 Taif Shah
Taif Shah Zahir Shah2
Zahir Shah2 Nafeesa Yasmeen
Nafeesa Yasmeen Zulqarnain Baloch
Zulqarnain Baloch- 1Faculty of Life Science and Technology, Kunming University of Science and Technology, Kunming, China
- 2College of Veterinary Sciences, The University of Agriculture Peshawar, Peshawar, Pakistan
- 3College of Veterinary Medicine, South China Agricultural University, Guangzhou, China
Coronavirus disease-2019 (COVID-19), caused by SARS-CoV-2, is an infectious disease that poses severe threats to global public health and significant economic losses. The COVID-19 global burden is rapidly increasing, with over 246.53 million COVID-19 cases and 49.97 million deaths reported in the WHO 2021 report. People with compromised immunity, such as tuberculosis (TB) patients, are highly exposed to severe COVID-19. Both COVID-19 and TB diseases spread primarily through respiratory droplets from an infected person to a healthy person, which may cause pneumonia and cytokine storms, leading to severe respiratory disorders. The COVID-19-TB coinfection could be fatal, exacerbating the current COVID-19 pandemic apart from cellular immune deficiency, coagulation activation, myocardial infarction, and other organ dysfunction. This study aimed to assess the pathogenesis of SARS-CoV-2-Mycobacterium tuberculosis coinfections. We provide a brief overview of COVID19-TB coinfection and discuss SARS-CoV-2 host cellular receptors and pathogenesis. In addition, we discuss M. tuberculosis host cellular receptors and pathogenesis. Moreover, we highlight the impact of SARS-CoV-2 on TB patients and the pathological pathways that connect SARS-CoV-2 and M. tuberculosis infection. Further, we discuss the impact of BCG vaccination on SARS-CoV-2 cases coinfected with M. tuberculosis, as well as the diagnostic challenges associated with the coinfection.
Introduction
Coronaviruses (CoVs) are positive single-stranded RNA viruses that cause respiratory and gastrointestinal tract infections in mammals, such as humans, amphibians, and birds. They are important members of the Coronaviridae subfamily, Orthocoronavirinae. The International Committee on Taxonomy of Viruses has classified CoVs into four important genera: α-CoV, β-CoV, γ-CoV, and Δ-CoV, which cause human diseases. All the identified seven human CoVs (HCoVs), HCoV-NL-63, HCoV-229E, HCoV-OC-43, HCoV-HKU-1, SARS-CoV, MERS-CoV, and SARS-CoV-2 cause respiratory diseases in humans. The emergence of β-CoVs (SARS-CoV in 2002 and MERS-CoV in 2012) demonstrated the emerging possibility of new pathogenic CoVs in humans via zoonotic transmission (1, 2). The pathogenic CoV, SARS-CoV-2, causes coronavirus disease 2019 (COVID-19), first reported in Wuhan, China, in late 2019, resulting in severe global public health issues and substantial economic losses (3–5). Bats act as a reservoir for CoVs; therefore, it is speculated that the SARS-CoV-2 originated in horseshoe bats before transmitting to humans via an unknown intermediate host (6). Because of its high contagiousness and the presence of asymptomatic human carriers, this pathogen spreads rapidly across the globe, claiming human lives and obstructing social and economic activity (4, 7). According to the WHO 2021 report, over 246.53 million COVID-19 cases and 49.97 million deaths are associated with this disease worldwide (8). The United States has the highest COVID-19 prevalence, with 45.63 million cases and a death rate of approximately 0.74 million, followed by India with 34.29 million cases and 0.46 million deaths, Brazil with 21.80 million cases and 0.61 million deaths, and the United Kingdom with 9.02 million cases and approximately 0.14 million deaths (8).
It is true that the COVID-19 pandemic has dominated both the printed and electronic media and scientific literature. However, other infectious diseases such as tuberculosis (TB), a contagious and airborne bacterial infection caused by Mycobacterium tuberculosis, should not be neglected. TB has been an ancient threat to public health since the prehistoric ages and is one of the top 10 causes of human death (9). Further, TB is the second leading infection after COVID-19. According to the WHO 2021 report, nine out of ten people infected with TB belong to the 30 countries with the highest TB burden, including India, China, Indonesia, the Philippines, Pakistan, Nigeria, etc. (10). WHO data from more than 200 countries shows that the number of people who died from TB increased from 1.4 million to 1.5 million (2019–2020). Aside from the increased global death rate, the number of newly diagnosed TB cases reported to the local governments fell from 7.1 million to 5.8 million in the same period, which is an 18% decrease from 2012, with 1.3 million TB deaths (9). This report further stated that among the 9.9 million TB infections last year, 4.1 million were either undiagnosed or not reported to the state in 2020 (9). TB affects males and females of all ages, with adult men bearing the greatest burden, accounting for approximately 56% of all TB cases in 2020, while women and children accounted for 33% and 11%. A higher proportion of TB cases among adult men consistently shows that TB affects adult men more than females, which is most likely due to men’s having a higher detection and reporting rate than females (9).
People affected by chronic respiratory, metabolic, or cardiovascular diseases carry a higher risk of severe COVID-19 infection (11, 12). The COVID-19 signs and symptoms are almost identical to those of TB and other influenza infections. Therefore, SARS-CoV-2 coinfection with other viruses (13), bacteria, and fungi (14, 15) frequently impedes COVID-19 prevention, diagnosis, and control strategies. Both COVID-19 and TB target the human respiratory tract, particularly the lungs, and are transmitted via aerosol droplets from an infected person to a healthy one. Evidence shows that COVID-19 patients coinfected with TB (COVID-19-TB) have a higher risk of death than a single pathogen (9, 15). The COVID-19 pandemic has already challenged public health care systems and impaired TB services for TB patients, increasing their morbidity and mortality globally. M. tuberculosis interacts with other pathogens such as HIV in coinfection to impair the host’s defenses (16). However, the synergism between SARS-CoV-2 and M. tuberculosis is yet unclear. Thus, a comprehensive investigation of COVID-19-TB coinfection’s impact, synergism, and pathogenesis is of great clinical importance (8, 9, 11). There have been few clinical studies on COVID-19-TB coinfection (15, 17, 18), and some of the published case reports and cohort studies have serious flaws. First, the sample sizes are small, and most of the studies were carried out in low-TB-burden countries with poorly described clinical features. The second problem is a lack of understanding of previous comorbidities like diabetes, hypertension, obesity, etc. Sometimes, it is difficult to confirm whether TB was known before or after the COVID-19 diagnosis. Despite these flaws, studies have concluded that active TB makes a patient more vulnerable to severe COVID-19. However, it is worth noting that various social conditions, history of diseases, comorbidities, and limited access to healthcare influence the prognosis of TB patients to COVID-19 coinfection.
The current study aimed to discuss the impact of COVID-19-TB coinfection, the SARS-CoV-2 host cellular receptors (ACE2, auxiliary, and alternative to ACE2 receptors), and pathogenesis. We also discuss M. tuberculosis host cellular receptors (TLRs, NLRs, CLRs, scavenger receptors) and pathogenesis. In addition, we highlight the impact of COVID-19 on TB patients and the pathological pathways that link SARS-CoV-2 and M. tuberculosis coinfection. Further, we discuss the impact of BCG vaccination on coinfection and the diagnostic problems associated with COVID-19-TB coinfection.
SARS-CoV-2 and M. tuberculosis Host Cellular Receptors and Pathogenesis
SARS-CoV-2 ACE2, Auxiliary and Alternative to ACE2 SARS-CoV-2 Receptors and Pathogenesis
The SARS-CoV-2 S protein comprises two subunits, S1 and S2, that aid in viral attachment and entry into the host cell cytoplasm. Subunit S1 has a receptor-binding domain that promotes host receptor binding, whereas subunit S2 directly fuses the virus with the host membrane (7, 19). The S1 attaches to the angiotensin-converting enzyme 2 (ACE2) peptidase on the target cell surface and invades via clathrin-mediated endocytosis (20–22). Infection of susceptible cell lines (19, 23) and transgenic mice expressing human ACE2 (24) demonstrated that the ACE2 protein in human cells primarily acts as a CoV receptor. Several other researchers have confirmed these findings and reported that ACE2 depleted Vero-E6 inhibited SARS-CoV-2 infection (25), Huh-7 (hepatocyte cell line) (25, 26), Caco-2 (immortal human cell line) (27), and Calu-3 (pulmonary cancerous cell line) (28, 29). On the other hand, SARS-CoV-2 can infect ACE2-deficient cells (30), and the reason behind this infection might be mutations in the spike protein (31). ACE2 exists in two forms, i.e., cell membrane-bound and soluble, which are released after cleavage by the metallopeptidase domain 17 (ADAM17). According to a recent finding, soluble ACE2 forms a complex with the S of SARS-CoV-2 and vasopressin proteins to promote infection (32). SARS-CoV-2 first infects respiratory epithelial and alveolar cells (33), followed by infecting and replicating in ciliated mucus-secreting bronchial epithelial cells (also called type-2 pneumocytes of the lungs) (34), macrophages (35), intestines, heart, kidneys, blood, liver, and brain (7). COVID-19 infection increased ACE2 expression (threefold) in respiratory epithelial cells (36), owing to the fact that the ACE2 gene promotes interferon (IFN) production (36). Despite discovering SARS-CoV-2 genomic mRNA in airway epithelial cells that express ACE2 (36, 37), a correlation between SARS-CoV-2 infection and ACE2 expression at a single-cell level needs further investigation to reveal the viral pathogenesis mechanism. Because SARS-CoV-2 damages multiple organs, it is also possible that other host cellular factors aid in virus replication and transmission in vivo. Several SARS-CoV-2 receptors act as cofactors, allowing the virus to enter the host cell cytoplasm. For example, heparan-sulfate polysaccharide expressed on host cell surfaces binds to SARS-CoV-2 spike protein (38, 39), indicating that heparan-sulfate depletion may reduce SARS-CoV-2 attachment and infection (39). Like other influenza viruses, SARS-CoV-2 may use heparan-sulfate to bind to the host surface, increasing viral interactions with other host cellular receptors for entry and pathogenesis (40). This is because many influenza viruses attach to heparan-sulfate due to in vitro adaptation to cell culture. Future research should determine whether SARS-CoV-2 binding is a natural viral capability to heparan-sulfate on the host cell. In addition to heparan-sulfate, scavenger receptor B-1 is another host cellular receptor that facilitates the uptake of high-density lipoprotein (HDL). According to the evidence, SARS-CoV-2 S1 shows an affinity for host HDL, and the increased HDL in cells increases viral attachment and infiltration (41). Increased HDL levels may overexpress the SCARB1 gene (which encodes scavenger receptor B1), resulting in severe SARS-CoV-2 infection. Conversely, SCARB1 knockdown cells showed reduced infection, indicating that scavenger receptor B-1 facilitates cellular uptake of HDL-bounded SARS-CoV-2. It has been confirmed that SARS-CoV-2 is distinguished from SARS-CoV (42) due to the presence of a unique furin cleavage site at the S1 and S2 domains. Two separate studies (43, 44) found that the polybasic motif at the S1/S2 C terminal binds directly to the neuropilin-1 receptor to promote SARS-CoV-2 pathogenesis. It has been reported that scavenger receptor B-1 and neuropilin-1 receptors may overexpress ACE2 to promote viral entry into the host cell cytoplasm (41, 43, 44), suggesting that scavenger receptor B-1 and neuropilin-1 act as cofactors that enhance SARS-CoV-2 attachment and penetration via ACE2 receptors. In the absence of ACE2, several other potential candidate receptors allow SARS-CoV-2 attachment and penetration into host cells. In a study, receptors such as host cellular tyrosine-protein kinase (30), low-density lipoprotein, and C-type lectin receptors (CLRs) (45) showed strong affinity for the SARS-CoV-2 spike. Depleting these host receptors may reduce infection, whereas overexpression of these proteins in ACE2-knockout cell lines induced SARS-CoV-2 infection, indicating that these cellular receptors and ACE2 have a similar function. Basigin (BSG), also known as CD147 or EMMPRIN, is an alternative widely expressed putative cellular receptor on the human cell surface for SARS-CoV-2 attachment and entry (46), though a study failed to confirm this finding (47). A study reported that human BSG expression in mice allowed for severe COVID-19 infection because SARS-CoV-2 could not infect mice via ACE2 (23), implying that BSG may be an alternative to ACE2. SARS-CoV-2 invades and enters the human lungs and avoids detection by the host immune system, infecting and replicating in ciliated mucus-secreting epithelial type-2 pneumocytes (34) and alveolar macrophages (35).
M. tuberculosis TLRs, NLRs, CLRs, and Scavenger Receptors and Pathogenesis
Like SARS-CoV-2, M. tuberculosis invades and replicates in ciliated mucus-secreting epithelial type-2 pneumocytes and alveolar macrophages (48, 49) using host pattern recognition receptors (PRRs), complement receptors (CRs), toll-like receptors (TLRs), CD14 receptors, dendritic cell-specific ICAM-3-grabbing-non-integrin-1 (DC-SIGN), Fcγ receptors, mannose receptors, and scavenger receptors (49, 50). Alveolar macrophages phagocytosed M. tuberculosis before being transferred to lysosomes for destruction (48, 49, 51). A successful mycobacterial infection depends on its encounter with the host cell factors, particularly alveolar macrophages. Mycobacteria are gram-positive and have been classified as acid-fast bacilli due to the presence of a lipid-rich cell wall. The physical features of pathogens and host factors significantly affect mycobacterial pathogenesis (49, 52). Many immunological peculiarities are attributed to glycolipid layers in the mycobacterial wall, including lipoarabinomannan and mycolic acid. M. tuberculosis uses various surface molecules (52–54) to bind host surface receptors, including surfactant proteins, Fcγ receptors, CRs, CD14 receptors, and macrophage mannose receptors (51). Several Fcγ receptors bind immunoglobulin-G-opsonized M. tuberculosis, allowing the bacteria to enter the host cell, promoting phagosome-lysosome fusion and reactive oxygen intermediates (55, 56). One of the most intriguing aspects of M. tuberculosis pathogenesis is the fate of macrophages. M. tuberculosis communicates with macrophages via its various receptors. In vivo responses of macrophages to M. tuberculosis receptors are poorly understood, while in vitro studies show that specific receptors may influence the macrophage response. For example, Fcγ receptor binding produces reactive oxygen intermediates and phagosome-lysosome fusion, whereas M. tuberculosis binding via CR3 or mannose receptor inhibits the respiratory burst and prevents phagosome maturation almost identical to the endosome (57), indicating that mycobacteria can interact and penetrate the target cell cytoplasm via several receptors. After inhaling M. tuberculosis, the bacterium activates the alternative pathway and is opsonized by the CR3 within the alveolus, enhancing phagocytosis by alveolar macrophage CR1 or CR3 protein (58, 59). Studies revealed that CR3-deficient mice infected with M. tuberculosis showed similar bacteria burden, granuloma formation, and survival rate (60, 61), demonstrating CR3 redundancy. In another study, cholesterol was essential for mycobacteria entry into alveolar macrophages because cholesterol-depleted cells inhibited bacterial infection, demonstrating that cholesterol probably mediates the phagosome-associated tryptophan-aspartate proteins, preventing phagosome maturation into phagolysosome (56, 62).
Host surface PRRs, including TLRs, CLRs, NOD-like receptors (NLRs), scavenger receptors (such as MARCO, CD36, and MSR1), aryl hydrocarbon receptors, CD14, and AIM2-like receptors, recognize M. tuberculosis pathogen-associated molecular patterns (PAMPs) during phagocytosis (63). Much research has been done on mycobacteria-derived PAMPs with TLRs on the host cell surface. In TB patients, TLRs are responsible for recruiting MYD88, TIR adaptor-inducing interferon (TRIF), Toll/IL-1, and TRIF. TLRs have been classified into two groups: endosome localized (TLR3, TLR9, TLR7, TLR8) or surface localized (TLR1, TLR6, TLR2, TLR5, TLR4, TLR10), which help in the recognition of bacterial surface antigens, particularly the LPS layer (64). After pathogen infection, TLR recruits different adapter molecules to relay signals for activating signaling pathways like NF-κB, MAPK, PI3K, and Akt, further inducing pro-inflammatory cytokines and type-1 IFN. TLR expression and activation are useful indicators of the immune response in TB patients (65). Mice lacking the MYD88 signaling adaptor molecule are extremely vulnerable to M. tuberculosis infection (66), implying the importance of MYD88 against M. tuberculosis infection (66). In another experiment, TLR2 deficient mice showed defective granuloma formation after M. tuberculosis infection and were much more susceptible to infection than WT mice. People with TLR2 genetic polymorphisms were more susceptible to pulmonary tuberculosis (67), whereas TLR4, TLR7, and TLR8 polymorphisms were more susceptible and severe to TB in Asians, particularly Indians (68), implying that blocking phagosome-lysosome fusion increased phagocytosis in addition to a weak immune response (69). Mice lacking TLR9 expression die soon after M. tuberculosis infection (70). Conversely, a study found that only MyD88 (not TLR2/4/9) was a key factor for macrophage activation in TB patients (71). A study also discovered that cytokines could be produced during M. tuberculosis infection using TLR or caspase-1 (71). M. tuberculosis expresses various lipoproteins to recognize TLRs on the host cell. For example, lipoprotein lpqH recognizes TLR2 (72), while TLR3 promotes an IL10 response via the PI3K/AKT signaling pathway in TB patients (73). The leucine-responsive regulatory protein of M. tuberculosis regulates the TLR2-mediated PI3K/AKT pathway, inhibiting inflammatory cytokine production and de-regulating macrophage antigen presentation (74). Previously, it was discovered that the M. tuberculosis-secreted proteins Mce3E and PtpA target MAPK and NF-κB pathways to modulate TLR signaling (75, 76). Mycobacterial phagocytosis into macrophages was also promoted by activating the signaling pathways ERK and MAPK, which are likely to be involved in mycobacterial pathogenesis (77). More research is required to investigate the potential molecular interactions of TLR activating signaling pathways with M. tuberculosis receptors. Apart from TLRs, several intracellular NLRs, i.e., NOD1, NLRC4, NOD2, and NLRP3, recognize bacterial components and activate inflammatory pathways against invading pathogens (78). In response, M. tuberculosis can escape from phagosomes via an ESAT-6 system-1 (ESX-1)-associated pathway (79). In contrast, a study found that NOD2 deficient mice increased susceptibility to M. tuberculosis infection (80). Simultaneously, activation of NOD2 in alveolar macrophages with muramyl-dipeptide derived from the mycobacterial cell wall prevents bacterial movement of autophagy-related polypeptides to the autophagosome, implying the importance of PRR in autophagy (81). In another study, three NOD2 gene polymorphisms in African American people were linked to TB susceptibility (82). It has also been reported that the NOD2 single-nucleotide polymorphism showed increased susceptibility to TB (83). Many recent studies have looked into the roles of other NLRs, particularly the NLRP3 inflammasome, during M. tuberculosis infection. M. tuberculosis ESAT-6 system activates the NLRP3 inflammasome in macrophages, resulting in the production of IL1-β and pyroptosis (84). The NLRP3 gene, an adaptor protein, and caspase-1 mediate immune responses against M. tuberculosis infection in mice. In the presence of NLRP3, caspase-1, and PYCARD, M. tuberculosis-infected macrophages showed induced IL1-β, indicating that IL1-β production makes mice more susceptible to M. tuberculosis in vitro. However, in vivo, NLRP3, Casp-1 depleted, and WT mice produced the same amount of IL1-β during M. tuberculosis infection (85). Thus, M. tuberculosis may activate the inflammasomes to promote persistent TB. Despite discovering the preventive role of novel NLRs against M. tuberculosis, more research is needed to explore the NLR-induced signaling pathways. Further, it is unknown whether M. tuberculosis effectors interact with NLR domains (e.g., the PYD domain) to regulate signaling pathways. In the future, it will be fascinating to investigate the potential underlying regulatory mechanisms in host cells that control NLR signaling pathways in TB patients. Based on phylogeny and structure, CLRs have been classified into 17 groups, including collectins, endocytic, selectin receptors, proteoglycans, and phagocytic receptors. The CLRs identified include mannose receptor, mincle, DC-SIGN, dectin-1-3/macrophage, dendritic cell immune receptor (DCIR), and collectin CL-L1, CL-K1 are all critical factors for mycobacterial invasion. Important immune regulators are CLRs that bind to M. tuberculosis carbohydrates, lipids, or proteins. In addition, CLRs directly recognize M. tuberculosis mannose-capped lipoarabinomannan (ManLAM) and cord factors and are essential immune modulators in TB patients (86), apart from promoting mycobacterial phagocytosis by alveolar macrophages (87). Mycobacteria that target DC-SIGN generate intracellular signals to stimulate dendritic cells to produce IL10, implying that the mycobacteria-targeted DC-SIGN suppresses the host immune response during infection (88). Trehalose 6, 6′-dimycolate, and cord factor are lipid-rich cell wall components in virulent mycobacteria, which act as a macrophage-inducible CLR in humans (89). Evidence shows that Dectin1 and TLR2 can regulate pro-inflammatory macrophages against invading mycobacteria (90). In another study, Dectin1 contributes to M. tuberculosis susceptibility in mice (91). In contrast, Dectin2 acts as a receptor for M. tuberculosis ManLAM to prevent infection (92). Surprisingly, both Dectin-3 and Mincle are required for the host immune response, with Dectin-3 stimulating Mincle expression and thus amplifying the mincle-mediated immune response against M. tuberculosis (93, 94). Collectin CL-LK was a novel soluble CLR capable of binding to M. tuberculosis ManLAM (95). In addition, Dectin-2 and DCIR modulate immune responses against M. tuberculosis by sustaining IFN-1 signaling in dendritic cells (96). More research on human CLRs recognizing M. tuberculosis components and their interactions with other immune cells is needed to gain insight into M. tuberculosis-induced innate immunity.
Impact of SARS-CoV-2 Pandemic on TB Patients
The Global TB Network reported COVID-19-TB coinfection in several countries (15, 97, 98). Both pulmonary and extrapulmonary TB (disseminated TB: evidence of TB in bone, central nervous system, gastrointestinal and genitourinary tracts, larynx, lymph nodes, peritoneal, pleural, spinal cord, etc.) patients have been found coinfected with COVID-19 (15, 99). Eight studies reported 80 COVID-19-TB coinfected humans from nine countries, with Italy reporting the highest cases (51%) of active pulmonary TB (100). Some of the COVID-19-TB coinfections reported in different countries are shown in Table 1. Belgium, Spain, Brazil, Singapore, France, Switzerland, Italy, India, Russia, and China reported COVID-19-TB coinfections. Due to the recent COVID-19 rise, we can expect increased COVID-19-TB infections in people of all races, ages, and genders. According to clinical evidence, COVID-19 occurs with or without TB, i.e., before, after, or during concurrent TB (126). COVID-19-TB coinfection was more common in migrants and men, i.e., > 80% of male cases (98, 127). Both COVID-19 and TB are highly infectious diseases (128); for example, a single COVID-19 patient may infect approximately 2.5 people in only five days, whereas an active pulmonary TB patient may infect upto 15 people per year (may be due to long incubation period) (129). The reported predisposing factors in COVID-19-TB patients were comparable to those in TB patients without COVID-19. Diabetes, kidney failure, liver disease, and smoking are comorbidities for COVID-19-TB infections (15). A study found that 41% of the COVID-19-TB patients were smokers, 31% were unemployed, and 20% had a history of alcohol use (98, 127). According to human studies, COVID-19 in TB patients is more common, particularly in high-TB burden countries, such as India, Vietnam, etc. (130), while Brazil and Argentina, with high COVID-19 cases, experienced varying degrees of healthcare system disruption (131). Similarly, COVID-19-TB cases within a country differ depending on people’s socioeconomic status and disease preventive measures. In addition, several COVID-19-TB risk factors, such as age, malnutrition, and comorbidities such as pre-respiratory disorders, diabetes, etc., have been identified (126). Studies also revealed that older and younger people, with or without pre-existing clinical complications, a previous history of TB or lung injury, are at risk of COVID-19 infection (132). A clinical study revealed a similar dysregulated immunological response in COVID-19-TB patients, implying that coinfection poses a dual risk of disease worsening (132). In addition, poor hygiene, overcrowding, and other autoimmune diseases are risk factors for developing both diseases (133, 134). A study that developed a model of pathogen dissemination showed that high-risk influenza patients are at high risk of M. tuberculosis infection (135). The WHO reported that the COVID-19 social and economic losses could be more severe in the highest TB burden regions (136). Another serious issue of COVID-19 is TB nature, patient’s long treatment (usually 6–24 months), poor treatment outcomes (i.e., drug-resistant TB), drug discontinuation, and other stringent pandemic isolation measures (137). Treatment discontinuation risks and other issues confronting TB clinical trials in COVID-19 cases have raised concerns (138). To address these issues, self-administered anti-TB therapy monitoring with digital technology or video-assisted administration has been recommended (139). Hypoxemia, respiratory disorders, glucose abnormalities, prolonged hospitalization, superimposed bacterial infection, and multiple organ failures have been reported in COVID-19-TB cases (98, 140).
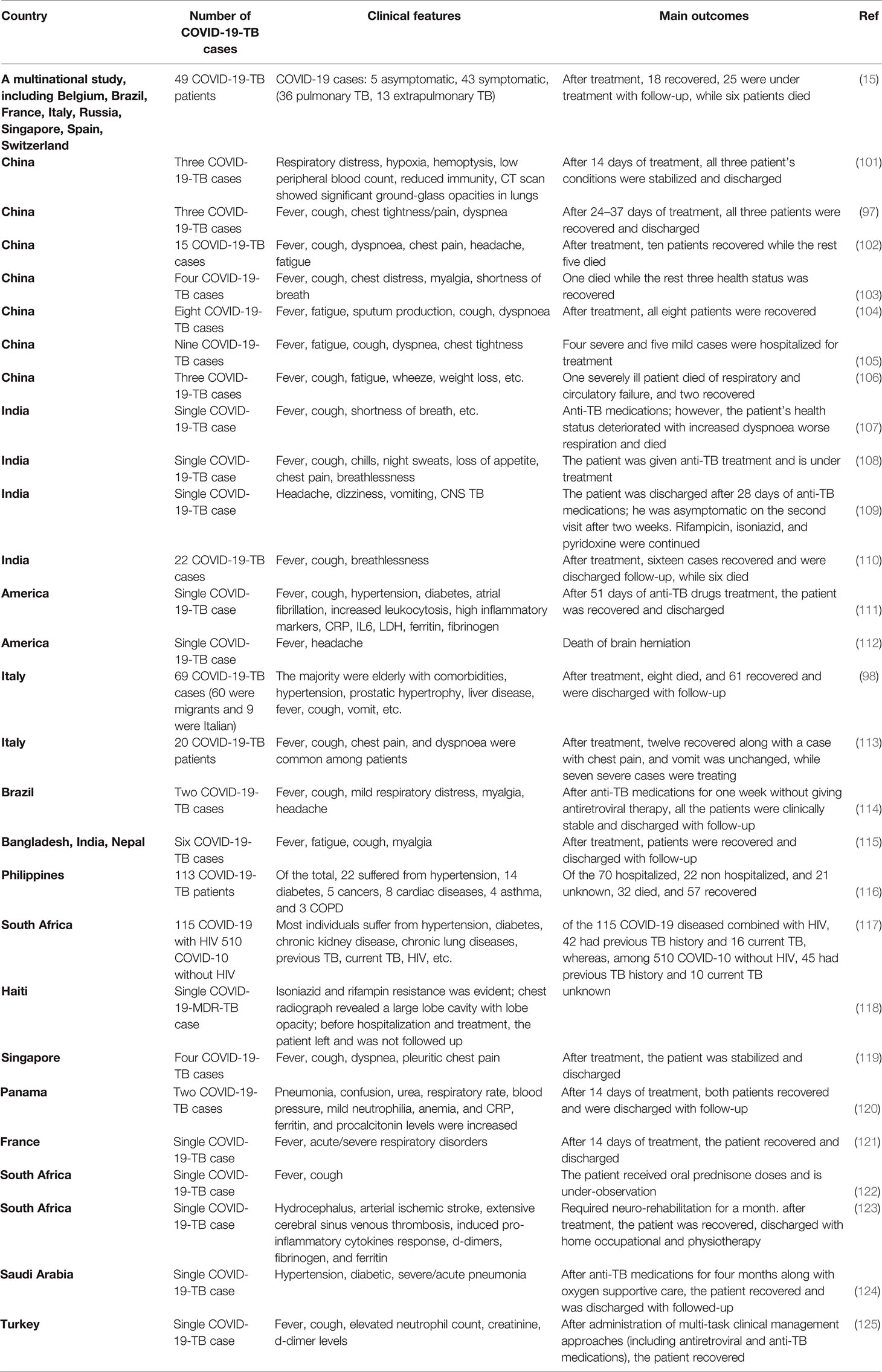
Table 1 Studies assessing the impact of the COVID-19 pandemic on TB patients.
COVID-19 and TB are the two leading causes of death among respiratory diseases (12, 141). In a cohort of 49 COVID-19-TB cases from eight countries, i.e., Brazil, Singapore, Russia, Spain, Switzerland, Belgium, France, and Italy (15), 26 patients were detected as TB positive before COVID-19, and COVID-19 was diagnosed in 14 patients before TB treatment. Of the total, 42 patients had active pulmonary TB, and seven developed TB complications. Similar findings were confirmed by a study conducted in India (110). In a similar study conducted in Sondalo Hospital, Italy, clinical, laboratory, and radiological characteristics showed that SARS-CoV-2 infected 20 of the 24 hospitalized TB patients. Four patients received only hydroxychloroquine at the time of hospitalization. A single TB case later coinfected with COVID-19 died, whereas the remaining 19 people experienced severe medical outcomes such as pneumonia (113). In a case-control study from Shenyang Primary-Care Hospital, China, 13 out of 36 COVID-19 people tested positive for TB (142), implying that M. tuberculosis infection increases host susceptibility to SARS-CoV-2 infections. Therefore, routine TB coinfection diagnosis in COVID-19 cases is advised. Moreover, a large-scale clinical research study should evaluate the negative impact of severe TB on COVID-19 coinfection. The mortality rate was higher in aged people (> 70 years old) and COVID-19-TB patients, whereas the migrants had lower mortality rates, which may be attributed to their younger age and the absence of clinical comorbidities (15, 98). COVID-19 is associated with higher mortality among TB patients (98, 140) who acquired COVID-19 through nosocomial transmission. According to a meta-analysis, COVID-19 coinfection increases the risk of TB patient death (1.4 times) (143). The findings of 69 patients from eight countries (98) suggest a COVID-19-TB fatality rate of 11.6% and 14.3% (15). Mortality is likely to occur in old COVID-19-TB patients having medical comorbidities. For example, 5% of the old cases with comorbidities in Italy (113). According to evidence, migrants had lower mortality rates and comorbidities, most likely due to their younger age (144). However, patients with severe TB or MDR-TB could experience increased mortality, particularly younger individuals (144). A longitudinal cohort study looked at the risk of COVID-19 patient deaths when they were also infected with TB, and it was discovered that the COVID-19-TB death rate was higher (2.17 times) than the single COVID-19 death rate. In comparison, nearly 25% of COVID-19-TB patients recovered, which was lower than single COVID-19-infected individuals (116), highlighting the need to prioritize routine TB testing for COVID-19 patients. To understand this coinfection interaction, the Global TB Network and WHO jointly launched a study on TB and COVID-19 patients. COVID-19-TB clinical comorbidities have been described in 597 cases studied from 132 centers in 36 different countries/states (145).
Studies have also elucidated the molecular interactions of the host with COVID-19-TB, and it was found that this viral-bacterial coinfection worsens respiratory disorders (12, 141). Clinical manifestations such as hemoptysis, cough, weakness, and fever are common among people suffering from COVID-19-TB, making an accurate diagnosis difficult. As COVID-19-TB causes an unbalanced inflammatory response in severely infected individuals; therefore, understanding the molecular interactions between SARS-CoV-2-M. tuberculosis and their host will be critical for developing anti-COVID-19-TB therapeutic agents (141). A study also examined the immunological status in COVID-19-TB cases, i.e., increased C-reactive protein (CRP), d-dimers, ferritin, neutrophils, lymphocytes, cytokine storm, and chemokines have been linked to COVID-19 severity and patient mortality (146, 147). The bronchoalveolar fluid from severe/mild COVID-19 patients showed increased chemokine (C-C motif ligand-2: CCL-2) and CCL-7, attracting the CCL2-associated monocytes. In addition, severe COVID-19 patients revealed increased mononuclear phagocyte counts (accounting for 80% of total bronchoalveolar fluid cells) compared to 60% in mild cases and 40% in healthy controls (147, 148). Phagocyte activation can induce pro-inflammatory cytokines (cytokine storm), which increases alveolar epithelial infection (149). Increased pro-inflammatory cytokine, IL6, IL1-β, and IP10 expression in COVID-19 patients promotes neutrophil proliferation and infiltration into the lung for injury (150, 151). Severe TB patients showed increased pulmonary immune cell (macrophages, dendritic cells, etc.) responses. These immune cells overproduce cytokines like IL1, IL10, IL18, IFNα, and IL6 (152, 153). In brief, COVID-19 coinfection with TB promotes cytokine storms that cause multiple organ injuries, particularly to the lungs, heart, or liver (5, 150, 151). These two pathogens induce cytokine storms with similar features but different magnitudes and outcomes. For example, M. tuberculosis infection elicits a lower immune response than SARS-CoV-2. In addition, cytokine storm causes respiratory distress in COVID-19 patients, whereas it causes long-term organ damage/failure in chronic cases. In addition to cytokine storm, lymphocytopenia (decreased lymphocyte counts) is another notable feature of the immune response in COVID-19 and TB patients. Lymphocyte counts of less than 1.5×109 per liter have been observed in severe COVID-19 cases (154, 155). After penetration, SARS-CoV-2 may directly invade lymphocytes and destroy them. The damaged lymphatic system in severe COVID-19 patients further promotes lymphocytopenia (156). Increased pro-inflammatory cytokine levels of IL6 and TNFα can also cause lymphocytopenia in severe COVID-19 patients (156). Moreover, T lymphocyte dysregulation was observed in TB cases. For example, a study also reported that the host defense response is dependent on CD4+ cell responses against M. tuberculosis infection (157, 158). A low CD4+ cell count increases the risk of TB reactivation, resulting in severe radiological lesions or patient death (48). Immunosuppression caused by CD4+ cell depletion in SARS-CoV-2-TB cases may increase the disease severity and patient morbidity. Therefore, prolonged clinical consideration should be given to all the above concerns.
Pathological Pathways That Connect SARS-CoV-2 and M. tuberculosis Coinfection
Since the first SARS-CoV-2 outbreak in Wuhan city (133), there has been little data on M. tuberculosis coinfection, probably due to M. tuberculosis’s prolonged incubation from exposure to symptoms (134, 159). Based on the population study findings and the published data about the etiology of COVID-19 and TB, it is possible to discuss some aspects of COVID-19-TB coinfection (160). Both COVID-19 and TB have airborne transmission primarily affecting the lungs and share the same social determinants and symptoms (Figure 1). However, SARS-CoV-2 and M. tuberculosis present significant differences in their pathogenesis, and learning about their interactions with the host may aid in the development of new COVID-19-TB treatment strategies. SARS-CoV-2 and M. tuberculosis may act synergistically in infected host cells (141). During latent TB infection, M. tuberculosis interacts with the pulmonary microenvironment and induces immune responses (141). Concrete evidence suggests that SARS-CoV-2 may cause hostile pro-inflammatory responses, including IL2, IL1-β, IL4, IL6, IL10, IFNγ, and TNFα in the infected cells (133). In addition, several stimuli probably add up in COVID-19-TB coinfection, leading to a cytokine storm. The necrosis and pyroptosis of the lung may cause damage-associated molecular pattern dispersion. SARS-CoV-2 presents a much more aggressive pyroptosis and promotes immunopathology and tissue damage (161, 162). Pulmonary alveoli are like battlefields for both SARS-CoV-2 and M. tuberculosis. M. tuberculosis silently infiltrates the lungs and avoids an exaggerated host immune response. In the case of mild infection, individual immune responses successfully eliminate both pathogens (163). Sometimes, the damaged lungs of TB patients influence local immunity and make the host more susceptible to COVID-19 and other airborne pathogens (15). It has been shown that SARS-CoV-2 aggravates the pulmonary TB status, causing latent TB to become active, which further deteriorates lung function (164). Further, inflammatory cytokine responses play a vital role in host resistance to M. tuberculosis infection as revealed in murine models (165), which was also validated in TB patients with mutations (blocking) in the IFNγ and IL12 signaling pathways (165, 166). According to the findings of a cohort of 49 COVID-19 TB patients, the possibility that SARS-CoV-2 infection may increase the occurrence of TB coinfection (15). A meta-analysis showed that patients with a TB history are not more likely to contract COVID-19; however, TB may increase the risk of contracting COVID-19 (143). Active TB patients in an Italian hospital had higher COVID-19 coinfection and clinical characteristics (113). COVID-19 coinfection worsened TB status and increased death in a group of 69 patients (98). In contrast, 20 COVID-19-TB coinfected people had benign clinical manifestations, with only one death. A chest X-ray revealed that TB lesions had not been aggravated; only four of the patients had recently developed pneumonia. In a quantitative study conducted in Belarus, 844 COVID-19 confirmed patients admitted to hospitals were tested for TB. Of the total, 47 patients had TB and were resistant to rifampicin (167). A study including 36 COVID-19 cases from China discovered that a history of pulmonary TB increases a patient’s vulnerability to severe SARS-CoV-2 infection (142), implying that TB status should be routinely checked in COVID-19 cases (135). According to findings from a developed pathogen model, populations at high risk of contracting SARS-CoV-2 may have a higher M. tuberculosis prevalence (135).
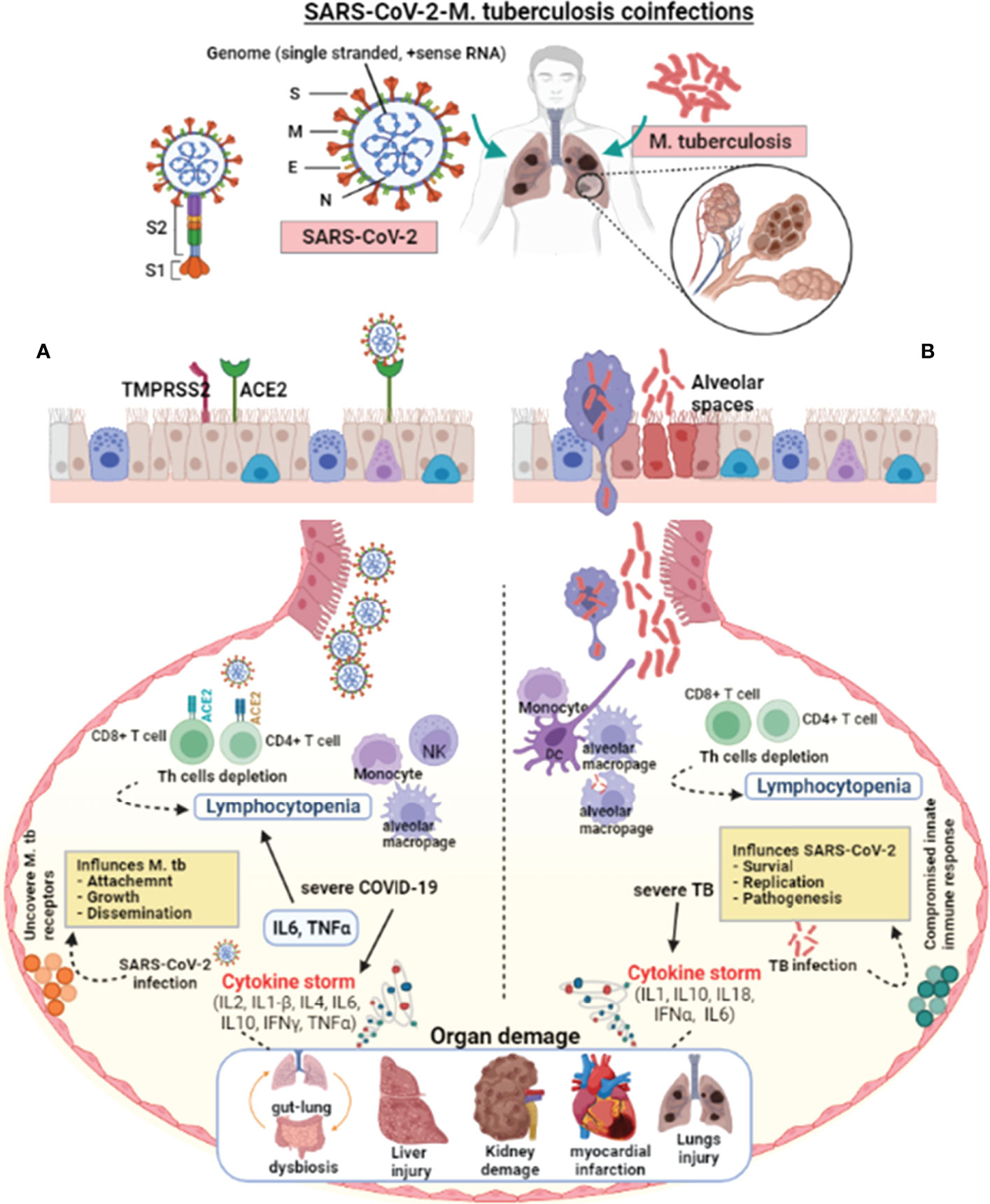
Figure 1 Pathophysiological effect of SARS-CoV-2 and M. tuberculosis on the host cell. SARS-CoV-2 enters the host via aerosol, travels to the alveoli, and interacts with the host’s innate immune cells. SARS-CoV-2 and M. tuberculosis-infected alveolar macrophages secrete cytokines to activate other immune cells, i.e., monocytes, macrophages, CD4+, CD8+ lymphocytes, neutrophils, dendritic cells, and natural killer cells to the infected site. (A) In severe COVID-19 infections, the exuberant pro-inflammatory cytokine response may result in lung injury. The lungs of severely infected COVID-19 patients showed an elevated immune response, which resulted in pneumonia, respiratory distress, lung fibrosis, and lymphocytopenia (decreased lymphocyte count). SARS-CoV-2 virulence factors interact with the host lungs, eliciting an immune response. These interactions may weaken the innate immune response, leading to increased mycobacterial attachment, growth, and dissemination. (B) In severe TB infection, activated lymphocytes produce excessive pro-inflammatory cytokines response called cytokine storms. Infection with M. tuberculosis causes symptomatic TB in people who have weakened immune systems or are immune-compromised. Cytokine storm-mediated inflammation causes multiple organ dysfunctions. M. tuberculosis infection and colonization may predispose the lungs to SARS-CoV-2 by down-regulating the host immune responses, allowing virus survival, growth, and pathogenesis. The suppressed host immune response in COVID-19-TB coinfection may cause exacerbated TB. In addition, reactivation of latent to active TB indicates that SARS-CoV-2 infection can exacerbate M. tuberculosis pathogenesis.
Impact of BCG on COVID-19 Patients
Previous immunological studies have shown that BCG vaccination prevents TB, DNA, and RNA viral infections, such as herpes and influenza viruses, resulting in lower morbidity and mortality (168, 169). The BCG vaccine stimulates the host immune response to produce antibodies that protect against TB infection and prevent the spread of the invaded M. tuberculosis (170). BCG vaccination in adults may increase non-specific pro-inflammatory cytokines IL1-β and IL6 against other bacterial pathogens (171) by stimulating CD4+ and CD8+ cells against non-targeted antigens, modulating lymphocyte responses against secondary infections. BCG administration also promotes innate immune cell responses, including monocytes, natural killer cells, and alveolar macrophages, increasing host resistance to future bacterial/viral infections (172, 173). For example, BCG vaccination induces a protective humoral immune response against the negative-strand RNA pneumovirus that causes respiratory disorders (174). Based on previous research, the BCG vaccination may boost the host’s immunity to reduce the severity of COVID-19 (175). Subsequently, increased pro-inflammatory cytokines TNFα, IL1-β, IL6, IFNγ, alveolar macrophages, T-lymphocytes, and antibody titers have been observed in BCG vaccinated people (168). Based on the host immune responses, BCG vaccination may prevent or reduce SARS-CoV-2 infection, particularly in BCG vaccinated children (176, 177). Although these findings are hypothetical, a nationwide BCG program may reduce COVID-19 severity and mortality; however, clinical trials on large datasets are recommended for BCG’s efficacy against the COVID-19 pandemic. Following BCG administration, healthy people will experience an immune response to SARS-CoV-2 infection, likely reducing viral loads, inhibiting viral replication, reducing pro-inflammatory cytokine responses, and inducing lymphocytopenia (176). Australia’s Muldron Children’s Research Institute started a phase III trial to determine if a healthcare worker’s BCG immunization program affects SARS-CoV-2 infection. National BCG vaccination programs in countries like Asia, Africa, and America have resulted in fewer COVID-19 cases (177, 178), necessitating further investigation to reveal the BCG connection with mild COVID-19 infections. BCG vaccination most probably stimulates innate immune responses against SARS-CoV-2. Following BCG vaccination, epigenetic programming trains monocytes and natural killer cells to clear many viral infections, particularly SARS-CoV-2 (176). More clinical data is required to understand the trained innate immune response and the adverse effects of potential innate and adaptive immunity in BCG-vaccinated COVID-19 cases. However, increased immunosuppression and cytokine storms were linked to severe complications in COVID-19 patients (179). Given the increased global TB and SARS-CoV-2 burden, particularly in developing and underdeveloped countries, BCG vaccination that could aid in the fight against both TB and COVID-19 would be highly desirable. Previously, recombinant adenoviral vectors were designed to boost patient immunity with humoral and cellular immune response enhancement (180). Adults with or without prior BCG vaccination should receive ChAdOx1-85A (181). In addition to BCG vaccination, scientists are struggling to develop an effective SARS-CoV-2 therapeutic agent to treat severe cases, particularly people who have pre-comorbidities. Isoniazid, ethambutol, pyrazinamide, and rifampin drugs are used to treat resistant TB (98, 140), whereas bedaquiline, clofazimine, levofloxacin, and linezolid can be combined with pyrazinamide for the treatment of MDR-TB cases. COVID19-TB coinfection can be treated with azithromycin, hydroxychloroquine (an anti-rheumatic drug), and protease inhibitors (lopinavir, ritonavir, darunavir, etc.) (15, 98). Moreover, enoxaparin and parnaparine anticoagulants may prevent the formation of blood clots (15).
Diagnostic Challenges and Management of COVID-19-TB Cases
Many governments imposed lockdowns to prevent SARS-CoV-2 transmission (182). People were forced to stay indoors, which affected their lives and health. Because of the similarity in symptoms between COVID-19 and TB, most people probably experience delayed TB with almost similar symptoms to COVID-19. In addition, the COVID-19-TB coinfection may have discouraged people from getting a diagnosis even if they experienced disease symptoms. These undiagnosed people were mostly from the region with lower socioeconomic status, struggling to make ends meet and eat. As a result, their pre-existing misery would have been exacerbated by the additional fear of quarantine. Furthermore, COVID-19-TB transmission could be a major concern due to close interactions at home.
The implementation of city lockdowns delayed TB diagnosis among those seeking medical advice, as non-emergency services were suspended in most parts of the world. Along with the restriction on accessing government and private clinics, laboratories were primarily dedicated to processing COVID-19 patient specimens (183), resulting in decreased COVID-19-TB detection and notification, as demonstrated by the 2020 Global TB Report. According to this report, the patient reporting rate was reduced by 25% in the three highest TB burden countries (Indonesia, India, and the Philippines) in only six months (January to June 2020) compared to 2019. COVID-19 has disrupted TB services globally, particularly in countries with the highest TB prevalence. For example, data from sixteen countries showed that COVID-19 disrupted TB services, particularly during the first four months of the pandemic (184). According to reports, deaths from TB are expected to increase by almost 13% in the coming years (185), which is probably a significant setback in the global fight against global TB. Due to COVID-19 restrictions, patients who have already been diagnosed with TB will also suffer. Sputum microscopy and bacterial culture growth primarily used for pulmonary TB follow-up patients were lost due to lockdowns. As a result, those with anti-TB therapy failure or multi-drug resistance to TB may have continuous health deterioration (183). Counseling and patient motivation are required to cope with TB clinical consequences and long-term anti-TB medication. Both COVID-19 and TB primarily affect the human lungs; however, TB symptoms shortly appear after COVID-19 infection (186, 187). Given the clinical complications of COVID-19 and TB, including cough, fever, breathing problems, and lung lesions (186, 187), proper diagnostic facilities are encouraged to avoid the misdiagnosis of one disease over the other. Tuberculin tests are less expensive and more widely used, whereas IGRAs are rarely used to diagnose TB (188). However, due to host immunity against M. tuberculosis or the BCG vaccine (189), a potential error in disease diagnosis may exist in immune-compromised people (190). Increased age, lymphocytopenia, and immunosuppressive therapies have also been linked to false-negative IGRAs (181), resulting in a false TB diagnosis. Furthermore, excessive inflammatory marker production may interfere with the IGRA testing, and increased CRP levels may confound the false-negative QuantiFERON-TB Gold In-Tube Test (191). In another study, severe COVID-19 patients had a lower peripheral lymphocyte count, natural killer cells, high CRP levels, and pro-inflammatory cytokines IL6, IL8, TNFα, IL2-R, IL1-β, etc. This unusual cytokine production may destroy host immune responses, i.e., low lymphocyte infiltration into infected lungs and multiple organ damage (192). Because SARS-CoV-2 is a novel CoV and has no specific treatment (5), suspected people must be tested accurately to prevent virus transmission. Apart from traditional diagnostic testing for detecting viral antigens or antiviral antibodies, the nucleic acid detection method has been developed for routine testing (193). To identify COVID-19 clinical manifestations (such as fever, dyspnea, cough), radiological features, and rapid virus transmission among people (194), doctors may encounter difficulty in differential diagnosis or overlook TB. In addition, scientists are struggling to develop an effective SARS-CoV-2 therapeutic agent to treat severe cases, particularly people who have pre-comorbidities. A lack of TB-specific radiological findings is likely to be missed due to the non-specific COVID-19-TB features. Another reason could be immunomodulators (a class of drugs that target pathways to reduce immune response) in mild/severe COVID-19 cases that may reactivate latent TB in endemic areas (107). Based on clinical, radiological, and laboratory findings, a patient in Russia developed pulmonary TB in the infiltration phase and COVID-19, which was complicated by spontaneous pneumothorax of the left lung. A TB specialist, an infectious diseases specialist, and a thoracic surgeon discussed the case condition. Due to his severe condition, he was given detoxification, bronchiolitis, and antibiotics (azithromycin, ceftriaxone, levofloxacin). Following TB confirmation, the patient was started on anti-TB antibiotics; rifampicin, ethambutol, and pyrazinamide. The chest drain was removed from the left pleural cavity, and lung function quickly recovered. The postoperative wound area had residual subcutaneous emphysema. Two nucleic acid tests for SARS-CoV-2 yielded negative results, and the patient was transferred to a TB hospital for further management (195).
Access to COVID-19 and TB diagnostic testing is a critical first step toward reducing disease transmission. As a result, there is a case to improve access to COVID-19 and TB testing through the implementation of simultaneous testing, particularly in countries with a high TB burden, to reduce the impact of the current pandemic on TB patients. Several countries, including Indonesia (196), South Africa (197), Nigeria (198), and India (199), began concurrent diagnostic testing for COVID-19 and TB during the pandemic. Notably, India’s Ministry of Health and Family Welfare issued guidelines to reduce the impact of the COVID-19 pandemic on TB control and management, including COVID-19 screening for TB confirmed patients or TB screening for COVID-19 confirmed patients (199). FIND, a non-profit global alliance, develops low-cost diagnostic tests for COVID-19 and TB. The project allows for rapid integrated testing for COVID-19 and TB for improved detection of both diseases. Rapid antigen testing for COVID-19 or TB symptoms assists in patient management by identifying coinfection hotspots. In addition, the private healthcare sector collaborated with the Joint Effort for the Elimination of TB (JEET) to improve access to affordable TB and COVID-19 testing services and educate people about coinfection precautions and symptoms and treatment. These experiences have demonstrated the importance of concurrent COVID-19 and TB testing in increasing access to coinfection diagnostics. The difficulties encountered during the simultaneous COVID-19-TB testing were primarily related to staff deficiencies and a lack of personal protective equipment. Huge gaps exist in the area of simultaneous COVID-19-TB testing, for example, the development of integrated COVID-19-TB testing for the same sample would increase the integrated system’s cost-efficiency. Other key areas that require additional research include evaluating a community for simultaneous testing and the cost-effectiveness of simultaneous testing (200). Future efforts should include the development and validation of COVID-19 and TB-related symptom-screening apps, considering factors like geography, age, risk factors, and research into the optimal sampling strategy for COVID-19-TB testing.
Conclusions and Future Perspectives
In the current scenario, TB coinfection should always be suspected in COVID-19 patients, whether or not they exhibit specific respiratory symptoms”– the data presented above indicate that TB coinfection contributes to COVID-19 severity and worse outcomes. Thus, rather than all patients, COVID-19 and TB coinfection may be suspected in severe TB patients, those resistant to therapy, or those from countries with a high prevalence of TB infection.
The COVID-19 pandemic has significantly affected human life and substantially lost the global economy. Available data shows a high mortality rate among COVID-19-TB coinfected individuals. SARS-CoV-2-M. tuberculosis coinfections impair host immunity. It is reasonable to assume that COVID-19 and TB harmful synergism contribute to severe clinical manifestations, most likely affecting patients, causing respiratory disorders via cytokine-mediated responses and an increased risk of latent TB reactivation.
To avoid COVID-19 coinfection, severe TB patients should only be hospitalized. Despite increasing COVID-19-TB cases, comprehensive global clinical trials are required to investigate the potential effect of COVID-19 on TB cases deeply. The current COVID-19 cases undermine the WHO goal of reducing the global TB burden. To lessen the impact of the COVID-19 pandemic on TB patients, consistent practices are needed to highlight the global TB burden while taking urgent steps to support and organize innovative TB control programs and minimize hospital visits to prevent COVID-19 coinfection (201). TB patients’ follow-up in their homes should be encouraged to ensure proper anti-TB medications. In addition, the implementation of smartphone technology might be an excellent choice for assessing and monitoring anti-TB medications. Collaboration of WHO anti-TB programs with local governments in developing countries is also recommended to prevent COVID-19-TB coinfection. Increasing testing capacity helped the healthcare system in high-risk areas. Implementing effective strategies to identify new TB hotspots and ensure continuous anti-TB drug transportation and distribution will be beneficial, especially in high-risk areas (202). Advanced preventive measures can also be taken to control COVID-19-TB coinfection. Despite the COVID-19 pandemic’s social and economic challenges, proper healthcare systems, effective social policies, and equal health facilities should be provided to control and prevent TB coinfection.
Author Contributions
XX, ZB, and TS performed the original draft preparation and revision. ZS, and NY made suggestions for the writing of the manuscript and revision. All authors actively contributed to the article and approved the submitted version.
Funding
This study was supported by grants from the major science and technology special project of Yunnan Province, No. 2019ZF004.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Cui J, Li F, Shi ZL. Origin and Evolution of Pathogenic Coronaviruses. Nat Rev Microbiol (2019) 17:181–92. doi: 10.1038/s41579-018-0118-9
2. Liu DX, Liang JQ, Fung TS, Coronavirus-229E H. -OC43, -NL63, and -HKU1 (Coronaviridae). Encyclopedia Virol (2021) 428–40. doi: 10.1016/B978-0-12-809633-8.21501-X
3. Shah T, Shah Z, Yasmeen N, Ma ZR. Psychological Impact of the COVID-19 Pandemic on Chinese Population: An Online Survey. World J Clin Cases (2021) 9:9500–8. doi: 10.12998/wjcc.v9.i31.9500
4. WHO. Director-General’s Opening Remarks at the Media Briefing on COVID-19(2020) (Accessed November 01, 2021).
5. Shah T, Shah Z, Xia K, Baloch Z. Therapeutic Mechanisms and Impact of Traditional Chinese Medicine on COVID-19 and Other Influenza Diseases. Pharmacol Res (2021) 2:100029. doi: 10.1016/j.prmcm.2021.100029
6. Wacharapluesadee S, Tan CW, Maneeorn P, Duengkae P, Zhu F, Joyjinda Y, et al. Evidence for SARS-CoV-2 Related Coronaviruses Circulating in Bats and Pangolins in Southeast Asia. Nat Commun (2021) 12:972. doi: 10.1038/s41467-021-21240-1
7. Baggen J, Vanstreels E, Jansen S, Daelemans D. Cellular Host Factors for SARS-CoV-2 Infection. Nat Microbiol (2021) 6:1219–32. doi: 10.1038/s41564-021-00958-0
10. UN. Global Perspective Human Stories; COVID-19 Caused Rise in TB Deaths for First Time in a Decade, Gains ‘Reversed’ (2021) (Accessed 27 Dec, 2021).
11. Callender LA, Curran M, Bates SM, Mairesse M, Weigandt J, Betts CJ. The Impact of Pre-Existing Comorbidities and Therapeutic Interventions on COVID-19. Front Immunol (2020) 11:1991. doi: 10.3389/fimmu.2020.01991
12. Song WM, Zhao JY, Zhang QY, Liu SQ, Zhu XH, An QQ, et al. COVID-19 and Tuberculosis Coinfection: An Overview of Case Reports/Case Series and Meta-Analysis. Front Med (Lausanne) (2021) 8:657006. doi: 10.3389/fmed.2021.657006
13. Lai CC, Wang CY, Hsueh PR. Co-Infections Among Patients With COVID-19: The Need for Combination Therapy With non-Anti-SARS-CoV-2 Agents? J Microbiol Immunol Infect (2020) 53:505–12. doi: 10.1016/j.jmii.2020.05.013
14. Rawson TM, Moore LSP, Zhu N, Ranganathan N, Skolimowska K, Gilchrist M, et al. Bacterial and Fungal Coinfection in Individuals With Coronavirus: A Rapid Review To Support COVID-19 Antimicrobial Prescribing. Clin Infect Dis (2020) 71:2459–68. doi: 10.1093/cid/ciaa530
15. Tadolini M, Codecasa LR, García-García JM, Blanc FX, Borisov S, Alffenaar JW, et al. Active Tuberculosis, Sequelae and COVID-19 Co-Infection: First Cohort of 49 Cases. Eur Respir J (2020) 56:2001398. doi: 10.1183/13993003.01398-2020
16. Bell LCK, Noursadeghi M. (2018). Pathogenesis of HIV-1 and Mycobacterium Tuberculosis Co-Infection. Nature Rev Microbiol 16. 80–90. doi: 10.1038/nrmicro.2017.128
17. Opoka LM, Wyrostkiewicz D, Winek J, Błasińska K, Miłkowska-Dymanowska J, Szturmowicz M. SARS-CoV-2 Lung Disease in a Patient With Pulmonary Sarcoidosis - Case Report. Adv Respir Med (2020) 88:620–5. doi: 10.5603/ARM.a2020.0199
18. Martínez Orozco JA, Sánchez Tinajero Á., Becerril Vargas E, Delgado Cueva AI, Reséndiz Escobar H, Vázquez Alcocer E, et al. COVID-19 and Tuberculosis Coinfection in a 51-Year-Old Taxi Driver in Mexico City. Am J Case Rep (2020) 21:e927628. doi: 10.12659/AJCR.927628
19. Hoffmann M, Kleine-Weber H, Schroeder S, Krüger N, Herrler T, Erichsen S, et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell (2020) 181:271–80.e8. doi: 10.1016/j.cell.2020.02.052
20. Wang Y, Wang Y, Chen Y, Qin Q. Unique Epidemiological and Clinical Features of the Emerging 2019 Novel Coronavirus Pneumonia (COVID-19) Implicate Special Control Measures. J Med Virol (2020) 92:568–76. doi: 10.1002/jmv.25748
21. Lan J, Ge J, Yu J, Shan S, Zhou H, Fan S, et al. Structure of the SARS-CoV-2 Spike Receptor-Binding Domain Bound to the ACE2 Receptor. Nature (2020) 581:215–20. doi: 10.1038/s41586-020-2180-5
22. Yan R, Zhang Y, Li Y, Xia L, Guo Y, Zhou Q. Structural Basis for the Recognition of SARS-CoV-2 by Full-Length Human ACE2. Science (2020) 367:1444–8. doi: 10.1126/science.abb2762
23. Zhou P, Yang XL, Wang XG, Hu B, Zhang L, Zhang W, et al. A Pneumonia Outbreak Associated With a New Coronavirus of Probable Bat Origin. Nature (2020) 579:270–3. doi: 10.1038/s41586-020-2012-7
24. Bao L, Deng W, Huang B, Gao H, Liu J, Ren L, et al. The Pathogenicity of SARS-CoV-2 in Hace2 Transgenic Mice. Nature (2020) 583:830–3. doi: 10.1038/s41586-020-2312-y
25. Schneider WM, Luna JM, Hoffmann HH, Sánchez-Rivera FJ, Leal AA, Ashbrook AW, et al. Genome-Scale Identification of SARS-CoV-2 and Pan-Coronavirus Host Factor Networks. Cell (2021) 184:120–32.e14. doi: 10.1016/j.cell.2020.12.006
26. Hoffmann HH, Sánchez-Rivera FJ, Schneider WM, Luna JM, Soto-Feliciano YM, Ashbrook AW, et al. Functional Interrogation of a SARS-CoV-2 Host Protein Interactome Identifies Unique and Shared Coronavirus Host Factors. Cell Host Microbe (2021) 29:267–80.e5. doi: 10.1016/j.chom.2020.12.009
27. Gordon DE, Jang GM, Bouhaddou M, Xu J, Obernier K, White KM, et al. A SARS-CoV-2 Protein Interaction Map Reveals Targets for Drug Repurposing. Nature (2020) 583:459–68. doi: 10.1038/s41586-020-2286-9
28. Biering SB, Sarnik SA, Wang E, Zengel JR, Sathyan V, Nguyenla X, et al. Genome-Wide, Bidirectional CRISPR Screens Identify Mucins as Critical Host Factors Modulating SARS-CoV-2 Infection. bioRxiv (2021) 2021:04. doi: 10.1101/2021.04.22.440848
29. Rebendenne A, Roy P, Bonaventure B, Chaves Valadão AL, Desmarets L, Rouillé Y, et al. Bidirectional Genome-Wide CRISPR Screens Reveal Host Factors Regulating SARS-CoV-2, MERS-CoV and Seasonal Coronaviruses. bioRxiv (2021) 2021:05.19.444823. doi: 10.1101/2021.05.19.444823
30. Wang S, Qiu Z, Hou Y, Deng X, Xu W, Zheng T, et al. AXL Is a Candidate Receptor for SARS-CoV-2 That Promotes Infection of Pulmonary and Bronchial Epithelial Cells. Cell Res (2021) 31:126–40. doi: 10.1038/s41422-020-00460-y
31. Puray-Chavez M, LaPak KM, Schrank TP, Elliott JL, Bhatt DP, Agajanian MJ, et al. Systematic Analysis of SARS-CoV-2 Infection of an ACE2-Negative Human Airway Cell. Cell Rep (2021) 36:109364. doi: 10.1016/j.celrep.2021.109364
32. Yeung ML, Teng JLL, Jia L, Zhang C, Huang C, Cai JP, et al. Soluble ACE2-Mediated Cell Entry of SARS-CoV-2 via Interaction With Proteins Related to the Renin-Angiotensin System. Cell (2021) 184:2212–28.e12. doi: 10.1016/j.cell.2021.02.053
33. Harrison AG, Lin T, Wang P. Mechanisms of SARS-CoV-2 Transmission and Pathogenesis. Trends Immunol (2020) 41:1100–15. doi: 10.1016/j.it.2020.10.004
34. Mason RJ. Pathogenesis of COVID-19 From a Cell Biology Perspective. Eur Respir J 55 (2020) 55:2000607. doi: 10.1183/13993003.00607-2020
35. Abassi Z, Knaney Y, Karram T, Heyman SN. The Lung Macrophage in SARS-CoV-2 Infection: A Friend or a Foe? Front Immunol (2020) 11:1312. doi: 10.3389/fimmu.2020.01312
36. Chua RL, Lukassen S, Trump S, Hennig BP, Wendisch D, Pott F, et al. COVID-19 Severity Correlates With Airway Epithelium-Immune Cell Interactions Identified by Single-Cell Analysis. Nat Biotechnol (2020) 38:970–9. doi: 10.1038/s41587-020-0602-4
37. Bost P, Giladi A, Liu Y, Bendjelal Y, Xu G, David E, et al. Host-Viral Infection Maps Reveal Signatures of Severe COVID-19 Patients. Cell (2020) 181:1475–88.e12. doi: 10.1016/j.cell.2020.05.006
38. Liu L, Chopra P, Li X, Bouwman KM, Tompkins SM, Wolfert MA, et al. Heparan Sulfate Proteoglycans as Attachment Factor for SARS-CoV-2. ACS Cent Sci (2021) 7:1009–18. doi: 10.1021/acscentsci.1c00010
39. Clausen TM, Sandoval DR, Spliid CB, Pihl J, Perrett HR, Painter CD, et al. SARS-CoV-2 Infection Depends on Cellular Heparan Sulfate and ACE2. Cell (2020) 183:1043–57.e15. doi: 10.1016/j.cell.2020.09.033
40. Cagno V, Tseligka ED, Jones ST, Tapparel C. Heparan Sulfate Proteoglycans and Viral Attachment: True Receptors or Adaptation Bias? Viruses (2019) 11:596. doi: 10.3390/v11070596
41. Wei C, Wan L, Yan Q, Wang X, Zhang J, Yang X, et al. (2020). HDL-Scavenger Receptor B Type 1 Facilitates SARS-CoV-2 Entry. Nat Metab 2. 1391–400. doi: 10.1038/s42255-020-00324-0
42. Bradsher RW, Monson TP, Steele RW. Brain Abscess Due to Nocardia Caviae. Report of a Fatal Outcome Associated With Abnormal Phagocyte Function. Am J Clin Pathol (1982) 78:124–7. doi: 10.1093/ajcp/78.1.124
43. Daly JL, Simonetti B, Klein K, Chen KE, Williamson MK, Antón-Plágaro C, et al. Neuropilin-1 Is a Host Factor for SARS-CoV-2 Infection. Science (2020) 370:861–5. doi: 10.1126/science.abd3072
44. Cantuti-Castelvetri L, Ojha R, Pedro LD, Djannatian M, Franz J, Kuivanen S, et al. Neuropilin-1 Facilitates SARS-CoV-2 Cell Entry and Infectivity. Science (2020) 370:856–60. doi: 10.1126/science.abd2985
45. Zhu S, Liu Y, Zhou Z, Zhang Z, Xiao X, Liu Z, et al. Genome-Wide CRISPR Activation Screen Identifies Candidate Receptors for SARS-CoV-2 Entry. Sci China Life Sci (2021) 4:701–17. doi: 10.1007/s11427-021-1990-5
46. Wang K, Chen W, Zhang Z, Deng Y, Lian JQ, Du P, et al. CD147-Spike Protein Is a Novel Route for SARS-CoV-2 Infection to Host Cells. Signal Transduct Target Ther (2020) 5:283. doi: 10.1038/s41392-020-00426-x
47. Shilts J, Crozier TWM, Greenwood EJD, Lehner PJ, Wright GJ. No Evidence for Basigin/CD147 as a Direct SARS-CoV-2 Spike Binding Receptor. Sci Rep (2021) 11:413. doi: 10.1038/s41598-020-80464-1
48. Tapela K, Ochieng’ Olwal C, Quaye O. Parallels in the Pathogenesis of SARS-CoV-2 and M. Tuberculosis: A Synergistic or Antagonistic Alliance? Future Microbiol (2020) 15:1691–5. doi: 10.2217/fmb-2020-0179
49. Shah T, Shah Z, Baloch Z, Cui X. The Role of Microbiota in Respiratory Health and Diseases, Particularly in Tuberculosis. BioMed Pharmacother (2021) 143:112108. doi: 10.1016/j.biopha.2021.112108
50. Hossain MM, Norazmi MN. Pattern Recognition Receptors and Cytokines in Mycobacterium Tuberculosis Infection–the Double-Edged Sword? BioMed Res Int (2013) 2013:179174. doi: 10.1155/2013/179174
51. Leopold Wager CM, Arnett E, Schlesinger LS. Mycobacterium Tuberculosis and Macrophage Nuclear Receptors: What We Do and Don’t Know. Tuberculosis (Edinb) (2019) 11698–106. doi: 10.1016/j.tube.2019.04.016
52. Shah T, Hayat A, Jadoon A, Ahmad S, Bahadar K. Molecular Detection of Multi Drug Resistant Tuberculosis (MDR-TB) in MDR-TB Patients’ Attendant in North Western Pakistan PAFMJ (2017) 67:982–7.
53. Vinod V, Vijayrajratnam S, Vasudevan AK, Biswas RS. The Cell Surface Adhesins of Mycobacterium Tuberculosis. Microbiol Res (2020) 232:126392. doi: 10.1016/j.micres.2019.126392
54. Bisht D, Meena LS. Adhesion Molecules Facilitate Host-Pathogen Interaction & Mediate Mycobacterium Tuberculosis Pathogenesis. Indian J Med Res (2019) 150:23–32. doi: 10.4103/ijmr.IJMR_2055_16
55. Hussain Bhat K, Mukhopadhyay S. Macrophage Takeover and the Host-Bacilli Interplay During Tuberculosis. Future Microbiol (2015) 10:853–72. doi: 10.2217/fmb.15.11
56. Zhai W, Wu F, Zhang Y, Fu Y, Liu Z. The Immune Escape Mechanisms of Mycobacterium Tuberculosis. Int J Mol Sci 20 (2019) 20:340. doi: 10.3390/ijms20020340
57. Rajaram MVS, Arnett E, Azad AK, Guirado E, Ni B, Gerberick AD, et al. M. Tuberculosis-Initiated Human Mannose Receptor Signaling Regulates Macrophage Recognition and Vesicle Trafficking by Fcrγ-Chain, Grb2, and SHP-1. Cell Rep (2017) 21:126–40. doi: 10.1016/j.celrep.2017.09.034
58. Ferguson JS, Weis JJ, Martin JL, Schlesinger LS. Complement Protein C3 Binding to Mycobacterium Tuberculosis is Initiated by the Classical Pathway in Human Bronchoalveolar Lavage Fluid. Infect Immun (2004) 72:2564–73. doi: 10.1128/IAI.72.5.2564-2573.2004
59. Bohlson SS, Strasser JA, Bower JJ, Schorey JS. Role of Complement in Mycobacterium Avium Pathogenesis: In Vivo and In Vitro Analyses of the Host Response to Infection in the Absence of Complement Component C3. Infect Immun (2001) 69:7729–35. doi: 10.1128/IAI.69.12.7729-7735.2001
60. Turner J, Torrelles JB. Mannose-Capped Lipoarabinomannan in Mycobacterium Tuberculosis Pathogenesis. Pathog Dis 76 (2018) 76: fty026. doi: 10.1093/femspd/fty026
61. Sasindran SJ, Torrelles JB. Mycobacterium Tuberculosis Infection and Inflammation: What is Beneficial for the Host and for the Bacterium? Front Microbiol (2011) 2:2. doi: 10.3389/fmicb.2011.00002
62. Kumar GA, Jafurulla M, Chattopadhyay A. The Membrane as the Gatekeeper of Infection: Cholesterol in Host-Pathogen Interaction. Chem Phys Lipids (2016) 199:179–85. doi: 10.1016/j.chemphyslip.2016.02.007
63. Liu CH, Liu H, Ge B. Innate Immunity in Tuberculosis: Host Defense vs Pathogen Evasion. Cell Mol Immunol (2017) 14:963–75. doi: 10.1038/cmi.2017.88
64. Kawai T, Akira S. The Role of Pattern-Recognition Receptors in Innate Immunity: Update on Toll-Like Receptors. Nat Immunol (2010) 11:373–84. doi: 10.1038/ni.1863
65. Saraav I, Singh S, Sharma S. Outcome of Mycobacterium Tuberculosis and Toll-Like Receptor Interaction: Immune Response or Immune Evasion? Immunol Cell Biol (2014) 92:741–6. doi: 10.1038/icb.2014.52
66. Sugawara I, Yamada H, Mizuno S, Takeda K, Akira S. Mycobacterial Infection in MyD88-Deficient Mice. Microbiol Immunol (2003) 47:841–7. doi: 10.1111/j.1348-0421.2003.tb03450.x
67. Drennan MB, Nicolle D, Quesniaux VJ, Jacobs M, Allie N, Mpagi J, et al. Toll-Like Receptor 2-Deficient Mice Succumb to Mycobacterium Tuberculosis Infection. Am J Pathol (2004) 164:49–57. doi: 10.1016/S0002-9440(10)63095-7
68. Najmi N, Kaur G, Sharma SK, Mehra NK. Human Toll-Like Receptor 4 Polymorphisms TLR4 Asp299Gly and Thr399Ile Influence Susceptibility and Severity of Pulmonary Tuberculosis in the Asian Indian Population. Tissue Antigens (2010) 76:102–9. doi: 10.1111/j.1399-0039.2010.01481.x
69. Lai YF, Lin TM, Wang CH, Su PY, Wu JT, Lin MC, et al. Functional Polymorphisms of the TLR7 and TLR8 Genes Contribute to Mycobacterium Tuberculosis Infection. Tuberculosis (Edinb) (2016) 98:125–31. doi: 10.1016/j.tube.2016.03.008
70. Carvalho NB, Oliveira FS, Durães FV, de Almeida LA, Flórido M, Prata LO, et al. Toll-Like Receptor 9 Is Required for Full Host Resistance to Mycobacterium Avium Infection But Plays No Role in Induction of Th1 Responses. Infect Immun (2011) 79:1638–46. doi: 10.1128/IAI.01030-10
71. Hölscher C, Reiling N, Schaible UE, Hölscher A, Bathmann C, Korbel D, et al. Containment of Aerogenic Mycobacterium Tuberculosis Infection in Mice Does Not Require MyD88 Adaptor Function for TLR2, -4 and -9. Eur J Immunol (2008) 38:680–94. doi: 10.1002/eji.200736458
72. Pecora ND, Gehring AJ, Canaday DH, Boom WH, Harding CV. Mycobacterium Tuberculosis LprA Is a Lipoprotein Agonist of TLR2 That Regulates Innate Immunity and APC Function. J Immunol (2006) 177:422–9. doi: 10.4049/jimmunol.177.1.422
73. Bai W, Liu H, Ji Q, Zhou Y, Liang L, Zheng R, et al. TLR3 Regulates Mycobacterial RNA-Induced IL-10 Production Through the PI3K/AKT Signaling Pathway. Cell Signal (2014) 26:942–50. doi: 10.1016/j.cellsig.2014.01.015
74. Liu Y, Li JY, Chen ST, Huang HR, Cai H. The Rlrp of Mycobacterium Tuberculosis Inhibits Proinflammatory Cytokine Production and Downregulates APC Function in Mouse Macrophages via a TLR2-Mediated PI3K/Akt Pathway Activation-Dependent Mechanism. Cell Mol Immunol (2016) 13:729–46. doi: 10.1038/cmi.2015.58
75. Wang J, Li BX, Ge PP, Li J, Wang Q, Gao GF, et al. Mycobacterium Tuberculosis Suppresses Innate Immunity by Coopting the Host Ubiquitin System. Nat Immunol (2015) 16:237–45. doi: 10.1038/ni.3096
76. Li J, Chai QY, Zhang Y, Li BX, Wang J, Qiu XB, et al. Mycobacterium Tuberculosis Mce3E Suppresses Host Innate Immune Responses by Targeting ERK1/2 Signaling. J Immunol (2015) 194:3756–67. doi: 10.4049/jimmunol.1402679
77. Yang H, Liu H, Chen H, Mo H, Chen J, Huang X, et al. G Protein-Coupled Receptor160 Regulates Mycobacteria Entry Into Macrophages by Activating ERK. Cell Signal (2016) 28:1145–51. doi: 10.1016/j.cellsig.2016.05.022
78. Kim YK, Shin JS, Nahm MH. NOD-Like Receptors in Infection, Immunity, and Diseases. Yonsei Med J (2016) 57:5–14. doi: 10.3349/ymj.2016.57.1.5
79. Simeone R, Bobard A, Lippmann J, Bitter W, Majlessi L, Brosch R, et al. Phagosomal Rupture by Mycobacterium Tuberculosis Results in Toxicity and Host Cell Death. PLos Pathog (2012) 8:e1002507. doi: 10.1371/journal.ppat.1002507
80. Divangahi M, Mostowy S, Coulombe F, Kozak R, Guillot L, Veyrier F, et al. NOD2-Deficient Mice Have Impaired Resistance to Mycobacterium Tuberculosis Infection Through Defective Innate and Adaptive Immunity. J Immunol (2008) 181:7157–65. doi: 10.4049/jimmunol.181.10.7157
81. Juárez E, Carranza C, Hernández-Sánchez F, León-Contreras JC, Hernández-Pando R, Escobedo D, et al. NOD2 Enhances the Innate Response of Alveolar Macrophages to Mycobacterium Tuberculosis in Humans. Eur J Immunol (2012) 42:880–9. doi: 10.1002/eji.201142105
82. Austin CM, Ma X, Graviss EA. Common Nonsynonymous Polymorphisms in the NOD2 Gene are Associated With Resistance or Susceptibility to Tuberculosis Disease in African Americans. J Infect Dis (2008) 197:1713–6. doi: 10.1086/588384
83. Pan H, Dai Y, Tang S, Wang J. Polymorphisms of NOD2 and the Risk of Tuberculosis: A Validation Study in the Chinese Population. Int J Immunogenet (2012) 39:233–40. doi: 10.1111/j.1744-313X.2011.01079.x
84. Mishra BB, Moura-Alves P, Sonawane A, Hacohen N, Griffiths G, Moita LF, et al. Mycobacterium Tuberculosis Protein ESAT-6 is a Potent Activator of the NLRP3/ASC Inflammasome. Cell Microbiol (2010) 12:1046–63. doi: 10.1111/j.1462-5822.2010.01450.x
85. McElvania Tekippe E, Allen IC, Hulseberg PD, Sullivan JT, McCann JR, Sandor M, et al. Granuloma Formation and Host Defense in Chronic Mycobacterium Tuberculosis Infection Requires PYCARD/ASC But Not NLRP3 or Caspase-1. PLos One (2010) 5:e12320. doi: 10.1371/journal.pone.0012320
86. Goyal S, Klassert TE, Slevogt H. C-Type Lectin Receptors in Tuberculosis: What We Know. Med Microbiol Immunol (2016) 205:513–35. doi: 10.1007/s00430-016-0470-1
87. Kang PB, Azad AK, Torrelles JB, Kaufman TM, Beharka A, Tibesar E, et al. The Human Macrophage Mannose Receptor Directs Mycobacterium Tuberculosis Lipoarabinomannan-Mediated Phagosome Biogenesis. J Exp Med (2005) 202:987–99. doi: 10.1084/jem.20051239
88. Geurtsen J, Chedammi S, Mesters J, Cot M, Driessen NN, Sambou T, et al. Identification of Mycobacterial Alpha-Glucan as a Novel Ligand for DC-SIGN: Involvement of Mycobacterial Capsular Polysaccharides in Host Immune Modulation. J Immunol (2009) 183:5221–31. doi: 10.4049/jimmunol.0900768
89. Matsunaga I, Moody DB. Mincle is a Long Sought Receptor for Mycobacterial Cord Factor. J Exp Med (2009) 206:2865–8. doi: 10.1084/jem.20092533
90. Yadav M, Schorey JS. The Beta-Glucan Receptor Dectin-1 Functions Together With TLR2 to Mediate Macrophage Activation by Mycobacteria. Blood (2006) 108:3168–75. doi: 10.1182/blood-2006-05-024406
91. Marakalala MJ, Guler R, Matika L, Murray G, Jacobs M, Brombacher F, et al. The Syk/CARD9-Coupled Receptor Dectin-1 is Not Required for Host Resistance to Mycobacterium Tuberculosis in Mice. Microbes Infect (2011) 13:198–201. doi: 10.1016/j.micinf.2010.10.013
92. Yonekawa A, Saijo S, Hoshino Y, Miyake Y, Ishikawa E, Suzukawa M, et al. Dectin-2 Is a Direct Receptor for Mannose-Capped Lipoarabinomannan of Mycobacteria. Immunity (2014) 41:402–13. doi: 10.1016/j.immuni.2014.08.005
93. Miyake Y, Masatsugu OH, Yamasaki S. C-Type Lectin Receptor MCL Facilitates Mincle Expression and Signaling Through Complex Formation. J Immunol (2015) 194:5366–74. doi: 10.4049/jimmunol.1402429
94. Wilson GJ, Marakalala MJ, Hoving JC, van Laarhoven A, Drummond RA, Kerscher B, et al. The C-Type Lectin Receptor CLECSF8/CLEC4D is a Key Component of Anti-Mycobacterial Immunity. Cell Host Microbe (2015) 17:252–9. doi: 10.1016/j.chom.2015.01.004
95. Troegeler A, Lugo-Villarino G, Hansen S, Rasolofo V, Henriksen ML, Mori K, et al. Collectin CL-LK Is a Novel Soluble Pattern Recognition Receptor for Mycobacterium Tuberculosis. PLos One (2015) 10:e0132692. doi: 10.1371/journal.pone.0132692
96. Troegeler A, Mercier I, Cougoule C, Pietretti D, Colom A, Duval C, et al. C-Type Lectin Receptor DCIR Modulates Immunity to Tuberculosis by Sustaining Type I Interferon Signaling in Dendritic Cells. Proc Natl Acad Sci U S A (2017) 114:E540–9. doi: 10.1073/pnas.1613254114
97. He G, Wu J, Shi J, Dai J, Gamber M, Jiang X, et al. COVID-19 in Tuberculosis Patients: A Report of Three Cases. J Med Virol (2020) 92:1802–6. doi: 10.1002/jmv.25943
98. Motta I, Centis R, D’Ambrosio L, García-García JM, Goletti D, Gualano G, et al. Tuberculosis, COVID-19 and Migrants: Preliminary Analysis of Deaths Occurring in 69 Patients From Two Cohorts. Pulmonology (2020) 26:233–40. doi: 10.1016/j.pulmoe.2020.05.002
99. Sadanshiv M, George AA, Mishra AK, Kuriakose CK. Rifampicin-Induced Immune Allergic Reaction. Trop Doct (2018) 48:156–9. doi: 10.1177/0049475517724689
100. Mishra A, George AA, Sahu KK, Lal A, Abraham G. Tuberculosis and COVID-19 Co-Infection: An Updated Review. Acta BioMed (2020) 92:e2021025. doi: 10.23750/abm.v92i1.10738
101. Liu C, Yu Y, Fleming J, Wang T, Shen S, Wang Y, et al. Severe COVID-19 Cases With a History of Active or Latent Tuberculosis. Int J Tuberc Lung Dis (2020) 24:747–9. doi: 10.5588/ijtld.20.0163
102. Liu J, Zhang S, Wu Z, Shang Y, Dong X, Li G, et al. Clinical Outcomes of COVID-19 in Wuhan, China: A Large Cohort Study. Ann Intensive Care (2020) 10:99. doi: 10.1186/s13613-020-00706-3
103. Chen T, Dai Z, Mo P, Li X, Ma Z, Song S, et al. Clinical Characteristics and Outcomes of Older Patients With Coronavirus Disease 2019 (COVID-19) in Wuhan, China: A Single-Centered, Retrospective Study. J Gerontol A Biol Sci Med Sci (2020) 75:1788–95. doi: 10.1093/gerona/glaa089
104. Du RH, Liang LR, Yang CQ, Wang W, Cao TZ, Li M, et al. Predictors of Mortality for Patients With COVID-19 Pneumonia Caused by SARS-CoV-2: A Prospective Cohort Study. Eur Respir J 55 (2020) 55:2000524. doi: 10.1183/13993003.00524-2020
105. Li X, Xu S, Yu M, Wang K, Tao Y, Zhou Y, et al. Risk Factors for Severity and Mortality in Adult COVID-19 Inpatients in Wuhan. J Allergy Clin Immunol (2020) 146:110–8. doi: 10.1016/j.jaci.2020.04.006
106. Yao Z, Chen J, Wang Q, Liu W, Zhang Q, Nan J, et al. Three Patients With COVID-19 and Pulmonary Tuberculosis, Wuhan, China, January-February 2020. Emerg Infect Dis (2020) 26:2755–8. doi: 10.3201/eid2611.201536
107. Kumar DR, Bhattacharya DB, Meena DV, Soneja DM, Wig DN. COVID-19 and TB Co-Infection - ‘Finishing Touch’ in Perfect Recipe to ‘Severity’ or ‘Death’. J Infect (2020) 81:e39–40. doi: 10.1016/j.jinf.2020.06.062
108. Yadav S, Rawal G. The Case of Pulmonary Tuberculosis With COVID-19 in an Indian Male-a First of Its Type Case Ever Reported From South Asia. Pan Afr Med J (2020) 36:374. doi: 10.11604/pamj.2020.36.374.24260
109. Ata F, Yousaf Q, Veliyankodan Parambil J, Parengal J, Mohamedali MG, Yousaf Z. A 28-Year-Old Man From India With SARS-Cov-2 and Pulmonary Tuberculosis Co-Infection With Central Nervous System Involvement. Am J Case Rep (2020) 21:e926034. doi: 10.12659/AJCR.926034
110. Gupta N, Ish P, Gupta A, Malhotra N, Caminero JA, Singla R, et al. A Profile of a Retrospective Cohort of 22 Patients With COVID-19 and Active/Treated Tuberculosis. Eur Respir J 56 (2020) 56:2003408. doi: 10.1183/13993003.03408-2020
111. Garg N, Lee YI. Reactivation of TB With Severe COVID-19. Chest (2020) 158:A777. doi: 10.1016/j.chest.2020.08.724
112. Freij BJ, Gebara BM, Tariq R, Wang AM, Gibson J, El-Wiher N, et al. Fatal Central Nervous System Co-Infection With SARS-CoV-2 and Tuberculosis in a Healthy Child. BMC Pediatr (2020) 20:429. doi: 10.1186/s12887-020-02308-1
113. Stochino C, Villa S, Zucchi P, Parravicini P, Gori A, Raviglione MC. Clinical Characteristics of COVID-19 and Active Tuberculosis Co-Infection in an Italian Reference Hospital. Eur Respir J 56 (2020) 56:2003408. doi: 10.1183/13993003.01708-2020
114. Gadelha Farias LAB, Gomes Moreira AL, Austregésilo Corrêa E, Landim de Oliveira Lima CA, Lopes IMP, de Holanda PEL, et al. Case Report: Coronavirus Disease and Pulmonary Tuberculosis in Patients With Human Immunodeficiency Virus: Report of Two Cases. Am J Trop Med Hyg (2020) 103:1593–6. doi: 10.4269/ajtmh.20-0737
115. Yousaf Z, Khan AA, Chaudhary HA, Mushtaq K, Parengal J, Aboukamar M, et al. Cavitary Pulmonary Tuberculosis With COVID-19 Coinfection. IDCases (2020) 22:e00973. doi: 10.1016/j.idcr.2020.e00973
116. Sy KTL, Haw NJL, Uy J. Previous and Active Tuberculosis Increases Risk of Death and Prolongs Recovery in Patients With COVID-19. Infect Dis (Lond) (2020) 52:902–7. doi: 10.1080/23744235.2020.1806353
117. Davies MA. HIV and Risk of COVID-19 Death: A Population Cohort Study From the Western Cape Province, South Africa. medRxiv (2020) 3: 20145185. doi: 10.1101/2020.07.02.20145185
118. Vilbrun SC, Mathurin L, Pape JW, Fitzgerald D, Walsh KF. Case Report: Multidrug-Resistant Tuberculosis and COVID-19 Coinfection in Port-Au-Prince, Haiti. Am J Trop Med Hyg (2020) 103:1986–8. doi: 10.4269/ajtmh.20-0851
119. Tham SM, Lim WY, Lee CK, Loh J, Premkumar A, Yan B, et al. Four Patients With COVID-19 and Tuberculosis, Singapore, April-May 2020. Emerg Infect Dis (2020) 26:2764–6. doi: 10.3201/eid2611.202752
120. Rivas N, Espinoza M, Loban A, Luque O, Jurado J, Henry-Hurtado N, et al. Case Report: COVID-19 Recovery From Triple Infection With Mycobacterium Tuberculosis, HIV, and SARS-CoV-2. Am J Trop Med Hyg (2020) 103:1597–9. doi: 10.4269/ajtmh.20-0756
121. Lopinto J, Teulier M, Milon A, Voiriot G, Fartoukh M. Severe Hemoptysis in Post-Tuberculosis Bronchiectasis Precipitated by SARS-CoV-2 Infection. BMC Pulm Med (2020) 20:244. doi: 10.1186/s12890-020-01285-6
122. Goussard P, Solomons RS, Andronikou S, Mfingwana L, Verhagen LM, Rabie H. COVID-19 in a Child With Tuberculous Airway Compression. Pediatr Pulmonol (2020) 55:2201–3. doi: 10.1002/ppul.24927
123. Essajee F, Solomons R, Goussard P, Van Toorn R. Child With Tuberculous Meningitis and COVID-19 Coinfection Complicated by Extensive Cerebral Sinus Venous Thrombosis. BMJ Case Rep 13 (2020) 13:e238597. doi: 10.1136/bcr-2020-238597
124. Faqihi F, Alharthy A, Noor A, Balshi A, Balhamar A, Karakitsos D. COVID-19 in a Patient With Active Tuberculosis: A Rare Case-Report. Respir Med Case Rep (2020) 31:101146. doi: 10.1016/j.rmcr.2020.101146
125. Çınar OE, Sayınalp B, Aladağ Karakulak E, Avşar Karataş A, Velet M, İnkaya A, et al. Convalescent (Immune) Plasma Treatment in a Myelodysplastic COVID-19 Patient With Disseminated Tuberculosis. Transfus Apher Sci (2020) 59:102821. doi: 10.1016/j.transci.2020.102821
126. Visca D, Ong CWM, Tiberi S, Centis R, D’Ambrosio L, Chen B, et al. Tuberculosis and COVID-19 Interaction: A Review of Biological, Clinical and Public Health Effects. Pulmonology (2021) 27:151–65. doi: 10.1016/j.pulmoe.2020.12.012
127. Sahu KK, Lal A, Mishra AK. An Update on CT Chest Findings in Coronavirus Disease-19 (COVID-19). Heart Lung (2020) 49:442–3. doi: 10.1016/j.hrtlng.2020.03.007
128. Lal A, Mishra AK, Sahu KK, Davaro R. Tuberculous Cold Abscess Eroding Iliac Bone. Rev Esp Patol (2020) 53:71–2. doi: 10.1016/j.patol.2019.08.002
129. Prompetchara E, Ketloy C, Palaga T. Immune Responses in COVID-19 and Potential Vaccines: Lessons Learned From SARS and MERS Epidemic. Asian Pac J Allergy Immunol (2020) 38:1–9. doi: 10.12932/AP-200220-0772
130. Dong E, Du H, Gardner L. An Interactive Web-Based Dashboard to Track COVID-19 in Real Time. Lancet Infect Dis (2020) 20:533–4. doi: 10.1016/S1473-3099(20)30120-1
131. Hale T, Angrist N, Goldszmidt R, Kira B, Petherick A, Phillips T, et al. A Global Panel Database of Pandemic Policies (Oxford COVID-19 Government Response Tracker). Nat Hum Behav (2021) 5:529–38. doi: 10.1038/s41562-021-01079-8
132. Hogan AB, Jewell BL, Sherrard-Smith E, Vesga JF, Watson OJ, Whittaker C, et al. Potential Impact of the COVID-19 Pandemic on HIV, Tuberculosis, and Malaria in Low-Income and Middle-Income Countries: A Modelling Study. Lancet Glob Health (2020) 8:e1132–41. doi: 10.1016/S2214-109X(20)30288-6
133. Guan WJ, Ni ZY, Hu Y, Liang WH, Ou CQ, He JX, et al. Clinical Characteristics of Coronavirus Disease 2019 in China. N Engl J Med (2020) 382:1708–20. doi: 10.1056/NEJMoa2002032
134. WHO. Addressing the Needs of Vulnerable Populations. (2020) Regional Office for Europe. Available at: https://www.euro.who.int/__data/assets/pdf_file/0003/446340/Factsheet-May-2020-Vulnerable-populations-during-COVID-19-response-eng.pdf.
135. Hotchkiss JR, Strike DG, Crooke PS. Pathogen Transmission and Clinic Scheduling. Emerg Infect Dis (2006) 12:159–62. doi: 10.3201/eid1201.050349
136. Harding E. WHO Global Progress Report on Tuberculosis Elimination. Lancet Respir Med (2020) 8:19. doi: 10.1016/S2213-2600(19)30418-7
137. Amimo F, Lambert B, Magit A. What Does the COVID-19 Pandemic Mean for HIV, Tuberculosis, and Malaria Control? Trop Med Health (2020) 48:32. doi: 10.1186/s41182-020-00219-6
138. Rusen ID. Challenges in Tuberculosis Clinical Trials in the Face of the COVID-19 Pandemic: A Sponsor’s Perspective. Trop Med Infect Dis 5 (2020) 5:86. doi: 10.3390/tropicalmed5020086
139. Manyazewal T, Woldeamanuel Y, Blumberg HM, Fekadu A, Marconi VC. The Fight to End Tuberculosis Must Not Be Forgotten in the COVID-19 Outbreak. Nat Med (2020) 26:811–2. doi: 10.1038/s41591-020-0917-1
140. Sahu KK, Mishra AK, Martin K, Chastain I. COVID-19 and Restrictive Lung Disease: A Deadly Combo to Trip Off the Fine Balance. Monaldi Arch Chest Dis 90 (2020) 90. doi: 10.4081/monaldi.2020.1346
141. Mousquer GT, Peres A, Fiegenbaum M. Pathology of TB/COVID-19 Co-Infection: The Phantom Menace. Tuberculosis (Edinb) (2021) 126:102020. doi: 10.1016/j.tube.2020.102020
142. Chen Y, Wang Y, Fleming J, Yu Y, Gu Y, Liu C, et al. Active or Latent Tuberculosis Increases Susceptibility to COVID-19 and Disease Severity. medRxiv (2020) 2020:3. doi: 10.1101/2020.03.10.20033795
143. Gao Y, Liu M, Chen Y, Shi S, Geng J, Tian J. Association Between Tuberculosis and COVID-19 Severity and Mortality: A Rapid Systematic Review and Meta-Analysis. J Med Virol (2021) 93:194–6. doi: 10.1002/jmv.26311
144. Boulle A, Davies M, Hussey H, Ismail M, Morden E, Vundle Z, et al. Risk Factors for Coronavirus Disease 2019 (COVID-19) Death in a Population Cohort Study From the Western Cape Province, South Africa. Clin Infect Dis (2021) 73:e2005–15. doi: 10.1093/cid/ciaa1198
145. Casco N, Jorge AL, Palmero D, Alffenaar JW, Fox G, Ezz W, et al. TB and COVID-19 Co-Infection: Rationale and Aims of a Global Study. Int J Tuberc Lung Dis (2021) 25:78–80. doi: 10.5588/ijtld.20.0786
146. Chen G, Wu D, Guo W, Cao Y, Huang D, Wang H, et al. Clinical and Immunological Features of Severe and Moderate Coronavirus Disease 2019. J Clin Invest (2020) 130:2620–9. doi: 10.1172/JCI137244
147. Merad M, Martin JC. Pathological Inflammation in Patients With COVID-19: A Key Role for Monocytes and Macrophages. Nat Rev Immunol (2020) 20:355–62. doi: 10.1038/s41577-020-0331-4
148. Liao M, Liu Y, Yuan J, Wen Y, Xu G, Zhao J, et al. The Landscape of Lung Bronchoalveolar Immune Cells in COVID-19 Revealed by Single-Cell RNA Sequencing. MedRxiv (2020) 20026690. doi: 10.1101/2020.02.23.20026690
149. Coperchini F, Chiovato L, Croce L, Magri F, Rotondi M. The Cytokine Storm in COVID-19: An Overview of the Involvement of the Chemokine/Chemokine-Receptor System. Cytokine Growth Factor Rev (2020) 53:25–32. doi: 10.1016/j.cytogfr.2020.05.003
150. Liu B, Li M, Zhou Z, Guan X, Xiang Y. Can We Use Interleukin-6 (IL-6) Blockade for Coronavirus Disease 2019 (COVID-19)-Induced Cytokine Release Syndrome (CRS)? J Autoimmun (2020) 111:102452. doi: 10.1016/j.jaut.2020.102452
151. Xie P, Ma W, Tang H, Liu D. Severe COVID-19: A Review of Recent Progress With a Look Toward the Future. Front Public Health (2020) 8:189. doi: 10.3389/fpubh.2020.00189
152. Etna MP, Giacomini E, Severa M, Coccia EM. Pro- and Anti-Inflammatory Cytokines in Tuberculosis: A Two-Edged Sword in TB Pathogenesis. Semin Immunol (2014) 26:543–51. doi: 10.1016/j.smim.2014.09.011
153. Flesch IE, Kaufmann SH. Role of Cytokines in Tuberculosis. Immunobiology (1993) 189:316–39. doi: 10.1016/S0171-2985(11)80364-5
154. Zheng M, Gao Y, Wang G, Song G, Liu S, Sun D, et al. Functional Exhaustion of Antiviral Lymphocytes in COVID-19 Patients. Cell Mol Immunol (2020) 17:533–5. doi: 10.1038/s41423-020-0402-2
155. Zhao Q, Meng M, Kumar R, Wu Y, Huang J, Deng Y, et al. Lymphopenia Is Associated With Severe Coronavirus Disease 2019 (COVID-19) Infections: A Systemic Review and Meta-Analysis. Int J Infect Dis (2020) 96:131–5. doi: 10.1016/j.ijid.2020.04.086
156. Tan L, Wang Q, Zhang D, Ding J, Huang Q, Tang YQ, et al. Lymphopenia Predicts Disease Severity of COVID-19: A Descriptive and Predictive Study. Signal Transduct Target Ther (2020) 5:33. doi: 10.1038/s41392-020-0148-4
157. Al-Aska A, Al-Anazi AR, Al-Subaei SS, Al-Hedaithy MA, Barry MA, Somily AM, et al. CD4+ T-Lymphopenia in HIV Negative Tuberculous Patients at King Khalid University Hospital in Riyadh, Saudi Arabia. Eur J Med Res (2011) 16:285–8. doi: 10.1186/2047-783X-16-6-285
158. Rahman S, Gudetta B, Fink J, Granath A, Ashenafi S, Aseffa A, et al. Compartmentalization of Immune Responses in Human Tuberculosis: Few CD8+ Effector T Cells But Elevated Levels of FoxP3+ Regulatory T Cells in the Granulomatous Lesions. Am J Pathol (2009) 174:2211–24. doi: 10.2353/ajpath.2009.080941
159. Dheda K, Gumbo T, Gandhi NR, Murray M, Theron G, Udwadia Z, et al. Global Control of Tuberculosis: From Extensively Drug-Resistant to Untreatable Tuberculosis. Lancet Respir Med (2014) 2:321–38. doi: 10.1016/S2213-2600(14)70031-1
160. Yasri S, Wiwanitkit V. Tuberculosis and Novel Wuhan Coronavirus Infection: Pathological Interrelationship. Indian J Tuberc (2020) 67:264. doi: 10.1016/j.ijtb.2020.02.004
161. Ehlers S, Schaible UE. The Granuloma in Tuberculosis: Dynamics of a Host-Pathogen Collusion. Front Immunol (2012) 3:411. doi: 10.3389/fimmu.2012.00411
162. Yang M. Cell Pyroptosis, a Potential Pathogenic Mechanism of 2019-Ncov Infection. Available at SSRN 3527420 (2020) 3527420. doi: 10.2139/ssrn.3527420
163. Sakurai A, Sasaki T, Kato S, Hayashi M, Tsuzuki SI, Ishihara T, et al. Natural History of Asymptomatic SARS-CoV-2 Infection. N Engl J Med (2020) 383:885–6. doi: 10.1056/NEJMc2013020
164. Oei W, Nishiura H. The Relationship Between Tuberculosis and Influenza Death During the Influenza (H1N1) Pandemic From 1918-19. Comput Math Methods Med (2012) 2012:124861. doi: 10.1155/2012/124861
165. Mayer-Barber KD, Sher A. Cytokine and Lipid Mediator Networks in Tuberculosis. Immunol Rev (2015) 264:264–75. doi: 10.1111/imr.12249
166. Harris J, Keane J. How Tumour Necrosis Factor Blockers Interfere With Tuberculosis Immunity. Clin Exp Immunol (2010) 161:1–9. doi: 10.1111/j.1365-2249.2010.04146.x
167. Sereda Y, Korotych O, Klimuk D, Zhurkin D, Solodovnikova V, Grzemska M, et al. Tuberculosis Co-Infection Is Common in Patients Requiring Hospitalization for COVID-19 in Belarus: Mixed-Methods Study. Int J Environ Res Public Health 19 (2022) 19:4370. doi: 10.3390/ijerph19074370
168. Moorlag S, Arts RJW, van Crevel R, Netea MG. Non-Specific Effects of BCG Vaccine on Viral Infections. Clin Microbiol Infect (2019) 25:1473–8. doi: 10.1016/j.cmi.2019.04.020
169. Salem A, Nofal A, Hosny D. Treatment of Common and Plane Warts in Children With Topical Viable Bacillus Calmette-Guerin. Pediatr Dermatol (2013) 30:60–3. doi: 10.1111/j.1525-1470.2012.01848.x
170. Roland M. The Immunopathology of Tuberculosis, the Mode of Action of the Bacillus Calmette–Guérin, of the Tuberculin and of the Immunotherapy. BBRJ (2019) 3:67–76. doi: 10.4103/bbrj.bbrj_5_19
171. Covián C, Fernández-Fierro A, Retamal-Díaz A, Díaz FE, Vasquez AE, Lay MK, et al. BCG-Induced Cross-Protection and Development of Trained Immunity: Implication for Vaccine Design. Front Immunol (2019) 10:2806. doi: 10.3389/fimmu.2019.02806
172. Kleinnijenhuis J, Quintin J, Preijers F, Benn CS, Joosten LA, Jacobs C, et al. Long-Lasting Effects of BCG Vaccination on Both Heterologous Th1/Th17 Responses and Innate Trained Immunity. J Innate Immun (2014) 6:152–8. doi: 10.1159/000355628
173. Netea MG, Joosten LA, Latz E, Mills KH, Natoli G, Stunnenberg HG, et al. Trained Immunity: A Program of Innate Immune Memory in Health and Disease. Sci 352 (2016) 22:aaf1098. doi: 10.1126/science.aaf1098
174. Soto JA, Gálvez NMS, Rivera CA, Palavecino CE, Céspedes PF, Rey-Jurado E, et al. Recombinant BCG Vaccines Reduce Pneumovirus-Caused Airway Pathology by Inducing Protective Humoral Immunity. Front Immunol (2018) 9:2875. doi: 10.3389/fimmu.2018.02875
175. WHO. Bacille Calmette-Guérin (BCG) Vaccination and COVID-19. (2020). Available at: https://www.euro.who.int/__data/assets/pdf_file/0003/446340/Factsheet-May-2020-Vulnerable-populations-during-COVID-19-response-eng.pdf.
176. O’Neill LAJ, Netea MG. BCG-Induced Trained Immunity: Can it Offer Protection Against COVID-19? Nat Rev Immunol (2020) 20:335–7. doi: 10.1038/s41577-020-0337-y
177. Urashima M, Otani K, Hasegawa Y, Akutsu T, Vaccination BCG. And Mortality of COVID-19 Across 173 Countries: An Ecological Study. Int J Environ Res Public Health 17 (2020) 17:5589. doi: 10.3390/ijerph17155589
178. Wickramasinghe D, Wickramasinghe N, Kamburugamuwa SA, Arambepola C, Samarasekera DN. Correlation Between Immunity From BCG and the Morbidity and Mortality of COVID-19. Trop Dis Travel Med Vaccines (2020) 6:17. doi: 10.1186/s40794-020-00117-z
179. Mehta P, McAuley DF, Brown M, Sanchez E, Tattersall RS, Manson JJ. COVID-19: Consider Cytokine Storm Syndromes and Immunosuppression. Lancet (2020) 395:1033–4. doi: 10.1016/S0140-6736(20)30628-0
180. Folegatti PM, Ewer KJ, Aley PK, Angus B, Becker S, Belij-Rammerstorfer S, et al. Safety and Immunogenicity of the ChAdOx1 Ncov-19 Vaccine Against SARS-CoV-2: A Preliminary Report of a Phase 1/2, Single-Blind, Randomised Controlled Trial. Lancet (2020) 396:467–78. doi: 10.1016/S0140-6736(20)31604-4
181. Crisan-Dabija R, Grigorescu C, Pavel CA, Artene B, Popa IV, Cernomaz A, et al. Tuberculosis and COVID-19: Lessons From the Past Viral Outbreaks and Possible Future Outcomes. Can Respir J (2020) 2020:1401053. doi: 10.1155/2020/1401053
182. Meo SA, Abukhalaf AA, Alomar AA, AlMutairi FJ, Usmani AM, Klonoff DC. Impact of Lockdown on COVID-19 Prevalence and Mortality During 2020 Pandemic: Observational Analysis of 27 Countries. Eur J Med Res (2020) 25:56. doi: 10.1186/s40001-020-00456-9
183. Kant S, Tyagi R. The Impact of COVID-19 on Tuberculosis: Challenges and Opportunities. Ther Adv Infect Dis (2021) 8:20499361211016973. doi: 10.1177/20499361211016973
184. Migliori GB, Thong PM, Akkerman O, Alffenaar JW, Álvarez-Navascués F, Assao-Neino MM, et al. Worldwide Effects of Coronavirus Disease Pandemic on Tuberculosis Services, January-April 2020. Emerg Infect Dis (2020) 26:2709–12. doi: 10.3201/eid2611.203163
186. Golli AL, Niţu MF, Turcu F, Popescu M, Ciobanu-Mitrache L, Olteanu M. Tuberculosis Remains a Public Health Problem in Romania. Int J Tuberc Lung Dis (2019) 23:226–31. doi: 10.5588/ijtld.18.0270
187. Raoult D, Zumla A, Locatelli F, Ippolito G, Kroemer G. Coronavirus Infections: Epidemiological, Clinical and Immunological Features and Hypotheses. Cell Stress (2020) 4:66–75. doi: 10.15698/cst2020.04.216
188. Di L, Li Y. The Risk Factor of False-Negative and False-Positive for T-SPOT.TB in Active Tuberculosis. J Clin Lab Anal 32 (2018) 32:e22273. doi: 10.1002/jcla.22273
189. Lu LL, Smith MT, Yu KKQ, Luedemann C, Suscovich TJ, Grace PS, et al. (2019). IFN-γ-Independent Immune Markers of Mycobacterium Tuberculosis Exposure. Nat Med 25:977–87. doi: 10.1038/s41591-019-0441-3
190. Yamasue M, Komiya K, Usagawa Y, Umeki K, Nureki SI, Ando M, et al. Factors Associated With False Negative Interferon-γ Release Assay Results in Patients With Tuberculosis: A Systematic Review With Meta-Analysis. Sci Rep (2020) 10:1607. doi: 10.1038/s41598-020-58459-9
191. Kwon YS, Kim YH, Jeon K, Jeong BH, Ryu YJ, Choi JC, et al. Factors That Predict Negative Results of QuantiFERON-TB Gold In-Tube Test in Patients With Culture-Confirmed Tuberculosis: A Multicenter Retrospective Cohort Study. PLos One (2015) 10:e0129792. doi: 10.1371/journal.pone.0129792
192. Zhang W, Zhao Y, Zhang F, Wang Q, Li T, Liu Z, et al. The Use of Anti-Inflammatory Drugs in the Treatment of People With Severe Coronavirus Disease 2019 (COVID-19): The Perspectives of Clinical Immunologists From China. Clin Immunol (2020) 214:108393. doi: 10.1016/j.clim.2020.108393
193. Li JY, You Z, Wang Q, Zhou ZJ, Qiu Y, Luo R, et al. The Epidemic of 2019-Novel-Coronavirus (2019-Ncov) Pneumonia and Insights for Emerging Infectious Diseases in the Future. Microbes Infect (2020) 22:80–5. doi: 10.1016/j.micinf.2020.02.002
194. Salehi S, Abedi A, Balakrishnan S, Gholamrezanezhad A. Coronavirus Disease 2019 (COVID-19): A Systematic Review of Imaging Findings in 919 Patients. AJR Am J Roentgenol (2020) 215:87–93. doi: 10.2214/AJR.20.23034
195. Starshinova A, Guglielmetti L, Rzhepishevska O, Ekaterincheva O, Zinchenko Y, Kudlay D. Diagnostics and Management of Tuberculosis and COVID-19 in a Patient With Pneumothorax (Clinical Case). J Clin Tuberc Other Mycobact Dis (2021) 24:100259. doi: 10.1016/j.jctube.2021.100259
196. XINHUANET.com. Indonesia Steps Up Battle Against Tuberculosis Amid COVID-19 Pandemic (2020) (Accessed May 10, 2022).
197. Devidiscourse, Provinces Experimenting With Combining TB and COVID-19 Screening (2020) (Accessed December 20, 2021).
198. WHO-Africa. Kaduna State and WHO Scale Up COVID-19 and TB Search With Mobile Testing in Communities (2020) (Accessed December 20, 2021).
199. Government-of-India, Government of India Ministry of Health and Family Welfare. Rapid Response Plan to Mitigate Impact of COVID-19 Pandemic on TB Epidemic and National TB Elimination Program (NTEP) Activities in India-Reg (2020) (Accessed December 20, 2021).
200. Ruhwald M, Hannay E, Sarin S, Kao K, Sen R, Chadha S. Considerations for Simultaneous Testing of COVID-19 and Tuberculosis in High-Burden Countries. Lancet Glob Health (2022) 10:e465–6. doi: 10.1016/S2214-109X(22)00002-X
201. Moran A, Mphahlele M, Mvusi L, Dlamini C, Ahmedov S, AlMossawi HJ, et al. Learning From Tuberculosis: COVID-19 Highlights the Need for More Robust Infection Control Policy. J Glob Health (2020) 10:020328. doi: 10.7189/jogh.10.020328
Keywords: COVID-19, tuberculosis, coinfection, SARS-CoV-2-M. tuberculosis pathogenesis, BCG vaccination
Citation: Shah T, Shah Z, Yasmeen N, Baloch Z and Xia X (2022) Pathogenesis of SARS-CoV-2 and Mycobacterium tuberculosis Coinfection. Front. Immunol. 13:909011. doi: 10.3389/fimmu.2022.909011
Received: 31 March 2022; Accepted: 23 May 2022;
Published: 16 June 2022.
Edited by:
Tengchuan Jin, University of Science and Technology of China, ChinaReviewed by:
Alexandru Burlacu, Grigore T. Popa University of Medicine and Pharmacy, RomaniaMonira Sarmin, International Centre for Diarrhoeal Disease Research (ICDDR), Bangladesh
Copyright © 2022 Shah, Shah, Yasmeen, Baloch and Xia. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zulqarnain Baloch, em5iYWxvb2NoQHlhaG9vLmNvbQ==; Xueshan Xia, b2xpdmVyeGlhMjAwMEBhbGl5dW4uY29t