 Kareem Awad
Kareem Awad Amany Sayed Maghraby1,2
Amany Sayed Maghraby1,2
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
SYSTEMATIC REVIEW article
Front. Immunol., 04 July 2022
Sec. Microbial Immunology
Volume 13 - 2022 | https://doi.org/10.3389/fimmu.2022.912899
This article is part of the Research TopicA year in review: Discussions in Microbial ImmunologyView all 7 articles
Introduction: Metabolic reprogramming in immune cells is diverse and distinctive in terms of complexity and flexibility in response to heterogeneous pathogenic stimuli. We studied the carbohydrate metabolic changes in immune cells in different types of infectious diseases. This could help build reasonable strategies when understanding the diagnostics, prognostics, and biological relevance of immune cells under alternative metabolic burdens.
Methods: Search and analysis were conducted on published peer-reviewed papers on immune cell metabolism of a single pathogen infection from the four known types (bacteria, fungi, parasites, and viruses). Out of the 131 selected papers based on the PIC algorithm (pathogen type/immune cell/carbohydrate metabolism), 30 explored immune cell metabolic changes in well-studied bacterial infections, 17 were on fungal infections of known medical importance, and 12 and 57 were on parasitic and viral infections, respectively.
Results and Discussion: While carbohydrate metabolism in immune cells is signaled by glycolytic shift during a bacterial or viral infection, it is widely evident that effector surface proteins are expressed on the surface of parasites and fungi to modulate metabolism in these cells.
Conclusions: Carbohydrate metabolism in immune cells can be categorized according to the pathogen or the disease type. Accordingly, this classification can be used to adopt new strategies in disease diagnosis and treatment.
Immune cells are flexible in terms of their ability to dynamically reprogram their cellular metabolism. This flexibility can overcome diverse conditions of nutrients and oxygen availability in multiple tissues and contain significant metabolic stresses from their micro-environmental conditions (1– 2). A clear example is a lymphocyte that can turn from a relatively static cell to another involved in growth and proliferation that produces considerable cytokine storms or effector molecules (3). Moreover, metabolic programming will also affect specific levels of metabolites that consequently result in different immune cell functionalities (1, 4, 5). These changes in metabolite levels include a rich and diverse set of post-translational modified forms of enzymes or products of glycolysis and tricarboxylic acid (TCA) cycle in immune cells that have not been fully understood yet (6). These modified enzymes or products result in different alterations of immune cell phenotypes and consequently, modulate immune responses and inflammation in different contexts (6). Beyond the utilization of specific metabolic pathways, metabolic reprogramming in immune cells also repurposes their oxidation–reduction system, as shown through the role of reactive oxygen species (ROS) in the proliferation and functionality of cytotoxic and humoral immune cells as well as monocytes and macrophages (MQs) (7).
The immune system encompasses populations of various cells that are substantially dynamic in terms of their proliferation, survival, and functions in response to different immunological stimuli and share the ability to rapidly respond to infection, trauma, and other perturbations (1, 3). The need for energy of such cellular functions is essential with glucose metabolism being the major carbon fuel source through two distinct pathways: glycolysis as a first pathway producing pyruvate and adenosine triphosphate (ATP) and TCA as an immediately following pathway (4, 8). Metabolic reprogramming mainly happens through specific receptors and the tuning of growth-factor cytokine signaling, as well as nutrients and neuronal regulation that are all dynamic factors connected to the existence of a pathogen (9, 10).
Pathogens such as bacteria, fungi, parasites, and viruses have developed sophisticated tools to manipulate their victims’ cells for their proliferation and existence, so the normal structures and functions of essential mammalian host organelles such as endosomes, Golgi apparatus, mitochondria or even their membrane components or nuclear envelopes are targeted by microbial machines (11). Here, we analyzed how the immune cell metabolism is manipulated by pathogens including bacteria, fungi, parasites, and viruses, with a focus on the interaction with specific immune cell metabolic pathways.
Metabolomic analysis of infected cells provides insights to the formation and consequences of the common metabolic pathway changes (12). These include metabolites essential for glycolysis, amino acid and fatty acid synthesis, and uncommon metabolic changes including those using xenobiotics such as the sugar substitute erythritol and chemicals like phenol red (12).
Pathogens exert particular mechanisms to alter metabolic pathways to continue their life cycle and such mechanisms represent candidate therapeutic targets to treat infections (12–14). Metabolomic approaches are widely used to study changes in cellular metabolism (15–18) among which is the use of magnetic resonance spectroscopy, mass spectrometry, or gas and liquid chromatography to concomitantly detect various metabolite alterations (15–18). These approaches have widely allowed for the determination of infections or tumor-induced alterations in metabolism. For example, the levels of metabolites involved in glycolysis and Krebs cycle remarkably increased as detected by liquid chromatography-tandem mass spectrometry when the human cytomegalovirus (HCMV) progresses into an infection in human fibroblasts (15).
Similar metabolomic approaches, as well as these metabolites, have been used to identify novel proteins and lipid molecules during hepatitis C virus (HCV) infection in cultured cells (18). These included two fatty-acid oxidation enzymes hydroxyacyl-CoA dehydrogenase beta subunit (HADHB) and dodecenoyl coenzyme A delta isomerase that have demonstrated differential regulation both in vitro and in HCV-infected patients with coincidence with cytopathic effect in cell culture and significant liver lesion in HCV infected patients (18). In vitro analysis by 1H NMR and then 31P NMR of extracts from malignant tumors indicated an initial reduction in phosphocholine, glycerophosphocholine, and myo-inositol, and then phosphoethanolamine and glycerophosphoethanolamine, respectively (17). In keeping with these results, as measured by 1H NMR, inhibition of the hypoxia-induced factor 1 (HIF-1) with PX-478 as an experimental anticancer agent gave rise to a considerable change in total choline in vivo (17).
Here, we summarized the changes in carbohydrate metabolomic immune cells associating infections with various pathogens with the aim to identify pathogen/disease-specific carbohydrate metabolic immune cell signatures of potential diagnostic and/or prognostic relevance.
A literature search of medical research electronic databases was conducted for peer-reviewed articles (Figure 1). These included Medline (PubMed), produced by the National Library of Medicine, Washington, USA; Embase (Scopus), produced by Elsevier, Amsterdam, the Netherlands; the Cochrane Central trials register, produced by John Wiley & Sons, Ltd.; and Clarivate, produced by Thomson Reuters, USA. This research was done in the time period 2018–2022. The key search terms used were T cell, monocyte MQ, immune, CD+4 –T, inflammation immune cell,NK, and innate and adaptive immune cell metabolism in combination with various pathogens including bacteria, fungi, parasites (including protozoa and helminths), viruses, and COVID-19.

Figure 1 PRISMA flow diagram of the systematic review.
Titles and abstracts were carefully screened and full-text peer-reviewed published articles were retrieved. Data were extracted based on the PIC algorithm (pathogen type/immune cell/carbohydrate metabolism). Accordingly, we subtracted information from studies on single infections in one immune cell model while those with multiple infections were excluded. Also, publications on sugar metabolic pathways only in immune cells were included, while other metabolic pathways in either somatic cells or immune cells were excluded, unless exceptionally showing relation to glucose metabolism.
Out of the 131 reviewed papers, 30, 17, 12, and 57 have explored immune cell metabolic changes accompanying bacterial, fungal, parasitic, and viral infections, respectively. Current perspectives on immune cell specific metabolism due to the current COVID-19 pandemic were from 18 publications out of the total 57 manuscripts related to viruses (Table 1). A total of 18 reviewed papers introduced and defined immunometabolism and the scientific methods used in its study. The reviewed bacterial pathogens were Legionella pneumophila, Mycobacterium species, Pseudomonas aeruginosa, and Salmonella enterica. The reviewed fungi were Candida albicans and entomopathogenic fungi (EPF). The included protozoa parasites were Entamoeba histolytica, Theileria annulata, and Toxoplasma gondii, whereas the helminth parasites were Fasciola hepatica and Schistosoma mansoni. The reviewed viruses were DNA viruses such as adenovirus E4ORF, varicella-zoster virus (VZV), Epstein–Barr virus (EBV), and HCMV. RNA viruses such as coxsackie virus B3, dengue virus (DENV), human immunodeficiency virus (HIV), HCV, poliovirus, influenza virus, yellow fever, and West Nile were also discussed. Virus-induced cytokines were briefly argued.
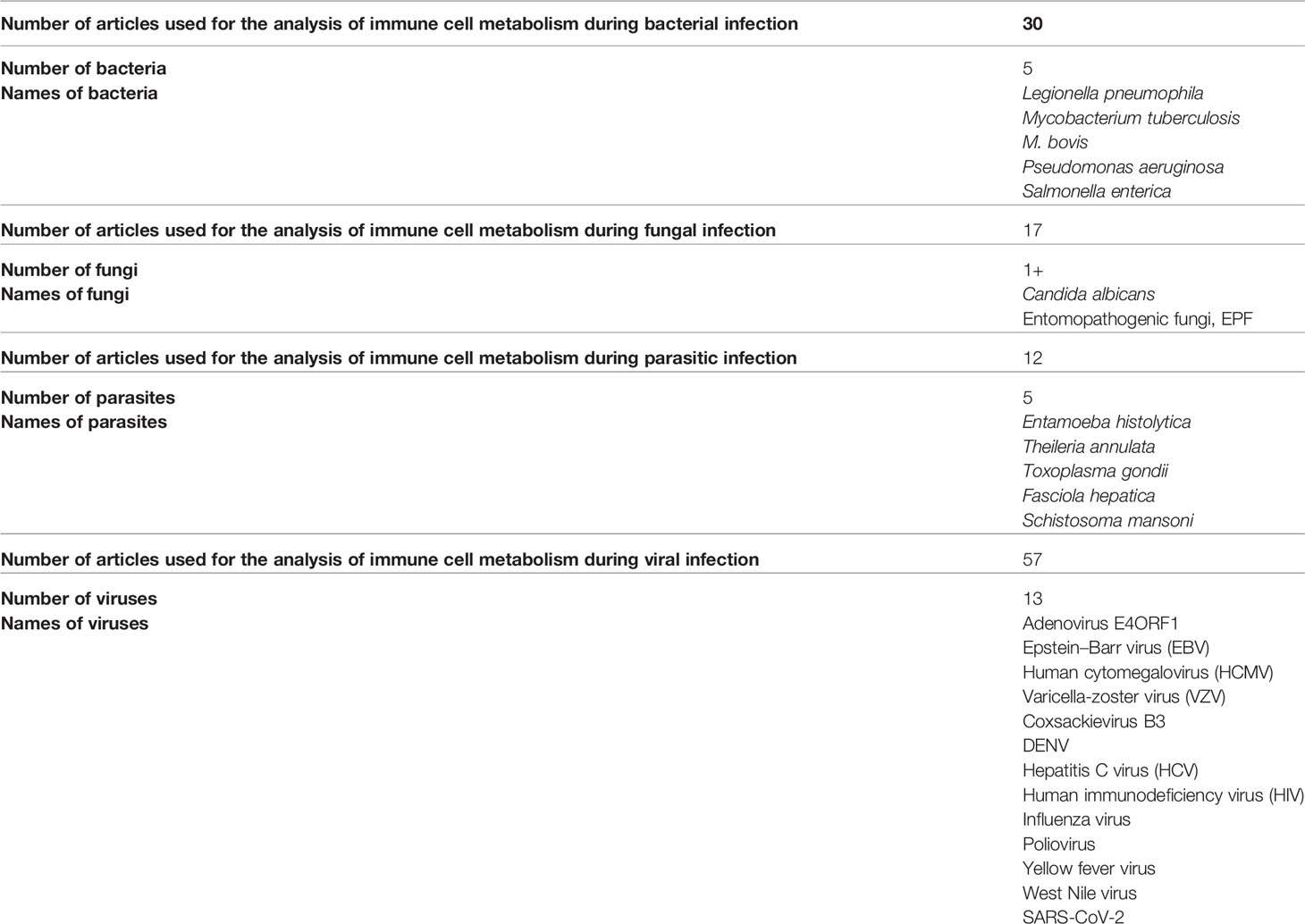
Table 1 Number of analyzed articles and reviewed diseases in this review.
The effect of bacterial pathogens on the infected host’s metabolism had been long identified in animals and plants (19–21); however, understanding bacterial modifications, particularly of immune cell metabolism, can be important in developing host-directed immune therapies. This drives current attention to the analysis of bacterial-induced metabolites, which manipulate immune cell proliferation, differentiation, and function.
A frequent cause of severe pneumonia in humans is Legionella pneumophila, which is used as a model for exploring immune responses to intravacuolar bacteria (22). Remodeling of the TCA cycle is a metabolic adaptation during MQ polarization as recently shown for succinate, which regulates the pro-inflammatory IL-1β- hypoxia-inducible factor-1α (HIF-1α) axis (23). During L. pneumophila infection, type I and II interferons (IFNs) are master regulators of gene expression and stimulators of alveolar MQ-intrinsic immune responses that inhibit bacterial growth during the course of the disease (22). Type I or II IFN-dependent production of immune responsive gene 1, the mediator of the bactericidal itaconic acid, has shown control on infection in MQ and also inhibits pro-inflammatory cytokine expression and mitochondrial respiration (22–24). In both human and mouse immune cells, itaconate modulates the MQ metabolism and effector functions by inhibiting the succinate dehydrogenase (SDH)-mediated oxidation and thus, controlling the succinate levels and function (23–25). So, during L. pneumophila infection, mitochondrial respiration can be modulated during MQ activation because of the change of itaconate levels (Figure 2).
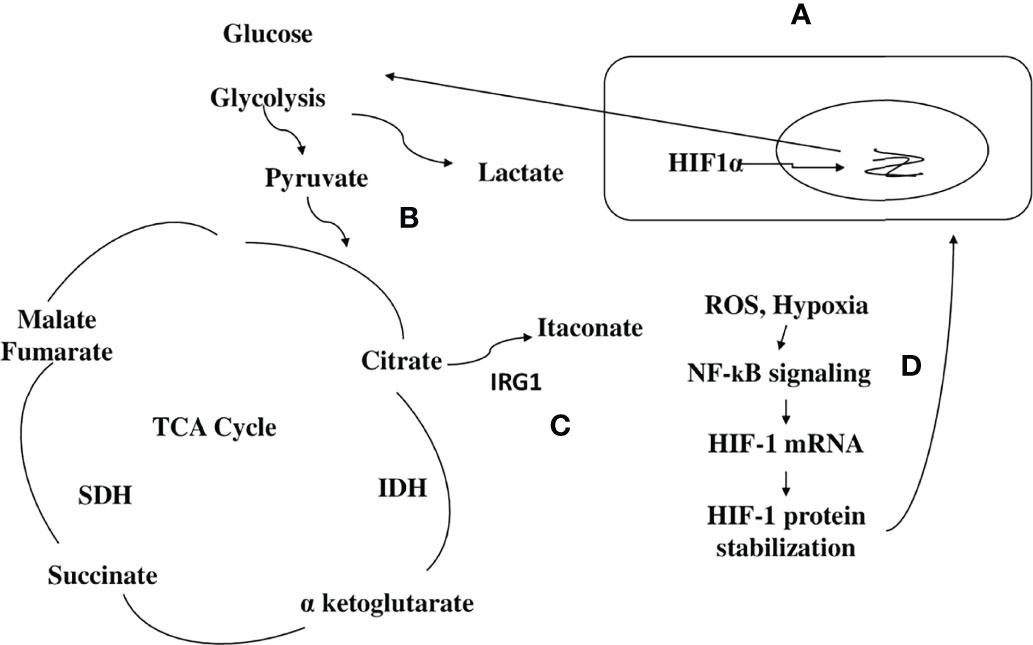
Figure 2 Scheme hypothesizing how itaconate regulates TCA flow in monocytes/macrophages infected with a bacterial pathogen. (1) (A) During bacterial infection, activated macrophages (MQs) accumulate stabilized HIF-1α, which subsequently translocates to the nucleus, initiating the transcription of genes related to glycolysis. (B) Activated MQs and other surrounding immune cells such as induced T cells have shown high glycolysis during infection that is accompanied by Warburg-effect metabolism (accumulation of lactate). (C) Immune responsive gene 1 (IRG1) links cellular metabolism with immune defense by catalyzing itaconic acid production in mouse and human macrophages, and itaconate modulates MQ metabolism and effector functions by inhibiting succinate dehydrogenase (SDH)-mediated oxidation of succinate and thus, controls succinate levels and function. (D) Instead of producing ATP, this metabolic switch repurposes succinate oxidation in the mitochondria to release bactericidal ROS catalyzed by SDH and isocitrate dehydrogenase (IDH). An organism like Pseudomonas aeruginosa can also sense ROS, which promotes an acquired change, the biofilm formation.
Tuberculosis (TB) granulomas are agglomerations of induced and uninduced MQs, neutrophils, T cells, and other immune cells (26). Several unique metabolic modifications occur to modulate the behavior of immune cells within the granuloma, potentially boosting bacterial persistence against immunopathology (26). Growth inhibition has been demonstrated in Mycobacterium tuberculosis, expressing isocitrate lyase bacteria by itaconic acid in human and mouse MQs that is significantly reversed by Irg1 gene silencing (25). This suggests that IRG1 links cellular metabolism with immune defense by catalyzing itaconic acid production in mouse and human MQs (Figure 2) (25).
A unique opportunity to study the influence of a bacterial infection on glucose metabolism within immune cells has been provided with the involvement of many metabolically active immune cells during different stages of TB infection (27). Apparently, what characterize cancer cells and virus-infected cells are not far from what might also be called bacterial Warburg-effect type of metabolism, which highly dominates during TB infection among different immune cells (25). Activated M1 MQ and other surrounding immune cells such as induced T cells have shown high glycolysis during TB infection that is accompanied by Warburg-effect metabolism due to the accumulation of imported glucose (27).
High-resolution nuclear magnetic resonance spectroscopy detected an increase in lactate as TB progressed in the lungs of infected guinea pig evidencing the pathway of glycolysis during infection (28). Lactate detection is an indicator for the Warburg-effect metabolism in cancer or low oxygen demand metabolism type (29). Lactate release is common in mycobacterial infection across species as relatively high amounts of lactate have been detected in the serum of M. bovis and M. tuberculosis-infected mice and cattle (30, 31). Lactate activates an MQ phenotype characterized by increased activity of arginase 1 (32) that was hypothesized to favor nitric oxide (NO) inhibition, a key antimicrobial response. Substantially, lactate metabolism and signaling would be valuable host-directed therapy targets for TB treatment; however, crucial scientific effort is still needed to better understand the role of lactate in the host–pathogen interaction throughout the TB infection (33, 34).
A lesson is introduced by the Drosophila melanogaster, that is, short-chain fatty acids produced by fermentation of bacterial carbohydrates and other bacterial metabolites are sensed by specific G-protein surface receptors of enteroendocrine cells and enterocytes, or captured and converted by enterocytes into metabolically active molecules such as acetyl-CoA (35, 36). This modulates the host carbohydrate and lipid utilization systemically in the intestine by stimulating the release of enteroendocrine peptides into the systemic circulation that activates other signaling cascades (35, 36).
The bacterial pathogen Pseudomonas aeruginosa, which causes cystic fibrosis, exploits metabolic elements such as succinate and itaconate, to induce metabolic and transcriptomic modulations in immune cells that increase its capacity to cause stubborn diseases (37). P. aeruginosa, through the process of catabolite repression, preferentially consumes succinate compared to other carbon sources (38). Metabolic reprogramming in activated MQs switches to aerobic glycolysis instead of oxidative phosphorylation pathways in the mitochondria (39). Instead of producing ATP, this metabolic switch repurposes succinate oxidation in the mitochondria to release bactericidal ROS catalyzed by SDH and isocitrate dehydrogenase (IDH) (Figure 2) (39).
This enzymatic reaction is potentiated by a biochemical process called anaplerosis that favors the accumulation and synthesis of succinates from foreign metabolites, such as environmental glutamine (39, 40). Thus, P. aeruginosa provides itself with a favored substrate, succinate. Excess succinate is with time, compensated by the TCA cycle intermediate, itaconate, that inhibits MQ SDH and blocks glycolysis by altering the enzymatic function of both glyceraldehyde 3-phosphate dehydrogenase and aldolase (41, 42). Itaconate, in this way, functions as a major immunoregulatory molecule that resolves inflammation by modulating MQ metabolism. In this context, early infection by P. aeruginosa that is accompanied by succinate increase compared to a late or chronic infection that is accompanied by itaconate compensation should be considered when designing therapeutic strategies for this infection.
In another considerable mechanism, P. aeruginosa alters its immunometabolism in response to itaconate, which induces bacterial membrane stress, resulting in the upregulation of extracellular polysaccharides (EPS) and downregulation of lipopolysaccharides (LPS) (43). This process favors itaconate assimilation and the formation of a biofilm on the one side and on the other side, EPS, inducing itaconate production by myeloid cells and skewing the host immune response to one permissive chronic infection (43).
Another modulation of immunometabolism by P. aeruginosa is that it requires glucose transport via glucose transporter 1 (Glut1) in MQ as demonstrated in a sepsis mouse model where increased glucose uptake in MQs post-infection and lipopolysaccharide (LPS) production have been shown (44, 45). The activated serine-threonine protein kinase Akt1 which is essential for salmonella infection of MQs, phosphorylates the E3 ligase Mdm2, which ubiquitinates p53 and thus, may lead to increased glucose flux (46).
Although immunometabolism alterations in response to bacterial infection show a similar trend in experiments done in mouse and human cells, differences have been reported in comparable studies (25, 47). Intracellular itaconic acid concentration has demonstrated almost double production in mouse immune cells compared to human primary MQs (25). Another difference between mouse and human immune cells is the production of NO in mouse MQs that increases inflammatory conditions (47). NO inhibits aconitase, the enzyme producing the itaconic acid precursor cis-aconitate (48).
Fungal effectors have often been recognized for their diversity, while current research has focused on effector biology, highlighting that different fungi utilize similar mechanisms for creating a more harmonious host environment, among which is manipulating the host immune metabolism (49, 50). Host immune responses have been suppressed by bioactive metabolites from fungi that act as immune modulators (49, 50). When considering fungi of human medical importance such as Candida albicans, different proteomic methods have detected enzymes at the cell surface of this fungus that are involved in cell wall glycan biosynthesis (for example, Uridine diphosphate (UDP)–glucose pyrophosphorylase) and may show immune cell modulation (51, 52). It is worth mentioning that C. albicans, in low glucose conditions, effectively utilizes various fermentable and non-fermentable carbon sources to remain and grow. Utilization of alternative carbon sources highly impacts the need to understand all these pathways as potential and attractive curative host targets (53).
Metabolic reprogramming has also been shown in the C. albicans infection of monocytes/MQs as killed hyphae and yeast induce gene expression that regulates the glycolytic pathway to a higher extent (Figure 3A) (54–56). Activation of MQ using C. albicans yeasts led to an increase in cellular respiration compared with the LPS/IFNγ-activation of MQs (57). C. albicans also reprograms NK cells toward higher glycolysis, which is required for proper lytic function of this cell type (Figure 3B) (58). This metabolic shift in immune cells toward higher glycolysis by C. albicans associated this infection to developed point mutations and metastasis in several studies (59, 60). It is noteworthy that the increase in glycolysis and glycolytic gene expression such as phosphofructokinase and pyruvate kinase were reported to have been highly expressed in MQs of both infected mouse and human cells (61, 62). Although high glycolysis could be needed by C. albicans to proliferate and escape immune mechanisms in the host, it also results in increased lactate that is thought to be an important carbon source for C. albicans in humans, where it is produced by activated inflammatory MQs that have switched to Warburg-effect metabolism (Figure 3A) (63, 64).
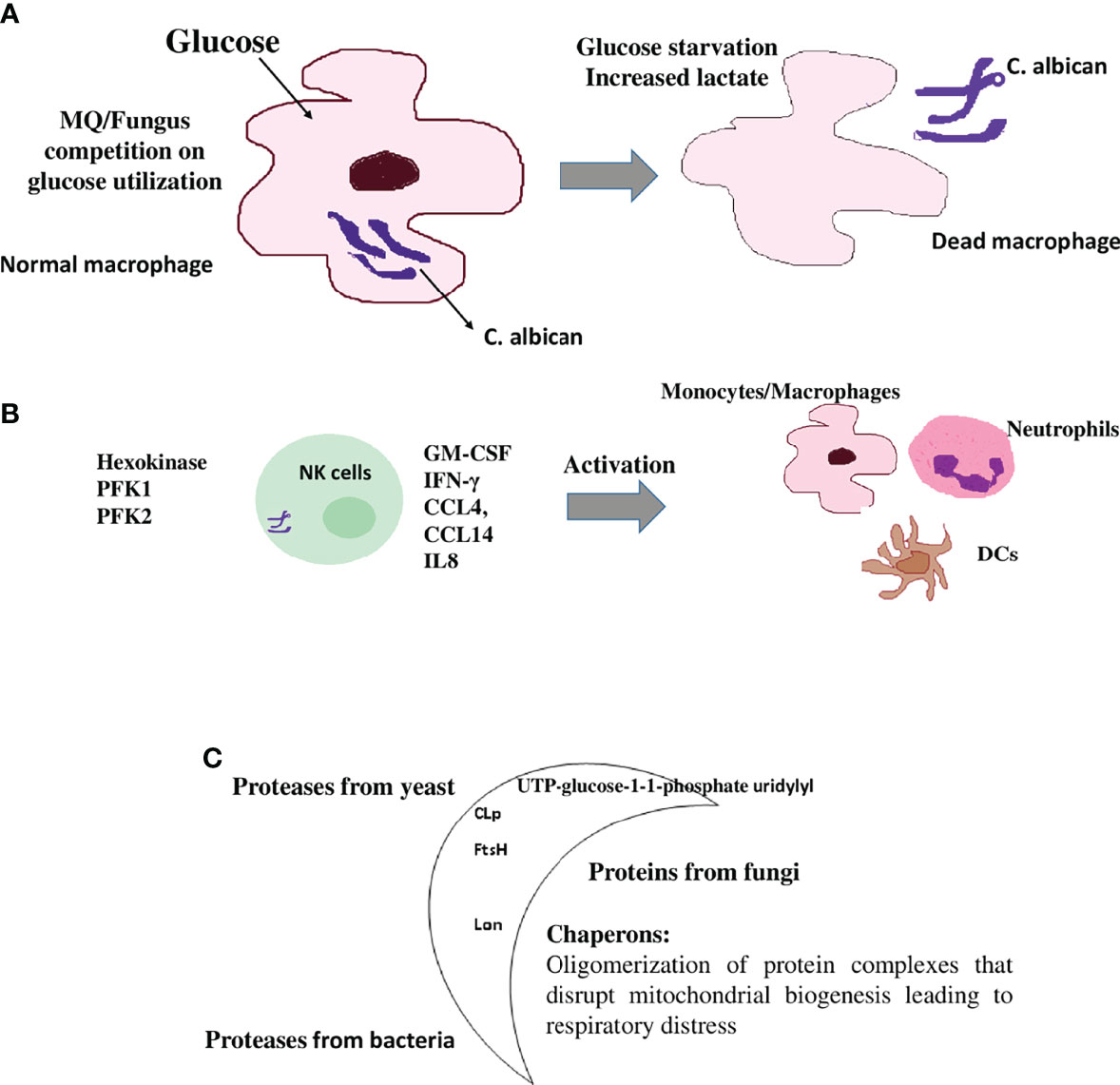
Figure 3 Immunometabolic changes in immune cells upon fungal and parasitic infection (A) MQs are metabolically inflexible and depend on glucose as a carbon source. The discussed Candida albicans fungus switches from yeast to filamentous hyphae, once taken by MQs. Fungal hyphae reactivate glycolysis and compete with MQs for glucose, which leads to depletion of glucose available to this immune cell by the fungus. MQ is killed by glucose starvation (56). High glycolysis is a metabolic shift in MQs during C. albicans infection. High glycolysis also results in increased lactate (Warburg-effect metabolism status) that is also utilized by C. albicans. (B) Natural killer (NK) cells respond to C. albicans pathogenII with the upregulation of genes involved in immune signaling and release of inflammatory cytokines that are involved in the activation of other immune cells as well as a shift to aerobic glycolysis. These include IFN-γ, GM-CSF, CCL4 (MIP-1β), IL8 (involved in the recruitment and activation of neutrophils), CCL14 (monocytes activator), XCL1 (dendritic cells activator), and the glycolytic enzymes hexokinase 2, phosphofructokinase1 (PFK1), phosphofructokinase fructose-2,6-bisphosphate (PFK2), and lactatedehydrogenase. (C) The moonlighting proteins: multifunctional proteins act as chaperones and are widely involved during the pathogenesis of infection through cell surface presentation. Some pathogenic bioactive metabolites are immune modulators and have been implicated in suppressing the host immune response. For example, in bacteria, the production of FtsH and CLp proteases, which are responsible for infectivity and virulence of a number of bacterial pathogens. In C. albicans, UTP-glucose-1-1-phosphate uridylyl transferase, an enzyme involved in cell wall glycans biosynthesis, has been detected at the cell surface of this fungus and shows immune cell modulation. In helminths and protozoan parasites, the synthesis of glycoconjugates and glycan-binding proteins such as the mitochondrial Lon protease are examples of these immune modulators.
Modulating innate and humoral responses is crucial for EPF; yet, any manipulation that is detrimental to the host could result in displacement of EPF and opportunistic microbes causing sepsis (49). Immune modulators could burden the host’s guards and facilitate fungal colonization and biomass generation with the production of copious conidia needed for propagation and proliferation (65). Suppressing the host immune machinery saves the EPF’s required energy for the sake of colonization and growth rather than containing the host immune responses as shown in both human and mouse cells (8). Note that many known human immune suppressant medicines such as myriocin and cyclosporine originated from bioactive EPF (8). However, studies that can show the role of these metabolites in modulating metabolism in immune cells are lacking.
Parasites are larger organisms that secrete metabolites able to interact and modulate immune cell metabolism in both human and animal cell models, while taking in account the mutual signaling among the complex network of parasites including two or more hosts (66–68). Despite their complexity, they depend on their hosts that often, for long periods, supply a rich environment that supports their growth, maturation, and reproduction (66–69). The protozoan parasite E. histolytica adopts strategies for immune evasion including suppression of IFN-γ production and other immunomodulatory mechanisms (66, 69). Reported metabolic alterations include the elimination of immune cells and other immune modulators as well as reactive nitrogen and oxygen species to overcome the attack from the immune system (66, 69). On the other hand, the protozoan parasite T. gondii can oppose genetic repression of its glucose transporter by increasing glutamine-derived carbon flux through the Krebs cycle and by synchronously activating gluconeogenesis, producing enough ATP required for the parasite replication and host-cell invasion (68).
As multicellular parasites, helminths promptly associate with their cell surface to the human host surroundings of various host ligand proteins such as plasma or extracellular matrix proteins as well as blood and immune cells (67, 68). An example of a complex biological matrix component that is constantly exposed to the host immune system is the outer tegumental coat of F. hepatica (FhTeg), a multicellular helminth that causes fasciolosis in humans (68). This FhTeg consists of glycolytic motifs and active glycoproteins that modulate immune cells (67). Among these proteins are a number of immune modulators such as tetraspanin CD63 membrane surface receptor, which is involved in the innate immune cell polarization, differentiation, and adhesion (70). Another molecule is TRIL (TLR4 interactor with leucine-rich repeats) that can bind to LPS and could hinder LPS activation of mast cells and dendritic cells (DCs) (50, 51). F. hepatica nardilysin-like molecules may associate with T-cell imbalance or prevent the activation and binding of TNFα-converting enzyme (71). S. mansoni is another helminth parasite that secretes components that can condition immune cells such as omega-1, a glycosylated T2 ribonuclease (Rnase) that conditions DCs to prime T helper 2 responses (68).
The common feature of helminths and protozoan parasites to respond to and counteract changes in their macro- and micro-environments, as well as to synthesize glycan-binding proteins and glycoconjugates for their protection, is broadly involved during the pathogenesis of infection through cell surface presentation of the so-called moonlighting proteins (72). The divergent extensiveness of these proteins in almost all organisms raises a question about the reason for their existence. One elucidation is the multifunctionality of these proteins. For instance, human lysyl hydroxylase 3 is an enzyme that catalyzes a few stages in the metabolic pathway and is also responsible for three consecutive steps through hydroxylysine-linked carbohydrate formation in post-translational modifications of collagen (73).
Another example is the economic usage of the mitochondrial Lon protease as one molecule, which, based on the environmental status and the needs of the cell, converts between chaperone functions and protein degradation (74). This phenomenon could have evolved during the formation of unique protein organizations that happened without changes to previous primary functions or structures of the questioned protein, or through the adaptability of the surface of unused molecules as new catalytic or binding sites for other diverse ligands (75). These multifunctional proteins could have been preserved during the course of cell evolution, as long as this alternative function does not obstruct the original enzymatic function (Figure 3C) (72, 75).
Direct interaction between parasites and metabolic behaviors of specific immune cells has been rarely studied in extracted cells from animal models (76, 77). The apicomplexan parasite T. annulata infection of MQ cell lines in normoxic conditions has driven its host cell to perform Warburg effect-like glycolysis which has been shown to be HIF-1α-dependent and is suggested as a way to overcome toxic levels of oxidative stress that originated from a Theileria infection and then resulted to uncontrolled proliferation of the host cell (76). In a separate study, IL-10, which is produced by MQ in various immune occasions and is important in controlling immune-mediated pathology, is produced by murine MQ derived from the bone marrow and exposed to S. mansoni cercariae secretory/excretory products in a mechanism linked to altered glycolysis, Krebs cycle, and oxidative phosphorylation and is involved the production of TLR2 and TLR4. However, there are concerns on the increase in glucose uptake and glycolytic kinase activity, and not the production of lactate (77).
Metabolic alterations caused by virus infections are not limited to the infected cells but rather expand to other host cell types that respond to the infection and impact both the pattern of human and experimental animal viral diseases (78). These metabolic changes are necessary for tasks leading to virus replication, a process, which in a very short time, demands a considerable amount of energy (78, 79). Experiments on different viruses have demonstrated species- and time-related infected cells’ metabolic patterns obviously because of the different productivities when it comes to virus-type specificity or the infection dynamics reflected in the host cell type-specific metabolism (2, 5, 80–82). Moreover, there have been discussions on the changes in metabolic dynamics in different human and mouse immune cells on the infection with Epstein–Barr virus (EBV), flu, and human immune deficiency virus (HIV) and are reported with glycolytic activity, for example, with the metabolite phosphoenolpyruvate (PEP) activity and the intracellular redox state (82). It has been shown that the different immune cells derived from human or animal subjects express different metabolic patterns such as in the reduced activity of the TCA cycle in MQs and DCs, higher glycolysis in M1 MQs, TCA cycle-based M2 MQ metabolism, decreased oxidative phosphorylation activity in monocyte-derived DCs, and the central role of the mechanistic target of rapamycin (mTOR) in natural killer (NK) cells (83). These patterns can be shifted, relying on the new human body’s status observed during viral infection (84).
It has been shown that Myc glycolytic targets are activated by the E4ORF1 protein of adenovirus to induce a Warburg effect-like status that converts glucose into nucleotides supporting replication of the virus (15). The Warburg effect has been reported in human and animal studies on viral infections as metabolism is signaled by higher glycolysis flow, rather than employing the more economical Krebs cycle for the production of energy (85). This results in increased lactate levels while TCA is kept in lower flow than in normal conditions (83). It is highly probable that reprogramming of the cellular genetic materials and protein translation machinery, as a result of the divergence and convergence of cellular and viral oncogenes targeting crucial protein mechanistic networks, is necessary for evolved pathological consequences (83).
Viral metabolomic studies were first adopted to characterize alterations in the host cellular metabolism caused by the HCMV infection of a human cell model (15, 85). These studies demonstrated that HCMV infection drives changes to specific metabolic signaling pathways that are necessary for the production and replication of the virus (15, 85).
Similar to carbohydrate metabolism, evidence on fatty-acid metabolic changes in the immune cells upon viral infections has been shown on many occasions and suggests an intermetabolic cooperation in glycolytic and fatty-acid pathways (86–89). HCMV also provides a model that crucially explores the characteristic features of sugar and lipid cellular cooperative metabolism to enhance its life cycle across species and how truncation of virus replication can be achieved by suppressing this cooperation (90). It has been further indicated that the shift in metabolism can eventually be an interest for the virus by helping the survival of infected cells (79). Still, targeting these modulated metabolic pathways can prevent the development of infection (85). For example, inhibition of the fatty-acid synthase (FAS) employing drugs such as C75 (trans-4-carboxy-5-octyl-3-methylene-butyrolactone) that shows specificity for HCMV infected cells or 5-(tetradecyloxy)-2-furoic acid (TOFA), which inhibits acetyl-CoA carboxylase enzymatic activity (85). It has also been proven that the inhibition of FAS has suppressed replication of many double-stranded viruses such as EBV (91) and VZV (92).
Like DNA viruses, FAS repression has also demonstrated decreased virus replications in human immune cells such as in DENV (93), HCV (94), influenza virus (85), poliovirus (95), West Nile virus, and yellow fever virus (93). DENV infection has progressed to inflammation and metabolic abnormalities in mice and induced immune cell deterioration on the liver and spleen that were encountered upon stabilization of mast cells (96). Suggested attractive metabolic targets such as the glycolysis of the first enzyme hexokinase or glucose transporter 1 have shown upregulation following DENV infection (97). Viruses have different capabilities to adapt to each cell’s idiosyncratic environment, therefore, virus-induced metabolic programs are overall, cell type-specific (78, 98). Cells infected by RNA viruses such as HIV, HBV, and HCV have demonstrated alterations in the profile of their cellular metabolism (18, 99–101).
It becomes obvious that the metabolic components of the immune system, including innate and humoral cells, are crucial for the immune responses and can shift their behaviors based on either the specific viral invader or conditions in the human host (1, 83). HIV infection, for example, favors MQs and CD4+ T cells in which metabolism is promoted. The activation of divergent metabolic pathways shapes the metabolic environment of the cell and facilitates the viral replication cycle to support infection (101). It is a well-established dogma that CD4+ T cell activation is joined by the reprogramming of specific metabolic pathways, which includes a turn from quiescent cell oxidative metabolism to intensified aerobic glycolysis (92). Activated human monocytes extremely alter their metabolic phenotype from oxidative metabolism to the glycolytic pathway (102). Exceptionally, it was shown that oxidative metabolism was dominated, unlike the reduction in the uptake of glucose and glycolysis as the human U1 monocytic cell line was induced with HIV (103). An recent understanding of glucose metabolism and HIV infection was established, demonstrating that increased cellular permissiveness to HIV-1 infection was a result of CD4+ T cell increased expression of Glut1 in human cultures (104, 105). The process is partially mediated by the PI3K-Akt-mTOR cell survival pathway, which can be targeted in a therapy model in HIV-1 infection (99, 106).
Generally, the relationship between HIV pathogenesis in humans and immune cell metabolism is proposed as immune cells excessively uptake glucose, which could result in hyper induction of immune cells that progresses to complications of HIV infection (107). Still, the fact that our capacity to understand the exact influence of viral infection on the innate and humoral metabolism is limited to the little research effort done in this research area, what may not prevent us to hint on the specific differences between DNA and RNA viruses’ mechanisms to promote infection in the human host (108).
In another context, IFNs are potent modulators of the basic cellular processes (109). For example, PI3K/AKT-dependent uptake of glucose is stimulated by type I IFNs, which suggests an antiviral state as the inhibition of this glycolysis pathway enhances the replication of coxsackievirus B3 in vitro (110). Increased glycolysis is often accompanied by a hypoxia-inducible factor 1α independent, decreased oxidative consumption, a Warburg-effect state, which is as shown in experimental mice crucial to successfully prime CD4+ and CD8+ T cells (111). Type II IFN activation of MQ induces a break in the Krebs cycle flux compared to a high flow of glycolysis (110). This is accompanied by citrate and succinate accumulation (112). Citrate can drive the production of mitochondrial ROS, which holds a preserved role against a number of pathogens and apoptosis inducers (112, 113).
From these analyzed studies, it may appear that viral infections modulate the metabolic statuses in immune cells derived from either human or experimental animals, and a shift to a higher glycolytic activity is a common feature of this modulated metabolism (15, 82–86).
In the frame of a lethal circumstance such as SARS-CoV-2 viral infection, immune cells activate their metabolic reprogramming machinery to evoke this inflamed status and consequences. The ground of metabolic reprogramming during COVID-19 proposes a chance for metabolites with immunomodulatory properties to be interrogated as possible therapeutics to overcome the infection accompanied hyperinflammatory response (114). Using a murine model of SARS-CoV-2, human COVID-19 phenotypes have been mirrored by metabolic repression of oxidative phosphorylation and the TCA cycle in many organs with splenic atrophy, lymphopenia, and neutrophilia (115). One interesting finding in patients with severe COVID-19 is that monocytes/MQs circulated in the blood and are not involved in the generation of cytokine storm (116). It was explained that circulating DCs and monocytes/MQs in patients with severe COVID-19 are repressed by circulating tryptophan metabolites as a result of modulated immunometabolism (116).
On the other hand, the glycolytic shift into lactate has been evidenced by increasing disease severity showing an increase in lactate dehydrogenase activity in immune cells (116). Peripheral blood monocytes obtained from COVID-19 patients have demonstrated an increase in the respiratory mitochondrial electron transport chain (ETC) that has a crucial role in the generation of ROS and the cellular ATP molecules, which could propose a critical role in reprogramming immune cell metabolism to generate a critical immune response in COVID-19 patients (117, 118).
Obesity and factors related to it, such as increased fat intake and obesity-associated chronic inflammation as well as accompanied paradox attenuated innate immune response within the pulmonary compartment, may be crucial determinants in the host interaction with COVID-19 (119). Thus, adjustment of the immune cells’ metabolic potential and bioenergetics can centrally function in the innate immune response to this novel pathogen (119). In this concern, diet and nutrition may influence the outcome of COVID-19, while events such as patients with metabolic problems can change the whole infection progress (120).
In addition, age-related metabolic dysfunction in the innate immune cells due to alteration in glucose metabolism in the immune cells may predispose older individuals to severe outcomes during COVID-19 (121). To note, patients that are at a higher risk for the complications of the disease are usually diagnosed with other metabolic disorders, such as obesity and type II diabetes (122).
There is growing evidence is that some variants of coronavirus can evade immune reactions provoked by previous virus exposure and vaccination, raising attention and worry on some emerging mutations and variants that can manipulate normal metabolic status in immune cells (123).
The lung is the initial infection site and lung failure is a severe complication of SARS-CoV-2 infection (124). Therefore, when evaluating clinical outcomes and mortality as a result of this infection, immune metabolic signatures in the lower respiratory tract of COVID-19 patients are important (124). A common immunomodulation feature observed in COVID-19 patients is the failure to induce significant type I IFN responses in whole blood or peripheral blood mononuclear cells (PBMCs) in concurrence with consistently modulated metabolic pathways including the upregulation of oxidative phosphorylation and downregulation of fructose and mannose metabolism and other glycan degradation (Figure 4A) (124). This leads to increased mortality because of the infection (124).
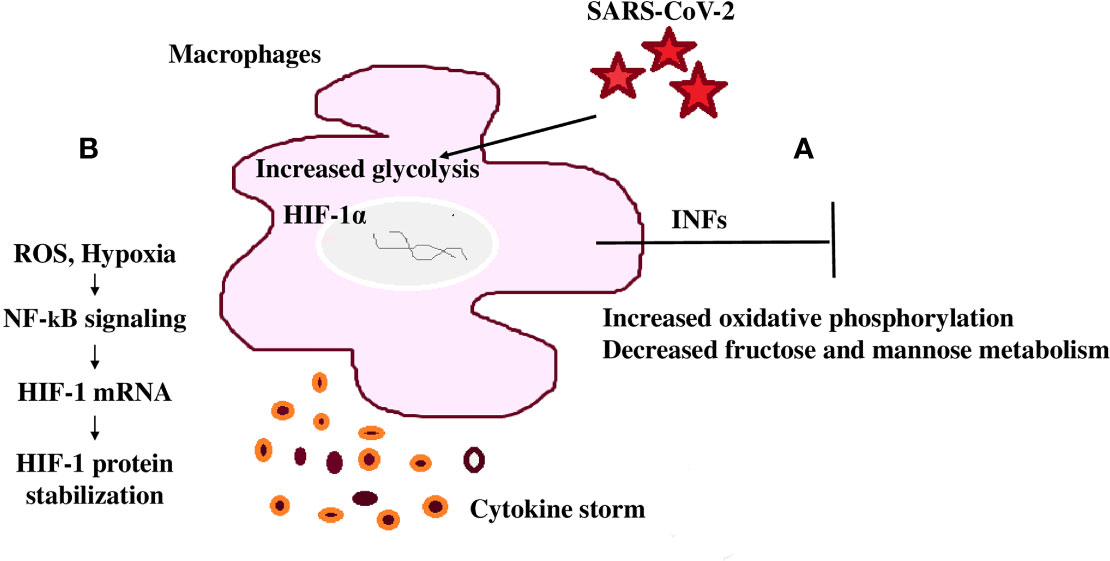
Figure 4 SARS-CoV-2 mediated immunomodulation contribution to mortality (A) COVID-19 patients share a common immunomodulatory failure to induce significant type I IFN responses in whole blood or PBMCs. This happens in concurrence with other modulated metabolic pathways including the upregulation of oxidative phosphorylation and downregulation of mannose and fructose metabolism. (B) SARS-CoV-2 spike protein induces monocytes and macrophages and increases glycolytic metabolism associated with the production of inflammatory cytokines. This response is dependent on hypoxia-inducible factor-1α (HIF-1α); SARS-CoV-2 infection triggers mitochondrial ROS production that stabilizes HIF-1α and consequently, activates glycolysis. HIF-1α-induced changes in monocyte metabolism directly inhibit T cell response that causes progression to epithelial cell COVID-19 mortality.
Monocytes undergo metabolic reprogramming and produce inflammatory cytokines when stimulated with SARS-CoV-2, highlighting the importance of this modulation in the induction of the cytokine storm (124). Stimulation of monocytes with recombinant SARS-CoV-2 spike protein subunit 1 showed a dose-dependent increase in glycolytic metabolism associated with the production of inflammatory cytokines (124, 125). This response is dependent on HIF-1α and evidenced through the inhibition of glycolysis and cytokine production by metformin or by 2-deoxyglucose or glucose deprivation (124). In this way, SARS-CoV-2 spike protein induces a pro-inflammatory immunometabolic response in monocytes that is linked to hyperinflammation during COVID-19 (124).
T cells play an essential and persisting role in the recovery and protection from acute SARS-CoV-2 infection (126). SARS-CoV-2 infection triggers mitochondrial ROS production, which induces stabilization of HIF-1α and consequently promotes glycolysis. HIF-1α-induced changes in monocyte metabolism directly inhibit T cell response and reduce epithelial cell survival progressing COVID-19 mortality (Figure 4B) (125).
It is noteworthy that monocyte-linked severe COVID-19 has been proven in human patients, as well as during experimental infections in different animal models (127–131).
There is growing attention recently toward immunometabolism and its functionality in the frame of infection (1, 16). More precisely, the concept of manipulating glycolysis and oxidative phosphorylation significantly to help prevent disease progression to possibly affect innate and humoral immune cell sustainability and functionality (1, 16). This could result in the development of novel, tolerable immunotherapeutics in the future; early control of the infection or good prognosis of infection; or in the adoption of more preventive strategies based on our understanding of the pathogen/human metabolic mutual interaction (1, 12, 14, 16).
The interplay between host metabolism and bacteria, and the manipulation of the host metabolism by signaling pathways from these bacteria, or induced from the immune cells themselves or from the interaction of the immune cells and other pathogens, are quite of interest and suggests that either targeting or stimulating these signaling pathways can be of importance in fighting the bacterial pathogen (22, 24). Carbohydrate metabolism in the immune cells during TB is signaled by the glycolytic shift while with P. aeruginosa and S. enterica infections, activation is required of specific signals such as Glut1 or Akt1, which may help glucose transport that may in turn stimulate a glucose flux, suggesting that targeting the pathogens’ metabolism can be declared as a potential strategy for an effective remedy for human diseases of both bacterial infection and/or metabolic origin (36). The deep understanding of the challenge facing cell metabolism during infection by bacteria and how these metabolic burdens control the infection outcome is generally still limited. Challenges remain in this regard, in terms of the choice of suitable infection models or established cell lines or even the lack of consistency in the current methodological approaches as recently reviewed (46). It is widely evident that effector surface proteins are expressed on the surface of larger pathogens such as fungi and parasites as discussed using EPF and protozoa or helminths, respectively, to modulate metabolism in these cells, enhancing immune evasion by binding and activating host enzymatic cascades as well as the adhesion and binding of the pathogen to the extracellular matrix and basement membranes of the host cells (72). These proteins may not only be considered possible drug targets or vaccine candidates, but rather possible bioactive human medicines (65). Viral infections are able to demonstrate, beyond this hyper glycolytic activity, different strategies such as the inclusion of oncogenes, expression of modulatory metabolites, or the utilization of possible cooperation of different pathways (15, 84, 86, 90). Viral and cellular oncogenes can diverge and converge to develop critical networks of functional proteins that reprogram the cellular DNA and protein translational machinery, therefore targeting these oncogenes can be useful to prevent unwanted outcomes (12).
Taking into account that the metabolic reprogramming developed by viruses is cell-specific, according to the ability of each virus to compel host cells to adopt new distinctive settings (78). In this sense, targeting the modulatory metabolites or any cellular processes resulted from the interaction between different metabolic pathways for the sake of viral utilization or life cycle preservation can curtail virus replication specifically (85, 86, 90). IFNs are the potent modulators of basic cellular processes during viral infection (109, 110). Modulating, enhancing, or suppressing their signaling cascades may suggest an antiviral state (109).
A common feature during bacterial and viral infections is the upregulation of glycolysis signified by the lactate increase and disturbed TCA cycles signified by higher levels of succinate and other metabolically active intermediates such as itaconic acid. Furthermore, many glycolytic and TCA enzymes are detected on the surface of many fungi and parasites such as pyruvate carboxylase, pyruvate decarboxylase, SDH, and phosphoglycerate kinase, suggesting an unknown role of proteins and products secreted from these organisms in controlling carbohydrate metabolic cycles. Efforts taken to widen the understanding of both the metabolic new status of the pathogen-associated changes in carbohydrate metabolic pathways and signals involved in the pathogen–host interaction to determine specific pathogen signals and metabolites, would show future promise in terms of the design and usage of new vaccines or therapeutics and could help in building a new classification that can be used to adopt new strategies in diagnosis and treatment.
We may also conclude that SARS-CoV-2 can induce metabolic reprogramming in human immune cells and this could result to COVID-19 mortality in some patients; thus, metabolism in immune cells can possibly serve as a potential therapeutic approach to target COVID-19 in a cell-specific manner.
The original contributions presented in the study are included in the article/supplementary material. Further inquiries can be directed to the corresponding authors.
MB conceptualized, structured, and edited the manuscript. KA gathered scientific materials, and analyzed and wrote the manuscript. AM edited and contributed to the immune-metabolism in bacteria section. DA-E edited and contributed to the section on viruses’ immunometabolism. All authors revised and approved the final manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Pearce EL and Pearce EJ. Metabolic Pathways in Immune Cell Activation and Quiescence. Immunity; (2013) 38:633–43. doi: 10.1016/j.immuni.2013.04.005
2. Motawi TK, Shahin NN, Maghraby AS, Kirschfink M, Abd-Elshafy DN, Awad K, et al. H1N1 Infection Reduces Glucose Level in Human U937 Monocytes Culture. Viral Immunol (2020) 33:384–90. doi: 10.1089/vim.2019.0163
3. Loftus RM, Finlay DK. Immunometabolism: Cellular Metabolism Turns Immune Regulator. J Biol Chem (2016) 291:1–10. doi: 10.1074/jbc.R115.693903
4. Biswas SK, Mantovani A. Orchestration of Metabolism by Macrophages. Cell Metab (2012) 15:432–7. doi: 10.1016/j.cmet.2011.11.013
5. Motawi TK, Shahin NN, Awad K, Maghraby AS, Abd-Elshafy DN, Bahgat MM. Glycolytic and Immunological Alterations in Human U937 Monocytes in Response to H1N1 Infection. IUBMB Life (2020) 72:2481–98. doi: 10.1002/iub.2378
6. Muri J, Kopf M. Redox Regulation of Immunometabolism. Nat Rev Immunol (2021) 21:363–81. doi: 10.1038/s41577-020-00478-8
7. Diskin C, Ryan TAJ, O'Neill LAJ. Modification of Proteins by Metabolites in Immunity. Immunity; (2021) 54:19–31. doi: 10.1016/j.immuni.2020.09.014
8. Minhas PS, Latif-Hernandez A, McReynolds MR, Durairaj AS, Wang Q, Rubin A, et al. Restoring Metabolism of Myeloid Cells Reverses Cognitive Decline in Ageing. Nature; (2021) 590:122–8. doi: 10.1038/s41586-020-03160-0
9. Buck MD, O'Sullivan D, Pearce EL. T Cell Metabolism Drives Immunity. J Exp Med (2015) 212:1345–60. doi: 10.1084/jem.20151159
10. Schiller M, Ben-Shaanan TL, Rolls A. Neuronal Regulation of Immunity: Why, How and Where? Nat Rev Immunol (2021) 21:20–36. doi: 10.1038/s41577-020-0387-1
11. Kellermann M, Scharte F, Hensel M. Manipulation of Host Cell Organelles by Intracellular Pathogens. Int J Mol Sci (2021) 22:6484. doi: 10.3390/ijms22126484
12. Delgado T, Sanchez EL, Camarda R, Lagunoff M. Global Metabolic Profiling of Infection by an Oncogenic Virus: KSHV Induces and Requires Lipogenesis for Survival of Latent Infection. PLoS Pathog (2012) 8:e1002866. doi: 10.1371/journal.ppat.1002866
13. Combs JA, Monk CH, Harrison MAA, Norton EB, Morris CA, Sullivan DE, et al. Inhibiting Cytomegalovirus Replication Through Targeting the Host Electron Transport Chain. Antiviral Res (2021) 194:105159. doi: 10.1016/j.antiviral.2021.105159
14. Saraceni C, Birk J. A Review of Hepatitis B Virus and Hepatitis C Virus Immunopathogenesis. J Clin Transl Hepatol (2021) 9:409–18. doi: 10.14218/JCTH.2020.00095
15. Munger J, Bajad SU, Coller HA, Shenk T, Rabinowitz JD, et al. Dynamics of the Cellular Metabolome During Human Cytomegalovirus Infection. PLoS Pathog (2006) 2:e132. doi: 10.1371/journal.ppat.0020132
16. Carbone F, Bruzzaniti S, Fusco C, Colamatteo A, Micillo T, De Candia P, et al. Metabolomics, Lipidomics, and Immunometabolism. Methods Mol Biol (2021) 2285:319–28. doi: 10.1007/978-1-0716-1311-5_24
17. Weljie AM, Jirik FR. Hypoxia-Induced Metabolic Shifts in Cancer Cells: Moving Beyond the Warburg Effect. Int J Biochem Cell Biol (2001) 43:981–9. doi: 10.1016/j.biocel.2010.08.009
18. Diamond DL, Syder AJ, Jacobs JM, Sorensen CM, Walters KN, Proll SC, et al. Temporal Proteome and Lipidome Profiles Reveal Hepatitis C Virus-Associated Reprogramming of Hepatocellular Metabolism and Bioenergetics. PLoS Pathog (2010) 6:e1000719. doi: 10.1371/journal.ppat.1000719
19. Beisel WR. Metabolic Response to Infection. Annu Rev Med (1975) 26:9–20. doi: 10.1146/annurev.me.26.020175.000301
21. Fan J, Chen C, Brlansky RH, Gmitter FJ, Li ZG. Changes in Carbohydrate Metabolism in Citrus Sinensis Infected With ‘Candidatus Liberibacter Asiaticus’. Plant Pathol (2010) 59:1037–43. doi: 10.1111/j.1365-3059.2010.02328.x
22. Naujoks J, Tabeling C, Dill BD, Hoffmann C, Brown AS, Kunze M, et al. IFNs Modify the Proteome of Legionella-Containing Vacuoles and Restrict Infection via IRG1-Derived Itaconic Acid. PLoS Pathog (2016) 12:e1005408. doi: 10.1371/journal.ppat.1005408
23. Lampropoulou V, Sergushichev A, Bambouskova M, Nair S, Vincent EE, Loginicheva E, et al. Itaconate Links Inhibition of Succinate Dehydrogenase With Macrophage Metabolic Remodeling and Regulation of Inflammation. Cell Metab (2016) 24:158–66. doi: 10.1016/j.cmet.2016.06.004
24. Lacey CA, Ponzilacqua-Silva B, Chambers CA, Dadelahi AS, Skyberg JA. MyD88-Dependent Glucose Restriction and Itaconate Production Control Brucella Infection. Infect Immun (2021) 89:e0015621. doi: 10.1128/IAI.00156-21
25. Michelucci A, Cordes T, Ghelfi J, Pailot A, Reiling N, Goldmann O, et al. Immune-Responsive Gene 1 Protein Links Metabolism to Immunity by Catalyzing Itaconic Acid Production. Proc Nat Acad Sci USA (2013) 110:7820–5. doi: 10.1073/pnas.1218599110
26. Qualls JE, Murray PJ. Immunometabolism Within the Tuberculosis Granuloma: Amino Acids, Hypoxia, and Cellular Respiration. Semin Immunopathol (2016) 38:139–52. doi: 10.1007/s00281-015-0534-0
27. Shi L, Salamon H, Eugenin EA, Pine R, Cooper A, Gennaro ML. Infection With Mycobacterium Tuberculosis Induces the Warburg Effect in Mouselungs. Sci Rep (2015) 5:18176. doi: 10.1038/srep18176
28. Somashekar BS, Amin AG, Rithner CD, Troudt J, Basaraba R, Izzo A, et al. Metabolic Profiling of Lung Granuloma in Mycobacterium Tuberculosis Infected Guinea Pigs: Ex Vivo 1H Magic Angle Spinning NMR Studies. J Proteome Res (2011) 10:4186–95. doi: 10.1021/pr2003352
29. Palsson-McDermott EM, O’Neill LA. The Warburg Effect Then and Now: From Cancer to Inflammatory Diseases. Bioessays; (2013) 35:965–73. doi: 10.1002/bies.201300084
30. Shin JH, Yang JY, Jeon BY, Yoon YJ, Cho SN, Kang YH, et al. 1h-NMR-Based Metabolomic Profiling in Mice Infected With Mycobacterium Tuberculosis. J Proteome Res (2011) 10:2238–47. doi: 10.1021/pr101054m
31. Chen Y, Wu J, Tu L, Xiong X, Hu X, Huang J, et al. 1h-NMR Spectroscopy Revealed Mycobacterium Tuberculosis Caused Abnormal Serum Metabolic Profile of Cattle. PLoS One (2013) 8:e74507. doi: 10.1371/journal.pone.0074507
32. Colegio OR, Chu NQ, Szabo AL, Chu T, Rhebergen AM, Jairam V, et al. Functional Polarization of Tumour-Associated Macrophages by Tumor-Derived Lactic Acid. Nature; (2014) 513:559–63. doi: 10.1038/nature13490
33. Kiran D, Basaraba RJ. Lactate Metabolism and Signaling in Tuberculosis and Cancer: A Comparative Review. Front Cell Infect Microbiol (2021) 11:624607. doi: 10.3389/fcimb.2021.624607
34. Sheedy FJ, Divangahi M. Targeting Immunometabolism in Host Defence Against Mycobacterium Tuberculosis. Immunol; (2021) 162:145–59. doi: 10.1111/imm.13276
35. Shi L, Eugenin EA, Subbian S. Immunometabolism in Tuberculosis. Front Immunol (2016) 21:7–150. doi: 10.3389/fimmu.2016.00150
36. Wong AC, Vanhove AS, Watnick PI. The Interplay Between Intestinal Bacteria and Host Metabolism in Health and Disease: Lessons From Drosophila Melanogaster. Dis Model Mech (2016) 9:271–8. doi: 10.1242/dmm.023408
37. Riquelme SA, Prince A. Pseudomonas Aeruginosa Consumption of Airway Metabolites Promotes Lung Infection. Pathogens (2021) 10:957. doi: 10.3390/pathogens10080957
38. Sonnleitner E, Abdou L, Haas D. Small RNA as Global Regulator of Carbon Catabolite Repression in Pseudomonas Aeruginosa. Proc Natl Acad Sci USA (2009) 106:21866–71. doi: 10.1073/pnas.0910308106
39. Mills EL, Kelly B, Logan A, Costa ASH, Varma M, Bryant CE, et al. Succinate Dehydrogenase Supports Metabolic Repurposing of Mitochondria to Drive Inflammatory Macrophages. Cell; (2016) 167:457–470.e13. doi: 10.1016/j.cell.2016.08.064
40. Tannahill GM, Curtis AM, Adamik J, Palsson-McDermott EM, McGettrick AF, Goel G, et al. Succinate is an Inflammatory Signal That Induces IL-1beta Through HIF-1alpha. Nature (2013) 496:238–42. doi: 10.1038/nature11986
41. Qin W, Qin K, Zhang Y, Jia W, Chen Y, Cheng B, et al. S-Glycosylation-Based Cysteine Profiling Reveals Regulation of Glycolysis by Itaconate. Nat Chem Biol (2019) 15:983–91. doi: 10.1038/s41589-019-0323-5
42. Liao ST, Han C, Xu DQ, Fu XW, Wang JS, Kong LY, et al. 4-Octyl Itaconate Inhibits Aerobic Glycolysis by Targeting GAPDH to Exert Anti-Inflammatory Effects. Nat Commun (2019) 10:1–11. doi: 10.1038/s41467-019-13078-5
43. Sebastián AR, Liimatta K, Wong Fok Lung T, Fields B, Ahn D, Chen D, et al. Pseudomonas Aeruginosa Utilizes Host-Derived Itaconate to Redirect its Metabolism to Promote Biofilm Formation. Cell Metab (2020) 31:1091–106.e6. doi: 10.1016/j.cmet.2020.04.017
44. Barghouthi S, Everett KD, Speert DP. Nonopsonic Phagocytosis of Pseudomonas Aeruginosa Requires Facilitated Transport of D-Glucose by Macrophages. J Immunol (1995) 154:3420–8.
45. Gamelli RL, Liu H, He LK, Hofmann CA. Augmentations of Glucose Uptake and Glucose Transporter-1 in Macrophages Following Thermal Injury and Sepsis in Mice. J Leukoc Biol (1996) 59:639–47. doi: 10.1002/jlb.59.5.639
46. Eisenreich W, Heesemann J, Rudel T, Goebel W. Metabolic Host Responses to Infection by Intracellular Bacterial Pathogens. Front Cell Infect Microbiol (2013) 3:24. doi: 10.3389/fcimb.2013.00024
47. Schroder K, Irvine KM, Taylor MS, Bokil NJ, Le Cao KA, Masterman KA, et al. Conservation and Divergence in Toll-Like Receptor 4-Regulated Gene Expression in Primary Human Versus Mouse Macrophages. Proc Natl Acad Sci USA (2012) 109:E944–53. doi: 10.1073/pnas.1110156109
48. Gardner PR, Costantino G, Szabó C, Salzman AL. Nitric Oxide Sensitivity of the Aconitases. J Biol Chem (1997) 272:25071–6. doi: 10.1074/jbc.272.40.25071
49. Pal S, St Leger RJ, Wu LP. Fungal Peptide Destruxin A Plays a Specific Role in Suppressing the Innate Immune Response in Drosophila Melanogaster. J Bio Chem (2007) 282:8969–77. doi: 10.1074/jbc.M605927200
50. Vey A, Matha V, Dumas C. Effects of the Peptide Mycotoxin Destruxin E on Insect Haemocytes and on Dynamics and Efficiency of the Multicellular Immune Reaction. J Invert Path (2002) 80:177–87. doi: 10.1016/S0022-2011(02)00104-0
51. Pitarch A, Sánchez M, Nombela C, Gil C. Sequential Fractionation and Two-Dimensional Gel Analysis Unravels the Complexity of the Dimorphic Fungus Candida Albicans Cell Wall Proteome. Mol Cell Proteomics (2002) 1:967–82. doi: 10.1074/mcp.M200062-MCP200
52. Urban C, Sohn K, Lottspeich F, Brunner H, Rupp S. Identification of Cell Surface Determinants in Candida Albicans Reveals Tsa1p, a Protein Differentially Localized in the Cell. FEBS Lett (2003) 544:228–35. doi: 10.1016/S0014-5793(03)00455-1
53. Padder SA, Ramzan A, Tahir I, Rehman RU, Shah AH. Metabolic Flexibility and Extensive Adaptability Governing Multiple Drug Resistance and Enhanced Virulence in Candida Albicans. Crit Rev Microbiol (2021) 2:1–20. doi: 10.1080/1040841X.2021.1935447
54. Pellon A, Sadeghi Nasab SD, Moyes DL. New Insights in Candida Albicans Innate Immunity at the Mucosa: Toxins, Epithelium, Metabolism, and Beyond. Front Cell Infect Microbiol (2020) 10:81. doi: 10.3389/fcimb.2020.00081
55. Domínguez-Andrés J, Arts RJW, Ter Horst R, Gresnigt MS, Smeekens SP, Ratter JM, et al. Rewiring Monocyte Glucose Metabolism via C-Type Lectin Signaling Protects Against Disseminated Candidiasis. PLoS Pathog (2017) 13:e1006632. doi: 10.1371/journal.ppat.1006632
56. Traven A, Naderer T. Central Metabolic Interactions of Immune Cells and Microbes: Prospects for Defeating Infections. EMBO Rep (2019) 20:e47995. doi: 10.15252/embr.201947995
57. El Kasmi KC, Stenmark KR. Contribution of Metabolic Reprogramming to Macrophage Plasticity and Function. Semin Immunol (2015) 27:267–75. doi: 10.1016/j.smim.2015.09.001
58. Hellwig D, Voigt J, Bouzani M, Löffler J, Albrecht-Eckardt D, Weber M, et al. Candida Albicans Induces Metabolic Reprogramming in Human NK Cells and Responds to Perforin With a Zinc Depletion Response. Front Microbiol (2016) 7:750. doi: 10.3389/fmicb.2016.00750
59. Chung LM, Liang JA, Lin CL, Sun LM, Kao CH. Cancer Risk in Patients With Candidiasis: A Nationwide Population-Based Cohort Study. Oncotarget (2017) 8:63562–73. doi: 10.18632/oncotarget.18855
60. Perera M, Al-Hebshi NN, Perera I, Ipe D, Ulett GC, Speicher DJ, et al. A Dysbiotic Mycobiome Dominated by Candida Albicans is Identified Within Oral Squamous-Cell Carcinomas. J Oral Microbiol (2017) 9:1385369. doi: 10.1080/20002297.2017.1385369
61. Barelle CJ. Niche-Specific Regulation of Central Metabolic Pathways in a Fungal Pathogen. Cell Microbiol (2006) 8:961–71. doi: 10.1111/j.1462-5822.2005.00676.x
62. Thewes S. In Vivo and Ex Vivo Comparative Transcriptional Profiling of Invasive and non-Invasive Candida Albicans Isolates Identifies Genes Associated With Tissue Invasion. Mol Microbiol (2007) 63:1606–28. doi: 10.1111/j.1365-2958.2007.05614.x
63. Ballou ER, Avelar GM, Childers DS, Mackie J, Bain JM, Wagener J, et al. Lactate Signalling Regulates Fungal Beta-Glucan Masking and Immune Evasion. Nat Microbiol (2016) 2:16238. doi: 10.1038/nmicrobiol.2016.238
64. Tucey TM, Verma J, Harrison PF, Snelgrove SL, Lo TL, Scherer AK, et al. Glucose Homeostasis is Important for Immune Cell Viability During Candida Challenge and Host Survival of Systemic Fungal Infection. Cell Metab (2018) 27:988–1006.e7. doi: 10.1016/j.cmet.2018.03.019
65. Molnár I, Gibson DM, Krasnoff SB. Secondary Metabolites From Entomopathogenic Hypocrealean Fungi. Nat Prod Rep (2010) 27:1241–75. doi: 10.1039/c001459c
66. Nakada-Tsukui K, Nozaki T. Immune Response of Amebiasis and Immune Evasion by Entamoeba Histolytica. Front Immunol (2016) 7:175. doi: 10.3389/fimmu.2016.00175
67. Ravidà A, Cwiklinski K, Aldridge AM, Clarke P, Thompson R, Gerlach JQ, et al. Fasciola Hepatica Surface Tegument: Glycoproteins at the Interface of Parasite and Host. Mol Cell Proteomics (2016) 15:3139–53. doi: 10.1074/mcp.M116.059774
68. Carvalho L, Sun J, Kane C, Marshall F, Krawczyk C, Pearce EJ. Review Series on Helminths, Immune Modulation and the Hygiene Hypothesis: Mechanisms Underlying Helminth Modulation of Dendritic Cell Function. Immunol (2009) 126:28–34. doi: 10.1111/j.1365-2567.2008.03008.x
69. Uribe-Querol E, Rosales C. Immune Response to the Enteric Parasite Entamoeba Histolytica. Physiol (Bethesda) (2020) 35:244–60. doi: 10.1152/physiol.00038.2019
70. Tippett E, Cameron PU, Marsh M, Crowe SM. Characterization of Tetraspanins CD9, CD53, CD63, and CD81 in Monocytes and Macrophages in HIV-1 Infection. J Leukoc Biol (2013) 93:913–20. doi: 10.1189/jlb.0812391
71. Aldridge A, O’Neill SM. Fasciola Hepatica Tegumental Antigens Induce Anergic Like T-Cells via Dendritic Cells in a Mannose Receptor Dependent Manner. Eur J Immunol (2016) 46:1180–92. doi: 10.1002/eji.201545905
72. Karkowska-Kuleta J, Kozik A. Moonlighting Proteins as Virulence Factors of Pathogenic Fungi, Parasitic Protozoa and Multicellular Parasites. Mol Oral Microbiol (2014) 29:6–270. doi: 10.1111/omi.12078
73. Wang C, Luosujärvi H, Heikkinen J, Risteli M, Uitto L, Myllylä R. The Third Activity for Lysyl Hydroxylase 3: Galactosylation of Hydroxylysyl Residues in Collagens In Vitro. Matrix Biol (2002) 21:559–66. doi: 10.1016/S0945-053X(02)00071-9
74. Suzuki CK, Rep M, van Dijl JM, Suda K, Grivell LA, Schatz G. ATP-Dependent Proteases That Also Chaperone Protein Biogenesis. Trends Biochem Sci (1997) 22:118–23. doi: 10.1016/S0968-0004(97)01020-7
75. Jeffery CJ. Mass Spectrometry and the Search for Moonlighting Proteins. Mass Spectrom Rev (2005) 24:772–82. doi: 10.1002/mas.20041
76. Metheni M, Echebli N, Chaussepied M, Ransy C, Chéreau C, Jensen K, et al. The Level of H2O2 Type Oxidative Stress Regulates Virulence of Theileria-Transformed Leukocytes. Cell Microbiol (2014) 16:269–79. doi: 10.1111/cmi.12218
77. Sanin DE, Prendergast CT, Mountford AP. IL-10 Production in Macrophages is Regulated by a TLR-Ddriven CREB-Mediated Mechanism That is Linked to Genes Involved in Cell Metabolism. J Immunol (2015) 195:1218–32. doi: 10.4049/jimmunol.1500146
78. Sumbria D, Berber E, Mathayan M, Rouse BT, et al. Virus Infections and Host Metabolism-can We Manage the Interactions? Front Immunol (2021) 11:594963. doi: 10.3389/fimmu.2020.594963
79. Miyake-Stoner SJ, O'Shea CC. Metabolism Goes Viral. Cell Metab (2014) 19:549–50. doi: 10.1016/j.cmet.2014.03.022
80. Ritter JB, Wahl AS, Freund S, Genzel Y, Reichl U. Metabolic Effects of Influenza Virus Infection in Cultured Animal Cells: Intra- and Extracellular Metabolite Profiling. BMC Syst Biol (2010) 13:4–61. doi: 10.1186/1752-0509-4-61
81. Strumillo ST, Kartavykh D, de Carvalho FF Jr, Cruz NC, de Souza Teodoro AC, Sobhie Diaz R, et al. Host-Virus Interaction and Viral Evasion. Cell Biol Int (2021) 45:1124–47. doi: 10.1002/cbin.11565
82. Palmer CS, Cherry CL, Sada-Ovalle I, Singh A, Crowe SM. Glucose Metabolism in T Cells and Monocytes: New Perspectives in HIV Pathogenesis. E Bio Med (2016) 6:31–41. doi: 10.1016/j.ebiom.2016.02.012
83. González Plaza JJ, Hulak N, Kausova G, Zhumadilov Z, Akilzhanova A. Role of Metabolism During Viral Infections, and Crosstalk With the Innate Immune System. Intractable Rare Dis Res (2016) 5:90–6. doi: 10.5582/irdr.2016.01008
84. Thai M, Graham NA, Braas D, Nehil M, Komisopoulou E, Kurdistani SK, et al. Adenovirus E4ORF1-Induced MYC Activation Promotes Host Cell Anabolic Glucose Metabolism and Virus Replication. Cell Metab (2014) 19:694–70. doi: 10.1016/j.cmet.2014.03.009
85. Munger J, Bennett BD, Parikh A, Feng XJ, McArdle J, Rabitz HA, et al. Systems-Level Metabolic Flux Profiling Identifies Fatty Acid Synthesis as a Target for Antiviral Therapy. Nat Biotechnol (2008) 26:1179–86. doi: 10.1038/nbt.1500
86. DeVito SR, Ortiz-Riaño E, Martínez-Sobrido L, Munger J. Cytomegalovirus-Mediated Activation of Pyrimidine Biosynthesis Drives UDP-Sugar Synthesis to Support Viral Protein Glycosylation. Proc Nat Acad Sci USA (2014) 111:18019–24. doi: 10.1073/pnas.1415864111
87. Sankaran S, Guadalupe M, Reay E, George MD, Flamm J, Prindiville T, et al. Gut Mucosal T Cell Responses and Gene Expression Correlate With Protection Against Disease in Long-Term HIV-1-Infected non Progressors. Proc Natl Acad Sci USA (2005) 102:9860–5. doi: 10.1073/pnas.0503463102
88. Blanc M, Hsieh WY, Robertson KA, Watterson S, Shui G, Lacaze P, et al. Host Defense Against Viral Infection Involves Interferon Mediated Down Regulation of Sterol Biosynthesis. PLoS Biol (2011) 9:e1000598. doi: 10.1371/journal.pbio.1000598
89. Miguel A, Vazguez-Calvo A, Caridi F, Saiz J, Sobrino F. Lipid Involvement in Viral Infections: Present and Future Perspectives for the Design of Antiviral Strategies. In Tech Croatia (2013), 291–322. doi: 10.5772/51068
90. Kotzamanis K, Angulo A, Ghazal P. Infection Homeostasis: Implications for Therapeutic and Immune Programming of Metabolism in Controlling Infection. Med Microbiol Immunol (2015) 204:395–407. doi: 10.1007/s00430-015-0402-5
91. Li Y, Webster-Cyriaque J, Tomlinson CC, Yohe M, Kenney S. Fatty Acid Synthase Expression is Induced by the Epstein-Barr Virus Immediate-Early Protein BRLF1 and is Required for Lytic Viral Gene Expression. J Virol (2004) 78:4197–206. doi: 10.1128/JVI.78.8.4197-4206.2004
92. Namazue J, Kato T, Okuno T, Shiraki K, Yamanishi K. Evidence for Attachment of Fatty Acid to Varicella-Zoster Virus Glycoproteins and Effect of Cerulenin on the Maturation of Varicella-Zoster Virus Glycoproteins. Intervirology; (1989) 30:268–77. doi: 10.1159/000150102
93. Heaton NS, Perera R, Berger KL, Khadka S, Lacount DJ, Kuhn RJ, et al. Dengue Virus Nonstructural Protein 3 Redistributes Fatty Acid Synthase to Sites of Viral Replication and Increases Cellular Fatty Acid Synthesis. Proc Natl Acad Sci USA (2010) 107:17345–50. doi: 10.1073/pnas.1010811107
94. Yang W, Hood BL, Chadwick SL, Liu S, Watkins SC, Luo G, et al. Fatty Acid Synthase is Up-Regulated During Hepatitis C Virus Infection and Regulates Hepatitis C Virus Entry and Production. Hepatol; (2008) 48:1396–403. doi: 10.1002/hep.22508
95. Guinea R, Carrasco L. Phospholipid Biosynthesis and Poliovirus Genome Replication, Two Coupled Phenomena. EMBO J (1990) 9:2011–6. doi: 10.1002/j.1460-2075.1990.tb08329.x
96. Morrison J, Rathore APS, Mantri CK, Aman SAB, Nishida A, St John AL, et al. Transcriptional Profiling Confirms the Therapeutic Effects of Mast Cell Stabilization in a Dengue Disease Model. J Virol (2017) 91:18. doi: 10.1128/JVI.00617-17
97. Fontaine KA, Sanchez EL, Camarda R, Lagunoff M. Dengue Virus Induces and Requires Glycolysis for Optimal Replication. J Virol (2015) 89:2358–66. doi: 10.1128/JVI.02309-14
98. Vastag L, Koyuncu E, Grady SL, Shenk TE, Rabinowitz JD. Divergent Effects of Human Cytomegalovirus and Herpes Simplex Virus-1 on Cellular Metabolism. PLoS Pathog (2011) 7:e1002124. doi: 10.1371/journal.ppat.1002124
99. Palmer CS, Crowe SM. The Role of Glucose and Lipid Metabolism in the Pathogenesis of HIV-1 Infection. Curr Trends Immunol (2012) 13:37–50.
100. Zhou L, He R, Fang P, Li M, Yu H, Wang Q, et al. Hepatitis B Virus Rigs the Cellular Metabolome to Avoid Innate Immune Recognition. Nat Commun (2021) 12:98. doi: 10.1038/s41467-020-20316-8
101. Sáez-Cirión A, Sereti I. Immunometabolism and HIV-1 Pathogenesis: Food for Thought. Nat Rev Immunol (2021) 21:5–19. doi: 10.1038/s41577-020-0381-7
102. Dietl K, Renner K, Dettmer K, Timischl B, Eberhart K, Dorn C, et al. Lactic Acid and Acidification Inhibit TNF Secretion and Glycolysis of Human Monocytes. J Immunol (2010) 184:1200–9. doi: 10.4049/jimmunol.0902584
103. Hollenbaugh JA, Munger J, Kim B. Metabolite Profiles of Human Immunodeficiency Virus Infected CD4+T Cells and Macrophages Using LC–MS/MS Analysis. Virol; (2011) 415:153–9. doi: 10.1016/j.virol.2011.04.007
104. Loisel-Meyer S, Swainson L, Craveiro M, Oburoglu L, Mongellaz C, Costa C, et al. Glut1-Mediated Glucose Transport Regulates HIV Infection. Proc Nat Acad Sci USA (2012) 109:2549–54. doi: 10.1073/pnas.1121427109
105. Sorbara LR, Maldarelli F, Chamoun G, Schilling B, Chokekijcahi S, Staudt L, et al. Human Immunodeficiency Virus Type 1 Infection of H9 Cells Induces Increased Glucose Transporter Expression. J Virol (1996) 70:7275–9. doi: 10.1128/jvi.70.10.7275-7279.1996
106. Dzobo K. The Role of Viruses in Carcinogenesis and Molecular Targeting: From Infection to Being a Component of the Tumor Microenvironment. OMICS; (2021) 25:358–71. doi: 10.1089/omi.2021.0052
107. Dagenais-Lussier X, Mouna A, Routy JP, Tremblay C, Sekaly RP, El-Far M, et al. Current Topics in HIV-1 Pathogenesis: The Emergence of Deregulated Immuno-Metabolism in HIV-Infected Subjects. Cyto G F Rev (2015) 26:603–13. doi: 10.1016/j.cytogfr.2015.09.001
108. Speckhart K, Williams JM, Tsai B. How DNA and RNA Viruses Exploit Host Chaperones to Promote Infection? Viruses; (2021) 13:958. doi: 10.3390/v13060958
109. Galani IE, Rovina N, Lampropoulou V, Triantafyllia V, Manioudaki M, Pavlos E, et al. Untuned Antiviral Immunity in COVID-19 Revealed by Temporal Type I/III Interferon Patterns and Flu Comparison. Nat Immunol (2021) 22:32–40. doi: 10.1038/s41590-020-00840-x
110. Fritsch SD, Weichhart T. Effects of Interferons and Viruses on Metabolism. Front Immunol (2016) 7:630. doi: 10.3389/fimmu.2016.00630
111. Pantel A, Teixeira A, Haddad E, Wood EG, Steinman RM, Longhi MP, et al. Direct Type I IFN But Not MDA5/TLR3 Activation of Dendritic Cells is Required for Maturation and Metabolic Shift to Glycolysis After Poly IC Stimulation. PLoS Biol (2014) 12:e1001759. doi: 10.1371/journal.pbio.1001759
112. Chouchani ET, Pell VR, Gaude E, Aksentijević D, Sundier SY, Robb EL, et al. Ischaemic Accumulation of Succinate Controls Reperfusion Injury Through Mitochondrial ROS. Nature; (2014) 515:431–5. doi: 10.1038/nature13909
113. Paiva CN, Bozza MT. Are Reactive Oxygen Species Always Detrimental to Pathogens? Antioxid Redox Signal (2014) 20:1000–37. doi: 10.1089/ars.2013.5447
114. O'Carroll SM, O'Neill LAJ. Targeting Immunometabolism to Treat COVID-19. Immunother Adv (2021) 1:tab013. doi: 10.1093/immadv/ltab013
115. Li S, Ma F, Yokota T, Garcia G Jr, Palermo A, Wang Y. Metabolic Reprogramming and Epigenetic Changes of Vital Organs in SARS-CoV-2-Induced Systemic Toxicity. JCI Insight (2021) 6:e145027. doi: 10.1172/jci.insight.145027
116. Kumar V. How Could We Forget Immunometabolism in SARS-CoV2 Infection or COVID-19? Int Rev Immunol (2021) 40:72–107. doi: 10.1080/08830185.2020.1840567
117. Gardinassi LG, Souza COS, Sales-Campos H, Fonseca SG. Immune and Metabolic Signatures of COVID-19 Revealed by Transcriptomics Data Reuse. Front Immunol (2020) 11:1636. doi: 10.3389/fimmu.2020.01636
118. Zhao R-Z, Jiang S, Zhang L, Yu ZB. Mitochondrial Electron Transport Chain, ROS Generation and Uncoupling (Review). Int J Mol Med (2019) 44:3–15. doi: 10.3892/ijmm.2019.4188
119. Gleeson LE, Roche HM, Sheedy FJ. Obesity, COVID-19 and Innate Immunometabolism. Br J Nutr (2021) 125:628–32. doi: 10.1017/S0007114520003529
120. Berber E, Sumbria D, Rouse BT. Could Targeting Immunometabolism be a Way to Control the Burden of COVID-19 Infection? Microbes Infect (2021) 23:104780. doi: 10.1016/j.micinf.2021.104780
121. Pence BD. Aging and Monocyte Immunometabolism in COVID-19. Aging (Albany NY) (2021) 13:9154–5. doi: 10.18632/aging.202918
122. Batabyal R, Freishtat N, Hill E, Rehman M, Freishtat R, Koutroulis I. Metabolic Dysfunction and Immunometabolism in COVID-19 Pathophysiology and Therapeutics. Int J Obes (Lond) (2021) 45:1163–9. doi: 10.1038/s41366-021-00804-7
123. Callaway E. Fast-Spreading COVID Variant can Elude Immune Responses. Nature; (2021) 589:500–1. doi: 10.1038/d41586-021-00121-z
124. Cory TJ, Emmons RS, Yarbro JR, Davis KL, Pence BD. Metformin Suppresses Monocyte Immunometabolic Activation by SARS-CoV-2 Spike Protein Subunit 1. Front Immunol (2021) 12:733921. doi: 10.3389/fimmu.2021.733921
125. Codo AC, Davanzo GG, Monteiro LB, de Souza GF, Muraro SP, Virgilio-da-Silva JV, et al. Elevated Glucose Levels Favor SARS-CoV-2 Infection and Monocyte Response Through a HIF-1α/Glycolysis-Dependent Axis. Cell Metab (2020) 32:437–46.e5. doi: 10.2139/ssrn.3606770
126. Hurme A, Jalkanen P, Heroum J, Liedes O, Vara S, Melin M, et al. Long-Lasting T Cell Responses in BNT162b2 COVID-19 mRNA Vaccinees and COVID-19 Convalescent Patients. Front Immunol (2022) 13:869990. doi: 10.3389/fimmu.2022.869990
127. Winkler ES, Bailey AL, Kafai NM, Nair S, McCune BT, Yu J, et al. SARS-CoV-2 Infection of Human ACE2-Transgenic Mice Causes Severe Lung Inflammation and Impaired Function. Nat Immunol (2020) 21:1327–35. doi: 10.1038/s41590-020-0778-2
128. Fahlberg MD, Blair RV, Doyle-Meyers LA, Midkiff CC, Zenere G, Russell-Lodrigue KE, et al. Cellular Events of Acute, Resolving or Progressive COVID-19 in SARS-CoV-2 Infected non-Human Primates. Nat Commun (2020) 11:6078. doi: 10.1038/s41467-020-19967-4
129. Bao L, Deng W, Huang B, Gao H, Liu J, Ren L, et al. The Pathogenicity of SARS-CoV-2 in Hace2 Transgenic Mice. Nature; (2020) 583:830–3. doi: 10.1038/s41586-020-2312-y
130. Sia SF, Yan LM, Chin AWH, Fung K, Choy KT, Wong AYL, et al. Pathogenesis and Transmission of SARS-CoV-2 in Golden Hamsters. Nature; (2020) 583:834–8. doi: 10.1038/s41586-020-2342-5
Keywords: carbohydrate metabolism, immune cells, pathogens, COVID-19, glucose
Citation: Awad K, Maghraby AS, Abd-Elshafy DN and Bahgat MM (2022) Carbohydrates Metabolic Signatures in Immune Cells: Response to Infection. Front. Immunol. 13:912899. doi: 10.3389/fimmu.2022.912899
Received: 05 April 2022; Accepted: 01 June 2022;
Published: 04 July 2022.
Edited by:
Ulrich Emil Schaible, Research Center Borstel (LG), GermanyReviewed by:
Pramod Kumar Gupta, Bhabha Atomic Research Centre (BARC), IndiaCopyright © 2022 Awad, Maghraby, Abd-Elshafy and Bahgat. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kareem Awad, a2FyZWVtLmF3YWRAd2ViLmRl; Mahmoud Mohamed Bahgat, bW0ucmlhZEBucmMuc2NpLmVn, bWJhaGdhdHJpYWRAeWFob28uY29t
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.