
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol., 25 July 2022
Sec. Cancer Immunity and Immunotherapy
Volume 13 - 2022 | https://doi.org/10.3389/fimmu.2022.920059
This article is part of the Research TopicCombinational Immunotherapy of Cancer: Novel Targets, Mechanisms, and StrategiesView all 85 articles
 Chunmei Zhang
Chunmei Zhang Ning Liu*
Ning Liu*Ovarian cancer (OC) is one of the most common malignancies that causes death in women and is a heterogeneous disease with complex molecular and genetic changes. Because of the relatively high recurrence rate of OC, it is crucial to understand the associated mechanisms of drug resistance and to discover potential target for rational targeted therapy. Cell death is a genetically determined process. Active and orderly cell death is prevalent during the development of living organisms and plays a critical role in regulating life homeostasis. Ferroptosis, a novel type of cell death discovered in recent years, is distinct from apoptosis and necrosis and is mainly caused by the imbalance between the production and degradation of intracellular lipid reactive oxygen species triggered by increased iron content. Necroptosis is a regulated non-cysteine protease–dependent programmed cell necrosis, morphologically exhibiting the same features as necrosis and occurring via a unique mechanism of programmed cell death different from the apoptotic signaling pathway. Pyroptosis is a form of programmed cell death that is characterized by the formation of membrane pores and subsequent cell lysis as well as release of pro-inflammatory cell contents mediated by the abscisin family. Studies have shown that ferroptosis, necroptosis, and pyroptosis are involved in the development and progression of a variety of diseases, including tumors. In this review, we summarized the recent advances in ferroptosis, necroptosis, and pyroptosis in the occurrence, development, and therapeutic potential of OC.
Ovarian cancer (OC) is one kind of gynecologic malignancies with high mortality (1) and annually increased incidence (2, 3), which seriously threatens women’s life and health. OC is prevalent in middle-aged and elderly women. There is often no obvious clinical manifestation in the early stage. About 70% of patients first present with abdominal distention, ascites, and abdominal pain (4). Therefore, more than 75% of patients with OC present with advanced stages at the time of first or confirmed diagnosis (5, 6). In clinical settings, OC is featured with insidious onset, lack of early diagnostic markers, high malignancy, easy metastasis, and poor prognosis (7, 8). Currently, surgery combined with platinum and paclitaxel-based chemotherapy is the mainstream treatment for OC (7, 9). Although surgery and chemotherapy have significantly improved the prognosis of patients with OC in recent years, the 5-year survival rate of patients with advanced OC remains low (10, 11) because most patients with OC are advanced at the time of diagnosis and some patients with OC develop chemoresistance later following treatment (12, 13). Therefore, the search for potential biomarkers and therapeutic targets is of great clinical importance for early screening of patients with OC and improving the prognosis of patients with OC.
Cell death is a life phenomenon and an irreversible life process of cells. Cell death plays an indispensable role in the biological process of maintaining the normative homeostasis of the body and inhibiting the rapid proliferation of tumor cells (14, 15). Cell death includes regulated cell death (RCD) and accidental cell death (ACD) (16–18). RCD is a genetically determined form of active and ordered cell death that plays an important role in the maintenance of homeostasis (19, 20). Currently, the common types of RCD include apoptosis, necroptosis, ferroptosis, autophagy, and pyroptosis (21). Ferroptosis, a newly discovered non-apoptotic form of cell death, is essentially iron ion–dependent RCD. Necroptosis is mainly mediated by cytokines (TNF-α, IFN-α, and IFN-γ), Toll-like receptors (TLR3, TLR4, and TLR9), and nucleic acid (DNA and RNA) receptors. RIPK1/3 and MLKL are involved in the development of necroptosis, with MLKL being the key molecule (22, 23). Pyroptosis, which is a pathological form of suicide of cells distinct from apoptosis, is mainly mediated by Caspase-1 and Caspase-4/5/11. Pyroptotic signaling pathway mainly includes classical pyroptosis pathway and non-classical pyroptosis pathway, with inflammatory vesicle production and Gasdermin D (GSDMD) activation as the hallmarks of pyroptosis pathway (24, 25). Gasdermin B (GSDMB) is highly expressed in inflammatory bowel disease and contributes to the progression of inflammation by disrupting epithelial barrier function and promoting the development of ferroptosis (26). Necroptosis is mainly mediated by cytokines (TNF-α, IFN-α, and IFN-γ), Toll-like receptors (TLR3, TLR4, and TLR9), and nucleic acid (DNA and RNA) receptors. RIPK1/3 and MLKL are involved in the development of necroptosis, with MLKL being the key molecule (22, 23). Necroptosis is induced by cigarette smoke exposure and is increased in the lungs of patients with chronic obstructive pulmonary disease (COPD) and patients with experimental COPD. Inhibition of necroptosis attenuated cigarette smoke-induced airway inflammation, airway remodeling, and emphysema (27). Recently, accumulating evidence has showed that ferroptosis, necroptosis, and pyroptosis play an important role in tumor development. Expression levels of ZBP1 are significantly increased in necrotic tumors. In addition, ZBP1 deficiency blocked necroptosis and significantly inhibited tumor metastasis during breast cancer development (28). In breast cancer, DRD2 promotes M1 polarization of macrophages and triggers GSDME-executed pyroptosis that regulates the tumor microenvironment and inhibits tumor malignant progression (29). All these pieces of evidence highlight the important roles of ferroptosis, necroptosis, and pyroptosis in the progression and metastasis of human malignancies.
RCD in malignancies has been extensively studied and more and more pieces of evidence reveals that ferroptosis, necroptosis, and pyroptosis are highly involved the development, progression, and regression of OC (30, 31). In this paper, we reviewed the molecular mechanisms of ferroptosis, necroptosis, and pyroptosis and their regulatory roles in OC, providing a new perspective on the pathogenesis and targeted therapy of OC and exploring their potential as potential therapeutic targets for death.
Programmed cell death plays an important biological effect in maintaining the homeostasis of the organism. As a novel mode of cell death, ferroptosis was first described in 2012. The small-molecule inhibitor erastin was found to induce a unique mode of cell death in ras mutant cells that could not be rescued by apoptosis inhibitors and necrosis inhibitors but was reversed by the iron ion chelator deferoxamine. Later, this novel mode of cell death was named ferroptosis (32). Ferroptosis is an iron- and ROS-dependent mode of cell death, which is characterized by major cytological changes including reduction or loss of mitochondrial cristae, rupture of the outer mitochondrial membrane, and mitochondrial membrane ruffling (33, 34). All these above changes are caused to loss of selective permeability of the cell membrane due to the occurrence of peroxidation of lipid components of the cell membrane and oxidative stress (35, 36). In addition, different physiological conditions and pathological stresses have been found to induce tissue ferroptosis (37, 38). Ferroptosis is gradually recognized as an adaptive process by which the body eliminates malignant cells by removing cells damaged by nutritional deficiency, infection, or stress (39–41). Thus, ferroptosis has an inhibitory effect on tumorigenesis under normal conditions, and abnormalities in the oxidative stress pathway are an important cause of ferroptosis. Although tumor cells are in a constant state of excessive oxidative stress, they are less likely to develop ferroptosis, which is mainly dependent on their own antioxidant system (42, 43). In-depth studies based on the mechanisms of ferroptosis occurrence and regulation in tumor cells are of great clinical importance for the formation of new strategies for tumor therapy.
Phospholipid hyperoxidation of polyunsaturated fatty acids in the cytoplasmic membrane has been shown to be the most important driver of ferroptosis (44). The proportion of polyunsaturated fatty acids in lipids determines the ease with which lipid peroxidation occurs in cells. Acyl-coa synthetase long-chain family member 4 (ACSL4) and lysophosphatidylcholine acyltransferase 3 (LPCAT3) are key enzymes that regulate polyunsaturated fatty acid synthesis in phospholipid membranes, whereas the inhibition of both ACSL4 and LPCAT3 promotes ferroptosis resistance (45). There are two types of intracellular lipid peroxidation, namely, non-enzymatic and enzymatic lipid peroxidation. Non-enzymatic lipid peroxidation, also known as lipid autoxidation, is mainly a free radical-mediated chain reaction in which hydroxyl radicals generated by the Fenton reaction oxidize polyunsaturated fatty acids to lipid hydroperoxides (46). In contrast, enzymatic lipid peroxidation is mainly a process of direct oxidation of free polyunsaturated fatty acids to various types of lipid hydroperoxides catalyzed by lipoxygenase (LOX) (47, 48). Lipid hydroperoxides are catalyzed by iron ions to generate alkoxy radicals, which participate in the next lipid peroxidation chain reaction, ultimately leading to cell death (49). The exact mechanism by which lipid peroxidation leads to cellular ferroptosis is still not well understood. It could be due to the formation of structural lipid gaps, similar to protein gaps in necrosis and focal death. It is also possible that the depletion of polyunsaturated fatty acids causes structural changes in the fluidity of the cell membrane as well as an increase in membrane permeability, ultimately leading to loss of membrane integrity (50). In addition, lipid peroxides can be broken down into toxic aldehydes, which can further enhance the lipid peroxidation of ferroptosis by promoting protein inactivation through cross-linking reactions (51, 52). The mechanisms of amino acids and lipid metabolism in ferroptosis were displayed in Figure 1. Iron ions are important catalysts for lipid peroxidation reactions. Intracellular uptake, release, and storage of iron ions are all important regulators of ferroptosis. Inhibition of nitrogen fixation 1 (NFS1), which provides sulfur from cysteine for the synthesis of iron-sulfur clusters, activates the iron starvation response by simultaneously increasing transferrin receptor (TFRC) expression and degrading ferritin heavy chain 1 (FTH1), causing an increase in free iron ions, thereby making cells sensitive to ferroptosis activator (53–55). Overactivated heme oxygenase 1 increases intracellular free iron content and enhances ferroptosis effect by degrading hemoglobin into free iron, biliverdin, and carbon monoxide (56–58). Nuclear receptor coactivator 4 (NCOA4) recognizes intracellular after ferritin recognition, and ferritin transfers stored ferric ions to the autophagosome for degradation, which, in turn, releases ferric ions into the cytoplasm to become free iron, a process also known as iron autophagy (59, 60). In addition, genes such as nuclear receptor coactivator (NRF2) and heat shock protein B1 have been found to affect the sensitivity of cells to ferroptosis inducers by regulating intracellular iron ion metabolism (61, 62). This shows that iron ion metabolism is a potential regulatory point for the induction of cellular ferroptosis. The mechanisms of iron metabolism in ferroptosis were displayed in Figure 2. In addition to iron metabolism and lipid peroxidation responses, a wide range of intracellular antioxidant mechanisms also plays an important role in regulating ferroptosis sensitivity (63, 64). Glutathione (GSH), the most important intracellular antioxidant metabolite, requires cysteine as a raw material for its synthesis, and cells can endogenously synthesize cysteine via the transsulfuration pathway to resist ferroptosis (65, 66). In addition, myristoylation modification of ferroptosis inhibitory protein 1 leads to a nicotinamide adenine dinucleotide (NADH)–dependent decrease in coenzyme Q, which acts as a radical-trapping antioxidant to inhibit lipid peroxide proliferation (67, 68).
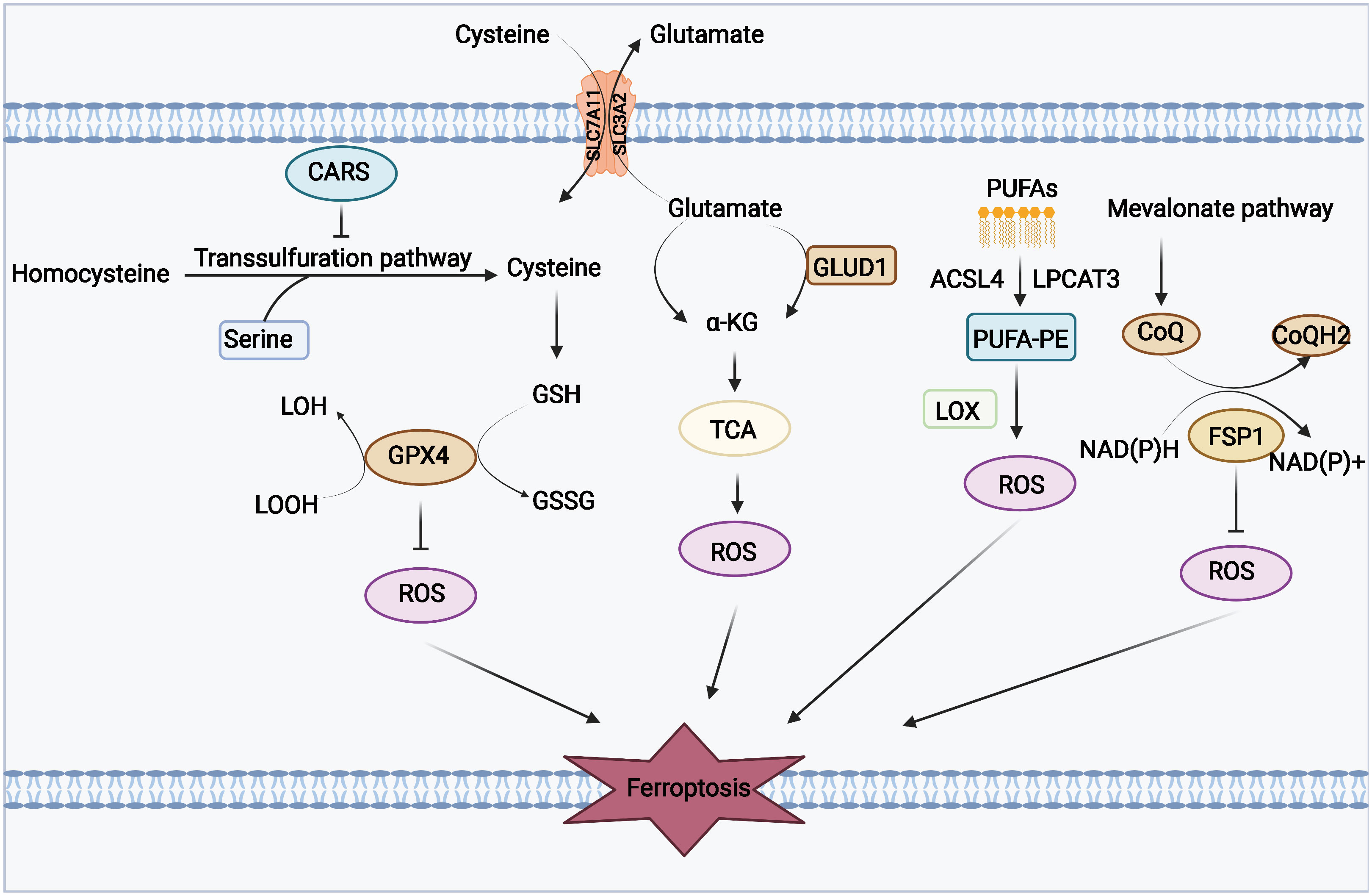
Figure 1 Mechanisms of amino acids and lipid metabolism in ferroptosis. Cysteine can be transported into the cell, whereas glutamate can be transported out of the cell by the Xc-system. Cysteine can be used to synthesize glutathione to maintain the balance of the redox state, and it can also be synthesized through the transsulfurization pathway blocked by CARS. Glutamate can be converted to a-KG by transaminase or GLUD1 pathway and participate in TCA, thereby generating ROS. PUFAs derived from cell membranes can be catalyzed by ACSL4 and LPCAT to PUFA-PE, and PUFA-PE can be peroxidized by the LOX family. FSP1 and coenzyme Q also play an important role in the antioxidant system of coenzyme Q.
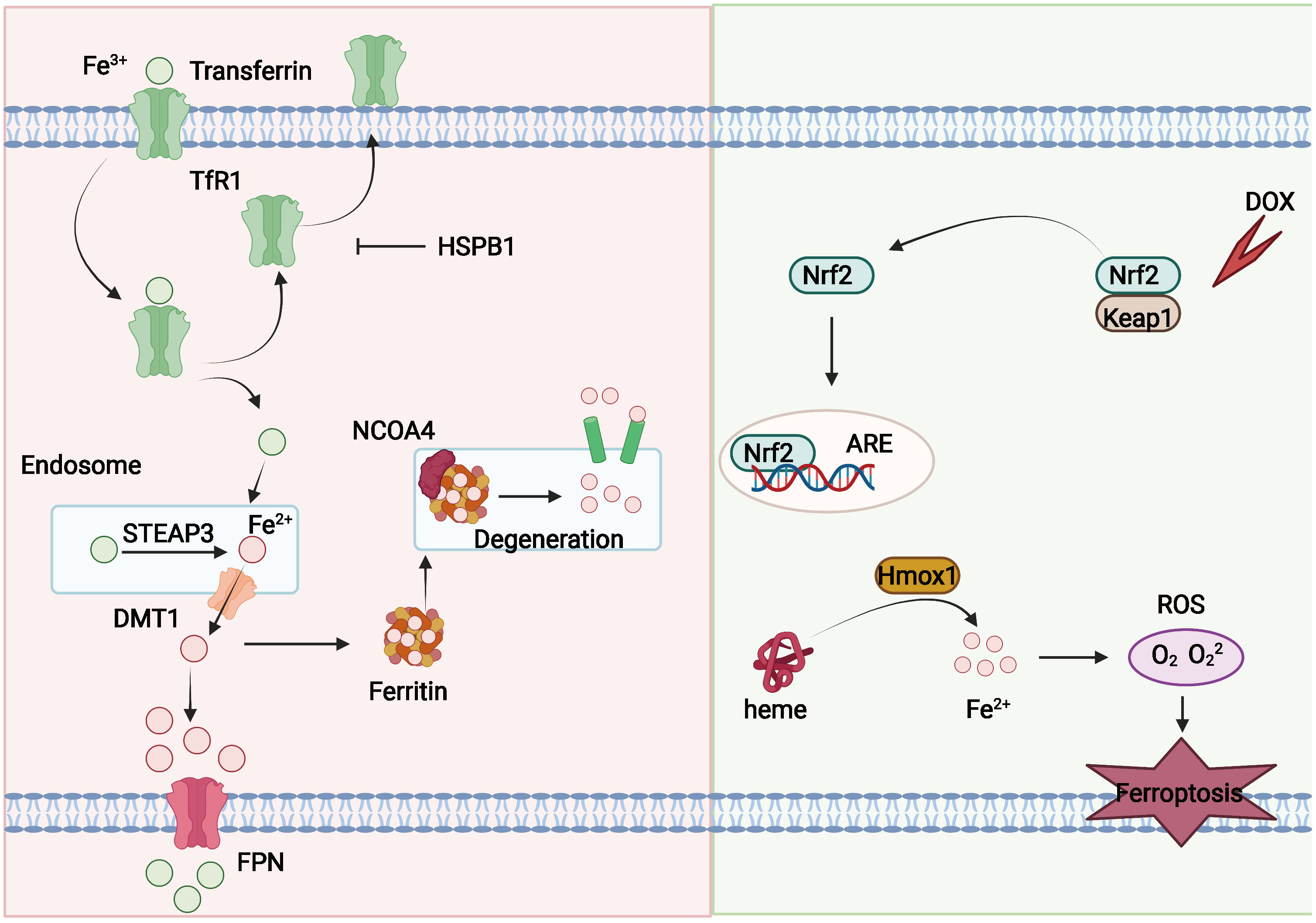
Figure 2 Mechanisms of iron metabolism in ferroptosis. Fe3+ can couple to transferrin and enter the intercellular milieu mediated by TfR1. Transferrin can be recycled and exported extracellularly and blocked by HSPB1. Fe3+ is reduced to Fe2+ by DMT1 in endosomes, and Fe2+ can be transported into the cytoplasm. Fe2+ can be released from ferritin through NCOA4-mediated ferritin phagocytosis, and part of Fe2+ can be exported outside the cell and oxidized by FPN. In addition, DOX can also induce ferroptosis. Cardiac output of DOX activates the Keap1/Nrf2 pathway, and Nrf2 further activates the downstream protein Hmox1 and prompts it to oxidize heme and release iron, leading to ferroptosis.
Necroptosis, also known as programmed necrosis, is a necrosis-like form of cell death that relies on receptor interacting serine/threonine protein kinase 1 (RIPK1), RIPK3, and mixed lineage kinase domain–like pseudokinase (MLKL) and does not depend on cysteine-aspartic proteases 1 (caspase-1) (23, 69, 70). Necrosis was once thought to be passive and non-programmed, but recent studies have revealed that cell necrosis is an active and modifiable process (71). In the absence or inhibition of caspase-8 or Fas-associating protein with a novel death domain (FADD), cells induced by tumor necrosis factor α (TNF-α) still die, and the cell death morphology resembles that of necrotic cells (72–74), gradually revealing a caspase-independent cell death similar to necrosis. Degterev et al. first described the role of the small-molecule Nec-1 in regulating cell necrosis, updating, for the first time, the concept of unregulated necrosis to cells that can be regulated by Nec-1 necroptosis (16, 75). Since then, necroptosis has been defined as programmed necrosis, whereby cells undergoing necroptosis have their cell membranes ruptured, releasing intracellular material that can stimulate a variety of cells (including macrophages, fibroblasts, and endothelial cells) to participate in the intrinsic immune response and exacerbate the inflammatory response by releasing inflammatory cytokines, leading to a dual role for necroptosis in different physiological or pathological processes (22, 23).
The potential mechanism of necroptosis is shown in Figure 3. The programmed cell death pattern is driven by RIPK1 through its kinase function, including through the formation of complex iia leading to apoptosis and complex iib leading to necroptosis (18, 76, 77). After TNF-α interacts with TNFR, TNFR1 starts to recruit the downstream protein molecules TNFR1- associated death domain protein (TRADD), RIPK1, TRAF2/5, and linear ubiquitin chain assembly complex (LU⁃BAC) proteins to form complex I, in which RIPK1 is polyubiquitinated and activates nuclear RIPK1 polyubiquitinates and activates the nuclear factor κB (NF-κB) and mitogen-activated protein kinase (MAPK) signaling pathways, inhibiting caspase-8 activation and promoting cell survival (72, 78–80). If TNF-α recruits TRADD, FADD, pro–caspase-8, and RIPK1 to form complex iia, then complex iia promotes activation of caspase-8, and activated caspase-8 undergoes apoptosis by activating caspase-3 (73, 81, 82). When caspase-8 is inhibited or its activity level is relatively low, RIPK1 recruits RIPK3 and both recruit MLKL through the RIP homotypic interaction motif (RHIM) to form complex iib, also known as necrosome (83, 84). MLKL phosphorylation causes oligomerization and membrane localization. Oligomerized MLKL has the ability to bind directly to lipids, allowing polymerized MLKL to form membrane permeable pores, disrupt cell membrane integrity, and undergo necroptosis (71, 85). However, it inhibits the formation of complex iib to inhibit necroptosis, so the role of RIPK1 in cells can be determined by targeting the drug to determine whether the cells survive or undergo necroptosis (74, 86, 87).
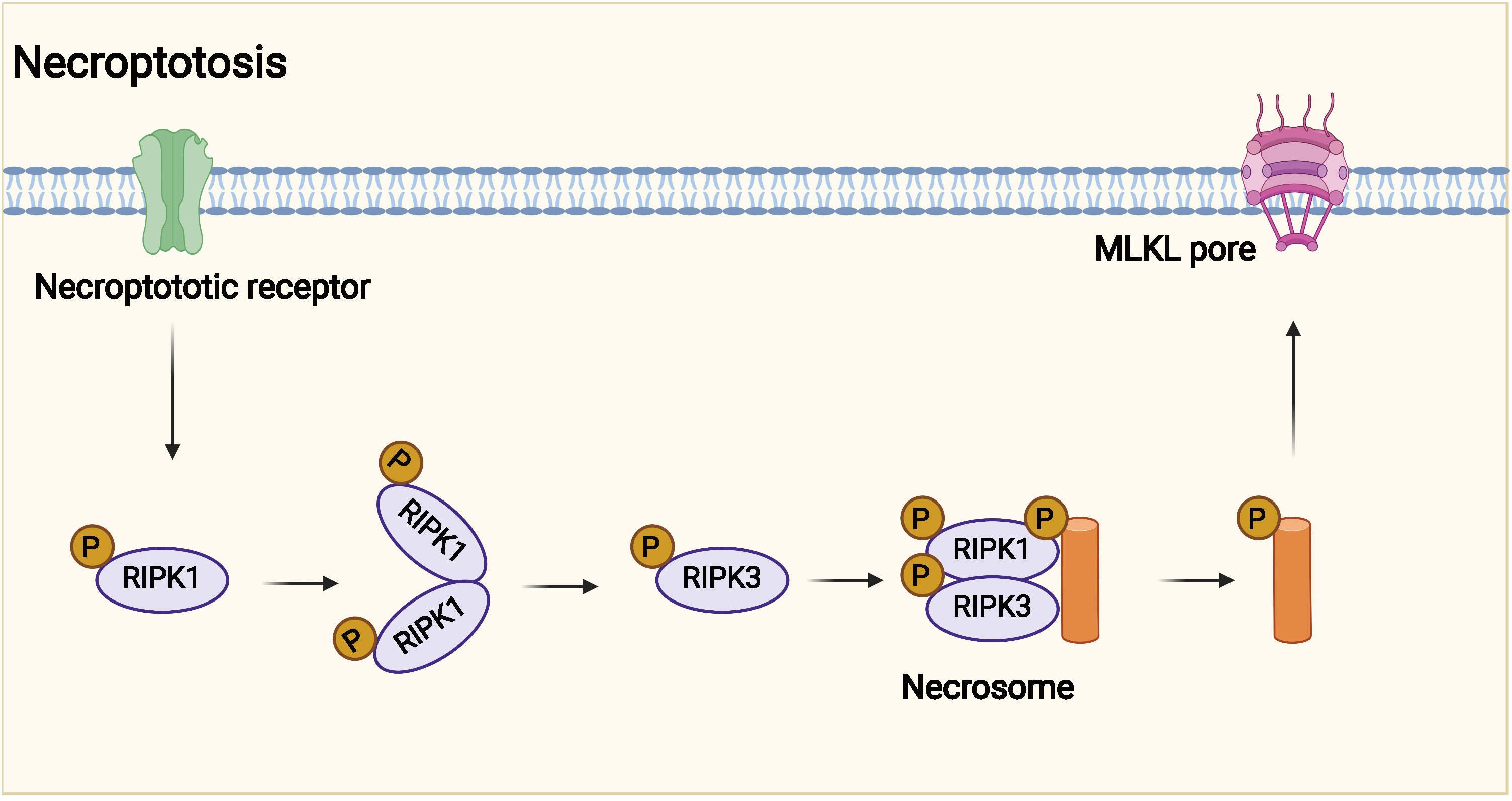
Figure 3 Potential mechanism of necroptosis. Necroptotic death may have evolved into the innate immune mechanism that complements apoptosis to eliminate pathogens. Necroptosis is affected by receptor interacting protein kinase 3 (RIPK3) and mixed lineage kinase domain-like protein (MLKL).
Pyroptosis is a caspase-1–mediated mode of programmed cell death (88, 89) characterized by rapid plasma membrane rupture followed by the release of cellular contents and pro-inflammatory substances such as IL, which triggers an inflammatory cascade response that results in cellular damage (24, 90). As early as 1992, Zychlinsky et al. observed experimentally that Shigella fowleri could induce lytic death in infected host macrophages (91). In 2001, Cookson et al. showed that this form of death was caspase-1 activity–dependent, unlike caspase-3 activity–dependent apoptosis (92). They first defined focal cell death as a caspase-1–dependent form of cell death. Furthermore, Shao et al. showed that cell scorch death can also be induced by the activation of caspase-4/5/11 by intracytoplasmic LPS and that activated caspase-4/5/11 ultimately induces cell scorch death through cleavage of Gasdermin family proteins. Therefore, they defined cell scorch death as programmed cell necrosis mediated by the Gasdermin family (93, 94). When cell death occurs, the nucleus is condensed, chromatin DNA is randomly broken and degraded, numerous pores appear in the cell membrane, the cell membrane loses its ability to regulate the entry and exit of substances, the cell loses internal and external ionic balance, osmotic swelling occurs, and the membrane ruptures, releasing cell contents and other active substances, stimulating the body’s immune response, and recruiting more inflammatory cells. This stimulates the body’s immune response, recruits more inflammatory cells, and amplifies the inflammatory response.
The potential mechanism of pyroptosis is shown in Figure 4. The assembly of the inflammasome is the initiating step of the classical pathway of pyroptosis, a complex of intracellular macromolecular proteins necessary for inflammation to occur and capable of recognizing dangerous signaling molecules such as bacteria and viruses. The inflammasome is mainly composed of pattern recognition receptors (prrs), apoptosis-associated speck-like protein (ASC), and pro–caspase-1 (95–98). Among them, prrs are receptor proteins responsible for the recognition of different intracellular signaling stimuli, which are mainly composed of nucleotide-binding oligomerization domain–like receptor protein 1 (NLRP1) and NLRP3; nucleotide ASC is an articulated protein that consists mainly of the N-terminal pyrin domain (PYD), caspase activation, and recruitment domain (CARD) (95, 99). Pro–caspase-1 is an effector molecule that is activated to specifically cleave GSDMD. Danger-signaling sensors after the recognition of the danger signaling molecule by NLRP1, NLRP3, or AIM2 bind to the PYD structural domain at the N-terminal end of the bridging protein through its N-terminal PYD structural domain, and ASC then recruits caspase-1 through the interaction of the CARDCARD structural domain to complete the assembly of the inflammasome (100, 101). The inflammasome acts by activating the effector molecule pro–caspase-1 to form active caspase-1. Activated caspase-1 is able to specifically cleave the GSDMD to generate the N-terminal and C-terminal ends, and the N-terminal end of the GSDMD binds to cell membrane phospholipids, causing many small pores to form in the cell membrane (102, 103). The integrity of the cell is disrupted, and the water flows inward, leading to cell swelling and rupture, releasing intracellular inflammatory substances, and inducing pyroptosis (104). In addition, caspase-1 also promotes the maturation of IL-18 and IL-1β precursors, which are cleaved into active IL-18 and IL-1β (90, 105, 106) and secreted through the cell membrane pores to the outside of the cell, recruiting more inflammatory cells and causing an inflammatory waterfall response. This caspase-1–mediated cell death is called the classical pathway of pyroptosis. The non-classical pathway of pyroptosis is mainly mediated by caspase-4, caspase-5, and caspase-11. Upon stimulation of cells by bacterial LPS, caspase-4, caspase-5, and caspase-11 bind directly to and are activated by bacterial LPS (40, 107). Activated caspase-4, caspase-5, and caspase-11 specifically cleave the GSDMD, relieving intramolecular inhibition of the GSDMD N structural domain (108), and the GSDMD N terminus binds to cell membrane phospholipids, causing cell membrane pore formation, cell swelling, rupture, and induction of pyroptosis (108). The GSDMD N terminus also amplifies the inflammatory response by activating the NLRP3 inflammasome, which, in turn, activates caspase-1 (109), which stimulates the maturation of IL-18 and IL-1β precursors and secretes IL-18 and IL-1β extracellularly (110).
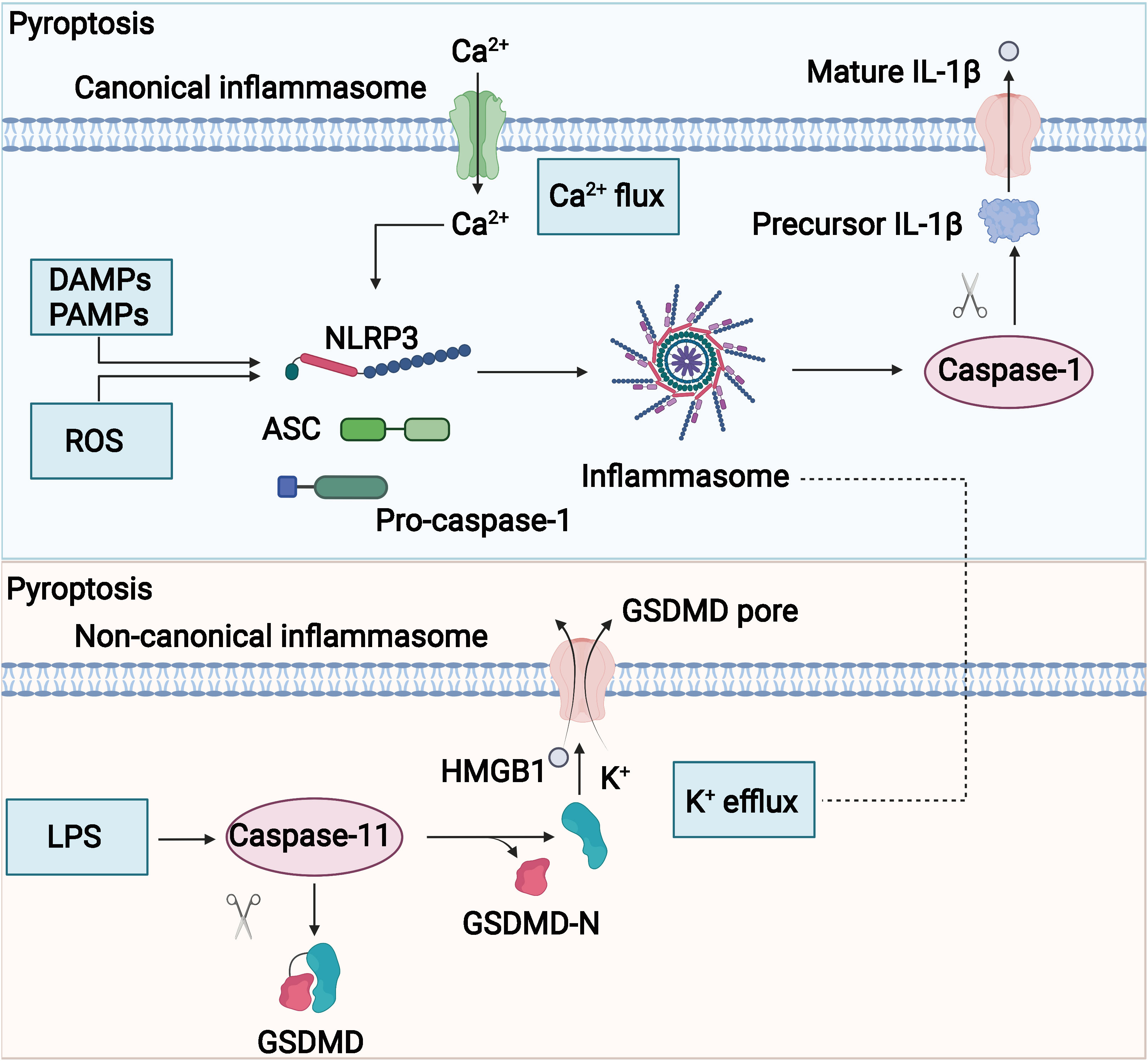
Figure 4 Potential mechanism of pyroptosis. The molecular mechanisms of pyrolysis mainly include canonical and noncanonical signaling.
Ferroptosis is a novel form of RCD induced by iron-dependent lipid peroxidation damage, which is morphologically, genetically, and molecularly different from other cell death modalities such as apoptosis, autophagy, and necrosis. The relationship between ferroptosis and tumors is extremely close, and there are numerous studies to design and develop ferroptosis-based anticancer drugs, and ferroptosis is expected to be a novel therapeutic approach for tumors. The interactive dialogue between triple-negative breast cancer (TNBC) cells and tumor-associated macrophages (tams) promotes the sustained activation of HLF in tumor cells through the IL-6–TGF-β1 axis. Subsequently, hepatic leukemia factor (HLF) promotes resistance to ferroptosis in TNBC cells via GGT1, ultimately promoting malignant tumor progression (1). The current design of corresponding compounds targeting key molecules in ferroptosis can effectively inhibit tumor progression with significant clinical translational implications. Mimetic drugs composed of small-molecule inducers of ferroptosis, erastin, and RSL3 with BH3 were effective in synergistically killing U251 cells and inhibiting malignant progression of glioblastoma (111). Nanocatalytic activity leads to simultaneous inhibition of GPX4/GSH and FSP1/coq10h2 pathways and synergizes with the GPX4 inactivation function of RSL3 to cause significant ferroptosis damage and thus inhibit malignant progression of triple-negative breast cancer (112). Depletion of PSTK leads to inactivation of GSH peroxide 4 (GPX4) and inhibition of selenocysteine and cysteine synthesis. GSH metabolism is disrupted due to inhibition of selenocysteine and cysteine synthesis, which enhances the induction of ferroptosis after targeted chemotherapy, leading to malignant progression of hepatocellular liver cancer (113). The use of PSTK inhibitor-punicalin together with sorafenib for the treatment of HCC in vitro and in vivo exhibits synergistic effects.
Research on targeted tumor therapy based on necroptosis is currently underway, suggesting that necroptosis will provide a new strategy for tumor treatment. Disintegrin and Metalloprotease 17 (ADAM17) was identified as a novel important regulator of necroptosis whose activity could significantly affect the role of TNFR1-dependent tumor cell induction of endothelial cell death, tumor cell extravasation, and subsequent metastatic seeding (114). Furthermore, mediated TNFR1 extracellular domain shedding and subsequent processing by the γ-secretase complex are key enzymatic steps in the induction of TNF-induced necrotic apoptosis.ADAM17 may serve as an important target as an anti-metastatic and advanced cancer therapy. RIPK3 may act as a tumor suppressor to inhibit malignant progression of malignant mesothelioma through induction of necrotic apoptosis, whereas RIPK3 DNA epigenetic silencing of methylation impairs necroptosis and leads to chemoresistance as well as poorer prognosis in malignant mesothelioma (115). Tsc1/mTOR has a critical role in suppressing RIPK3 expression and activation in intestinal epithelial cells through TRIM11-mediated ubiquitination and autophagy-dependent degradation. mTOR can act on RIPK3 to enhance the expression and activation of RIPK3 by TNF and microbial pathogen-associated molecular pattern (PAMP)–induced necroptosis. mTOR/RIPK3/necroptosis axis is a driver of intestinal inflammation and cancer (116).
Various components of the scorch pathway are associated with tumorigenesis, invasion, and metastasis, and studies on scorch death have opened up new frontiers in tumor therapy. maternal embryonic leucine zipper kinase (MELK) expression is elevated in lung adenocarcinoma (LUAD) and promotes the malignant progression of LUAD cells by regulating the PLK1-CDC25C-CDK1 signaling pathway to promote proliferation and inhibit apoptosis-mediated cell scorch (16, 117). GSDME-mediated cell scorch death promotes colorectal cancer progression by releasing HMGB1, which induces tumor cell proliferation and PCNA expression through the extracellular regulated protein kinases 1/2 (ERK1/2) pathway (118). Circneil3, a circulating RNA, can act as a sponge by directly binding to mir-1184 and thereby releasing the inhibitory effect of miR-1184. The inhibition of PIF1 by mir-1184 ultimately induces DNA damage and triggers AIM2 inflammasome activation-mediated cell scorching (119). Mediating the cell scorch-induced circneil3/mir-1184/PIF1 regulatory axis may be a promising clinical therapeutic strategy for lung cancer.
Accumulating evidence has identified the crosstalk between ferroptosis, necroptosis, and pyroptosis. A number of factors including NEK7, Tom20, and caspase-1 have been found to mediate the crosstalk between these programmed cell death pathways. For instance, necroptosis and pyroptosis are both able to promote cell lysis. Z-DNA binding protein 1 (ZBP1) has been found to promote pyroptosis and necroptosis upon sensing infection with fungus (120). A well-established cell marker for apoptosis, Bcl-2, is found to regulate pyroptosis and necroptosis by targeting BH3-like domains in GSDMD and MLKL (121). Moreover, caspase-8 plays a key role in switching from necroptosis topyroptosis (122). Moreover, the key molecular regulator for ferroptosis, iron, could promote excessive reactive oxygen species (ROS) production and mediate crosstalk between ferroptosis and necroptosis (123). In the model of myocardial fibrosis, it has been reported that mixed-lineage kinase 3 (MLK3) modulates pyroptosis and ferroptosis via distant signaling pathways (124). Interestingly, non-coding RNAs have also been reported to play a key role in the crosstalk between necroptosis, ferroptosis, and pyroptosis (125). All these pieces of evidence highlight the correlation between necroptosis, pyroptosis, and ferroptosis.
Apoptosis used to be considered the predominant means of programmed cell death in tumor cells that decide the proliferation rate of cells. Lately, mounting evidence has showed that other types of programmed cell death including ferroptosis, necroptosis, and pyroptosis are highly involved in a variety of cell processes of OC cells, such as chemoresistance and immune response.
In brief, the regulatory role of ferroptosis related genes in OC progression was displayed in Table 1. Studies have shown that elevated intracellular iron levels are closely associated with OC. FPN was decreased, TFR1 and TF were increased, and iron levels were elevated in high-grade plasma cytotic OC tissues compared with normal ovarian tissues (141). Genetic models of OC initiating cells also exhibit reduced iron efflux pumps and upregulated expression of iron transport–related proteins. This suggests that intracellular iron levels are elevated in OC cells early in the development of OC. Li et al. (126) found that ferrous ammonium citrate (FAC) promoted intracellular iron expression levels in OC cells and inhibited OC cell proliferation, induced apoptosis, promoted inflammatory responses, and inhibited the reduction of lipid peroxides. Inhibition of GPX4 modulated intracellular iron homeostasis and lipid peroxide reduction, induced ferroptosis, and exerted anti-cancer effects.
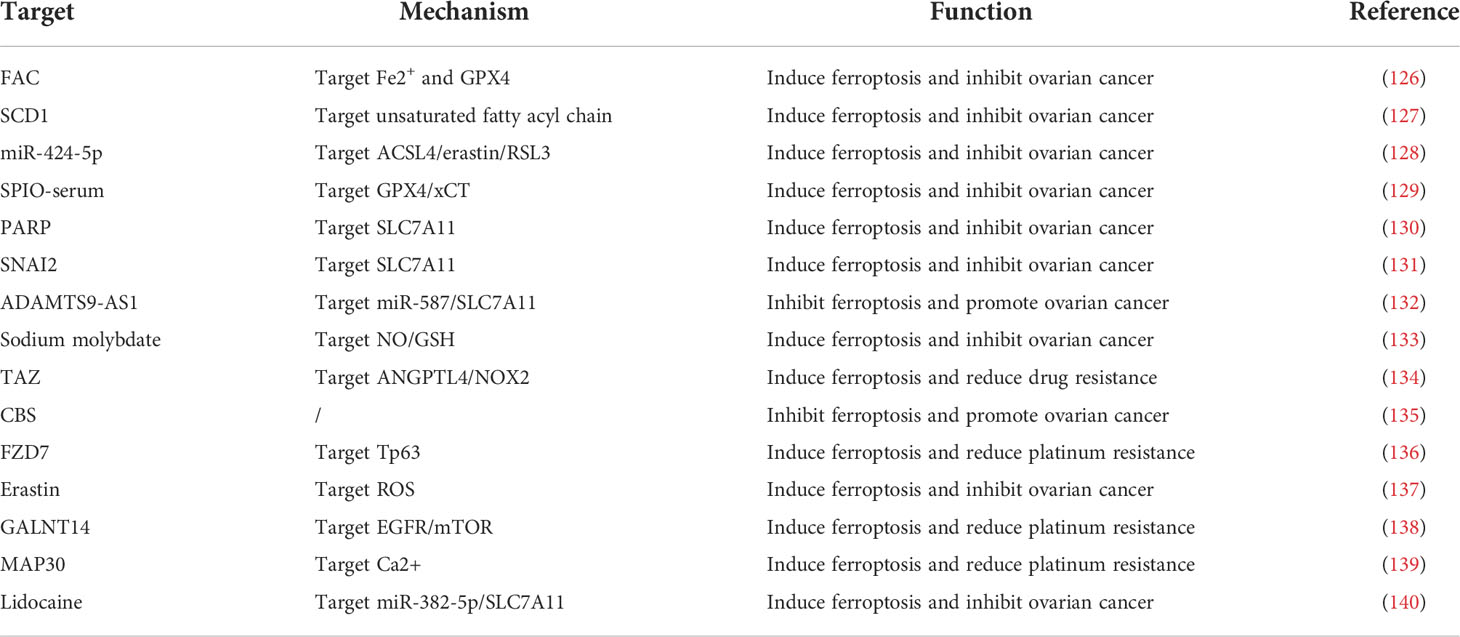
Table 1 The regulatory role of ferroptosis related genes in ovarian cancer progression.
In ferroptosis, lipid peroxidation driven by ROS plays an important role in the ferroptosis pathway. Tesfay et al. (127) found that steroyl coa desaturase (SCD1) was highly expressed in OC tissues, cell lines, and genetic models of OC stem cells. Inhibition of SCD1 significantly reduced unsaturated fatty acyl chains and increased long-chain saturated ceramides in membrane phospholipids and enhanced the anti-tumor effects of ferroptosis inducers in OC cell lines and in situ xenograft models in mice.
OC is associated with abnormal expression of many genes. Ma et al. (128) demonstrated that upregulation of mir-424-5p inhibited ACSL4 expression by directly binding to the 3′-untranslated region (UTR) of ACSL4, thereby reducing erastin- and RSL3-induced ferroptosis and ultimately inhibiting the malignant progression of OC. Mutation of the p53 gene alone caused migration of mouse oviductal epithelial cells; when p53 mutation combined with K-ras activation occurred, mouse oviductal epithelial cells were transformed into tumor cells (142). Zhang et al. (129) found that superparamagnetic iron oxide (SPIO)-serum effectively induced lipid peroxidation and produced large amounts of toxic ROS and promoted the down-regulation of GPX4 and xct, leading to iron-dependent oxidative death. These effects could be reversed using the iron chelator DFO and the lipid peroxidation inhibitor Fer-1. In addition, p53 contributed to promote SPIO-serum–induced ferroptosis in OC cells. Inhibition of PARP downregulates the expression of the cystine transporter protein SLC7A11 in a p53-dependent manner, which, in turn, leads to reduced GSH biosynthesis and promotes lipid peroxidation and ferroptosis (130). SNAI2 inhibits the malignant progression of OC by binding to the promoter region of SLC7A11 and thereby inducing the onset of ferroptosis in OC cells (131). Cai et al. (132) found that ADAMTS9-AS1 can inhibit the malignant progression of OC by regulating the mir-587/SLC7A11 axis to attenuate ferroptosis process in OC and promote proliferation, migration, and invasion of OC cells, leading to malignant progression of OC.
Numerous studies have confirmed that elevated intracellular GSH levels and high expression of related metabolic enzymes are closely associated with drug resistance in OC. Mao et al. (133) found that sodium molybdate induced elevated pools of unstable iron in OC cells and induced GSH depletion by mediating nitric oxide (NO) production and further promoted ferroptosis in OC cells. In addition, NO induces mitochondrial damage through inhibition of mitochondrial aconitase activity, ATP production, and mitochondrial membrane potential, leading to apoptosis in OC cells. Yang et al. (134) found that transcriptional coactivator with PDZ-binding motif (TAZ) removed conferred ferroptosis resistance, whereas overexpression of TAZS89A promoted cellular susceptibility to ferroptosis, and lower TAZ levels were an important reason for reduced ferroptosis susceptibility in chemotherapy-resistant relapsed OC TAZ that can promote OC cell sensitivity to ferroptosis by regulating ANGPTL4 and activating NOX2 entry and exit. Liu et al. (143) established erastin-tolerant cell lines and found that the cell lines could still maintain GSH levels, suggesting the existence of other intracellular pathways for cystine synthesis. The assay revealed high expression of CBS, a key enzyme of the transsulfuration pathway, suggesting that the erastin-tolerant cell line provides cystine to the cells through the upregulated transsulfuration pathway. Verschoor et al. (144) treated an OC cell model using an Xc-system inhibitor and a transsulfuration pathway inhibitor and found that intracellular GSH levels were significantly reduced after transsulfuration pathway inhibition, suggesting that the transsulfuration. Chakraborty et al. (135) found that the expression level of CBS in the transsulfuration pathway was elevated in a few OC cell lines and that CBS gene silencing inhibited cell migration and invasion of OC cells.
The mechanism of action of platinum drug is based on the generation of intracellular ROS, eventually leading to cellular damage and death. Wang et al. (136) found that platinum-resistant cells and tumors exhibited Frizzled 7 (FZD7) expression and that knockdown of FZD7 increased platinum sensitivity and delayed tumorigenesis. In contrast, overexpression of FZD7 activated the oncogenic factor Tp63, which promoted upregulation of the metabolic pathway, leading to platinum resistance in OC cells. Chen et al. (137) found that erastin induced ferroptosis and increased ROS levels, thereby enhancing the cytotoxic effects of cisplatin. Erastin synergistically with cisplatin significantly inhibited OC cell growth. polypeptide N-acetylgalactosaminyltransferase (GALNT14) promoted mtor by modifying EGFR. The combination of mtor inhibitor and cisplatin resulted in a cumulative effect on cell death (138). Santos et al. (145) speculated that reversal of drug resistance by interfering with cysteine metabolism was suggested by the results of a study suggesting that selenium-containing salicin could contribute to a reduction in GSH levels and that an inhibitory effect on CBS was inhibited. Designing a nanodrug could be a new strategy to improve OC treatment.
The mechanism of ferroptosis-related drugs remained to be investigated, but they may be a new pathway to improve OC treatment through the ferroptosis pathway. Chan et al. (139) reported that MAP30 induces an increase in intracellular Ca2+ ion concentration, which triggers ROS-mediated cancer cell death through apoptosis and ferroptosis. Natural MAP30 may be used as a non-toxic supplement to enhance chemotherapy in patients with OC with peritoneal metastases. Sun et al. (140) found that lidocaine could inhibit the ferroptosis process in OC cells by enhancing the expression of mir-382-5p in cells, which, in turn, inhibited the ferroptosis process in OC cells by targeting SLC7A11. Lidocaine treatment inhibited tumor growth of OC in vivo.
In addition, many other authors have explored the expression levels of ferroptosis-related genes and proteins in patients with OC and the correlation with patient prognosis by bioinformatics analysis. The results of all analyses are presented in Table 2.

Table 2 Ferroptosis related genes in patients with ovarian cancer.
In brief, the regulatory role of necroptosis related genes in OC progression was displayed in Table 3. Hahne et al. (156) explored the ability of the PI3K/AKT inhibitor AEZS-126 alone and in combination with rapamycin to selectively target OC cell proliferation and survival in vitro. They found by validation that AEZS-126 exhibited anti-cytotoxicity in an in vitro model of OC and that the primary mechanism was the regulation of the necroptotic apoptotic process in OC cells. Mccabe et al. (157)found that inhibitor of apoptosis protein (IAP) plus cystein inhibitor (IZ) treatment selectively induced TNFα-dependent death in several anti-apoptotic cell lines and patient xenografts. Qiu et al. (158) found that upregulation of CD40 may be relatively common in low-grade serous carcinomas (lgscs) and that CD40 activation induced RIP1-dependent, necrosis-like cell death in LGSC cells. Dey et al. (159)found that BMI1 in OC was able to participate in the PINK1-PARK2–dependent mitochondrial pathway and induce a novel non-apoptotic, necroptosis-mediated cell death pattern. Necroptosis enhances the phosphorylation of the downstream substrate MLKL by activating the RIPK1-RIPK3 complex. In addition, inhibition of caspase-8 was found to significantly inhibit NF-κB signaling and lead to necrotic cell death by stabilizing RIPK1 expression (160). Blocking NF-κB signaling and depleting cIAP using SMAC mimics could further render these cells susceptible to necroptosis killing. Increasing caspase-8 expression in vivo may be an important tool to improve the prognosis of patients with OC.

Table 3 The regulatory role of necroptosis related genes in ovarian cancer progression.
In addition, studies have shown that targeting necroptosis may also promote prognosis in patients with OC. Wu et al. found that luteal-phase progesterone (P4) binds to P4 receptors (prs) and via the TNF-a/RIPK1/RIPK3/MLKL pathway in the oviductal epithelium of Trp53M−/− mice, and immortalized human p53-deficient bacterial hair epithelium induces necrotic apoptosis (161), which may be a potential mechanism for progesterone to prevent OC onset. MLKL may be a novel pro-necrotic apoptotic target of ceramide in OC models, and knockdown of MLKL with small interfering RNA (siRNA) significantly abrogated ceramide nanoliposomes (CNL)–induced cell death (162). As a SMAC (second mitochondria-derived activator of caspase) mimetic, DEBIO 1143 was able to reverse resistance to carboplatin by targeting cIAP1, XIAP, and caspase-9 and inducing apoptosis or necroptosis depending on the cell line. Chefetz et al. (164) identified two selective inhibitors of the ALDH1A family (ALDH1Ai) and found that they preferentially killed CD133+ OC stem cell-like cells (CSC). ALDH1Ai induced mitochondrial uncoupling proteins and reduced oxidative phosphorylation to induce necrotizing CSC death. In addition, they found that ALDH1Ai was highly synergistic with chemotherapy, significantly reducing tumor initiation capacity and increasing tumor eradication in vivo. Chen et al. (165) found that excellent tumor ablation could be achieved by combining treatment with cus-mns2 nanoflowers and 808-nm NIR laser. Cusmns2 nanoflowers could be used as a promising multifunctional nanotheranostic agent for MRI and as a photothermal/photodynamic cancer therapy agent through necroptosis. Liu et al. (166) found that berberine (BBR) could significantly inhibit the proliferative capacity of OC cells in a dose- and time-dependent manner. Combined treatment with BBR and DDP significantly promoted the proportion of necrotic apoptosis in OC cells and had a significant effect on OC cell proliferation and induction of G0/G1 cell cycle arrest. Combined treatment with BBR and DDP significantly increased OC cell death through induction of apoptosis and necroptosis, thereby enhancing the anticancer effects of chemotherapeutic agents. In addition, both ROS-mediated apoptosis and necroptosis could be involved in cisplatin-induced cell death. Therefore, RIP1 can act as a tumor suppressor that promotes the anticancer effects of chemotherapeutic agents such as cisplatin.
In brief, the regulatory role of pyroptosis-related genes in OC progression was displayed in Table 4. The mechanisms of inflammatory cells in the tumor microenvironment are gradually being understood with the advancement of research. Recent studies have shown that the occurrence and development of OC are closely associated with elevated levels of various inflammatory factors. Qiao et al. (169) found that knockdown of caspase-4 or GSDMD in OC cells significantly inhibited the killing activity of a-NETA cells, suggesting that a-NETA may play a biological role by regulating the cell scorching pathway. Zhang et al. (170) found that nobiletin, a prospective food-derived phytochemical derived from citrus fruits, could induce apoptosis and trigger ROS-mediated cell scorch death by regulating autophagy in OC cells, thereby inhibiting the malignant progression of OC. Liang et al. (168) found that serpentine, a derivative of coumarin, significantly inhibited OC cell growth and induced OC cell death by regulating OC cell apoptosis, cell scorching, and autophagic processes, with good therapeutic promise. HOXA transcript at the distal tip (HOTTIP) was able to increase AKT2 expression and inhibit ASK1/JNK signaling through negative regulation of mir-148a-3p, which, in turn, led to OC cell proliferation and NLRP1 inflammasome-mediated cell scorching process, resulting in OC malignant progression (171).

Table 4 The regulatory role of pyroptosis-related targets in ovarian cancer progression.
In addition, many other authors have explored the expression levels offerroptosis-related genes and proteins in patients with OC and the correlation with patient prognosis by bioinformatics analysis. The results of all analyses are presented in Table 5.

Table 5 Pyroptosis-related genes in patients with ovarian cancer.
Programmed cell death is a hot issue in biological and medical research, and targeting the cell death process is a common approach in tumor therapy. However, current compounds that induce programmed death are only effective against certain tumor cells, and different types of cancers seem to have different sensitivities to programmed death (177–179). Efforts to understand the sensitivity of different tissue tumors to programmed death are important for the practice of clinical application of programmed death in tumor therapy. Regarding how programmed death is precisely induced in vivo, the key regulators of programmed death that have been identified provide important therapeutic targets. Currently, the commonly used anti-tumor drugs in clinical practice have disadvantages such as poor selectivity, toxic side effects, and the tendency to develop drug resistance, which seriously limit their efficacy. According to the characteristics of TCM, studying the effect of TCM and its specific components on programmed tumor death, discovering effective anti-tumor components, and combining multi-disciplinary and multi-disciplinary approaches to target tumor sites by loading programmed death inducers, reactants of programmed death process, or TCM preparations through nanotechnology, so that drug concentrations accumulate at tumor sites, may bring new options for cancer treatment based on programmed death (178, 180, 181). The exploration of Chinese medicine to intervene in programmed death is still in its infancy, but there are already studies, showing extraordinary research prospects by loading Chinese medicine with nanotechnology for tumor treatment (182).
Ferroptosis has become a hot research topic in tumor; the pathway network between iron metabolism, Fenton reaction, Xc-system, and GPX4 has been initially established (183, 184); and other important related pathways (including transsulfur pathway) and ferroptosis-related drugs need to be further investigated. Current chemotherapy regimens for OC are still dominated by platinum and paclitaxel drugs, but the prognosis of patients with advanced OC remains bleak. An in-depth investigation of the link between ferroptosis pathway and OC will facilitate the search for a new chemotherapeutic regimen. Ferroptosis inducers and inhibitors are expected to be used effectively and rationally in the future, thus improving the precision of treatment for OC and other tumors. As a newly discovered mode of programmed cell death, necroptosis is closely related to a variety of case-physiological processes (185, 186), and most of the studies related to necroptosis are at the stage of basic experiments. Necroptosis plays an opposing role in anti-tumor; on the one hand, it can inhibit the proliferation and migration of tumor cells; on the other hand, it can play a pro-tumor growth role and participate in early tumor formation. Further in-depth study on the molecular mechanism of necroptosis pathway and the relationship between upstream and downstream signaling molecules of related signaling pathways, to explore its role in OC in different modes and to find corresponding target drugs, is one of the future directions to improve the therapeutic effect of OC. Cell scorch is a programmed cell death mediated mainly by inflammatory caspases (177). NLRP3 inflammatory vesicles activate caspase-1, which, in turn, causes pro–IL-1β and pro–IL-18 to form mature IL-1β and IL-18 and trigger cellular inflammation (187, 188). At the same time, caspase-1 cleaves the downstream factor abscisicin D. These actions create active pores in the cell membrane and lead to the onset of cellular scorching. The occurrence and development of OC are closely related to the inflammatory response, so the cell scorching caused by inflammatory vesicles/factors may play an important role in OC. A variety of Chinese herbal components and formulations have regulatory effects on cell scorch, and with further research, Chinese medicine may be used to regulate cell scorch in the prevention and treatment of OC.
In conclusion, this article reviews the progress of research on ferroptosis, necroptosis, or pyroptosis in the development of OC and after prognosis and treatment. Nevertheless, the exact roles of ferroptosis, necroptosis, and pyroptosis in OC remain to be fully elucidated. It is important to investigate the molecular mechanisms and physiopathological roles of ferroptosis, necroptosis, and pyroptosis and to specifically design the corresponding drug therapy for OC.
Original draft preparation, allocation, supplementation, and editing: CZ. Revision: NL and CZ. All authors have read and agreed to the published version of the manuscript.
This work was supported by the internal fund of Shenjing Hospital of China Medical University (Grant No. M0797).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
OC, ovarian cancer; RCD, regulated cell death; ACD, accidental cell death; TLRs, Toll-like receptors; GSDMB, Gasdermin B; ACSL4, acyl-coa synthetase long-chain family member 4; LPCAT3, lysophosphatidylcholine acyltransferase 3; LOX, lipoxygenase; NFS1, nitrogen fixation 1; TFRC, transferrin receptor; FTH1, ferritin heavy chain 1; NCOA4, nuclear receptor coactivator 4; NADH, nicotinamide adenine dinucleotide; RIPK1, serine/threonine protein kinase 1; MLKL, mixed lineage kinase domain–like pseudokinase; caspase-1, cysteine-aspartic proteases 1; FADD, Fas-associating protein with a novel death domain; TNF-α, tumor necrosis factor α; TRADD, TNFR1-associated death domain protein; LU-BAC, linear ubiquitin chain assembly complex; NF-κB, nuclear factor κB; MAPK, mitogen-activated protein kinase; RHIM, RIP homotypic interaction motif; PRRs, pattern recognition receptors; ASC, apoptosis-associated speck-like protein; NLRP1, nucleotide-binding oligomerization domain–like receptor protein 1; PYD, pyrin domain; CARD, caspase activation and recruitment domain; TAMs, tumor-associated macrophages; GPX4, glutathione peroxide 4; PAMP, pathogen-associated molecular pattern; LUAD, lung adenocarcinoma; FAC, ferrous ammonium citrate; FZD7, Frizzled 7; IAP, inhibitor of apoptosis protein; LGSCs, low-grade serous carcinomas; CNL, ceramide nanoliposomes; BBR, berberine.
1. Li H, Yang P, Wang J, Zhang J, Ma Q, Jiang Y, et al. HLF regulates ferroptosis, development and chemoresistance of triple-negative breast cancer by activating tumor cell-macrophage crosstalk. J Hematol Oncol (2022) 15:2. doi: 10.1186/s13045-021-01223-x
2. Fillon M. Opportunistic salpingectomy may reduce ovarian cancer risk. CA Cancer J Clin (2022) 72:97–9. doi: 10.3322/caac.21716
3. Torre LA, Trabert B, DeSantis CE, Miller KD, Samimi G, Runowicz CD, et al. Ovarian cancer statistics, 2018. CA Cancer J Clin (2018) 68:284–96. doi: 10.3322/caac.21456
4. Gardner GJ, Chi DS. Recurrent ovarian cancer - sculpting a promising future with surgery. N Engl J Med (2021) 385:2187–8. doi: 10.1056/NEJMe2116353
5. Kosaka M, Mizutani T, Ishiki H. What is the optimal treatment for vulnerable older women with ovarian cancer? JAMA Oncol (2021) 7:1725–6. doi: 10.1001/jamaoncol.2021.4125
6. Kurian AW, Abrahamse P, Bondarenko I, Hamilton AS, Deapen D, Gomez SL, et al. Association of genetic testing results with mortality among women with breast cancer or ovarian cancer. J Natl Cancer Inst (2022) 114:245–53. doi: 10.1093/jnci/djab151
7. Kuroki L, Guntupalli SR. Treatment of epithelial ovarian cancer. BMJ (2020) 371:m3773. doi: 10.1136/bmj.m3773
8. Nameki R, Chang H, Reddy J, Corona RI, Lawrenson K. Transcription factors in epithelial ovarian cancer: histotype-specific drivers and novel therapeutic targets. Pharmacol Ther (2021) 220:107722. doi: 10.1016/j.pharmthera.2020.107722
9. Wang Z, Meng F, Zhong Z. Emerging targeted drug delivery strategies toward ovarian cancer. Adv Drug Delivery Rev (2021) 178:113969. doi: 10.1016/j.addr.2021.113969
10. Baert T, Ferrero A, Sehouli J, O'Donnell DM, Gonzalez-Martin A, Joly F, et al. The systemic treatment of recurrent ovarian cancer revisited. Ann Oncol (2021) 32:710–25. doi: 10.1016/j.annonc.2021.02.015
11. Ledermann JA. Front-line therapy of advanced ovarian cancer: new approaches. Ann Oncol (2017) 28:viii46–50. doi: 10.1093/annonc/mdx452
12. Chiappa M, Guffanti F, Bertoni F, Colombo I, Damia G. Overcoming PARPi resistance: Preclinical and clinical evidence in ovarian cancer. Drug Resist Update (2021) 55:100744. doi: 10.1016/j.drup.2021.100744
13. Mirza MR, Coleman RL, Gonzalez-Martin A, Moore KN, Colombo N, Ray-Coquard I, et al. The forefront of ovarian cancer therapy: update on PARP inhibitors. Ann Oncol (2020) 31:1148–59. doi: 10.1016/j.annonc.2020.06.004
14. Zeng Q, Ma X, Song Y, Chen Q, Jiao Q, Zhou L. Targeting regulated cell death in tumor nanomedicines. Theranostics (2022) 12:817–41. doi: 10.7150/thno.67932
15. Tummers B, Green DR. The evolution of regulated cell death pathways in animals and their evasion by pathogens. Physiol Rev (2022) 102:411–54. doi: 10.1152/physrev.00002.2021
16. Zamzami N, Hirsch T, Dallaporta B, Petit PX, Kroemer G. Mitochondrial implication in and programmed cell death: apoptosis and necrosis. J Bioenerg Biomembr (1997) 29(2):185–93. doi: 10.1023/A:1022694131572
17. Christgen S, Tweedell RE, Kanneganti TD. Programming inflammatory cell death for therapy. Pharmacol Ther (2022) 232:108010. doi: 10.1016/j.pharmthera.2021.108010
18. Moujalled D, Strasser A, Liddell JR. Molecular mechanisms of cell death in neurological diseases. Cell Death Differ (2021) 28:2029–44. doi: 10.1038/s41418-021-00814-y
19. Dionisio PA, Amaral JD, Rodrigues CMP. Oxidative stress and regulated cell death in parkinson's disease. Ageing Res Rev (2021) 67:101263. doi: 10.1016/j.arr.2021.101263
20. Koren E, Fuchs Y. Modes of regulated cell death in cancer. Cancer Discov (2021) 11:245–65. doi: 10.1158/2159-8290.CD-20-0789
21. Bertheloot D, Latz E, Franklin BS. Necroptosis, pyroptosis and apoptosis: an intricate game of cell death. Cell Mol Immunol (2021) 18:1106–21. doi: 10.1038/s41423-020-00630-3
22. Verdonck S, Nemegeer J, Vandenabeele P, Maelfait J. Viral manipulation of host cell necroptosis and pyroptosis. Trends Microbiol (2021) 30:593–605. doi: 10.1016/j.tim.2021.11.011
23. Roberts JZ, Crawford N, Longley DB. The role of ubiquitination in apoptosis and necroptosis. Cell Death Differ (2022) 29:272–84. doi: 10.1038/s41418-021-00922-9
24. Yu P, Zhang X, Liu N, Tang L, Peng C, Chen X. Pyroptosis: mechanisms and diseases. Signal Transduct Target Ther (2021) 6:128. doi: 10.1038/s41392-021-00507-5
25. Wu D, Wang S, Yu G, Chen X. Cell death mediated by the pyroptosis pathway with the aid of nanotechnology: Prospects for cancer therapy. Angew Chem Int Ed Engl (2021) 60:8018–34. doi: 10.1002/anie.202010281
26. Rana N, Privitera G, Kondolf HC, Bulek K, Lechuga S, De Salvo C, et al. GSDMB is increased in IBD and regulates epithelial restitution/repair independent of pyroptosis. Cell (2022) 185:283–298.e217. doi: 10.1016/j.cell.2021.12.024
27. Lu Z, Van Eeckhoutte HP, Liu G, Nair PM, Jones B, Gillis CM, et al. Necroptosis signaling promotes inflammation, airway remodeling, and emphysema in chronic obstructive pulmonary disease. Am J Respir Crit Care Med (2021) 204:667–81. doi: 10.1164/rccm.202009-3442OC
28. Baik JY, Liu Z, Jiao D, Kwon HJ, Yan J, Kadigamuwa C, et al. ZBP1 not RIPK1 mediates tumor necroptosis in breast cancer. Nat Commun (2021) 12:2666. doi: 10.1038/s41467-021-23004-3
29. Tan Y, Sun R, Liu L, Yang D, Xiang Q, Li L, et al. Tumor suppressor DRD2 facilitates M1 macrophages and restricts NF-kappaB signaling to trigger pyroptosis in breast cancer. Theranostics (2021) 11:5214–31. doi: 10.7150/thno.58322
30. Chen J, Wei Z, Fu K, Duan Y, Zhang M, Li K, et al. Non-apoptotic cell death in ovarian cancer: Treatment, resistance and prognosis. BioMed Pharmacother (2022) 150:112929. doi: 10.1016/j.biopha.2022.112929
31. Liu T, Hou M, Li M, Qiu C, Cheng L, Zhu T, et al. Pyroptosis: A developing foreland of ovarian cancer treatment. Front Oncol (2022) 12:828303. doi: 10.3389/fonc.2022.828303
32. Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell (2012) 149:1060–72. doi: 10.1016/j.cell.2012.03.042
33. Rodriguez R, Schreiber SL, Conrad M. Persister cancer cells: Iron addiction and vulnerability to ferroptosis. Mol Cell (2022) 82:728–40. doi: 10.1016/j.molcel.2021.12.001
34. Yao X, Li W, Fang, Xiao C, Wu X, Li M, et al. Emerging roles of energy metabolism in ferroptosis regulation of tumor cells. Adv Sci (Weinh) (2021) 8:e2100997. doi: 10.1002/advs.202100997
35. Ajoolabady A, Aslkhodapasandhokmabad H, Libby P, Tuomilehto J, Lip GYH, Penninger JM, et al. Ferritinophagy and ferroptosis in the management of metabolic diseases. Trends Endocrinol Metab (2021) 32:444–62. doi: 10.1016/j.tem.2021.04.010
36. Chen X, Kang R, Kroemer G, Tang D. Ferroptosis in infection, inflammation, and immunity. J Exp Med (2021) 218(6):e2021051. doi: 10.1084/jem.20210518
37. Wang ZL, Yuan L, Li W, Li JY. Ferroptosis in parkinson's disease: glia-neuron crosstalk. Trends Mol Med (2022) 28:258–69. doi: 10.1016/j.molmed.2022.02.003
38. Chen X, Kang R, Kroemer G, Tang D. Organelle-specific regulation of ferroptosis. Cell Death Differ (2021) 28:2843–56. doi: 10.1038/s41418-021-00859-z
39. Ebrahimi N, Adelian S, Shakerian S, Afshinpour M, Chaleshtori SR, Rostami N, et al. Crosstalk between ferroptosis and the epithelial-mesenchymal transition: Implications for inflammation and cancer therapy. Cytokine Growth Factor Rev (2022) 64:33–45. doi: 10.1016/j.cytogfr.2022.01.006
40. Yang Z, Shi J, Chen L, Fu C, Shi D, Qu H. Role of pyroptosis and ferroptosis in the progression of atherosclerotic plaques. Front Cell Dev Biol (2022) 10:811196. doi: 10.3389/fcell.2022.811196
41. Chen J, Li X, Ge C, Min J, Wang F. The multifaceted role of ferroptosis in liver disease. Cell Death Differ (2022) 29:467–80. doi: 10.1038/s41418-022-00941-0
42. Lei G, Zhuang L, Gan B. Targeting ferroptosis as a vulnerability in cancer. Nat Rev Cancer (2022) 22:381–96. doi: 10.1038/s41568-022-00459-0
43. Zhang C, Liu X, Jin S, Chen Y, Guo R. Ferroptosis in cancer therapy: a novel approach to reversing drug resistance. Mol Cancer (2022) 21:47. doi: 10.1186/s12943-022-01530-y
44. Friedmann Angeli JP, Xavier da Silva TN, Schilling B. CD8(+) T cells PUF(A)ing the flames of cancer ferroptotic cell death. Cancer Cell (2022) 40:346–8. doi: 10.1016/j.ccell.2022.03.003
45. Shui S, Zhao Z, Wang H, Conrad M, Liu G. Non-enzymatic lipid peroxidation initiated by photodynamic therapy drives a distinct ferroptosis-like cell death pathway. Redox Biol (2021) 45:102056. doi: 10.1016/j.redox.2021.102056
46. Saito Y. Lipid peroxidation products as a mediator of toxicity and adaptive response - the regulatory role of selenoprotein and vitamin e. Arch Biochem Biophys (2021) 703:108840. doi: 10.1016/j.abb.2021.108840
47. Foret MK, Lincoln R, Do Carmo S, Cuello AC, Cosa G. Connecting the "Dots": From free radical lipid autoxidation to cell pathology and disease. Chem Rev (2020) 120:12757–87. doi: 10.1021/acs.chemrev.0c00761
48. Stoyanovsky DA, Tyurina YY, Shrivastava I, Bahar I, Tyurin VA, Protchenko O, et al. Iron catalysis of lipid peroxidation in ferroptosis: Regulated enzymatic or random free radical reaction? Free Radic Biol Med (2019) 133:153–61. doi: 10.1016/j.freeradbiomed.2018.09.008
49. Li Y, Yang J, Gu G, Guo X, He C, Sun J, et al. Pulmonary delivery of theranostic nanoclusters for lung cancer ferroptosis with enhanced Chemodynamic/Radiation synergistic therapy. Nano Lett (2022) 22:963–72. doi: 10.1021/acs.nanolett.1c03786
50. Lu S, Wang XZ, He C, Wang L, Liang SP, Wang CC, et al. ATF3 contributes to brucine-triggered glioma cell ferroptosis via promotion of hydrogen peroxide and iron. Acta Pharmacol Sin (2021) 42:1690–702. doi: 10.1038/s41401-021-00700-w
51. Feng Z, Qin Y, Huo F, Jian Z, Li X, Geng J, et al. NMN recruits GSH to enhance GPX4-mediated ferroptosis defense in UV irradiation induced skin injury. Biochim Biophys Acta Mol Basis Dis (2022) 1868:166287. doi: 10.1016/j.bbadis.2021.166287
52. Zhu ZY, Liu YD, Gong Y, Jin W, Topchiy E, Turdi S, et al. Mitochondrial aldehyde dehydrogenase (ALDH2) rescues cardiac contractile dysfunction in an APP/PS1 murine model of alzheimer's disease via inhibition of ACSL4-dependent ferroptosis. Acta Pharmacol Sin (2022) 43:39–49. doi: 10.1038/s41401-021-00635-2
53. Alvarez SW, Sviderskiy VO, Terzi EM, Papagiannakopoulos T, Moreira AL, Adams S, et al. NFS1 undergoes positive selection in lung tumours and protects cells from ferroptosis. Nature (2017) 551:639–43. doi: 10.1038/nature24637
54. Wei XB, Jiang WQ, Zeng JH, Huang LQ, Ding HG, Jing YW, et al. Exosome-derived lncRNA NEAT1 exacerbates sepsis-associated encephalopathy by promoting ferroptosis through regulating miR-9-5p/TFRC and GOT1 axis. Mol Neurobiol (2022) 59:1954–69. doi: 10.1007/s12035-022-02738-1
55. Zhang R, Pan T, Xiang Y, Zhang M, Xie H, Liang Z, et al. Curcumenol triggered ferroptosis in lung cancer cells via lncRNA H19/miR-19b-3p/FTH1 axis. Bioact Mater (2022) 13:23–36. doi: 10.1016/j.bioactmat.2021.11.013
56. Shi H, Hou B, Li H, Zhou H, Du B. Cyclophosphamide induces the ferroptosis of tumor cells through heme oxygenase-1. Front Pharmacol (2022) 13:839464. doi: 10.3389/fphar.2022.839464
57. Yang S, Ouyang J, Lu Y, Harypursat V, Chen Y. A dual role of heme oxygenase-1 in tuberculosis. Front Immunol (2022) 13:842858. doi: 10.3389/fimmu.2022.842858
58. Yang Y, Lin Y, Wang M, Yuan K, Wang Q, Mu P, et al. Targeting ferroptosis suppresses osteocyte glucolipotoxicity and alleviates diabetic osteoporosis. Bone Res (2022) 10:26. doi: 10.1038/s41413-022-00198-w
59. He J, Li Z, Xia P, Shi A, FuChen X, Zhang J, et al. Ferroptosis and ferritinophagy in diabetes complications. Mol Metab (2022) 60:101470. doi: 10.1016/j.molmet.2022.101470
60. Santana-Codina N, Gikandi A, Mancias JD. The role of NCOA4-mediated ferritinophagy in ferroptosis. Adv Exp Med Biol (2021) 1301:41–57. doi: 10.1007/978-3-030-62026-4_4
61. Liu B, Li X, Wang D, Yu Y, Lu D, Chen L, et al. CEMIP promotes extracellular matrix-detached prostate cancer cells survival by inhibiting ferroptosis. Cancer Sci (2022) 113:2056–70. doi: 10.1111/cas.15356
62. Wu X, Qin K, Iroegbu CD, Xiang K, Peng J, Guo J, et al. Genetic analysis of potential biomarkers and therapeutic targets in ferroptosis from coronary artery disease. J Cell Mol Med (2022) 26:2177–90. doi: 10.1111/jcmm.17239
63. Zhou Y, Lin W, Rao T, Zheng J, Zhang T, Zhang M, et al. Ferroptosis and its potential role in the nervous system diseases. J Inflammation Res (2022) 15:1555–74. doi: 10.2147/JIR.S351799
64. Homma T, Kobayashi S, Conrad M, Konno H, Yokoyama C, Fujii J. Nitric oxide protects against ferroptosis by aborting the lipid peroxidation chain reaction. Nitric Oxide (2021) 115:34–43. doi: 10.1016/j.niox.2021.07.003
65. Huang C, Zhan L. Network pharmacology identifies therapeutic targets and the mechanisms of glutathione action in ferroptosis occurring in oral cancer. Front Pharmacol (2022) 13:851540. doi: 10.3389/fphar.2022.851540
66. Li Q, Su R, Bao X, Cao K, Du Y, Wang N, et al. Glycyrrhetinic acid nanoparticles combined with ferrotherapy for improved cancer immunotherapy. Acta Biomater (2022) 144:109–20. doi: 10.1016/j.actbio.2022.03.030
67. Pallotti F, Bergamini C, Lamperti C, Fato R. The roles of coenzyme q in disease: Direct and indirect involvement in cellular functions. Int J Mol Sci (2021) 23:128. doi: 10.3390/ijms23010128
68. Gan B. Mitochondrial regulation of ferroptosis. J Cell Biol (2021) 220(9):e202105043. doi: 10.1083/jcb.202105043
69. Yan J, Wan P, Choksi S, Liu ZG. Necroptosis and tumor progression. Trends Cancer (2022) 8:21–7. doi: 10.1016/j.trecan.2021.09.003
70. Wang B, Cui Y, Zhang Q, Wang S, Xu S. Selenomethionine alleviates LPS-induced JNK/NLRP3 inflammasome-dependent necroptosis by modulating miR-15a and oxidative stress in chicken lungs. Metallomics (2021) 13(8):mfab048. doi: 10.1093/mtomcs/mfab048
71. Wang Y, Kanneganti TD. From pyroptosis, apoptosis and necroptosis to PANoptosis: A mechanistic compendium of programmed cell death pathways. Comput Struct Biotechnol J (2021) 19:4641–57. doi: 10.1016/j.csbj.2021.07.038
72. Teh CE, Preston SP, Robbins AK, Stutz MD, Cooney J, Clark MP, et al. Caspase-8 has dual roles in regulatory T cell homeostasis balancing immunity to infection and collateral inflammatory damage. Sci Immunol (2022) 7:eabn8041. doi: 10.1126/sciimmunol.abn8041
73. Li X, Li F, Zhang X, Zhang H, Zhao Q, Li M, et al. Caspase-8 auto-cleavage regulates programmed cell death and collaborates with RIPK3/MLKL to prevent lymphopenia. Cell Death Differ (2022). doi: 10.1038/s41418-022-00938-9
74. Yin H, Guo X, Chen Y, Zeng Y, Mo X, Hong S, et al. TAB2 deficiency induces dilated cardiomyopathy by promoting RIPK1-dependent apoptosis and necroptosis. J Clin Invest (2022) 132(4):e152297. doi: 10.1172/JCI152297
75. Rosenbaum DM, Degterev A, David J, Rosenbaum PS, Roth S, Grotta JC, et al. Necroptosis, a novel form of caspase-independent cell death, contributes to neuronal damage in a retinal ischemia-reperfusion injury model. J Neurosci Res (2010) 88:1569–76. doi: 10.1002/jnr.22314
76. Zhang R, Kang R, Tang D. The STING1 network regulates autophagy and cell death. Signal Transduct Target Ther (2021) 6:208. doi: 10.1038/s41392-021-00613-4
77. Meng Y, Sandow JJ, Czabotar PE, Murphy JM. The regulation of necroptosis by post-translational modifications. Cell Death Differ (2021) 28:861–83. doi: 10.1038/s41418-020-00722-7
78. Nicole L, Sanavia T, Cappellesso R, Maffeis V, Akiba J, Kawahara A, et al. Necroptosis-driving genes RIPK1, RIPK3 and MLKL-p are associated with intratumoral CD3(+) and CD8(+) T cell density and predict prognosis in hepatocellular carcinoma. J Immunother Cancer (2022) 10(3):e004031. doi: 10.1136/jitc-2021-004031
79. Mohanty S, Yadav P, Lakshminarayanan H, Sharma P, Vivekanandhan A, Karunagaran D. RETRA induces necroptosis in cervical cancer cells through RIPK1, RIPK3, MLKL and increased ROS production. Eur J Pharmacol (2022) 920:174840. doi: 10.1016/j.ejphar.2022.174840
80. Xu D, Zou C, Yuan J. Genetic regulation of RIPK1 and necroptosis. Annu Rev Genet (2021) 55:235–63. doi: 10.1146/annurev-genet-071719-022748
81. Peng T, Tao X, Xia Z, Hu S, Xue J, Zhu Q, et al. Pathogen hijacks programmed cell death signaling by arginine ADPR-deacylization of caspases. Mol Cell (2022) 82:1806–20.e8. doi: 10.1016/j.molcel.2022.03.010
82. Woznicki JA, Saini N, Flood P, Rajaram S, Lee CM, Stamou P, et al. TNF-alpha synergises with IFN-gamma to induce caspase-8-JAK1/2-STAT1-dependent death of intestinal epithelial cells. Cell Death Dis (2021) 12:864. doi: 10.1038/s41419-021-04151-3
83. Karlowitz R, van Wijk SJL. Surviving death: emerging concepts of RIPK3 and MLKL ubiquitination in the regulation of necroptosis. FEBS J (2021). doi: 10.1111/febs.16255
84. Sethi A, Horne CR, Fitzgibbon C, Wilde K, Davies KA, Garnish SE, et al. Membrane permeabilization is mediated by distinct epitopes in mouse and human orthologs of the necroptosis effector, MLKL. Cell Death Differ (2022). doi: 10.1038/s41418-022-00965-6
85. Martens S, Bridelance J, Roelandt R, Vandenabeele P, Takahashi N. MLKL in cancer: more than a necroptosis regulator. Cell Death Differ (2021) 28:1757–72. doi: 10.1038/s41418-021-00785-0
86. Shlomovitz I, Erlich Z, Arad G, Edry-Botzer L, Zargarian S, Cohen H, et al. Proteomic analysis of necroptotic extracellular vesicles. Cell Death Dis (2021) 12:1059. doi: 10.1038/s41419-021-04317-z
87. Yu Z, Jiang N, Su W, Zhuo Y. Necroptosis: A novel pathway in neuroinflammation. Front Pharmacol (2021) 12:701564. doi: 10.3389/fphar.2021.701564
88. Liu L, Li H, Hu D, Wang Y, Shao W, Zhong J, et al. Insights into N6-methyladenosine and programmed cell death in cancer. Mol Cancer (2022) 21:32. doi: 10.1186/s12943-022-01508-w
89. Bauernfried S, Hornung V. Human NLRP1: From the shadows to center stage. J Exp Med (2022) 219(1):e20211405. doi: 10.1084/jem.20211405
90. Hou J, Hsu JM, Hung MC. Molecular mechanisms and functions of pyroptosis in inflammation and antitumor immunity. Mol Cell (2021) 81:4579–90. doi: 10.1016/j.molcel.2021.09.003
91. Zychlinsky A, Prevost MC, Sansonetti PJ. Shigella flexneri induces apoptosis in infected macrophages. Nature (1992) 358:167–9. doi: 10.1038/358167a0
92. Cookson BT, Brennan MA. Pro-inflammatory programmed cell death. Trends Microbiol (2001) 9:113–4. doi: 10.1016/S0966-842X(00)01936-3
93. Shi J, Zhao Y, Wang Y, Gao W, Ding J, Li P, et al. Inflammatory caspases are innate immune receptors for intracellular LPS. Nature (2014) 514:187–92. doi: 10.1038/nature13683
94. Shi J, Zhao Y, Wang K, Shi X, Wang Y, Huang H, et al. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature (2015) 526:660–5. doi: 10.1038/nature15514
95. Diaz-Garcia E, Garcia-Tovar S, Alfaro E, Jaureguizar A, Casitas R, Sanchez-Sanchez B, et al. Inflammasome activation: A keystone of proinflammatory response in obstructive sleep apnea. Am J Respir Crit Care Med (2022) 205:1337–48. doi: 10.1164/rccm.202106-1445OC
96. Hsu CG, Chavez CL, Zhang C, Sowden M, Yan C, Berk BC. The lipid peroxidation product 4-hydroxynonenal inhibits NLRP3 inflammasome activation and macrophage pyroptosis. Cell Death Differ (2022). doi: 10.1038/s41418-022-00966-5
97. Xu T, Yu W, Fang H, Wang Z, Chi Z, Guo X, et al. Ubiquitination of NLRP3 by gp78/Insig-1 restrains NLRP3 inflammasome activation. Cell Death Differ (2022). doi: 10.1038/s41418-022-00947-8
98. Ross C, Chan AH, Pein JBv, Maddugoda MP, Boucher D, Schroder K. Inflammatory caspases: Toward a unified model for caspase activation by inflammasomes. Annu Rev Immunol (2022) 40:249–69. doi: 10.1146/annurev-immunol-101220-030653
99. Camilli G, Bohm M, Piffer AC, Lavenir R, Williams DL, Neven B, et al. Beta-glucan-induced reprogramming of human macrophages inhibits NLRP3 inflammasome activation in cryopyrinopathies. J Clin Invest (2020) 130:4561–73. doi: 10.1172/JCI134778
100. Lee S, Ishitsuka A, Noguchi M, Hirohama M, Fujiyasu Y, Petric PP, et al. Influenza restriction factor MxA functions as inflammasome sensor in the respiratory epithelium. Sci Immunol (2019) 4(40):eaau4643. doi: 10.1126/sciimmunol.aau4643
101. Stancu IC, Cremers N, Vanrusselt H, Couturier J, Vanoosthuyse A, Kessels S, et al. Aggregated tau activates NLRP3-ASC inflammasome exacerbating exogenously seeded and non-exogenously seeded tau pathology in vivo. Acta Neuropathol (2019) 137:599–617. doi: 10.1007/s00401-018-01957-y
102. Zhang P, Liu Y, Hu L, Huang K, Hong M, Wang Y, et al. NLRC4 inflammasome-dependent cell death occurs by a complementary series of three death pathways and determines lethality in mice. Sci Adv (2021) 7:eabi9471. doi: 10.1126/sciadv.abi9471
103. Wu J, Raman A, Coffey NJ, Sheng X, Wahba J, Seasock MJ, et al. The key role of NLRP3 and STING in APOL1-associated podocytopathy. J Clin Invest (2021) 131(20):e136329. doi: 10.1172/JCI136329
104. Ding B, Sheng J, Zheng P, Li C, Li D, Cheng Z, et al. Biodegradable upconversion nanoparticles induce pyroptosis for cancer immunotherapy. Nano Lett (2021) 21:8281–9. doi: 10.1021/acs.nanolett.1c02790
105. de Carvalho Ribeiro M, Szabo G. Role of the inflammasome in liver disease. Annu Rev Pathol (2022) 17:345–65. doi: 10.1146/annurev-pathmechdis-032521-102529
106. Shao S, Chen C, Shi G, Zhou Y, Wei Y, Fan N, et al. Therapeutic potential of the target on NLRP3 inflammasome in multiple sclerosis. Pharmacol Ther (2021) 227:107880. doi: 10.1016/j.pharmthera.2021.107880
107. Liu J, Du S, Kong Q, Zhang X, Jiang S, Cao X, et al. HSPA12A attenuates lipopolysaccharide-induced liver injury through inhibiting caspase-11-mediated hepatocyte pyroptosis via PGC-1alpha-dependent acyloxyacyl hydrolase expression. Cell Death Differ (2020) 27:2651–67. doi: 10.1038/s41418-020-0536-x
108. Lee BL, Stowe IB, Gupta A, Kornfeld OS, Roose-Girma M, Anderson K, et al. Caspase-11 auto-proteolysis is crucial for noncanonical inflammasome activation. J Exp Med (2018) 215:2279–88. doi: 10.1084/jem.20180589
109. Gao W, Li Y, Liu X, Wang S, Mei P, Chen Z, et al. TRIM21 regulates pyroptotic cell death by promoting gasdermin d oligomerization. Cell Death Differ (2022) 29:439–50. doi: 10.1038/s41418-021-00867-z
110. Petrovski G, Ayna G, Majai G, Hodrea J, Benko S, Madi A, et al. Phagocytosis of cells dying through autophagy induces inflammasome activation and IL-1beta release in human macrophages. Autophagy (2011) 7:321–30. doi: 10.4161/auto.7.3.14583
111. Moujalled D, Southon AG, Saleh E, Brinkmann K, Ke F, Iliopoulos M, et al. BH3 mimetic drugs cooperate with temozolomide, JQ1 and inducers of ferroptosis in killing glioblastoma multiforme cells. Cell Death Differ (2022). doi: 10.1038/s41418-022-00977-2
112. Li K, Lin C, Li M, Xu K, He Y, Mao Y, et al. Multienzyme-like reactivity cooperatively impairs glutathione peroxidase 4 and ferroptosis suppressor protein 1 pathways in triple-negative breast cancer for sensitized ferroptosis therapy. ACS Nano (2022) 16:2381–98. doi: 10.1021/acsnano.1c08664
113. Chen Y, Li L, Lan J, Cui Y, Rao X, Zhao J, et al. CRISPR screens uncover protective effect of PSTK as a regulator of chemotherapy-induced ferroptosis in hepatocellular carcinoma. Mol Cancer (2022) 21:11. doi: 10.1186/s12943-021-01466-9
114. Bolik J, Krause F, Stevanovic M, Gandrass M, Thomsen I, Schacht SS, et al. Inhibition of ADAM17 impairs endothelial cell necroptosis and blocks metastasis. J Exp Med (2022) 219(1):e20201039. doi: 10.1084/jem.20201039
115. Tan Y, Sementino E, Cheung M, Peri S, Menges CW, Kukuyan AM, et al. Somatic epigenetic silencing of RIPK3 inactivates necroptosis and contributes to chemoresistance in malignant mesothelioma. Clin Cancer Res (2021) 27:1200–13. doi: 10.1158/1078-0432.CCR-18-3683
116. Xie Y, Zhao Y, Shi L, Li W, Chen K, Li M, et al. Gut epithelial TSC1/mTOR controls RIPK3-dependent necroptosis in intestinal inflammation and cancer. J Clin Invest (2020) 130:2111–28. doi: 10.1172/JCI133264
117. Tang Q, Li W, Zheng X, Ren L, Liu J, Li S, et al. MELK is an oncogenic kinase essential for metastasis, mitotic progression, and programmed death in lung carcinoma. Signal Transduct Target Ther (2020) 5:279. doi: 10.1038/s41392-020-00288-3
118. Tan G, Huang C, Chen J, Zhi F. HMGB1 released from GSDME-mediated pyroptotic epithelial cells participates in the tumorigenesis of colitis-associated colorectal cancer through the ERK1/2 pathway. J Hematol Oncol (2020) 13:149. doi: 10.1186/s13045-020-00985-0
119. Zhang T, Wu DM, Luo PW, Liu T, Han R, Deng SH, et al. CircNEIL3 mediates pyroptosis to influence lung adenocarcinoma radiotherapy by upregulating PIF1 through miR-1184 inhibition. Cell Death Dis (2022) 13:167. doi: 10.1038/s41419-022-04561-x
120. Banoth B, Tuladhar S, Karki R, Sharma BR, Briard B, Kesavardhana S, et al. ZBP1 promotes fungi-induced inflammasome activation and pyroptosis, apoptosis, and necroptosis (PANoptosis). J Biol Chem (2020) 295:18276–83. doi: 10.1074/jbc.RA120.015924
121. Shi CS, Kehrl JH. Bcl-2 regulates pyroptosis and necroptosis by targeting BH3-like domains in GSDMD and MLKL. Cell Death Discovery (2019) 5:151. doi: 10.1038/s41420-019-0230-2
122. Fritsch M, Gunther SD, Schwarzer R, Albert MC, Schorn F, Werthenbach JP, et al. Caspase-8 is the molecular switch for apoptosis, necroptosis and pyroptosis. Nature (2019) 575:683–7. doi: 10.1038/s41586-019-1770-6
123. Zhou B, Zhang JY, Liu XS, Chen HZ, Ai YL, Cheng K, et al. Tom20 senses iron-activated ROS signaling to promote melanoma cell pyroptosis. Cell Res (2018) 28:1171–85. doi: 10.1038/s41422-018-0090-y
124. Wang J, Deng B, Liu Q, Huang Y, Chen W, Li J, et al. Pyroptosis and ferroptosis induced by mixed lineage kinase 3 (MLK3) signaling in cardiomyocytes are essential for myocardial fibrosis in response to pressure overload. Cell Death Dis (2020) 11:574. doi: 10.1038/s41419-020-02777-3
125. Liu Y, Chen Q, Zhu Y, Wang T, Ye L, Han L, et al. Non-coding RNAs in necroptosis, pyroptosis and ferroptosis in cancer metastasis. Cell Death Discovery (2021) 7(1):210. doi: 10.1038/s41420-021-00596-9
126. Li D, Zhang M, Chao H. Significance of glutathione peroxidase 4 and intracellular iron level in ovarian cancer cells-"utilization" of ferroptosis mechanism. Inflammation Res (2021) 70:1177–89. doi: 10.1007/s00011-021-01495-6
127. Tesfay L, Paul BT, Konstorum A, Deng Z, Cox AO, Lee J, et al. Stearoyl-CoA desaturase 1 protects ovarian cancer cells from ferroptotic cell death. Cancer Res (2019) 79:5355–66. doi: 10.1158/0008-5472.CAN-19-0369
128. Ma LL, Liang L, Zhou D, Wang SW. Tumor suppressor miR-424-5p abrogates ferroptosis in ovarian cancer through targeting ACSL4. Neoplasma (2021) 68:165–73. doi: 10.4149/neo_2020_200707N705
129. Zhang Y, Xia M, Zhou Z, Hu X, Wang J, Zhang M, et al. p53 promoted ferroptosis in ovarian cancer cells treated with human serum incubated-superparamagnetic iron oxides. Int J Nanomed (2021) 16:283–96. doi: 10.2147/IJN.S282489
130. Hong T, Lei G, Chen X, Li H, Zhang X, Wu N, et al. PARP inhibition promotes ferroptosis via repressing SLC7A11 and synergizes with ferroptosis inducers in BRCA-proficient ovarian cancer. Redox Biol (2021) 42:101928. doi: 10.1016/j.redox.2021.101928
131. Jin Y, Chen L, Li L, Huang G, Huang H, Tang C. SNAI2 promotes the development of ovarian cancer through regulating ferroptosis. Bioengineered (2022) 13:6451–63. doi: 10.1080/21655979.2021.2024319
132. Cai L, Hu X, Ye L, Bai P, Jie Y, Shu K. Long non-coding RNA ADAMTS9-AS1 attenuates ferroptosis by targeting microRNA-587/solute carrier family 7 member 11 axis in epithelial ovarian cancer. Bioengineered (2022) 13:8226–39. doi: 10.1080/21655979.2022.2049470
133. Mao G, Xin D, Wang Q, Lai D. Sodium molybdate inhibits the growth of ovarian cancer cells via inducing both ferroptosis and apoptosis. Free Radic Biol Med (2022) 182:79–92. doi: 10.1016/j.freeradbiomed.2022.02.023
134. Yang WH, Huang Z, Wu J, Ding CC, Murphy SK, Chi JT. A TAZ-ANGPTL4-NOX2 axis regulates ferroptotic cell death and chemoresistance in epithelial ovarian cancer. Mol Cancer Res (2020) 18:79–90. doi: 10.1158/1541-7786.MCR-19-0691
135. Chakraborty PK, Xiong X, Mustafi SB, Saha S, Dhanasekaran D, Mandal NA, et al. Role of cystathionine beta synthase in lipid metabolism in ovarian cancer. Oncotarget (2015) 6:37367–84. doi: 10.18632/oncotarget.5424
136. Wang Y, Zhao G, Condello S, Huang H, Cardenas H, Tanner EJ, et al. Frizzled-7 identifies platinum-tolerant ovarian cancer cells susceptible to ferroptosis. Cancer Res (2021) 81:384–99. doi: 10.1158/0008-5472.CAN-20-1488
137. Cheng Q, Bao L, Li M, Chang K, Yi X. Erastin synergizes with cisplatin via ferroptosis to inhibit ovarian cancer growth in vitro and in vivo. J Obstet Gynaecol Res (2021) 47:2481–91. doi: 10.1111/jog.14779
138. Li HW, Liu MB, Jiang X, Song T, Feng SX, Wu JY, et al. GALNT14 regulates ferroptosis and apoptosis of ovarian cancer through the EGFR/mTOR pathway. Future Oncol (2022) 18:149–61. doi: 10.2217/fon-2021-0883
139. Chan DW, Yung MM, Chan YS, Xuan Y, Yang H, Xu D, et al. MAP30 protein from momordica charantia is therapeutic and has synergic activity with cisplatin against ovarian cancer in vivo by altering metabolism and inducing ferroptosis. Pharmacol Res (2020) 161:105157. doi: 10.1016/j.phrs.2020.105157
140. Sun D, Li YC, Zhang XY. Lidocaine promoted ferroptosis by targeting miR-382-5p /SLC7A11 axis in ovarian and breast cancer. Front Pharmacol (2021) 12:681223. doi: 10.3389/fphar.2021.681223
141. Basuli D, Tesfay L, Deng Z, Paul B, Yamamoto Y, Ning G, et al. Iron addiction: a novel therapeutic target in ovarian cancer. Oncogene (2017) 36:4089–99. doi: 10.1038/onc.2017.11
142. Quartuccio SM, Karthikeyan S, Eddie SL, Lantvit DD, Ó hAinmhire E, Modi DA, et al. Mutant p53 expression in fallopian tube epithelium drives cell migration. Int J Cancer (2015) 137:1528–38. doi: 10.1002/ijc.29528
143. Liu N, Lin X, Huang C. Activation of the reverse transsulfuration pathway through NRF2/CBS confers erastin-induced ferroptosis resistance. Br J Cancer (2020) 122:279–92. doi: 10.1038/s41416-019-0660-x
144. Verschoor ML, Singh G. Ets-1 regulates intracellular glutathione levels: key target for resistant ovarian cancer. Mol Cancer (2013) 12:138. doi: 10.1186/1476-4598-12-138
145. Santos I, Ramos C, Mendes C, Sequeira CO, Tome CS, Fernandes DGH, et al. Targeting glutathione and cystathionine beta-synthase in ovarian cancer treatment by selenium-chrysin polyurea dendrimer nanoformulation. Nutrients (2019) 11:2523. doi: 10.3390/11102523.
146. Yang L, Tian S, Chen Y, Miao C, Zhao Y, Wang R, et al. Ferroptosis-related gene model to predict overall survival of ovarian carcinoma. J Oncol (2021) 2021:6687391. doi: 10.1155/2021/6687391
147. Zhang J, Xi J, Huang P, Zeng S. Comprehensive analysis identifies potential ferroptosis-associated mRNA therapeutic targets in ovarian cancer. Front Med (Lausanne) (2021) 8:644053. doi: 10.3389/fmed.2021.644053
148. Ye Y, Dai Q, Li S, He J, Qi H. A novel defined risk signature of the ferroptosis-related genes for predicting the prognosis of ovarian cancer. Front Mol Biosci (2021) 8:645845. doi: 10.3389/fmolb.2021.645845
149. Yu Z, He H, Chen Y, Ji Q, Sun M. A novel ferroptosis related gene signature is associated with prognosis in patients with ovarian serous cystadenocarcinoma. Sci Rep (2021) 11:11486. doi: 10.1038/s41598-021-90126-5
150. You Y, Fan Q, Huang J, Wu Y, Lin H, Zhang Q. Ferroptosis-related gene signature promotes ovarian cancer by influencing immune infiltration and invasion. J Oncol (2021) 2021:9915312. doi: 10.1155/2021/9915312
151. Li XX, Xiong L, Wen Y, Zhang ZJ. Comprehensive analysis of the tumor microenvironment and ferroptosis-related genes predict prognosis with ovarian cancer. Front Genet (2021) 12:774400. doi: 10.3389/fgene.2021.774400
152. Feng S, Yin H, Zhang K, Shan M, Ji X, Luo S, et al. Integrated clinical characteristics and omics analysis identifies a ferroptosis and iron-metabolism-related lncRNA signature for predicting prognosis and therapeutic responses in ovarian cancer. J Ovarian Res (2022) 15:10. doi: 10.1186/s13048-022-00944-y
153. Wang H, Cheng Q, Chang K, Bao L, Yi X. Integrated analysis of ferroptosis-related biomarker signatures to improve the diagnosis and prognosis prediction of ovarian cancer. Front Cell Dev Biol (2021) 9:807862. doi: 10.3389/fcell.2021.807862
154. Peng J, Hao Y, Rao B, Zhang Z. A ferroptosis-related lncRNA signature predicts prognosis in ovarian cancer patients. Transl Cancer Res (2021) 10:4802–16. doi: 10.21037/tcr-21-1152
155. Zheng J, Guo J, Wang Y, Zheng Y, Zhang K, Tong J. Bioinformatic analyses of the ferroptosis-related lncRNAs signature for ovarian cancer. Front Mol Biosci (2021) 8:735871. doi: 10.3389/fmolb.2021.735871
156. Hahne JC, Kurz A, Meyer SR, Dietl J, Engel JB, Honig A. Anti-tumour activity of phosphoinositide-3-kinase antagonist AEZS-126 in models of ovarian cancer. Arch Gynecol Obstet (2015) 291:131–41. doi: 10.1007/s00404-014-3389-z
157. McCabe KE, Bacos K, Lu D, Delaney JR, Axelrod J, Potter MD, et al. Triggering necroptosis in cisplatin and IAP antagonist-resistant ovarian carcinoma. Cell Death Dis (2014) 5:e1496. doi: 10.1038/cddis.2014.448
158. Qiu X, Klausen C, Cheng JC, Leung PC. CD40 ligand induces RIP1-dependent, necroptosis-like cell death in low-grade serous but not serous borderline ovarian tumor cells. Cell Death Dis (2015) 6:e1864. doi: 10.1038/cddis.2015.229
159. Dey A, Mustafi SB, Saha S, Kumar Dhar Dwivedi S, Mukherjee P, Bhattacharya R. Inhibition of BMI1 induces autophagy-mediated necroptosis. Autophagy (2016) 12:659–70. doi: 10.1080/15548627.2016.1147670
160. Hernandez L, Kim MK, Noonan AM, Sagher E, Kohlhammer H, Wright G, et al. A dual role for Caspase8 and NF-kappaB interactions in regulating apoptosis and necroptosis of ovarian cancer, with correlation to patient survival. Cell Death Discov (2015) 1:15053. doi: 10.1038/cddiscovery.2015.53
161. Wu NY, Huang HS, Chao TH, Chou HM, Fang C, Qin CZ, et al. Progesterone prevents high-grade serous ovarian cancer by inducing necroptosis of p53-defective fallopian tube epithelial cells. Cell Rep (2017) 18:2557–65. doi: 10.1016/j.celrep.2017.02.049
162. Zhang X, Kitatani K, Toyoshima M, Ishibashi M, Usui T, Minato J, et al. Ceramide nanoliposomes as a MLKL-dependent, necroptosis-inducing, chemotherapeutic reagent in ovarian cancer. Mol Cancer Ther (2018) 17:50–9. doi: 10.1158/1535-7163.MCT-17-0173
163. Thibault B, Genre L, Le Naour A, Broca C, Mery E, Vuagniaux G, et al. DEBIO 1143, an IAP inhibitor, reverses carboplatin resistance in ovarian cancer cells and triggers apoptotic or necroptotic cell death. Sci Rep (2018) 8:17862. doi: 10.1038/s41598-018-35860-z
164. Chefetz I, Grimley E, Yang K, Hong L, Vinogradova EV, Suciu R, et al. A pan-ALDH1A inhibitor induces necroptosis in ovarian cancer stem-like cells. Cell Rep (2019) 26:3061–3075.e3066. doi: 10.1016/j.celrep.2019.02.032
165. Chen W, Wang X, Zhao B, Zhang R, Xie Z, He Y, et al. CuS-MnS2 nano-flowers for magnetic resonance imaging guided photothermal/photodynamic therapy of ovarian cancer through necroptosis. Nanoscale (2019) 11:12983–9. doi: 10.1039/C9NR03114F
166. Liu L, Fan J, Ai G, Liu J, Luo N, Li C, et al. Berberine in combination with cisplatin induces necroptosis and apoptosis in ovarian cancer cells. Biol Res (2019) 52:37. doi: 10.1186/s40659-019-0243-6
167. Zheng XL, Yang JJ, Wang YY, Li Q, Song YP, Su M, et al. RIP1 promotes proliferation through G2/M checkpoint progression and mediates cisplatin-induced apoptosis and necroptosis in human ovarian cancer cells. Acta Pharmacol Sin (2020) 41:1223–33. doi: 10.1038/s41401-019-0340-7
168. Liang J, Zhou J, Xu Y, Huang X, Wang X, Huang W, et al. Osthole inhibits ovarian carcinoma cells through LC3-mediated autophagy and GSDME-dependent pyroptosis except for apoptosis. Eur J Pharmacol (2020) 874:172990. doi: 10.1016/j.ejphar.2020.172990
169. Qiao L, Wu X, Zhang J, Liu L, Sui X, Zhang R, et al. Alpha-NETA induces pyroptosis of epithelial ovarian cancer cells through the GSDMD/caspase-4 pathway. FASEB J (2019) 33:12760–7. doi: 10.1096/fj.201900483RR
170. Zhang R, Chen J, Mao L, Guo Y, Hao Y, Deng Y, et al. Nobiletin triggers reactive oxygen species-mediated pyroptosis through regulating autophagy in ovarian cancer cells. J Agric Food Chem (2020) 68:1326–36. doi: 10.1021/acs.jafc.9b07908
171. Tan C, Liu W, Zheng ZH, Wan XG. LncRNA HOTTIP inhibits cell pyroptosis by targeting miR-148a-3p/AKT2 axis in ovarian cancer. Cell Biol Int (2021) 45:1487–97. doi: 10.1002/cbin.11588
172. Ye Y, Dai Q, Qi H. A novel defined pyroptosis-related gene signature for predicting the prognosis of ovarian cancer. Cell Death Discovery (2021) 7:71. doi: 10.1038/s41420-021-00451-x
173. Berkel C, Cacan E. Differential expression and copy number variation of gasdermin (GSDM) family members, pore-forming proteins in pyroptosis, in normal and malignant serous ovarian tissue. Inflammation (2021) 44:2203–16. doi: 10.1007/s10753-021-01493-0
174. Zhou M, Li B, Liu J, Hong L. Genomic, immunological, and clinical characterization of pyroptosis in ovarian cancer. J Inflammation Res (2021) 14:7341–58. doi: 10.2147/JIR.S344554
175. Zhang Z, Xu Z, Yan Y. Role of a pyroptosis-related lncRNA signature in risk stratification and immunotherapy of ovarian cancer. Front Med (Lausanne) (2021) 8:793515. doi: 10.3389/fmed.2021.793515
176. Cao X, Zhang Q, Zhu Y, Huo X, Bao J, Su M. Derivation, comprehensive analysis, and assay validation of a pyroptosis-related lncRNA prognostic signature in patients with ovarian cancer. Front Oncol (2022) 12:780950. doi: 10.3389/fonc.2022.780950
177. Cai Z, Yuan S, Luan X, Feng J, Deng L, Zuo Y, et al. Pyroptosis-related inflammasome pathway: A new therapeutic target for diabetic cardiomyopathy. Front Pharmacol (2022) 13:842313. doi: 10.3389/fphar.2022.842313
178. Qi X, Li Q, Che X, Wang Q, Wu G. Application of regulatory cell death in cancer: Based on targeted therapy and immunotherapy. Front Immunol (2022) 13:837293. doi: 10.3389/fimmu.2022.837293
179. Zhao J, Jiang P, Guo S, Schrodi SJ, He D. Apoptosis, autophagy, NETosis, necroptosis, and pyroptosis mediated programmed cell death as targets for innovative therapy in rheumatoid arthritis. Front Immunol (2021) 12:809806. doi: 10.3389/fimmu.2021.809806
180. Park Y, Kim MJ, Choi Y, Kim NH, Kim L, Hong SPD, et al. Role of mass spectrometry-based serum proteomics signatures in predicting clinical outcomes and toxicity in patients with cancer treated with immunotherapy. J Immunother Cancer (2022) 10(3):e00356. doi: 10.1136/jitc-2021-003566
181. Huang J, Chang Z, Lu Q, Chen X, Najafi M. Nobiletin as an inducer of programmed cell death in cancer: a review. Apoptosis (2022) 27:297–310. doi: 10.1007/s10495-022-01721-4
182. Zhang S, Wang J, Kong Z, Sun X, He Z, Sun B, et al. Emerging photodynamic nanotherapeutics for inducing immunogenic cell death and potentiating cancer immunotherapy. Biomaterials (2022) 282:121433. doi: 10.1016/j.biomaterials.2022.121433
183. Zuo YB, Zhang YF, Zhang R, Tian JW, Lv XB, Li R, et al. Ferroptosis in cancer progression: Role of noncoding RNAs. Int J Biol Sci (2022) 18:1829–43. doi: 10.7150/ijbs.66917
184. Yang J, Hu S, Bian Y, Yao J, Wang D, Liu X, et al. Targeting cell death: Pyroptosis, ferroptosis, apoptosis and necroptosis in osteoarthritis. Front Cell Dev Biol (2021) 9:789948. doi: 10.3389/fcell.2021.789948
185. Xu W, Huang Y. Regulation of inflammatory cell death by phosphorylation. Front Immunol (2022) 13:851169. doi: 10.3389/fimmu.2022.851169
186. Zhuang J, Xie L, Zheng L. A glimpse of programmed cell death among bacteria, animals, and plants. Front Cell Dev Biol (2021) 9:790117. doi: 10.3389/fcell.2021.790117
187. Liu X, Wan L. Molecular insights into the biochemical functions and signalling mechanisms of plant NLRs. Mol Plant Pathol (2022) 23:772–80. doi: 10.1111/mpp.13195
Keywords: ovarian cancer, ferroptosis, necroptosis, pyroptosis, malignant progression
Citation: Zhang C and Liu N (2022) Ferroptosis, necroptosis, and pyroptosis in the occurrence and development of ovarian cancer. Front. Immunol. 13:920059. doi: 10.3389/fimmu.2022.920059
Received: 14 April 2022; Accepted: 27 June 2022;
Published: 25 July 2022.
Edited by:
Yubin Li, University of Pennsylvania, United StatesReviewed by:
Sanjima Pal, McGill University, CanadaCopyright © 2022 Zhang and Liu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ning Liu, bmluZ2xpdUBjbXUuZWR1LmNu
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.