
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol., 26 May 2022
Sec. NK and Innate Lymphoid Cell Biology
Volume 13 - 2022 | https://doi.org/10.3389/fimmu.2022.914266
This article is part of the Research TopicInnate Lymphoid Cells: Characterization and ClassificationView all 16 articles
 Michela Calvi1
Michela Calvi1 Clara Di Vito2
Clara Di Vito2 Alessandro Frigo1
Alessandro Frigo1 Sara Trabanelli3,4
Sara Trabanelli3,4 Camilla Jandus3,4
Camilla Jandus3,4 Domenico Mavilio1,2*
Domenico Mavilio1,2*Innate lymphoid cells (ILCs) were firstly described by different independent laboratories in 2008 as tissue-resident innate lymphocytes mirroring the phenotype and function of T helper cells. ILCs have been subdivided into three distinct subgroups, ILC1, ILC2 and ILC3, according to their cytokine and transcriptional profiles. Subsequently, also Natural Killer (NK) cells, that are considered the innate counterpart of cytotoxic CD8 T cells, were attributed to ILC1 subfamily, while lymphoid tissue inducer (LTi) cells were attributed to ILC3 subgroup. Starting from their discovery, significant advances have been made in our understanding of ILC impact in the maintenance of tissue homeostasis, in the protection against pathogens and in tumor immune-surveillance. However, there is still much to learn about ILC ontogenesis especially in humans. In this regard, NK cell developmental intermediates which have been well studied and characterized prior to the discovery of helper ILCs, have been used to shape a model of ILC ontogenesis. Herein, we will provide an overview of the current knowledge about NK cells and helper ILC ontogenesis in humans. We will also focus on the newly disclosed circulating ILC subsets with killing properties, namely unconventional CD56dim NK cells and cytotoxic helper ILCs, by discussing their possible role in ILC ontogenesis and their contribution in both physiological and pathological conditions.
Starting from 2008, several independent laboratories around the world identified new players of innate immunity in both humans and mice (1–4). These cells, named innate lymphoid cells (ILCs), are a heterogeneous group of lymphocytes lacking recombination-activating gene (RAG)-dependent rearranged antigen-specific receptors.
ILCs originate from the common lymphoid progenitor (CLP) and require the common γ chain of the interleukin (IL)-2 receptor and the transcriptional repressor ID2 for their development (2–4). They are considered the innate counterpart of adaptive T lymphocytes: ILCs share with T cells the transcription factors governing their differentiation and the same cytokines in response to inflammatory insults (5, 6), which allows the classification of ILCs into different subsets. Indeed, the ILC1, ILC2 and ILC3 subsets produce T helper (Th)1-, Th2- and Th17/22-cytokines, respectively. Furthermore, given their phenotypic, developmental and functional similarities, Natural Killer (NK) cells, the innate counterpart of cytotoxic T lymphocytes, are now grouped together with ILC1s, whereas lymphoid tissue inducer (LTi) cells, belong now to group 3 ILCs (5, 7, 8).
Despite these similarities, ILCs arise from distinct developmental pathways and display unique epigenetic and transcriptional programs with respect to T cells, thus suggesting nonredundant roles of ILCs in immunity (9).
Differently from NK cells, that are mainly circulating lymphocytes, helper ILCs are primarily tissue resident cells and have been found in both lymphoid and non-lymphoid tissues. Indeed, they are particularly enriched at the mucosal surfaces of several organs, such as gut, lungs and skin, where they play a pivotal role in tissue homeostasis and disease, by promoting immune responses, inflammation, tissue repair and tolerance to commensal microbiota (10–12). Despite being a rare population in peripheral blood (PB), several lines of evidence indicate that circulating helper ILCs are characterized by a unique pattern of cytokine receptors, thus suggesting that they are not exclusively tissue resident (9, 13). Moreover, given their innate nature, ILCs represent one of the primary sources of pro- and anti-inflammatory cytokines during the early stages of the immune responses (14–17).
Although progresses have been made in understanding the role of ILCs in the maintenance of tissue homeostasis, in immune-defense and in tumor immune-surveillance, there is still much to learn concerning ILCs, especially in humans.
In this review we will summarize the principal features of ILCs, focusing mainly on circulating subsets. Moreover, we will provide an overview on ILC development in humans. In this context, we will focus on the newly disclosed circulating ILC subsets with cytotoxic properties, namely CD56dimCD16neg NK cells and cytotoxic helper ILCs, by discussing their possible role in ILC ontogenesis and their contribution in both physiological and pathological conditions.
ILCs are subdivided into three main groups based on the cytokine production, genetic signature and transcription factors involved in their development (Figure 1A).
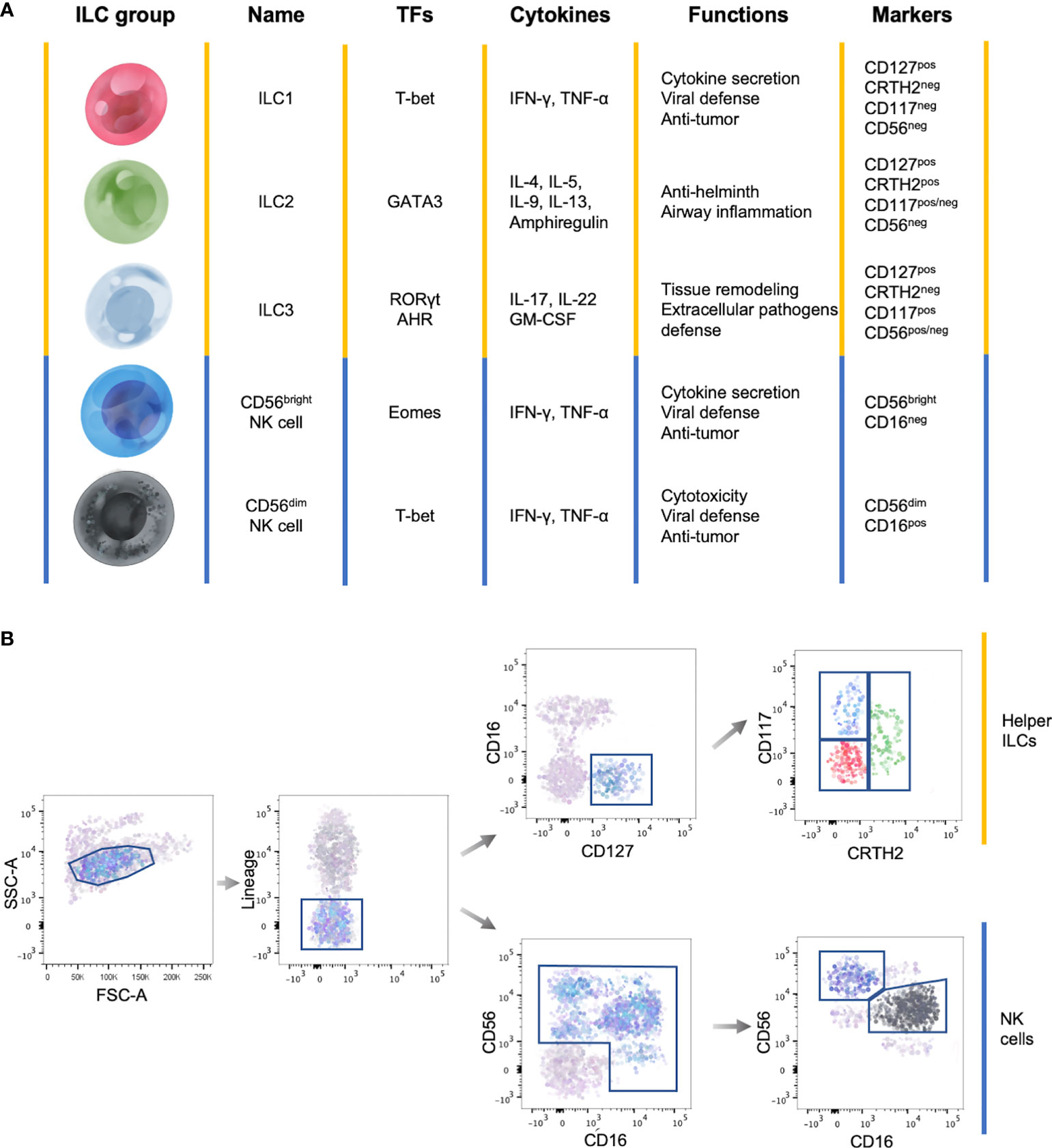
Figure 1 General features of ILC subsets. (A) Summary table showing the main transcription factors (TFs) governing the development, the cytokine production, the functions and marker expression in the different ILC subsets. (B) Representative flow cytometry gating strategy to identify circulating ILC subsets among Lineageneg lymphocytes. Yellow lines identify helper ILCs, while the blue lines identify NK cells.
Total circulating ILCs, which comprises helper ILCs and NK cells, are identified as lineage (CD3, CD14, CD15, CD19, CD20, CD33, CD34, CD203c and FcϵRI) negative lymphocytes (Figure 1B) (17). This lineage markers can be used also to identify tissue resident ILCs, with the exception of human tonsil ILC3s which express CD33 (18). Total helper ILCs are defined as CD16neg cells that constitutively express the IL-7 receptor-α chain (CD127) and the different subsets are identified according to the expression of CRTH2, cKit (CD117) and CD56: ILC1s are CRTH2negCD117negCD56neg, ILC2s are CRTH2posCD117pos/negCD56neg and ILC3s CRTH2negCD117posCD56pos/neg (Figure 1B) (5, 17, 19). However, it has been shown that ILC2s, in some cases, can express CD56 (20). In addition, among lineage negative cells, NK cell subsets are identified according to the differential expression of CD56 and CD16 (Figure 1B) (21).
Despite the phenotype of the different ILC subsets is well defined, their precise distribution across organs and species needs to be refined. Indeed, ILC subset distribution and cytokine production profiles are impacted by environmental cues, that enable a prompt response in case of inflammatory insults without the de novo recruitment of ILC subsets (22, 23).
NK cells and ILC1s have been grouped together in group 1 ILCs. Indeed, both subsets are characterized by the production of interferon-γ (IFN-γ) and tumor necrosis factor α (TNF-α) in response to IL-12 and IL-18 (5, 7, 24). Moreover, IL-15 is required for the differentiation, homeostasis, and function of both NK cells and ILC1s (25). Similarly to NK cells, ILC1s express natural cytotoxicity receptors (NCRs) (26) and the T-box transcription factor expressed in T cells (T-bet) (25, 27). In addition, both ILC1s and NK cells require the expression of HOBIT TFs, encoded by Zfp683, for their differentiation and functional programs (28–30).
Despite their overlapping features, NK cells and ILC1s are two phenotypically and functionally distinct immune cell subsets. Indeed, while ILC1s require T-bet for their development, NK cells need T-bet expression only for maturation and require the expression of the transcription factor Eomesodermin (Eomes) for differentiation (25, 27). Moreover, ILC1s are fundamentally tissue-resident lymphocytes, whereas NK cells can circulate across lymphoid organs via the bloodstream and lymphatic system to act as immune sentinels. Indeed, NK cells are unique in their ability to recognize and kill virally infected and malignantly transformed cells, through the balance of the signaling that NK cells receive from their repertoire of activating and inhibitory NK cell receptors (31, 32).
Circulating NK cells comprise two main subsets, a cytotoxic CD56dim and a regulatory CD56bright NK cell subset. CD56dim NK cell subset accounts for up to the 90% of circulating NK cells. They show high baseline perforin expression and are endowed with cytotoxic abilities against target cells not expressing or downregulating the major histocompatibility complex (MHC) class I molecules on their surface. They preferentially produce cytokines in response to direct target cell interactions rather than via monocyte-derived cytokines stimulation (33). On the other hand, CD56bright NK cells represent the 10% of circulating NK cells, while they are enriched in peripheral and lymphoid tissues (34). Differently from CD56dim NK cells, and similarly to ILC1s, CD56bright NK cells are poorly cytotoxic, can rapidly secrete cytokines, including IFN-γ, following stimulation by monocyte-derived cytokines (35).
Group 2 ILCs are involved in several processes, including lipid metabolism, protection against parasites and accumulate during type 2 inflammation in the airways (36). Among all ILCs, ILC2s express the highest level of the transcription factor GATA3 and are characterized by the production of Th2-associated cytokines, such as IL-4, IL-5, IL-9, IL-13 and amphiregulin (AREG) in response to IL-25, IL-33 and thymic stromal lymphopoietin (TSLP). Hence, they are considered the innate counterpart of Th2 cells (37–39). Of note, several lines of evidence indicate that ILC2s can produce higher levels of cytokine than T cells, thus suggesting their primary role in the innate local immunity against infections in different organs (40). Indeed, they have been described in a variety of human tissues, including tonsils, bone marrow (BM), spleen, skin, adenoids, adipose tissue, lung lymph nodes (LNs) (41).
ILC2s are characterized by the expression of the transcription factor BCL11B which controls their identity (42), the prostaglandin D2 receptor 2 (CRTH2), the IL-33 receptor (IL1RL1 also referred as ST2) and by variable levels of c-Kit (43), all involved in ILC2 localization and function (43–46). More recently, it has been shown that ILC2s are further characterized by the expression of the killer cell lectin-like receptor subfamily G member 1 (KLRG1), a co-inhibitory receptor previously reported in T and NK cells that binds to the members of the cadherin family and is upregulated during infection in response to IL-25 (20, 47).
Group 3 ILCs, consisting of ILC3s as well as LTis, share features with Th17 cells, including the expression of the transcription factor retinoic acid orphan receptor isoform γt (RORγt), required for their development and function, and the aryl hydrocarbon receptor (AHR) (7, 48). Group 3 ILCs are capable of producing IL-17, IL-22 and granulocyte-macrophage colony-stimulating factor (GM-CSF) in response to IL-23, IL-1β, or natural cytotoxicity receptor ligands (NCR-L), thus mirroring Th17 response (5, 49). Regardless of the functional association with ILC3s, LTis are considered a separate ILC lineage (49, 50). Indeed, LTis are involved in the secondary lymphoid organ formation during embryogenesis and adulthood and in their restoration following infection (51), whereas ILC3s mainly contribute to the immune responses against specific extracellular pathogens and in the maintenance of tissue homeostasis at mucosal sites, where they are mainly localized (48).
ILC3s can be further subdivided according to the expression of the NCR NKp44: human NKp44pos ILC3s, largely co-expressing NKp46, are the majority in adult tonsil and intestine and represent an exclusive source of IL-22, while NKp44neg ILC3s are the major population in fetal LNs and preferentially express IL-17 transcripts (48).
Differently from tissues, ILC3s are under-represented in the circulating lymphocyte pool. However, recently, a subpopulation of cells phenotypically resembling to NKp44neg ILC3s has been described in cord and adult PB. However, most peripheral-blood CD127posCD117pos ILCs express low levels of RORγt, do not produce any of the ILC cytokine signatures following stimulation with IL-1β and IL-23 and their transcriptome is different from that of mature ILC3s present in secondary lymphoid organs (52, 53). Circulating CD127posCD117pos ILCs are instead multi-potent ILC precursors (ILCPs) that retain the ability to give rise to functionally mature helper ILC subsets, as well as to Eomespos NK cells, after in vitro culture with appropriate cytokine mix or after transfer in vivo into immunodeficient mice (49, 53).
ILC-poiesis has been a topic of ardent research in the last several years. Although many issues remain to be disclosed, including the transcriptional regulators that dictate the choice of mature ILC subset fate, the ILC differentiation in mouse has been deeply investigated, thanks to genetically modified rodent models. On the contrary, the study of the human ILC differentiation is still in its infancy because of different reasons. Firstly, the lack of comparable genetic and tracing tools that are available in animal models of lymphoid development. Secondly, the heterogeneity of the studies conducted in terms of definition of progenitor, identification of lineages based solely on cytokine secretion or on few surrogate markers.
Despite these limitations, over the past years a map of human ILC development has been under construction (Figure 2) by taking advantage of both murine ILC-developmental model and the pre-existing model of human NK cell development, based on the identification and characterization of human NK cell developmental intermediates (NKDIs) prior to the discovery of helper ILCs (54).
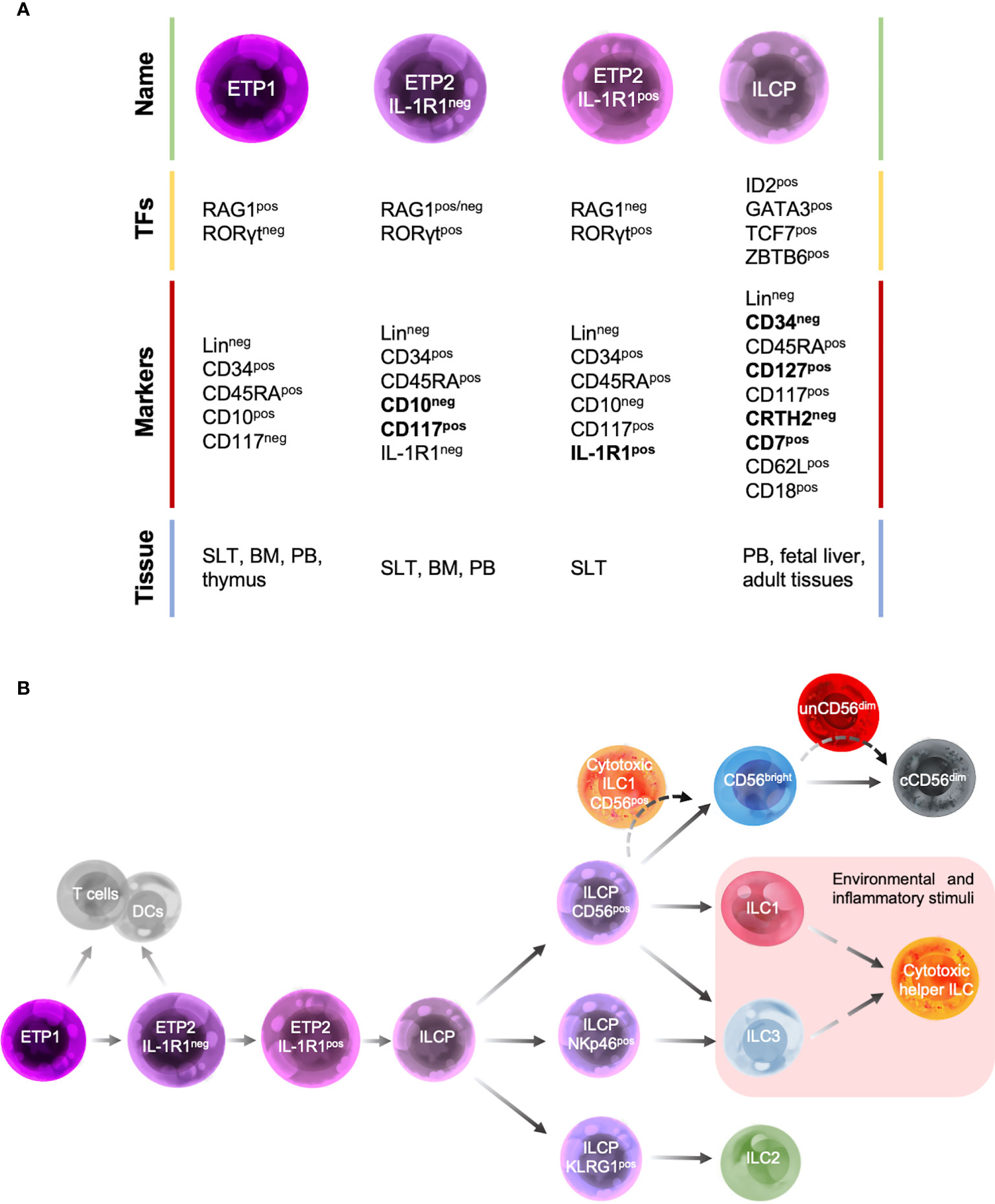
Figure 2 Human ILC developmental stages. (A) Summary table of the main transcription factors (TFs), marker expression and tissue localization of common precursors/progenitors. (B) Schematic representation of different stages of human ILC-poiesis starting from the early tonsillar progenitors (ETPs), that still retain the ability to give rise to T cells and dendritic cells (DCs), to ILC precursor (ILCPs) that branches into progenitors with restricted differentiation potential and give rise to different mature ILC subsets. Dashed lines indicate the hypothetic developmental pathways of unconventional cytotoxic ILC subsets.
Since their discovery in 1970s, NKDIs have been well characterized in both mice and humans as well as the site for NK cell development.
Human NK cells were originally thought to develop strictly within the BM (33, 55). Indeed, CD34posCD45RApos precursors with ex vivo potential for NK cell differentiation were firstly identified in the BM, and then were also found in PB and in extramedullary tissues, including thymus, secondary lymphoid tissues (SLTs), liver, and uterus, at steady state conditions (56–58). Noteworthy, IL-15-responsive CD34posCD45RApos precursors comprise a relatively higher proportion of total CD34pos progenitor cells (5-10%) in the blood compared to bone marrow (<1%), and in SLT they comprise the major subset of CD34pos progenitor cells (>90%) (59, 60). In this regard, several lines of evidence demonstrated that CD34posCD45RApos precursors originate in the BM and traffic to extramedullary tissues where later stages of NK cell differentiations can take place, giving rise to tissue-specific and functionally distinct mature NK cell subsets. In particular, tonsils, spleen, and lymph nodes are considered those SLTs hosting the main extra-medullary sites of NK cell development and maturation (57, 59–62).
Originally, five main sequential stages of NK cell maturation were identified: NK cell progenitors (NKPs, stage 1), pre-NK cells (stage 2), immature NK cells (iNK, stage 3) (63–65) and the mature CD56bright (stage 4) and CD56dim (stage 5) NK cell subsets [reviewed in (62)].
Briefly, NKPs and pre-NK cells still express CD34 and retain the ability to differentiate into T cells, dendritic cells (DCs) and other ILCs. Subsequently, the expression of CD122 (IL-2Rβ), together with the downregulation of CD34, marks the irreversible fate decision into NK cell lineage. The commitment of NKPs towards pre-NK cells also required the acquisition of CD117 expression (64, 66). Recently, the stage 2 has been further subdivided into two additional stages: the IL-1R1neg stage 2a, mainly enriched in SLTs and PB, that still retains the ability to give rise to T cells and DCs, and the IL-1R1pos stage 2b, with a commitment restricted to the generation of ILCs, including NK cells (62, 67). IL-1R1pos stage 2b give rise to iNKs whose features, including AHR, CD127, RORγt, IL-1R1, and IL-22 expression, mirror those of Group 3 ILCs (5, 68, 69). Indeed, it is not yet clear if iNKs and ILC3s are entirely overlapping at least in their phenotypic characteristics and further investigation is needed.
The final transition of iNK into mature NK cell subsets is marked by the appearance of CD56 expression and the main functional properties, including cytokine secretion (i.e. TNF, IL-8, GM-CSF, CXCL12 and IL-13, together with IL-22), IFN-γ release and then cytolytic activity (58, 70–72). Two distinct stages 4 CD56bright NK cells have been described: the NKp80neg stage 4a cells, that, despite the bright expression of CD56, are more similar to iNKs cells due to their higher expression of transcription factors RORγt and AHR, their higher production of IL-22 and their preferential localization in SLTs, the NKp80pos stage 4b cells, expressing higher levels of T-bet and Eomes and producing IFN-γ (73). Subsequently, through the acquisition of CD16, Killer Immunoglobulin-like receptors (KIRs) and cytotoxic granules, the fully mature CD56dim NK cells, endowed with cytolytic potential and able to perform antibody-dependent cell-mediated cytotoxicity (ADCC) are generated.
Despite the process of NK cell-poiesis is well defined, with the identification and characterization of other subtypes of ILCs, the old model of NK cell ontogenesis needs to be reassessed and refined in the context of new data about helper ILCs (74).
Murine ILC differentiation is regulated by a wide range of transcription factors, including Id2, Nfil3, Zbtb16, Tcf7, Gata3, Ets1, and Tox [reviewed in (75)].
Two distinct progenitors downstream of the murine CLP have been identified, each with restricted ILC potential. These include the CXCR6posα4β7posCD127pos α-lymphoid precursor (αLP) and the LinnegThy1.2negCD127negα4β7pos early innate lymphoid progenitor (EILCP) that along with the other downstream progenitors are most prevalent in murine BM (54, 76). The transcription factor Nfil3 is required for the generation of αLP. Indeed, Nfil3 knock out mouse models lack mature ILCs, including NK cells (76–78).
On the other hand, EILCPs, requires the transcription factor Tcf7 (79). Given the drastic reduction of αLPs together with EILCPs in Tcf7-deficient mice, it has been also proposed that αLP could constitute an intermediate stage of maturation between CLP and EILCP (76).
Two additional EILCP subsets with different commitment potential have been identified: the early-stage EILCPs (EILP1s), that can give rise to DCs as well as cytotoxic and helper ILCs, and the committed EILPs (cEILCP) expressing TCF-1 and losing the ability to differentiate in DCs (80).
The EILCP can further differentiate in Id2-dependent LinnegId2posIL-7RposIL-2Rαnegα4β7pos common helper-like ILC progenitors (CHILPs), which can give rise to helper-like ILCs (ILC1s, ILC2s, and ILC3s) and LTis but not to NK cells (79, 81) or in Id2-independent NK1.1negIL-2Rβpos NKPs (82, 83).
CHILPs can be separated into two different subsets based on the expression of Zbtb16. The Zbtb16neg common ILC precursor (CILCP) which can no longer produce LTi cells (83, 84) and the Zbtb16pos ILC precursor (ILCP) capable of potentially giving rise to conventional NK cells, ILC1s and ILC2s, but with a reduced ability to differentiate into ILC3s (83–86).
According to NK cell developmental model, the most immature ILC progenitors identified in humans are mainly localized in SLTs and were originally described as stage 1 and stage 2 NKIDs and are now defined as early tonsillar progenitors (ETPs) in the context of ILC-poiesis (Figure 2A). In particular, ETPs are subdivided into LinnegCD34posCD10posCD117neg ETP1 and LinnegCD34posCD10negCD117pos ETP2 (54, 63). Both ETP1s and ETP2s are multipotent and could also give rise to T cells and DCs in vitro (54, 63). ETP2s are heterogeneous in terms of IL-1R1 expression. IL-1R1neg ETP2s have a residual T-cell and DC potential, whereas IL-1R1pos ETP2s are ILC restricted. These population are also characterized by a unique transcription factor signature: ETP1 are RAG1pos and express low levels of ID2 and RORγt, IL-1R1neg ETP2s are ID2posRORγtpos and retain a low expression of RAG1, whereas IL-1R1pos ETP2s are ID2posRORγtposRAG1neg (Figures 2A, B) (67). Hence, IL-1R1posETP2s are the earliest committed human CILCP identified to date.
In addition, a LinnegCD34negCD7posCD127posCD117posCRTH2neg ILC progenitor (ILCP), with phenotypic features that overlap those of stage 3 NKIDs endowed with a restricted potential for ILC generation, has been recently identified in human cord and adult PB as well as fetal liver and several adult tissues. Upon in vitro culture with an appropriate cytokine environment or after transfer in vivo to immunodeficient mice, these human ILCPs demonstrate their potential for generating all mature helper- and cytotoxic-ILCs (53).
Consistent with their differentiation potential, ILCPs express high levels of transcription factors that have been shown to be essential for mouse ILC development, such as ID2, GATA3, TCF-7 and ZBTB16 (Figure 2A). In contrast, moderate to low levels of the lineage-determining transcription factors RORγt, T-bet, Eomes, cytokine receptors (including IL-1R1), and signature cytokines have been found (53). ILCPs show a migratory profile including the expression of L-selectin (CD62L) and β2-integrin (CD18), which would allow these cells to populate the mucosal tissues where they terminally differentiate into mature ILCs in response to local and environmental triggers, during both homeostatic and inflammatory conditions (54, 87, 88). Consistent with this idea, ILCPs were found at mucosal sites where they mature (53).
In light of this evidence, ILCPs might be the equivalents of naïve CD4pos T cells. Indeed, they both express CD45RA and CD62L and the development of ILC subsets from ILCP parallels that of CD4pos T cell subsets from naive CD4pos T cells, with similar polarizing cytokines and transcription factors being required for their differentiation, although the differentiation of naïve CD4pos T cell subsets also depends on TCR signaling and CD28 co-stimulation (22).
Different ILCP populations with a restricted differentiation potential have been recently described and can be identified based on the expression of CD56, NKp46 and KLRG1. CD56pos ILCPs show a restricted potential for NK cells, ILC1s and ILC3s, NKp46pos ILCP predominantly differentiate into ILC3s, whereas KLRG1pos ILC precursors mostly develop into ILC2s (Figure 2B) (88, 89).
Adding complexity to this scenario there is the so called ILC plasticity phenomenon. Indeed, it has been demonstrated in both humans and mice, that ILC subsets can switch into another subset depending on the presence of cytokines and NOTCH ligands in their environment. This process is regulated by a complex network of transcription factors. Briefly, ILC2s and ILC3s transdifferentiate into ILC1s in response to IL-1β and IL-12, whereas IL-1β and IL-23 can drive the plasticity of ILC1s and ILC2s towards ILC3s. Despite ILC2s lack the expression of IL-23 receptor, IL-1 β is known to induce ILC2s responsiveness to IL-23 by STAT3 phosphorylation (90). The transdifferentiation of ILC2s into ILC1s or ILC3s can be reversed by IL-4. ILC2s requires TGF-β in addition to IL-1β and IL-23 to differentiate into ILC3s (22). Moreover, NK cells, in a TGF-β-rich tumor environment, transdifferentiate into ILC1-like cells devoid of cytotoxic activity (91, 92). Likely, the plasticity of ILCs is the mechanism of tissue-resident ILCs to dynamically adapt to a given stimulus, such as an inflamed state (23).
Until recently, NK cells were considered the only cytotoxic innate lymphocytes, being functionally associated with CD8 T cells (7). However, owing to the highly plastic nature of ILCs, increasing evidence shows that beside their ability to transdifferentiate between helper subpopulations, these cells can also acquire cytotoxic capacities upon defined environmental conditions (93). Indeed, it has been recently reported that, upon exposure to cytokine cocktails, ILC3s or ILC1s, isolated from human tonsils, secondary lymphoid organs (e.g., spleen) and intestinal tissues of humanized mice, give rise to cytotoxic lymphocytes resembling stage 4a NK cells (94, 95). In particular, hallmark NK cell genes, such as NCAM-1, KLRD1, KLRC1, CD2, CD226 have been reported to be significantly upregulated in ILC3s or ILC1s exposed to IL-12/IL-15, and to be paralleled by the acquisition of cell surface expression of at least some of these markers (e.g. NKG2A, NKG2C, CD2). Moreover, these cells show a weak IFN-γ secretion in response to K562, but an efficient perforin- and granzyme-dependent cytotoxicity, primarily mediated by the EomesposT-betpos fraction of stimulated ILC3s. Nevertheless, these cytotoxic responses remain weaker and of slower kinetics as compared to that of conventional NK cells.
Phenotypically, the distinction between human cytotoxic helper ILCs and NK cells is complicated by the fact that many markers are shared, including CD56, CD161, NKp44 and NKp46. The same stands for mouse ILCs and NK cells that share the expression of NKp46 and NK1.1. Possible discriminators to distinguish cytotoxic helper ILCs, or helper ILCs in general, from NK cells include CD127, which is constitutively expressed by both human and murine helper ILC subsets but not by terminally-differentiated CD56dim NK cells (67) and only at intermediate levels on human peripheral blood CD56bright NK cells (96). Noteworthy, in mice, ILCs with cytotoxic potential were also described within NK1.1posCD49apos cells, lacking CD127 expression (26).
Another marker enabling the discrimination between helper ILCs and NK cells might be the inhibitory receptor CD200R1. The expression of CD200R1 has been described to be specific for ILCs in human blood and tonsils (20), and in mice (84, 97). However, intestinal NK cells have been recently described to express CD200R1, although at lower levels as compared to conventional ILCs (95). Inversely, CD200R1 is poorly expressed by intraepithelial ILC1s in the small intestine, lamina propria and visceral adipose tissue (98). Overall, these results highlight the difficulty to identify a universal marker enabling ultimate discrimination between helper ILCs, including cytotoxic ones, and NK cells. Nevertheless, despite potential overlapping phenotypes and functions, the distinct anatomic distribution of cytotoxic helper ILCs and NK cells argues for complementary roles in the protection against infections or emerging malignancies.
Evidence of in vivo ability of helper ILCs to acquire cytotoxic features has been recently shown in different districts, as detailed in the next paragraphs.
In the context of NK cell development, an additional NK cell subset has been recently identified. This subset, named unconventional NK cells (unCD56dim), displays a CD56dimCD16neg/CD56lowCD16low phenotype. Barely detectable in the PB, this NK cell subset is mainly enriched in the BM (99, 100). On the other hand, their presence in extramedullary tissues has not been investigated so far.
UnCD56dim NK cell subset expresses surface markers of mature NK cells, such as NKG2D and NKp30 (99). Moreover, this NK cell subset is equipped with lytic molecules, thus suggesting its putative role in mediating cytotoxic responses.
However, their phenotype and transcriptional profile suggest that unCD56dim NK cells are a bona fide NK cell subset distinct from activated CD56dim that underwent CD16 shedding mediated by the metalloproteinase-17 (101). Indeed, compared to CD56dim NK cells, unCD56dim NK cells show higher levels of CD27, whose expression was described to decline during NK cell development in mice, and lower levels of markers of terminally differentiated and licensed NK cells, namely CD57 and KIRs, which are acquired at late stages of NK cell differentiation (100). Moreover, unCD56dim NK cells display higher levels of CD25, CD122 and CD127, the receptor chains for IL-2, IL-15 and IL-7 cytokines, which play a major role in controlling NK cell development, homeostasis, survival and activation (100). The chemokine receptor expression pattern of unCD56dim is consistent with a less differentiated phenotype compared to CD56dim NK cells. Indeed, they are characterized by undetectable CX3CR1 expression, which is usually acquired during NK cell development, and by a higher expression of CXCR4 compared to the other conventional NK cell subsets, in line with their preferential BM localization (100).
In the context of the lymphopenic environment of patients affected by hematologic malignancies and which underwent haploidentical hematopoietic stem cell transplantation (haplo-HSCT), others and we reported that unCD56dim NK cells are by far the largest subset of NK cells immune-reconstituting in the first 2-4 weeks after the transplant, compensating the low frequency of the conventional cytotoxic CD56dim NK cells (99, 102). These data, together with the transcriptional and phenotypic characteristics of unCD56dim NK cells, intermediate between that of CD56bright and CD56dim NK cells, suggest that this subset could represent an additional or alternative stage of NK cell differentiation that drives the NK cell maturation process (Figure 2B) (99, 100).
In vitro experimental evidence also suggests that unCD56dim NK cells possess multifunctional ability and superior effector-functions. Indeed, despite poorly present under homeostatic conditions in the PB, they are endowed with potent cytotoxicity, significantly higher than that of CD56dim, and an IFN-γ producing capability comparable to that of CD56bright in response to cytokine stimulation (99, 100, 103). On the contrary, immune-reconstituting unCD56dim NK cells, highly expanded early after haplo-HSCT, are anergic due to a high expression of CD94/NKG2A, an inhibitory receptor involved in NK cell differentiation and education, thus further supporting the assumption of unCD56dim NK cells as a distinct NK cell subset and highlighting their key role in NK cell development. Moreover, this observation allowed us to develop a phase II clinical trial (ONC-2020-001) by using an anti-NKG2A humanized monoclonal antibody (humZ270 mAb, IPH2201, monalizumab, AstraZeneca) to block this inhibitory checkpoint, unleashing alloreactive unCD56dim NK cells, thus potentially improving the clinical outcome of haplo-HSCT early after transplant (99, 104, 105).
The ex vivo analysis of human tonsil ILCs has shown the existence of a population of CD94pos cells, that co-expressed CD200R1, while negative for CD16, NKp80 and KIRs. In terms of transcriptomic profiles, these cells cluster close to NKp44pos ILC3s, sharing with them the expression of RORγt, but being distinct in terms of cytotoxic gene expression (e.g., Eomes, Granzymes, Granulysin) (106). This gene expression pattern has been also correlated with direct cytotoxic activity against the target cell line K562, which is predominant for the CD94posNKp44neg ILC3 subpopulation. Additional evidence of the in vivo generation of cytotoxic helper ILCs in human inflamed tonsils has been recently reported by combining bulk and single cell RNA sequencing (scRNAseq), flow- and mass-cytometry studies (107). In particular, it has been demonstrated that ILC3s and ILC1s reside at the terminal ends of a differentiation spectrum. Between these two extremes, four ILC3-ILC1 intermediates, with a NKp44posCD56pos phenotype, emerge by RNA velocity analyses. These intermediates are characterized by the gradual acquisition of genes expressed in conventional NK cells, such as KLRD1, KLRC2, GZMB and GZMK, loss of RORG, CD200R1, KIT, IL7R and upregulation of Tbx21 and IKZF3.
We recently identified a CD56-expressing subset of circulating ILCs with high cytotoxic potential that belong to conventional ILC1s, being characterized by the lack of lineage markers, the expression of CD127 and the absence of CD117 and CRTH2 (108). RNA-sequencing analysis revealed a transcriptional profile closer to ILCPs and NK cells rather than ILC1s. These CD56posILC1-like cells showed distinct nutrient uptake and mitochondrial activity in comparison to helper ILCs and NK cells, low expression of NKp46 and the capacity to produce IL-8, beside IFN-γ, when stimulated with IL-12, IL-15 and IL-18. Through comparison with NK developmental intermediates in terms of phenotype (73), by verifying their presence in patients with severe combined immunodeficiency, in human fetal tissues and during immune reconstitution in humanized mice, and by assessing their capacity to differentiate into conventional ILCs/NKs when cultured on OP9 mouse stromal cells, we concluded that they are related to the developmental stage 4a of NK cells. The cytotoxic machinery of CD56posILC1-like ILCs comprised DNAM-1, NKp30, NKp80 and TRAIL, as well as the ability to produce perforin, granzyme A, B, K, M and granulysin. Functionally, CD56posILC1-like ILCs can kill both the MHC-Ineg K562 cell line and MHC-Ipos cell lines, such as BJAB and U937, in accordance with the absence of KIRs. Their cytotoxic capacity is dependent on the expression of NKp30, NKp80 and TRAIL, since the addition of specific blocking antibodies can inhibit their killing ability. We further investigated their presence and function in acute myeloid leukemia (AML), a hematologic malignancy characterized by a dysfunctional helper ILC compartment (109, 110). The data obtained demonstrated that in AML patients at diagnosis, the cytotoxic machinery of CD56posILC1-like ILCs is completely impaired, resulting in their inability to kill target cells, either K562 or autologous blasts. On the other hand, AML patients that achieved remission showed a completely restored function of CD56posILC1-like cells. Interestingly, CD56posILC1-like ILCs but not conventional NK cells from AML patients at diagnosis had a reduced expression of TRAIL, NKp80 and granulysin, thus suggesting that despite their relatedness they are distinct populations able to differentially react to the microenvironment. Further studies are needed to determine whether this population is an intermediate between helper ILCs and NK cells, or a specific cytotoxic circulating helper ILC subset.
In a recent study aimed at creating a high-dimensional tissue map of human NK cells across multiple tissues, cytotoxic helper ILCs have been also identified (111). Indeed, CD56posCD3neg cells expressing CD127, CD56 and CD161 have been found to be enriched in the intestine, as well as in lung-draining lymph nodes and mesenteric lymph nodes. These cells secreted IFN-γ upon stimulation and upregulated CD107a when co-cultured with MHC-Ineg targets, suggesting that they might represent either immature NK cells or tissue-resident cytotoxic helper ILC3s, present at selected anatomical sites. These cells might be key in maintaining intestinal homeostasis by cytokine secretion. In that regard, intraepithelial cytotoxic ILCs, very much resembling NK cells, have also been identified by others in the intestinal mucosa (26). These cells are characterized by the expression of CD56, Eomes, T-bet, variable levels of NKp44 and CD103, low levels of CD94 and CD127. This phenotype has not been observed in other anatomic locations, suggesting a selective localization of this subset in the mucosal epithelium of the gut. In vitro characterization of these cells revealed that they are able to respond to IL-12 and IL-15 stimulation by secreting IFN-γ, to produce Granzyme B and to express CD107a when exposed to cell targets. Studies in mice showed that these intraepithelial cytotoxic ILCs rely on Nfil3- and Tbx21-transcription factors, while IL-15 is dispensable for their development, suggesting that, at least in mice, they are distinct from conventional NK cells. The investigation of their putative role in gut inflammation revealed that their frequency is increased in patients with Crohn’s disease as well as in in vivo models of colitis, arguing for a direct involvement in pathology. In a more recent study, amplified CD94posCD127pos ILCs has been reported in the intestinal lamina propria in adults, but not in the epithelium, as opposed to the previous study (95). scRNAseq analysis of healthy and Chron’s disease patients’ tissue specimens revealed the existence of two clusters of lamina propria CD94posCD127pos cells, with cytotoxic attributes. These cells express Eomes and CD200R1, but lacked CD16 expression. Heterogeneity was observed in terms of CD161, CD117, CD18, cytotoxic molecules (e.g., granzymes and perforin) and granulysin expression. Specifically, the granulysinhigh perforinhigh subpopulation is highly amplified in Chron’s disease patients, arguing for its induction during the inflammatory process. This hypothesis is supported by the observation that CD94posCD127pos ILCs are absent in fetal intestine, where instead NK cells are abundant. The activation of these CD94posCD127pos ILC1-like ILCs in patients with bowel inflammation might have opposite effects: sustain inflammation on the one hand and fulfill bactericidal activities on the other hand. However, additional experimental investigations are needed to confirm this hypothesis. Given the localization of ILC3s at mucosal barriers, these in vitro observations argue for a potential role of cytokine-induced NK-cell like ILC3s in providing cytotoxic protection at mucosal sites, where NK cells are low abundant. If and how this NK cell-like activity of ILC3s could be exploited in vaccination settings against viruses or cancer remains to be studied (94).
A very recent work, investigating the immune phenotype of ILCs in hepatocellular carcinoma by scRNAseq, has demonstrated the existence of a cluster of liver-resident ILC1s endowed with cytotoxic potential (112). This cluster is characterized by the expression of cytolytic effector genes, including FGFBP2, FCGR3A, CX3CR1, GZMB, GZMH, and PRF1. Moreover, these cytotoxic ILC1s are mainly enriched in non-tumor tissues, while in tumor samples ILC1s are characterized by higher levels of exhaustion markers, such as LAG3, thus suggesting that ILC1s could undergo functional conversion during liver cancer progression. Further, a high accessibility to the granzyme C gene locus and high GrzmC transcripts were recently observed in ILC1s purified from murine liver and salivary glands. Granzyme Cpos ILC1 could be differentiated from ILC precursors, are ontogenetically distinct from NK cells and do not convert into ILC2 or ILC3. Granzyme C expression is dependent on T-bet, while sustained TGF-β signaling is required for the maintenance of granzyme Cpos ILC1 in the salivary gland, but not in the liver. Using the PyMT breast carcinoma model, the authors show that these cells expand and contribute to tumor anti-tumor functions in a TGF-β-dependent manner. If a similar ILC1 subset exists in humans remains to be tested (113).
Overall, cytotoxic helper ILCs have been so far identified within the ILC1 and/or the ILC3/ILCP subsets, but not within ILC2 compartment (Figure 2B). However, CD56pos (89), CD94pos (20) and NKG2Dpos (114) ILC2s have been reported in different settings, either in vitro upon cytokine exposure or in vivo in human peripheral blood. It remains to be verified if these cells have direct cytotoxic functions, like their cytotoxic ILC1 and ILC3 counterparts.
Since their discovery, many efforts have been done to characterize the origin, the function and identity of different ILC subsets. The knowledge regarding ILC biology is continuing to expand and includes the identification and characterization of progenitors, the refinement of mature ILC identities as well as the definition of additional ILC subsets. However, it is of utmost importance to understand if these novel ILC subsets coincide to additional developmental intermediate stages or they are the result of ILC plasticity to adapt to environmental stimuli. Furthermore, emerging evidence highlights the existence of circulating and tissue-resident helper ILCs endowed with cytotoxic potential. These cells, with a phenotype resembling to ILC1s and/or ILC3s/Ps but not ILC2s, most likely are a consequence of environmental and/or inflammatory triggers and could provide early innate defenses against different pathogens, particularly in mucosal tissues, where NK cells are underrepresented.
In the present review, we gave an overview of the current knowledge of ILC biology, mainly focusing on their developmental process. We further summarized the possible developmental pathways of the unconventional cytotoxic ILC subsets recently identified (Figure 2B).
Nevertheless, further studies are also needed to deeper characterize the pathways of human ILC development and to understand the differences and similarities with murine ILC-poiesis. In this context, studying the immune-reconstitution of ILC subsets after HSCT certainly represents an important strategy to shedding light on the in vivo ILC developmental trajectories at least in periphery. Moreover, the understanding of ILC-poiesis and homeostatic mechanisms driving donor-derived ILC immune-reconstitution as well as determining the acquisition of cytotoxic features, could be of clinical utility. Indeed, it will allow the development of protocols to ameliorate the HSCT outcome based either on adoptive ILC transfer therapies of ex vivo generated alloreactive ILCs or on systemic cytokine infusion/blocking antibodies to boost in vivo ILC effector-functions. Moreover, given the important role of helper ILCs in tissue immune-surveillance, these novel therapeutic options will find application in the management of solid as well as hematologic cancers and of inflammatory disorders.
MC, CDV, AF, ST, CJ, and DM wrote and critically reviewed the manuscript. AF and MC draw the figures. All authors gave the final approval to the manuscript.
This work was supported by Associazione Italiana per la Ricerca sul Cancro (IG 2018-21567 to DM), Intramural Research Funding of Istituto Clinico Humanitas (to DM), the Italian Ministry of Health (Bando Ricerca Finalizzata PE-2016-02363915 to DM), SNSF PRIMA fellowship (PR900P3_17972729 to CJ), the Swiss Cancer Research Foundation (KFS 5250-02-2021 to CJ) and the Geneva Cancer League (GCL, 2007 to CJ). MC is a recipient of the Leonelli AIRC fellowship (26580). MC and AF are recipients of competitive fellowships awarded from the PhD program of Experimental Medicine from University of Milan. ST is recipient of a Dr Henri Dubois-Ferrière Dinu Lipatti Foundation research fellowship. We also thank the financial support from Fondazione Romeo ed Enrica Invernizzi.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Kiessling R, Klein E, Pross H, Wigzell H. “Natural” Killer Cells in the Mouse. Ii. Cytotoxic Cells With Specificity for Mouse Moloney Leukemia Cells. Characteristics of the Killer Cell. Eur J Immunol (1975) 5(2):117–21. doi: 10.1002/eji.1830050209
2. Mebius RE, Rennert P, Weissman IL. Developing Lymph Nodes Collect Cd4+Cd3- Ltbeta+ Cells That Can Differentiate to Apc, Nk Cells, and Follicular Cells But Not T or B Cells. Immunity (1997) 7(4):493–504. doi: 10.1016/s1074-7613(00)80371-4
3. Spits H, Di Santo JP. The Expanding Family of Innate Lymphoid Cells: Regulators and Effectors of Immunity and Tissue Remodeling. Nat Immunol (2011) 12(1):21–7. doi: 10.1038/ni.1962
4. Vivier E. The Discovery of Innate Lymphoid Cells. Nat Rev Immunol (2021) 21(10):616. doi: 10.1038/s41577-021-00595-y
5. Spits H, Artis D, Colonna M, Diefenbach A, Di Santo JP, Eberl G, et al. Innate Lymphoid Cells–a Proposal for Uniform Nomenclature. Nat Rev Immunol (2013) 13(2):145–9. doi: 10.1038/nri3365
6. Vivier E, van de Pavert SA, Cooper MD, Belz GT. The Evolution of Innate Lymphoid Cells. Nat Immunol (2016) 17(7):790–4. doi: 10.1038/ni.3459
7. Vivier E, Artis D, Colonna M, Diefenbach A, Di Santo JP, Eberl G, et al. Innate Lymphoid Cells: 10 Years on. Cell (2018) 174(5):1054–66. doi: 10.1016/j.cell.2018.07.017
8. Shih HY, Sciume G, Poholek AC, Vahedi G, Hirahara K, Villarino AV, et al. Transcriptional and Epigenetic Networks of Helper T and Innate Lymphoid Cells. Immunol Rev (2014) 261(1):23–49. doi: 10.1111/imr.12208
9. Ercolano G, Wyss T, Salome B, Romero P, Trabanelli S, Jandus C. Distinct and Shared Gene Expression for Human Innate Versus Adaptive Helper Lymphoid Cells. J Leukoc Biol (2020) 108(2):723–37. doi: 10.1002/JLB.5MA0120-209R
10. Panda SK, Colonna M. Innate Lymphoid Cells in Mucosal Immunity. Front Immunol (2019) 10:861. doi: 10.3389/fimmu.2019.00861
11. Mjosberg J, Spits H. Human Innate Lymphoid Cells. J Allergy Clin Immunol (2016) 138(5):1265–76. doi: 10.1016/j.jaci.2016.09.009
12. Ebbo M, Crinier A, Vely F, Vivier E. Innate Lymphoid Cells: Major Players in Inflammatory Diseases. Nat Rev Immunol (2017) 17(11):665–78. doi: 10.1038/nri.2017.86
13. Gasteiger G, Fan X, Dikiy S, Lee SY, Rudensky AY. Tissue Residency of Innate Lymphoid Cells in Lymphoid and Nonlymphoid Organs. Science (2015) 350(6263):981–5. doi: 10.1126/science.aac9593
14. Mebius RE. Organogenesis of Lymphoid Tissues. Nat Rev Immunol (2003) 3(4):292–303. doi: 10.1038/nri1054
15. Tait Wojno ED, Artis D. Innate Lymphoid Cells: Balancing Immunity, Inflammation, and Tissue Repair in the Intestine. Cell Host Microbe (2012) 12(4):445–57. doi: 10.1016/j.chom.2012.10.003
16. Diefenbach A, Colonna M, Koyasu S. Development, Differentiation, and Diversity of Innate Lymphoid Cells. Immunity (2014) 41(3):354–65. doi: 10.1016/j.immuni.2014.09.005
17. Trabanelli S, Gomez-Cadena A, Salome B, Michaud K, Mavilio D, Landis BN, et al. Human Innate Lymphoid Cells (Ilcs): Toward a Uniform Immune-Phenotyping. Cytometry B Clin Cytom (2018) 94(3):392–9. doi: 10.1002/cyto.b.21614
18. Cella M, Otero K, Colonna M. Expansion of Human Nk-22 Cells With Il-7, Il-2, and Il-1beta Reveals Intrinsic Functional Plasticity. Proc Natl Acad Sci USA (2010) 107(24):10961–6. doi: 10.1073/pnas.1005641107
19. Sonnenberg GF, Mjosberg J, Spits H, Artis D. Snapshot: Innate Lymphoid Cells. Immunity (2013) 39(3):622–e1. doi: 10.1016/j.immuni.2013.08.021
20. Nagasawa M, Heesters BA, Kradolfer CMA, Krabbendam L, Martinez-Gonzalez I, de Bruijn MJW, et al. Correction: Klrg1 and Nkp46 Discriminate Subpopulations of Human Cd117(+)Crth2(-) Ilcs Biased Toward Ilc2 or Ilc3. J Exp Med (2019) 216(9):2221–2. doi: 10.1084/jem.2019049007302019c
21. Fan YY, Yang BY, Wu CY. Phenotypically and Functionally Distinct Subsets of Natural Killer Cells in Human Pbmcs. Cell Biol Int (2008) 32(2):188–97. doi: 10.1016/j.cellbi.2007.08.025
22. Bal SM, Golebski K, Spits H. Plasticity of Innate Lymphoid Cell Subsets. Nat Rev Immunol (2020) 20(9):552–65. doi: 10.1038/s41577-020-0282-9
23. Murphy JM, Ngai L, Mortha A, Crome SQ. Tissue-Dependent Adaptations and Functions of Innate Lymphoid Cells. Front Immunol (2022) 13:836999. doi: 10.3389/fimmu.2022.836999
24. Juelke K, Romagnani C. Differentiation of Human Innate Lymphoid Cells (Ilcs). Curr Opin Immunol (2016) 38:75–85. doi: 10.1016/j.coi.2015.11.005
25. Daussy C, Faure F, Mayol K, Viel S, Gasteiger G, Charrier E, et al. T-Bet and Eomes Instruct the Development of Two Distinct Natural Killer Cell Lineages in the Liver and in the Bone Marrow. J Exp Med (2014) 211(3):563–77. doi: 10.1084/jem.20131560
26. Fuchs A, Vermi W, Lee JS, Lonardi S, Gilfillan S, Newberry RD, et al. Intraepithelial Type 1 Innate Lymphoid Cells Are a Unique Subset of Il-12- and Il-15-Responsive Ifn-Gamma-Producing Cells. Immunity (2013) 38(4):769–81. doi: 10.1016/j.immuni.2013.02.010
27. Gordon SM, Chaix J, Rupp LJ, Wu J, Madera S, Sun JC, et al. The Transcription Factors T-Bet and Eomes Control Key Checkpoints of Natural Killer Cell Maturation. Immunity (2012) 36(1):55–67. doi: 10.1016/j.immuni.2011.11.016
28. Post M, Cuapio A, Osl M, Lehmann D, Resch U, Davies DM, et al. The Transcription Factor Znf683/Hobit Regulates Human Nk-Cell Development. Front Immunol (2017) 8:535. doi: 10.3389/fimmu.2017.00535
29. Friedrich C, Taggenbrock R, Doucet-Ladeveze R, Golda G, Moenius R, Arampatzi P, et al. Effector Differentiation Downstream of Lineage Commitment in Ilc1s Is Driven by Hobit Across Tissues. Nat Immunol (2021) 22(10):1256–67. doi: 10.1038/s41590-021-01013-0
30. Yomogida K, Bigley TM, Trsan T, Gilfillan S, Cella M, Yokoyama WM, et al. Hobit Confers Tissue-Dependent Programs to Type 1 Innate Lymphoid Cells. Proc Natl Acad Sci USA (2021) 118(50):e2117965118. doi: 10.1073/pnas.2117965118
31. Lanier LL. Nk Cell Recognition. Annu Rev Immunol (2005) 23:225–74. doi: 10.1146/annurev.immunol.23.021704.115526
32. Vivier E, Raulet DH, Moretta A, Caligiuri MA, Zitvogel L, Lanier LL, et al. Innate or Adaptive Immunity? The Example of Natural Killer Cells. Science (2011) 331(6013):44–9. doi: 10.1126/science.1198687
33. Caligiuri MA. Human Natural Killer Cells. Blood (2008) 112(3):461–9. doi: 10.1182/blood-2007-09-077438
34. Melsen JE, Lugthart G, Lankester AC, Schilham MW. Human Circulating and Tissue-Resident Cd56(Bright) Natural Killer Cell Populations. Front Immunol (2016) 7:262. doi: 10.3389/fimmu.2016.00262
35. Cooper MA, Fehniger TA, Caligiuri MA. The Biology of Human Natural Killer-Cell Subsets. Trends Immunol (2001) 22(11):633–40. doi: 10.1016/s1471-4906(01)02060-9
36. Kim BS, Artis D. Group 2 Innate Lymphoid Cells in Health and Disease. Cold Spring Harb Perspect Biol (2015) 7(5):a016337. doi: 10.1101/cshperspect.a016337
37. Neill DR, Wong SH, Bellosi A, Flynn RJ, Daly M, Langford TK, et al. Nuocytes Represent a New Innate Effector Leukocyte That Mediates Type-2 Immunity. Nature (2010) 464(7293):1367–70. doi: 10.1038/nature08900
38. Moro K, Kabata H, Tanabe M, Koga S, Takeno N, Mochizuki M, et al. Interferon and Il-27 Antagonize the Function of Group 2 Innate Lymphoid Cells and Type 2 Innate Immune Responses. Nat Immunol (2016) 17(1):76–86. doi: 10.1038/ni.3309
39. Monticelli LA, Osborne LC, Noti M, Tran SV, Zaiss DM, Artis D. Il-33 Promotes an Innate Immune Pathway of Intestinal Tissue Protection Dependent on Amphiregulin-Egfr Interactions. Proc Natl Acad Sci USA (2015) 112(34):10762–7. doi: 10.1073/pnas.1509070112
40. Fonseca W, Lukacs NW, Elesela S, Malinczak CA. Role of Ilc2 in Viral-Induced Lung Pathogenesis. Front Immunol (2021) 12:675169. doi: 10.3389/fimmu.2021.675169
41. Meininger I, Carrasco A, Rao A, Soini T, Kokkinou E, Mjosberg J. Tissue-Specific Features of Innate Lymphoid Cells. Trends Immunol (2020) 41(10):902–17. doi: 10.1016/j.it.2020.08.009
42. Califano D, Cho JJ, Uddin MN, Lorentsen KJ, Yang Q, Bhandoola A, et al. Transcription Factor Bcl11b Controls Identity and Function of Mature Type 2 Innate Lymphoid Cells. Immunity (2015) 43(2):354–68. doi: 10.1016/j.immuni.2015.07.005
43. Hochdorfer T, Winkler C, Pardali K, Mjosberg J. Expression of C-Kit Discriminates Between Two Functionally Distinct Subsets of Human Type 2 Innate Lymphoid Cells. Eur J Immunol (2019) 49(6):884–93. doi: 10.1002/eji.201848006
44. Oyesola OO, Duque C, Huang LC, Larson EM, Fruh SP, Webb LM, et al. The Prostaglandin D2 Receptor Crth2 Promotes Il-33-Induced Ilc2 Accumulation in the Lung. J Immunol (2020) 204(4):1001–11. doi: 10.4049/jimmunol.1900745
45. Stier MT, Zhang J, Goleniewska K, Cephus JY, Rusznak M, Wu L, et al. Il-33 Promotes the Egress of Group 2 Innate Lymphoid Cells From the Bone Marrow. J Exp Med (2018) 215(1):263–81. doi: 10.1084/jem.20170449
46. Hernandez DC, Juelke K, Muller NC, Durek P, Ugursu B, Mashreghi MF, et al. An in Vitro Platform Supports Generation of Human Innate Lymphoid Cells From Cd34(+) Hematopoietic Progenitors That Recapitulate Ex Vivo Identity. Immunity (2021) 54(10):2417–32.e5. doi: 10.1016/j.immuni.2021.07.019
47. Huang Y, Guo L, Qiu J, Chen X, Hu-Li J, Siebenlist U, et al. Il-25-Responsive, Lineage-Negative Klrg1(Hi) Cells Are Multipotential 'Inflammatory' Type 2 Innate Lymphoid Cells. Nat Immunol (2015) 16(2):161–9. doi: 10.1038/ni.3078
48. Montaldo E, Juelke K, Romagnani C. Group 3 Innate Lymphoid Cells (Ilc3s): Origin, Differentiation, and Plasticity in Humans and Mice. Eur J Immunol (2015) 45(8):2171–82. doi: 10.1002/eji.201545598
49. An Z, Flores-Borja F, Irshad S, Deng J, Ng T. Pleiotropic Role and Bidirectional Immunomodulation of Innate Lymphoid Cells in Cancer. Front Immunol (2019) 10:3111. doi: 10.3389/fimmu.2019.03111
50. Ishizuka IE, Chea S, Gudjonson H, Constantinides MG, Dinner AR, Bendelac A, et al. Single-Cell Analysis Defines the Divergence Between the Innate Lymphoid Cell Lineage and Lymphoid Tissue-Inducer Cell Lineage. Nat Immunol (2016) 17(3):269–76. doi: 10.1038/ni.3344
51. van de Pavert SA. Lymphoid Tissue Inducer (Lti) Cell Ontogeny and Functioning in Embryo and Adult. BioMed J (2021) 44(2):123–32. doi: 10.1016/j.bj.2020.12.003
52. Bar-Ephraim YE, Cornelissen F, Papazian N, Konijn T, Hoogenboezem RM, Sanders MA, et al. Cross-Tissue Transcriptomic Analysis of Human Secondary Lymphoid Organ-Residing Ilc3s Reveals a Quiescent State in the Absence of Inflammation. Cell Rep (2017) 21(3):823–33. doi: 10.1016/j.celrep.2017.09.070
53. Lim AI, Li Y, Lopez-Lastra S, Stadhouders R, Paul F, Casrouge A, et al. Systemic Human Ilc Precursors Provide a Substrate for Tissue Ilc Differentiation. Cell (2017) 168(6):1086–100.e10. doi: 10.1016/j.cell.2017.02.021
54. Scoville SD, Freud AG, Caligiuri MA. Cellular Pathways in the Development of Human and Murine Innate Lymphoid Cells. Curr Opin Immunol (2019) 56:100–6. doi: 10.1016/j.coi.2018.11.003
55. Colucci F, Caligiuri MA, Di Santo JP. What Does It Take to Make a Natural Killer? Nat Rev Immunol (2003) 3(5):413–25. doi: 10.1038/nri1088
56. Chinen H, Matsuoka K, Sato T, Kamada N, Okamoto S, Hisamatsu T, et al. Lamina Propria C-Kit+ Immune Precursors Reside in Human Adult Intestine and Differentiate Into Natural Killer Cells. Gastroenterology (2007) 133(2):559–73. doi: 10.1053/j.gastro.2007.05.017
57. Moroso V, Famili F, Papazian N, Cupedo T, van der Laan LJ, Kazemier G, et al. Nk Cells Can Generate From Precursors in the Adult Human Liver. Eur J Immunol (2011) 41(11):3340–50. doi: 10.1002/eji.201141760
58. Vacca P, Vitale C, Montaldo E, Conte R, Cantoni C, Fulcheri E, et al. Cd34+ Hematopoietic Precursors Are Present in Human Decidua and Differentiate Into Natural Killer Cells Upon Interaction With Stromal Cells. Proc Natl Acad Sci USA (2011) 108(6):2402–7. doi: 10.1073/pnas.1016257108
59. Freud AG, Becknell B, Roychowdhury S, Mao HC, Ferketich AK, Nuovo GJ, et al. A Human Cd34(+) Subset Resides in Lymph Nodes and Differentiates Into Cd56bright Natural Killer Cells. Immunity (2005) 22(3):295–304. doi: 10.1016/j.immuni.2005.01.013
60. Yu J, Freud AG, Caligiuri MA. Location and Cellular Stages of Natural Killer Cell Development. Trends Immunol (2013) 34(12):573–82. doi: 10.1016/j.it.2013.07.005
61. Male V, Hughes T, McClory S, Colucci F, Caligiuri MA, Moffett A. Immature Nk Cells, Capable of Producing Il-22, Are Present in Human Uterine Mucosa. J Immunol (2010) 185(7):3913–8. doi: 10.4049/jimmunol.1001637
62. Di Vito C, Mikulak J, Mavilio D. On the Way to Become a Natural Killer Cell. Front Immunol (2019) 10:1812. doi: 10.3389/fimmu.2019.01812
63. Freud AG, Yokohama A, Becknell B, Lee MT, Mao HC, Ferketich AK, et al. Evidence for Discrete Stages of Human Natural Killer Cell Differentiation in Vivo. J Exp Med (2006) 203(4):1033–43. doi: 10.1084/jem.20052507
64. Freud AG, Caligiuri MA. Human Natural Killer Cell Development. Immunol Rev (2006) 214:56–72. doi: 10.1111/j.1600-065X.2006.00451.x
65. Grzywacz B, Kataria N, Sikora M, Oostendorp RA, Dzierzak EA, Blazar BR, et al. Coordinated Acquisition of Inhibitory and Activating Receptors and Functional Properties by Developing Human Natural Killer Cells. Blood (2006) 108(12):3824–33. doi: 10.1182/blood-2006-04-020198
66. McClory S, Hughes T, Freud AG, Briercheck EL, Martin C, Trimboli AJ, et al. Evidence for a Stepwise Program of Extrathymic T Cell Development Within the Human Tonsil. J Clin Invest (2012) 122(4):1403–15. doi: 10.1172/JCI46125
67. Scoville SD, Mundy-Bosse BL, Zhang MH, Chen L, Zhang X, Keller KA, et al. A Progenitor Cell Expressing Transcription Factor Rorgammat Generates All Human Innate Lymphoid Cell Subsets. Immunity (2016) 44(5):1140–50. doi: 10.1016/j.immuni.2016.04.007
68. Hughes T, Becknell B, Freud AG, McClory S, Briercheck E, Yu J, et al. Interleukin-1beta Selectively Expands and Sustains Interleukin-22+ Immature Human Natural Killer Cells in Secondary Lymphoid Tissue. Immunity (2010) 32(6):803–14. doi: 10.1016/j.immuni.2010.06.007
69. Cella M, Fuchs A, Vermi W, Facchetti F, Otero K, Lennerz JK, et al. A Human Natural Killer Cell Subset Provides an Innate Source of Il-22 for Mucosal Immunity. Nature (2009) 457(7230):722–5. doi: 10.1038/nature07537
70. Lanier LL, Le AM, Phillips JH, Warner NL, Babcock GF. Subpopulations of Human Natural Killer Cells Defined by Expression of the Leu-7 (Hnk-1) and Leu-11 (Nk-15) Antigens. J Immunol (1983) 131(4):1789–96.
71. Loza MJ, Perussia B. Final Steps of Natural Killer Cell Maturation: A Model for Type 1-Type 2 Differentiation? Nat Immunol (2001) 2(10):917–24. doi: 10.1038/ni1001-917
72. Ahn YO, Blazar BR, Miller JS, Verneris MR. Lineage Relationships of Human Interleukin-22-Producing Cd56+ Rorgammat+ Innate Lymphoid Cells and Conventional Natural Killer Cells. Blood (2013) 121(12):2234–43. doi: 10.1182/blood-2012-07-440099
73. Freud AG, Keller KA, Scoville SD, Mundy-Bosse BL, Cheng S, Youssef Y, et al. Nkp80 Defines a Critical Step During Human Natural Killer Cell Development. Cell Rep (2016) 16(2):379–91. doi: 10.1016/j.celrep.2016.05.095
74. Scoville SD, Freud AG, Caligiuri MA. Modeling Human Natural Killer Cell Development in the Era of Innate Lymphoid Cells. Front Immunol (2017) 8:360. doi: 10.3389/fimmu.2017.00360
75. Serafini N, Vosshenrich CA, Di Santo JP. Transcriptional Regulation of Innate Lymphoid Cell Fate. Nat Rev Immunol (2015) 15(7):415–28. doi: 10.1038/nri3855
76. Seillet C, Mielke LA, Amann-Zalcenstein DB, Su S, Gao J, Almeida FF, et al. Deciphering the Innate Lymphoid Cell Transcriptional Program. Cell Rep (2016) 17(2):436–47. doi: 10.1016/j.celrep.2016.09.025
77. Yu X, Wang Y, Deng M, Li Y, Ruhn KA, Zhang CC, et al. The Basic Leucine Zipper Transcription Factor Nfil3 Directs the Development of a Common Innate Lymphoid Cell Precursor. Elife (2014) 3:e04406. doi: 10.7554/eLife.04406
78. Guia S, Narni-Mancinelli E. Helper-Like Innate Lymphoid Cells in Humans and Mice. Trends Immunol (2020) 41(5):436–52. doi: 10.1016/j.it.2020.03.002
79. Harly C, Cam M, Kaye J, Bhandoola A. Development and Differentiation of Early Innate Lymphoid Progenitors. J Exp Med (2018) 215(1):249–62. doi: 10.1084/jem.20170832
80. Yang Q, Li F, Harly C, Xing S, Ye L, Xia X, et al. Tcf-1 Upregulation Identifies Early Innate Lymphoid Progenitors in the Bone Marrow. Nat Immunol (2015) 16(10):1044–50. doi: 10.1038/ni.3248
81. Harly C, Kenney D, Ren G, Lai B, Raabe T, Yang Q, et al. The Transcription Factor Tcf-1 Enforces Commitment to the Innate Lymphoid Cell Lineage. Nat Immunol (2019) 20(9):1150–60. doi: 10.1038/s41590-019-0445-7
82. Renoux VM, Zriwil A, Peitzsch C, Michaelsson J, Friberg D, Soneji S, et al. Identification of a Human Natural Killer Cell Lineage-Restricted Progenitor in Fetal and Adult Tissues. Immunity (2015) 43(2):394–407. doi: 10.1016/j.immuni.2015.07.011
83. Xu W, Cherrier DE, Chea S, Vosshenrich C, Serafini N, Petit M, et al. An Id2(Rfp)-Reporter Mouse Redefines Innate Lymphoid Cell Precursor Potentials. Immunity (2019) 50(4):1054–68.e3. doi: 10.1016/j.immuni.2019.02.022
84. Walker JA, Clark PA, Crisp A, Barlow JL, Szeto A, Ferreira ACF, et al. Polychromic Reporter Mice Reveal Unappreciated Innate Lymphoid Cell Progenitor Heterogeneity and Elusive Ilc3 Progenitors in Bone Marrow. Immunity (2019) 51(1):104–18.e7. doi: 10.1016/j.immuni.2019.05.002
85. Constantinides MG, McDonald BD, Verhoef PA, Bendelac A. A Committed Precursor to Innate Lymphoid Cells. Nature (2014) 508(7496):397–401. doi: 10.1038/nature13047
86. Klose CSN, Flach M, Mohle L, Rogell L, Hoyler T, Ebert K, et al. Differentiation of Type 1 Ilcs From a Common Progenitor to All Helper-Like Innate Lymphoid Cell Lineages. Cell (2014) 157(2):340–56. doi: 10.1016/j.cell.2014.03.030
87. Bar-Ephraim YE, Koning JJ, Burniol Ruiz E, Konijn T, Mourits VP, Lakeman KA, et al. Cd62l Is a Functional and Phenotypic Marker for Circulating Innate Lymphoid Cell Precursors. J Immunol (2019) 202(1):171–82. doi: 10.4049/jimmunol.1701153
88. Mazzurana L, Czarnewski P, Jonsson V, Wigge L, Ringner M, Williams TC, et al. Tissue-Specific Transcriptional Imprinting and Heterogeneity in Human Innate Lymphoid Cells Revealed by Full-Length Single-Cell Rna-Sequencing. Cell Res (2021) 31(5):554–68. doi: 10.1038/s41422-020-00445-x
89. Chen L, Youssef Y, Robinson C, Ernst GF, Carson MY, Young KA, et al. Cd56 Expression Marks Human Group 2 Innate Lymphoid Cell Divergence From a Shared Nk Cell and Group 3 Innate Lymphoid Cell Developmental Pathway. Immunity (2018) 49(3):464–76.e4. doi: 10.1016/j.immuni.2018.08.010
90. Golebski K, Ros XR, Nagasawa M, van Tol S, Heesters BA, Aglmous H, et al. Il-1beta, Il-23, and Tgf-Beta Drive Plasticity of Human Ilc2s Towards Il-17-Producing Ilcs in Nasal Inflammation. Nat Commun (2019) 10(1):2162. doi: 10.1038/s41467-019-09883-7
91. Gao Y, Souza-Fonseca-Guimaraes F, Bald T, Ng SS, Young A, Ngiow SF, et al. Tumor Immunoevasion by the Conversion of Effector Nk Cells Into Type 1 Innate Lymphoid Cells. Nat Immunol (2017) 18(9):1004–15. doi: 10.1038/ni.3800
92. Cortez VS, Ulland TK, Cervantes-Barragan L, Bando JK, Robinette ML, Wang Q, et al. Smad4 Impedes the Conversion of Nk Cells Into Ilc1-Like Cells by Curtailing Non-Canonical Tgf-Beta Signaling. Nat Immunol (2017) 18(9):995–1003. doi: 10.1038/ni.3809
93. Krabbendam L, Bernink JH, Spits H. Innate Lymphoid Cells: From Helper to Killer. Curr Opin Immunol (2021) 68:28–33. doi: 10.1016/j.coi.2020.08.007
94. Raykova A, Carrega P, Lehmann FM, Ivanek R, Landtwing V, Quast I, et al. Interleukins 12 and 15 Induce Cytotoxicity and Early Nk-Cell Differentiation in Type 3 Innate Lymphoid Cells. Blood Adv (2017) 1(27):2679–91. doi: 10.1182/bloodadvances.2017008839
95. Krabbendam L, Heesters BA, Kradolfer CMA, Haverkate NJE, Becker MAJ, Buskens CJ, et al. Cd127+ Cd94+ Innate Lymphoid Cells Expressing Granulysin and Perforin Are Expanded in Patients With Crohn's Disease. Nat Commun (2021) 12(1):5841. doi: 10.1038/s41467-021-26187-x
96. Romagnani C, Juelke K, Falco M, Morandi B, D'Agostino A, Costa R, et al. Cd56brightcd16- Killer Ig-Like Receptor- Nk Cells Display Longer Telomeres and Acquire Features of Cd56dim Nk Cells Upon Activation. J Immunol (2007) 178(8):4947–55. doi: 10.4049/jimmunol.178.8.4947
97. Weizman OE, Adams NM, Schuster IS, Krishna C, Pritykin Y, Lau C, et al. Ilc1 Confer Early Host Protection at Initial Sites of Viral Infection. Cell (2017) 171(4):795–808.e12. doi: 10.1016/j.cell.2017.09.052
98. McFarland AP, Yalin A, Wang SY, Cortez VS, Landsberger T, Sudan R, et al. Multi-Tissue Single-Cell Analysis Deconstructs the Complex Programs of Mouse Natural Killer and Type 1 Innate Lymphoid Cells in Tissues and Circulation. Immunity (2021) 54(6):1320–37.e4. doi: 10.1016/j.immuni.2021.03.024
99. Roberto A, Di Vito C, Zaghi E, Mazza EMC, Capucetti A, Calvi M, et al. The Early Expansion of Anergic Nkg2a(Pos)/Cd56(Dim)/Cd16(Neg) Natural Killer Represents a Therapeutic Target in Haploidentical Hematopoietic Stem Cell Transplantation. Haematologica (2018) 103(8):1390–402. doi: 10.3324/haematol.2017.186619
100. Stabile H, Nisti P, Morrone S, Pagliara D, Bertaina A, Locatelli F, et al. Multifunctional Human Cd56 Low Cd16 Low Natural Killer Cells Are the Prominent Subset in Bone Marrow of Both Healthy Pediatric Donors and Leukemic Patients. Haematologica (2015) 100(4):489–98. doi: 10.3324/haematol.2014.116053
101. Lajoie L, Congy-Jolivet N, Bolzec A, Gouilleux-Gruart V, Sicard E, Sung HC, et al. Adam17-Mediated Shedding of Fcgammariiia on Human Nk Cells: Identification of the Cleavage Site and Relationship With Activation. J Immunol (2014) 192(2):741–51. doi: 10.4049/jimmunol.1301024
102. Stabile H, Nisti P, Peruzzi G, Fionda C, Pagliara D, Brescia PL, et al. Reconstitution of Multifunctional Cd56(Low)Cd16(Low) Natural Killer Cell Subset in Children With Acute Leukemia Given Alpha/Beta T Cell-Depleted Hla-Haploidentical Haematopoietic Stem Cell Transplantation. Oncoimmunology (2017) 6(9):e1342024. doi: 10.1080/2162402X.2017.1342024
103. Vulpis E, Stabile H, Soriani A, Fionda C, Petrucci MT, Mariggio E, et al. Key Role of the Cd56(Low)Cd16(Low) Natural Killer Cell Subset in the Recognition and Killing of Multiple Myeloma Cells. Cancers (Basel) (2018) 10(12):473. doi: 10.3390/cancers10120473
104. Zaghi E, Calvi M, Marcenaro E, Mavilio D, Di Vito C. Targeting Nkg2a to Elucidate Natural Killer Cell Ontogenesis and to Develop Novel Immune-Therapeutic Strategies in Cancer Therapy. J Leukoc Biol (2019) 105(6):1243–51. doi: 10.1002/JLB.MR0718-300R
105. Locatelli F, Pende D, Mingari MC, Bertaina A, Falco M, Moretta A, et al. Cellular and Molecular Basis of Haploidentical Hematopoietic Stem Cell Transplantation in the Successful Treatment of High-Risk Leukemias: Role of Alloreactive Nk Cells. Front Immunol (2013) 4:15. doi: 10.3389/fimmu.2013.00015
106. Krabbendam L, Heesters BA, Kradolfer CMA, Spits H, Bernink JH. Identification of Human Cytotoxic Ilc3s. Eur J Immunol (2021) 51(4):811–23. doi: 10.1002/eji.202048696
107. Cella M, Gamini R, Secca C, Collins PL, Zhao S, Peng V, et al. Subsets of Ilc3-Ilc1-Like Cells Generate a Diversity Spectrum of Innate Lymphoid Cells in Human Mucosal Tissues. Nat Immunol (2019) 20(8):980–91. doi: 10.1038/s41590-019-0425-y
108. Salome B, Gomez-Cadena A, Loyon R, Suffiotti M, Salvestrini V, Wyss T, et al. Cd56 as a Marker of an Ilc1-Like Population With Nk Cell Properties That Is Functionally Impaired in Aml. Blood Adv (2019) 3(22):3674–87. doi: 10.1182/bloodadvances.2018030478
109. Trabanelli S, Curti A, Lecciso M, Salome B, Riether C, Ochsenbein A, et al. Cd127+ Innate Lymphoid Cells Are Dysregulated in Treatment Naive Acute Myeloid Leukemia Patients at Diagnosis. Haematologica (2015) 100(7):e257–60. doi: 10.3324/haematol.2014.119602
110. Munneke JM, Bjorklund AT, Mjosberg JM, Garming-Legert K, Bernink JH, Blom B, et al. Activated Innate Lymphoid Cells Are Associated With a Reduced Susceptibility to Graft-Versus-Host Disease. Blood (2014) 124(5):812–21. doi: 10.1182/blood-2013-11-536888
111. Dogra P, Rancan C, Ma W, Toth M, Senda T, Carpenter DJ, et al. Tissue Determinants of Human Nk Cell Development, Function, and Residence. Cell (2020) 180(4):749–63.e13. doi: 10.1016/j.cell.2020.01.022
112. He Y, Luo J, Zhang G, Jin Y, Wang N, Lu J, et al. Single-Cell Profiling of Human Cd127(+) Innate Lymphoid Cells Reveals Diverse Immune Phenotypes in Hepatocellular Carcinoma. Hepatology (2022). doi: 10.1002/hep.32444
113. Nixon BG, Chou C, Krishna C, Dadi S, Michel AO, Cornish AE, et al. Cytotoxic Granzyme C-Expressing Ilc1s Contribute to Antitumor Immunity and Neonatal Autoimmunity. Sci Immunol (2022) 7(70):eabi8642. doi: 10.1126/sciimmunol.abi8642
Keywords: innate lymphoid cells (ILCs), natural killer (NK) cells, ILC-poiesis, cytotoxicity, unconventional subsets, inflammation, cancer
Citation: Calvi M, Di Vito C, Frigo A, Trabanelli S, Jandus C and Mavilio D (2022) Development of Human ILCs and Impact of Unconventional Cytotoxic Subsets in the Pathophysiology of Inflammatory Diseases and Cancer. Front. Immunol. 13:914266. doi: 10.3389/fimmu.2022.914266
Received: 06 April 2022; Accepted: 28 April 2022;
Published: 26 May 2022.
Edited by:
Rachel Golub, Université Paris Diderot, FranceReviewed by:
Marina Cella, Washington University in St. Louis, United StatesCopyright © 2022 Calvi, Di Vito, Frigo, Trabanelli, Jandus and Mavilio. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Domenico Mavilio, ZG9tZW5pY28ubWF2aWxpb0B1bmltaS5pdA==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.