 Roberta Rovito
Roberta Rovito Matteo Augello
Matteo Augello Assaf Ben-Haim
Assaf Ben-Haim Valeria Bono
Valeria Bono Antonella d’Arminio Monforte
Antonella d’Arminio Monforte Giulia Marchetti
Giulia Marchetti- Clinic of Infectious Diseases and Tropical Medicine, Azienda Socio Sanitaria Territoriale (ASST) Santi Paolo e Carlo, Department of Health Sciences, University of Milan, Milan, Italy
Two years into Coronavirus Disease 2019 (COVID-19) pandemic, a comprehensive characterization of the pathogenesis of severe and critical forms of COVID-19 is still missing. While a deep dysregulation of both the magnitude and functionality of innate and adaptive immune responses have been described in severe COVID-19, the mechanisms underlying such dysregulations are still a matter of scientific debate, in turn hampering the identification of new therapies and of subgroups of patients that would most benefit from individual clinical interventions. Here we review the current understanding of viral and host factors that contribute to immune dysregulation associated with COVID-19 severity in the attempt to unfold and broaden the comprehension of COVID-19 pathogenesis and to define correlates of protection to further inform strategies of targeted therapeutic interventions.
1 Introduction
Since the end of 2019, the Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2, previously referred to as 2019-nCoV) was identified as the etiological agent of Coronavirus Disease 2019 (COVID-19) (1). SARS-CoV-2 is an enveloped virus, positive-sense single-stranded RNA, of the Coronaviridae family (2), the third of zoonotic transmission causing a large outbreak in humans. Indeed, SARS-CoV emerged in South China in 2002 (3), the Middle Eastern respiratory syndrome coronavirus (MERS-CoV) in Saudi Arabia in 2012 (4), and SARS-CoV-2 in the Chinese province of Hubei in 2019. COVID-19 is characterized by a broad spectrum of clinical manifestations, ranging from flu-like symptoms to systemic inflammation and multi-organ failure (5–10). Severe COVID-19 occurs in 14% of infected individuals, whereas critical COVID-19 in 5% (11).
SARS-CoV-2 is primarily transmitted by a direct, person-to-person transmission via the respiratory route, whereby the virus is suspended either on droplets or, less commonly, on aerosols (12). SARS-CoV-2 primarily infects epithelial cells in the airways via angiotensin-converting enzyme 2 (ACE2), its primary target receptor. The binding is followed by a proteolytic activation at the plasma membrane by the transmembrane protease serine 2 (TMPRSS2) or at the endosomal membrane by cathepsin L (13). The infection of the lower respiratory tract is followed by apoptosis, as a consequence of the viral replicative cycle, and by recognition of the viral infection by tissue-resident immune cells, as well as a first wave of inflammation due to macrophage and neutrophils activation that attempt to control the infection. This is followed by the recruitment of other immune cells from the bloodstream, further exacerbating inflammation, and eventually leading to the cytokine storm (14, 15). This represents the basis of pulmonary pathology, characterized by alveolar damage, oedema, inflammatory infiltrates, and fibrin deposition, resulting in the development of bilateral interstitial pneumonia (16–18). Worsening of this clinical picture involves manifestation of acute respiratory distress syndrome (ARDS), vascular damage, thrombosis, disseminated intravascular coagulation (CID), and multi-organ damage (7, 19).
ACE2 is also widely expressed in many other tissues, including the oral and nasal mucosa, nasopharynx, stomach, small intestine, colon, skin, lymph nodes, thymus, bone marrow, spleen, liver, kidney, testis, and brain (20), as well as in the endothelial and smooth muscle cells of these organs. In agreement with these findings, several studies detected the presence of SARS-CoV-2 in multiple organs of COVID-19 patients, including the brain, intestine, pharynx, heart, liver, kidneys, testicles, and blood, demonstrating its multi-organ tropism (21–23). How the virus disseminates onto these organs is still not fully understood.
Since the beginning of the pandemic, many definitions of COVID-19 severity have been employed, each focusing on different clinical aspects of the disease or referring to diverse laboratory and instrumental cut-offs, therefore hindering the reliable comparison of different studies and, in turn, the recognition of specific factors associated with a poor prognosis (24–32). To date, the most used classification system is the one developed by the World Health Organization (WHO), which distinguishes between four degrees of disease severity, based on clinical indicators: mild, moderate, severe, and critical (33), as summarized in Table 1.
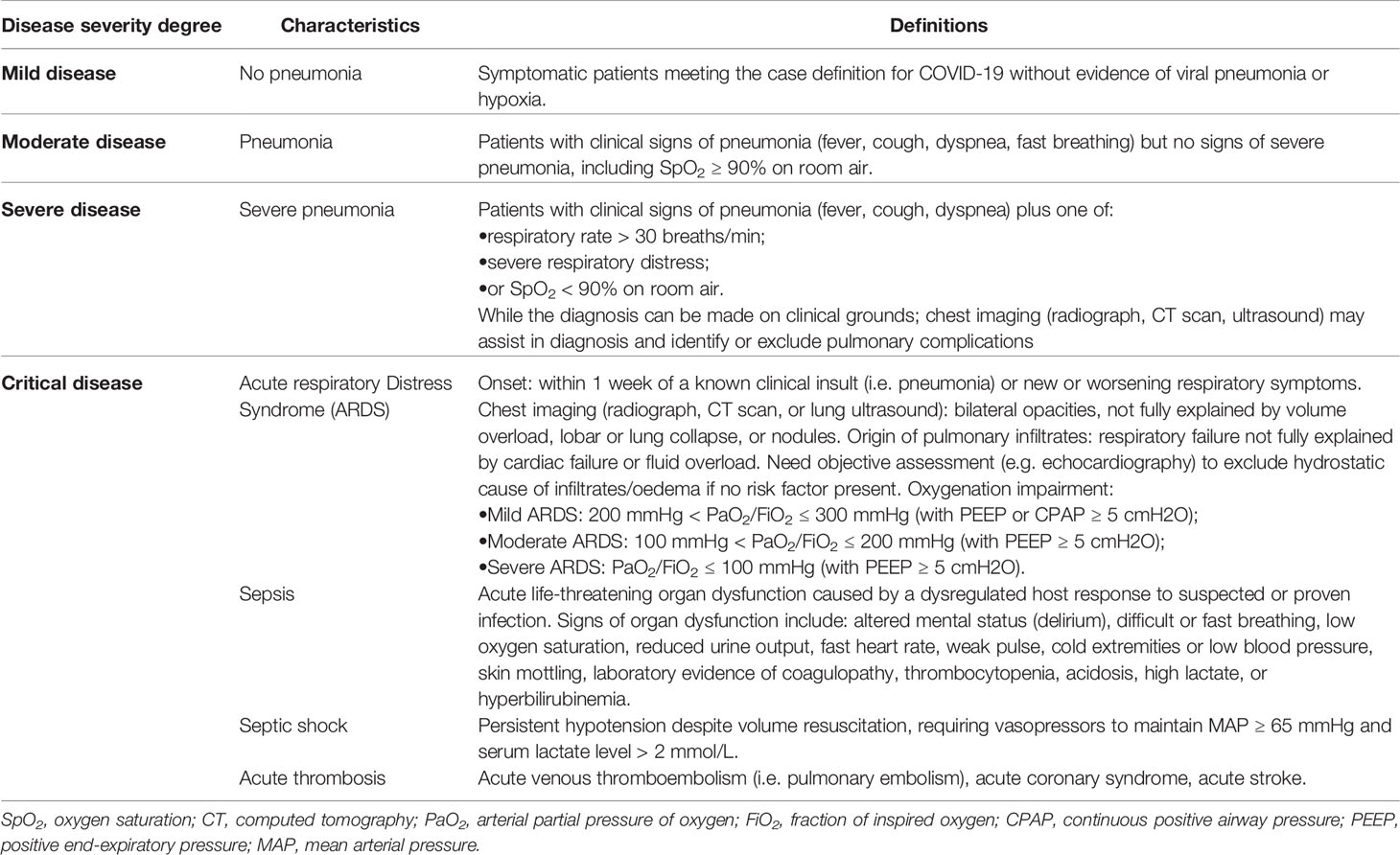
Table 1 WHO COVID-19 disease severity classification in adults.
Several clinical conditions have been associated with an increased risk of severe COVID-19, and include older age, chronic lung disease, cardiovascular disease, obesity, diabetes mellitus, liver disease, end-stage renal disease, and immunocompromise. However, the specific pathogenetic factors that dictate whether an individual will develop the severe form of the disease still remain a matter of scientific debate. The present narrative review article will shed light on our current understanding of the immune dysregulation associated to COVID-19 severity and the underlying viral and host pathogenetic mechanisms.
2 Immune Dysregulation in Severe COVID-19
The ultimate goal of the immune response is to clear the pathogen and develop a memory so that at the second encounter, the pathogen can be quickly cleared. The development of an antigen-specific memory immune response is a multifactorial process that begins with the initial viral detection by pathogen recognition receptors (PRRs), e.g. Toll-like receptors (TLRs), to initiate the IFN response. The initiation of the innate machinery may occur already a few hours post-infection and, while controlling the viral replication, tries to prime the adaptive immune response to produce cytotoxic and memory cells. The immune system of severe COVID-19 patients present a peculiar array of alterations compared to mild ones, which will be briefly described as follows (Figure 1).
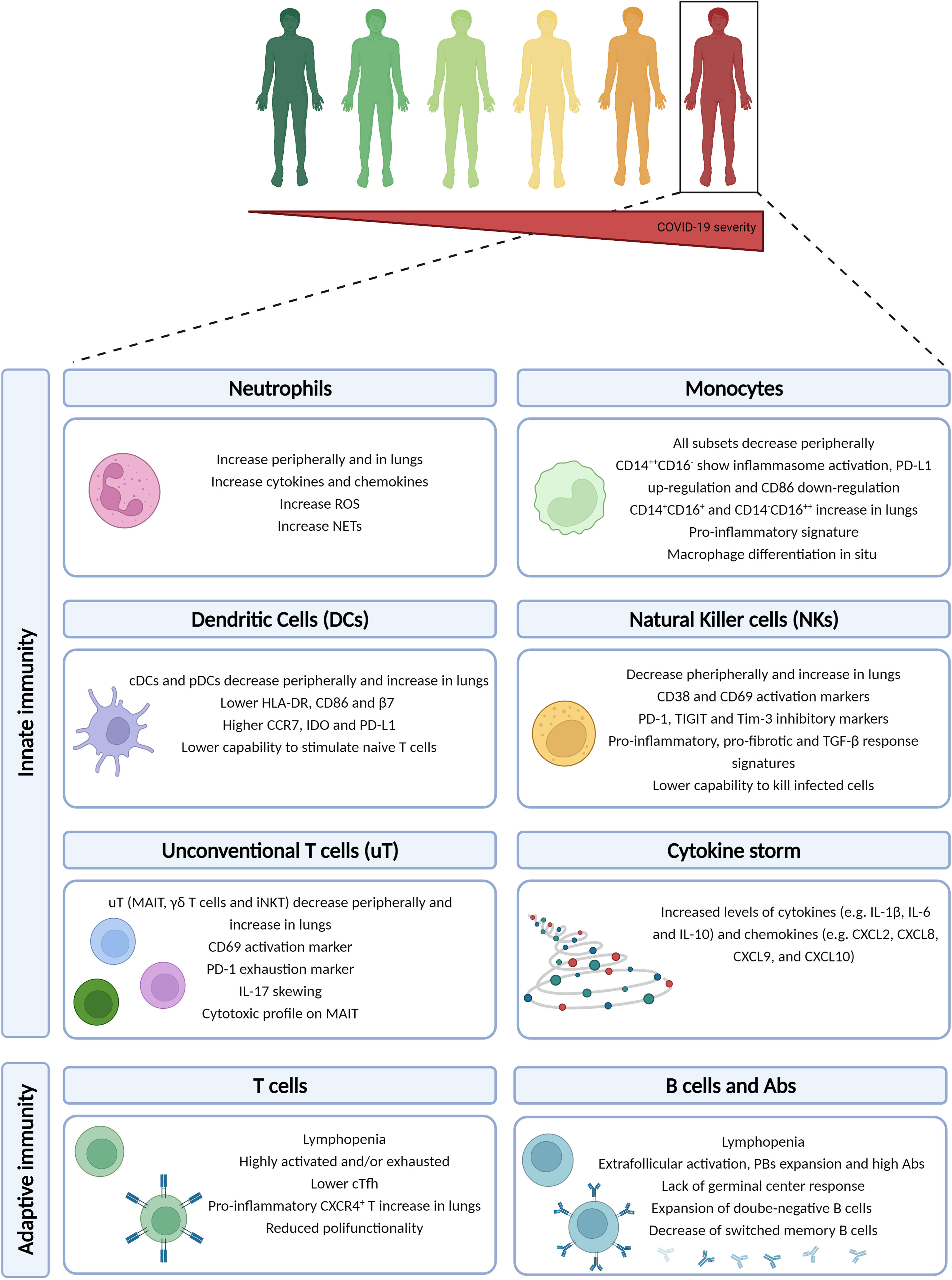
Figure 1 Immune dysregulation in COVID-19 severe patients. The severe/critical form of COVID-19 disease is associated to a multi-layered immune dysregulation involving both innate and adaptive immune responses. Created with BioRender.com.
2.1 Innate Immunity
2.1.1 Cytokine Storm
An exaggerated inflammatory response is the main feature of severe and critical COVID-19 that was soon well documented by several reports (34–36). In general, serum levels of IL-2R, IL-4, IL-6, TNF-α, IL-1RA, IL-1β, IFN-γ are elevated (37), in conjunction with increased levels of chemokines such as CCL2, CCL8, CXCL2, CXCL8, CXCL9, and CXCL16 (38). Some of these cytokines (IL-6, IL-10) and chemokines (CXCL9, CXCL10) were found to be significantly higher in severe patients when compared to milder ones (39). IL-6 has a pivotal role in driving the hyperinflammatory response (35, 40, 41), and it is an independent predictor of patient survival (42). Increased CXCL2, CXCL8, IL-6, and IL-1β are consistent with neutrophilia that is often seen in COVID-19 patients, particularly in those who are severely ill (34, 36, 43, 44). The cytokine storm has been suggested to be characterized by a positive feedback loop. Indeed, the initial wave of cytokines may induce a form of inflammatory cell death (i.e. PANoptosis, composed of pyroptosis, apoptosis, and necrosis), that further induces the release of cytokines, eventually leading to the cytokine storm (45, 46). The PANoptosis seems to be associated to the concomitant increase of IFN-γ and TNF-α. A recent study showed that the change in cytokines and chemokines occurs within the first few days from symptoms onset (47), suggesting that the dysregulation may occur very early in the disease course. Finally, the overall increase of cytokines is usually followed by elevated clinical laboratory parameters such as C reactive protein (CRP), ferritin, and D-dimer, which are well-established acute phase proteins, as well as alanine aminotransferase (ALT), aspartate aminotransferase (AST), and lactate dehydrogenase (LDH), that are markers of tissue damage (43, 48).
Soon after the pandemic started, the cytokine storm became the hallmark of severe COVID-19. However, the cytokine storm is an important feature also in other respiratory infections. For instance, influenza virus infection, which can result in a broad range of clinical manifestations, is associated to a remarkable pro-inflammatory response that, in absence of suitable anti-inflammatory responses, leads to the establishment of the cytokine storm (49–52). Similarly to COVID-19, the cytokine storm mainly occurs in severe cases suggesting a common need to give more insights into the underlying mechanisms to guide future targeted therapeutic interventions.
2.1.2 Neutrophils
Neutrophils are crucial early mediators against infections, as they can rapidly clear pathogens. In particular, neutrophils can employ various intracellular and extracellular mechanisms to control infections, i.e. phagocytosis, the release of cytokines and chemokines, production of reactive oxygen species (ROS), and release of neutrophil extracellular traps (NETs). NETs consist of an extracellular mixture of chromatin, microbicidal molecules, and oxidant enzymes in an attempt to control the infection. However, when these processes are dysregulated, the tissue damage due to hyperinflammation may take over. In the context of COVID-19, neutrophils’ amount and functionality have been associated to disease severity, with an increase at the local and systemic level (48, 53, 54). In particular, a substantial increase in neutrophils has been shown in severe compared to mild COVID-19 (55); besides, they had a more activated phenotype and were characterized by NETosis (54, 56). Indeed, in serum of COVID-19 patients various elements of NET have been identified, e.g. citrullinated histone H3 and myeloperoxidase (MPO) (57). Interestingly, when serum of COVID-19 patients is incubated with neutrophils isolated from healthy individuals in vitro, the release of DNA-bound MPO enzyme and extracellular chromatin structures with neutrophils elastase were observed, suggesting a remarkable NET trigger (57). The ability of NETs to exacerbate inflammation and microvascular thrombosis is in line with the pro-thrombotic events observed in severe COVID-19 patients. Finally, severe patients were characterized by occurrence of pro- and pre-neutrophils precursors, as well as a transcriptional status of dysfunctional and immunosuppressed neutrophils (58).
2.1.3 Monocytes
The second major player during the first wave of innate response is represented by the monocyte subset. Monocytes are quickly recruited at the site of inflammation and can differentiate into macrophages or monocyte-derived dendritic cells (DCs) (59). Monocytes are further divided into immature classical (CD14++CD16-), differentiated inflammatory transitional (CD14+CD16+) and non-classical (CD14-CD16++) subsets.
In general, the peripheral blood of COVID-19 patients is characterized by a reduction of the various monocytes subsets, and such reduction increases with severity (60). Additionally, the classical CD14++CD16- monocytes of severe COVID-19 patients showed inflammasome activation, as evidenced by caspase-1/ASC-speck activation (61). However, the down-regulation of the co-stimulatory CD86 with the concurrent up-regulation of the inhibitory PD-L1 molecule in classical monocytes coincided with a reduced capability to stimulate naïve T cells in vitro (62), suggesting that the enhanced pro-inflammatory status eventually may lead to dysfunction. Furthermore, the reduction of transitional and non-classical monocytes in peripheral blood is paired with selective recruitment in the lungs and with an increase of peripheral inflammatory markers such as IL-6 (60). The peripheral reduction and local increase of monocytes were more marked in those patients with superinfections in the lungs (60). Furthermore, CD16hiCD14lo/- monocytes enriched in the lungs were characterized by expression of HLA-DR, suggesting non-classical monocyte-derived macrophages differentiation in situ (60), whereas the remaining circulating monocytes of severe patients are characterized by lower expression of HLA-DR (63, 64). Other studies have shown an abundant pool of pro-inflammatory monocyte-derived macrophages in the lungs (48), expressing CXCL10+CCL2+ with a pro-inflammatory transcriptomic signature composed of markers such as STAT1, IFNGR1, IFNGR2, NFKB1 and IL1B (65). These pro-inflammatory mediators can contribute to the immunopathology of the lungs.
2.1.4 Dendritic Cells
Dendritic cells (DCs) consist of two main types, i.e. conventional or myeloid DCs (cDCs), i.e. CD1c+CD141+, and type I IFN-producing plasmacytoid DCs (pDCs), i.e. CD123hi. Within cDCs other subsets may be distinguishable, aimed at cross-presenting antigens to CD8 T cells (cDC1), initiating Th response (cDC2), or sharing features of cDC2 and monocytes (DC3) (62, 66). Therefore, DCs may have a pivotal role in the response against SARS-CoV-2 infection as they act at the bridge between innate and adaptive response.
Overall, SARS-CoV-2 infection induces a peripheral reduction of both subsets (67, 68), with a parallel influx in lungs and lymph nodes (48, 68). The immunophenotype of the DCs subset with regard to the homing and activation patterns were also altered. In particular, a lower percentage of the homing receptor integrin-β7+ (β7) and CD86+ DCs, as well as a higher percentage of the C-C chemokine receptor type 7+ (CCR7) and of the marker of immune tolerance and suppression indoleamine 2,3-dyoxigenase (IDO) were observed in COVID-19 patients. Importantly, the alterations initially observed have been shown to persist up to 7 months from infection, regardless of hospitalization (68). The overall reduction of DCs observed during SARS-CoV-2 infection is more pronounced in severe COVID-19 patients and refers to both populations, cDCs, and pDCs (60, 64, 67, 69), regardless of whether they presented superinfections in the lungs (60). Severe COVID-19 patients presented selective recruitment in lungs in particular of cDC2 (60). Additionally, within the DC3 subset, an increase of CD163+CD14+ was observed in severe COVID-19 (62).
Finally, the reduction of DCs in the peripheral blood of COVID-19 patients is accompanied by altered functionality (64, 67, 69). For example, cDCs and pDCs are impaired in their capacity of producing cytokines in response to TLR stimulation (64), and in their capability to stimulate naïve T cells in vitro, thus priming the adaptive immune responses, which correlated with disease severity (62). Such impairment coincided with lower levels of HLA-DR, CD86, and increased levels of the inhibitory PD-L1 molecule (62, 64, 67, 69).
2.1.5 Natural Killer Cells
Natural killer (NK) cells are divided into cytokine-producing (i.e. CD56bright) and cytotoxic (i.e. CD56dim) NK cells. Despite NK cells have mainly antiviral and antitumoral functions, they can also control fibrogenesis.
Overall, a feature of COVID-19 is the reduction of NK cells at the periphery with a parallel increase in the lungs (48, 70, 71), with an expression of both activation markers, such as CD38 and CD69 (70, 72), and inhibitory receptors, such as NKG2A, PD-1, CD39, TIGIT, and Tim-3 (70, 71, 73), particularly in severe patients. Importantly, NK cells of severe COVID-19 patients showed a distinct gene expression signature compared to those with a milder form of disease, e.g. an IFN-α signature (71). Furthermore, in parallel with such re-distribution, the immunophenotype was altered, with enrichment of inflammatory and proliferating cytotoxic CD56dim NK cells (71), as well as adaptive CD57+ NK cells expressing high levels of perforin and NKG2C (70, 74).
Additionally, with re-distribution and immunophenotype alteration comes functional impairment. Indeed, normal NK cells from healthy donors were able to reduce viral proteins when co-cultured with SARS-CoV-2-infected lung fibroblasts in vitro, as well as to reduce the expression of pro-fibrotic genes such as COL1A and ACTA2 (71). Whereas, NK cells from severe COVID-19 patients were not able to reduce viral proteins nor to suppress pro-fibrotic genes (71). Indeed, NK cells of severe COVID-19 patients showed an expression signature that is typical of pulmonary NK cells of patients with lung fibrosis (71). Similar findings were shown in another study of NK cells co-culture with SARS-CoV-2 infected cells in vitro, from both healthy and COVID-19 donors: NK cells from healthy donors killed SARS-CoV-2 infected cells in a dose-dependent manner, whereas this did not occur in NK cells from severe COVID-19 patients, in whom lower NK cells counts also associated to a slower decline of viral load (75). Interestingly, TGF-β was identified as having a putative role in such impairment as NK cells showed a remarkable TGF-β response signature, characterized by downregulation of T-bet and integrin-β2, that prevent the proper NK cells binding to SARS-CoV-2 infected cells, granule exocytosis and cell-mediated cytotoxicity (75). Interestingly, incubation of NK cells from healthy individuals with plasma of severe COVID-19 patients in vitro induced impairments in NK cells, which is in line with the positive correlation between reduction and impairment of NK cells with the systemic hyperinflammation (71, 75). Therefore, hyperactivation and hyperinflammation are important mechanisms of NK cells impairment, as described in the settings of chronic stimulation (76).
2.2 Unconventional T Cells
Unconventional T (uT) cells represent about 10% of circulating T cells and are a major component of the mucosal immune system, exhibiting both innate and adaptive features. These immune cells express invariant TCRs that recognize nonpeptide antigens in an MHC-unrestricted manner and can promptly respond upon activation with the production of cytokines, e.g. IFN-γ, TNF-α, and IL-17A, and cytotoxic activity without undergoing clonal expansion and differentiation into effector cells. This heterogeneous population encompasses three main lineages, i.e., mucosa-associated invariant T (MAIT), γδT, and invariant natural killer T (iNKT) cells, which are engaged in mucosal homeostasis as well as in antitumor and antimicrobial immunity (77).
MAIT cells, Vδ2+ γδT cells as well as iNKT cells appear to be greatly depleted in the peripheral blood of COVID-19 patients in a severity-dependent manner (78–83). Residual circulating uT cells show a significant increase in the expression of both activation (78, 80, 81, 84) and exhaustion (78) markers, i.e. CD69 and PD-1, respectively. CD69 expression on MAIT cells has shown to be more pronounced in patients with detectable SARS-CoV-2 viremia (80), as well as in those with a more severe disease (80, 81, 84), suggesting a striking association between MAIT cells activation and worse clinical outcome. On the contrary, another study found a positive correlation between CD69 expression on MAIT and iNKT cells at the time of admission and the PaO2/FiO2 ratio, suggesting a possible beneficial role of early MAIT and iNKT cells activation in severe COVID-19 (78). Circulating uT cells of COVID-19 patients produce less IFN-γ in the backdrop of more IL-17A, a pro-inflammatory and pro-fibrotic cytokine (78), whereas MAIT cells additionally showed an increased Granzyme B production, with a direct correlation between MAIT cytotoxic activity and severity of disease (80, 81).
The reduction of circulating uT cells is paralleled by increased proportions of MAIT (78, 80) and γδT cells (85), expressing high levels of activation and exhaustion markers in endotracheal aspirates, bronchoalveolar fluid, and pleural effusions of COVID-19 patients, suggesting recruitment of these cells in inflamed lungs. This is further supported by a high expression of MAIT cells chemoattractant such as CXCL10 in airway fluids (78), as well as a reduced expression of chemokine receptors involved in lung homing, e.g. CXCR3 or CCR6, on circulating MAIT cells. Finally, as shown for circulating MAIT cells, also local cells show increased production of IL-17A and Granzyme B in the backdrop of decreased IFN-γ (80, 81). The pro-inflammatory IL-18 produced by monocyte/macrophages has been described as one of the main driving factors for uT cells activation and cytotoxicity. Indeed, a positive correlation between plasmatic IL-18 and CD69 on MAIT and iNKT cells, as well as Granzyme B by MAIT cells, was described (78, 81).
Taken together, these data suggest recruitment of uT cells to the lungs as well as an excessive response with heightened cytotoxicity and skewing toward IL-17A production, which can contribute to lungs inflammation and fibrosis. Interestingly, the similarities of MAIT cells alterations between COVID-19 and obese patients (86–88), could partially explain the poorer COVID-19 outcome in people with obesity (89).
2.3 Adaptive Immunity
The clinical aggravation of COVID-19 occurs approximately one week from symptoms onset, which corresponds to the temporal bridging between innate and adaptive immunity (7, 9, 90), suggesting dysfunction in one or more steps of the adaptive immunity priming. Lymphopenia is a common finding amongst patients with SARS-CoV-2 infection that correlates with increased severity (10, 34, 36, 91). Pathological studies have also documented lymphocytes depletion in secondary lymphoid organs specimens (92, 93). Therefore, the use of lymphopenia and neutrophilia as a combined marker (neutrophil to lymphocyte ratio, NLR) has been suggested crucial in assessing disease severity (94, 95).
2.3.1 Immunophenotypes
Deep immune profile analysis of hospitalized COVID-19 patients showed a highly heterogeneous adaptive response and identified two main distinct immunophenotypes: immunophenotype 1, associated with severity, characterized by highly activated CD4 T cells, fewer circulating T follicular helper (cTfh) cells, and hyperactivated or exhausted CD8 T cells; immunophenotype 2, associated with a more favorable outcome, including less activated CD4 T cells, Tbet+ effector CD4 and CD8 T cells, and proliferating B cells (96). The activated features of phenotype 1 have been described in several reports (39, 63), as well as the exhaustion features (36, 41, 48, 97–101). Exhaustion is a multifactorial and multifaceted state characterized by expression of various inhibitory receptors (e.g. PD-1, Tim-3, CTLA-4, and TIGIT), as well as low proliferative capacity, reduced cytokines production and cytotoxic potential of immune cells. It is plausible that immune exhaustion in COVID-19 is a consequence of the general prolonged activation and inflammation. Furthermore, CD4 T cells differentiation into various subsets, e.g. Th1 and Th2, has been shown to have an important role in influencing the clinical outcome of infections such as HIV (102). In the context of COVID-19, despite the general lymphopenia, an incorrect Th polarization has been shown to be associated with outcome in several studies (103–105). In particular, a polarization toward Th2 was associated with disease severity and with a worse outcome. This may be due to the intrinsic features of such subsets. Indeed, the Th1 subset coordinates the antiviral response by means of CTLs and NKs activation; whereas the Th2 subset activates cells such as eosinophils and basophils, thus tends to inhibit the Th1 response.
2.3.2 Antigen-Specific T Cell Responses
The antigen-specific T cell response is highly heterogeneous, both in the CD4 and CD8 T cells subsets. Indeed, virus-specific T cells can differentiate into a broad range of subpopulations with different effector functions such as: direct antiviral activity via cytokines secretion and recruitment of many immune cells (i.e. T helper cells, Th); cytotoxicity (i.e. CD8 T cells); support of B cells maturation and antibody production (i.e. T follicular helper cells, Tfh). The goal of the adaptive immune response is to clear the infection and build a memory to rapidly intervene upon secondary encounter with the pathogen. Memory T cells differentiate from naïve T cells into central memory T cells (TCM), effector memory T cells (TEM), and terminally differentiated effector memory T cells (TEMRA).
In response to SARS-CoV-2 infection, antigen-specific T cells are generally produced (106), as early as two days post-symptoms onset, with CD4 T cells frequencies overcoming those of CD8 T cells (107, 108). The presence of SARS-CoV-2-specific CD4 and CD8 T cells has been associated with milder disease (106, 107, 109). Furthermore, the functional properties were altered in severe COVID-19 patients, as showed by a lower percentage of polyfunctional CD4 T cells producing simultaneously the classical Th1 cytokines, i.e. IFN-γ, TNF-α, and IL-2 (110). The importance of SARS-CoV-2-specific T cells is further supported by the infection resolution in a COVID-19 patient who did not produce neutralizing antibodies but had instead antigen-specific CD4 and CD8 T cells (107). Interestingly, in critical/deceased COVID-19 patients, it has been shown that SARS-CoV-2-specific T cell responses occur and are comparable to that of moderate patients (111), though not able to clear the infection and/or limit the systemic damage.
SARS-CoV-2-specific T cells can migrate into the lungs as part of COVID-19 pathogenesis. An increased proportion of inflammatory CXCR4+ CD4 and CD8 T cells in the lungs of severe COVID-19 patients was shown (109), whereas in the lungs of moderate patients, an increased proportion of resident memory T cells was observed (48). Additionally, in a study of scRNA-seq on nasopharyngeal and lung samples, a transcriptional signature characterized by strong interactions between epithelial cells and hyperactivated T cells was described, pointing to a potential direct contribution of hyperactivated immune cells on epithelial cells damage (53). These data suggest that inflammatory and hyperactivated T cells are involved in immunopathology.
Finally, contrasting data exist on regulatory T cells (Treg). Despite the generally low levels of SARS-CoV-2-specific T cells described in COVID-19 patients, some studies described an increase of Treg frequencies in severe COVID-19 patients (97, 109, 112), whereas others did not find such association (34).
2.3.3 Antigen-Specific B Cell and Humoral Responses
Humoral response is characterized by a first phase of short-lived low-affinity antibody-secreting plasmablasts (PBs), followed by a second phase of long-lived plasma cells and high-affinity memory B cells formed upon the germinal center response to generate long-lived plasma cells and high-affinity memory B cells. Therefore, PBs produced during the acute phase tend to quickly disappear, but upon re-infection memory B cells can rapidly form IgG-secreting PBs.
Though the majority of studies focus on T cell and antibody responses, perturbations in the B cell compartment associated with COVID-19 severity have been described. Severe COVID-19 patients showed extrafollicular B cells activation with expansion of PBs that inevitably produce high titers of antibodies, similarly to what was reported in autoimmune diseases, which strongly correlates with the level of proinflammatory cytokines (96, 113). The lack of a positive correlation between the titer of neutralizing antibodies and disease severity has been shown by several reports (114, 115), though others have found reduced levels of SARS-CoV-2-specific peripheral IgA, IgG, and IgG1 in severe/critical COVID-19 patients (116). In particular, such populations are characterized by CD19+CD27+CD38hi antibody-producing cells, as well as CD11+ activated naïve B cells that differentiate in effector B cells lacking naïve (IgD) and memory (CD27) markers, i.e. CD19+CD27-CD38-CD24-IgD-CD11c+CD21-, also called double-negative B cells (113, 117). The expansion of B cells lacking memory markers has been described in several studies and has been referred to as atypical memory B cells (CD21loCD27-CD10-) (118). Additionally, a decrease of switched memory B cells was suggested as independent risk factor for mortality in COVID-19 patients (116), whereas proliferating memory B cells were not associated with severity (96). Importantly, the antibody-secreting cells produce high levels of antibodies that can have neutralizing features in severe COVID-19 patients, as shown in several studies (63, 119, 120). However, many severe COVID-19 patients fail to develop the germinal center reaction, as shown in studies of deceased COVID-19 patients where a lack of germinal centers response in spleen and lymph nodes, as well as a reduction of Tfh cells, was demonstrated (121). Indeed, this is further supported by the association of cTfh frequency with a reduced severity (107). Importantly, COVID-19 patients with somatic mutations in memory B cells presented a quick recovery from SARS-CoV-2 infection (122, 123).
The mechanisms underlying the lack of disease control in the backdrop of high levels of neutralizing antibodies still need further clarification, however, the aforementioned data suggest that an accurate B cell maturation, e.g. involving somatic hypermutation, is pivotal for the development of cells with high-affinity and breadth.
Finally, coordinated immune responses made by the three arms of the adaptive response, i.e. CD4 T cells, CD8 T cells, and antibodies, were shown to be associated with milder disease, whereas uncoordinated responses were more often seen in aged and severely ill individuals (107).
3 Underlying Mechanisms of Immune Dysregulation
In the following sections, we will review the mechanisms underlying the immune dysregulation described in severe COVID-19 patients. In particular, viral and host factors, as well as the role of microbiota (Figure 2).
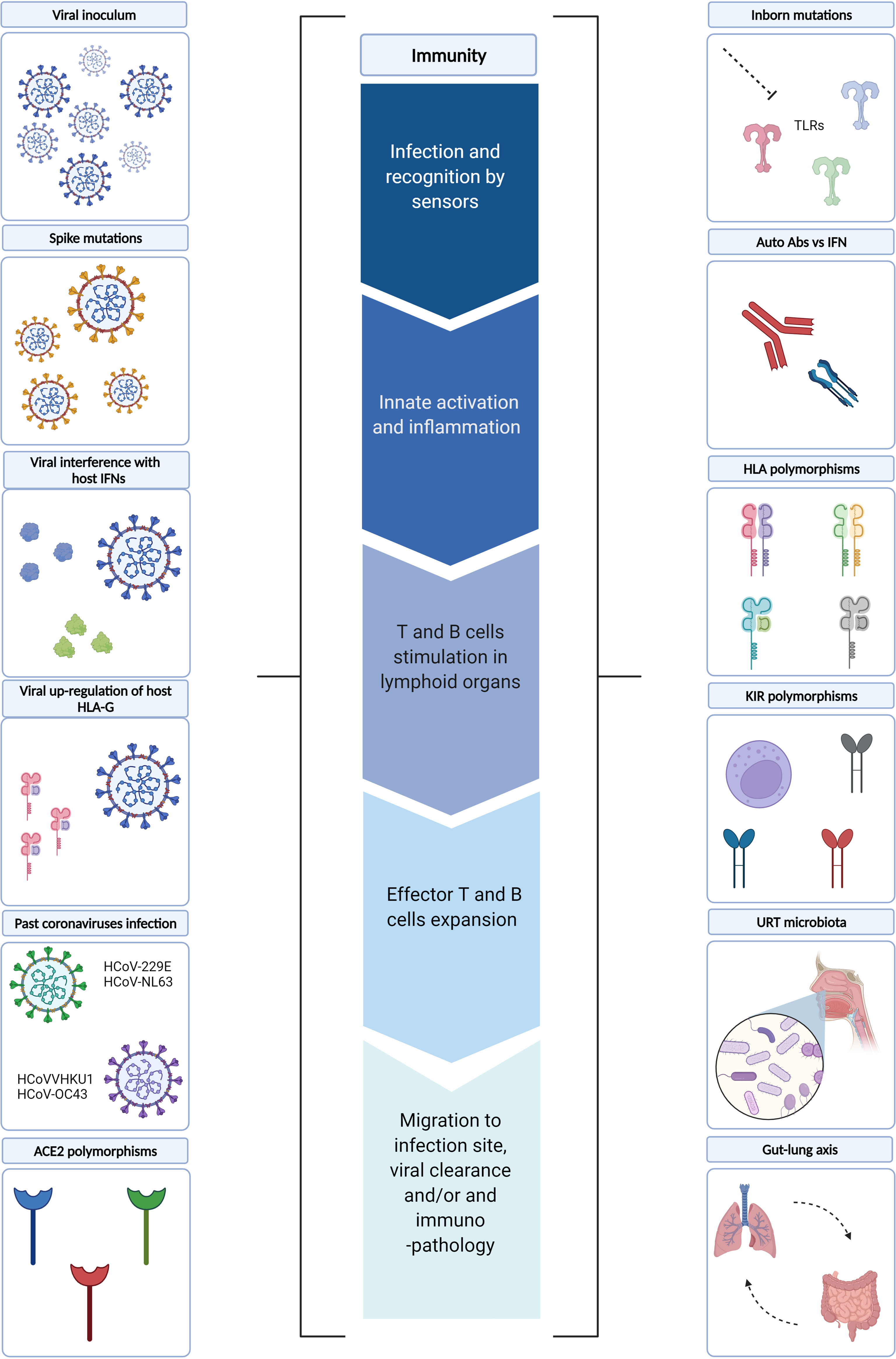
Figure 2 Underlying mechanisms of immune dysregulation. Several viral and host factors have been described as influencing one or more steps of the immune response during SARS-CoV-2 infection. In particular, the early phases of the immune response may be influenced by i) viral factors, such as viral inoculum, Spike mutations and viral interference of host IFN pathways, as well as ii) host factors, such as ACE2 polymorphisms and URT microbiota. Whereas viral up-regulation of host HLA-G, inborn host mutations, auto-reactive Abs vs IFN, HLA and KIR polymorphisms, past coronavirus infections may influence the later phases of immune responses. In this context, gut-lung axis perturbations may further fuel systemic inflammation. TLRs, Toll-like receptors; IFN, Interferon; Abs: Antibodies; HLA, Human Leukocyte Antigens; KIRs, Killer Cell Immunoglobulin-like Receptors; URT, Upper Respiratory Tract; ACE2, Angiotensin-converting Enzyme 2. Created with BioRender.com.
3.1 Viral Factors
The SARS-CoV-2 viral load an individual is infected with, i.e. viral inoculum, certainly represent a first viral factor that may potentially shape the immune responses. Given the severity of the disease, human challenge studies aimed at evaluating disease severity in relation to the initial viral inoculum are lacking; however, several animal studies suggest interesting associations. In a murine model of mouse-adapted SARS-CoV-2 intranasal infection, a dose-dependent increase in morbidity and mortality was observed (124). Furthermore, in a Syrian Hamster model of SARS-CoV-2 infection, the severity of pneumonia increased with viral inoculum when administered intranasally, whereas the oral administration yielded mild pneumonia (125). The use of animal models allows to exclude all variables that may influence the outcome, e.g. genetic background, immunity, and environment. In real-life settings, this cannot occur, and the studies evaluating SARS-CoV-2 viral load in relation to COVID-19 outcome quantify such parameter at hospitalization and/or at symptoms onset, i.e. several days from priming. Therefore, several mechanisms attempting to control a high viral inoculum may have already taken place. This may explain the apparently contrasting data among studies evaluating the association of viral load in nasopharyngeal swabs with COVID-19 outcome. Indeed, some reports have found an association with disease severity (24–28), whereas others did not find a strong association (29, 30), or did not even find it (31, 32).
Importantly, SARS-CoV-2 genome, proteins, or virus-like particles have been detected in many other compartments than the respiratory tract (21–23, 126, 127). How the virus spreads in the other organs is not fully understood, even though there is some evidence that the viral presence in the bloodstream, i.e. referred to as SARS-CoV-2 RNAemia, may be a first critical step. Whether the RNAemia corresponds to infectious viral particles is still under discussion. A study showed a direct cytopathic effect upon Vero-E6 cells infection with the plasma of an immunocompromised patient, and these cells produced virions (127). In the same study, infectious SARS-CoV-2 virus has been isolated from several tissues, i.e. heart and kidney (127). Another study visualized SARS-CoV-2 virions in centrifuged plasma pellets by means of several imaging-based approaches in a cohort of SARS-CoV-2 infected individuals with different degrees of severity, none of them were reported to be immunocompromised (128). Therefore, these data suggest that SARS-CoV-2 RNAemia could be partially explained by SARS-CoV-2 viremia. Despite the viral control in immunocompromised patients is highly impaired compared to immunocompetent individuals, similar mechanisms of replication-competent SARS-CoV-2 spreading may occur in those immunocompetent patients with multi-organ involvement. The detection of SARS-CoV-2 genome in the bloodstream occurs in a relatively small fraction of infected individuals (7, 24, 129, 130); its levels were consistently associated with poor outcomes and were mainly observed within critically ill patients (127, 128, 130–132). SARS-CoV-2 viral load measured in plasma is usually lower than the load measured in the respiratory tract and can be detected as early as the first week from disease onset (130, 133, 134), suggesting that in some patients can be an early event in the pathogenesis. The low levels of plasmatic SARS-CoV-2 may be a strong limiting factor to the isolation of sufficient replication-competent viruses. Interestingly, whereas a strong positive correlation is observed in SARS-CoV-2 viral load measured in different districts of the respiratory tract, their correlation with RNAemia significantly drops (130, 135). However, those individuals with high viral loads both in the lungs and in the plasma succumb rapidly (127). These different mechanisms may underly compartmentalized immune responses (135), and potential organ-specific evolution, as shown in immunocompromised patients (127).
Finally, SARS-CoV-2, as any other RNA virus, is characterized by high mutation and recombination rates (136), with heterogeneous consequences in the viral proteins. Each mutation may confer a new/improved skill that, together with those of the other mutations, confer at the new strain the typical characteristics of a Variant of Concern (VOC) that negatively impacts public health. To date, five VOCs have been identified by the World Health Organization (WHO), i.e. B.1.1.7 (Alpha), B.1.351 (Beta), P.1 (Gamma), B.1.617.2 (Delta), and B.1.1.529 (Omicron). Some of the accumulated mutations possess important immune evasion features that could influence the immune dysregulation observed in COVID-19 patients, e.g. the mutation E484K in the RBD portion of the Spike protein, present in the Beta and Gamma VOCs. In in vitro and in deep mutational scanning (DMS) studies E484K has shown to induce evasion both from monoclonal treatment and humoral response (137, 138). Additionally, the most recent Omicron VOC, which is heavily mutated, with 15 out of 30 mutations located in the RBD portion, contains various mutations that could individually escape neutralizing Abs, i.e. K417N, G446S, E484A, and Q493R (139). This may occur in the backdrop of mutations that confer an advantage on transmissibility, e.g. the mutation N501Y, present in Alpha, Beta, and Gamma VOCs, which increases the affinity of the RBD for ACE2, thus increasing transmissibility (140). It has also been shown both in vitro and in animal models that such mutation increases the rate of virus replication (141). In the Omicron VOC, the additional Q498R mutation makes the binding affinity to ACE2 even stronger (142). The Spike mutation E484K present in the Beta and Gamma VOCs also increases the affinity of the RBD for ACE2 (143). Additionally, the mutation P618H, present in Alpha and Omicron, can greatly enhance spike cleavage, thus increasing transmission (142). Interestingly, a study of replication competency in human ex vivo explant cultures has shown that Omicron replicates faster than all the other VOCs in the bronchus but less efficiently in the lung, which can partially explain the reduced severity in the backdrop of an enhanced transmissibility (144).
Viruses have developed countless countermeasures to directly influence the diverse aspects of human antiviral responses. The SARS-CoV-2 attempt to interfere with immunity starts with the innate response, which is the first line of defense aimed at containing viral replication and dissemination while priming the adaptive immune response. Several SARS-CoV-2 proteins have been described as affecting the IFN response. However, the most characterized so far are the non-structural protein 1 (nsp1) and ORF6, both of which have already been described as virulence factors in SARS-CoV infection (145, 146). In SARS-CoV-2 infection, they act via different mechanisms. Nsp1 reduces translation by means of the interaction with the 40S subunit of the ribosome, leading to lower interferons (IFNs) and interferon-stimulated genes (ISGs) products synthesis while protecting the viral translation (147, 148). ORF6 blocks the IRF3 and STAT1/2 nuclear translocation by means of interaction with export factors such as Rae1 and Nup98 (149, 150). Interestingly, a study evaluating the gene expression profile of several cell lines upon infection with different SARS-CoV-2 multiplicities of infection (MOIs), showed that low-MOI infection yielded a classical signature of reduced IFNs in the backdrop of increased chemokines, whereas high-MOI infection maintained high levels of IFN (38). These data may suggest that the action of viral antagonists of interferon signalling may be ineffective in conditions of high-MOI, as a high viral burden may be able to activate the usual innate antiviral mechanisms. Whether this occurs in vivo remains to be determined. Furthermore, as mentioned earlier, one of the hallmarks of severe COVID-19, as well as of severe influenza virus infection, is the establishment of the cytokine storm. Previously, much attention was put on the highly pathogenetic avian influenza virus H5N1 due to the zoonotic transmission and high mortality rate. Interestingly, the avian H5N1 virus could replicate more in myeloid cells compared to the human adapted virus, and the higher RNAs induced a striking cytokine production thus leading to a more severe outcome in murine models (151). This was attributed to internal viral genes such as polymerase and the non-structural proteins.
One of the mechanisms by which NK cells are activated is the “missing-self” mode by which infected cells down-regulate self-antigens. In particular, activating NK receptors recognize pathogens-induced markers, whereas inhibiting NK receptors bind to self-antigens such as the non-classical HLA class I molecule HLA-E. It has been shown that SARS-CoV-2 encodes a peptide, i.e. non-structural protein 13 (Nsp13), that binds to HLA-E stabilizing the complex on the surface of infected cells. However, instead of binding the inhibitory NKG2A receptor, it prevents such binding, thus leading to the activation of NK cells (152). However, whether it represents an advantage for the pathogen in viral infections such as CMV, characterized by an expansion of adaptive NKG2C+ NK cells, still remains to be elucidated (153).
An additional evasion mechanism employed by several viruses is the up-regulation of HLA-G in the host cell. HLA-G is a non-classical HLA Class I antigen that has immune inhibitory features via receptor signalling and exists in various forms, i.e. membrane-bound (HLA-G 1-4) and soluble (sHLA-G 5-7). Several polymorphisms have been described, some of which are associated with a higher expression (154), therefore, with a potential heightened inhibitory capacity. Upon SARS-CoV infection of lung epithelial Calu-3 cells, an upregulation of HLA-G was observed, whereas this did not occur when infecting with MERS-CoV (155). Unfortunately, very few reports exist on its potential clinical impact on SARS-CoV-2 infection and it certainly merits further evaluation. sHLA-G was found increased in severe COVID-19 patients compared to controls (156, 157); however, when followed up for a short period of time, an increase of sHLA-G was associated with a better outcome, most likely reducing the neutrophils adhesion (156). Moreover, in a study following the dynamic of HLA-G expression on various immune cells in a COVID-19 patient up to the convalescence phase, a high-low-high pattern of HLA-G was observed, most likely reflecting the SARS-CoV-2 infection dynamics (158).
3.2 Host Factors
3.2.1 Smoke
Interestingly, a pre-existing pulmonary damage may play a role in COVID-19 severity. One of the most common habits that can underly pulmonary damage is smoke. Indeed, a worse outcome has been observed in smoking individuals (159). Smoke may also contribute to disease severity by impairing local immune responses and epithelial regenerative capacity. It has been shown in vitro that acute exposure of airway epithelium to cigarette smoke allows for more severe proximal SARS-CoV-2 induced airway epithelial damage by reducing the mucosal innate immune responses and the proliferation of airways basal stem cells (160).
3.2.2 Sex and Age
Finally, soon after the pandemic started, sex and age were the first risk factors to be identified, i.e. worse outcomes in males and in elderly (161–163). These factors could be partially ascribed to intrinsic immune features. First of all, it is known that, overall, the immune responses elicited by females are somewhat enhanced than those elicited by males (164). Interestingly, in a murine model of SARS-CoV infection, estrogens were identified as a receptor signalling key for protection in females (165). Additionally, there might be genetic factors more frequently observed in males. For example, 90% of severe COVID-19 patients with type I IFN autoantibodies were reported to be male (166). Furthermore, COVID-19 has been inversely correlated with the frequency of naïve T cells (96, 107), and the pool of naïve T cells remarkably declines over time (167). The decline of naïve T cells observed with age is more pronounced in males compared to females and is associated to a male-specific decline in B-cell specific loci (168).
3.2.3 Host Genetic Factors
A number of host genetic factors may differentially shape the initial viral binding, recognition, and immune activation. In particular, various polymorphisms of the ACE2 gene have been associated with disease severity as these may change the binding affinity to the Spike protein of SARS-CoV-2 (169). Furthermore, individuals with autoantibodies against IFN-I (166, 170), or inborn mutations resulting in defective genes related to IFN-I, e.g. TLR3, TLR7, and IRF7 (171–173), presented a worse outcome. The importance of an early immune activation is supported by the counter-productive action of early corticosteroid treatment, as it reduces the viral-induced danger signals necessary to activate the immune system (174).
After the initial viral recognition, viral antigens need to be processed and presented to immune cells via the Major Histocompatibility (MHC) molecules, coded by the human leukocyte antigens (HLAs) genes. An in-silico analysis of viral peptide-MCH class I binding affinity highlighted two HLA alleles with potential implications for COVID-19 severity (175). In particular, HLA-B*46:01 presented the fewest predicted binding peptides for SARS-CoV-2, suggesting that individuals carrying such allele may be more likely to develop severity, as shown for SARS-CoV infection (176). Conversely, HLA-B*15:03 showed a high capacity to present conserved peptides, i.e. shared among other coronaviruses, suggesting a potential cross-protection. In an Italian cohort, the HLA-DRB1*15:01, HLA-DQB1*06:02, and HLA-B*27:07 alleles were associated with severe disease (177). Furthermore, NK cells express a variety of killer cell immunoglobulin-like receptors (KIR), which are highly polymorphic glycoproteins that induce inhibitory or activating downstream signals. Usually, upon recognition of the self-HLA molecules, inhibitory KIRs are involved in suppressing NK cells, whereas the down-regulation of the self-HLA, upon viral infections, induces activation of NK cells. In the context of SARS-CoV-2, the KIR AA genotype was associated to a severe COVID-19 disease, in particular, the KIR2DS4 was the most significantly associated with severity, followed by KIR3DL1. Interestingly, the AA genotype encodes for various inhibitory KIRs and only one activating KIR (178).
As mentioned earlier, IL-6 is a key cytokine involved in the cytokine storm, i.e. a hallmark of severe COVID-19. Polymorphisms of IL-6 gene account for differences in plasmatic cytokine levels: the GG genotype (-174G/C) has been previously associated with higher circulating IL-6 (179). Interestingly, IL-6 polymorphisms have been described to affect disease course and response to therapy in several viral infections: the low IL-6-producing CC genotype has been associated with more severe symptoms during respiratory syncytial virus (RSV) infection (180), whereas the high IL-6-producing genotypes, GG or GC, were associated with sustained virologic response in patients with HIV/HCV co-infection treated with pegylated interferon-α (181).
In the context of COVID-19, contrasting data exist on the topic: some studies did not show an association between the abovementioned IL-6 polymorphism with COVID-19 severity, whereas others did find an association (182–184). Interestingly, polymorphisms in other cytokines have been associated to COVID-19 severity. In particular, the AA TNF-α genotype is associated with a more severe COVID-19 disease (185). Furthermore, polymorphisms in other cytokines and cytokine receptors such as IL-17 and IL10RB have been suggested as having a potential influence on COVID-19 progression (186, 187). While the whole genetic background and the immune signature of different populations are likely to influence the role these polymorphisms have on COVID-19 outcome, it appears that carriers of cytokine/cytokine receptors high-producing genotypes may negatively impact COVID-19 severity most likely through the enhancement of the pro-inflammatory milieu. Whether the simultaneous presence of high-producing polymorphisms of different cytokines may have a synergistic effect on the cytokine storm in severe COVID-19 still remains to be determined.
3.2.4 Vitamin D
To date, the role of Vitamin D on COVID-19 severity is still controversial. Indeed, some studies found an association between Vitamin D and COVID-19 severity, whereas others did not find it (188–191). However, there is no doubt that vitamin D exerts important functions in antiviral immunity, as shown in various viral models. For instance, in a murine model of influenza virus infection, the daily supplementation of a high dose of 25-hydroxyvitamin D3 reduced the production of pro-inflammatory cytokines and improved the outcome (192). Whereas in humans, a meta-analysis study showed that the daily or weekly administration of Vitamin D reduced the incidence of acute respiratory tract infections, and that the protective effect was stronger in those individuals with a baseline level of Vitamin D below 25nmol/L (193). For these reasons, despite the controversial data, the role of Vitamin D in COVID-19 severity and outcome is still under evaluation, mostly for its potential therapeutic effect. Overall, it appeared that Vitamin D supplementation started during SARS-CoV-2 infection has limited to no effect on the disease course, whereas when administered regularly for a longer period of time before infection it may have a positive effect (188, 194–196), most likely reflecting the kinetic of vitamin D, that requires some time to reach a functional concentration (197). New insights into the mechanisms underlying the abovementioned associations are necessary to finally understand the actual role of Vitamin D in the pathogenesis of COVID-19.
3.2.5 Exosomes
During viral infections, extracellular vesicles of 30-100 nm are normally produced from a broad array of cells and, in certain cases, can present viral antigens to the host immune system. A study evaluating exosomes isolated from plasma of COVID-19 patients has shown that exosomes of mild patients (which were of B cell, DC, and monocyte/macrophages origin) contained a higher number of SARS-CoV-2 antigens and were able to induce CD4 T cell activation compared to exosomes isolated from severe COVID-19 patients, suggesting that exosomes can contribute to prime adaptive immune responses, thus reducing the severity of disease (198).
3.2.6 Previous Infection With Seasonal Coronaviruses
Seasonal coronaviruses share a certain degree of homology with SARS-CoV-2 and are frequently found in the population. Therefore, one of the aspects long debated is whether past seasonal coronavirus infection may confer any degree of cross-protection during SARS-CoV-2 infection, thus leading to less severe disease. The etiologic agents of the seasonal common cold are either alphacoronaviruses, i.e. HCoV-229E and HCoV-NL63, or betacoronaviruses, i.e. HCoVHKU1 and HCoV-OC43; SARS-CoV, MERS-CoV, and SARS-CoV-2 belong to the betacoronavirus genera (199).
Interestingly, several SARS-CoV-2-specific T cell studies reported that 20-50% of individuals never exposed to SARS-CoV-2 had significant T cell reactivity, mainly CD4 T cells (106, 200, 201), and all authors speculated that this phenomenon may have been due to preexisting memory responses against human “common cold” coronaviruses (HCoVs). Subsequently, this phenomenon was experimentally confirmed in a study evaluating the antigen-specific CD4 T cells response after stimulation with HCoV epitopes homologous to SARS-CoV-2 in unexposed donors and SARS-CoV-2 convalescent (202). The same trend was observed for memory B cells and humoral response. In particular, in a study comparing serum antibodies and memory B cell responses to coronavirus spike proteins in pre-pandemic and convalescent SARS-CoV-2 donors, cross-reactive memory B cells were present in pre-pandemic samples, whereas cross-reactive antibodies were barely detectable (203). Another study comparing the humoral response of children and adults in pre-pandemic era and SARS-CoV-2 convalescent sera showed that up to 40% of children had cross-reactive Abs, and their levels correlated with anti-HCoVs (204). Additionally, a notable increase of anti-HCoVs antibodies in convalescent sera was observed, suggesting a potential activation of cross-reactive memory B cells (204). The big proportion of children bearing anti-HCoVs antibodies underlies the higher proportion of infections with HCoVs compared to adults and may be a partial explanation behind the lower likelihood of children to develop severe COVID-19 (205).
However, there are conflicting reports on whether such cross-reactivity gets translated into a protective immunity, thus leading to less severe disease. Indeed, it has been shown that cross-reactive anti-HCoVs antibodies do not confer protection against SARS-CoV-2 infection nor to hospitalizations, though they are boosted upon SARS-CoV-2 infection (206, 207). On the contrary, another study showed that recent HCoVs infections are associated with less severe COVID-19 (208).
4 The Role of Microbiota
4.1 Upper Respiratory Tract Microbiota
The respiratory tract is the site of initial SARS-CoV-2 infection, which occurs in the context of a local microbiota that has been described to have a role in infection susceptibility and disease severity. Distinct alterations of the upper respiratory tract (URT) microbiota have been reported in SARS-CoV-2 infection: reduced microbial diversity (135, 209–215); depletion of commensal bacteria capable of controlling the constitutive production of type I and type III interferons (e.g. Corynebacterium, Streptococcus, Dolosigranulum, Fusobacterium periodonticum (135, 211, 214, 216)); increase of potential pathogen microbes (e.g. Pseudomonaceae, Salmonella, Serratia, Haemophilus influenzae, Moraxella catarrhalis, Prevotella, Veillonella, Staphylococcus, Peptostreptococcus, Clostridium) (135, 209–212). Such perturbations have been shown to be more pronounced in severely ill patients (135, 209, 211–213, 215, 216). As previous studies reported a role of Fusobacterium periodonticum in the metabolism of sialic acids (217), which can facilitate binding and viral entry (218, 219), depletion of Fusobacterium periodonticum in the upper respiratory tract microbiota may increase susceptibility to SARS-CoV-2 infection (216). Furthermore, reduction of microbial diversity, as well as depletion of Corynebacterium in nasopharyngeal microbiota, have been associated with lower levels of mucosal cytokines (IL-33, IFN-λ3, and IFN-γ) that may be important for viral control, and heightened levels of the chemokine CCL2, which has a role in recruiting pro-inflammatory monocytes into infected tissues (135). Similarly, enrichment of Staphylococcus in URT microbiota is positively correlated with nasopharyngeal viral load and levels of systemic inflammatory cytokines (IL-6 and TNF-α) (135).
Taken together, these observations suggest that disruption of the upper airway’s microbial homeostasis features COVID-19 in a severity-dependent fashion and is associated with dysregulation of local and systemic immune responses.
4.2 Lung Dysbiosis
Although classically assumed to be sterile, lungs harbor their own microbiota, albeit consisting of less abundant microbial populations as compared to gut microbiota (220). Due to the technical difficulties in low airways sampling, only a few studies have examined the lower respiratory tract microbiota in SARS-CoV-2 infection, hindering the possibility to draw firm conclusions. An observational study evaluating the lung microbiota in post-mortem lung biopsies from 20 fatal cases of COVID-19 showed that lung microbiota in these deceased patients was dominated by Acinetobacter, Chryseobacterium, Burkholderia, Brevundimonas, Sphingobium, and Enterobacteriaceae; besides, the lung fungal microbiota was dominated by Criptococcus (221). Another study displayed that lung microbiota of mechanically ventilated COVID-19 patients is enriched with oral commensals, such as Mycoplasma salivarium, Prevotella oris, and Candida albicans; further, deceased patients had higher total bacterial loads in their bronchoalveolar lavage (BAL) and a striking enrichment with Mycoplasma salivarium than patients who survived (222). As it has been previously shown that enrichment of the lower airway microbiota with oral commensals associate with a pro-inflammatory state in several diseases (223, 224), it is plausible that enrichment of lung microbiota with Mycoplasma salivarium in COVID-19 patients contributes to fueling lung and systemic inflammation, thus entailing a worse clinical outcome. A further study reported that lung microbiome of intubated COVID-19 patients is characterized by a low diversity of microbial communities and enriched with common respiratory pathogens (Staphylococcus, Stenotrophomonas), oral commensals (Corynebacterium, Prevotella) or gut-derived microbes (Enterococcus and Enterobacteriaceae such as Escherichia and Klebsiella) (215). Finally, a study based on BAL single-cell transcriptomic of COVID-19 patients in an intensive care unit (ICU) showed lung microbiota to be enriched with oral bacterial commensals, including Mycoplasma salivarium and Prevotella spp., in physical association with immune cells (i.e. following infection or internalization), which show high levels of inflammatory markers, suggesting a role of such bacteria in directly contributing to lung inflammatory response in severe COVID-19 (225). Thus, the enrichment of the lung microbiota with oral commensals in critically ill COVID-19 patients, which could be mainly explained by mechanical ventilation-associated aspiration, may also exacerbate inflammation in SARS-CoV-2 infection, as reported in other infectious and non-infectious diseases (223, 224).
4.3 Gut Barrier Dysfunction and Microbial Translocation
Disruption of the gut mucosal integrity with subsequent microbial translocation, i.e. leakage of intestinal microbes (bacteria and fungi) as well as their products from the gut to the systemic circulation, has been described in diverse infectious and non-infectious diseases as a factor fuelling systemic inflammation (226–228).
Severe COVID-19 patients presented high plasma levels of zonulin (229, 230), a mediator of tight junctions permeability that regulate the intestinal epithelial paracellular pathway inducing the disassembly of the protein ZO-1 from the tight junctional complex (227, 231, 232), and occludin (229), a tight junction structural protein. However, levels of intestinal fatty-acid binding protein (I-FABP), a marker of enterocyte apoptosis, are not heightened (229, 233, 234), suggesting that the alteration of the gut barrier in severe COVID-19 is due to an increased tight junctions’ permeability rather than to enterocyte death. Disruption in several intestinal metabolic pathways has also been displayed: plasma citrulline levels, an established marker of gut function (235), are lower in severe COVID-19 patients as compared to mild ones and controls (229), pointing to enterocyte dysfunction as an additional mechanism of gut leakage in these subjects.
Individuals with severe SARS-CoV-2 infection also show high circulating levels of lipopolysaccharide (LPS) (230), LPS binding protein (LBP) (229, 233, 234, 236), and β-glucan (229),as well as lower levels of EndoCAb-IgM (neutralizing antibodies against LPS endotoxin core antigen) (233), suggesting bacterial and fungal translocation in the systemic circulation. Bacterial proteins belonging to pro-inflammatory bacteria enriched in the gut microbiome of COVID-19 patients, such as Burkholderia spp., Pseudomonas spp., Bifidobacterium longum, have also been detected in blood samples of these subjects (236). Likewise, markers of microbial-mediated myeloid inflammation, i.e. sCD14, sCD163 (monocyte inflammation markers), and MPO (myeloperoxidase; neutrophil inflammation marker) are heightened in severe COVID-19 (229). Furthermore, plasma LPS positively correlates with zonulin (230), advocating that microbial translocation in COVID-19 is a direct consequence of gut barrier disruption.
The abovementioned markers of intestinal barrier permeability and microbial translocation have also been shown to correlate with high levels of several systemic inflammation and immune activation markers, pointing to a role of gut mucosal integrity disruption and microbial translocation in exacerbating systemic inflammation and thus contributing to disease severity (229, 236).
Interestingly, detectable plasma LPS, i.e. endotoxemia, has also been demonstrated to be associated with intra-hospital thrombotic complications in COVID-19 patients (230), suggesting that LPS may promote a prothrombotic state, further supported by the observation of higher levels of TIRAP phosphorylation in platelets (230), pointing to a TLR4-mediated activation by LPS (237). Additionally, plasma levels of LBP have been found heightened throughout the hospital stay in COVID-19 patients with elevated markers of cardiac involvement (N-terminal pro-B-type natriuretic peptide, troponin T, troponin I) and positively correlated to levels of inflammasome activity markers, which are associated with cardiac involvement too, suggesting that gut leakage with subsequent LPS-driven priming of the NLRP3 inflammasome may have a role in the pathogenesis of cardiac involvement during COVID-19 (234).
Intestinal barrier disruption during SARS-CoV-2 infection might be explained by several mechanisms: a direct viral infection of intestinal epithelial cells (238); down-regulation of ACE2 after viral infection of enterocytes (239); systemic inflammation sustained by cytokine storm (226); IL-6-mediated vascular damage (240); intestinal inflammation sustained by gut homing of T cells, as suggested by high plasma levels of CCL25, a gut homing marker, ligand for the chemokine receptor CCR9 (234, 241), and gut dysbiosis with subsequent mucosal inflammation (226, 242). The resulting increase in gut barrier permeability facilitates the passage of microbes and microbial products from the gut to the bloodstream, thus boosting systemic inflammation, which in turn promotes further gut leakiness, fostering a pro-inflammatory vicious cycle that can ultimately contribute to COVID-19 severity. Interestingly, well-established risk factors for severe COVID-19, like obesity and diabetes, are known to be associated with impairment of the gut barrier function and disturbances of the intestinal microbiota (243–245); hence, such alterations might contribute, at least in part, to the severity of disease in these subjects.
4.4 Gut Dysbiosis
As mentioned above, alteration of the gut microbiota, i.e. gut dysbiosis, is one of the mechanisms that can contribute to the intestinal barrier dysfunction observed in severe COVID-19 (242). Several alterations of the gut microbiota have been reported in SARS-CoV-2 infection: decreased microbial diversity (242, 246–248); decreased Firmicutes/Bacteroidetes ratio (242); depletion of beneficial butyrate-producing bacteria with anti-inflammatory and immunomodulatory potential (e.g. Ruminococcaceae, Lachnospiraceae, Fusicatenibacter, Anaerostipes, Agathobacter, Eubacterium hallii, Clostridium butyricum, Clostridium leptum, Eubacterium rectale, Bifidobacterium bifidum, Bifidobacterium adolescentis, Bifidobacterium pseudocatenulatum, Alistipes onderdonkii) (242, 246, 248–251); enrichment of potential pathogen microbes (e.g. Streptococcus, Rothia, Veillonella, Erysipelatoclostridium, Actinomyces, Enterococcus, Enterobacteriaceae, Clostridium ramosum, Clostridium hathewayi, Clostridium innocuum, Coprobacilllus, Bacteroides nordii, Burkholderia contaminans, Bifidobacterium longum, Blautia) (236, 242, 246, 248–251); overgrowth of opportunistic fungal pathogens (e.g. Candida albicans, Candida auris, Aspergillus flavus, Aspergillus niger) (252); skewing of microbial metabolic pathways (i.e. enhancement of glycolysis, fermentation, methionine biosynthesis, vitamin B12 biosynthesis, teichoic acid biosynthesis, and tryptophane catabolism) (229, 236).
It has been hypothesized that down-regulation of ACE2 on SARS-CoV-2-infected enterocytes could be the link between COVID-19 and gut dysbiosis (239). Indeed, it is known that ACE2 down-regulation in small intestine epithelial cells reduces the expression of the sodium-dependent neutral amino acid transporter B (0)AT1 (253), which in turn disturbs tryptophan absorption. As a result, the deficiency of tryptophan and its metabolite nicotinamide decreases the activity of the mTOR pathway, which regulates the expression of antimicrobial peptides in the intestinal mucosa, with subsequent alteration of the gut microbiota ecology (254).
Of note, most of these gut microbiota perturbations were found to be associated with higher levels of inflammatory and pro-thrombotic markers as well as more pronounced in severe and critical COVID-19 patients, suggesting that gut dysbiosis is involved in the magnitude of COVID-19 severity possibly via modulation of gut barrier permeability as well as host inflammatory and immune responses (229, 236, 242, 247–251).
4.5 Gut-Lung Axis
Although gut and lungs are anatomically distinct, complex pathways involving their microbial communities, i.e. microbiota, and immune cells, configure bidirectional interactions between intestinal and respiratory mucosa, known as gut-lung axis (255), which are believed to be involved in several physiologic and pathologic conditions (256, 257). Gut-lung axis perturbations involving gut barrier dysfunction with microbial translocation, gut dysbiosis, and lung dysbiosis, have also been implicated in COVID-19 immunopathogenesis and severity of disease (258).
The observation that the lung microbiota of SARS-CoV-2-infected patients can be enriched with bacteria that are typical of the gut microbiota, along with the evidence of gut barrier dysfunction and microbial translocation, suggests that translocation of microbes from the gut to the lungs via the bloodstream may occur in COVID-19, as previously described in mouse models of sepsis and in humans with ARDS (259), pointing to a role of the gut-lung axis in fueling lung/systemic inflammation and, in turn, disease severity.
5 Conclusions
In COVID-19 the majority of individuals do not have significant complications suggesting that there is a series of favorable events that successfully clear the virus. The factors that dictate whether a patient will develop the severe/critical form are still a matter of scientific debate. The comparison of pauci-symptomatic/asymptomatic individuals with symptomatic patients would be extremely helpful to define these factors. However, few studies managed to include a group of pauci-symptomatic/asymptomatic patients, thus limiting comparative assessments versus symptomatic and severe phenotypes. Additionally, heterogeneity in the definition of disease severity as well as in measurements of immune response further increase variability among studies. What is more, whereas at the beginning of the pandemic, the entire population was naïve to SARS-CoV-2 infection, i.e. no underlying immunity, two years after the beginning of the pandemic, the majority of subjects are now experienced to SARS-CoV-2 through either natural infection, vaccination or both, hence further hindering the identification of correlates of protection. Undoubtfully, while the activation and recruitment of the immune cells mentioned in the present article are initially part of the defensive response against SARS-CoV-2 infection, there seems to be a specific turning point in the natural course of the infection, in which the same immune pathways take a pathogenetic flavor, which overrides any later immune attempt to control the infection. The common thread towards a deleterious response appears to be the excessive activation, the skewing towards “non-useful” immune phenotypes, as well as exhaustion, altogether contributing to the pathological inflammatory and fibrotic process in severe COVID-19, eventually leading to ARDS and post-COVID fibrotic pulmonary sequelae. In the backdrop of a broad knowledge of the immune features associated with COVID-19 severity, very few underlying mechanisms have been identified so far. The identification of the factors underlying immune dysregulation in severe COVID-19 is crucial to understanding COVID-19 pathogenesis, defining early correlates of protection, and informing strategies of targeted therapeutic interventions.
Author Contributions
RR: contributed to paper conceptualization, structured and wrote the review, created figures; MA: wrote the manuscript and created figures; AB-H: contributed to literature search and manuscript writing; VB: contributed to literature search and manuscript writing and figure design; ADM: edited the manuscript; GM: paper conceptualization and writing and revision of manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by grants from Fondazione Cariplo in collaboration with Regione Lombardia and Fondazione Umberto Veronesi (Rif. 2020-1355 and 2020-1376) and Fondazione di Comunità Milano.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We are grateful to all COVID-19 patients that inspired us to try to unfold the complexities underlying the severe forms of the disease. The authors acknowledge the support of the APC central fund of the University of Milan and the Fondo di Specialitá 2022 of the Clinic of Infectious Diseases and Tropical Medicine, University of Milan
References
1. Zhu N, Zhang D, Wang W, Li X, Yang B, Song J, et al. A Novel Coronavirus From Patients With Pneumonia in China, 2019. N Engl J Med (2020) 382(8):727–33. doi: 10.1056/NEJMoa2001017
2. Gorbalenya AE, Baker SC, Baric RS, de Groot RJ, Drosten C, Gulyaeva AA, et al. The Species Severe Acute Respiratory Syndrome-Related Coronavirus: Classifying 2019-Ncov and Naming it SARS-CoV-2. Nat Microbiol (2020) 5(4):536–44. doi: 10.1038/s41564-020-0695-z
3. Peiris JS, Guan Y, Yuen KY. Severe Acute Respiratory Syndrome. Nat Med (2004) 10(12 Suppl):S88–97. doi: 10.1038/nm1143
4. Zaki AM, van Boheemen S, Bestebroer TM, Osterhaus AD, Fouchier RA. Isolation of a Novel Coronavirus From a Man With Pneumonia in Saudi Arabia. N Engl J Med (2012) 367(19):1814–20. doi: 10.1056/NEJMoa1211721
5. Chan JF-W, Yuan S, Kok K-H, To KK-W, Chu H, Yang J, et al. A Familial Cluster of Pneumonia Associated With the 2019 Novel Coronavirus Indicating Person-to-Person Transmission: A Study of a Family Cluster. Lancet (2020) 395(10223):514–23. doi: 10.1016/S0140-6736(20)30154-9
6. Bajema KL, Oster AM, Mcgovern OL, Lindstrom S, Stenger MR, Anderson TC, et al. Persons Evaluated for 2019 Novel Coronavirus — United States. In: MMWR Morbidity and Mortality Weekly Report, vol. 69(6). (2020) 9(6):166–70. doi: 10.15585/mmwr.mm6906e1
7. Huang C, Wang Y, Li X, Ren L, Zhao J, Hu Y, et al. Clinical Features of Patients Infected With 2019 Novel Coronavirus in Wuhan, China. Lancet (2020) 395(10223):497–506. doi: 10.1016/S0140-6736(20)30183-5
8. Chen N, Zhou M, Dong X, Qu J, Gong F, Han Y, et al. Epidemiological and Clinical Characteristics of 99 Cases of 2019 Novel Coronavirus Pneumonia in Wuhan, China: A Descriptive Study. Lancet (2020) 395(10223):507–13. doi: 10.1016/S0140-6736(20)30211-7
9. Wang D, Hu B, Hu C, Zhu F, Liu X, Zhang J, et al. Clinical Characteristics of 138 Hospitalized Patients With 2019 Novel Coronavirus–Infected Pneumonia in Wuhan, China. JAMA (2020) 323(11):1061. doi: 10.1001/jama.2020.1585
10. Yang X, Yu Y, Xu J, Shu H, Xia JA, Liu H, et al. Clinical Course and Outcomes of Critically Ill Patients With SARS-CoV-2 Pneumonia in Wuhan, China: A Single-Centered, Retrospective, Observational Study. Lancet Respir Med (2020) 8(5):475–81. doi: 10.1016/S2213-2600(20)30079-5
11. Wu Z, Mcgoogan JM. Characteristics of and Important Lessons From the Coronavirus Disease 2019 (COVID-19) Outbreak in China. JAMA (2020) 323(13):1239. doi: 10.1001/jama.2020.2648
12. Meyerowitz EA, Richterman A, Gandhi RT, Sax PE. Transmission of SARS-CoV-2: A Review of Viral, Host, and Environmental Factors. Ann Intern Med (2021) 174(1):69–79. doi: 10.7326/M20-5008
13. Hoffmann M, Kleine-Weber H, Schroeder S, Krüger N, Herrler T, Erichsen S, et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell (2020) 181(2):271–80. doi: 10.1016/j.cell.2020.02.052
14. Shi Y, Wang Y, Shao C, Huang J, Gan J, Huang X, et al. COVID-19 Infection: The Perspectives on Immune Responses. Cell Death Differ (2020) 27(5):1451–4. doi: 10.1038/s41418-020-0530-3
15. Cao X. COVID-19: Immunopathology and its Implications for Therapy. Nat Rev Immunol (2020) 20(5):269–70. doi: 10.1038/s41577-020-0308-3
16. Ackermann M, Verleden SE, Kuehnel M, Haverich A, Welte T, Laenger F, et al. Pulmonary Vascular Endothelialitis, Thrombosis, and Angiogenesis in Covid-19. N Engl J Med (2020) 383(2):120–8. doi: 10.1056/NEJMoa2015432
17. Aguiar D, Lobrinus JA, Schibler M, Fracasso T, Lardi C. Inside the Lungs of COVID-19 Disease. Int J Leg Med (2020) 134(4):1271–4. doi: 10.1007/s00414-020-02318-9
18. Pernazza A, Mancini M, Rullo E, Bassi M, De Giacomo T, Rocca CD, et al. Early Histologic Findings of Pulmonary SARS-CoV-2 Infection Detected in a Surgical Specimen. Virchows Arch (2020) 477(5):743–8. doi: 10.1007/s00428-020-02829-1
19. Xu Z, Shi L, Wang Y, Zhang J, Huang L, Zhang C, et al. Pathological Findings of COVID-19 Associated With Acute Respiratory Distress Syndrome. Lancet Respir Med (2020) 8(4):420–2. doi: 10.1016/S2213-2600(20)30076-X
20. Hamming I, Timens W, Bulthuis ML, Lely AT, Navis G, van Goor H. Tissue Distribution of ACE2 Protein, the Functional Receptor for SARS Coronavirus. A First Step in Understanding SARS Pathogenesis. J Pathol (2004) 203(2):631–7. doi: 10.1002/path.1570
21. Puelles VG, Lütgehetmann M, Lindenmeyer MT, Sperhake JP, Wong MN, Allweiss L, et al. Multiorgan and Renal Tropism of SARS-CoV-2. New Engl J Med (2020) 383(6):590–2. doi: 10.1056/NEJMc2011400
22. Liu J, Li Y, Liu Q, Yao Q, Wang X, Zhang H, et al. SARS-CoV-2 Cell Tropism and Multiorgan Infection. Cell Discov (2021) 7(1):17. doi: 10.1038/s41421-021-00249-2
23. Cheung CCL, Goh D, Lim X, Tien TZ, Lim JCT, Lee JN, et al. Residual SARS-CoV-2 Viral Antigens Detected in GI and Hepatic Tissues From Five Recovered Patients With COVID-19. Gut (2022) 71(1):226–9. doi: 10.1136/gutjnl-2021-324280
24. Zheng S, Fan J, Yu F, Feng B, Lou B, Zou Q, et al. Viral Load Dynamics and Disease Severity in Patients Infected With SARS-CoV-2 in Zhejiang Province, China, January-March 2020: Retrospective Cohort Study. BMJ (2020) 369:m1443. doi: 10.1136/bmj.m1443
25. Liu Y, Yang Y, Zhang C, Huang F, Wang F, Yuan J, et al. Clinical and Biochemical Indexes From 2019-Ncov Infected Patients Linked to Viral Loads and Lung Injury. Sci China Life Sci (2020) 63(3):364–74. doi: 10.1007/s11427-020-1643-8
26. Trunfio M, Venuti F, Alladio F, Longo BM, Burdino E, Cerutti F, et al. Diagnostic SARS-CoV-2 Cycle Threshold Value Predicts Disease Severity, Survival, and Six-Month Sequelae in COVID-19 Symptomatic Patients. Viruses (2021) 13(2):281. doi: 10.3390/v13020281
27. El Zein S, Chehab O, Kanj A, Akrawe S, Alkassis S, Mishra T, et al. SARS-CoV-2 Infection: Initial Viral Load (iVL) Predicts Severity of Illness/Outcome, and Declining Trend of iVL in Hospitalized Patients Corresponds With Slowing of the Pandemic. PloS One (2021) 16(9):e0255981. doi: 10.1371/journal.pone.0255981
28. Liu Y, Yan L-M, Wan L, Xiang T-X, Le A, Liu J-M, et al. Viral Dynamics in Mild and Severe Cases of COVID-19. Lancet Infect Dis (2020) 20(6):656–7. doi: 10.1016/S1473-3099(20)30232-2
29. Jacot D, Greub G, Jaton K, Opota O. Viral Load of SARS-CoV-2 Across Patients and Compared to Other Respiratory Viruses. Microbes Infect (2020) 22(10):617–21. doi: 10.1016/j.micinf.2020.08.004
30. Salto-Alejandre S, Berastegui-Cabrera J, Camacho-Martínez P, Infante-Domínguez C, Carretero-Ledesma M, Crespo-Rivas JC, et al. SARS-CoV-2 Viral Load in Nasopharyngeal Swabs Is Not an Independent Predictor of Unfavorable Outcome. Sci Rep (2021) 11(1):12931. doi: 10.1038/s41598-021-92400-y
31. Zou L, Ruan F, Huang M, Liang L, Huang H, Hong Z, et al. SARS-CoV-2 Viral Load in Upper Respiratory Specimens of Infected Patients. New Engl J Med (2020) 382(12):1177–9. doi: 10.1056/NEJMc2001737
32. Cocconcelli E, Castelli G, Onelia F, Lavezzo E, Giraudo C, Bernardinello N, et al. Disease Severity and Prognosis of SARS-CoV-2 Infection in Hospitalized Patients Is Not Associated With Viral Load in Nasopharyngeal Swab. Front Med (Lausanne). (2021) 8:714221. doi: 10.3389/fmed.2021.714221
33. (WHO) WHO. Living Guidance for Clinical Management of COVID-19. Available at: https://www.who.int/publications/i/item/WHO-2019-nCoV-clinical-2021-2.
34. Qin C, Zhou L, Hu Z, Zhang S, Yang S, Tao Y, et al. Dysregulation of Immune Response in Patients With Coronavirus 2019 (COVID-19) in Wuhan, China. Clin Infect Dis (2020) 71(15):762–8. doi: 10.1093/cid/ciaa248
35. Moore JB, June CH. Cytokine Release Syndrome in Severe COVID-19. Science (2020) 368(6490):473–4. doi: 10.1126/science.abb8925
36. Chen G, Wu D, Guo W, Cao Y, Huang D, Wang H, et al. Clinical and Immunological Features of Severe and Moderate Coronavirus Disease 2019. J Clin Invest (2020) 130(5):2620–9. doi: 10.1172/JCI137244
37. Mazzoni A, Salvati L, Maggi L, Annunziato F, Cosmi L. Hallmarks of Immune Response in COVID-19: Exploring Dysregulation and Exhaustion. Semin Immunol (2021) 55:101508. doi: 10.1016/j.smim.2021.101508
38. Blanco-Melo D, Nilsson-Payant BE, Liu WC, Uhl S, Hoagland D, Møller R, et al. Imbalanced Host Response to SARS-CoV-2 Drives Development of COVID-19. Cell (2020) 181(5):1036–45.e9. doi: 10.1016/j.cell.2020.04.026
39. Tincati C, Cannizzo ES, Giacomelli M, Badolato R, d'Arminio Monforte A, Marchetti G. Heightened Circulating Interferon-Inducible Chemokines, and Activated Pro-Cytolytic Th1-Cell Phenotype Features Covid-19 Aggravation in the Second Week of Illness. Front Immunol (2020) 11:580987. doi: 10.3389/fimmu.2020.580987
40. Giamarellos-Bourboulis EJ, Netea MG, Rovina N, Akinosoglou K, Antoniadou A, Antonakos N, et al. Complex Immune Dysregulation in COVID-19 Patients With Severe Respiratory Failure. Cell Host Microbe (2020) 27(6):992–1000.e3. doi: 10.1016/j.chom.2020.04.009
41. Mazzoni A, Salvati L, Maggi L, Capone M, Vanni A, Spinicci M, et al. Impaired Immune Cell Cytotoxicity in Severe COVID-19 is IL-6 Dependent. J Clin Invest (2020) 130(9):4694–703. doi: 10.1172/JCI138554
42. Del Valle DM, Kim-Schulze S, Huang HH, Beckmann ND, Nirenberg S, Wang B, et al. An Inflammatory Cytokine Signature Predicts COVID-19 Severity and Survival. Nat Med (2020) 26(10):1636–43. doi: 10.1038/s41591-020-1051-9
43. Tang Q, Liu Y, Fu Y, Di Z, Xu K, Tang B, et al. A Comprehensive Evaluation of Early Potential Risk Factors for Disease Aggravation in Patients With COVID-19. Sci Rep (2021) 11(1):8062. doi: 10.1038/s41598-021-87413-6
44. Vardhana SA, Wolchok JD. The Many Faces of the Anti-COVID Immune Response. J Exp Med (2020) 217(6):e20200678. doi: 10.1084/jem.20200678
45. Diamond MS, Kanneganti TD. Innate Immunity: The First Line of Defense Against SARS-CoV-2. Nat Immunol (2022) 23(2):165–76. doi: 10.1038/s41590-021-01091-0
46. Karki R, Sharma BR, Tuladhar S, Williams EP, Zalduondo L, Samir P, et al. Synergism of TNF-α and IFN-γ Triggers Inflammatory Cell Death, Tissue Damage, and Mortality in SARS-CoV-2 Infection and Cytokine Shock Syndromes. Cell (2021) 184(1):149–68.e17. doi: 10.1016/j.cell.2020.11.025
47. Kleymenov DA, Bykonia EN, Popova LI, Mazunina EP, Gushchin VA, Kolobukhina LV, et al. A Deep Look Into COVID-19 Severity Through Dynamic Changes in Blood Cytokine Levels. Front Immunol (2021) 12:771609. doi: 10.3389/fimmu.2021.771609
48. Liao M, Liu Y, Yuan J, Wen Y, Xu G, Zhao J, et al. Single-Cell Landscape of Bronchoalveolar Immune Cells in Patients With COVID-19. Nat Med (2020) 26(6):842–4. doi: 10.1038/s41591-020-0901-9
49. Beigel JH, Farrar J, Han AM, Hayden FG, Hyer R, de Jong MD, et al. Avian influenza A (H5N1) Infection in Humans. N Engl J Med (2005) 353(13):1374–85. doi: 10.1056/NEJMra052211
50. Liu Q, Zhou YH, Yang ZQ. The Cytokine Storm of Severe Influenza and Development of Immunomodulatory Therapy. Cell Mol Immunol (2016) 13(1):3–10. doi: 10.1038/cmi.2015.74
51. Tisoncik JR, Korth MJ, Simmons CP, Farrar J, Martin TR, Katze MG. Into the Eye of the Cytokine Storm. Microbiol Mol Biol Rev (2012) 76(1):16–32. doi: 10.1128/MMBR.05015-11
52. Short KR, Kroeze EJBV, Fouchier RAM, Kuiken T. Pathogenesis of Influenza-Induced Acute Respiratory Distress Syndrome. Lancet Infect Dis (2014) 14(1):57–69. doi: 10.1016/S1473-3099(13)70286-X
53. Chua RL, Lukassen S, Trump S, Hennig BP, Wendisch D, Pott F, et al. COVID-19 Severity Correlates With Airway Epithelium-Immune Cell Interactions Identified by Single-Cell Analysis. Nat Biotechnol (2020) 38(8):970–9. doi: 10.1038/s41587-020-0602-4
54. Masso-Silva JA, Moshensky A, Lam MTY, Odish MF, Patel A, Xu L, et al. Increased Peripheral Blood Neutrophil Activation Phenotypes and Neutrophil Extracellular Trap Formation in Critically Ill Coronavirus Disease 2019 (COVID-19) Patients: A Case Series and Review of the Literature. Clin Infect Dis (2022) 74(3):479–89. doi: 10.1093/cid/ciab437
55. Cunha LL, Perazzio SF, Azzi J, Cravedi P, Riella LV. Remodeling of the Immune Response With Aging: Immunosenescence and Its Potential Impact on COVID-19 Immune Response. Front Immunol (2020) 11:1748. doi: 10.3389/fimmu.2020.01748
56. Aschenbrenner AC, Mouktaroudi M, Krämer B, Oestreich M, Antonakos N, Nuesch-Germano M, et al. Disease Severity-Specific Neutrophil Signatures in Blood Transcriptomes Stratify COVID-19 Patients. Genome Med (2021) 13(1):7. doi: 10.1186/s13073-020-00823-5
57. Zuo Y, Yalavarthi S, Shi H, Gockman K, Zuo M, Madison JA, et al. Neutrophil Extracellular Traps in COVID-19. JCI Insight (2020) 5(11):e138999. doi: 10.1172/jci.insight.138999
58. Schulte-Schrepping J, Reusch N, Paclik D, Baßler K, Schlickeiser S, Zhang B, et al. Severe COVID-19 Is Marked by a Dysregulated Myeloid Cell Compartment. Cell (2020) 182(6):1419–40.e23. doi: 10.1016/j.cell.2020.08.001
59. Coillard A, Segura E. In vivo Differentiation of Human Monocytes. Front Immunol (2019) 10:1907. doi: 10.3389/fimmu.2019.01907
60. Sánchez-Cerrillo I, Landete P, Aldave B, Sánchez-Alonso S, Sánchez-Azofra A, Marcos-Jiménez A, et al. COVID-19 Severity Associates With Pulmonary Redistribution of CD1c+ DCs and Inflammatory Transitional and Nonclassical Monocytes. J Clin Invest (2020) 130(12):6290–300. doi: 10.1172/JCI140335
61. Lage SL, Amaral EP, Hilligan KL, Laidlaw E, Rupert A, Namasivayan S, et al. Persistent Oxidative Stress and Inflammasome Activation in CD14. Front Immunol (2021) 12:799558. doi: 10.3389/fimmu.2021.799558
62. Winheim E, Rinke L, Lutz K, Reischer A, Leutbecher A, Wolfram L, et al. Impaired Function and Delayed Regeneration of Dendritic Cells in COVID-19. PloS Pathog (2021) 17(10):e1009742. doi: 10.1371/journal.ppat.1009742
63. Kuri-Cervantes L, Pampena MB, Meng W, Rosenfeld AM, Ittner CAG, Weisman AR, et al. Comprehensive Mapping of Immune Perturbations Associated With Severe COVID-19. Sci Immunol (2020) 5(49):eabd7114. doi: 10.1126/sciimmunol.abd7114
64. Arunachalam PS, Wimmers F, Mok CKP, Perera RAPM, Scott M, Hagan T, et al. Systems Biological Assessment of Immunity to Mild Versus Severe COVID-19 Infection in Humans. Science (2020) 369(6508):1210–20. doi: 10.1126/science.abc6261
65. Zhang F, Mears JR, Shakib L, Beynor JI, Shanaj S, Korsunsky I, et al. IFN-γ and TNF-α Drive a CXCL10+ CCL2+ Macrophage Phenotype Expanded in Severe COVID-19 Lungs and Inflammatory Diseases With Tissue Inflammation. Genome Med (2021) 13(1):64. doi: 10.1186/s13073-021-00881-3
66. Cabeza-Cabrerizo M, Cardoso A, Minutti CM, Pereira da Costa M, Reis e Sousa C. Dendritic Cells Revisited. Annu Rev Immunol (2021) 39:131–66. doi: 10.1146/annurev-immunol-061020-053707
67. Kvedaraite E, Hertwig L, Sinha I, Ponzetta A, Hed Myrberg I, Lourda M, et al. Major Alterations in the Mononuclear Phagocyte Landscape Associated With COVID-19 Severity. Proc Natl Acad Sci USA (2021) 118(6):e2018587118. doi: 10.1101/2020.08.25.20181404
68. Pérez-Gómez A, Vitallé J, Gasca-Capote C, Gutierrez-Valencia A, Trujillo-Rodriguez M, Serna-Gallego A, et al. Dendritic Cell Deficiencies Persist Seven Months After SARS-CoV-2 Infection. Cell Mol Immunol (2021) 18(9):2128–39. doi: 10.1101/2021.03.18.436001
69. Laing AG, Lorenc A, Del Molino Del Barrio I, Das A, Fish M, Monin L, et al. A Dynamic COVID-19 Immune Signature Includes Associations With Poor Prognosis. Nat Med (2020) 26(10):1623–35. doi: 10.1038/s41591-020-1038-6
70. Maucourant C, Filipovic I, Ponzetta A, Aleman S, Cornillet M, Hertwig L, et al. Natural Killer Cell Immunotypes Related to COVID-19 Disease Severity. Sci Immunol (2020) 5(50):eabd6832. doi: 10.1126/sciimmunol.abd6832
71. Krämer B, Knoll R, Bonaguro L, ToVinh M, Raabe J, Astaburuaga-García R, et al. Early IFN-α Signatures and Persistent Dysfunction are Distinguishing Features of NK Cells in Severe COVID-19. Immunity (2021) 54(11):2650–69. doi: 10.1016/j.immuni.2021.09.002
72. Liu C, Martins AJ, Lau WW, Rachmaninoff N, Chen J, Imberti L, et al. Time-Resolved Systems Immunology Reveals a Late Juncture Linked to Fatal COVID-19. Cell (2021) 184(7):1836–57.e22. doi: 10.1016/j.cell.2021.02.018
73. Demaria O, Carvelli J, Batista L, Thibult ML, Morel A, André P, et al. Identification of Druggable Inhibitory Immune Checkpoints on Natural Killer Cells in COVID-19. Cell Mol Immunol (2020) 17(9):995–7. doi: 10.1038/s41423-020-0493-9
74. Varchetta S, Mele D, Oliviero B, Mantovani S, Ludovisi S, Cerino A, et al. Unique Immunological Profile in Patients With COVID-19. Cell Mol Immunol (2021) 18(3):604–12. doi: 10.1038/s41423-020-00557-9
75. Witkowski M, Tizian C, Ferreira-Gomes M, Niemeyer D, Jones TC, Heinrich F, et al. Untimely TGFβ Responses in COVID-19 Limit Antiviral Functions of NK Cells. Nature (2021) 600(7888):295–301. doi: 10.1038/s41586-021-04142-6
76. Merino A, Zhang B, Dougherty P, Luo X, Wang J, Blazar BR, et al. Chronic Stimulation Drives Human NK Cell Dysfunction and Epigenetic Reprograming. J Clin Invest. (2019) 129(9):3770–85. doi: 10.1172/JCI125916
77. Godfrey DI, Uldrich AP, McCluskey J, Rossjohn J, Moody DB. The Burgeoning Family of Unconventional T Cells. Nat Immunol (2015) 16(11):1114–23. doi: 10.1038/ni.3298
78. Jouan Y, Guillon A, Gonzalez L, Perez Y, Boisseau C, Ehrmann S, et al. Phenotypical and Functional Alteration of Unconventional T Cells in Severe COVID-19 Patients. J Exp Med (2020) 217(12):e20200872. doi: 10.1084/jem.20200872
79. Rijkers G, Vervenne T, van der Pol P. More Bricks in the Wall Against SARS-CoV-2 Infection: Involvement of γ9δ2 T Cells. Cell Mol Immunol (2020) 17(7):771–2. doi: 10.1038/s41423-020-0473-0
80. Parrot T, Gorin JB, Ponzetta A, Maleki KT, Kammann T, Emgård J, et al. Mait Cell Activation and Dynamics Associated with Covid-19 Disease Severity. Sci Immunol (2020) 5(51):eabe1670. doi: 10.1126/sciimmunol.abe1670
81. Flament H, Rouland M, Beaudoin L, Toubal A, Bertrand L, Lebourgeois S, et al. Outcome of SARS-CoV-2 Infection is Linked to MAIT Cell Activation and Cytotoxicity. Nat Immunol (2021) 22(3):322–35. doi: 10.1038/s41590-021-00870-z
82. Carissimo G, Xu W, Kwok I, Abdad MY, Chan YH, Fong SW, et al. Whole Blood Immunophenotyping Uncovers Immature Neutrophil-to-VD2 T-Cell Ratio as an Early Marker for Severe COVID-19. Nat Commun (2020) 11(1):5243. doi: 10.1038/s41467-020-19080-6
83. Zhang JY, Wang XM, Xing X, Xu Z, Zhang C, Song JW, et al. Single-Cell Landscape of Immunological Responses in Patients With COVID-19. Nat Immunol (2020) 21(9):1107–18. doi: 10.1038/s41590-020-0762-x
84. Youngs J, Provine NM, Lim N, Sharpe HR, Amini A, Chen YL, et al. Identification of Immune Correlates of Fatal Outcomes in Critically Ill COVID-19 Patients. PloS Pathog (2021) 17(9):e1009804. doi: 10.1371/journal.ppat.1009804
85. Cerapio JP, Perrier M, Pont F, Tosolini M, Laurent C, Bertani S, et al. Single-Cell RNAseq Profiling of Human γδ T Lymphocytes in Virus-Related Cancers and COVID-19 Disease. Viruses (2021) 13(11):2212. doi: 10.3390/v13112212
86. Brien AO, Kedia-Mehta N, Tobin L, Veerapen N, Besra GS, Shea DO, et al. Targeting Mitochondrial Dysfunction in MAIT Cells Limits IL-17 Production in Obesity. Cell Mol Immunol (2020) 17(11):1193–5. doi: 10.1038/s41423-020-0375-1
87. Carolan E, Tobin LM, Mangan BA, Corrigan M, Gaoatswe G, Byrne G, et al. Altered Distribution and Increased IL-17 Production by Mucosal-Associated Invariant T Cells in Adult and Childhood Obesity. J Immunol (2015) 194(12):5775–80. doi: 10.4049/jimmunol.1402945
88. O'Brien A, Loftus RM, Pisarska MM, Tobin LM, Bergin R, Wood NAW, et al. Obesity Reduces Mtorc1 Activity in Mucosal-Associated Invariant T Cells, Driving Defective Metabolic and Functional Responses. J Immunol (2019) 202(12):3404–11. doi: 10.4049/jimmunol.1801600
89. Popkin BM, Du S, Green WD, Beck MA, Algaith T, Herbst CH, et al. Individuals With Obesity and COVID-19: A Global Perspective on the Epidemiology and Biological Relationships. Obes Rev (2020) 21(11):e13128. doi: 10.1111/obr.13128
90. Zheng Y, Xiong C, Liu Y, Qian X, Tang Y, Liu L, et al. Epidemiological and Clinical Characteristics Analysis of COVID-19 in the Surrounding Areas of Wuhan, Hubei Province in 2020. Pharmacol Res (2020) 157:104821. doi: 10.1016/j.phrs.2020.104821
91. Tan L, Wang Q, Zhang D, Ding J, Huang Q, Tang Y-Q, et al. Lymphopenia Predicts Disease Severity of COVID-19: A Descriptive and Predictive Study. Signal Transduct Targeted Ther (2020) 5(1):33. doi: 10.1038/s41392-020-0148-4
92. Liu Q, Shi Y, Cai J, Duan Y, Wang R, Zhang H, et al. Pathological Changes in the Lungs and Lymphatic Organs of 12 COVID-19 Autopsy Cases. Natl Sci Rev (2020) 7(12):1868–78. doi: 10.1093/nsr/nwaa247
93. Xiang Q, Feng Z, Diao B, Tu C, Qiao Q, Yang H, et al. SARS-CoV-2 Induces Lymphocytopenia by Promoting Inflammation and Decimates Secondary Lymphoid Organs. Front Immunol (2021) 12:661052. doi: 10.3389/fimmu.2021.661052
94. Lagunas-Rangel FA. Neutrophil-To-Lymphocyte Ratio and Lymphocyte-to-C-Reactive Protein Ratio in Patients With Severe Coronavirus Disease 2019 (COVID-19): A Meta-Analysis. J Med Virol (2020) 92(10):1733–4. doi: 10.1002/jmv.25819
95. Gelzo M, Cacciapuoti S, Pinchera B, De Rosa A, Cernera G, Scialò F, et al. Prognostic Role of Neutrophil to Lymphocyte Ratio in COVID-19 Patients: Still Valid in Patients That Had Started Therapy? Front Public Health (2021) 9:664108. doi: 10.3389/fpubh.2021.664108
96. Mathew D, Giles JR, Baxter AE, Oldridge DA, Greenplate AR, Wu JE, et al. Deep Immune Profiling of COVID-19 Patients Reveals Distinct Immunotypes With Therapeutic Implications. Science (2020) 369(6508):eabc8511. doi: 10.1126/science.abc8511
97. De Biasi S, Meschiari M, Gibellini L, Bellinazzi C, Borella R, Fidanza L, et al. Marked T Cell Activation, Senescence, Exhaustion and Skewing Towards TH17 in Patients With COVID-19 Pneumonia. Nat Commun (2020) 11(1):3434. doi: 10.1038/s41467-020-17292-4
98. Diao B, Wang C, Tan Y, Chen X, Liu Y, Ning L, et al. Reduction and Functional Exhaustion of T Cells in Patients With Coronavirus Disease 2019 (COVID-19). Front Immunol (2020) 11:827. doi: 10.3389/fimmu.2020.00827
99. Zheng M, Gao Y, Wang G, Song G, Liu S, Sun D, et al. Functional Exhaustion of Antiviral Lymphocytes in COVID-19 Patients. Cell Mol Immunol (2020) 17(5):533–5. doi: 10.1038/s41423-020-0402-2
100. Song J-W, Zhang C, Fan X, Meng F-P, Xu Z, Xia P, et al. Immunological and Inflammatory Profiles in Mild and Severe Cases of COVID-19. Nat Commun (2020) 11(1):3410. doi: 10.1038/s41467-020-17240-2
101. Zheng H-Y, Zhang M, Yang C-X, Zhang N, Wang X-C, Yang X-P, et al. Elevated Exhaustion Levels and Reduced Functional Diversity of T Cells in Peripheral Blood may Predict Severe Progression in COVID-19 Patients. Cell Mol Immunol (2020) 17(5):541–3. doi: 10.1038/s41423-020-0401-3
102. Calarota SA, Weiner DB. Enhancement of Human Immunodeficiency Virus Type 1-DNA Vaccine Potency Through Incorporation of T-Helper 1 Molecular Adjuvants. Immunol Rev (2004) 199:84–99. doi: 10.1111/j.0105-2896.2004.00150.x
103. Gil-Etayo FJ, Suàrez-Fernández P, Cabrera-Marante O, Arroyo D, Garcinuño S, Naranjo L, et al. T-Helper Cell Subset Response Is a Determining Factor in COVID-19 Progression. Front Cell Infect Microbiol (2021) 11:624483. doi: 10.3389/fcimb.2021.624483
104. Roncati L, Nasillo V, Lusenti B, Riva G. Signals Of T H 2 Immune Response From Covid-19 Patients Requiring Intensive Care. Ann Hematol (2020) 99(6):1419–20. Signals of T. doi: 10.1007/s00277-020-04066-7
105. Pavel AB, Glickman JW, Michels JR, Kim-Schulze S, Miller RL, Guttman-Yassky E. Th2/Th1 Cytokine Imbalance Is Associated With Higher COVID-19 Risk Mortality. Front Genet (2021) 12:706902. doi: 10.3389/fgene.2021.706902
106. Grifoni A, Weiskopf D, Ramirez SI, Mateus J, Dan JM, Moderbacher CR, et al. Targets of T Cell Responses to SARS-CoV-2 Coronavirus in Humans With COVID-19 Disease and Unexposed Individuals. Cell (2020) 181(7):1489–501.e15. doi: 10.1016/j.cell.2020.05.015
107. Rydyznski Moderbacher C, Ramirez SI, Dan JM, Grifoni A, Hastie KM, Weiskopf D, et al. Antigen-Specific Adaptive Immunity to SARS-CoV-2 in Acute COVID-19 and Associations With Age and Disease Severity. Cell (2020) 183(4):996–1012.e19. doi: 10.1016/j.cell.2020.09.038
108. Schulien I, Kemming J, Oberhardt V, Wild K, Seidel LM, Killmer S, et al. Characterization of Pre-Existing and Induced SARS-CoV-2-Specific CD8. Nat Med (2021) 27(1):78–85. doi: 10.1038/s41591-020-01143-2
109. Neidleman J, Luo X, George AF, McGregor M, Yang J, Yun C, et al. Distinctive Features of SARS-CoV-2-Specific T Cells Predict Recovery From Severe COVID-19. Cell Rep (2021) 36(3):109414. doi: 10.1016/j.celrep.2021.109414
110. Schub D, Klemis V, Schneitler S, Mihm J, Lepper PM, Wilkens H, et al. High Levels of SARS-CoV-2-Specific T Cells With Restricted Functionality in Severe Courses of COVID-19. JCI Insight (2020) 5(20):e142167. doi: 10.1172/jci.insight.142167
111. Thieme CJ, Anft M, Paniskaki K, Blazquez-Navarro A, Doevelaar A, Seibert FS, et al. Robust T Cell Response Toward Spike, Membrane, and Nucleocapsid SARS-CoV-2 Proteins Is Not Associated With Recovery in Critical COVID-19 Patients. Cell Rep Med (2020) 1(6):100092. doi: 10.1016/j.xcrm.2020.100092
112. Galván-Peña S, Leon J, Chowdhary K, Michelson DA, Vijaykumar B, Yang L, et al. Profound Treg Perturbations Correlate With COVID-19 Severity. Proc Natl Acad Sci USA (2021) 118(37):e2111315118. doi: 10.1073/pnas.2111315118
113. Woodruff MC, Ramonell RP, Nguyen DC, Cashman KS, Saini AS, Haddad NS, et al. Extrafollicular B Cell Responses Correlate With Neutralizing Antibodies and Morbidity in COVID-19. Nat Immunol (2020) 21(12):1506–16. doi: 10.1038/s41590-020-00814-z
114. Piccoli L, Park YJ, Tortorici MA, Czudnochowski N, Walls AC, Beltramello M, et al. Mapping Neutralizing and Immunodominant Sites on the SARS-CoV-2 Spike Receptor-Binding Domain by Structure-Guided High-Resolution Serology. Cell (2020) 183(4):1024–42. doi: 10.1016/j.cell.2020.09.037
115. Robbiani DF, Gaebler C, Muecksch F, Lorenzi JCC, Wang Z, Cho A, et al. Convergent Antibody Responses to SARS-CoV-2 in Convalescent Individuals. Nature (2020) 584(7821):437–42. doi: 10.1038/s41586-020-2456-9
116. Çölkesen F, Kepenek Kurt E, Vatansev H, Korkmaz C, Yücel F, Yıldız E, et al. Memory B Cells and Serum Immunoglobulins Are Associated With Disease Severity and Mortality in Patients With COVID-19. Postgrad Med J (2022):140540. doi: 10.1136/postgradmedj-2021-140540
117. Cervantes-Díaz R, Sosa-Hernández VA, Torres-Ruíz J, Romero-Ramírez S, Cañez-Hernández M, Pérez-Fragoso A, et al. Severity of SARS-CoV-2 Infection Is Linked to Double-Negative (CD27- IgD -) B cell subset numbers. Inflammation Res (2022) 71(1):131–40. doi: 10.1007/s00011-021-01525-3
118. Oliviero B, Varchetta S, Mele D, Mantovani S, Cerino A, Perotti CG, et al. Expansion of Atypical Memory B Cells Is a Prominent Feature of COVID-19. Cell Mol Immunol (2020) 17(10):1101–3. doi: 10.1038/s41423-020-00542-2
119. Dan JM, Mateus J, Kato Y, Hastie KM, Yu ED, Faliti CE, et al. Immunological Memory to SARS-CoV-2 Assessed for Up to 8 Months After Infection. Science (2021) 371(6529):eabf4063. doi: 10.1126/science.abf4063
120. Long QX, Liu BZ, Deng HJ, Wu GC, Deng K, Chen YK, et al. Antibody Responses to SARS-CoV-2 in Patients With COVID-19. Nat Med (2020) 26(6):845–8. doi: 10.1038/s41591-020-0897-1
121. Kaneko N, Kuo HH, Boucau J, Farmer JR, Allard-Chamard H, Mahajan VS, et al. Loss of Bcl-6-Expressing T Follicular Helper Cells and Germinal Centers in COVID-19. Cell (2020) 183(1):143–57.e13. doi: 10.1016/j.cell.2020.08.025
122. Gaebler C, Wang Z, Lorenzi JCC, Muecksch F, Finkin S, Tokuyama M, et al. Evolution of Antibody Immunity to SARS-CoV-2. Nature (2021) 591(7851):639–44. doi: 10.1038/s41586-021-03207-w
123. Chen Y, Zuiani A, Fischinger S, Mullur J, Atyeo C, Travers M, et al. Quick COVID-19 Healers Sustain Anti-SARS-CoV-2 Antibody Production. Cell (2020) 183(6):1496–507.e16. doi: 10.1016/j.cell.2020.10.051
124. Leist SR, Dinnon KH, Schäfer A, Tse LV, Okuda K, Hou YJ, et al. A Mouse-Adapted SARS-CoV-2 Induces Acute Lung Injury and Mortality in Standard Laboratory Mice. Cell (2020) 183(4):1070–85. doi: 10.1016/j.cell.2020.09.050
125. Lee AC, Zhang AJ, Chan JF, Li C, Fan Z, Liu F, et al. Oral SARS-CoV-2 Inoculation Establishes Subclinical Respiratory Infection With Virus Shedding in Golden Syrian Hamsters. Cell Rep Med (2020) 1(7):100121. doi: 10.1016/j.xcrm.2020.100121
126. Trypsteen W, Van Cleemput J, Snippenberg WV, Gerlo S, Vandekerckhove L. On the Whereabouts of SARS-CoV-2 in the Human Body: A Systematic Review. PloS Pathog (2020) 16(10):e1009037. doi: 10.1371/journal.ppat.1009037
127. Van Cleemput J, van Snippenberg W, Lambrechts L, Dendooven A, D'Onofrio V, Couck L, et al. Organ-Specific Genome Diversity of Replication-Competent SARS-CoV-2. Nat Commun (2021) 12(1):6612. doi: 10.1038/s41467-021-26884-7
128. Jacobs JL, Bain W, Naqvi A, Staines B, Castanha PMS, Yang H, et al. Severe Acute Respiratory Syndrome Coronavirus 2 Viremia Is Associated With Coronavirus Disease 2019 Severity and Predicts Clinical Outcomes”.. Clin Infect Dis (2021) 74(9):1525–33. doi: 10.1093/cid/ciab686
129. Wang W, Xu Y, Gao R, Lu R, Han K, Wu G, et al. Detection of SARS-CoV-2 in Different Types of Clinical Specimens. JAMA (2020) 323(18):1843–44. doi: 10.1001/jama.2020.3786
130. Fajnzylber J, Regan J, Coxen K, Corry H, Wong C, Rosenthal A, et al. SARS-CoV-2 Viral Load Is Associated With Increased Disease Severity and Mortality. Nat Commun (2020) 11(1):5493. doi: 10.1038/s41467-020-19057-5
131. Chen X, Zhao B, Qu Y, Chen Y, Xiong J, Feng Y, et al. Detectable Serum Severe Acute Respiratory Syndrome Coronavirus 2 Viral Load (RNAemia) Is Closely Correlated With Drastically Elevated Interleukin 6 Level in Critically Ill Patients With Coronavirus Disease 2019. Clin Infect Dis (2020) 71(8):1937–42. doi: 10.1093/cid/ciaa449
132. Li H, Gu X, Gong F, Xu J, Wang Y, Ruan S, et al. Risk Factors of Viral RNAaemia and Its Association With Clinical Prognosis Among Patients With Severe COVID-19. Chest (2021) 159(4):1382–6. doi: 10.1016/j.chest.2020.11.071
133. Wang Y, Zhang L, Sang L, Ye F, Ruan S, Zhong B, et al. Kinetics of Viral Load and Antibody Response in Relation to COVID-19 Severity. J Clin Invest. (2020) 130(10):5235–44. doi: 10.1172/JCI138759
134. Li Y, Schneider AM, Mehta A, Sade-Feldman M, Kays KR, Gentili M, et al. SARS-CoV-2 Viremia Is Associated With Distinct Proteomic Pathways and Predicts COVID-19 Outcomes. J Clin Invest (2021) 131(13):e148635. doi: 10.1172/JCI148635
135. Smith N, Goncalves P, Charbit B, Grzelak L, Beretta M, Planchais C, et al. Distinct Systemic and Mucosal Immune Responses During Acute SARS-CoV-2 Infection. Nat Immunol (2021) 22(11):1428–39. doi: 10.1038/s41590-021-01028-7
136. Yang H, Rao Z. Structural Biology of SARS-CoV-2 and Implications for Therapeutic Development. Nat Rev Microbiol (2021) 19(11):685–700. doi: 10.1038/s41579-021-00630-8
137. Baum A, Fulton BO, Wloga E, Copin R, Pascal KE, Russo V, et al. Antibody Cocktail to SARS-CoV-2 Spike Protein Prevents Rapid Mutational Escape Seen With Individual Antibodies. Science (2020) 369(6506):1014–8. doi: 10.1126/science.abd0831
138. Greaney AJ, Starr TN, Gilchuk P, Zost SJ, Binshtein E, Loes AN, et al. Complete Mapping of Mutations to the SARS-CoV-2 Spike Receptor-Binding Domain That Escape Antibody Recognition. Cell Host Microbe (2021) 29(1):44–57. doi: 10.1016/j.chom.2020.11.007
139. Cao Y, Wang J, Jian F, Xiao T, Song W, Yisimayi A, et al. Omicron Escapes the Majority of Existing SARS-CoV-2 Neutralizing Antibodies. Nature (2022) 602(7898):657–63. doi: 10.1038/d41586-021-03796-6
140. Davies NG, Abbott S, Barnard RC, Jarvis CI, Kucharski AJ, Munday JD, et al. Estimated Transmissibility and Impact of SARS-CoV-2 Lineage B.1.1.7 in England. Science (2021) 372(6538):eabg3055. doi: 10.1126/science.abg3055
141. Liu Y, Liu J, Plante KS, Plante JA, Xie X, Zhang X, et al. The N501Y Spike Substitution Enhances SARS-CoV-2 Infection and Transmission. Nature (2022) 602(7896):294–9. doi: 10.1038/s41586-021-04245-0
142. CDC, Centers for Disease Control and Prevention,Science Brief: Omicron (B.1.1.529) (2022). Available at: https://www.cdc.gov/coronavirus/2019-ncov/science/science-briefs/scientific-brief-omicron-variant.html.
143. Barton MI, MacGowan SA, Kutuzov MA, Dushek O, Barton GJ, van der Merwe PA. Effects of Common Mutations in the SARS-CoV-2 Spike RBD and its Ligand, the Human ACE2 Receptor on Binding Affinity and Kinetics. Elife (2021) 26:10. doi: 10.7554/eLife.70658
144. Hui KPY, Ho JCW, Cheung MC, Ng KC, Ching RHH, Lai KL, et al. SARS-CoV-2 Omicron Variant Replication in Human Bronchus and Lung Ex Vivo. Nature (2022) 603(7902):715–20. doi: 10.1038/s41586-022-04479-6
145. Nakagawa K, Makino S. Mechanisms of Coronavirus Nsp1-Mediated Control of Host and Viral Gene Expression. Cells (2021) 10(2):300. doi: 10.3390/cells10020300
146. Frieman M, Yount B, Heise M, Kopecky-Bromberg SA, Palese P, Baric RS. Severe Acute Respiratory Syndrome Coronavirus ORF6 Antagonizes STAT1 Function by Sequestering Nuclear Import Factors on the Rough Endoplasmic Reticulum/Golgi Membrane. J Virol (2007) 81(18):9812–24. doi: 10.1128/JVI.01012-07
147. Vazquez C, Swanson SE, Negatu SG, Dittmar M, Miller J, Ramage HR, et al. SARS-CoV-2 Viral Proteins NSP1 and NSP13 Inhibit Interferon Activation Through Distinct Mechanisms. PloS One (2021) 16(6):e0253089. doi: 10.1371/journal.pone.0253089
148. Banerjee AK, Blanco MR, Bruce EA, Honson DD, Chen LM, Chow A, et al. SARS-CoV-2 Disrupts Splicing, Translation, and Protein Trafficking to Suppress Host Defenses. Cell (2020) 183(5):1325–39.e21. doi: 10.1016/j.cell.2020.10.004
149. Kato K, Ikliptikawati DK, Kobayashi A, Kondo H, Lim K, Hazawa M, et al. Overexpression of SARS-CoV-2 Protein ORF6 Dislocates RAE1 and NUP98 From the Nuclear Pore Complex. Biochem Biophys Res Commun (2021) 536: 59–66. doi: 10.1016/j.bbrc.2020.11.115
150. Kimura I, Konno Y, Uriu K, Hopfensperger K, Sauter D, Nakagawa S, et al. Sarbecovirus ORF6 Proteins Hamper Induction of Interferon Signaling. Cell Rep (2021) 34(13):108916. doi: 10.1016/j.celrep.2021.108916
151. Li H, Bradley KC, Long JS, Frise R, Ashcroft JW, Hartgroves LC, et al. Internal Genes of a Highly Pathogenic H5N1 Influenza Virus Determine High Viral Replication in Myeloid Cells and Severe Outcome of Infection in Mice. PloS Pathog (2018) 14(1):e1006821. doi: 10.1371/journal.ppat.1006821
152. Hammer Q, Dunst J, Christ W, Picarazzi F, Wendorff M, Momayyezi P, et al. SARS-CoV-2 Nsp13 Encodes for an HLA-E-Stabilizing Peptide That Abrogates Inhibition of NKG2A-Expressing NK Cells. Cell Rep (2022) 38(10):110503. doi: 10.1016/j.celrep.2022.110503
153. Hammer Q, Rückert T, Borst EM, Dunst J, Haubner A, Durek P, et al. Peptide-Specific Recognition of Human Cytomegalovirus Strains Controls Adaptive Natural Killer Cells. Nat Immunol (2018) 19(5):453–63. doi: 10.1038/s41590-018-0082-6
154. Chen XY, Yan WH, Lin A, Xu HH, Zhang JG, Wang XX. The 14 Bp Deletion Polymorphisms in HLA-G Gene Play an Important Role in the Expression of Soluble HLA-G in Plasma. Tissue Antigens (2008) 72(4):335–41. doi: 10.1111/j.1399-0039.2008.01107.x
155. Josset L, Menachery VD, Gralinski LE, Agnihothram S, Sova P, Carter VS, et al. Cell Host Response to Infection With Novel Human Coronavirus EMC Predicts Potential Antivirals and Important Differences With SARS Coronavirus. mBio (2013) 4(3):e00165–13. doi: 10.1128/mBio.00165-13
156. Bortolotti D, Gentili V, Rizzo S, Schiuma G, Beltrami S, Spadaro S, et al. Increased sHLA-G Is Associated With Improved COVID-19 Outcome and Reduced Neutrophil Adhesion. Viruses (2021) 13(9):1855. doi: 10.3390/v13091855
157. Al-Bayatee NT, Ad'hiah AH. Soluble HLA-G is Upregulated in Serum of Patients With Severe COVID-19. Hum Immunol (2021) 82(10):726–32. doi: 10.1016/j.humimm.2021.07.007
158. Zhang S, Gan J, Chen BG, Zheng D, Zhang JG, Lin RH, et al. Dynamics of Peripheral Immune Cells and Their HLA-G and Receptor Expressions in a Patient Suffering From Critical COVID-19 Pneumonia to Convalescence. Clin Transl Immunol (2020) 9(5):e1128. doi: 10.1002/cti2.1128
159. Hou H, Li Y, Zhang P, Wu J, Shi L, Xu J, et al. Smoking Is Independently Associated With an Increased Risk for COVID-19 Mortality: A Systematic Review and Meta-Analysis Based on Adjusted Effect Estimates. Nicotine Tob Res (2021) 23(11):1947–51. doi: 10.1093/ntr/ntab112
160. Purkayastha A, Sen C, Garcia G, Langerman J, Shia DW, Meneses LK, et al. Direct Exposure to SARS-CoV-2 and Cigarette Smoke Increases Infection Severity and Alters the Stem Cell-Derived Airway Repair Response. Cell Stem Cell (2020) 27(6):869–75.e4. doi: 10.1016/j.stem.2020.11.010
161. Li J, Huang DQ, Zou B, Yang H, Hui WZ, Rui F, et al. Epidemiology of COVID-19: A Systematic Review and Meta-Analysis of Clinical Characteristics, Risk Factors, and Outcomes. J Med Virol (2021) 93(3):1449–58. doi: 10.1002/jmv.26424
162. d'Arminio Monforte A, Tavelli A, Bai F, Tomasoni D, Falcinella C, Castoldi R, et al. The Importance of Patients' Case-Mix for the Correct Interpretation of the Hospital Fatality Rate in COVID-19 Disease. Int J Infect Dis (2020) 100:67–74. doi: 10.1016/j.ijid.2020.09.037
163. D'Arminio Monforte A, Tavelli A, Bai F, Tomasoni D, Falcinella C, Castoldi R, et al. Declining Mortality Rate of Hospitalised Patients in the Second Wave of the COVID-19 Epidemics in Italy: Risk Factors and the Age-Specific Patterns. Life (Basel) 2021 11(9):979. doi: 10.3390/life11090979
164. Ghosh S, Klein RS. Sex Drives Dimorphic Immune Responses to Viral Infections. J Immunol (2017) 198(5):1782–90. doi: 10.4049/jimmunol.1601166
165. Channappanavar R, Fett C, Mack M, Ten Eyck PP, Meyerholz DK, Perlman S. Sex-Based Differences in Susceptibility to Severe Acute Respiratory Syndrome Coronavirus Infection. J Immunol (2017) 198(10):4046–53. doi: 10.4049/jimmunol.1601896
166. Bastard P, Rosen LB, Zhang Q, Michailidis E, Hoffmann H-H, Zhang Y, et al. Autoantibodies Against Type I IFNs in Patients With Life-Threatening COVID-19. Science (2020) 370(6515):eabd4585. doi: 10.1126/science.abd4585
167. Briceño O, Lissina A, Wanke K, Afonso G, von Braun A, Ragon K, et al. Reduced Naïve CD8(+) T-Cell Priming Efficacy in Elderly Adults. Aging Cell (2016) 15(1):14–21. doi: 10.1111/acel.12384
168. Márquez EJ, Chung CH, Marches R, Rossi RJ, Nehar-Belaid D, Eroglu A, et al. Sexual-Dimorphism in Human Immune System Aging. Nat Commun (2020) 11(1):751. doi: 10.1038/s41467-020-14396-9
169. Suryamohan K, Diwanji D, Stawiski EW, Gupta R, Miersch S, Liu J, et al. Human ACE2 Receptor Polymorphisms and Altered Susceptibility to SARS-CoV-2. Commun Biol (2021) 4(1):475. doi: 10.1038/s42003-021-02030-3
170. Zhou W, Wang W. Auto-Antibodies Against Type I IFNs are Associated With Severe COVID-19 Pneumonia. Signal Transduct Targeted Ther (2021) 6(1):96. doi: 10.1038/s41392-021-00514-6
171. Zhang Q, Bastard P, Liu Z, Le Pen J, Moncada-Velez M, Chen J, et al. Inborn Errors of Type I IFN Immunity in Patients With Life-Threatening COVID-19. Science (2020) 370(6515):eabd4570. doi: 10.1126/science.abd4570
172. Fallerini C, Daga S, Mantovani S, Benetti E, Picchiotti N, Francisci D, et al. Association of Toll-Like Receptor 7 Variants With Life-Threatening COVID-19 Disease in Males: Findings From a Nested Case-Control Study. Elife (2021), 10. doi: 10.7554/eLife.67569
173. van der Made CI, Simons A, Schuurs-Hoeijmakers J, van den Heuvel G, Mantere T, Kersten S, et al. Presence of Genetic Variants Among Young Men With Severe COVID-19. JAMA (2020) 324(7):663–73. doi: 10.1001/jama.2020.13719
174. Matthay MA, Wick KD. Corticosteroids, COVID-19 Pneumonia, and Acute Respiratory Distress Syndrome. J Clin Invest. (2020) 130(12):6218–21. doi: 10.1172/JCI143331
175. Nguyen A, David JK, Maden SK, Wood MA, Weeder BR, Nellore A, et al. Human Leukocyte Antigen Susceptibility Map for Severe Acute Respiratory Syndrome Coronavirus 2. J Virol (2020) 94(13):e00510–20. doi: 10.1128/JVI.00510-20
176. Lin M, Tseng HK, Trejaut JA, Lee HL, Loo JH, Chu CC, et al. Association of HLA Class I With Severe Acute Respiratory Syndrome Coronavirus Infection. BMC Med Genet (2003) 4:9. doi: 10.1186/1471-2350-4-9
177. Novelli A, Andreani M, Biancolella M, Liberatoscioli L, Passarelli C, Colona VL, et al. HLA Allele Frequencies and Susceptibility to COVID-19 in a Group of 99 Italian Patients. HLA (2020) 96(5):610–4. doi: 10.1111/tan.14047
178. Hajeer A, Jawdat D, Massadeh S, Aljawini N, Abedalthagafi MS, Arabi YM, et al. Association of KIR Gene Polymorphisms With COVID-19 Disease. Clin Immunol (2022) 234:108911. doi: 10.1016/j.clim.2021.108911
179. Rana BK, Flatt SW, Health DD, Pakiz B, Quintana EL, Natarajan L, et al. The IL-6 Gene Promoter SNP and Plasma IL-6 in Response to Diet Intervention. Nutrients (2017) 9(6):552. doi: 10.3390/nu9060552
180. Doyle WJ, Casselbrant ML, Li-Korotky HS, Doyle AP, Lo CY, Turner R, et al. The Interleukin 6 -174 C/C Genotype Predicts Greater Rhinovirus Illness. J Infect Dis (2010) 201(2):199–206. doi: 10.1086/649559
181. Nattermann J, Vogel M, Berg T, Danta M, Axel B, Mayr C, et al. Effect of the Interleukin-6 C174G Gene Polymorphism on Treatment of Acute and Chronic Hepatitis C in Human Immunodeficiency Virus Coinfected Patients. Hepatology (2007) 46(4):1016–25. doi: 10.1002/hep.21778
182. Kerget F, Kerget B. Frequency of Interleukin-6 Rs1800795 (-174G/C) and Rs1800797 (-597G/A) Polymorphisms in COVID-19 Patients in Turkey Who Develop Macrophage Activation Syndrome. Jpn J Infect Dis (2021) 74(6):543–8. doi: 10.7883/yoken.JJID.2021.046
183. Chen T, Lin YX, Zha Y, Sun Y, Tian J, Yang Z, et al. A Low-Producing Haplotype of Interleukin-6 Disrupting CTCF Binding Is Protective Against Severe COVID-19. mBio (2021) 12(5):e0137221. doi: 10.1128/mBio.01372-21
184. Gong B, Huang L, He Y, Xie W, Yin Y, Shi Y, et al. A Genetic Variant in IL-6 Lowering its Expression Is Protective for Critical Patients With COVID-19. Signal Transduct Target Ther (2022) 7(1):112. doi: 10.1038/s41392-022-00923-1
185. Saleh A, Sultan A, Elashry MA, Farag A, Mortada MI, Ghannam MA, et al. Association of TNF-α G-308 a Promoter Polymorphism With the Course and Outcome of COVID-19 Patients. Immunol Invest. (2022) 51(3):546–57. doi: 10.1080/08820139.2020.1851709
186. Nishida N, Sugiyama M, Kawai Y, Naka I, Iwamoto N, Suzuki T, et al. Genetic Association of IL17 and the Importance of ABO Blood Group Antigens in Saliva to COVID-19. Sci Rep (2022) 12(1):3854. doi: 10.1038/s41598-022-07856-3
187. Karakas Celik S, Cakmak Genc G, Dursun A. A Bioinformatic Approach to Investigating Cytokine Genes and Their Receptor Variants in Relation to COVID-19 Progression. Int J Immunogenet. (2021) 48(2):211–8. doi: 10.1111/iji.12522
188. Parant F, Bouloy J, Haesebaert J, Bendim'red L, Goldet K, Vanhems P, et al. Vitamin D and COVID-19 Severity in Hospitalized Older Patients: Potential Benefit of Prehospital Vitamin D Supplementation. Nutrients (2022) 14(8):1641. doi: 10.3390/nu14081641
189. Pereira M, Dantas Damascena A, Galvão Azevedo LM, de Almeida Oliveira T, da Mota Santana J. Vitamin D Deficiency Aggravates COVID-19: Systematic Review and Meta-Analysis. Crit Rev Food Sci Nutr (2022) 62(5):1308–16. doi: 10.1080/10408398.2020.1841090
190. Barassi A, Pezzilli R, Mondoni M, Rinaldo RF, Cozzolino M, et al. Vitamin D in Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2) Patients With non-Invasive Ventilation Support. Panminerva Med (2021). doi: 10.23736/S0031-0808.21.04277-4
191. Bassatne A, Basbous M, Chakhtoura M, El Zein O, Rahme M, El-Hajj Fuleihan G. The Link Between COVID-19 and VItamin D (VIVID): A Systematic Review and Meta-Analysis. Metabolism (2021) 119:154753. doi: 10.1016/j.metabol.2021.154753
192. Hayashi H, Okamatsu M, Ogasawara H, Tsugawa N, Isoda N, Matsuno K, et al. Oral Supplementation of the Vitamin D Metabolite 25(Oh)D. Nutrients. (2020) 12(7):2000. doi: 10.3390/nu12072000
193. Martineau AR, Jolliffe DA, Hooper RL, Greenberg L, Aloia JF, Bergman P, et al. Vitamin D Supplementation to Prevent Acute Respiratory Tract Infections: Systematic Review and Meta-Analysis of Individual Participant Data. BMJ (2017) 356:i6583. doi: 10.1136/bmj.i6583
194. Murai IH, Fernandes AL, Sales LP, Pinto AJ, Goessler KF, Duran CSC, et al. Effect of a Single High Dose of Vitamin D3 on Hospital Length of Stay in Patients With Moderate to Severe COVID-19: A Randomized Clinical Trial. JAMA (2021) 325(11):1053–60. doi: 10.1001/jama.2020.26848
195. Leaf DE, Ginde AA. Vitamin D3 to Treat COVID-19: Different Disease, Same Answer. JAMA (2021) 325(11):1047–8. doi: 10.1001/jama.2020.26850
196. Annweiler G, Corvaisier M, Gautier J, Dubée V, Legrand E, Sacco G, et al. Vitamin D Supplementation Associated to Better Survival in Hospitalized Frail Elderly COVID-19 Patients: The GERIA-COVID Quasi-Experimental Study. Nutrients (2020) 12(11):3377. doi: 10.3390/nu12113377
197. Fassio A, Adami G, Rossini M, Giollo A, Caimmi C, Bixio R, et al. Pharmacokinetics of Oral Cholecalciferol in Healthy Subjects With Vitamin D Deficiency: A Randomized Open-Label Study. Nutrients (2020) 12(6):1553. doi: 10.3390/nu12061553
198. Pesce E, Manfrini N, Cordiglieri C, Santi S, Bandera A, Gobbini A, et al. Exosomes Recovered From the Plasma of COVID-19 Patients Expose SARS-CoV-2 Spike-Derived Fragments and Contribute to the Adaptive Immune Response. Front Immunol (2021) 12:785941. doi: 10.3389/fimmu.2021.785941
199. Waterlow NR, van Leeuwen E, Davies NG, Flasche S, Eggo RM, Group CC-W. How Immunity From and Interaction With Seasonal Coronaviruses can Shape SARS-CoV-2 Epidemiology. Proc Natl Acad Sci USA (2021) 118(49):e2108395118. doi: 10.1073/pnas.2108395118
200. Le Bert N, Tan AT, Kunasegaran K, Tham CYL, Hafezi M, Chia A, et al. SARS-CoV-2-Specific T Cell Immunity in Cases of COVID-19 and SARS, and Uninfected Controls. Nature (2020) 584(7821):457–62. doi: 10.1038/s41586-020-2550-z
201. Braun J, Loyal L, Frentsch M, Wendisch D, Georg P, Kurth F, et al. SARS-CoV-2-Reactive T Cells in Healthy Donors and Patients With COVID-19. Nature (2020) 587(7833):270–4. doi: 10.1038/s41586-020-2598-9
202. Mateus J, Grifoni A, Tarke A, Sidney J, Ramirez SI, Dan JM, et al. Selective and Cross-Reactive SARS-CoV-2 T Cell Epitopes in Unexposed Humans. Science (2020) 370(6512):89–94. doi: 10.1126/science.abd3871
203. Song G, He WT, Callaghan S, Anzanello F, Huang D, Ricketts J, et al. Cross-Reactive Serum and Memory B-Cell Responses to Spike Protein in SARS-CoV-2 and Endemic Coronavirus Infection. Nat Commun (2021) 12(1):2938. doi: 10.1038/s41467-021-23074-3
204. Shrwani K, Sharma R, Krishnan M, Jones T, Mayora-Neto M, Cantoni D, et al. Detection of Serum Cross-Reactive Antibodies and Memory Response to SARS-CoV-2 in Prepandemic and Post-COVID-19 Convalescent Samples. J Infect Dis (2021) 224(8):1305–15. doi: 10.1093/infdis/jiab333
205. Brodin P. Why is COVID-19 So Mild in Children? Acta Paediatr (2020) 109(6):1082–3. doi: 10.1111/apa.15271
206. Gombar S, Bergquist T, Pejaver V, Hammarlund NE, Murugesan K, Mooney S, et al. SARS-CoV-2 Infection and COVID-19 Severity in Individuals With Prior Seasonal Coronavirus Infection. Diagn Microbiol Infect Dis (2021) 100(2):115338. doi: 10.1016/j.diagmicrobio.2021.115338
207. Anderson EM, Goodwin EC, Verma A, Arevalo CP, Bolton MJ, Weirick ME, et al. Seasonal Human Coronavirus Antibodies Are Boosted Upon SARS-CoV-2 Infection But Not Associated With Protection. Cell (2021) 184(7):1858–64. doi: 10.1016/j.cell.2021.02.010
208. Sagar M, Reifler K, Rossi M, Miller NS, Sinha P, White LF, et al. Recent Endemic Coronavirus Infection is Associated With Less-Severe COVID-19. J Clin Invest (2021) 131(1):e143380. doi: 10.1172/JCI143380
209. Rueca M, Fontana A, Bartolini B, Piselli P, Mazzarelli A, Copetti M, et al. Investigation of Nasal/Oropharyngeal Microbial Community of COVID-19 Patients by 16S rDNA Sequencing. Int J Environ Res Public Health (2021) 18(4):2174. doi: 10.3390/ijerph18042174
210. Zhang H, Ai JW, Yang W, Zhou X, He F, Xie S, et al. Metatranscriptomic Characterization of Coronavirus Disease 2019 Identified a Host Transcriptional Classifier Associated With Immune Signaling. Clin Infect Dis (2021) 73(3):376–85. doi: 10.1093/cid/ciaa663
211. Mostafa HH, Fissel JA, Fanelli B, Bergman Y, Gniazdowski V, Dadlani M, et al. Metagenomic Next-Generation Sequencing of Nasopharyngeal Specimens Collected From Confirmed and Suspect COVID-19 Patients. mBio (2020) 11(6):e01969–20. doi: 10.1128/mBio.01969-20
212. Ventero MP, Cuadrat RRC, Vidal I, Andrade BGN, Molina-Pardines C, Haro-Moreno JM, et al. Nasopharyngeal Microbial Communities of Patients Infected With SARS-CoV-2 That Developed COVID-19. Front Microbiol (2021) 12:637430. doi: 10.3389/fmicb.2021.637430
213. Ventero MP, Moreno-Perez O, Molina-Pardines C, Paytuví-Gallart A, Boix V, Escribano I, et al. Nasopharyngeal Microbiota as an Early Severity Biomarker in COVID-19 Hospitalised Patients. J Infect (2022) 84(3):329–36. doi: 10.1016/j.jinf.2021.12.030
214. Engen PA, Naqib A, Jennings C, Green SJ, Landay A, Keshavarzian A, et al. Nasopharyngeal Microbiota in SARS-CoV-2 Positive and Negative Patients. Biol Proced Online. (2021) 23(1):10. doi: 10.1186/s12575-021-00148-6
215. Merenstein C, Liang G, Whiteside SA, Cobián-Güemes AG, Merlino MS, Taylor LJ, et al. Signatures of COVID-19 Severity and Immune Response in the Respiratory Tract Microbiome. mBio (2021) 12(4):e0177721. doi: 10.1128/mBio.01777-21
216. Nardelli C, Gentile I, Setaro M, Di Domenico C, Pinchera B, Buonomo AR, et al. Nasopharyngeal Microbiome Signature in COVID-19 Positive Patients: Can We Definitively Get a Role to. Front Cell Infect Microbiol (2021) 11:625581. doi: 10.3389/fcimb.2021.625581
217. Yoneda S, Loeser B, Feng J, Dmytryk J, Qi F, Merritt J. Ubiquitous Sialometabolism Present Among Oral Fusobacteria. PloS One (2014) 9(6):e99263. doi: 10.1371/journal.pone.0099263
218. Morniroli D, Giannì ML, Consales A, Pietrasanta C, Mosca F. Human Sialome and Coronavirus Disease-2019 (COVID-19) Pandemic: An Understated Correlation? Front Immunol (2020) 11:1480. doi: 10.3389/fimmu.2020.01480
219. Nguyen L, McCord KA, Bui DT, Bouwman KM, Kitova EN, Elaish M, et al. Sialic Acid-Containing Glycolipids Mediate Binding and Viral Entry of SARS-CoV-2. Nat Chem Biol (2022) 18(1):81–90. doi: 10.1038/s41589-021-00924-1
220. Man WH, de Steenhuijsen Piters WA, Bogaert D. The Microbiota of the Respiratory Tract: Gatekeeper to Respiratory Health. Nat Rev Microbiol (2017) 15(5):259–70. doi: 10.1038/nrmicro.2017.14
221. Fan J, Li X, Gao Y, Zhou J, Wang S, Huang B, et al. The Lung Tissue Microbiota Features of 20 Deceased Patients With COVID-19. J Infect (2020) 81(3):e64–e7. doi: 10.1016/j.jinf.2020.06.047
222. Sulaiman I, Chung M, Angel L, Tsay JJ, Wu BG, Yeung ST, et al. Microbial Signatures in the Lower Airways of Mechanically Ventilated COVID-19 Patients Associated With Poor Clinical Outcome. Nat Microbiol (2021) 6(10):1245–58. doi: 10.1038/s41564-021-00961-5
223. Tsay JJ, Wu BG, Sulaiman I, Gershner K, Schluger R, Li Y, et al. Lower Airway Dysbiosis Affects Lung Cancer Progression. Cancer Discov (2021) 11(2):293–307. doi: 10.1158/2159-8290.CD-20-0263
224. Sulaiman I, Wu BG, Li Y, Scott AS, Malecha P, Scaglione B, et al. Evaluation of the Airway Microbiome in Nontuberculous Mycobacteria Disease. Eur Respir J (2018) 52(4):10. doi: 10.1183/13993003.00810-2018
225. Lloréns-Rico V, Gregory AC, Van Weyenbergh J, Jansen S, Van Buyten T, Qian J, et al. Clinical Practices Underlie COVID-19 Patient Respiratory Microbiome Composition and its Interactions With the Host. Nat Commun (2021) 12(1):6243. doi: 10.1038/s41467-021-26500-8
226. Camilleri M. Leaky Gut: Mechanisms, Measurement and Clinical Implications in Humans. Gut (2019) 68(8):1516–26. doi: 10.1136/gutjnl-2019-318427
227. Fasano A. All Disease Begins in the (Leaky) Gut: Role of Zonulin-Mediated Gut Permeability in the Pathogenesis of Some Chronic Inflammatory Diseases. F1000Res (2020) 9:9:F1000. doi: 10.12688/f1000research.20510.1
228. Marchetti G, Tincati C, Silvestri G. Microbial Translocation in the Pathogenesis of HIV Infection and AIDS. Clin Microbiol Rev (2013) 26(1):2–18. doi: 10.1128/CMR.00050-12
229. Giron LB, Dweep H, Yin X, Wang H, Damra M, Goldman AR, et al. Plasma Markers of Disrupted Gut Permeability in Severe COVID-19 Patients. Front Immunol (2021) 12:686240. doi: 10.3389/fimmu.2021.779064
230. Oliva A, Cammisotto V, Cangemi R, Ferro D, Miele MC, De Angelis M, et al. Low-Grade Endotoxemia and Thrombosis in COVID-19. Clin Transl Gastroenterol (2021) 12(6):e00348. doi: 10.14309/ctg.0000000000000348
231. Fasano A. Zonulin, Regulation of Tight Junctions, and Autoimmune Diseases. Ann NY Acad Sci (2012) 1258:25–33. doi: 10.1111/j.1749-6632.2012.06538.x
232. Clemente MG, De Virgiliis S, Kang JS, Macatagney R, Musu MP, Di Pierro MR, et al. Early Effects of Gliadin on Enterocyte Intracellular Signalling Involved in Intestinal Barrier Function. Gut (2003) 52(2):218–23. doi: 10.1136/gut.52.2.218
233. Oliva A, Miele MC, Di Timoteo F, De Angelis M, Mauro V, Aronica R, et al. Persistent Systemic Microbial Translocation and Intestinal Damage During Coronavirus Disease-19. Front Immunol (2021) 12:708149. doi: 10.3389/fimmu.2021.708149
234. Hoel H, Heggelund L, Reikvam DH, Stiksrud B, Ueland T, Michelsen AE, et al. Elevated Markers of Gut Leakage and Inflammasome Activation in COVID-19 Patients With Cardiac Involvement. J Intern Med (2021) 289(4):523–31. doi: 10.1111/joim.13178
235. Fragkos KC, Forbes A. Citrulline as a Marker of Intestinal Function and Absorption in Clinical Settings: A Systematic Review and Meta-Analysis. United Eur Gastroenterol J (2018) 6(2):181–91. doi: 10.1177/2050640617737632
236. Sun Z, Song ZG, Liu C, Tan S, Lin S, Zhu J, et al. Gut Microbiome Alterations and Gut Barrier Dysfunction Are Associated With Host Immune Homeostasis in COVID-19 Patients. BMC Med (2022) 20(1):24. doi: 10.1186/s12916-021-02212-0
237. Faraday N, Schunke K, Saleem S, Fu J, Wang B, Zhang J, et al. Cathepsin G-Dependent Modulation of Platelet Thrombus Formation In Vivo by Blood Neutrophils. PloS One (2013) 8(8):e71447. doi: 10.1371/journal.pone.0071447
238. Lamers MM, Beumer J, van der Vaart J, Knoops K, Puschhof J, Breugem TI, et al. SARS-CoV-2 Productively Infects Human Gut Enterocytes. Science (2020) 369(6499):50–4. doi: 10.1126/science.abc1669
239. Zhang H, Shao B, Dang Q, Chen Z, Zhou Q, Luo H, et al. Pathogenesis and Mechanism of Gastrointestinal Infection With COVID-19. Front Immunol (2021) 12:674074. doi: 10.3389/fimmu.2021.674074
240. Alsaffar H, Martino N, Garrett JP, Adam AP. Interleukin-6 Promotes a Sustained Loss of Endothelial Barrier Function via Janus Kinase-Mediated STAT3 Phosphorylation and De Novo Protein Synthesis. Am J Physiol Cell Physiol (2018) 314(5):C589–602. doi: 10.1152/ajpcell.00235.2017
241. Wurbel MA, McIntire MG, Dwyer P, Fiebiger E. CCL25/CCR9 Interactions Regulate Large Intestinal Inflammation in a Murine Model of Acute Colitis. PloS One (2011) 6(1):e16442. doi: 10.1371/journal.pone.0016442
242. Moreira-Rosário A, Marques C, Pinheiro H, Araújo JR, Ribeiro P, Rocha R, et al. Gut Microbiota Diversity and C-Reactive Protein Are Predictors of Disease Severity in COVID-19 Patients. Front Microbiol (2021) 12:705020. doi: 10.3389/fmicb.2021.705020
243. Amar J, Chabo C, Waget A, Klopp P, Vachoux C, Bermúdez-Humarán LG, et al. Intestinal Mucosal Adherence and Translocation of Commensal Bacteria at the Early Onset of Type 2 Diabetes: Molecular Mechanisms and Probiotic Treatment. EMBO Mol Med (2011) 3(9):559–72. doi: 10.1002/emmm.201100159
244. Massier L, Chakaroun R, Tabei S, Crane A, Didt KD, Fallmann J, et al. Adipose Tissue Derived Bacteria Are Associated With Inflammation in Obesity and Type 2 Diabetes. Gut (2020) 69(10):1796–806. doi: 10.1136/gutjnl-2019-320118
245. Patterson E, Ryan PM, Cryan JF, Dinan TG, Ross RP, Fitzgerald GF, et al. Gut Microbiota, Obesity and Diabetes. Postgrad Med J (2016) 92(1087):286–300. doi: 10.1136/postgradmedj-2015-133285
246. Gu S, Chen Y, Wu Z, Gao H, Lv L, Guo F, et al. Alterations of the Gut Microbiota in Patients With Coronavirus Disease 2019 or H1N1 Influenza. Clin Infect Dis (2020) 71(10):2669–78. doi: 10.1093/cid/ciaa709
247. Chen Y, Gu S, Lu H, Shi D, Guo J, Wu WR, et al. Six-Month Follow-Up of Gut Microbiota Richness in Patients With COVID-19. Gut (2022) 71(1):222–5. doi: 10.1136/gutjnl-2021-324090
248. Mazzarelli A, Giancola ML, Farina A, Marchioni L, Rueca M, Gruber CEM, et al. 16s rRNA Gene Sequencing of Rectal Swab in Patients Affected by COVID-19. PloS One (2021) 16(2):e0247041. doi: 10.1371/journal.pone.0247041
249. Tang L, Gu S, Gong Y, Li B, Lu H, Li Q, et al. Clinical Significance of the Correlation Between Changes in the Major Intestinal Bacteria Species and COVID-19 Severity. Eng (Beijing). (2020) 6(10):1178–84. doi: 10.1016/j.eng.2020.05.013
250. Yeoh YK, Zuo T, Lui GC, Zhang F, Liu Q, Li AY, et al. Gut Microbiota Composition Reflects Disease Severity and Dysfunctional Immune Responses in Patients With COVID-19. Gut (2021) 70(4):698–706. doi: 10.1136/gutjnl-2020-323020
251. Zuo T, Zhang F, Lui GCY, Yeoh YK, Li AYL, Zhan H, et al. Alterations in Gut Microbiota of Patients With COVID-19 During Time of Hospitalization. Gastroenterology (2020) 159(3):944–55.e8. doi: 10.1053/j.gastro.2020.05.048
252. Zuo T, Zhan H, Zhang F, Liu Q, Tso EYK, Lui GCY, et al. Alterations in Fecal Fungal Microbiome of Patients With COVID-19 During Time of Hospitalization Until Discharge. Gastroenterology (2020) 159(4):1302–10. doi: 10.1053/j.gastro.2020.06.048
253. Singer D, Camargo SM, Ramadan T, Schäfer M, Mariotta L, Herzog B, et al. Defective Intestinal Amino Acid Absorption in Ace2 Null Mice. Am J Physiol Gastrointest Liver Physiol (2012) 303(6):G686–95. doi: 10.1152/ajpgi.00140.2012
254. Hashimoto T, Perlot T, Rehman A, Trichereau J, Ishiguro H, Paolino M, et al. ACE2 Links Amino Acid Malnutrition to Microbial Ecology and Intestinal Inflammation. Nature (2012) 487(7408):477–81. doi: 10.1038/nature11228
255. Dang AT, Marsland BJ. Microbes, Metabolites, and the Gut-Lung Axis. Mucosal Immunol (2019) 12(4):843–50. doi: 10.1038/s41385-019-0160-6
256. Enaud R, Prevel R, Ciarlo E, Beaufils F, Wieërs G, Guery B, et al. The Gut-Lung Axis in Health and Respiratory Diseases: A Place for Inter-Organ and Inter-Kingdom Crosstalks. Front Cell Infect Microbiol (2020) 10:9. doi: 10.3389/fcimb.2020.00009
257. Sencio V, Machado MG, Trottein F. The Lung-Gut Axis During Viral Respiratory Infections: The Impact of Gut Dysbiosis on Secondary Disease Outcomes. Mucosal Immunol (2021) 14(2):296–304. doi: 10.1038/s41385-020-00361-8
258. de Oliveira GLV, Oliveira CNS, Pinzan CF, de Salis LVV, Cardoso CRB. Microbiota Modulation of the Gut-Lung Axis in COVID-19. Front Immunol (2021) 12:635471. doi: 10.3389/fimmu.2021.635471
Keywords: SARS-CoV-2, COVID-19, severity, immune dysregulation, biomarker, immunity, gut-lung axis, microbiota
Citation: Rovito R, Augello M, Ben-Haim A, Bono V, d’Arminio Monforte A and Marchetti G (2022) Hallmarks of Severe COVID-19 Pathogenesis: A Pas de Deux Between Viral and Host Factors. Front. Immunol. 13:912336. doi: 10.3389/fimmu.2022.912336
Received: 04 April 2022; Accepted: 02 May 2022;
Published: 10 June 2022.
Edited by:
Federica Pulvirenti, Accademic Hospital Policlinico Umberto, ItalyReviewed by:
Sudipta Kumar Sinha, Indian Institute of Technology Ropar, IndiaMarcos Lopez-Hoyos, Marqués de Valdecilla Health Research Institute (IDIVAL), Spain
Copyright © 2022 Rovito, Augello, Ben-Haim, Bono, d’Arminio Monforte and Marchetti. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Roberta Rovito, roberta.rovito@unimi.it