 Xuan Lu
Xuan Lu Yun-Mei Yang1,2
Yun-Mei Yang1,2 Yuan-Qiang Lu
Yuan-Qiang Lu
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 18 July 2022
Sec. Inflammation
Volume 13 - 2022 | https://doi.org/10.3389/fimmu.2022.917293
This article is part of the Research Topic Immunosenescence after Sepsis View all 6 articles
Progressive immune dysfunction associated with aging is known as immunosenescence. The age-related deterioration of immune function is accompanied by chronic inflammation and microenvironment changes. Immunosenescence can affect both innate and acquired immunity. Sepsis is a systemic inflammatory response that affects parenchymal organs, such as the respiratory system, cardiovascular system, liver, urinary system, and central nervous system, according to the sequential organ failure assessment (SOFA). The initial immune response is characterized by an excess release of inflammatory factors, followed by persistent immune paralysis. Moreover, immunosenescence was found to complement the severity of the immune disorder following sepsis. Furthermore, the immune characteristics associated with sepsis include lymphocytopenia, thymus degeneration, and immunosuppressive cell proliferation, which are very similar to the characteristics of immunosenescence. Therefore, an in-depth understanding of immunosenescence after sepsis and its subsequent effects on the organs may contribute to the development of promising therapeutic strategies. This paper focuses on the characteristics of immunosenescence after sepsis and rigorously analyzes the possible underlying mechanism of action. Based on several recent studies, we summarized the relationship between immunosenescence and sepsis-related organs. We believe that the association between immunosenescence and parenchymal organs might be able to explain the delayed consequences associated with sepsis.
Aging is a complex phenomenon characterized by the progressive loss of physical functions and numerous age-related diseases, such as neurological disorders, cardiovascular diseases, renal insufficiency, and chronic obstructive pulmonary disease (COPD) (1). Three main characteristics are associated with senescent cells: apoptotic resistance, cell proliferation arrest, and senescence-associated secretory phenotype (SASP) (2). Immunosenescence refers to the age-related deterioration of the immune system. The concept of immunosenescence was proposed as early as 1964 (3). The most prominent characteristics include reduced adaptive immunity and resistance to infection and an increased risk of autoimmune disorders (4). Additionally, immunosenescence is regulated by factors like excessive chronic inflammation and alterations in the microenvironment (5). Immunosenescence may lead to a vicious cycle of an imbalance between an impaired immune system and the indirect regulation of parenchymal tissues and organs (6).
Sepsis is an intricate, heterogeneous, and highly fatal syndrome (7), which is responsible for life-threatening organ dysfunction due to the immune regulation disorder. The third international consensus definition of sepsis and septic shock (Sepsis 3.0) recommended the sequential organ failure assessment (SOFA) to assess sepsis and hence predict the subsequent prognosis (8). While the old definitions of sepsis greatly emphasized infection, Sepsis 3.0 focused on the dysregulation of the body’s response to infection and organ dysfunction. Furthermore, the organ damage scored by SOFA focuses on organs like lungs, heart, liver, kidneys, and brain. Surviving sepsis is associated with chronic, long-term consequences in host protective immunity (9). Additionally, researchers observed that most of the survivors suffered from issues like nervous system disturbances and cognitive dysfunction throughout their life span (10). Since several similarities are found between immunosuppression after sepsis and immunosenescence, researchers hypothesized that these two factors might be associated with the progressive failure of immune functions (11, 12). In sepsis, an increase in the number of myeloid-derived suppressor cells (MDSCs) was associated with the regulation of the function of other immune cells, and excessive inflammation was blocked (13, 14). It has been suggested that MDSCs play a paradoxical role in sepsis: these cells may increase the production of proinflammatory cytokine during emergency myelogenesis and be also potently immunosuppressive (15). MDSCs may induce immunosenescence in the remodeled immune system (16). Therefore, we were interested in analyzing the effect of immunosenescence on sepsis, including the effect on the parenchymal organs. Here we review the possible relationship between septic injury-related organs and immunosenescence and analyze the possible mechanisms of immunosenescence after sepsis, which may shed some light on the delayed consequences of sepsis.
The pathogenesis associated with immunosenescence is multifactorial, which affects both innate and adaptive immunity. Immunosenescence involves the generation, migration, proliferation, differentiation, and activation stages of immune cells (16). The life-threatening consequences include a declined response to antigen stimulation, an increased response to chronic inflammation, a reduction in the defense to pathogenic microbial invasion, and a decrease in gene mutation identification and clearance ability in cells (16). These could increase the risk and severity of infectious diseases, autoimmune diseases, and cancer (17). At the cellular level, the senescence of immune cells includes atrophy and failure of normal cellular functions. The prominent features associated with the detection of senescence in immune cells are shown in Figure 1. They include the following: 1) Cell cycle inhibition: downstream signaling of p53/p21CIP and p16INK4a/pRB pathways and inhibition of cyclin-dependent kinases (18). 2) Telomere attrition: the shortening of telomere length, wherein the telomerase activity is impaired (19). 3) SASP: it refers to the secretion of a wide range of soluble factors by senescent cells, such as chemokines, pro-inflammatory cytokines, growth factors, and proteases (20). 4) An excess accumulation of beta-galactosidase (SA-β-gal) in the lysosomes results in pH changes in lysosomes (21). 5) Mitochondrial dysfunction: Different immune cells express different mitochondrial phenotypes after aging and are affected by oxidative stress (22). 6) DNA damage: The persistence of DNA damage can lead to apoptosis or senescence (23). 7) Proteostasis: a gradual loss of unfolded protein response with aging, which results in proteostasis (24).
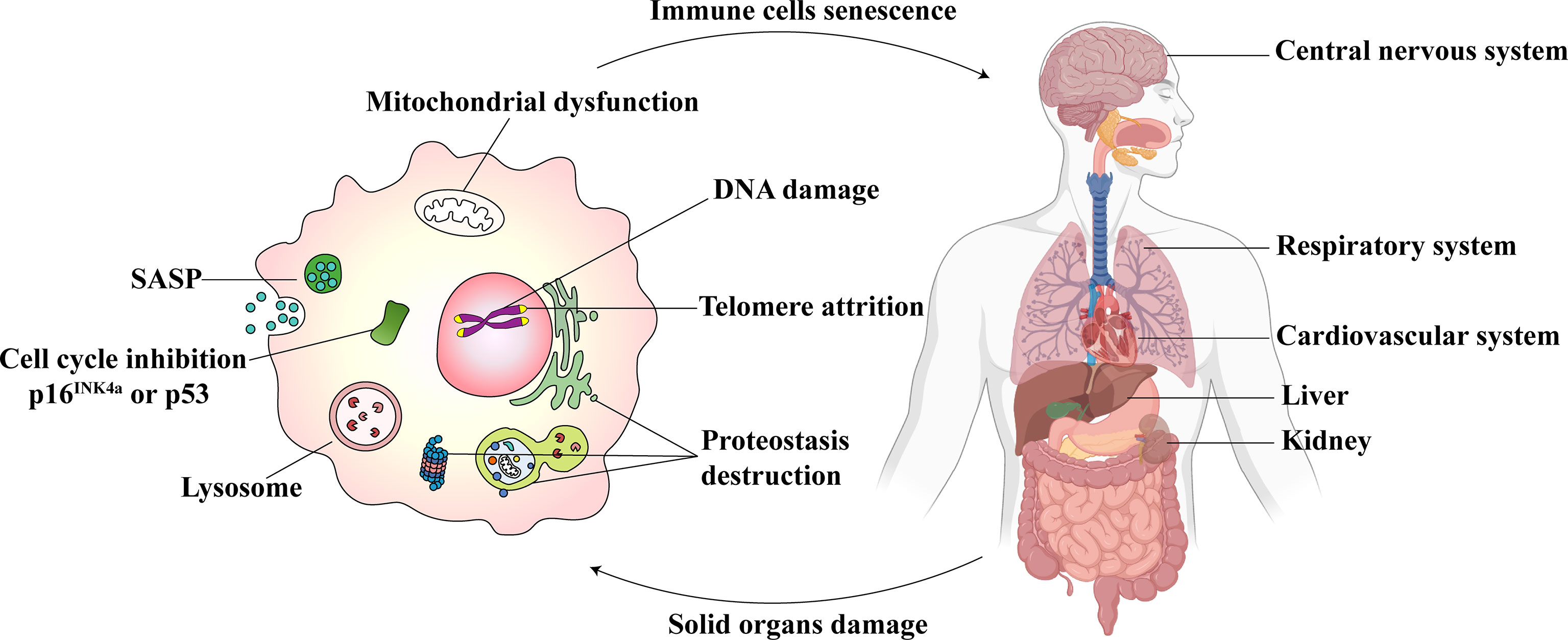
Figure 1 Immunosenescence and parenchymal organ damage complement each other. The senescent immune cells (Left) show excessive oxidative stress in the mitochondria. The dysfunction, in turn, leads to DNA damage, telomere attrition, and proteostasis destruction. The cell cycle is inhibited. The senescence-associated secretory phenotype (SASP) is released, and lysosomal SA-β-gal accumulates. Immunosenescence could influence the central nervous system, respiratory system, cardiovascular system, liver, and kidneys (Right). At the same time, the damage to these parenchymal organs may also cause immune senescence.
Sepsis could lead to an unbalanced immune response. According to Sepsis 3.0, this phenomenon is associated with excessive inflammation and immunosuppression in the body (8). A persistent immune stimulation is characterized by invading pathogens and release of damage-associated molecular patterns (DAMPs). DAMPs activate pattern recognition receptors, which can detect the pathogen-associated molecular patterns (PAMPs), triggering a vicious cycle of continuous activation and dysfunction of the immune system (25) (Figure 2). In the initial stage, many pro-inflammatory factors are released, such as TNF-α, IL-1β, IL-6, IL-12, and IL-18. Further on, researchers observed that the patients with sepsis suffered profound immunosuppression. An altered immune response includes an intense “storm of inflammatory factors,” which act as immunosenescence accelerators (12). Immunosenescence is a dynamic process that diminishes immune system functions (26). It leads to a low-grade chronic inflammation called “inflammaging” and was found to reduce the ability to trigger an effective antibody and cellular response (27, 28). Therefore, chronic inflammation may be the result or an influencing factor of immunosenescence.
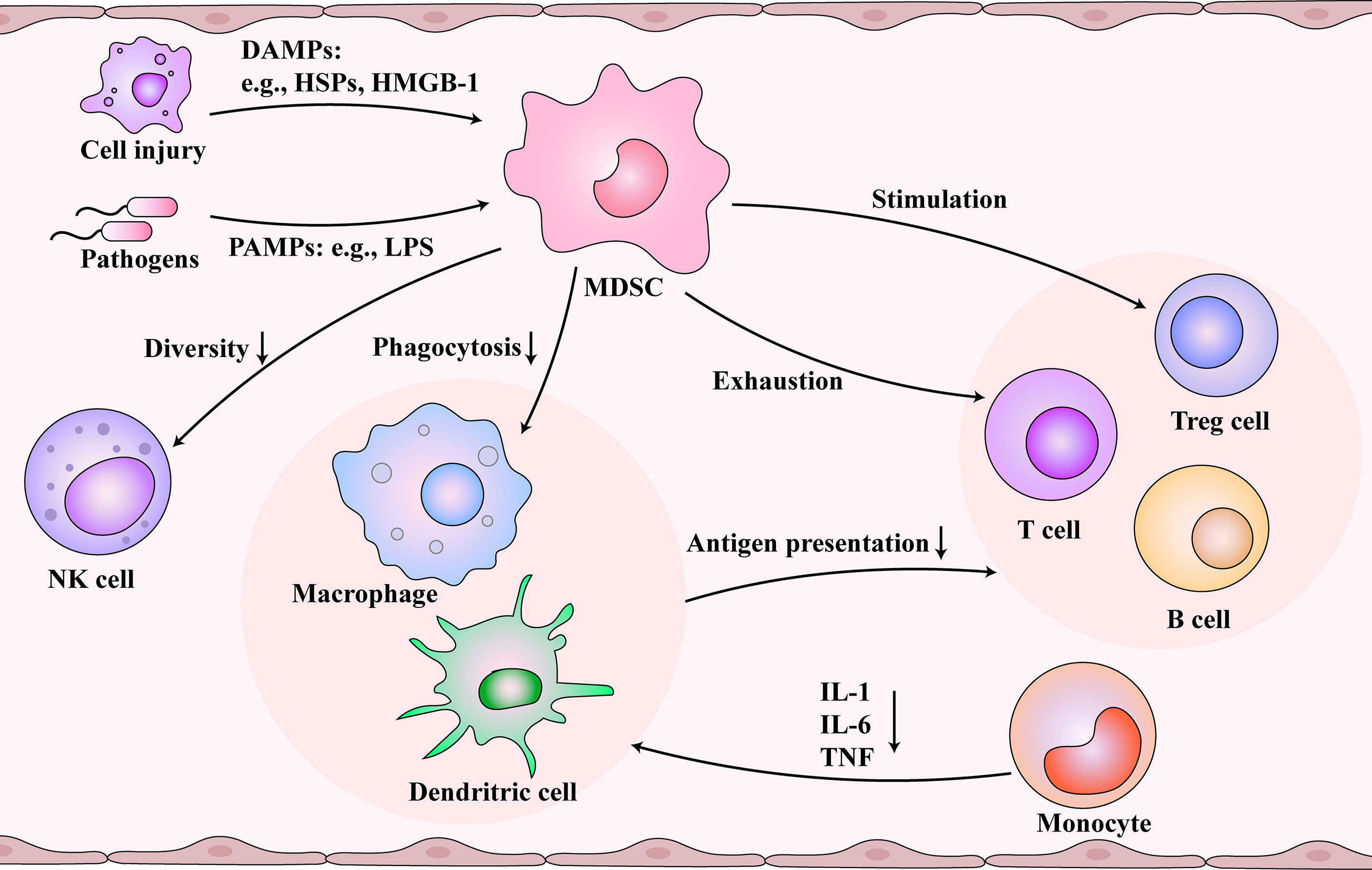
Figure 2 Immunosuppression and immunosenescence caused by myeloid-derived suppressor cells (MDSCs) after sepsis. Damaged cells and pathogens release damage-associated molecular patterns (DAMPs) and pathogen-associated molecular patterns (PAMPs), respectively, which are picked up by the immune system. During immunosuppression and immunosenescence stage of sepsis, MDSCs are activated and inhibit the function of dendritic cells (DCs) and macrophages. At the same time, MDSCs reduce the diversity of NK cells. Monocytes differentiate into DCs and macrophages, but MDSCs change the cytokines secreted by them. The functions of the T cells and B cells are inhibited by damaged antigen-presenting cells, controlled by MDSCs, and regulated by regulatory T (Treg) cells. Immunosenescence and immunosuppression form a vicious cycle.
MDSCs are a heterogeneous immature myeloid cell population that originates from hematopoietic stem cells and undergo the myeloid progenitor cell developmental stage in the bone marrow (29). In many pathological conditions, the accumulation of MDSCs occurs after an impaired myelopoietic maturation of granulocytes, monocytes, macrophages, and dendritic cells (30). Emergency myelogenesis in the bone marrow is triggered when peripheral inflammatory factors stimulate the secretion of colony-stimulating factors and chemokines (31). Gabrilovich et al. proposed a two-stage model that the expansion and activation of MDSCs require 1) emergency myelopoiesis drove by exogenous stimulus like infection and 2) pathological myeloid activation induced by the release of DAMPs or PAMPs from organ injury and secondary infections (32, 33). MDSCs are associated with prolonged immunosuppression in sepsis (34). Meanwhile, the number of MDSCs increases with age (35–37). Next, we investigated the immunosuppression caused by MDSCs, which may be associated with immunosenescence. MDSCs and aging demonstrate similar effects on immune cells, which might be essential for understanding immunosenescence after sepsis (Figure 2). MDSCs are essential for eliciting a macrophage immune response to inflammation (38). MDSCs inhibit the function of DCs and macrophages (39). The antigen presentation and phagocytosis function of macrophages in the blood are weakened with age (40). In monocytes, IL-1, IL-6, and TNF are reduced (41), and several defects in monocyte surface molecules, including CD14 and CD71, have also been observed in sepsis patients (42). Also, aging could inhibit the diversity of natural killer (NK) cells (43). MDSCs could reduce the secretion of cytokines and the cytotoxicity of NK cells (44, 45). Also, the effect of MDSCs and aging on T cells are profound. The cytotoxicity associated with CD8+ T cells is inhibited by reducing the number of IL-2 and INF-γ. Additionally, MDSCs promote T cell depletion, which is consistent with the decrease in the number of T cells with increasing age (46). Studies also found that the responsiveness of T cells to different external injuries decreases with age (47). In addition, MDSCs can stimulate the production of Treg cells and further inhibit the function of T cells (39, 48). In sepsis, B cells develop toward exhaustion. Both MDSCs and senescence contribute to the decline of B lymphocytes, including proliferation and antibody production (49, 50). The expression of MHC II decreases, and the secretion of IL-10 increases (51).
After sepsis, MDSCs increase the expression of arginase 1 (ARG1), which lowers the arginine concentration in the microenvironment (52). Also, MDSCs activation stimulates the expression of indoleamine-pyrrole 2,3-dioxygenase (IDO), which leads to tryptophan deficiency (53). As a result of the lack of both proteins, protein synthesis is inhibited, and T cell proliferation is affected. MDSCs also demonstrate the potential to enhance oxidative stress. The expression of NADPH oxidase 2 (NOX2) and inducible nitric oxide synthase (iNOS) is increased (54). The production of NO and ROS compounds oxidizes tyrosine residues on the T cell receptor (TCR), resulting in reduced anti-specific stimulation of T cells (55). Reactive oxygen species (ROS) may induce immunosenescence (56). In addition, MDSCs can also inhibit T cell function through the PD1/PDL1 axis, resulting in immunosuppression (48).
The mechanism of immunosenescence after sepsis is a hot research topic, and several recent studies suggested many different possibilities. Telomeres, located at the end of chromosomes, control cell aging and regulate gene expression. Telomere shortening has become a putative causative event of cellular aging. Sepsis proved to cause telomere shortening with decreased telomerase activity in both mice and humans (57). Also, telomere shortening may be related to the increased oxidative stress after sepsis (58). Previous studies showed a negative correlation between the level of oxidative stress and telomere length (59).
A significant manifestation of immunosuppression is the reprogramming of immune cells, including the epigenetic regulation of gene expression through histone modification and DNA methylation (DNAm) (60). Histone acetylation of lysine residues supports transcription, while their methylation leads to the formation of active euchromatin or silent heterochromatin (61). The methylation of histone-3 lysine-27 (H3K27) and histone-3 lysine-4 (H3K4) are associated with the inhibition and activation of transcription, respectively. Lipopolysaccharide (LPS)-tolerant macrophages show an increased tumor-suppressive histone modification of H3K9 me2 in promoter regions encoding IL-1β and TNF genes. Similar results were obtained in LPS-tolerant PBMCs in sepsis patients (62). In patients with sepsis, monocytes and macrophages can also be reprogrammed. Additionally, DNAm is linked to age (63, 64). Age-related methylation in the nuclei and mitochondria DNA is considered a senescence biomarker (65). For example, methylation in the CpG islands of gene ELOVL2 is highly age-related (66, 67). Extensive research demonstrated that DNAm could be used to explore the outcome of immunosenescence (68–70).
The regulation of immune checkpoints is essential in both immunosuppression and immunosenescence after sepsis. The programmed cell death-protein 1 (PD-1), programmed death-ligand 1 (PD-L1), and cytotoxic T lymphocyte-associated protein (CTLA-4) are currently the focus of immune checkpoint proteins. Increased expression of PD-1 is associated with an increased incidence and mortality of secondary nosocomial infection after septic shock and decreased T cell proliferation (71). In vitro experiments demonstrated that blocking PD-1 and PD-L1 pathways could improve lymphocyte apoptosis and regulate immune function (72). PD-L1 expression of neutrophils and monocytes and PD-1 expression of CD8+ T cells and NK cells resulted in impaired phagocytosis (73). The immune checkpoint inhibitor (ICI) of PD-1 could improve the survival rate in septic mice (74). Similarly, anti-CTLA-4 antibody therapy reduced sepsis-induced spleen apoptosis and improved survival (75). Although a controversy still exists about whether PD-1 antagonists can alleviate aging, many studies showed that ICIs are also meaningful for immunosenescence. A recent meta-analysis was conducted on the efficacy of ICI in older and younger patients. ICIs improved overall survival in both young and old patients with a cut-off age of 65-70 years (76). However, more than one study showed that ICI is more effective in older patients with metastatic melanoma than in younger patients (77–79). Due to the heterogeneity of different ICIs and different diseases, the research on the effect of ICIs on immunosenescence is still in progress.
A decline in the T cell output is a characteristic of immunosenescence (80). T cell senescence differs from anergy and depletion in that it is an irreversible process (81, 82). T cell senescence has been demonstrated in tumors and has been shown to occur in inflammaging (83, 84). Here, we explore the mechanism of T cell senescence following sepsis-associated organ injury (Figure 3). Impaired glucose homeostasis may occur in sepsis (85). Furthermore, the disruption of glucose metabolism activates the PI3K-AkT-mTOR signaling pathways in senescent T cells (86, 87). Studies showed that cyclic adenosine monophosphate (cAMP) production can activate the cAMP-PKA-CREB pathway, which could induce senescence in T cells (88). In addition, ERK1/2 and p38 pathways are activated by cAMP (89). The P38 arrests cell cycle progression by activating p53, p21, and p16 (90). Also, organ damage caused by sepsis produces DAMPs which activate the cGAS-STING pathway (91). At the same time, the activated NFκB pathway leads to DNA damage and SASP secretion and causes mitochondrial oxidative stress, resulting in mitochondrial dysfunction (92).
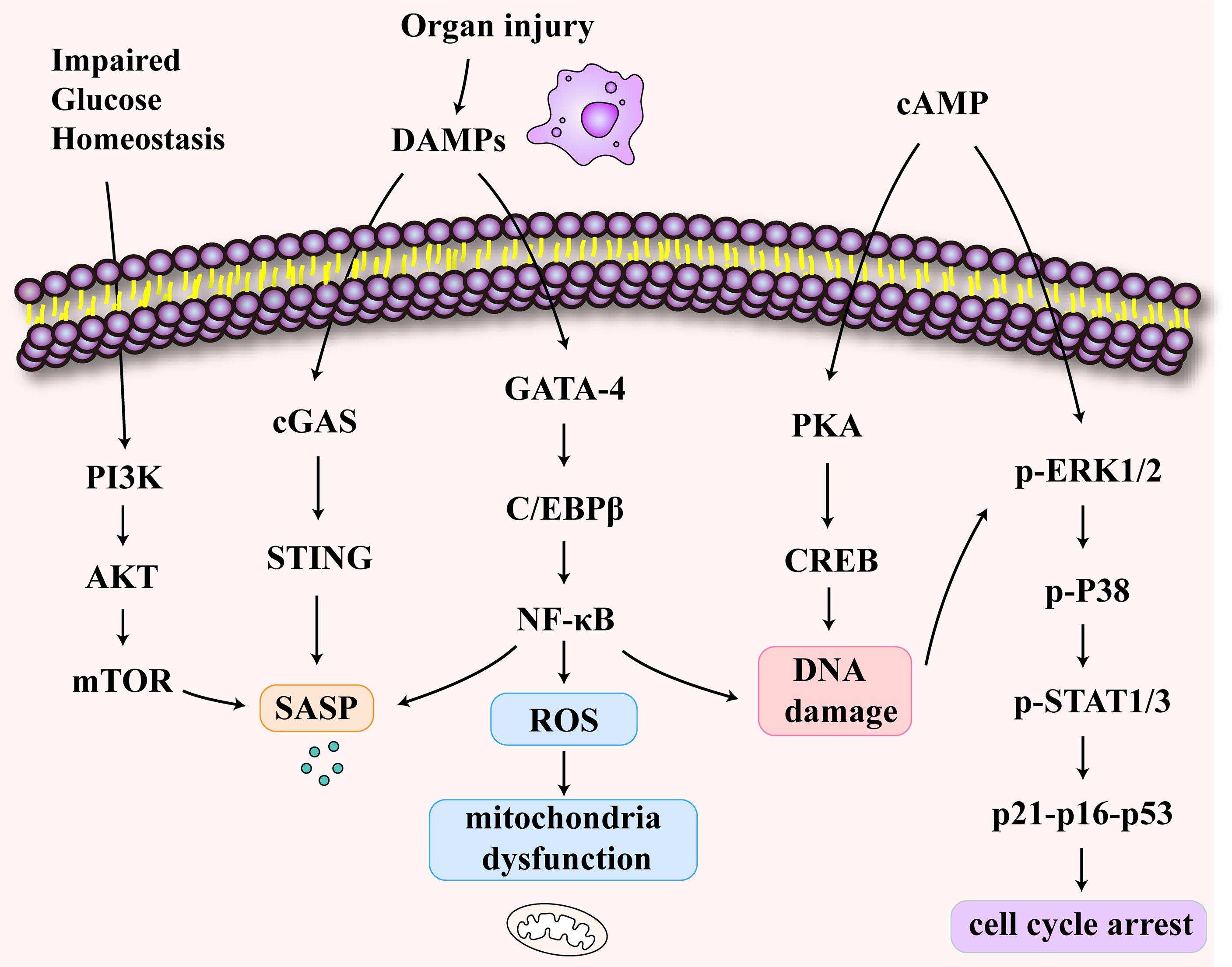
Figure 3 The signaling pathways of T cell senescence following sepsis-associated organ injury. The disruption of glucose metabolism activates the PI3K-AkT-mTOR signaling pathways in senescent T cells. The increase of cyclic adenosine monophosphate (cAMP) activates cAMP-PKA-CREB pathway, ERK1/2 and p38 pathways. These pathways may induce DNA damage and cell cycle arrest. The release of damage-associated molecular patterns (DAMPs) activates cGAS-STING pathway and NFκB pathway which may be related to SASP secretion and mitochondrial dysfunction.
Immunosenescence affects immune organs, including the spleen, bone marrow, and thymus, thus accelerating the degeneration of the immune system (93). It is considered a comprehensive remodeling of the immune system and the microenvironment (94). Researchers observed that with aging, the immune system could drive systematic age-related changes in solid non-immune organs (95). This alteration can result in the loss of tissue homeostasis and cause significant damage. As immunosenescence progresses, the homeostasis of innate and adaptive immune cells in solid organs is disrupted, and the body’s susceptibility to infectious diseases and cancer increases (96). In addition to detecting aging markers, p16, p21, and SASP in the liver, kidneys, lungs, and other solid organs of immunosenescent mice, tissue damage is also observed in these organs. The aging and damage of these organs will eventually lead to a shortened lifespan. The extent of inflammation can predict the possibility of cardiovascular diseases, neurodegenerative disorders, and weakness (97). Sepsis induces damage to the organs, and growing evidence exists linking sepsis and immunosenescence. Immunosenescence may occur after sepsis and could complement organ damage (Figure 1). Therefore, exploring the effect of immunosenescence on solid organs is of great significance.
Different individualistic factors like lifestyle, long-term environmental exposure, gene factors, and systemic diseases can be used as regulatory factors of lung dysfunction, resulting in lung remodeling, impaired respiratory function, and increased susceptibility to lung diseases (98–100). When sepsis occurs, the lungs are most likely to be damaged first (101). Acute lung injury caused by sepsis, including acute respiratory distress syndrome, is one of the most common causes of death in patients with sepsis (102).
Oxidative stress-induced by immunosenescence may lead to structural and functional abnormalities in cilia (103). The activation of PKCϵ by oxidation can reduce the airway’s ciliary beat frequency and curtail the ciliary clearance function (104). The barrier function of airway epithelial cells (AECs) decreases with immunosenescence (105), and SASP secreted by senescent cells can promote senescence of AECs (106). Senescent epithelial cells can increase airway susceptibility (107). An immunological crosstalk occurs between AECs and dendritic cells (DCs). The DCs of the elderly secrete pro-inflammatory factor, TNF-α, and affects the function of primary bronchial epithelial cells, making the elderly more susceptible to chronic airway inflammation (108). DC’s activation and maturation are inhibited, and antigen presentation ability decreases in the elderly (109). In addition to its association with DCs, AECs also exhibit a synergistic effect on the airway macrophages. AECs and airway macrophages can interact to engulf the particles or pathogens (110). A study on senescent lung macrophages showed that the proliferation ability of macrophages was decreased, the production of cytokines was impaired, and the phagocytosis and killing of bacteria and the clearance effect on apoptotic neutrophils were weakened (40, 111–113).
In addition, the impaired immune function caused by immunosenescence affects the adaptive immune system of the lungs. Senescence decreases the number of B cells, transforming them from naive B cells into antigen-expressing B cells (114). Many reasons for this decrease exist, including cytokine secretion and metabolism. Studies showed that a decrease in IL-10 in old-aged mice can reduce B cells (115). Tryptophan metabolism is influenced by aging, and a decrease in tryptophan concentration inhibits B cell development (116). Thymic involution may cause T cell shifts. The distribution of immune cells might vary, and the lungs might endure pro-inflammatory metastases after immunosenescence (117). The dysfunction of T cells also includes a decrease in the number of memory and naive T cells (118). Activating thymic T-regulatory cells (Tregs) associated with Th17 and persistent chronic inflammation leads to an increase in age-related lung autoimmunity. Tregs are further activated by the release of inflammatory factors (119).
Approximately 70% of sepsis patients demonstrate the possibility of sepsis-associated encephalopathy (120). Sepsis-related encephalopathy causes acute organ dysfunction and many long-term mental problems. For example, immunosenescence is associated with neurodegenerative diseases like delayed onset Alzheimer’s disease and Parkinson’s disease (PD) (121). Intestinal flora affects neural development, regulates behavior, and promotes neurological disorders (122). The location and accumulation of gut microbes are associated with immunosenescence (123). As a result, the intestinal barrier’s adhesion and leakage of microorganisms and microbial byproducts are increased (124, 125). Changes in the microbial-gut-brain axis led to an increase in circulating inflammatory factors (126). Short-chain fatty acids produced by the gut flora activate brain microglia, which cause neuroinflammation and neuron damage (127).
The blood-brain barrier (BBB) is a highly selective barrier between blood and brain tissue that limits the uncontrolled diffusion of cells and molecules from the blood into the central nervous system and regulates the entry of nutrients (128). In the case of sepsis, potentially harmful proteins can penetrate the brain through the compromised BBB (129, 130). Reduced expression of tight junction proteins inhibits endothelial cell communication, increases the BBB permeability, and decreases the BBB repair response (131). Next, P-GP and LRP-1 are essential efflux transporters that mediate amyloid-β peptide (Aβ) clearance in brain tissues (132). The expression of P-GP and LRP-1 proteins decreases with immunosenescence, leading to the accumulation of Aβ in the brain (133–135). Also, it affects the function of the glucose transporter 1 (GLUT1) on BBB vascular endothelial cells, leading to reduced glucose uptake by the brain and a reduced BBB function (136, 137). It can be speculated that immunosenescence results in increased permeability and decreased function of BBB, resulting in some potentially harmful proteins entering the brain, accumulation of Aβ, and reduced regulation of nutrient intake.
The choroid plexus (CP) is a part of the BBB, wherein almost all types of immune cells can be found, and uninterrupted crosstalk occurs through TNFα and IFNγ released by the central nervous system (138). An increase in the Th1/Th2 ratio during aging leads to an increased expression of the chemokine, CCL11, which may be associated with cognitive impairment (139). Also, an increase in CCL11 can transform the microglia into an inflammatory state (140). Thus, immunosenescence may disrupt the intrinsic balance of immune cells in the CP, leading to central nervous system dysfunction.
Microglia cells are innate immune cells in the brain that generally remain stable and in a low replication rate. Senescent microglia cells exhibit significantly smaller and less branched dendrites, exhibit slower acute responses, lower motility, and cell mobility (141). Several studies showed that aging microglia show higher proliferation and promote the production of pro-inflammatory cytokines (142). However, chemotaxis and Aβ scavenging ability attenuate, and the protective function of the central nervous system decreases. In addition, the long-term activation of microglia results in chronic inflammation of the central nervous system, which increases oxygen free radicals, mitochondrial damage, and possibly the death of neurons (142). These may further be associated with the damage of Triggering Receptor Expressed on Myeloid Cells 2-DNAX activation protein 12 (TREM2-DAP12) and CX3Cl1-CX3Cr1 axes (143). TREM2-DAP12 axes could promote microglial cell activation and phagocytosis (144). CX3Cl1-CX3Cr1 refers to an important communication channel between neurons and microglia (145). Immunosenescence disrupts both of these pathways leading to the development of neurodegenerative diseases (146). Oxidative stress-related mechanisms prior to Aβ oligomerization trigger microglia senescence and malnutrition (147). Immune receptors such as major histocompatibility complex (MHC) II, CD68, CD14, CD11, and TLRs are upregulated by aging microglial cells (148, 149). In senescent microglia and macrophages, lipid messenger prostaglandin E2 (PGE2) signaling promotes glucose conversion to glycogen through its EP2 receptor. At the same time, a decreased glucose flux and weakened mitochondrial respiration leads to maladaptive pro-inflammatory responses (150).
The phagocytosis and chemotaxis abilities of macrophages are inhibited by senescence (151). When induced by accumulated Aβ, macrophages switch from a regulatory and anti-inflammatory M2 phenotype to a pro-inflammatory M1 phenotype, producing IL-1B and TNF-α (152). Next, macrophages that interact with microglial cells and amyloid plaques are thought to be inflammatory activators that produce inflammatory cytokines and ROS, leading to neuronal loss and apoptosis (121). Therefore, immunosenescence can be manifested as the destruction of inherent barriers and the release of pro-inflammatory mediators by the interaction of microglial cells and neurons, leading to the occurrence and development of diseases.
Heart failure and immunosenescence are mutually reinforcing processes. Chronic heart failure (CHF) is characterized by high levels of cytokines, such as IL-6, in an inflammatory state, which may also induce immune aging (153). Moreover, the more intense T cell differentiation in CHF patients, the more serious is the senescence degree (154). Additionally, the telomere length of lymphocytes is reduced by 40% in CHF (155).
Patients with sepsis may experience local myocardial ischemia or infarction secondary to coronary artery disease (156). In myocardial infarction, inflammatory signaling is a crucial reparative pathway in regulating the deposition and metabolism of extracellular matrix proteins (157, 158). Macrophage-derived matrix metalloproteinase 9 (MMP-9) can directly or indirectly improve wound healing and remodeling after myocardial infarction (159). Immunosuppression delayed granulation tissue formation and decreased collagen deposition after myocardial infarction in elderly mice to prevent myocardial remodeling (160). Studies showed that T cell metabolic failure caused by abnormal mitochondrial function could also serve as an accelerating factor of cardiovascular changes and affect organ function and life span (97). Mitochondrial transcription factor A (TFAM) deficiency could accelerate T cell senescence. Blocking of TNF-α signal transduction can partially reduce premature senescence in TFAM deficient T cells (97). Transplantation of senescent T cells into immune-deficient mice accelerated angiotensin (Ang) II-induced cardiovascular fibrosis (161). In aging patients, reducing B lymphocytes and their subsets in peripheral blood associated with chronic cardiac insufficiency paralleled the altered expression of miRNA miR-181c involved in lymphogenesis (162). These findings suggest that immune-aging-related miRNAs might play an essential role in age-related cardiac insufficiency.
Acute kidney injury (AKI) is one of the most common complications associated with sepsis in the intensive care unit (ICU) (163). However, the pathophysiological mechanism of sepsis-related to renal injury remains unclear. Innate immune cells of the kidney and white blood cells in the peripheral blood circulation are associated with AKI and chronic kidney disease (CKD) (164, 165). In other words, CKD can lead to accelerated aging of the immune system and increase AKI risk (166) Analysis proved that the loss of renal function is related to the dysregulation of circulating T cells. Thymus output was significantly reduced in patients with end-stage renal disease (ESRD), and the susceptibility of naive T cells to apoptosis was increased, equivalent to 20 to 30 years of T cell aging (167). T cells also demonstrate a highly variable maturation phenotype in children with kidney disease, especially in patients who have been treated with immunosuppressive drugs (168). This suggests that a uremic environment may lead to premature senescence of T cells, which is manifested by the decrease of naive T cells and the increase of CD8+ TEMRA cells and pro-inflammatory monocytes (169). Also, similar immune changes were positively correlated with the duration of renal dialysis (170). In a multi-ethnic population-based cohort study, an algorithm for predicting aging based on DNA methylation percentage (DNAm-based age) confirmed that immunosenescence was an essential factor influencing kidney disease (171). Senescent T cells may lead to renal fibrosis and promote the expression of inflammatory factors and the production of superoxide in the kidneys. The senescent T cells could stimulate inflammatory cytokine expression and oxidative stress in Ang II-treated renal epithelial cells (161). Age-dependent tertiary lymphoid tissue formation can promote intra-renal inflammation (172). Researchers confirmed that CD153 and CD30 signaling may interact with CD153+PD-1+CD4+ senescence-associated T cells (SAT) and CD30+ T-bet+ age-related B cells, resulting in tertiary lymphoid tissue generation in chronically inflammatory organs, thereby promoting renal inflammation and fibrosis (173). In addition to ESRD, CD8+ T cell depletion occurs as early as CKD3 in diabetes mellitus, showing the characteristics of aggravated immunosenescence (174). Moreover, the occurrence and development of kidney diseases might be associated with cardiovascular diseases. Clinical studies demonstrated that telomere shortening of leukocytes in patients with CHF may be associated with renal dysfunction (175).
Liver dysfunction after sepsis can be a risk factor for multiple organ dysfunction and death caused by sepsis. The liver acts as an immune organ. In sepsis, the liver-mediated immune response can be responsible for removing bacteria and toxins but can also lead to inflammation, immunosuppression, and organ dysfunction (176). Also, liver damage may be present in the early stages of sepsis (177). However, only a few studies are found on immunosenescence during liver injury, and immunosenescence has been found in several liver diseases. The mean telomere length of white blood cells was shorter in patients with cirrhosis than in controls (178). In liver transplant recipients, lymphocytes expressed more mature phenotypic markers than controls. Also, the telomeres of lymphocytes became shorter (179). Several innate immune cells in the liver, such as resident macrophages, known as Kupffer cells, are the most representative immune cells in the liver (180). As the liver ages, pro-inflammatory cells accumulate, and the secretion of IL-6 by Kupffer cells increases (181). Some studies showed that the telomere length of Kupffer cells decreased while the number increased, and the phagocytosis ability was enhanced (182). Very few studies exist on the influence of aging on liver immunity, and further studies are needed.
As a manifestation of immune dysfunction, immunosenescence plays a critical role in the occurrence and development of sepsis and affects the function of solid organs. We reviewed the current research on the effects of immune aging on septic-related solid organs. We found that immunosenescence and organ damage were mutually reinforcing processes that are linked and promote each other. At the same time, we summarized the role of MDSCs in sepsis and immunosenescence and analyzed its regulatory mechanisms. These may contribute to delayed outcomes after sepsis. The suggestion of immunosenescence in sepsis exhibits similarities to the immunosuppression stage of sepsis. However, the relationship between immunosenescence and sepsis is still undefined, and more evidence is required to confirm it.
XL conceived and completed the manuscript. Y-QL and Y-MY supervised and revised the manuscript. All authors contributed to the article and approved the submitted version.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Fahy GM, Brooke RT, Watson JP, Good Z, Vasanawala SS, Maecker H, et al. Reversal of epigenetic aging and immunosenescent trends in humans. Aging Cell (2019) 18(6):e13028. doi: 10.1111/acel.13028
2. Coppé JP, Patil CK, Rodier F, Sun Y, Muñoz DP, Goldstein J, et al. Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PloS Biol (2008) 6(12):2853–68. doi: 10.1371/journal.pbio.0060301
3. Walford RL. The Immunologic Theory of Aging. Gerontologist (1964) 4:195–7. doi: 10.1093/geront/4.4.195
4. Santoro A, Bientinesi E, Monti D. Immunosenescence and inflammaging in the aging process: age-related diseases or longevity? Ageing Res Rev (2021) 71:101422. doi: 10.1016/j.arr.2021.101422
5. Franceschi C, Garagnani P, Vitale G, Capri M, Salvioli S. Inflammaging and 'Garb-aging'. Trends Endocrinol Metab (2017) 28(3):199–212. doi: 10.1016/j.tem.2016.09.005
6. Coppé JP, Desprez PY, Krtolica A, Campisi J. The senescence-associated secretory phenotype: the dark side of tumor suppression. Annu Rev Pathol (2010) 5:99–118. doi: 10.1146/annurev-pathol-121808-102144
7. Angus DC, van der Poll T. Severe sepsis and septic shock. N Engl J Med (2013) 369(9):840–51. doi: 10.1056/NEJMra1208623
8. Singer M, Deutschman CS, Seymour CW, Shankar-Hari M, Annane D, Bauer M, et al. The Third International Consensus Definitions for Sepsis and Septic Shock (Sepsis-3). Jama (2016) 315(8):801–10. doi: 10.1001/jama.2016.0287
9. Stoller J, Halpin L, Weis M, Aplin B, Qu W, Georgescu C, et al. Epidemiology of severe sepsis: 2008-2012. J Crit Care (2016) 31(1):58–62. doi: 10.1016/j.jcrc.2015.09.034
10. Hall MJ, Williams SN, DeFrances CJ, Golosinskiy A. Inpatient care for septicemia or sepsis: a challenge for patients and hospitals. NCHS Data Brief (2011) 62):1–8.
11. Monneret G, Gossez M, Venet F. Sepsis and immunosenescence: closely associated in a vicious circle. Aging Clin Exp Res (2021) 33(3):729–32. doi: 10.1007/s40520-019-01350-z
12. Martin S, Perez A, Aldecoa C. Sepsis and Immunosenescence in the Elderly Patient: A Review. Front Med (Lausanne) (2017) 4:20. doi: 10.3389/fmed.2017.00020
13. Darden DB, Bacher R, Brusko MA, Knight P, Hawkins RB, Cox MC, et al. Single-Cell RNA-seq of Human Myeloid-Derived Suppressor Cells in Late Sepsis Reveals Multiple Subsets With Unique Transcriptional Responses: A Pilot Study. Shock (2021) 55(5):587–95. doi: 10.1097/SHK.0000000000001671
14. Cheng L, Wang J, Li X, Xing Q, Du P, Su L, et al. Interleukin-6 induces Gr-1+CD11b+ myeloid cells to suppress CD8+ T cell-mediated liver injury in mice. PloS One (2011) 6(3):e17631. doi: 10.1371/journal.pone.0017631
15. Cuenca AG, Delano MJ, Kelly-Scumpia KM, Moreno C, Scumpia PO, Laface DM, et al. A paradoxical role for myeloid-derived suppressor cells in sepsis and trauma. Mol Med (2011) 17(3-4):281–92. doi: 10.2119/molmed.2010.00178
16. Salminen A, Kaarniranta K, Kauppinen A. Immunosenescence: the potential role of myeloid-derived suppressor cells (MDSC) in age-related immune deficiency. Cell Mol Life Sci (2019) 76(10):1901–18. doi: 10.1007/s00018-019-03048-x
17. Shanley DP, Aw D, Manley NR, Palmer DB. An evolutionary perspective on the mechanisms of immunosenescence. Trends Immunol (2009) 30(7):374–81. doi: 10.1016/j.it.2009.05.001
18. Bruce JL, Hurford RK Jr., Classon M, Koh J, Dyson N. Requirements for cell cycle arrest by p16INK4a. Mol Cell (2000) 6(3):737–42. doi: 10.1016/S1097-2765(00)00072-1
19. Aubert G, Baerlocher GM, Vulto I, Poon SS, Lansdorp PM. Collapse of telomere homeostasis in hematopoietic cells caused by heterozygous mutations in telomerase genes. PloS Genet (2012) 8(5):e1002696. doi: 10.1371/journal.pgen.1002696
20. Pantsulaia I, Ciszewski WM, Niewiarowska J. Senescent endothelial cells: Potential modulators of immunosenescence and ageing. Ageing Res Rev (2016) 29:13–25. doi: 10.1016/j.arr.2016.05.011
21. Dimri GP, Lee X, Basile G, Acosta M, Scott G, Roskelley C, et al. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc Natl Acad Sci USA (1995) 92(20):9363–7. doi: 10.1073/pnas.92.20.9363
22. Conte M, Martucci M, Chiariello A, Franceschi C, Salvioli S. Mitochondria, immunosenescence and inflammaging: a role for mitokines? Semin Immunopathol (2020) 42(5):607–17. doi: 10.1007/s00281-020-00813-0
23. Ou HL, Schumacher B. DNA damage responses and p53 in the aging process. Blood (2018) 131(5):488–95. doi: 10.1182/blood-2017-07-746396
24. Martínez G, Duran-Aniotz C, Cabral-Miranda F, Vivar JP, Hetz C. Endoplasmic reticulum proteostasis impairment in aging. Aging Cell (2017) 16(4):615–23. doi: 10.1111/acel.12599
25. Deutschman CS, Tracey KJ. Sepsis: current dogma and new perspectives. Immunity (2014) 40(4):463–75. doi: 10.1016/j.immuni.2014.04.001
26. Paolisso G, Barbieri M, Bonafè M, Franceschi C. Metabolic age modelling: the lesson from centenarians. Eur J Clin Invest (2000) 30(10):888–94. doi: 10.1046/j.1365-2362.2000.00729.x
27. Fulop T, Larbi A, Dupuis G, Le Page A, Frost EH, Cohen AA, et al. Immunosenescence and Inflamm-Aging As Two Sides of the Same Coin: Friends or Foes? Front Immunol (2017) 8:1960. doi: 10.3389/fimmu.2017.01960
28. Franceschi C, Salvioli S, Garagnani P, de Eguileor M, Monti D, Capri M. Immunobiography and the Heterogeneity of Immune Responses in the Elderly: A Focus on Inflammaging and Trained Immunity. Front Immunol (2017) 8:982. doi: 10.3389/fimmu.2017.00982
29. Millrud CR, Bergenfelz C, Leandersson K. On the origin of myeloid-derived suppressor cells. Oncotarget (2017) 8(2):3649–65. doi: 10.18632/oncotarget.12278
30. Boettcher S, Manz MG. Sensing and translation of pathogen signals into demand-adapted myelopoiesis. Curr Opin Hematol (2016) 23(1):5–10. doi: 10.1097/MOH.0000000000000201
31. Loftus TJ, Mohr AM, Moldawer LL. Dysregulated myelopoiesis and hematopoietic function following acute physiologic insult. Curr Opin Hematol (2018) 25(1):37–43. doi: 10.1097/MOH.0000000000000395
32. Gabrilovich DI, Ostrand-Rosenberg S, Bronte V. Coordinated regulation of myeloid cells by tumours. Nat Rev Immunol (2012) 12(4):253–68. doi: 10.1038/nri3175
33. Gabrilovich DI, Nagaraj S. Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol (2009) 9(3):162–74. doi: 10.1038/nri2506
34. Venet F, Monneret G. Advances in the understanding and treatment of sepsis-induced immunosuppression. Nat Rev Nephrol (2018) 14(2):121–37. doi: 10.1038/nrneph.2017.165
35. Verschoor CP, Johnstone J, Millar J, Dorrington MG, Habibagahi M, Lelic A, et al. Blood CD33(+)HLA-DR(-) myeloid-derived suppressor cells are increased with age and a history of cancer. J Leukoc Biol (2013) 93(4):633–7. doi: 10.1189/jlb.0912461
36. Alves AS, Ishimura ME, Duarte YAO, Bueno V. Parameters of the Immune System and Vitamin D Levels in Old Individuals. Front Immunol (2018) 9:1122. doi: 10.3389/fimmu.2018.01122
37. Enioutina EY, Bareyan D, Daynes RA. A role for immature myeloid cells in immune senescence. J Immunol (2011) 186(2):697–707. doi: 10.4049/jimmunol.1002987
38. Ortega-Gómez A, Perretti M, Soehnlein O. Resolution of inflammation: an integrated view. EMBO Mol Med (2013) 5(5):661–74. doi: 10.1002/emmm.201202382
39. Ost M, Singh A, Peschel A, Mehling R, Rieber N, Hartl D. Myeloid-Derived Suppressor Cells in Bacterial Infections. Front Cell Infect Microbiol (2016) 6:37. doi: 10.3389/fcimb.2016.00037
40. Jackaman C, Tomay F, Duong L, Abdol Razak NB, Pixley FJ, Metharom P, et al. Aging and cancer: The role of macrophages and neutrophils. Ageing Res Rev (2017) 36:105–16. doi: 10.1016/j.arr.2017.03.008
41. Munoz C, Carlet J, Fitting C, Misset B, Blériot JP, Cavaillon JM. Dysregulation of in vitro cytokine production by monocytes during sepsis. J Clin Invest (1991) 88(5):1747–54. doi: 10.1172/JCI115493
42. Williams MA, White SA, Miller JJ, Toner C, Withington S, Newland AC, et al. Granulocyte-macrophage colony-stimulating factor induces activation and restores respiratory burst activity in monocytes from septic patients. J Infect Dis (1998) 177(1):107–15. doi: 10.1086/513802
43. Gayoso I, Sanchez-Correa B, Campos C, Alonso C, Pera A, Casado JG, et al. Immunosenescence of human natural killer cells. J Innate Immun (2011) 3(4):337–43. doi: 10.1159/000328005
44. Hoechst B, Voigtlaender T, Ormandy L, Gamrekelashvili J, Zhao F, Wedemeyer H, et al. Myeloid derived suppressor cells inhibit natural killer cells in patients with hepatocellular carcinoma via the NKp30 receptor. Hepatology (2009) 50(3):799–807. doi: 10.1002/hep.23054
45. Zhu J, Huang X, Yang Y. Myeloid-derived suppressor cells regulate natural killer cell response to adenovirus-mediated gene transfer. J Virol (2012) 86(24):13689–96. doi: 10.1128/JVI.01595-12
46. Arnold CR, Wolf J, Brunner S, Herndler-Brandstetter D, Grubeck-Loebenstein B. Gain and loss of T cell subsets in old age–age-related reshaping of the T cell repertoire. J Clin Immunol (2011) 31(2):137–46. doi: 10.1007/s10875-010-9499-x
47. Quinn KM, Fox A, Harland KL, Russ BE, Li J, Nguyen THO, et al. Age-Related Decline in Primary CD8(+) T Cell Responses Is Associated with the Development of Senescence in Virtual Memory CD8(+) T Cells. Cell Rep (2018) 23(12):3512–24. doi: 10.1016/j.celrep.2018.05.057
48. Ruan WS, Feng MX, Xu J, Xu YG, Song CY, Lin LY, et al. Early Activation of Myeloid-Derived Suppressor Cells Participate in Sepsis-Induced Immune Suppression via PD-L1/PD-1 Axis. Front Immunol (2020) 11:1299. doi: 10.3389/fimmu.2020.01299
49. Kennedy DE, Knight KL. Inhibition of B Lymphopoiesis by Adipocytes and IL-1-Producing Myeloid-Derived Suppressor Cells. J Immunol (2015) 195(6):2666–74. doi: 10.4049/jimmunol.1500957
50. Dunn-Walters DK. The ageing human B cell repertoire: a failure of selection? Clin Exp Immunol (2016) 183(1):50–6. doi: 10.1111/cei.12700
51. Gustave CA, Gossez M, Demaret J, Rimmelé T, Lepape A, Malcus C, et al. Septic Shock Shapes B Cell Response toward an Exhausted-like/Immunoregulatory Profile in Patients. J Immunol (2018) 200(7):2418–25. doi: 10.4049/jimmunol.1700929
52. Bronte V, Serafini P, Mazzoni A, Segal DM, Zanovello P. L-arginine metabolism in myeloid cells controls T-lymphocyte functions. Trends Immunol (2003) 24(6):302–6. doi: 10.1016/S1471-4906(03)00132-7
53. Yu J, Du W, Yan F, Wang Y, Li H, Cao S, et al. Myeloid-derived suppressor cells suppress antitumor immune responses through IDO expression and correlate with lymph node metastasis in patients with breast cancer. J Immunol (2013) 190(7):3783–97. doi: 10.4049/jimmunol.1201449
54. Redd PS, Ibrahim ML, Klement JD, Sharman SK, Paschall AV, Yang D, et al. SETD1B Activates iNOS Expression in Myeloid-Derived Suppressor Cells. Cancer Res (2017) 77(11):2834–43. doi: 10.1158/0008-5472.CAN-16-2238
55. Nagaraj S, Gupta K, Pisarev V, Kinarsky L, Sherman S, Kang L, et al. Altered recognition of antigen is a mechanism of CD8+ T cell tolerance in cancer. Nat Med (2007) 13(7):828–35. doi: 10.1038/nm1609
56. Sizzano F, Collino S, Cominetti O, Monti D, Garagnani P, Ostan R, et al. Evaluation of Lymphocyte Response to the Induced Oxidative Stress in a Cohort of Ageing Subjects, including Semisupercentenarians and Their Offspring. Mediators Inflamm (2018) 2018:7109312. doi: 10.1155/2018/7109312
57. Harley CB, Futcher AB, Greider CW. Telomeres shorten during ageing of human fibroblasts. Nature (1990) 345(6274):458–60. doi: 10.1038/345458a0
58. Oliveira NM, Rios ECS, de Lima TM, Victorino VJ, Barbeiro H, Pinheiro da Silva F, et al. Sepsis induces telomere shortening: a potential mechanism responsible for delayed pathophysiological events in sepsis survivors? Mol Med (2017) 22:886–91. doi: 10.2119/molmed.2016.00225
59. Fritsch RD, Shen X, Illei GG, Yarboro CH, Prussin C, Hathcock KS, et al. Abnormal differentiation of memory T cells in systemic lupus erythematosus. Arthritis Rheum (2006) 54(7):2184–97. doi: 10.1002/art.21943
60. Johnson ND, Conneely KN. The role of DNA methylation and hydroxymethylation in immunosenescence. Ageing Res Rev (2019) 51:11–23. doi: 10.1016/j.arr.2019.01.011
61. Vachharajani V, Liu T, McCall CE. Epigenetic coordination of acute systemic inflammation: potential therapeutic targets. Expert Rev Clin Immunol (2014) 10(9):1141–50. doi: 10.1586/1744666X.2014.943192
62. El Gazzar M, Yoza BK, Chen X, Garcia BA, Young NL, McCall CE. Chromatin-specific remodeling by HMGB1 and linker histone H1 silences proinflammatory genes during endotoxin tolerance. Mol Cell Biol (2009) 29(7):1959–71. doi: 10.1128/MCB.01862-08
63. Alisch RS, Barwick BG, Chopra P, Myrick LK, Satten GA, Conneely KN, et al. Age-associated DNA methylation in pediatric populations. Genome Res (2012) 22(4):623–32. doi: 10.1101/gr.125187.111
64. Bell JT, Tsai PC, Yang TP, Pidsley R, Nisbet J, Glass D, et al. Epigenome-wide scans identify differentially methylated regions for age and age-related phenotypes in a healthy ageing population. PloS Genet (2012) 8(4):e1002629. doi: 10.1371/journal.pgen.1002629
65. D'Aquila P, Giordano M, Montesanto A, De Rango F, Passarino G, Bellizzi D. Age-and gender-related pattern of methylation in the MT-RNR1 gene. Epigenomics (2015) 7(5):707–16. doi: 10.2217/epi.15.30
66. Gopalan S, Carja O, Fagny M, Patin E, Myrick JW, McEwen LM, et al. Trends in DNA Methylation with Age Replicate Across Diverse Human Populations. Genetics (2017) 206(3):1659–74. doi: 10.1534/genetics.116.195594
67. Marttila S, Kananen L, Häyrynen S, Jylhävä J, Nevalainen T, Hervonen A, et al. Ageing-associated changes in the human DNA methylome: genomic locations and effects on gene expression. BMC Genomics (2015) 16(1):179. doi: 10.1186/s12864-015-1381-z
68. Horvath S. DNA methylation age of human tissues and cell types. Genome Biol (2013) 14(10):R115. doi: 10.1186/gb-2013-14-10-r115
69. Larson AR, Dresser KA, Zhan Q, Lezcano C, Woda BA, Yosufi B, et al. Loss of 5-hydroxymethylcytosine correlates with increasing morphologic dysplasia in melanocytic tumors. Mod Pathol (2014) 27(7):936–44. doi: 10.1038/modpathol.2013.224
70. Horvath S, Pirazzini C, Bacalini MG, Gentilini D, Di Blasio AM, Delledonne M, et al. Decreased epigenetic age of PBMCs from Italian semi-supercentenarians and their offspring. Aging (Albany NY) (2015) 7(12):1159–70. doi: 10.18632/aging.100861
71. Guignant C, Lepape A, Huang X, Kherouf H, Denis L, Poitevin F, et al. Programmed death-1 levels correlate with increased mortality, nosocomial infection and immune dysfunctions in septic shock patients. Crit Care (2011) 15(2):R99. doi: 10.1186/cc10112
72. Chang K, Svabek C, Vazquez-Guillamet C, Sato B, Rasche D, Wilson S, et al. Targeting the programmed cell death 1: programmed cell death ligand 1 pathway reverses T cell exhaustion in patients with sepsis. Crit Care (2014) 18(1):R3. doi: 10.1186/cc13176
73. Patera AC, Drewry AM, Chang K, Beiter ER, Osborne D, Hotchkiss RS. Frontline Science: Defects in immune function in patients with sepsis are associated with PD-1 or PD-L1 expression and can be restored by antibodies targeting PD-1 or PD-L1. J Leukoc Biol (2016) 100(6):1239–54. doi: 10.1189/jlb.4HI0616-255R
74. Brahmamdam P, Inoue S, Unsinger J, Chang KC, McDunn JE, Hotchkiss RS. Delayed administration of anti-PD-1 antibody reverses immune dysfunction and improves survival during sepsis. J Leukoc Biol (2010) 88(2):233–40. doi: 10.1189/jlb.0110037
75. Inoue S, Bo L, Bian J, Unsinger J, Chang K, Hotchkiss RS. Dose-dependent effect of anti-CTLA-4 on survival in sepsis. Shock (2011) 36(1):38–44. doi: 10.1097/SHK.0b013e3182168cce
76. Nishijima TF, Muss HB, Shachar SS, Moschos SJ. Comparison of efficacy of immune checkpoint inhibitors (ICIs) between younger and older patients: A systematic review and meta-analysis. Cancer Treat Rev (2016) 45:30–7. doi: 10.1016/j.ctrv.2016.02.006
77. Bastholt L, Schmidt H, Bjerregaard JK, Herrstedt J, Svane IM. Age favoured overall survival in a large population-based Danish patient cohort treated with anti-PD1 immune checkpoint inhibitor for metastatic melanoma. Eur J Cancer (2019) 119:122–31. doi: 10.1016/j.ejca.2019.06.022
78. Kugel CH 3rd, Douglass SM, Webster MR, Kaur A, Liu Q, Yin X, et al. Age Correlates with Response to Anti-PD1, Reflecting Age-Related Differences in Intratumoral Effector and Regulatory T-Cell Populations. Clin Cancer Res (2018) 24(21):5347–56. doi: 10.1158/1078-0432.CCR-18-1116
79. Perier-Muzet M, Gatt E, Péron J, Falandry C, Amini-Adlé M, Thomas L, et al. Association of Immunotherapy With Overall Survival in Elderly Patients With Melanoma. JAMA Dermatol (2018) 154(1):82–7. doi: 10.1001/jamadermatol.2017.4584
80. Lian J, Yue Y, Yu W, Zhang Y. Immunosenescence: a key player in cancer development. J Hematol Oncol (2020) 13(1):151. doi: 10.1186/s13045-020-00986-z
81. Larbi A, Fulop T. From "truly naïve" to "exhausted senescent" T cells: when markers predict functionality. Cytometry A (2014) 85(1):25–35. doi: 10.1002/cyto.a.22351
82. Zhao Y, Shao Q, Peng G. Exhaustion and senescence: two crucial dysfunctional states of T cells in the tumor microenvironment. Cell Mol Immunol (2020) 17(1):27–35. doi: 10.1038/s41423-019-0344-8
83. Sitkovsky MV, Kjaergaard J, Lukashev D, Ohta A. Hypoxia-adenosinergic immunosuppression: tumor protection by T regulatory cells and cancerous tissue hypoxia. Clin Cancer Res (2008) 14(19):5947–52. doi: 10.1158/1078-0432.CCR-08-0229
84. Quinn KM, Palchaudhuri R, Palmer CS, La Gruta NL. The clock is ticking: the impact of ageing on T cell metabolism. Clin Transl Immunol (2019) 8(11):e01091. doi: 10.1002/cti2.1091
85. Horn P, Metzing UB, Steidl R, Romeike B, Rauchfuß F, Sponholz C, et al. Chemerin in peritoneal sepsis and its associations with glucose metabolism and prognosis: a translational cross-sectional study. Crit Care (2016) 20:39. doi: 10.1186/s13054-016-1209-5
86. Callender LA, Carroll EC, Beal RWJ, Chambers ES, Nourshargh S, Akbar AN, et al. Human CD8(+) EMRA T cells display a senescence-associated secretory phenotype regulated by p38 MAPK. Aging Cell (2018) 17(1):e12675. doi: 10.1111/acel.12675
87. Mittelbrunn M, Kroemer G. Hallmarks of T cell aging. Nat Immunol (2021) 22(6):687–98. doi: 10.1038/s41590-021-00927-z
88. Vang T, Torgersen KM, Sundvold V, Saxena M, Levy FO, Skålhegg BS, et al. Activation of the COOH-terminal Src kinase (Csk) by cAMP-dependent protein kinase inhibits signaling through the T cell receptor. J Exp Med (2001) 193(4):497–507. doi: 10.1084/jem.193.4.497
89. Wang W, Chen JX, Liao R, Deng Q, Zhou JJ, Huang S, et al. Sequential activation of the MEK-extracellular signal-regulated kinase and MKK3/6-p38 mitogen-activated protein kinase pathways mediates oncogenic ras-induced premature senescence. Mol Cell Biol (2002) 22(10):3389–403. doi: 10.1128/MCB.22.10.3389-3403.2002
90. Lanna A, Gomes DC, Muller-Durovic B, McDonnell T, Escors D, Gilroy DW, et al. A sestrin-dependent Erk-Jnk-p38 MAPK activation complex inhibits immunity during aging. Nat Immunol (2017) 18(3):354–63. doi: 10.1038/ni.3665
91. Schuliga M, Read J, Knight DA. Ageing mechanisms that contribute to tissue remodeling in lung disease. Ageing Res Rev (2021) 70:101405. doi: 10.1016/j.arr.2021.101405
92. Garrabou G, Morén C, López S, Tobías E, Cardellach F, Miró O, et al. The effects of sepsis on mitochondria. J Infect Dis (2012) 205(3):392–400. doi: 10.1093/infdis/jir764
93. Globerson A, Effros RB. Ageing of lymphocytes and lymphocytes in the aged. Immunol Today (2000) 21(10):515–21. doi: 10.1016/S0167-5699(00)01714-X
94. Pawelec G. Immunosenescence: impact in the young as well as the old? Mech Ageing Dev (1999) 108(1):1–7. doi: 10.1016/s0047-6374(99)00010-x
95. Yousefzadeh MJ, Flores RR, Zhu Y, Schmiechen ZC, Brooks RW, Trussoni CE, et al. An aged immune system drives senescence and ageing of solid organs. Nature (2021) 594(7861):100–5. doi: 10.1038/s41586-021-03547-7
96. Chen Y, Klein SL, Garibaldi BT, Li H, Wu C, Osevala NM, et al. Aging in COVID-19: Vulnerability, immunity and intervention. Ageing Res Rev (2021) 65:101205. doi: 10.1016/j.arr.2020.101205
97. Desdín-Micó G, Soto-Heredero G, Aranda JF, Oller J, Carrasco E, Gabandé-Rodríguez E, et al. T cells with dysfunctional mitochondria induce multimorbidity and premature senescence. Science (2020) 368(6497):1371–6. doi: 10.1126/science.aax0860
98. Meyer KC. Management of interstitial lung disease in elderly patients. Curr Opin Pulm Med (2012) 18(5):483–92. doi: 10.1097/MCP.0b013e3283541337
99. Sicard D, Haak AJ, Choi KM, Craig AR, Fredenburgh LE, Tschumperlin DJ. Aging and anatomical variations in lung tissue stiffness. Am J Physiol Lung Cell Mol Physiol (2018) 314(6):L946–l955. doi: 10.1152/ajplung.00415.2017
100. Ribeiro Júnior G, de Souza Xavier Costa N, Belotti L, Dos Santos Alemany AA, Amato-Lourenço LF, da Cunha PG, et al. Diesel exhaust exposure intensifies inflammatory and structural changes associated with lung aging in mice. Ecotoxicol Environ Saf (2019) 170:314–23. doi: 10.1016/j.ecoenv.2018.11.139
101. Park I, Kim M, Choe K, Song E, Seo H, Hwang Y, et al. Neutrophils disturb pulmonary microcirculation in sepsis-induced acute lung injury. Eur Respir J (2019) 53(3):1800786. doi: 10.1183/13993003.00786-2018
102. Lagu T, Rothberg MB, Shieh MS, Pekow PS, Steingrub JS, Lindenauer PK. Hospitalizations, costs, and outcomes of severe sepsis in the United States 2003 to 2007. Crit Care Med (2012) 40(3):754–61. doi: 10.1097/CCM.0b013e318232db65
103. Ho JC, Chan KN, Hu WH, Lam WK, Zheng L, Tipoe GL, et al. The effect of aging on nasal mucociliary clearance, beat frequency, and ultrastructure of respiratory cilia. Am J Respir Crit Care Med (2001) 163(4):983–8. doi: 10.1164/ajrccm.163.4.9909121
104. Bailey KL, Kharbanda KK, Katafiasz DM, Sisson JH, Wyatt TA. Oxidative stress associated with aging activates protein kinase Cepsilon, leading to cilia slowing. Am J Physiol Lung Cell Mol Physiol (2018) 315(5):L882–90. doi: 10.1152/ajplung.00033.2018
105. Ghadially R, Brown BE, Sequeira-Martin SM, Feingold KR, Elias PM. The aged epidermal permeability barrier. Structural, functional, and lipid biochemical abnormalities in humans and a senescent murine model. J Clin Invest (1995) 95(5):2281–90. doi: 10.1172/JCI117919
106. Parikh P, Wicher S, Khandalavala K, Pabelick CM, Britt RD Jr., Prakash YS. Cellular senescence in the lung across the age spectrum. Am J Physiol Lung Cell Mol Physiol (2019) 316(5):L826–l842. doi: 10.1152/ajplung.00424.2018
107. Yuan L, Du X, Tang S, Wu S, Wang L, Xiang Y, et al. ITGB4 deficiency induces senescence of airway epithelial cells through p53 activation. FEBS J (2019) 286(6):1191–203. doi: 10.1111/febs.14749
108. Prakash S, Agrawal S, Vahed H, Ngyuen M, BenMohamed L, Gupta S, et al. Dendritic cells from aged subjects contribute to chronic airway inflammation by activating bronchial epithelial cells under steady state. Mucosal Immunol (2014) 7(6):1386–94. doi: 10.1038/mi.2014.28
109. Jaiswal AK, Yadav J, Makhija S, Sandey M, Suryawanshi A, Mitra AK, et al. Short Palate, Lung, and Nasal Epithelial Clone1 (SPLUNC1) level determines steroid-resistant airway inflammation in aging. Am J Physiol Lung Cell Mol Physiol (2021) 322(1):L102–L115. doi: 10.1152/ajplung.00315.2021
110. Lloyd CM, Marsland BJ. Lung Homeostasis: Influence of Age, Microbes, and the Immune System. Immunity (2017) 46(4):549–61. doi: 10.1016/j.immuni.2017.04.005
111. Canan CH, Gokhale NS, Carruthers B, Lafuse WP, Schlesinger LS, Torrelles JB, et al. Characterization of lung inflammation and its impact on macrophage function in aging. J Leukoc Biol (2014) 96(3):473–80. doi: 10.1189/jlb.4A0214-093RR
112. Albright JM, Dunn RC, Shults JA, Boe DM, Afshar M, Kovacs EJ. Advanced Age Alters Monocyte and Macrophage Responses. Antioxid Redox Signal (2016) 25(15):805–15. doi: 10.1089/ars.2016.6691
113. Angelidis I, Simon LM, Fernandez IE, Strunz M, Mayr CH, Greiffo FR, et al. An atlas of the aging lung mapped by single cell transcriptomics and deep tissue proteomics. Nat Commun (2019) 10(1):963. doi: 10.1038/s41467-019-08831-9
114. Johnson SA, Rozzo SJ, Cambier JC. Aging-dependent exclusion of antigen-inexperienced cells from the peripheral B cell repertoire. J Immunol (2002) 168(10):5014–23. doi: 10.4049/jimmunol.168.10.5014
115. Malinina A, Dikeman D, Westbrook R, Moats M, Gidner S, Poonyagariyagorn H, et al. IL10 deficiency promotes alveolar enlargement and lymphoid dysmorphogenesis in the aged murine lung. Aging Cell (2020) 19(4):e13130. doi: 10.1111/acel.13130
116. van Beek AA, Hugenholtz F, Meijer B, Sovran B, Perdijk O, Vermeij WP, et al. Frontline Science: Tryptophan restriction arrests B cell development and enhances microbial diversity in WT and prematurely aging Ercc1(-/Δ7) mice. J Leukoc Biol (2017) 101(4):811–21. doi: 10.1189/jlb.1HI0216-062RR
117. Aoshiba K, Nagai A. Chronic lung inflammation in aging mice. FEBS Lett (2007) 581(18):3512–6. doi: 10.1016/j.febslet.2007.06.075
118. Onyema OO, Decoster L, Njemini R, Forti LN, Bautmans I, De Waele M, et al. Shifts in subsets of CD8+ T-cells as evidence of immunosenescence in patients with cancers affecting the lungs: an observational case-control study. BMC Cancer (2015) 15:1016. doi: 10.1186/s12885-015-2013-3
119. Sun L, Hurez VJ, Thibodeaux SR, Kious MJ, Liu A, Lin P, et al. Aged regulatory T cells protect from autoimmune inflammation despite reduced STAT3 activation and decreased constraint of IL-17 producing T cells. Aging Cell (2012) 11(3):509–19. doi: 10.1111/j.1474-9726.2012.00812.x
120. Tauber SC, Djukic M, Gossner J, Eiffert H, Brück W, Nau R. Sepsis-associated encephalopathy and septic encephalitis: an update. Expert Rev Anti Infect Ther (2021) 19(2):215–31. doi: 10.1080/14787210.2020.1812384
121. Cao W, Zheng H. Peripheral immune system in aging and Alzheimer's disease. Mol Neurodegener (2018) 13(1):51. doi: 10.1186/s13024-018-0284-2
122. Golomb SM, Guldner IH, Zhao A, Wang Q, Palakurthi B, Aleksandrovic EA, et al. Multi-modal Single-Cell Analysis Reveals Brain Immune Landscape Plasticity during Aging and Gut Microbiota Dysbiosis. Cell Rep (2020) 33(9):108438. doi: 10.1016/j.celrep.2020.108438
123. Licastro F, Candore G, Lio D, Porcellini E, Colonna-Romano G, Franceschi C, et al. Innate immunity and inflammation in ageing: a key for understanding age-related diseases. Immun Ageing (2005) 2:8. doi: 10.1186/1742-4933-2-8
124. Mitchell EL, Davis AT, Brass K, Dendinger M, Barner R, Gharaibeh R, et al. Reduced Intestinal Motility, Mucosal Barrier Function, and Inflammation in Aged Monkeys. J Nutr Health Aging (2017) 21(4):354–61. doi: 10.1007/s12603-016-0725-y
125. Odamaki T, Kato K, Sugahara H, Hashikura N, Takahashi S, Xiao JZ, et al. Age-related changes in gut microbiota composition from newborn to centenarian: a cross-sectional study. BMC Microbiol (2016) 16:90. doi: 10.1186/s12866-016-0708-5
126. Scott KA, Ida M, Peterson VL, Prenderville JA, Moloney GM, Izumo T, et al. Revisiting Metchnikoff: Age-related alterations in microbiota-gut-brain axis in the mouse. Brain Behav Immun (2017) 65:20–32. doi: 10.1016/j.bbi.2017.02.004
127. Sampson TR, Debelius JW, Thron T, Janssen S, Shastri GG, Ilhan ZE, et al. Gut Microbiota Regulate Motor Deficits and Neuroinflammation in a Model of Parkinson's Disease. Cell (2016) 167(6):1469–1480.e12. doi: 10.1016/j.cell.2016.11.018
128. Daneman R, Prat A. The blood-brain barrier. Cold Spring Harb Perspect Biol (2015) 7(1):a020412. doi: 10.1101/cshperspect.a020412
129. Montagne A, Barnes SR, Sweeney MD, Halliday MR, Sagare AP, Zhao Z, et al. Blood-brain barrier breakdown in the aging human hippocampus. Neuron (2015) 85(2):296–302. doi: 10.1016/j.neuron.2014.12.032
130. Barichello T, Generoso JS, Collodel A, Petronilho F, Dal-Pizzol F. The blood-brain barrier dysfunction in sepsis. Tissue Barriers (2021) 9(1):1840912. doi: 10.1080/21688370.2020.1840912
131. Lee P, Kim J, Williams R, Sandhir R, Gregory E, Brooks WM, et al. Effects of aging on blood brain barrier and matrix metalloproteases following controlled cortical impact in mice. Exp Neurol (2012) 234(1):50–61. doi: 10.1016/j.expneurol.2011.12.016
132. Storck SE, Hartz AMS, Bernard J, Wolf A, Kachlmeier A, Mahringer A, et al. The concerted amyloid-beta clearance of LRP1 and ABCB1/P-gp across the blood-brain barrier is linked by PICALM. Brain Behav Immun (2018) 73:21–33. doi: 10.1016/j.bbi.2018.07.017
133. van Assema DM, Lubberink M, Boellaard R, Schuit RC, Windhorst AD, Scheltens P, et al. P-glycoprotein function at the blood-brain barrier: effects of age and gender. Mol Imaging Biol (2012) 14(6):771–6. doi: 10.1007/s11307-012-0556-0
134. Hoffman JD, Parikh I, Green SJ, Chlipala G, Mohney RP, Keaton M, et al. Age Drives Distortion of Brain Metabolic, Vascular and Cognitive Functions, and the Gut Microbiome. Front Aging Neurosci (2017) 9:298. doi: 10.3389/fnagi.2017.00298
135. Donahue JE, Flaherty SL, Johanson CE, Duncan JA 3rd, Silverberg GD, Miller MC, et al. RAGE, LRP-1, and amyloid-beta protein in Alzheimer's disease. Acta Neuropathol (2006) 112(4):405–15. doi: 10.1007/s00401-006-0115-3
136. Bonte S, Vandemaele P, Verleden S, Audenaert K, Deblaere K, Goethals I, et al. Healthy brain ageing assessed with 18F-FDG PET and age-dependent recovery factors after partial volume effect correction. Eur J Nucl Med Mol Imaging (2017) 44(5):838–49. doi: 10.1007/s00259-016-3569-0
137. Ding F, Yao J, Rettberg JR, Chen S, Brinton RD. Early decline in glucose transport and metabolism precedes shift to ketogenic system in female aging and Alzheimer's mouse brain: implication for bioenergetic intervention. PloS One (2013) 8(11):e79977. doi: 10.1371/journal.pone.0079977
138. Schwartz M, Deczkowska A. Neurological Disease as a Failure of Brain-Immune Crosstalk: The Multiple Faces of Neuroinflammation. Trends Immunol (2016) 37(10):668–79. doi: 10.1016/j.it.2016.08.001
139. Baruch K, Ron-Harel N, Gal H, Deczkowska A, Shifrut E, Ndifon W, et al. CNS-specific immunity at the choroid plexus shifts toward destructive Th2 inflammation in brain aging. Proc Natl Acad Sci USA (2013) 110(6):2264–9. doi: 10.1073/pnas.1211270110
140. Baruch K, Schwartz M. CNS-specific T cells shape brain function via the choroid plexus. Brain Behav Immun (2013) 34:11–6. doi: 10.1016/j.bbi.2013.04.002
141. Damani MR, Zhao L, Fontainhas AM, Amaral J, Fariss RN, Wong WT. Age-related alterations in the dynamic behavior of microglia. Aging Cell (2011) 10(2):263–76. doi: 10.1111/j.1474-9726.2010.00660.x
142. Regen F, Hellmann-Regen J, Costantini E, Reale M. Neuroinflammation and Alzheimer's Disease: Implications for Microglial Activation. Curr Alzheimer Res (2017) 14(11):1140–8. doi: 10.2174/1567205014666170203141717
143. Mecca C, Giambanco I, Donato R, Arcuri C. Microglia and Aging: The Role of the TREM2-DAP12 and CX3CL1-CX3CR1 Axes. Int J Mol Sci (2018) 19(1):318. doi: 10.3390/ijms19010318
144. Poliani PL, Wang Y, Fontana E, Robinette ML, Yamanishi Y, Gilfillan S, et al. TREM2 sustains microglial expansion during aging and response to demyelination. J Clin Invest (2015) 125(5):2161–70. doi: 10.1172/JCI77983
145. Limatola C, Lauro C, Catalano M, Ciotti MT, Bertollini C, Di Angelantonio S, et al. Chemokine CX3CL1 protects rat hippocampal neurons against glutamate-mediated excitotoxicity. J Neuroimmunol (2005) 166(1-2):19–28. doi: 10.1016/j.jneuroim.2005.03.023
146. Krasemann S, Madore C, Cialic R, Baufeld C, Calcagno N, El Fatimy R, et al. The TREM2-APOE Pathway Drives the Transcriptional Phenotype of Dysfunctional Microglia in Neurodegenerative Diseases. Immunity (2017) 47(3):566–581.e9. doi: 10.1016/j.immuni.2017.08.008
147. Sutherland GT, Chami B, Youssef P, Witting PK. Oxidative stress in Alzheimer's disease: Primary villain or physiological by-product? Redox Rep (2013) 18(4):134–41. doi: 10.1179/1351000213Y.0000000052
148. Landreth GE, Reed-Geaghan EG. Toll-like receptors in Alzheimer's disease. Curr Top Microbiol Immunol (2009) 336:137–53. doi: 10.1007/978-3-642-00549-7_8
149. Hart AD, Wyttenbach A, Perry VH, Teeling JL. Age related changes in microglial phenotype vary between CNS regions: grey versus white matter differences. Brain Behav Immun (2012) 26(5):754–65. doi: 10.1016/j.bbi.2011.11.006
150. Minhas PS, Latif-Hernandez A, McReynolds MR, Durairaj AS, Wang Q, Rubin A, et al. Restoring metabolism of myeloid cells reverses cognitive decline in ageing. Nature (2021) 590(7844):122–8. doi: 10.1038/s41586-020-03160-0
151. Rieger AM, Hanington PC, Belosevic M, Barreda DR. Control of CSF-1 induced inflammation in teleost fish by a soluble form of the CSF-1 receptor. Fish Shellfish Immunol (2014) 41(1):45–51. doi: 10.1016/j.fsi.2014.03.035
152. De la Fuente M, Medina S, Del Rio M, Ferrández MD, Hernanz A. Effect of aging on the modulation of macrophage functions by neuropeptides. Life Sci (2000) 67(17):2125–35. doi: 10.1016/S0024-3205(00)00799-2
153. Moro-García MA, Echeverría A, Galán-Artímez MC, Suárez-García FM, Solano-Jaurrieta JJ, Avanzas-Fernández P, et al. Immunosenescence and inflammation characterize chronic heart failure patients with more advanced disease. Int J Cardiol (2014) 174(3):590–9. doi: 10.1016/j.ijcard.2014.04.128
154. Xydonas S, Parissis J, Lioni L, Kapsimali V, Psarra E, Farmakis D, et al. Immunosenescence in patients with chronic systolic heart failure. J Cardiovasc Med (Hagerstown) (2016) 17(8):624–30. doi: 10.2459/JCM.0000000000000372
155. van der Harst P, van der Steege G, de Boer RA, Voors AA, Hall AS, Mulder MJ, et al. Telomere length of circulating leukocytes is decreased in patients with chronic heart failure. J Am Coll Cardiol (2007) 49(13):1459–64. doi: 10.1016/j.jacc.2007.01.027
156. Merx MW, Weber C. Sepsis and the heart. Circulation (2007) 116(7):793–802. doi: 10.1161/CIRCULATIONAHA.106.678359
157. Szentes V, Gazdag M, Szokodi I, Dézsi CA. The Role of CXCR3 and Associated Chemokines in the Development of Atherosclerosis and During Myocardial Infarction. Front Immunol (2018) 9:1932. doi: 10.3389/fimmu.2018.01932
158. Liehn EA, Postea O, Curaj A, Marx N. Repair after myocardial infarction, between fantasy and reality: the role of chemokines. J Am Coll Cardiol (2011) 58(23):2357–62. doi: 10.1016/j.jacc.2011.08.034
159. Meschiari CA, Jung M, Iyer RP, Yabluchanskiy A, Toba H, Garrett MR, et al. Macrophage overexpression of matrix metalloproteinase-9 in aged mice improves diastolic physiology and cardiac wound healing after myocardial infarction. Am J Physiol Heart Circ Physiol (2018) 314(2):H224–h235. doi: 10.1152/ajpheart.00453.2017
160. Bujak M, Kweon HJ, Chatila K, Li N, Taffet G, Frangogiannis NG. Aging-related defects are associated with adverse cardiac remodeling in a mouse model of reperfused myocardial infarction. J Am Coll Cardiol (2008) 51(14):1384–92. doi: 10.1016/j.jacc.2008.01.011
161. Pan XX, Wu F, Chen XH, Chen DR, Chen HJ, Kong LR, et al. T-cell senescence accelerates angiotensin II-induced target organ damage. Cardiovasc Res (2021) 117(1):271–83. doi: 10.1093/cvr/cvaa032
162. Seeger T, Haffez F, Fischer A, Koehl U, Leistner DM, Seeger FH, et al. Immunosenescence-associated microRNAs in age and heart failure. Eur J Heart Fail (2013) 15(4):385–93. doi: 10.1093/eurjhf/hfs184
163. Bellomo R, Kellum JA, Ronco C, Wald R, Martensson J, Maiden M, et al. Acute kidney injury in sepsis. Intensive Care Med (2017) 43(6):816–28. doi: 10.1007/s00134-017-4755-7
164. Takaori K, Nakamura J, Yamamoto S, Nakata H, Sato Y, Takase M, et al. Severity and Frequency of Proximal Tubule Injury Determines Renal Prognosis. J Am Soc Nephrol (2016) 27(8):2393–406. doi: 10.1681/ASN.2015060647
165. Krebs CF, Paust HJ, Krohn S, Koyro T, Brix SR, Riedel JH, et al. Autoimmune Renal Disease Is Exacerbated by S1P-Receptor-1-Dependent Intestinal Th17 Cell Migration to the Kidney. Immunity (2016) 45(5):1078–92. doi: 10.1016/j.immuni.2016.10.020
166. Betjes MG. Immune cell dysfunction and inflammation in end-stage renal disease. Nat Rev Nephrol (2013) 9(5):255–65. doi: 10.1038/nrneph.2013.44
167. Betjes MG, Langerak AW, van der Spek A, de Wit EA, Litjens NH. Premature aging of circulating T cells in patients with end-stage renal disease. Kidney Int (2011) 80(2):208–17. doi: 10.1038/ki.2011.110
168. George RP, Mehta AK, Perez SD, Winterberg P, Cheeseman J, Johnson B, et al. Premature T Cell Senescence in Pediatric CKD. J Am Soc Nephrol (2017) 28(1):359–67. doi: 10.1681/ASN.2016010053
169. Yang TO, Chuang YF, Chiu YL. T-cell aging in end-stage renal disease: an evolving story with CMV. Med Microbiol Immunol (2019) 208(3-4):281–7. doi: 10.1007/s00430-019-00596-8
170. Chiu YL, Shu KH, Yang FJ, Chou TY, Chen PM, Lay FY, et al. A comprehensive characterization of aggravated aging-related changes in T lymphocytes and monocytes in end-stage renal disease: the iESRD study. Immun Ageing (2018) 15:27. doi: 10.1186/s12979-018-0131-x
171. Matias-Garcia PR, Ward-Caviness CK, Raffield LM, Gao X, Zhang Y, Wilson R, et al. DNAm-based signatures of accelerated aging and mortality in blood are associated with low renal function. Clin Epigenet (2021) 13(1):121. doi: 10.1186/s13148-021-01082-w
172. Sato Y, Yanagita M. Resident fibroblasts in the kidney: a major driver of fibrosis and inflammation. Inflammation Regener (2017) 37:17. doi: 10.1186/s41232-017-0048-3
173. Sato Y, Oguchi A, Fukushima Y, Masuda K, Toriu N, Taniguchi K, et al. CD153-CD30 signaling promotes age-dependent tertiary lymphoid tissue expansion and kidney injury. J Clin Inves (2021) 132(2):e146071. doi: 10.1172/JCI146071
174. Chiu YL, Tsai WC, Hung RW, Chen IY, Shu KH, Pan SY, et al. Emergence of T cell immunosenescence in diabetic chronic kidney disease. Immun Ageing (2020) 17:31. doi: 10.1186/s12979-020-00200-1
175. van der Harst P, Wong LS, de Boer RA, Brouilette SW, van der Steege G, Voors AA, et al. Possible association between telomere length and renal dysfunction in patients with chronic heart failure. Am J Cardiol (2008) 102(2):207–10. doi: 10.1016/j.amjcard.2008.03.040
176. Yan J, Li S, Li S. The role of the liver in sepsis. Int Rev Immunol (2014) 33(6):498–510. doi: 10.3109/08830185.2014.889129
177. Kramer L, Jordan B, Druml W, Bauer P, Metnitz PG. Incidence and prognosis of early hepatic dysfunction in critically ill patients–a prospective multicenter study. Crit Care Med (2007) 35(4):1099–104. doi: 10.1097/01.CCM.0000259462.97164.A0
178. Calado RT, Brudno J, Mehta P, Kovacs JJ, Wu C, Zago MA, et al. Constitutional telomerase mutations are genetic risk factors for cirrhosis. Hepatology (2011) 53(5):1600–7. doi: 10.1002/hep.24173
179. Gelson W, Hoare M, Vowler S, Shankar A, Gibbs P, Akbar AN, et al. Features of immune senescence in liver transplant recipients with established grafts. Liver Transpl (2010) 16(5):577–87. doi: 10.1002/lt.22033
180. Luci C, Bourinet M, Leclère PS, Anty R, Gual P. Chronic Inflammation in Non-Alcoholic Steatohepatitis: Molecular Mechanisms and Therapeutic Strategies. Front Endocrinol (Lausanne) (2020) 11:597648. doi: 10.3389/fendo.2020.597648
181. Maeso-Díaz R, Ortega-Ribera M, Fernández-Iglesias A, Hide D, Muñoz L, Hessheimer AJ, et al. Effects of aging on liver microcirculatory function and sinusoidal phenotype. Aging Cell (2018) 17(6):e12829. doi: 10.1111/acel.12829
Keywords: sepsis, immune aging, immunosenescence, immunosuppression, myeloid-derived suppressor cells
Citation: Lu X, Yang Y-M and Lu Y-Q (2022) Immunosenescence: A Critical Factor Associated With Organ Injury After Sepsis. Front. Immunol. 13:917293. doi: 10.3389/fimmu.2022.917293
Received: 11 April 2022; Accepted: 22 June 2022;
Published: 18 July 2022.
Edited by:
Philippe Saas, INSERM U1098 Interactions Hôte-Greffon-Tumeur & Ingénierie Cellulaire et Génique, FranceReviewed by:
Lyle Leonard Moldawer, University of Florida, United StatesCopyright © 2022 Lu, Yang and Lu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yuan-Qiang Lu, bHV5dWFucWlhbmdAemp1LmVkdS5jbg==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.