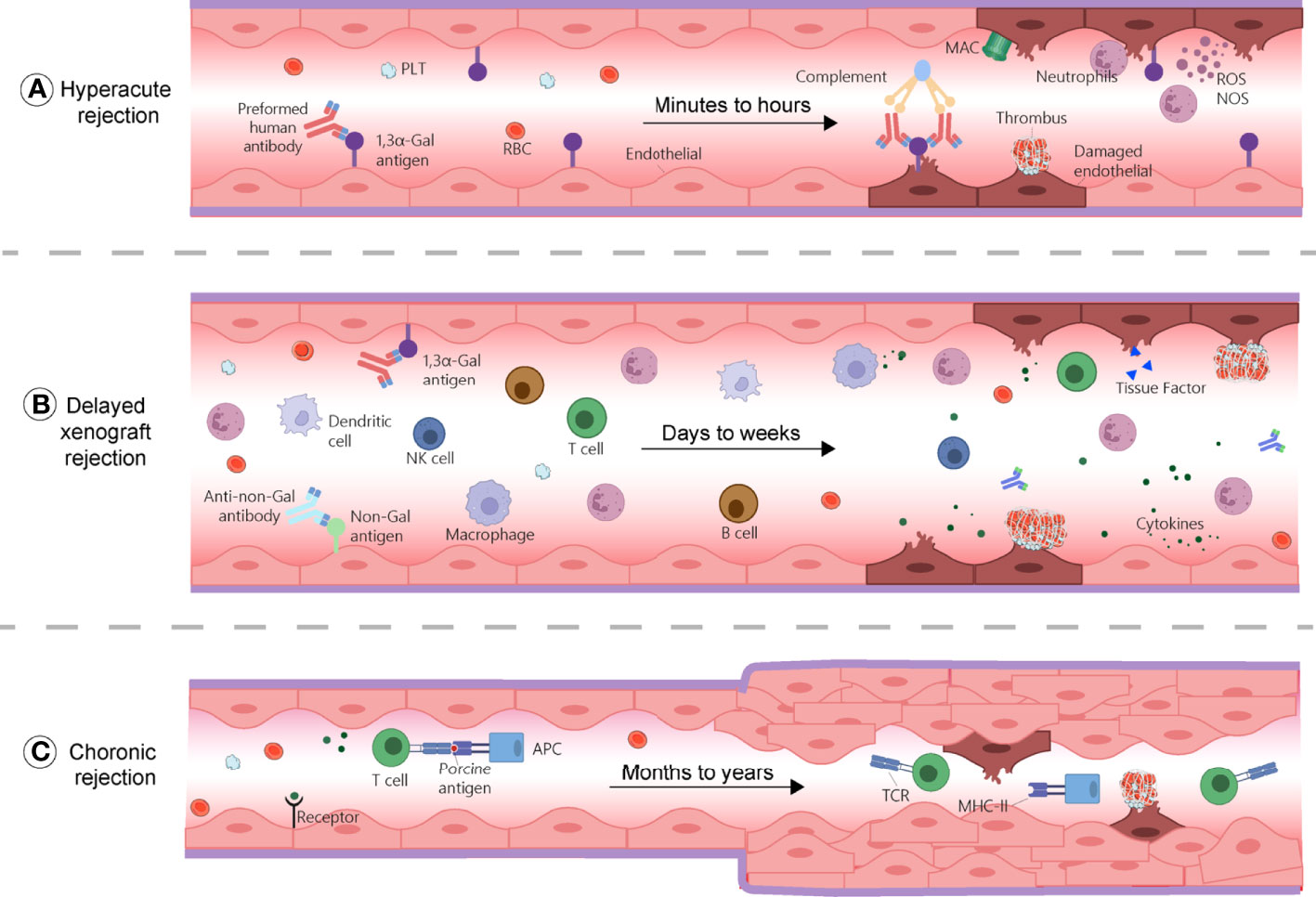
Figure 1 Mechanisms of rejections during xenotransplantation. (A) Hyperacute rejection occurs within minutes to hours and is caused by the binding of the host’s pre-existing antibodies to α-Gal antigens on the graft, which results in complement activation and membrane attacking complex (MAC) formation. This reaction causes endothelial cells lysis, fibrinoid occlusion, and vasculature destruction. Neutrophils, through the production of ROS and NOS also contribute to this process. (B) Delayed xenograft rejection (DXR) occurs within days to weeks and include acute humoral xenograft rejection (AHXR), cellular xenograft rejection, and coagulation dysregulation. AHXR is antibody-mediated and involve non-Gal antibodies and α-Gal antibodies reactivity against non-Gal epitopes and α-Gal of the graft. Various innate and adaptive immune cells, proinflammatory cytokines, and coagulation dysregulation contribute to rejection, resulting in massive deposition of immunoglobins, fibrin, endothelial cell lysis, and interstitial bleeding. (C) Chronic rejection occurs within months to years. Xenoantigens are surveyed by host APCs and presented to T cells, leading to their activation and triggering inflammatory cascades, characterized by thrombotic microangiopathy, proliferation of the graft vascular endothelial cells, vessel narrowing, and interstitial fibrosis. APC, antigen presenting cell; MAC, membrane attacking complex; MHC-II, major histocompatibility complex class II; NK cell, natural killer cell; NOS, nitric oxide species; PLT, platelet; RBC, red blood cells; ROS, reactive oxygen species; TCR, T cell receptor.
Two ways are usually taken to prevent HAR. One consists in knocking out the α1,3GT gene in pigs (GTKO pigs) (23, 24), while the other relies on inhibiting complement activation by inducing the expression of human complement-regulatory proteins, i.e., hCD46, hCD55, and hCD59, on pig cells (25). Kuwaki et al. reported that the elimination of the α-Gal epitopes successfully averted HAR in baboons receiving hearts from GTKO pigs (n = 8) and increased the pig heart survival by 2–6 months (median, 78 d) (26). In a study using pig liver transplant to NHPs, genetically engineered expression of hCD55 or of hCD46 combined with GTKO was associated with a survival time of seven to nine days while the wild-type (WT) liver graft did not extend over three days (27, 28). Combination with GTKO, hCD46 or hCD55 expression reduced the early graft failure (signs of early transplantation failure within three days of transplantation) to 7% compared to 43% with GTKO organs (29).
Although these measures allow the grafts to survive beyond 24 hours, graft failure can still result from antibody recovery, through a mechanism termed acute humoral xenograft rejection (AHXR), acute vascular rejection, or delayed xenograft rejection (DXR), as explained below (20).
Delayed xenograft rejection refers to post-HAR and is also called acute humoral xenograft rejection from a mechanistic prospect, or acute vascular xenograft rejection (AVXR) from a pathophysiological prospect. AHXR or AVXR define xenograft injuries occurring within the vasculature and involving antibodies, while complements play a minor role during this type of rejection (21). In other articles, DXR may refer to antibody- and complement-independent cellular xenograft rejection (CXR) (30). Although there are clear differences between the mechanisms underlying these different terms, these are often used interchangeably and there is no international standard. Some efforts should be considered to achieve internationally recognized definitions and descriptions of these different concepts. Below, we discuss the concepts of AHXR and CXR in separate sections.
Provided that a xenograft does not fail due to HAR, a second step consist in overcoming AHXR, which causes immunological destruction within a few days to a few weeks (Figure 1B) (14). The histological characteristics of AHXR are focal ischemia and diffuse intravascular coagulation mediated by both humoral and cellar immune responses provoking endothelial cell activation and exaggerated inflammation (31, 32). Lin et al. reported that anti-Gal antibodies removal from the blood of baboons prevents AHXR of porcine organs transgenic for human decay-accelerating factor and CD59, demonstrating that Gal-specific antibodies were implicated in AHXR, in addition to HAR (33). However, antibody-meditated rejection still occurred during transplantation of GTKO pigs’ kidneys or hearts into NHPs, and eventually led to graft failure over the course of several days (34, 35). These results suggest that non-Gal antigens also contribute to AHXR (34, 35).The presence in the recipient of pre-existing antibodies against non-Gal epitopes, such as carbohydrate N-glycolyl neuraminic sialic acid (Neu5Gc), glycan SDa, defining the blood group of the same name, was also implicated in rejection (Figure 2) (36–38). Zhu et al. identified Neu5Gc as a crucial non-Gal xenoantigen in 2002 (36), while the SDa blood group was discovered in 1967 (39). Neu5Ac is hydroxylated to produce Neu5Gc by the cytidine monophosphate-N-acetylneuraminic acid hydroxylase (CMAH) present in pigs and other animals, but not in humans (40). The gene encoding this enzyme has been inactivated during primate evolution (41). Double knockout (DKO) of the genes responsible for Gal and NeuGc synthesis in mouse reduced significantly xenoreactive antibody-mediated complement-dependent cytotoxicity of human sera towards mouse tissues, compared to wild-type mouse tissues. The gene encoding the Beta-1,4-N-acetyl-galactosaminyltransferase 2 (β4GALNT2) is responsible for the SDa positive blood group. Its inactivation can significantly diminish porcine xenoantigenicity and reduce the effects of human and NHP non-Gal antibodies (42). Therefore, Neu5Gc and SDa represent key targets for clinical xenotransplantation. Other non-Gal antigens such as Gabarapl1 (GABA type A receptor-associated protein like1), and COX-2 can also induce antibody-mediated rejection (43). In addition, shared common epitopes of human leucocyte antigen (HLA) and swine leucocyte antigen class I (SLA-I) could lead to cross-reactivity between human and porcine (44). Some studies showed that human CD8+T cells were capable of recognizing SLA-I and elicited immune responses and anti-HLA class II antibodies in patients could cross-react with SLA-II (45–48). Immunoengineering of the vascular endothelium to silence SLA expression might be feasible to reduce the immunogenicity (49–52).
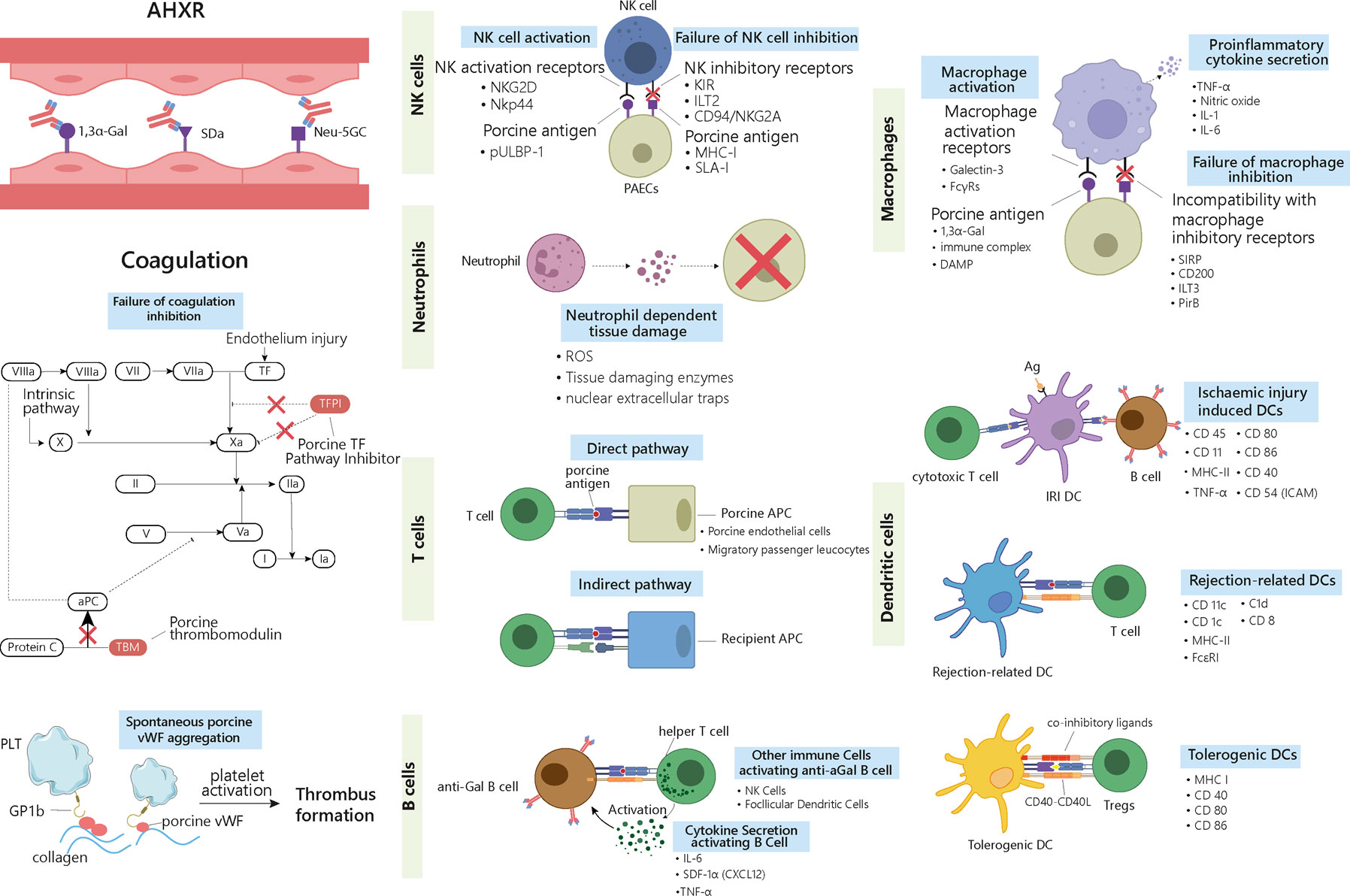
Figure 2 Delayed xenograft rejection: AHXR, cellular xenograft rejection and coagulation dysregulation. The antibodies involved in AHXR are mainly directed against 1,3α-Gal epitopes and non-Gal epitopes such as Neu-5GC and SDa blood group. During cellular responses, ligands on the graft cells activate recipients’ NK cells and macrophages via different activating receptors. Meanwhile, graft cells fail to deliver inhibitory signals to the activated cells and enhance their proinflammatory and cytotoxic properties. Neutrophils can generate ROS, tissue-digesting enzyme, and NETs upon activation by graft cells, which causes tissue damage. Three types of DCs are involved in graft rejection. IRI-related DCs activate both cytotoxic T cells and B cells, thus triggering both cell- and antibody-mediated rejection. Rejection-related DCs promote acute and chronic rejection by activating T cells. Tolerogenic DCs suppress immune rejection by inducing Tregs. The different surface markers characterizing the different immune cells are listed in the right-hand side of the cells. T cells are involved in rejection through both direct and indirect pathways. In the direct pathway, porcine APCs present antigens and activate host’s T cells. In the indirect pathway, the graft antigens are presented by the host’s APCs. B cells are activated by T helper cells and secreted cytokines. Antibodies released by activated B cells that have differentiated into plasma cells contribute to graft rejection. AHXR and cellular xenograft rejection are both accompanied with coagulation dysregulation, where porcine TFPI cannot fully inhibit factor Xa and fails to inactivate TF. Porcine TBM also fails to regulate protein C. Porcine vWF can aggregate spontaneously and activate the host’s platelets through GP1b receptors. Altogether these reactions lead to the formation of thrombus in the graft vessels. AHXR, acute humoral xenograft rejection; Ag, antigen; aPC, activate protein C; DC, dendritic cell; IRI: ischemia-reperfusion injury; NET, nuclear extracellular traps; TBM, thrombomodulin; TF, tissue factor; Tregs, T regulatory cells; vWF, von Willebrand Factor.
The binding IgM and IgG to non-Gal antigens triggers the onset of AHXR by activating the complement cascade, which is a key actor of antibody-mediated damages (53). Besides, humoral responses against endothelial epitopes can also cause activation and damage of the vascular endothelium through other mechanisms, for example antibody-dependent cell-mediated cytotoxicity (ADCC) and inflammation. Further, neutrophils may activate porcine endothelial cells (pECs) (54). Natural killer (NK) cells (55) and macrophages (56) are also agents of AHXR, but the exact pathogenesis involving these cells remains unclear. The histological characteristics of AHXR include extensive interstitial bleeding, infarction, necrosis, thrombosis, neutrophil infiltration, and massive deposition of immunoglobins, complements, fibrin and platelets (34). These manifestations are similar to those found in HAR (14).
Genome editing tools, such as zinc-finger nucleases, transcription activator-like effector nucleases (TALEN), and CRISPR (clustered, regularly interspaced, short palindromic repeats)/Cas9 (CRISPR-associated protein 9), have paved the way for significant breakthroughs in dealing with AXHR by creating DKO (GGTA1/CMAH or GGTA1/B4GALNT2) pigs, or triple knockout (TKO, GGTA1/CMAH/B4GALNT2) pigs, which had been first generated in the USA (57–60). Cells from GGTA1/CMAH pigs were associated with reduced affinity of the human antibodies compared with cells from GTKO pigs (59). Moreover, the absence of Neu5Gc and Gal epitopes from murine xenogeneic cells has been proven to dampen the immune reaction provoked by the pre-formed antibodies contained in human serum (61). These data suggest that the deletion of the Neu5Gc epitopes could help increase the survival time of xenografts. In vitro evidence showed that the reactivity of pre-existing antibodies against the xenografts could be reduced by inactivating the B4GalNT2 gene (60). This result indicates that TKO pig organs can promote major development for transplantation into human compared with GTKO and DKO xenografts, but still TKO pig organs were not sufficient to achieve prolonged graft survival, limiting by complement-mediated coagulopathies (62). It should also be noted that a fourth xenoantigen seems to be exposed when the Neu5Gc epitopes are absent, and that baboons and other Old World monkeys are more likely to reject this fourth xenoantigen than humans are (63). Therefore, TKO pig heart or kidney transplantation in baboons may appear more problematic than they would be in a clinical trial involving humans. Due to the cross-reactivity between HLA-specific antibodies and SLA, modifying SLA genes may help further reduce the human immunoglobulin binding against pig cells. Two inactivating mutational approaches targeting either the light β-chain or a conserved region in the heavy α-chain of SLA-I have been successfully utilized in the pig genome to generate pigs with no or limited expression of SLA (45). PBMC from these pigs induced almost no proliferation of purified human CD8+ T cells (45). However, complete removal of SLA in pig genomes may not be an ideal solution due to logistical, immunological, and infectious consequences of SLA deletion (64). Instead, for highly HLA sensitive recipients, an ideal organ-source pig might be one with site-specific mutations to eliminate cross-reacting antibody binding.
If both HAR and AHXR are overcome, but immunosuppressive therapy is insufficient, CXR may occur and lead to graft rejection within days or weeks following transplantation (65, 66). CXR can be mediated by the innate and/or the adaptive immune system, and may involve NK cells, macrophages, neutrophils, dendritic cells (DCs), T cells, and B cells (Figure 2).
NK cells mediate xenograft rejection by direct NK cell cytotoxicity (NKCC) or ADCC. Upon direct contact with the target cells, NK cells deploy cytotoxic functions by engaging a series of activating and inhibitory receptors and ligands (53). Recent research has revealed a mechanism whereby NK cells adhere to and transmigrate through the porcine endothelium by interacting with an as yet to define porcine ligand, via CD49, integrins including CD11a/CD18 and CD11b/CD18, and CD99 (67). NKG2D and NKp44 are activating receptors that can bind porcine pULBP-1 and an unidentified ligand, respectively (68). Their engagement initiates the release of lytic granules containing cytotoxic proteins such as perforin and serine esterase, which lead to the lysis of the donor’s endothelial cells (69). Inhibitory receptors such as killer-cell immunoglobulin receptors (KIRs), immunoglobulin-like transcript 2 (ILT2), and CD94/NKG2A heterodimers mainly recognize MHC-I molecules (70, 71). Low expression or absence of MHC-I molecules on porcine cells leads to reduced inhibitory signals transduced to the NK cells, which triggers their activation and subsequent lysis of the porcine cells (72, 73). Stable expression of transgenic human leukocyte antigen (HLA)-Cw3 and/or G, and/or E, on porcine cells could protect the xenograft from human NK cytotoxicity (74).
Besides direct cytotoxicity, NK cells can also employ complement-independent ADCC to destroy the graft (75). Preformed natural antibodies against α-Gal carbohydrates or Neu5Gc can bind to the pECs with their Fab portion. Fc receptors (FcRs), including CD16 (FcγRIIIa) on NK cells, recognize the Fc portion of the antibodies, triggering a signalling cascade that causes degranulation (76). In addition, CD16 recognizes induced antibodies against SLA-I, which can mediate ADCC (77). Furthermore, NK cells can promote the production of non-Gal antibodies against the graft in a T-independent manner, by interacting with splenic marginal zone B cells via CD40/CD154 interaction (78).
The role of NK cells in CXR needs further elucidation to prolong xenotransplant survival. To date, most knowledge comes from in vitro studies and animal models. Future in vivo studies on pig-to-NHP transplants are needed to clarify the role of NK cells in xenograft rejection.
Macrophages carry out diverse functions, ranging from phagocytosis, cytokine production, antigen presentation, to tissue repair. Like for other innate immune cells, Toll-like receptors on the surface of the macrophages can recognize nonself molecules such as danger-associated molecular patterns (DAMPs) arising from injured xenogeneic cells (79–82), pathogen-associated molecular patterns (PAMPs), polysaccharides, and polynucleotides (83). Under the synergistic effect of Toll-like receptors activation and interferon γ (IFN-γ), macrophages are licensed to process and present xenoantigens, promote the differentiation of pro-inflammatory T helper 1 (Th1) and T helper 17 (Th17) by producing interleukin-12 (IL-12), and exert direct cytotoxicity by producing proinflammatory cytokines such as tumour necrosis factor α (TNFα), IL-1, IL-6, and nitric oxide (84, 85). Interspecies incompatibility of CD47 was also reported to contribute significantly to macrophage-mediated rejection of xenogeneic cells. That is, in a human macrophage-like cell line, porcine CD47 does not stimulate the inhibitory receptor signal-regulatory protein α (SIRPα), while soluble human CD47-Fc fusion protein does, which inhibits porcine cell phagocytosis (86). Human macrophages were found to phagocytose porcine red blood cells independently of the presence of antibodies or complement activation, even in setups where the α-Gal epitopes were absent from the porcine cells (87).
In CXR, macrophages mainly act by promoting T-cells mediated rejection (88–90). Feng et al. used IL-10/Fc to treat mice before or after transplantation of a pancreatic islet xenograft, and found that the T cell effector functions were inhibited, with reduced IFN-γ and IL-4 expression probably due to the inhibition of IL-12 production by macrophages (91). In whole organ xenografts, Lin et al. showed that the macrophages might be the cause of rejections occurring within 3–6 days. In their study, hamster hearts were transplanted into genetically engineered T cell-deficient rats depleted or not of NK cells, which demonstrated that in absent of T and NK cells the graft was still rejected. The spleen of the recipient and the rejected organs were predominantly infiltrated by macrophages (92). Since abundant evidence shows that macrophages play a role in xenograft rejection, the regulation of their activity might enhance the survival of future xenografts.
There are at least three mechanisms whereby activated neutrophils can induce tissue damage: (i) ROS generation; (ii) release of tissue-digesting enzymes; and (iii) nuclear extracellular traps (NETs) formation (93).
Human neutrophils can directly recognize pECs, and subsequently upregulate adhesion molecules and render porcine cells more vulnerable by exposing them to NK cell-mediated cell lysis (94). Specific recognition pathways were thought to be responsible for the adhesion of human neutrophils to porcine endothelium, as under flow conditions, adhesion occurred independently of the presence of α-Gal or ICAM-1 (95). Mohanna et al. demonstrated that neutrophils can be activated directly by porcine aortic endothelial cells, which subsequently causes a transient rise of calcium flux triggering the production of reactive oxygen metabolites and inflammatory cytokines (54). Cardozo et al. further demonstrated that the adhesion of human neutrophils to pECs can be facilitated by a soluble chemotactic factor produced by pECs (96). Activated neutrophils also exert cellular damages by generating superoxides via NADPH oxidase activity (97).
Apart from ROS generation, leukocyte proteases have been implicated in neutrophil-mediated graft tissue damage, through disruption of endothelial cell junctional complexes (98). For instance, after acute ischemia/reperfusion insults in liver, neutrophil elastase (NE) breaks down graft’s homeostatic barriers by degrading the extracellular matrix (ECM) components, including collagen, elastin, and fibronectin (99).
“NETosis”, a program for production of neutrophil extracellular traps (NETs), is a unique process whereby neutrophils induce inflammation and cell death in porcine grafts. NETs are mainly composed of antibacterial peptides, histones, and serine proteases that accumulate in the lung in both experimental and clinical primary graft dysfunction (PGD). Disruption of NETs with DNase I reduces lung injury (100).
The most efficient antigen presenting cells (APCs) in activating T cells and initiating immune tolerance are the DCs. Based on different surface markers, DCs are generally divided into three subsets: (i) DCs involved in ischemia-reperfusion injury (IRI) and expressing C1d, CD8α, CD11c, CD40, CD45, CD54 (ICAM), CD80, CD86, MHC-II, and TNFα, but are negative for CD4 and CD205 (101); when activated by antigens released during ischemia-reperfusion, these DCs can trigger both cellular and antibody-mediated rejection, resulting in harmful antibodies secretion by activated B cells and killing of donor cells by cytotoxic T cells (102); (ii) rejection-related DCs, promoting acute and chronic rejection via different interactions with T cells and are characterized by the expression of CD11c, MHC-II, CD1c and FcϵRI (103); (iii) tolerogenic DCs, suppressing the rejection process by dampening the T cell effector functions and promoting T regulatory cells (Tregs) activity; these DCs express significantly lower levels of MHC, T cell co-stimulatory molecules, such as CD40, and CD80/86, and inhibitory ligands, such as programmed death ligand-1 (PD-L1) and death-inducing ligands, reflecting their non-phagocytic profile (104, 105). In response to specific signals such as DAMPs, host DCs can acquire a rejection-related phenotype, which can further evolve towards a tolerogenic phenotype upon treatment with rapamycin, IL-10, vitamin D, or low-dose granulocyte-macrophage colony-stimulation factor (GM-CSF). Tolerogenic DCs reduce CD4+ T cell activation and impair CD8+ T cell functions, which helps suppressing graft rejection (102). Manna et al. reported that DCs activation by the porcine aortic endothelial cells could be blocked by a pre-treatment of the DCs with antibodies specific for the human leukocyte function-associated antigen-1 or CD54 (106). However, the exact mechanisms underlying DCs participation to the rejection process still need to be clarified in order to develop anti-rejection drugs.
The role of T cells in cellular rejection during pig-to-baboon xenotransplantation has been demonstrated, although the relevant studies were only few (107, 108). Upon xenotransplantation, T cells are activated by both direct and indirect pathways (109). In the direct pathway, porcine APCs expressing CD80/86 constitutively, such as pECs and migratory passenger leukocytes, can directly prime primate T cells (110). Simultaneously, primate TCRs interact with SLA-I/II peptide complexes, resulting in T cell-mediated cytotoxicity against the porcine vascular endothelium. In the indirect pathway, T cell activation occurs through the presentation of porcine peptides by the hosts’ APCs. T cell activation requires antigen recognition through the TCR coupled with costimulatory signals (111), involving CD40-CD154 and/or CD80/CD86-CD28 interactions (112). Different drugs targeting these costimulatory pathways could be administrated in xenotransplantation, as for example anti-CD40 mAb and CTLA4Ig, as discussed below. In addition, abrogation of SLA-I expression has been proved to silence T-cell and NK cell-mediated cell lysis (113, 114).
One percent of total circulating IgGs, and 1–4% of total IgM in the human serum are directed against α-Gal epitopes (115). Previous studies established that the cells producing anti-Gal antibodies reside mainly in the spleen, and to a lesser degree in lymph nodes and bone marrow (116). This location matches that of a recently described splenic B cell subtype characterized by a Mac1+ B1b-like phenotype (117). Antibody production results from the interaction between B cells and other immune cells, including T cells, NK cells, and follicular DCs. Immunization of α1,3GT KO mice with pig cell membranes induces clonal expansion of anti-Gal B cells that can present antigen to T helper lymphocytes via MHC-II and provide CD40 co-stimulation, causing cytokine production by the activated CD4+ T cells. This process provides activated B cells with the helper signals necessary to their proliferation and maturation in germinal centres, resulting in production of high-affinity anti-Gal antibodies (118). Another study suggested that marginal zone B cells can produce xenoantibodies after receiving help from NK cells, independently of T cell help (78). Moreover, the follicular DCs, expressing the complement receptors 1 and 2, can activate α-Gal-reactive B cells by presenting α-Gal immune complexes (119).
As B cells are the main sources of elicited anti-porcine antibodies, they represent an important target to overcome AHXR. Efficient depletion of circulating and secondary lymphoid organ-resident B cells by anti-CD20 antibody at the time of transplant prevents anti-pig humoral responses and resulting graft injury, and significantly delays or prevents the systemic dysregulation of the coagulation pathway and thrombotic microangiopathy (120, 121). Zhao et al. reported that anti-high mobility group box protein 1 (HMGB1)-neutralizing antibody prolonged xenograft survival, and dampened tissue damage and immune cell infiltration by suppressing xenoreactive B cell responses (122).
Coagulation dysregulation was first described by Ierino in the late 1990s. Both AHXR and cellular xenograft rejection are accompanied with coagulation dysregulation, which results in the development of thrombotic microangiopathy in the graft. Antibody-mediated and cellular rejections cause endothelium injury, exposing tissue factor (TF) and collagen. The binding of TF to activated factor VII (FVIIa) initiates thrombin generation, converting fibrinogen into fibrin. Simultaneously, sub-endothelial collagen triggers the accumulation and activation of platelets (123). Importantly, this process is enhanced by molecular incompatibilities between the primate and porcine coagulation homeostatic systems. The porcine tissue factor pathway inhibitor (TFPI) cannot fully inhibit the factor Xa in primates and fails to inactivate TF (124). In addition, the porcine thrombomodulin (TBM) is unable to regulate the primate thrombin, and thus, fails to activate the protein C (Figure 2) (125). Another incompatibility lies between the primate platelet lycoprotein 1b (GP1b) and the porcine von Willebrand Factor (pvWF). pvWF can spontaneously aggregate without shear stress and activate primate platelets through the GP1b receptor (126). The subsequent graft vessel thrombosis caused by fibrin deposition and platelet aggregation eventually leads to ischemic injury (Figure 2) (26, 127). Approaches to tackle coagulation dysregulation include the transgenic expression of human complement proteins (hCRP) and coagulation proteins such as human TBM by the donor porcine organs (128).
Chronic rejection usually occurs several months to years after organ transplantation. It has similar histopathological characteristics to those found in allotransplantation and are mainly related to thrombotic microangiopathy, characterized by the proliferation of graft vascular endothelial cells, vessel narrowing, interstitial fibrosis, which ultimately, result in graft failure (Figure 1C) (129). Since there are only few long-term survivors to xenotransplantation, the mechanism of chronic rejection has not been sufficiently documented, but it is almost certainly related to long-term, low-amplitude immune responses.
Current research indicates that chronic rejection involves both immune and non-immune factors. Molecular incompatibilities between the porcine and the NHP coagulation factors may play a vital role (130). Mohiuddi et al. reported that gene-editing of pig heart (GTKOhTg.hCD46.hTBM), alongside with anti-CD40 monoclonal antibody treatment, allowed for successful survival of a graft for 236 days in baboons. This result indicates a crucial role of human TBM expression combined with anti-CD40 treatment not only for the long-term survival of the graft, but also to avoid thrombotic microangiopathy and other coagulation-related problems (131). Another study demonstrated that transgenic expression of human CD39 (a major vascular nucleotidase that converts ATP and ADP into AMP, further degraded into anti-thrombotic and anti-inflammatory adenosine) in mouse significantly prevents thrombotic events in the heart graft and improves the duration of graft survival from three days in WT mice, to six days in the transgenic mice (132).
Another study by Kim et al. achieved a long-term survival of 499 days with pig-to-rhesus macaque renal xenografts by depleting CD4+ T cells (46), indicating that these cells are responsible for chronic rejection. Similar to allotransplantation, the host’s MHC class II molecules recognize porcine xenoantigens and present them to the host’s CD4+ T cells, leading to their activation (109). Yet, the mechanisms involving the CD4+ T cells in chronic rejection are poorly understood. Besides, a sustained inflammatory response is still a key challenge to achieve successful grafts. Future studies should explore the roles of inflammatory cytokines such as IL-6, TNF-α, IL-17, and their inhibitors to uncover therapeutic targets (133, 134).
Since 2009, porcine models with new genetic modifications have been constantly implemented to improve molecular compatibilities. As gene editing techniques such as zinc finger nucleases, TALEN, and CRIPSR/Cas9 genome editing system improve, the production of multiple-gene edited pigs has become easier and faster (135). This section of the review will focus on the mechanisms and usage of common immunosuppressants in xenotransplantation area (Figure 3).
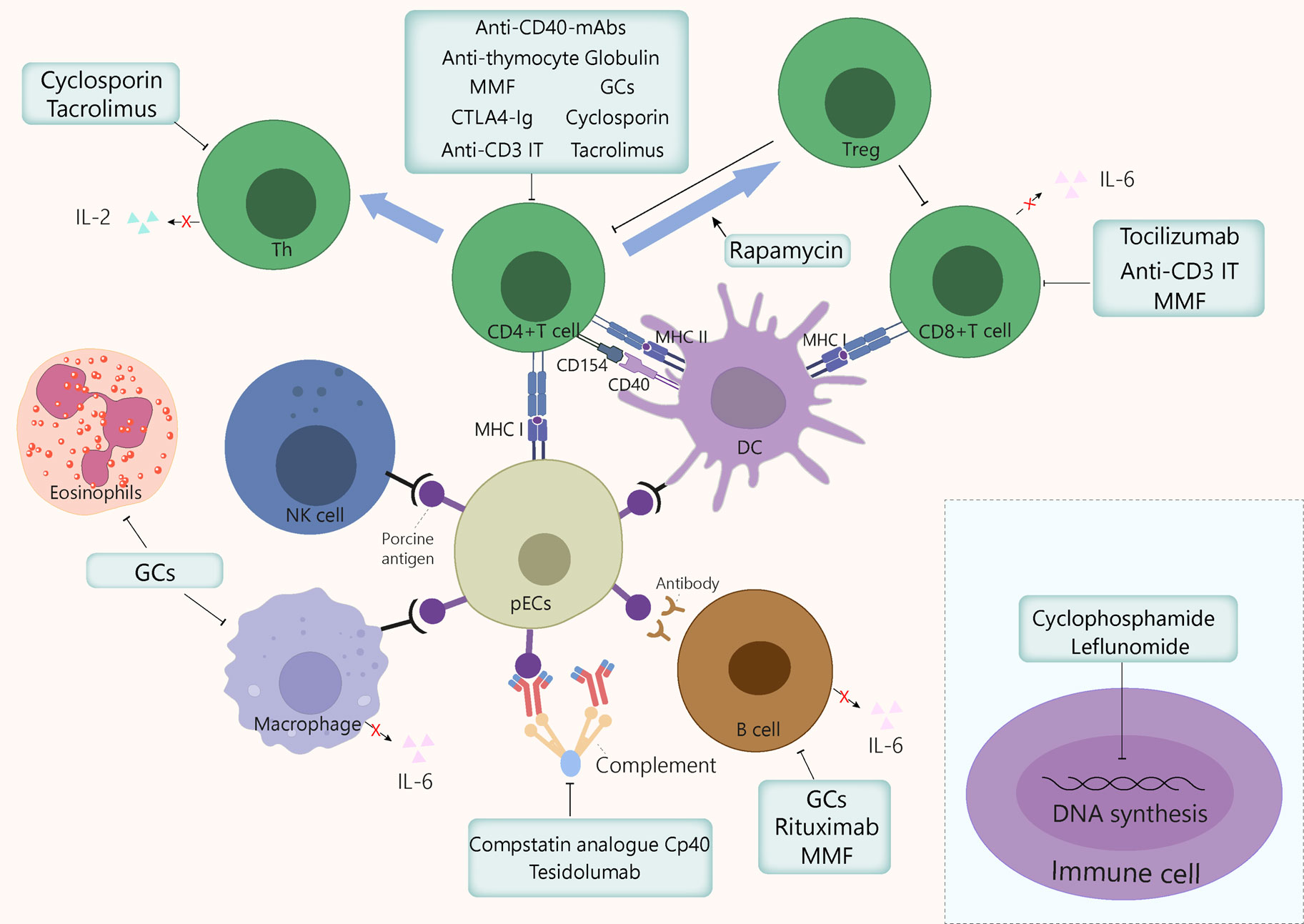
Figure 3 Mechanism of action of the immunosuppressants commonly used in xenotransplantation. GCs exert anti-inflammatory and immunosuppressive effects by inhibiting macrophages, eosinophils, T cells, and to a lesser extent, B cells, by binding to GC receptors in cytoplasm. Cyclosporin binds to cyclophilin, then this drug-immunophilin complex binds to calcineurin, which subsequently prevents Th cell activation and IL-2 production, which eventually inhibits T cell clonal proliferation. Tacrolimus inhibits T cells proliferation by binding to FKBP, which inhibits several transcription factors involved in the production of proinflammatory cytokines. Cyclophosphamide blocks DNA alkylation in various cell types, leading to programmed cell death induction and preventing cell division. Leflunomide inhibits the synthesis of pyrimidines, thus arresting cell cycle in S phase. Mycophenolate mofetil prevents T and B cell proliferation by specifically inhibiting a purine pathway required for lymphocyte division. Polyclonal anti-thymocyte globulins are mainly directed against T cells. However, other immune cells sharing common surface antigens with T cells can also be affected to a lesser extent. Monoclonal antibodies target specific cytokine pathways (e.g., IL-6Rα) or cell surface markers, such as CD3, for anti-C3 IT, or CD20, for rituximab. The IL-6 receptor inhibitor tocilizumab reduces systemic inflammation and inhibits of CD8+ T cell and B cell differentiation. Anti-CD3 IT can deplete CD3+ T cells transiently and reduces the number of T cells in circulation and in lymph nodes. Rituximab is a B cell-depleting drug that targets CD20. Rapamycin exerts immunosuppressive and anti-proliferative effects of T cells by inhibiting the activation of S6K1 and PI3 kinase signalling. CTLA-4Ig and anti-CD40mAb target the costimulatory pathways CD80/86:CD28 and CD154:CD40, respectively, thereby dampening T cell activation. The compstatin analogue Cp40 and Tesidolumab inhibits complement C3 and C5 respectively, thereby reducing complement activities. FKBP, FK506 binding protein; GCs, glucocorticoids; IT, immunotoxin; mAb, monoclonal antibody; MMF, mycophenolate mofetil, pECs, porcine endothelial cells.
Immunosuppressive agents are used commonly in treatments for transplantation rejection. Conventional immunosuppressive therapy, for example, corticosteroids, tacrolimus, and cyclophosphamide (136–138), when used at high dosages, may delay graft failure. In non-human concordant models for kidney or liver xenografts, long-term survival can be achieved with conventional immunosuppressive agents (139). In 2000, a co-stimulation blockade-based immunosuppressive therapy was introduced to xenotransplantation by Buhler et al. (140), and has proven more successful than common therapies.
Glucocorticoids (GCs) belong to the steroid family and were discovered in the 1940s (141). GCs have been used as first-line medication during the induction and maintenance phases after transplantation to prevent acute rejection. In pig-to-primate xenotransplantation, immunosuppressive GC regimens have also been used in most trials (142–144). In an islet xenotransplantation study, all monkeys experienced a normalization of all diabetic and glycemic parameters within four days of GCs administration, suggesting a reversal of diabetes mellitus (145). In another pre-clinical study, the longest survival time (78 days) for a porcine renal xenograft was obtained by applying GCs combined with other immunosuppressants (146).
The 17-hydroxy, 21-carbon steroid configuration characterizing the GC molecules is required for their activity and binding to the GC receptors (GCRs). Changes in this configuration can alter the pharmacodynamic specificities of the GCs (147). GCs’ major anti-inflammatory effects result from interferences between activated GCRs and proinflammatory transcription factors such as nuclear factor-κB and activator protein-1 (148). The immunosuppressive mechanisms triggered by GCs include: (i) T cell depletion via inhibition of IL-2, which prevents Th1 differentiation and subsequently results in T cell apoptosis; (ii) prevention of B cell clonal expansion through inhibition of the production of IL-2 and related peptides, which reduces antibody production; (iii) induction of eosinophil apoptosis either directly or through IL-5 inhibition (147, 149); (iv) downregulation of the Fc receptors and MHC class II molecules on macrophage surface via proinflammatory cytokine inhibition, e.g., IL-1 and TNF-α, and prostaglandins (150); (v) widespread decrease of inflammatory responses in the host by induction of lipocortin-1 (annexin-1) synthesis; (vi) acceleration of lymphocytes apoptosis and abrogation of alloimmune responses to third-party antigens, such as allergens and autoantigens. In conclusion, all immune cells that express GCRs can be modulated by GCs, and as a result, lose abilities such as migration or phagocytosis.
The exact dosage of GCs for induction and maintenance phases varies between different institutions. Methylprednisolone doses vary from 1 to 15 mg/kg, for a duration varying from four days to continuous treatment (143, 146). A metanalysis showed that long-term GCs usage may increase the risk of infections for the hosts (151). Pulse dose steroids are now favoured for treating acute allograft rejection but are less crucial in maintenance immunosuppressive therapy. According to some guidelines for clinical practice, GCs could be discontinued early at first week post transplantation for patients with low immunological risk who receive depleting antibodies as induction therapy (152).
Two common calcineurin inhibitors (CNIs), cyclosporin and tacrolimus, inhibit the dephosphorylation of nuclear factor of activated T cells (NFAT) by calcineurin, preventing its nuclear translocation and subsequent calcineurin-dependent gene transcription (153, 154). This inhibition results in decreased T cell maturation and lymphokine production, including that of IL-2.
Cyclosporin is a cyclic polypeptide consisting of 11 amino acids, most of which are hydrophobic. The discovery of cyclosporin in the early 1980s had a huge impact on the transplantation field by decreasing drastically the rate of acute rejection. This drug binds to cyclophilin; the drug-immunophilin complex then binds to calcineurin, which prevents T cell activation and IL-2 production, thus inhibiting T cell clonal proliferation. Cardiac xenografts treated with steroids and cyclosporin achieved a survival of 77 days, with no signs of hyperacute rejection or cyclosporin-induced malignancies (155). In baboon-to-monkey liver xenotransplantation, two monkeys survived for 91 and 1076 days, respectively, with cyclosporin administered after transplantation, for example, at doses of 3 to 8 mg/kg/day (139), or 20 mg/kg intravenously two hour before transplantation followed by oral administration on next three days (143). In a cardiac xenograft transplantation, the highest cyclosporin blood levels (around 1000 ng/ml) correlated with the highest graft survival rate of the host animals (142).
Tacrolimus, a 23-membered macrolide lactone, was isolated for the first in 1987 from Streptomyces tsukubaensis (156). It inhibits T cell proliferation by binding FK506 binding protein (FKBP) (157), which inhibits calcineurin by binding it specifically and competitively (158). Subsequently, NFAT nuclear translocation is inhibited, which provokes the downregulation of downstream genes encoding cytokines, including TNF-α, IL-2, IL-3, IL-4, CD40L, IFNγ, and GM-CSF (158, 159). This tacrolimus-induced cascade finally leads to reduced T cell proliferation. Tacrolimus was first approved for liver transplantation in 1994, and since, its used has been extended to become the backbone of immunosuppressive therapy after solid organ transplantation. Later, it has been used for induction and maintenance immunosuppressive therapy, usually in combination with GCs that are then rapidly de-escalated (160). Tacrolimus effectively prevents acute rejection and leads to lower rejection rates and longer rejection-free periods (161, 162). In pig-to-rat islet xenotransplantation model, tacrolimus also exerts a noticeable immunosuppressive effect (163, 164). The oral bioavailability of tacrolimus ranges from 5 to 67% (mean value of 27%), and its half-life ranges from 3.5 to 40.5 hours (165). Protocols with tacrolimus constantly evolve (166). In a pre-clinical study on pig-to-NHP islet xenotransplantation, tacrolimus was orally administered daily from day −3 to up to day 56 to achieve stable levels (3–6 ng/mL) (164). In another study, tacrolimus was injected intramuscularly twice daily at a dose of 0.05 mg/kg from day –2 and for up to 6 months after transplantation (167). The preferred administration route remains oral rather than sublingual or intramuscular (168). The adverse effects associated with this drug are mainly nephrotoxicity and neurotoxicity, which can be mild to severe. To avoid adverse effects, it is important to maintain tacrolimus at a stable dose. Because the CYP3A5 genotype is associated with a remarkable impact on tacrolimus pharmacokinetics, it should be considered in the dosing algorithm of this drug (169). Moreover, some research attempted to develop machine-learning models to predict tacrolimus dose stability, which might provide more accurate approaches to achieve personalized medicine in clinics (170).
Cyclophosphamide (CYC) is widely used to prevent transplant rejection and graft-vs-host complications (171). It is a nitrogen mustard drug that affects DNA alkylation in a non-cell cycle phase-specific manner, and is toxic for all human cells to various degrees (172). The active form of CYC inhibits protein synthesis via DNA and RNA crosslinking, leading to programmed cell death and prevention of cell division (173).
The immunosuppressive effect of CYC mainly relies on direct deletion of the host’s mature T cells that are highly proliferating and reactive to the donor’s antigens (174). It also has the capacity to deplete Tregs to counteract immunosuppression in cancer, decrease the production of T cell growth factors, e.g., type I interferons, and precondition host T cells for donor cells, hence attenuating rejection (175). Regimens with CYC in xenotransplantation have proven effective in some cases (146, 176). In pig-to-rhesus corneal transplantation, intravenous injection of CYC followed by pig bone marrow cell transplantation reduced inflammatory cell infiltration (177). CYC is applied typically as a continuous treatment administered orally or intravenously in pulses, with doses ranging from 10 to 40 mg/kg (146, 178). Intermittent intravenous rather than daily oral CYC has been used to minimise bladder and gonadal toxicity. Another side effect of CYC is myelosuppression, which causes leukopenia and neutropenia and can lead to severe and sometimes fatal infections, including viral infections (179).
The mammalian target of rapamycin (mTOR) signalling pathway has important functions in cell growth and metabolism regulation (180). Rapamycin can bind a 12-kDa FK506-binding protein (FKBP12) to form a gain-of-function complex that acts as an allosteric inhibitor of mammalian TOR complex 1 (mTORC1) (181). Rapamycin exerts its immunosuppressive and anti-proliferative properties via the inhibition of S6K1, a serine/threonine kinase activated by a variety of agonists (182, 183). In rat-to-mouse islet transplantation, rapamycin could induce Treg-mediated tolerance (184). Moreover, Singh et al. verified that in baboons, treatment with rapamycin increases CD4+ Tregs induction from naïve CD4+ T cells, thereby suppressing anti-porcine xenogeneic response in vitro (185). Furthermore, in both allo- and xenotransplantation, graft recipients treated with IL-17-neutralizing antibodies showed the highest percentage of Tregs (186, 187). An example of reported schedule for treatment with rapamycin consists of 0.2 mg/kg during the first three days post-transplantation, followed by treatment every other day until day 14 (184).
Leflunomide inhibits the dihydro-orotate dehydrogenase, a critical rate-limiting enzyme for pyrimidine synthesis. Therefore, it arrests cell cycle progression from S to G2 phase (188). The literature regarding the role of leflunomide in xenotransplantation is limited but indicates that this drug inhibits rat-to-mouse cardiac xenograft rejection by supressing NF-κB signalling pathway and adaptive immune responses (189).
Mycophenolate mofetil is the semisynthetic morpholinoethyl ester of mycophenolate acid, which prevents T and B cell proliferation by specifically inhibiting a purine pathway required for lymphocyte division (190). MMF, usually administered intravenously at a dose of 20mg/kg twice per day, has been mainly applied together with other immunosuppressants as maintenance regimen and achieved considerable long-term survival of xenograft (the longest reported to be 945 days) in cardiac xenotransplantation (191–194).
Monoclonal antibodies (mAbs) are widely used in clinics and experiments. Most have cell-specific immune-modulatory properties directed for example at CD3+ T cells, which are particularly pathogenic in the context of solid organ transplant rejection (195). Recent studies suggested that in the absence of irradiation or chronic immunosuppressive drugs, renal tolerance can be stably established in primates (18–20) by using an anti-CD3 immunotoxins (ITs) that ablate T cells transiently (145, 196, 197). T cell numbers in blood and lymph nodes could be reduced to 1% of their initial values following anti-CD3 IT depletion, which established long-term tolerance towards mismatched renal allografts (145). Anti-CD3 ITs administered two hours pre-transplantation at a dose of 100 μg/kg and again on the day following transplantation has demonstrated efficacy (143). However, this schedule should be further verified in the future.
Subsequently, monoclonal antibodies were generated against specific cytokines that play a role in immune cell-mediated toxicity and tissue damage. IL-6 is induced by inflammation and contributes to CD8+ T cell and B cell differentiation (198). In addition, it is a crucial factor in systemic inflammation and endothelial cell survival after xenotransplantation. More recently, Zhao et al. proposed that IL-6 may promote coagulation and inflammation during xenotransplantation (198). Furthermore, Ezzelarab et al. uncovered that biologics inhibiting the IL-6 pathway (e.g., Tocilizumab) could mitigate systemic inflammation in xenograft recipients (SIXR) and may be required to prevent coagulation dysregulation after xenotransplantation (199). Tocilizumab is a biological that blocks human IL-6Rα, and has been considered to reduce inflammation by inactivating the STAT3 pathway acting downstream of IL-6Rα. Tocilizumab was also found to delay the revascularisation of xeno-islets in a pig-to-NHP model (200). Another case report brought exciting results on the use of tocilizumab in combination with other immunosuppressants, which allowed to achieve a 136 day-pig kidney survival (201). However, another recent research by Zhang et al. reported that serum IL-6 increased in baboons receiving tocilizumab before xenotransplantation. This increase could be detrimental to the survival of the pig xenograft by promoting IL-6 binding to pig IL-6R and subsequent pig cell activation (202). Thus, more clinical trials are needed to determine whether tocilizumab is beneficial or detrimental to xenotransplantation. The dose of tocilizumab was consistently 10 mg/kg in all reported cases, with a treatment schedule usually starting on days -1, 7 and 14, followed by administration every two weeks (198).
Another monoclonal antibody used in transplantation is the chimeric anti-CD20mAb rituximab that leads to B cell depletion (203). In addition of being an effective treatment for post-transplant lymphoproliferative disorders, Rituximab can serve as treatment for acute rejection, as some evidence suggested that it could stop the progression towards chronic antibody-mediated rejection (204). Its mechanism of action may be explained by its impact on B cell modulation of the T cell responses, and its long-term effects on plasma cell development (205). In combination with other immunosuppressants, anti-CD20mAb was reported to achieve a 136 day-pig kidney survival (201). An example of reported schedule for treatment with rituximab in transplantation is 19 mg/kg at days -14, -7, 0 and 7 (206).
Polyclonal anti-thymocyte globulins (ATGs) are antibodies obtained by injecting animals, usually rabbits, with human lymphoid cells such as B lymphoblasts, peripheral T cells, or thymocytes, and then harvesting and processing the sera to purified the immunoglobulins (207). ATGs are predominantly directed against T cells, but other immune cells sharing surface antigens with T cells can also be targeted to lesser degrees, as for example B cells, monocytes, and neutrophils. ATG primary mechanism of action consist in promoting lymphocyte depletion through T cell activation-induced apoptosis and complement-dependent lysis (208). In a preclinical study, ATGs were shown to improve engraftment and survival of neonatal porcine xenoislets (209). They can also extend pig kidney survival when combined with other immunosuppressants (201). ATGs are usually prescribed before transplantation at a dose of 10 mg/kg on day –3 (201), or at a dose of 5 mg/kg on days –2 and –1 (206).
T cell activation requires co-stimulation via engagement of CD28 on the T cell with CD80/86 on the APC. Cytotoxic T lymphocyte–associated protein 4 (CTLA4) is a competitive inhibitor of CD80/86 that downregulates T cell responses (210). This T cell suppressive activity served to engineer a human IgG heavy chains coupled with CTLA4 to create a fusion antibody able to prevent graft rejection (211). The new generation CTLA4Ig, belatacept, displayed a significantly higher affinity for CD80/86 in a pre-clinical renal transplantation model in primate (212), and showed greater efficacy in modulating adaptive immune responses (213, 214). Belatacept proved able to decrease the antigraft humoral immune response in intracerebral transplantation of mesencephalic pig xenografts into primates (215). In vivo, CTLA4Ig is able to dampen T cell-dependent immune responses and prolong long-term xeno- and allograft survival (216–218). Levisetti et al. reported that two out of five CTLA4Ig-treated monkeys showed prolonged graft survival, while the humoral responses were suppressed in all treated animals (219). Buerck et al. and his group generated a novel transgenic (tg) pig line expressing the CTLA-4Ig analogue LEA29Y and demonstrated that transplanted INSLEA29Y-tg porcine neonatal porcine islet-like clusters (NPICCs) displayed normal beta cell function and survived from rapid T lymphocyte-mediated rejection during 30-day observation period. However, the long-term effect regarding xenograft rejection still remained unknown (220). In another pig to baboon clinical trial, the blockade of CD28-B7 costimulation pathway using human CTLA4Ig has been shown unsuccessful to prevent xenograft rejection, making the role of CTLA4Ig controversial (221).
The interaction between CD154 on activated T cells and CD40 on APCs results in CD80/86 upregulation on APCs, enhancing another component of T cell co-stimulation (222). CD154 is also found on platelets, and not surprisingly drugs targeting CD154 are associated with an increased risk of thrombosis in primate (223). Thus, the focus of drug development has been changed to target CD40 and several anti-CD40mAbs are under development. Blockade of CD40/CD154 signaling by anti-CD40mAb was shown to prolong graft survival and suppress xenograft rejection (192, 224). Among these, a fully humanized anti-CD40mAb, iscalimab, appears to be a promising candidate in transplantation (225). Pre-clinical application using immunosuppressive anti-CD40mAb 2C10R4 combined with tacrolimus in pig-to-NHP islet xenotransplantation was effective in prolonging islet graft survival (164). In pig-to-mouse islet xenotransplantation, short-term administration of the anti-CD40mAb MR-1 and the anti-LFA-1mAb increased the survival of neonatal porcine islets (226). Interestingly, the short-term use of MR-1 alone prolonged porcine islet graft survival and promoted CD4+ Tregs recruitment into the graft and secondary lymphoid tissues (227). Consistently, lower numbers of CD4+ Tregs increased the risk of rejection in cardiac xenotransplantation in a pig-to-NHP model (228). For treatment, anti-CD40mAb was reported to be infused intravenously at a dose of 20–50 mg/kg on days –4, 0, 4, 7, 10 and 14 of transplantation, followed by weekly infusion for three months, and biweekly fusion thereafter (164). Alternatively, anti-CD40mAb could be used at a dose of 50 mg/kg on days –1, 0, 5, 9, and 14 (206).
As discussed above, complement activation is involved at every stage of xenograft rejection. Thus, a complementary approach is to administer agents that either deplete or inhibit complement activation. Many interventions have been introduced to prevent complement-mediated injuries during xenotransplantation (229, 230). Among these, cobra venom factor extends graft survival significantly in allotransplantation, albeit it only has a temporary effect (231). C1-esterase inhibitor has been reported to be active in NHPs and was recommended to replace cobra venom factor as complement inhibitor (232, 233). The compstatin analogue Cp40, a newly developed potent inhibitor of complement C3, inhibits leukocytes adhesion and neutrophils attachment to porcine endothelium (230). Moreover, Cp40 inhibits pECs and leukocytes activation. It reduces the levels of adhesion molecules such as E-selectin, ICAM-1, ICAM-2, and VCAM-1 on pECs, and of the integrin CD11b on neutrophils, paving the way for future therapeutic interventions targeting complement activities (230). Schmitz et al. reported that Cp40 could significantly prolong median allograft survival time in an NHP model. Normal kidney function was maintained at 50% in Cp40-treated primates after the last day of treatment (234). In another case report, Tibetan macaques receiving liver xenografts with immunosuppressors, including Cp40, did not exhibit severe coagulation disorders or immune rejection (235). Another complement inhibitor, the anti-C5 antibody Tesidolumab, has been recently reported to reduce early antibody-mediated rejection and prolong survival in renal xenotransplantation (236). Cp40 was used at a dose of 2 mg/kg three times daily, on day 2 prior to kidney transplantation and day 14 after kidney transplantation (234). Yet, the most appropriate dosages need to be determined more precisely. Tesidolumab was given at a dose of 30 mg/kg on the day of transplant, followed by weekly intravenous injection at 10 mg/kg for seven weeks, at which point anti-C5 was discontinued.
Currently we are able to create new genetic modifications of the porcine genome (over 40 genetic variants to date), hoping to achieve better survival of the xenografts. The multiplex creation of a GGTA1/CMAH/B4GalNT2 KO pig has shown the ability to reduce antibody mediated rejection in humans. There are also other extensive genome engineered pigs which have greater compatibility with the human immune system. For example, the generation of porcine endogenous retroviruses (PERVs) KO·3KO·9TG (hCD46, hCD55, hCD59, hTHBD, hTFPI, hCD39, hB2M, HLA-E and hCD47) pig enhances the pigs’ immunological compatibility and blood-coagulation compatibility with humans (64, 237–240). Recombinant expression of human complement regulatory molecules hCD59 and hDAF on porcine articular chondrocytes could also prevent humoral rejection in cartilage repair (241). By using CRISPR/Cas9 system, Sake et al. tried to abrogate MHC-I expression on xenografts to silence T-cell and NK cell-mediated cell lysis (64). Four genetic pigs died within the first days due to weakness, and the remaining two piglets developed acute fevers at an age of 3-4 weeks leading to sudden death (64). Xie et al. succeeded in alleviating antibody-mediated rejection using Gabarapl1 knockdowns in primary porcine aortic endothelial cells (PAECs) (238). While in another pre-clinical trial, the inhibition of COX-2 expression decreased PAECs death from 20% to 7% after 2 hours, making COX-2 inhibitors a candidate for therapeutic targeting to protect vascular endothelial cells in xenotransplantation (43). More is not always better, extensive genetic engineering can lead to congenital malformations/decrease animal viability (242, 243). What exact genetic modifications do we need in the organ-source pig should be fully considered in the future.
It is noteworthy that researchers are investigating alternative options. One of them is to grow complex tissues or organs using the body’s own regenerative capacity. Masano et al. tried to use a xenogeneic animal as an in vivo bioreactor to promote regeneration of a liver graft and successfully acquired fully regenerated small liver grafts under appropriate immunosuppressive therapy (244). This alternative option has fewer ethical concerns, but before it can be considered further, more research are needed to reduce complications and tested in larger animal models.
In recent years, a number of pig-to-NHP preclinical xenotransplantation studies have been performed with various organs. While HAR has been alleviated owing to gene-editing technologies, DXR and chronic rejection remain urgent issues to be solved. Another concern focuses on PERVs transmission, which is a major hurdle to the clinical use of pig cells, tissues, and organs for treatment of organ failure in humans. However, it is still uncertain whether PERVs is pathogenic to humans, or if it could recombine with hERVs to form new viruses. Genetic engineering techniques such as CRIPSR/Cas9 genome editing system could prevent their activation or delete them from the pig cells (245, 246). Further, the ethical issues around xenotransplantation have not been sufficiently discussed. Strict medical and ethical guidelines and regulations are needed before clinical applications can be tried on selected patients. Beyond the common ethical and technical issues shared by the different areas of xenotransplantation, there are also organ-specific barriers that are briefly addressed below.
Islet xenotransplantation is a promising alternative approach to Type 1 Diabetes (T1D) treatment and has achieved long-term normoglycemia in porcine-to-primate studies (247–249). Based on preliminary studies in NHPs, the first case of clinical islet xenotransplantation to human can be traced back to 1994 (250). Currently, clinical trials using islet xenotransplantation are developing more rapidly than those in other xenotransplantation areas. In 2014, a clinical trial using islet xenotransplantation under regulatory framework was registered at ClinicalTrial.gov (251). This was followed by Phase I/IIa and IIb clinical trials using encapsulated neonatal porcine islets in xenotransplantation performed in Argentina (252), which resulted in a mean transplant estimated function of approximately 0.5, with transplants maintained for more than two years, and a significant reduction in the number of unaware hypoglycemia episodes. Surely, there are still many problems to be solved for islet xenotransplantation before reaching their clinical use, including physiological function consistency, immune rejection, islet loss, and prevention of PERVs infection (253). Immune rejections included HAR, mediated by Gal and non-Gal antigens (254), instant blood-mediated inflammatory reaction (IBMIR) that may have provoked 60–80% of islet loss (255), and CD4+ T cell-mediated cellular rejection that plays a major role in islet destruction (256). Strategies to alleviate rejection mainly include islet encapsulation and gene editing technology. Recent clinical trials mainly focused on encapsulating the neonatal porcine islets in different high molecular compounds (257). While cell encapsulation technology can potentially shield the islets from the host’s immune rejection at initial stage, long-term therapeutic efficacy is still a challenge (258). Another hot spot of research is to attempt different transplantation sites, including the portal vein, subrenal capsule, subcutaneously, the muscle, spleen, gastric submucosal space and peritoneum, depending on the islet volume and its naked or encapsulated status, in order to reduce islet loss (259, 260). There is still no optimal site for transplantation, but the peritoneal cavity is favoured in clinical trials.
Liver xenotransplantation from chimpanzee to human was first held in the 1960s (261). More trials were carried out in the 1990s (262), which remained ultimately unsuccessful. The transplanted patients either died from sepsis due excessive immunosuppression, or from hepatic failure with clear rejection. These failures terminated the attempts of liver xenotransplantation in clinical application. Until today, the research has mainly focused on pig liver xenotransplantation to NHPs. Shah et al. (263) have recorded, so far, the longest survival time after xenotransplantation of a pig livers to NHPs, which was of 25 days, using with an α1,3-galactosyltransferase knockout miniature swine as a donor. Two major problems must be overcome in liver xenotransplantation: lethal thrombocytopenia and antibody-mediated rejection (AMR) targeting antigens such as α1,3GT, N- glycolylneuraminic acid and β4GALNT2 (235). Moreover, hepatic cold-induced injuries are also a serious concern (264). AMR has now been reduced by using gene editing technology including CRISPR/Cas9, TALEN, and other genome editing and transgenic methods (254). To solve hepatic cold‐induced injuries, Li et al. succeeded to increase porcine hepatocyte viability by optimizing spheroid cold storage conditions under four different cold storage solutions (265). Despite these efforts, the graft survival is limited by either the development of a thrombotic microangiopathy and/or consumptive coagulopathy (266, 267). Cross-species thromboregulation becomes more complicated in case of liver xenotransplantation because the liver produces most coagulation factors. Current preclinical studies are dedicated to elucidating the immunobiology behind platelet activation, aggregation, and phagocytosis, especially during interactions between platelets and liver sinusoidal endothelial cells, hepatocytes, and Kupffer cells (28, 268). We believe that if the severe and immediate thrombocytopenia could be prevented, pig liver xenotransplantation could be used as a bridge towards allotransplantation.
The first attempt of pig-to-NHP cardiac xenotransplantation (CXTx) started in the mid-1980s (269). Currently, it is the standard model to conduct preclinical xenotransplantations. A major breakthrough came with the introduction of the genetic deletion of the α1,3GT gene in 2003, which reduces HAR to a large extent. The longest survival of heterotopic heart xenograft, reaching up to 945 days, has been achieve in baboons, using cardiac xenografts from GTKO.hCD46.hTBM pigs, with ATG and anti-CD20 antibody treatments, followed by maintenance with MMF and high-dose anti-CD40 immunosuppressive regimen (193). The first clinical trial of pig to human CXTx was carried out with genetically modified pig heart transplanted into a 57-year-old man in the USA in 2022, and the patient survived for two months without signs of rejection, while the cause of death is unknown (270). Despite many breakthroughs on different aspects of xenotransplantation in recent years, there are still barriers to be overcome before large scale clinical CXTx can be conducted, including immunological barriers, perioperative cardiac xenograft dysfunction (PCXD), detrimental xenograft growth, and PERV infection. PCXD is unique to orthotopic CXTx and has not been observed in heterotopic CXTx. It can cause xenograft failure within the first 48 hours (194). The exact mechanism of PCXD is unclear but may stem in incompatibilities between porcine and primate plasma, the latter carrying non-Gal antibodies (271). Cold non-ischemic continuous perfusion of the donor’s heart with STEEN solution (a buffered extracellular solution) appears to be an effective way to alleviate PCXD (233, 272). The detrimental xenograft overgrowth occurring after CXTx leads to diastolic dysfunction and congestive liver damage. The overgrowth could be inhibited by lowering baboons’ blood pressure to match that in pigs’ heart, by reducing the use of cortisone early, or by using temsirolimus, as in a particular study (233). This latter strategy has not tested by other researchers. The relevant ethical issues around CXTx have not been completely defined yet. Strict medical and ethical guidelines and regulations are still needed before proceeding towards clinical application to selected patients.
Kidney xenotransplantation has a long and largely unsuccessful history. The first clinical trial was carried out in 1905, when slices of rabbit kidneys were inserted into a child, who died 16 days later due to pulmonary congestion. Reemtsma et al. transplanted pairs of chimpanzee kidneys into six patients in 1964, with the longest survival reaching nine months (273). Later attempts using monkeys and baboons as source of kidneys were even less successful. In majority, the deaths occurring in these clinical studies were related to either rejections or infections. These disappointing results terminated the clinical application of kidney xenotransplantation. However, researchers have re-explored this possibility later, due to shortage of available kidneys, and pig-to-NHP xenotransplantation has now become a standard experimental model. Similar to CXTx, rapid progress has been made since 2005, with the availability of genetically engineered pigs (274). In 2015, two groups reported the survival of life-supporting genetically engineered pig kidneys for > 4 months, maintained by a treatment involving new immunosuppressive agents blocking T cell co-stimulation (201). Based on this progress, a new clinical trial with two GTKO porcine kidneys transplanted to a brain-dead patient on a ventilator has been carried out in 2021. The kidneys were connected outside of the body to blood vessels on the patient’s legs and monitored over a period of 72 hours (275). No HAR and no transmission of porcine retroviruses were detected, and the kidneys produced variable amounts of urine, but creatinine clearance was not recovered. Although this attempt surely brought substantial information and improvement for kidney xenotransplantation, there is still a long way to make kidney xenotransplantation possible in humans. In this case, the host was brain dead and artificially maintained, and therefore cannot been considered a living body. This trial is close to a clinical trial, but it still cannot be considered as such. We would consider this attempt as a bridge between xenotransplantation trials in animals and clinical trials in humans. Organ-specific problems linked to kidney xenotransplantation include hypovolemia syndrome, erythropoietin function-associated anaemia, and rapid growth of the pig kidneys after transplantation (199). The primate organisms are not aware of the fluid loss occurring during hypovolemia syndrome, which may result from a dysfunction of the renin-angiotensinogen system. This could be avoided by conserving the native kidneys in situ (276). It is difficult to assess whether the pig erythropoietin functions adequately in primates, but pigs genetically engineered to produce human erythropoietin may solve this issue (277). Kidneys transplanted from a strain of pig grow early and rapidly in primates. This phenomenon may result from an innate factor, and could also be solved by knocking out the gene encoding the growth hormone receptors of the pigs (278). Finally, the inclusion criteria for the selection of patient candidate to kidney xenotransplantation is more difficult than in other areas, as dialysis represents a therapeutic option for these patients. Therefore, until proven safer and more efficient, kidney xenotransplantation cannot be considered.
The field of xenotransplantation has been progressing rapidly with many breakthrough achievements in recent years. However, there are still several problems ahead of its use in clinical practice: (i) although porcine-to-human xenotransplantation of kidneys and hearts have been carried out, it is not known when xenotransplantation of liver, small intestine, and even pancreas will become possible. (ii) NHPs are phylogenetically close to humans and share many physiological, anatomical, immunological, and neurological similarities, making them excellent experimental models for research (279). However, there are still differences between species, and it is not clear to which extent studies in NHPs can fully predict xenorejection and the clinical outcome in humans. (iii) Until now, the survival of transplanted organs on the long term largely depends on high doses of different immunosuppressants, which would expose the recipients to high infection risks and other side effects. Although more and more genetically engineered pigs are created by gene-editing technologies, the question remains as to whether it will become possible to achieve long-time survival without immunosuppressants, by reducing the immunogenicity of the transgenic donors. Ethical aspects of xenotransplantation have been discussed for many years (280), with particular considerations on issues related to the risks for patient with xenograft do develop and propagate porcine infections (281). (iv) While CRISPR/Cas9 genome editing can remove the PERV gene from pig, the risk of xenozoonosis with other roseoloviruses remains (282). For example, porcine cytomegalovirus was the cause of a significant reduction of the survival time of the transplanted pig organs (283). However, such concerns can also be appropriately handled with modern drug therapies, selective breeding, and genetic modification.
As allotransplantation is restricted due to cell and organ shortage, xenograft provides an alternative source of tissues, and xenotransplantation may represent the next revolution in medicine. More patients with end-organ failure would undoubtedly benefit from breaking the immunological barriers to xenotransplantation in near future.
YW, CC and SD conceived the idea. QZho and TL wrote the manuscript. KW and QZha prepared the figures. CC, KW, ZG, CC and YW revised the manuscript. All authors contributed to the article and approved the submitted version.
This study was supported by an NSFC grant (No. 81802504), the International Innovation Cooperation Project of Sichuan Science and Technology Bureau (2022YTH0005 and 2019YFS0439), the Science and Technology Innovation Project of Chengdu, China (No. 2021-YF05-00225-SN), a Sichuan Medical Association grant (No. Q19037) to YW, the Science and Technology Bureau of Sichuan Province for outstanding young talent (No. 2020JDJQ0067) to QZho, and the Pelotonia Postdoc Fellowship and an OSU Department of Radiation-Oncology Basic Research seed grant to CC.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Jawoniyi O, Gormley K, McGleenan E, Noble HR. Organ donation and transplantation: Awareness and roles of healthcare professionals-a systematic literature review. J Clin Nurs (2018) 27(5-6):e726–38. doi: 10.1111/jocn.14154
2. Pedersen PB, Henriksen DP, Brabrand M, Lassen AT. Prevalence of organ failure and mortality among patients in the emergency department: a population-based cohort study. BMJ Open (2019) 9(10):e032692. doi: 10.1136/bmjopen-2019-032692
3. Lone NI, Walsh TS. Impact of intensive care unit organ failures on mortality during the five years after a critical illness. Am J Respir Crit Care Med (2012) 186(7):640–7. doi: 10.1164/rccm.201201-0059OC
4. Jones, Ben. Keeping kidneys. Bull World Health Organisation (2012) 90(10):718–9. doi: 10.2471/BLT.12.021012
5. D. Working Group on Incentives for Living, Matas AJ, Satel S, Munn S, Richards JR, Tan-Alora A, et al. Danguilan: Incentives for organ donation: proposed standards for an internationally acceptable system. Am J Transplant (2012) 12(2):306–12. doi: 10.1111/j.1600-6143.2011.03881.x
6. Ravitsky V. Incentives for postmortem organ donation: ethical and cultural considerations. J Med Ethics (2013) 39(6):380–1. doi: 10.1136/medethics-2013-101322
7. Shafran D, Kodish E, Tzakis A. Organ shortage: the greatest challenge facing transplant medicine. World J Surg (2014) 38(7):1650–7. doi: 10.1007/s00268-014-2639-3
8. Gerber Y, Weston SA, Redfield MM, Chamberlain AM, Manemann SM, Jiang R, et al. A contemporary appraisal of the heart failure epidemic in Olmsted county, Minnesota, 2000 to 2010. JAMA Intern Med (2015) 175(6):996–1004. doi: 10.1001/jamainternmed.2015.0924
9. Cooper DK, Ezzelarab MB, Hara H, Iwase H, Lee W, Wijkstrom M, et al. The pathobiology of pig-to-primate xenotransplantation: a historical review. Xenotransplantation (2016) 23(2):83–105. doi: 10.1111/xen.12219
10. Carvalho-Oliveira M, Valdivia E, Blasczyk R, Figueiredo C. Immunogenetics of xenotransplantation. Int J Immunogenet (2021) 48(2):120–34. doi: 10.1111/iji.12526
11. Cooper DK, Human PA, Lexer G, Rose AG, Rees J, Keraan M, et al. Effects of cyclosporin and antibody adsorption on pig cardiac xenograft survival in the baboon. J Heart Transplant (1988) 7(3):238–46.
12. Galili U. The alpha-gal epitope and the anti-gal antibody in xenotransplantation and in cancer immunotherapy. Immunol Cell Biol (2005) 83(6):674–86. doi: 10.1111/j.1440-1711.2005.01366.x
13. Huai G, Qi P, Yang H, Wang Y. Characteristics of alpha-gal epitope, anti-gal antibody, alpha1,3 galactosyltransferase and its clinical exploitation (Review). Int J Mol Med (2016) 37(1):11–20. doi: 10.3892/ijmm.2015.2397
14. Cooper DKC, Ekser B, Tector AJ. Immunobiological barriers to xenotransplantation. Int J Surg (2015) 23(Pt B):211–6. doi: 10.1016/j.ijsu.2015.06.068
15. Galili U. Natural anti-carbohydrate antibodies contributing to evolutionary survival of primates in viral epidemics? Glycobiology (2016) 26(11):1140–50. doi: 10.1093/glycob/cww088
16. Zeyland J, Lipinski D, Slomski R. The current state of xenotransplantation. J Appl Genet (2015) 56(2):211–8. doi: 10.1007/s13353-014-0261-6
17. Yang YG, Sykes M. Xenotransplantation: current status and a perspective on the future. Nat Rev Immunol (2007) 7(7):519–31. doi: 10.1038/nri2099
18. Kobayashi T, Cooper DK. Anti-gal, alpha-gal epitopes, and xenotransplantation. Subcell Biochem (1999) 32:229–57. doi: 10.1007/978-1-4615-4771-6_10
19. Platt JL, Fischel RJ, Matas AJ, Reif SA, Bolman RM, Bach FH. Immunopathology of hyperacute xenograft rejection in a swine-to-primate model. Transplantation (1991) 52(2):214–20. doi: 10.1097/00007890-199108000-00006
20. Schuurman HJ, Cheng J, Lam T. Pathology of xenograft rejection: a commentary. Xenotransplantation (2003) 10(4):293–9. doi: 10.1034/j.1399-3089.2003.02092.x
21. Ierino FL, Sandrin MS. Spectrum of the early xenograft response: from hyperacute rejection to delayed xenograft injury. Crit Rev Immunol (2007) 27(2):153–66. doi: 10.1615/critrevimmunol.v27.i2.30
22. Rose AG, Cooper DK, Human PA, Reichenspurner H, Reichart B. Histopathology of hyperacute rejection of the heart: experimental and clinical observations in allografts and xenografts. J Heart Lung Transplant (1991) 10(2):223–34.
23. Phelps CJ, Koike C, Vaught TD, Boone J, Wells KD, Chen SH, et al. Production of alpha 1,3-galactosyltransferase-deficient pigs. Science (2003) 299(5605):411–4. doi: 10.1126/science.1078942
24. Lai L, Kolber-Simonds D, Park KW, Cheong HT, Greenstein JL, Im GS, et al. Production of alpha-1,3-galactosyltransferase knockout pigs by nuclear transfer cloning. Science (2002) 295(5557):1089–92. doi: 10.1126/science.1068228
25. Petersen B, Carnwath JW, Niemann H. The perspectives for porcine-to-human xenografts. Comp Immunol Microbiol Infect Dis (2009) 32(2):91–105. doi: 10.1016/j.cimid.2007.11.014
26. Kuwaki K, Tseng YL, Dor FJ, Shimizu A, Houser SL, Sanderson TM, et al. Heart transplantation in baboons using alpha1,3-galactosyltransferase gene-knockout pigs as donors: initial experience. Nat Med (2005) 11(1):29–31. doi: 10.1038/nm1171
27. Yeh H, Machaidze Z, Wamala I, Fraser JW, Navarro-Alvarez N, Kim K, et al. Increased transfusion-free survival following auxiliary pig liver xenotransplantation. Xenotransplantation (2014) 21(5):454–64. doi: 10.1111/xen.12111
28. Ekser B, Burlak C, Waldman JP, Lutz AJ, Paris LL, Veroux M, et al. Immunobiology of liver xenotransplantation. Expert Rev Clin Immunol (2012) 8(7):621–34. doi: 10.1586/eci.12.56
29. Azimzadeh AM, Kelishadi SS, Ezzelarab MB, Singh AK, Stoddard T, Iwase H, et al. 3rd: Early graft failure of GalTKO pig organs in baboons is reduced by expression of a human complement pathway-regulatory protein. Xenotransplantation (2015) 22(4):310–6. doi: 10.1111/xen.12176
30. Xia G, Ji P, Rutgeerts O, Waer M. Natural killer cell- and macrophage mediated discordant guinea pig–>rat xenograft rejection in the absence of complement, xenoantibody and T cell immunity. Transplantation (2000) 70(1):86–93.
31. Gollackner B, Goh SK, Qawi I, Buhler L, Robson SC. Acute vascular rejection of xenografts: Roles of natural and elicited xenoreactive antibodies in activation of vascular endothelial cells and induction of procoagulant activity. Transplantation (2004) 77(11):1735–41. doi: 10.1097/01.TP.0000131167.21930.B8
32. Ekser B, Cooper DK. Overcoming the barriers to xenotransplantation: prospects for the future. Expert Rev Clin Immunol (2010) 6(2):219. doi: 10.1586/eci.09.81
33. Lin SS, Hanaway MJ, Gonzalez-Stawinski GV, Lau CL, Parker W, Davis RD, et al. The role of anti-Galalpha1-3Gal antibodies in acute vascular rejection and accommodation of xenografts. Transplantation (2000) 70(12):1667–74. doi: 10.1097/00007890-200012270-00002
34. Chen G, Qian H, Starzl T, Sun H, Garcia B, Wang X, et al. Acute rejection is associated with antibodies to non-gal antigens in baboons using gal-knockout pig kidneys. Nat Med (2005) 11(12):1295–8. doi: 10.1038/nm1330
35. Byrne GW, Stalboerger PG, Davila E, Heppelmann CJ, Gazi MH, McGregor HC, et al. Proteomic identification of non-gal antibody targets after pig-to-primate cardiac xenotransplantation. Xenotransplantation (2008) 15(4):268–76. doi: 10.1111/j.1399-3089.2008.00480.x
36. Zhu A, Hurst R. Anti-n-glycolylneuraminic acid antibodies identified in healthy human serum. Xenotransplantation (2002) 9(6):376–381. doi: 10.1034/j.1399-3089.2002.02138.x
37. Byrne G, Ahmad-Villiers S, Du Z, Mcgregor C. B4GALNT2 and xenotransplantation: A newly appreciated xenogeneic antigen. Xenotransplantation (2018) 25(Pt 1):e12394. doi: 10.1111/xen.12394
38. Bouhours D, Pourcel C, Bouhours JE. Simultaneous expression by porcine aorta endothelial cells of glycosphingolipids bearing the major epitope for human xenoreactive antibodies (Gal alpha 1-3Gal), blood group h determinant and n-glycolylneuraminic acid. Glycoconj J (1996) 13(6):947–53. doi: 10.1007/BF01053190
39. Renton PH, Howell P, Ikin EW, Giles CM. Anti-sd^a, a new blood group antibody. Vox Sanguinis (2010) 13(6):493–501. doi: 10.1111/j.1423-0410.1967.tb03796.x
40. Paul A, Padler-Karavani V. Evolution of sialic acids: Implications in xenotransplant biology. Xenotransplantation (2018) 25(6):e12424. doi: 10.1111/xen.12424
41. Varki A. Loss of n-glycolylneuraminic acid in humans: Mechanisms, consequences, and implications for hominid evolution. Am J Phys Anthropol (2001) 33:54–69. doi: 10.1002/ajpa.10018
42. Estrada JL, Martens G, Li P, Adams A, Newell KA, Ford ML, et al. Evaluation of human and non-human primate antibody binding to pig cells lacking GGTA1/CMAH/β4GalNT2 genes. Xenotransplantation (2015) 22(3):194–202. doi: 10.1111/xen.12161
43. Thomas A, Hawthorne WJ, Burlak C. Xenotransplantation literature update, November/December 2019. Xenotransplantation (2020) 27(1):e12582. doi: 10.1111/xen.12582
44. Martens GR, Reyes LM, Li P, Butler JR, Ladowski JM, Estrada JL, et al. Humoral reactivity of renal transplant-waitlisted patients to cells from GGTA1/CMAH/B4GalNT2, and SLA class I knockout pigs. Transplantation (2017) 101(4):e86–92. doi: 10.1097/tp.0000000000001646
45. Fischer K, Rieblinger B, Hein R, Sfriso R, Zuber J, Fischer A, et al. Viable pigs after simultaneous inactivation of porcine MHC class I and three xenoreactive antigen genes GGTA1, CMAH and B4GALNT2. Xenotransplantation (2020) 27(1):e12560. doi: 10.1111/xen.12560
46. Kim SC, Mathews DV, Breeden CP, Higginbotham LB, Ladowski J, Martens G, et al. Long-term survival of pig-to-rhesus macaque renal xenografts is dependent on CD4 T cell depletion. Am J Transplant (2019) 19(8):2174–85. doi: 10.1111/ajt.15329
47. Díaz Varela I, Sánchez Mozo P, Centeno Cortés A, Alonso Blanco C, Valdés Cañedo F. Cross-reactivity between swine leukocyte antigen and human anti-HLA-specific antibodies in sensitized patients awaiting renal transplantation. J Am Soc Nephrol (2003) 14(10):2677–83. doi: 10.1097/01.asn.0000088723.07259.cf
48. Ladowski JM, Houp J, Hauptfeld-Dolejsek V, Javed M, Hara H, Cooper DKC. Aspects of histocompatibility testing in xenotransplantation. Transpl Immunol (2021) 67:101409. doi: 10.1016/j.trim.2021.101409
49. Figueiredo C, Carvalho Oliveira M, Chen-Wacker C, Jansson K, Höffler K, Yuzefovych Y, et al. Immunoengineering of the vascular endothelium to silence MHC expression during normothermic ex vivo lung perfusion. Hum Gene Ther (2019) 30(4):485–96. doi: 10.1089/hum.2018.117
50. Ladowski JM, Hara H, Cooper DKC. The role of SLAs in xenotransplantation. Transplantation (2021) 105(2):300–7. doi: 10.1097/tp.0000000000003303
51. Yuzefovych Y, Valdivia E, Rong S, Hack F, Rother T, Schmitz J, et al. Genetic engineering of the kidney to permanently silence MHC transcripts during ex vivo organ perfusion. Front Immunol (2020) 11:265. doi: 10.3389/fimmu.2020.00265
52. Hammer SE, Ho CS, Ando A, Rogel-Gaillard C, Charles M, Tector M, et al. Importance of the major histocompatibility complex (Swine leukocyte antigen) in swine health and biomedical research. Annu Rev Anim Biosci (2020) 8:171–98. doi: 10.1146/annurev-animal-020518-115014
53. Vadori M, Cozzi E. The immunological barriers to xenotransplantation. Tissue Antigens (2015) 86(4):239–53. doi: 10.1111/tan.12669
54. Al-Mohanna F, Saleh S, Parhar RS, Khabar K, Collison K. Human neutrophil gene expression profiling following xenogeneic encounter with porcine aortic endothelial cells: the occult role of neutrophils in xenograft rejection revealed. J Leukoc Biol (2005) 78(1):51–61. doi: 10.1189/jlb.0904494
55. Baumann BC, Forte P, Hawley RJ, Rieben R, Schneider M, Seebach JD. Lack of galactose-alpha-1,3-galactose expression on porcine endothelial cells prevents complement-induced lysis but not direct xenogeneic NK cytotoxicity. J Immunol (2004) 172(10):6460–7. doi: 10.4049/jimmunol.172.10.6460
56. Fox A, Mountford J, Braakhuis A, Harrison LC. Innate and adaptive immune responses to nonvascular xenografts: evidence that macrophages are direct effectors of xenograft rejection. J Immunol (2001) 166(3):2133–40. doi: 10.4049/jimmunol.166.3.2133
57. Lutz AJ, Li P, Estrada JL, Sidner RA, Chihara RK, Downey SM, et al. Double knockout pigs deficient in n-glycolylneuraminic acid and galactose alpha-1,3-galactose reduce the humoral barrier to xenotransplantation. Xenotransplantation (2013) 20(1):27–35. doi: 10.1111/xen.12019
58. Adams AB, Kim SC, Martens GR, Ladowski JM, Estrada JL, Reyes LM, et al. Xenoantigen deletion and chemical immunosuppression can prolong renal xenograft survival. Ann Surg (2018) 268(4):564–73. doi: 10.1097/sla.0000000000002977
59. Burlak C, Paris LL, Lutz AJ, Sidner RA, Estrada J, Li P, et al. Reduced binding of human antibodies to cells from GGTA1/CMAH KO pigs. Am J Transplant (2014) 14(8):1895–900. doi: 10.1111/ajt.12744
60. Estrada JL, Martens G, Li P, Adams A, Newell KA, Ford ML, et al. Evaluation of human and non-human primate antibody binding to pig cells lacking GGTA1/CMAH/beta4GalNT2 genes. Xenotransplantation (2015) 22(3):194–202. doi: 10.1111/xen.12161
61. Basnet NB, Ide K, Tahara H, Tanaka Y, Ohdan H. Deficiency of nglycolylneuraminic acid and Galalpha1-3Galbeta1-4GlcNAc epitopes in xenogeneic cells attenuates cytotoxicity of human natural antibodies. Xenotransplantation (2010) 17(6):440–8. doi: 10.1111/j.1399-3089.2010.00610.x
62. Nomura S, Ariyoshi Y, Watanabe H, Pomposelli T, Takeuchi K, Garcia G, et al. Transgenic expression of human CD47 reduces phagocytosis of porcine endothelial cells and podocytes by baboon and human macrophages. Xenotransplantation (2020) 27(1):e12549. doi: 10.1111/xen.12549
63. Yamamoto T, Hara H, Iwase H, Jagdale A, Bikhet MH, Morsi MA, et al. The final obstacle to successful pre-clinical xenotransplantation? Xenotransplantation (2020) 27(5):e12596. doi: 10.1111/xen.12596
64. Sake HJ, Frenzel A, Lucas-Hahn A, Nowak-Imialek M, Hassel P, Hadeler KG, et al. Possible detrimental effects of beta-2-microglobulin knockout in pigs. Xenotransplantation (2019) 26(6):e12525. doi: 10.1111/xen.12525
65. H. Auchincloss, Sachs DH. Xenogeneic transplantation. Annu Rev Immunol (1998) 16:433–70. doi: 10.1146/annurev.immunol.16.1.433
66. Sykes M, Lee LA, Sachs DH. Xenograft tolerance. Immunol Rev (1994) 141:245–76. doi: 10.1111/j.1600-065x.1994.tb00880.x
67. Schneider MK, Ghielmetti M, Rhyner DM, Antsiferova MA, Seebach JD. Human leukocyte transmigration across Galalpha(1,3)Gal-negative porcine endothelium is regulated by human CD18 and CD99. Transplantation (2009) 87(4):491–9. doi: 10.1097/TP.0b013e318195fb8d
68. Lilienfeld BG, Garcia-Borges C, Crew MD, Seebach JD. Porcine UL16-binding protein 1 expressed on the surface of endothelial cells triggers human NK cytotoxicity through NKG2D. J Immunol (2006) 177(4):2146–52. doi: 10.4049/jimmunol.177.4.2146
69. Matter-Reissmann UB, Forte P, Schneider MK, Filgueira L, Groscurth P, Seebach JD. Xenogeneic human NK cytotoxicity against porcine endothelial cells is perforin/granzyme b dependent and not inhibited by bcl-2 overexpression. Xenotransplantation (2002) 9(5):325–37. doi: 10.1034/j.1399-3089.2002.01074.x
70. Iwaszko M, Bogunia-Kubik K. Clinical significance of the HLA-e and CD94/NKG2 interaction. Arch Immunol Ther Exp (Warsz) (2011) 59(5):353–67. doi: 10.1007/s00005-011-0137-y
71. Crinier A, Narni-Mancinelli E, Ugolini S, Vivier E. SnapShot: Natural killer cells. Cell (2020) 180(6):1280–1280 e1. doi: 10.1016/j.cell.2020.02.029
72. Sullivan JA, Oettinger HF, Sachs DH, Edge AS. Analysis of polymorphism in porcine MHC class I genes: alterations in signals recognized by human cytotoxic lymphocytes. J Immunol (1997) 159(5):2318–26.
73. Long EO, Kim HS, Liu D, Peterson ME, Rajagopalan S. Controlling natural killer cell responses: integration of signals for activation and inhibition. Annu Rev Immunol (2013) 31:227–58. doi: 10.1146/annurev-immunol-020711-075005
74. Forte P, Baumann BC, Weiss EH, Seebach JD. HLA-e expression on porcine cells: protection from human NK cytotoxicity depends on peptide loading. Am J Transplant (2005) 5(9):2085–93. doi: 10.1111/j.1600-6143.2005.00987.x
75. Ritschl P, Ebner S, Fabritius C, Resch T, Kotsch, Katja. The role of natural killer cells in humoral rejection. Transplant: Off J Transplant Soc (2015) 99(7):1335–40. doi: 10.1097/TP.0000000000000757
76. Ochoa MC, Minute L, Rodriguez I, Garasa S, Perez-Ruiz E, Inoges S, et al. Antibody-dependent cell cytotoxicity: immunotherapy strategies enhancing effector NK cells. Immunol Cell Biol (2017) 95(4):347–55. doi: 10.1038/icb.2017.6
77. Puga Yung G, Schneider MKJ, Seebach JD. The role of NK cells in pig-to-Human xenotransplantation. J Immunol Res (2017) 2017:4627384. doi: 10.1155/2017/4627384
78. Li S, Yan Y, Yuan L, Bullens DM, Billiau AD. Rapidly induced, T-cell independent xenoantibody production is mediated by marginal zone b cells and requires help from NK cells. Blood (2008) 110(12):3926–35. doi: 10.1182/blood-2007-01-065482
79. Nishinakamura H, Itoh T, Kumano K, Hata Y, Kodama, Shohta. Islet-derived damage-associated molecular pattern molecule contributes to immune responses following microencapsulated neonatal porcine islet xenotransplantation in mice. Xenotransplantation (2016) 23(5):393–404. doi: 10.1111/xen.12253
80. Wu H, Ma J, Wang P, Corpuz TM, Panchapakesan U, Wyburn KR, et al. HMGB1 contributes to kidney ischemia reperfusion injury. J Am Soc Nephrol (2010) 21(11):1878–90. doi: 10.1681/ASN.2009101048
81. Chen J, Gao H, Chen LL, Wang X, Mou L. A potential role of TLR2 in xenograft rejection of porcine iliac endothelial cells: An in vitro study. Xenotransplantation (2019) 26(5):e12526. doi: 10.1111/xen.12526
82. Chung H, Hong SJ, Choi SW, Koo JY, Park CG. High mobility group box 1 secretion blockade results in the reduction of early pancreatic islet graft loss. Biochem Biophys Res Commun (2019) 514(3):1081–6. doi: 10.1016/j.bbrc.2019.05.003
83. Cadili A, Kneteman N. The role of macrophages in xenograft rejection. Transplant Proc (2008) 40(10):3289–93. doi: 10.1016/j.transproceed.2008.08.125
84. Hibbs JB, Taintor RR, Vavrin Z, Rachlin EM. NO: A cytotoxic acivated-macrophage effector molecule. Biochem Biophys Res Commun (1988) 157(1):87–94. doi: 10.1016/S0006-291X(88)80015-9
85. El-Ouaghlidi A, Jahr H, Pfeiffer G, Hering BJ, Brandhorst D, Brandhorst H, et al. Cytokine mRNA expression in peripheral blood cells of immunosuppressed human islet transplant recipients. J Mol Med (1999) 77(1):115. doi: 10.1007/s001090050315
86. Ide K, Wang H, Tahara H, Liu J, Wang X, , Toshimasa A, et al. Role for CD47-SIRPα signaling in xenograft rejection by macrophages. Proc Natl Acad Sci United States America (2007). doi: 10.1073/pnas.0609661104
87. Ide K, Ohdan H, Kobayashi T, Hara H, Ishiyama K, Asahara T. Antibody- and complement-independent phagocytotic and cytolytic activities of human macrophages toward porcine cells. Xenotransplantation (2005) 12(3):181–8. doi: 10.1111/j.1399-3089.2005.00222.x
88. Morris CF, Simeonovic CJ, Fung MC, Wilson JD, Hapel AJ. Intragraft expression of cytokine transcripts during pig proislet xenograft rejection and tolerance in mice. J Immunol (1995) 154(5):2470–82.
89. Loudovaris T, Mandel TE, Charlton B. CD4+ T cell mediated destruction of xenografts within cell-impermeable membranes in the absence of CD8+ T cells and b cells. Transplantation (1996) 61(12):1678–84. doi: 10.1097/00007890-199606270-00003
90. Fox A, Koulmanda M, Mandel TE, van Rooijen N, Harrison LC. Evidence that macrophages are required for T-cell infiltration and rejection of fetal pig pancreas xenografts in nonobese diabetic mice. Transplantation (1998) 66(11):1407–16. doi: 10.1097/00007890-199812150-00002
91. Feng X, Zheng XX, Yi S, Lehnert AM, Strom TB, O'Connell PJ. IL-10/Fc inhibits macrophage function and prolongs pancreatic islet xenograft survival. Transplantation (1999) 68(11):1775–83. doi: 10.1097/00007890-199912150-00023
92. Lin Y, Vandeputte M, Waer M. Natural killer cell- and macrophage-mediated rejection of concordant xenografts in the absence of T and b cell responses. J Immunol (1997) 158(12):5658–67.
93. Wang HT, Maeda A, Sakai R, Lo PC, Takakura C, Jiaravuthisan P, et al. Human CD31 on porcine cells suppress xenogeneic neutrophil-mediated cytotoxicity via the inhibition of NETosis. Xenotransplantation (2018) 25(5):e12396. doi: 10.1111/xen.12396
94. al-Mohanna F, Collison K, Parhar R, Kwaasi A, Meyer B, Saleh S, et al. Activation of naive xenogeneic but not allogeneic endothelial cells by human naive neutrophils: a potential occult barrier to xenotransplantation. Am J Pathol (1997) 151(1):111–20.
95. Sheikh S, Parhar R, Al-Mohanna F. Rapid static adhesion of human naive neutrophil to naive xenoendothelium under physiologic flow is independent of Galalpha1,3-gal structures. J Leukoc Biol (2002) 71(6):932–40. doi: 10.1189/jlb.71.6.932
96. Cardozo LA, Rouw DB, Ambrose LR, Midulla M, Florey O, Haskard DO, et al. The neutrophil: the unnoticed threat in xenotransplantation? Transplantation (2004) 78(12):1721–8. doi: 10.1097/01.tp.0000147341.40485.b4
97. Loukogeorgakis SP, van den Berg MJ, Sofat R, Nitsch D, Charakida M, Haiyee B, et al. Role of NADPH oxidase in endothelial ischemia/reperfusion injury in humans. Circulation (2010) 121(21):2310–6. doi: 10.1161/CIRCULATIONAHA.108.814731
98. Moll T, Dejana E, Vestweber D. In vitro degradation of endothelial catenins by a neutrophil protease. J Cell Biol (1998) 140(2):403–7. doi: 10.1083/jcb.140.2.403
99. Uchida Y, Freitas MC, Zhao D, Busuttil RW, Kupiec-Weglinski JW. The protective function of neutrophil elastase inhibitor in liver ischemia/reperfusion injury. Transplantation (2010) 89(9):1050–6. doi: 10.1097/TP.0b013e3181d45a98
100. Sayah DM, Mallavia B, Liu F, Ortiz-Munoz G, Caudrillier A, DerHovanessian A, et al. Neutrophil extracellular traps are pathogenic in primary graft dysfunction after lung transplantation. Am J Respir Crit Care Med (2015) 191(4):455–63. doi: 10.1164/rccm.201406-1086OC
101. Dong X, Swaminathan S, Bachman LA, Croatt AJ, Nath KA, Griffin MD. Resident dendritic cells are the predominant TNF-secreting cell in early renal ischemia-reperfusion injury. Kidney Int (2007) 71(7):619–28. doi: 10.1038/sj.ki.5002132
102. Lin J, Wang H, Liu C, Cheng A, Deng Q, Zhu H, et al. Dendritic cells: Versatile players in renal transplantation. Front Immunol (2021) 12:654540. doi: 10.3389/fimmu.2021.654540
103. Jongbloed SL, Kassianos AJ, McDonald KJ, Clark GJ, Ju X, Angel CE, et al. Human CD141+ (BDCA-3)+ dendritic cells (DCs) represent a unique myeloid DC subset that cross-presents necrotic cell antigens. J Exp Med (2010) 207(6):1247–60. doi: 10.1084/jem.20092140
104. Marin E, Cuturi MC, Moreau A. Tolerogenic dendritic cells in solid organ transplantation: Where do we stand? Front Immunol (2018) 9:274. doi: 10.3389/fimmu.2018.00274
105. Zahorchak AF, Macedo C, Hamm DE, Butterfield LH, Metes DM, Thomson AW. High PD-L1/CD86 MFI ratio and IL-10 secretion characterize human regulatory dendritic cells generated for clinical testing in organ transplantation. Cell Immunol (2018) 323:9–18. doi: 10.1016/j.cellimm.2017.08.008
106. Manna PP, Duffy B, Olack B, Lowell J, Mohanakumar T. Activation of human dendritic cells by porcine aortic endothelial cells: transactivation of naïve T cells through costimulation and cytokine generation. Transplantation (2001) 72(9):1563–71. doi: 10.1097/00007890-200111150-00015
107. Hisashi Y, Yamada K, Kuwaki K, Tseng YL, Dor FJ, Houser SL, et al. Rejection of cardiac xenografts transplanted from alpha1,3-galactosyltransferase gene-knockout (GalT-KO) pigs to baboons. Am J Transplant (2008) 8(12):2516–26. doi: 10.1111/j.1600-6143.2008.02444.x
108. Davila E, Byrne GW, LaBreche PT, McGregor HC, Schwab AK, Davies WR, et al. T-Cell responses during pig-to-primate xenotransplantation. Xenotransplantation (2006) 13(1):31–40. doi: 10.1111/j.1399-3089.2005.00258.x
109. Scalea J, Hanecamp I, Robson SC, Yamada K. T-Cell-mediated immunological barriers to xenotransplantation. Xenotransplantation (2012) 19:1399–3089. doi: 10.1111/j.1399-3089.2011.00687.x
110. Griesemer A, Yamada K, Sykes M. Xenotransplantation: immunological hurdles and progress toward tolerance. Immunol Rev (2014) 258(1):241. doi: 10.1111/imr.12152
111. Samy KP, Butler JR, Ping L, Cooper D, Burcin E. The role of costimulation blockade in solid organ and islet xenotransplantation. J Immunol Res (2017) 2017:8415205. doi: 10.1155/2017/8415205
112. Mardomi A, Mohammadi N, Khosroshahi HT, Abediankenari S. An update on potentials and promises of T cell co:ignaling molecules in transplantation. J Cell Physiol (2019) 235(5):4183–97. doi: 10.1002/jcp.29369
113. Carvalho Oliveira M, Valdivia E, Verboom M, Yuzefovych Y, Sake HJ, Pogozhykh O, et al. Generating low immunogenic pig pancreatic islet cell clusters for xenotransplantation. J Cell Mol Med (2020) 24(9):5070–81. doi: 10.1111/jcmm.15136
114. Hein R, Sake HJ, Pokoyski C, Hundrieser J, Brinkmann A, Baars W, et al. Triple (GGTA1, CMAH, B2M) modified pigs expressing an SLA class i(low) phenotype-effects on immune status and susceptibility to human immune responses. Am J Transplant (2020) 20(4):988–98. doi: 10.1111/ajt.15710
115. Galili U. Evolution and pathophysiology of the human natural anti-α-galactosyl IgG (anti-gal) antibody. Springer Semin Immunopathol (1993) 15(2–3):155–71. doi: 10.1007/BF00201098
116. Xu Y, Yang YG, Ohdan H, Ryan D, Harper D, Wu C, et al. Characterization of anti-gal antibody-producing cells of baboons and humans. Transplantation (2006) 81(6):940–8. doi: 10.1097/01.tp.0000203300.87272.a3
117. Ohdan H, Swenson KG, Kruger Gray HS, Yang YG, Xu Y, Thall AD, et al. Mac-1-negative b-1b phenotype of natural antibody-producing cells, including those responding to gal alpha 1,3Gal epitopes in alpha 1,3-galactosyltransferase-deficient mice. J Immunol (2000) 165(10):5518–29. doi: 10.4049/jimmunol.165.10.5518
118. Tanemura M, Yin D, Chong AS, Galili U. Differential immune responses to alpha-gal epitopes on xenografts and allografts: implications for accommodation in xenotransplantation. J Clin Invest (2000) 105(3):301–10. doi: 10.1172/jci7358
119. Shimizu I, Kawahara T, Haspot F, Bardwell PD, Carroll MC, Sykes M. B-cell extrinsic CR1/CR2 promotes natural antibody production and tolerance induction of anti-alphaGAL-producing b-1 cells. Blood (2007) 109(4):1773–81. doi: 10.1182/blood-2006-02-002386
120. Gonzalez-Stawinski GV, Davis RD Jr. Rituximab as monotherapy for elicited xenoreactive antibody responses. J Heart Lung Transplant (2006) 25(12):1462–6. doi: 10.1016/j.healun.2006.09.008
121. Alwayn IP, Xu Y, Basker M, Wu C, Buhler L, Lambrigts D, et al. Effects of specific anti-b and/or anti-plasma cell immunotherapy on antibody production in baboons: depletion of CD20- and CD22-positive b cells does not result in significantly decreased production of anti-alphaGal antibody. Xenotransplantation (2001) 8(3):157–71. doi: 10.1034/j.1399-3089.2001.008003157.x
122. Li JH, Zhao B, Zhu XH, Wang L, Zou HJ, Chen S, et al. Blockade of extracellular HMGB1 suppresses xenoreactive b cell responses and delays acute vascular xenogeneic rejection. Am J Transplant (2015) 15(8):2062–74. doi: 10.1111/ajt.13275
123. Furie B, Furie BC. Mechanisms of thrombus formation. N Engl J Med (2008) 359(9):938–49. doi: 10.1056/NEJMra0801082
124. Cowan PJ, d'Apice AJ. Complement activation and coagulation in xenotransplantation. Immunol Cell Biol (2009) 87(3):203–8. doi: 10.1038/icb.2008.107
125. Roussel JC, Moran CJ, Salvaris EJ, Nandurkar HH, D"Apice AJF, Cowan PJ. Pig thrombomodulin binds human thrombin but is a poor cofactor for activation of human protein c and TAFI. Am J Transplant (2010) 8(6):1101–12. doi: 10.1111/j.1600-6143.2008.02210.x
126. Pareti FI, Mazzucato M, Bottini E, Mannucci and PM. Interaction of porcine von willebrand factor with the platelet glycoproteins ib and IIb/IIIa complex. Br J Haematol (1992) 82(1):81–6. doi: 10.1111/j.1365-2141.1992.tb04597.x
127. Bühler LB, Basker M, Alwayn IPJ, Goepfert C, Kitamura H, Kawai T, et al. Coagulation and thrombotic disorders associated with pig organ and hematopoietic cell transplantation in nonhuman primates. Transplantation (2000) 70(9):1323–31. doi: 10.1097/00007890-200011150-00010
128. Rosales IA, Colvin RB. The pathology of solid organ xenotransplantation叉叉. Curr Opin Organ Transplant (2019) 24(5):535–42. doi: 10.1097/MOT.0000000000000681
129. Lin CC, Ezzelarab M, Shapiro R, Ekser B, Long C, Hara H, et al. Recipient tissue factor expression is associated with consumptive coagulopathy in pig-to-primate kidney xenotransplantation. Am J Transplant (2010) 10(7):1556–68. doi: 10.1111/j.1600-6143.2010.03147.x
130. Cowan PJ. Coagulation and the xenograft endothelium. Xenotransplantation (2007) 14(1):7–12. doi: 10.1111/j.1399-3089.2006.00368.x
131. Mohiuddin MM, Singh AK, Corcoran PC, Hoyt RF, Thomas ML 3rd, Ayares D, et al. Genetically engineered pigs and target-specific immunomodulation provide significant graft survival and hope for clinical cardiac xenotransplantation. J Thorac Cardiovasc Surg (2014) 148(3):1106–13; discussion 1113-4. doi: 10.1016/j.jtcvs.2014.06.002
132. Dwyer KM, Robson SC, Nandurkar HH, Campbell DJ, Gock H, Murray-Segal LJ, et al. Thromboregulatory manifestations in human CD39 transgenic mice and the implications for thrombotic disease and transplantation. J Clin Invest (2004) 113(10):1440–6. doi: 10.1172/jci19560
133. Cooper DC, Iwase H, Wang L, Yamamoto T, Li Q, Li J, et al. Bringing home the bacon: Update on the state of kidney xenotransplantation. Blood Purif (2018) (45):254–9. doi: 10.1159/000485163
134. Zhao Y, Cooper D, Wang H, Chen P, He C, Cai Z, et al. Potential pathological role of pro-Inflammatory cytokines (IL TNF│ and IL) in xenotransplantation. Xenotransplantation (2019) 26(3):e12502. doi: 10.1111/xen.12502
135. Naeimi Kararoudi M, Hejazi SS, Elmas E, Hellström M, Naeimi Kararoudi M, Padma AM, et al. Clustered regularly interspaced short palindromic Repeats/Cas9 gene editing technique in xenotransplantation. Front Immunol (2018) 9:1711. doi: 10.3389/fimmu.2018.01711
136. Cozzi E, Vial C, Ostlie D, Farah B, Friend PJ. Maintenance triple immunosuppression with cyclosporin a, mycophenolate sodium and steroids allows prolonged survival of primate recipients of hDAF porcine renal xenografts. Xenotransplantation (2010) 10(4):300–10. doi: 10.1034/j.1399-3089.2003.02014.x
137. Mcgregor C, Davies WR, Oi K, Teotia SS, Schirmer JM, Risdahl JM, et al. Cardiac xenotransplantation: Recent preclinical progress with 3-month median survival. J Thorac Cardiovasc Surg (2005) 130(3):844.e1–9. doi: 10.1016/j.jtcvs.2005.04.017
138. Zhou Q, Zhang Q, Wang K, Huang T, Deng S, Wang Y, et al. Anti-rheumatic drug-induced hepatitis b virus reactivation and preventive strategies for hepatocellular carcinoma. Pharmacol Res (2022) 178:106181. doi: 10.1016/j.phrs.2022.106181
139. Zhong R, Tucker J, Zhang Z, Wall W, Grant D, Quan D, et al. The long-term survival of baboon-to-monkey kidney and liver xenografts. Xenotransplantation (2003) 10(5):398–409. doi: 10.1034/j.1399-3089.2003.02054.x
140. Buhler L, Awwad M, Basker M, Gojo S, Watts A, Treter S, et al. High-dose porcine hematopoietic cell transplantation combined with CD40 ligand blockade in baboons prevents an induced anti-pig humoral response. Transplantation (2000) 69(11):2296–304. doi: 10.1097/00007890-200006150-00013
141. Vandewalle J, Luypaert A, De Bosscher K, Libert C. Therapeutic mechanisms of glucocorticoids. Trends Endocrinol Metab (2018) 29(1):42–54. doi: 10.1016/j.tem.2017.10.010
142. Rijkelijkhuizen JK, Bouwman E, van der Burg MP, Ringers J, Ossevoort MA, Kuhn EM, et al. Successful suppression of the early rejection of pig islets in monkeys. Cell Transplant (2000) 9(6):909–12. doi: 10.1177/096368970000900618
143. Contreras JL, Eckhoff DE, Cartner S, Bilbao G, Ricordi C, Neville DM Jr., et al. Long-term functional islet mass and metabolic function after xenoislet transplantation in primates. Transplantation (2000) 69(2):195–201. doi: 10.1097/00007890-200001270-00001
144. Thomas FT, Ricordi C, Contreras JL, Hubbard WJ, Jiang XL, Eckhoff DE, et al. Reversal of naturally occuring diabetes in primates by unmodified islet xenografts without chronic immunosuppression. Transplantation (1999) 67(6):846–54. doi: 10.1097/00007890-199903270-00011
145. Hu H, Stavrou S, Cairns Baker B, Tornatore C, Scharff J, Okunieff P, et al. Depletion of T lymphocytes with immunotoxin retards the progress of experimental allergic encephalomyelitis in rhesus monkeys. Cell Immunol (1997) 177(1):26–34. doi: 10.1006/cimm.1997.1096
146. Cozzi E, Bhatti F, Schmoeckel M, Chavez G, Smith KG, Zaidi A, et al. Long-term survival of nonhuman primates receiving life-supporting transgenic porcine kidney xenografts. Transplantation (2000) 70(1):15–21.
147. Strehl C, Spies CM, Buttgereit F. Pharmacodynamics of glucocorticoids. Clin Exp Rheumatol (2011) 29(5 Suppl 68):S13.
148. Hayashi R, Wada H, Ito K, Adcock IM. Effects of glucocorticoids on gene transcription. Eur J Pharmacol (2004) 500(1-3):51–62. doi: 10.1016/j.ejphar.2004.07.011
149. Fauci AS, Dale DC, Balow JE. Glucocorticosteroid therapy: mechanisms of action and clinical considerations. Ann Intern Med (1976) 84(3):304–15. doi: 10.7326/0003-4819-84-3-304
150. Becker DE. Basic and clinical pharmacology of glucocorticosteroids. Anesth Prog (2013) 60(1):25–32. doi: 10.2344/0003-3006-60.1.25
151. Gheith OA, Nematalla AH, Bakr MA, Refaie A, Shokeir AA, Ghoneim MA. Steroid avoidance reduce the cost of morbidities after live-donor renal allotransplants: a prospective, randomized, controlled study. Exp Clin Transplant (2011) 9(2):121–7.
152. Chadban SJ, Ahn C, Axelrod DA, Foster BJ, Kasiske BL, Kher V, et al. Summary of the kidney disease: Improving global outcomes (KDIGO) clinical practice guideline on the evaluation and management of candidates for kidney transplantation. Transplantation (2020) 104(4):708–14. doi: 10.1097/tp.0000000000003137
153. Noble S, Markham A. Cyclosporin. a review of the pharmacokinetic properties, clinical efficacy and tolerability of a microemulsion-based formulation (Neoral). Drugs (1995) 50(5):924–41. doi: 10.2165/00003495-199550050-00009
154. Peters DH, Fitton A, Plosker GL, Faulds D. Tacrolimus. a review of its pharmacology, and therapeutic potential in hepatic and renal transplantation. Drugs (1993) 46(4):746–94. doi: 10.2165/00003495-199346040-00009
155. Michler RE, Mcmanus RP, Smith CR, Sadeghi AN, Marboe CC, Reemtsma K, et al. Prolongation of primate cardiac xenograft survival with cyclosporin. Transplantation (1987) 44(5):632–6. doi: 10.1097/00007890-198711000-00007
156. Pritchard DI. Sourcing a chemical succession for cyclosporin from parasites and human pathogens. Drug Discovery Today (2005) 10(10):688–91. doi: 10.1016/s1359-6446(05)03395-7
157. Thomson AW, Bonham CA, Zeevi A. Mode of action of tacrolimus (FK506): molecular and cellular mechanisms. Ther Drug Monit (1995) 17(6):584–91. doi: 10.1097/00007691-199512000-00007
158. Wiederrecht G, Lam E, Hung S, Martin M, Sigal N. The mechanism of action of FK-506 and cyclosporin a. Ann N Y Acad Sci (1993) 696:9–19. doi: 10.1111/j.1749-6632.1993.tb17137.x
159. Mochizuki M, Masuda K, Sakane T, Ito K, Kogure M, Sugino N, et al. A clinical trial of FK506 in refractory uveitis. Am J Ophthalmol (1993) 115(6):763–9. doi: 10.1016/s0002-9394(14)73645-1
160. Randomised trial comparing tacrolimus (FK506) and cyclosporin in prevention of liver allograft rejection. European FK506 multicentre liver study group. Lancet (1994) 344(8920):423–8.
161. Griffith BP, Bando K, Hardesty RL, Armitage JM, Keenan RJ, Pham SM, et al. A prospective randomized trial of FK506 versus cyclosporin after human pulmonary transplantation. Transplantation (1994) 57(6):848–51. doi: 10.1097/00007890-199403270-00013
162. Guethoff S, Meiser BM, Groetzner J, Eifert S, Grinninger C, Ueberfuhr P, et al. Ten-year results of a randomized trial comparing tacrolimus versus cyclosporin a in combination with mycophenolate mofetil after heart transplantation. Transplantation (2013) 95(4):629–34. doi: 10.1097/TP.0b013e318277e378
163. Krook H, Wennberg L, Hagberg A, Song Z, Groth CG, Korsgren O. Immunosuppressive drugs in islet xenotransplantation: a tool for gaining further insights in the mechanisms of the rejection process. Transplantation (2002) 74(8):1084–9. doi: 10.1097/00007890-200210270-00005
164. Shin JS, Kim JM, Min BH, Yoon IH, Kim HJ, Kim JS, et al. Pre-clinical results in pig-to-non-human primate islet xenotransplantation using anti-CD40 antibody (2C10R4)-based immunosuppression. Xenotransplantation (2018) 25(1):e12356. doi: 10.1111/xen.12356
165. Venkataramanan R, Jain A, Warty VS, Abuelmagd K, Alessiani M, Lever J, et al. Pharmacokinetics of FK 506 in transplant patients. Transplant Proc (1991) 23(6):2736–40.
166. Song Z, Wennberg L, Zhang J, Sundberg B, Bari S, Groth CG, et al. Protective effect of FK-506 in pig-to-rat islet xenotransplantation is abrogated by prednisolone. Transplant Proc (2000) 32(5):1025. doi: 10.1016/s0041-1345(00)01096-4
167. Yoon CH, Choi SH, Choi HJ, Lee HJ, Kang HJ, Kim JM, et al. Long-term survival of full-thickness corneal xenografts from α1,3-galactosyltransferase gene-knockout miniature pigs in non-human primates. Xenotransplantation (2020) 27(1):e12559. doi: 10.1111/xen.12559
168. Jiang T, Acosta D. An in vitro model of cyclosporin-induced nephrotoxicity. Fundam Appl Toxicol (1993) 20(4):486–95. doi: 10.1006/faat.1993.1059
169. Birdwell KA, Decker B, Barbarino JM, Peterson JF, Stein CM, Sadee W, et al. Clinical pharmacogenetics implementation consortium (CPIC) guidelines for CYP3A5 genotype and tacrolimus dosing. Clin Pharmacol Ther (2015) 98(1):19–24. doi: 10.1002/cpt.113
170. Boughton O, Borgulya G, Cecconi M, Fredericks S, Moreton-Clack M, MacPhee IA. A published pharmacogenetic algorithm was poorly predictive of tacrolimus clearance in an independent cohort of renal transplant recipients. Br J Clin Pharmacol (2013) 76(3):425–31. doi: 10.1111/bcp.12076
171. Emadi A, Jones RJ, Brodsky RA. Cyclophosphamide and cancer: golden anniversary. Nat Rev Clin Oncol (2009) 6(11):638–47. doi: 10.1038/nrclinonc.2009.146
172. Hengstler JG, Hengst A, Fuchs J, Tanner B, Pohl J, Oesch F. Induction of DNA crosslinks and DNA strand lesions by cyclophosphamide after activation by cytochrome P450 2B1. Mutat Res (1997) 373(2):215–23. doi: 10.1016/s0027-5107(96)00200-x
173. Colvin OM. An overview of cyclophosphamide development and clinical applications. Curr Pharm Design (1999) 5(8):555–60.
174. Eto M, Mayumi H, Tomita Y, Yoshikai Y, Nishimura Y, Nomoto K. Sequential mechanisms of cyclophosphamide-induced skin allograft tolerance including the intrathymic clonal deletion followed by late breakdown of the clonal deletion. J Immunol (1990) 145(5):1303–10.
175. Ahlmann M, Hempel G. The effect of cyclophosphamide on the immune system: implications for clinical cancer therapy. Cancer Chemother Pharmacol (2016) 78(4):1–11. doi: 10.1007/s00280-016-3152-1
176. Schmoeckel M, Bhatti FN, Zaidi A, Cozzi E, Waterworth PD, Tolan MJ, et al. Orthotopic heart transplantation in a transgenic pig-to-primate model. Transplantation (1998) 65(12):1570–7. doi: 10.1097/00007890-199806270-00006
177. Jie Y, Liu L, Pan Z, Wang L. Survival of pig-to-rhesus corneal xenografts prolonged by prior donor bone marrow transplantation. Mol Med Rep (2013) 7(3):869–74. doi: 10.3892/mmr.2013.1294
178. Ashton-Chess J, Roussel JC, Manez R, Ruiz C, Moreau A, Cozzi E, et al. Cellular participation in delayed xenograft rejection of hCD55 transgenic pig hearts by baboons. Xenotransplantation (2003) 10(5):446–53. doi: 10.1034/j.1399-3089.2003.00018.x
179. Cavallasca JA, Costa CA, Maliandi Mdel R, Contini LE, Fernandez de Carrera E, Musuruana JL. Severe infections in patients with autoimmune diseases treated with cyclophosphamide. Reumatol Clin (2015) 11(4):221–3. doi: 10.1016/j.reuma.2014.09.003
180. Wei Z, Liu X, Cheng C, Yu W, Yi P. Metabolism of amino acids in cancer. Front Cell Dev Biol (2020) 8:603837. doi: 10.3389/fcell.2020.603837
181. Li J, Kim SG, Blenis J. Rapamycin: one drug, many effects. Cell Metab (2014) 19(3):373–9. doi: 10.1016/j.cmet.2014.01.001
182. Chung J, Kuo CJ, Crabtree GR, Blenis J. Rapamycin-FKBP specifically blocks growth-dependent activation of and signaling by the 70 kd S6 protein kinases. Cell (1992) 69(7):1227–36. doi: 10.1016/0092-8674(92)90643-q
183. Kuo CJ, Chung J, Fiorentino DF, Flanagan WM, Blenis J, Crabtree GR. Rapamycin selectively inhibits interleukin-2 activation of p70 S6 kinase. Nature (1992) 358(6381):70–3. doi: 10.1038/358070a0
184. Muller YD, Mai G, Morel P, Serre-Beinier V, Gonelle-Gispert C, Yung GP, et al. Anti-CD154 mAb and rapamycin induce T regulatory cell mediated tolerance in rat-to-mouse islet transplantation. PloS One (2010) 5(4):e10352. doi: 10.1371/journal.pone.0010352
185. Singh AK, Horvath KA, Mohiuddin MM. Rapamycin promotes the enrichment of CD4(+)CD25(hi)FoxP3(+) T regulatory cells from naïve CD4(+) T cells of baboon that suppress antiporcine xenogenic response in vitro. Transplant Proc (2009) 41(1):418–21. doi: 10.1016/j.transproceed.2008.10.079
186. Itoh S, Kimura N, Axtell RC, Velotta JB, Gong Y, Wang X, et al. Interleukin-17 accelerates allograft rejection by suppressing regulatory T cell expansion. Circulation (2011) 124(11 Suppl):S187–96. doi: 10.1161/circulationaha.110.014852
187. Chai H, Yang L, Gao L, Guo Y, Li H, Fan X, et al. Decreased percentages of regulatory T cells are necessary to activate Th1-Th17-Th22 responses during acute rejection of the peripheral nerve xenotransplantation in mice. Transplantation (2014) 98(7):729–37. doi: 10.1097/tp.0000000000000319
188. Strand V, Cohen, Stanley. Treatment of active rheumatoid arthritis with leflunomide compared with placebo and methotrexate. Arch Internal Med (1999) 159(21):2542–50. doi: 10.1001/archinte.159.21.2542
189. Ma Y, Xie B, Guo J, Chen Y, Zhong M, Lin Q, et al. Leflunomide inhibits rat-to-Mouse cardiac xenograft rejection by suppressing adaptive immune cell response and NF-κB signaling activation. Cell Transplant (2021) 30:9636897211054503. doi: 10.1177/09636897211054503
190. Chen W, Lin J. Mycophenolate for the treatment of primary sjogren's syndrome. J Transl Int Med (2020) 8(3):146–9. doi: 10.2478/jtim-2020-0023
191. Mohiuddin MM, Singh AK, Corcoran PC, Hoyt RF, Thomas ML 3rd, Lewis BG, et al. One-year heterotopic cardiac xenograft survival in a pig to baboon model. Am J Transplant (2014) 14(2):488–9. doi: 10.1111/ajt.12562
192. Mohiuddin MM, Singh AK, Corcoran PC, Hoyt RF, Thomas ML 3rd, Lewis BG, et al. Role of anti-CD40 antibody-mediated costimulation blockade on non-gal antibody production and heterotopic cardiac xenograft survival in a GTKO. hCD46Tg Pig-to-baboon Model Xenotransplantation (2014) 21(1):35–45. doi: 10.1111/xen.12066
193. Mohiuddin MM, Singh AK, Corcoran PC, Thomas Iii ML, Clark T, Lewis BG, et al. Chimeric 2C10R4 anti-CD40 antibody therapy is critical for long-term survival of GTKO.hCD46.hTBM pig-to-primate cardiac xenograft. Nat Commun (2016) 7:11138. doi: 10.1038/ncomms11138
194. Mohiuddin MM, Reichart B, Byrne GW, McGregor CGA. Current status of pig heart xenotransplantation. Int J Surg (2015) 23(Pt B):234–9. doi: 10.1016/j.ijsu.2015.08.038
195. Monoclonal antibodies. In: LiverTox: Clinical and research information on drug-induced liver injury. Bethesda (MD): National Institute of Diabetes and Digestive and Kidney Diseases.
196. Thomas JM, Neville DM, Contreras JL, Eckhoff DE, Meng G, Lobashevsky AL, et al. Preclinical studies of allograft tolerance in rhesus monkeys: a novel anti-CD3-immunotoxin given peritransplant with donor bone marrow induces operational tolerance to kidney allografts. Transplantation (1997) 64(1):124–35. doi: 10.1097/00007890-199707150-00022
197. Neville DM Jr., Scharff J, Hu HZ, Rigaut K, Shiloach J, Slingerland W, et al. A new reagent for the induction of T-cell depletion, anti-CD3-CRM9. J Immunother Emphasis Tumor Immunol (1996) 19(2):85–92. doi: 10.1097/00002371-199603000-00001
198. Zhao Y, Cooper DKC, Wang H, Chen P, He C, Cai Z, et al. Potential pathological role of pro-inflammatory cytokines (IL-6, TNF-α, and IL-17) in xenotransplantation. Xenotransplantation (2019) 26(3):e12502. doi: 10.1111/xen.12502
199. Iwase H, Hara H, Ezzelarab M, Li T, Zhang Z, Gao B, et al. Immunological and physiological observations in baboons with life-supporting genetically engineered pig kidney grafts. Xenotransplantation (2017) 24(2). doi: 10.1111/xen.12293
200. Min BH, Shin JS, Kim JM, Kang SJ, Kim HJ, Yoon IH, et al. Delayed revascularization of islets after transplantation by IL-6 blockade in pig to non-human primate islet xenotransplantation model. Xenotransplantation 25(1) (2018) 25(1):e12374. doi: 10.1111/xen.12374
201. Iwase H, Liu H, Wijkstrom M, Zhou H, Singh J, Hara H, et al. Pig kidney graft survival in a baboon for 136 days: longest life-supporting organ graft survival to date. Xenotransplantation (2015) 22(4):302–9. doi: 10.1111/xen.12174
202. Zhang G, Iwase H, Wang L, Yamamoto T, Jagdale A, Ayares D, et al. Is interleukin-6 receptor blockade (tocilizumab) beneficial or detrimental to pig-to-baboon organ xenotransplantation? Am J Transplant (2020) 20(4):999–1013. doi: 10.1111/ajt.15712
203. Du FH, Mills EA, Mao-Draayer Y. Next-generation anti-CD20 monoclonal antibodies in autoimmune disease treatment. Auto Immun Highlights (2017) 8(1):12. doi: 10.1007/s13317-017-0100-y
204. Macklin PS, Morris PJ, Knight SR. A systematic review of the use of rituximab for the treatment of antibody-mediated renal transplant rejection. Transplant Rev (Orlando) (2017) 31(2):87–95. doi: 10.1016/j.trre.2017.01.002
205. Barnett AN, Hadjianastassiou VG, Mamode N. Rituximab in renal transplantation. Transpl Int (2013) 26(6):563–75. doi: 10.1111/tri.12072
206. Chan JL, Singh AK, Corcoran PC, Thomas ML, Lewis BG, Ayares DL, et al. Encouraging experience using multi-transgenic xenografts in a pig-to-baboon cardiac xenotransplantation model. Xenotransplantation 24(6) (2017) 24(6):e12330. doi: 10.1111/xen.12330
208. Zand MS, Vo T, Huggins J, Felgar R, Liesveld J, Pellegrin T, et al. Polyclonal rabbit antithymocyte globulin triggers b-cell and plasma cell apoptosis by multiple pathways. Transplantation (2005) 79(11):1507–15. doi: 10.1097/01.tp.0000164159.20075.16
209. Qimeng G, Robert D, Zachary F, Michael M, Brian E, Paul S, et al. Anti-thymoglobulin induction improves neonatal porcine xenoislet engraftment and survival. Xenotransplantation (2021) 28(6):e12713. doi: 10.1111/xen.12713
210. Wang Y, Wang Y, Ren Y, Zhang Q, Yi P, Cheng C. Metabolic modulation of immune checkpoints and novel therapeutic strategies in cancer. Semin Cancer Biol (2022). doi: 10.1016/j.semcancer.2022.02.010
211. Vanhove B, Poirier N, Soulillou JP, Blancho G. Selective costimulation blockade with antagonist anti-CD28 therapeutics in transplantation. Transplantation (2019) 103(9):1783–9. doi: 10.1097/TP.0000000000002740
212. Larsen CP, Pearson TC, Adams AB, Tso P, Shirasugi N, Strobert E, et al. Rational development of LEA29Y (belatacept), a high-affinity variant of CTLA4-ig with potent immunosuppressive properties. Am J Transplant (2005) 5(3):443–53. doi: 10.1111/j.1600-6143.2005.00749.x
213. Linsley PS, Brady W, Urnes M, Grosmaire LS, Damle NK, Ledbetter JA. CTLA-4 is a second receptor for the b cell activation antigen B7. J Exp Med (1991) 174(3):561–9. doi: 10.1084/jem.174.3.561
214. Walunas TL, Lenschow DJ, Bakker CY, Linsley PS, Freeman GJ, Green JM, et al. CTLA-4 can function as a negative regulator of T cell activation. Immunity (1994) 1(5):405–13. doi: 10.1016/1074-7613(94)90071-x
215. Aron Badin R, Vadori M, Vanhove B, Nerriere-Daguin V, Naveilhan P, Neveu I, et al. Cell therapy for parkinson's disease: A translational approach to assess the role of local and systemic immunosuppression. Am J Transplant (2016) 16(7):2016–29. doi: 10.1111/ajt.13704
216. Linsley PS, Wallace PM, Johnson J, Gibson MG, Greene JL, Ledbetter JA, et al. Immunosuppression in vivo by a soluble form of the CTLA-4 T cell activation molecule. Science (1992) 257(5071):792–5. doi: 10.1126/science.1496399
217. Lenschow DJ, Zeng Y, Thistlethwaite JR, Montag A, Brady W, Gibson MG, et al. Long-term survival of xenogeneic pancreatic islet grafts induced by CTLA4lg. Science (1992) 257(5071):789–92. doi: 10.1126/science.1323143
218. Turka LA, Linsley PS, Lin H, Brady W, Leiden JM, Wei RQ, et al. T-Cell activation by the CD28 ligand B7 is required for cardiac allograft rejection in vivo. Proc Natl Acad Sci U.S.A. (1992) 89(22):11102–5. doi: 10.1073/pnas.89.22.11102
219. Levisetti MG, Padrid PA, Szot GL, Mittal N, Meehan SM, Wardrip CL, et al. Immunosuppressive effects of human CTLA4Ig in a non-human primate model of allogeneic pancreatic islet transplantation. J Immunol (1997) 159(11):5187–91.
220. Buerck LW, Schuster M, Oduncu FS, Baehr A, Mayr T, Guethoff S, et al. LEA29Y expression in transgenic neonatal porcine islet-like cluster promotes long-lasting xenograft survival in humanized mice without immunosuppressive therapy. Sci Rep (2017) 7(1):3572. doi: 10.1038/s41598-017-03913-4
221. Yamamoto T, Hara H, Foote J, Wang L, Li Q, Klein EC, et al. Life-supporting kidney xenotransplantation from genetically engineered pigs in baboons: A comparison of two immunosuppressive regimens. Transplantation (2019) 103(10):2090–104. doi: 10.1097/tp.0000000000002796
222. Cooper DKC, Hara H, Iwase H, Yamamoto T, Jagdale A, Kumar V, et al. Clinical pig kidney xenotransplantation: How close are we? J Am Soc Nephrol (2020) 31(1):12–21. doi: 10.1681/ASN.2019070651
223. Sidiropoulos PI, Boumpas DT. Lessons learned from anti-CD40L treatment in systemic lupus erythematosus patients. Lupus (2004) 13(5):391–7. doi: 10.1191/0961203304lu1032oa
224. Bottino R, Knoll MF, Graeme-Wilson J, Klein EC, Ayares D, Trucco M, et al. Safe use of anti-CD154 monoclonal antibody in pig islet xenotransplantation in monkeys. Xenotransplantation (2017) 24(1). doi: 10.1111/xen.12283
225. Espié P, He Y, Koo P, Sickert D, Dupuy C, Chokoté E, et al. First-in-human clinical trial to assess pharmacokinetics, pharmacodynamics, safety, and tolerability of iscalimab, an anti-CD40 monoclonal antibody. Am J Transplant (2020) 20(2):463–73. doi: 10.1111/ajt.15661
226. Arefanian H, Tredget EB, Rajotte RV, Gill RG, Korbutt GS, Rayat GR. Short-term administrations of a combination of anti-LFA-1 and anti-CD154 monoclonal antibodies induce tolerance to neonatal porcine islet xenografts in mice. Diabetes (2010) 59(4):958–66. doi: 10.2337/db09-0413
227. Yoon IH, Chung H, Kim HJ, Nam HY, Shin JS, Kim YH, et al. Peri-graft porcine-specific CD4(+) FoxP3(+) regulatory T cells by CD40-CD154 blockade prevented the rejection of porcine islet graft in diabetic mice. Xenotransplantation (2019) 26(5):e12533. doi: 10.1111/xen.12533
228. Singh AK, Chan JL, DiChiacchio L, Hardy NL, Corcoran PC, Lewis BGT, et al. Cardiac xenografts show reduced survival in the absence of transgenic human thrombomodulin expression in donor pigs. Xenotransplantation (2019) 26(2):e12465. doi: 10.1111/xen.12465
229. Miyagawa S, Yamamoto A, Matsunami K, Wang D, Takama Y, Ueno T, et al. Complement regulation in the GalT KO era. Xenotransplantation (2010) 17(1):11–25. doi: 10.1111/j.1399-3089.2010.00569.x
230. Kourtzelis I, Ferreira A, Mitroulis I, Ricklin D, Bornstein SR, Waskow C, et al. Complement inhibition in a xenogeneic model of interactions between human whole blood and porcine endothelium. Horm Metab Res (2015) 47(1):36–42. doi: 10.1055/s-0034-1390452
231. Kobayashi T, Taniguchi S, Neethling FA, Rose AG, Hancock WW, Ye Y, et al. Delayed xenograft rejection of pig-to-baboon cardiac transplants after cobra venom factor therapy. Transplantation (1997) 64(9):1255–61. doi: 10.1097/00007890-199711150-00005
232. Bikhet M, Iwase H, Yamamoto T, Jagdale A, Foote JB, Ezzelarab M, et al. What therapeutic regimen will be optimal for initial clinical trials of pig organ transplantation? Transplantation (2021) 105(6):1143–55. doi: 10.1097/tp.0000000000003622
233. Längin M, Mayr T, Reichart B, Michel S, Buchholz S, Guethoff S, et al. Consistent success in life-supporting porcine cardiac xenotransplantation. Nature (2018) 564(7736):430–3. doi: 10.1038/s41586-018-0765-z
234. Schmitz R, Fitch ZW, Schroder PM, Choi AY, Manook M, Yoon J, et al. C3 complement inhibition prevents antibody-mediated rejection and prolongs renal allograft survival in sensitized non-human primates. Nat Commun (2021) 12(1):5456. doi: 10.1038/s41467-021-25745-7
235. Zhang Z, Li X, Zhang H, Zhang X, Chen H, Pan D, et al. Cytokine profiles in Tibetan macaques following α-1,3-galactosyltransferase-knockout pig liver xenotransplantation. Xenotransplantation (2017) 24(5). doi: 10.1111/xen.12321
236. Adams AB, Lovasik BP, Faber DA, Burlak C, Breeden C, Estrada JL, et al. Anti-C5 antibody tesidolumab reduces early antibody-mediated rejection and prolongs survival in renal xenotransplantation. Ann Surg (2021) 274(3):473–80. doi: 10.1097/sla.0000000000004996
237. Yue Y, Xu W, Kan Y, Zhao HY, Zhou Y, Song X, et al. Extensive germline genome engineering in pigs. Nat BioMed Eng (2021) 5(2):134–43. doi: 10.1038/s41551-020-00613-9
238. Xie C, Qu Z, Hara H, Dai W, Wang X, Pan D, et al. Downregulation of Gabarapl1 significantly attenuates antibody binding to porcine aortic endothelial cells. Xenotransplantation (2019) 26(6):e12537. doi: 10.1111/xen.12537
239. Chen P, Zhao Y, Gao H, Huang J, Lu Y, Song J, et al. Selective inhibition of cyclooxygenase-2 protects porcine aortic endothelial cells from human antibody-mediated complement-dependent cytotoxicity. Xenotransplantation (2019) 26(6):e12536. doi: 10.1111/xen.12536
240. Fischer K, Kraner-Scheiber S, Petersen B, Rieblinger B, Buermann A, Flisikowska T, et al. Efficient production of multi-modified pigs for xenotransplantation by 'combineering', gene stacking and gene editing. Sci Rep (2016) 6:29081. doi: 10.1038/srep29081
241. Sommaggio R, Bello-Gil D, Perez-Cruz M, Brokaw JL, Manez R, Costa C. Genetic engineering strategies to prevent the effects of antibody and complement on xenogeneic chondrocytes. Eur Cell Mater (2015) 30:258–70. doi: 10.22203/ecm.v030a18
242. Riella L, Bromberg JS. LITERATURE watch: implications for transplantation. Am J Transplant (2013) 13(1):3. doi: 10.1111/ajt.12072
243. Oldani G, Peloso A, Lacotte S, Meier R, Toso C. Xenogeneic chimera-generated by blastocyst complementation-as a potential unlimited source of recipient-tailored organs. Xenotransplantation (2017) 24(4). doi: 10.1111/xen.12327
244. Masano Y, Yagi S, Miyachi Y, Okumura S, Kaido T, Haga H, et al. Auxiliary xenotransplantation as an in vivo bioreactor-development of a transplantable liver graft from a tiny partial liver. Xenotransplantation (2019) 26(6):e12545. doi: 10.1111/xen.12545
245. Niu D, Wei HJ, Lin L, George H, Wang T, Lee IH, et al. Inactivation of porcine endogenous retrovirus in pigs using CRISPR-Cas9. Science (2017) 357(6357):1303–7. doi: 10.1126/science.aan4187
246. Ramsoondar J, Vaught T, Ball S, Mendicino M, Monahan J, Jobst P, et al. Production of transgenic pigs that express porcine endogenous retrovirus small interfering RNAs. Xenotransplantation (2009) 16(3):164–80. doi: 10.1111/j.1399-3089.2009.00525.x
247. Cardona K, Milas Z, Strobert E, Cano J, Jiang W, Safley SA, et al. Engraftment of adult porcine islet xenografts in diabetic nonhuman primates through targeting of costimulation pathways. Am J Transplant (2007) 7(10):2260–8. doi: 10.1111/j.1600-6143.2007.01933.x
248. Thompson P, Badell IR, Lowe M, Cano J, Song M, Leopardi F, et al. Islet xenotransplantation using gal-deficient neonatal donors improves engraftment and function. Am J Transplant (2011) 11(12):2593–602. doi: 10.1111/j.1600-6143.2011.03720.x
249. van der Windt DJ, Bottino R, Casu A, Campanile N, Smetanka C, He J, et al. Long-term controlled normoglycemia in diabetic non-human primates after transplantation with hCD46 transgenic porcine islets. Am J Transplant (2009) 9(12):2716–26. doi: 10.1111/j.1600-6143.2009.02850.x
250. Groth CG, Korsgren O, Tibell A, Tollemar J, Möller E, Bolinder J, et al. Transplantation of porcine fetal pancreas to diabetic patients. Lancet (1994) 344(8934):1402–4. doi: 10.1016/s0140-6736(94)90570-3
251. Matsumoto S, Tan P, Baker J, Durbin K, Tomiya M, Azuma K, et al. Clinical porcine islet xenotransplantation under comprehensive regulation. Transplant Proc (2014) 46(6):1992–5. doi: 10.1016/j.transproceed.2014.06.008
252. Yang HK, Yoon KH. Current status of encapsulated islet transplantation. J Diabetes Complications (2015) 29(5):737–43. doi: 10.1016/j.jdiacomp.2015.03.017
253. Denner J. Recent progress in xenotransplantation, with emphasis on virological safety. Ann Transplant (2016) 21:717–27. doi: 10.12659/aot.900531
254. Ekser B, Ezzelarab M, Hara H, van der Windt DJ, Wijkstrom M, Bottino R, et al. Clinical xenotransplantation: the next medical revolution? Lancet (2012) 379(9816):672–83. doi: 10.1016/s0140-6736(11)61091-x
255. Hu Q, Liu Z, Zhu H. Pig islets for islet xenotransplantation: current status and future perspectives. Chin Med J (Engl) (2014) 127(2):370–7. doi: 10.3760/cma.j.issn.0366-6999.20132030
256. Zhu HT, Wang WL, Yu L, Wang B. Pig-islet xenotransplantation: recent progress and current perspectives. Front Surg 1 7 (2014) 1. doi: 10.3389/fsurg.2014.00007
257. Moulay S, Hadj-Ziane AZ, Canselier JP. Microemulsion breakdown by pervaporation technique: effect of the alkyl chain length of n-alkanol, a cosurfactant of the microemulsion. J Colloid Interface Sci (2007) 311(2):556–61. doi: 10.1016/j.jcis.2007.02.069
258. Sacks D, Baxter B, Campbell BCV, Carpenter JS, Cognard C, Dippel D, et al. Multisociety consensus quality improvement revised consensus statement for endovascular therapy of acute ischemic stroke. Int J Stroke (2018) 13(6):612–32. doi: 10.1177/1747493018778713
259. Echeverri GJ, McGrath K, Bottino R, Hara H, Dons EM, van der Windt DJ, et al. Endoscopic gastric submucosal transplantation of islets (ENDO-STI): technique and initial results in diabetic pigs. Am J Transplant (2009) 9(11):2485–96. doi: 10.1111/j.1600-6143.2009.02815.x
260. Rajab A. Islet transplantation: alternative sites. Curr Diabetes Rep (2010) 10(5):332–7. doi: 10.1007/s11892-010-0130-6
261. Starzl TE, Marchioro TL, Faris TD, McCardle RJ, Iwaski Y. Avenues of future research in homotransplantation of the liver with particular reference to hepatic supportive procedures, antilymphocyte serum, and tissue typing. Am J Surg (1966) 112(3):391–400. doi: 10.1016/0002-9610(66)90209-1
262. Wv A, Ak A, Ts B, Vw B. Baboon-to-human liver transplantation. New York, NY, United States: Lancet. (1993) 341(8853):1158
263. Shah JA, Navarro-Alvarez N, DeFazio M, Rosales IA, Elias N, Yeh H, et al. A bridge to somewhere: 25-day survival after pig-to-Baboon liver xenotransplantation. Ann Surg (2016) 263(6):1069–71. doi: 10.1097/sla.0000000000001659
264. Forneris N, Levy H, Burlak C. Xenotransplantation literature update, July/August 2019. Xenotransplantation (2019) 26(5):e12561. doi: 10.1111/xen.12561
265. Li Y, Chen HS, Shaheen M, Joo DJ, Amiot BP, Rinaldo P, et al. Cold storage of porcine hepatocyte spheroids for spheroid bioartificial liver. Xenotransplantation (2019) 26(4):e12512. doi: 10.1111/xen.12512
266. Ekser B, Long C, Echeverri GJ, Hara H, Ezzelarab M, Lin CC, et al. Impact of thrombocytopenia on survival of baboons with genetically modified pig liver transplants: clinical relevance. Am J Transplant (2010) 10(2):273–85. doi: 10.1111/j.1600-6143.2009.02945.x
267. Ekser B, Klein E, He J, Stolz DB, Echeverri GJ, Long C, et al. Genetically-engineered pig-to-baboon liver xenotransplantation: histopathology of xenografts and native organs. PloS One (2012) 7(1):e29720. doi: 10.1371/journal.pone.0029720
268. Neil A, Barclay, Timo K, van den, Berg. The interaction between signal regulatory protein alpha (SIRPot) and CD47: Structure, function, and therapeutic target. Annu Rev Immunol (2014) 32:25–50. doi: 10.1146/annurev-immunol-032713-120142
269. Lexer G, Cooper DK, Rose AG, Wicomb WN, Rees J, Keraan M, et al. Hyperacute rejection in a discordant (pig to baboon) cardiac xenograft model. J Heart Transplant (1986) 5(6):411–8. doi: 10.3389/fsurg.2014.00007
270. Reardon S. First pig-to-human heart transplant: what can scientists learn? Nature (2022) 601(7893):305–6. doi: 10.1038/d41586-022-00111-9
271. Wang L, Cooper DKC, Burdorf L, Wang Y, Iwase H. Overcoming coagulation dysregulation in pig solid organ transplantation in nonhuman primates: Recent progress. Transplantation (2018) 102(7):1050–8. doi: 10.1097/tp.0000000000002171
272. Steen S, Paskevicius A, Liao Q, Sjöberg T. Safe orthotopic transplantation of hearts harvested 24 hours after brain death and preserved for 24 hours. Scand Cardiovasc J (2016) 50(3):193–200. doi: 10.3109/14017431.2016.1154598
273. Reemtsma K, Mccracken BH, Schlegel JU, Pearl MA, Creech O. Renal heterotransplantation in man*. Ann Surg (1964) 35(3):384–410. doi: 10.1097/00000658-196409000-00006
274. Yamada K, Yazawa K, Shimizu A, Iwanaga T, Hisashi Y, Nuhn M, et al. Marked prolongation of porcine renal xenograft survival in baboons through the use of alpha1,3-galactosyltransferase gene-knockout donors and the cotransplantation of vascularized thymic tissue. Nat Med (2005) 11(1):32–4. doi: 10.1038/nm1172
275. Porrett PM, Orandi BJ, Kumar V, Houp J, Anderson D, Cozette Killian A, et al. First clinical-grade porcine kidney xenotransplant using a human decedent model. Am J Transplant (2022) 22(4):1037–53. doi: 10.1111/ajt.16930
276. Iwase H, Yamamoto T, Cooper DKC. Episodes of hypovolemia/dehydration in baboons with pig kidney transplants: A new syndrome of clinical importance? Xenotransplantation (2019) 26(2):e12472. doi: 10.1111/xen.12472
277. Iwase H, Klein EC, Cooper DK. Physiologic aspects of pig kidney transplantation in nonhuman primates. Comp Med (2018) 68(5):332–40. doi: 10.30802/aalas-cm-17-000117
278. Iwase H, Ball S, Adams K, Eyestone W, Walters A, Cooper DKC. Growth hormone receptor knockout: Relevance to xenotransplantation. Xenotransplantation (2021) 28(2):e12652. doi: 10.1111/xen.12652
279. Zhang XL, Pang W, Hu XT, Li JL, Yao YG, Zheng YT. Experimental primates and non-human primate (NHP) models of human diseases in China: current status and progress. Zool Res (2014) 35(06):3–20. doi: 10.13918/j.issn.2095-8137.2014.6.447
280. Smetanka C, Cooper DK. The ethics debate in relation to xenotransplantation. Rev Sci Tech (2005) 24(1):335–42. doi: 10.20506/rst.24.1.1574
281. Hurst DJ, Padilla LA, Walters W, Hunter JM, Cooper DKC, Eckhoff DM, et al. Paediatric xenotransplantation clinical trials and the right to withdraw. J Med Ethics (2020) 46(5):311–5. doi: 10.1136/medethics-2019-105668
282. Denner J, Bigley TM, Phan TL, Zimmermann C, Zhou X, Kaufer BB. Comparative analysis of roseoloviruses in humans, pigs, mice, and other species. Viruses 11(12) (2019) 11(12):1108. doi: 10.3390/v11121108
Keywords: Xenotransplantation, hyperacute rejection, delayed xenograft rejection, chronic rejection, glucocorticoids, immunosuppressants
Citation: Zhou Q, Li T, Wang K, Zhang Q, Geng Z, Deng S, Cheng C and Wang Y (2022) Current status of xenotransplantation research and the strategies for preventing xenograft rejection. Front. Immunol. 13:928173. doi: 10.3389/fimmu.2022.928173
Received: 25 April 2022; Accepted: 07 July 2022;
Published: 28 July 2022.
Edited by:
Lisha Mou, Shenzhen Second People’s Hospital, ChinaReviewed by:
Sabine Hammer, University of Veterinary Medicine Vienna, AustriaCopyright © 2022 Zhou, Li, Wang, Zhang, Geng, Deng, Cheng and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Chunming Cheng, Q2h1bm1pbmcuY2hlbmdAb3N1bWMuZWR1; Yi Wang, d195aTIwMjJAMTYzLmNvbQ==
†These authors share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.