 Xiaomin Zhang
Xiaomin Zhang Lingling Zhu
Lingling Zhu Hui Zhang4
Hui Zhang4 Yang Xiao
Yang Xiao- 1Department of Hematology, Jinshazhou Hospital of Guangzhou University of Chinese Medicine, Guangzhou, China
- 2Institute of Clinical Medicine College, Guangzhou University of Chinese Medicine, Guangzhou, China
- 3Cancer Center, Integrated Hospital of Traditional Chinese Medicine, Southern Medical University, Guangzhou, China
- 4School of Medicine, Jishou University, Jishou, China
- 5Department of Hematology, Shenzhen Qianhai Shekou Pilot Free Trade Zone Hospital, Shenzhen, China
Chimeric antigen receptor T (CAR-T) cell therapy represents a major breakthrough in cancer treatment, and it has achieved unprecedented success in hematological malignancies, especially in relapsed/refractory (R/R) B cell malignancies. At present, CD19 and BCMA are the most common targets in CAR-T cell therapy, and numerous novel therapeutic targets are being explored. However, the adverse events related to CAR-T cell therapy might be serious or even life-threatening, such as cytokine release syndrome (CRS), CAR-T-cell-related encephalopathy syndrome (CRES), infections, cytopenia, and CRS-related coagulopathy. In addition, due to antigen escape, the limited CAR-T cell persistence, and immunosuppressive tumor microenvironment, a considerable proportion of patients relapse after CAR-T cell therapy. Thus, in this review, we focus on the progress and challenges of CAR-T cell therapy in hematological malignancies, such as attractive therapeutic targets, CAR-T related toxicities, and resistance to CAR-T cell therapy, and provide some practical recommendations.
Introduction
Traditionally, the treatment of hematological malignancies mainly includes chemotherapy, radiotherapy, and hematopoietic stem cell transplantation (HSCT). However, with advances in tumor immunology, immune targeted therapy, such as monoclonal antibodies, bispecific antibodies, antibody-drug conjugates, and chimeric antigen receptor T (CAR-T) cell therapy, has opened a new avenue for the treatment of malignancies. In particular, CAR-T cell therapy has revolutionized the treatment of hematological malignancies and achieved unprecedented responses in recent years, especially in relapsed/refractory (R/R) B-cell acute lymphocytic leukemia (B-ALL), non-Hodgkin lymphoma (NHL), and multiple myeloma (MM). At present, there are six CAR-T cell products approved by the US Food and Drug Administration (FDA) for the treatment of R/R B cell malignancies, including tisagenlecleucel (Kymriah; Novartis), axicabtagene ciloleucel (Yescarta; Gilead), brexucabtagene autoleucel (Tecartus; Gilead), lisocabtagene maraleucel (Breyanzi; Bristol Myers Squibb), idecabtagene vicleucel (Abecma; Bristol Myers Squibb and Bluebird Bio), and ciltacabtagene autoleucel (Carvykti; Legend and Janssen), which target the most common target antigens CD19 and B cell maturation antigen (BCMA) (Table 1 landmark clinical trials). In addition, there are a considerable number of novel targets which are being explored.
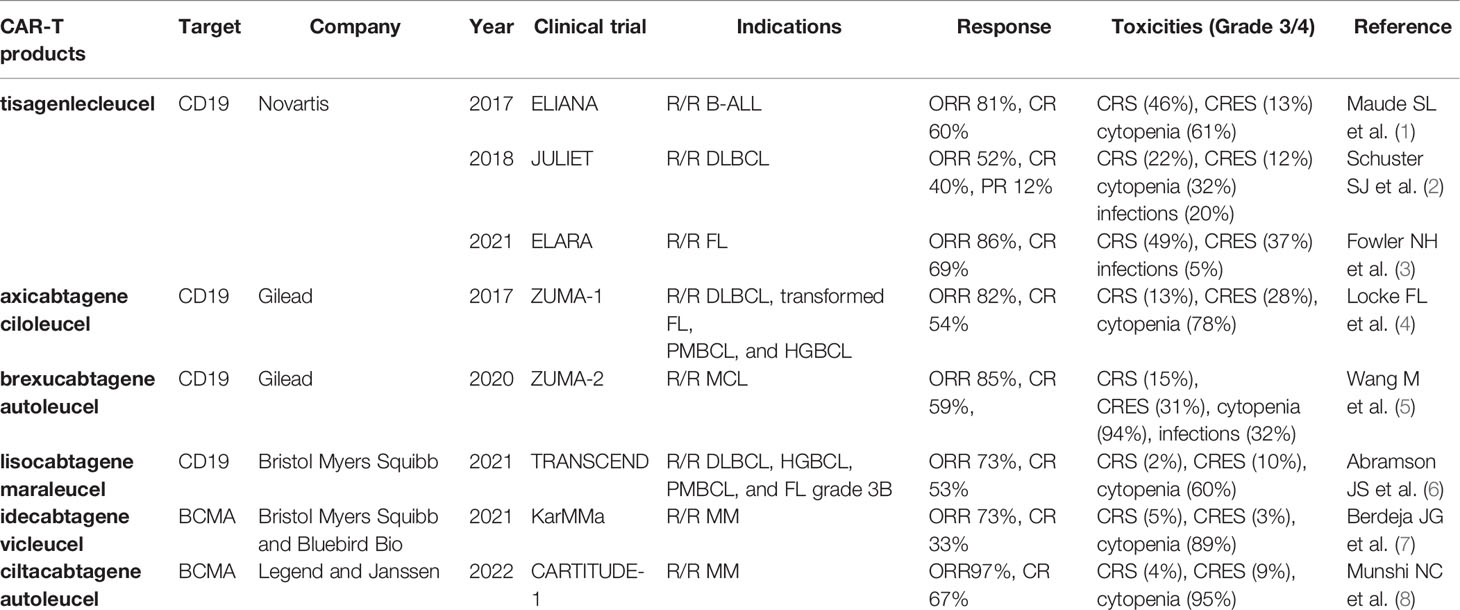
Table 1 Landmark clinical trials of FDA-approved CAR-T cell products.
However, despite the remarkable breakthrough of CAR-T therapy in B cell malignancies, severe toxicities associated with CAR-T cell therapy may compromise its efficacy and could even progress into life-threatening conditions, such as multiple organ dysfunction, sepsis, and disseminated intravascular coagulation (DIC). Cytokine release syndrome (CRS) is the most common complication, which is a systemic inflammatory response induced by the overactivation of CAR-T cells and endogenous immune cells, such as macrophages and dendritic cells. Its manifestations are diverse and partially similar with infections. Moreover, severe CRS is correlated with the increased risk of CAR-T-cell-related encephalopathy syndrome (CRES) and coagulopathy (9). Thus, clarifying their underlying mechanisms could facilitate the prevention and management of these adverse events. In addition, the resistance after CAR T-cell therapy cannot be ignored.
Thus, this review aims to introduce the advances and challenges in CAR-T cell therapy, such as attractive therapeutic targets, toxicities related to CAR-T cell therapy, and resistance to CAR-T cell therapy, and explore their underlying mechanisms and effective treatment strategies in order to facilitate the application and management of CAR-T cell therapy.
Overview of CAR-T Cell Therapy
To manufacture CAR-T cells, T cells are collected from peripheral blood of patients or donors and then genetically engineered in vitro to express chimeric antigen receptor (CAR). Thus, CAR-T cells recognize specific surface antigens on tumor cells without antigen processing and presentation, which indicates that antigen recognition by CAR-T cells is independently of major histocompatibility complex (MHC) restriction. After genetic modifications, they undergoextensive expansion in vitro. Then the patients receive lymphodepleting chemotherapy to make room for these adoptive CAR-T cells, and subsequently these genetically engineered CAR-T cells are re-infused into the patients. These CAR-T cells specifically recognize target antigens and rapidly proliferate to exert anti-tumor effects in vivo.
The CAR structure consists of an extracellular antigen-recognition domain, a transmembrane domain, and an intracellular signaling domain. The extracellular domain, a single-chain variable fragment (scFv), is able to specifically recognize tumor surface antigens. Typically, tumor antigens are categorized into tumor-associated antigens (TAAs) and tumor specific antigens (TSAs), and most of they are TAAs. Once TAAs are identified by scFv, CAR-T cells are activated and transmit activation signals to the intracellular domain. The first-generation CAR construct contains an antigen-recognition domain scFv and an intracellular CD3ζ activation domain. Due to the absence of costimulatory signals, they exhibit the limited proliferative capacity and anti-tumor effects. The second-generation CAR construct adds a costimulatory domain, such as CD28, 4-1BB, OX40, or ICOS, which enables themselves to possess the better proliferative capacity and release more cytokines. Currently, these commercial CAR-T cell products both utilize the second-generation CAR construct. The third-generation CAR construct encompasses two distinct costimulatory molecules, such as CD28 and 4-1BB. The fourth-generation CAR construct, also named TRUCK or armored CAR, is additionally modified to secrete cytokines or express suicide genes, such as IL-7, IL-12, IL-15, IL-21, and iCaspase-9 (10, 11) (Figure 1). The fourth-generation CAR-T cells may be more effective in eliminating tumor cells by activating the endogenous immune responses. However, the characteristics of fourth-generation CAR-T cells are largely unknown. In addition, the 2020 American Society of Hematology (ASH) annual meeting announced two studies about FasT CAR-T cells, including CD19-CD22 FasT dual-targeting CAR-T cells (GC022F) in B-ALL patients and BCMA-CD19 FasT dual-targeting CAR-T cells (GC012F) in R/R MM patients (12, 13). These FasT CAR-T products were manufactured in 24 to 36 hours and showed superior efficacy in preliminary studies, indicating that they might be more suitable for rapidly progressive B cell malignancies.
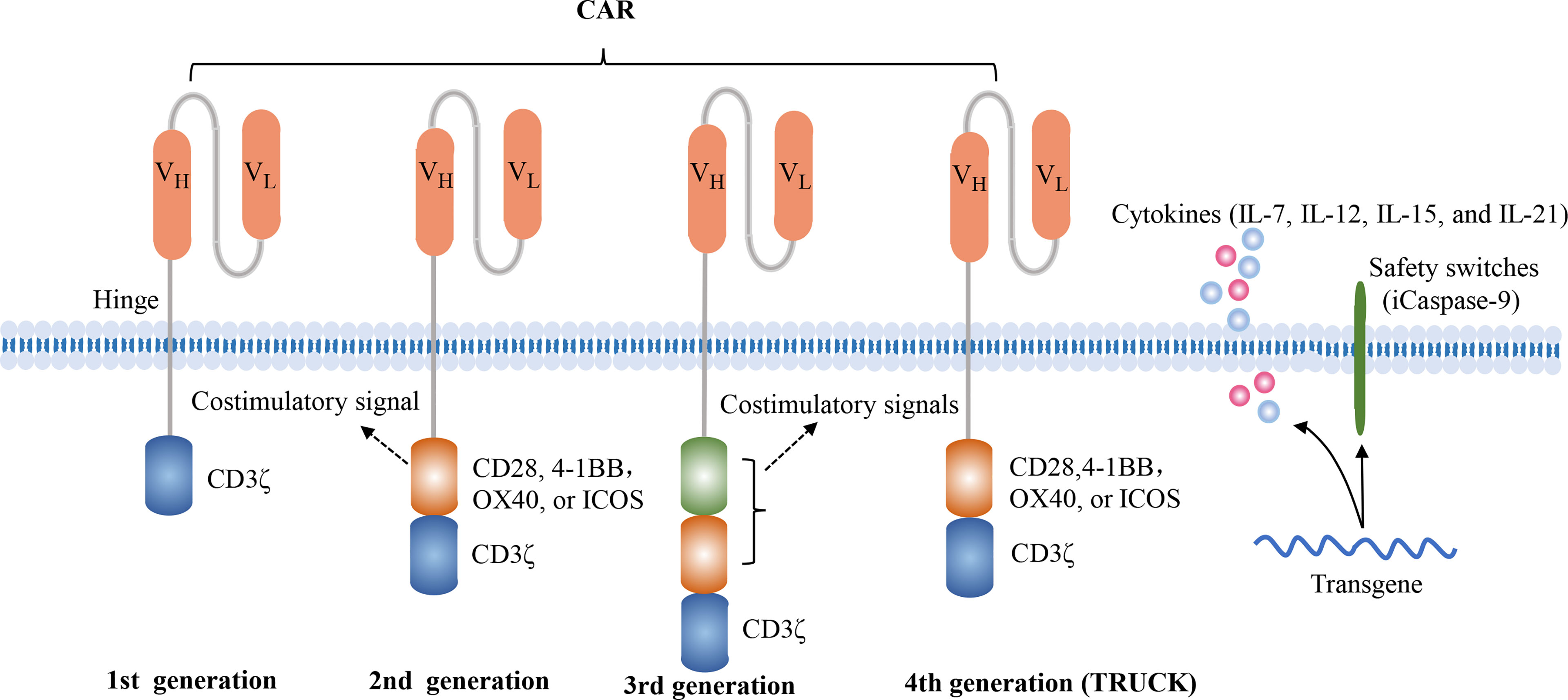
Figure 1 Four generations of CAR constructs. The first-generation of CAR consists of the antigen recognition domain scFv and an intracellular T cell activation domain CD3ζ. The second-generation CAR adds a costimulatory molecule, such as CD28, 4-1BB, OX40 or ICOS, which enables the T cells to obtain a superior proliferative capacity and secrete large amounts of cytokines. The third-generation CAR contains two distinct costimulatory domains, such as CD28 and 4-1BB. The fourth-generation of CAR, also known as TRUCK or armored CAR, is additionally equipped with safety switches or engineered to secrete cytokines in order to regulate the persistence or the function of CAR-T cells, such as iCaspase-9, IL-7, IL-15, IL-21.
At present, all commercial CAR-T cell products are manufactured using autologous T lymphocytes, but high manufacturing costs, relatively longer manufacturing cycle, and decreased number and function of lymphocytes after multiline chemotherapies have restricted their further application. However, it seems that universal CAR-T (UCAR-T) cells which are derived from healthy donors are able to overcome these limitations. The large-scale production of UCAR-T cells makes them “off-the-shelf” products, which could reduce manufacturing costs and increase their accessibility. Unfortunately, these allogeneic UCAR-T cells could induce graft versus host disease (GVHD) (14). Furthermore, the host immune system is able to reject these donor-derived UCAR-T cells and impairs their persistence. In addition, due to the excellent natural killing functions of NK cells and their abundant sources, such as NK92 cell line, cord blood, peripheral blood, and induced pluripotent stem cells, as well as no induction of GVHD, CAR-NK cells are currently being explored (15).
Attractive Targets for CAR-T Cell Therapy in Hematological Malignancies
Currently, CD19 and BCMA are the most common targets in CAR-T cell therapy. Although anti-CD19 CAR-T cell therapy and anti-BCMA CAR-T cell therapy have achieved outstanding outcomes in B cell malignancies, relapse after CAR-T cell therapy is frequently observed. In addition, due to the antigenic heterogeneity of acute myeloid leukemia (AML) as well as the lack of CD19 expression in Hodgkin lymphoma (HL) and T cell malignancies, a number of potential targets are currently being investigated (Figure 2).
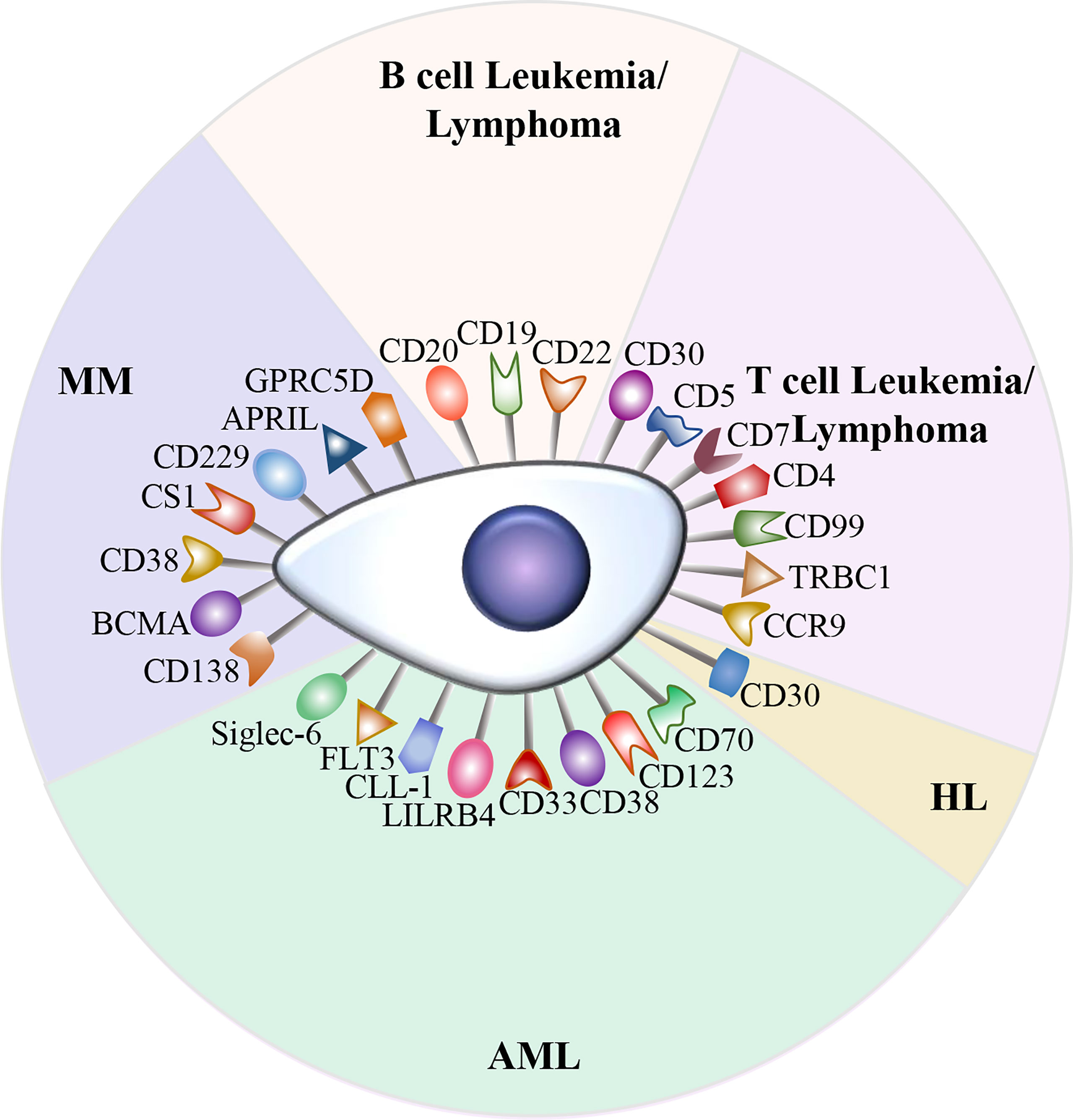
Figure 2 Potential therapeutic targets in hematological malignancies. A variety of attractive targets for CAR-T cell therapy in hematological malignancies, including T and B cell leukemia/lymphoma, HL, AML, and MM.
Targets for CAR-T Cell Therapy in B Cell Lymphoblastic Leukemia/Lymphoma
The CD19 is one of the most important target antigens in B cell malignancies, including B-ALL and NHL. In recent years, anti-CD19 CAR-T cell therapy has achieved rapid and durable responses in patients with R/R B-ALL and NHL (1–6, 16), and has dramatically altered the therapeutic landscape of B cell malignancies. Until now, four anti-CD19 CAR-T cell products have been approved by FDA for the treatment of R/R B-ALL and NHL (17). Despite the outstanding clinical results of anti-CD19 CAR-T therapy, CD19 antigen loss is frequently observed (18). Thus, the alternative targets for CAR-T cell therapy in R/R B-ALL and NHL have been explored.
CD20 is overexpressed in over 90% of B cell lymphomas and identified as an attractive target for CD20 positive B cell lymphomas, and the anti-CD20 monoclonal antibody rituximab has showed the excellent effect on NHL over the past few years. In an early clinical trial, the overall response rate (ORR) of anti-CD20 CAR-T cell therapy in DLBCL patients was 86% (19). In subsequent phase 1 and 2 trials, the anti-CD20 CAR-T cells were administrated to 17 R/R NHL patients, and 54.5% of patients achieved complete remissions (CRs) and 12 patients remained in remission with a median follow-up of 20 months (20). To prevent antigen escape, the combination of anti-CD19 and anti-CD20 CAR-T cells for the treatment of R/R DLBCL was investigated, and this combinational therapy was demonstrated to be safe and feasible (21). CD22 is highly expressed on most B cell malignancies, including B-ALL and DLBCL (22, 23). In particular, it is restrictively expressed on normal B cells and not expressed on hematopoietic stem cells, so it is an ideal target for CAR-T cell therapy in R/R B-ALL and DLBCL. In several clinical trials, the anti-CD22 CAR-T cell therapy has shown excellent efficacy in R/R B-ALL and R/R DLBCL patients who have failed in previous anti-CD19 CAR-T cell therapy (24–26). In addition, the humanized anti-CD22 CAR- T cells exhibits potent activity against leukemia cells with low CD22 expression (27).
Targets for CAR-T Cell Therapy in T Cell Lymphoblastic Leukemia/Lymphoma
Patients with R/R T-cell acute lymphoblastic leukemia (T-ALL) and T cell lymphomas often have poor prognosis. Compared with the outstanding clinical outcomes of anti-CD19 CAR-T cell therapy in B cell malignancies, the efficacy and safety CAR-T cell therapy in T cell malignancies are largely unknown and under investigation. CD7 is highly expressed in 95% of T-ALL patients and represents a desirable target for the treatment of T-ALL (28). In an open-label and single-arm clinical trial, 2 R/R T-ALL patients were treated with allogeneic anti-CD7 CAR-T cell therapy. One patient remained in remission for over 1 year, while the other relapsed 48 days after CAR-T cell infusion (28). In another phase I clinical trial, 20 R/R T-ALL patients received donor-derived anti-CD7 CAR-T cell infusion, and 90% of participants achieved CRs (29). In addition, a case study reported that an 11-year-old T-ALL patient who didn’t respond to induction failure was treated with autologous anti-CD7 CAR-T cell therapy, and he achieved remission on day 17 and subsequently underwent HSCT (30). CD5 is expressed in approximately 85% of T cell malignancies, such as T-cell lymphoblastic lymphoma (T-LBL) and peripheral T-cell lymphoma (PTCL). A recent study has demonstrated that anti-CD5 CAR-T cells effectively eliminated malignant T cells (31). In a phase I clinical trial, a refractory T-LBL patient with central nervous system (CNS) infiltration received the anti-CD5 CAR-T cell therapy and achieved CR within 4 weeks (32). Additionally, anti-CD4 CAR-T cells showed superior activity against T cell malignancies in preclinical studies (33). However, all three targets CD4, CD5 and CD7 are expressed on normal T cells, so targeting them may result in the depletion of normal T cells and the fratricide of CAR-T cells (34). Malignant T cells express T cell receptor β-chain constant domains 1 (TRBC1), so anti-TRBC1 CAR-T cells are able to selectively eliminate TRBC1 positive malignant T cells. Importantly, it could retain a majority of normal T cells in vivo (35). CD99 is highly expressed in newly diagnosed T-ALL patients, and it represents a novel target for T-ALL (36). Furthermore, chemokine receptor CCR9 is expressed in over 70% of T-ALL patients, and only on less than 5% of normal T cells. In addition, it is correlated with multidrug resistance and poor prognosis. Thus, it represents an ideal target for CCR9 positive T-ALL. In preclinical studies, anti-CCR9 CAR-T cells exhibited potent anti-leukemic activity and were resistant to fratricide (37).
In addition, CD30 is highly expressed in anaplastic large-cell lymphoma (ALCL) and is variably expressed in PTCL subtypes. In particular, CD30 is restrictively expressed on normal T cells. Thus, CD30 is an ideal target for these lymphoma subtypes, and the anti-CD30 antibody-drug conjugate brentuximab vedotin (BV) has shown a high response rate in newly diagnosed PTCL patients (38, 39). Given the encouraging clinical efficacy of BV, the anti-CD30 CAR-T cells were developed and exhibited remarkable cytotoxicity against CD30 positive lymphomas in preclinical studies (40, 41).
Targets for CAR-T Cell Therapy in Hodgkin Lymphoma
Anti-CD19 CAR-T cell therapy has shown excellent results in R/R B cell NHL. However, HL lacks the expression of CD19. Interestingly, CD30 is universally expressed in classical HL. Currently, several clinical trials have been carried out to evaluate the safety and efficacy of anti-CD30 CAR-T cell therapy in R/R HL (42–45). In a clinical trial from China, 5 of 6 HL patients achieved CRs after the infusion of the third-generation anti-CD30 CAR-T cells, and the long-term remission lasted over 24 months in 3 patients (45). In another phase 1/2 clinical trial, 27 patients were treated with the anti-CD30 CAR-T cells and 67% of patients achieved CRs within 6 weeks, but 63% of patients experienced disease progression with a median follow-up of 9.5 months (46). In addition, the expression of CD30 in HL was down-regulated after anti-CD30 CAR-T cell therapy (47).
Targets for CAR-T Cell Therapy in Acute Myeloid Leukemia
AML is the most common acute adult leukemia. Unfortunately, due to antigenic heterogeneity, the CAR-T cell therapy in AML has not achieved the same success as ALL. Recently, CD123, CD33, CD38, CD70, C-type lectin-like molecule-1(CLL-1), leukocyte immunoglobulin-like receptor-B4 (LILRB4), FMS-like tyrosine kinase 3 (FLT3) and sialic acid-binding immunoglobulin-like lectin 6 (Siglec-6) have been explored. CD123 and CD33 are highly expressed on leukemic stem cells in over 80% of AML patients, but they are expressed on hematopoietic stem cells as well (48). Accordingly, targeting them could increase the risk of long-term myelosuppression (49). To decrease hematological toxicity, a rapidly switchable universal anti-CD123 CAR-T cells were prepared (50, 51). In a small study, 3 R/R AML patients were treated with this universal anti-CD123 CAR-T cells, and all of them achieved a clinical response with the rapid hematologic recovery after the withdrawal of switch-mediated co-stimulation (52). In another phase 1 trial, 3 R/R AML patients received autologous anti-CD33 CAR-T cell infusion. Unfortunately, all of them died from disease progression (53). CD38 is expressed on most AML blast cells, and anti-CD38 CAR-T cell therapy was demonstrated to be effective in relapsed AML after allogeneic HSCT (54). CLL-1, which has been identified as an myeloid cell surface marker, is overexpressed on leukemic stem cells (55). Importantly, it is absent on hematopoietic stem cells. The CLL-1-targeted CAR-T cells specifically eliminated CLL-1 positive leukemia in preclinical studies (56, 57). CD70 is expressed on AML blasts but not on normal myeloid cells, making it a promising target for the treatment of AML (58, 59). Currently, the safety and efficacy of anti-CD70 CAR-T cell therapy are under investigation. In addition, LILRB4 is highly expressed on monocytic AML cells, and it is an attractive target for monocytic AML (60). Siglec-6 is expressed in approximately 60% of AML patients and absent on normal hematopoietic stem and progenitor cells. In preclinical studies, Siglec-6 CAR T cells effectively eliminated AML blasts in an AML mouse xenotransplantation model (61). Thus, it could serve as a well-validated target for CAR-T cell therapy in AML. In addition, nucleophosmin 1 (NPM1) mutations have been observed in 30%-35% of AML patients, and they are considered to be initiating mutations in leukemic cells. In preclinical mouse models, CAR-T cells targeting a nucleophosmin neoepitope which is presented by HLA-A2 exhibited potent specific anti-leukemia activity (62).
FLT3 is a transmembrane tyrosine kinase expressed on malignant blasts in approximately 30% of AML patients. FLT3 mutations include point mutations and an internal tandem duplication (ITD), and FLT3‐ITD is correlated with poor prognosis. In preclinical studies, the FLT3-targeted CAR-T cells successfully eliminated FLT3 positive AML cells (63, 64), and the FLT3 inhibitor crenolanib promoted their anti-tumor effects (64). Unfortunately, FLT3 is also expressed on normal hematopoietic stem and progenitor cells, so the FLT3-targeted CAR-T cells may affect normal hematopoiesis (64, 65).
Targets for CAR-T Cell Therapy in Multiple Myeloma
MM remains an incurable plasma cell malignancy. With the application of novel agents, such as proteasome inhibitors, immunomodulatory drugs, and anti-CD38 monoclonal antibodies, MM patients have significantly improved survival outcomes (66). However, almost all MM patients inevitably relapse. BCMA is highly selectively expressed on malignant plasma cells, so it represents one of the most promising therapeutic targets for MM. At present, anti-BCMA CAR-T cell therapy has been demonstrated to be effective in R/R MM and achieved unprecedented responses (7, 8, 67–69), and two anti-BCMA CAR-T cell products, idecabtagene vicleucel and ciltacabtagene autoleucel, have been approved by the FDA for the treatment of R/R MM (7, 70). Furthermore, the anti-BCMA CAR-T cell therapy is effective in R/R MM patients with extramedullary disease (71–73). However, some MM patients still relapse after anti-BCMA CAR-T therapy, and BCMA expression is downregulated under therapeutic pressure. Therefore, new target antigens are required (74).
Currently, several potential target antigens have been investigated, such as CD38, CD138, CD229, SLAMF7, a proliferation-inducing ligand (APRIL), and G protein-coupled receptor, class C group 5 member D (GPRC5D). CD138 is highly expressed on MM cells and promote their survival and proliferation. In a preclinical study, anti-CD138 CAR-T cells effectively eliminated MM cells (75). In a small clinical trial, 5 patients received anti-CD138 CAR-T cell therapy, and 4 of them had a clinical response and remained stable for at least three months (76). CD38 is not only highly expressed on MM cells, but also expressed on hematopoietic cells and activated lymphocytes cells (77). Unfortunately, although the anti-CD38 CAR-T cells exhibited significant anti-tumor effects in mouse models, they impaired normal hematopoietic cells and lymphocytes (78). Clinically, CD38 is frequently combined with other targets, such as BCMA and CD138, to produce bispecific CAR-T cells, thereby reducing the risk of antigen escape (79, 80). CD229 is a surface antigen highly expressed on MM cells (81). The anti-CD229 CAR-T cells effectively eliminated MM cells in preclinical studies (82). SLAMF7, also known as CS1, is highly expressed in over 95% of MM patients. Similar to CD38, SLAMF7 is also expressed on normal lymphocytes, including activated T cells, NK cells, and B cells (83). Thus, SLAMF7 CAR-T cells could kill normal lymphocytes and increase the risk of CAR-T cell fratricide (84). Currently, the clinical trial of SLAMF7 CAR-T cells is ongoing (85). APRIL is a natural ligand which could directly bind to BCMA and transmembrane activator and CAML interactor (TACI). Thus, APRIL-targeted CAR-T cells recognize both BCMA and TACI expressed on MM cells, which may decrease the risk of antigen escape (86), and preserving its trimeric conformation could improve the anti-tumor activities (87). In addition, TGPRC5D is expressed on more than 50% of CD138 positive malignant plasma cells in bone marrow of MM patients, which also represents a potential target for the treatment of MM (88).
Toxicities Related to CAR-T Cell Therapy and Their Underlying Mechanisms
Although CAR-T cell therapy has achieved great success in hematological malignancies, the adverse events related to CAR-T cell therapy remain to be a major challenge, such as CRS, CRES, B cell aplasia, cytopenia, and CRS-related coagulopathy. Without active and effective interventions, these complications might be life-threatening. In order to effectively manage these complications, it is important to explore their underlying mechanisms and recognize them in early stages.
Cytokine Release Syndrome
CRS is one of the most common toxicities of CAR-T cell therapy. The incidence of CRS depends on a variety of factors, including disease characteristics, CAR structure, tumor burden, and CAR-T cell doses (89). The clinical manifestations of CRS are diverse, but they are frequently characterized by fever, fatigue, myalgia, poor appetite, hypoxia, hypotension, and even organ dysfunction. If left untreated, it might rapidly progress into life-threatening conditions, such as hemodynamic instability and multiple organ dysfunction. However, the recent study revealed that the patients with ≥ grade 2 CRS had higher rates of remission and longer progression-free survival (PFS) compared with those with < grade 2 CRS, which indicates that appropriate CRS might facilitate the efficacy of CAR-T therapy (90). Because CRS are primarily mediated by IL-6, IL-6 receptor antagonist tocilizumab is mainly recommended to relieve the clinical symptoms of CRS. According to different CRS grading, different treatment regimens are adopted. The symptomatic treatment and the supportive treatment are indicated for grade 1 CRS. Tocilizumab and corticosteroids are recommended for grade 3 and 4 CRS as well as grade 2 CRS accompanied by severe symptoms. Moreover, IL-1 is another important cytokine involved in CRS and CRES, and IL-1 receptor antagonist anakinra has been demonstrated to ameliorate both CRS and CRES (91–95). Furthermore, GM-CSF deficiency or inhibition not only can alleviate CRS and CRES by inhibiting the local infiltration of myeloid cells and T cells, but also enhance the anti-tumor effects of CAR-T cells (96, 97). Besides, the severity of CRS is positively associated with the patient’s tumor burden (89). To reduce tumor burden, traditional chemotherapy and radiotherapy could serve as the effective bridging strategies before CAR-T cell infusion.
The detailed mechanisms of CRS remain incompletely understood. After recognizing target antigens, CAR-T cells are rapidly activated and secrete a large amount of granzyme, perforin, IFN-γ, and TNF-α. Perforin forms pores on tumor cell membrane and allows granzyme B to enter tumor cells. Granzyme B activates GSDME which is widely expressed on CD19 positive malignant B cells, resulting in tumor cell pyroptosis and the release of danger associated molecular patterns (DAMPs), such as high-mobility group box 1 (HMGB1) (98–101). Then DAMPs recruit and activate endogenous innate immune cells, such as macrophages and dendritic cells, thereby amplifying inflammatory responses and increasing the release of cytokines, including IL-1β and IL-6. Currently, it has been demonstrated that macrophages and monocytes rather than CAR-T cells are the major sources of these cytokines and contribute to CRS (91, 92, 102, 103). In addition, the CD40 and CD40 ligand (CD40L) interactions between CAR-T cells and host antigen-presenting cells (APCs) as well as tumor cells also play an important role in immune activation and the release of cytokines (104–106). CD40 is expressed on multiple APCs, including B cells, macrophages, dendritic cells (DCs), and monocytes, and highly expressed in a variety of hematological malignancies, such as NHL, AML, MM (105–108). CD40L is highly expressed on activated T cells, including CAR-T cells (104, 109). The contact-dependent CD40/CD40L interactions enhance the antigen presentation of APCs and CD40 positive malignancies and promote their secretion of cytokines, such as IL-1β, IL-6, and TNF-α (110, 111) (Figure 3). In addition, because endothelial cells act as conditional innate immune cells and express CD40, the CD40/CD40L interactions also participate in the activation of endothelial cells.
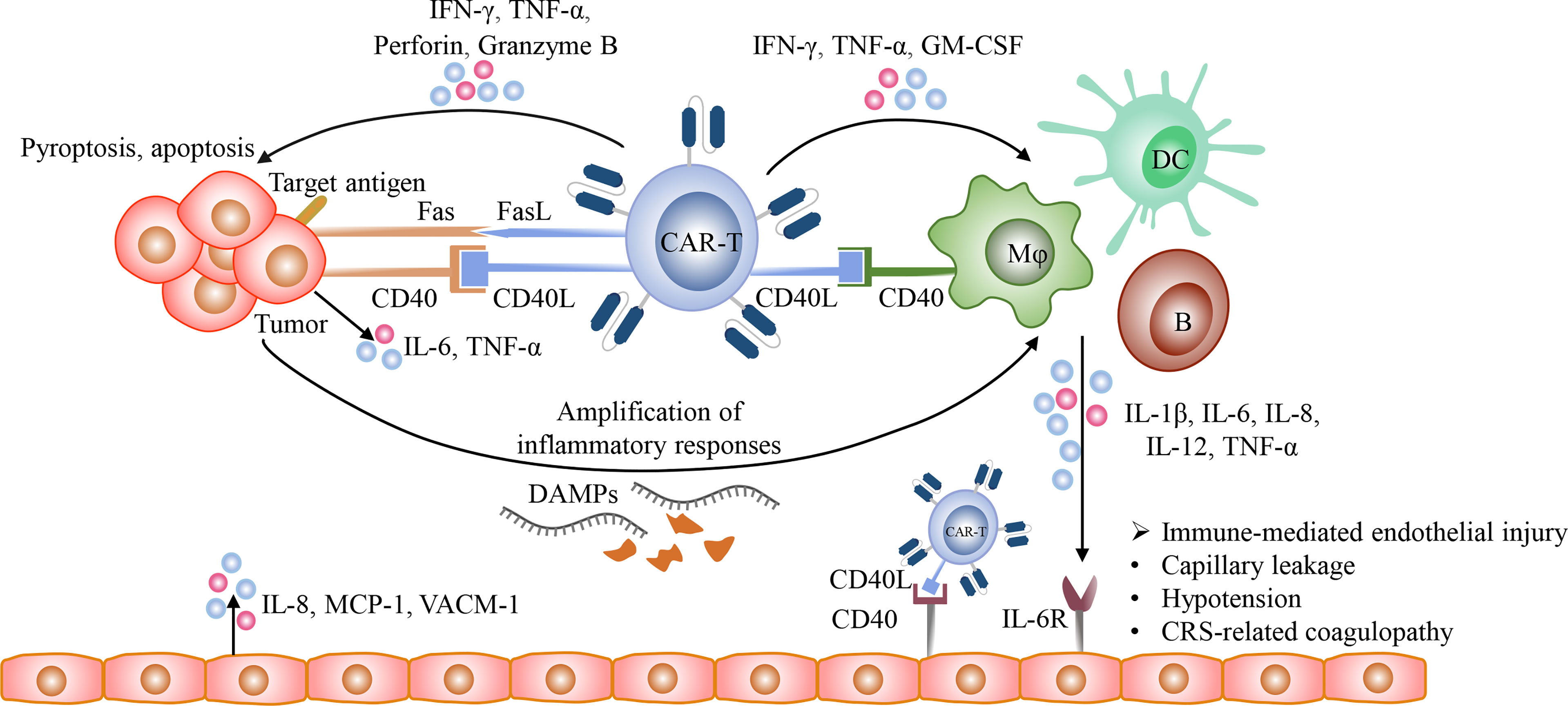
Figure 3 The mechanisms of CRS. After the recognition of target antigens, CAR T-cells rapidly proliferate and release multiple cytotoxic molecules, such as granzyme, perforin, IFN-γ, and TNF-α, and upregulate the expression of CD40L and Fas ligand (FasL), and eventually induce pyroptosis and apoptosis of tumor cells. Besides, the CD40/CD40L interactions between tumor cells and CAR T-cells promote Fas-mediated apoptosis. Then the lysed tumor cells release large amounts of DAMPs, such as HMGB1, which could activate innate immune cells, including macrophages and dendritic cells, further amplifying inflammatory responses. In addition, the CD40/CD40L interactions participate in the activation of various immune cells, including T cells, B cells, macrophages, dendritic cells, and conditional innate immune cells such as endothelial cells. The activated CAR-T cells with the increased expression of CD40L could activate macrophages and endothelial cells and promote their production of pro-inflammatory cytokines in a CD40-dependent manner. The cytokines released from activated immune cells could bind to their receptors on endothelial cells and then mediate endothelial dysfunction, resulting in capillary leakage and the release of procoagulant factors.
Since lymphoma lesions are mostly localized, CRS in lymphoma exhibits some distinct manifestations, such as local CRS, which is characterized by local redness, swelling, and heat. Then, CAR-T cells persistently expand and overflow into the circulation, and experience redistribution as well as mediating tissue damage. Finally, inflammation subsides and the impaired tissues are gradually repaired (112). Thus, the early management of local CRS is helpful to reduce the occurrence of subsequent systemic CRS.
CAR-T-Cell-Related Encephalopathy Syndrome
CRES, also known as CAR-T cell-related neurotoxicity, is another common toxicity during CAR-T cell therapy. It usually occurs simultaneously with CRS or later than CRS. The manifestations of CRES include headache, dizziness, delirium, seizures and cerebral edema. Due to the lack of suitable animal models, the underlying pathological mechanisms of CRES are not fully understood. Severe CRS, high tumor burden, and excessive CAR-T cell expansion might be correlated with the increased risk of CRES. Currently, immune-mediated endothelial activation is a well-established mechanism involved in the occurrence of CRES (113–115). Upon the recognition of target antigens, CAR-T cells rapidly expand and secrete cytokines to activate endogenous immune cells, such as macrophages, which in turn release large amounts of cytokines and activate cerebral microvascular endothelial cells, eventually resulting in the disruption of tight junctions and the increased blood-brain barrier (BBB) permeability (114, 115). Then, the high concentrations of serum cytokines enter the BBB by passive diffusion, and the elevated levels of pro-inflammatory cytokines in CSF seem to be associated with CRES, including IL-1β, IL6, IL-8, IFN-γ, GM-CSF, MCP-1, and granzyme B (116–118). In addition, it has been demonstrated that T cells and macrophages, including CAR-T cells, could infiltrate into the CNS due to the disruption of the BBB (118–121). These infiltrated immune cells and cytokines could induce the activation of microglia which are brain-resident macrophages, further amplifying local inflammatory responses and eventually resulting in neurotoxicity (115, 119, 122–124). Thus, immune-mediated endothelial injury is a trigger factor for CRES (113–115, 125). As tocilizumab couldn’t cross the BBB, it exhibits the limited efficacy in the management of CRES. Given the increased CNS penetration of corticosteroids, it is recommended for the treatment of CRES, and it could not affect the proliferation and the anti-tumor effects of CAR-T cells (126).
B/T Cell Aplasia and Infections
CD19 and CD20 are expressed on multiple differentiated B-lineage cells as well as malignant B cells, and represent attractive targets for CAR-T cell therapy in B-ALL and lymphoma. BCMA, a plasma cell-selective protein, is highly expressed on MM cells as well as mature B cells and normal plasma cells. Thus, CD19‐targeted, CD20‐targeted and BCMA‐targeted CAR T cells exhibit superior anti-tumor activity in B cell malignancies (1, 3, 67, 127), but they attack normal B cells as well, which could result in impaired humoral immunity, such as B cell aplasia and hypogammaglobulinemia (128–131). Moreover, lymphodepleting chemotherapy prior to CAR-T cell infusion could also impair host immunity. Due to impaired host immunity, these individuals are more susceptible to infections (132). It has been demonstrated that most of infection events occur during the first 30 days of CAR-T cell infusion, and the bacterial infection predominates, mainly including bloodstream infection and respiratory infection (133). In a phase 1/2 study, 31% of the patients who received the anti-CD19 CAR-T therapy experienced infections between day 31 and day 180 (134). Thus, the long term follow-up and the detection of gamma globulin levels might be helpful. To restore humoral immunity, immunoglobulin supplementation is essential for these immune-compromised individuals. In addition, the high-dose CAR-T cell infusion seems to be associated with the infections after CAR-T cell infusion (134, 135). Thus, CAR-T cells can be administrated in a dose-escalation regimen. Furthermore, CAR-T cell therapy increases the risk of HBV reactivation in patients with resolved HBV infections due to persistent B-cell aplasia, so antiviral prophylaxis and regular monitoring of the virus are recommended (128, 136, 137). Unfortunately, with the application of CAR-T cell therapy in T cell malignancies, T cell aplasia might be observed because a majority of target antigens are co-expressed on normal T cells (34).
However, it is difficult to differentiate between CRS and infections due to the similar clinical manifestations, such as pyrexia and the elevated levels of pro-inflammatory cytokines and C-reactive protein (CRP). Moreover, CRS is likely to occur simultaneously with infections. In order to avoid the life-threatening infections during CAR-T cell therapy, the early recognition and management of infections is important. Nevertheless, it is usually not timely to identify the infection by blood culture (133). Thus, the detection of special biomarkers or the establishment of a prediction model for infection is critical. IL-6 is one of the key cytokines involved in the infection-induced cytokine storm and CAR-T cell therapy-induced CRS. Typically, the elevation of serum IL-6 associated with CRS occurs within 3 weeks after CAR-T cell infusion, so the “double peaks of IL-6” is identified as one of the characteristics of life-threatening infections. Compared with blood culture, it seems that employing the “double peaks of IL-6” pattern to predict the life-threatening infection is faster (135). When infection is suspected, empiric anti-infective treatment should be initiated immediately once blood and sputum samples are collected for the detection of pathogenic microorganisms, especially in neutropenic patients. In addition, Herpesvirus infections have been occasionally observed in several clinical trials (138–140). To prevent herpesvirus infections, it’s recommended that acyclovir 400 mg should be prophylactically administrated twice daily from lymphodepletion chemotherapy to at least 6 months post CAR-T cell infusion (141).
Cytopenia
Cytopenia is frequently observed during CAR-T cell therapy and lasts for several days to months, including anemia, thrombocytopenia, and leukopenia, and the incidence of cytopenia range from 30% to 100% in clinical trials (2, 29, 72, 79, 142, 143). It has been demonstrated that cytopenia is associated with severe CRS (144–146). Under inflammatory conditions, CD40 is significantly up-regulated on granulocytic progenitor/precursor cells which also express low levels of CD40L, and the CD40/CD40L interactions between granulocytic progenitor/precursor cells significantly promote their own apoptosis (147). In addition, the pro-inflammatory cytokines, such as IL-1, TNF-α, and HMGB1, could suppress erythropoietin production (148, 149), and the activated macrophages could destroy erythrocytes (150). The limited hematopoietic capacity mediated by prior chemotherapy and HSCT might be involved in cytopenia as well (151–153). Furthermore, some target antigens are co-expressed on normal hematopoietic stem or progenitor cells, so CAR-T cells could directly mediate the destruction of hematopoietic cells (48, 49). Clinically, red blood cell and platelet transfusions and hematopoietic growth factors, such as granulocyte colony-stimulating factor (G-CSF) and thrombopoietin (TPO), as well as TPO receptor agonists and sirolimus, are able to ameliorate cytopenia (154, 155).
CRS-related Coagulopathy
As a newly identified toxicity, coagulopathy is frequently observed within 1 month after CAR-T cell infusion (156, 157). Its severity shows a positive correlation with CRS grade as well as the levels of IL-6 (9), so it’s also known as CRS-related coagulopathy. The recent studies have reported that the incidence of coagulopathy during CAR-T therapy is approximately 50% (9, 156). There are multiple abnormal coagulation parameters in patients with CRS-related coagulopathy, mainly characterized by the elevated levels of D-dimer, the increased fibrinogen degradation products, and the decreased levels of fibrinogen as well as the prolonged prothrombin time. The progress of CRS-related coagulopathy can be divided into three stages, including hypercoagulable stage, consumptive hypo-coagulable stage, and hyperfibrinolysis stage (9). At hypercoagulable stage, the patients mainly present with excessive micro-thrombosis, which can be treated with anticoagulant drugs, such as low-molecular-weight heparin. At consumptive hypo-coagulable stage, the individuals exhibit bleeding, accompanied by the decreased fibrinogen levels and the prolonged APTT and PT, so replacement treatment is required. At hyperfibrinolysis stage, the fibrinogen level is substantially decreased and the level of D-Dimer is significantly up-regulated. Once the fibrinogen level in plasma is lower than 1.5 g/L, the fibrinogen concentrate or cryoprecipitate replacement should be administrated (Table 2 toxicities related to CAR-T cell therapy) Given that cytokine storm plays an essential role in CRS-related coagulopathy, the early and effective management of CRS might be helpful to reduce the incidence of coagulopathy. Without timely and effective intervention, a part of patients with coagulopathy may further develop disseminated intravascular coagulation (DIC), accompanied by the poor prognosis (9, 156, 158).
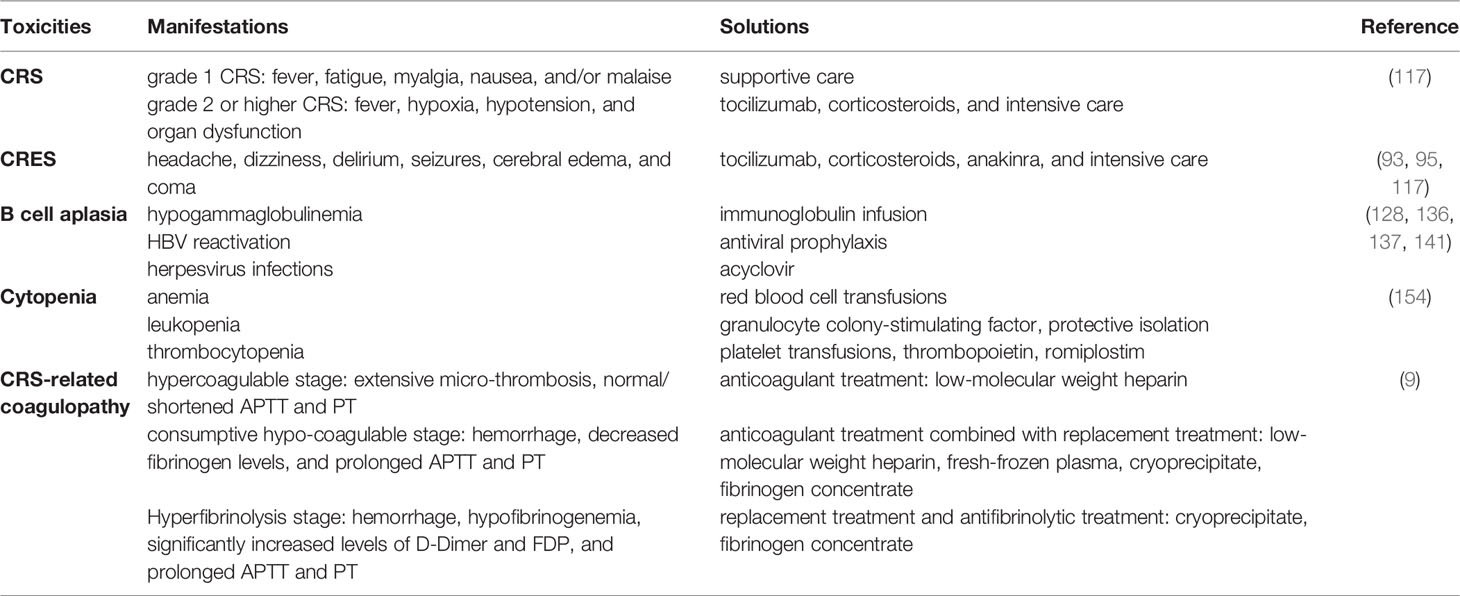
Table 2 Toxicities related to CAR-T cell therapy and effective solutions.
The mechanisms of CRS-related coagulopathy remain unclear. The activated platelets, monocytes, and endothelial cells as well as the CD40/CD40L interactions between them may collectively contribute to CRS-related coagulopathy. The CD40L expressed on activated CAR-T cells induces platelet activation in a CD40-independent manner in blood circulation (159). The activated platelets are prone to the form monocyte-platelet aggregates (MPAs) and are highly express CD40L, which could induce the expression of tissue factor (TF) in monocytes and endothelial cells through the direct interaction with CD40 (159–162). TF could activate the extrinsic coagulation cascade, and monocytes are the major sources of TF. Besides the CD40/CD40L interactions, there are also a variety of inducers could stimulate monocytes to upregulate the expression of TF, such as C-reactive protein (CRP), IL-1β and TNF-α (163–165). In addition, the CD40/CD40L interactions between them promote the excessive production of cytokines as well, such as IL-1β, TNF-α, and IL-6. High levels of cytokines further mediate endothelial injury and result in the release of TF, the procoagulant particles Weibel-Palade bodies (WPBs), and von Willebrand factor (VWF) as well as the exposure of the collagen fibers. The exposed collagen fibers trigger intrinsic coagulation pathway. In addition, cytokines IL-6, TNF-α, and IFN-γ can directly inhibit the production and activity of ADAMTS13, which contributes to the elevated levels VWF in blood and promote platelet adhesion and aggregation (166, 167). Moreover, serious liver damage induced by CAR-T cell therapy influences the production of coagulation factors, and some patients with hematological malignancies had be in a hypercoagulable state prior to CAR-T cell therapy (168) (Figure 4).
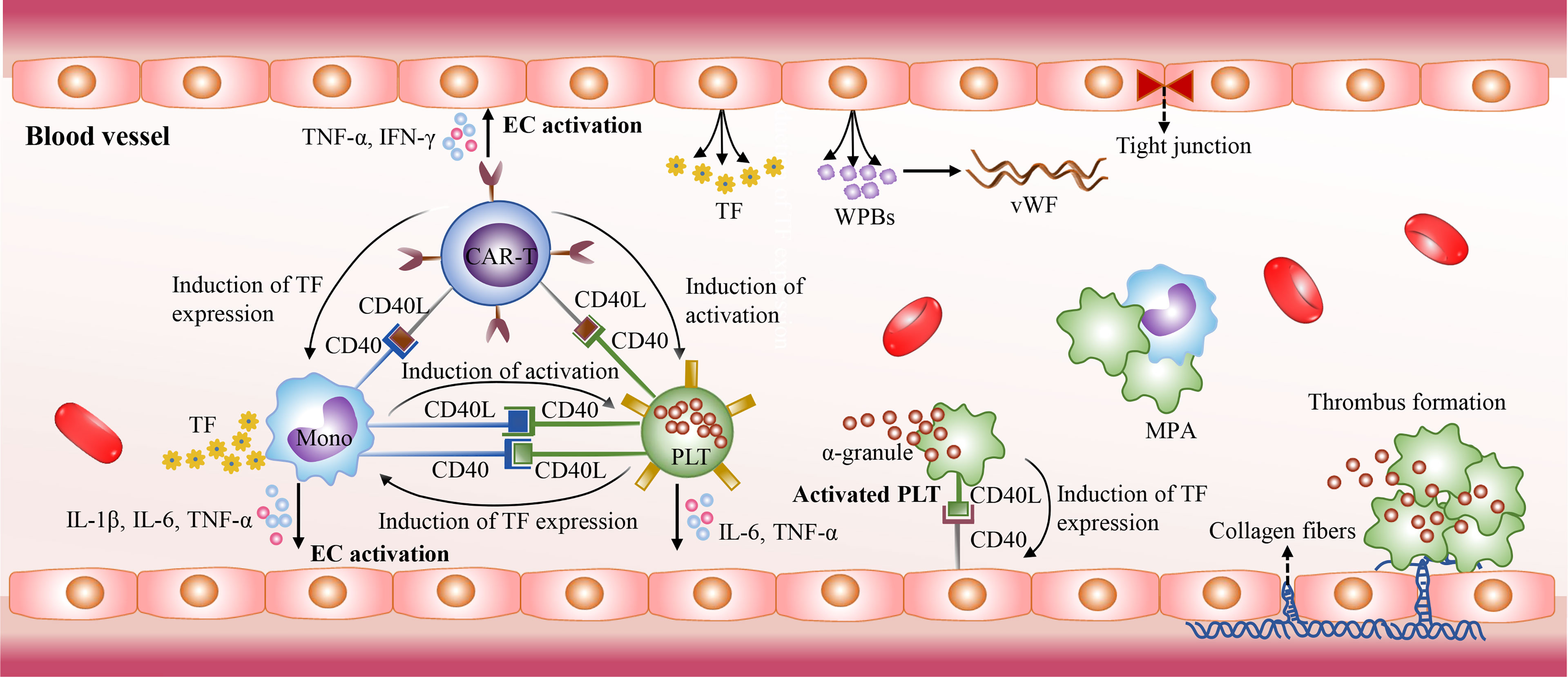
Figure 4 The mechanisms of CRS-related coagulopathy. The CD40/CD40L interactions also play an essential role in CRS-related coagulopathy. The activated CAR-T cells with high CD40L expression mediate platelet activation in a CD40-independent manner. Then the activated platelets express high levels of CD40L. It could stimulate endothelial cell activation, and induce the expression of TF in monocytes and endothelial cells through direct interaction with CD40. Then TF triggers the extrinsic coagulation cascade. In addition, the CD40/CD40L interactions stimulate the excessive release of cytokines, such as IL-1β, TNF-α, and IL-6. High levels of cytokines further mediate endothelial injury and promote the release of TF and Weibel-Palade bodies (WPBs). The WPBs contains von Willebrand factor (VWF) which plays an essential role in platelet adhesion and aggregation. Due to endothelial injury, collagen fibers are exposed and activate intrinsic coagulation pathway.
Resistance to CAR-T Cell Therapy and Potential Effective Strategies
However, with the widespread application of CAR-T cell therapy in R/R B-cell malignancies, a considerable proportion of patients relapse after CAR-T cell therapy. There are multiple factors which contribute to relapse after CAR-T cell therapy, including antigen escape, the limited CAR-T cell persistence, and immunosuppressive tumor microenvironment. There are some therapeutic strategies to overcoming the resistance to CAR-T cell therapy, including the application of bispecific or armored CAR-T cells, optimizing the CAR structure, combining CAR-T cell therapy with other approaches, such as small‐molecule drugs, localized radiotherapy, and oncolytic viruses.
Overcoming Antigen Escape
Although CAR-T cell therapy has made impressive achievements in R/R B cell malignancies, a majority of patients still relapse (143). One of the primary mechanisms of relapse after CAR-T cell therapy is antigen loss (169, 170). The antigen mutations under therapeutic pressure of CAR-T cell therapy are the most common mechanisms of antigen loss, including splice variants, lineage switching, and biallelic mutations (171–173). In addition to antigen mutations, the lower antigen density on the surface of induced by the endocytosis of CAR-T cells could promote tumor immune escape as well (174). Last but not least, a study has reported that the anti-CD19 CAR was incidentally transferred into a leukemic B cell during CAR-T cell manufacturing and then bound to the CD19 epitope on leukemic blasts, so “epitope-masking” prevented leukemic blasts from being recognized by anti-CD19 CAR-T cells (175).
Targeting distinct antigens is one of the most effective approaches to overcome antigen-negative relapse. The dual-targeting CAR-T cells which could recognize two distinct target antigens have been demonstrated to reduce the risk of antigen-negative relapse, such as the bispecific CAR-T cells in B cell lymphoma/leukemia and the APRIL-based CAR-T cells targeting both BCMA and TACI in MM (86, 176–180). The multi-targeted CAR-T cells have also been explored. It has been demonstrated that the tri-specific CD19-CD20-CD22-targeting CAR-T cells could rapidly eliminate B cell lymphoma in a preclinical study (181), and BAFF ligand-based CAR-T cells simultaneously target three receptors, including BAFF-R, BCMA, and TACI (182). In addition to simultaneously targeting different antigens, increasing immunogenicity of target cells might be a feasible strategy. For example, small molecule γ-secretase inhibitors could reduce the shedding of BCMA and promote the recognition of MM cells by CAR-T cells (183).
The γδ T cells (γδ T) are a small population of peripheral blood cytotoxic T cells, which express both T cell receptors (TCRs) and natural killer receptors (NKRs), and involved in anti-tumor immunity. In particular, NKRs expressed on γδ T cells play a major role in tumor cell recognition in hematological malignancies (184–186). Thus, besides antigen recognition mediated by the scFvs, γδ CAR-T cells could also recognize antigen-negative leukemia cells via NKRs in an MHC-independent manner (187). Moreover, γδ T cells did not induce graft-versus-host disease (GVHD) in allogeneic and HLA-haploidentical hematopoietic stem cell transplantation, which indicates that γδ T cells don’t trigger alloreactivity (188, 189). Thus, they are more suitable for the development of universal CAR-T cells.
Regulating the Persistence of CAR-T Cells
The short-term persistence of CAR-T cells limits their anti-tumor efficacy and may result in antigen-positive relapse. There are multiple strategies to improve the persistence of CAR-T cells, such as optimizing CAR-T cell construct, utilizing memory T cells, and rationally designing the ratio of CD4 to CD8 CAR-T cells (190). To date, CD28 and 4-1BB are the most common co-stimulatory molecules in CAR-T cell products. However, it has been demonstrated that 4-1BB co-stimulation could ameliorate CAR-T cell exhaustion compared with CD28 co-stimulation (191, 192). Remarkably, combining CD28 and 4-1BB could simultaneously augment the anti-tumor effects and increase the persistence of CAR-T cells (193–196). In addition, the CAR-T cell structure can be optimized by the fully humanized CARs. The humanized CAR-T cells could avoid the rejection by the host immune system, and they were still effective in R/R patients who have failed in prior murine CAR-T cell therapy (68, 197). CD4+ T cells exhibit developmental plasticity and can directly kill tumor cells (198), but they eliminate tumor cells at the slower rate and release the lower levels of Granzyme B than CD8+ T cells. Thus, CD4+ CAR-T cells exhibit a superior persistence (199–202), and the ratio of CD4/CD8 CAR-T cells may influence the therapeutic efficacy. Currently, CD4/CD8 CAR-T cells at a 1:1 ratio have been demonstrated to exert excellent anti-tumor effects (200).
CAR-T cells have been considered as “living drugs”, but they lack the precise regulation. Given that the excessive expansion of CAR-T cells could lead to the life-threatening CRS, so it is necessary to regulate the expansion and persistence of CAR-T cells to mitigate unexpected or severe toxicities through the addition of the safety switches. The well-known inducible caspase 9 (iCasp9) suicide gene and the small molecule control systems have been explored (203, 204). In small molecule control systems, the FDA-approved small molecule drugs act as the key switches to specifically regulate antigen recognition or deplete CAR-T cells, such as lenalidomide, methotrexate, alemtuzumab, rituximab, and cetuximab (31, 63, 205–208), as well as orthogonal IL-2 (205, 206). The above-mentioned monoclonal antibodies mediate the depletion of CAR-T cells through antibody-dependent cell-mediated cytotoxicity (ADCC) or complement-dependent cytotoxicity (CDC) (63, 209). Nevertheless, the depletion of CAR-T cells is slow in this strategy, which may be not suitable for patients with severe toxicities.
Improving Anti-Tumor Efficacy of CAR-T Cell Therapy Through Combinatorial Strategies
The low activity of CAR-T cells could also limit the efficacy of CAR-T cell therapy. Multiple immune-stimulatory molecules, including certain cytokines or co-stimulatory molecules, have been demonstrated to play an important role in regulating the development and function of T cells, such as IL-7, IL-12, IL-15, IL-18, IL-21, and CD40L (109, 210–214). They could promote the robust expansion of the CAR-T cells and increase memory-phenotype CAR-T cells as well as improving their persistence (215–218). In addition to adding these exogenous cytokines, the genetic modifications to constitutively express these immune-stimulatory molecules or their receptors could also improve the function of CAR-T cells (10, 213, 219, 220). These fourth-generation CAR-T cells have showed the improved anti-tumor activities by stimulating the activation of themselves or endogenous immune cells in a autocrine or paracrine manner (11, 220–222).
To improve the efficacy of CAR-T cell therapy, combining CAR-T cell therapy with small‐molecule drugs appears to be promising and may produce synergistic effects. The selective inhibitors of nuclear export selinexor, lenalidomide and carfilzomib have been approved for the treatment of MM (223, 224). Intriguingly, combining them with CAR-T cells also achieved encouraging outcomes, with the improved cytotoxic activity and cytokine production of CAR-T cells (225, 226). In particular, the recent clinical studies showed that the R/R MM patients resistant to anti-BCMA CAR-T cell therapy could also benefit from selinexor-based regimens and carfilzomib-based regimens (227, 228), and a study reported that anti-BCMA CAR-T cell therapy combined with lenalidomide was effective in the R/R MM patients who had previously relapsed after anti-BCMA CAR-T cell therapy (229). Ibrutinib, a well-known Bruton’s tyrosine kinase inhibitor, has been approved for the treatment of CLL and MCL. Importantly, Ibrutinib not only improved CAR T cell-anti-tumor efficacy in both preclinical and clinical studies, but also reduced the risk of severe CRS (230–233). In addition, it has been demonstrated that demethylating agents azacitidine and decitabine could enhance cytotoxic effect of CAR-T cells as well (234–236). Besides, CAR-T cell therapy in combination with inhibitors of antiapoptotic proteins could overcome the resistance induced by antiapoptotic proteins (237). However, CAR-T cell therapy combined with small‐molecule drugs is still in its infancy, and numerous combinational strategies are being explored.
Additionally, the localized radiotherapy could serve as a well-tolerated and effective bridging strategy between the leukapheresis and CAR-T cell infusion for lymphoma or MM patients with bulky disease (238–240). On the one hand, this combinational therapy could prevent disease progression and reduce tumor burden; On the other hand, it may sensitize the CAR-T cells through the abscopal effect, which may be associated with the upregulation of intratumoral chemokines and cytokines, the release of neo-antigens, and the activation of endogenous immune cells (241–245). Nonetheless, the optimal irradiation dose and fractionation remain to be identified.
Overcoming Immunosuppressive Microenvironment
While directly targeting tumor cells is important, it is also critical to overcome the immunosuppressive tumor microenvironment. Although tumor microenvironment is believed to play a relatively minor role in drug resistance in hematological malignancies, MM, leukemia, and lymphoma microenvironment also contains tumor supportive components, such as stromal cells, myeloid-derived suppressor cells, regulatory T-cells, tumor-associated macrophages, and tumor-associated neutrophils (246–250), which interact closely with malignant cells and promote their survival as well as immune escape (250–253). In addition, these immunosuppressive components impair the cytotoxic effects of CAR-T cells and result in CAR-T cell exhaustion (248, 249). Therefore, it is also necessary to overcome the immunosuppressive microenvironment in hematological malignancies. In addition to armored CAR-T cells, CAR-T cell therapy in combination with checkpoint blockades or oncolytic viruses also appears to be an appealing strategy.
The PD-1/PD-L1 pathway plays a major role in T cell exhaustion and represents a major mechanism of tumor immune escape. Thus, blockage of PD-1/PD-L1 interaction could promote the immune system to fight against cancer cells, and PD-1 blockade has achieved tremendous success in diverse tumor types in recent years, especially in lymphoma (254, 255). PD-1 blockade is usually administrated in combination with conventional chemotherapy or other immunotherapies. Currently, multiple studies have explored the combination therapy with CAR-T cells and PD-1 blockade (Table 3 combinatorial strategies with CAR-T cell therapy). In a clinical trial enrolled 11 NHL patients, 45.5% of patients achieved CRs after this combinatorial therapy, and the toxicities were well-tolerable (256, 257). The mechanisms may be mainly attributed to the decreased CAR-T cell exhaustion (258–260). In addition, the CAR-T cells which could secrete the PD-1 blocking scFv have been developed. The preclinical study demonstrated that the efficacy of such CAR-T cells was equally effective or superior to the combinational therapy of CAR-T cells and PD-1 inhibitor (261). TIM-3 is another inhibitory immune checkpoint, and the combination of TIM-3 blockade with CAR-T cells exerts synergistic anti-tumor activity as well (262).
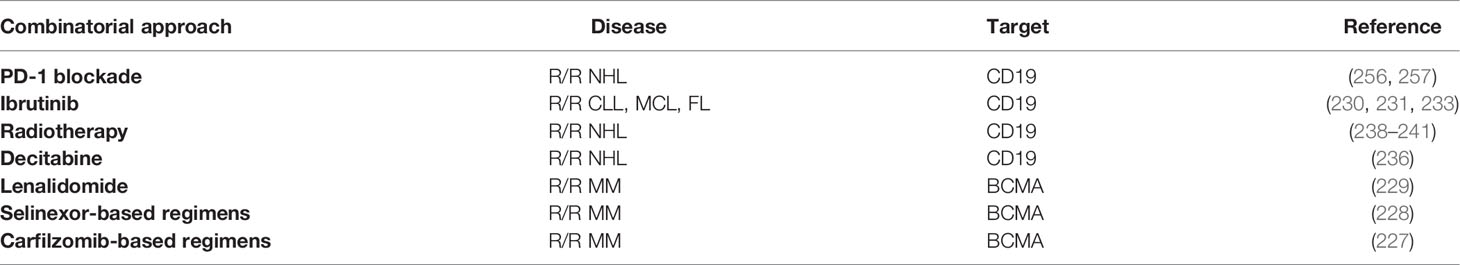
Table 3 Combinatorial strategies with CAR-T cell therapy reported in clinical studies.
The combination of CAR-T cell therapy and oncolytic viruses is an innovative strategy to overcome immunosuppressive microenvironment. The virus-infected tumor cells which carry pathogen-associated molecular patterns (PAMPs) could recruit host immune cells and thereby promote the recognition of TAAs by the host immune system and the oncolytic viruses also can be genetically modified with immune-stimulatory molecules to enhance the anti-tumor activity of CAR-T cells (263, 264). Besides, oncolytic viruses directly lyse tumor cells and result in the release of TAAs and damage-associated molecular patterns (DAMPs), which could increase tumor immunogenicity and activate APCs through pattern recognition receptors (PRRs), eventually activating tumor-specific T cells (265–267).
In addition, the armored CAR-T cells which express the immune-regulatory molecules, such as IL-15, IL-18, CD40L as well as TGF-β dominant-negative receptor II, are able to remodel the tumor microenvironment (211, 219, 220, 268). Oncometabolites in the tumor microenvironment could inhibit the metabolism and function of CAR-T cells, so suppressing the accumulation oncometabolites is a potential therapeutic option. Kynurenine (Kyn) is an oncometabolite which exists in various hematopoietic malignancies, such as lymphoma and leukemia, and the enzyme kynurenine catalyzes the degradation of Kyn. Thus, the anti-CD19 CAR-T cells were genetically modified with the enzyme kynurenine gene, and they exhibited the improved anti-tumor activity against Nalm6-GL cells in the immunosuppressive tumor microenvironment with high Kyn (269). Interestingly, some target antigens are co-expressed on immunosuppressive cells in tumor microenvironment, so targeting these antigens can simultaneously eliminate malignant cells and immunosuppressive cells. For example, anti-CD123 CAR-T cells target both malignant cells and TAMs in HL (270).
Conclusions
There are many challenges and opportunities presented by CAR-T cell therapy. With the identification of novel therapeutic targets and the optimization of CAR constructs, CAR-T cell therapy will have broader clinical applications, beyond hematological malignancies. However, with the rapid commercialization of CAR-T cell therapy, it poses a significant challenge for the management of CAR-T cell therapy, such as the toxicities associated with CAR-T therapy and relapse after CAR T-cell therapy. Therefore, exploring their underlying mechanisms and overcoming these limitations will help R/R patients gain more benefits from this promising. Currently, multiple combinatorial approaches with CAR-T cell therapy are being explored and seem to be promising immunotherapy. In addition, UCAR-T cells and CAR-NK cells also show great potential in cancer treatment due to their low manufacturing costs and off-the-shelf availability.
Author Contributions
YX, XZ, and LZ designed the manuscript. XZ, HZ, and SC drafted the manuscript and created the figures. XZ and YX revised the manuscript. All authors read and approved the final manuscript.
Funding
This work was supported by the National Natural Science Foundation of China (Grant No. 81973583 and 81873426), and Guangdong Basic and Applied Basic Research Foundation (2021A1515011575).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Maude SL, Laetsch TW, Buechner J, Rives S, Boyer M, Bittencourt H, et al. Tisagenlecleucel in Children and Young Adults With B-Cell Lymphoblastic Leukemia. N Engl J Med (2018) 378(5):439–48. doi: 10.1056/NEJMoa1709866
2. Schuster SJ, Bishop MR, Tam CS, Waller EK, Borchmann P, McGuirk JP, et al. Tisagenlecleucel in Adult Relapsed or Refractory Diffuse Large B-Cell Lymphoma. N Engl J Med (2019) 380:45–56. doi: 10.1056/NEJMoa1804980
3. Fowler NH, Dickinson M, Dreyling M, Martinez-Lopez J, Kolstad A, Butler J, et al. Tisagenlecleucel in Adult Relapsed or Refractory Follicular Lymphoma: The Phase 2 ELARA Trial. Nat Med (2021) 28(2):325–32. doi: 10.1038/s41591-021-01622-0
4. Locke FL, Ghobadi A, Jacobson CA, Miklos DB, Lekakis LJ, Oluwole OO, et al. Long-Term Safety and Activity of Axicabtagene Ciloleucel in Refractory Large B-Cell Lymphoma (ZUMA-1): A Single-Arm, Multicentre, Phase 1-2 Trial. Lancet Oncol (2019) 20(1):31–42. doi: 10.1016/S1470-2045(18)30864-7
5. Wang M, Munoz J, Goy A, Locke FL, Jacobson CA, Hill BT, et al. KTE-X19 CAR T-Cell Therapy in Relapsed or Refractory Mantle-Cell Lymphoma. N Engl J Med (2020) 382(14):1331–42. doi: 10.1056/NEJMoa1914347
6. Abramson JS, Palomba ML, Gordon LI, Lunning MA, Wang M, Arnason J, et al. Lisocabtagene Maraleucel for Patients With Relapsed or Refractory Large B-Cell Lymphomas (TRANSCEND NHL 001): A Multicentre Seamless Design Study. Lancet (2020) 396(10254):839–52. doi: 10.1016/S0140-6736(20)31366-0
7. Berdeja JG, Madduri D, Usmani SZ, Jakubowiak A, Agha M, Cohen AD, et al. Ciltacabtagene Autoleucel, a B-Cell Maturation Antigen-Directed Chimeric Antigen Receptor T-Cell Therapy in Patients With Relapsed or Refractory Multiple Myeloma (CARTITUDE-1): A Phase 1b/2 Open-Label Study. Lancet (2021) 398(10297):314–24. doi: 10.1016/S0140-6736(21)00933-8
8. Munshi NC, Anderson LD Jr, Shah N, Madduri D, Berdeja J, Lonial S, et al. Idecabtagene Vicleucel in Relapsed and Refractory Multiple Myeloma. N Engl J Med (2021) 384(8):705–16. doi: 10.1056/NEJMoa2024850
9. Jiang H, Liu L, Guo T, Wu Y, Ai L, Deng J, et al. Improving the Safety of CAR-T Cell Therapy by Controlling CRS-Related Coagulopathy. Ann Hematol (2019) 98(7):1721–32. doi: 10.1007/s00277-019-03685-z
10. Lanitis E, Rota G, Kosti P, Ronet C, Spill A, Seijo B, et al. Optimized Gene Engineering of Murine CAR-T Cells Reveals the Beneficial Effects of IL-15 Coexpression. J Exp Med (2021) 218(2):e20192203. doi: 10.1084/jem.20192203
11. Duan D, Wang K, Wei C, Feng D, Liu Y, He Q, et al. The BCMA-Targeted Fourth-Generation CAR-T Cells Secreting IL-7 and CCL19 for Therapy of Refractory/Recurrent Multiple Myeloma. Front Immunol (2021) 12:609421. doi: 10.3389/fimmu.2021.609421
12. Yang J, Jiang P, Zhang X, Li J, Wu Y, Xu L, et al. Successful 24-Hours Manufacture of Anti-CD19/CD22 Dual Chimeric Antigen Receptor (CAR) T Cell Therapy for B-Cell Acute Lymphoblastic Leukemia (B-ALL). Blood (2020) 136 (Supplement 1):2–3. doi: 10.1182/blood-2020-136866
13. Jiang H, Dong B, Gao L, Liu L, Ge J, He A, et al. Clinical Results of a Multicenter Study of the First-in-Human Dual BCMA and CD19 Targeted Novel Platform Fast CAR-T Cell Therapy for Patients with Relapsed/Refractory Multiple Myeloma. Blood (2020) 136 (Supplement 1):25–6. doi: 10.1182/blood-2020-136866
14. Hu Y, Wang J, Wei G, Yu J, Luo Y, Shi J, et al. A Retrospective Comparison of Allogenic and Autologous Chimeric Antigen Receptor T Cell Therapy Targeting CD19 in Patients With Relapsed/Refractory Acute Lymphoblastic Leukemia. Bone Marrow Transplant (2019) 54(8):1208–17. doi: 10.1038/s41409-018-0403-2
15. Liu E, Marin D, Banerjee P, Macapinlac HA, Thompson P, Basar R, et al. Use of CAR-Transduced Natural Killer Cells in CD19-Positive Lymphoid Tumors. N Engl J Med (2020) 382(6):545–53. doi: 10.1056/NEJMoa1910607
16. Jacobson CA, Chavez JC, Sehgal AR, William BM, Munoz J, Salles G, et al. Axicabtagene Ciloleucel in Relapsed or Refractory Indolent non-Hodgkin Lymphoma (ZUMA-5): A Single-Arm, Multicentre, Phase 2 Trial. Lancet Oncol (2022) 23(1):91–103. doi: 10.1016/S1470-2045(21)00591-X
17. Zelenetz AD, Gordon LI, Chang JE, Christian B, Abramson JS, Advani RH, et al. NCCN Guidelines® Insights: B-Cell Lymphomas, Version 5.2021. J Natl Compr Canc Netw (2021) 19(11):1218–30. doi: 10.6004/jnccn.2021.0054
18. Pan J, Yang JF, Deng BP, Zhao XJ, Zhang X, Lin YH, et al. High Efficacy and Safety of Low Dose CD19 Directed CAR-T Cell Therapy in 51 Refractory or Relapsed B Acute Lymphoblastic Leukemia Patients. Leukemia (2017) 31:2587–93. doi: 10.1038/leu.2017.145
19. Wang Y, Zhang WY, Han QW, Liu Y, Dai HR, Guo YL, et al. Effective Response and Delayed Toxicities of Refractory Advanced Diffuse Large B-Cell Lymphoma Treated by CD20-Directed Chimeric Antigen Receptor-Modified T Cells. Clin Immunol (2014) 155(2):160–75. doi: 10.1016/j.clim.2014.10.002
20. Zhang WY, Liu Y, Wang Y, Wang CM, Yang QM, Zhu HL, et al. Long-Term Safety and Efficacy of CART-20 Cells in Patients With Refractory or Relapsed B-Cell non-Hodgkin Lymphoma: 5-Years Follow-Up Results of the Phase I and IIa Trials. Signal Transduct Target Ther (2017) 2:17054. doi: 10.1038/sigtrans.2017.54
21. Sang W, Shi M, Yang J, Cao J, Xu L, Yan D, et al. Phase II Trial of Co-Administration of CD19- and CD20-Targeted Chimeric Antigen Receptor T Cells for Relapsed and Refractory Diffuse Large B Cell Lymphoma. Cancer Med (2020) 9(16):5827–38. doi: 10.1002/cam4.3259
22. Olejniczak SH, Stewart CC, Donohue K, Czuczman MS. A Quantitative Exploration of Surface Antigen Expression in Common B-Cell Malignancies Using Flow Cytometry. Immunol Invest (2006) 35(1):93–114. doi: 10.1080/08820130500496878
23. Haso W, Lee DW, Shah NN, Stetler-Stevenson M, Yuan CM, Pastan IH, et al. Anti-CD22-Chimeric Antigen Receptors Targeting B-Cell Precursor Acute Lymphoblastic Leukemia. Blood (2013) 121(7):1165–74. doi: 10.1182/blood-2012-06-438002
24. Pan J, Niu Q, Deng B, Liu S, Wu T, Gao Z, et al. CD22 CAR T-Cell Therapy in Refractory or Relapsed B Acute Lymphoblastic Leukemia. Leukemia (2019) 33(12):2854–66. doi: 10.1038/s41375-019-0488-7
25. Fry TJ, Shah NN, Orentas RJ, Stetler-Stevenson M, Yuan CM, Ramakrishna S, et al. CD22-Targeted CAR T Cells Induce Remission in B-ALL That is Naive or Resistant to CD19-Targeted CAR Immunotherapy. Nat Med (2018) 24(1):20–8. doi: 10.1038/nm.4441
26. Zhu H, Deng H, Mu J, Lyu C, Jiang Y, Deng Q. Anti-CD22 CAR-T Cell Therapy as a Salvage Treatment in B Cell Malignancies Refractory or Relapsed After Anti-CD19 CAR-T Therapy. Onco Targets Ther (2021) 14:4023–37. doi: 10.2147/OTT.S312904
27. Tan Y, Cai H, Li C, Deng B, Song W, Ling Z, et al. A Novel Full-Human CD22-CAR T Cell Therapy With Potent Activity Against CD22 Low B-ALL. Blood Cancer J (2021) 11(4):71. doi: 10.1038/s41408-021-00465-9
28. Li S, Wang X, Yuan Z, Liu L, Luo L, Li Y, et al. Eradication of T-ALL Cells by CD7-Targeted Universal CAR-T Cells and Initial Test of Ruxolitinib-Based CRS Management. Clin Cancer Res (2021) 27(5):1242–6. doi: 10.1158/1078-0432.CCR-20-1271
29. Pan J, Tan Y, Wang G, Deng B, Ling Z, Song W, et al. Donor-Derived CD7 Chimeric Antigen Receptor T Cells for T-Cell Acute Lymphoblastic Leukemia: First-In-Human, Phase I Trial. J Clin Oncol (2021) 39(30):3340–51. doi: 10.1200/JCO.21.00389
30. Xie L, Ma L, Liu S, Chang L, Wen F. Chimeric Antigen Receptor T Cells Targeting CD7 in a Child With High-Risk T-Cell Acute Lymphoblastic Leukemia. Int Immunopharmacol (2021) 96:107731. doi: 10.1016/j.intimp.2021.107731
31. Wada M, Zhang H, Fang L, Feng J, Tse CO, Zhang W, et al. Characterization of an Anti-CD5 Directed CAR T-Cell Against T-Cell Malignancies. Stem Cell Rev Rep (2020) 16(2):369–84. doi: 10.1007/s12015-019-09937-9
32. Feng J, Xu H, Cinquina A, Wu Z, Chen Q, Zhang P, et al. Treatment of Aggressive T Cell Lymphoblastic Lymphoma/leukemia Using Anti-CD5 CAR T Cells. Stem Cell Rev Rep (2021) 17(2):652–61. doi: 10.1007/s12015-020-10092-9
33. Pinz K, Liu H, Golightly M, Jares A, Lan F, Zieve GW, et al. Preclinical Targeting of Human T-Cell Malignancies Using CD4-Specific Chimeric Antigen Receptor (CAR)-Engineered T Cells. Leukemia (2016) 30(3):701–7. doi: 10.1038/leu.2015.311
34. Alcantara M, Tesio M, June CH, Houot R. CAR T-Cells for T-Cell Malignancies: Challenges in Distinguishing Between Therapeutic, Normal, and Neoplastic T-Cells. Leukemia (2018) 32(11):2307–15. doi: 10.1038/s41375-018-0285-8
35. Maciocia PM, Wawrzyniecka PA, Philip B, Ricciardelli I, Akarca AU, Onuoha SC, et al. Targeting the T cell Receptor β-Chain Constant Region for Immunotherapy of T Cell Malignancies. Nat Med (2017) 23(12):1416–23. doi: 10.1038/nm.4444
36. Shi J, Zhang Z, Cen H, Wu H, Zhang S, Liu J, et al. CAR T Cells Targeting CD99 as an Approach to Eradicate T-Cell Acute Lymphoblastic Leukemia Without Normal Blood Cells Toxicity. J Hematol Oncol (2021) 14(1):162. doi: 10.1186/s13045-021-01178-z
37. Maciocia PM, Wawrzyniecka PA, Maciocia NC, Burley A, Karpanasamy T, Devereaux S, et al. Anti-CCR9 Chimeric Antigen Receptor T Cells for T Cell Acute Lymphoblastic Leukemia. Blood (2022), blood.2021013648. doi: 10.1182/blood.2021013648
38. Horwitz S, O'Connor OA, Pro B, Illidge T, Fanale M, Advani R, et al. Brentuximab Vedotin With Chemotherapy for CD30-Positive Peripheral T-Cell Lymphoma (ECHELON-2): A Global, Double-Blind, Randomised, Phase 3 Trial. Lancet (2019) 393:229–40. doi: 10.1016/S0140-6736(18)32984-2
39. Fanale MA, Horwitz SM, Forero-Torres A, Bartlett NL, Advani RH, Pro B, et al. Five-Year Outcomes for Frontline Brentuximab Vedotin With CHP for CD30-Expressing Peripheral T-Cell Lymphomas. Blood (2018) 131(19):2120–4. doi: 10.1182/blood-2017-12-821009
40. Guercio M, Orlando D, Di Cecca S, Sinibaldi M, Boffa I, Caruso S, et al. CD28.OX40 Co-Stimulatory Combination is Associated With Long In Vivo Persistence and High Activity of CAR.CD30 T-Cells. Haematologica (2021) 106(4):987–99. doi: 10.3324/haematol.2019.231183
41. Wu Y, Chen D, Lu Y, Dong SC, Ma R, Tang WY, et al. A New Immunotherapy Strategy Targeted CD30 in Peripheral T-Cell Lymphomas: CAR-Modified T-Cell Therapy Based on CD30 mAb. Cancer Gene Ther (2022) 29(2):167–77. doi: 10.1038/s41417-021-00295-8
42. Wang CM, Wu ZQ, Wang Y, Guo YL, Dai HR, Wang XH, et al. Autologous T Cells Expressing CD30 Chimeric Antigen Receptors for Relapsed or Refractory Hodgkin Lymphoma: An Open-Label Phase I Trial. Clin Cancer Res (2017) 23(5):1156–66. doi: 10.1158/1078-0432.CCR-16-1365
43. Ramos CA, Grover NS, Beaven AW, Lulla PD, Wu MF, Ivanova A, et al. Anti-CD30 CAR-T Cell Therapy in Relapsed and Refractory Hodgkin Lymphoma. J Clin Oncol (2020) 38(32):3794–804. doi: 10.1200/JCO.20.01342
44. Ramos CA, Ballard B, Zhang H, Dakhova O, Gee AP, Mei Z, et al. Clinical and Immunological Responses After CD30-Specific Chimeric Antigen Receptor-Redirected Lymphocytes. J Clin Invest (2017) 127(9):3462–71. doi: 10.1172/JCI94306
45. Wang D, Zeng C, Xu B, Xu JH, Wang J, Jiang LJ, et al. Anti-CD30 Chimeric Antigen Receptor T Cell Therapy for Relapsed/Refractory CD30+ Lymphoma Patients. Blood Cancer J (2020) 10(1):8. doi: 10.1038/s41408-020-0274-9
46. Voorhees TJ, Zhao B, Oldan J, Hucks G, Khandani A, Dittus C, et al. Pretherapy Metabolic Tumor Volume is Associated With Response to CD30 CAR T Cells in Hodgkin Lymphoma. Blood Adv (2022) 6(4):1255–63. doi: 10.1182/bloodadvances.2021005385
47. Kim DH, Vega F. Relapsed Classic Hodgkin Lymphoma With Decreased CD30 Expression After Brentuximab and Anti-CD30 CAR-T Therapies. Blood (2022) 139(6):951. doi: 10.1182/blood.2021013881
48. Nguyen DH, Ball ED, Varki A. Myeloid Precursors and Acute Myeloid Leukemia Cells Express Multiple CD33-Related Siglecs. Exp Hematol (2006) 34:728–35. doi: 10.1016/j.exphem.2006.03.003
49. Baroni ML, Sanchez Martinez D, Gutierrez Aguera F, Roca Ho H, Castella M, Zanetti SR, et al. 41BB-Based and CD28-Based CD123-Redirected T-Cells Ablate Human Normal Hematopoiesis In Vivo. J Immunother Cancer (2020) 8(1):e000845. doi: 10.1136/jitc-2020-000845
50. Loff S, Dietrich J, Meyer JE, Riewaldt J, Spehr J, von Bonin M, et al. Rapidly Switchable Universal CAR-T Cells for Treatment of CD123-Positive Leukemia. Mol Ther Oncolytics (2020) 17:408–20. doi: 10.1016/j.omto.2020.04.009
51. Meyer JE, Loff S, Dietrich J, Spehr J, Jurado Jiménez G, von Bonin M, et al. Evaluation of Switch-Mediated Costimulation in Trans on Universal CAR-T Cells (UniCAR) Targeting CD123-Positive AML. Oncoimmunology (2021) 10(1):1945804. doi: 10.1080/2162402X.2021.1945804
52. Wermke M, Kraus S, Ehninger A, Bargou RC, Goebeler ME, Middeke JM, et al. Proof of Concept for a Rapidly Switchable Universal CAR-T Platform With UniCAR-T-CD123 in Relapsed/Refractory AML. Blood (2021) 137(22):3145–8. doi: 10.1182/blood.2020009759
53. Tambaro FP, Singh H, Jones E, Rytting M, Mahadeo KM, Thompson P, et al. Autologous CD33-CAR-T Cells for Treatment of Relapsed/Refractory Acute Myelogenous Leukemia. Leukemia (2021) 35(11):3282–6. doi: 10.1038/s41375-021-01232-2
54. Cui Q, Qian C, Xu N, Kang L, Dai H, Cui W, et al. CD38-Directed CAR-T Cell Therapy: A Novel Immunotherapy Strategy for Relapsed Acute Myeloid Leukemia After Allogeneic Hematopoietic Stem Cell Transplantation. J Hematol Oncol (2021) 14(1):82. doi: 10.1186/s13045-021-01092-4
55. Bakker AB, van den Oudenrijn S, Bakker AQ, Feller N, van Meijer M, Bia JA, et al. C-Type Lectin-Like Molecule-1: A Novel Myeloid Cell Surface Marker Associated With Acute Myeloid Leukemia. Cancer Res (2004) 64:8443–50. doi: 10.1158/0008-5472.CAN-04-1659
56. Wang J, Chen S, Xiao W, Li W, Wang L, Yang S, et al. CAR-T Cells Targeting CLL-1 as an Approach to Treat Acute Myeloid Leukemia. J Hematol Oncol (2018) 11:7. doi: 10.1186/s13045-017-0553-5
57. Lin G, Zhang Y, Yu L, Wu D. Cytotoxic Effect of CLL-1 CAR-T Cell Immunotherapy With PD-1 Silencing on Relapsed/Refractory Acute Myeloid Leukemia. Mol Med Rep (2021) 23(3):208. doi: 10.3892/mmr.2021.11847
58. Sauer T, Parikh K, Sharma S, Omer B, Sedloev D, Chen Q, et al. CD70-Specific CAR T Cells Have Potent Activity Against Acute Myeloid Leukemia Without HSC Toxicity. Blood (2021) 138(4):318–30. doi: 10.1182/blood.2020008221
59. Riether C, Pabst T, Höpner S, Bacher U, Hinterbrandner M, Banz Y, et al. Targeting CD70 With Cusatuzumab Eliminates Acute Myeloid Leukemia Stem Cells in Patients Treated With Hypomethylating Agents. Nat Med (2020) 26(9):1459–67. doi: 10.1038/s41591-020-0910-8
60. John S, Chen H, Deng M, Gui X, Wu G, Chen W, et al. A Novel Anti-LILRB4 CAR-T Cell for the Treatment of Monocytic AML. Mol Ther (2018) 26(10):2487–95. doi: 10.1016/j.ymthe.2018.08.001
61. Jetani H, Navarro-Bailón A, Maucher M, Frenz S, Verbruggen C, Yeguas A, et al. Siglec-6 is a Novel Target for CAR T-Cell Therapy in Acute Myeloid Leukemia. Blood (2021) 138(19):1830–42. doi: 10.1182/blood.2020009192
62. Xie G, Ivica NA, Jia B, Li Y, Dong H, Liang Y, et al. CAR-T Cells Targeting a Nucleophosmin Neoepitope Exhibit Potent Specific Activity in Mouse Models of Acute Myeloid Leukaemia. Nat BioMed Eng (2021) 5(5):399–413. doi: 10.1038/s41551-020-00625-5
63. Sommer C, Cheng HY, Nguyen D, Dettling D, Yeung YA, Sutton J, et al. Allogeneic FLT3 CAR T Cells With an Off-Switch Exhibit Potent Activity Against AML and Can Be Depleted to Expedite Bone Marrow Recovery. Mol Ther (2020) 28(10):2237–51. doi: 10.1016/j.ymthe.2020.06.022
64. Jetani H, Garcia-Cadenas I, Nerreter T, Thomas S, Rydzek J, Meijide JB, et al. CAR T-Cells Targeting FLT3 Have Potent Activity Against FLT3-ITD+ AML and Act Synergistically With the FLT3-Inhibitor Crenolanib. Leukemia (2018) 32(5):1168–79. doi: 10.1038/s41375-018-0009-0
65. Kikushige Y, Yoshimoto G, Miyamoto T, Iino T, Mori Y, Iwasaki H, et al. Human Flt3 is Expressed at the Hematopoietic Stem Cell and the Granulocyte/Macrophage Progenitor Stages to Maintain Cell Survival. J Immunol (2008) 180:7358–67. doi: 10.4049/jimmunol.180.11.7358
66. Rajkumar SV, Dimopoulos MA, Palumbo A, Blade J, Merlini G, Mateos MV, et al. International Myeloma Working Group Updated Criteria for the Diagnosis of Multiple Myeloma. Lancet Oncol (2014) 15(12):e538–48. doi: 10.1016/S1470-2045(14)70442-5
67. Shah N, Chari A, Scott E, Mezzi K, Usmani SZ. B-Cell Maturation Antigen (BCMA) in Multiple Myeloma: Rationale for Targeting and Current Therapeutic Approaches. Leukemia (2020) 34(4):985–1005. doi: 10.1038/s41375-020-0734-z
68. Wang D, Wang J, Hu G, Wang W, Xiao Y, Cai H, et al. A Phase 1 Study of a Novel Fully Human BCMA-Targeting CAR (CT103A) in Patients With Relapsed/Refractory Multiple Myeloma. Blood (2021) 137(21):2890–901. doi: 10.1182/blood.2020008936
69. Li C, Cao W, Que Y, Wang Q, Xiao Y, Gu C, et al. A Phase I Study of Anti-BCMA CAR T Cell Therapy in Relapsed/Refractory Multiple Myeloma and Plasma Cell Leukemia. Clin Transl Med (2021) 11(3):e346. doi: 10.1002/ctm2.346
70. Sharma P, Kanapuru B, George B, Lin X, Xu Z, Bryan WW, et al. FDA Approval Summary: Idecabtagene Vicleucel for Relapsed or Refractory Multiple Myeloma. Clin Cancer Res (2022) 28(9): 1759–64. doi: 10.1158/1078-0432.CCR-21-3803
71. Deng H, Liu M, Yuan T, Zhang H, Cui R, Li J, et al. Efficacy of Humanized Anti-BCMA CAR T Cell Therapy in Relapsed/Refractory Multiple Myeloma Patients With and Without Extramedullary Disease. Front Immunol (2021) 12:720571. doi: 10.3389/fimmu.2021.720571
72. Que Y, Xu M, Xu Y, Almeida VDF, Zhu L, Wang Z, et al. Anti-BCMA CAR-T Cell Therapy in Relapsed/Refractory Multiple Myeloma Patients With Extramedullary Disease: A Single Center Analysis of Two Clinical Trials. Front Immunol (2021) 12:755866. doi: 10.3389/fimmu.2021.755866
73. Oriol A, Abril L, Torrent A, Ibarra G, Ribera JM. The Role of Idecabtagene Vicleucel in Patients With Heavily Pretreated Refractory Multiple Myeloma. Ther Adv Hematol (2021) 12:20406207211019622. doi: 10.1177/20406207211019622
74. van de Donk NWCJ, Usmani SZ, Yong K. CAR T-Cell Therapy for Multiple Myeloma: State of the Art and Prospects. Lancet Haematol (2021) 8(6):e446–61. doi: 10.1016/S2352-3026(21)00057-0
75. Sun C, Mahendravada A, Ballard B, Kale B, Ramos C, West J, et al. Safety and Efficacy of Targeting CD138 With a Chimeric Antigen Receptor for the Treatment of Multiple Myeloma. Oncotarget (2019) 10:2369–83. doi: 10.18632/oncotarget.26792
76. Guo B, Chen M, Han Q, Hui F, Dai H, Zhang W, et al. CD138-Directed Adoptive Immunotherapy of Chimeric Antigen Receptor (CAR)-Modified T Cells for Multiple Myeloma. J Cell Immunother (2016) 2:28–35. doi: 10.1016/j.jocit.2014.11.001
77. van de Donk NW, Janmaat ML, Mutis T, Lammerts van Bueren JJ, Ahmadi T, Sasser AK, et al. Monoclonal Antibodies Targeting CD38 in Hematological Malignancies and Beyond. Immunol Rev (2016) 270(1):95–112. doi: 10.1111/imr.12389
78. Drent E, Groen RW, Noort WA, Themeli M, Lammerts van Bueren JJ, Parren PW, et al. Pre-Clinical Evaluation of CD38 Chimeric Antigen Receptor Engineered T Cells for the Treatment of Multiple Myeloma. Haematologica (2016) 101:616–25. doi: 10.3324/haematol.2015.137620
79. Tang Y, Yin H, Zhao X, Jin D, Liang Y, Xiong T, et al. High Efficacy and Safety of CD38 and BCMA Bispecific CAR-T in Relapsed or Refractory Multiple Myeloma. J Exp Clin Cancer Res (2022) 41(1):2. doi: 10.1186/s13046-021-02214-z
80. Mei H, Li C, Jiang H, Zhao X, Huang Z, Jin D, et al. A Bispecific CAR-T Cell Therapy Targeting BCMA and CD38 in Relapsed or Refractory Multiple Myeloma. J Hematol Oncol (2021) 14(1):161. doi: 10.1186/s13045-021-01170-7
81. Atanackovic D, Panse J, Hildebrandt Y, Jadczak A, Kobold S, Cao Y, et al. Surface Molecule CD229 as a Novel Target for the Diagnosis and Treatment of Multiple Myeloma. Haematologica (2011) 96(10):1512–20. doi: 10.3324/haematol.2010.036814
82. Radhakrishnan SV, Luetkens T, Scherer SD, Davis P, Vander Mause ER, Olson ML, et al. CD229 CAR T Cells Eliminate Multiple Myeloma and Tumor Propagating Cells Without Fratricide. Nat Commun (2020) 11(1):798. doi: 10.1038/s41467-020-14619-z
83. Hsi ED, Steinle R, Balasa B, Szmania S, Draksharapu A, Shum BP, et al. CS1, a Potential New Therapeutic Antibody Target for the Treatment of Multiple Myeloma. Clin Cancer Res (2008) 14:2775–84. doi: 10.1158/1078-0432.CCR-07-4246
84. Gogishvili T, Danhof S, Prommersberger S, Rydzek J, Schreder M, Brede C, et al. SLAMF7-CAR T Cells Eliminate Myeloma and Confer Selective Fratricide of SLAMF7(+) Normal Lymphocytes. Blood (2017) 130:2838–47. doi: 10.1182/blood-2017-04-778423
85. Prommersberger S, Reiser M, Beckmann J, Danhof S, Amberger M, Quade-Lyssy P, et al. CARAMBA: A First-in-Human Clinical Trial With SLAMF7 CAR-T Cells Prepared by Virus-Free Sleeping Beauty Gene Transfer to Treat Multiple Myeloma. Gene Ther (2021) 28(9):560–71. doi: 10.1038/s41434-021-00254-w
86. Lee L, Draper B, Chaplin N, Philip B, Chin M, Galas-Filipowicz D, et al. An APRIL-Based Chimeric Antigen Receptor for Dual Targeting of BCMA and TACI in Multiple Myeloma. Blood (2018) 131(7):746–58. doi: 10.1182/blood-2017-05-781351
87. Schmidts A, Ormhøj M, Choi BD, Taylor AO, Bouffard AA, Scarfò I, et al. Rational Design of a Trimeric APRIL-Based CAR-Binding Domain Enables Efficient Targeting of Multiple Myeloma. Blood Adv (2019) 3(21):3248–60. doi: 10.1182/bloodadvances.2019000703
88. Smith EL, Harrington K, Staehr M, Masakayan R, Jones J, Long TJ, et al. GPRC5D is a Target for the Immunotherapy of Multiple Myeloma With Rationally Designed CAR T Cells. Sci Transl Med (2019) 11(485):eaau7746. doi: 10.1126/scitranslmed.aau7746
89. Obstfeld AE, Frey NV, Mansfield K, Lacey SF, June CH, Porter DL, et al. Cytokine Release Syndrome Associated With Chimeric-Antigen Receptor T-Cell Therapy: Clinicopathological Insights. Blood (2017) 130(23):2569–72. doi: 10.1182/blood-2017-08-802413
90. Dong R, Jiang S, Chen Y, Ma Y, Sun L, Xing C, et al. Prognostic Significance of Cytokine Release Syndrome in B Cell Hematological Malignancies Patients After Chimeric Antigen Receptor T Cell Therapy. J Interferon Cytokine Res (2021) 41(12):469–76. doi: 10.1089/jir.2021.0057
91. Norelli M, Camisa B, Barbiera G, Falcone L, Purevdorj A, Genua M, et al. Monocyte-Derived IL-1 and IL-6 are Differentially Required for Cytokine-Release Syndrome and Neurotoxicity Due to CAR T Cells. Nat Med (2018) 24(6):739–48. doi: 10.1038/s41591-018-0036-4
92. Giavridis T, van der Stegen SJC, Eyquem J, Hamieh M, Piersigilli A, Sadelain M. CAR T Cell-Induced Cytokine Release Syndrome is Mediated by Macrophages and Abated by IL-1 Blockade. Nat Med (2018) 24(6):731–8. doi: 10.1038/s41591-018-0041-7
93. Wehrli M, Gallagher K, Chen YB, Leick MB, McAfee SL, El-Jawahri AR, et al. Single-Center Experience Using Anakinra for Steroid-Refractory Immune Effector Cell-Associated Neurotoxicity Syndrome (ICANS). J Immunother Cancer (2022) 10(1):e003847. doi: 10.1136/jitc-2021-003847
94. Mehta P, Cron RQ, Hartwell J, Manson JJ, Tattersall RS. Silencing the Cytokine Storm: The Use of Intravenous Anakinra in Haemophagocytic Lymphohistiocytosis or Macrophage Activation Syndrome. Lancet Rheumatol (2020) 2(6):e358–67. doi: 10.1016/S2665-9913(20)30096-5
95. Strati P, Ahmed S, Kebriaei P, Nastoupil LJ, Claussen CM, Watson G, et al. Clinical Efficacy of Anakinra to Mitigate CAR T-Cell Therapy-Associated Toxicity in Large B-Cell Lymphoma. Blood Adv (2020) 4(13):3123–7. doi: 10.1182/bloodadvances.2020002328
96. Sterner RM, Sakemura R, Cox MJ, Yang N, Khadka RH, Forsman CL, et al. GM-CSF Inhibition Reduces Cytokine Release Syndrome and Neuroinflammation But Enhances CAR-T Cell Function in Xenografts. Blood (2019) 133(7):697–709. doi: 10.1182/blood-2018-10-881722
97. Sachdeva M, Duchateau P, Depil S, Poirot L, Valton J. Granulocyte-Macrophage Colony-Stimulating Factor Inactivation in CAR T-Cells Prevents Monocyte-Dependent Release of Key Cytokine Release Syndrome Mediators. J Biol Chem (2019) 294(14):5430–7. doi: 10.1074/jbc.AC119.007558
98. Liu Y, Fang Y, Chen X, Wang Z, Liang X, Zhang T, et al. Gasdermin E-Mediated Target Cell Pyroptosis by CAR T Cells Triggers Cytokine Release Syndrome. Sci Immunol (2020) 5(43):eaax7969. doi: 10.1126/sciimmunol.aax7969
99. Gong T, Liu L, Jiang W, Zhou R. DAMP-Sensing Receptors in Sterile Inflammation and Inflammatory Diseases. Nat Rev Immunol (2020) 20(2):95–112. doi: 10.1038/s41577-019-0215-7
100. Broz P, Pelegrín P, Shao F. The Gasdermins, a Protein Family Executing Cell Death and Inflammation. Nat Rev Immunol (2020) 20(3):143–57. doi: 10.1038/s41577-019-0228-2
101. Deng T, Tang C, Zhang G, Wan X. DAMPs Released by Pyroptotic Cells as Major Contributors and Therapeutic Targets for CAR-T-Related Toxicities. Cell Death Dis (2021) 12(1):129. doi: 10.1038/s41419-021-03428-x
102. Singh N, Hofmann TJ, Gershenson Z, Levine BL, Grupp SA, Teachey DT. Monocyte Lineage-Derived IL-6 Does Not Affect Chimeric Antigen Receptor T-Cell Function. Cytotherapy (2017) 19(7):867–80. doi: 10.1016/j.jcyt.2017.04.001
103. Hao Z, Li R, Meng L, Han Z, Hong Z. Macrophage, the Potential Key Mediator in CAR-T Related CRS. Exp Hematol Oncol (2020) 9:15. doi: 10.1186/s40164-020-00171-5
104. Elgueta R, Benson MJ, de Vries VC, Wasiuk A, Guo Y, Noelle RJ. Molecular Mechanism and Function of CD40/CD40L Engagement in the Immune System. Immunol Rev (2009) 229(1):152–72. doi: 10.1111/j.1600-065X.2009.00782.x
105. Aldinucci D, Poletto D, Nanni P, Degan M, Rupolo M, Pinto A, et al. CD40L Induces Proliferation, Self-Renewal, Rescue From Apoptosis, and Production of Cytokines by CD40-Expressing AML Blasts. Exp Hematol (2002) 30(11):1283–92. doi: 10.1016/s0301-472x(02)00921-9
106. Qi C, Tian S, Wang J, Ma H, Qian K, Zhang X. Co-Expression of CD40/CD40L on XG1 Multiple Myeloma Cells Promotes IL-6 Autocrine Function. Cancer Invest (2015) 33(1):6–15. doi: 10.3109/07357907.2014.988340
107. Pellat-Deceunynck C, Amiot M, Robillard N, Wijdenes J, Bataille R. CD11a-CD18 and CD102 Interactions Mediate Human Myeloma Cell Growth Arrest Induced by CD40 Stimulation. Cancer Res (1996) 56:1909–16.
108. Teoh G, Tai YT, Urashima M, Shirahama S, Matsuzaki M, Chauhan D, et al. CD40 Activation Mediates P53-Dependent Cell Cycle Regulation in Human Multiple Myeloma Cell Lines. Blood (2000) 95(3):1039–46. doi: 10.1182/blood.V95.3.1039.003k02_1039_1046
109. Tang T, Cheng X, Truong B, Sun L, Yang X, Wang H. Molecular Basis and Therapeutic Implications of CD40/CD40L Immune Checkpoint. Pharmacol Ther (2021) 219:107709. doi: 10.1016/j.pharmthera.2020.107709
110. Bullock TNJ. CD40 Stimulation as a Molecular Adjuvant for Cancer Vaccines and Other Immunotherapies. Cell Mol Immunol (2022) 19(1):14–22. doi: 10.1038/s41423-021-00734-4
111. van Mierlo GJ, den Boer AT, Medema JP, van der Voort EI, Fransen MF, Offringa R, et al. CD40 Stimulation Leads to Effective Therapy of CD40(-) Tumors Through Induction of Strong Systemic Cytotoxic T Lymphocyte Immunity. Proc Natl Acad Sci USA (2002) 99(8):5561–6. doi: 10.1073/pnas.082107699
112. Wei J, Liu Y, Wang C, Zhang Y, Tong C, Dai G, et al. The Model of Cytokine Release Syndrome in CAR T-Cell Treatment for B-Cell non-Hodgkin Lymphoma. Signal Transduct Target Ther (2020) 5(1):134. doi: 10.1038/s41392-020-00256-x
113. Gust J, Taraseviciute A, Turtle CJ. Neurotoxicity Associated With CD19-Targeted CAR-T Cell Therapies. CNS Drugs (2018) 32(12):1091–101. doi: 10.1007/s40263-018-0582-9
114. Mackall CL, Miklos DB. CNS Endothelial Cell Activation Emerges as a Driver of CAR T Cell-Associated Neurotoxicity. Cancer Discov (2017) 7(12):1371–3. doi: 10.1158/2159-8290.CD-17-1084
115. Gust J, Hay KA, Hanafi LA, Li D, Myerson D, Gonzalez-Cuyar LF, et al. Endothelial Activation and Blood-Brain Barrier Disruption in Neurotoxicity After Adoptive Immunotherapy With CD19 CAR-T Cells. Cancer Discov (2017) 7(12):1404–19. doi: 10.1158/2159-8290.CD-17-0698
116. Gust J, Ponce R, Liles WC, Garden GA, Turtle CJ. Cytokines in CAR T Cell-Associated Neurotoxicity. Front Immunol (2020) 11:577027. doi: 10.3389/fimmu.2020.577027
117. Morris EC, Neelapu SS, Giavridis T, Sadelain M. Cytokine Release Syndrome and Associated Neurotoxicity in Cancer Immunotherapy. Nat Rev Immunol (2022) 22(2):85–96. doi: 10.1038/s41577-021-00547-6
118. Deng Q, Han G, Puebla-Osorio N, Ma MCJ, Strati P, Chasen B, et al. Characteristics of anti-CD19 CAR T cell infusion products associated with efficacy and toxicity in patients with large B cell lymphomas. Nat Med (2020) 26(12):1878–87. doi: 10.1038/s41591-020-1061-7
119. Santomasso BD, Park JH, Salloum D, Riviere I, Flynn J, Mead E, et al. Clinical and Biological Correlates of Neurotoxicity Associated With CAR T-Cell Therapy in Patients With B-Cell Acute Lymphoblastic Leukemia. Cancer Discov (2018) 8(8):958–71. doi: 10.1158/2159-8290.CD-17-1319
120. Taraseviciute A, Tkachev V, Ponce R, Turtle CJ, Snyder JM, Liggitt HD, et al. Chimeric Antigen Receptor T Cell-Mediated Neurotoxicity in Nonhuman Primates. Cancer Discov (2018) 8(6):750–63. doi: 10.1158/2159-8290.CD-17-1368
121. Hu Y, Sun J, Wu Z, Yu J, Cui Q, Pu C, et al. Predominant Cerebral Cytokine Release Syndrome in CD19-Directed Chimeric Antigen Receptor-Modified T Cell Therapy. J Hematol Oncol (2016) 9(1):70. doi: 10.1186/s13045-016-0299-5
122. Gomez-Nicola D, Valle-Argos B, Nieto-Sampedro M. Blockade of IL-15 Activity Inhibits Microglial Activation Through the NFkappaB, P38, and ERK1/2 Pathways, Reducing Cytokine and Chemokine Release. Glia (2010) 58(3):264–76. doi: 10.1002/glia.20920
123. Shiomi A, Usui T. Pivotal Roles of GM-CSF in Autoimmunity and Inflammation. Mediat Inflammation (2015) 2015:568543. doi: 10.1155/2015/568543
124. Ponomarev ED, Shriver LP, Maresz K, Pedras-Vasconcelos J, Verthelyi D, Dittel BN. GM-CSF Production by Autoreactive T Cells is Required for the Activation of Microglial Cells and the Onset of Experimental Autoimmune Encephalomyelitis. J Immunol (2007) 178(1):39–48. doi: 10.4049/jimmunol.178.1.39
125. Galea I. The Blood-Brain Barrier in Systemic Infection and Inflammation. Cell Mol Immunol (2021) 18(11):2489–501. doi: 10.1038/s41423-021-00757-x
126. Liu S, Deng B, Yin Z, Pan J, Lin Y, Ling Z, et al. Corticosteroids do Not Influence the Efficacy and Kinetics of CAR-T Cells for B-Cell Acute Lymphoblastic Leukemia. Blood Cancer J (2020) 10(2):15. doi: 10.1038/s41408-020-0280-y
127. Sanchez L, Dardac A, Madduri D, Richard S, Richter J. B-Cell Maturation Antigen (BCMA) in Multiple Myeloma: The New Frontier of Targeted Therapies. Ther Adv Hematol (2021) 12:2040620721989585. doi: 10.1177/2040620721989585
128. Li P, Zhou L, Ye S, Zhang W, Wang J, Tang X, et al. Risk of HBV Reactivation in Patients With Resolved HBV Infection Receiving Anti-CD19 Chimeric Antigen Receptor T Cell Therapy Without Antiviral Prophylaxis. Front Immunol (2021) 12:638678. doi: 10.3389/fimmu.2021.638678
129. Spiegel JY, Craig J, Claire GK, Frank MJ, Muffly L, Shiraz P, et al. Immune Reconstitution and Infectious Complications Following Axicabtagene Ciloleucel Therapy for Large B-Cell Lymphoma. Blood Adv (2021) 5(1):143–55. doi: 10.1182/bloodadvances.2020002732
130. Hartmann J, Schüßler-Lenz M, Bondanza A, Buchholz CJ. Clinical Development of CAR T Cells-Challenges and Opportunities in Translating Innovative Treatment Concepts. EMBO Mol Med (2017) 9(9):1183–97. doi: 10.15252/emmm.201607485
131. Pfeiffer A, Thalheimer FB, Hartmann S, Frank AM, Bender RR, Danisch S, et al. In Vivo Generation of Human CD19-CAR T Cells Results in B-Cell Depletion and Signs of Cytokine Release Syndrome. EMBO Mol Med (2018) 10(11):e9158. doi: 10.15252/emmm.201809158
132. Wei J, Zhao J, Han M, Meng F, Zhou J. SARS-CoV-2 Infection in Immunocompromised Patients: Humoral Versus Cell-Mediated Immunity. J Immunother Cancer (2020) 8(2):e000862. doi: 10.1136/jitc-2020-000862
133. Park JH, Romero FA, Taur Y, Sadelain M, Brentjens RJ, Hohl TM, et al. Cytokine Release Syndrome Grade as a Predictive Marker for Infections in Patients With Relapsed or Refractory B-Cell Acute Lymphoblastic Leukemia Treated With Chimeric Antigen Receptor T Cells. Clin Infect Dis (2018) 67(4):533–40. doi: 10.1093/cid/ciy152
134. Hill JA, Li D, Hay KA, Green ML, Cherian S, Chen X, et al. Infectious Complications of CD19-Targeted Chimeric Antigen Receptor-Modified T-Cell Immunotherapy. Blood (2018) 131(1):121–30. doi: 10.1182/blood-2017-07-793760
135. Luo H, Wang N, Huang L, Zhou X, Jin J, Li C, et al. Inflammatory Signatures for Quick Diagnosis of Life-Threatening Infection During the CAR T-Cell Therapy. J Immunother Cancer (2019) 7(1):271. doi: 10.1186/s40425-019-0767-x
136. Han L, Zhou J, Zhou K, Zhu X, Zhao L, Fang B, et al. Safety and Efficacy of CAR-T Cell Targeting BCMA in Patients With Multiple Myeloma Coinfected With Chronic Hepatitis B Virus. J Immunother Cancer (2020) 8(2):e000927. doi: 10.1136/jitc-2020-000927
137. Wang D, Mao X, Que Y, Xu M, Cheng Y, Huang L, et al. Viral Infection/Reactivation During Long-Term Follow-Up in Multiple Myeloma Patients With Anti-BCMA CAR Therapy. Blood Cancer J (2021) 11(10):168. doi: 10.1038/s41408-021-00563-8
138. Strati P, Varma A, Adkins S, Nastoupil LJ, Westin J, Hagemeister FB, et al. Hematopoietic Recovery and Immune Reconstitution After Axicabtagene Ciloleucel in Patients With Large B-Cell Lymphoma. Haematologica (2021) 106(10):2667–72. doi: 10.3324/haematol.2020.254045
139. Yang C, Lei W, Xie H, Wu G, Wei J, Liang A, et al. Sustained Remission of Relapsed or Refractory Mantle Cell Lymphoma After 4-1bb-Based CD19-Directed CAR-T Therapy. Onco Targets Ther (2020) 13:12163–8. doi: 10.2147/OTT.S280535
140. Xu J, Chen LJ, Yang SS, Sun Y, Wu W, Liu YF, et al. Exploratory Trial of a Biepitopic CAR T-Targeting B Cell Maturation Antigen in Relapsed/Refractory Multiple Myeloma. Proc Natl Acad Sci USA (2019) 116(19):9543–51. doi: 10.1073/pnas.1819745116
141. Hill JA, Seo SK. How I Prevent Infections in Patients Receiving CD19-Targeted Chimeric Antigen Receptor T Cells for B-Cell Malignancies. Blood (2020) 136(8):925–35. doi: 10.1182/blood.2019004000
142. Neelapu SS, Locke FL, Bartlett NL, Lekakis LJ, Miklos DB, Jacobson CA, et al. Axicabtagene Ciloleucel CAR T-Cell Therapy in Refractory Large B-Cell Lymphoma. N Engl J Med (2017) 377(26):2531–44. doi: 10.1056/NEJMoa1707447
143. Raje N, Berdeja J, Lin Y, Siegel D, Jagannath S, Madduri D, et al. Anti-BCMA CAR T-Cell Therapy Bb2121 in Relapsed or Refractory Multiple Myeloma. N Engl J Med (2019) 380(18):1726–37. doi: 10.1056/NEJMoa1817226
144. Rejeski K, Perez A, Sesques P, Hoster E, Berger C, Jentzsch L, et al. CAR-HEMATOTOX: A Model for CAR T-Cell-Related Hematologic Toxicity in Relapsed/Refractory Large B-Cell Lymphoma. Blood (2021) 138(24):2499–513. doi: 10.1182/blood.2020010543
145. Juluri KR, Wu V, Voutsinas JM, Hou J, Hirayama AV, Mullane E, et al. Severe Cytokine Release Syndrome is Associated With Hematologic Toxicity Following CD19 CAR T-Cell Therapy. Blood Adv (2022) 6(7):2055–68. doi: 10.1182/bloodadvances.2020004142
146. Jain T, Knezevic A, Pennisi M, Chen Y, Ruiz JD, Purdon TJ, et al. Hematopoietic Recovery in Patients Receiving Chimeric Antigen Receptor T-Cell Therapy for Hematologic Malignancies. Blood Adv (2020) 4(15):3776–87. doi: 10.1182/bloodadvances.2020002509
147. Mavroudi I, Papadaki V, Pyrovolaki K, Katonis P, Eliopoulos AG, Papadaki HA. The CD40/CD40 Ligand Interactions Exert Pleiotropic Effects on Bone Marrow Granulopoiesis. J Leukoc Biol (2011) 89(5):771–83. doi: 10.1189/jlb.0610330
148. Dulmovits BM, Tang Y, Papoin J, He M, Li J, Yang H, et al. HMGB1-Mediated Restriction of EPO Signaling Contributes to Anemia of Inflammation. Blood (2022) 139(21): 3181–93. doi: 10.1182/blood.2021012048
149. Jelkmann W. Proinflammatory Cytokines Lowering Erythropoietin Production. J Interferon Cytokine Res (1998) 18(8):555–119. doi: 10.1089/jir.1998.18.555
150. Ganz T. Anemia of Inflammation. N Engl J Med (2019) 381(12):1148–57. doi: 10.1056/NEJMra1804281
151. Schubert ML, Dietrich S, Stilgenbauer S, Schmitt A, Pavel P, Kunz A, et al. Feasibility and Safety of CD19 Chimeric Antigen Receptor T Cell Treatment for B Cell Lymphoma Relapse After Allogeneic Hematopoietic Stem Cell Transplantation. Biol Blood Marrow Transplant (2020) 26(9):1575–80. doi: 10.1016/j.bbmt.2020.04.025
152. Fried S, Avigdor A, Bielorai B, Meir A, Besser MJ, Schachter J, et al. Early and Late Hematologic Toxicity Following CD19 CAR-T Cells. Bone Marrow Transplant (2019) 54(10):1643–50. doi: 10.1038/s41409-019-0487-3
153. Jain MD, Davila ML. Concise Review: Emerging Principles From the Clinical Application of Chimeric Antigen Receptor T Cell Therapies for B Cell Malignancies. Stem Cells (2018) 36(1):36–44. doi: 10.1002/stem.2715
154. Baur R, Jitschin R, Kharboutli S, Stoll A, Völkl S, Büttner-Herold M, et al. Thrombopoietin Receptor Agonists for Acquired Thrombocytopenia Following Anti-CD19 CAR-T-Cell Therapy: A Case Report. J Immunother Cancer (2021) 9(7):e002721. doi: 10.1136/jitc-2021-002721
155. Xing L, Wang Y, Liu H, Gao S, Shao Q, Yue L, et al. Case Report: Sirolimus Alleviates Persistent Cytopenia After CD19 CAR-T-Cell Therapy. Front Oncol (2021) 11:798352. doi: 10.3389/fonc.2021.798352
156. Wang Y, Qi K, Cheng H, Cao J, Shi M, Qiao J, et al. Coagulation Disorders After Chimeric Antigen Receptor T Cell Therapy: Analysis of 100 Patients With Relapsed and Refractory Hematologic Malignancies. Biol Blood Marrow Transplant (2020) 26(5):865–75. doi: 10.1016/j.bbmt.2019.11.027
157. Johnsrud A, Craig J, Baird J, Spiegel J, Muffly L, Zehnder J, et al. Incidence and Risk Factors Associated With Bleeding and Thrombosis Following Chimeric Antigen Receptor T-Cell Therapy. Blood Adv (2021) 5(21):4465–75. doi: 10.1182/bloodadvances.2021004716
158. Shao M, Yu Q, Teng X, Guo X, Wei G, Xu H, et al. CRS-Related Coagulopathy in BCMA Targeted CAR-T Therapy: A Retrospective Analysis in a Phase I/II Clinical Trial. Bone Marrow Transplant (2021) 56(7):1642–50. doi: 10.1038/s41409-021-01226-9
159. Danese S, de la Motte C, Reyes BM, Sans M, Levine AD, Fiocchi C. Cutting Edge: T Cells Trigger CD40-Dependent Platelet Activation and Granular RANTES Release: A Novel Pathway for Immune Response Amplification. J Immunol (2004) 172:2011–5. doi: 10.4049/jimmunol.172.4.2011
160. Lindmark E, Tenno T, Siegbahn A. Role of Platelet P-Selectin and CD40 Ligand in the Induction of Monocytic Tissue Factor Expression. Arterioscler Thromb Vasc Biol (2000) 20(10):2322–8. doi: 10.1161/01.atv.20.10.2322
161. Hottz ED, Azevedo-Quintanilha IG, Palhinha L, Teixeira L, Barreto EA, Pão CRR, et al. Platelet Activation and Platelet-Monocyte Aggregate Formation Trigger Tissue Factor Expression in Patients With Severe COVID-19. Blood (2020) 136(11):1330–41. doi: 10.1182/blood.2020007252
162. Henn V, Slupsky JR, Gräfe M, Anagnostopoulos I, Förster R, Müller-Berghaus G, et al. CD40 Ligand on Activated Platelets Triggers an Inflammatory Reaction of Endothelial Cells. Nature (1998) 391(6667):591–4. doi: 10.1038/35393
163. Cermak J, Key NS, Bach RR, Balla J, Jacob HS, Vercellotti GM. C-Reactive Protein Induces Human Peripheral Blood Monocytes to Synthesize Tissue Factor. Blood (1993) 82(2):513–20. doi: 10.1182/blood.V82.2.513.513
164. Khan MM, Liu Y, Khan ME, Gilman ML, Khan ST, Bromberg M, et al. Upregulation of Tissue Factor in Monocytes by Cleaved High Molecular Weight Kininogen is Dependent on TNF-Alpha and IL-1beta. Am J Physiol Heart Circ Physiol (2010) 298(2):H652–8. doi: 10.1152/ajpheart.00825.2009
165. Mach F, Schönbeck U, Bonnefoy JY, Pober JS, Libby P. Activation of Monocyte/Macrophage Functions Related to Acute Atheroma Complication by Ligation of CD40: Induction of Collagenase, Stromelysin, and Tissue Factor. Circulation (1997) 96(2):396–9. doi: 10.1161/01.cir.96.2.396
166. Bernardo A, Ball C, Nolasco L, Moake JF, Dong JF. Effects of Inflammatory Cytokines on the Release and Cleavage of the Endothelial Cell-Derived Ultralarge Von Willebrand Factor Multimers Under Flow. Blood (2004) 104(1):100–6. doi: 10.1182/blood-2004-01-0107
167. Cao WJ, Niiya M, Zheng XW, Shang DZ, Zheng XL. Inflammatory Cytokines Inhibit ADAMTS13 Synthesis in Hepatic Stellate Cells and Endothelial Cells. J Thromb Haemost (2008) 6(7):1233–5. doi: 10.1111/j.1538-7836.2008.02989.x
168. Ikezoe T. Advances in the Diagnosis and Treatment of Disseminated Intravascular Coagulation in Haematological Malignancies. Int J Hematol (2021) 113(1):34–44. doi: 10.1007/s12185-020-02992-w
169. Guo Z, Tu S, Yu S, Wu L, Pan W, Chang N, et al. Preclinical and Clinical Advances in Dual-Target Chimeric Antigen Receptor Therapy for Hematological Malignancies. Cancer Sci (2021) 112(4):1357–68. doi: 10.1111/cas.14799
170. Neelapu SS, Ghobadi A, Jacobson CA, Miklos DB, Lekakis LJ, Oluwole O, et al. 2-Year Follow-Up and High-Risk Subset Analysis of Zuma-1, the Pivotal Study of Axicabtagene Ciloleucel (Axi-Cel) in Patients With Refractory Large B Cell Lymphoma. Biol Blood Marrow Tr (2019) 25(3):S65. doi: 10.1016/j.bbmt.2018.12.148
171. Orlando EJ, Han X, Tribouley C, Wood PA, Leary RJ, Riester M, et al. Genetic Mechanisms of Target Antigen Loss in CAR19 Therapy of Acute Lymphoblastic Leukemia. Nat Med (2018) 24(10):1504–6. doi: 10.1038/s41591-018-0146-z
172. Jacoby E, Nguyen SM, Fountaine TJ, Welp K, Gryder B, Qin H, et al. CD19 CAR Immune Pressure Induces B-Precursor Acute Lymphoblastic Leukaemia Lineage Switch Exposing Inherent Leukaemic Plasticity. Nat Commun (2016) 7:12320. doi: 10.1038/ncomms12320
173. Samur MK, Fulciniti M, Aktas Samur A, Bazarbachi AH, Tai YT, Prabhala R, et al. Biallelic Loss of BCMA as a Resistance Mechanism to CAR T Cell Therapy in a Patient With Multiple Myeloma. Nat Commun (2021) 12(1):868. doi: 10.1038/s41467-021-21177-5
174. Hamieh M, Dobrin A, Cabriolu A, van der Stegen SJC, Giavridis T, Mansilla-Soto J, et al. CAR T Cell Trogocytosis and Cooperative Killing Regulate Tumour Antigen Escape. Nature (2019) 568(7750):112–6. doi: 10.1038/s41586-019-1054-1
175. Ruella M, Xu J, Barrett DM, Fraietta JA, Reich TJ, Ambrose DE, et al. Induction of Resistance to Chimeric Antigen Receptor T Cell Therapy by Transduction of a Single Leukemic B Cell. Nat Med (2018) 24(10):1499–503. doi: 10.1038/s41591-018-0201-9
176. Zanetti SR, Velasco-Hernandez T, Gutierrez-Agüera F, Díaz VM, Romecín PA, Roca-Ho H, et al. A Novel and Efficient Tandem CD19- and CD22-Directed CAR for B Cell ALL. Mol Ther (2022) 30(2):550–63. doi: 10.1016/j.ymthe.2021.08.033
177. Tong C, Zhang Y, Liu Y, Ji X, Zhang W, Guo Y, et al. Optimized Tandem CD19/CD20 CAR-Engineered T Cells in Refractory/Relapsed B-Cell Lymphoma. Blood (2020) 136(14):1632–44. doi: 10.1182/blood.2020005278
178. Shah NN, Johnson BD, Schneider D, Zhu F, Szabo A, Keever-Taylor CA, et al. Bispecific Anti-CD20, Anti-CD19 CAR T Cells for Relapsed B Cell Malignancies: A Phase 1 Dose Escalation and Expansion Trial. Nat Med (2020) 26(10):1569–75. doi: 10.1038/s41591-020-1081-3
179. Wei G, Zhang Y, Zhao H, Wang Y, Liu Y, Liang B, et al. CD19/CD22 Dual-Targeted CAR T-Cell Therapy for Relapsed/Refractory Aggressive B-Cell Lymphoma: A Safety and Efficacy Study. Cancer Immunol Res (2021) 9(9):1061–70. doi: 10.1158/2326-6066.CIR-20-0675
180. Dai H, Wu Z, Jia H, Tong C, Guo Y, Ti D, et al. Bispecific CAR-T Cells Targeting Both CD19 and CD22 for Therapy of Adults With Relapsed or Refractory B Cell Acute Lymphoblastic Leukemia. J Hematol Oncol (2020) 13(1):30. doi: 10.1186/s13045-020-00856-8
181. Schneider D, Xiong Y, Wu D, Hu P, Alabanza L, Steimle B, et al. Trispecific CD19-CD20-CD22-Targeting duoCAR-T Cells Eliminate Antigen-Heterogeneous B Cell Tumors in Preclinical Models. Sci Transl Med (2021) 13(586):eabc6401. doi: 10.1126/scitranslmed.abc6401
182. Wong DP, Roy NK, Zhang K, Anukanth A, Asthana A, Shirkey-Son NJ, et al. A BAFF Ligand-Based CAR-T Cell Targeting Three Receptors and Multiple B Cell Cancers. Nat Commun (2022) 13(1):217. doi: 10.1038/s41467-021-27853-w
183. Pont MJ, Hill T, Cole GO, Abbott JJ, Kelliher J, Salter AI, et al. Gamma-Secretase Inhibition Increases Efficacy of BCMA-Specific Chimeric Antigen Receptor T Cells in Multiple Myeloma. Blood (2019) 134(19):1585–97. doi: 10.1182/blood.2019000050
184. Correia DV, Lopes A, Silva-Santos B. Tumor Cell Recognition by γδ T Lymphocytes: T-Cell Receptor vs. NK-Cell Receptors. Oncoimmunology (2013) 2(1):e22892. doi: 10.4161/onci.22892
185. Lança T, Correia DV, Moita CF, Raquel H, Neves-Costa A, Ferreira C, et al. The MHC Class Ib Protein ULBP1 is a Nonredundant Determinant of Leukemia/Lymphoma Susceptibility to Gammadelta T-Cell Cytotoxicity. Blood (2010) 115(12):2407–11. doi: 10.1182/blood-2009-08-237123
186. Pistoia V, Tumino N, Vacca P, Veneziani I, Moretta A, Locatelli F, et al. Human γδ T-Cells: From Surface Receptors to the Therapy of High-Risk Leukemias. Front Immunol (2018) 9:984. doi: 10.3389/fimmu.2018.00984
187. Rozenbaum M, Meir A, Aharony Y, Itzhaki O, Schachter J, Bank I, et al. Gamma-Delta CAR-T Cells Show CAR-Directed and Independent Activity Against Leukemia. Front Immunol (2020) 11:1347. doi: 10.3389/fimmu.2020.01347
188. Airoldi I, Bertaina A, Prigione I, Zorzoli A, Pagliara D, Cocco C, et al. γδ T-Cell Reconstitution After HLA-Haploidentical Hematopoietic Transplantation Depleted of TCR-αβ+/CD19+ Lymphocytes. Blood (2015) 125(15):2349–58. doi: 10.1182/blood-2014-09-599423
189. Handgretinger R, Schilbach K. The Potential Role of γδ T Cells After Allogeneic HCT for Leukemia. Blood (2018) 131(10):1063–72. doi: 10.1182/blood-2017-08-752162
190. McLellan AD, Ali Hosseini Rad SM. Chimeric Antigen Receptor T Cell Persistence and Memory Cell Formation. Immunol Cell Biol (2019) 97(7):664–74. doi: 10.1111/imcb.12254
191. Long AH, Haso WM, Shern JF, Wanhainen KM, Murgai M, Ingaramo M, et al. 4-1BB Costimulation Ameliorates T Cell Exhaustion Induced by Tonic Signaling of Chimeric Antigen Receptors. Nat Med (2015) 21(6):581–90. doi: 10.1038/nm.3838
192. Li G, Boucher JC, Kotani H, Park K, Zhang Y, Shrestha B, et al. 4-1BB Enhancement of CAR T Function Requires NF-κb and TRAFs. JCI Insight (2018) 3(18):e121322. doi: 10.1172/jci.insight.121322
193. Zhao Z, Condomines M, van der Stegen SJC, Perna F, Kloss CC, Gunset G, et al. Structural Design of Engineered Costimulation Determines Tumor Rejection Kinetics and Persistence of CAR T Cells. Cancer Cell (2015) 28(4):415–28. doi: 10.1016/j.ccell.2015.09.004
194. Roselli E, Boucher JC, Li G, Kotani H, Spitler K, Reid K, et al. 4-1BB and Optimized CD28 Co-Stimulation Enhances Function of Human Mono-Specific and Bi-Specific Third-Generation CAR T Cells. J Immunother Cancer (2021) 9(10):e003354. doi: 10.1136/jitc-2021-003354
195. Weinkove R, George P, Dasyam N, McLellan AD. Selecting Costimulatory Domains for Chimeric Antigen Receptors: Functional and Clinical Considerations. Clin Transl Immunol (2019) 8(5):e1049. doi: 10.1002/cti2.1049
196. Ramos CA, Rouce R, Robertson CS, Reyna A, Narala N, Vyas G, et al. In Vivo Fate and Activity of Second- Versus Third-Generation CD19-Specific CAR-T Cells in B Cell Non-Hodgkin's Lymphomas. Mol Ther (2018) 26(12):2727–37. doi: 10.1016/j.ymthe.2018.09.009
197. Myers RM, Li Y, Barz Leahy A, Barrett DM, Teachey DT, Callahan C, et al. Humanized CD19-Targeted Chimeric Antigen Receptor (CAR) T Cells in CAR-Naive and CAR-Exposed Children and Young Adults With Relapsed or Refractory Acute Lymphoblastic Leukemia. J Clin Oncol (2021) 39(27):3044–55. doi: 10.1200/JCO.20.03458
198. Taniuchi I. CD4 Helper and CD8 Cytotoxic T Cell Differentiation. Annu Rev Immunol (2018) 36:579–601. doi: 10.1146/annurev-immunol-042617-053411
199. Agarwal S, Hanauer JDS, Frank AM, Riechert V, Thalheimer FB, Buchholz CJ. In Vivo Generation of CAR T Cells Selectively in Human CD4+ Lymphocytes. Mol Ther (2020) 28(8):1783–94. doi: 10.1016/j.ymthe.2020.05.005
200. Turtle CJ, Hanafi LA, Berger C, Gooley TA, Cherian S, Hudecek M, et al. CD19 CAR-T Cells of Defined CD4+:CD8+ Composition in Adult B Cell ALL Patients. J Clin Invest (2016) 126(6):2123–38. doi: 10.1172/JCI85309
201. Liadi I, Singh H, Romain G, Rey-Villamizar N, Merouane A, Adolacion JR, et al. Individual Motile CD4(+) T Cells Can Participate in Efficient Multikilling Through Conjugation to Multiple Tumor Cells. Cancer Immunol Res (2015) 3(5):473–82. doi: 10.1158/2326-6066.CIR-14-0195
202. Melenhorst JJ, Chen GM, Wang M, Porter DL, Chen C, Collins MA, et al. Decade-Long Leukaemia Remissions With Persistence of CD4+ CAR T Cells. Nature (2022) 602(7897):503–9. doi: 10.1038/s41586-021-04390-6
203. Diaconu I, Ballard B, Zhang M, Chen Y, West J, Dotti G, et al. Inducible Caspase-9 Selectively Modulates the Toxicities of CD19-Specific Chimeric Antigen Receptor-Modified T Cells. Mol Ther (2017) 25(3):580–92. doi: 10.1016/j.ymthe.2017.01.011
204. Wu CY, Roybal KT, Puchner EM, Onuffer J, Lim WA. Remote Control of Therapeutic T Cells Through a Small Molecule-Gated Chimeric Receptor. Science (2015) 350(6258):aab4077. doi: 10.1126/science.aab4077
205. Aspuria PJ, Vivona S, Bauer M, Semana M, Ratti N, McCauley S, et al. An Orthogonal IL-2 and IL-2rβ System Drives Persistence and Activation of CAR T Cells and Clearance of Bulky Lymphoma. Sci Transl Med (2021) 13(625):eabg7565. doi: 10.1126/scitranslmed.abg7565
206. Zhang Q, Hresko ME, Picton LK, Su L, Hollander MJ, Nunez-Cruz S, et al. A Human Orthogonal IL-2 and IL-2rβ System Enhances CAR T Cell Expansion and Antitumor Activity in a Murine Model of Leukemia. Sci Transl Med (2021) 13(625):eabg6986. doi: 10.1126/scitranslmed.abg6986
207. Jan M, Scarfò I, Larson RC, Walker A, Schmidts A, Guirguis AA, et al. Reversible ON- and OFF-Switch Chimeric Antigen Receptors Controlled by Lenalidomide. Sci Transl Med (2021) 13(575):eabb6295. doi: 10.1126/scitranslmed.abb6295
208. Park S, Pascua E, Lindquist KC, Kimberlin C, Deng X, Mak YSL, et al. Direct Control of CAR T Cells Through Small Molecule-Regulated Antibodies. Nat Commun (2021) 12(1):710. doi: 10.1038/s41467-020-20671-6
209. Paszkiewicz PJ, Fräßle SP, Srivastava S, Sommermeyer D, Hudecek M, Drexler I, et al. Targeted Antibody-Mediated Depletion of Murine CD19 CAR T Cells Permanently Reverses B Cell Aplasia. J Clin Invest (2016) 126(11):4262–72. doi: 10.1172/JCI84813
210. Cieri N, Camisa B, Cocchiarella F, Forcato M, Oliveira G, Provasi E, et al. IL-7 and IL-15 Instruct the Generation of Human Memory Stem T Cells From Naive Precursors. Blood (2013) 121(4):573–84. doi: 10.1182/blood-2012-05-431718
211. Avanzi MP, Yeku O, Li X, Wijewarnasuriya DP, van Leeuwen DG, Cheung K, et al. Engineered Tumor-Targeted T Cells Mediate Enhanced Anti-Tumor Efficacy Both Directly and Through Activation of the Endogenous Immune System. Cell Rep (2018) 23(7):2130–41. doi: 10.1016/j.celrep.2018.04.051
212. Ma A, Koka R, Burkett P. Diverse Functions of IL-2, IL-15, and IL-7 in Lymphoid Homeostasis. Annu Rev Immunol (2006) 24:657–79. doi: 10.1146/annurev.immunol.24.021605.090727
213. Shum T, Omer B, Tashiro H, Kruse RL, Wagner DL, Parikh K, et al. Constitutive Signaling From an Engineered IL7 Receptor Promotes Durable Tumor Elimination by Tumor-Redirected T Cells. Cancer Discov (2017) 7(11):1238–47. doi: 10.1158/2159-8290.CD-17-0538
214. Cui W, Liu Y, Weinstein JS, Craft J, Kaech SM. An Interleukin-21-Interleukin-10-STAT3 Pathway is Critical for Functional Maturation of Memory CD8+ T Cells. Immunity (2011) 35(5):792–805. doi: 10.1016/j.immuni.2011.09.017
215. Battram AM, Bachiller M, Lopez V, Fernández de Larrea C, Urbano-Ispizua A, Martín-Antonio B. IL-15 Enhances the Persistence and Function of BCMA-Targeting CAR-T Cells Compared to IL-2 or IL-15/IL-7 by Limiting CAR-T Cell Dysfunction and Differentiation. Cancers (Basel) (2021) 13(14):3534. doi: 10.3390/cancers13143534
216. Zhou J, Jin L, Wang F, Zhang Y, Liu B, Zhao T. Chimeric Antigen Receptor T (CAR-T) Cells Expanded With IL-7/IL-15 Mediate Superior Antitumor Effects. Protein Cell (2019) 10(10):764–9. doi: 10.1007/s13238-019-0643-y
217. Alizadeh D, Wong RA, Yang X, Wang D, Pecoraro JR, Kuo CF, et al. IL15 Enhances CAR-T Cell Antitumor Activity by Reducing Mtorc1 Activity and Preserving Their Stem Cell Memory Phenotype. Cancer Immunol Res (2019) 7(5):759–72. doi: 10.1158/2326-6066.CIR-18-0466
218. Loschinski R, Böttcher M, Stoll A, Bruns H, Mackensen A, Mougiakakos D. IL-21 Modulates Memory and Exhaustion Phenotype of T-Cells in a Fatty Acid Oxidation-Dependent Manner. Oncotarget (2018) 9(17):13125–38. doi: 10.18632/oncotarget.24442
219. Curran KJ, Seinstra BA, Nikhamin Y, Yeh R, Usachenko Y, van Leeuwen DG, et al. Enhancing Antitumor Efficacy of Chimeric Antigen Receptor T Cells Through Constitutive CD40L Expression. Mol Ther (2015) 23(4):769–78. doi: 10.1038/mt.2015.4
220. Kuhn NF, Purdon TJ, van Leeuwen DG, Lopez AV, Curran KJ, Daniyan AF, et al. CD40 Ligand-Modified Chimeric Antigen Receptor T Cells Enhance Antitumor Function by Eliciting an Endogenous Antitumor Response. Cancer Cell (2019) 35(3):473–488.e6. doi: 10.1016/j.ccell.2019.02.006
221. Hoyos V, Savoldo B, Quintarelli C, Mahendravada A, Zhang M, Vera J, et al. Engineering CD19-Specific T Lymphocytes With Interleukin-15 and a Suicide Gene to Enhance Their Anti-Lymphoma/Leukemia Effects and Safety. Leukemia (2010) 24(6):1160–70. doi: 10.1038/leu.2010.75
222. Štach M, Ptáčková P, Mucha M, Musil J, Klener P, Otáhal P. Inducible Secretion of IL-21 Augments Anti-Tumor Activity of Piggybac-Manufactured Chimeric Antigen Receptor T Cells. Cytotherapy (2020) 22(12):744–54. doi: 10.1016/j.jcyt.2020.08.005
223. Azmi AS, Uddin MH, Mohammad RM. The Nuclear Export Protein XPO1-From Biology to Targeted Therapy. Nat Rev Clin Oncol (2021) 18(3):152–69. doi: 10.1038/s41571-020-00442-4
224. Syed YY. Selinexor: First Global Approval. Drugs (2019) 79(13):1485–94. doi: 10.1007/s40265-019-01188-9
225. Wang S, Sellner L, Wang L, Sauer T, Neuber B, Gong W, et al. Combining Selective Inhibitors of Nuclear Export (SINEs) With Chimeric Antigen Receptor (CAR) T Cells for CD19-positive Malignancies. Oncol Rep (2021) 46(2):170. doi: 10.3892/or.2021.8121
226. Works M, Soni N, Hauskins C, Sierra C, Baturevych A, Jones JC, et al. Anti-B-Cell Maturation Antigen Chimeric Antigen Receptor T Cell Function Against Multiple Myeloma Is Enhanced in the Presence of Lenalidomide. Mol Cancer Ther (2019) 1818(12):2246–57. doi: 10.1158/1535-7163.MCT-18-1146
227. Parrondo RD, Sam K, Rasheed A, Alegria V, Sher T, Roy V, et al. Subsequent Anti-Myeloma Therapy After Idecabtagene Vicleucel Treatment in Patients With Relapsed/Refractory Multiple Myeloma: A Single Center Analysis. Blood Cancer J (2022) 12(4):66. doi: 10.1038/s41408-022-00662-0
228. Chari A, Vogl DT, Jagannath S, Jasielec J, Unger TJ, DeCastro A, et al. Selinexor-Based Regimens for the Treatment of Myeloma Refractory to Chimeric Antigen Receptor T Cell Therapy. Br J Haematol (2020) 189(4):e126–30. doi: 10.1111/bjh.16550
229. Zhao G, Wei R, Feng L, Wu Y, He F, Xiao M, et al. Lenalidomide Enhances the Efficacy of Anti-BCMA CAR-T Treatment in Relapsed/Refractory Multiple Myeloma: A Case Report and Revies of the Literature. Cancer Immunol Immunother (2022) 71(1):39–44. doi: 10.1007/s00262-021-02959-8
230. Gauthier J, Hirayama AV, Purushe J, Hay KA, Lymp J, Li DH, et al. Feasibility and Efficacy of CD19-Targeted CAR T Cells With Concurrent Ibrutinib for CLL After Ibrutinib Failure. Blood (2020) 135(19):1650–60. doi: 10.1182/blood.2019002936
231. Liu M, Deng H, Mu J, Li Q, Pu Y, Jiang Y, et al. Ibrutinib Improves the Efficacy of Anti-CD19-CAR T-Cell Therapy in Patients With Refractory non-Hodgkin Lymphoma. Cancer Sci (2021) 112(7):2642–51. doi: 10.1111/cas.14915
232. Liu M, Wang X, Li Z, Zhang R, Mu J, Jiang Y, et al. Synergistic Effect of Ibrutinib and CD19 CAR-T Cells on Raji Cells In Vivo and In Vitro. Cancer Sci (2020) 111(11):4051–60. doi: 10.1111/cas.14638
233. Gill SI, Frey NV, Hexner E, Metzger S, O'Brien M, Hwang WT, et al. Anti-CD19 CAR T Cells in Combination With Ibrutinib for the Treatment of Chronic Lymphocytic Leukemia. Blood Adv (2022). doi: bloodadvances.2022007317 doi: 10.1182/bloodadvances.2022007317
234. You L, Han Q, Zhu L, Zhu Y, Bao C, Yang C, et al. Decitabine-Mediated Epigenetic Reprograming Enhances Anti-Leukemia Efficacy of CD123-Targeted Chimeric Antigen Receptor T-Cells. Front Immunol (2020) 11:1787. doi: 10.3389/fimmu.2020.01787
235. El Khawanky N, Hughes A, Yu W, Myburgh R, Matschulla T, Taromi S, et al. Demethylating Therapy Increases Anti-CD123 CAR T Cell Cytotoxicity Against Acute Myeloid Leukemia. Nat Commun (2021) 12(1):6436. doi: 10.1038/s41467-021-26683-0
236. Li S, Xue L, Wang M, Qiang P, Xu H, Zhang X, et al. Decitabine Enhances Cytotoxic Effect of T Cells With an Anti-CD19 Chimeric Antigen Receptor in Treatment of Lymphoma. Onco Targets Ther (2019) 12:5627–38. doi: 10.2147/OTT.S198567
237. Holthof LC, van der Schans JJ, Katsarou A, Poels R, Gelderloos AT, Drent E, et al. Bone Marrow Mesenchymal Stromal Cells Can Render Multiple Myeloma Cells Resistant to Cytotoxic Machinery of CAR T Cells Through Inhibition of Apoptosis. Clin Cancer Res (2021) 27(13):3793–803. doi: 10.1158/1078-0432.CCR-20-2188
238. Pinnix CC, Gunther JR, Dabaja BS, Strati P, Fang P, Hawkins MC, et al. Bridging Therapy Prior to Axicabtagene Ciloleucel for Relapsed/Refractory Large B-Cell Lymphoma. Blood Adv (2020) 4(13):2871–83. doi: 10.1182/bloodadvances.2020001837
239. Wright CM, LaRiviere MJ, Baron JA, Uche C, Xiao Y, Arscott WT, et al. Bridging Radiation Therapy Before Commercial Chimeric Antigen Receptor T-Cell Therapy for Relapsed or Refractory Aggressive B-Cell Lymphoma. Int J Radiat Oncol Biol Phys (2020) 108(1):178–88. doi: 10.1016/j.ijrobp.2020.05.014
240. Sim AJ, Jain MD, Figura NB, Chavez JC, Shah BD, Khimani F, et al. Radiation Therapy as a Bridging Strategy for CAR T Cell Therapy With Axicabtagene Ciloleucel in Diffuse Large B-Cell Lymphoma. Int J Radiat Oncol Biol Phys (2019) 105(5):1012–21. doi: 10.1016/j.ijrobp.2019.05.065
241. Smith EL, Mailankody S, Staehr M, Wang X, Senechal B, Purdon TJ, et al. BCMA-Targeted CAR T-Cell Therapy Plus Radiotherapy for the Treatment of Refractory Myeloma Reveals Potential Synergy. Cancer Immunol Res (2019) 7(7):1047–53. doi: 10.1158/2326-6066.CIR-18-0551
242. DeSelm C, Palomba ML, Yahalom J, Hamieh M, Eyquem J, Rajasekhar VK, et al. Low-Dose Radiation Conditioning Enables CAR T Cells to Mitigate Antigen Escape. Mol Ther (2018) 26(11):2542–52. doi: 10.1016/j.ymthe.2018.09.008
243. Pocaterra A, Catucci M, Mondino A. Adoptive T Cell Therapy of Solid Tumors: Time to Team Up With Immunogenic Chemo/Radiotherapy. Curr Opin Immunol (2021) 74:53–9. doi: 10.1016/j.coi.2021.10.004
244. Spiotto M, Fu YX, Weichselbaum RR. The Intersection of Radiotherapy and Immunotherapy: Mechanisms and Clinical Implications. Sci Immunol (2016) 1(3):EAAG1266. doi: 10.1126/sciimmunol.aag1266
245. Sridharan V, Margalit DN, Lynch SA, Severgnini M, Zhou J, Chau NG, et al. Definitive Chemoradiation Alters the Immunologic Landscape and Immune Checkpoints in Head and Neck Cancer. Br J Cancer (2016) 115(2):252–60. doi: 10.1038/bjc.2016.166
246. Asimakopoulos F, Hope C, Johnson MG, Pagenkopf A, Gromek K, Nagel B. Extracellular Matrix and the Myeloid-in-Myeloma Compartment: Balancing Tolerogenic and Immunogenic Inflammation in the Myeloma Niche. J Leukoc Biol (2017) 102(2):265–75. doi: 10.1189/jlb.3MR1116-468R
247. Pimenta DB, Varela VA, Datoguia TS, Caraciolo VB, Lopes GH, Pereira WO. The Bone Marrow Microenvironment Mechanisms in Acute Myeloid Leukemia. Front Cell Dev Biol (2021) 9:764698. doi: 10.3389/fcell.2021.764698
248. Epperly R, Gottschalk S, Velasquez MP. A Bump in the Road: How the Hostile AML Microenvironment Affects CAR T Cell Therapy. Front Oncol (2020) 10:262. doi: 10.3389/fonc.2020.00262
249. Holthof LC, Mutis T. Challenges for Immunotherapy in Multiple Myeloma: Bone Marrow Microenvironment-Mediated Immune Suppression and Immune Resistance. Cancers (Basel) (2020) 12(4):988. doi: 10.3390/cancers12040988
250. Autio M, Leivonen SK, Brück O, Karjalainen-Lindsberg ML, Pellinen T, Leppä S. Clinical Impact of Immune Cells and Their Spatial Interactions in Diffuse Large B-Cell Lymphoma Microenvironment. Clin Cancer Res (2022) 28(4):781–92. doi: 10.1158/1078-0432.CCR-21-3140
251. Kline J, Godfrey J, Ansell SM. The Immune Landscape and Response to Immune Checkpoint Blockade Therapy in Lymphoma. Blood (2020) 135(8):523–33. doi: 10.1182/blood.2019000847
252. Crespo J, Sun H, Welling TH, Tian Z, Zhou W. T Cell Anergy, Exhaustion, Senescence, and Stemness in the Tumor Microenvironment. Curr Opin Immunol (2013) 25:214–21. doi: 10.1016/j.coi.2012.12.003
253. Dhakal B, Hari PN, Usmani SZ, Hamadani M. Chimeric Antigen Receptor T Cell Therapy in Multiple Myeloma: Promise and Challenges. Bone Marrow Transplant (2021) 56(1):9–19. doi: 10.1038/s41409-020-01023-w
254. Hatic H, Sampat D, Goyal G. Immune Checkpoint Inhibitors in Lymphoma: Challenges and Opportunities. Ann Transl Med (2021) 9(12):1037. doi: 10.1038/s41409-020-01023-w
255. Song MK, Park BB, Uhm J. Understanding Immune Evasion and Therapeutic Targeting Associated With PD-1/PD-L1 Pathway in Diffuse Large B-Cell Lymphoma. Int J Mol Sci (2019) 20(6):1326. doi: 10.3390/ijms20061326
256. Cao Y, Lu W, Sun R, Jin X, Cheng L, He X, et al. Anti-CD19 Chimeric Antigen Receptor T Cells in Combination With Nivolumab are Safe and Effective Against Relapsed/Refractory B-Cell non-Hodgkin Lymphoma. Front Oncol (2019) 9:767. doi: 10.3389/fonc.2019.00767
257. Wang J, Deng Q, Jiang YY, Zhang R, Zhu HB, Meng JX, et al. CAR-T 19 Combined With Reduced-Dose PD-1 Blockade Therapy for Treatment of Refractory Follicular Lymphoma: A Case Report. Oncol Lett (2019) 18(5):4415–20. doi: 10.3892/ol.2019.10783
258. Song W, Zhang M. Use of CAR-T Cell Therapy, PD-1 Blockade, and Their Combination for the Treatment of Hematological Malignancies. Clin Immunol (2020) 214:108382. doi: 10.1016/j.clim.2020.108382
259. John LB, Kershaw MH, Darcy PK. Blockade of PD-1 Immunosuppression Boosts CAR T-Cell Therapy. Oncoimmunology (2013) 2(10):e26286. doi: 10.4161/onci.26286
260. Zhang R, Deng Q, Jiang YY, Zhu HB, Wang J, Zhao MF. Effect and Changes in PD-1 Expression of CD19 CAR-T Cells From T Cells Highly Expressing PD-1 Combined With Reduced-Dose PD-1 Inhibitor. Oncol Rep (2019) 41(6):3455–63. doi: 10.3892/or.2019.7096
261. Rafiq S, Yeku OO, Jackson HJ, Purdon TJ, van Leeuwen DG, Drakes DJ, et al. Targeted Delivery of a PD-1-Blocking scFv by CAR-T Cells Enhances Anti-Tumor Efficacy In Vivo. Nat Biotechnol (2018) 36(9):847–56. doi: 10.1038/nbt.4195
262. Kenderian SS, Ruella M, Shestova O, Klichinsky M, Kim M, Porter DL, et al. Identification of PD1 and TIM3 As Checkpoints That Limit Chimeric Antigen Receptor T Cell Efficacy in Leukemia. Biol Blood Marrow Transplant (2016) 22(3):S19–21. doi: 10.1016/j.bbmt.2015.11.291
263. Wenthe J, Naseri S, Labani-Motlagh A, Enblad G, Wikström KI, Eriksson E, et al. Boosting CAR T-Cell Responses in Lymphoma by Simultaneous Targeting of CD40/4-1BB Using Oncolytic Viral Gene Therapy. Cancer Immunol Immunother (2021) 70(10):2851–65. doi: 10.1007/s00262-021-02895-7
264. Eriksson E, Milenova I, Wenthe J, Ståhle M, Leja-Jarblad J, Ullenhag G, et al. Shaping the Tumor Stroma and Sparking Immune Activation by CD40 and 4-1BB Signaling Induced by an Armed Oncolytic Virus. Clin Cancer Res (2017) 23(19):5846–57. doi: 10.1158/1078-0432.CCR-17-0285
265. Jiang H, Fueyo J. Healing After Death: Antitumor Immunity Induced by Oncolytic Adenoviral Therapy. Oncoimmunology (2014) 3(7):e947872. doi: 10.4161/21624011
266. Workenhe ST, Mossman KL. Rewiring Cancer Cell Death to Enhance Oncolytic Viro-Immunotherapy. Oncoimmunology (2013) 2(12):e27138. doi: 10.4161/onci.27138
267. Wenthe J, Naseri S, Hellström AC, Wiklund HJ, Eriksson E, Loskog A. Immunostimulatory Oncolytic Virotherapy for Multiple Myeloma Targeting 4-1BB and/or CD40. Cancer Gene Ther (2020) 27(12):948–59. doi: 10.1038/s41417-020-0176-9
268. Alabanza LM, Xiong Y, Vu B, Webster B, Wu D, Hu P, et al. Armored BCMA CAR T Cells Eliminate Multiple Myeloma and Are Resistant to the Suppressive Effects of TGF-β. Front Immunol (2022) 13:832645. doi: 10.3389/fimmu.2022.832645
269. Yang Q, Hao J, Chi M, Wang Y, Xin B, Huang J, et al. Superior Antitumor Immunotherapy Efficacy of Kynureninase Modified CAR-T Cells Through Targeting Kynurenine Metabolism. Oncoimmunology (2022) 11(1):2055703. doi: 10.1080/2162402X.2022.2055703
Keywords: CAR-T cell, hematological malignancies, CAR-T related toxicities, antigen escape, immunosuppressive tumor microenvironment, combinatorial therapy
Citation: Zhang X, Zhu L, Zhang H, Chen S and Xiao Y (2022) CAR-T Cell Therapy in Hematological Malignancies: Current Opportunities and Challenges. Front. Immunol. 13:927153. doi: 10.3389/fimmu.2022.927153
Received: 23 April 2022; Accepted: 16 May 2022;
Published: 10 June 2022.
Edited by:
Katy Rezvani, University of Texas MD Anderson Cancer Center, United StatesReviewed by:
Katherina Psarra, Evaggelismos General Hospital, GreeceAli Roghanian, University of Southampton, United Kingdom
Copyright © 2022 Zhang, Zhu, Zhang, Chen and Xiao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yang Xiao, amR4aWFvMTExQDE2My5jb20=