 Emma Boberg
Emma Boberg Julie Weidner
Julie Weidner Carina Malmhäll
Carina Malmhäll Jenny Calvén
Jenny Calvén Carmen Corciulo
Carmen Corciulo Madeleine Rådinger
Madeleine Rådinger
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Immunol. , 03 June 2022
Sec. Molecular Innate Immunity
Volume 13 - 2022 | https://doi.org/10.3389/fimmu.2022.915906
This article is part of the Research Topic The Role of Innate Immune and Resident Cells in Allergic Diseases and their Potential in Therapeutic Intervention View all 9 articles
The alarmin cytokine interleukin (IL)-33 plays an important proinflammatory role in type 2 immunity and can act on type 2 innate lymphoid cells (ILC2s) and type 2 T helper (TH2) cells in eosinophilic inflammation and asthma. The mechanistic target of rapamycin (mTOR) signaling pathway drives immune responses in several inflammatory diseases, but its role in regulating bone marrow responses to IL-33 is unclear. The aim of this study was to determine the role of the mTORC1 signaling pathway in IL-33-induced bone marrow ILC2 responses and its impact on IL-33-induced eosinophilia. Wild-type mice were intranasally exposed to IL-33 only or in combination with the mTORC1 inhibitor, rapamycin, intraperitoneally. Four groups were included in the study: saline-treated (PBS)+PBS, rapamycin+PBS, PBS+IL-33 and rapamycin+IL-33. Bronchoalveolar lavage fluid (BALF), serum and bone marrow cells were collected and analyzed by differential cell count, enzyme-linked immunosorbent assay and flow cytometry. IL-33 induced phosphorylation of the mTORC1 protein rpS6 in bone marrow ILC2s both ex vivo and in vivo. The observed mTOR signal was reduced by rapamycin treatment, indicating the sensitivity of bone marrow ILC2s to mTORC1 inhibition. IL-5 production by ILC2s was reduced in cultures treated with rapamycin before stimulation with IL-33 compared to IL-33 only. Bone marrow and airway eosinophils were reduced in mice given rapamycin before IL-33-exposure compared to mice given IL-33 only. Bone marrow ILC2s responded to IL-33 in vivo with increased mTORC1 activity and rapamycin treatment successfully decreased IL-33-induced eosinophilic inflammation, possibly by inhibition of IL-5-producing bone marrow ILC2s. These findings highlight the importance of investigating specific cells and proinflammatory pathways as potential drivers of inflammatory diseases, including asthma.
While type 2 inflammation and eosinophilia are typically associated with allergic asthma, they can also be present during non-allergic asthma. The mechanisms behind non-allergic asthma are not fully understood, though it is believed to be caused by an altered airway epithelium response to environmental factors. Interleukin (IL)-33 is released from the airway epithelium when triggered by environmental factors, including allergens (1). Thus, IL-33, a member of the IL-1 family, is an important inflammatory driver in eosinophilic inflammation and asthma (2–5). IL-33 signaling is involved in both allergic and non-allergic eosinophilic inflammation. This includes type 2 cytokine production by type 2 innate lymphoid cells (ILC2s) and T helper 2 (TH2) cells, both of which express the IL-33 receptor ST2 (6–11). Mouse lung ILC2s play a central role in worsening antigen-induced inflammatory responses mediated by IL-33 (10). Higher expression of IL-33 is associated with asthma severity, and IL-33 is considered a promising therapeutic target for eosinophilic asthma (2, 12, 13). Indeed, patients with eosinophilic asthma display elevated serum levels of IL-33 compared to non-eosinophilic phenotypes (14).
Eosinophils develop from progenitor cells in the bone marrow, and IL-5 is crucial for cell migration, terminal differentiation, and proliferation (7, 15, 16). ILC2s are potent producers of type 2 cytokines, including IL-5, and play important roles in several diseases, including asthma (2, 17–19). We have previously identified bone marrow ILC2s as an early source of IL-5 after IL-33 challenge, demonstrating that bone marrow ILC2s are important mediators central to eosinophil development (20). In addition, we have shown in two different IL-33-dependent murine models of allergic airway inflammation, i.e. papain and house dust mite (HDM), that bone marrow ILC2s are IL-33-responsive cells (9, 21). Furthermore, we demonstrated that the adaptive immune system was dispensable during both papain challenge and IL-33-induced eosinophilic airway inflammation, exhibiting ILC2s as potent sources of type 2 cytokines driving the eosinophilic inflammation. However, the mechanisms regulating the inflammatory properties of bone marrow ILC2s in IL-33-induced inflammation are still poorly understood.
The mechanistic target of rapamycin (mTOR) signaling pathway regulates many cellular processes including cell metabolism, migration, differentiation and cytokine responses (22, 23). The mTOR signaling pathway is implicated in several inflammatory diseases (23–27) and rapamycin, an inhibitor of mTOR Complex 1 (mTORC1), can suppress eosinophil differentiation in both allergic and non-allergic airway inflammation (28–30). Zhang et al. reported that eosinophil infiltration in mouse lung was impaired after treatment with rapamycin and other mTOR inhibitors in an ovalbumin-induced asthma model (31). Furthermore, the same group showed increased mTOR levels in serum from children with asthma exacerbations compared to patients in asthma remission (31). Another murine study reported decreased eosinophilia when rapamycin was administered together with HDM, but worsened inflammation when rapamycin was administered during ongoing eosinophilic inflammation (28). Thus, the effects of mTORC1 inhibition on eosinophilic inflammation remain unclear.
Accumulating evidence indicates that ILCs have tissue-specific functions (32). ILC2s exhibit different phenotypes and functions at different localizations, and single-cell profiling identified ILC2 subsets that expressed distinct activating receptors (33). Salmond et al. reported an important role for the mTOR signaling pathway in the inflammatory functions of lung ILC2s and CD4+ TH2 cells during IL-33-induced eosinophilic inflammation (30). However, the role of mTOR in the regulation and function of bone marrow ILC2s remains elusive. Thus, the overall aim of this study was to determine the role of the mTORC1 signaling pathway in vivo in response to IL-33 in bone marrow ILC2s. We further investigated whether mTORC1 inhibition, by rapamycin treatment, modifies inflammatory properties of bone marrow ILC2s in IL-33-induced eosinophilic inflammation.
Wild-type (WT) mice, C57BL/6J were purchased from Charles River (Sulzfeld, Germany) or obtained via in-house breeding (University of Gothenburg, Sweden). Male mice used in all experiments were 10 – 12 weeks old, housed in pathogen-free conditions and given food and water ad libitum. All animal experiments were approved by the Gothenburg County Regional Ethical Committee (permit numbers 126/14 and 2459/19).
WT mice were given 1 µg recombinant murine (rm) IL-33 (PeproTech, Rocky Hill, NJ, USA) intranasally (i.n.) every other day for a total of 5 days. Rapamycin doses (2 mg/kg, R0395, Sigma-Aldrich, St. Louis, MO, USA, dissolved in dimethyl sulfoxide) were administered intraperitoneally (i.p.) one hour before each i.n. IL-33 administration. Control mice received phosphate buffered saline (PBS) vehicle i.n. and i.p., where the i.p. injection contained the same concentration of dimethyl sulfoxide for mice receiving rapamycin. Four groups were included in the study: PBS+PBS, rapamycin+PBS, PBS+IL-33, and rapamycin+IL-33, where the former in each group describes the i.p. dosing and the latter describes the i.n. dosing.
Serum, bronchoalveolar lavage fluid (BALF) and bone marrow cells were collected 24 h after the final exposure. Blood was obtained by puncturing the heart. BALF was collected by instilling 0.25 mL of PBS through the tracheal cannula, followed by gentle aspiration and a second lavage of 0.25 mL. BALF cells were processed for differential cell count analysis and cell-free BALF and serum were processed for mediator analysis by enzyme-linked immunosorbent assay (ELISA). Bone marrow cells were obtained from left and right femurs flushed with 5 mL wash buffer each (2% fetal bovine serum in 1xPBS) and filtered through a 100 µm cell strainer (CellTrics®, Sysmex, Goerlitz, Germany). Bone marrow cells were then processed for differential cell count, ex vivo stimulations and flow cytometry. The red blood cells were lysed using red blood cell lysis buffer (0.1 mM EDTA in distilled water/0.8% NH4Cl, Sigma-Aldrich/Merck Chemicals) and incubated for 10 min on ice.
Concentrations of serum IL-5 and BALF CCL24/eotaxin-2 were measured using mouse ELISA DuoSets (R&D Systems, Minneapolis, MN) according to the manufacturer’s instructions. The BM Chemiluminescence ELISA (POD) substrate kit (Roche Diagnostics GmbH, Roche Applied Science, Mannheim, Germany) was used for detection of signal and luminescence was measured on a Varioskan™ LUX multimode microplate reader (ThermoFisher Scientific, Vantaa, Finland). Samples below the detection limit were set to zero.
Approximately 10,000-50,000 cells were collected via cytospin (425 x g, 6 min, Shandon Cytospin 3 centrifuge) and stained with Hemacolor® Rapid stain (Merck, Darmstadt, Germany) according to the manufacturer’s protocol. Eosinophils were assessed under an Axioplan 2 microscope (Carl Zeiss Jena GmbH).
To determine if the mTORC1 signaling pathway was activated in ILC2s and TH cells, bone marrow cells from naïve WT mice were incubated for one hour with rapamycin (50 ng/mL). Cells were stimulated with rmIL-33 (100 ng/mL) for 15 minutes. Complete cell medium was used as a control, containing RPMI-1640 (HyClone™; GE Healthcare Life Sciences, South Logan, UT, USA), 10% fetal bovine serum (Sigma-Aldrich), 2 mM L-glutamine (HyClone), 100 U/mL penicillin, 100 μg/mL streptomycin (HyClone), 1 mM sodium pyruvate (Sigma-Aldrich). Phosphorylated ribosomal protein rpS6 (pRPS6), an mTORC1 target, was measured by intracellular flow cytometry.
A total of 4 x 106 bone marrow cells from naïve WT mice were incubated for one hour with rapamycin (50 ng/mL), followed by 3 h stimulation with rmIL-33 (100 ng/mL). Monensin (BD GolgiStop™, BD Biosciences, Erembodegem, Belgium) was added to all samples (4 μL GolgiStop™/6 mL media) before IL-33 stimulation. The frequency of IL-5+ ILC2s was measured by intracellular flow cytometry.
Bone marrow cells were resuspended in 2% mouse serum (Dako, Glostrup, Denmark) and surface antibodies were added (30 min, 4°C, in the dark). For ex vivo experiments, cells were also stained with viability dye (Live/Dead™Fixable Aqua stain, Invitrogen, Life Technologies Corp, Eugene, Oregon, USA). Cells were washed, then fixed (BD CellFix™, BD Biosciences) for 15 min in the dark at room temperature and washed before intracellular staining or direct analysis. For intracellular staining, monensin (4 µL GolgiStop™/6 mL buffer) was added to all solutions prior to fixation.
After fixation, cells were washed and permeabilized using 0.1% saponin (Sigma-Aldrich) in Hank’s balanced salt solution. Anti-IL-5 antibodies or isotype control antibodies (Table S1) were added and incubated 40 min (in the dark, at room temperature) followed by washing and flow cytometric analysis.
Bone marrow cells were resuspended in 2% mouse serum (Dako) and surface antibodies were added (30 min, 4°C). Cells were washed and fixed (BD Cytofix™, BD Biosciences) for 10 min in the dark at 37°C. Cells were washed and permeabilized using ice-cold Phosflow™ Perm Buffer II (BD Biosciences) followed by 30 min incubation on ice. Cells were washed, stained for intracellular anti-S6 (60 min in the dark, at room temperature, Table S1), and washed again before analysis.
Flow cytometric analysis was performed using a BD FACSVerse™ flow cytometer running BD FACSuite™ version 1.0.6. Collected data were analyzed by FlowJo™ software (BD Biosciences). Eosinophil progenitors and mature eosinophils were defined as SSCloCD45+CD34+CD125+ and SSChiCD45+CD34-CD125loCCR3+Siglec-F+ respectively. TH cells were defined as SSCloCD45+CD3+CD4+CD8-B220-. Lineage-negative cells were defined as CD3-CD45R/B220-CD11b-TER-119-Ly-G6/Gr1-CD11c-CD19-NK-1.1-FceR1-. ILC2s were defined as SSCloLin-CD45+CD127+CD25+ST2+. Antibodies used are listed in Table S1. The ST2 expression on ILC2s, chemokine receptor 3 (CCR3) expression on SSChiCD45+CD34-CD125lo, and the intensity of IL-5 and pRPS6 in ILC2s were estimated by mean fluorescence intensity (MFI) values. Relative MFI (rMFI) equals MFI of monoclonal antibody divided by MFI of corresponding fluorescence minus one value.
Data are expressed as mean ± SEM and Graphpad Prism 8 Software (Graphpad Software Inc., La Jolla, CA, USA) was used for statistical analysis. Paired Student’s t-test was used for the ex vivo experiments, and Mann-Whitney U test was used for in vivo comparisons. Statistical significance was defined as *p<0.05, **p<0.01, ***p<0.001 and ****p<0.0001.
To determine the role of rapamycin in eosinophilia, we administered rapamycin to C57BL/6 male mice before inducing eosinophilic inflammation by IL-33 (Figure 1A). As expected, IL-33 induced airway and bone marrow eosinophilia (Figures 1B, C) and elevated CCL24/eotaxin-2 levels (Figure 1D) in BALF compared to control groups. Reduced levels of airway and bone marrow eosinophils and lower concentrations of CCL24/eotaxin-2 were observed in IL-33-exposed mice given rapamycin versus IL-33-exposure alone. Analysis of bone marrow eosinophils by flow cytometry revealed an increase of mature eosinophils in mice exposed to rmIL-33 compared to control mice given PBS. Mice treated with rapamycin before IL-33-exposure exhibited normal levels of mature eosinophils, i.e., similar numbers as control mice (Figure 1E). Furthermore, the expression of the chemokine receptor CCR3, which is highly expressed on eosinophils and a receptor for eotaxins, was analyzed on eosinophils. The CCR3 expression on mature eosinophils was significantly reduced in the rapamycin-treated and IL-33-exposed group, suggesting impaired recruitment of eosinophils to the airways (Figure 1F). No difference in eosinophil progenitor numbers or expression of the IL5Rα were seen among the treatment groups (Figures 1G, S1).
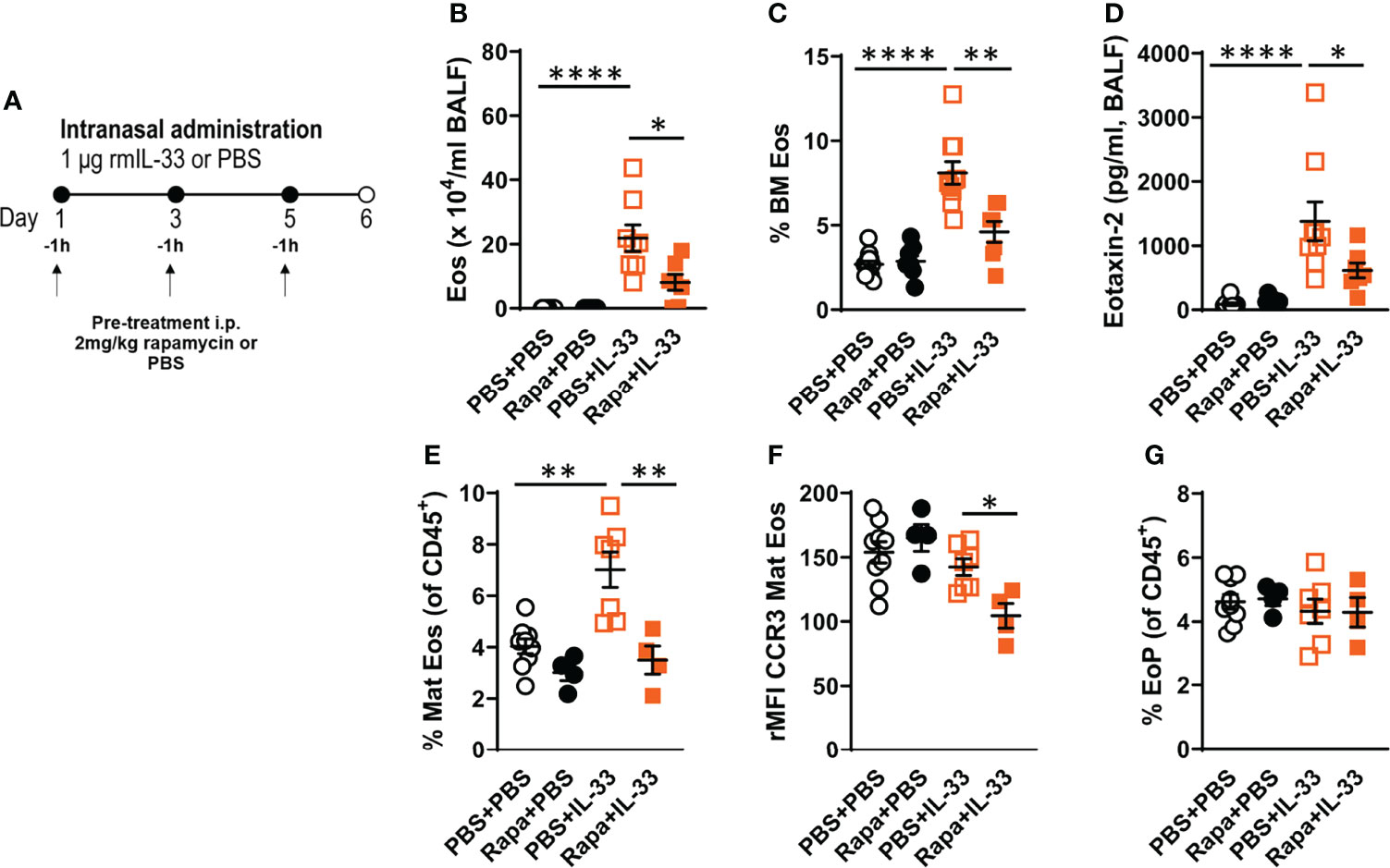
Figure 1 Rapamycin reduced IL-33-induced eosinophilia in BALF and bone marrow. (A) C57BL/6 male mice received rapamycin or PBS i.p. one hour before intranasal challenges of rmIL-33 or PBS. Analysis was performed 24 h after the final challenge. (B) Number of eosinophils in BALF and (C) bone marrow assessed by differential cell count. (D) CCL24/Eotaxin-2 concentration in BALF quantified by ELISA. (E) Number of mature eosinophils among all CD45+ leukocytes in the bone marrow. (F) CCR3 expression on mature eosinophils measured by relative mean fluorescence intensity (rMFI). (G) Number of eosinophil progenitors among all CD45+ leukocytes in the bone marrow. Data are representative of 1 – 3 independent experiments (4 – 12/group) and displayed as mean ± SEM. Mann-Whitney U test. *P < 0.05, **P < 0 .01 and ****P < 0.0001. Eos, Eosinophil; BM, Bone marrow; Mat Eos, Mature eosinophil; EoP, Eosinophil progenitor; Rapa, Rapamycin; i.p, intraperitoneal; i.n., intranasal; PBS+PBS, PBS i.p. and PBS i.n; Rapa+PBS, Rapa i.p. and PBS i.n.; PBS+IL-33, PBS i.p. and IL-33 i.n.; Rapa+IL-33, Rapa i.p. and IL-33 i.n.
We have previously demonstrated that during IL-33-driven inflammation, bone marrow ILC2s produce IL-5, a critical cytokine for eosinophil differentiation (20, 21). In the current study, we found a decrease in IL-5-producing bone marrow ILC2s in cultures treated with rapamycin before rmIL-33 compared to cultures stimulated with IL-33 only (Figures 2A, B). Additionally, rMFI revealed decreased overall intensity of IL-5 in ILC2s after pre-treatment with rapamycin (Figure 2C). In vivo, we found a lower concentration of IL-5 in serum from mice pre-treated with rapamycin before IL-33 exposure compared to mice treated with PBS before IL-33 exposure (Figure 2D).
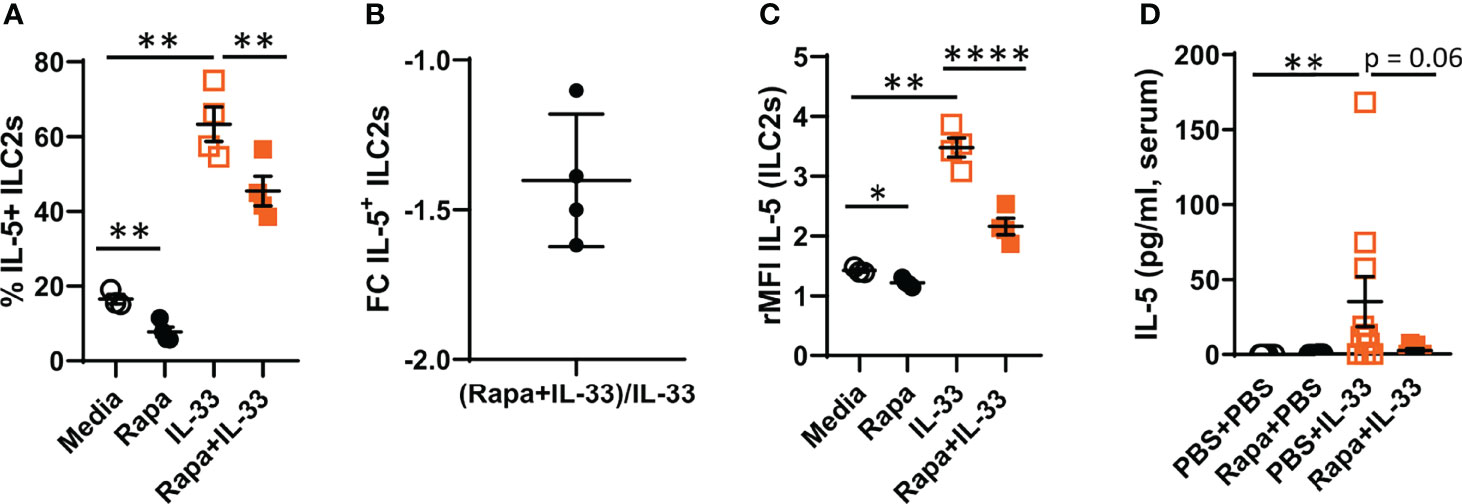
Figure 2 Decreased IL-5 production by bone marrow ILC2s after rapamycin treatment and IL-33 stimulation ex vivo. (A) Number of IL-5+ ILC2 cells. (B) Number of IL-5+ ILC2s in rapamycin+IL-33 and PBS+IL-33 mice shown as fold change (FC). (C) IL-5 intensity in ILC2s measured by relative mean fluorescence intensity (rMFI). (D) Concentration of IL-5 in serum. Data are representative of 1 – 3 independent experiments (3 – 10/group). Paired Student’s t-test (A ,C), Mann-Whitney U test (D). *P < 0.05, **P < 0.01 and ****P < 0.0001. Rapa, Rapamycin; i.p, intraperitoneal; i.n., intranasal; PBS+PBS, PBS i.p. and PBS i.n; Rapa+PBS, Rapa i.p. and PBS i.n.; PBS+IL-33, PBS i.p. and IL-33 i.n.; Rapa+IL-33, Rapa i.p. and IL-33 i.n.
To determine the mTORC1 activity in bone marrow ILC2s after IL-33 stimulation, we measured phosphorylation of the known mTORC1 target rpS6 by intracellular flow cytometry. The gating strategy for bone marrow pRPS6+ILC2s is shown in Figure 3A. IL-33-stimulation of bone marrow cultures induced phosphorylation of rpS6+ ILC2s compared to controls (Figure 3B). Rapamycin inhibited the basal pRPS6+ ILC2s signal compared to media only (Figure 3B). The overall intensity of pRPS6 in bone marrow ILC2s was increased after IL-33 stimulation compared to controls. This effect was inhibited in cultures treated with rapamycin in addition to IL-33 (Figure 3C). Because ILCs share some immune functions with T cells, we also examined mTORC1 activity in bone marrow TH cells compared to ILC2s. IL-33 did not induce rpS6 phosphorylation in bone marrow TH cells compared to controls (Figure 3D). Additionally, controls showed that only a small percentage of TH cells were pRPS6+ at baseline and that rapamycin further reduced this basal activity (Figures 3D, E).
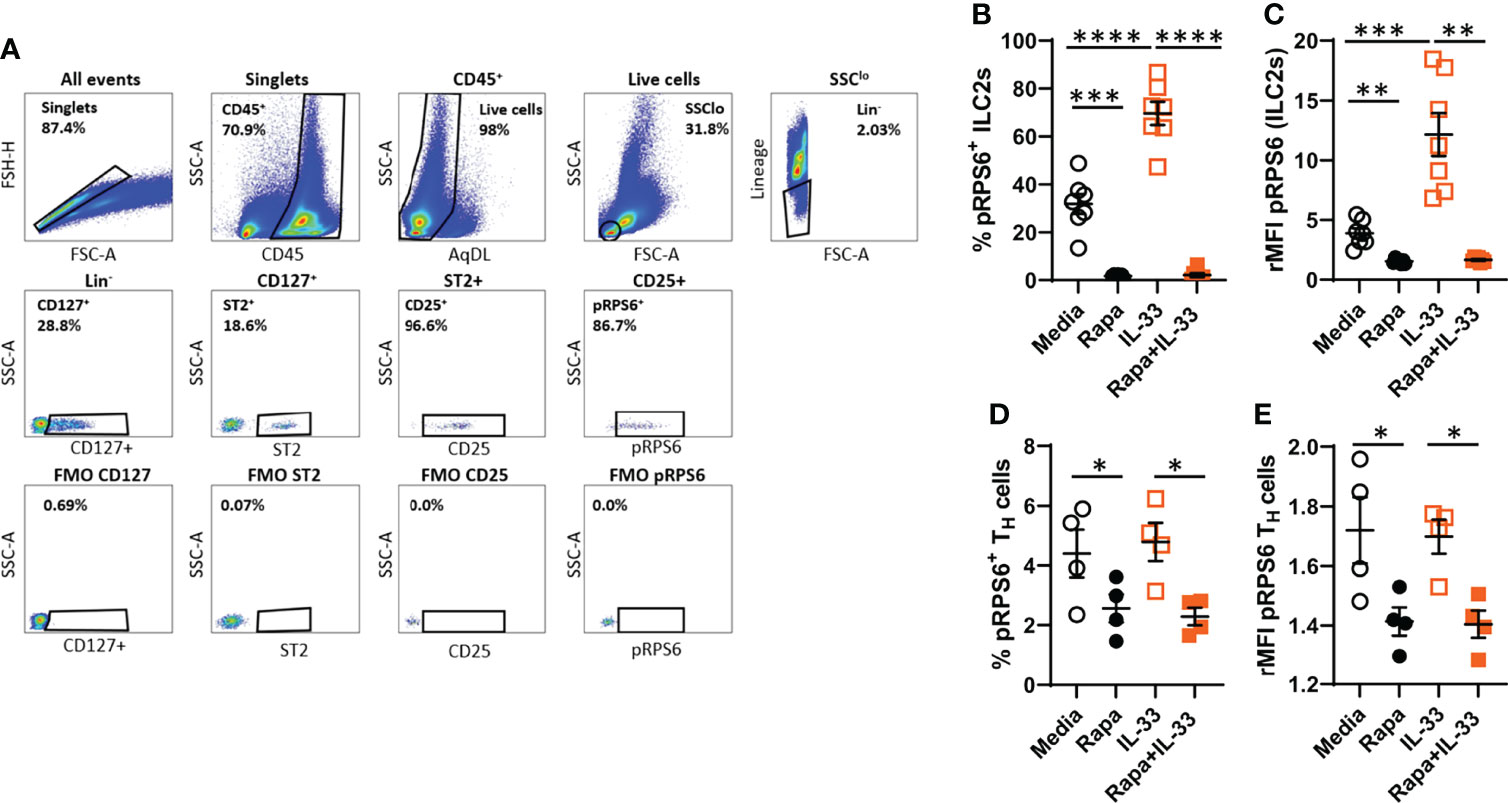
Figure 3 IL-33 induced high mTORC1 activity in bone marrow ILC2s ex vivo. (A) Gating strategy for quantifying mTOR activity in ILC2s measured by pRPS6 after IL-33 stimulation of cultured bone marrow cultures. (B) Number of pRPS6+ ILC2s and (C) pRPS6 intensity in ILC2s measured by relative mean fluorescence intensity (rMFI). (D) Number of pRPS6+ TH cells and (E) pRPS6 intensity in TH cells measured rMFI. Data are representative of 1 – 2 independent experiments (4 – 7/group). Paired Student’s t-test. *P < 0.05, **P < 0 .01, ***P < 0.001 and ****P < 0.0001. Rapa, Rapamycin.
After demonstrating the induction of mTORC1 activity in bone marrow ILC2s after IL-33 stimulation and the reduction of mTORC1 activity by rapamycin treatment, we investigated this mechanism in ILC2s in vivo. The number of ILC2s was similar in all experiment groups (Figure 4A), though an increase in pRPS6+ ILC2s was observed in mice challenged with IL-33 compared to controls (Figure 4B). A reduction of pRPS6+ ILC2s was observed in mice pre-treated with rapamycin before IL-33 exposure. The intensity of the pRPS6 signal was increased in IL-33-exposed mice compared to controls (Figure 4C) and decreased in mice treated with rapamycin in addition to IL-33 (Figures 4C, D).
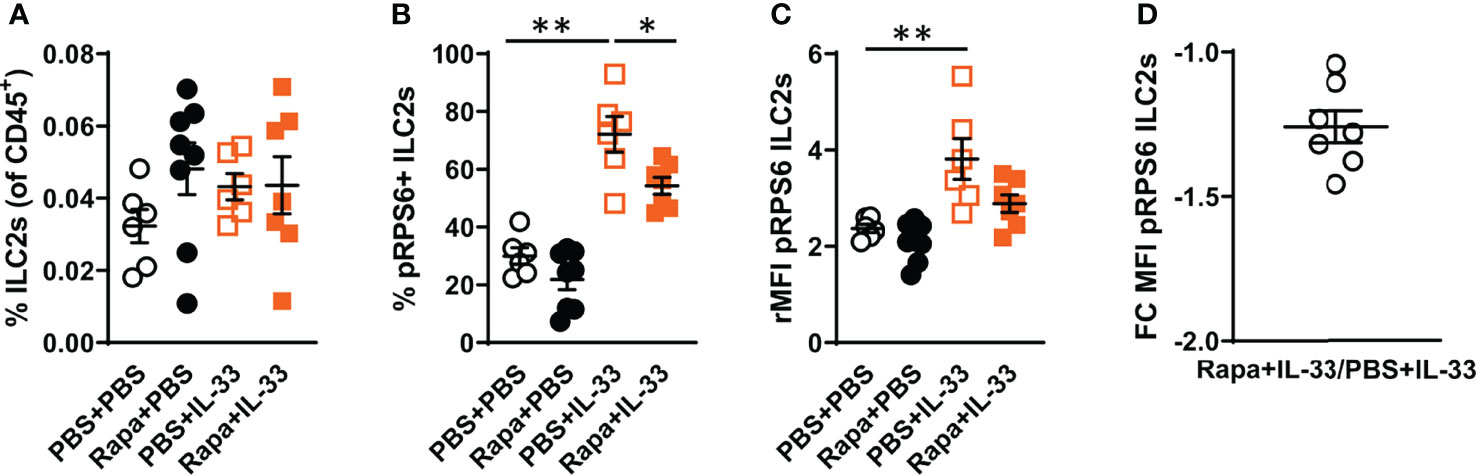
Figure 4 IL-33 induced high mTORC1 activity in bone marrow ILC2s in vivo. (A) Number of ILC2s among all CD45+ leukocytes in the bone marrow. (B) Number of pRPS6+ ILC2s and (C) pRPS6 intensity in ILC2s measured by relative mean fluorescence intensity (rMFI) in mice exposed to IL-33+rapamycin or PBS. (D) Fold change (FC) rMFI pRPS6 in mice exposed to IL-33+rapamycin and PBS+IL-33. Data are representative of 2 independent experiments (6 – 8/group). Mann-Whitney U test. *P < 0.05 and **P < 0.01. Rapa, Rapamycin; i.p, intraperitoneal; i.n., intranasal; PBS+PBS, PBS i.p. and PBS i.n; Rapa+PBS, Rapa i.p. and PBS i.n.; PBS+IL-33, PBS i.p. and IL-33 i.n.; Rapa+IL-33, Rapa i.p. and IL-33 i.n.
It has been shown that IL-33 induces mTOR activation through ST2 signaling (30). Therefore, we investigated the effect of rapamycin on ST2 expression on TH cells, mature eosinophils and ILC2s in vivo. The number of ST2+ TH cells was increased in mice exposed to IL-33 and decreased in mice exposed to rapamycin in addition to IL-33 (Figure 5A). Additionally, rapamycin treatment reduced ST2 expression on TH cells in control mice given saline. In contrast to TH cells, rapamycin treatment did not affect the ST2 expression on eosinophils or ILC2s (Figures 5B, C). Mice exposed to IL-33 showed an increase in ST2+ mature eosinophils and ST2 expression on ILC2s (Figures 5B, C).
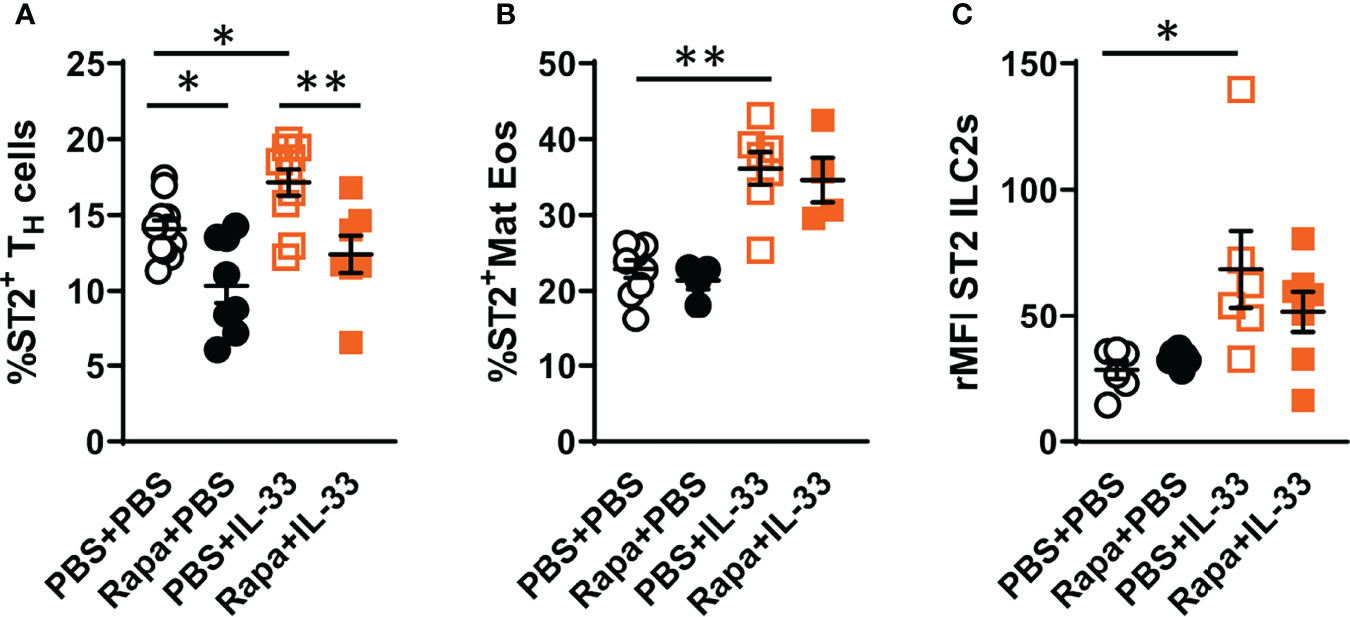
Figure 5 Decreased ST2 expression on bone marrow TH cells after rapamycin treatment and IL-33 challenge. (A) Number of ST2+ TH cells and (B) mature eosinophils. (C) ST2 expression on ILC2s shown as relative mean fluorescence intensity (rMFI). Data are representative of 1 – 3 independent experiments (4 – 11/group). Mann-Whitney U test. *P < 0.05 and **P < 0.01. TH, T helper cell; ST2=IL-33 receptor; Mat Eos, Mature eosinophil; Rapa, Rapamycin; i.p, intraperitoneal; i.n., intranasal; PBS+PBS, PBS i.p. and PBS i.n; Rapa+PBS, Rapa i.p. and PBS i.n.; PBS+IL-33, PBS i.p. and IL-33 i.n.; Rapa+IL-33, Rapa i.p. and IL-33 i.n.
In this study, the mTORC1 signaling pathway in bone marrow ILC2s was investigated using an IL-33-induced in vivo model. Furthermore, the mTORC1 inhibitor, rapamycin, was used to investigate how the treatment affected proinflammatory properties of bone marrow ILC2s in vivo. We show for the first time, at the single-cell level, that IL-33 promoted high mTORC1 activity, as measured by the phosphorylation of the known mTORC1 target rpS6 in bone marrow ILC2s both ex vivo and in vivo. Moreover, this activity was reduced by rapamycin treatment.
There is substantial evidence that IL-5 is a critical cytokine acting on numerous processes in eosinophil biology, driving eosinophilic diseases. Eosinophils develop from CD34+ progenitor cells in the bone marrow, where IL-5 promotes terminal eosinophil differentiation and trafficking of bone marrow eosinophils to the airways during different inflammatory conditions (16, 34–36). IL-33 stimulation of bone marrow cultures from naïve WT mice revealed an increased IL-5 production by bone marrow ILC2s and confirmed our previous studies (9, 20, 21). In the current study, we also demonstrated for the first time that the mTORC1 inhibitor rapamycin significantly reduced the number of IL-5+ ILC2s in bone marrow cultures stimulated with IL-33. Furthermore, rapamycin also exhibited an effect under basal conditions, as demonstrated by decreased IL-5 production in bone marrow ILC2s in mice given saline.
Extensive evidence indicates that ILCs have tissue-specific functions, and more research is needed to better understand the properties of ILC2s in different tissues during various inflammatory conditions. A lower IL-5 production by lung ILC2s has been reported after rapamycin treatment (30). The same study reported the absence of pRPS6 in sorted lung ILC2s stimulated with IL-33 and rapamycin in vitro compared to ILC2s stimulated with IL-33 only (30). We found that rapamycin inhibited IL-5 production by bone marrow ILC2s, thus decreasing the eosinophilic inflammation induced by IL-33. Furthermore, our study demonstrated, in vivo, that bone marrow ILC2s act similarly to lung ILC2s during eosinophilic inflammation driven by IL-33.
Assessment of bone marrow eosinophils revealed a decrease in mature eosinophils in IL-33-exposed mice pre-treated with rapamycin compared to mice given IL-33 only. We have previously shown that ILC2s produce large amounts of IL-5 in response to IL-33 which is in contrast to eosinophil progenitors that produce low levels of IL-5 (20). Moreover, we further demonstrate that rapamycin treatment had no effect on IL-5Rα expression on eosinophil progenitors which remained the same in all treatment groups. Together, these data indicate that the lower IL-5 production by bone marrow ILC2s observed in the present study may explain the decreased number of mature eosinophils in mice treated with rapamycin. Our data suggest that the mTORC1 signaling pathway is critical for eosinophilic inflammation and type 2 responses mediated by IL-5-producing ILC2s in the bone marrow. A decrease in airway and mature bone marrow eosinophils in mice treated with rapamycin and IL-33, compared to IL-33 only, indicates that rapamycin may affect the maturation of eosinophils in the bone marrow.
The chemokine receptor CCR3 is highly expressed on eosinophils and contributes to the accumulation and activation of eosinophils (15). Expression of CCR3 on mature eosinophils in the bone marrow remained unchanged in mice exposed to IL-33 compared to control mice, but was reduced in IL-33-exposed mice pre-treated with rapamycin compared to mice given IL-33 only. A reduction in mRNA levels of CCR3 in in vitro differentiated bone marrow eosinophils after rapamycin treatment have previously been reported, thus suggesting a cell-intrinsic effect of rapamycin (29). In addition to its role in eosinophil maturation, IL-5 primes eosinophils for migration to sites of inflammation by upregulating CCR3 on eosinophils. Besides a potential cell-intrinsic effect on eosinophils from rapamycin treatment, the decrease in IL-5+ ILC2s in bone marrow cultures may indicate that rapamycin affected eosinophil recruitment to the airways by downregulating CCR3. Moreover, a decrease in the CCR3 ligand CCL24/eotaxin-2 was observed in the airways in mice exposed to rapamycin in addition to IL-33 compared to IL-33 only, further suggesting that the recruitment might be affected. Taken together, reduced expression of CCR3 on eosinophils and CCL24/eotaxin-2 in the airways could potentially explain the decrease in airway eosinophilia observed in mice given rapamycin in addition to IL-33.
Our findings confirm previously reported reduced airway eosinophilia after rapamycin treatment in IL-33-exposed mice (30). Furthermore, several studies have also reported a decrease in eosinophils in the airways of mice treated with rapamycin in OVA-induced allergic eosinophilic inflammation (29, 31, 37). One study reported paradoxical effects of rapamycin in HDM-induced airway eosinophilia (28). In that study, rapamycin decreased eosinophilia when administered together with HDM, but increased eosinophilia when administered after an established HDM-induced inflammation. These findings highlight the complexity of mTOR activation and the need for further investigation into how rapamycin affects eosinophilic inflammation.
Several immune cells express ST2, including ILC2s, TH cells and eosinophils (38, 39). We have previously shown that bone marrow ILC2s, TH cells and mature eosinophils all respond to IL-33 by upregulating the ST2 receptor in mice exposed to IL-33, papain or HDM compared to control mice (9, 20, 21). Moreover, it has been proposed that IL-33 induces mTOR activation through ST2 signaling in experiments performed on a murine TH2 clone (30). However, little is known about how mTOR inhibition affects ST2 expression on immune cells in the bone marrow. In the present study, we demonstrated a decrease in ST2+ TH cells in mice pre-treated with rapamycin in both IL-33-exposed mice and control mice, whereas ST2 expression was not affected on mature eosinophils or ILC2s in any of the examined groups. Further kinetic studies are needed to investigate the clinical impact of decreased ST2 expression after rapamycin treatment. Kinetic studies would also reveal whether ST2 expression on eosinophils and ILC2s varies at other time points or if the altered ST2 expression after rapamycin treatment is unique for TH cells. In contrast to ILC2s which all express ST2, approximately 15% of all bone marrow TH cells of control mice expressed ST2. The difference in ST2 expression may explain the absence of rpS6 phosphorylation after IL-33 stimulation in TH cells. A detailed characterisation of T cell subsets was not addressed in the current study. Thus, in future studies, it would be interesting to determine the effects of rapamycin treatment on ST2 expression in both TH2 cells and T regulatory cells and assess the downstream effects during IL-33-induced inflammation.
Clinical trials of anti-IL-33 therapies for asthma are already underway. For patients who do not respond to currently available pharmacological treatments, it is imperative to identify new disease mechanisms during IL-33 inflammation (40, 41). Indeed, future studies that target both mTOR signaling pathways are warranted to deepen the mechanistic understanding of how the mTOR signaling pathways regulate bone marrow ILC2s during IL-33-driven inflammation.
To conclude, we demonstrated for the first time that bone marrow ILC2s responded to IL-33 in vivo with increased mTORC1 activity. Rapamycin treatment decreased IL-33-induced eosinophilic airway inflammation, possibly by inhibiting IL-5-producing bone marrow ILC2s. Collectively, our data support previous studies demonstrating beneficial effects of rapamycin treatment during eosinophilic inflammation. The mTORC1 signaling pathway might be a disease driver and a future therapeutic target in inflammatory diseases including asthma.
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
The animal study was reviewed and approved by Gothenburg County Regional Ethical Committee (permit numbers 126/14 and 2459/19).
EB, JW, CM and MR designed experiments. EB, JW, CM, JC and CC performed experiments. EB analyzed data. EB, JW, CM and MR wrote the manuscript. EB, JW, CM, JC, CC and MR revised the manuscript. EB, JW, CM, JC, CC and MR approved the final manuscript. All authors contributed to the article and approved the submitted version.
This research was funded by grants from the Swedish Research Council (202001815), the Swedish Heart and Lung Foundation (20190378) and the VBG Group Herman Krefting Foundation for Asthma and Allergy Research.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The authors would like to thank Reghan Borer for reviewing the manuscript.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2022.915906/full#supplementary-material
1. Rodriguez-Rodriguez N, Gogoi M, McKenzie ANJ. Group 2 Innate Lymphoid Cells: Team Players in Regulating Asthma. Annu Rev Immun (2021) 39:167–98. doi: 10.1146/annurev-immunol-110119-091711
2. Christianson CA, Goplen NP, Zafar I, Irvin C, Good JT Jr, Rollins DR, et al. Persistence of Asthma Requires Multiple Feedback Circuits Involving Type 2 Innate Lymphoid Cells and Il-33. J Allergy Clin Immunol (2015) 136(1):59–68.e14. doi: 10.1016/j.jaci.2014.11.037
3. Kearley J, Buckland KF, Mathie SA, Lloyd CM. Resolution of Allergic Inflammation and Airway Hyperreactivity Is Dependent Upon Disruption of the T1/St2-Il-33 Pathway. Am J Respir Crit Care Med (2009) 179(9):772–81. doi: 10.1164/rccm.200805-666OC
4. Iijima K, Kobayashi T, Hara K, Kephart GM, Ziegler SF, McKenzie AN, et al. Il-33 and Thymic Stromal Lymphopoietin Mediate Immune Pathology in Response to Chronic Airborne Allergen Exposure. J Immunol (2014) 193(4):1549–59. doi: 10.4049/jimmunol.1302984
5. Mitchell PD, Salter BM, Oliveria JP, El-Gammal A, Tworek D, Smith SG, et al. Il-33 and Its Receptor St2 After Inhaled Allergen Challenge in Allergic Asthmatics. Int Arch Allergy Immunol (2018) 176(2):133–42. doi: 10.1159/000488015
6. Oboki K, Ohno T, Kajiwara N, Arae K, Morita H, Ishii A, et al. Il-33 Is a Crucial Amplifier of Innate Rather Than Acquired Immunity. Proc Natl Acad Sci USA (2010) 107(43):18581–6. doi: 10.1073/pnas.1003059107
7. Johnston LK, Hsu CL, Krier-Burris RA, Chhiba KD, Chien KB, McKenzie A, et al. Il-33 Precedes Il-5 in Regulating Eosinophil Commitment and Is Required for Eosinophil Homeostasis. J Immunol (2016) 197(9):3445–53. doi: 10.4049/jimmunol.1600611
8. Kurowska-Stolarska M, Kewin P, Murphy G, Russo RC, Stolarski B, Garcia CC, et al. Il-33 Induces Antigen-Specific Il-5+ T Cells and Promotes Allergic-Induced Airway Inflammation Independent of Il-4. J Immunol (2008) 181(7):4780–90. doi: 10.4049/jimmunol.181.7.4780
9. Boberg E, Johansson K, Malmhäll C, Calvén J, Weidner J, Rådinger M. Interplay Between the Il-33/St2 Axis and Bone Marrow Ilc2s in Protease Allergen-Induced Il-5-Dependent Eosinophilia. Front Immunol (2020) 11:1058. doi: 10.3389/fimmu.2020.01058
10. Sjöberg LC, Nilsson AZ, Lei Y, Gregory JA, Adner M, Nilsson GP. Interleukin 33 Exacerbates Antigen Driven Airway Hyperresponsiveness, Inflammation and Remodeling in a Mouse Model of Asthma. Sci Rep (2017) 7(1):4219. doi: 10.1038/s41598-017-03674-0
11. Warren KJ, Poole JA, Sweeter JM, DeVasure JM, Dickinson JD, Peebles RS Jr, et al. Neutralization of Il-33 Modifies the Type 2 and Type 3 Inflammatory Signature of Viral Induced Asthma Exacerbation. Respir Res (2021) 22(1):206. doi: 10.1186/s12931-021-01799-5
12. Préfontaine D, Lajoie-Kadoch S, Foley S, Audusseau S, Olivenstein R, Halayko AJ, et al. Increased Expression of Il-33 in Severe Asthma: Evidence of Expression by Airway Smooth Muscle Cells. J Immunol (2009) 183(8):5094–103. doi: 10.4049/jimmunol.0802387
13. Momen T, Ahanchian H, Reisi M, Shamsdin SA, Shahsanai A, Keivanfar M. Comparison of Interleukin-33 Serum Levels in Asthmatic Patients With a Control Group and Relation With the Severity of the Disease. Int J Prev Med (2017) 8:65. doi: 10.4103/ijpvm.IJPVM_179_16
14. Gasiuniene E, Janulaityte I, Zemeckiene Z, Barkauskiene D, Sitkauskiene B. Elevated Levels of Interleukin-33 Are Associated With Allergic and Eosinophilic Asthma. Scand J Immunol (2019) 89(5):e12724. doi: 10.1111/sji.12724
15. Rothenberg ME, Hogan SP. The Eosinophil. Annu Rev Immunol (2006) 24:147–74. doi: 10.1146/annurev.immunol.24.021605.090720
16. Robinson DS, Damia R, Zeibecoglou K, Molet S, North J, Yamada T, et al. Cd34(+)/Interleukin-5ralpha Messenger Rna+ Cells in the Bronchial Mucosa in Asthma: Potential Airway Eosinophil Progenitors. Am J Respir Cell Mol Biol (1999) 20(1):9–13. doi: 10.1165/ajrcmb.20.1.3449
17. Klose CS, Artis D. Innate Lymphoid Cells as Regulators of Immunity, Inflammation and Tissue Homeostasis. Nat Immunol (2016) 17(7):765–74. doi: 10.1038/ni.3489
18. Salimi M, Barlow JL, Saunders SP, Xue L, Gutowska-Owsiak D, Wang X, et al. A Role for Il-25 and Il-33-Driven Type-2 Innate Lymphoid Cells in Atopic Dermatitis. J Exp Med (2013) 210(13):2939–50. doi: 10.1084/jem.20130351
19. Jackson DJ, Makrinioti H, Rana BM, Shamji BW, Trujillo-Torralbo MB, Footitt J, et al. Il-33-Dependent Type 2 Inflammation During Rhinovirus-Induced Asthma Exacerbations in Vivo. Am J Respir Crit Care Med (2014) 190(12):1373–82. doi: 10.1164/rccm.201406-1039OC
20. Johansson K, Malmhall C, Ramos-Ramirez P, Radinger M. Bone Marrow Type 2 Innate Lymphoid Cells: A Local Source of Interleukin-5 in Interleukin-33-Driven Eosinophilia. Immunology (2018) 153(2):268–78. doi: 10.1111/imm.12842
21. Boberg E, Johansson K, Malmhäll C, Weidner J, Rådinger M. House Dust Mite Induces Bone Marrow Il-33-Responsive Ilc2s and T(H) Cells. Int J Mol Sci (2020) 21(11):3751. doi: 10.3390/ijms21113751
22. Weichhart T, Hengstschlager M, Linke M. Regulation of Innate Immune Cell Function by Mtor. Nat Rev Immunol (2015) 15(10):599–614. doi: 10.1038/nri3901
23. Laplante M, Sabatini DM. Mtor Signaling in Growth Control and Disease. Cell (2012) 149(2):274–93. doi: 10.1016/j.cell.2012.03.017
24. Lyons JJ, Milner JD. The Clinical and Mechanistic Intersection of Primary Atopic Disorders and Inborn Errors of Growth and Metabolism. Immunol Rev (2019) 287(1):135–44. doi: 10.1111/imr.12727
25. Ma CA, Stinson JR, Zhang Y, Abbott JK, Weinreich MA, Hauk PJ, et al. Germline Hypomorphic Card11 Mutations in Severe Atopic Disease. Nat Genet (2017) 49(8):1192–201. doi: 10.1038/ng.3898
26. Zhang M, Iwata S, Sonomoto K, Ueno M, Fujita Y, Anan J, et al. Mtor Activation in Cd8+ Cells Contributes to Disease Activity of Rheumatoid Arthritis and Increases Therapeutic Response to Tnf Inhibitors. Rheumatol (Oxford) (2021). doi: 10.1093/rheumatology/keab834
27. Fang L, Roth M, S'Ng CT, Tamm M, Han B, Hoang BX. Zinc Salicylate Reduces Airway Smooth Muscle Cells Remodelling by Blocking Mtor and Activating P21((Waf1/Cip1)). J Nutr Biochem (2021) 89:108563. doi: 10.1016/j.jnutbio.2020.108563
28. Fredriksson K, Fielhaber JA, Lam JK, Yao X, Meyer KS, Keeran KJ, et al. Paradoxical Effects of Rapamycin on Experimental House Dust Mite-Induced Asthma. PLos One (2012) 7(5):e33984. doi: 10.1371/journal.pone.0033984
29. Hua W, Liu H, Xia LX, Tian BP, Huang HQ, Chen ZY, et al. Rapamycin Inhibition of Eosinophil Differentiation Attenuates Allergic Airway Inflammation in Mice. Respirology (2015) 20(7):1055–65. doi: 10.1111/resp.12554
30. Salmond RJ, Mirchandani AS, Besnard AG, Bain CC, Thomson NC, Liew FY. Il-33 Induces Innate Lymphoid Cell-Mediated Airway Inflammation by Activating Mammalian Target of Rapamycin. J Allergy Clin Immunol (2012) 130(5):1159–66.e6. doi: 10.1016/j.jaci.2012.05.018
31. Zhang Y, Jing Y, Qiao J, Luan B, Wang X, Wang L, et al. Activation of the Mtor Signaling Pathway Is Required for Asthma Onset. Sci Rep (2017) 7(1):4532. doi: 10.1038/s41598-017-04826-y
32. Meininger I, Carrasco A, Rao A, Soini T, Kokkinou E, Mjösberg J. Tissue-Specific Features of Innate Lymphoid Cells. Trends Immunol (2020) 41(10):902–17. doi: 10.1016/j.it.2020.08.009
33. Ricardo-Gonzalez RR, Van Dyken SJ, Schneider C, Lee J, Nussbaum JC, Liang HE, et al. Tissue Signals Imprint Ilc2 Identity With Anticipatory Function. Nat Immunol (2018) 19(10):1093–9. doi: 10.1038/s41590-018-0201-4
34. Johnston LK, Bryce PJ. Understanding Interleukin 33 and Its Roles in Eosinophil Development. Front Med (Lausanne) (2017) 4:51. doi: 10.3389/fmed.2017.00051
35. Johansson AK, Sergejeva S, Sjostrand M, Lee JJ, Lotvall J. Allergen-Induced Traffic of Bone Marrow Eosinophils, Neutrophils and Lymphocytes to Airways. Eur J Immunol (2004) 34(11):3135–45. doi: 10.1002/eji.200425043
36. Rosenberg HF, Phipps S, Foster PS. Eosinophil Trafficking in Allergy and Asthma. J Allergy Clin Immunol (2007) 119(6):1303–10. doi: 10.1016/j.jaci.2007.03.048
37. Zhu C, Xia L, Li F, Zhou L, Weng Q, Li Z, et al. Mtor Complexes Differentially Orchestrates Eosinophil Development in Allergy. Sci Rep (2018) 8(1):6883. doi: 10.1038/s41598-018-25358-z
38. Cayrol C, Girard JP. Il-33: An Alarmin Cytokine With Crucial Roles in Innate Immunity, Inflammation and Allergy. Curr Opin Immunol (2014) 31:31–7. doi: 10.1016/j.coi.2014.09.004
39. Liew FY, Girard JP, Turnquist HR. Interleukin-33 in Health and Disease. Nat Rev Immunol (2016) 16(11):676–89. doi: 10.1038/nri.2016.95
40. Porsbjerg CM, Sverrild A, Lloyd CM, Menzies-Gow AN, Bel EH. Anti-Alarmins in Asthma: Targeting the Airway Epithelium With Next-Generation Biologics. Eur Respir J (2020) 56(5):2000260. doi: 10.1183/13993003.00260-2020
Keywords: innate lymphoid cells, interleukin-33, rapamycin, interleukin-5, eosinophils, bone marrow, mTOR, asthma
Citation: Boberg E, Weidner J, Malmhäll C, Calvén J, Corciulo C and Rådinger M (2022) Rapamycin Dampens Inflammatory Properties of Bone Marrow ILC2s in IL-33-Induced Eosinophilic Airway Inflammation. Front. Immunol. 13:915906. doi: 10.3389/fimmu.2022.915906
Received: 08 April 2022; Accepted: 10 May 2022;
Published: 03 June 2022.
Edited by:
Peisong Gao, Johns Hopkins University, United StatesReviewed by:
Jorg Hermann Fritz, McGill University, CanadaCopyright © 2022 Boberg, Weidner, Malmhäll, Calvén, Corciulo and Rådinger. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Madeleine Rådinger, bWFkZWxlaW5lLnJhZGluZ2VyQGd1LnNl
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.