 Xiaoting Zhou
Xiaoting Zhou Yanghong Ni
Yanghong Ni Xiao Liang
Xiao Liang Yi Lin
Yi Lin Xia Zhao
Xia Zhao
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 15 September 2022
Sec. Cancer Immunity and Immunotherapy
Volume 13 - 2022 | https://doi.org/10.3389/fimmu.2022.915094
This article is part of the Research Topic Combinational Immunotherapy of Cancer: Novel Targets, Mechanisms, and Strategies View all 85 articles
Immune checkpoint blockade (ICB) has rapidly transformed the treatment paradigm for various cancer types. Multiple single or combinations of ICB treatments have been approved by the US Food and Drug Administration, providing more options for patients with advanced cancer. However, most patients could not benefit from these immunotherapies due to primary and acquired drug resistance. Thus, a better understanding of the mechanisms of ICB resistance is urgently needed to improve clinical outcomes. Here, we focused on the changes in the biological functions of CD8+ T cells to elucidate the underlying resistance mechanisms of ICB therapies and summarized the advanced coping strategies to increase ICB efficacy. Combinational ICB approaches and individualized immunotherapies require further in-depth investigation to facilitate longer-lasting efficacy and a more excellent safety of ICB in a broader range of patients.
The emergence of immune checkpoint blockade (ICB) has brought the oncology field to a new stage, offering renewed hope for patients with advanced cancer. Over the past decades, ICB, as one of the representative cancer immunotherapies, has produced the broadest impact on cancer treatment (1). ICB, including programmed cell death protein 1 (PD-1), programmed cell death ligand 1 (PD-L1), and cytotoxic T lymphocyte antigen 4 (CTLA-4) monoclonal antibodies, have shown antitumor efficacies in multiple advanced solid tumors since the initial approval of CTLA-4 inhibitors for metastatic melanoma in 2011 by the US Food and Drug Administration (FDA) (2). There are currently three main classes of ICB approved by the FDA in the treatment of various solid tumors, including six drugs targeting the programmed cell death protein 1 (PD-1)/programmed cell death ligand 1 (PD-L1) checkpoint (nivolumab, pembrolizumab, cemiplimab, avelumab, durvalumab, atezolizumab), anti-CTLA-4 checkpoint (ipilimumab), and recently approved anti-LAG-3 (relatlimab) (3).
Unfortunately, most patients suffer primary resistance and do not respond to anti-PD-1/PD-L1 treatments. The limited efficacy of anti-PD1/PDL1 may be attributed to a range of mechanisms involving the whole immune response process. The most straightforward reasons for primary resistance are insufficient tumor immunogenicity, poor CD8+ T-cell infiltration, and irreversible T-cell exhaustion. Moreover, some patients with the initial response develop resistance or relapse eventually, which is called acquired resistance (2, 4). The mechanisms accounting for either form of resistance are intricate and complex, which have not been fully cleared up yet. Golnaz Morad et al. systematically divided the factors that affect ICB response into host-intrinsic factors, including tumor cells, non-tumor cells, age, gender, obesity, and gut microbiota, and host-extrinsic factors such as environmental exposures, social pressure, and unhealthy lifestyles. According to their discussion, the role of host systemic and environmental factors should be noted in the study of ICB response (5). Similarly, Aldea et al. overviewed the tumor cell–intrinsic mechanisms and stromal mechanisms. Of note, the different locations of metastasis can lead to an opposite response to ICB (6). Bagchi et al. reviewed the mechanism of ICB resistance from primary and acquired resistance perspectives. Most cancer cell–intrinsic factors contribute to the primary resistance, for instance, the expression intensity of ICB biomarkers, tumor mutation burden, and epigenetic variations. However, the mechanisms of acquired resistance are not well understood, and some common mechanisms may be shared by both types of resistance (7). Genetic mutations are common during the process of tumor progression. Kobayashi et al. summarized six signaling pathways related to ICB resistance. Understanding these could provide potential combinational options for immunotherapy and molecular-targeted therapies. In addition, as a consequence of activating oncogenic drivers or in response to external stimuli, alteration in phenotype plasticity is another integral approach exploited by tumor cells to avoid immune surveillance, thus getting resistance to immunotherapy (8, 9). Based on the analysis of a panel of syngeneic melanoma mouse models, a melanocytic plasticity signature was uncovered to predict the response to ICB and the outcome of patients, implicating the core of plasticity in ICB resistance (10). Novel strategies targeting tumor cell plasticity could be beneficial for patients receiving immunotherapy (11).
A mounting number of preclinical and clinical studies are ongoing to reveal the mechanisms underlying immune checkpoint inhibitor resistance and offer abundant clues for potential combined therapeutic strategies (12, 13). Combination strategies, promising to solve the restrictions of anti-PD-1/PD-L1 treatment, include a combination with traditional chemotherapy and radiotherapy, other immune checkpoint inhibitors, CAR T therapy agonists of the costimulatory molecule, antiangiogenic agents, oncogenic pathway–targeted therapy, microbiota-centered interventions, and metabolic and epigenetic regulation (14–19). Overall, the higher response rates elicited by combination regimens are associated with boosting multiple phases in the cancer-immunity cycle.
This review will discuss the mechanisms underlying ICB resistance, focusing on the changes in the biological function of CD8+ T cells. We then highlight existing and emerging strategies to overcome resistance to ICB and boost immunotherapy in preclinical and clinical studies.
As is well known, CD8+ cytotoxic T lymphocytes (CTLs) play a significant role in antitumor immunotherapy because they are directly lethal to cancer cells. The central theme of ICB immunotherapy lies in the generation or reactivation of this population of cells (20). Antitumor immunity can be described briefly as antigen presentation cells (APCs), such as dendritic cells (DCs), internalize and process tumor-associated antigens (TAAs) in peripheral tissue; then, DCs migrate to lymph nodes and present tumor-peptide-major histocompatibility complexes to naïve CD8+ T cells (21). Meanwhile, mature DCs provide the second signal to naïve CD8+ T cells by upregulating CD80 and CD86. Upon these efficient stimulations, naïve CD8+ T cells differentiate into CTLs. Eventually, CTLs infiltrate lesion sites and kill cancer cells (22). Effective immunotherapy depends mainly on CD8+ T cells as well as their successful activation (23). Therefore, we focused on the immune response procedures, especially changes in the biological function of CD8+ T cells, for a deeper understanding of the mechanisms of immunotherapy resistance in ICB.
Drug resistance occurs in blocking the different phases of a cancer immunity cycle, from tumor-specific antigen recognition to presentation, from T-cell activation to recruitment. Overall, the mechanisms of resistance to ICB (Figure 1) can be summarized as the (1) failure of antigen recognition; (2) deficiency of antigen presentation; (3) poor CD8+ T-cell infiltration; (4) inhibited activity of CD8+ T cells; (5) exhaustion of CD8+ T cells; and (6) insensitivity to CTL mediated killing.
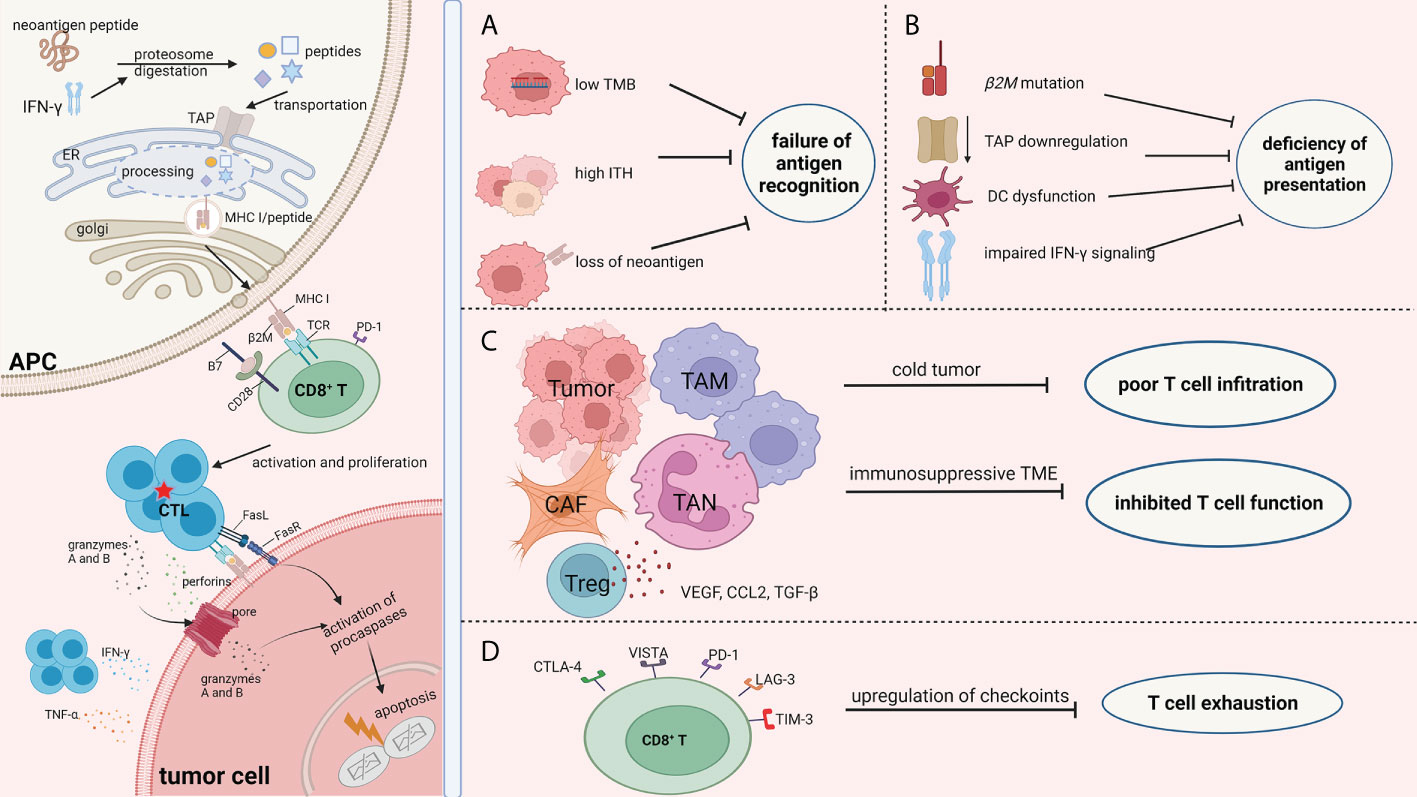
Figure 1 Mechanisms of ICB resistance from the perspective of immune response process. The success of ICB immunotherapy lies in the generation and/or reactivation of the population of CTL cells, which are also the central theme of immunotherapy. The left part of the picture depicts the normal immune response procedure which involves antigen processing and presentation, CD8+T cell priming, and the efficient killing of tumor cells by CTLs. Failure of immunotherapy occurs when the different phases of the cancer immunity cycle are compromised and blocked. There are numerous factors that decrease the effect of the antitumor immunity during the fight between tumor cells and immune cells. Regardless of the complexity of the immunotherapy resistance mechanisms, the consequence of these factors can be summarized as (A) failure of antigen recognition; (B) deficiency of antigen presentation; (C) poor CD8+ T cells infiltration and inhibited activity of CD8+ T cells; and (D) exhaustion of CD8+ T cells. Therefore, we focused on the immune response procedures, especially changes in biological function of CD8+T cells, with an aim to better understand the resistance mechanisms of ICB. The picture was created with BioRender.com. APC, antigen presentation cell; TAP, transporters associated with neoantigen presentation; ER, endoplasmic reticulum; MHC I, major histocompatibility complex class I; TCR, T cell receptor; CTL, cytotoxic T lymphocytes; TMB, tumor mutation burden; ITH, intra-tumor heterogeneity; DC, dendritic cell; TAM, tumor associated macrophages; CAF, cancer associated fibroblasts; TAN, tumor associated neutrophil; CTLA-4, cytotoxic T-lymphocyte antigen 4; VISTA, V-domain Ig suppressor of T cell activation; LAG-3, lymphocyte activation gene‐3; PD-1, programmed cell death protein -1; TIM-3, T-cell immunoglobulin mucin-3.
The immune recognition of tumor cells depends on the HLA-presented antigenic peptide. During cancer progression, gene mutation occurs within cancer cells, resulting in the accumulation of mutated peptides. These neo-peptides are also termed neoantigens because they are different from self-antigens and can be immunogenic most of the time (24). Thus, increased expression of neoantigens within the tumor site can enhance antitumor immunity.
The concept of tumor mutation burden (TMB) has been introduced and utilized as a critical indicator to define tumor antigenicity and evaluate the clinical response to ICB (25). A considerable positive correlation was observed between TMB and the objective remission rate, with a correlation coefficient of 0.7 (26). Non-small lung cancer and melanoma have shown higher TMB and a better response to PD-1 inhibition. Conversely, sarcoma, prostate cancer, and ovarian cancer display lower TMB as well as primary resistance to PD-1inhibition (26, 27). Patients with high TMB (defined as “greater than or equal to 10mut/mb”) were shown to have dramatically higher objective remission rates when treated with pembrolizumab (29%) than patients with low TMB treated with pembrolizumab (6%) in a clinical trial (NCT02628067) (28). On the other hand, tumors with microsatellite instability (MSI) phenotypes, or those with genetic defects in DNA repair enzymes, which is also called DNA mismatch repair deficiency (dMMR), display high mutation loads and more significant response to checkpoint inhibition immunotherapy (29). TMB alone is not a specific determinant of treatment efficacy. Differences in analytical methods, such as different sequencing coverage and depth, lead to differences in sensitivity and specificity when estimating TMB (30, 31). In fact, the durable efficacy of pembrolizumab was still obtained in patients with malignant rhabdoid tumors whose TMB was very low (31). Although high TMB plays a significant role in tumor response to ICB, the prediction of ICB response is far more than TMB estimation.
High intratumor heterogeneity (ITH) can also result in the ineffective recognition of tumor-specific neoantigen and decrease T-cell response to different subclones of tumor cells (32). Pan-cancer analysis indicated that a higher ITH level of tumors was associated with worse survival (33). Wolff et al. demonstrated that low intratumor heterogeneity was a prognosticator of overall survival (OS; p = 0.046) but not TMB (p = 0.16), which suggested that tumors with high ITH were able to escape the immune system despite having high neoantigens (34). McGranahan et al. studied the impact of neoantigen load and neoantigen intratumor heterogeneity on OS in patients who were diagnosed with lung adenocarcinoma (LUAD) and lung squamous cell carcinoma (LUSC). No significant correlation between neoantigen load and neoantigen intratumor heterogeneity with OS in LUSC was discovered, even though the neoantigen burden of LUSC was equally high as LUAD, suggesting the importance of ITH (35).
The loss of neoantigens disturbs the recognition of tumor cells by T cells and causes resistance to ICB. Anagnostou et al. analyzed the data of NSCLC patients who developed required drug resistance after initial response. They discovered 7–18 assumed neoantigens in the resistant tumors. The mechanism of neoantigen loss lies in the deletion of chromosomal regions and the abolition of tumor subclones. The loss of neoantigens was correlated with changes in T-cell receptor clonality (36).
In summary, low TMB and/or high ITH, as well as neoantigen loss, can impact the antigen recognition by CTLs, causing primary or secondary drug resistance to ICBs. In general, tumors with elevated neoantigen expression at the onset of malignant cell cloning will respond better to ICB (37).
The activation of CD8+ T cells depends on the combination of the T-cell receptor (TCR) and major histocompatibility complex class I (MHC I) molecules (38). MHC I molecule–related neoantigen presentation is modulated by multiple proteins. Beta-2 microglobulin (β2M) is responsible for stabilizing MHC I molecules and promoting antigenic peptide loading (39). The mutations of β2M have been found in patients who have acquired resistance to ICBs. For example, in relapse melanoma patients with acquired resistance to pembrolizumab, it was found that a truncating mutation of β2M exists in biopsy analysis, leading to the loss of MHC I molecule expression (40). Point mutation, deletion, and the loss of heterozygosity (LOH) were also detected in metastatic melanoma tissues. The degree of β2M LOH was tripled in non-responders (approximately 30%) when compared with responders (approximately 10%) and was correlated with inferior OS (41). Apart from melanoma, the links between β2M alteration and acquired resistance have been reported in lung cancer (42), gastrointestinal adenocarcinoma (43), and colorectal cancer with a microsatellite instability–high (MSI-H) phenotype (44).
Reduced human leukocyte antigen (HLA) class I gene expression may lead to decreased antigen presentation, thus promoting immune evasion (45). There are up to six different HLA class I alleles in the genome. Highly polymorphic HLA class I genes, including HLA-A, HLA-B, and HLA-C, are responsible for encoding MHC I molecules (46). Eric et al. presented that resistance to KRAS G12D–specific T cell transfer therapy occurred in a patient with metastatic colorectal carcinoma after 9 months. The mechanism of this immunotherapy resistance lies in the deletion of chromosome HLA-C*08:02 in the resistant lesions. Since the existence of the HLA-C*08:02 allele was necessary for KRAS G12D neoantigen presentation and recognition by T cells, its loss directly caused immune evasion (47).
Transporters associated with neoantigen presentation (TAP) are critical players in the MHC I antigen presentation pathway. TAP is a heterodimer consisting of TAP1 and TAP2, both of which are required for peptide translocation (48). The loss or downregulation of TAP in cancers may result in immune evasion and is often associated with an unfavorable prognosis (49, 50). Zhang et al. reported that TAP deficiency resulted in resistance to anti-PD-1, while the efficacy was enhanced in patients lacking both TAP and the non-classical MHC I molecule Qa-1b. The results suggested that the immune microenvironment can be altered by inhibiting Qa-1b, especially in the case of defective antigen processing (51). The accumulation of presentation defects may, in turn, lead to a reduced recognition of malignant cells by tumor-specific T cells.
The interruption of IFN-γ signaling, which facilitates MHC I molecule expression on the cell surface in normal conditions, influences neoantigen presentation. Specifically, IFN-γ is an essential signaling molecule for immune-proteasome formation during the degradation of intracellular proteins (52). The loss of IFN-γ signal causes reduced antigen presentation through compromising the coordinated upregulation of the antigen processing procedure (53). Decreased expression of elements in the MHC I antigen presentation pathway can usually be reversed by IFN-γ treatment (53, 54).
The dysfunction of DCs, the most potent antigen-presenting cells, plays a critical role in ICB resistance (55). The deletion of atypical chemokine receptor 4 (ACKR4) in colorectal tumor cells but not stromal cells inhibited the migration of DCs to tumor-draining lymph nodes and impaired antigen presentation. In addition, the knockdown of ACKR4 reduced tumor cells’ sensitivity to ICB (56). High enrichment of myeloid dendritic cells in lung cancer tissues shows an immune activation state, and those patients may benefit from ICB treatment (57). Cytotoxic T-lymphocyte antigen 4 (CTLA4) has a higher affinity to CD80/86 than CD28. CTLA4-positive Treg cells impair the maturation of DCs by binding to CD80/86 and inhibit costimulatory signals (58). Antigen presentation by immature DC or CD80/86 low-expressed DC was unable to stimulate CD8+ T cells potently, resulting in CD8+ T cells being anergic with low proliferation and insufficient to produce cytokines (59).
Different tumor types exhibit various tumor-associated T-cell infiltration densities. The immune landscape of tumors can be divided into three types (1): hot tumor. It is characterized by the enrichment of T cells and their infiltration into tumor tissues, such as lung cancer and melanoma (60) (2). Cold tumor, such as prostate cancer (61) and brain cancer (62), features fewer T cells in the tumor parenchyma or stroma (63). (3) “Immune excluded” tumor. Immune cells do not infiltrate the parenchyma of these tumors, even though there is an abundance of immune cells (64). Compared to hot tumors, the latter two phenotypes rarely respond to ICB immunotherapy, which results in primary drug resistance (65). The infiltration of CD8+ T cells into the tumor tissues can be considered a good prognostic parameter for lung cancer and is associated with lymphocyte motility (66).
Genetic alterations within tumor cells have unfavorable effects on T-cell infiltration. PTEN loss was associated with reduced T-cell density, lower T-cell expansion, and poor response to PD-1 inhibited therapy in melanoma. Mechanically, the absence of PTEN in tumor cells enhances the level of immunosuppressive cytokines, including CCL2 and VEGF, causing less T-cell infiltration and inhibiting autophagy as well, thereby impairing CTL-mediated cell killing (67). BRAF mutations are common in melanoma (50%) (68), thyroid papillary cancers (approximately 35%) (69), and colorectal cancers (5%–10%) (70). The biopsy analysis of metastatic melanoma patients showed that selectively inhibiting BRAF with PLX4720 or GSK2118436 induced abundant CD8+ T cells in tumors, which provided powerful support for combining BRAF inhibitors with immunotherapy (71). Skoulidis and colleagues showed that STK11/LKB1 mutation is associated with less expression of PD-L1 and decreased infiltrative CTL density, resulting in primary resistance to PD-1-based immunotherapies in both human and murine STK11/LKB1-deficient lung adenocarcinoma (72). Additionally, the loss of TET2, which encodes ten-eleven translocation (TET) DNA dioxygenase, is correlated with reduced Th1-type chemokine generation, including CXCL9, CXCL10, and CXCL11, with the downregulated expression of PD-L1 and impaired T-cell attraction to tumor tissues, leading to immune escape and resistance to anti-PD-L1 therapy in the B16-OVA melanoma tumor model (73). NSCLC patients with EGFR mutations demonstrated an inadequate response to anti-PD-1 therapy than those with the EGFR wild type. EGFR mutation is associated with a reduction in PD-L1 expression, a deficiency in T-cell infiltration, and a decrease in TMB (74).
The elevated vascular endothelial growth factor (VEGF) within the tumor and the consequent aberrant vascular system with high interstitial pressure impair the recruitment of immune cells, correlated with decreased penetration of immune checkpoint inhibitors and increased drug resistance. VEGF inhibits T lymphocyte infiltration within the tumor microenvironment (TME) by suppressing NF-κB signals (75). Tumor-intrinsic STING signaling facilitates BRCA-1 mutated ovarian cancer cells’ resistance to both PD-L1 and CTLA-4 therapies by upregulating VEGF-A (76). In addition to VEGF, increased C-C motif chemokine ligand 2 (CCL2) was found to be correlated with primary resistance to ICB. CCL2 contributes to insensitivity to ICB by recruiting monocytes and reducing CD8+ T-cell infiltration in pancreatic tumors. The poor efficacy of anti-PD-1 therapy can be reversed by CCL2 inhibition or monocyte neutralization (77). Meanwhile, transforming growth factor-beta (TGF-β) produced by cancer-associated fibroblasts (CAFs) was capable of preventing T cells from entering tumor tissue (78). The results from the transcriptional analysis of 298 metastatic urothelial carcinoma samples suggested that the enhanced TGF-β in CAFs was related to poor CD8+ T-cell infiltration within tumor parenchyma and weak response to atezolizumab (79). Aside from CAFs, tumor-associated macrophages (TAMs) play an essential role in excluding T-cell infiltration from tumor sites. Interactions between CD8+ T cells and TAMs are durable (at least 20 min), resulting in slowed CD8+ T-cell motility (66).
The TME is infiltrated by diverse innate and adaptive immune cells. The complex crosstalk between immune cells and tumor cells determines the immune status and the implementation of T-cell function, thus facilitating or inhibiting the tumor response to ICB (Figure 2). With the progression of tumors, the TME becomes progressively immunosuppressive. Immunosuppressive cells as well as their products facilitate tumor immune evasion and inevitable resistance to checkpoint inhibitors.
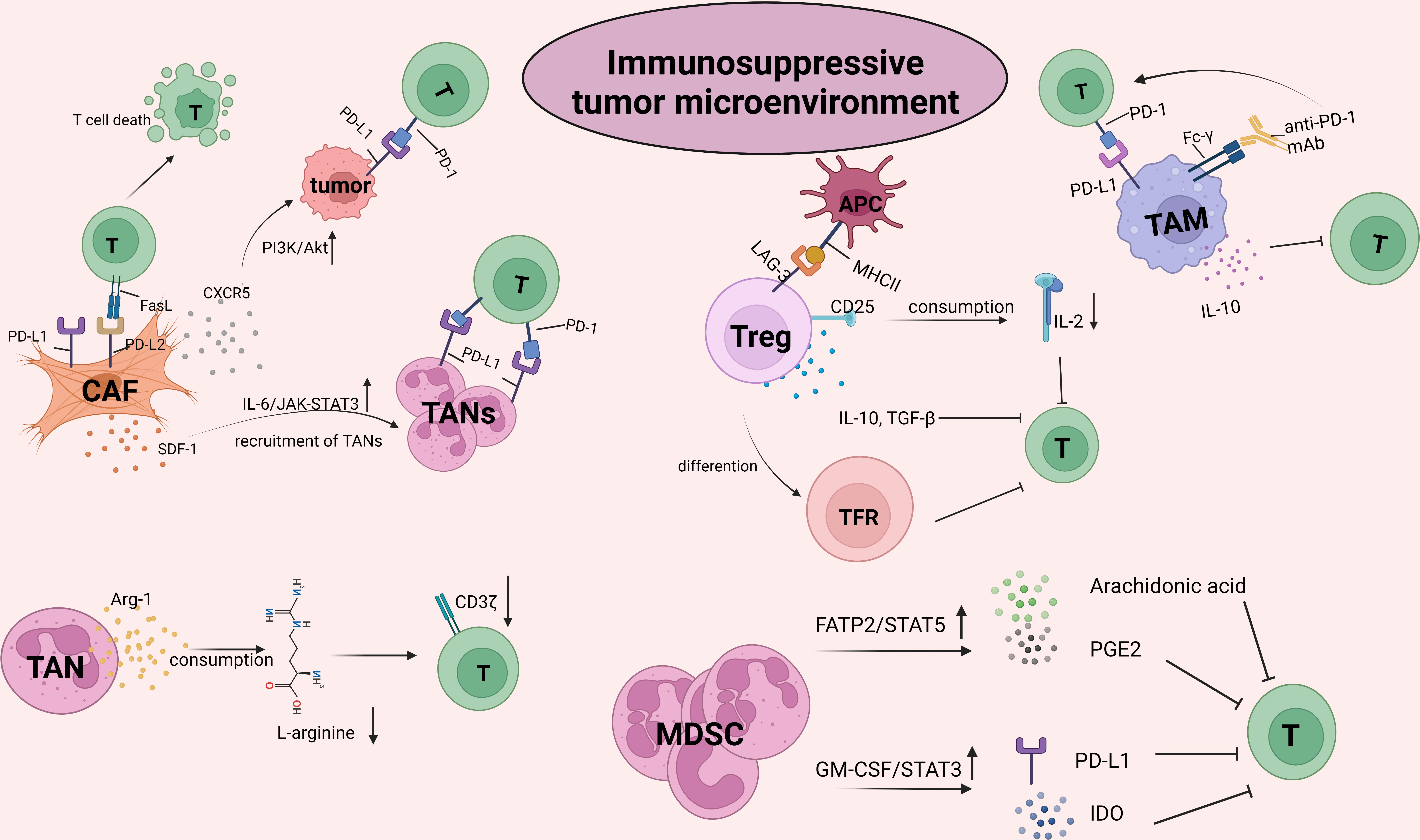
Figure 2 The crosstalk between CD8+T cells and the other suppressive cells within tumor microenvironment (TME). TME is infiltrated by different types of innate and adaptive immune cells. The complex crosstalk between these immune cells and tumor cells determines the immune status and the implementation of T cell function, thus to facilitate or inhibit the tumor response to ICBs. With the progression of malignant cells, immune cells within TME, for example, macrophages and neutrophils, are educated into pro-tumor cells. As such, TME becomes progressively immunosuppressive. Immunosuppressive cells inhibit the activity of T cells by upregulating immune checkpoints, capturing anti-PD-1 antibodies and secreting pro-tumor soluble factors such as arg-1, IL-10, TGF-β, promoting tumor immune evasion and resulting in resistance to checkpoint inhibitors. The picture was created with BioRender.com. CAF, cancer associated fibroblasts; TAN, Tumor associated neutrophil; TAM, Tumor associated macrophage; MDSC, myeloid-derived suppressor cell; PGE2, prostaglandin E2; GM-CSF, granulocyte-macrophages colony-stimulating factor; IDO, indoleamine 2,3-dioxygenase; TFR, follicle-regulating T cell.
Tumor-associated neutrophils (TANs) are one of the critical characteristics of ICB resistance. Immunosuppressive neutrophils from blood and tumors are commonly named granulocyte–myeloid-derived suppressor cells (G-MDSCs) or polymorphonuclear MDSC (PMN-MDSC) (80). Neutrophil-enriched breast tumors display a required resistance to ICB, suggesting a direct suppressive effect on CTLs mediated by TANs (81). In colorectal cancer, the non-response group shows increased levels of MDSC infiltration than the response group treated with anti-PD-1 (82). Consistent with this, a smaller amount of MDSC was found to be linked with a more robust response to ipilimumab in melanoma patients (83). TANs can attenuate the activity of CD8+ T cells by secreting various mediators. One of the essential pathways participating in the immunosuppressive activity of MDSCs is STAT-1-dependent signaling. IFNγ-mediated signals generated by activated T cells can stimulate STAT-1, which subsequently induces the increased expression of immunosuppressive cytokines in MDSCs, such as arginase 1 (Arg-1) (84). Arg-1 results in the downregulation of the CD3ζ chain of T cells by L-arginine exhaustion, suppressing T-cell proliferation and function (85). In addition, the overexpression of fatty acid transporter protein 2 (FATP2) mediated by STAT5 signaling was associated with the enhanced uptake of arachidonic acid and the release of prostaglandin E2 (PGE2) in MDSCs (86). The interaction between tumor cells and MDSCs also plays a critical role in modulating the function of MDSCs. It is reported that MC38 cells secrete the granulocyte–macrophage colony-stimulating factor (GM-CSF) that binds with GM-CSF-R on MDSCs. The combination activates the STAT3 signal within MDSCs, which increases the immunosuppressive effect of MDSC by upregulating indoleamine 2,3-dioxygenase (IDO) and PD-L1, as well as FATP2 (87, 88). The combination of ICB and FATP2 inhibitors delays tumor progression and decreases the expression of PD-L1 on CD8+ T cells (86, 88).
TAMs also significantly contribute to ICB resistance by inducing immunosuppressive interactions within the TME. Notably, TAMs are one of the most enriched immune cells in TME and are involved in both immune stimulation and immunosuppression (89). There are two distinct functional groups of the TAM population, M1 cells (the antitumor macrophages) and M2 cells (the pro-tumor macrophages) (90). Phenotypes can be reversed dynamically between M1 and M2 mediated by cytokines and signals, which is called polarization (91). Firstly, TAMs attenuate T-cell activity by capturing ICB antibodies (mainly of the IgG1 subclass) through Fc-γ receptors, leading to ICB resistance. By using an in vivo image to monitor the activity of anti-PD-1 in real time, Arlauckas et al. proved that the anti-PD-1 monoclonal antibody (mAbs) could efficiently bind PD-1+ tumor-infiltrating CD8+ T cells initially after treatment. Nevertheless, this combination is transient because anti-PD-1 monoclonal antibody are removed by PD-1- TAMs from the T-cell surface within minutes. Measures to block Fc/FcγR binding inhibit the transfer of anti-PD-1 mAbs from CD8+ T cells to macrophages in vivo, thereby strengthening the therapeutic effect of anti-PD-1 (92). Secondly, TAM reduces ICI efficacy by directly impeding the antitumor capacity of CD8+ T cells. It was found that TAMs directly or indirectly suppress CD8+ T cells by secreting IL-10 (93). IL-10 inhibits CD8+ T cells primarily by increasing N-glycan branching, thus upregulating the antigenic threshold needed for T-cell activation (94). Thirdly, TAM suppresses T-cell activity by expressing alternative immune checkpoints against ICI efficacy. On one hand, the majority of PD-L1+ TAMs are M2 cells, constituting the major TAM population in advanced tumors (95). Thus, high expression of the inhibitory checkpoint on TAMs is inherently a crucial immunosuppressive factor in the TME. On the other hand, PD-L1 expression on TAMs plays a regulatory role during the interplay of TAMs presenting antigenic peptides to homologous effector T cells, which may restrict T-cell superactivation (96).
Under normal conditions, fibroblasts have a low proliferative capacity and metabolic state and are present in a relatively quiescent state in most tissues (97). However, within the TME, tumor cells can promote fibroblast activation by secreting growth factors such as TGFβ, platelet-derived growth factor (PDGF), and fibroblast growth factor (FGF) (98, 99). The CAF-mediated inhibition of T-cell cytotoxic function can be achieved by the upregulation of immune checkpoint molecules. CAFs from melanoma patient biopsies showed the elevated expression of PD-L1 and PD-L2, which directly abrogated CD8+ T-cell function (100). It is suggested that enhanced expression of PD-L2 in CAFs results in antigen-specific T-cell death through PD-L2 and Fas ligand engagement, protecting tumor cells from immune destruction (101). Interestingly, some CAFs also participate in antigen presentation and thus can directly kill activated CD8+ T cells via the involvement of PD-L2 and Fas ligands (101). PD-L1 and PD-L2 were simultaneously upregulated in CAFs in pancreatic cancer patients. Meanwhile, the CAFs facilitate inhibitory immune checkpoint receptor expression in proliferating T cells. However, the underlying mechanism is not fully understood (102). Apart from upregulating the immune checkpoint directly, CAFs can also indirectly increase the level of immune checkpoint molecules on malignant cells and other cells within the TME. Hepatocellular carcinoma– derived CAFs were demonstrated to recruit neutrophils by secreting SDF1a and facilitating neutrophils’ activation via IL-6-JAK-STAT3 signaling. Then, the activated neutrophils upregulated the expression of PD-L1 and exerted a suppressive effect on T-cell immunity (103). CAF-derived CXCL5 is a potent cytokine, which mediates the upregulation of PD-L1 in a PI3K/AKT-dependent pathway within tumor cell lines, including B16, CT26, A375, and HCT116 (104). As such, it is essential to notice that the CAF-mediated dysfunction of CD8+ T cells is not limited to a direct interplay of these two cell types.
Regulatory T lymphocytes (Tregs) are of vital importance in tumor progression and their resistance to immunotherapy. Increased infiltration of Tregs has been generally perceived as a biomarker of poor clinical outcomes such as high death hazards and decreased survival (105, 106). Tregs were initially identified as CD4+ T cells with increased expression of CD25 (α chain for the IL-2 receptor). FoxP3 was then characterized as a specific marker and major regulator for the maintenance of the immunosuppressive functions of Treg cells (107, 108). Once activated, T cells begin to produce IL-2, which is essential for the sustained proliferation and activation of T cells (109). CD25 has a high affinity to IL-2. Tregs consume IL-2 by upregulating CD25, limiting the sustained activation and proliferation of effector T cells (110). Ren et al. reported that impaired T-cell immunity caused by IL-2 signaling obstruction could be restored by using a low-affinity IL-2 conjugated with anti-PD-1 (PD-1-laIL-2). PD-1-laIL-2, with a higher affinity to PD-1+CD8+ T cells than to peripheral Treg cells, was able to amplify the dysfunctional tumor-specific CD8+ T cells potently, thus overcoming tumor resistance to ICB (111). Moreover, Tregs suppress T-cell activity by upregulating the expression level of immune checkpoints. Activated Tregs can express lymphocyte activation gene‐3 (LAG-3). CD4+CD25highFoxp3+LAG-3+ T cells possess robust inhibitory activity by releasing cytokines, including IL-10 and TGF-β1, without IL-2 (112). It has been proven that Tregs can differentiate into follicle-regulating T (TFR) cells with PD-1 expression, which inhibit the germinal center response (113). TFR cells are distinguished by the coexpression of CXCR5 and GITR2,5 or the transcription factors FOXP3 and BCL-6 (114, 115). TFR cells show advantageous suppressive capacity and in vivo persistence compared to conventional regulatory T cells, reducing the effect of an-PD-1 (116). Interestingly, Zappasodi et al. explored the role of a non-conventional subset of CD4+FOXP3-PD-1high T cells and found that this population of cells expresses a TFR-like phenotype and could limit the functions of the T-cell effector. However, in contrast to regulatory T cells, CD4+FOXP3-PD-1high T cells were helpful for B-cell activation (117).
T-cell exhaustion is characterized by an impaired tumor cell–killing function, the persistent and upregulated expression of inhibitory receptors, and the diverse transcriptional states of normal effector T cells or memory T cells. It is a status of T-cell dysfunction (118). Increased expression of immune checkpoints was reported to be associated with acquired resistance to ICB. Ntrk1 has been proven to induce the upregulation of PD-L1 in mesenchymal Kras/p53 mutant lung cancer cells by stimulating Jak/Stat signaling, leading to the exhaustion of CD8+ T cells within the TME (119). Enhanced expression of T-cell immunoglobulin mucin-3 (Tim-3) was observed in lung cancer patients who progressed after initially responding to anti-PD-1 therapy (120). The coexpression of PD-1 and Tim-3 in T cells was linked with an exhausted phenotype in head and neck squamous cell carcinoma (HNSCC) patients. Mechanically, the upregulated expression level of Tim-3 in T lymphocytes is dependent on the activation of the PI3K/Akt signaling pathway (121). Several checkpoints were coexpressed in TILs isolated from an ovarian tumor mouse model, including PD-1, CTLA-4, and lymphocyte activation gene-3 (LAG-3). The efficacy of single-agent blockade can be impaired by the compensatory enhancement of the other checkpoint molecules, resulting in poor response and resistance (122). With early PD-1 expression and late LAG-3/B- and T-cell lymphocyte attenuator (BTLA) expression, T cells gradually acquire the coexpression of these checkpoint receptors (123). The V-domain Ig suppressor of T-cell activation (VISTA) is another checkpoint of T cells. In melanoma patients with the initial response to anti-PD-1, the density of VISTA-positive T cells was significantly upregulated after treatment, which led to disease progression (124). Increased expression of these inhibitory coreceptors is associated with TCR signaling dysfunction and represents the initiation of negative regulatory signaling, leading to T-cell exhaustion and dysfunction (125). However, exhaustion does not mean the end of T cells’ fate, and their function can be restored by blocking those overexpressed signals mentioned above.
It is a consensus that CTLs kill tumor cells through two major pathways: granzymes A and B–mediated granule exocytosis and Fas/FasL conjugation–mediated apoptosis induction. Moreover, activated CTLs also secrete cytotoxic cytokines, including interferon-γ (IFN-γ) and tumor necrosis factor-α (TNF-α), to elicit cytotoxicity in tumor cells (126). From this perspective, the sensitive response of tumor cells to cytotoxic factors released by CTLs is vital in preventing immune evasion (127). On one hand, IFN-γ is quite essential for T cells’ penetration into tumors. The effects of antigen-specific immunotherapy depend, to some extent, on tumor sensitivity to IFN-γ (128). The IFN-γ receptor (IFNGR) consists of two subunits, IFNGR1 and IFNGR2. The binding of IFN-γ to its receptor results in the activation of JAK1 and JAK2, which subsequently phosphorylates and dimerizes transcription factor STAT1. STAT1 homodimers then enter the nucleus, binding to specific promoters and initiating the transcription of IFN-γ-regulated genes (129). On the other hand, the release of IFN-γ also mediates the expression of PD-L1 and MHC class I molecules, which may be beneficial for anti-PD-L1 therapy (130).
The dysfunction of the IFN-γ signaling pathway was associated with the primary resistance to ipilimumab therapy in melanoma patients (131). The mutation of JAK1/JAK2 results in PD-L1 depletion and insensitivity to IFN-γ, ultimately causing the primary resistance to anti-PD-1 treatment in melanoma and colorectal cancer patients (40, 132). The depletion of the IFNGR1 gene in B16 tumor cells suppressed IFN-γ mediated apoptosis and decreased the antitumor effects of anti-CTLA-4 therapy in a mouse model (131). However, the impact of additional IFN-γ pathway genomic alterations other than JAK1 and JAK2 on acquired drug resistance to ICB needs to be further investigated. Of note, the correlations between TNF mutations and survival were not discovered in any type of cancer by Cancer Genome Atlas (TCGA) analysis, indicating that although TNF acts as another cytotoxic factor, its effect is not as sufficient as IFN-γ (133).
In accordance with the aforementioned proposed biological mechanisms of non-response to ICB, studies on potential therapeutic strategies addressing resistance mechanisms would be ideal for providing specific insights to improve clinical outcomes. Basically, strategies to reverse ICB tolerance are currently being explored (Table 1), which can be outlined as (1) releasing tumor antigens; (2) enhancing antigen presentation; (3) promoting T-cell infiltration; (4) reversing T-cell exhaustion; and (5) CD8+ T-cell stimulation.
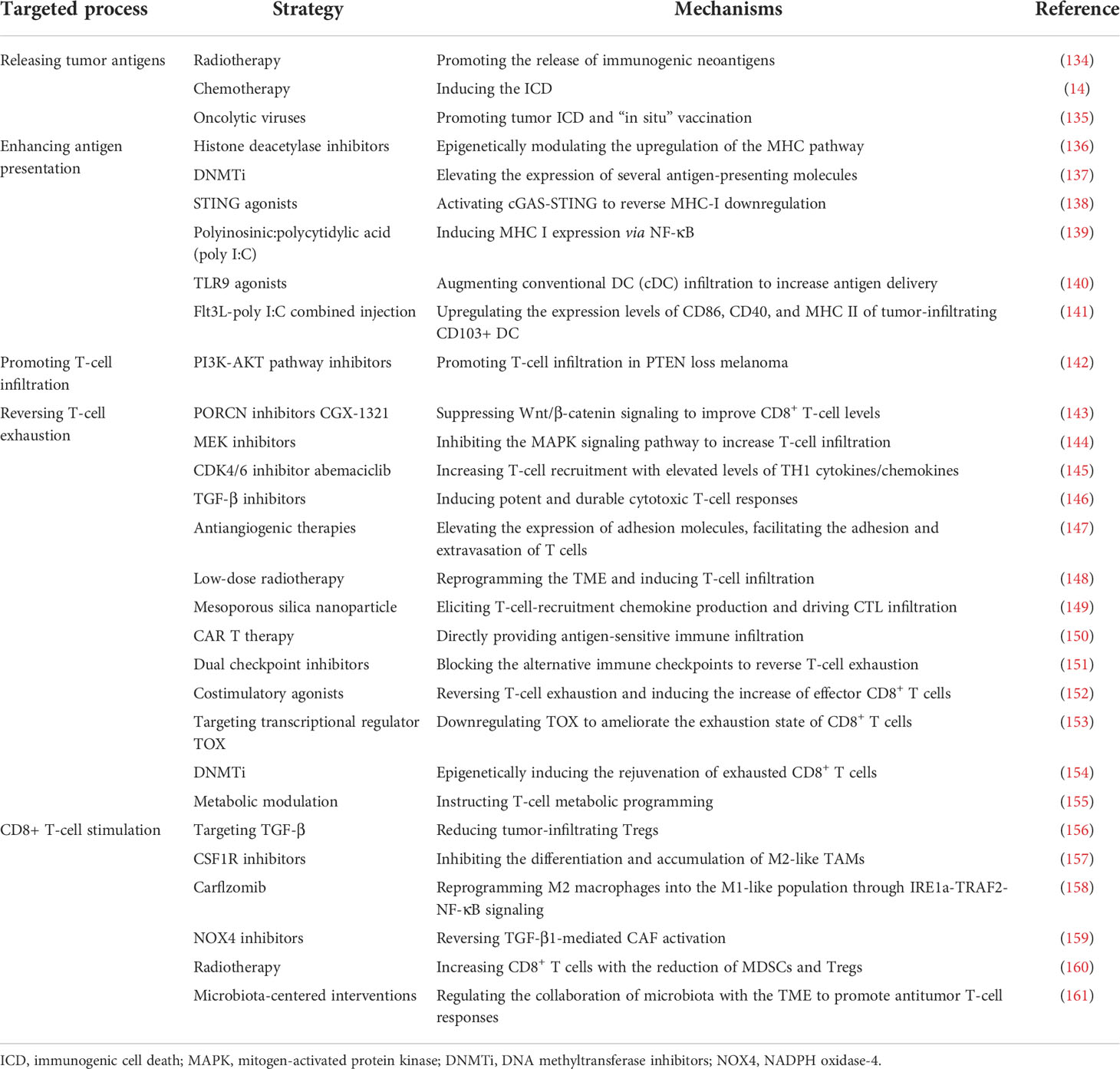
Table 1 Potential combination strategies to improve the antitumor effect of programmed cell death protein 1 (PD-1)/programmed cell death ligand 1 blockade.
Low TMB and weak or unresponsive neoantigens contribute to the failure of antigen recognition, resulting in ICB resistance. Thus, elevating the release of tumor antigens appears to be a potentially effective approach to reversing ICB resistance (Figure 3).
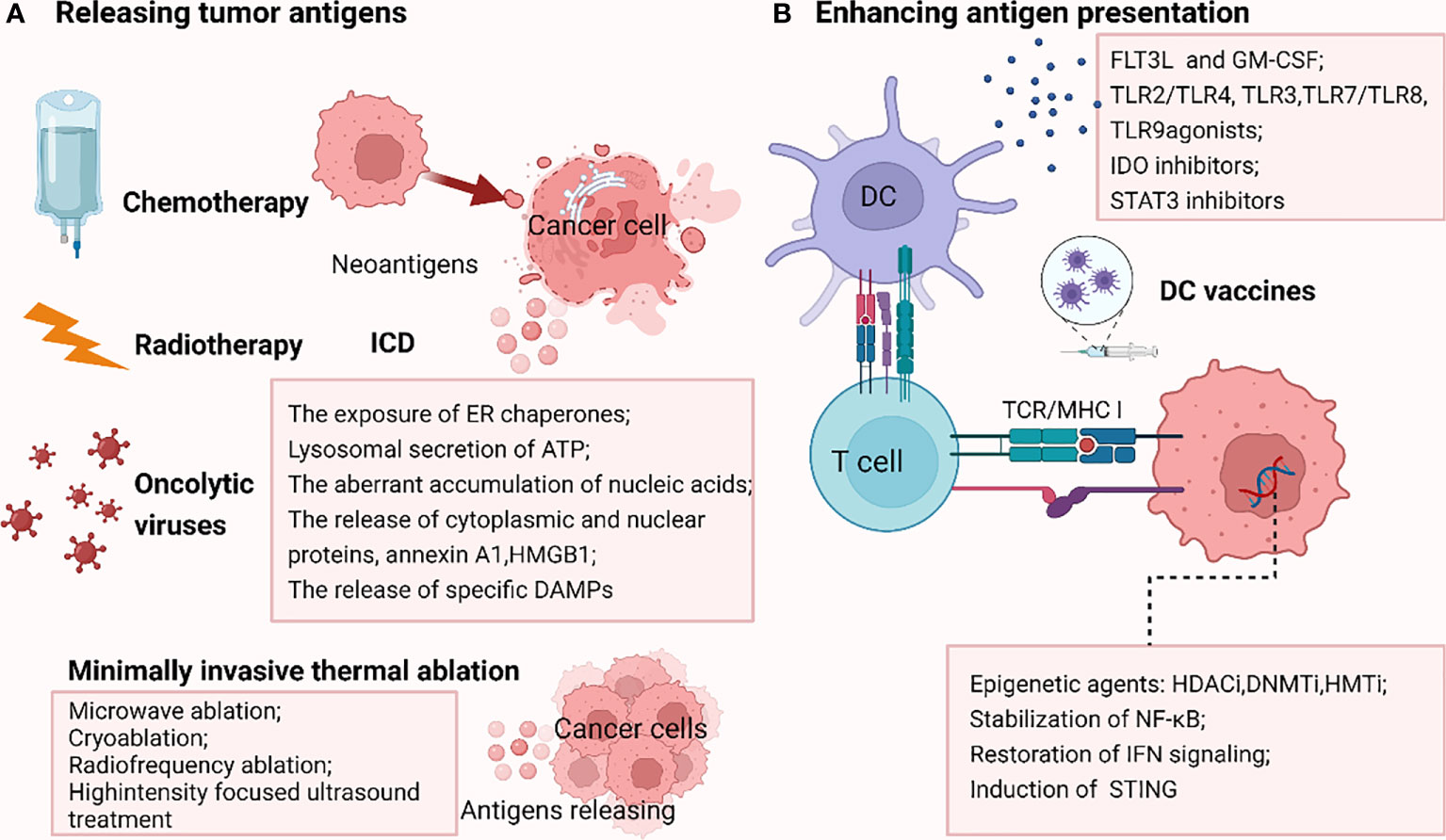
Figure 3 Strategies reversing PD-1/PDL1 blockade by releasing tumor antigens (A) and enhancing antigen presentation (B). A.Chemotherapy, radiotherapy and oncolytic viruses could promote the immunogenic cell death (ICD), enhancing the liberation of immunogenic neoantigens, thus increasing the antigenicity in tumors resistant to ICB due to the failure of antigen recognition. In addition, some minimally invasive thermal ablation treatments lead to antigens release as well. (B) DNMTi, HDACi, HMTi epigenetically modulate the upregulation of MHC pathway. Stabilization of NF-κB, restoration of IFN signaling and induction of stimulator of interferon genes (STING) also reverse MHC-I downregulation. Besides, stimulation factors including cytokines such as FLT3L (FMS-like tyrosine kinase 3 ligand) and GM-CSF (granulocyte–macrophage colony-stimulating factor), Toll-like receptor (TLR2/TLR4, TLR3, TLR7/TLR8, TLR9) agonists, IDO inhibitors and STAT3 inhibitors could augment the infiltration, activation, and effector function of conventional DCs (cDCs), thus increasing antigen delivery. DC vaccines are also important tools boosting antigen presentation. The picture was created with BioRender.com. ICD, immunogenic cell death; STING, stimulator of interferon genes; FLT3L, FMS-like tyrosine kinase 3 ligand; GM-CSF, granulocyte–macrophage colony-stimulating factor; TLR, Toll-like receptor; IDO, indoleamine- (2,3)-dioxygenase; DC, dendritic cell.
Radiotherapy, as one of the most effective cytotoxic treatments, especially for localized solid cancers, has been considered to cause antitumor immune response apart from causing DNA damage to irradiated cancer cells (15). The abscopal response, originally described in 1953, referring to the shrinkage of tumors outside the irradiated area, has long been thought to involve the mechanisms of the immune response (162). Interestingly, this infrequently occurring abscopal effect could be strengthened by the addition of immunotherapy, which is, in turn, enhanced by radiotherapy (163). Increasing preclinical studies on radiotherapy combined with immunomodulators support the potential role of radiotherapy as an effective immune adjuvant (164). Mechanistically, radiation promotes the release of immunogenic neoantigens, known as TAAs, which play a vital role in in situ vaccination (134). Both in vitro and in vivo studies revealed that the irradiation effectively upregulates cancer testis antigens in the background of necrotic and apoptotic tumor cells and debris, followed with the promotion of the immunological recognition of the tumor (165).
Chemotherapy agents are the conventional treatment for various malignancies. As is known, cytotoxic chemotherapy primarily exerts an antitumor effect by blocking cell division (166). Apart from tumor debulking, chemotherapeutic agents have been demonstrated to promote immunogenic cell death (ICD), which is featured by the exposure of endoplasmic reticulum (ER) chaperones; lysosomal-secreting ATP; the aberrant accumulation of nucleic acids; the release of cytoplasmic and nuclear proteins such as high-mobility group box 1 (HMGB1), annexin A1; and the release of specific damage-associated molecular patterns (DAMPs) (14). Overall, this increasing antigenicity leads to on-target immunostimulatory effects in cancer (167). Recently, a bioresponsive doxorubicin (DOX)-based nanogel has been engineered to directionally release the loaded drugs after being internalized into the TME. These chemoimmunotherapies are promising to conquer the challenges of current ICB-based immunotherapy and provide a paradigm for developing immunomodulatory nanomedicines (168). Data from 12 NSCLC patients suggested that multiple non-mutated neoantigens released from cisplatin-induced apoptotic tumor cells elicited CD8+ or CD4+ Teff cell responses, which could notably be promoted by anti-PD-1 therapy, correlating with OS (167). Recent trial data on chemotherapy combined with PD-1/L1 inhibitors demonstrate the clinical benefit in patients with NSCLC, triple-negative breast cancer, gastric cancer, and HCC (166, 169, 170).
Oncolytic viruses (OVs) are another selective approach to promoting the release of antigens (171). Similarly, OVs induce tumor ICD and “in situ” vaccination. Subsequently, these soluble TAAs from dying tumor cells facilitate both innate and adaptive antitumor immune responses. Researchers found that in a model of disseminated lung cancer resistant to PD-1 immunotherapy, intratumoral virotherapy elicits CD8+ T-cell responses against a set of cancer-specific neoepitopes, overcoming systemic resistance to PD-1 immunotherapy (135). However, different OVs are not capable of inducing ICD equally (172). Thus, incorporating ICD-related DAMP genes seems to be a further attractive option to enhance immunogenicity. In this way, OVs function as engineering platforms for combination immunotherapy. Still, challenges exist in allowing OVs to arrive at the directed primary and metastatic tumor position to perform systematic therapeutic effects (173).
Hopefully, many novel strategies for promoting tumor antigen release are under study. Minimally invasive thermal ablation treatments such as microwave ablation, cryoablation, radiofrequency ablation, or highintensity focused ultrasound treatment are the common selective therapies for patients with inoperable tumors. Interestingly, these local applications of extreme temperatures lead to the release of antigens from the necrotic tumor lesion, enhancing the activation of the tumor-specific immune response. However, the effect of single thermal ablation is too limited, and appropriate immunomodulators are required for promoting an effective therapeutical systemic antitumor immune response (174–176). Recently, a novel tumor microenvironment ROS/GSH dual-responsive nanoplatform consisting of chemophotodynamic therapy and synergistical control-release PTX has been designed to induce the release of DAMPs after tumor cell pyroptosis, boosting the curative effect of anti-PD-1 treatment in a CT26 tumor model (177).
The deficiency of antigen presentation represents another major challenge in ICB therapy, which is caused by multiple factors as stated above, including MHC I defects, β2M/HLA gene loss, deficient IFN signaling, and dysfunctional DCs (178). Aiming at these abnormalities is a promising strategy to improve the responsiveness to ICB regimens (Figure 3).
The epigenetic control of immune resistance has been implicated as associated with an overall loss of antigen presentation via the loss of antigen expression or downregulation of MHC I (179). Histone deacetylases (HDACs) are one class of epigenetic regulators, comprising four families (class I, IIa, IIb, and IV). HDACs appear to have crucial roles in both innate and adaptive immune responses. HDAC1 and HDAC2 have been reported to negatively mediate antigen presentation by inhibiting the main transcriptional regulator of MHC class II genes (180). Accordingly, histone deacetylase inhibitors (HDACis) can epigenetically modulate the upregulation of the MHC pathway, facilitating the immune targeting of cancer cells (136). Four HDACis (e.g., romidepsin, belinostat, vorinostat, and panobinostat) have been approved by the FDA for lymphoma and/or multiple myeloma treatment. In both colon and ovarian cancer cell lines, HDACi treatment promoted increased antigen processing and antigen presentation (181). The efficacy of combining HDACi with PD-1 inhibitors has been evaluated in multiple preclinical cancer models, including melanoma, ovarian cancer, breast cancer, and lung cancer, showing great promise (136, 182, 183). Other epigenetic agents such as DNA methyltransferase inhibitors (DNMTis) as well as histone methyltransferase inhibitors (HMTis) have also been indicated to improve antigen presentation by elevating the expression of several antigen-presenting molecules, thus enhancing the recognition and activation of immune cells (137). Based on these exciting preclinical results, a combination of DNMTi or/and HDACi with ICB has undergone clinical trials in advanced colorectal cancer (NCT02512172), non-small cell lung cancer (NCT01928576, NCT00387465), head and neck cancer (NCT03019003), and gastrointestinal cancers (NCT03812796) (184).
Apart from the epigenetic modification of MHC I antigen presentation, targeting pathways associated with MHC I expression has been described to reverse MHC I downregulation and boost immunotherapy efficacy. Potential therapeutic strategies include the stabilization of NF-κB, restoration of IFN signaling, and induction of stimulator of interferon genes (STING) (138, 139). Notably, the effects of NF-κB and IFNs are pro- or antitumorigenic in different stages and types of tumors. Accordingly, both negative and positive regulators of NF-κB and IFNs have been reported to upregulate MHC I expression (185).
Several strategies to augment conventional DC (cDC) infiltration, activation, or effective function have been proposed to increase antigen delivery and enhance the efficacy of ICB. The stimulation factors include Toll-like receptor (TLR2/TLR4, TLR3, TLR7/TLR8, TLR9) agonists, IDO (indoleamine- (2, 3)-dioxygenase) inhibitors, and STAT3 inhibitor cytokines such as GM-CSF, and FLT3L (FMS-like tyrosine kinase 3 ligand) (186). For example, combining pembrolizumab with a synthetic CpG oligonucleotide TLR9 agonist, SD-101, exhibited greater clinical efficacy than PD-1 blockade alone in a phase Ib trial, which was associated with elevated tumor-infiltrating DC characteristics (140). Similarly, Flt3L-poly I:C combined injection significantly induced the upregulating expression levels of CD86, CD40, and MHC II of tumor-infiltrating CD103+ DC and promoted DC immunogenic function, eventually enhancing antitumor responses synergized with anti-PD-L1 Ab treatment in BRAF-mutant and B16 melanoma mouse models (141). Nanomaterials have recently been applied in facilitating the tumor antigen presentation of DCs. A cationic nanoscale metal–organic framework (nMOF) was designed to exert the effects of local immunogenic photodynamic therapy treatment and CpG stimulation, enhancing antigen presentation and synergizing with ICB to induce tumor regression in a breast cancer model (187). Moreover, “next-generation” DC vaccines, essential tools for anticancer therapy, have been suggested to be a desirable combinatorial counterpart for ICB, especially in tumors with low mutational burden (188).
As a robust prognostic biomarker, tumor-infiltrating lymphocytes are influenced by multiple mechanisms, including genetic alterations within tumor cells, aberrant vasculature, and elevated immunosuppressive factors like TGF-β (12, 146, 189, 190). Low lymphocyte infiltration mainly accounts for the limited efficacy of ICB in many tumors, especially in the immune-infiltrated and -excluded phenotypes (191). Hence, promoting T-cell infiltration via targeting these factors provides an outlook on the future for improving ICB effectiveness (Figure 4).
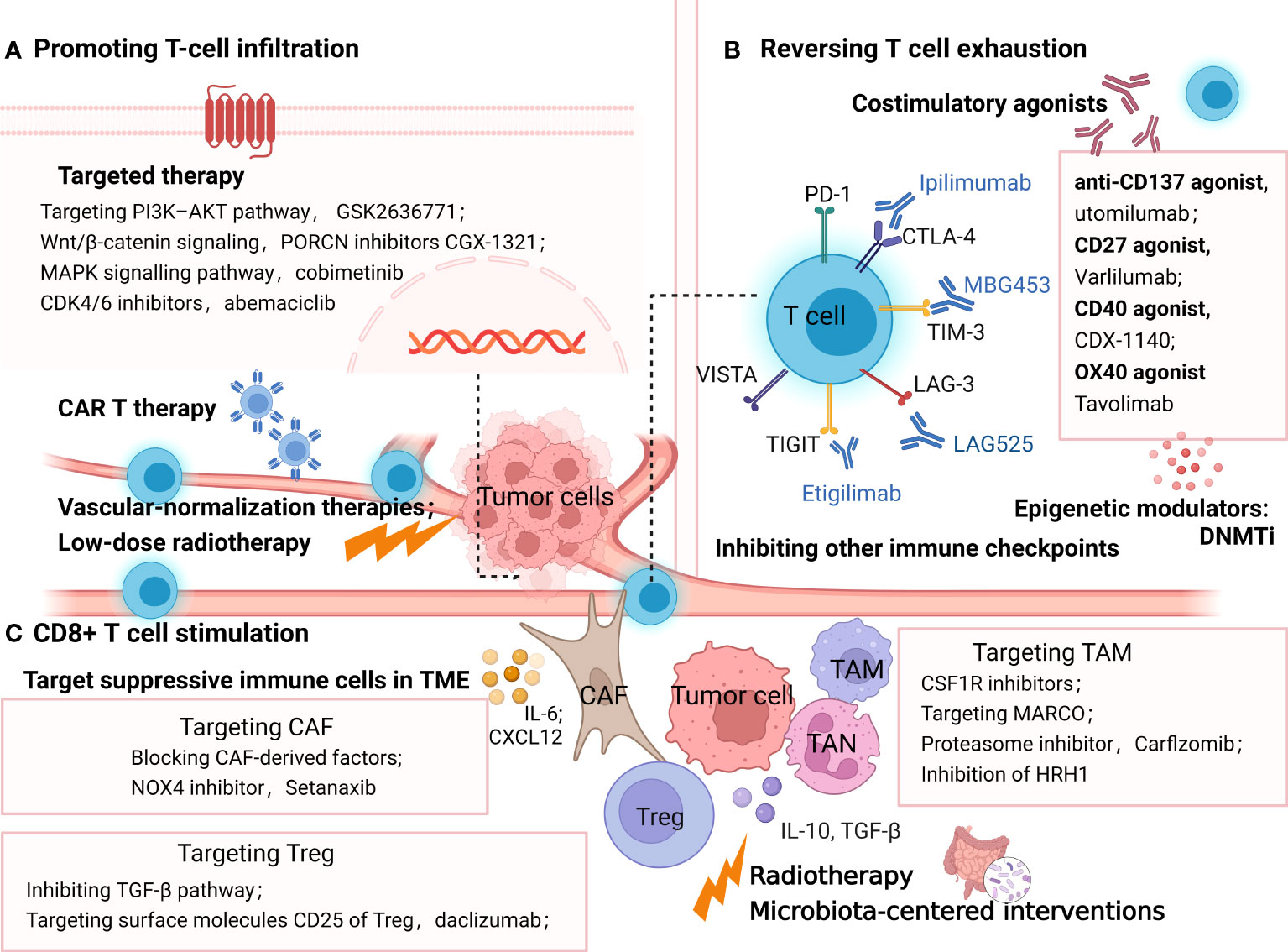
Figure 4 Strategies overcoming resistance to PD-1/PDL1 by promoting T-cell infiltration (A), reversing T cell exhaustion (B), and CD8+ T cell stimulation (C). (A) methods promoting T-cell infiltration include targeted therapy, vascular-normalization therapies, CAR T therapy and low-dose radiotherapy; (B) treatment options to reinvigorate of T cell exhaustion include blocking the alternative immune checkpoints, targeting co-stimulatory receptors, inhibiting soluble immune suppressive mediators and epigenetically coordinating exhausted CD8+ T (Tex) cells. (C) strategies targeting immune-suppressive cells in TME such as TAM, Treg and CAF to stimulate T cells. In addition, radiotherapy and microbiota-centered interventions also reprogram the immunosuppressive TME, promoting antitumor T-cell responses. The picture was created with BioRender.com. CAR, chimeric antigen receptor, Treg, regulatory T lymphocytes; DC, dendritic cell; TAM, tumor associated macrophages; CAF, cancer associated fibroblasts; MARCO, macrophage receptor with collagenous structure; HRH1, histamine and histamine receptor H1.
mRNA nanoparticles reactivating the tumor suppressor PTEN have been proven to significantly elicit antitumor immune responses and restore the therapeutic effect of ICB in PTEN-null prostate cancer and a PTEN-mutated melanoma model by promoting CD8+ T-cell infiltration (190). Furthermore, a drug candidate D18 could suppress the downregulation of PTEN expression by increasing KDM5A abundance, which also potentialized the efficacy of various ICBs in multiple tumor models (192). Moreover, targeting the PI3K-AKT pathway downstream of PTEN is a selective approach to elevate tumor-infiltrating T cells. For example, the PI3Kb inhibitor GSK2636771 sensitized PTEN-null melanomas to both CTLA-4 and PD-1 inhibitors and promoted T-cell infiltration to enhance the antitumor activity in vivo (142). Wnt/β-catenin signaling is another tumor-intrinsic pathway associated with poor spontaneous T-cell infiltration. Many inhibitors targeting WNT signaling have been developed to restore T-cell infiltration and reestablish anticancer immunity with ICB. In ovarian cancers, a typical “cold” immune phenotype, PORCN inhibitors CGX-1321 suppressing Wnt/β-catenin signaling, has been confirmed to improve CD8+ T-cell levels in the omentum TME (143). Other Wnt signaling inhibitors such as the anti-FZD7 antibody, β-catenin inhibitor DCR-BCAT, DKK1 inhibitor, and WNT inhibitor have been suggested to exert immunomodulatory effects as well (193). Furthermore, clinical trials combining Wnt inhibitor and ICB are ongoing, including DKN-01 (DKK1 antibody) plus pembrolizumab (NCT02013154) and PORCN inhibitor WNT974 combined with spartalizumab (NCT01351103) (194, 195).
The mitogen-activated protein kinase (MAPK) signaling pathway, another oncogenic signaling pathway associated with shaping tumor immunogenicity, has been proposed to be a promising target combined with ICB therapies (12). In a preclinical model of BRAF(V600)-mutated metastatic melanoma, antiPD1 therapy in combination with BRAF and MEK inhibitors contributed to complete tumor regression with increasing T-cell infiltration into tumors (144). Similarly, it has been reported in colon cancer (the CT26 model) that MEK inhibition promotes the accumulation of TIL by preventing the death of CD8+ T cells triggered by chronic TCR stimulation (196). Clinical studies of MAPK signaling inhibitors plus ICB have shown encouraging results. In BRAF V600–mutated melanoma patients, treatment with the combination of atezolizumab (anti-PD-L1) plus vemurafenib (BRAF inhibitor) + cobimetinib (MEK inhibitor) promoted 71.8% objective responses (a complete response rate of 20%). Meanwhile, the run-in of cobimetinib and vemurafenib contributed to the increase of circulating proliferating CD4+ T-helper cells (197).
Cyclin-dependent kinases 4 and 6 (CDK4/6) inhibition has been highlighted to exert antitumor immune response via promoting antigen presentation and enhancing CD8+ T-cell infiltration (145). The FDA-approved CDK4/6 inhibitor abemaciclib has shown preclinical synergistic antitumor effects with PD-1 inhibitor in breast cancer mouse models, the ID8 murine ovarian cancer model, and the colon adenocarcinoma murine model, which depends on increased T-cell recruitment with elevated levels of TH1 cytokines/chemokines (198–200).
Immunosuppressive cytokine TGFβ has received growing attention in cancer immunotherapy for its ability to block the antitumor immune response by limiting T- cell infiltration (201). Preclinical models suggested that coinhibiting TGF-β and PD-L1 induced potent and durable cytotoxic T-cell responses, transforming tumors from an excluded to an inflamed phenotype (146, 202). Strategies targeting TGF-β are under development, including the TGF-βRI kinase inhibitor galunisertib, neutralizing antibodies against the mature TGF-β cytokines, antibodies against TGF-βRII, and soluble TGF-β receptor traps, some of which are undergoing clinical trials in combination with anti-PD1 antibodies (203, 204).
As previously described, VEGF-induced immunosuppression inhibits T lymphocyte infiltration in the TME, hampering the therapeutic effect of ICB. In several earlier preclinical studies, vascular-normalization therapies have been proven to facilitate the transformation of the immunosuppressive TME toward an immune-supportive phenotype (205), which manifests as the aggregation of antitumor T cells and DC maturation inside tumors (206). In addition, the process of increased T lymphocyte infiltration induced by antiangiogenic therapies was partly associated with the elevated expression of adhesion molecules (intercellular adhesion molecule–1, vascular cell adhesion molecule-1), which facilitated the adhesion and extravasation of T cells (147). In preclinical mouse models and clinical trials, antiangiogenic agents significantly improved immunotherapy outcomes (205, 207). The various antiangiogenic therapeutic agents mainly consist of anti-VEGFA monoclonal antibodies such as bevacizumab, inhibitors of angiopoietin-2, and VEGFR tyrosine kinase inhibitors (TKIs) such as sorafenib (207). Some of them are presently undergoing clinical trials combining with ICB, receiving more significant clinical benefits than monotherapy in some early data (19).
In addition to the combination of targeted therapies mentioned above, low-dose radiotherapy has been reported to reprogram the TME and induce T-cell infiltration in mouse models of immune-desert tumors (148). Meanwhile, in “inflamed” human tumors, the preexistent intratumoral T cells not only survived radiotherapy but also acquired improved antitumor effects with the increasing production of IFN-γ (208).
It is also noteworthy that biomaterials at the nanoscale have been explored to establish a T-cell-inflamed TME and overcome resistance to ICB. Mesoporous silica nanoparticles were reported to elicit T-cell-recruitment chemokine production and drive CTL infiltration in multiple tumor models resistant to PD-1 antibodies (149). A supramolecular gold nanorod has been reported to reprogram the TME and improve TILs, significantly augmenting ICB therapy, which depends on the hyperthermal activation of ICD and genome editing of PD-L1 (209).
Moreover, chimeric antigen receptor (CAR) T cells may be a direct approach to provide antigen-sensitive immune infiltrates, implying a new opportunity for patients with less immunogenic or “noninflamed” tumors. CAR-T therapy could target T cells directly to tumor cells by genetically modifying T cells (210). Since the initial proposition of CAR-T in 1989, its antitumor efficacy and persistence have been improved due to altering the construction in the advanced generations of CAR-T. Based on these remarkable clinical responses, the FDA has approved four anti-CD19 CAR T-cell products and one anti-BCMA CAR T-cell therapy in different hematological cancers (211). However, the clinical efficacy of CAR T cells in the solid tumor has shown much less satisfactory results. One of the major obstacles includes the fact that PD-1-mediated immunosuppression leads to the poor persistence and dysfunctions of CAR T cells (150). Therefore, ICB and CAR T-cell combination therapy holds promise to refresh the immune system and enhance therapeutic efficacy. A synergy effect has been reported in the combination of PD-1 blockade and CAR-T cell therapy (212). In a transgenic Her-2 recipient mice model, anti-PD-1 antibody combined with CAR T cells showed the enhanced activation and proliferation of anti-Her-2 T cells, with the significant regression of established tumor (213). Other preclinical studies have shown the synergistic antitumor activity of combination therapies in thyroid cancers (214) and pleural mesothelioma (215). Some encouraging clinical results suggested the safety, low toxicity, and clinical responses of combinatorial treatment. One case report demonstrated five patients with diffuse large B-cell lymphoma who endured progression/relapse post-CART19/20 therapy received anti-PD-1 treatment (sintilimab or camrelizumab). Three of five patients had objective responses, including two complete responses and one partial response (216). Similarly, E. A. Chong et al. reported that in 12 B-cell lymphoma patients who were relapsing after or refractory to CD19-directed CAR T-cell therapy, anti-PD1 ICB (pembrolizumab) treatment showed safety and clinical responses (217). Based on these promising preclinical results, a series of one-half of clinical trials exploring the combination immunotherapy of CAR T cells and PD-1 blockade agents for multiple malignancies are under investigation, including relapsed/refractory Hodgkin lymphoma (NCT04134325), classical Hodgkin lymphoma (NCT05352828), relapsed/refractory B-cell lymphoma (NCT04539444), HER2-positive sarcoma (NCT04995003), and glioblastoma (NCT03726515). Some early results of clinical trials suggested the safety and promising efficacy of this combination in patients with malignant pleural disease (218), relapsed/refractory (r/r) diffuse large B-cell lymphoma (219), and relapsed/refractory aggressive B-cell non-Hodgkin lymphoma (220). However, minimal response with no meaningful durability has also been reported in two relapsed, refractory (R/R) B-cell non-Hodgkin lymphoma patients receiving the combination therapy of bispecific CAR T cells and PD-1 inhibitors (221). Therefore, further research is needed to confirm the therapeutic efficacy and optimal administration method of this combination treatment.
As stated above, T-cell exhaustion is characterized by the increased expression of suppressive cytokines and inhibitory receptors, including PD-1, CTLA, LAG-3, TIM-3, VISTA and ITIM domain (TIGIT), hierarchical decreased cytokine production (IL-2, TNF, IFNγ), and reduced proliferative capacity, with underlying distinct epigenetic states (222, 223). Accordingly, upcoming treatment options to overcome ICB resistance by the reinvigoration of T-cell exhaustion (Figure 4) include blocking the alternative immune checkpoints, targeting costimulatory receptors, inhibiting soluble immune- suppressive mediators, and epigenetically coordinating exhausted CD8+ T (Tex) cells (224–226).
Combining blockade treatments against multiple inhibitory receptors or combining checkpoint inhibitors with costimulatory agonists is a promising way to reinvigorate exhausted CD8+ T cells. Desirable therapeutic outcomes have been indicated in the preclinical and clinical studies of many tumors (227). Alternative targeting IRs include anti-TIM-3(MBG453), anti-LAG-3(LAG525), anti-TIGIT (etigilimab), anti-VISTA (JNJ-61,610,588), and anti-B7-H3 (enoblituzumab) (228–231). Accordingly, a wide range of combination strategies are undergoing research in various malignancies both preclinically and clinically. For instance, ipilimumab (anti-CTLA-4) plus nivolumab (anti-PD-1) is the most well-studied immuno-oncology (IO) combination showing comparatively better efficacy in multiple advanced tumors. It has become the earliest dual ICB treatment that received FDA approval in September 2015 for the first-line therapy of metastatic melanoma. Currently, this combination has been approved for the treatment of advanced renal cell carcinoma (RCC), metastatic colorectal cancer with MMR/MSI-H aberrations, PD-L1-positive (≥1%) metastatic NSCLC, and HCC as well. Noteworthily, the increasing incidence and intensity of the adverse events have been reported in the combining blockade, which suggest the importance of further studies (151). Costimulatory agonists are another good choice for reversing T-cell exhaustion in treating ICB. For example, the anti-CD137 agonist utomilumab has been shown to induce the increase of effector CD8+ T cells and improve survival in synergy with ICB in an ovarian cancer model (232). Recently, a growing number of agonist antibodies targeting immune costimulatory receptors are in clinical development for cancer indications, such as CD27 agonist varlilumab (CDX−1127) and CD40 agonist CDX−1140, OX40 agonist tavolimab (MEDI0562). Although none have been approved to date, combination approaches are still full of therapeutic potential (152).
Pauken et al. demonstrated that PD-1 blockade alone minimally remodeled the Tex epigenetic landscape. Hence, epigenetic modifiers, or T-cell epigenomic engineering with checkpoint blockade, may help reacquire durable immune memory against tumors (233). The transcriptional regulator TOX has recently been highlighted to be involved in programming CD8+ T-cell exhaustion transcriptionally and epigenetically, which is associated with plenty of transcription-factor networks downstream of TCR signaling (225). The knockdown of TOX ameliorated the exhaustion state of CD8+ T cells, enhancing the response to ICB treatment in an HCC mouse model (234), suggesting a new strategy to maximize immunotherapeutic efficacy by the downregulation of TOX expression. Interestingly, coblocking PD-1 and TIGIT could reinvigorate TOX-expressing PD-1highCD8+ TILs with better therapeutic outcomes in bladder cancer patients (153). Other modulators of the epigenetic landscape stated above, such as DNMTi, have also been found to induce the rejuvenation of exhausted CD8+ T cells, synergizing with a PD-1 inhibitor in a prostate adenocarcinoma mouse model (154).
Metabolic insufficiency play a crucial function in modulating T-cell exhaustion, implicating that metabolic modulation is a selective way to rejuvenate exhausted T cells, eliciting superior antitumor immunity (17, 155). In addition, ICB has been demonstrated to exert an inhibitory effect on immune cells’ metabolism and suppress glycolysis while increasing FAO and lipolysis. Therefore, the combinations of ICB with metabolic interventions appear to be ideal opportunities to improve antitumor effects via reversing immune metabolic dysfunctions (235). Many metabolic interventions have been exploited, such as enhancing mitochondrial fitness, enforcing fatty acid oxidation, and ameliorating ER stress (236). For example, in a B16 melanoma mouse model, metformin combined with anti-PD-1 therapy promoted increasing tumor clearance with an elevated intratumoral T-cell function. In addition, this reinvigoration of T cells mediated by metformin is associated with modulating the oxygen tension of the TME (237).
Various elements of the TME, including TANs, TAMs, CAFs, and Tregs, play critical immune-suppressive roles in mediating resistance to ICB. Correspondingly, therapies combined with ICB and strategies targeting these immune-suppressive cells appear to overcome resistance and improve clinical outcomes (Figure 4).
As is known, Tregs mediate tumor resistance against ICB in multiple ways, including upregulating the expression of other immune checkpoints including LAG-3, TIM-3, GITR, TIGIT, and VISTA; secreting high levels of TGF-β; and increasing the activation of the PI3K signaling pathway (238, 239). In glioblastoma, a typical immunologically ‘cold’ tumor, the suppressive Treg cells were converted toward CD4 effector T cells by an agonistic antibody (αGITR), which promoted the cure rates in GBM models combined with PD1 antibodies (240). Similar results have been reported in the coblockade of PD-1 and other immune checkpoints (241, 242). Importantly, this combined immunotherapy needs to be adapted to the specific immune environment for each tumor type. Targeting TGF-β is another appealing approach to reducing tumor-infiltrating Tregs and improving response to ICB treatment. R. Ravi et al. invented bifunctional antibody–ligand traps (Y-traps), simultaneously inhibiting the TGF-β pathway and CTLA-4 or PD-L1. This engineered antibody (a-CTLA4TGFβRIIecd and a-PDL1-TGFβRIIecd) significantly counteracted Tregs and restored beneficial TH1 cells in the TME, exhibiting superior antitumor efficacy than either the CTLA-4 antibody or PD-L1 antibodies in human melanoma (A375)–bearing NSG mice (156). Other strategies such as daclizumab, targeting the surface molecules CD25 of Treg, have been experimented both preclinically and clinically. Daclizumab administration reprogrammed Tregs. However, it also diminished activated Teff, showing no augmentation of T-cell responses in metastatic melanoma patients (243). Obviously, Treg-silencing strategies coupled with ICB require a deeper investigation of the crosstalk between the TME and Tregs.
As a vital source of PD-1, TAM has been demonstrated to hinder ICB efficacy by capturing ICB antibodies, secreting inhibitory cytokines, and expressing coinhibitory molecules. TAM-centered strategies are promising treatments to improve the efficacy of ICB agents (244, 245). CSF1R inhibitors enhanced the therapeutic efficacy of PD1 blockade by inhibiting the differentiation and accumulation of M2-like TAMs in melanoma models (157). Another monoclonal antibody targeting MARCO (macrophage receptor with collagenous structure) has also been reported to switch the TAM phenotype and boost checkpoint therapy effectively in melanoma tumor–bearing mice, which notably was induced by activating NK-cell-mediated killing other than T- cell-directed immunotherapy (246). Carfilzomib, a proteasome inhibitor approved by the FDA to treat relapsed/refractory multiple myeloma patients, has been supported to reprogram M2 macrophages into an M1-like population through IRE1a-TRAF2-NF-κB signaling and synergize with PD-1 inhibitors to reduce tumor growth in an autochthonous lung cancer model (158). Intriguingly, a recent study revealed that the high expression of histamine and histamine receptor H1 (HRH1) attenuated response to immunotherapies via polarizing TAMs toward an M2-like immunosuppressive phenotype. Hence, the HRH1 knockout or inhibition of HRH1 on macrophages with antihistamines reshaped the transcriptomic landscape of immune cells and blocked immune resistance when combined with anti-PD-1 treatment in mammary tumor and colon cancer mice models. In agreement with these results, the clinical data suggested that preexisting allergy or high histamine levels contributed to the inadequate immunotherapy responses in cancer patients (247). The similar antitumor properties of histamine dihydrochloride have been proven in MC-38 colon carcinoma and EL-4 lymphoma mouse model (248). However, in the murine cholangiocarcinoma (CAA) model, TAM blockade by anti-CSF1R failed to reduce CCA growth due to the compensatory infiltration of G-MDSCs. Meanwhile, the dual inhibition of TAMs and G-MDSCs was sufficient to enhance the efficiency of the PD-1 inhibitor in the orthotopic mouse model of CCA. Notably, the response rate to the ICB monotherapy of CAA patients is only 5.8% (249). Thus, targeting these immunosuppressive elements, particularly TAMs, is significant in potentiating PD-1 blockade.
Targeting CAF in the suppressive TME would be another valuable option to improve immunotherapy efficacy. Specifically, the targeted strategies include depleting CAF, interrupting their tumor-promoting ability, blocking CAF activation, and reverting CAF to a quiescent state (250). The inhibition of fibroblast activation protein (FAP)–positive CAF has disappointing results in metastatic colorectal cancer patients, possibly due to off-target effects (251). In recent years, single-cell RNA sequencing has characterized the heterogeneity of CAF in multiple tumor types, which suggests that targeting the subtype of CAF therapy may require a more nuanced approach (252). Blocking CAF-derived factors such as IL-6 and CXCL12 has been demonstrated to increase the accumulation of T cells and boost response to ICB in the models of multiple cancers (253). The ROS-producing enzyme NADPH oxidase-4 (NOX4) inhibition has been demonstrated as a well-studied approach to reversing TGF-β1-mediated CAF activation and promoting the transformation into a quiescent fibroblast-like phenotype (254). Using the NOX inhibitor GKT137831 (setanaxib) with immunotherapy can improve clinical outcomes in CAF-rich solid tumor models, indicating that reversing myofibroblastic CAFs to ‘normalized’ by setanaxib may be a considerable way to resensitize CAF-rich tumors to ICB, such as head and neck, colorectal, esophageal, and pancreatic cancers (255).
Apart from aiming at a specific group of cells or cytokines, radiotherapy is an appealing approach to shifting the immunosuppressive TME in the presence of immunotherapy. Combinatorial therapy has been shown to significantly increase CD8+ T cells by reducing MDSCs and Tregs, compared with RT or immunotherapy alone (160, 256). However, the immunosuppression effect of RT was known as well. Those irradiated cells that died of apoptosis could release anti-inflammatory cytokines such as TGF-β and adenosine to reduce tumor tolerance (257). Therefore, the definition of the optimum dose, appropriate fraction, and suitable target site of RT is fundamental (258).
Microbiota-centered interventions have recently gained growing attention for the engagement of the gut microbiome in primary and acquired resistance to ICB in different tumors such as melanoma, RCC, NSCLC, pancreatic ductal adenocarcinoma, and colon cancer (18, 259). Studies have proposed that regulating the collaboration of microbiota with the TME could contribute to metabolic changes, promoting antitumor T-cell responses and ameliorating anti-PD-1 blockade resistance (161). B. Routy et al. revealed that Akkermansia muciniphila and Enterococcus hirae are the primary factors in eliciting immunological changes, increasing CCR9+CXCR3+CD4+ T lymphocytes, which rely on interleukin-12 (18). Deep mechanisms accounting for the immunomodulatory effects of the gut microbiome remain to be explored. Nevertheless, manipulating the gut ecosystem is a profitable strategy to facilitate a better immune response (260). The specific interventions include supplementation with probiotics, the transfer of the fecal microbial content, microbiome-based metabolite therapy, and the depletion of the unfavorable bacterial taxa by proper oral antibiotics as well as dietary interventions, some of which have been evaluated in early phase clinical studies (261, 262). Intriguingly, researchers found that orally supplementing camu-camu, a polyphenol-rich berry, could circumvent anti-PD-1 resistance by reprogramming the TME in a microbiome-dependent way (263).
Based on the importance of chemotherapy in traditional cancer treatment and the beneficial immunomodulating effects of chemotherapy in the map of PD1/PDL1 therapy, chemotherapy has been the most widely used combination strategy approved in various indications so far and chemoimmunotherapy has become a standard of treatment for some cancer patients. The FDA granted pembrolizumab plus chemotherapy (pemetrexed and platinum) as the first-line therapy for advanced non-squamous NSCLC based on the clinical trial KEYNOTE-021 in 2017. Later in 2018, pembrolizumab plus carboplatin and either paclitaxel or nab-paclitaxel were approved as the first-line treatment of metastatic squamous NSCLC based on the results of KEYNOTE-407. On the strength of a series of successes in clinical trials, the approval of pembrolizumab plus chemotherapy covers more tumors, including gastroesophageal junction cancer (KEYNOTE-811), advanced triple-negative breast cancer (KEYNOTE-355), and esophageal cancer (KEYNOTE-590) (264–266). Meanwhile, anti-PD-L1-based chemoimmunotherapy such as atezolizumab plus chemotherapy and durvalumab combined with platinum plus etoposide treatment, has also received approval from the FDA in different tumors (170, 267). There is currently a rapidly growing number of clinical trials assessing chemoimmunotherapeutic regimens with the PD-1/PD-L1 inhibitor in clinical development but have not yet been approved by the FDA (166). The dose and sequence of administration require further evaluation to maximize the benefits of immunogenic chemotherapy.
Based on the above-mentioned preclinical data suggesting the potential synergistic effect of combining radiotherapy with anti−PD−1/PD−L1, a mounting number of translations into clinical trials are ongoing, most of which are still in phase I or II. In addition, the majority of radioimmunotherapy regimens are based on stereotactic body radiotherapy (SBRT). For instance, in PEMBRO-RT, a multicenter randomized phase 2 study of 92 patients with advanced NSCLC, patients who received SBRT (three doses of 8 Gy) before pembrolizumab showed improved trends in OS, progression-free survival (PFS), and objective response rate (ORR) compared with the non-irradiated group (268). However, in a single-center, randomized, phase II trial (NCT02684253) for patients with metastatic or recurrent HNSCC, nivolumab plus SBRT showed no improvement in response compared with nivolumab single arm (269). Further research is needed to explore the best radioimmunotherapy options, including the dose, volume, fractionation, and sequence.
The combination of ipilimumab (anti-CTLA-4) and nivolumab (anti-PD-1) is the first FDA-approved dual ICB treatment based on the results of CheckMate-067, CheckMate-069, and CheckMate-142 (151, 270). This combination is currently applied for the treatment of melanoma, RCC, HCC, PD-L1-positive NSCLC, MSI-H/dMMR colorectal cancer, and malignant pleural mesothelioma (3). Moreover, the FDA recently approved the first fixed-dose combination of nivolumab (Opdivo) and relatlimab (LAG-3 inhibitor) for unresectable or metastatic melanoma patients based on an appealing result from the phase-II/III RELATIVITY-047 trial. This trial demonstrated that the relatlimab–nivolumab combination yielded a progression-free survival rate of 10.1 months compared with 4.6 months in nivolumab monotherapy without new safety problems (271). Combinations of PD-1/PD-L1 blockers with other ICB are still in clinical trials. For instance, another CTLA-4 targeted monoclonal antibody, tremelimumab plus durvalumab, has entered phase 3 clinical trials in various malignancies, including small-cell lung cancer, high-risk urothelial carcinoma, advanced colorectal cancer, and advanced gastric and gastroesophageal junction adenocarcinoma, some of which received unsatisfactory results. No additional benefit was shown in combination (272–275). The severity and incidence of immune-related adverse events (irAEs), including colitis, thyroiditis, pneumonitis, and hypophysitis, have also been reported in the coblockade of PD-1/PD-L1 and CTLA-4 patients (276). In the primary analysis of the phase 2 CITYSCAPE trial, the TIGIT inhibitor tiragolumab plus atezolizumab (anti-PD-L1) showed improvement in PFS (stratified HR, 0.58; 95% CI, 0.38–0.89) in PD-L1-positive NSCLC patients (277).
Preclinical and clinical studies have verified the synergetic effect of the angiogenesis inhibitor with anti−PD−1/PD−L1. Based on studies 309/KEYNOTE-775 (NCT03517449) and KEYNOTE581 (NCT02811861), lenvatinib plus pembrolizumab has been approved by the FDA in the treatment of advanced endometrial carcinoma and advanced RCC (278). The KEYNOTE-426 study revealed that patients receiving pembrolizumab plus axitinib gained statistically significant PFS, OS, and ORR improvement compared with sunitinib monotherapy, which promoted the approval of pembrolizumab plus axitinib as the first-line therapy for advanced RCC (279). In 2018, based on the IMpower150 trial (NCT02366143), atezolizumab with chemotherapy and bevacizumab was approved for the first-line treatment of metastatic non-squamous NSCLC (280). Additionally, atezolizumab combined with bevacizumab was approved in 2020 for unresectable hepatocellular carcinoma on the basis of the IMbrave150 trial (NCT03434379) (281). Moreover, the FDA approved axitinib plus avelumab (based on JAVELIN Renal 101) and cabozantinib plus nivolumab (based on CheckMate-9ER) for RCC initial-line treatment as well (282, 283).
Noteworthily, plenty of clinical trials are exploring the combination strategies of angiogenesis inhibitors and anti-PD-1/PD-L1 at present. The preliminary data of some combinations demonstrated favorable therapeutic effects such as camrelizumab plus apatinib in advanced triple-negative breast cancer (NCT03394287), advanced cervical cancer (NCT03816553), and advanced HCC (NCT03463876) and sintilimab plus anlotinib in advanced NSCLC (NCT03628521) and PD-L1-positive recurrent or metastatic cervical cancer (284). Subsequent phase 3 trials are necessary to confirm the effectiveness of these combination regimens.
Apart from angiogenesis inhibitors, various targeted therapies combined with anti-PD-1/PD-L1 are undergoing clinical trials, such as nivolumab plus erlotinib (EGFR) in NSCLC patients (NCT01454102), tislelizumab plus pamiparib (PARP) in solid tumor patients (NCT02660034), cobimetinib (MEK) plus atezolizumab in colorectal cancer patients (NCT02788279), nivolumab plus copanlisib (PI3K) in lymphoma and solid tumor patients (NCT03502733), and pembrolizumab plus abemaciclib (CDK4/6) in NSCLC and breast cancer patients (NCT02779751). Altogether, most clinical trials are still in phase I or II. Further research is needed to explore the efficacy of anti-PD-1/PD-L1-based combined strategies in phase 3 trials.
ICB has revolutionized the field of cancer treatment. However, the initial wave of success on ICB is challenged by primary and acquired resistance. The number of patients benefiting from ICB is limited. Thus, a more detailed map of resistant mechanisms is reasonably necessary to develop coping strategies to improve clinical outcomes. Firstly, in this context, we primarily focus on the changes in the biological functions of CD8+ T cells to elucidate the underlying resistance mechanisms of ICB therapies. Based on the mechanical studies of both tumoral and systemic changes in the immune system, dozens of combinational regimens have been proposed, some of which exhibit potent antitumor activities in preclinical and clinical studies. Secondly, chemotherapy, VEGF/VEGFR-targeted therapy, and CTLA4-targeted treatment have been shown to be the most promising combinational options with anti-PD-1/PD-L1 therapy. They have great potential to improve the efficacy of ICB treatment in the condition of drug resistance. Nevertheless, only a tiny number of combinational strategies have been approved by the FDA, including anti-PD-1/PD-L1 plus chemotherapy, angiogenesis inhibitor, anti-CTLA-4, and anti-LAG-3 (Table 2). Overall, with a more profound elucidation of ICB resistance mechanisms, more novel clues of combinational strategies will emerge. Additional effort is needed to overcome barriers, including the occurrence of irAEs, the assessment of predictive biomarkers, and the definition of administration regimens such as dosage, timing, and sequence.

Table 2 Approved combination strategies with the PD-1/PDL1 inhibitor.
XTZ and YN jointly contributed to the first draft of the article, tables, and figures. XL provided assistance in making figures. YL, BA, and XH revised the manuscript. XZ conceived the presented idea, revised the manuscript again, and approved the final version. All authors approved this manuscript for publication.
This work was supported by the National Natural Science Foundation of China (No. 81902662), the National Natural Science Foundation of China (No. 81821002), Sichuan Science and Technology Program 2021YJ0011, and Sichuan Science and Technology Program 2018YJ0609.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Postow MA, Callahan MK, Wolchok JD. Immune checkpoint blockade in cancer therapy. J Clin Oncol (2015) 33(17):1974–82. doi: 10.1200/JCO.2014.59.4358
2. Sharma P, Hu-Lieskovan S, Wargo JA, Ribas A. Primary, adaptive, and acquired resistance to cancer immunotherapy. Cell (2017) 168(4):707–23. doi: 10.1016/j.cell.2017.01.017
3. Vaddepally RK, Kharel P, Pandey R, Garje R, Chandra AB. Review of indications of fda-approved immune checkpoint inhibitors per nccn guidelines with the level of evidence. Cancers (Basel) (2020) 12(3):738. doi: 10.3390/cancers12030738
4. Schoenfeld AJ, Hellmann MD. Acquired resistance to immune checkpoint inhibitors. Cancer Cell (2020) 37(4):443–55. doi: 10.1016/j.ccell.2020.03.017
5. Morad G, Helmink BA, Sharma P, Wargo JA. Hallmarks of response, resistance, and toxicity to immune checkpoint blockade. Cell (2021) 184(21):5309–37. doi: 10.1016/j.cell.2021.09.020
6. Aldea M, Andre F, Marabelle A, Dogan S, Barlesi F, Soria J-C. Overcoming resistance to tumor-targeted and immune-targeted therapies. Cancer Discovery (2021) 11(4):874–99. doi: 10.1158/2159-8290.CD-20-1638
7. Bagchi S, Yuan R, Engleman EG. Immune checkpoint inhibitors for the treatment of cancer: Clinical impact and mechanisms of response and resistance. Annu Rev Pathol (2021) 16:223–49. doi: 10.1146/annurev-pathol-042020-042741
8. Li J, Stanger BZ. How tumor cell dedifferentiation drives immune evasion and resistance to immunotherapy. Cancer Res (2020) 80(19):4037–41. doi: 10.1158/0008-5472.CAN-20-1420
9. Arozarena I, Wellbrock C. Phenotype plasticity as enabler of melanoma progression and therapy resistance. Nat Rev Cancer (2019) 19(7):377–91. doi: 10.1038/s41568-019-0154-4
10. Perez-Guijarro E, Yang HH, Araya RE, El Meskini R, Michael HT, Vodnala SK, et al. Multimodel preclinical platform predicts clinical response of melanoma to immunotherapy. Nat Med (2020) 26(5):781–91. doi: 10.1038/s41591-020-0818-3
11. Horn LA, Fousek K, Palena C. Tumor plasticity and resistance to immunotherapy. Trends Cancer (2020) 6(5):432–41. doi: 10.1016/j.trecan.2020.02.001
12. Kalbasi A, Ribas A. Tumour-intrinsic resistance to immune checkpoint blockade. Nat Rev Immunol (2020) 20(1):25–39. doi: 10.1038/s41577-019-0218-4
13. Horvath L, Thienpont B, Zhao L, Wolf D, Pircher A. Overcoming immunotherapy resistance in non-small cell lung cancer (Nsclc) - novel approaches and future outlook. Mol Cancer (2020) 19(1):141. doi: 10.1186/s12943-020-01260-z
14. Galluzzi L, Humeau J, Buque A, Zitvogel L, Kroemer G. Immunostimulation with chemotherapy in the era of immune checkpoint inhibitors. Nat Rev Clin Oncol (2020) 17(12):725–41. doi: 10.1038/s41571-020-0413-z
15. Schaue D, McBride WH. Opportunities and challenges of radiotherapy for treating cancer. Nat Rev Clin Oncol (2015) 12(9):527–40. doi: 10.1038/nrclinonc.2015.120
16. Morandi F, Airoldi I. Hla-G and other immune checkpoint molecules as targets for novel combined immunotherapies. Int J Mol Sci (2022) 23(6):2925. doi: 10.3390/ijms23062925
17. Franco F, Jaccard A, Romero P, Yu YR, Ho PC. Metabolic and epigenetic regulation of T-cell exhaustion. Nat Metab (2020) 2(10):1001–12. doi: 10.1038/s42255-020-00280-9
18. Routy B, Le Chatelier E, Derosa L, Duong CPM, Alou MT, Daillere R, et al. Gut microbiome influences efficacy of pd-1-Based immunotherapy against epithelial tumors. Science (2018) 359(6371):91–7. doi: 10.1126/science.aan3706
19. Yi M, Jiao D, Qin S, Chu Q, Wu K, Li A. Synergistic effect of immune checkpoint blockade and anti-angiogenesis in cancer treatment. Mol Cancer (2019) 18(1):60. doi: 10.1186/s12943-019-0974-6
20. Melief CJM. Mutation-specific T cells for immunotherapy of gliomas. N Engl J Med (2015) 372(20):1956–8. doi: 10.1056/NEJMcibr1501818
21. Huang Y, Shah S, Qiao L. Tumor resistance to Cd8+ T cell-based therapeutic vaccination. Arch Immunol Ther Exp (Warsz) (2007) 55(4):205–17. doi: 10.1007/s00005-007-0029-3
22. Lanzavecchia A, Sallusto F. Progressive differentiation and selection of the fittest in the immune response. Nat Rev Immunol (2002) 2(12):982–7. doi: 10.1038/nri959
23. Sade-Feldman M, Yizhak K, Bjorgaard SL, Ray JP, de Boer CG, Jenkins RW, et al. Defining T cell states associated with response to checkpoint immunotherapy in melanoma. Cell (2018) 175(4):998–1013.e20. doi: 10.1016/j.cell.2018.10.038
24. Li W, Sun T, Li M, He Y, Li L, Wang L, et al. Gnifdb: A neoantigen intrinsic feature database for glioma. Database (Oxford) (2022) 2022:baac004. doi: 10.1093/database/baac004
25. Strickler JH, Hanks BA, Khasraw M. Tumor mutational burden as a predictor of immunotherapy response: Is more always better? Clin Cancer Res (2021) 27(5):1236–41. doi: 10.1158/1078-0432.CCR-20-3054
26. Yarchoan M, Hopkins A, Jaffee EM. Tumor mutational burden and response rate to pd-1 inhibition. N Engl J Med (2017) 377(25):2500–1. doi: 10.1056/NEJMc1713444
27. Larkin J, Chiarion-Sileni V, Gonzalez R, Grob JJ, Cowey CL, Lao CD, et al. Combined nivolumab and ipilimumab or monotherapy in untreated melanoma. N Engl J Med (2015) 373(1):23–34. doi: 10.1056/NEJMoa1504030
28. Marabelle A, Fakih M, Lopez J, Shah M, Shapira-Frommer R, Nakagawa K, et al. Association of tumour mutational burden with outcomes in patients with advanced solid tumours treated with pembrolizumab: Prospective biomarker analysis of the multicohort, open-label, phase 2 keynote-158 study. Lancet Oncol (2020) 21(10):1353–65. doi: 10.1016/S1470-2045(20)30445-9
29. von Loga K, Woolston A, Punta M, Barber LJ, Griffiths B, Semiannikova M, et al. Extreme intratumour heterogeneity and driver evolution in mismatch repair deficient gastro-oesophageal cancer. Nat Commun (2020) 11(1):139. doi: 10.1038/s41467-019-13915-7
30. Strickler JH, Hanks BA, Khasraw M. Tumor mutational burden as a predictor of immunotherapy response: Is more always better? Clin Cancer Res (2021) 27(5):1236–41. doi: 10.1158/1078-0432.CCR-20-3054
31. Henon C, Blay JY, Massard C, Mir O, Bahleda R, Dumont S, et al. Long lasting major response to pembrolizumab in a thoracic malignant rhabdoid-like Smarca4-deficient tumor. Ann Oncol (2019) 30(8):1401–3. doi: 10.1093/annonc/mdz160
32. Gejman RS, Chang AY, Jones HF, DiKun K, Hakimi AA, Schietinger A, et al. Rejection of immunogenic tumor clones is limited by clonal fraction. Elife (2018) 7:e41090. doi: 10.7554/eLife.41090
33. Andor N, Graham TA, Jansen M, Xia LC, Aktipis CA, Petritsch C, et al. Pan-cancer analysis of the extent and consequences of intratumor heterogeneity. Nat Med (2016) 22(1):105–13. doi: 10.1038/nm.3984
34. Wolf Y, Bartok O, Patkar S, Eli GB, Cohen S, Litchfield K, et al. Uvb-induced tumor heterogeneity diminishes immune response in melanoma. Cell (2019) 179(1):219–235.e21. doi: 10.1016/j.cell.2019.08.032
35. McGranahan N, Furness AJ, Rosenthal R, Ramskov S, Lyngaa R, Saini SK, et al. Clonal neoantigens elicit T cell immunoreactivity and sensitivity to immune checkpoint blockade. Science (2016) 351(6280):1463–9. doi: 10.1126/science.aaf1490
36. Anagnostou V, Smith KN, Forde PM, Niknafs N, Bhattacharya R, White J, et al. Evolution of neoantigen landscape during immune checkpoint blockade in non-small cell lung cancer. Cancer Discovery (2017) 7(3):264–76. doi: 10.1158/2159-8290.CD-16-0828
37. Jamal-Hanjani M, Wilson GA, McGranahan N, Birkbak NJ, Watkins TBK, Veeriah S, et al. Tracking the evolution of non-Small-Cell lung cancer. N Engl J Med (2017) 376(22):2109–21. doi: 10.1056/NEJMoa1616288
38. Blum JS, Wearsch PA, Cresswell P. Pathways of antigen processing. Annu Rev Immunol (2013) 31:443–73. doi: 10.1146/annurev-immunol-032712-095910
39. Hulpke S, Tampé R. The mhc I loading complex: A multitasking machinery in adaptive immunity. Trends Biochem Sci (2013) 38(8):412–20. doi: 10.1016/j.tibs.2013.06.003
40. Zaretsky JM, Garcia-Diaz A, Shin DS, Escuin-Ordinas H, Hugo W, Hu-Lieskovan S, et al. Mutations associated with acquired resistance to pd-1 blockade in melanoma. N Engl J Med (2016) 375(9):819–29. doi: 10.1056/NEJMoa1604958
41. Sade-Feldman M, Jiao YJ, Chen JH, Rooney MS, Barzily-Rokni M, Eliane J-P, et al. Resistance to checkpoint blockade therapy through inactivation of antigen presentation. Nat Commun (2017) 8(1):1136. doi: 10.1038/s41467-017-01062-w
42. Gettinger S, Choi J, Hastings K, Truini A, Datar I, Sowell R, et al. Impaired hla class I antigen processing and presentation as a mechanism of acquired resistance to immune checkpoint inhibitors in lung cancer. Cancer Discovery (2017) 7(12):1420–35. doi: 10.1158/2159-8290.CD-17-0593
43. Busch E, Ahadova A, Kosmalla K, Bohaumilitzky L, Pfuderer PL, Ballhausen A, et al. Mutations are linked to a distinct metastatic pattern and a favorable outcome in microsatellite-unstable stage iv gastrointestinal cancers. Front Oncol (2021) 11:669774. doi: 10.3389/fonc.2021.669774
44. Gurjao C, Liu D, Hofree M, AlDubayan SH, Wakiro I, Su M-J, et al. Intrinsic resistance to immune checkpoint blockade in a mismatch repair-deficient colorectal cancer. Cancer Immunol Res (2019) 7(8):1230–6. doi: 10.1158/2326-6066.CIR-18-0683
45. Kalbasi A, Ribas A. Antigen presentation keeps trending in immunotherapy resistance. Clin Cancer Res (2018) 24(14):3239–41. doi: 10.1158/1078-0432.CCR-18-0698
46. Sivapalan L, Anagnostou V. Genetic variation in antigen presentation and cancer immunotherapy. Immunity (2022) 55(1):3–6. doi: 10.1016/j.immuni.2021.12.010
47. Tran E, Robbins PF, Lu Y-C, Prickett TD, Gartner JJ, Jia L, et al. T-Cell transfer therapy targeting mutant kras in cancer. N Engl J Med (2016) 375(23):2255–62. doi: 10.1056/NEJMoa1609279
48. Saveanu L, Carroll O, Hassainya Y, van Endert P. Complexity, contradictions, and conundrums: Studying post-proteasomal proteolysis in hla class I antigen presentation. Immunol Rev (2005) 207:42–59. doi: 10.1111/j.0105-2896.2005.00313.x
49. Cabrera CM, Jiménez P, Cabrera T, Esparza C, Ruiz-Cabello F, Garrido F. Total loss of mhc class I in colorectal tumors can be explained by two molecular pathways: Beta2-microglobulin inactivation in msi-positive tumors and Lmp7/Tap2 downregulation in msi-negative tumors. Tissue Antigens (2003) 61(3):211–9. doi: 10.1034/j.1399-0039.2003.00020.x
50. Vitale M, Rezzani R, Rodella L, Zauli G, Grigolato P, Cadei M, et al. Hla class I antigen and transporter associated with antigen processing (Tap1 and Tap2) down-regulation in high-grade primary breast carcinoma lesions. Cancer Res (1998) 58(4):737–42.
51. Zhang X, Sabio E, Krishna C, Ma X, Wang J, Jiang H, et al. Qa-1 modulates resistance to anti-Pd-1 immune checkpoint blockade in tumors with defects in antigen processing. Mol Cancer Res (2021) 19(6):1076–84. doi: 10.1158/1541-7786.MCR-20-0652
52. Vigneron N, Ferrari V, Van den Eynde BJ, Cresswell P, Leonhardt RM. Cytosolic processing governs tap-independent presentation of a critical melanoma antigen. J Immunol (2018) 201(7):1875–88. doi: 10.4049/jimmunol.1701479
53. Romero JM, Jiménez P, Cabrera T, Cózar JM, Pedrinaci S, Tallada M, et al. Coordinated downregulation of the antigen presentation machinery and hla class I/Beta2-microglobulin complex is responsible for hla-abc loss in bladder cancer. Int J Cancer (2005) 113(4):605–10. doi: 10.1002/ijc.20499
54. López-Albaitero A, Nayak JV, Ogino T, Machandia A, Gooding W, DeLeo AB, et al. Role of antigen-processing machinery in the in vitro resistance of squamous cell carcinoma of the head and neck cells to recognition by ctl. J Immunol (2006) 176(6):3402–9. doi: 10.4049/jimmunol.176.6.3402
55. Okada M, Shimizu K, Iyoda T, Ueda S, Shinga J, Mochizuki Y, et al. Pd-L1 expression affects neoantigen presentation. iScience (2020) 23(6):101238. doi: 10.1016/j.isci.2020.101238
56. Wangmo D, Premsrirut PK, Yuan C, Morris WS, Zhao X, Subramanian S. Ackr4 in tumor cells regulates dendritic cell migration to tumor-draining lymph nodes and T-cell priming. Cancers (Basel) (2021) 13(19):5021. doi: 10.3390/cancers13195021
57. Liu X, Shang X, Li J, Zhang S. The prognosis and immune checkpoint blockade efficacy prediction of tumor-infiltrating immune cells in lung cancer. Front Cell Dev Biol (2021) 9:707143. doi: 10.3389/fcell.2021.707143
58. Walker LSK, Sansom DM. The emerging role of Ctla4 as a cell-extrinsic regulator of T cell responses. Nat Rev Immunol (2011) 11(12):852–63. doi: 10.1038/nri3108
59. Maeda Y, Nishikawa H, Sugiyama D, Ha D, Hamaguchi M, Saito T, et al. Detection of self-reactive Cd8+T cells with an anergic phenotype in healthy individuals. Science (2014) 346(6216):1536–40. doi: 10.1126/science.aaa1292
60. Narayanan S, Vicent S, Ponz-Sarvisé M. Pdac as an immune evasive disease: Can 3d model systems aid to tackle this clinical problem? Front Cell Dev Biol (2021) 9:787249. doi: 10.3389/fcell.2021.787249
61. Annels NE, Simpson GR, Denyer M, Arif M, Coffey M, Melcher A, et al. Oncolytic reovirus-mediated recruitment of early innate immune responses reverses immunotherapy resistance in prostate tumors. Mol Ther Oncolytics (2021) 20:434–46. doi: 10.1016/j.omto.2020.09.010
62. Salemizadeh Parizi M, Salemizadeh Parizi F, Abdolhosseini S, Vanaei S, Manzouri A, Ebrahimzadeh F. Myeloid-derived suppressor cells (Mdscs) in brain cancer: Challenges and therapeutic strategies. Inflammopharmacology (2021) 29(6):1613–24. doi: 10.1007/s10787-021-00878-9
63. Gajewski TF, Woo S-R, Zha Y, Spaapen R, Zheng Y, Corrales L, et al. Cancer immunotherapy strategies based on overcoming barriers within the tumor microenvironment. Curr Opin Immunol (2013) 25(2):268–76. doi: 10.1016/j.coi.2013.02.009
64. Mühlberger M, Janko C, Unterweger H, Friedrich RP, Friedrich B, Band J, et al. Functionalization of T lymphocytes with citrate-coated superparamagnetic iron oxide nanoparticles for magnetically controlled immune therapy. Int J Nanomedicine (2019) 14:8421–32. doi: 10.2147/IJN.S218488
65. Chen DS, Mellman I. Elements of cancer immunity and the cancer-immune set point. Nature (2017) 541(7637):321–30. doi: 10.1038/nature21349
66. Peranzoni E, Lemoine J, Vimeux L, Feuillet V, Barrin S, Kantari-Mimoun C, et al. Macrophages impede Cd8 T cells from reaching tumor cells and limit the efficacy of anti-Pd-1 treatment. Proc Natl Acad Sci U.S.A. (2018) 115(17):E4041–E50. doi: 10.1073/pnas.1720948115
67. Peng W, Chen JQ, Liu C, Malu S, Creasy C, Tetzlaff MT, et al. Loss of pten promotes resistance to T cell-mediated immunotherapy. Cancer Discovery (2016) 6(2):202–16. doi: 10.1158/2159-8290.CD-15-0283
68. Karoulia Z, Gavathiotis E, Poulikakos PI. New perspectives for targeting raf kinase in human cancer. Nat Rev Cancer (2017) 17(11):676–91. doi: 10.1038/nrc.2017.79
69. Kimura ET, Nikiforova MN, Zhu Z, Knauf JA, Nikiforov YE, Fagin JA. High prevalence of braf mutations in thyroid cancer: Genetic evidence for constitutive activation of the Ret/Ptc-Ras-Braf signaling pathway in papillary thyroid carcinoma. Cancer Res (2003) 63(7):1454–7.
70. Rajagopalan H, Bardelli A, Lengauer C, Kinzler KW, Vogelstein B, Velculescu VE. Tumorigenesis: Raf/Ras oncogenes and mismatch-repair status. Nature (2002) 418(6901):934. doi: 10.1038/418934a
71. Wilmott JS, Long GV, Howle JR, Haydu LE, Sharma RN, Thompson JF, et al. Selective braf inhibitors induce marked T-cell infiltration into human metastatic melanoma. Clin Cancer Res (2012) 18(5):1386–94. doi: 10.1158/1078-0432.CCR-11-2479
72. Skoulidis F, Goldberg ME, Greenawalt DM, Hellmann MD, Awad MM, Gainor JF, et al. Mutations and pd-1 inhibitor resistance in -mutant lung adenocarcinoma. Cancer Discovery (2018) 8(7):822–35. doi: 10.1158/2159-8290.CD-18-0099
73. Xu Y-P, Lv L, Liu Y, Smith MD, Li W-C, Tan X-M, et al. Tumor suppressor Tet2 promotes cancer immunity and immunotherapy efficacy. J Clin Invest (2019) 129(10):4316–31. doi: 10.1172/JCI129317
74. Dong Z-Y, Zhang J-T, Liu S-Y, Su J, Zhang C, Xie Z, et al. Egfr mutation correlates with uninflamed phenotype and weak immunogenicity, causing impaired response to pd-1 blockade in non-small cell lung cancer. Oncoimmunology (2017) 6(11):e1356145. doi: 10.1080/2162402X.2017.1356145
75. Huang H, Langenkamp E, Georganaki M, Loskog A, Fuchs PF, Dieterich LC, et al. Vegf suppresses T-lymphocyte infiltration in the tumor microenvironment through inhibition of nf-κb-Induced endothelial activation. FASEB J (2015) 29(1):227–38. doi: 10.1096/fj.14-250985
76. Bruand M, Barras D, Mina M, Ghisoni E, Morotti M, Lanitis E, et al. Cell-autonomous inflammation of Brca1-deficient ovarian cancers drives both tumor-intrinsic immunoreactivity and immune resistance Via sting. Cell Rep (2021) 36(3):109412. doi: 10.1016/j.celrep.2021.109412
77. Li X, He G, Liu J, Yan M, Shen M, Xu L, et al. Ccl2-mediated monocytes regulate immune checkpoint blockade resistance in pancreatic cancer. Int Immunopharmacol. (2022) 106:108598. doi: 10.1016/j.intimp.2022.108598
78. Tauriello DVF, Palomo-Ponce S, Stork D, Berenguer-Llergo A, Badia-Ramentol J, Iglesias M, et al. Tgfβ drives immune evasion in genetically reconstituted colon cancer metastasis. Nature (2018) 554(7693):538–43. doi: 10.1038/nature25492
79. Mariathasan S, Turley SJ, Nickles D, Castiglioni A, Yuen K, Wang Y, et al. Tgfβ attenuates tumour response to pd-L1 blockade by contributing to exclusion of T cells. Nature (2018) 554(7693):544–8. doi: 10.1038/nature25501
80. Shaul ME, Fridlender ZG. The dual role of neutrophils in cancer. Semin Immunol (2021) 57:101582. doi: 10.1016/j.smim.2021.101582
81. Kim IS, Gao Y, Welte T, Wang H, Liu J, Janghorban M, et al. Immuno-subtyping of breast cancer reveals distinct myeloid cell profiles and immunotherapy resistance mechanisms. Nat Cell Biol (2019) 21(9):1113–26. doi: 10.1038/s41556-019-0373-7
82. Chen J, Sun H-W, Yang Y-Y, Chen H-T, Yu X-J, Wu W-C, et al. Reprogramming immunosuppressive myeloid cells by activated T cells promotes the response to anti-Pd-1 therapy in colorectal cancer. Signal Transduct. Target Ther (2021) 6(1):4. doi: 10.1038/s41392-020-00377-3
83. Meyer C, Cagnon L, Costa-Nunes CM, Baumgaertner P, Montandon N, Leyvraz L, et al. Frequencies of circulating mdsc correlate with clinical outcome of melanoma patients treated with ipilimumab. Cancer Immunol Immunother. (2014) 63(3):247–57. doi: 10.1007/s00262-013-1508-5
84. Movahedi K, Guilliams M, Van den Bossche J, Van den Bergh R, Gysemans C, Beschin A, et al. Identification of discrete tumor-induced myeloid-derived suppressor cell subpopulations with distinct T cell-suppressive activity. Blood (2008) 111(8):4233–44. doi: 10.1182/blood-2007-07-099226
85. Liu C-Y, Wang Y-M, Wang C-L, Feng P-H, Ko H-W, Liu Y-H, et al. Population alterations of l-arginase- and inducible nitric oxide synthase-expressed Cd11b+/Cd14-/Cd15+/Cd33+ myeloid-derived suppressor cells and Cd8+ T lymphocytes in patients with advanced-stage non-small cell lung cancer. J Cancer Res Clin Oncol (2010) 136(1):35–45. doi: 10.1007/s00432-009-0634-0
86. Veglia F, Tyurin VA, Blasi M, De Leo A, Kossenkov AV, Donthireddy L, et al. Fatty acid transport protein 2 reprograms neutrophils in cancer. Nature (2019) 569(7754):73–8. doi: 10.1038/s41586-019-1118-2
87. Thorn M, Guha P, Cunetta M, Espat NJ, Miller G, Junghans RP, et al. Tumor-associated gm-csf overexpression induces immunoinhibitory molecules Via Stat3 in myeloid-suppressor cells infiltrating liver metastases. Cancer Gene Ther (2016) 23(6):188–98. doi: 10.1038/cgt.2016.19
88. Adeshakin AO, Liu W, Adeshakin FO, Afolabi LO, Zhang M, Zhang G, et al. Regulation of ros in myeloid-derived suppressor cells through targeting fatty acid transport protein 2 enhanced anti-Pd-L1 tumor immunotherapy. Cell Immunol (2021) 362:104286. doi: 10.1016/j.cellimm.2021.104286
89. DeNardo DG, Ruffell B. Macrophages as regulators of tumour immunity and immunotherapy. Nat Rev Immunol (2019) 19(6):369–82. doi: 10.1038/s41577-019-0127-6
90. Orlikowsky T, Dannecker GE, Wang Z, Horowitz H, Niethammer D, Hoffmann MK. Activation or destruction of T cells. Via Macrophages. Pathobiol. (1999) 67(5-6):298–301. doi: 10.1159/000028084
91. Mantovani A, Sozzani S, Locati M, Allavena P, Sica A. Macrophage polarization: Tumor-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends Immunol (2002) 23(11):549–55. doi: 10.1016/S1471-4906(02)02302-5
92. Arlauckas SP, Garris CS, Kohler RH, Kitaoka M, Cuccarese MF, Yang KS, et al. In vivo imaging reveals a tumor-associated macrophage-mediated resistance pathway in anti-Pd-1 therapy. Sci Transl Med (2017) 9(389):eaal3604. doi: 10.1126/scitranslmed.aal3604
93. Ruffell B, Chang-Strachan D, Chan V, Rosenbusch A, Ho CMT, Pryer N, et al. Macrophage il-10 blocks Cd8+ T cell-dependent responses to chemotherapy by suppressing il-12 expression in intratumoral dendritic cells. Cancer Cell (2014) 26(5):623–37. doi: 10.1016/j.ccell.2014.09.006
94. Smith LK, Boukhaled GM, Condotta SA, Mazouz S, Guthmiller JJ, Vijay R, et al. Interleukin-10 directly inhibits Cd8 T cell function by enhancing n-glycan branching to decrease antigen sensitivity. Immunity (2018) 48(2):299–312.e5. doi: 10.1016/j.immuni.2018.01.006
95. Zerdes I, Wallerius M, Sifakis EG, Wallmann T, Betts S, Bartish M, et al. Stat3 activity promotes programmed-death ligand 1 expression and suppresses immune responses in breast cancer. Cancers (Basel) (2019) 11(10):1479. doi: 10.3390/cancers11101479
96. Singhal S, Stadanlick J, Annunziata MJ, Rao AS, Bhojnagarwala PS, O'Brien S, et al. Human tumor-associated Monocytes/Macrophages and their regulation of T cell responses in early-stage lung cancer. Sci Transl Med (2019) 11(479):eaat1500. doi: 10.1126/scitranslmed.aat1500
97. Darby IA, Laverdet B, Bonté F, Desmoulière A. Fibroblasts and myofibroblasts in wound healing. Clin Cosmet Investig Dermatol (2014) 7:301–11. doi: 10.2147/CCID.S50046
98. Kuzet S-E, Gaggioli C. Fibroblast activation in cancer: When seed fertilizes soil. Cell Tissue Res (2016) 365(3):607–19. doi: 10.1007/s00441-016-2467-x
99. Gardner H, Strehlow D, Bradley L, Widom R, Farina A, de Fougerolles A, et al. Global expression analysis of the fibroblast transcriptional response to tgfbeta. Clin Exp Rheumatol (2004) 22(3 Suppl 33):S47–57.
100. Khalili JS, Liu S, Rodríguez-Cruz TG, Whittington M, Wardell S, Liu C, et al. Oncogenic Braf(V600e) promotes stromal cell-mediated immunosuppression Via induction of interleukin-1 in melanoma. Clin Cancer Res (2012) 18(19):5329–40. doi: 10.1158/1078-0432.CCR-12-1632
101. Lakins MA, Ghorani E, Munir H, Martins CP, Shields JD. Cancer-associated fibroblasts induce antigen-specific deletion of Cd8 T cells to protect tumour cells. Nat Commun (2018) 9(1):948. doi: 10.1038/s41467-018-03347-0
102. Gorchs L, Fernández Moro C, Bankhead P, Kern KP, Sadeak I, Meng Q, et al. Human pancreatic carcinoma-associated fibroblasts promote expression of Co-inhibitory markers on Cd4 and Cd8 T-cells. Front Immunol (2019) 10:847. doi: 10.3389/fimmu.2019.00847
103. Cheng Y, Li H, Deng Y, Tai Y, Zeng K, Zhang Y, et al. Cancer-associated fibroblasts induce Pdl1+ neutrophils through the Il6-Stat3 pathway that foster immune suppression in hepatocellular carcinoma. Cell Death Dis (2018) 9(4):422. doi: 10.1038/s41419-018-0458-4
104. Li Z, Zhou J, Zhang J, Li S, Wang H, Du J. Cancer-associated fibroblasts promote pd-L1 expression in mice cancer cells Via secreting Cxcl5. Int J Cancer (2019) 145(7):1946–57. doi: 10.1002/ijc.32278
105. Curiel TJ, Coukos G, Zou L, Alvarez X, Cheng P, Mottram P, et al. Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat Med (2004) 10(9):942–9. doi: 10.1038/nm1093
106. Ma GF, Miao Q, Liu YM, Gao H, Lian JJ, Wang YN, et al. High Foxp3 expression in tumour cells predicts better survival in gastric cancer and its role in tumour microenvironment. Br J Cancer (2014) 110(6):1552–60. doi: 10.1038/bjc.2014.47
107. Shimizu J, Yamazaki S, Takahashi T, Ishida Y, Sakaguchi S. Stimulation of Cd25(+)Cd4(+) regulatory T cells through gitr breaks immunological self-tolerance. Nat Immunol (2002) 3(2):135–42. doi: 10.1038/ni759
108. Bennett CL, Christie J, Ramsdell F, Brunkow ME, Ferguson PJ, Whitesell L, et al. The immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome (Ipex) is caused by mutations of Foxp3. Nat Genet (2001) 27(1):20–1. doi: 10.1038/83713
109. Zhang Y, Maksimovic J, Huang B, De Souza DP, Naselli G, Chen H, et al. Cord blood Cd8 T cells have a natural propensity to express il-4 in a fatty acid metabolism and caspase activation-dependent manner. Front Immunol (2018) 9:879. doi: 10.3389/fimmu.2018.00879
110. Pereira LMS, Gomes STM, Ishak R, Vallinoto ACR. Regulatory T cell and forkhead box protein 3 as modulators of immune homeostasis. Front Immunol (2017) 8:605. doi: 10.3389/fimmu.2017.00605
111. Ren Z, Zhang A, Sun Z, Liang Y, Ye J, Qiao J, et al. Selective delivery of low-affinity il-2 to pd-1+ T cells rejuvenates antitumor immunity with reduced toxicity. J Clin Invest (2022) 132(3):e153604. doi: 10.1172/JCI153604
112. Camisaschi C, Casati C, Rini F, Perego M, De Filippo A, Triebel F, et al. Lag-3 expression defines a subset of Cd4(+)Cd25(High)Foxp3(+) regulatory T cells that are expanded at tumor sites. J Immunol (2010) 184(11):6545–51. doi: 10.4049/jimmunol.0903879
113. Linterman MA, Pierson W, Lee SK, Kallies A, Kawamoto S, Rayner TF, et al. Foxp3+ follicular regulatory T cells control the germinal center response. Nat Med (2011) 17(8):975–82. doi: 10.1038/nm.2425
114. Sage PT, Francisco LM, Carman CV, Sharpe AH. The receptor pd-1 controls follicular regulatory T cells in the lymph nodes and blood. Nat Immunol (2013) 14(2):152–61. doi: 10.1038/ni.2496
115. Vanderleyden I, Fra-Bido SC, Innocentin S, Stebegg M, Okkenhaug H, Evans-Bailey N, et al. Follicular regulatory T cells can access the germinal center independently of Cxcr5. Cell Rep (2020) 30(3):611–619.e4. doi: 10.1016/j.celrep.2019.12.076
116. Eschweiler S, Clarke J, Ramírez-Suástegui C, Panwar B, Madrigal A, Chee SJ, et al. Intratumoral follicular regulatory T cells curtail anti-Pd-1 treatment efficacy. Nat Immunol (2021) 22(8):1052–63. doi: 10.1038/s41590-021-00958-6
117. Zappasodi R, Budhu S, Hellmann MD, Postow MA, Senbabaoglu Y, Manne S, et al. Non-conventional inhibitory Cd4foxp3pd-1 T cells as a biomarker of immune checkpoint blockade activity. Cancer Cell (2018) 33(6):1017–1032.e7. doi: 10.1016/j.ccell.2018.05.009
119. Konen JM, Rodriguez BL, Fradette JJ, Gibson L, Davis D, Minelli R, et al. Ntrk1 promotes resistance to pd-1 checkpoint blockade in mesenchymal Kras/P53 mutant lung cancer. Cancers (Basel) (2019) 11(4):462. doi: 10.3390/cancers11040462
120. Koyama S, Akbay EA, Li YY, Herter-Sprie GS, Buczkowski KA, Richards WG, et al. Adaptive resistance to therapeutic pd-1 blockade is associated with upregulation of alternative immune checkpoints. Nat Commun (2016) 7:10501. doi: 10.1038/ncomms10501
121. Shayan G, Srivastava R, Li J, Schmitt N, Kane LP, Ferris RL. Adaptive resistance to anti-Pd1 therapy by Tim-3 upregulation is mediated by the Pi3k-akt pathway in head and neck cancer. Oncoimmunology (2017) 6(1):e1261779. doi: 10.1080/2162402X.2016.1261779
122. Huang R-Y, Francois A, McGray AR, Miliotto A, Odunsi K. Compensatory upregulation of pd-1, lag-3, and ctla-4 limits the efficacy of single-agent checkpoint blockade in metastatic ovarian cancer. Oncoimmunology (2017) 6(1):e1249561. doi: 10.1080/2162402X.2016.1249561
123. Thommen DS, Schreiner J, Müller P, Herzig P, Roller A, Belousov A, et al. Progression of lung cancer is associated with increased dysfunction of T cells defined by coexpression of multiple inhibitory receptors. Cancer Immunol Res (2015) 3(12):1344–55. doi: 10.1158/2326-6066.CIR-15-0097
124. Kakavand H, Jackett LA, Menzies AM, Gide TN, Carlino MS, Saw RPM, et al. Negative immune checkpoint regulation by vista: A mechanism of acquired resistance to anti-Pd-1 therapy in metastatic melanoma patients. Mod Pathol (2017) 30(12):1666–76. doi: 10.1038/modpathol.2017.89
125. Bauer C, Kühnemuth B, Duewell P, Ormanns S, Gress T, Schnurr M. Prevailing over T cell exhaustion: New developments in the immunotherapy of pancreatic cancer. Cancer Lett (2016) 381(1):259–68. doi: 10.1016/j.canlet.2016.02.057
126. Farhood B, Najafi M, Mortezaee K. Cd8 cytotoxic T lymphocytes in cancer immunotherapy: A review. J Cell Physiol (2019) 234(6):8509–21. doi: 10.1002/jcp.27782
127. Kearney CJ, Vervoort SJ, Hogg SJ, Ramsbottom KM, Freeman AJ, Lalaoui N, et al. Tumor immune evasion arises through loss of tnf sensitivity. Sci Immunol (2018) 3(23):eaar3451. doi: 10.1126/sciimmunol.aar3451
128. Dominiecki ME, Beatty GL, Pan Z-K, Neeson P, Paterson Y. Tumor sensitivity to ifn-gamma is required for successful antigen-specific immunotherapy of a transplantable mouse tumor model for hpv-transformed tumors. Cancer Immunol Immunother (2005) 54(5):477–88. doi: 10.1007/s00262-004-0610-0
129. Ikeda H, Old LJ, Schreiber RD. The roles of ifn gamma in protection against tumor development and cancer immunoediting. Cytokine Growth Factor Rev (2002) 13(2):95–109. doi: 10.1016/S1359-6101(01)00038-7
130. Bach EA, Aguet M, Schreiber RD. The ifn gamma receptor: A paradigm for cytokine receptor signaling. Annu Rev Immunol (1997) 15:563–91. doi: 10.1146/annurev.immunol.15.1.563
131. Gao J, Shi LZ, Zhao H, Chen J, Xiong L, He Q, et al. Loss of ifn-Γ pathway genes in tumor cells as a mechanism of resistance to anti-Ctla-4 therapy. Cell (2016) 167(2):397–404.e9. doi: 10.1016/j.cell.2016.08.069
132. Shin DS, Zaretsky JM, Escuin-Ordinas H, Garcia-Diaz A, Hu-Lieskovan S, Kalbasi A, et al. Primary resistance to pd-1 blockade mediated by Jak1/2 mutations. Cancer Discovery (2017) 7(2):188–201. doi: 10.1158/2159-8290.CD-16-1223
133. Vredevoogd DW, Kuilman T, Ligtenberg MA, Boshuizen J, Stecker KE, de Bruijn B, et al. Augmenting immunotherapy impact by lowering tumor tnf cytotoxicity threshold. Cell (2019) 178(3):585–599.e15. doi: 10.1016/j.cell.2019.06.014
134. Herrera FG, Bourhis J, Coukos G. Radiotherapy combination opportunities leveraging immunity for the next oncology practice. CA Cancer J Clin (2017) 67(1):65–85. doi: 10.3322/caac.21358
135. Woller N, Gurlevik E, Fleischmann-Mundt B, Schumacher A, Knocke S, Kloos AM, et al. Viral infection of tumors overcomes resistance to pd-1-Immunotherapy by broadening neoantigenome-directed T-cell responses. Mol Ther (2015) 23(10):1630–40. doi: 10.1038/mt.2015.115
136. Turner TB, Meza-Perez S, Londono A, Katre A, Peabody JE, Smith HJ, et al. Epigenetic modifiers upregulate mhc ii and impede ovarian cancer tumor growth. Oncotarget (2017) 8(27):44159–70. doi: 10.18632/oncotarget.17395
137. Mazzone R, Zwergel C, Mai A, Valente S. Epi-drugs in combination with immunotherapy: A new avenue to improve anticancer efficacy. Clin Epigenet (2017) 9:59. doi: 10.1186/s13148-017-0358-y
138. Lv M, Chen M, Zhang R, Zhang W, Wang C, Zhang Y, et al. Manganese is critical for antitumor immune responses Via cgas-sting and improves the efficacy of clinical immunotherapy. Cell Res (2020) 30(11):966–79. doi: 10.1038/s41422-020-00395-4
139. Kalbasi A, Tariveranmoshabad M, Hakimi K, Kremer S, Campbell KM, Funes JM, et al. Uncoupling interferon signaling and antigen presentation to overcome immunotherapy resistance due to Jak1 loss in melanoma. Sci Transl Med (2020) 12(565):eabb0152. doi: 10.1126/scitranslmed.abb0152
140. Ribas A, Medina T, Kummar S, Amin A, Kalbasi A, Drabick JJ, et al. Sd-101 in combination with pembrolizumab in advanced melanoma: Results of a phase ib, multicenter study. Cancer Discovery (2018) 8(10):1250–7. doi: 10.1158/2159-8290.CD-18-0280
141. Salmon H, Idoyaga J, Rahman A, Leboeuf M, Remark R, Jordan S, et al. Expansion and activation of Cd103(+) dendritic cell progenitors at the tumor site enhances tumor responses to therapeutic pd-L1 and braf inhibition. Immunity (2016) 44(4):924–38. doi: 10.1016/j.immuni.2016.03.012
142. Peng W, Chen JQ, Liu C, Malu S, Creasy C, Tetzlaff MT, et al. Loss of pten promotes resistance to T cell-mediated immunotherapy. Cancer Discovery (2016) 6(2):202–16. doi: 10.1158/2159-8290.CD-15-0283
143. Wall JA, Meza-Perez S, Scalise CB, Katre A, Londoño AI, Turbitt WJ, et al. Manipulating the Wnt/β-catenin signaling pathway to promote anti-tumor immune infiltration into the tme to sensitize ovarian cancer to icb therapy. Gynecol. Oncol (2021) 160(1):285–94. doi: 10.1016/j.ygyno.2020.10.031
144. Ribas A, Lawrence D, Atkinson V, Agarwal S, Miller WH Jr., Carlino MS, et al. Combined braf and mek inhibition with pd-1 blockade immunotherapy in braf-mutant melanoma. Nat Med (2019) 25(6):936–40. doi: 10.1038/s41591-019-0476-5
145. Goel S, DeCristo MJ, Watt AC, BrinJones H, Sceneay J, Li BB, et al. Cdk4/6 inhibition triggers anti-tumour immunity. Nature (2017) 548(7668):471–5. doi: 10.1038/nature23465
146. Mariathasan S, Turley SJ, Nickles D, Castiglioni A, Yuen K, Wang Y, et al. Tgfbeta attenuates tumour response to pd-L1 blockade by contributing to exclusion of T cells. Nature (2018) 554(7693):544–8. doi: 10.1038/nature25501
147. Schmittnaegel M, Rigamonti N, Kadioglu E, Cassara A, Wyser Rmili C, Kiialainen A, et al. Dual angiopoietin-2 and vegfa inhibition elicits antitumor immunity that is enhanced by pd-1 checkpoint blockade. Sci Transl Med (2017) 9(385):eaak9670. doi: 10.1126/scitranslmed.aak9670
148. Klug F, Prakash H, Huber PE, Seibel T, Bender N, Halama N, et al. Low-dose irradiation programs macrophage differentiation to an Inos(+)/M1 phenotype that orchestrates effective T cell immunotherapy. Cancer Cell (2013) 24(5):589–602. doi: 10.1016/j.ccr.2013.09.014
149. Sun M, Gu P, Yang Y, Yu L, Jiang Z, Li J, et al. Mesoporous silica nanoparticles inflame tumors to overcome anti-Pd-1 resistance through Tlr4-nfkappab axis. J Immunother Cancer (2021) 9(6):e002508. doi: 10.1136/jitc-2021-002508
150. Grosser R, Cherkassky L, Chintala N, Adusumilli PS. Combination immunotherapy with car T cells and checkpoint blockade for the treatment of solid tumors. Cancer Cell (2019) 36(5):471–82. doi: 10.1016/j.ccell.2019.09.006
151. Rotte A. Combination of ctla-4 and pd-1 blockers for treatment of cancer. J Exp Clin Cancer Res (2019) 38(1):255. doi: 10.1186/s13046-019-1259-z
152. Mayes PA, Hance KW, Hoos A. The promise and challenges of immune agonist antibody development in cancer. Nat Rev Drug Discovery (2018) 17(7):509–27. doi: 10.1038/nrd.2018.75
153. Han HS, Jeong S, Kim H, Kim HD, Kim AR, Kwon M, et al. Tox-expressing terminally exhausted tumor-infiltrating Cd8(+) T cells are reinvigorated by Co-blockade of pd-1 and tigit in bladder cancer. Cancer Lett (2021) 499:137–47. doi: 10.1016/j.canlet.2020.11.035
154. Ghoneim HE, Fan Y, Moustaki A, Abdelsamed HA, Dash P, Dogra P, et al. De novo epigenetic programs inhibit pd-1 blockade-mediated T cell rejuvenation. Cell (2017) 170(1):142–57.e19. doi: 10.1016/j.cell.2017.06.007
155. Buck MD, O'Sullivan D, Klein Geltink RI, Curtis JD, Chang CH, Sanin DE, et al. Mitochondrial dynamics controls T cell fate through metabolic programming. Cell (2016) 166(1):63–76. doi: 10.1016/j.cell.2016.05.035
156. Ravi R, Noonan KA, Pham V, Bedi R, Zhavoronkov A, Ozerov IV, et al. Bifunctional immune checkpoint-targeted antibody-ligand traps that simultaneously disable tgfbeta enhance the efficacy of cancer immunotherapy. Nat Commun (2018) 9(1):741. doi: 10.1038/s41467-017-02696-6
157. Neubert NJ, Schmittnaegel M, Bordry N, Nassiri S, Wald N, Martignier C, et al. T Cell-induced Csf1 promotes melanoma resistance to Pd1 blockade. Sci Transl Med (2018) 10(436):eaan3311. doi: 10.1126/scitranslmed.aan3311
158. Zhou Q, Liang J, Yang T, Liu J, Li B, Li Y, et al. Carfilzomib modulates tumor microenvironment to potentiate immune checkpoint therapy for cancer. EMBO Mol Med (2022) 14(1):e14502. doi: 10.15252/emmm.202114502
159. Cheng Y, Li H, Deng Y, Tai Y, Zeng K, Zhang Y, et al. Cancer-associated fibroblasts induce Pdl1+ neutrophils through the Il6-Stat3 pathway that foster immune suppression in hepatocellular carcinoma. Cell Death Dis (2018) 9(4):422. doi: 10.1038/s41419-018-0458-4
160. Ji D, Song C, Li Y, Xia J, Wu Y, Jia J, et al. Combination of radiotherapy and suppression of tregs enhances abscopal antitumor effect and inhibits metastasis in rectal cancer. J Immunother Cancer (2020) 8(2):e000826. doi: 10.1136/jitc-2020-000826
161. Derosa L, Routy B, Desilets A, Daillère R, Terrisse S, Kroemer G, et al. Microbiota-centered interventions: The next breakthrough in immuno-oncology? Cancer Discovery (2021) 11(10):2396–412. doi: 10.1158/2159-8290.Cd-21-0236
162. Mole RH. Whole body irradiation; radiobiology or medicine? Br J Radiol (1953) 26(305):234–41. doi: 10.1259/0007-1285-26-305-234
163. Ngwa W, Irabor OC, Schoenfeld JD, Hesser J, Demaria S, Formenti SC. Using immunotherapy to boost the abscopal effect. Nat Rev Cancer (2018) 18(5):313–22. doi: 10.1038/nrc.2018.6
164. Takahashi J, Nagasawa S. Immunostimulatory effects of radiotherapy for local and systemic control of melanoma: A review. Int J Mol Sci (2020) 21(23):9324. doi: 10.3390/ijms21239324
165. Sharma A, Bode B, Wenger RH, Lehmann K, Sartori AA, Moch H, et al. Gamma-radiation promotes immunological recognition of cancer cells through increased expression of cancer-testis antigens in vitro and in vivo. PloS One (2011) 6(11):e28217. doi: 10.1371/journal.pone.0028217
166. Salas-Benito D, Perez-Gracia JL, Ponz-Sarvise M, Rodriguez-Ruiz ME, Martinez-Forero I, Castanon E, et al. Paradigms on immunotherapy combinations with chemotherapy. Cancer Discovery (2021) 11(6):1353–67. doi: 10.1158/2159-8290.CD-20-1312
167. Grimaldi A, Cammarata I, Martire C, Focaccetti C, Piconese S, Buccilli M, et al. Combination of chemotherapy and pd-1 blockade induces T cell responses to tumor non-mutated neoantigens. Commun Biol (2020) 3(1):85. doi: 10.1038/s42003-020-0811-x
168. Ma X, Yang S, Zhang T, Wang S, Yang Q, Xiao Y, et al. Bioresponsive immune-Booster-Based prodrug nanogel for cancer immunotherapy. Acta Pharm Sin B (2022) 12(1):451–66. doi: 10.1016/j.apsb.2021.05.016
169. Mathew M, Enzler T, Shu CA, Rizvi NA. Combining chemotherapy with pd-1 blockade in nsclc. Pharmacol Ther (2018) 186:130–7. doi: 10.1016/j.pharmthera.2018.01.003
170. Schmid P, Rugo HS, Adams S, Schneeweiss A, Barrios CH, Iwata H, et al. Atezolizumab plus nab-paclitaxel as first-line treatment for unresectable, locally advanced or metastatic triple-negative breast cancer (Impassion130): Updated efficacy results from a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol (2020) 21(1):44–59. doi: 10.1016/S1470-2045(19)30689-8
171. Russell L, Peng KW, Russell SJ, Diaz RM. Oncolytic viruses: Priming time for cancer immunotherapy. BioDrugs (2019) 33(5):485–501. doi: 10.1007/s40259-019-00367-0
172. Bommareddy PK, Shettigar M, Kaufman HL. Integrating oncolytic viruses in combination cancer immunotherapy. Nat Rev Immunol (2018) 18(8):498–513. doi: 10.1038/s41577-018-0014-6
173. Twumasi-Boateng K, Pettigrew JL, Kwok YYE, Bell JC, Nelson BH. Oncolytic viruses as engineering platforms for combination immunotherapy. Nat Rev Cancer (2018) 18(7):419–32. doi: 10.1038/s41568-018-0009-4
174. Takaki H, Cornelis F, Kako Y, Kobayashi K, Kamikonya N, Yamakado K. Thermal ablation and immunomodulation: From preclinical experiments to clinical trials. Diagn Interv Imaging (2017) 98(9):651–9. doi: 10.1016/j.diii.2017.04.008
175. Xing R, Gao J, Cui Q, Wang Q. Strategies to improve the antitumor effect of immunotherapy for hepatocellular carcinoma. Front Immunol (2021) 12:783236. doi: 10.3389/fimmu.2021.783236
176. Harari A, Graciotti M, Bassani-Sternberg M, Kandalaft LE. Antitumour dendritic cell vaccination in a priming and boosting approach. Nat Rev Drug Discovery (2020) 19(9):635–52. doi: 10.1038/s41573-020-0074-8
177. Xiao Y, Zhang T, Ma X, Yang QC, Yang LL, Yang SC, et al. Microenvironment-responsive prodrug-induced pyroptosis boosts cancer immunotherapy. Adv Sci (Weinh) (2021) 8(24):e2101840. doi: 10.1002/advs.202101840
178. Sade-Feldman M, Jiao YJ, Chen JH, Rooney MS, Barzily-Rokni M, Eliane JP, et al. Resistance to checkpoint blockade therapy through inactivation of antigen presentation. Nat Commun (2017) 8(1):1136. doi: 10.1038/s41467-017-01062-w
179. Gomez S, Tabernacki T, Kobyra J, Roberts P, Chiappinelli KB. Combining epigenetic and immune therapy to overcome cancer resistance. Semin Cancer Biol (2020) 65:99–113. doi: 10.1016/j.semcancer.2019.12.019
180. Falkenberg KJ, Johnstone RW. Histone deacetylases and their inhibitors in cancer, neurological diseases and immune disorders. Nat Rev Drug Discovery (2014) 13(9):673–91. doi: 10.1038/nrd4360
181. Siebenkas C, Chiappinelli KB, Guzzetta AA, Sharma A, Jeschke J, Vatapalli R, et al. Inhibiting DNA methylation activates cancer testis antigens and expression of the antigen processing and presentation machinery in colon and ovarian cancer cells. PloS One (2017) 12(6):e0179501. doi: 10.1371/journal.pone.0179501
182. Suraweera A, O'Byrne KJ, Richard DJ. Combination therapy with histone deacetylase inhibitors (Hdaci) for the treatment of cancer: Achieving the full therapeutic potential of hdaci. Front Oncol (2018) 8:92. doi: 10.3389/fonc.2018.00092
183. Zheng H, Zhao W, Yan C, Watson CC, Massengill M, Xie M, et al. Hdac inhibitors enhance T-cell chemokine expression and augment response to pd-1 immunotherapy in lung adenocarcinoma. Clin Cancer Res (2016) 22(16):4119–32. doi: 10.1158/1078-0432.CCR-15-2584
184. Chen X, Pan X, Zhang W, Guo H, Cheng S, He Q, et al. Epigenetic strategies synergize with pd-L1/Pd-1 targeted cancer immunotherapies to enhance antitumor responses. Acta Pharm Sin B (2020) 10(5):723–33. doi: 10.1016/j.apsb.2019.09.006
185. Cornel AM, Mimpen IL, Nierkens S. Mhc class I downregulation in cancer: Underlying mechanisms and potential targets for cancer immunotherapy. Cancers (Basel) (2020) 12(7):1760. doi: 10.3390/cancers12071760
186. Wculek SK, Cueto FJ, Mujal AM, Melero I, Krummel MF, Sancho D. Dendritic cells in cancer immunology and immunotherapy. Nat Rev Immunol (2020) 20(1):7–24. doi: 10.1038/s41577-019-0210-z
187. Ni K, Luo T, Lan G, Culbert A, Song Y, Wu T, et al. A nanoscale metal-organic framework to mediate photodynamic therapy and deliver cpg oligodeoxynucleotides to enhance antigen presentation and cancer immunotherapy. Angew Chem Int Ed Engl (2020) 59(3):1108–12. doi: 10.1002/anie.201911429
188. Garg AD, Coulie PG, Van den Eynde BJ, Agostinis P. Integrating next-generation dendritic cell vaccines into the current cancer immunotherapy landscape. Trends Immunol (2017) 38(8):577–93. doi: 10.1016/j.it.2017.05.006
189. Huang Y, Kim BYS, Chan CK, Hahn SM, Weissman IL, Jiang W. Improving immune-vascular crosstalk for cancer immunotherapy. Nat Rev Immunol (2018) 18(3):195–203. doi: 10.1038/nri.2017.145
190. Lin YX, Wang Y, Ding J, Jiang A, Wang J, Yu M, et al. Reactivation of the tumor suppressor pten by mrna nanoparticles enhances antitumor immunity in preclinical models. Sci Transl Med (2021) 13(599):eaba9772. doi: 10.1126/scitranslmed.aba9772
191. Liu YT, Sun ZJ. Turning cold tumors into hot tumors by improving T-cell infiltration. Theranostics (2021) 11(11):5365–86. doi: 10.7150/thno.58390
192. Wang L, Gao Y, Zhang G, Li D, Wang Z, Zhang J, et al. Enhancing Kdm5a and tlr activity improves the response to immune checkpoint blockade. Sci Transl Med (2020) 12(560):eaax2282. doi: 10.1126/scitranslmed.aax2282
193. Li X, Xiang Y, Li F, Yin C, Li B, Ke X. Wnt/Beta-catenin signaling pathway regulating T cell-inflammation in the tumor microenvironment. Front Immunol (2019) 10:2293. doi: 10.3389/fimmu.2019.02293
194. Klempner SJ, Bendell JC, Villaflor VM, Tenner LL, Stein SM, Rottman JB, et al. Safety, efficacy, and biomarker results from a phase ib study of the anti-Dkk1 antibody dkn-01 in combination with pembrolizumab in advanced esophagogastric cancers. Mol Cancer Ther (2021) 20(11):2240–9. doi: 10.1158/1535-7163.MCT-21-0273
195. Ganesh S, Shui X, Craig KP, Park J, Wang W, Brown BD, et al. Rnai-mediated beta-catenin inhibition promotes T cell infiltration and antitumor activity in combination with immune checkpoint blockade. Mol Ther (2018) 26(11):2567–79. doi: 10.1016/j.ymthe.2018.09.005
196. Ebert PJR, Cheung J, Yang Y, McNamara E, Hong R, Moskalenko M, et al. Map kinase inhibition promotes T cell and anti-tumor activity in combination with pd-L1 checkpoint blockade. Immunity (2016) 44(3):609–21. doi: 10.1016/j.immuni.2016.01.024
197. Sullivan RJ, Hamid O, Gonzalez R, Infante JR, Patel MR, Hodi FS, et al. Atezolizumab plus cobimetinib and vemurafenib in braf-mutated melanoma patients. Nat Med (2019) 25(6):929–35. doi: 10.1038/s41591-019-0474-7
198. Heckler M, Ali LR, Clancy-Thompson E, Qiang L, Ventre KS, Lenehan P, et al. Inhibition of Cdk4/6 promotes Cd8 T-cell memory formation. Cancer Discovery (2021) 11(10):2564–81. doi: 10.1158/2159-8290.CD-20-1540
199. Deng J, Wang ES, Jenkins RW, Li S, Dries R, Yates K, et al. Cdk4/6 inhibition augments antitumor immunity by enhancing T-cell activation. Cancer Discovery (2018) 8(2):216–33. doi: 10.1158/2159-8290.CD-17-0915
200. Zhang QF, Li J, Jiang K, Wang R, Ge JL, Yang H, et al. Cdk4/6 inhibition promotes immune infiltration in ovarian cancer and synergizes with pd-1 blockade in a b cell-dependent manner. Theranostics (2020) 10(23):10619–33. doi: 10.7150/thno.44871
201. Grauel AL, Nguyen B, Ruddy D, Laszewski T, Schwartz S, Chang J, et al. Tgfbeta-blockade uncovers stromal plasticity in tumors by revealing the existence of a subset of interferon-licensed fibroblasts. Nat Commun (2020) 11(1):6315. doi: 10.1038/s41467-020-19920-5
202. Chen X, Wang L, Li P, Song M, Qin G, Gao Q, et al. Dual tgf-beta and pd-1 blockade synergistically enhances mage-A3-Specific Cd8(+) T cell response in esophageal squamous cell carcinoma. Int J Cancer (2018) 143(10):2561–74. doi: 10.1002/ijc.31730
203. Batlle E, Massague J. Transforming growth factor-beta signaling in immunity and cancer. Immunity (2019) 50(4):924–40. doi: 10.1016/j.immuni.2019.03.024
204. Lind H, Gameiro SR, Jochems C, Donahue RN, Strauss J, Gulley JM, et al. Dual targeting of tgf-beta and pd-L1 Via a bifunctional anti-Pd-L1/Tgf-Betarii agent: Status of preclinical and clinical advances. J Immunother Cancer (2020) 8(1):e000433. doi: 10.1136/jitc-2019-000433
205. Huinen ZR, Huijbers EJM, van Beijnum JR, Nowak-Sliwinska P, Griffioen AW. Anti-angiogenic agents - overcoming tumour endothelial cell anergy and improving immunotherapy outcomes. Nat Rev Clin Oncol (2021) 18(8):527–40. doi: 10.1038/s41571-021-00496-y
206. Zhao Y, Ting KK, Li J, Cogger VC, Chen J, Johansson-Percival A, et al. Targeting vascular endothelial-cadherin in tumor-associated blood vessels promotes T-Cell-Mediated immunotherapy. Cancer Res (2017) 77(16):4434–47. doi: 10.1158/0008-5472.CAN-16-3129
207. Fukumura D, Kloepper J, Amoozgar Z, Duda DG, Jain RK. Enhancing cancer immunotherapy using antiangiogenics: Opportunities and challenges. Nat Rev Clin Oncol (2018) 15(5):325–40. doi: 10.1038/nrclinonc.2018.29
208. Arina A, Beckett M, Fernandez C, Zheng W, Pitroda S, Chmura SJ, et al. Tumor-reprogrammed resident T cells resist radiation to control tumors. Nat Commun (2019) 10(1):3959. doi: 10.1038/s41467-019-11906-2
209. Tang H, Xu X, Chen Y, Xin H, Wan T, Li B, et al. Reprogramming the tumor microenvironment through second-near-Infrared-Window photothermal genome editing of pd-L1 mediated by supramolecular gold nanorods for enhanced cancer immunotherapy. Adv Mater (2021) 33(12):e2006003. doi: 10.1002/adma.202006003
210. Depil S, Duchateau P, Grupp SA, Mufti G, Poirot L. 'Off-the-Shelf' allogeneic car T cells: Development and challenges. Nat Rev Drug Discovery (2020) 19(3):185–99. doi: 10.1038/s41573-019-0051-2
211. Gumber D, Wang LD. Improving car-T immunotherapy: Overcoming the challenges of T cell exhaustion. EBioMedicine (2022) 77:103941. doi: 10.1016/j.ebiom.2022.103941
212. Song W, Zhang M. Use of car-T cell therapy, pd-1 blockade, and their combination for the treatment of hematological malignancies. Clin Immunol (2020) 214:108382. doi: 10.1016/j.clim.2020.108382
213. John LB, Devaud C, Duong CPM, Yong CS, Beavis PA, Haynes NM, et al. Anti-Pd-1 antibody therapy potently enhances the eradication of established tumors by gene-modified T cells. Clin Cancer Res (2013) 19(20):5636–46. doi: 10.1158/1078-0432.Ccr-13-0458
214. Gray KD, McCloskey JE, Vedvyas Y, Kalloo OR, Eshaky SE, Yang Y, et al. Pd1 blockade enhances Icam1-directed car T therapeutic efficacy in advanced thyroid cancer. Clin Cancer Res (2020) 26(22):6003–16. doi: 10.1158/1078-0432.Ccr-20-1523
215. Cherkassky L, Morello A, Villena-Vargas J, Feng Y, Dimitrov DS, Jones DR, et al. Human car T cells with cell-intrinsic pd-1 checkpoint blockade resist tumor-mediated inhibition. J Clin Invest (2016) 126(8):3130–44. doi: 10.1172/jci83092
216. Wang C, Shi F, Liu Y, Zhang Y, Dong L, Li X, et al. Anti-Pd-1 antibodies as a salvage therapy for patients with diffuse Large b cell lymphoma who Progressed/Relapsed after Cart19/20 therapy. J Hematol Oncol (2021) 14(1):106. doi: 10.1186/s13045-021-01120-3
217. Chong EA, Alanio C, Svoboda J, Nasta SD, Landsburg DJ, Lacey SF, et al. Pembrolizumab for b-cell lymphomas relapsing after or refractory to Cd19-directed car T-cell therapy. Blood (2022) 139(7):1026–38. doi: 10.1182/blood.2021012634
218. Wang Z, Li N, Feng K, Chen M, Zhang Y, Liu Y, et al. Phase I study of car-T cells with pd-1 and tcr disruption in mesothelin-positive solid tumors. Cell Mol Immunol (2021) 18(9):2188–98. doi: 10.1038/s41423-021-00749-x
219. Osborne W, Chen R, Jonnaert M, Khokhar NZ, Peddareddigari VGR, Pule M, et al. Phase 1/2 study of Auto3 the first bicistronic chimeric antigen receptor (Car) targeting Cd19 and Cd22 followed by an anti-Pd1 in patients with Relapsed/Refractory (R/R) diffuse Large b cell lymphoma (Dlbcl): Results of cohort 1 and 2 of the Alexander study. Blood (2019) 134(Supplement_1):246. doi: 10.1182/blood-2019-122724
220. Hirayama AV, Gauthier J, Hay KA, Sheih A, Turtle CJ. Efficacy and toxicity of Jcar014 in combination with durvalumab for the treatment of patients with Relapsed/Refractory aggressive b-cell non-Hodgkin lymphoma. Blood (2018) 132(Suppl_1):1680. doi: 10.1182/blood-2018-99-116745
221. Zurko J, Chaney K, Astle JM, Johnson BD, Hari P, Shah NN. Pd-1 blockade after bispecific Lv20.19 car T modulates car T-cell immunophenotype without meaningful clinical response. Haematologica. (2021) 106(10):2788–90. doi: 10.3324/haematol.2021.278461
222. Thommen DS, Schumacher TN. T Cell dysfunction in cancer. Cancer Cell (2018) 33(4):547–62. doi: 10.1016/j.ccell.2018.03.012
223. Blank CU, Haining WN, Held W, Hogan PG, Kallies A, Lugli E, et al. Defining 'T cell exhaustion'. Nat Rev Immunol (2019) 19(11):665–74. doi: 10.1038/s41577-019-0221-9
224. Zarour HM. Reversing T-cell dysfunction and exhaustion in cancer. Clin Cancer Res (2016) 22(8):1856–64. doi: 10.1158/1078-0432.Ccr-15-1849
225. Khan O, Giles JR, McDonald S, Manne S, Ngiow SF, Patel KP, et al. Tox transcriptionally and epigenetically programs Cd8(+) T cell exhaustion. Nature (2019) 571(7764):211–8. doi: 10.1038/s41586-019-1325-x
226. Wherry EJ, Kurachi M. Molecular and cellular insights into T cell exhaustion. Nat Rev Immunol (2015) 15(8):486–99. doi: 10.1038/nri3862
227. Tabana Y, Moon TC, Siraki A, Elahi S, Barakat K. Reversing T-cell exhaustion in immunotherapy: A review on current approaches and limitations. Expert Opin Ther Targets (2021) 25(5):347–63. doi: 10.1080/14728222.2021.1937123
228. Schoffski P, Tan DSW, Martin M, Ochoa-de-Olza M, Sarantopoulos J, Carvajal RD, et al. Phase I/Ii study of the lag-3 inhibitor ieramilimab (Lag525) +/- anti-Pd-1 spartalizumab (Pdr001) in patients with advanced malignancies. J Immunother Cancer (2022) 10(2):e003776. doi: 10.1136/jitc-2021-003776
229. Curigliano G, Gelderblom H, Mach N, Doi T, Tai D, Forde PM, et al. Phase I/Ib clinical trial of sabatolimab, an anti-Tim-3 antibody, alone and in combination with spartalizumab, an anti-Pd-1 antibody, in advanced solid tumors. Clin Cancer Res (2021) 27(13):3620–9. doi: 10.1158/1078-0432.CCR-20-4746
230. Mettu NB, Ulahannan SV, Bendell JC, Garrido-Laguna I, Strickler JH, Moore KN, et al. A phase 1a/B open-label, dose-escalation study of etigilimab alone or in combination with nivolumab in patients with locally advanced or metastatic solid tumors. Clin Cancer Res (2022) 28(5):882–92. doi: 10.1158/1078-0432.CCR-21-2780
231. Benzon B, Zhao SG, Haffner MC, Takhar M, Erho N, Yousefi K, et al. Correlation of B7-H3 with androgen receptor, immune pathways and poor outcome in prostate cancer: An expression-based analysis. Prostate Cancer Prostatic Dis (2017) 20(1):28–35. doi: 10.1038/pcan.2016.49
232. Wei H, Zhao L, Hellstrom I, Hellstrom KE, Guo Y. Dual targeting of Cd137 Co-stimulatory and pd-1 Co-inhibitory molecules for ovarian cancer immunotherapy. Oncoimmunology (2014) 3:e28248. doi: 10.4161/onci.28248
233. Pauken KE, Sammons MA, Odorizzi PM, Manne S, Godec J, Khan O, et al. Epigenetic stability of exhausted T cells limits durability of reinvigoration by pd-1 blockade. Science (2016) 354(6316):1160–5. doi: 10.1126/science.aaf2807
234. Wang X, He Q, Shen H, Xia A, Tian W, Yu W, et al. Tox promotes the exhaustion of antitumor Cd8(+) T cells by preventing Pd1 degradation in hepatocellular carcinoma. J Hepatol (2019) 71(4):731–41. doi: 10.1016/j.jhep.2019.05.015
235. Qorraj M, Bruns H, Bottcher M, Weigand L, Saul D, Mackensen A, et al. The pd-1/Pd-L1 axis contributes to immune metabolic dysfunctions of monocytes in chronic lymphocytic leukemia. Leukemia (2017) 31(2):470–8. doi: 10.1038/leu.2016.214
236. Ho PC, Bihuniak JD, Macintyre AN, Staron M, Liu X, Amezquita R, et al. Phosphoenolpyruvate is a metabolic checkpoint of anti-tumor T cell responses. Cell (2015) 162(6):1217–28. doi: 10.1016/j.cell.2015.08.012
237. Scharping NE, Menk AV, Whetstone RD, Zeng X, Delgoffe GM. Efficacy of pd-1 blockade is potentiated by metformin-induced reduction of tumor hypoxia. Cancer Immunol Res (2017) 5(1):9–16. doi: 10.1158/2326-6066.CIR-16-0103
238. Ohue Y, Nishikawa H. (Treg) cells in cancer: Can treg cells be a new therapeutic target? Cancer Sci (2019) 110(7):2080–9. doi: 10.1111/cas.14069
239. Whiteside TL. The role of regulatory T cells in cancer immunology. Immunotargets Ther (2015) 4:159–71. doi: 10.2147/ITT.S55415
240. Amoozgar Z, Kloepper J, Ren J, Tay RE, Kazer SW, Kiner E, et al. Targeting treg cells with gitr activation alleviates resistance to immunotherapy in murine glioblastomas. Nat Commun (2021) 12(1):2582. doi: 10.1038/s41467-021-22885-8
241. Hung AL, Maxwell R, Theodros D, Belcaid Z, Mathios D, Luksik AS, et al. Tigit and pd-1 dual checkpoint blockade enhances antitumor immunity and survival in gbm. Oncoimmunology (2018) 7(8):e1466769. doi: 10.1080/2162402X.2018.1466769
242. Liu J, Yuan Y, Chen W, Putra J, Suriawinata AA, Schenk AD, et al. Immune-checkpoint proteins vista and pd-1 nonredundantly regulate murine T-cell responses. Proc Natl Acad Sci U.S.A. (2015) 112(21):6682–7. doi: 10.1073/pnas.1420370112
243. Jacobs JF, Punt CJ, Lesterhuis WJ, Sutmuller RP, Brouwer HM, Scharenborg NM, et al. Dendritic cell vaccination in combination with anti-Cd25 monoclonal antibody treatment: A phase I/Ii study in metastatic melanoma patients. Clin Cancer Res (2010) 16(20):5067–78. doi: 10.1158/1078-0432.CCR-10-1757
244. Santoni M, Romagnoli E, Saladino T, Foghini L, Guarino S, Capponi M, et al. Triple negative breast cancer: Key role of tumor-associated macrophages in regulating the activity of anti-Pd-1/Pd-L1 agents. Biochim Biophys Acta Rev Cancer (2018) 1869(1):78–84. doi: 10.1016/j.bbcan.2017.10.007
245. Gordon SR, Maute RL, Dulken BW, Hutter G, George BM, McCracken MN, et al. Pd-1 expression by tumour-associated macrophages inhibits phagocytosis and tumour immunity. Nature (2017) 545(7655):495–9. doi: 10.1038/nature22396
246. Eisinger S, Sarhan D, Boura VF, Ibarlucea-Benitez I, Tyystjarvi S, Oliynyk G, et al. Targeting a scavenger receptor on tumor-associated macrophages activates tumor cell killing by natural killer cells. Proc Natl Acad Sci U.S.A. (2020) 117(50):32005–16. doi: 10.1073/pnas.2015343117
247. Li H, Xiao Y, Li Q, Yao J, Yuan X, Zhang Y, et al. The allergy mediator histamine confers resistance to immunotherapy in cancer patients Via activation of the macrophage histamine receptor H1. Cancer Cell (2022) 40(1):36–52.e9. doi: 10.1016/j.ccell.2021.11.002
248. Grauers Wiktorin H, Nilsson MS, Kiffin R, Sander FE, Lenox B, Rydstrom A, et al. Histamine targets myeloid-derived suppressor cells and improves the anti-tumor efficacy of pd-1/Pd-L1 checkpoint blockade. Cancer Immunol Immunother (2019) 68(2):163–74. doi: 10.1007/s00262-018-2253-6
249. Loeuillard E, Yang J, Buckarma E, Wang J, Liu Y, Conboy C, et al. Targeting tumor-associated macrophages and granulocytic myeloid-derived suppressor cells augments pd-1 blockade in cholangiocarcinoma. J Clin Invest (2020) 130(10):5380–96. doi: 10.1172/JCI137110
250. Freeman P, Mielgo A. Cancer-associated fibroblast mediated inhibition of Cd8+ cytotoxic T cell accumulation in tumours: Mechanisms and therapeutic opportunities. Cancers (Basel) (2020) 12(9):2687. doi: 10.3390/cancers12092687
251. Narra K, Mullins SR, Lee HO, Strzemkowski-Brun B, Magalong K, Christiansen VJ, et al. Phase ii trial of single agent Val-boropro (Talabostat) inhibiting fibroblast activation protein in patients with metastatic colorectal cancer. Cancer Biol Ther (2007) 6(11):1691–9. doi: 10.4161/cbt.6.11.4874
252. Kieffer Y, Hocine HR, Gentric G, Pelon F, Bernard C, Bourachot B, et al. Single-cell analysis reveals fibroblast clusters linked to immunotherapy resistance in cancer. Cancer Discovery (2020) 10(9):1330–51. doi: 10.1158/2159-8290.CD-19-1384
253. Feig C, Jones JO, Kraman M, Wells RJ, Deonarine A, Chan DS, et al. Targeting Cxcl12 from fap-expressing carcinoma-associated fibroblasts synergizes with anti-Pd-L1 immunotherapy in pancreatic cancer. Proc Natl Acad Sci U.S.A. (2013) 110(50):20212–7. doi: 10.1073/pnas.1320318110
254. Hanley CJ, Mellone M, Ford K, Thirdborough SM, Mellows T, Frampton SJ, et al. Targeting the myofibroblastic cancer-associated fibroblast phenotype through inhibition of Nox4. J Natl Cancer Inst (2018) 110(1):10920. doi: 10.1093/jnci/djx121
255. Ford K, Hanley CJ, Mellone M, Szyndralewiez C, Heitz F, Wiesel P, et al. Nox4 inhibition potentiates immunotherapy by overcoming cancer-associated fibroblast-mediated Cd8 T-cell exclusion from tumors. Cancer Res (2020) 80(9):1846–60. doi: 10.1158/0008-5472.CAN-19-3158
256. Deng L, Liang H, Burnette B, Beckett M, Darga T, Weichselbaum RR, et al. Irradiation and anti-Pd-L1 treatment synergistically promote antitumor immunity in mice. J Clin Invest (2014) 124(2):687–95. doi: 10.1172/JCI67313
257. Mondini M, Levy A, Meziani L, Milliat F, Deutsch E. Radiotherapy-immunotherapy combinations - perspectives and challenges. Mol Oncol (2020) 14(7):1529–37. doi: 10.1002/1878-0261.12658
258. Teng F, Kong L, Meng X, Yang J, Yu J. Radiotherapy combined with immune checkpoint blockade immunotherapy: Achievements and challenges. Cancer Lett (2015) 365(1):23–9. doi: 10.1016/j.canlet.2015.05.012
259. Shui L, Yang X, Li J, Yi C, Sun Q, Zhu H. Gut microbiome as a potential factor for modulating resistance to cancer immunotherapy. Front Immunol (2019) 10:2989. doi: 10.3389/fimmu.2019.02989
260. Simpson RC, Shanahan E, Scolyer RA, Long GV. Targeting the microbiome to overcome resistance. Cancer Cell (2021) 39(2):151–3. doi: 10.1016/j.ccell.2021.01.016
261. Routy B, Gopalakrishnan V, Daillere R, Zitvogel L, Wargo JA, Kroemer G. The gut microbiota influences anticancer immunosurveillance and general health. Nat Rev Clin Oncol (2018) 15(6):382–96. doi: 10.1038/s41571-018-0006-2
262. Derosa L, Routy B, Fidelle M, Iebba V, Alla L, Pasolli E, et al. Gut bacteria composition drives primary resistance to cancer immunotherapy in renal cell carcinoma patients. Eur Urol (2020) 78(2):195–206. doi: 10.1016/j.eururo.2020.04.044
263. Messaoudene M, Pidgeon R, Richard C, Ponce M, Diop K, Benlaifaoui M, et al. A natural polyphenol exerts antitumor activity and circumvents anti-Pd-1 resistance through effects on the gut microbiota. Cancer Discov (2022) 12(4):1070–87. doi: 10.1158/2159-8290.CD-21-0808
264. Cortes J, Cescon DW, Rugo HS, Nowecki Z, Im SA, Yusof MM, et al. Pembrolizumab plus chemotherapy versus placebo plus chemotherapy for previously untreated locally recurrent inoperable or metastatic triple-negative breast cancer (Keynote-355): A randomised, placebo-controlled, double-blind, phase 3 clinical trial. Lancet (2020) 396(10265):1817–28. doi: 10.1016/S0140-6736(20)32531-9
265. Sun JM, Shen L, Shah MA, Enzinger P, Adenis A, Doi T, et al. Pembrolizumab plus chemotherapy versus chemotherapy alone for first-line treatment of advanced oesophageal cancer (Keynote-590): A randomised, placebo-controlled, phase 3 study. Lancet (2021) 398(10302):759–71. doi: 10.1016/S0140-6736(21)01234-4
266. Chung HC, Bang YJ, S Fuchs C, Qin SK, Satoh T, Shitara K, et al. First-line Pembrolizumab/Placebo plus trastuzumab and chemotherapy in Her2-positive advanced gastric cancer: Keynote-811. Future Oncol (2021) 17(5):491–501. doi: 10.2217/fon-2020-0737
267. Paz-Ares L, Dvorkin M, Chen Y, Reinmuth N, Hotta K, Trukhin D, et al. Durvalumab plus platinum-etoposide versus platinum-etoposide in first-line treatment of extensive-stage small-cell lung cancer (Caspian): A randomised, controlled, open-label, phase 3 trial. Lancet (2019) 394(10212):1929–39. doi: 10.1016/S0140-6736(19)32222-6
268. Theelen W, Peulen HMU, Lalezari F, van der Noort V, de Vries JF, Aerts J, et al. Effect of pembrolizumab after stereotactic body radiotherapy vs pembrolizumab alone on tumor response in patients with advanced non-small cell lung cancer: Results of the pembro-rt phase 2 randomized clinical trial. JAMA Oncol (2019) 5(9):1276–82. doi: 10.1001/jamaoncol.2019.1478
269. McBride S, Sherman E, Tsai CJ, Baxi S, Aghalar J, Eng J, et al. Randomized phase ii trial of nivolumab with stereotactic body radiotherapy versus nivolumab alone in metastatic head and neck squamous cell carcinoma. J Clin Oncol (2021) 39(1):30–7. doi: 10.1200/JCO.20.00290
270. Hodi FS, Chesney J, Pavlick AC, Robert C, Grossmann KF, McDermott DF, et al. Combined nivolumab and ipilimumab versus ipilimumab alone in patients with advanced melanoma: 2-year overall survival outcomes in a multicentre, randomised, controlled, phase 2 trial. Lancet Oncol (2016) 17(11):1558–68. doi: 10.1016/S1470-2045(16)30366-7
271. Tawbi HA, Schadendorf D, Lipson EJ, Ascierto PA, Matamala L, Castillo Gutierrez E, et al. Relatlimab and nivolumab versus nivolumab in untreated advanced melanoma. N Engl J Med (2022) 386(1):24–34. doi: 10.1056/NEJMoa2109970
272. Goldman JW, Dvorkin M, Chen Y, Reinmuth N, Hotta K, Trukhin D, et al. Durvalumab, with or without tremelimumab, plus platinum-etoposide versus platinum-etoposide alone in first-line treatment of extensive-stage small-cell lung cancer (Caspian): Updated results from a randomised, controlled, open-label, phase 3 trial. Lancet Oncol (2021) 22(1):51–65. doi: 10.1016/S1470-2045(20)30539-8
273. Kelly RJ, Lee J, Bang YJ, Almhanna K, Blum-Murphy M, Catenacci DVT, et al. Safety and efficacy of durvalumab and tremelimumab alone or in combination in patients with advanced gastric and gastroesophageal junction adenocarcinoma. Clin Cancer Res (2020) 26(4):846–54. doi: 10.1158/1078-0432.CCR-19-2443
274. Rizvi NA, Cho BC, Reinmuth N, Lee KH, Luft A, Ahn MJ, et al. Durvalumab with or without tremelimumab vs standard chemotherapy in first-line treatment of metastatic non-small cell lung cancer: The mystic phase 3 randomized clinical trial. JAMA Oncol (2020) 6(5):661–74. doi: 10.1001/jamaoncol.2020.0237
275. Ferris RL, Haddad R, Even C, Tahara M, Dvorkin M, Ciuleanu TE, et al. Durvalumab with or without tremelimumab in patients with recurrent or metastatic head and neck squamous cell carcinoma: Eagle, a randomized, open-label phase iii study. Ann Oncol (2020) 31(7):942–50. doi: 10.1016/j.annonc.2020.04.001
276. Postow MA, Sidlow R, Hellmann MD. Immune-related adverse events associated with immune checkpoint blockade. N Engl J Med (2018) 378(2):158–68. doi: 10.1056/NEJMra1703481
277. Horvath L, Pircher A. Asco 2020 non-small lung cancer (Nsclc) personal highlights. Memo (2021) 14(1):66–9. doi: 10.1007/s12254-020-00673-2
278. Nocera L, Karakiewicz PI, Wenzel M, Tian Z, Shariat SF, Saad F, et al. Clinical outcomes and adverse events after first-line treatment in metastatic renal cell carcinoma: A systematic review and network meta-analysis. J Urol. (2022) 207(1):16–24. doi: 10.1097/JU.0000000000002252
279. Powles T, Plimack ER, Soulieres D, Waddell T, Stus V, Gafanov R, et al. Pembrolizumab plus axitinib versus sunitinib monotherapy as first-line treatment of advanced renal cell carcinoma (Keynote-426): Extended follow-up from a randomised, open-label, phase 3 trial. Lancet Oncol (2020) 21(12):1563–73. doi: 10.1016/S1470-2045(20)30436-8
280. Reck M, Mok TSK, Nishio M, Jotte RM, Cappuzzo F, Orlandi F, et al. Atezolizumab plus bevacizumab and chemotherapy in non-Small-Cell lung cancer (Impower150): Key subgroup analyses of patients with egfr mutations or baseline liver metastases in a randomised, open-label phase 3 trial. Lancet Respir Med (2019) 7(5):387–401. doi: 10.1016/S2213-2600(19)30084-0
281. Galle PR, Finn RS, Qin S, Ikeda M, Zhu AX, Kim TY, et al. Patient-reported outcomes with atezolizumab plus bevacizumab versus sorafenib in patients with unresectable hepatocellular carcinoma (Imbrave150): An open-label, randomised, phase 3 trial. Lancet Oncol (2021) 22(7):991–1001. doi: 10.1016/S1470-2045(21)00151-0
282. Motzer RJ, Robbins PB, Powles T, Albiges L, Haanen JB, Larkin J, et al. Avelumab plus axitinib versus sunitinib in advanced renal cell carcinoma: Biomarker analysis of the phase 3 javelin renal 101 trial. Nat Med (2020) 26(11):1733–41. doi: 10.1038/s41591-020-1044-8
283. Choueiri TK, Powles T, Burotto M, Escudier B, Bourlon MT, Zurawski B, et al. Nivolumab plus cabozantinib versus sunitinib for advanced renal-cell carcinoma. N Engl J Med (2021) 384(9):829–41. doi: 10.1056/NEJMoa2026982
Keywords: immune checkpoint blockade, combination therapy, T cell response, resistance mechanisms, immunotherapy
Citation: Zhou X, Ni Y, Liang X, Lin Y, An B, He X and Zhao X (2022) Mechanisms of tumor resistance to immune checkpoint blockade and combination strategies to overcome resistance. Front. Immunol. 13:915094. doi: 10.3389/fimmu.2022.915094
Received: 07 April 2022; Accepted: 19 August 2022;
Published: 15 September 2022.
Edited by:
Xuyao Zhang, Fudan University, ChinaReviewed by:
Chi-Ping Day, National Cancer Institute (NIH), United StatesCopyright © 2022 Zhou, Ni, Liang, Lin, An, He and Zhao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xia Zhao, eGlhLXpoYW9AMTI2LmNvbQ==
†These authors have contributed equally to this work and share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.