 Xianfeng Ouyang
Xianfeng Ouyang Yuping Gong
Yuping Gong
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol., 02 June 2022
Sec. Cancer Immunity and Immunotherapy
Volume 13 - 2022 | https://doi.org/10.3389/fimmu.2022.912526
This article is part of the Research TopicRNA Methylation and Tumor ImmunityView all 4 articles
Acute myeloid leukemia is the most common acute leukemia in adults, with accumulation of abundant blasts and impairment of hematogenic function. Despite great advances in diagnosis and therapy, the overall survival of patients with acute myeloid leukemia remains poor. Leukemia stem cells are the root cause of relapse and chemoresistance in acute myeloid leukemia. The tumor immune microenvironment is another trigger to induce recurrence and drug resistance. Understanding the underlying factors influencing leukemia stem cells and the tumor immune microenvironment is an urgent and unmet need. Intriguingly, N6-methyladenosine, the most widespread internal mRNA modification in eukaryotes, is found to regulate both leukemia stem cells and the tumor immune microenvironment. Methyltransferases and demethylases cooperatively make N6-methyladenosine modification reversible and dynamic. Increasing evidence demonstrates that N6-methyladenosine modification extensively participates in tumorigenesis and progression in various cancers, including acute myeloid leukemia. In this review, we summarize the current progress in studies on the functions of N6-methyladenosine modification in acute myeloid leukemia, especially in leukemia stem cells and the tumor immune microenvironment. We generalize the landscape of N6-methyladenosine modification in self-renewal of leukemia stem cells and immune microenvironment regulation, as well as in the initiation, growth, proliferation, differentiation, and apoptosis of leukemia cells. In addition, we further explore the clinical application of N6-methyladenosine modification in diagnosis, prognostic stratification, and effect evaluation. Considering the roles of N6-methyladenosine modification in leukemia stem cells and the tumor immune microenvironment, we propose targeting N6-methyladenosine regulators as one stone to kill two birds for acute myeloid leukemia treatment.
Acute myeloid leukemia (AML) is a hematologic malignancy characterized by abnormal proliferation of blasts or immature progenitor cells in the bone marrow, blood and other tissues with arrested differentiation and apoptosis disorder, generating suppression of normal hematopoiesis (1).
Over the last few decades, intensive induction chemotherapy, such as the combination of anthracycline and cytarabine, consisting of the “3+7” regimen, has been the standard scheme for fit patients (2). According to age, physical state, comorbidity, complications, and hematological and genetic prognostic indicators, current treatment approaches involve combination chemotherapy, hypomethylating agents (HMAs), BCL-2 inhibition, targeted therapy, immunotherapy, and/or hematopoietic stem cell transplantation (HSCT) (3, 4). In this model of treatment, complete remission rates can achieve 60-80% in younger patients and 40-60% in older patients (1, 5, 6).
The survival rates of patients with AML vary from person to person, which is attributed to differences in their ages and clinical and genomical prognostic characteristics. Nevertheless, most patients will eventually relapse (3). Leukemia stem cells (LSCs) are deemed to be resistant to chemotherapy, leading to disease recurrence (7). When transplanted into immunodeficient mice, LSCs have the ability to trigger the disease and can maintain self-renewal (8).
Immune evasion is another cause of the relapse of leukemia. It is widely known that the human immune system can identify and kill non-self components, such as leukemia cells (9). However, in fact, AML blasts, including LSCs, develop multiple mechanisms to escape host immune surveillance and eradication (10, 11). In addition to the feature of tumor initiation, immune evasion is an even more vital competence of cancer stem cells (CSCs) (12, 13). LSCs were first identified as CSCs employing combined immunodeficient mice (14). CSCs regulate the tumor immune microenvironment (TIME) via immune evasion, such as expressing immunosuppressive molecules and recruiting immunosuppressive cells (15). Likewise, the TIME can cooperate with CSCs to promote tumor progression (16–19).
Recently, research on the functions of RNA epigenetic modification in AML has grown vigorously. Above all, the N6-methyladenosine (m6A) RNA modification is of the greatest concern (20, 21). First, fat mass and obesity associated protein (FTO) was found to promote leukemia initiation and progression, and is considered to have carcinogenic activity. A variety of leukemia cells highly express FTO in several subtypes of AML (22). Subsequently, methyltransferase-like 3 (METTL3), methyltransferase-like 14 (METTL14), Wilm’s tumor 1-associated protein (WTAP), and AlkB homolog 5 (ALKBH5) were reported to be related to myeloid leukemia (23–25). Meanwhile, many studies have found that m6A participates in the regulation and maintenance of the stemness of CSCs (26–28). m6A modification is also pivotal for the self-renewal of LSCs (21). In addition, m6A modification also involves the modulation of the TIME in various cancers, as well as in AML (29–32).
In this review, we spark new ideas about m6A modification in LSCs and the TIME for better clinical treatment implications in AML.
Despite achieving complete remission after chemotherapy, AML patients ultimately die of recurrence due to chemotherapy-resistant LSCs (33–35). Lapidot et al. first identified AML-initiating cells as LSCs that had the ability to repopulate human leukemia in SCID mice. Analogous to normal hematopoietic stem cells (HSCs), CD34+CD38- is the typical phenotype of LSCs (14). In addition, LSCs can be present in CD34+CD38+ cellular compartments and even in the CD34- subpopulation (36, 37). In addition to the properties of self-renewal, proliferation and differentiation, LSCs are characterized by cell cycle quiescence, low energy requirements, hypomethylated state and chemoresistance (8, 38). Apart from CD34 and CD38, there are various cell-surface markers on LSCs. For instance, TIM-3, CLL-1, CD47, CD70/CD27, CD96, CD123, CD244, CD200 and CD93 (39–48).
T cell immunoglobulin and mucin protein 3 (TIM-3), which is expressed in AML cells, secretes its ligand Galectin-9 (Gal-9) to constitute a TIM-3/Gal-9 autocrine loop. The loop is capable of promoting the self-renewal of LSCs by activating both NF-kB and the Wnt/β-catenin signaling pathways (40).In addition to Gal-9, TIM-3 in AML cells produces soluble TIM-3 (sTIM-3), which can attenuate the release of IL-2. The cytokine IL-2, secreted by T cells, is pivotal to the activation of cytotoxic T cells and NK cells (49). Because of the expression of TIM-3 in various immune cells, such as T cells, NK cells, DCs and mast cells, Gal-9 secreted by LSCs can combine with TIM-3 in immune cells, giving rise to a suppressive tumor immune microenvironment. The accumulation of β-catenin in LSCs is essential for self-renewal and progression. Additionally, β-catenin even lures the expression of a variety of immune checkpoints, including TIM-3, to suppress the host immune system (50). Likewise, TIM-3 also activates the PI3K/mTOR signaling pathway, resulting in the accumulation of hypoxia-inducible factor 1-alpha (HIF1α), which can induce programmed cell death-ligand 1 (PD-L1) expression in tumor cells to escape adaptive immunity (51, 52).
CD47 is considered a marker of LSCs in AML. Elevated expression of CD47 on LSCs can restrain phagocytosis of macrophages, a kind of phagocyte expressing SIRPα. LSCs can initiate the signaling “Don’t eat me” through the CD47-SIRPα interaction to escape from host innate immune attack (42). CD200 is highly expressed in LSCs. Herbrich and his colleagues found that CD200+ AML cells could inhibit T cell cytokine secretion, change T cell composition and cell cycle, interrupt T cell metabolism, and weaken the macrophage response to AML cells. Furthermore, CD200 monoantibody therapy can counteract these effects, as foresaid (47). Poly-ADP-ribose polymerase 1(PARP1) in LSCs is able to suppress the expression of NKG2D ligand (NKG2DL), leading to evasion of NK cell killing (53).
Taken together, LSCs can take advantage of various mechanisms to escape from immunological surveillance in the tumor microenvironment (TME). In turn, immune cells in the TME can add further weight to promote and maintain the self-renewal and progression of LSCs. That CSCs can merely establish and self-renew in immunodeficient mice such as NOD/SCID mice is a most convincing example to elucidate that immune selection may play a crucial part in CSCs implantation and maintenance (15). Depreter and colleagues reported that immune dysregulation could be responsible for the initiation and maintenance of LSCs in pediatric AML (54). PD-1, CTLA-4, TIM-3 and LAG-3 overexpression in bone marrow T cells contributes to AML relapse after allogeneic HSCT. The function of these T cells in bone marrow is exhausted due to the declining ability to produce cytotoxic cytokines (55). Xu et al. found that TIGIT and PD-1 were simultaneously overexpressed on CD8+ T cells in the bone marrow of AML patients, giving rise to the immunosuppressive microenvironment (56). Additionally, cytokine signaling in the TIME also plays a role in the regulation of LSCs (57).
In brief, LSCs interact with the TIME in a variety of ways to enhance and maintain AML progression and chemoresistance, ultimately resulting in disease relapse.
It is well known that epigenetic modification plays pivotal roles in the genesis and development of all kinds of cancers, including AML. DNA methylation, histone modification, and chromatin remodeling are common epigenetic modifications. Recently, RNA modification has increasingly served as a hotspot of epigenetic studies. In particular, N6-methyladenosine (m6A) RNA modification, referring to methylation on the sixth atom of adenosine, is the most intriguing and most pervasive messenger RNA modification in the interior of eukaryotes (58). m6A RNA modification is dynamic and reversible under the control of m6A regulators and is divided into writers, erasers and readers by function. With rapid advances in high-throughput sequencing technology, studies on the function of m6A RNA modification in AML have made tremendous progress and triggered widespread interest, especially in LSCs maintenance and immune regulation.
Writers, namely, m6A methyltransferases, act as the transferrer of m6A to RNA adenosine in the form of a complex, including METTL3, METTL14, and WTAP. METTL3 interacts with METTL14 to constitute a heterodimer complex that is stable and difficult to interrupt. METTL3 has the catalytic function of adding m6A to RNA, while METTL14 is considered a scaffold to sustain the structure of METTL3 and to facilitate the binding of RNA. The METTL3-METTL14 complex coordinately induces RNA methylation (59). Despite being a WT-1-related protein, WTAP plays a role in m6A modification independent of WT1. In this process, WTAP is essential for localization of the METTL3-METTL14 complex in nuclear speckles and binding with target mRNAs (60). Despite its critical role in nuclear localization and recruitment of the METTL3-METTL14 complex, WTAP actually has no catalytic ability (61).
Furthermore, zinc finger CCCH-type containing 13 (ZC3H13), RNA binding motif protein 15/15B (RBM15/RBM15B) and vir-like m6A methyltransferase associated (VIRMA/KIAA1429) are also common writers. ZC3H13 functions as an anchor for WTAP-Virilizer-Hakai in nuclear speckles and regulates RNA m6A methylation, contributing to the self-renewal of mouse embryonic stem cells (62). RBM15 and RBM15B, with structural similarity and functional complementarity, possess peculiar domains that can bind the WTAP-METTL3 complex to target mRNA to induce m6A formation and can also bind the lncRNA X-inactive specific transcript (XIST) to facilitate XIST-mediated gene transcriptional silencing (63). VIRMA, also named KIAA1429, regulates preferential mRNA region-specific methylation in the 3′UTR and near the stop codon, by recruiting the catalytic complex METTL3/METTL14/WTAP (64).
Additionally, a series of new m6A writers have been discovered successively, for instance, methyltransferase-like 5 (METTL5), methyltransferase-like 16 (METTL16), NOP2/Sun RNA methyltransferase 2 (NSun2), phosphorylated CTD interacting factor 1 (PCIF1), zinc finger CCHC type containing 4 (ZCCHC4) and HAKAI. METTL5, as a heterodimer complex with TRMT112, cooperates with ZCCHC4 to deposit m6A on human 18S and 28S ribosome RNAs (rRNAs) (65). METTL16 participates in U6 small nuclear RNA (snRNA) methylation and contributes to controlling the homeostasis of S-adenosylmethionine (SAM), the methyl donor, by regulating the expression of MAT2A. Beyond that, METTL16 is also essential for common m6A RNA methylation modification (66). NSun2 is engaged in miRNA methylation to reduce the expression of miR-125b, resulting in cancer cell migration (67). PCIF1 exclusively acts as mammalian mRNA m6Am methyltransferase, contributing to suppressing translation activity (68).The type E3 ubiquitin ligase HAKAI, also called CBLL1, is essential for stabilization of the m6A-METTL-associated complex (MACOM), playing a critical role in m6A deposition in Drosophila (69).
Erasers, also called demethylases, are responsible for removing m6A methylation from the target mRNA. The coaction of methyltransferases and demethylases maintains m6A modification in a reversible and dynamic state. Two prominent erasers, FTO and ALKBH5, were successively discovered. FTO, belonging to the AlkB family of proteins, is the first to demethylate m6A in nuclear speckles of mRNA in an iron(II)- and α-KG-dependent manner, affecting the level of m6A (70). FTO predominantly targets nuclear m6A in a majority of cells and both cytoplasmic m6A and m6Am in mRNA (71). FTO can also affect the transcriptional level by regulating m6A demethylation in mRNA and influence translation by mediating m1A demethylation in tRNA. Furthermore, FTO can regulate the demethylation of m6A in U6 RNA and m6Am in snRNAs (71). As a first gene for non-syndromic human obesity, FTO is reported to participate in modulating adipogenesis via m6A demethylation (72).
ALKBH5, another demethylase belonging to the AlkB family, can exert catalytic effects to remove m6A from mRNA, regulating the abundance of m6A, mRNA export and RNA metabolism (73). Another member of the AlkB family, AlkB homolog 3 (ALKBH3), plays a central role in m1A and m3C demethylation in RNA and mediates m6A demethylation in tRNA to enhance protein translation (74).
Readers, known as m6A binding proteins, are indispensable for m6A to take effect. YTH domain family proteins (YTHDF1, YTHDF2 and YTHDF3) and YTH domain-containing proteins (YTHDC1 and YTHDC2), both belonging to the YT521-B homology (YTH) domain family, are widely known as m6A readers that recognize and bind to the m6A methylation site, affecting RNA fate and gene expression (75, 76). Until now, YTHDC2 and all YTH domain family proteins have been found to be located in the cytoplasm, while the uniquely known m6A binding protein of the YTH domain family, which is located in the cell nucleus, is YTHDC1 (76–78). YTHDF1 directly enhances the translational efficiency in the manner of m6A methylation by interacting with initiation factors (eIFs) and promoting ribosomes uptake of its target mRNA in the cytoplasm (79). YTHDF2 can selectively recognize m6A to impact protein translation and RNA metabolism, with the C-terminus binding to m6A- methylated mRNA and the N-terminus conveying the YTHDF2-mRNA complex to RNA degradation sites (80). YTHDF3, another cytoplasmic m6A reader of YTH domain family proteins, fascinates translation by collaborating with YTHDF1 but not interacting with eIFs and promotes degradation of methylated mRNAs together with YTHDF2, both in an m6A-dependent manner (81). YTHDC1, as the only reader in the nucleus, plays a part in transcription and gene expression by selectively binding to methylated RNA (82). YTHDC1 plays a vital role in directly regulating mRNA splicing in the form of accelerating exon inclusion in targeted mRNAs through enhancing SRSF3 but repressing SRSF10 RNA-binding ability (83). YTHDC1 is also engaged in nuclear export by promoting RNA binding to both SRSF3 and NXF1 in an m6A-dependent manner but independent of pre-mRNA splicing (84). YTHDC2, the only RNA helicase-containing m6A binding protein, can improve translation efficiency through m6A methylation in mRNA coding regions (CDS) and interaction with the small ribosomal subunit (85, 86). Furthermore, YTHDC2 can regulate the degradation of target mRNAs by collecting RNA decay factors (86).
In addition to the aforesaid m6A readers, which belong to the YTH domain family, there are two new families recognized as m6A binding proteins, the insulin-like growth factor 2 mRNA-binding protein (IGF2BP) family and the heterogeneous nuclear ribonucleoprotein (HNRNP) family. IGF2BPs, including IGF2BP1, IGF2BP2 and IGF2BP3, play a central role in enhancing the stability of mRNA and protein translation by recognizing m6A with K homology (KH) domains and can even interact with MYC to accelerate tumorigenesis in an m6A-dependent manner (87). HNRNPA2B1, one member of the HNRNP family, functions as a nuclear m6A ‘reader’ to directly bind the m6A mark, contributing to promoting pri-miRNA transcription by binding the microRNA microprocessor complex protein DGCR8 and regulating alternative splicing (88). HNRNPC and HNRNPG, two other members of the HNRNP family, both interact with target mRNAs by remodeling the RNA structure in an m6A-dependent way, changing the abundance and alternative splicing of target mRNAs (89, 90). The major roles of m6A regulators are summarized in Figure 1.

Figure 1 The functions of m6A RNA modification. m6A writers and erasers reversibly and dynamically regulate the m6A methylation modification. Readers can recognize and bind to the m6A methylation site to influence RNA fate, including nuclear exporting, splicing, stability, decay, and translation, as well as microRNA processing.
Currently, a variety of studies on the roles of m6A modification in leukemia have been conducted, displaying that m6A modification not only participates in the proliferation, colony formation, differentiation and apoptosis of leukemia cells but also maintains the pluripotency and self-renewal of LSCs.
METTL3 is indispensable for the development of AML cells due to the CAATT box binding protein CEBPZ in the transcriptional start site (TSS) binding chromatin, independent of METTL14. Promoter-bound METTL3 facilitates mRNA transcription and protein translation to maintain the growth of AML cells in an m6A-dependent manner. METTL3-knockdown cells exhibit distinct cell cycle arrest and marked differentiation, especially MLL-AF9-driven AML cells (91). METTL3 mRNA is highly expressed in human AML samples compared to other types of tumors. Increased METTL3 in leukemic cells is required for the maintenance of differentiation arrest of AML cells by enhancing transcripts of c-MYC, BCL-2 and PTEN and inactivating the PI3K/AKT pathway, accompanied by elevated m6A abundance. Loss of METTL3 in AML cells can induce apoptosis and enhance differentiation, impeding leukemic cell development (23). METTL3 also participates in regulating WTAP mRNA translation and protein stabilization to maintain WTAP protein homeostasis, which is recognized as an oncogenic protein in AML (92, 93). STM2457, a small-molecule METTL3 inhibition, can suppress the development and promote the differentiation and apoptosis of AML cells in vivo and in vitro. After treatment with STM2457, the CD93+ cell population, which is identified as leukemia stem cells, was significantly reduced, while the CD48+ intensity was apparently increased (94, 95). METTL3 inhibition annihilates LSCs, interrupting AML growth and propagation. We turn our attention to METTL3 as an active participant in the self-renewal of LSCs.
METTL14 plays a pivotal role in repressing AML differentiation and maintaining the self-renewal of LSCs by modulating the mRNA stability and translation of MYC and MYB in an m6A-dependent manner, while METTL14 expression is suppressed by SPI1, a master transcription factor (TF). In other words, METTL14 exerts an effect on AML initiation and development via the SPI1-METTL14-MYC/MYB signaling axis (24). METTL14 can also interact with chimeric mRNA to inhibit myeloid cell differentiation through complexes composed of fusion protein and MALAT1, a nuclear speckle-specific long noncoding RNA, depending on YTHDC1 and SRSF3 (96).
WTAP is engaged in promoting AML proliferation and differentiation arrest through the mammalian target of rapamycin mTOR signaling pathway, including its downstream effector p70 ribosomal subunit 6 kinase (pS6K), which functions as an oncogenic protein (93). WTAP is considered a poor prognostic factor by enhancing leukemogenesis, the cell cycle, and chemoresistance of AML cells based on m6A modification of MYC mRNA (97). Whether WTAP influences the self-renewal of LSCs remains elusive.
FTO, as an oncogenic protein, participates in promoting AML cell proliferation and transformation while hindering AML cell differentiation and apoptosis, especially in AML subtypes with MLL-rearrangements and PML-RARA, FLT3-ITD and NPM1 mutations. ASB2 and RARA, which play a part in hematopoietic cell differentiation, act as negative target genes of FTO in AML. FTO plays a carcinogenic role by inversely regulating ASB2 and RARA both at the RNA and protein levels in an m6A-dependent way (22). R-2-hydroxyglutarate (R-2HG), an antimetabolite generated by mutant isocitrate dehydrogenase 1/2 (IDH1/2), can target FTO to exert an anti-leukemia effect. By negatively regulating FTO, R-2HG inhibits MYC/CEBPA-related pathways and impedes lactate dehydrogenase B (LDHB) and phosphofructokinase platelet (PFKP) to abrogate aerobic glycolysis, both through a mechanism dependent on m6A (98, 99). In addition, FTO mediates the resistance to tyrosine kinase inhibitors (TKIs) in leukemia cells by regulating MERTK and BCL-2 through m6A demethylation (100). Upon FTO knockdown or treatment with the FTO inhibitors CS1 or CS2, the CD34+ LSC proportion was significantly decreased, indicating that FTO facilitates the self-renewal of LSCs by targeting MYC and CEBPA (101). The dramatically reduced number of CD34+CD38- LSCs in a PDX AML mouse model after treatment with the FTO inhibitor FB23-2 is another example demonstrating that FTO is associated with the self-renewal of LSCs (102).
ALKBH5 is reported to play key roles in the initiation of AML cells and self-renewal of LSCs in an m6A-dependent manner, predicting poor prognosis in AML patients. ALKBH5 directly regulates the mRNA stability of its target gene TACC3, which affects MYC and p21, contributing to leukemogenesis and the self-renewal of LSCs (25). ALKBH5 is affected by chromatin alterations attributed to decreased H3K9me3 and positively regulated by MYB and polymerase II (Pol II) in AML cells. KDM4C is the root cause of all these regulators influencing ALKBH5 expression, resulting in the progression of AML cells and the self-renewal of LSCs. ALKBH5 fine-tunes the stability of AXL mRNA depending on m6A modification mediated by YTHDF2, promoting the development and clonogenic potential of leukemia cells (103). Collectively, ALKBH5 plays essential roles in LSC self-renewal and maintenance through a variety of mechanisms.
YTHDF2, overexpressed in human AML samples, is considered to facilitate leukemia initiation and LSC maintenance by shortening the half-life of various m6A transcripts in favor of LSC functions and regulating mRNA decay in an m6A-dependent way. YTHDF2 depletion makes AML cells more susceptible to TNF, inducing increased apoptosis (104). The AML1/ETO-HIF1α loop participates in regulating YTHDF2 to promote leukemia cell development in t (8;21) AML cells. YTHDF2 downregulation can increase m6A abundance and TNF receptor superfamily member 1b (TNFRSF1b) expression, contributing to apoptosis (105). YTHDC1, a nuclear reader, plays a part in facilitating the initiation and maintenance of AML cells, especially blocking differentiation, by binding to m6A to form dynamic nuclear YTHDC1-m6A condensates (nYACs) mediated by liquid-liquid phase separation (LLPS). nYACs play a crucial role in maintaining the stability of the target mRNA MYC, which is directly regulated by YTHDC1 in an m6A-dependent pathway, by preventing RNA degradation by the polyA tail exosome targeting complex (PAXT) (106). In addition to proliferation and colony formation, YTHDC1 also accounts for LSC self-renewal by enhancing the stability of minichromosome maintenance 4 (MCM4), which contributes to DNA replication (107). IGF2BP1 expressed in AML cells promotes leukemogenesis and proliferation and maintains the stemness of leukemia cells by regulating HOXB4, MYB, and ALDH1A1, which are LSC phenotype-associated transcriptional and metabolic factors. IGF2BP1 depletion can reduce colony formation, impede leukemogenesis and proliferation, and postpone leukemia cell development in NSG mice. IGF2BP1 functions as an oncogenic protein and regulator of LSC self-renewal maintenance in an m6A-dependent manner (108). IGF2BPs interact with YBX1, an RNA-binding protein (RBP), to facilitate YBX1 stabilization of MYC and BCL-2 mRNA in an m6A-depentdent way to maintain AML cell development (109). Based on this finding, IGF2BPs indirectly take part in the initiation and maintenance of AML cells.
Taken together, m6A modification, mediated by writers, erasers or readers, is indispensable for AML cell initiation, proliferation, growth, differentiation and survival. Furthermore, m6A regulators play a critical role in LSC self-renewal, which is responsible for AML recurrence and chemoresistance.
The roles of m6A modification in the TIME have increasingly attracted extensive attention. METTL3 or METTL14 depletion promotes immune responses to anti-PD-1 therapy in colorectal cancer (CRC) by increasing CD8+ T cell infiltration, cytokine secretion and chemokine production in the TME. METTL3 and METTL14 expressed on tumor cells remodel the TIME by regulating immune cells, cytokines and chemokines through the IFN-c-Stat1-Irf1 pathway in an m6A-dependent manner (110). FTO enhances the expression of PD-L1 in colon cancer cells through m6A methylation modification but not the IFN-g signaling-dependent pathway (111). In addition, FTO represses T cell immune activation in the lung cancer cell lines and melanoma cells by regulating JunB and C/EBPb in an m6A-dependent way to accelerate tumor glycolysis. The FTO inhibitor Dac51 improves T cell infiltration and cytotoxic capacity cooperatively with an anti-PD-L1 antibody (112). ALKBH5 in tumor cells recruits immune suppressive cells, including myeloid-derived suppressor cells (MDSCs) and Tregs, and facilitates lactate accumulation to regulate the anti-PD-1 therapy response depending on m6A in melanoma. ALKBH5 inhibitor promotes anti-tumor immunity of anti-PD-1 (113). ALKBH5, induced by hypoxia in glioblastoma multiforme (GBM), is responsible for tumor-associated macrophage (TAM) recruitment, leading to a suppressive TIME by facilitating CCX8/IL-8 secretion (114).
At present, studies on m6A modification in the TIME of AML have been successively performed. FTO is reported to induce the expression of immune checkpoint leukocyte immunoglobulin-like receptor subfamily B 4 (LILRB4), which contributes to T cell suppression and tumor infiltration in AML, leading to immune evasion (115).Upon treatment with decitabine, FTO expression was significantly increased, resulting in decreased m6A abundance in AML cells. Meanwhile, the immune checkpoints LILRB4, PD-L1, PD-L2 and PD-1 in AML cells or T cells were also elevated, especially LILRB4, which was well above the others. With FTO knockdown or FTO inhibitors, LIRLB4 expression was dramatically reduced at both the RNA and protein levels through YTHDF2-mediated m6A modification (101). This may be a mechanism by which HMAs induce immune evasion and chemoresistance. After pretreatment with the FTO inhibitors CS1 or CS2, AML cells with high expression of LILRB4 were subsequently co-cultured with activated T cells, displaying that LILRB4 expression was significantly decreased and that the killing effect of T cells on AML cells was remarkably enhanced (101). Conceivably, FTO-associated m6A modification plays pivotal roles in immunoregulation in AML.
The gold nanorods GNRa-CSP12 regulates FTO and ALKBH5, which are both Fe2+-dependent demethylases, to impede the proliferation of AML cells by triggering ferroptosis in an m6A-dependent manner. Upon GNRa-CSP12 treatment, genes of immune checkpoint pathways are downregulated, which is ascribed to the decreased stability of SLC2A3, CD276, and PKM transcripts. GNRa-CSP12 can also overcome FTO-mediated TKI resistance. In addition, GNRa-CSP12 plays an important role in promoting the antileukemia efficacy of PD-L1 antibody by targeting FTO and ALKBH5 to activate T cell responses, such as the infiltration of CD4+ T cells and CD8+ T cells, as well as the cytotoxic cytokine IFN-γ in the TIME (116). This shows that m6A modification mediated by FTO and ALKBH5 participates in the regulation of the immune response in AML.
Given that genes involved in immune response processes are downregulated after YTHDF2 depletion in AML cells, YTHDF2 is perceived to mediate the immune response (104). The detailed mechanism by which YTHDF2 regulates the immune response in AML remains to be elucidated.
Du et al. divided m6A modification patterns into three distinct clusters based on 23 m6A regulators in 255 AML specimens derived from public databases, and calculated m6A scores with statistical software, indicating that different clusters were corresponded with different immune infiltration phenotypes in the TIME. They found that the expression of immune checkpoints, including PD-L1, PD-L2, MRP1, and MRP2, was remarkably higher in AML patients with lower m6A scores, implying that m6A modification takes part in antileukemia immunity. Additionally, m6A scores are positively correlated with immune response and better prognosis (117). In brief, m6A modification plays significant roles in regulating the TIME and affecting immunotherapy efficacy. m6A-related long noncoding RNAs (lncRNAs) are reported to influence the immune response in AML (118). It can be assumed that m6A indirectly modulates the immune response by interacting with lncRNAs.
Compared to solid tumors, studies on m6A modification in the TIME of AML are limited. However, it sheds further insight into future research to explore the impact of m6A on the immune microenvironment and to demonstrate why the therapeutic effect of immunotherapy alone is unsatisfactory in AML, paving the way to develop novel treatment strategies for AML patients. The outline of m6A modification in LSCs and the TIME is shown in Figure 2.
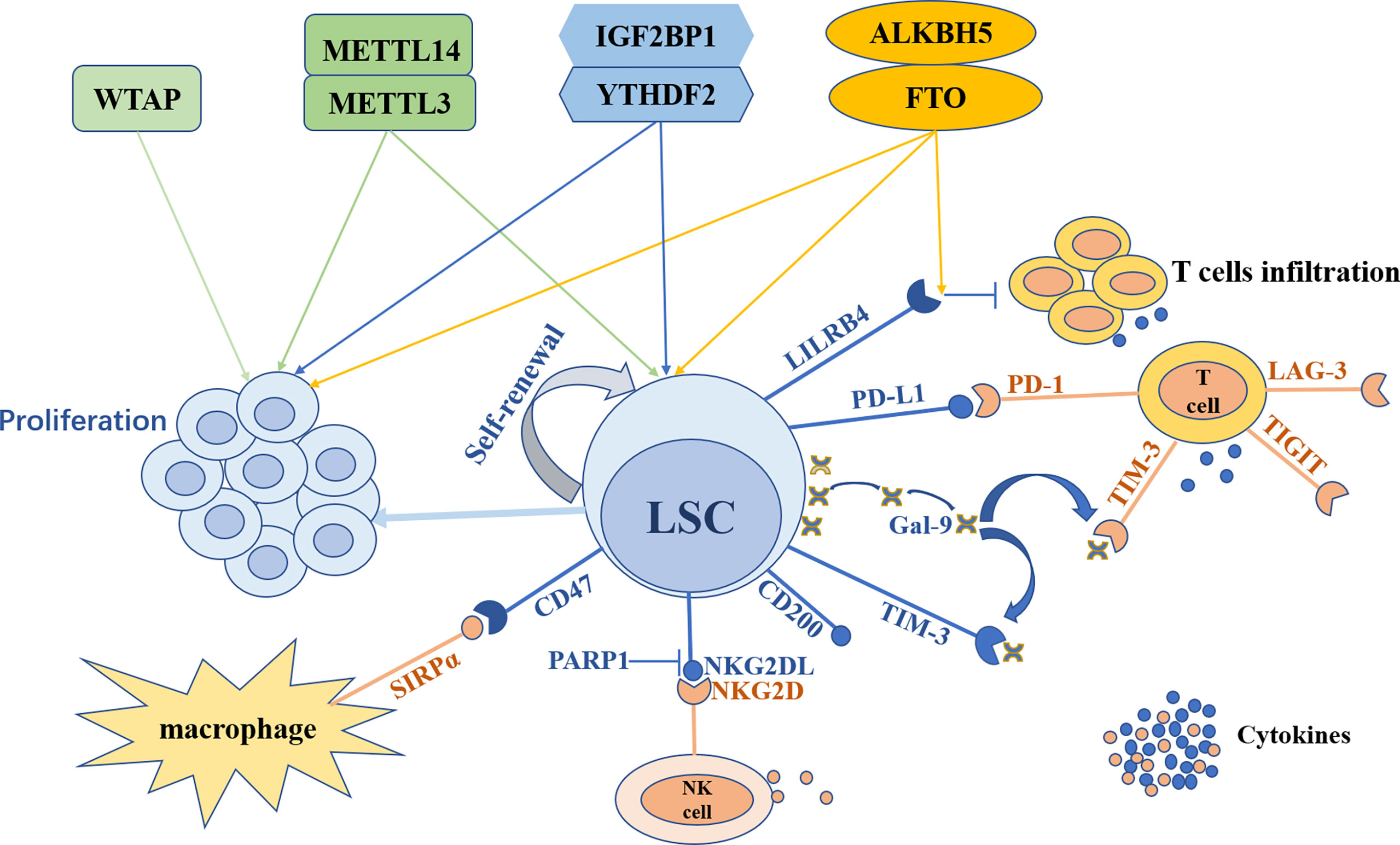
Figure 2 The outline of m6A modification in leukemia stem cells and the tumor immune microenvironment. Leukemia stem cells (LSCs) can maintain self-renewal and trigger leukemia cells proliferation. LSCs can escape from the surveillance and elimination of the immune system through various mechanisms. With the expression of TIM-3, LSCs secrete galectin-9 (Gal-9) to form TIM-3/Gal-9 autocrine loop. Meanwhile Gal-9 combines with TIM-3 expressed on T cells to inhibit T cells immunity. Furthermore, LSCs express PD-L1 to recognize and bind its receptor PD-1 on T cells to impede the killing of T cells. LSCs can also express LILRB4 and CD200 to suppress T cells activity. In addition, LSCs can express CD47 to combine with SIRPα on macrophages to escape from innate immune attacks and can suppress NKG2DL expression through PARP1 to avoid NK cells killing. m6A modification both participates in the regulation of LSCs and the TIME. METTL3, METTL14, FTO, ALKBH5, YTHDF2 and IGF2BP1 can both promote LSCs self-renewal and leukemia cells proliferation, while WTAP can only enhance leukemia cells proliferation. FTO regulates the expression of LILRB4 on LSCs to suppress T cells infiltration and cytokines secretion in an m6A-dependent way.
m6A regulators, such as METTL3, METTL14, WTAP, FTO, ALKBH5, and YTHDF2, are highly expressed in a variety of AML subtypes. The expression of METTL3 mRNAs is apparently increased in M0, while METTL14 is increased in M1 and M3 (92). MTTL14 is found to be much more highly expressed in AML with t (11q23), t (8;21), or t (15;17) (24). WTAP is significantly overexpressed in AML with FLT3-ITD and/or NPM1 mutations but not with t (15;17) and can induce chemoresistance during AML treatment (93, 97). Accompanied by complete remission (CR), WTAP expression is decreased (97). FTO is remarkably overexpressed in AML with t (15;17) and t (11q23), as well as FLT3-ITD and/or NPM1 mutations. Moreover, FTO is responsible for TKI resistance in leukemia cells, suggesting that targeting FTO may reverse TKI resistance (100). In AML carrying t (8,21), inv (16), and t (11q23), as well as normal karyotypes, ALKBH5 expression is relatively high (103). Genetic alterations of encoding genes, which are associated with the m6A writer complex, are negatively associated with prognosis in AML patients (23). WTAP is a poor prognostic risk factor in AML patients, resulting in a shorter overall survival (97). Similarly, ALKBH5 can also predict poor prognosis, and higher ALKBH5 expression is consistent with a high rate of relapse and chemoresistance (103). IGF2BP1 triggers chemoresistance through LSC phenotype-related regulators, including HOXB4, MYB, and ALDH1A1, and is correlated with prognosis (108). Based on these studies, m6A writers, erasers, and readers show promise as diagnostic and prognostic markers in AML patients.
m6A methylation plays vital roles in LSCs and the TIME, which can interplay with each other, leading to relapse and chemoresistance in AML. Many studies have confirmed that m6A writers, erasers, and readers are instrumental for myeloid leukemogenesis and maintaining LSC self-renewal. Meanwhile, FTO, ALKBH5 and YTHDF2 are reported to regulate the antileukemia immune response by altering the TIME (101, 104, 116). Hence, m6A regulators are expected to be potential therapeutic targets in AML. These studies shed new light on the treatment of AML by targeting m6A modification to eradicate LSCs and to increase the efficiency of immunotherapy.
At present, there have been several studies on targeting m6A for AML treatment. Small-molecule inhibitors of METTL3, STM2457 and UZH1a, can prohibit the proliferation of varying AML cell lines. STM2457 can also decrease the number of LSCs marked CD93+, weaken clonogenic capacity and accelerate apoptosis. Furthermore, STM2457 restrains the self-renewal of LSCs to hinder AML cell implantation and propagation in human PDX models (94, 95). This sheds further insight into a potential therapeutic window in which depletion of METTL14 expression can repress the self-renewal of LSCs while exerting a relatively weak effect in normal hematopoietic stem and progenitor cells (HSPCs). Upon treatment with all-trans retinoic acid (ATRA) or PMA, which are differentiation-inducing agents, the levels of METTL14 and m6A were also reduced (24). It is promising that the therapeutic regimen combing a METTL14 inhibitor with ATRA or ATO may have a synergistic effect.
Owing to the inverse effect on the differentiation of FTO, depletion of FTO consequently triggers ATRA-induced cell differentiation, significantly increasing the differentiated population of NB4 cells (22). Based on this conclusion, we can speculate that the therapeutic strategy combining with FTO inhibition with ATRA may improve the treatment effect of acute promyelocyte leukemia (APL). R-2HG plays an important role in anti-leukemia activity by targeting FTO through various mechanisms (98, 99). It provides a potential therapeutic window for combinational application with R-2HG, used as a nonspecific FTO inhibitor, and other agents, such as IDH inhibitor or HMAs or standard chemotherapy. To date, there are various FTO inhibitors with anti-leukemia activity in AML, including the natural compounds radicicol, Saikosaponin D, FB23-2, CS1 and CS2 (101, 102, 119, 120).Among them, FB23-2, CS1, and CS2 can reduce the numbers of LSCs. Furthermore, CS1 and CS2 can also surmount immune evasion induced by HMAs and make AML cells more susceptible to activated T cells (101). This striking finding paves the way to improve the therapeutic effect of AML or myelodysplastic syndromes (MDS) in combination with FTO inhibitors or anti-LILRB4 monoantibody based on HMAs.
ALKBH5 is engaged in the self-renewal and maintenance of LSCs, without influencing normal hematopoietic stem cells (HSCs), implying that ALKBH5 is expected to be a promising therapeutic target for LSC eradication (25, 103). To date, two low micromolar active ALKBH5 inhibitors have exhibited certain effects on the AML cell line HL-60 (121). Similar to ALKBH5, targeting YTHDF2 does not harm normal hematopoiesis, which makes YTHDF2 a feasible target to eliminate LSCs (104).
It’s worth noting that whether there are potential side effects to target m6A modulators in view of their important roles in normal biological process. The security of METTL3 inhibitor STM2457 has been demonstrated in vivo, with no apparent impact on body weight and normal hematopoiesis (95). Su and his colleagues have assessed the potential drug toxicity of FTO inhibitors, CS1 and CS2, in C57BL/6 mice. They found that the differences of complete blood count, Haemotoxylin and Eosin staining, whole body or organ weight between the drug-treated groups and control group are not significant (101).The toxicity of FTO inhibitor FB23-2 has been also evaluated in BALB/c mice. There are no obvious toxic side effects in FB23-2-treated group compared to control group, such as body weight loss, organ lesion and hematopoietic damage (102).The preclinical results showed that the potential side effects of therapy targeted m6A modulators is mild. But the safety of clinical application needs to be confirmed in more clinical trials.
Taking a broad view, targeting m6A opens a new door to treat AML by eliminating LSCs and regulating the immune microenvironment. Additionally, targeting m6A therapy is used in combination with other drugs, such as HMAs, ATRA, ATO or immune checkpoint inhibitors, to improve the therapeutic efficacy in AML patients. The roles of m6A modification in leukemia cells in AML are summarized in Table 1, and the roles of m6A modification in the immune microenvironment of AML are summarized in Table 2.
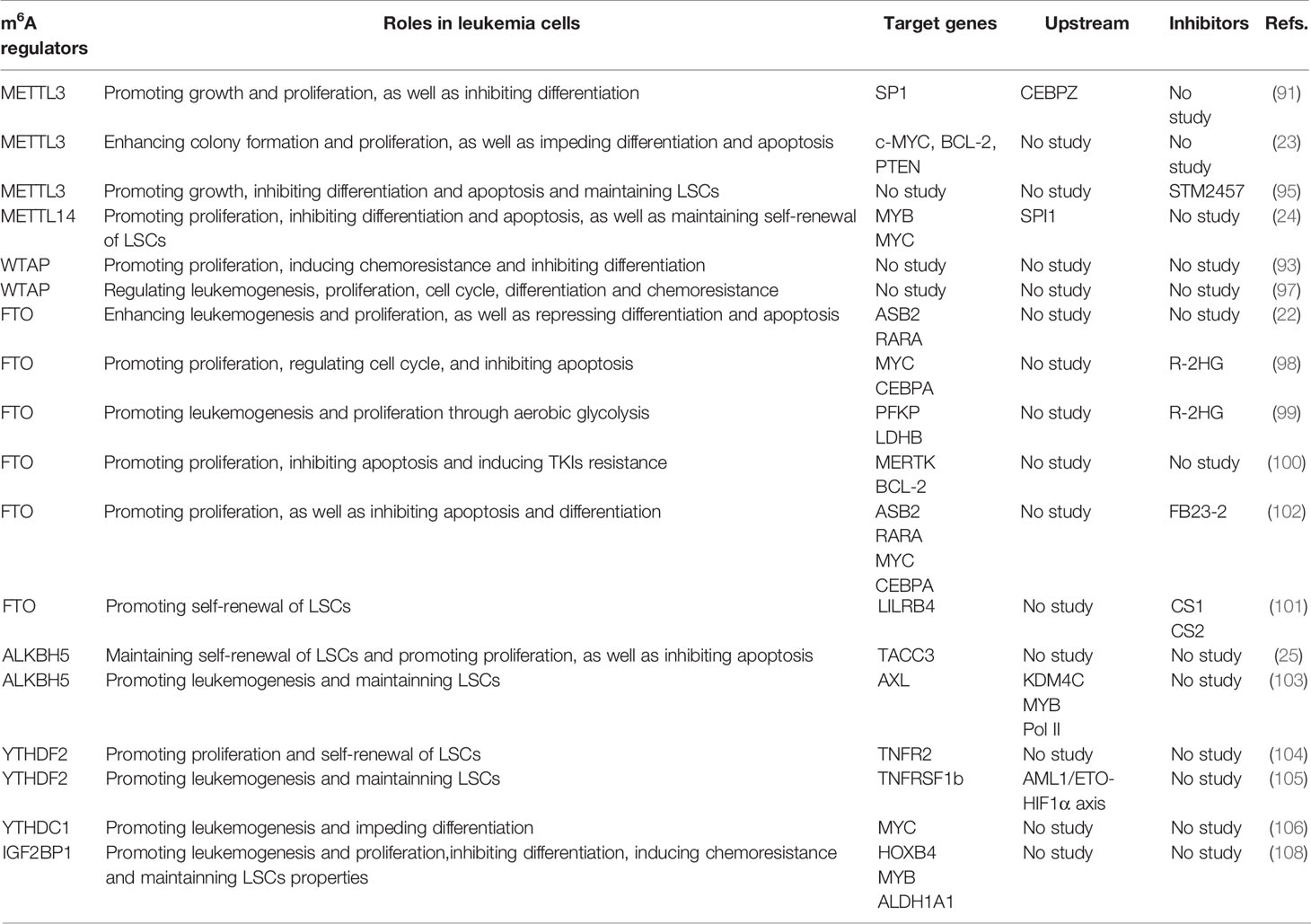
Table 1 The roles of m6A modification in leukemia cells in acute myeloid leukemia.

Table 2 The roles of m6A modification in the immune microenvironment of acute myeloid leukemia.
In summary, LSCs and the TIME cooperatively promote AML initiation and progression, contributing to relapse and chemoresistance in patients with AML through diverse mechanisms. m6A methylation, as a burgeoning RNA epigenetic modification, plays significant roles in leukemogenesis, proliferation, differentiation, apoptosis, and LSC self-renewal by regulating target mRNA stability and protein translation efficiency. To date, m6A writers (METTL3, METTL14, WTAP), erasers (FTO, ALKBH5), and readers (YTHDF2, YTHDC1, IGF2BP1) are considered oncogenic proteins in AML and are responsible for the maintenance of LSCs. METTL14, WTAP, and FTO are highly expressed in specific AML subtypes. Moreover, WTAP, ALKBH5, and IGF2BP1 are correlated with prognosis and chemoresistance. Based on the functions of m6A in AML, there have been some inhibitors targeting m6A, such as the METTL3 inhibitors STM2457 and UZH1a and the FTO inhibitors FB23-2, CS1 and CS2. With no or relatively less impact on HSCs, targeting METTL14, ALKBH5, or YTHDF2 is promising for eradicating LSCs and reducing relapse and chemoresistance in AML patients. In addition, combination inhibitors targeting m6A with other therapeutic agents, such as HMAs, ATRA, ATO, immune checkpoint blockade, and standard chemotherapy, are anticipated to be innovative therapeutic regimens. Therefore, m6A regulators can be regarded as novel biomarkers for AML diagnosis, prognosis, and response evaluation, as well as attractive targets to improve the therapeutic effect in AML patients. However, large-scale clinical studies are needed to demonstrate the efficacy and safety of targeting m6A in AML patients.
In addition, m6A modification also takes part in remodeling the TIME by influencing immune cell infiltration, cytokine secretion, and immune checkpoint expression. Among m6A regulators, FTO is well studied in immune regulation, mediating HMAs resistance and immune evasion in AML. To date, studies on the functions of m6A modification in the AML immune microenvironment, especially in the crosstalk between LSCs and the TIME, are just the tip of the iceberg. Therefore, more related studies are required to explore the roles and mechanisms.
Taken together, m6A modification not only maintains the stemness of leukemia cells but also regulate the immune microenvironment in AML. In view of this feature, targeting m6A is promising to kill two birds with one stone. It also provides a novel opportunity to refine diagnosis, prognosis stratification and therapeutic efficacy evaluation for AML patients. It sheds new light on precision medicine for AML patients.
XO and YG contributed to the design of the review. The manuscript was written by XO. All authors contributed to the article and approved the submitted version.
The work was supported by the Foundation of the Science and Technology Department of Sichuan Province (NO.2019YFS0026).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.

1. Döhner H, Weisdorf DJ, Bloomfield CD. Acute Myeloid Leukemia. N Engl J Med (2015) 373(12):1136–52. doi: 10.1056/NEJMra1406184
2. Mendez LM, Posey RR. Pandolfi PP(2019)The Interplay Between the Genetic and Immune Landscapes of AML: Mechanisms and Implications for Risk Stratification and Therapy. Front Oncol (2019) 9:1162. doi: 10.3389/fonc.2019.01162
3. Döhner H, Wei AH, Löwenberg B. Towards Precision Medicine for AML. Nat Rev Clin Oncol (2021) 18(9):577–90. doi: 10.1038/s41571-021-00509-w
4. Newell LF, Cook RJ. Advances in Acute Myeloid Leukemia. BMJ (2021) 375:n2026. doi: 10.1136/bmj.n2026
5. Döhner H, Estey EH, Amadori S, Appelbaum FR, Büchner T, Burnett AK, et al. Diagnosis and Management of Acute Myeloid Leukemia in Adults: Recommendations From an International Expert Panel, on Behalf of the European LeukemiaNet. Blood (2010) 115(3):453–74. doi: 10.1182/blood-2009-07-235358
6. Dombret H, Gardin C. An Update of Current Treatments for Adult Acute Myeloid Leukemia. Blood (2016) 127(1):53–61. doi: 10.1182/blood-2015-08-604520
8. Thomas D, Majeti R. Biology and Relevance of Human Acute Myeloid Leukemia Stem Cells. Blood (2017) 129(12):1577–85. doi: 10.1182/blood-2016-10-696054
9. Sun H, Li Y, Z-f Z, Ju Y, Li L, Zhang B-c, et al. Increase in Myeloid-Derived Suppressor Cells (MDSCs) Associated With Minimal Residual Disease (MRD) Detection in Adult Acute Myeloid Leukemia. Int J Hematol (2015) 102(5):579–86. doi: 10.1007/s12185-015-1865-2
10. Teague RM, Kline J. Immune Evasion in Acute Myeloid Leukemia: Current Concepts and Future Directions. J Immunother Cancer (2013) 1(13):1. doi: 10.1186/2051-1426-1-13
11. Taghiloo S, Asgarian-Omran H. Immune Evasion Mechanisms in Acute Myeloid Leukemia: A Focus on Immune Checkpoint Pathways. Crit Rev Oncol Hematol (2021) 157:103164. doi: 10.1016/j.critrevonc.2020.103164
12. Schatton T, Frank MH. Antitumor Immunity and Cancer Stem Cells. Ann N Y Acad Sci (2009) 1176:154–69. doi: 10.1111/j.1749-6632.2009.04568.x
13. Ma J, Frank MH. Tumor Initiation in Human Malignant Melanoma and Potential Cancer Therapies. Anticancer Agents Med Chem (2010) 10(2):131–6. doi: 10.2174/187152010790909254
14. Lapidot T, Sirard C, Vormoor J, Murdoch B, Hoang T, Caceres-Cortes J, et al. Dick Je(1994)A Cell Initiating Human Acute Myeloid Leukaemia After Transplantation Into SCID Mice. Nature (6464) 367:645–8. doi: 10.1038/367645a0
15. Tsuchiya H, Shiota G. Immune Evasion by Cancer Stem Cells. Regener Ther (2021) 17:20–33. doi: 10.1016/j.reth.2021.02.006
16. Zhang Z, Zhang C, Luo Y, Zhang G, Wu P, Sun N, et al. RNA N -Methyladenosine Modification in the Lethal Teamwork of Cancer Stem Cells and the Tumor Immune Microenvironment: Current Landscape and Therapeutic Potential. Clin Transl Med (2021) 11(9):e525. doi: 10.1002/ctm2.525
17. Lytle NK, Barber AG, Reya T. Stem Cell Fate in Cancer Growth, Progression and Therapy Resistance. Nat Rev Cancer (2018) 18(11):669–80. doi: 10.1038/s41568-018-0056-x
18. Lathia JD, Mack SC, Mulkearns-Hubert EE, Valentim CLL, Rich JN. Cancer Stem Cells in Glioblastoma. Genes Dev (2015) 29(12):1203–17. doi: 10.1101/gad.261982.115
19. Lathia JD, Heddleston JM, Venere M, Rich JN. Deadly Teamwork: Neural Cancer Stem Cells and the Tumor Microenvironment. Cell Stem Cell (2011) 8(5):482–5. doi: 10.1016/j.stem.2011.04.013
20. Pan Y, Ma P, Liu Y, Li W, Shu Y. Multiple Functions of M6a RNA Methylation in Cancer. J Hematol Oncol (2018) 11(1):48. doi: 10.1186/s13045-018-0590-8
21. Zheng X. Functions of RNA N-Methyladenosine Modification in Acute Myeloid Leukemia. Biomark Res (2021) 9(1):36. doi: 10.1186/s40364-021-00293-w
22. Li Z, Weng H, Su R, Weng X, Zuo Z, Li C, et al. FTO Plays an Oncogenic Role in Acute Myeloid Leukemia as a N-Methyladenosine RNA Demethylase. Cancer Cell (2017) 31(1):127–41. doi: 10.1016/j.ccell.2016.11.017
23. Vu LP, Pickering BF, Cheng Y, Zaccara S, Nguyen D, Minuesa G, et al. The N-Methyladenosine (M6a)-Forming Enzyme METTL3 Controls Myeloid Differentiation of Normal Hematopoietic and Leukemia Cells. Nat Med (2017) 23(11):1369–76. doi: 10.1038/nm.4416
24. Weng H, Huang H, Wu H, Qin X, Zhao BS, Dong L, et al. METTL14 Inhibits Hematopoietic Stem/Progenitor Differentiation and Promotes Leukemogenesis via mRNA M6a Modification. Cell Stem Cell (2018) 22(2):191–205.e9. doi: 10.1016/j.stem.2017.11.016
25. Shen C, Sheng Y, Zhu AC, Robinson S, Jiang X, Dong L, et al. RNA Demethylase ALKBH5 Selectively Promotes Tumorigenesis and Cancer Stem Cell Self-Renewal in Acute Myeloid Leukemia. Cell Stem Cell (2020) 27(1):64–80.e9. doi: 10.1016/j.stem.2020.04.009
26. Zhang C, Samanta D, Lu H, Bullen JW, Zhang H, Chen I, et al. Hypoxia Induces the Breast Cancer Stem Cell Phenotype by HIF-Dependent and ALKBH5-Mediated M6A-Demethylation of NANOG mRNA. Proc Natl Acad Sci USA (2016) 113(14):E2047–56. doi: 10.1073/pnas.1602883113
27. Zhang S, Zhao BS, Zhou A, Lin K, Zheng S, Lu Z, et al. M6a Demethylase ALKBH5 Maintains Tumorigenicity of Glioblastoma Stem-Like Cells by Sustaining FOXM1 Expression and Cell Proliferation Program. Cancer Cell (2017) 31(4):591–606.e6. doi: 10.1016/j.ccell.2017.02.013
28. Cui Q, Shi H, Ye P, Li L, Qu Q, Sun G, et al. M6a RNA Methylation Regulates the Self-Renewal and Tumorigenesis of Glioblastoma Stem Cells. Cell Rep (2017) 18(11):2622–34. doi: 10.1016/j.celrep.2017.02.059
29. Qin Q, Fang DL, Zhou W, Meng Y, Wei J. Classification and Immune Invasion Analysis of Breast Cancer Based on M6a Genes. Ann Transl Med (2021) 9(18):1418. doi: 10.21037/atm-21-3404
30. Lin Y, Wang S, Liu S, Lv S, Wang H, Li F. Identification and Verification of Molecular Subtypes With Enhanced Immune Infiltration Based on M6a Regulators in Cutaneous Melanoma. BioMed Res Int (2021) 2021:2769689. doi: 10.1155/2021/2769689
31. Chong W, Shang L, Liu J, Fang Z, Du F, Wu H, et al. M6a Regulator-Based Methylation Modification Patterns Characterized by Distinct Tumor Microenvironment Immune Profiles in Colon Cancer. Theranostics (2021) 11(5):2201–17. doi: 10.7150/thno.52717
32. Kumar S, Nagpal R, Kumar A, Ashraf MU, Bae Y-S. Immunotherapeutic Potential of M6a-Modifiers and MicroRNAs in Controlling Acute Myeloid Leukaemia. Biomedicines (2021) 9(6):690. doi: 10.3390/biomedicines9060690
33. Shlush LI, Mitchell A, Heisler L, Abelson S, Ng SWK, Trotman-Grant A, et al. Tracing the Origins of Relapse in Acute Myeloid Leukaemia to Stem Cells. Nature (2017) 547(7661):104–8. doi: 10.1038/nature22993
34. Ferrara F, Schiffer CA. Acute Myeloid Leukaemia in Adults. Lancet (2013) 381(9865):484–95. doi: 10.1016/S0140-6736(12)61727-9
35. Bahr C, Correia NC, Trumpp A. Stem Cells Make Leukemia Grow Again. EMBO J (2017) 36(18):2667–9. doi: 10.15252/embj.201797773
36. Kreso A, Dick JE. Evolution of the Cancer Stem Cell Model. Cell Stem Cell (2014) 14(3):275–91. doi: 10.1016/j.stem.2014.02.006
37. Moshaver B, Wouters RF, Kelder A, Ossenkoppele GJ, Westra GAH, Kwidama Z, et al. Relationship Between CD34/CD38 and Side Population (SP) Defined Leukemia Stem Cell Compartments in Acute Myeloid Leukemia. Leuk Res (2019) 81:27–34. doi: 10.1016/j.leukres.2019.04.004
38. Craddock C, Quek L, Goardon N, Freeman S, Siddique S, Raghavan M, et al. Azacitidine Fails to Eradicate Leukemic Stem/Progenitor Cell Populations in Patients With Acute Myeloid Leukemia and Myelodysplasia. Leukemia (2013) 27(5):1028–36. doi: 10.1038/leu.2012.312
39. Kikushige Y, Shima T, Takayanagi S-i, Urata S, Miyamoto T, Iwasaki H, et al. TIM-3 Is a Promising Target to Selectively Kill Acute Myeloid Leukemia Stem Cells. Cell Stem Cell (2010) 7(6):708–17. doi: 10.1016/j.stem.2010.11.014
40. Kikushige Y, Miyamoto T, Yuda J, Jabbarzadeh-Tabrizi S, Shima T, S-i T, et al. A TIM-3/Gal-9 Autocrine Stimulatory Loop Drives Self-Renewal of Human Myeloid Leukemia Stem Cells and Leukemic Progression. Cell Stem Cell (2015) 17(3):341–52. doi: 10.1016/j.stem.2015.07.011
41. van Rhenen A, van Dongen GAMS, Kelder A, Rombouts EJ, Feller N, Moshaver B, et al. The Novel AML Stem Cell Associated Antigen CLL-1 Aids in Discrimination Between Normal and Leukemic Stem Cells. Blood (2007) 110(7):2659–66. doi: 10.1182/blood-2007-03-083048
42. Majeti R, Chao MP, Alizadeh AA, Pang WW, Jaiswal S, Gibbs KD, et al. CD47 Is an Adverse Prognostic Factor and Therapeutic Antibody Target on Human Acute Myeloid Leukemia Stem Cells. Cell (2009) 138(2):286–99. doi: 10.1016/j.cell.2009.05.045
43. Riether C, Schürch CM, Bührer ED, Hinterbrandner M, Huguenin A-L, Hoepner S, et al. CD70/CD27 Signaling Promotes Blast Stemness and Is a Viable Therapeutic Target in Acute Myeloid Leukemia. J Exp Med (2017) 214(2):359–80. doi: 10.1084/jem.20152008
44. Hosen N, Park CY, Tatsumi N, Oji Y, Sugiyama H, Gramatzki M, et al. CD96 Is a Leukemic Stem Cell-Specific Marker in Human Acute Myeloid Leukemia. Proc Natl Acad Sci USA (2007) 104(26):11008–13. doi: 10.1073/pnas.0704271104
45. Jordan CT, Upchurch D, Szilvassy SJ, Guzman ML, Howard DS, Pettigrew AL, et al. The Interleukin-3 Receptor Alpha Chain Is a Unique Marker for Human Acute Myelogenous Leukemia Stem Cells. Leukemia (2000) 14(10):1777–84. doi: 10.1038/sj.leu.2401903
46. Zhang F, Liu X, Chen C, Zhu J, Yu Z, Xie J, et al. CD244 Maintains the Proliferation Ability of Leukemia Initiating Cells Through SHP-2/P27 Signaling. Haematologica (2017) 102(4):707–18. doi: 10.3324/haematol.2016.151555
47. Herbrich S, Baran N, Cai T, Weng C, Aitken MJL, Post SM, et al. Overexpression of CD200 Is a Stem Cell-Specific Mechanism of Immune Evasion in AML. J Immunother Cancer (2021) 9(7):e002968. doi: 10.1136/jitc-2021-002968
48. Iwasaki M, Liedtke M, Gentles AJ, Cleary ML. CD93 Marks a Non-Quiescent Human Leukemia Stem Cell Population and Is Required for Development of MLL-Rearranged Acute Myeloid Leukemia. Cell Stem Cell (2015) 17(4):412–21. doi: 10.1016/j.stem.2015.08.008
49. Gonçalves Silva I, Yasinska IM, Sakhnevych SS, Fiedler W, Wellbrock J, Bardelli M, et al. The Tim-3-Galectin-9 Secretory Pathway Is Involved in the Immune Escape of Human Acute Myeloid Leukemia Cells. EBioMedicine (2017) 22:44–57. doi: 10.1016/j.ebiom.2017.07.018
50. Perry JM, Tao F, Roy A, Lin T, He XC, Chen S, et al. Overcoming Wnt-β-Catenin Dependent Anticancer Therapy Resistance in Leukaemia Stem Cells. Nat Cell Biol (2020) 22(6):689–700. doi: 10.1038/s41556-020-0507-y
51. Prokhorov A, Gibbs BF, Bardelli M, Rüegg L, Fasler-Kan E, Varani L, et al. The Immune Receptor Tim-3 Mediates Activation of PI3 Kinase/mTOR and HIF-1 Pathways in Human Myeloid Leukaemia Cells. Int J Biochem Cell Biol (2015) 59:11–20. doi: 10.1016/j.biocel.2014.11.017
52. Barsoum IB, Smallwood CA, Siemens DR, Graham CH. A Mechanism of Hypoxia-Mediated Escape From Adaptive Immunity in Cancer Cells. Cancer Res (2014) 74(3):665–74. doi: 10.1158/0008-5472.CAN-13-0992
53. Paczulla AM, Rothfelder K, Raffel S, Konantz M, Steinbacher J, Wang H, et al. Absence of NKG2D Ligands Defines Leukaemia Stem Cells and Mediates Their Immune Evasion. Nature (2019) 572(7768):254–9. doi: 10.1038/s41586-019-1410-1
54. Depreter B, De Moerloose B, Vandepoele K, Uyttebroeck A, Van Damme A, Terras E, et al. Deciphering Molecular Heterogeneity in Pediatric AML Using a Cancer vs. Normal Transcriptomic Approach. Pediatr Res (2021) 89(7):1695–705. doi: 10.1038/s41390-020-01199-3
55. Noviello M, Manfredi F, Ruggiero E, Perini T, Oliveira G, Cortesi F, et al. Bone Marrow Central Memory and Memory Stem T-Cell Exhaustion in AML Patients Relapsing After HSCT. Nat Commun (2019) 10(1):1065. doi: 10.1038/s41467-019-08871-1
56. Xu L, Liu L, Yao D, Zeng X, Zhang Y, Lai J, et al. PD-1 and TIGIT Are Highly Co-Expressed on CD8 T Cells in AML Patient Bone Marrow. Front Oncol (2021) 11:686156. doi: 10.3389/fonc.2021.686156
57. Camacho V, McClearn V, Patel S, Welner RS. Regulation of Normal and Leukemic Stem Cells Through Cytokine Signaling and the Microenvironment. Int J Hematol (2017) 105(5):566–77. doi: 10.1007/s12185-017-2184-6
58. Desrosiers R, Friderici K, Rottman F. Identification of Methylated Nucleosides in Messenger RNA From Novikoff Hepatoma Cells. Proc Natl Acad Sci USA (1974) 71(10):3971–5. doi: 10.1073/pnas.71.10.3971
59. Wang X, Feng J, Xue Y, Guan Z, Zhang D, Liu Z, et al. Structural Basis of N(6)-Adenosine Methylation by the METTL3-METTL14 Complex. Nature (2016) 534(7608):575–8. doi: 10.1038/nature18298
60. Ping X-L, Sun B-F, Wang L, Xiao W, Yang X, Wang W-J, et al. Mammalian WTAP Is a Regulatory Subunit of the RNA N6-Methyladenosine Methyltransferase. Cell Res (2014) 24(2):177–89. doi: 10.1038/cr.2014.3
61. Schöller E, Weichmann F, Treiber T, Ringle S, Treiber N, Flatley A, et al. Interactions, Localization, and Phosphorylation of the M6a Generating METTL3-METTL14-WTAP Complex. RNA (2018) 24(4):499–512. doi: 10.1261/rna.064063.117
62. Wen J, Lv R, Ma H, Shen H, He C, Wang J, et al. Zc3h13 Regulates Nuclear RNA M6a Methylation and Mouse Embryonic Stem Cell Self-Renewal. Mol Cell (2018) 69(6):1028–1038.e6. doi: 10.1016/j.molcel.2018.02.015
63. Patil DP, Chen C-K, Pickering BF, Chow A, Jackson C, Guttman M, et al. M(6)A RNA Methylation Promotes XIST-Mediated Transcriptional Repression. Nature (2016) 537(7620):369–73. doi: 10.1038/nature19342
64. Yue Y, Liu J, Cui X, Cao J, Luo G, Zhang Z, et al. VIRMA Mediates Preferential M6a mRNA Methylation in 3'UTR and Near Stop Codon and Associates With Alternative Polyadenylation. Cell Discov (2018) 4:10. doi: 10.1038/s41421-018-0019-0
65. van Tran N, Ernst FGM, Hawley BR, Zorbas C, Ulryck N, Hackert P, et al. The Human 18S rRNA M6a Methyltransferase METTL5 Is Stabilized by TRMT112. Nucleic Acids Res (2019) 47(15):7719–33. doi: 10.1093/nar/gkz619
66. Pendleton KE, Chen B, Liu K, Hunter OV, Xie Y, Tu BP, et al. The U6 snRNA M6a Methyltransferase METTL16 Regulates SAM Synthetase Intron Retention. Cell (2017) 169(5):824–35.e14. doi: 10.1016/j.cell.2017.05.003
67. Yang L, Ma Y, Han W, Li W, Cui L, Zhao X, et al. Proteinase-Activated Receptor 2 Promotes Cancer Cell Migration Through RNA Methylation-Mediated Repression of miR-125b. J Biol Chem (2015) 290(44):26627–37. doi: 10.1074/jbc.M115.667717
68. Sendinc E, Valle-Garcia D, Dhall A, Chen H, Henriques T, Navarrete-Perea J, et al. PCIF1 Catalyzes M6am mRNA Methylation to Regulate Gene Expression. Mol Cell (2019) 75(3):620–30.e9. doi: 10.1016/j.molcel.2019.05.030
69. Bawankar P, Lence T, Paolantoni C, Haussmann IU, Kazlauskiene M, Jacob D, et al. Hakai Is Required for Stabilization of Core Components of the M6a mRNA Methylation Machinery. Nat Commun (2021) 12(1):3778. doi: 10.1038/s41467-021-23892-5
70. Jia G, Fu Y, Zhao X, Dai Q, Zheng G, Yang Y, et al. N6-Methyladenosine in Nuclear RNA Is a Major Substrate of the Obesity-Associated FTO. Nat Chem Biol (2011) 7(12):885–7. doi: 10.1038/nchembio.687
71. Wei J, Liu F, Lu Z, Fei Q, Ai Y, He PC, et al. Differential M6a, m6Am, and M1a Demethylation Mediated by FTO in the Cell Nucleus and Cytoplasm. Mol Cell (2018) 71(6):973–85.e5. doi: 10.1016/j.molcel.2018.08.011
72. Zhao X, Yang Y, Sun B-F, Shi Y, Yang X, Xiao W, et al. FTO-Dependent Demethylation of N6-Methyladenosine Regulates mRNA Splicing and Is Required for Adipogenesis. Cell Res (2014) 24(12):1403–19. doi: 10.1038/cr.2014.151
73. Zheng G, Dahl JA, Niu Y, Fedorcsak P, Huang C-M, Li CJ, et al. ALKBH5 Is a Mammalian RNA Demethylase That Impacts RNA Metabolism and Mouse Fertility. Mol Cell (2013) 49(1):18–29. doi: 10.1016/j.molcel.2012.10.015
74. Ueda Y, Ooshio I, Fusamae Y, Kitae K, Kawaguchi M, Jingushi K, et al. AlkB Homolog 3-Mediated tRNA Demethylation Promotes Protein Synthesis in Cancer Cells. Sci Rep (2017) 7:42271. doi: 10.1038/srep42271
75. Meyer KD, Jaffrey SR. Rethinking M(6)A Readers, Writers, and Erasers. In: Schekman R, editor. Annual Review of Cell and Developmental Biology, Vol 332017. California, United States: Annual Reviews (2017) p. 319–42.
76. Liao S, Sun H, Xu C. YTH Domain: A Family of N-6-Methyladenosine (M(6)A) Readers. Genomics Proteomics Bioinf (2018) 16(2):99–107. doi: 10.1016/j.gpb.2018.04.002
77. Xiang Y, Laurent B, Hsu C-H, Nachtergaele S, Lu Z, Sheng W, et al. RNA M6a Methylation Regulates the Ultraviolet-Induced DNA Damage Response. Nature (2017) 543(7646):573–6. doi: 10.1038/nature21671
78. Jain D, Puno MR, Meydan C, Lailler N, Mason CE, Lima CD, et al. Mutant Mice Uncover an Essential Meiotic Function for the Ancient RNA Helicase YTHDC2. eLife (2018) 7:e30919. doi: 10.7554/eLife.30919
79. Wang X, Zhao BS, Roundtree IA, Lu Z, Han D, Ma H, et al. N(6)-Methyladenosine Modulates Messenger RNA Translation Efficiency. Cell (2015) 161(6):1388–99. doi: 10.1016/j.cell.2015.05.014
80. Wang X, Lu Z, Gomez A, Hon GC, Yue Y, Han D, et al. N6-Methyladenosine-Dependent Regulation of Messenger RNA Stability. Nature (2014) 505(7481):117–20. doi: 10.1038/nature12730
81. Shi H, Wang X, Lu Z, Zhao BS, Ma H, Hsu PJ, et al. YTHDF3 Facilitates Translation and Decay of N-Methyladenosine-Modified RNA. Cell Res (2017) 27(3):315–28. doi: 10.1038/cr.2017.15
82. Xu C, Wang X, Liu K, Roundtree IA, Tempel W, Li Y, et al. Structural Basis for Selective Binding of M6a RNA by the YTHDC1 YTH Domain. Nat Chem Biol (2014) 10(11):927–9. doi: 10.1038/nchembio.1654
83. Xiao W, Adhikari S, Dahal U, Chen Y-S, Hao Y-J, Sun B-F, et al. Nuclear M(6)A Reader YTHDC1 Regulates mRNA Splicing. Mol Cell (2016) 61(4):507–19. doi: 10.1016/j.molcel.2016.01.012
84. Roundtree IA, Luo G-Z, Zhang Z, Wang X, Zhou T, Cui Y, et al. YTHDC1 Mediates Nuclear Export of N-Methyladenosine Methylated mRNAs. eLife (2017) 6:e31311. doi: 10.7554/eLife.31311
85. Mao Y, Dong L, Liu X-M, Guo J, Ma H, Shen B, et al. M6a in mRNA Coding Regions Promotes Translation via the RNA Helicase-Containing YTHDC2. Nat Commun (2019) 10(1):5332. doi: 10.1038/s41467-019-13317-9
86. Kretschmer J, Rao H, Hackert P, Sloan KE, Höbartner C, Bohnsack MT. The M6a Reader Protein YTHDC2 Interacts With the Small Ribosomal Subunit and the 5'-3' Exoribonuclease XRN1. RNA (2018) 24(10):1339–50. doi: 10.1261/rna.064238.117
87. Huang H, Weng H, Sun W, Qin X, Shi H, Wu H, et al. Recognition of RNA N-Methyladenosine by IGF2BP Proteins Enhances mRNA Stability and Translation. Nat Cell Biol (2018) 20(3):285–95. doi: 10.1038/s41556-018-0045-z
88. Alarcón CR, Goodarzi H, Lee H, Liu X, Tavazoie S, Tavazoie SF. HNRNPA2B1 Is a Mediator of M(6)A-Dependent Nuclear RNA Processing Events. Cell (2015) 162(6):1299–308. doi: 10.1016/j.cell.2015.08.011
89. Liu N, Dai Q, Zheng G, He C, Parisien M, Pan T. N(6)-Methyladenosine-Dependent RNA Structural Switches Regulate RNA-Protein Interactions. Nature (2015) 518(7540):560–4. doi: 10.1038/nature14234
90. Liu N, Zhou KI, Parisien M, Dai Q, Diatchenko L, Pan T. N6-Methyladenosine Alters RNA Structure to Regulate Binding of a Low-Complexity Protein. Nucleic Acids Res (2017) 45(10):6051–63. doi: 10.1093/nar/gkx141
91. Barbieri I, Tzelepis K, Pandolfini L, Shi J, Millán-Zambrano G, Robson SC, et al. Promoter-Bound METTL3 Maintains Myeloid Leukaemia by M6a-Dependent Translation Control. Nature (2017) 552(7683):126–31. doi: 10.1038/nature24678
92. Sorci M, Ianniello Z, Cruciani S, Larivera S, Ginistrelli LC, Capuano E, et al. METTL3 Regulates WTAP Protein Homeostasis. Cell Death Dis (2018) 9(8):796. doi: 10.1038/s41419-018-0843-z
93. Bansal H, Yihua Q, Iyer SP, Ganapathy S, Proia DA, Proia D, et al. WTAP Is a Novel Oncogenic Protein in Acute Myeloid Leukemia. Leukemia (2014) 28(5):1171–4. doi: 10.1038/leu.2014.16
94. Li J, Gregory RI. Mining for METTL3 Inhibitors to Suppress Cancer. Nat Struct Mol Biol (2021) 28(6):460–2. doi: 10.1038/s41594-021-00606-5
95. Yankova E, Blackaby W, Albertella M, Rak J, De Braekeleer E, Tsagkogeorga G, et al. Small-Molecule Inhibition of METTL3 as a Strategy Against Myeloid Leukaemia. Nature (2021) 593(7860):597–601. doi: 10.1038/s41586-021-03536-w
96. Chen Z-H, Chen T-Q, Zeng Z-C, Wang D, Han C, Sun Y-M, et al. Nuclear Export of Chimeric mRNAs Depends on an lncRNA-Triggered Autoregulatory Loop in Blood Malignancies. Cell Death Dis (2020) 11(7):566. doi: 10.1038/s41419-020-02795-1
97. Naren D, Yan T, Gong Y, Huang J, Zhang D, Sang L, et al. High Wilms' Tumor 1 Associating Protein Expression Predicts Poor Prognosis in Acute Myeloid Leukemia and Regulates M6a Methylation of MYC mRNA. J Cancer Res Clin Oncol (2021) 147(1):33–47. doi: 10.1007/s00432-020-03373-w
98. Su R, Dong L, Li C, Nachtergaele S, Wunderlich M, Qing Y, et al. R-2hg Exhibits Anti-Tumor Activity by Targeting FTO/m6A/MYC/CEBPA Signaling. Cell (2018) 172(1-2):90–105.e23. doi: 10.1016/j.cell.2017.11.031
99. Qing Y, Dong L, Gao L, Li C, Li Y, Han L, et al. R-2-Hydroxyglutarate Attenuates Aerobic Glycolysis in Leukemia by Targeting the FTO/m6A/PFKP/LDHB Axis. Mol Cell (2021) 81(5):922–39.e9. doi: 10.1016/j.molcel.2020.12.026
100. Yan F, Al-Kali A, Zhang Z, Liu J, Pang J, Zhao N, et al. A Dynamic N-Methyladenosine Methylome Regulates Intrinsic and Acquired Resistance to Tyrosine Kinase Inhibitors. Cell Res (2018) 28(11):1062–76. doi: 10.1038/s41422-018-0097-4
101. Su R, Dong L, Li Y, Gao M, Han L, Wunderlich M, et al. Targeting FTO Suppresses Cancer Stem Cell Maintenance and Immune Evasion. Cancer Cell (2020) 38(1):79–96.e11. doi: 10.1016/j.ccell.2020.04.017
102. Huang Y, Su R, Sheng Y, Dong L, Dong Z, Xu H, et al. Small-Molecule Targeting of Oncogenic FTO Demethylase in Acute Myeloid Leukemia. Cancer Cell (2019) 35(4):677–91.e10. doi: 10.1016/j.ccell.2019.03.006
103. Wang J, Li Y, Wang P, Han G, Zhang T, Chang J, et al. Leukemogenic Chromatin Alterations Promote AML Leukemia Stem Cells via a KDM4C-ALKBH5-AXL Signaling Axis. Cell Stem Cell (2020) 27(1):81–97.e8. doi: 10.1016/j.stem.2020.04.001
104. Paris J, Morgan M, Campos J, Spencer GJ, Shmakova A, Ivanova I, et al. Targeting the RNA M6a Reader YTHDF2 Selectively Compromises Cancer Stem Cells in Acute Myeloid Leukemia. Cell Stem Cell (2019) 25(1):137–148.e6. doi: 10.1016/j.stem.2019.03.021
105. Chen Z, Shao Y-L, Wang L-L, Lin J, Zhang J-B, Ding Y, et al. YTHDF2 Is a Potential Target of AML1/ETO-Hif1α Loop-Mediated Cell Proliferation in T(8;21) AML. Oncogene (2021) 40(22):3786–98. doi: 10.1038/s41388-021-01818-1
106. Cheng Y, Xie W, Pickering BF, Chu KL, Savino AM, Yang X, et al. N-Methyladenosine on mRNA Facilitates a Phase-Separated Nuclear Body That Suppresses Myeloid Leukemic Differentiation. Cancer Cell (2021) 39(7):958–972.e8. doi: 10.1016/j.ccell.2021.04.017
107. Sheng Y, Wei J, Yu F, Xu H, Yu C, Wu Q, et al. A Critical Role of Nuclear M6a Reader YTHDC1 in Leukemogenesis by Regulating MCM Complex-Mediated DNA Replication. Blood (2021) 138(26):2838–52. doi: 10.1182/blood.2021011707
108. Elcheva IA, Wood T, Chiarolanzio K, Chim B, Wong M, Singh V, et al. RNA-Binding Protein IGF2BP1 Maintains Leukemia Stem Cell Properties by Regulating HOXB4, MYB, and ALDH1A1. Leukemia (2020) 34(5):1354–63. doi: 10.1038/s41375-019-0656-9
109. Feng M, Xie X, Han G, Zhang T, Li Y, Li Y, et al. YBX1 Is Required for Maintaining Myeloid Leukemia Cell Survival by Regulating BCL2 Stability in an M6a-Dependent Manner. Blood (2021) 138(1):71–85. doi: 10.1182/blood.2020009676
110. Wang L, Hui H, Agrawal K, Kang Y, Li N, Tang R, et al. M A RNA Methyltransferases METTL3/14 Regulate Immune Responses to Anti-PD-1 Therapy. EMBO J (2020) 39(20):e104514. doi: 10.15252/embj.2020104514
111. Tsuruta N, Tsuchihashi K, Ohmura H, Yamaguchi K, Ito M, Ariyama H, et al. RNA N6-Methyladenosine Demethylase FTO Regulates PD-L1 Expression in Colon Cancer Cells. Biochem Biophys Res Commun (2020) 530(1):235–9. doi: 10.1016/j.bbrc.2020.06.153
112. Liu Y, Liang G, Xu H, Dong W, Dong Z, Qiu Z, et al. Tumors Exploit FTO-Mediated Regulation of Glycolytic Metabolism to Evade Immune Surveillance. Cell Metab (2021) 33(6):1221–1233.e11. doi: 10.1016/j.cmet.2021.04.001
113. Li N, Kang Y, Wang L, Huff S, Tang R, Hui H, et al. ALKBH5 Regulates Anti-PD-1 Therapy Response by Modulating Lactate and Suppressive Immune Cell Accumulation in Tumor Microenvironment. Proc Natl Acad Sci USA (2020) 117(33):20159–70. doi: 10.1073/pnas.1918986117
114. Dong F, Qin X, Wang B, Li Q, Hu J, Cheng X, et al. ALKBH5 Facilitates Hypoxia-Induced Paraspeckle Assembly and IL8 Secretion to Generate an Immunosuppressive Tumor Microenvironment. Cancer Res (2021) 81(23):5876–88. doi: 10.1158/0008-5472.CAN-21-1456
115. Deng M, Gui X, Kim J, Xie L, Chen W, Li Z, et al. LILRB4 Signalling in Leukaemia Cells Mediates T Cell Suppression and Tumour Infiltration. Nature (2018) 562(7728):605–9. doi: 10.1038/s41586-018-0615-z
116. Du Y, Han M, Cao K, Li Q, Pang J, Dou L, et al. Gold Nanorods Exhibit Intrinsic Therapeutic Activity via Controlling 6-Methyladenosine-Based Epitranscriptomics in Acute Myeloid Leukemia. ACS Nano (2021) 15(11):17689–704. doi: 10.1021/acsnano.1c05547
117. Du A, Wu X, Gao Y, Jiang B, Wang J, Zhang P, et al. M6a Regulator-Mediated Methylation Modification Patterns and Tumor Microenvironment Infiltration Characterization in Acute Myeloid Leukemia. Front Immunol (2021) 12:789914. doi: 10.3389/fimmu.2021.789914
118. Li D, Liang J, Cheng C, Guo W, Li S, Song W, et al. Identification of M6a-Related lncRNAs Associated With Prognoses and Immune Responses in Acute Myeloid Leukemia. Front Cell Dev Biol (2021) 9:770451. doi: 10.3389/fcell.2021.770451
119. Wang R, Han Z, Liu B, Zhou B, Wang N, Jiang Q, et al. Identification of Natural Compound Radicicol as a Potent FTO Inhibitor. Mol Pharm (2018) 15(9):4092–8. doi: 10.1021/acs.molpharmaceut.8b00522
120. Sun K, Du Y, Hou Y, Zhao M, Li J, Du Y, et al. Saikosaponin D Exhibits Anti-Leukemic Activity by Targeting FTO/m6A Signaling. Theranostics (2021) 11(12):5831–46. doi: 10.7150/thno.55574
Keywords: N6-methyladenosine, RNA methylation, acute myeloid leukemia, leukemia stem cells, tumor immune microenvironment
Citation: Ouyang X and Gong Y (2022) One Stone, Two Birds: N6-Methyladenosine RNA Modification in Leukemia Stem Cells and the Tumor Immune Microenvironment in Acute Myeloid Leukemia. Front. Immunol. 13:912526. doi: 10.3389/fimmu.2022.912526
Received: 04 April 2022; Accepted: 09 May 2022;
Published: 02 June 2022.
Edited by:
Guohui Wan, Sun Yat-sen University, ChinaReviewed by:
Takeshi Isoda, Tokyo Medical and Dental University, JapanCopyright © 2022 Ouyang and Gong. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yuping Gong, Z29uZ3l1cGluZzIwMTBAYWxpeXVuLmNvbQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.