 Manuel Rojas
Manuel Rojas Luke S. Heuer
Luke S. Heuer Weici Zhang
Weici Zhang Yi-Guang Chen
Yi-Guang Chen William M. Ridgway
William M. Ridgway
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 22 July 2022
Sec. Autoimmune and Autoinflammatory Disorders
Volume 13 - 2022 | https://doi.org/10.3389/fimmu.2022.918837
This article is part of the Research Topic Genetic Basis of Tolerance Induction Defects Underlying the Development of Autoimmune Pathologies View all 11 articles
Autoimmunity involves a loss of immune tolerance to self-proteins due to a combination of genetic susceptibility and environmental provocation, which generates autoreactive T and B cells. Genetic susceptibility affects lymphocyte autoreactivity at the level of central tolerance (e.g., defective, or incomplete MHC-mediated negative selection of self-reactive T cells) and peripheral tolerance (e.g., failure of mechanisms to control circulating self-reactive T cells). T regulatory cell (Treg) mediated suppression is essential for controlling peripheral autoreactive T cells. Understanding the genetic control of Treg development and function and Treg interaction with T effector and other immune cells is thus a key goal of autoimmunity research. Herein, we will review immunogenetic control of tolerance in one of the classic models of autoimmunity, the non-obese diabetic (NOD) mouse model of autoimmune Type 1 diabetes (T1D). We review the long (and still evolving) elucidation of how one susceptibility gene, Cd137, (identified originally via linkage studies) affects both the immune response and its regulation in a highly complex fashion. The CD137 (present in both membrane and soluble forms) and the CD137 ligand (CD137L) both signal into a variety of immune cells (bi-directional signaling). The overall outcome of these multitudinous effects (either tolerance or autoimmunity) depends upon the balance between the regulatory signals (predominantly mediated by soluble CD137 via the CD137L pathway) and the effector signals (mediated by both membrane-bound CD137 and CD137L). This immune balance/homeostasis can be decisively affected by genetic (susceptibility vs. resistant alleles) and environmental factors (stimulation of soluble CD137 production). The discovery of the homeostatic immune effect of soluble CD137 on the CD137-CD137L system makes it a promising candidate for immunotherapy to restore tolerance in autoimmune diseases.
Autoimmune diseases (ADs) are a chronic and clinically heterogeneous group of diseases affecting up to 5% of the world population (1, 2), and their incidence is rising (3). Different ADs share risk factors (e.g., environmental and genetic) and immunological mechanisms (4). A single autoimmune disease may manifest with autoantibodies of diverse organ specificities (i.e., latent polyautoimmunity) (5–7). Polymorphisms in HLA-DRB1, HLA-DQB1, CD226, PTPN22, STAT4, GPR103, TNFAIP3, and LRP1/STAT6 are associated with multiple ADs (8, 9), including systemic and organ-specific ADs (10). Therefore, the study of autoimmunity is complex and requires the analysis of multiple genes with diverse immunological effects.
A commonality among ADs is the failure to control peripheral autoreactive T cells, and most ADs exhibit dysfunctional T regulatory cells (Tregs) (11). This T cell population constitutively and highly expresses CD25 (IL-2 receptor α chain) (12), and more specifically, Tregs express the transcription factor Forkhead box P3 (FOXP3) (13–15). The clinical relevance of FOXP3 was demonstrated in patients with the immune dysregulation polyendocrinopathy enteropathy X-linked (IPEX) syndrome (16). More than 70 mutations in FOXP3 have been described in these patients (17), and they exhibit a high frequency of polyautoimmunity, such as autoimmune thyroid disease, autoimmune cytopenia, or type 1 diabetes (T1D) (18). Polymorphisms in other genes implicated in Treg function, such as IL2RA and CTLA4, have also been associated with the development of endocrinological and rheumatic ADs (19, 20). This evidence highlights the crucial role of Tregs in the disrupted immune homeostasis characteristic of autoimmunity.
The current management of ADs is centered on immunosuppression. Multiple non-specific immune-suppressive therapies are used to ameliorate autoreactivity/tissue damage (i.e., methotrexate, leflunomide). More recently, antibody-based therapies target specific molecules or cells involved in the immune response (i.e., anti-CD20 for depleting B cells) (21). However, these approaches have a major undesired effect: increased susceptibility to infections. Recently, new therapeutics focusing on Tregs have emerged. For example, administration of IL-2 in patients with systemic lupus erythematosus (SLE) ameliorated disease via the expansion of Tregs without an increased risk of infection, and low dose IL-2 therapy is being investigated in T1D (22–25). Restoring Treg function might treat autoimmunity while reducing the risk of life-threatening adverse effects. However, abnormal Treg function and conversion of Tregs to pathogenic Th17 cells are complications in Treg therapeutics (26–28).Thus, deeper knowledge of Treg biology is needed.
T1D is one of the most common ADs in children, characterized by the autoimmune destruction of insulin-producing β cells (29). T1D incidence is increasing rapidly, implying increasing environmental factors interacting with genetic risk loci (HLA and non-HLA genes) (29–31). Antigen-presenting cells (APCs) initiate pancreatic inflammation by producing inflammatory cytokines such as TNF-α (32, 33). The presentation of pancreatic antigens by APCs then leads to the activation of autoreactive CD4+ and CD8+ T cells, which perpetuate insulitis and the destruction of β cells (34, 35). Treg failure to maintain peripheral tolerance of these autoreactive T cells due to Treg dysfunction is critical in the persistence of inflammation and islet destruction (36).
Phase 1 clinical trials on early-onset T1D showed that the administration of autologous expanded CD4+CD25+CD127− Tregs was associated with a reduced requirement of exogenous insulin and preservation of β-cell function, with this effect persisting for up to 1 year after infusion without severe adverse reactions (37, 38). In a similar study, adult patients showed stable levels of C-peptide and insulin use for up to 2 years (39). However, this Treg strategy would necessitate periodic re-transfusions of Tregs to maintain the immune response, and autologous transplantations of Tregs may be difficult in low-income settings. In addition, these studies are in their infancy (i.e., phases 1 and 2), and the estimated magnitudes of the effect of these approaches were low. Thus, other strategies are needed to boost the peripheral Treg response to restore homeostasis.
The non-obese diabetic (NOD) mouse, which spontaneously develops autoimmune T1D, has long served as a model to delineate both genetic and immune mechanisms of T1D and its treatment. This model was established in 1980 by Makino et al. (40) and emerged from breeding a mouse strain that spontaneously developed cataracts (i.e., CTS strain) (41). Two groups of mice emerged: males with glucose intolerance but without glucosuria, later known as the non-obese non-diabetic (NON) strain, and females with polyuria, ketoacidosis, and glucosuria, subsequently known as the NOD strain (41). Histological examinations of NOD mice demonstrated lymphocyte infiltration in pancreatic islets (insulitis), as well as a decrease in the number of β-cells and islet size (Figure 1A) (40).
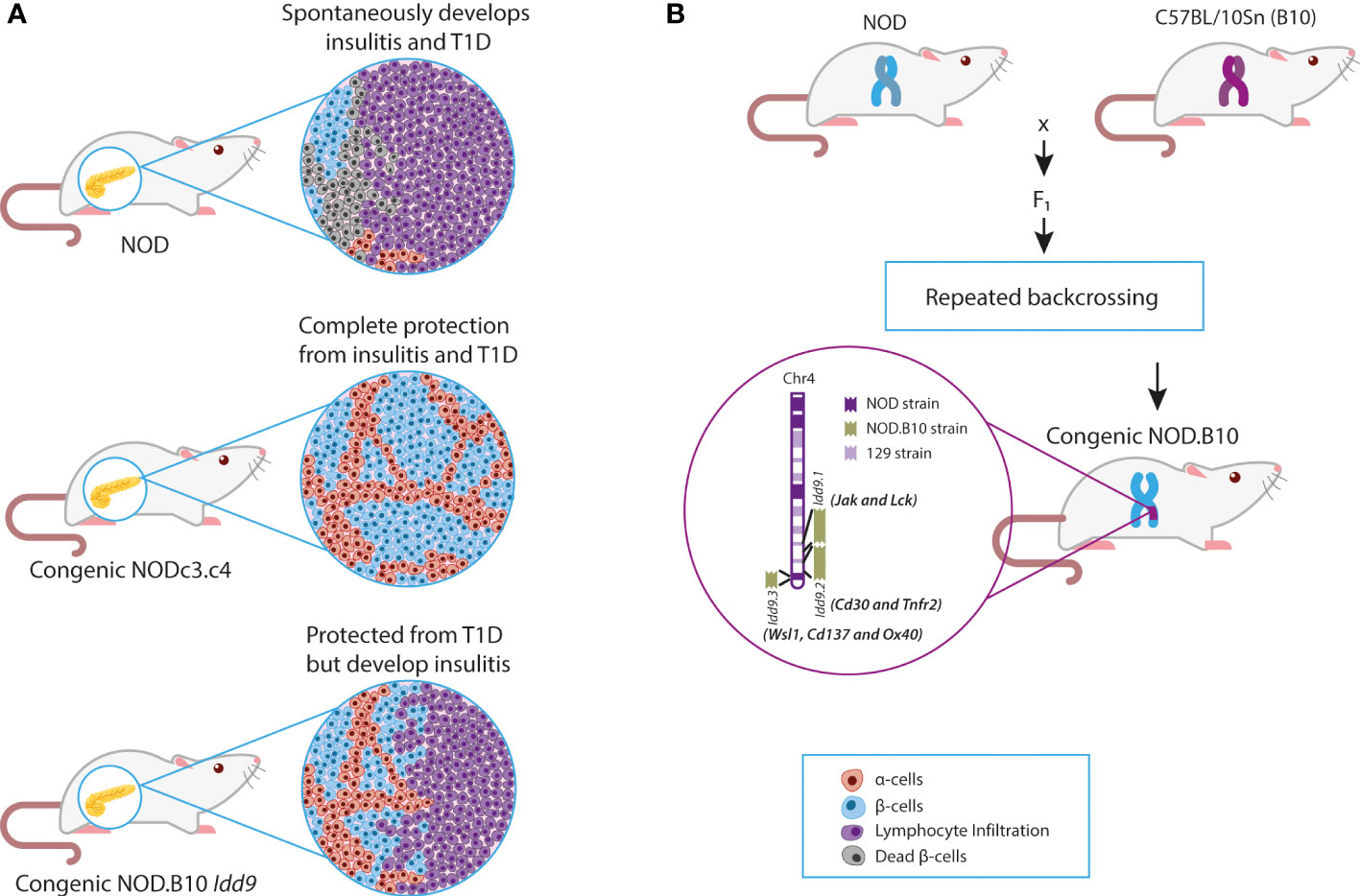
Figure 1 NOD and NOD Congenic mice. (A) Effects of congenic intervals on the clinical and histological phenotypes of NOD mice. (B) Breeding of NOD and B10 mice to produce congenic mice with Idd9 regions. Chr, Chromosome; NOD, Non-obese diabetic; T1D, Type 1 diabetes.
Typically, 80% of female NOD mice develop insulitis at three weeks and T1D at ~20 weeks (42). The H2g7 MHC haplotype essential for T1D development in NOD mice has the unique I-A allele (I-Ag7). I-Ag7 encodes histidine and serine at positions 56 and 57 instead of the two usually conserved proline and aspartic acid residues found in other mouse strains (43). The diabetogenic variants of the human class II HLA-DQβ homolog also have non-aspartic acid substitutions at residue 57 (44). The genetic association of both MHC class I and class II with disease supports the pathogenic role of CD8+ and CD4+ T cells in the destruction of β-cells in humans and mice (45–48). Multiple autoantigens are targeted by autoreactive T cells (e.g., GAD, insulin, or HSP) (49). However, while the MHC II I-Ag7 is a major susceptibility allele, it is not sufficient for the development of diabetes, as shown by complete T1D resistance in B10 mice expressing I-Ag7 molecules (50). In B6 congenic mice expressing I-Ag7, circulating T cells can react with the same β-cell autoantigens as in NOD mice; however, no autoimmunity results. These B6.G7 congenic mice confirm the importance of non-MHC genes in controlling autoimmunity in NOD genetic background (51, 52).
In addition to CD4+ and CD8+ T cell autoreactivity, Tregs are involved in suppressing the development of T1D in NOD mice. CD4+CD25+ Treg cell depletion at critical time points can accelerate T1D progression (53). Ablation of essential proliferative or co-stimulatory signals required for Treg cells, such as IL-2 or CD28, exacerbates T1D (54). NOD Treg quantity and functional capability are reduced, and increasing NOD Treg cell activity can prevent diabetes (55–57). These studies suggested that in addition to the crucial role of T cell autoreactivity, immunological pathways related to Tregs could be genetically determined in the NOD model. Further studies showed shared susceptibility genes affecting Treg function between mice and humans for T1D (e.g., IL2 and CTLA-4) (58–60). Thus, the study of NOD Treg function and control may allow the implementation of novel therapeutics in humans. These considerations highlight the significance of identifying non-HLA genes implicated in immune regulatory function, Treg function and development, and T1D pathogenesis (60). Identifying genes in the B6/B10 genetic background that can control autoimmunity has thus been a major goal in this field.
Before the advent of whole-genome sequencing, many non-HLA genomic regions associated with T1D were discovered by linkage analysis of the NOD genome (61–63). Identified genetic regions were confirmed to play a role in T1D pathogenesis through the construction of congenic mice (64). Congenic mice were constructed by introgression of resistant insulin-dependent diabetes (Idd) loci/regions onto the NOD background. Backcrossing of NOD with B6 (C57BL/6J), B10 (C57BL/10Sn), and other T1D resistant strains demonstrated that over 30 murine recessive Idd loci were associated with protection from spontaneous diabetes (65) (Figure 1B). These studies allowed the classification of Idd intervals into two groups: those that confer both insulitis and diabetes resistance and a second group that protects against T1D but has no effect on insulitis (62) (Figure 1A). For example, the Idd3 locus on chromosome 3 was implicated in the protection from insulitis and T1D, whereas the Idd4 locus on chromosome 11 did not protect from insulitis but prevented T1D (61). It suggested that genes within these regions exhibited differential effects on T1D development (i.e., T cell migration, cytotoxicity, or Treg function). The next step was to identify and confirm candidate genes within the introgressed genetic regions. This confirmation process ultimately has taken decades of work and the development of new technologies (e.g., whole-genome sequencing, CRISPR).
One of the first identified non-HLA candidate genes encodes interleukin 2 (IL-2). IL-2 is located in the Idd3 region and has profound effects on T cell and Treg function, and was thus a good candidate gene for T1D (66). NOD produces an altered IL-2 protein compared to the protective B10 allele, with a shortened tandem repeat sequence encoding a poly-glutamine stretch, plus an extra four amino acid insert, in the N-terminal coding region of IL-2 (62). These immunogenetic studies uncovered evidence of multiple genes with multiplicative effects on the immune response. For example, the Idd3/Idd5 double congenic mice, comprising the Il2 and Ctla4 candidate genes, were completely protected from T1D, whereas when studied alone, only ~20% and ~50% rates of protection were observed, respectively (60, 67, 68).
Genetic studies in the mouse were compared to human T1D genetic studies, and marked similarities were uncovered. The genetic architecture of mouse and human T1D is remarkably similar, with variants affecting multiple immune genes and pathways in common between both species, including IL-2, IL-2 receptor, CTLA-4, IL-10, the HLA region, PTPN22, and IL-7R (69, 70) For example, single nucleotide polymorphisms (SNPs) in the human homologous Il2 region were also associated with T1D susceptibility, identifying the IL-2 pathway as potentially shared in the pathogenesis of disease in both species (71). The NOD Il2 gene variant resulted in decreased production of IL-2, and elegant engineering of Il2 gene haplodeficency reproduced the NOD effect and resulted in functionally deficient Tregs (71). Low dose IL-2 therapy increased Tregs in mouse models, and this led to human trials of low dose IL-2; however, while this boosted human Treg numbers, it did not affect T1D outcome in initial trials (72). A variety of approaches have tried to optimize immune modulation effects via IL-2. IL-2 induced in vitro expansion of Tregs is one approach that was effective in NOD mice, tying the IL-2 immunogenetic effects to the enhancement of Treg deficiencies in T1D (73). Large numbers of Tregs are needed for human trials, and in vitro expansion may overcome some of the deficiencies of earlier Treg trials (74). Clinical trials in human T1D are ongoing with low-dose IL-2 therapy (75) and Treg therapy (76) which have built upon these earlier results.
CTLA-4 is another critical immune molecule with variants identified in mice and humans. The mouse locus (Idd5) was noted to overlie the orthologous human IDD12 locus (67). T1D susceptibility was subsequently mapped to a non-coding region of human CTLA-4 that resulted in lower levels of the CTLA-4 soluble splice variant; the mouse gene also demonstrated alterations in CLTA4 splicing (77). Human trials targeting CTLA-4 with a soluble form that blocks T cell activation appear promising (78). These therapies may be effective even though the human disease demonstrates remarkably different patterns of insulitis than the mouse, with much less exuberant immune infiltrates (79). The difference in β cell immune infiltration may explain why prevention of diabetes NOD is very easily achieved, whereas, in humans, prevention trials have until recently failed. One successful approach to the prevention of human T1D has been achieved using anti-CD3 antibodies, which preferentially target CD8 effector cells (80). Notably, anti-CD3 antibodies were discovered in NOD mice to reverse established disease (not simply prevent disease), demonstrating the usefulness of therapeutic trials of acute T1D in NOD mice. Overall, these examples illustrate the rich insights and potential therapies resulting from T1D immunogenetic studies. The latest large-scale study identified 78 genetic regions linked to T1D (including 36 novel loci) and confirmed the strong association with immune function and potential for clinical therapeutics (81). Thus much more work can be done to apply immunogenetic studies to novel therapeutic pathways.
Our labs have been investigating immunogenetic control of T1D, initially using NOD and NOD congenic mice, for over 20 years. In the rest of this review, we will detail the lengthy investigation of the immune effects of the Idd9 genetic region and our studies which demonstrated that Cd137 is the essential T1D susceptibility gene in this region. These studies have revealed many surprises about the function of an Idd gene in T1D immunology and have ultimately led to novel immunotherapy based on the immune function of CD137.
After identifying the Idd9 region in linkage studies, the Wicker group constructed congenic mice with the B10 Idd9 region introgressed onto the NOD background. The B10 Idd9 region prevented the onset of spontaneous diabetes in NOD mice (less than 5% of female mice developed T1D) (82). However, most mice still developed insulitis caused by T cells expressing CD30, producing high amounts of IL-4 (82) (Figure 1A). This confirmed that genes associated with lymphocyte infiltration were outside the Idd9 interval but suggested that some genes within this region halted autoimmunity.
This hypothesis was validated in double congenic mice comprising B6 (Idd3, Idd17, Idd10, and Idd18) from chromosome 3 and B10 (Idd9) regions (also known as the NOD.c3c4 strain). NOD.c3c4 mice were completely protected from diabetes, and only 10% of mice developed insulitis (82) (Figure 1A). This confirmed that spontaneous diabetes is a complex trait in which the epistasis of multiple genes (HLA and non-HLA) is critical for its development, but it also suggested that the Idd9 interval contained genes associated with T cell activation and modulation.
The Idd9 region, a 48 cM interval, was fine-mapped into three intervals (i.e., the Idd9.1, 9.2, and 9.3), with seven candidate genes (i.e., Jak1, Lck, Cd30, Tnfr2, Cd137, Wsl1, and Ox40) (66). The Wsl1, Cd137, and Ox40 were initially proposed as candidate genes within the Idd9.3 locus (82). However, B6 Wsl1 did not exhibit sequence variations compared to NOD, and Ox40 was subsequently found to be located outside of the Idd9.3 region and was excluded as a candidate gene (82). Thus, Cd137 remained the key candidate for T1D protection within the Idd9.3 locus. Jumping ahead 15 years, it was recently confirmed by using combined congenic mapping and nuclease-based gene targeting that Cd137 is the susceptibility gene within the Idd9.3 locus critical for modulation of T1D (82, 83).
Cd137 is located at 1.217-Mb of the Idd9 locus (i.e., Idd9.3) (84), and Idd9.3 conferred ~40% protection for T1D (82). Analysis of coding variants demonstrated two synonymous SNPs in NOD vs. B10 Cd137: a valine to alanine substitution at position 24 and leucine to proline substitution at position 211 (near the transmembrane domain). There is also alanine insertion in NOD between amino acids 174 and 175 (82) (Figure 2A). These structural modifications suggested that CD137(4-1BB) could be hypofunctional in NOD mice (82). Cd137 (4-1bb or Tnfrsf9) codes for two CD137 isoforms: membrane-bound (mCD137) and soluble (sCD137) forms (Figure 2B) (85). Membrane mCD137 is mostly found on CD4+ and CD8+ T cells, whereas the sCD137 is produced by Tregs (86). The ligand for both isoforms, CD137L, is coded by Tnfsf9 on chromosome 17 and is expressed on APCs and activated T cells (86).
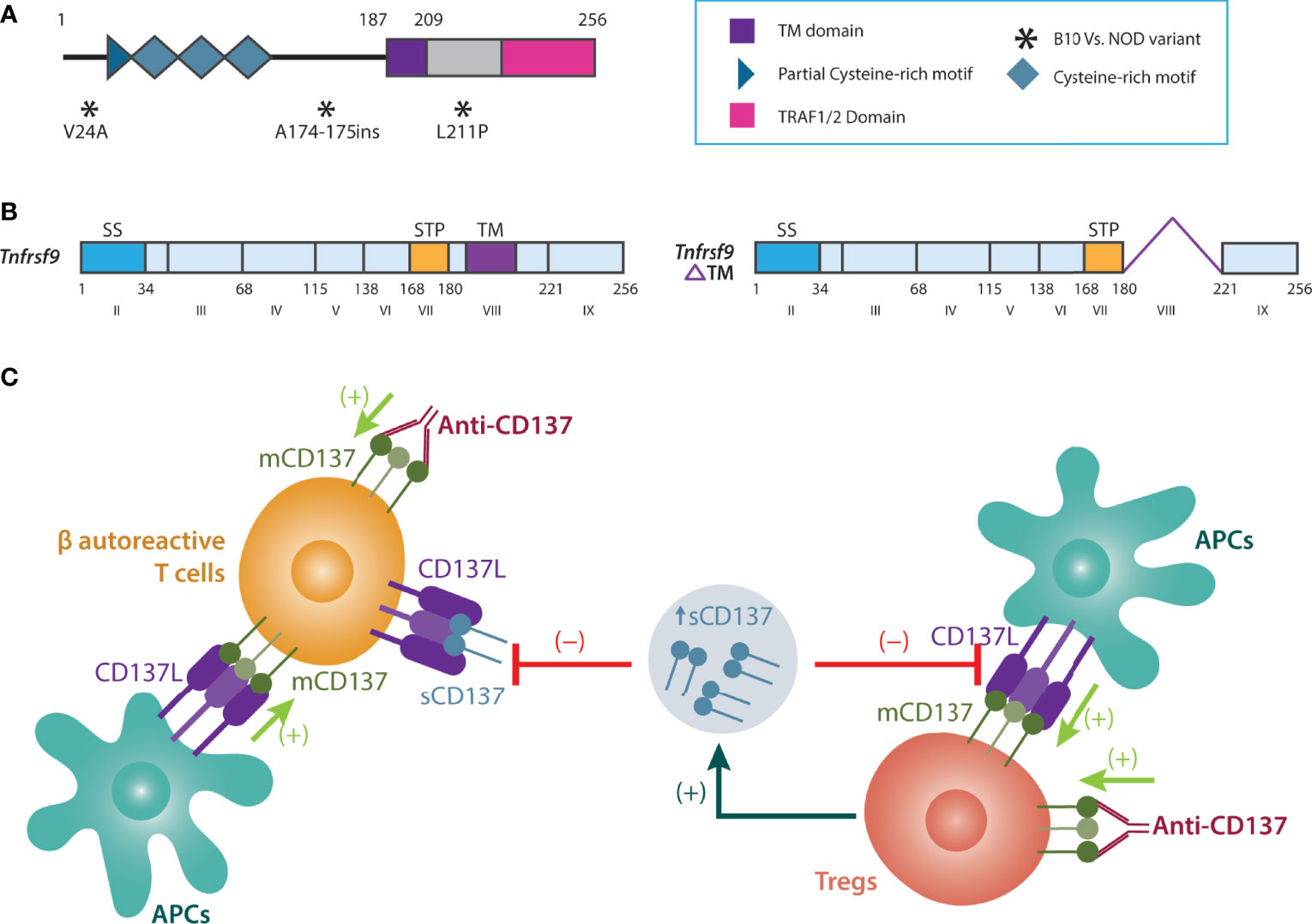
Figure 2 Biology and function of sCD137. (A) Non-synonymous SNPs of NOD vs. B6 Cd137. (B) membrane vs. soluble (alternatively spliced) CD137. (C) Tregs produce sCD137 with a dimeric structure. sCD137 induces altered CD137L signaling in APCs and autoreactive T cells (compared to membrane CD137), reducing inflammation and damage in the pancreas. In contrast to sCD137, although anti-CD137 antibodies activate Tregs (a strong immune regulatory effect), they may also increase autoreactive T cell survival and proliferation, thus perpetuating inflammation and autoimmunity. APCs, Antigen-presenting cells; CD137L, CD137 Ligand; mCD137, Membrane-bound CD137; NOD, Non-obese diabetic; sCD137, Soluble CD137; SS, signal sequence; STP, Ser/Thr/Pro-rich; T1D, Type 1 diabetes; TM, Transmembrane domain; Tregs, T regulatory cells.
Since the NOD Cd137 SNPs suggested that mCD137 was hypofunctional compared to the NOD.B10 strain (82), Cannons et al. (84) evaluated T cells activation and proliferation to test this hypothesis. They confirmed that NOD and NOD.B10 mice showed similar mCD137 expression after stimulation with anti-CD3 in Th1 and Th2 culture conditions. However, when T cells from NOD mice were costimulated with CD137L, they proliferated less and produced a reduced level of IL-2 than T cells from mice carrying the B10 allele of Cd137. This strongly suggested that the NOD SNPs lead to a hypo-functional mCD137 protein, which could play a role in T1D pathogenesis. Over the last 15 years, our work has begun to delineate the complex immune biology of CD137 and CD137L in T1D.
CD137 is a glycoprotein belonging to the TNF receptor superfamily, and the membrane form is expressed on activated CD4+ and CD8+ T lymphocytes (87), Tregs (88–94), and natural killer (NK) cells (95). CD137 is also constitutively expressed on a subset of Tregs (93). CD137L (4-1BBL), its ligand, belongs to the TNF superfamily and is expressed on activated APCs such as macrophages, B cells, and dendritic cells (96–98). Activated T cells also upregulate and express CD137L (99). mCD137 has no intrinsic enzymatic activity in its intracellular domain and functions by binding TRAF1 and TRAF2 adaptor proteins that enhance K63 polyubiquitination processes in the CD137 signalosome (100, 101). CD137L trimerization, in response to interaction with mCD137, causes mCD137 receptor clustering and TRAF-mediated activation of the ERK, JNK, p38, NF-kB, and MAPK intracellular signaling pathways, resulting in cell activation, proliferation, and T cell survival (102–109). Notably, signaling in the CD137:CD137L pathway is bidirectional: both the receptor and ligand signal into their respective cells (110, 111). This bidirectional signaling adds an additional layer of complexity to the analysis of the biological function of the pathway.
The effects of CD137 in T cell biology are diverse but with specific implications for inflammation and immune regulation. TCR-induced proliferation and cytokine production were enhanced after T cells were stimulated with agonistic anti-CD137 antibodies (also known as 3H3), independent of B7-CD28/CTLA-4 interactions (112, 113). mCD137 signaling results in NF-kB activation that promotes the expression of antiapoptotic genes encoding Bcl-xL and Bfl-1 (114, 115) and mitochondrial function and biogenesis, which improves T cell survival (116, 117).
mCD137 has a prominent role in CD8+ T-cell costimulation, influencing cytotoxicity in an IL-2-independent manner. Furthermore, CD8+ T cells produce a greater amount of IFN-γ after mCD137 activation (118). In vivo experiments showed that knockout mCD137/CD137L mice exhibited a reduced memory CD8+ T cell response to viruses (119–121) and decreased T cell survival (122). These findings pointed to a costimulatory involvement of mCD137 in long-lasting memory T-cell activation and enhancement of cytotoxicity and founded the basis for CD137-based therapies for cancer (85, 123–126). In contrast to these effects, when knockout mice were stimulated with CD3, T cells showed hyperresponsiveness, which indicated an additional immunosuppressive role of CD137 (105).
The expression of CD137L on APCs is increased at sites of inflammation in vivo (127, 128). Activating APCs by CD137L upregulated B7-1 and B7-2, and increased IL-6 and IL-12 secretion (127). CD137L is upregulated on activated T cells, and CD137L signaling is critical for CD8+ T cell survival via STAT3- and FAS-mediated pathways (129). CD4+ T cell activation can also be modulated by CD137L-expressing APCs (via APC CD137L signaling through T cell CD137) that stimulate IL-2 and IL-4 T cell production (112, 113).
This data established that CD137L on APCs affects the cytotoxic immune response and is critical for the survival of CD8+ and CD4+ T cells. This also confirmed that inhibition of mCD137 or CD137L might reduce inflammation via CD8+ T cells but may at the same time also affect CD4+CD25+CD137-expressing Tregs. Indeed, Cd137 is upregulated by Foxp3 (130). CD137 is expressed by Tregs infiltrating the islets in T1D, suggesting an immunoregulatory role for CD137+ Tregs (131). Thus attempting to modulate CD137 or CD137L action on T effector cells could potentially decrease immunosuppression via Tregs, illustrating the intricacies of this pathway and the potential for double-edged effects.
Type 1 regulatory T (Tr1) cells are another type of regulatory T cell characterized by the production of IL-10 and lack of constitutive Foxp3 expression (132). Despite the evidence of CD137L mRNA expression after stimulation (133), it is unknown whether these cells also exert their suppressive function by sCD137 or their role in NOD mice during T1D pathogenesis. Since Cd137 is upregulated by Foxp3 (130), Tr1 cells may not produce large quantities of sCD137. Further studies of this cell subset and their involvement in the mCD137/CD137L axis are warranted.
Agonistic anti-CD137 antibodies induced the proliferation of CD4+CD25+ Tregs with the maintenance of their suppressive activity (92). Interestingly, the effects of agonistic anti-CD137 antibodies are diverse and dependent on the target and the disease. Activating CD8+ T cells by anti-CD137 antibodies in cancer models leads to tumor cell elimination. In sharp contrast, in models of autoimmunity, e.g., murine models of SLE (134, 135), experimental autoimmune encephalomyelitis (136), collagen-induced arthritis (122, 137), Sjögren’s syndrome-like sialadenitis (138), and inflammatory bowel disease (139), anti-CD137 antibody treatment leads to immunoregulation and disease amelioration. For example, anti-CD137 administration in the SLE murine model reversed disease and reduced autoantibody production (i.e., dsDNA antibodies) and immune complex deposition (135). Induction of T cell anergy by anti-CD137 antibodies might play a role in some of these models (135, 140).
Since Cd137 was a candidate gene in T1D, we started our investigation of the role of CD137 in T1D with agonistic anti-CD137 antibodies. We showed that anti-CD137 antibodies in NOD mice prevented the development of T1D but did not ameliorate insulitis, which is consistent with the findings of residual insulitis in NOD congenic mice protected from T1D by the B10 Idd9.3 region (93). We found that anti-CD137 expanded CD4+CD25+ Tregs, and their transfer to NOD-scid mice completely prevented T1D (93). However, T1D progressed more rapidly when NOD-scid mice were treated with anti-CD137 after pathogenic CD4+ and CD8+ T cell transfer in the absence of Tregs. Therefore, in the absence of Tregs, mCD137 stimulation could potentially potentiate pancreatic destruction via CD8+ T cytotoxicity. This is similar to the effect of anti-CD137 administered in the context of autoimmune thyroiditis, which worsens the disease (141).
Due to this dual effect, activation of effector T cells in acute autoimmunity may prohibit the use of agonistic CD137 antibodies in clinical autoinflammatory states, including T1D, because activated T cells have upregulated mCD137 in these settings. In contrast, CD137 antibodies in non-inflammatory states (e.g., pre-diabetes) might prevent autoimmunity since it targets Tregs constitutively expressing mCD137 without activating T cell effector cells. This dual effect led us to look for alternate ways to therapeutically target the mCD137/CD137L pathway in T1D.
We turned our attention to sCD137, which is formed by alternative splicing (99, 142) (Figure 2B), and exists as a dimer (143). sCD137 was found in the supernatants of splenic and bone marrow-derived dendritic cells (144). Murine sCD137 differs from humans. In mice, only the exon coding for the transmembrane domain of CD137 is spliced out, whereas, in the latter, two splice variants are observed (145). sCD137 is preferentially secreted by CD4+ T cells, whereas CD8+ T cells express higher amounts of mCD137 (146). We found that the major source of sCD137 is CD4+CD25+CD137+ Tregs (94).
Spliced variants are critical for the modulation of immune response (147). The induction of alternative splicing is poorly understood but may occur as a response to environmental signals. In autoimmunity, splicing also occurs in the modulation of immune responses (147). Changes in the immunological environment (i.e., T cell autoreactivity and pro-inflammatory milieu) induce the production of sCD137 by Tregs. We have demonstrated that activating Treg cells increases the production of sCD137 by Tregs in mice and humans (148). Thus, inflammatory environmental changes may partly explain the origin of spliced variants of CD137 from Tregs as a homeostatic response to ameliorate inflammation. A similar process is seen with soluble CTLA4 (sCTLA4), a spliced variant of membrane-bound CTLA4 mainly produced by Foxp3+ Tregs (149). sCTLA4 suppresses early T-cell activation by preventing the interaction of CD80/CD86 with the costimulatory receptor CD28 (150). In addition, it inhibits IFN-α, IL-2, IL-7, and IL-13 production while activating TGF-β and IL-10 release (151). Silencing sCTLA-4 mRNA by RNA interference accelerated the onset of T1D in NOD mice and impaired the ability of Tregs to downregulate dendritic cell costimulation (149). Both spliced variants, sCTLA4 and sCD137, may be critical for effective Treg function in the pathogenesis of T1D.
What is the role of sCD137? Our hypothesis was that sCD137, similar to sCTLA4, functions as a negative feedback mechanism to downregulate immune response mediated by mCD137 and CD137L (85, 152). sCD137 reduces the production of IL-10 and IL-12 from activated splenocytes (146). In addition, T cell proliferation and IL-2 release were inhibited when sCD137 was administered to these cells (153). These initial reports clarified the effects of CD137 in different conditions (i.e., cancer and autoimmunity) and suggested that the sCD137 was the missing link in understanding the dual effects of the mCD137/CD137L axis.
To confirm the role of CD137 in the Idd9.3 locus, we evaluated the function (immunosuppressive effects) and quantity of CD137+ Treg cells in NOD.Idd9.3 congenic mice (94). When compared to NOD mice, the NOD.Idd9.3 strain had significantly higher percentages of CD4+CD25+CD137+Foxp3+ Tregs in the thymus and spleen, and the numbers increased with age. This supported the hypothesis that the hypofunctional NOD CD137 allele led to decreased Treg survival, consistent with the known effects of mCD137 on cell survival. CD137+ Tregs showed superior immunosuppression compared to CD4+CD25+CD137- Tregs, directly showing an effect of CD137 on Treg function. Thus, increased numbers of CD137 Tregs, mediated by the protective allele, led to increased overall suppressive capacity. Importantly, CD137+ Tregs showed suppressive capability in an independent contact assay. This supported our continued focus on the possible immunosuppressive role of sCD137 in T1D.
We first confirmed that sCD137 was mainly produced by CD4+CD25+CD137+ Tregs and in a higher amount in NOD.Idd9.3 congenic mice (94). Next, we demonstrated that sCD137 primarily exists as a ~55 kDa homodimer under non-reducing and a ~35 kDa monomer under reducing conditions (143). The existence of sCD137 as a dimer, rather than as a trimer as described for mCD137, suggested a structural reason for how sCD137 might suppress T cell function while mCD137 activated T cell function (143). Next, we showed that the administration of recombinant sCD137 to NOD mice prevented diabetes and reduced insulitis by preserving insulin+ islets (143). Since CD4+CD25+CD137+ Tregs inhibited T cells in a contact-independent manner (94), we evaluated the role of sCD137 in T cell inhibition. We demonstrated that sCD137 inhibited activated T cells by binding to CD137L (143). In addition, sCD137 can directly stop the proliferation of effector CD4+CD25-CD137- T cells in the absence of APCs, and without inducing cell death (143) (Figure 2C).
In addition to the crucial role of sCD137 in immunosuppression, additional reports suggest that mCD137, like other costimulatory molecules, has a nonredundant role in maintaining the pathogenic activity of β cell-autoreactive T cells in NOD mice. We found that, compared to wild-type mice, T1D development is reduced in NOD.Cd137-/- and their T cells are less capable of inducing T1D in NOD.Rag1-/- recipients (154). This, at first, seemed contradictory to our data on the immunoregulatory properties of CD137+ Tregs and sCD137. As sCD137 produced by Tregs is suppressive, evaluating the distinctive role of mCD137 in CD4+ and CD8+ T cells was crucial. Isolated T cells from NOD and NOD.Cd137-/- mice were transferred into NOD.Rag1-/- recipients. The T cell adoptive transfer studies revealed that CD137 expression in CD8+ T cells was required to develop T1D in NOD mice, but CD137 expression in CD4+ T cells was diabetes-protective (155). Specifically, CD137 expression in CD4+ Tregs is important for their T1D suppression function. We further demonstrated that CD137 cell-intrinsically stimulates the accumulation and proliferation of autoreactive CD8+ T lymphocytes within the islets, pointing to a role of mCD137 on the diabetogenic activity of CD8+ T cells. However, sCD137 suppressed the proliferation of CD8+ T cells. These experiments supported the concept that the T1D protection conferred by the Idd9.3 locus is mediated through the production of sCD137 by Tregs.
As the sCD137/CD137L interaction is implicated in the modulation of effector CD8+ T cells, clarifying the role of CD137L in the immunomodulation of T1D is essential. CD137L-deficient NOD mice were shown to exhibit less insulitis and delayed onset of T1D (156). Interestingly, CD137L expression on myeloid APCs appeared to be necessary for the survival of β-cell–autoreactive CD8+ T cells and T1D progression, but CD137L has no effect on the formation or homeostasis of Foxp3+ Tregs (156). It remains to be determined if mCD137 in Tregs modulates their function and whether Tregs capable of producing sCD137 but not mCD137 are sufficient to suppress T1D.
It is relatively easy to prevent T1D in NOD mice, and a much more stringent target is the reversal of actual acute T1D. Thus we treated NOD mice with new-onset T1D with recombinant sCD137 (148). This experiment confirmed that sCD137 could not only prevent T1D but also halt acute T1D and avert the development of end-stage diabetes. In effectively treated mice, β cell immunohistochemistry revealed considerable preservation of insulin+ β cells and a rise in insulin+ islets (148). In this setting, T cells showed downregulation of mTORC1, developed an anergic phenotype (reversed by IL-2), as well as the ability of sCD137 to suppress antigen-experienced and activated memory T cells. CD8+ effector memory cells also showed a reduction in the production of inflammatory cytokines in the presence of sCD137 (i.e., IFN-γ) (148).
In human pediatric T1D patients, we found low levels of sCD137 compared to non-diabetic age-matched controls during acute flares (hospital admission for hyperglycemia). We also confirmed that human Tregs were the primary source of sCD137 (148). Furthermore, human peripheral activated CD4+ T cells were inhibited by sCD137. These results were analogous to those in NOD congenic strains, supporting the notion that these murine models are useful and relevant to affecting the autoimmune phenomena driving human T1D. This evidence showed that sCD137 is associated with autoimmunity in T1D humans, and low sCD137 could be a biomarker in T1D. Further studies are required to confirm the role of sCD137 in reverting established destructive insulitis and the pathways associated with this phenomenon.
Surprisingly, sCD137 is reported to be increased in patients with rheumatoid arthritis (145, 157, 158), and multiple sclerosis (159); and the levels were directly correlated with the severity of the disease (157). Increased levels of sCD137 could be a homeostatic attempt by Tregs to modulate inflammation in these conditions. However, it also raises the possibility that in the presence of a substantial inflammatory substrate, the stoichiometric ratio of sCD137 to CD137L could be reduced, thus reducing the efficacy of sCD137 or possibly indicating that higher sCD137 doses would be required. New strategies improving the half-life and potency of sCD137 could be critical to enhancing their therapeutic effect in human autoimmunity.
Linkage studies and the construction of congenic mice allowed the identification of candidate genes with implications for the pathogenesis of T1D. The cumulative evidence suggests that Cd137 and its coding isoforms are crucial in the development of T1D, and the CD137-CD137L pathway is a good target for therapeutic modulation. Treg-generated sCD137 modulates the mCD137/CD137L axis, reduces insulitis, and halts T1D in the NOD mouse. The ability to effectively halt acute T1D with exogenous sCD137 is an exciting development with attractive therapeutic potential. Prevention studies in humans are difficult to implement, and those attempted so far have failed (160). Therefore, treating acute disease is a more appealing strategy, but the current landscape of approved therapeutics is limited. The use of antibodies to target the CD137-CD137L axis is appealing; however, while anti-CD137 antibodies are protective in some models of autoimmune diseases due to activation of Tregs, they can also enhance CD8+ T cell killing activity in the absence of Tregs. sCD137, on the other hand, only acts to suppress CD4+ and CD8+ T cell activation and may therefore be safer than an anti-CD137 based approach. In human studies, low levels of sCD137 during T1D flares, and the inhibition of activated CD8+ T cells in vitro after sCD137 stimulation, supports its further translational use. Soluble CD137 suppresses autoreactive CD8+ T cells through induction of anergy. However, little is known about the activity of sCD137 on innate immunity. The mechanistic role of sCD137 on CD137L-expressing myeloid APCs should be explored to determine if there will be lasting effects on innate immune function. In addition, it is unknown whether mCD137 on Tregs drives their differentiation to a more robust inhibitory phenotype. This could have therapeutic implications, particularly for the pharmacokinetics and pharmacodynamics of human sCD137.
MR, LH, WZ, YG, WR wrote, reviewed and revised the manuscript. All authors contributed to the article and approved the submitted version.
This work was supported by R01 DK107541 (Y-GC and WMR) and DK121747 (Y-GC).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
ADs, Autoimmune diseases; APCs, Antigen-presenting cells; CD, Cluster of differentiation; CD137L, CD137 Ligand; Foxp3, Forkhead box P3; Idd, Insulin-dependent diabetes loci; IPEX, Immune dysregulation polyendocrinopathy enteropathy X-linked; mCD137, Membrane-bound CD137; NK, Natural killer; NOD, Non-obese diabetic; NON, Non-obese non-diabetic; sCD137, Soluble CD137; sCTLA4, Soluble CTLA4; SLE, Systemic lupus erythematosus; SNPs, Single nucleotide polymorphisms; T1D, Type 1 diabetes; Tregs, T regulatory cells.
1. Anaya J-M, Shoenfeld Y, Rojas-Villarraga A, Levy RA, Cervera R eds. Autoimmunity: From bench to bedside. In: Autoimmunity: From Bench to Bedside. Bogota (Colombia: Rosario University Pres.
2. Rojas-Villarraga A, Amaya-Amaya J, Rodriguez-Rodriguez A, Mantilla RD, Anaya J-M. Introducing polyautoimmunity: Secondary autoimmune diseases no longer exist. Autoimm Dis (2012) 2012:254319. doi: 10.1155/2012/254319
3. Lerner A, Jeremias P, Matthias T. The world incidence and prevalence of autoimmune diseases is increasing. Int J Celiac Dis (2015) 3:151–5. doi: 10.12691/ijcd-3-4-8
4. Anaya J-M. The autoimmune tautology. a summary of evidence. Joint Bone Spine (2017) 84:251–3. doi: 10.1016/j.jbspin.2016.11.012
5. Diaz-Gallo L-M, Oke V, Lundström E, Elvin K, Ling Wu Y, Eketjäll S, et al. Four systemic lupus erythematosus subgroups, defined by autoantibodies status, differ regarding hla-Drb1 genotype associations and immunological and clinical manifestations. ACR Open Rheumatol (2021) 4(1):27-39. doi: 10.1002/acr2.11343
6. Pacheco Y, Barahona-Correa J, Monsalve DM, Acosta-Ampudia Y, Rojas M, Rodríguez Y, et al. Cytokine and autoantibody clusters interaction in systemic lupus erythematosus. J Transl Med (2017) 15:239. doi: 10.1186/s12967-017-1345-y
7. Botello A, Herrán M, Salcedo V, Rodríguez Y, Anaya J-M, Rojas M. Prevalence of latent and overt polyautoimmunity in autoimmune thyroid disease: A systematic review and meta-analysis. Clin Endocrinol (Oxf) (2020) 93:375–89. doi: 10.1111/cen.14304
8. Tomer Y, Dolan LM, Kahaly G, Divers J, D'Agostino RBJ, Imperatore G, et al. Genome wide identification of new genes and pathways in patients with both autoimmune thyroiditis and type 1 diabetes. J Autoimmun (2015) 60:32–9. doi: 10.1016/j.jaut.2015.03.006
9. Johar AS, Mastronardi C, Rojas-Villarraga A, Patel HR, Chuah A, Peng K, et al. Novel and rare functional genomic variants in multiple autoimmune syndrome and sjögren's syndrome. J Transl Med (2015) 13:173. doi: 10.1186/s12967-015-0525-x
10. Acosta-Herrera M, Kerick M, González-Serna D, Wijmenga C, Franke A, Gregersen PK, et al. Genome-wide meta-analysis reveals shared new loci in systemic seropositive rheumatic diseases. Ann Rheum Dis (2019) 78:311–9. doi: 10.1136/annrheumdis-2018-214127
11. Sakaguchi S, Mikami N, Wing JB, Tanaka A, Ichiyama K, Ohkura N. Regulatory T cells and human disease. Annu Rev Immunol (2020) 38:541–66. doi: 10.1146/annurev-immunol-042718-041717
12. Sakaguchi S, Sakaguchi N, Asano M, Itoh M, Toda M. Immunologic self-tolerance maintained by activated T cells expressing il-2 receptor alpha-chains (Cd25). breakdown of a single mechanism of self-tolerance causes various autoimmune diseases. J Immunol (1995) 155:1151–64.
13. Fontenot JD, Gavin MA, Rudensky AY. Foxp3 programs the development and function of Cd4+Cd25+ regulatory T cells. Nat Immunol (2003) 4:330–6. doi: 10.1038/ni904
14. Khattri R, Cox T, Yasayko S-A, Ramsdell F. An essential role for scurfin in Cd4+Cd25+ T regulatory cells. Nat Immunol (2003) 4:337–42. doi: 10.1038/ni909
15. Hori S, Nomura T, Sakaguchi S. Control of regulatory T cell development by the transcription factor Foxp3. Science (2003) 299:1057–61. doi: 10.1126/science.1079490
16. Jamee M, Zaki-Dizaji M, Lo B, Abolhassani H, Aghamahdi F, Mosavian M, et al. Clinical, immunological, and genetic features in patients with immune dysregulation, polyendocrinopathy, enteropathy, X-linked (Ipex) and ipex-like syndrome. J Allergy Clin Immunol Pract (2020) 8:2747–60.e7. doi: 10.1016/j.jaip.2020.04.070
17. Bacchetta R, Barzaghi F, Roncarolo M-G. From ipex syndrome to Foxp3 mutation: A lesson on immune dysregulation. Ann N Y Acad Sci (2018) 1417:5–22. doi: 10.1111/nyas.13011
18. Amaya-Uribe L, Rojas M, Azizi G, Anaya J-M, Gershwin ME. Primary immunodeficiency and autoimmunity: A comprehensive review. J Autoimmun (2019) 99:52–72. doi: 10.1016/j.jaut.2019.01.011
19. Todd JA, Walker NM, Cooper JD, Smyth DJ, Downes K, Plagnol V, et al. Robust associations of four new chromosome regions from genome-wide analyses of type 1 diabetes. Nat Genet (2007) 39:857–64. doi: 10.1038/ng2068
20. Okada Y, Wu D, Trynka G, Raj T, Terao C, Ikari K, et al. Genetics of rheumatoid arthritis contributes to biology and drug discovery. Nature (2014) 506:376–81. doi: 10.1038/nature12873
21. Barnas JL, Looney RJ, Anolik JH. B cell targeted therapies in autoimmune disease. Curr Opin Immunol (2019) 61:92–9. doi: 10.1016/j.coi.2019.09.004
22. He J, Zhang R, Shao M, Zhao X, Miao M, Chen J, et al. Efficacy and safety of low-dose il-2 in the treatment of systemic lupus erythematosus: A randomised, double-blind, placebo-controlled trial. Ann Rheum Dis (2020) 79:141–9. doi: 10.1136/annrheumdis-2019-215396
23. von Spee-Mayer C, Siegert E, Abdirama D, Rose A, Klaus A, Alexander T, et al. Low-dose interleukin-2 selectively corrects regulatory T cell defects in patients with systemic lupus erythematosus. Ann Rheum Dis (2016) 75:1407–15. doi: 10.1136/annrheumdis-2015-207776
24. Hartemann A, Bensimon G, Payan CA, Jacqueminet S, Bourron O, Nicolas N, et al. Low-dose interleukin 2 in patients with type 1 diabetes: A phase 1/2 randomised, double-blind, placebo-controlled trial. Lancet Diabetes Endocrinol (2013) 1:295–305. doi: 10.1016/S2213-8587(13)70113-X
25. Pham MN, von Herrath MG, Vela JL. Antigen-specific regulatory T cells and low dose of il-2 in treatment of type 1 diabetes. Front Immunol (2016) 6:651. doi: 10.3389/fimmu.2015.00651
26. Wan YY. Regulatory T cells: Immune suppression and beyond. Cell Mol Immunol (2010) 7:204–10. doi: 10.1038/cmi.2010.20
27. Zhou L, Lopes JE, Chong MMW, Ivanov II, Min R, Victora GD, et al. Tgf-Beta-Induced Foxp3 inhibits T(H)17 cell differentiation by antagonizing rorgammat function. Nature (2008) 453:236–40. doi: 10.1038/nature06878
28. Yang XO, Nurieva R, Martinez GJ, Kang HS, Chung Y, Pappu BP, et al. Molecular antagonism and plasticity of regulatory and inflammatory T cell programs. Immunity (2008) 29:44–56. doi: 10.1016/j.immuni.2008.05.007
29. Norris JM, Johnson RK, Stene LC. Type 1 diabetes–early life origins and changing epidemiology. Lancet Diabetes Endocrinol (2020) 8:226–38. doi: 10.1016/S2213-8587(19)30412-7
30. Mobasseri M, Shirmohammadi M, Amiri T, Vahed N, Hosseini Fard H, Ghojazadeh M. Prevalence and incidence of type 1 diabetes in the world: A systematic review and meta-analysis. Health Promotion Perspect (2020) 10:98–115. doi: 10.34172/hpp.2020.18
31. Bao YK, Weide LG, Ganesan VC, Jakhar I, McGill JB, Sahil S, et al. High prevalence of comorbid autoimmune diseases in adults with type 1 diabetes from the healthfacts database. J Diabetes (2019) 11:273–9. doi: 10.1111/1753-0407.12856
32. Dahlén E, Dawe K, Ohlsson L, Hedlund G. Dendritic cells and macrophages are the first and major producers of tnf-alpha in pancreatic islets in the nonobese diabetic mouse. J Immunol (1998) 160:3585–93.
33. Nikolic T, Geutskens SB, van Rooijen N, Drexhage HA, Leenen PJM. Dendritic cells and macrophages are essential for the retention of lymphocytes in (Peri)-insulitis of the nonobese diabetic mouse: A phagocyte depletion study. Lab Invest (2005) 85:487–501. doi: 10.1038/labinvest.3700238
34. Wållberg M, Cooke A. Immune mechanisms in type 1 diabetes. Trends Immunol (2013) 34:583–91. doi: 10.1016/j.it.2013.08.005
35. Turley S, Poirot L, Hattori M, Benoist C, Mathis D. Physiological beta cell death triggers priming of self-reactive T cells by dendritic cells in a type-1 diabetes model. J Exp Med (2003) 198:1527–37. doi: 10.1084/jem.20030966
36. ElEssawy B, Li XC. Type 1 diabetes and T regulatory cells. Pharmacol Res (2015) 98:22–30. doi: 10.1016/j.phrs.2015.04.009
37. Marek-Trzonkowska N, Mysliwiec M, Dobyszuk A, Grabowska M, Techmanska I, Juscinska J, et al. Administration of Cd4+Cd25highcd127- regulatory T cells preserves B-cell function in type 1 diabetes in children. Diabetes Care (2012) 35:1817–20. doi: 10.2337/dc12-0038
38. Marek-Trzonkowska N, Myśliwiec M, Dobyszuk A, Grabowska M, Derkowska I, Juścińska J, et al. Therapy of type 1 diabetes with Cd4(+)Cd25(High)Cd127-regulatory T cells prolongs survival of pancreatic islets - results of one year follow-up. Clin Immunol (2014) 153:23–30. doi: 10.1016/j.clim.2014.03.016
39. Bluestone JA, Buckner JH, Fitch M, Gitelman SE, Gupta S, Hellerstein MK, et al. Type 1 diabetes immunotherapy using polyclonal regulatory T cells. Sci Transl Med (2015) 7:315ra189. doi: 10.1126/scitranslmed.aad4134
40. Makino S, Kunimoto K, Muraoka Y, Mizushima Y, Katagiri K, Tochino Y. Breeding of a non-obese, diabetic strain of mice. Jikken Dobutsu (1980) 29:1–13. doi: 10.1538/expanim1978.29.1_1
41. Mullen Y. Development of the nonobese diabetic mouse and contribution of animal models for understanding type 1 diabetes. Pancreas (2017) 46:455–66. doi: 10.1097/MPA.0000000000000828
42. Ridgway WM. Dissecting genetic control of autoimmunity in nod congenic mice. Immunol Res (2006) 36:189–95. doi: 10.1385/IR:36:1:189
43. Acha-Orbea H, McDevitt HO. The first external domain of the nonobese diabetic mouse class ii I-a beta chain is unique. Proc Natl Acad Sci USA (1987) 84(8):2435–9. doi: 10.1073/pnas.84.8.2435
44. Todd JA, Acha-Orbea H, Bell JI, Chao N, Fronek Z, Jacob CO, et al. A molecular basis for mhc class ii–associated autoimmunity. Science (1988) 240(4855):1003–9. doi: 10.1126/science.3368786
45. Hamilton-Williams EE, Serreze DV, Charlton B, Johnson EA, Marron MP, Mullbacher A, et al. Transgenic rescue implicates Beta2-microglobulin as a diabetes susceptibility gene in nonobese diabetic (Nod) mice. Proc Natl Acad Sci USA (2001) 98:11533–8. doi: 10.1073/pnas.191383798
46. Nejentsev S, Howson JMM, Walker NM, Szeszko J, Field SF, Stevens HE, et al. Localization of type 1 diabetes susceptibility to the mhc class I genes hla-b and hla-a. Nature (2007) 450:887–92. doi: 10.1038/nature06406
47. Singer SM, Tisch R, Yang XD, McDevitt HO. An abd transgene prevents diabetes in nonobese diabetic mice by inducing regulatory T cells. Proc Natl Acad Sci USA (1993) 90:9566–70. doi: 10.1073/pnas.90.20.9566
48. Slattery RM, Kjer-Nielsen L, Allison J, Charlton B, Mandel TE, Miller JF. Prevention of diabetes in non-obese diabetic I-ak transgenic mice. Nature (1990) 345:724–6. doi: 10.1038/345724a0
49. Han S, Donelan W, Wang H, Reeves W, Yang L-J. Novel autoantigens in type 1 diabetes. Am J Trans Res (2013) 5:379–92.
50. Wicker LS, Miller BJ, Coker LZ, McNally SE, Scott S, Mullen Y, et al. Genetic control of diabetes and insulitis in the nonobese diabetic (Nod) mouse. J Exp Med (1987) 165:1639–54. doi: 10.1084/jem.165.6.1639
51. Koarada S, Wu Y, Olshansky G, Ridgway WM. Increased nonobese diabetic Th1:Th2 (Ifn-Gamma:Il-4) ratio is Cd4+ T cell intrinsic and independent of apc genetic background. J Immunol (2002) 169(11):6580–7. doi: 10.4049/jimmunol.169.11.6580
52. Koarada S, Wu Y, Ridgway WM. Increased entry into the ifn-gamma effector pathway by Cd4+ T cells selected by I-Ag7 on a nonobese diabetic versus C57bl/6 genetic background. J Immunol (2001) 167(3):1693–702. doi: 10.4049/jimmunol.167.3.1693
53. Ellis JS, Wan X, Braley-Mullen H. Transient depletion of Cd4+ Cd25+ regulatory T cells results in multiple autoimmune diseases in wild-type and b-Cell-Deficient nod mice. Immunology (2013) 139:179–86. doi: 10.1111/imm.12065
54. Pérol L, Lindner JM, Caudana P, Nunez NG, Baeyens A, Valle A, et al. Loss of immune tolerance to il-2 in type 1 diabetes. Nat Commun (2016) 7:13027. doi: 10.1038/ncomms13027
55. Salomon B, Lenschow DJ, Rhee L, Ashourian N, Singh B, Sharpe A, et al. B7/Cd28 costimulation is essential for the homeostasis of the Cd4+Cd25+ immunoregulatory T cells that control autoimmune diabetes. Immunity (2000) 12:431–40. doi: 10.1016/s1074-7613(00)80195-8
56. Wu AJ, Hua H, Munson SH, McDevitt HO. Tumor necrosis factor-alpha regulation of Cd4+Cd25+ T cell levels in nod mice. Proc Natl Acad Sci USA (2002) 99:12287–92. doi: 10.1073/pnas.172382999
57. D'Alise AM, Auyeung V, Feuerer M, Nishio J, Fontenot J, Benoist C, et al. The defect in T-cell regulation in nod mice is an effect on the T-cell effectors. Proc Natl Acad Sci USA (2008) 105:19857–62. doi: 10.1073/pnas.0810713105
58. Rainbow DB, Esposito L, Howlett SK, Hunter KM, Todd JA, Peterson LB, et al. Commonality in the genetic control of type 1 diabetes in humans and nod mice: Variants of genes in the il-2 pathway are associated with autoimmune diabetes in both species. Biochem Soc Trans (2008) 36:312–5. doi: 10.1042/BST0360312
59. Wicker LS, Clark J, Fraser HI, Garner VES, Gonzalez-Munoz A, Healy B, et al. Type 1 diabetes genes and pathways shared by humans and nod mice. J Autoimmun (2005) 25 Suppl:29–33. doi: 10.1016/j.jaut.2005.09.009
60. Ridgway WM, Peterson LB, Todd JA, Rainbow DB, Healy B, Burren OS, et al. Gene-gene interactions in the nod mouse model of type 1 diabetes. Adv Immunol (2008) 100:151–75. doi: 10.1016/S0065-2776(08)00806-7
61. Todd JA, Aitman TJ, Cornall RJ, Ghosh S, Hall JR, Hearne CM, et al. Genetic analysis of autoimmune type 1 diabetes mellitus in mice. Nature (1991) 351:542–7. doi: 10.1038/351542a0
62. Ghosh S, Palmer SM, Rodrigues NR, Cordell HJ, Hearne CM, Cornall RJ, et al. Polygenic control of autoimmune diabetes in nonobese diabetic mice. Nat Genet (1993) 4:404–9. doi: 10.1038/ng0893-404
63. McAleer MA, Reifsnyder P, Palmer SM, Prochazka M, Love JM, Copeman JB, et al. Crosses of nod mice with the related non strain. a polygenic model for iddm. Diabetes (1995) 44:1186–95. doi: 10.2337/diab.44.10.1186
64. Rogner UC, Avner P. Congenic mice: Cutting tools for complex immune disorders. Nat Rev Immunol (2003) 3:243–52. doi: 10.1038/nri1031
65. Driver JP, Serreze DV, Chen Y-G. Mouse models for the study of autoimmune type 1 diabetes: A nod to similarities and differences to human disease. Semin Immunopathol (2011) 33:67–87. doi: 10.1007/s00281-010-0204-1
66. Lyons PA, Armitage N, Argentina F, Denny P, Hill NJ, Lord CJ, et al. Congenic mapping of the type 1 diabetes locus, Idd3, to a 780-kb region of mouse chromosome 3: Identification of a candidate segment of ancestral DNA by haplotype mapping. Genome Res (2000) 10:446–53. doi: 10.1101/gr.10.4.446
67. Hill NJ, Lyons PA, Armitage N, Todd JA, Wicker LS, Peterson LB. Nod Idd5 locus controls insulitis and diabetes and overlaps the orthologous Ctla4/Iddm12 and Nramp1 loci in humans. Diabetes (2000) 49(10):1744–7. doi: 10.2337/diabetes.49.10.1744
68. Robles DT, Eisenbarth GS, Dailey NJM, Peterson LB, Wicker LS. Insulin autoantibodies are associated with islet inflammation but not always related to diabetes progression in nod congenic mice. Diabetes (2003) 52:882–6. doi: 10.2337/diabetes.52.3.882
69. Driver JP, Chen YG, Mathews CE. Comparative genetics: Synergizing human and nod mouse studies for identifying genetic causation of type 1 diabetes. Rev Diabetes Stud (2012) 9(4):169–87. doi: 10.1900/RDS.2012.9.169
70. Nyaga DM, Vickers MH, Jefferies C, Perry JK, O'Sullivan JM. The genetic architecture of type 1 diabetes mellitus. Mol Cell Endocrinol (2018) 477:70–80. doi: 10.1016/j.mce.2018.06.002
71. Yamanouchi J, Rainbow D, Serra P, Howlett S, Hunter K, Garner VE, et al. Interleukin-2 gene variation impairs regulatory T cell function and causes autoimmunity. Nat Genet (2007) 39(3):329–37. doi: 10.1038/ng1958
72. Long SA, Buckner JH, Greenbaum CJ. Il-2 therapy in type 1 diabetes: "Trials" and tribulations. Clin Immunol (2013) 149(3):324–31. doi: 10.1016/j.clim.2013.02.005
73. Izquierdo C, Ortiz AZ, Presa M, Malo S, Montoya A, Garabatos N, et al. Treatment of T1d Via optimized expansion of antigen-specific tregs induced by il-2/Anti-Il-2 monoclonal antibody complexes and Peptide/Mhc tetramers. Sci Rep (2018) 8(1):8106. doi: 10.1038/s41598-018-26161-6
74. Jarvis LB, Rainbow DB, Coppard V, Howlett SK, Georgieva Z, Davies JL, et al. Therapeutically expanded human regulatory T-cells are super-suppressive due to Hif1a induced expression of Cd73. Commun Biol (2021) 4(1):1186. doi: 10.1038/s42003-021-02721-x
75. Marcovecchio ML, Wicker LS, Dunger DB, Dutton SJ, Kopijasz S, Scudder C, et al. Interleukin-2 therapy of autoimmunity in diabetes (Itad): A phase 2, multicentre, double-blind, randomized, placebo-controlled trial. Wellcome Open Res (2020) 5:49. doi: 10.12688/wellcomeopenres.15697.1
76. Bettini M, Bettini ML. Function, failure, and the future potential of tregs in type 1 diabetes. Diabetes (2021) 70(6):1211–9. doi: 10.2337/dbi18-0058
77. Ueda H, Howson JM, Esposito L, Heward J, Snook H, Chamberlain G, et al. Association of the T-cell regulatory gene Ctla4 with susceptibility to autoimmune disease. Nature (2003) 423(6939):506–11. doi: 10.1038/nature01621
78. Rachid O, Osman A, Abdi R, Haik Y. Ctla4-ig (Abatacept): A promising investigational drug for use in type 1 diabetes. Expert Opin Investig Drugs (2020) 29(3):221–36. doi: 10.1080/13543784.2020.1727885
79. In't Veld P. Insulitis in human type 1 diabetes: A comparison between patients and animal models. Semin Immunopathol (2014) 36(5):569–79. doi: 10.1007/s00281-014-0438-4
80. Herold KC, Bundy BN, Long SA, Bluestone JA, DiMeglio LA, Dufort MJ, et al. An anti-Cd3 antibody, teplizumab, in relatives at risk for type 1 diabetes. N Engl J Med (2019) 381(7):603–13. doi: 10.1056/NEJMoa1902226
81. Robertson CC, Inshaw JRJ, Onengut-Gumuscu S, Chen WM, Santa Cruz DF, Yang H, et al. Fine-mapping, trans-ancestral and genomic analyses identify causal variants, cells, genes and drug targets for type 1 diabetes. Nat Genet (2021) 53(7):962–71. doi: 10.1038/s41588-021-00880-5
82. Lyons PA, Hancock WW, Denny P, Lord CJ, Hill NJ, Armitage N, et al. The nod Idd9 genetic interval influences the pathogenicity of insulitis and contains molecular variants of Cd30, Tnfr2, and Cd137. Immunity (2000) 13:107–15. doi: 10.1016/s1074-7613(00)00012-1
83. Forsberg MH, Foda B, Serreze DV, Chen Y-G. Combined congenic mapping and nuclease-based gene targeting for studying allele-specific effects of Tnfrsf9 within the Idd9.3 autoimmune diabetes locus. Sci Rep (2019) 9:4316. doi: 10.1038/s41598-019-40898-8
84. Cannons JL, Chamberlain G, Howson J, Smink LJ, Todd JA, Peterson LB, et al. Genetic and functional association of the immune signaling molecule 4-1bb (Cd137/Tnfrsf9) with type 1 diabetes. J Autoimmun (2005) 25:13–20. doi: 10.1016/j.jaut.2005.04.007
85. Luu K, Shao Z, Schwarz H. The relevance of soluble Cd137 in the regulation of immune responses and for immunotherapeutic intervention. J Leuk Biol (2020) 107:731–8. doi: 10.1002/JLB.2MR1119-224R
86. Itoh A, Ridgway WM. Targeting innate immunity to downmodulate adaptive immunity and reverse type 1 diabetes. ImmunoTargets Ther (2017) 6:31–8. doi: 10.2147/ITT.S117264
87. Pollok KE, Kim YJ, Zhou Z, Hurtado J, Kim KK, Pickard RT, et al. Inducible T cell antigen 4-1bb. Anal Expression Funct J Immunol (1993) 150:771–81.
88. Kwon BS, Weissman SM. Cdna sequences of two inducible T-cell genes. Proc Natl Acad Sci USA (1989) 86:1963–7. doi: 10.1073/pnas.86.6.1963
89. Gavin MA, Clarke SR, Negrou E, Gallegos A, Rudensky A. Homeostasis and anergy of Cd4(+)Cd25(+) suppressor T cells in vivo. Nat Immunol (2002) 3:33–41. doi: 10.1038/ni743
90. McHugh RS, Whitters MJ, Piccirillo CA, Young DA, Shevach EM, Collins M, et al. Cd4(+)Cd25(+) immunoregulatory T cells: Gene expression analysis reveals a functional role for the glucocorticoid-induced tnf receptor. Immunity (2002) 16:311–23. doi: 10.1016/s1074-7613(02)00280-7
91. Choi BK, Bae JS, Choi EM, Kang WJ, Sakaguchi S, Vinay DS, et al. 4-1bb-Dependent inhibition of immunosuppression by activated Cd4+Cd25+ T cells. J Leuk Biol (2004) 75:785–91. doi: 10.1189/jlb.1003491
92. Zheng G, Wang B, Chen A. The 4-1bb costimulation augments the proliferation of Cd4+Cd25+ regulatory T cells. J Immunol (2004) 173:2428–34. doi: 10.4049/jimmunol.173.4.2428
93. Irie J, Wu Y, Kachapati K, Mittler RS, Ridgway WM. Modulating protective and pathogenic Cd4+ subsets via Cd137 in type 1 diabetes. Diabetes (2007) 56:186–96. doi: 10.2337/db06-0793
94. Kachapati K, Adams DE, Wu Y, Steward CA, Rainbow DB, Wicker LS, et al. The B10 Idd9.3 locus mediates accumulation of functionally superior Cd137(+) regulatory T cells in the nonobese diabetic type 1 diabetes model. J Immunol (2012) 189:5001–15. doi: 10.4049/jimmunol.1101013
95. Melero I, Johnston JV, Shufford WW, Mittler RS, Chen L. Nk1.1 cells express 4-1bb (Cdw137) costimulatory molecule and are required for tumor immunity elicited by anti-4-1bb monoclonal antibodies. Cell Immunol (1998) 190:167–72. doi: 10.1006/cimm.1998.1396
96. Pollok KE, Kim YJ, Hurtado J, Zhou Z, Kim KK, Kwon BS. 4-1bb T-cell antigen binds to mature b cells and macrophages, and costimulates anti-Mu-Primed splenic b cells. Eur J Immunol (1994) 24:367–74. doi: 10.1002/eji.1830240215
97. Chitnis T, Khoury SJ. Role of costimulatory pathways in the pathogenesis of multiple sclerosis and experimental autoimmune encephalomyelitis. J Allergy Clin Immunol (2003) 112:837–49. doi: 10.1016/j.jaci.2003.08.025
98. Lee S-W, Park Y, So T, Kwon BS, Cheroutre H, Mittler RS, et al. Identification of regulatory functions for 4-1bb and 4-1bbl in myelopoiesis and the development of dendritic cells. Nat Immunol (2008) 9:917–26. doi: 10.1038/ni.1632
99. Goodwin RG, Din WS, Davis-Smith T, Anderson DM, Gimpel SD, Sato TA, et al. Molecular cloning of a ligand for the inducible T cell gene 4-1bb: A member of an emerging family of cytokines with homology to tumor necrosis factor. Eur J Immunol (1993) 23:2631–41. doi: 10.1002/eji.1830231037
100. Zapata JM, Perez-Chacon G, Carr-Baena P, Martinez-Forero I, Azpilikueta A, Otano I, et al. Cd137 (4-1bb) signalosome: Complexity is a matter of trafs. Front Immunol (2018) 9:2618. doi: 10.3389/fimmu.2018.02618
101. Etxeberria I, Glez-Vaz J, Teijeira Á, Melero I. New emerging targets in cancer immunotherapy: Cd137/4-1bb costimulatory axis. ESMO Open (2020) 4:e000733. doi: 10.1136/esmoopen-2020-000733
102. Cannons JL, Lau P, Ghumman B, DeBenedette MA, Yagita H, Okumura K, et al. 4-1bb ligand induces cell division, sustains survival, and enhances effector function of Cd4 and Cd8 T cells with similar efficacy. J Immunol (2001) 167:1313–24. doi: 10.4049/jimmunol.167.3.1313
103. Arch RH, Thompson CB. 4-1bb and Ox40 are members of a tumor necrosis factor (Tnf)-nerve growth factor receptor subfamily that bind tnf receptor-associated factors and activate nuclear factor kappab. Mol Cell Biol (1998) 18:558–65. doi: 10.1128/MCB.18.1.558
104. Jang IK, Lee ZH, Kim YJ, Kim SH, Kwon BS. Human 4-1bb (Cd137) signals are mediated by Traf2 and activate nuclear factor-kappa b. Biochem Biophys Res Commun (1998) 242:613–20. doi: 10.1006/bbrc.1997.8016
105. Cannons JL, Hoeflich KP, Woodgett JR, Watts TH. Role of the stress kinase pathway in signaling Via the T cell costimulatory receptor 4-1bb. J Immunol (1999) 163:2990–8.
106. Cannons JL, Choi Y, Watts TH. Role of tnf receptor-associated factor 2 and P38 mitogen-activated protein kinase activation during 4-1bb-Dependent immune response. J Immunol (2000) 165:6193–204. doi: 10.4049/jimmunol.165.11.6193
107. Wang C, Wen T, Routy J-P, Bernard NF, Sekaly RP, Watts TH. 4-1bbl induces tnf receptor-associated factor 1-dependent bim modulation in human T cells and is a critical component in the costimulation-dependent rescue of functionally impaired hiv-specific Cd8 T cells. J Immunol (2007) 179:8252–63. doi: 10.4049/jimmunol.179.12.8252
108. Sabbagh L, Pulle G, Liu Y, Tsitsikov EN, Watts TH. Erk-dependent bim modulation downstream of the 4-1bb-Traf1 signaling axis is a critical mediator of Cd8 T cell survival in vivo. J Immunol (2008) 180:8093–101. doi: 10.4049/jimmunol.180.12.8093
109. Sabbagh L, Andreeva D, Laramée GD, Oussa NAE, Lew D, Bisson N, et al. Leukocyte-specific protein 1 links tnf receptor-associated factor 1 to survival signaling downstream of 4-1bb in T cells. J Leuk Biol (2013) 93:713–21. doi: 10.1189/jlb.1112579
110. Kwon B. Cd137-Cd137 ligand interactions in inflammation. Immune Netw (2009) 9:84–9. doi: 10.4110/in.2009.9.3.84
111. Kwon B. Regulation of inflammation by bidirectional signaling through Cd137 and its ligand. Immune Netw (2012) 12:176–80. doi: 10.4110/in.2012.12.5.176
112. Hurtado JC, Kim YJ, Kwon BS. Signals through 4-1bb are costimulatory to previously activated splenic T cells and inhibit activation-induced cell death. J Immunol (1997) 158:2600–9.
113. DeBenedette MA, Shahinian A, Mak TW, Watts TH. Costimulation of Cd28- T lymphocytes by 4-1bb ligand. J Immunol (1997) 158:551–9.
114. Lee H-W, Park S-J, Choi BK, Kim HH, Nam K-O, Kwon BS. 4-1bb promotes the survival of Cd8+ T lymphocytes by increasing expression of bcl-xl and bfl-1. J Immunol (2002) 169:4882–8. doi: 10.4049/jimmunol.169.9.4882
115. Takahashi C, Mittler RS, Vella AT. Cutting edge: 4-1bb is a bona fide Cd8 T cell survival signal. J Immunol (1999) 162:5037–40.
116. Menk AV, Scharping NE, Rivadeneira DB, Calderon MJ, Watson MJ, Dunstane D, et al. 4-1bb costimulation induces T cell mitochondrial function and biogenesis enabling cancer immunotherapeutic responses. J Exp Med (2018) 215:1091–100. doi: 10.1084/jem.20171068
117. Teijeira A, Labiano S, Garasa S, Etxeberria I, Santamaría E, Rouzaut A, et al. Mitochondrial morphological and functional reprogramming following Cd137 (4-1bb) costimulation. Cancer Immunol Res (2018) 6:798–811. doi: 10.1158/2326-6066.CIR-17-0767
118. Shuford WW, Klussman K, Tritchler DD, Loo DT, Chalupny J, Siadak AW, et al. 4-1bb costimulatory signals preferentially induce Cd8+ T cell proliferation and lead to the amplification in vivo of cytotoxic T cell responses. J Exp Med (1997) 186:47–55. doi: 10.1084/jem.186.1.47
119. DeBenedette MA, Wen T, Bachmann MF, Ohashi PS, Barber BH, Stocking KL, et al. Analysis of 4-1bb ligand (4-1bbl)-Deficient mice and of mice lacking both 4-1bbl and Cd28 reveals a role for 4-1bbl in skin allograft rejection and in the cytotoxic T cell response to influenza virus. J Immunol (1999) 163:4833–41.
120. Tan JT, Whitmire JK, Ahmed R, Pearson TC, Larsen CP. 4-1bb ligand, a member of the tnf family, is important for the generation of antiviral Cd8 T cell responses. J Immunol (1999) 163:4859–68.
121. Kwon BS, Hurtado JC, Lee ZH, Kwack KB, Seo SK, Choi BK, et al. Immune responses in 4-1bb (Cd137)-deficient mice. J Immunol (2002) 168:5483–90. doi: 10.4049/jimmunol.168.11.5483
122. Foell JL, Diez-Mendiondo BI, Diez OH, Holzer U, Ruck P, Bapat AS, et al. Engagement of the Cd137 (4-1bb) costimulatory molecule inhibits and reverses the autoimmune process in collagen-induced arthritis and establishes lasting disease resistance. Immunology (2004) 113:89–98. doi: 10.1111/j.1365-2567.2004.01952.x
123. Melero I, Shuford WW, Newby SA, Aruffo A, Ledbetter JA, Hellström KE, et al. Monoclonal antibodies against the 4-1bb T-cell activation molecule eradicate established tumors. Nat Med (1997) 3:682–5. doi: 10.1038/nm0697-682
124. Taraban VY, Rowley TF, O'Brien L, Chan HTC, Haswell LE, Green MHA, et al. Expression and costimulatory effects of the tnf receptor superfamily members Cd134 (Ox40) and Cd137 (4-1bb), and their role in the generation of anti-tumor immune responses. Eur J Immunol (2002) 32:3617–27. doi: 10.1002/1521-4141(200212)32:12<3617::AID-IMMU3617>3.0.CO;2-M
125. May KFJ, Chen L, Zheng P, Liu Y. Anti-4-1bb monoclonal antibody enhances rejection of large tumor burden by promoting survival but not clonal expansion of tumor-specific Cd8+ T cells. Cancer Res (2002) 62:3459–65.
126. Martinez-Perez AG, Perez-Trujillo JJ, Garza-Morales R, Loera-Arias MJ, Saucedo-Cardenas O, Garcia-Garcia A, et al. 4-1bbl as a mediator of cross-talk between innate, adaptive, and regulatory immunity against cancer. Int J Mol Sci (2021) 22(12):1-13. doi: 10.3390/ijms22126210
127. Futagawa T, Akiba H, Kodama T, Takeda K, Hosoda Y, Yagita H, et al. Expression and function of 4-1bb and 4-1bb ligand on murine dendritic cells. Int Immunol (2002) 14:275–86. doi: 10.1093/intimm/14.3.275
128. Laderach D, Wesa A, Galy A. 4-1bb-ligand is regulated on human dendritic cells and induces the production of il-12. Cell Immunol (2003) 226:37–44. doi: 10.1016/j.cellimm.2003.11.003
129. Zhang B, Zhang Y, Niu L, Vella AT, Mittler RS. Dendritic cells and Stat3 are essential for Cd137-induced Cd8 T cell activation-induced cell death. J Immunol (2010) 184:4770–8. doi: 10.4049/jimmunol.0902713
130. Marson A, Kretschmer K, Frampton GM, Jacobsen ES, Polansky JK, MacIsaac KD, et al. Foxp3 occupancy and regulation of key target genes during T-cell stimulation. Nature (2007) 445:931–5. doi: 10.1038/nature05478
131. Chen Z, Herman AE, Matos M, Mathis D, Benoist C. Where Cd4+Cd25+ T reg cells impinge on autoimmune diabetes. J Exp Med (2005) 202:1387–97. doi: 10.1084/jem.20051409
132. Song Y, Wang N, Chen L, Fang L. Tr1 cells as a key regulator for maintaining immune homeostasis in transplantation. Front Immunol (2021) 12:671579. doi: 10.3389/fimmu.2021.671579
133. Gagliani N, Magnani CF, Huber S, Gianolini ME, Pala M, Licona-Limon P, et al. Coexpression of Cd49b and lag-3 identifies human and mouse T regulatory type 1 cells. Nat Med (2013) 19(6):739–46. doi: 10.1038/nm.3179
134. Sun Y, Chen HM, Subudhi SK, Chen J, Koka R, Chen L, et al. Costimulatory molecule-targeted antibody therapy of a spontaneous autoimmune disease. Nat Med (2002) 8:1405–13. doi: 10.1038/nm1202-796
135. Foell J, McCausland M, Burch J, Corriazzi N, Yan X-J, Suwyn C, et al. Cd137-mediated T cell Co-stimulation terminates existing autoimmune disease in sle-prone Nzb/Nzw F1 mice. Ann N Y Acad Sci (2003) 987:230–5. doi: 10.1111/j.1749-6632.2003.tb06052.x
136. Sun Y, Lin X, Chen HM, Wu Q, Subudhi SK, Chen L, et al. Administration of agonistic anti-4-1bb monoclonal antibody leads to the amelioration of experimental autoimmune encephalomyelitis. J Immunol (2002) 168:1457–65. doi: 10.4049/jimmunol.168.3.1457
137. Seo SK, Choi JH, Kim YH, Kang WJ, Park HY, Suh JH, et al. 4-1bb-Mediated immunotherapy of rheumatoid arthritis. Nat Med (2004) 10:1088–94. doi: 10.1038/nm1107
138. Zhou J, You BR, Yu Q. Agonist-induced 4-1bb activation prevents the development of sjögren's syndrome-like sialadenitis in non-obese diabetic mice. Biochim Biophys Acta Mol Basis Dis (2020) 1866:165605. doi: 10.1016/j.bbadis.2019.165605
139. Lee J, Lee E-N, Kim E-Y, Park H-J, Chang C-Y, Jung D-Y, et al. Administration of agonistic anti-4-1bb monoclonal antibody leads to the amelioration of inflammatory bowel disease. Immunol Lett (2005) 101:210–6. doi: 10.1016/j.imlet.2005.06.001
140. Mittler RS, Bailey TS, Klussman K, Trailsmith MD, Hoffmann MK. Anti-4-1bb monoclonal antibodies abrogate T cell-dependent humoral immune responses in vivo through the induction of helper T cell anergy. J Exp Med (1999) 190:1535–40. doi: 10.1084/jem.190.10.1535
141. Morris GP, Chen L, Kong Y-c. Cd137 signaling interferes with activation and function of Cd4+Cd25+ regulatory T cells in induced tolerance to experimental autoimmune thyroiditis. Cell Immunol (2003) 226:20–9. doi: 10.1016/j.cellimm.2003.11.002
142. Setareh M, Schwarz H, Lotz M. A mrna variant encoding a soluble form of 4-1bb, a member of the murine Ngf/Tnf receptor family. Gene (1995) 164:311–5. doi: 10.1016/0378-1119(95)00349-b
143. Kachapati K, Bednar KJ, Adams DE, Wu Y, Mittler RS, Jordan MB, et al. Recombinant soluble Cd137 prevents type one diabetes in nonobese diabetic mice. J Autoimmun (2013) 47:94–103. doi: 10.1016/j.jaut.2013.09.002
144. Wilcox RA, Chapoval AI, Gorski KS, Otsuji M, Shin T, Flies DB, et al. Cutting edge: Expression of functional Cd137 receptor by dendritic cells. J Immunol (2002) 168:4262–7. doi: 10.4049/jimmunol.168.9.4262
145. Michel J, Langstein J, Hofstädter F, Schwarz H. A soluble form of Cd137 (Ila/4-1bb), a member of the tnf receptor family, is released by activated lymphocytes and is detectable in sera of patients with rheumatoid arthritis. Eur J Immunol (1998) 28:290–5. doi: 10.1002/(SICI)1521-4141(199801)28:01<290::AID-IMMU290>3.0.CO;2-S
146. Shao Z, Sun F, Koh DR, Schwarz H. Characterisation of soluble murine Cd137 and its association with systemic lupus. Mol Immunol (2008) 45:3990–9. doi: 10.1016/j.molimm.2008.05.028
147. Yabas M, Elliott H, Hoyne GF. The role of alternative splicing in the control of immune homeostasis and cellular differentiation. Int J Mol Sci (2015) 17(1)1-21. doi: 10.3390/ijms17010003
148. Itoh A, Ortiz L, Kachapati K, Wu Y, Adams D, Bednar K, et al. Soluble Cd137 ameliorates acute type 1 diabetes by inducing T cell anergy. Front Immunol (2019) 10:2566. doi: 10.3389/fimmu.2019.02566
149. Gerold KD, Zheng P, Rainbow DB, Zernecke A, Wicker LS, Kissler S. The soluble ctla-4 splice variant protects from type 1 diabetes and potentiates regulatory T-cell function. Diabetes (2011) 60(7):1955–63. doi: 10.2337/db11-0130
150. Saverino D, Simone R, Bagnasco M, Pesce G. The soluble ctla-4 receptor and its role in autoimmune diseases: An update. Auto Immun Highlights (2010) 1(2):73–81. doi: 10.1007/s13317-010-0011-7
151. Simone R, Pesce G, Antola P, Rumbullaku M, Bagnasco M, Bizzaro N, et al. The soluble form of ctla-4 from serum of patients with autoimmune diseases regulates T-cell responses. BioMed Res Int (2014) 2014:215763. doi: 10.1155/2014/215763
152. Michel J, Schwarz H. Expression of soluble Cd137 correlates with activation-induced cell death of lymphocytes. Cytokine (2000) 12:742–6. doi: 10.1006/cyto.1999.0623
153. Hurtado JC, Kim SH, Pollok KE, Lee ZH, Kwon BS. Potential role of 4-1bb in T cell activation. comparison with the costimulatory molecule Cd28. J Immunol (1995) 155:3360–7.
154. Chen Y-G, Forsberg MH, Khaja S, Ciecko AE, Hessner MJ, Geurts AM. Gene targeting in nod mouse embryos using zinc-finger nucleases. Diabetes (2014) 63:68–74. doi: 10.2337/db13-0192
155. Forsberg MH, Ciecko AE, Bednar KJ, Itoh A, Kachapati K, Ridgway WM, et al. Cd137 plays both pathogenic and protective roles in type 1 diabetes development in nod mice. J Immunol (2017) 198:3857–68. doi: 10.4049/jimmunol.1601851
156. Foda BM, Ciecko AE, Serreze DV, Ridgway WM, Geurts AM, Chen Y-G. The Cd137 ligand is important for type 1 diabetes development but dispensable for the homeostasis of disease-suppressive Cd137(+) Foxp3(+) regulatory Cd4 T cells. J Immunol (2020) 204:2887–99. doi: 10.4049/jimmunol.1900485
157. Jung HW, Choi SW, Choi JIL, Kwon BS. Serum concentrations of soluble 4-1bb and 4-1bb ligand correlated with the disease severity in rheumatoid arthritis. Exp Mol Med (2004) 36:13–22. doi: 10.1038/emm.2004.2
158. Rioja I, Hughes FJ, Sharp CH, Warnock LC, Montgomery DS, Akil M, et al. Potential novel biomarkers of disease activity in rheumatoid arthritis patients: Cxcl13, Ccl23, transforming growth factor alpha, tumor necrosis factor receptor superfamily member 9, and macrophage colony-stimulating factor. Arthritis Rheum (2008) 58:2257–67. doi: 10.1002/art.23667
159. Liu G-Z, Gomes AC, Putheti P, Karrenbauer V, Kostulas K, Press R, et al. Increased soluble 4-1bb ligand (4-1bbl) levels in peripheral blood of patients with multiple sclerosis. Scand J Immunol (2006) 64:412–9. doi: 10.1111/j.1365-3083.2006.01796.x
Keywords: NOD, T1D (type 1 diabetes), t cell, treg cells, CD137, CD137L
Citation: Rojas M, Heuer LS, Zhang W, Chen Y-G and Ridgway WM (2022) The long and winding road: From mouse linkage studies to a novel human therapeutic pathway in type 1 diabetes. Front. Immunol. 13:918837. doi: 10.3389/fimmu.2022.918837
Received: 12 April 2022; Accepted: 27 June 2022;
Published: 22 July 2022.
Edited by:
Anette S. B. Wolff, Haukeland University Hospital, NorwayReviewed by:
Eddie A. James, Benaroya Research Institute, United StatesCopyright © 2022 Rojas, Heuer, Zhang, Chen and Ridgway. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: William M. Ridgway, d21yaWRnd2F5QHVjZGF2aXMuZWR1
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.