 Hannan Ai
Hannan Ai Bizhou Li
Bizhou Li Fanmei Meng
Fanmei Meng Yuncan Ai
Yuncan Ai
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Immunol. , 14 June 2022
Sec. Microbial Immunology
Volume 13 - 2022 | https://doi.org/10.3389/fimmu.2022.907646
This article is part of the Research Topic Insights in Microbial Immunology: 2021 View all 5 articles
Colon ascendens stent peritonitis (CASP) surgery induces a leakage of intestinal contents which may cause polymicrobial sepsis related to post-operative failure of remote multi-organs (including kidney, liver, lung and heart) and possible death from systemic syndromes. Mechanisms underlying such phenomena remain unclear. This article aims to elucidate the mechanisms underlying the CASP-model sepsis by analyzing real-world GEO data (GSE24327_A, B and C) generated from mice spleen 12 hours after a CASP-surgery in septic MyD88-deficient and wildtype mice, compared with untreated wildtype mice. Firstly, we identify and characterize 21 KO MyD88-associated signaling pathways, on which true key regulators (including ligands, receptors, adaptors, transducers, transcriptional factors and cytokines) are marked, which were coordinately, significantly, and differentially expressed at the systems-level, thus providing massive potential biomarkers that warrant experimental validations in the future. Secondly, we observe the full range of polymicrobial (viral, bacterial, and parasitic) sepsis triggered by the CASP-surgery by comparing the coordinated up- or down-regulations of true regulators among the experimental treatments born by the three data under study. Finally, we discuss the observed phenomena of “systemic syndrome”, “cytokine storm” and “KO MyD88 attenuation”, as well as the proposed hypothesis of “spleen-mediated immune-cell infiltration”. Together, our results provide novel insights into a better understanding of innate immune responses triggered by the CASP-model sepsis in both wildtype and MyD88-deficient mice at the systems-level in a broader vision. This may serve as a model for humans and ultimately guide formulating the research paradigms and composite strategies for the early diagnosis and prevention of sepsis.
Infectious diseases have led mortality rates worldwide. Sepsis is the prevalent cause of death in the intensive care unit (1–3). Sepsis occurs when the immune response to an infection becomes dysregulated, resulting in life-threatening organ dysfunction (4). Sepsis is scientifically termed as a systemic syndrome of persistent and overwhelming inflammation that occurs at the early stage of infection (4–6). It has been perceived that bacterial infection may cause sepsis (7–10); while viral infection may produce cytokine storm (11–13); and parasitic infection may also yield sepsis (10). It is well-known that Toll-like receptors (TLRs) including TLR2, TLR4 and TLR9 contribute to polymicrobial sepsis (14, 15). These TLRs play crucial roles in signaling via the common myeloid differentiation factor 88 (MyD88)-dependent pathways (5, 16–19). TLR4 also signals through MyD88-independent but TIR domain-containing adaptor inducing IFNβ-mediated transcription factor (Trif)-dependent pathway (10, 17, 18, 20). Based on the bench-experiments of multiplex immunoassay and flow cytometry analysis in deficient mice, the cytokine production, neutrophil migration, and phagocytic function have been suggested to play critical roles in the pathogenesis of sepsis (21–23). Cytokines and chemokines including IL-6 (24), IL-10 (7, 24), CD14 (17), to name a few, were suggested as potential biomarkers for early diagnosis (before organ dysfunction occurs) of sepsis (25). But due to evolutionary discordance between human and mouse innate immune signaling (26), these may not necessarily be true biomarkers (4).
The colon ascendens stent peritonitis (CASP) surgery induces a leakage of intestinal contents, causing polymicrobial sepsis related to post-operative failure of multi-organs (including heart, spleen, kidney, liver and lung) and possible death from syndromes (4). The CASP-model sepsis is a standardized approach to understanding pathogenesis of polymicrobial sepsis (8, 23, 24, 27–32). Deficient mice provide ideal models to investigate the roles of specific candidate biomarkers (24, 33). Transcriptomic analysis of mRNAs profiling on microarray, coupled with bioinformatics analysis, is powerful to decipher true key regulators through mapping them onto true KEGG pathways (34). In fact, KEGG pathways are manually curated based on pre-validated experimental and/or literature evidence (35, 36). The true key regulators identified by pathway enrichment analysis are rendered on KEGG pathways, which not only yields high efficacy of identification, but also offers true broader vision beyond traditional detection approaches (10, 24). As an imminent frontier in the field, top-down data-driven pathway enrichment analysis should become the advanced approach to investigate sepsis pathogenesis.
The real-world GSE24327 data were generated from samples recovered from spleens 12 hours after the CASP-model sepsis in MyD88-deficient and wildtype mice (24). The previous bench-experiments assessed the expression levels of regulators, including MFI (CD11b), CXCL10, MIP-2 (CXCL2), RANTES (CCL5), IL-17, IL-6, IL-10, IL-12, Ifng, Ifnb, IL-15, Ifit3, Ccl12, Rsad2 and IFNβ, by which a putative signaling pathway was determined, responsible for the innate immune responses triggered by the CASP-surgery (24). These data were not subjected to bioinformatics analysis like our perspectives and we are curious about additional occurences beyond the documented stories (24).
This study aims to elucidate mechanisms underlying the CASP-model sepsis at the systems-level through analyzing GSE24327 data (24) via top-down data-driven analysis. We discuss the observed phenomena of “systemic syndrome”, “cytokine storm”, and “KO MyD88 attenuation”, as well as the proposed hypothesis of “spleen-mediated immune-cell infiltration”. Our results provide novel insights into a better understanding of the CASP-model sepsis in mice, which may serve as a model for humans, to ultimately guide formulating the research paradigms and composite strategies for early diagnosis and prevention of severe sepsis.
We downloaded GSE24327 data from https://ncbi.nlm.nih.gov/geo/GSE24327. To select the significantly, differentially expressed genes (DEGs), we designed three subtypes of data according to the original bench-experiments (24): GSE24327_A (a. septic KO MyD88 vs. septic WT) for comparing septic null (Myd88–/–) with septic wildtype mice; GSE24327_B (b. septic KO MyD88 vs. untreated WT) for comparing septic null (Myd88–/–) with untreated wildtype mice; and GSE24327_C (c. septic WT vs. untreated WT) for comparing septic wildtype with untreated wildtype mice.
The PathwayKO package is available at https://github.com/allenaigit/pathwayko/. Briefly, the PathwayKO package is systems software, rather than a standalone application algorithm, which comprises multiple modules with diverse dependencies, allowing users to conduct integrated processes in a pipeline fashion, such as (I) preprocessing, (II) ROC-AUC calculating, (III) statistics analyzing, and (IV) visualizing. Users who might lack knowledge and/or experience would seek help from professionals and consult the offered users’ manual to understand our interpretations. It currently incorporates the state-of-the-art methods of pathway analysis, statistics analysis, and visualizing analysis. The non-topology-based methods of pathway analysis include SAFE (37), GSEA (38), GSA (39) and PADOG (40). The topology-based methods of pathway analysis include ROntoTools_PE (41), ROntoTools_pDIS (42) and SPIA (43). The statistics analysis methods include the changepoint package (44) and the pROC package (45). The visualizing analysis methods include the pROC package (45) and the Pathview package (34). Certain metrics are important for statistics comparison. The Youden’s best p-value (46) is employed to choose the best point on the ROC curve. AUC (with 95% CI, confidence interval) for a full area under the entire ROC-curve, and partial AUCs (pAUC_SP and pAUC_SE) for regions focusing on the 90–100% of specificity and sensitivity, in both original and corrected formats (47), are deployed to rank methods. The Adj.p-value is used to control the false discovery rate (FDR) for multiple testing (48, 49). Most importantly, the PathwayKO package has an advantage to conduct automatic selection of differentially expressed genes (DEGs) because it adapts the change-point method (44) based on HES threshold (50) that are automatically determined on the distribution of edge scores of each data, into which the fold-change and probability values of genes at the systems-level have been integrated.
To install the PathwayKO package, one needs to create and enter into a user directory (PathwayKO_demo_run), and type command lines:
$ cd/home/PathwayKO_demo_run
PathwayKO_demo_run$ R
> library(devtools)
> devtools::install_github(“allenaigit/pathwayko”)
To get started with the package, the user needs to run a demo:
$ cd/home/PathwayKO_demo_run
PathwayKO_demo_run$ R
> library(pathwayko)
> pathwayko_demo()
Batch-execution is conducted with parallel computations, which produces resulting output directories, each containing a group of desired output-files. The user completed a desired job by following instructions in the User’s manual, publicly available at https://github.com/allenaigit/pathwayko/tree/main/inst/docs/Users_manual.pdf.
We estimated the correlation between the expression level of key regulators and the infiltration level of core immune cells (CICs) (B, CD8+T, CD4+T, neutrophil, macrophage and dendritic cells) in human cancer cohorts by utilizing cohort database TIMER2.0 (https://cistrome.shinyapps.io/timer) (51).
Top-panel (Figure 1) displays the landscape of unbiased true DEGs automatically identified from GSE24327_A (408 DEGs), B (266 DEGs), and C (648 DEGs) data (Supplementary Tables S1–S3). Such true DEGs constitute putative regulatory networks, as shown by the landscapes in bottom-panel (Figure 1). Obviously, such true DEGs vary drastically among data, reflecting real-world variations among the bench-experimental treatments of (A), (B) and (C) under study. Note that Figure 1 only shows the landscapes of these true DEGs (named as Edge Score Histogram) and their putative regulatory networks (named as High-Edges plot) in a broader vision at the systems-level. Each landscape may comprise hundreds or thousands of DEGs, which are unreadable by humans. Among such true DEGs, key regulators will be specifically identified, precisely located, and differentially marked on target signaling pathways via the subsequent analyses throughout the current study.
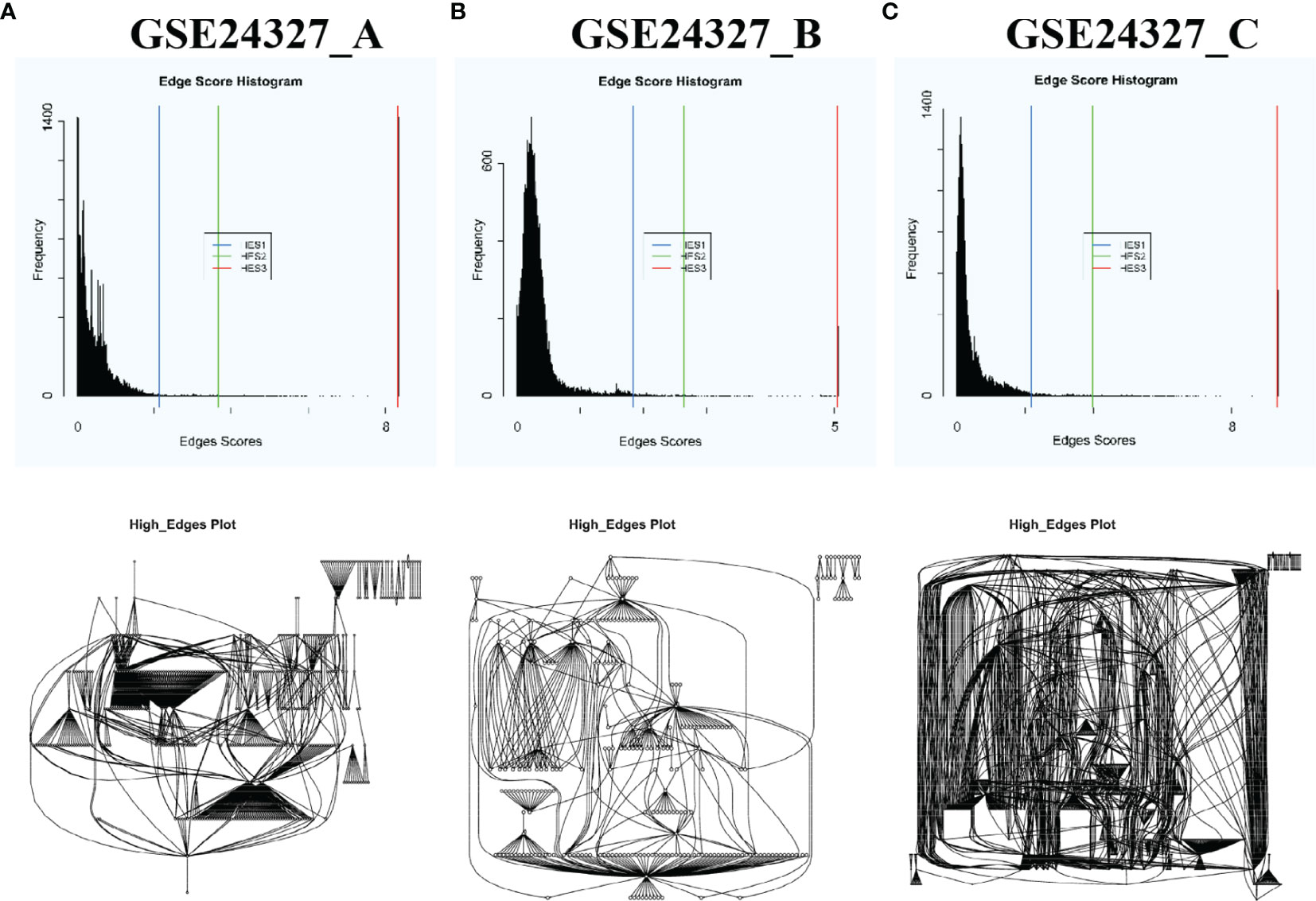
Figure 1 Automatic selection of DEGs with each HES1 threshold based on the distribution of edge scores and automatic construction of putative regulatory networks of DEGs of each data by using the PathwayKO package. Top-panel shows the landscape of true DEGs of each data. Bottom-panel displays the landscape of putative regulatory networks constituted by the true DEGs of each data. These landscapes drastically vary among data in a broader vision, reflecting real-world differences of the bench-experimental treatments: (A) septic KO MyD88 vs. septic wildtype (WT), (B) septic KO MyD88 vs. untreated WT, and (C) septic WT vs. untreated WT. See the main text.
Firstly, we compared three popular methods of pathway enrichment analysis via the PathwayKO package benchmarked on each data under study (Figure 2). The SPIA method (43) has an overall better performance over ROntoToosls_PE (41) and ROntoTools_PDIS (42) in terms of the ROC-curve-based statistics metrics, such as AUC (with 95% CI), pAUC_SP, pAUC_SE, and Youden’s best p-threshold (Figure 2). The SPIA method is thus chosen to conduct subsequent analyses throughout this study.
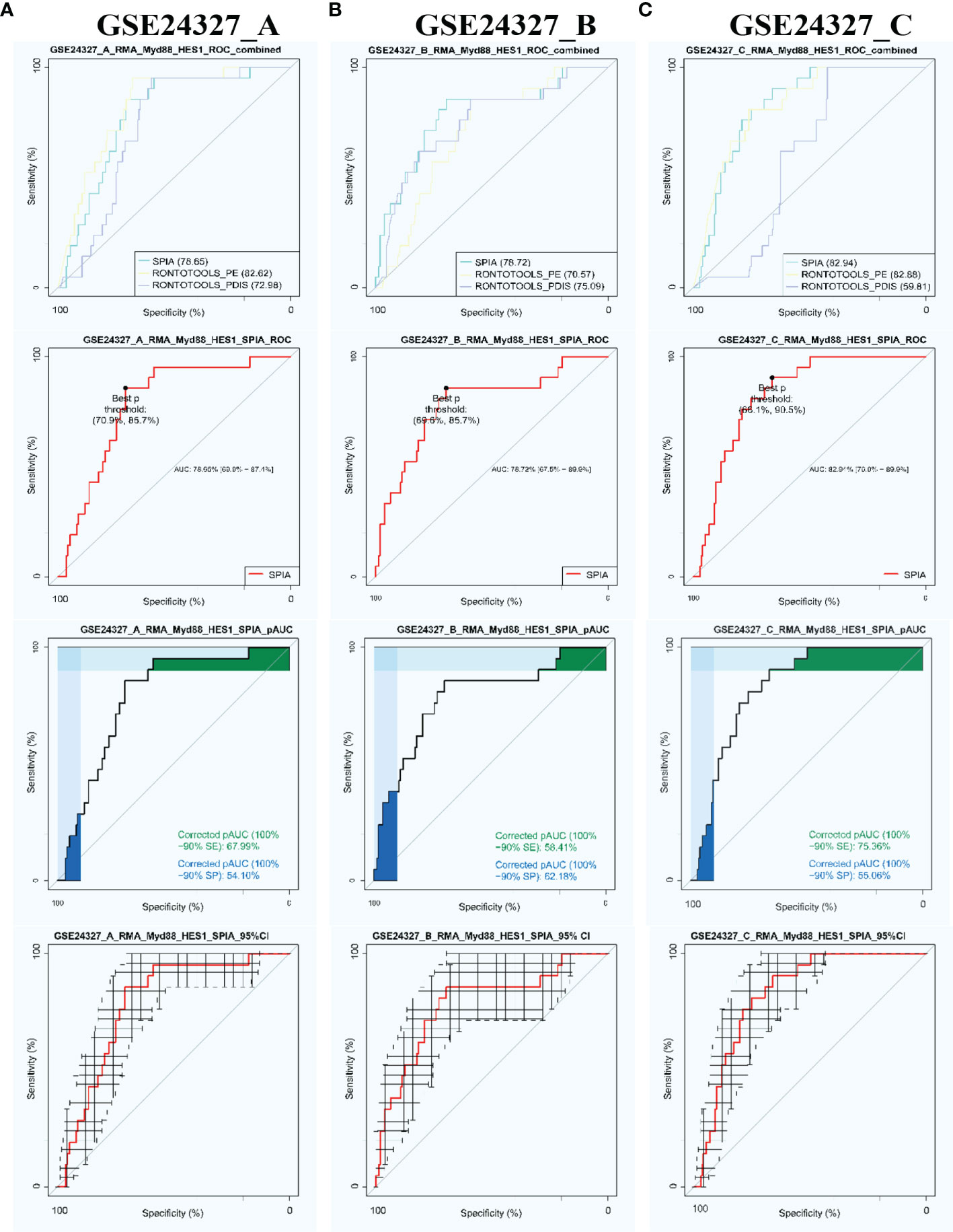
Figure 2 Automatic identification of target signaling pathways by using the PathwayKO package. The SPIA method demonstrates advantageous performance in view of the ROC-curve-based statistics metrics on each data. (A) GSE24327_A; (B) GSE24327_B; (C) GSE24327_C.
Secondly, we used the SPIA method via the PathwayKO package to identify target signaling pathways with each HES1 threshold from each data (see Figure 1). As a result, we automatically identified the target 21 KO MyD88-associated signaling pathways from each data (Table 1). All pathways are identified as significant (pGFdr <0.0001) from the (C) treatment (septic wildtype vs. untreated wildtype). Only 1 pathway is identified as insignificant (pGFdr >0.05) from the (A) treatment (septic KO MyD88 mice vs. septic wildtype mice). Only 2 pathways are identified as insignificant (pGFdr >0.05) from the (B) treatment (septic KO MyD88 mice vs. untreated wildtype mice). These findings suggest that such insignificant pathways are likely dependent on MyD88-deficiency, i.e., the KO MyD88 deficiency has attenuated signaling. For instance, the number of true DEGs on the Toll-like signaling pathway is remarkably decreased from 52 in (C) treatment to 50 in (B) treatment, and to 34 in (A) treatment (Table 1).
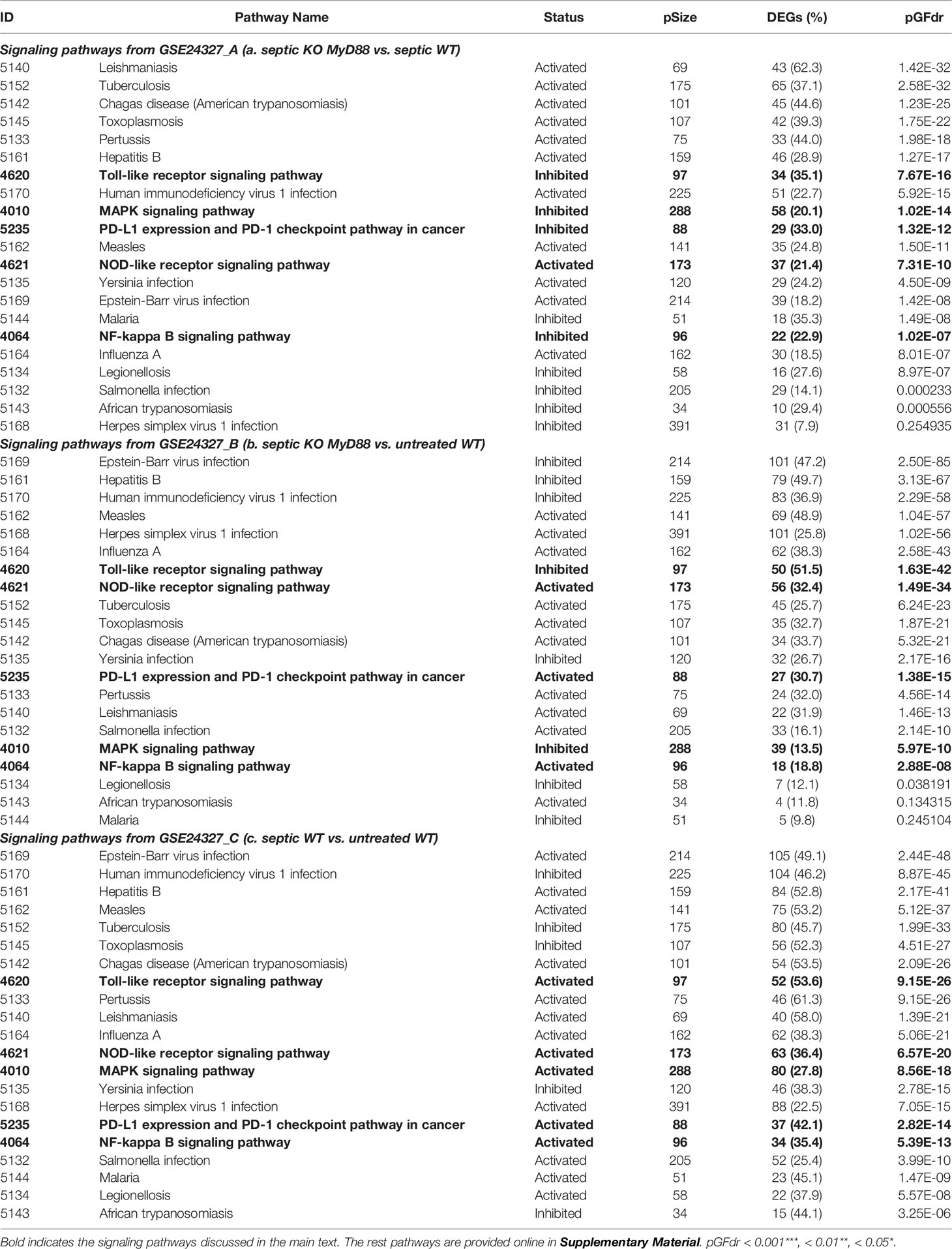
Table 1 Target MyD88-associated signaling pathways identified by SPIA.
Each of the above 21 target signaling pathways (Table 1) contains MyD88, which is impaired by KO MyD88 in deficient mice. We named the Toll-like receptor signaling pathway (mmu04620) as the basal target pathway that is solely responsible for the KO MyD88 phenotype (Figure 3). We also named other target pathways as composite target pathways because they embed the named basal signaling pathway (mmu04620) coupled with others. Some examples of common core pathways are highlighted (Figures 3–7), while the rest of infection-related pathways are provided online (Supplementary Figures S1–S16). Both are in the context of comparing innate immune responses, triggered 12 hours after the CASP-surgery, among the experimental treatments born by the three subtypes of data under study.
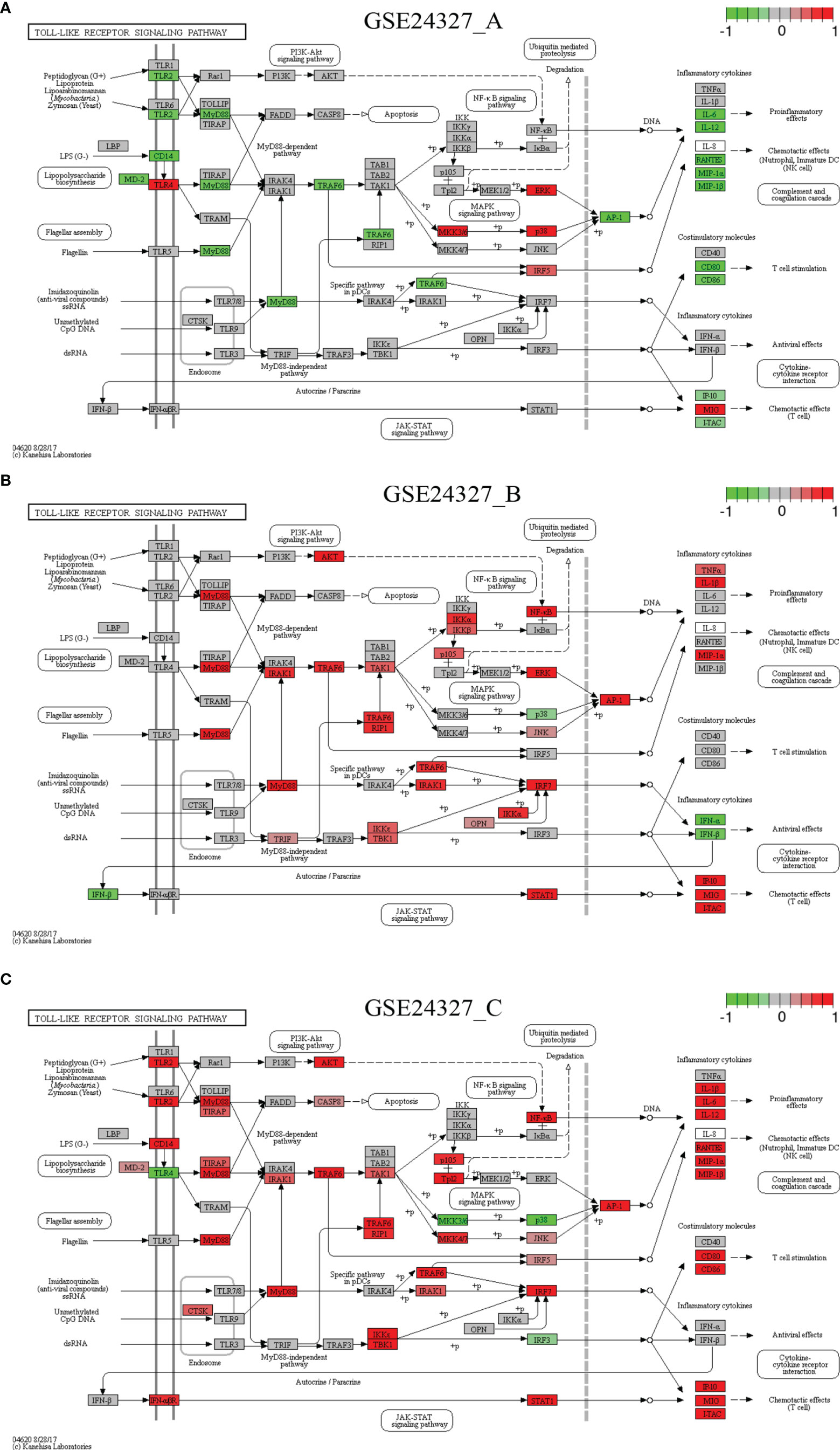
Figure 3 Comparison on the rendered Toll-like receptor signaling pathway (mmu04620). (A) GSE24327_A; (B) GSE24327_B; (C) GSE24327_C.
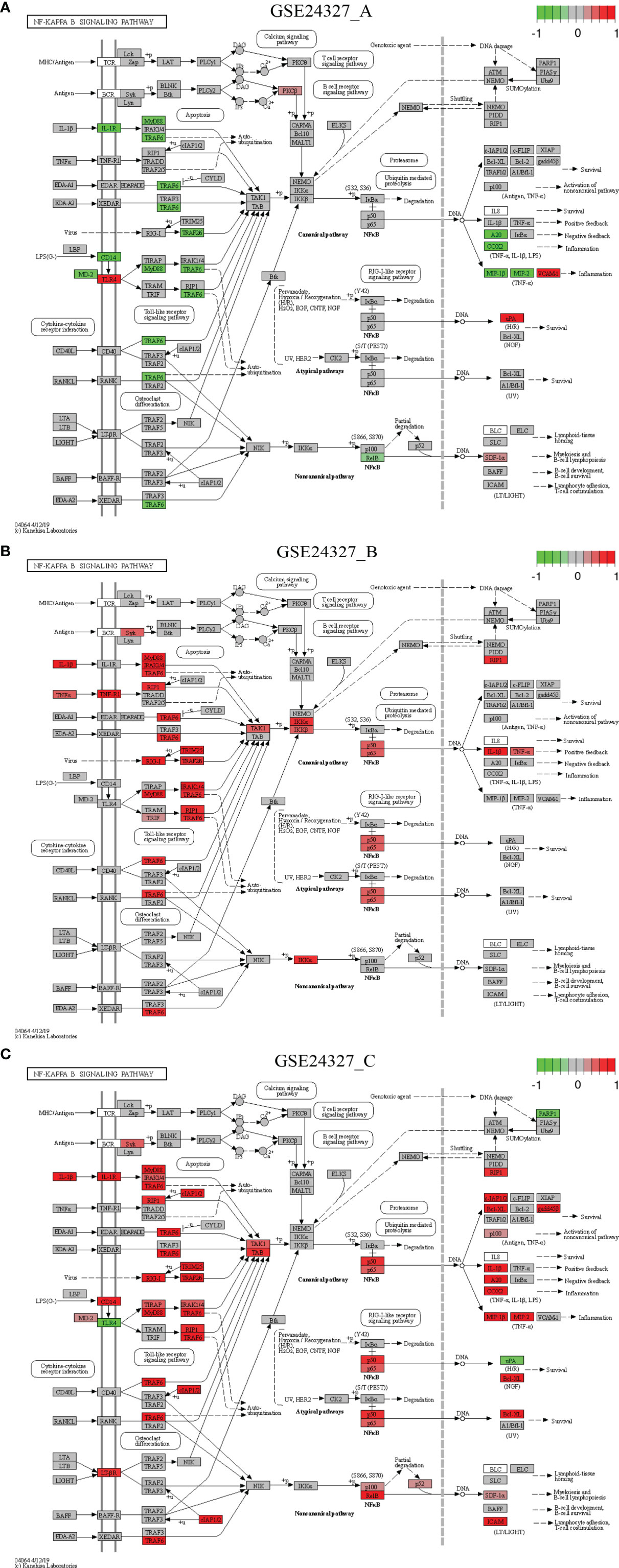
Figure 4 Comparison on the rendered NF-κB signaling pathway (mmu04064). (A) GSE24327_A; (B) GSE24327_B; (C) GSE24327_C.
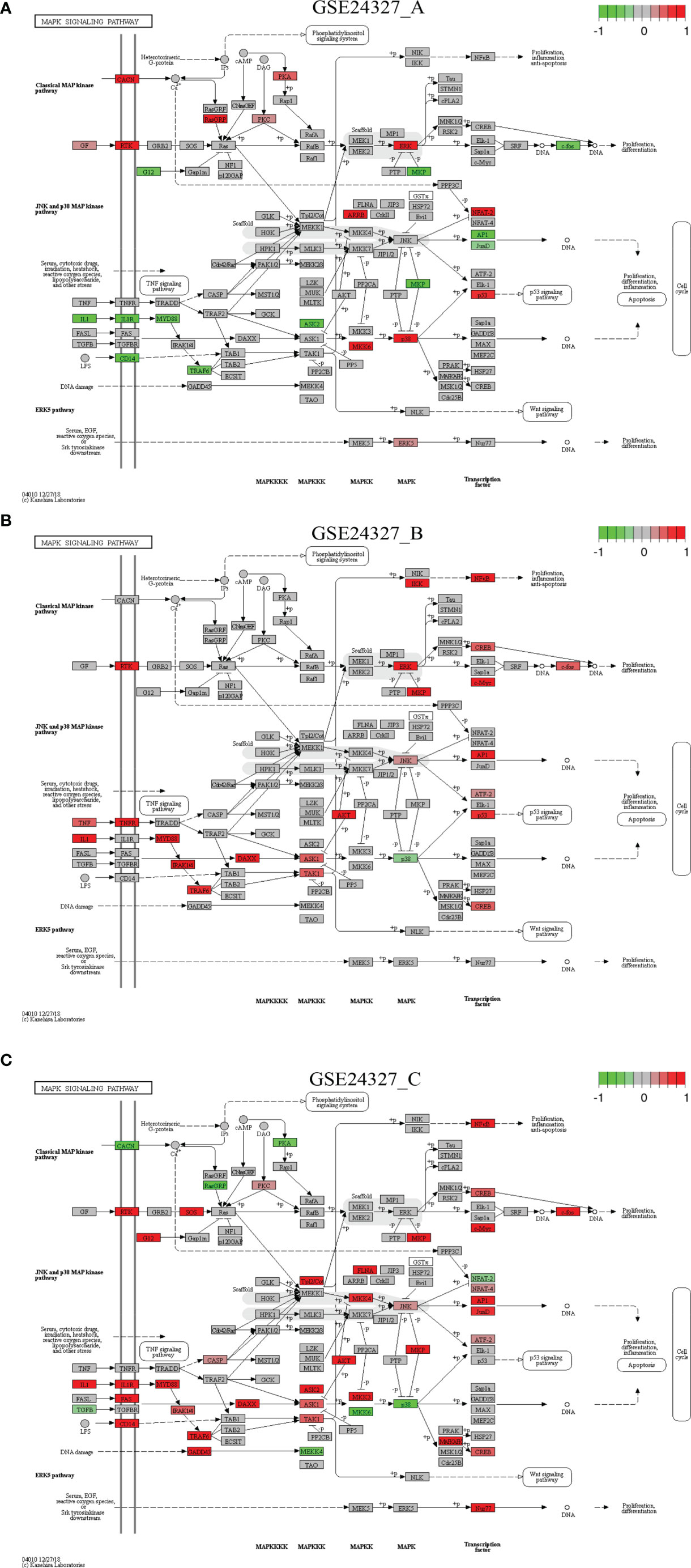
Figure 5 Comparison on the rendered MAPK signaling pathway (mmu04010). (A) GSE24327_A; (B) GSE24327_B; (C) GSE24327_C.
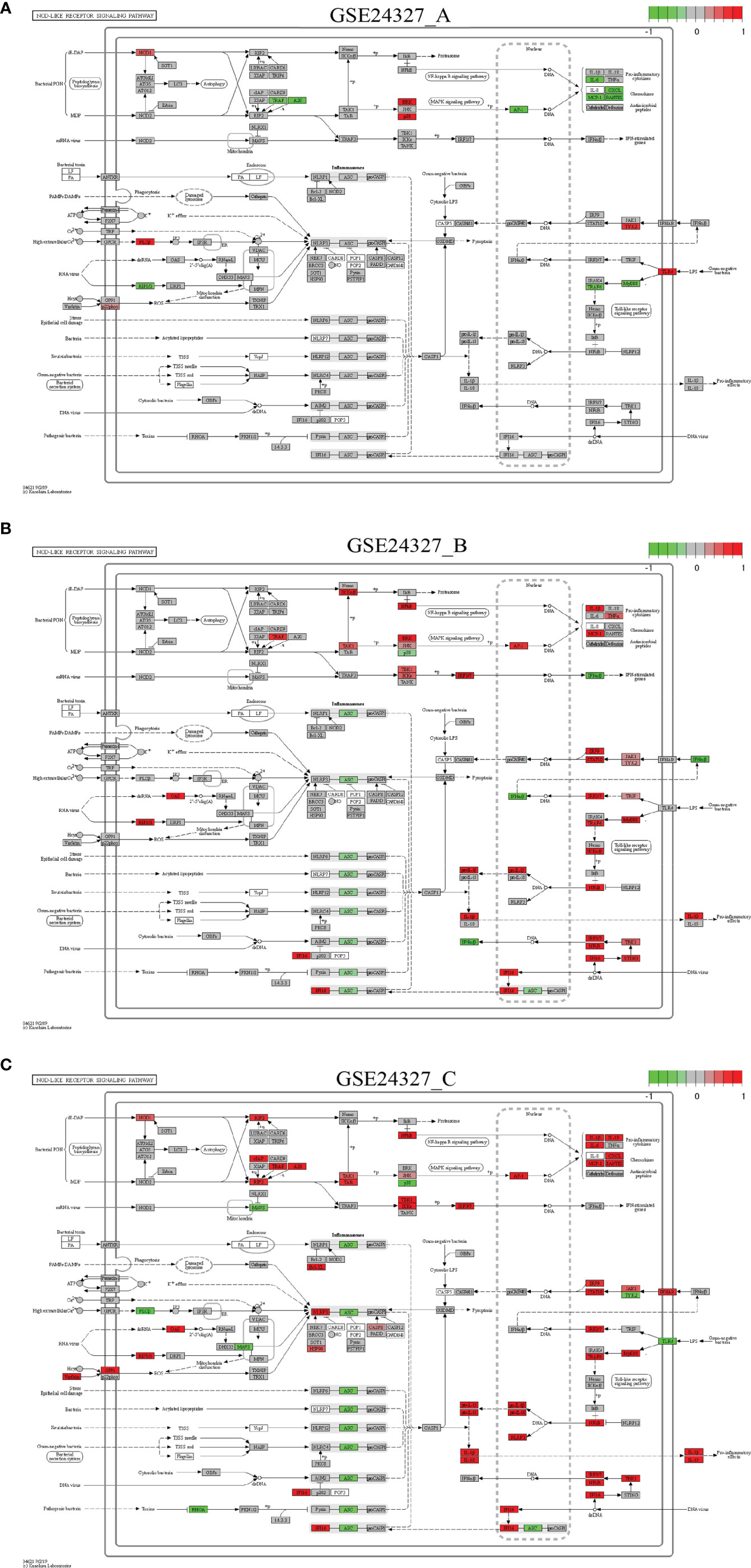
Figure 6 Comparison on the rendered NOD-like receptor signaling pathway (mmu04621). (A) GSE24327_A; (B) GSE24327_B; (C) GSE24327_C.
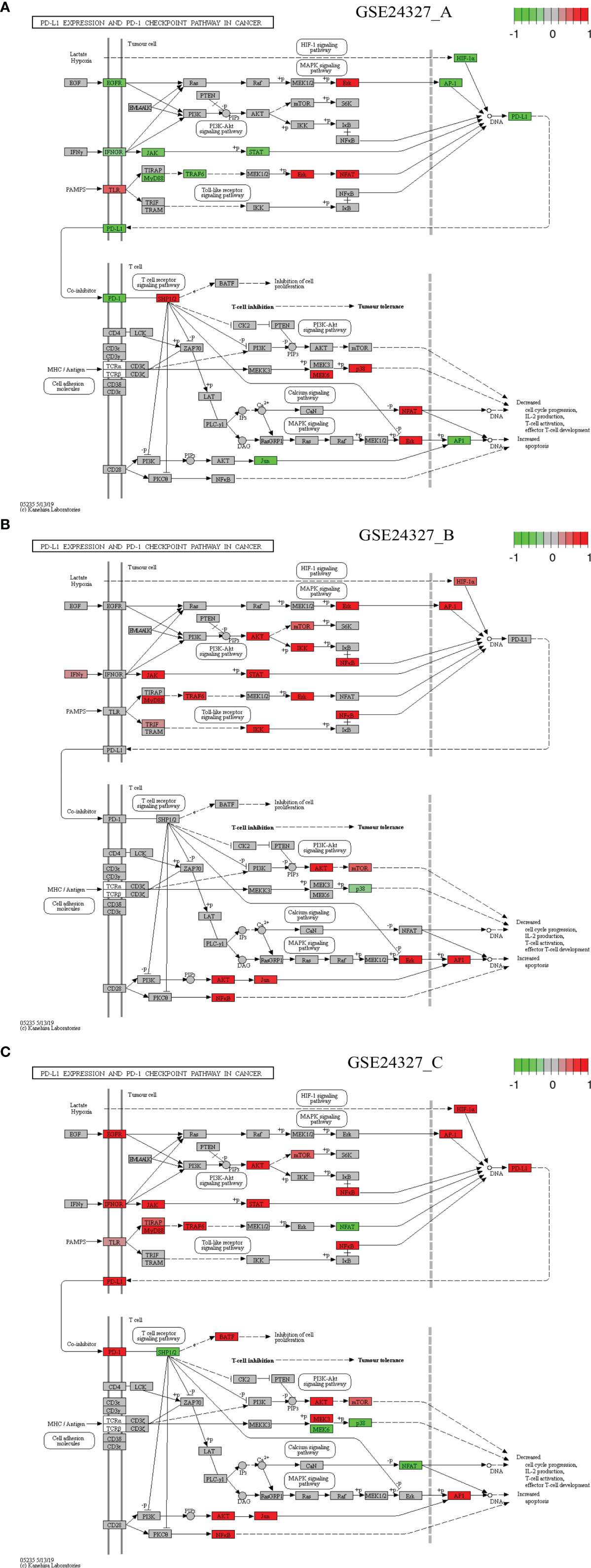
Figure 7 Comparison on the rendered PD-L1 expression and PD-1 checkpoint pathway in cancer (mmu05235). (A) GSE24327_A; (B) GSE24327_B; (C) GSE24327_C.
There are common features of comparisons on the target signaling pathways: 1) the (C) treatment provokes the strongest innate immune responses at the systems-level at 12 hours after the CASP-surgery, 2) the (A) or (B) treatment causes relatively weaker innate immune responses at the systems-level at 12 hours after the CASP-surgery, and 3) some regulators on the target signaling pathways are dependent on the KO MyD88-deficiency, but others are independent of it.
Case 1: The Toll-like receptor signaling pathway (mmu04620) is the basal target pathway that is solely responsible for the KO MyD88 phenotype (Figure 3). It is a significant pathway (pGFdr <0.0001) identified from all three data (see Table 1). This pathway comprises 97 critical genes, ofs which 34 in (A), 50 in (B) and 52 in (C) treatment (see Table 1) are true DEGs that are coordinately, significantly, differentially up- or down-regulated at 12 hours after the CASP-surgery.
Firstly, in the (C) treatment, MyD88 is up-regulated, where the majority of ligands, receptors, adaptors and transducers (MD-2, TLR1, TLR2, CD14, INFαβR, CTSK, TIRAP, AKT, CASP8, IRAK1, TRAF6, TAK1, RIP1, IKK3/ϵ, TBK1, IRF7, IRF5, JNK, MKK4/7, Tp12, p105 and NFκB) are up-regulated. Only a few (TLR4, IRF3, MKK3/6 and p38) are down-regulated. Therefore, the downstream effectors [AP-1 (Jun, c-Jun), IL-1β, IL-6, IL12, RANTES (CCL5), MIP-1α (CCL3), MIP-1β (CCL4), CD80, CD86, IP-10 (CXCL10), MIG (CXCL9) and ITAC] are collectively up-regulated at the systems-level.
Secondly, for instance, in the (A) treatment, the majority of regulators are down-regulated, except for the few (TLR4, IKK3/ϵ, ERK, p38, IRF5 and MIG) that remain up-regulated. In particular, members of the major axis (MyD88, MD-2, CD14, TLR2, TRAF6 and AP-1) are drastically down-regulated and the downstream effectors (IL-6, IL-12, RANTES, MIP-1α, MIP-1β, CD80, CD86, IP-10 and ITAC) are drastically attenuated at the systems-level. These data suggest that the KO MyD88-deficiency had attenuated the expression level of major inflammatory effectors (IL-6, IL-12, RANTES, MIP-1α, MIP-1β, CD80, CD86, IP-10 and ITAC) at the systems-level in deficient mice.
Case 2: The NF-κB signaling pathway (mmu04064) is a composite target pathway, which embeds the Toll-like signaling pathway, B-cell receptor signaling pathway, T-cell receptor signaling pathway, and RIG-I-like receptor signaling pathway (Figure 4). It is a significant (pGFdr <0.0001) target pathway identified from all three data (Table 1). Among 96 critical genes, 22 in (A), 18 in (B) and 34 in (C) treatment (see Table 1) are true DEGs that are coordinately, significantly, differentially up- or down-regulated at 12 hours after the CASP-surgery.
Firstly, in the (C) treatment, MyD88 is up-regulated, of which the majority of ligands, receptors, adaptors and transducers (IL-1β, IL-1R, MD-2, CD14, LT-βR, MyD88, TRAF6, Syk, IRAK1/4, RIP1, RIG-1, TIRAP, cIAP1/2, TRIM25, TRAF2/6, TAK1, TAB, p50, p52, p65, p100, RelB, RIP1, Bcl-XL, gadd45β, IL-1β, A20, COX2, MIP-1β (CCL4), MIP-2 (CXCL2), SDF-1α (CXCL12) and ICAM (CD54)) are up-regulated. Only a few (TLR4, uPA and PARP-1) are down-regulated. These key regulators are related to inflammation, survival, myeloiesis, B-cell lymphopoiesis, lymphocyte adhesion and T-cell co-stimulation at the systems-level.
Secondly, in the (A) treatment, the majority of regulators [MD-2, CD14, IL-1R, MyD88, TRAF6, RelB, A20, COX2, MIP-1β (CCL4) and MIP-2 (CXCL2)] are down-regulated except for a few [TLR4, PKCβ, uPA, VCNM1 and SDF-1α (CXCL12)] that are up-regulated. These regulators are related to inflammation, survival, myeloiesis and B-cell lymphopoiesis at the systems-level.
Case 3: The MAPK signaling pathway (mmu04010) is a composite target pathway, which embeds the Toll-like signaling pathway, TNF signaling pathway, and p53 signaling pathway (Figure 5). It is identified as a significant (pGFdr <0.0001) target pathway from all three data (Table 1). Among 288 critical genes, 58 in (A), 39 in (B) and 80 in (C) treatment (see Table 1) are true DEGs that are coordinately, significantly, differentially up- or down-regulated at 12 hours after the CASP-surgery. The classic MAPK pathway differs from the JNK/p38 MAPK pathway because the latter further incorporates the Toll-like signaling pathway, TNF signaling pathway, p53 signaling pathway and ERK5 signaling pathway.
Firstly, in the (C) treatment: 1) on the classic MAPK pathway, regulators (RTK, G12, SOS, PKC, MKP, NFκB, CREB, c-Myc and c-Fos) are up-regulated, while a few (CACN, RasGRP and PKA) are down-regulated, and 2) on the JNK/p38 MAPK pathway, the majority of regulators (IL1, IL1R, CD14, MyD88, FAS, IRAK1/4, TRAF6, GADD45, CASP, DAXX, TAK1, ASK1, ASK2, Tpl2/Co, AKT, MKK3, MKK4, FLNA, JNK, MKP, MAFKAFK, CREB, NFAT4, AP1, JunD, ATF-2 and Nur77) are up-regulated, while only a few (TGFB, MEKK4, MKK6, p38, and NFAT2) are down-regulated. These key regulators positively contribute to cell cycle regulation, such as proliferation, differentiation, inflammation, and anti-apoptosis at the systems-level.
Secondly, for instance, in the (A) treatment: 1) on the classic MAPK pathway, regulators (GF, RTK, CACN, RasGRP, PKA, PKC, and ERK) are up-regulated, while a few (G12, MKP and c-Fos) are down-regulated, and 2) on the JNK/p38 MAPK pathway, regulators (ARRB, MKK6, p38, p53, NFAT2, and ERK5) are up-regulated, while a few (IL1, IL1R, CD14, MyD88, TRAF6, ASK2, MKP, AP1, and JunD) are down-regulated at the systems-level.
Case 4: The NOD-like receptor signaling pathway (mmu04621) is a composite target pathway, which embeds the Toll-like signaling pathway, NFκB signaling pathway and MAPK signaling pathway (Figure 6). It is identified as a significant (pGFdr <0.0001) target pathway from all three data (Table 1). Among 173 critical genes, 37 in (A), 56 in (B), and 63 in (C) treatment (see Table 1) are true DEGs that are coordinately, significantly, differentially up- or down-regulated at 12 hours after the CASP-surgery.
Firstly, in the (C) treatment, MyD88 is up-regulated, around which the majority of regulators (IRF3/7, TRAF6, NFκB, JAK1, IFNAR, IRF9, STAT1/2, TBK1, IF116, NIRP3, AP-1, JNK, TAK1, TAB, IKKϵ, RIP2, cIAP, TRAF, A20, RIF2, NOD1, Bcl-XL, NLRP3, CASP8, HSP90, RIP1/3, OAS, GP91 and Visfatin) are up-regulated. Only a few regulators (TLR4, TYK2, ASC, MAYS, PLCβ, MAVS, RHOA and p38) are down-regulated. The downstream effectors [IL-1β, IL-18, pro-IL-1β, pro-IL-18, IL-6, CXCL, MCP-1 (CCL2) and RANTES (CCL5)] are accordingly up-regulated. Note that ASC means antibody secreting cells (18).
Secondly, in the (A) treatment, only a few regulators (MyD88, TRAF6, TRAF, A20, AP-1 and RIP1/3) are down-regulated, while a few (TLR4, TYK2, NOD1, ERK, p38, PLCβ and p22phox) are up-regulated. The downstream effectors (IL-6, CXCL, MCP-1 and RANTES) are accordingly down-regulated at the systems-level.
Case 5: The PD-L1 expression and PD-1 checkpoint pathway in cancer (mmu05235) is a composite target signaling pathway, which embeds the Toll-like signaling pathway, MAPK signaling pathway, PI3K-Akt signaling pathway, HIF-1 signaling pathway, and T-cell receptor signaling pathway (Figure 7). It is a significant (pGFdr <0.0001) target pathway identified from all three data (Table 1). Among 88 critical genes, 29 in (A), 27 in (B), and 37 in (C) treatment (see Table 1) are true DEGs that are coordinately, significantly, differentially up- or down-regulated at 12 hours after the CASP-surgery.
Firstly, in the (C) treatment: 1) on the Toll-like receptor signaling pathway, a few regulators (MyD88, TLR, TIRAP, TRAF6 and NFκB) are up-regulated, while only NFAT is down-regulated; 2) on the HIF-1 signaling pathway, HIF-1α is up-regulated; 3) on the MAPK signaling pathway, only EGFR and AP-1 are up-regulated; 4) on the PI3K-AKT signaling pathway, a few regulators (IFNGR, JAK, STAT, AKT, mTOR and NFκB) are up-regulated. All of these key regulators positively contribute to the increased production of PD-L1; and 5) on the T-cell receptor signaling pathway, the major regulators (PD-L1, PD-1, BATF, AKT, mTOR, NFκB, MEK3, JUN and AP-1) are up-regulated, while a few (SHP1/2, MEK6, p38 and NFAT) are down-regulated. All of these key regulators positively contribute to the increased apoptosis but decreased cell cycle progression, IL-2 production, T-cell activation, and effector T-cell development at the systems-level.
Secondly, in the (A) treatment: 1) on the Toll-like receptor signaling pathway, a few regulators (TLR, ERK and NFAT) are up-regulated, while a few (MyD88 and TRAF6) are down-regulated; 2) on the HIF-1 signaling pathway, only HIF-1α is down-regulated; 3) on the MAPK signaling pathway, only EGFR and AP-1 are down-regulated, while only ERK is up-regulated; and 4) on the PI3K-AKT signaling pathway, a few regulators (IFNGR, JAK and STAT) are down-regulated. All of these key regulators contribute to the decreased production of PD-L1 and 5) on the T-cell receptor signaling pathway, a few regulators (PD-L1, PD-1, JUN and AP-1) are down-regulated, while a few (SHP1/2, MEK6, p38 and NFAT) are up-regulated. All of these key regulators inversely contribute to the increased apoptosis and decreased cell cycle progression, IL-2 production, T-cell activation, and effector T-cell development at the systems-level.
Each infection-related signaling pathway embeds the basal Toll-like receptor signaling pathway coupled with more than one of the other composite signaling pathways (Supplementary Figures S1–S16). 1) The viral infection-related pathways include Epstein-Barr virus (Supplementary Figure S1), Human immunodeficiency virus 1 (Supplementary Figure S2), Hepatitis B (Supplementary Figure S3), Measles (Supplementary Figure S4), Influenza A (Supplementary Figure S5) and Herpes simplex virus 1 (Supplementary Figure S6); 2) The bacterial infection-related pathways include Tuberculosis (Supplementary Figure S7), Pertussis (Supplementary Figure S8), Yersinia (Supplementary Figure S9), Salmonella (Supplementary Figure S10) and Legionellosis (Supplementary Figure S11): and 3) The parasitic infection-related pathways include Toxoplasmosis (Supplementary Figure S12), Chagas disease (Supplementary Figure S13), Leishmaniasis (Supplementary Figure S14), Malaria (Supplementary Figure S15) and African trypanosomiasis (Supplementary Figure S16). The majority of these pathways are identified as significant (pGFdr <0.0001 or 0.05) from the (A) and (B) treatment, respectively; only one pathway (Herpes simplex virus 1) and two pathways (Malaria and African trypanosomiasis) are identified as insignificant (pGFdr >0.05) from the (A) and (B) treatment, respectively. All of these pathways are identified as significant (pGFdr <0.0001) from the (C) treatment that compared septic wildtype with untreated wildtype mice. These data suggest possible death from systemic syndromes, which increase the difficulty of early diagnosis and prevention of sepsis because it is unlikely that such systemic syndromes can be precisely attributed to a specific type (viral, bacterial or parasitic) of infections.
To focus on the roles of individual key regulators among the thousands of true DEGs (Supplementary Tables S1–S3), we further conducted the Venn diagram analysis to extract top 50-ranked DEGs intersected across GSE24327_A, B and C data (Table 2). As a result, there are 82 distinct core DEGs responsible for the immune-cell growth, activation and function (Table 2), including 1) ligands, receptors and adaptors (MyD88 and Lepr); 2) signal transducers (Jak1~Jak3, Stat3, Mapk1~Mapk3, Mapk8~Mapk14, Rela, Jun, Fos, Mmp9, Osmr and Pdgfra); 3) transcriptional factors (Nfkb1, Hif1a, Bcl3 and Eif3e); and 4) downstream effectors, such as colony-stimulating factors (Csf3r and Csf2rb), interleukins (Il1a, Il2ra, Il6ra and Il15ra), and chemokines (Ccr1~Ccr10, Cxcr1~Cxcr6, Cx3cr1, Ccr1l1 and Xcr1).
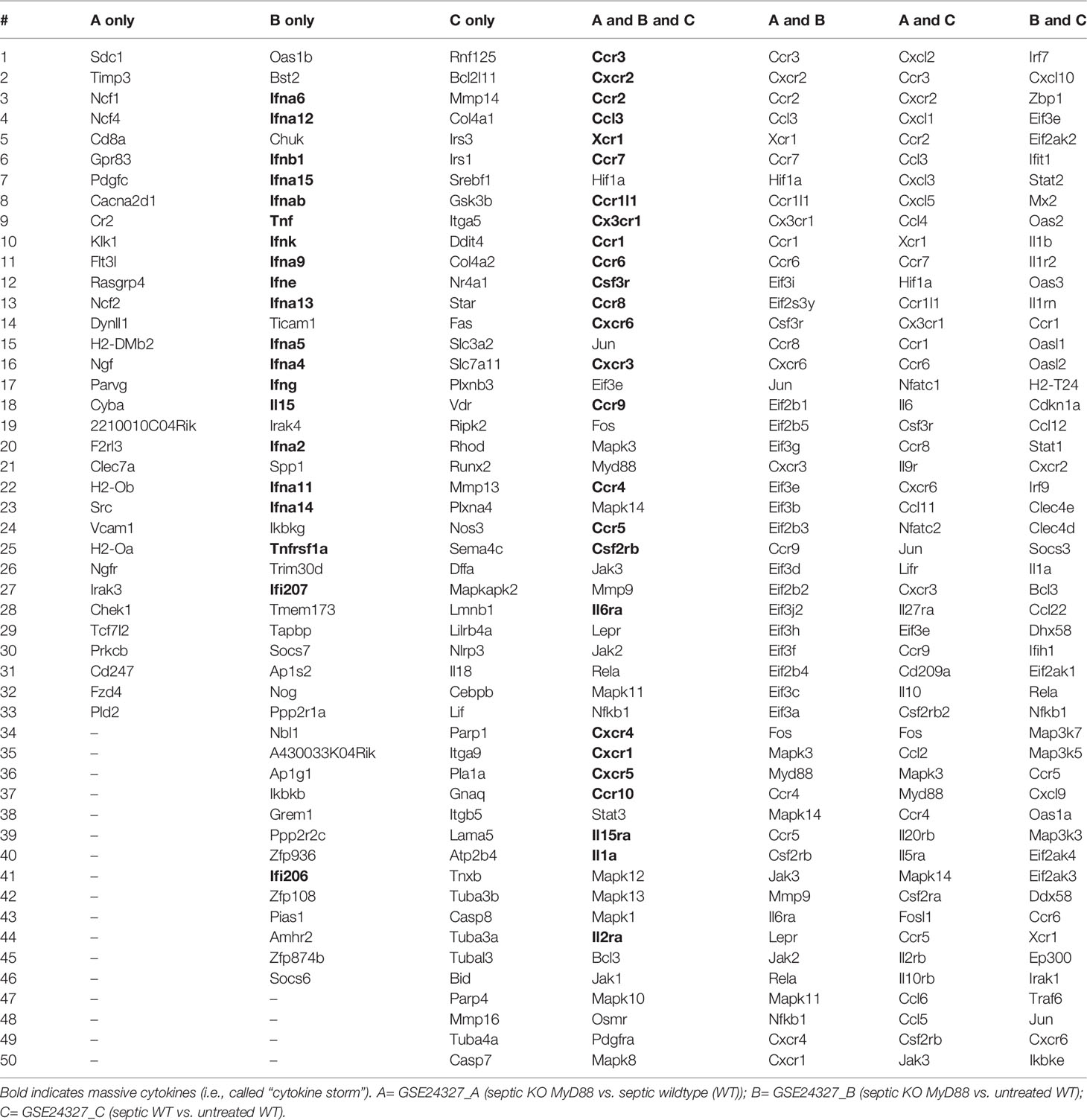
Table 2 Top 50-ranked DEGs intersected across GSE24327_A, B and C data.
Interestingly, the tumor necrosis factors (TNFs: Tnf and Tnfrsf1a) and the interferons (IFNs: Ifna2, Ifna4~Ifna6, Ifna9, Ifna11~Ifna15, Ifnab, Ifnb1, Ifne, Ifng, Ifnk, Ifi206, Ifi207) are categorized in the (B) treatment (septic KO MyD88 vs. untreated wildtype) (see Bonly in Table 2). In contrast, chemokines are intersected across all of the three data (see AandBandC in Table 2). It is well-known that TNFs stimulate immune-cell proliferation and activation; type I IFNs mediate antiviral immune responses, type II IFNs are responsible for antibacterial responses; and chemokines recruit different immune cells to the site of injury and/or infection. Taken together, our data suggest that the anti-viral (by Type I IFNs) and the anti-bacterial (by Type II IFNs) effects, as well as the recruitment (by chemokines) effects, have been strongly induced in spleen by the CASP-surgery on intestine just after 12 hours of the surgery, which strongly suggests a rapid progression of the CASP-model sepsis across remote organs in the body.
To examine possible infiltration of the core immune cells (CICs) (including B, CD4+T, CD8+T, Neutrophil, Macrophage and Dendritic cells) that would be recruited by the top 50-ranked regulators intersected across all of the three data (see Table 2), we searched them against multiple cancer cohorts, including colon (COAD), kidney (KIRC), liver (LIHC) and lung (LUAD) in database TIMER2.0 (51) at https://cistrome.shinyapps.io/timer. As a result, the expression level of each key regulator is positively and significantly (p<0.001) correlated with the infiltration level of CICs. Some examples are highlighted (Figures 8, 9), and more are provided online (Supplementary Figures S17–S20). These data suggest that the high expression of such regulators would recruit such infiltrating CICs at the systems-level. Our results imply that post-operative failure of remote multi-organs (colon, kidney, liver and lung) are likely due to the infiltration of such core immune cells that are recruited by the massive key regulators (including chemokines) and triggered by the CASP-model sepsis at the systems-level in the body (possibly progressing via the axis of intestine (colon)–heart–spleen–liver–kidney–lung, etc.).
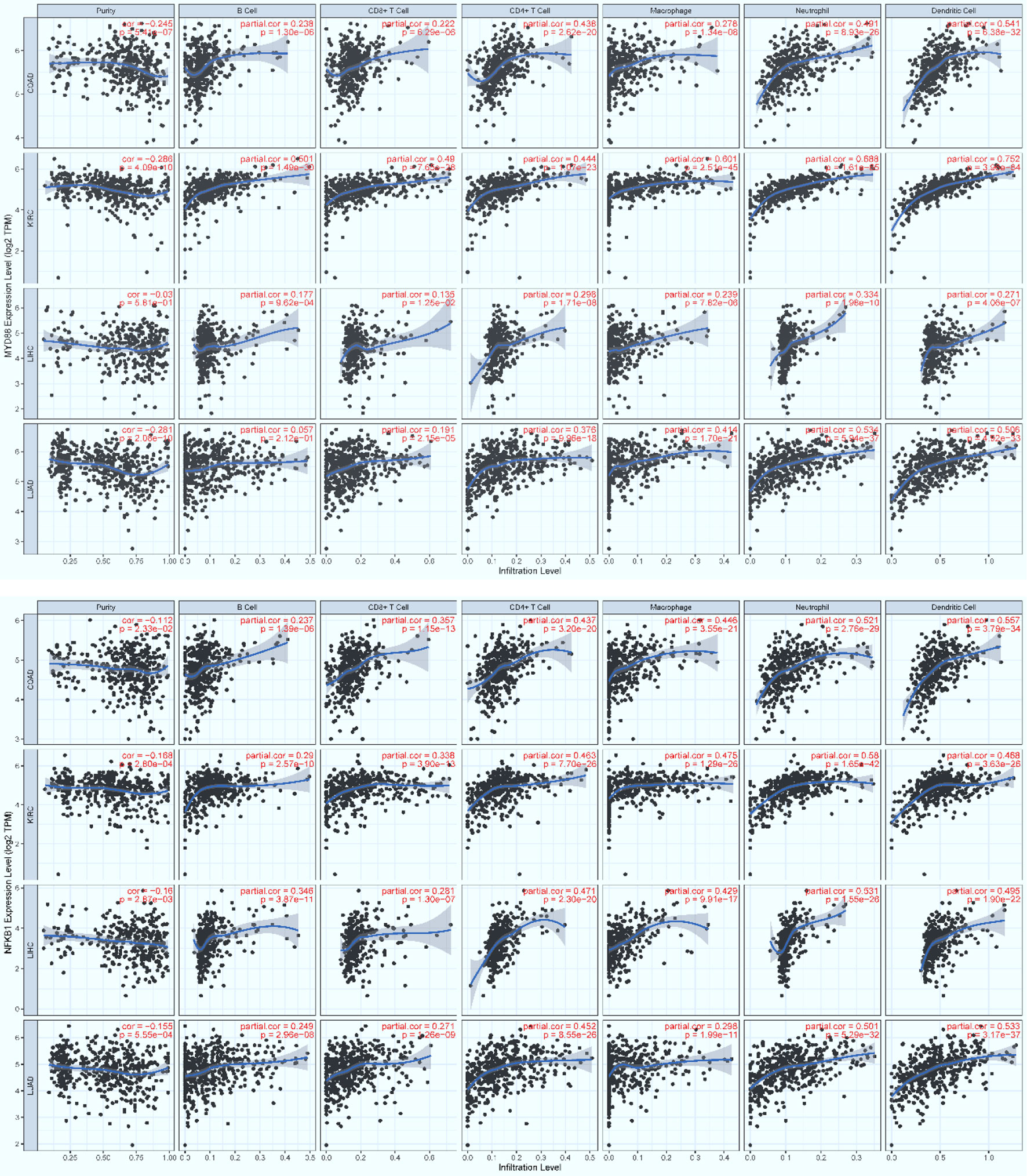
Figure 8 The positive, significant correlation between the infiltration level of core immune cells (X-axis) and the expression level of key regulators (Y-axis), MyD88 (top panel) and Nfkb1 (bottom panel). COAD: colon, KIRC: kidney, LIHC: liver, and LUAD: lung. p < 0.001***.
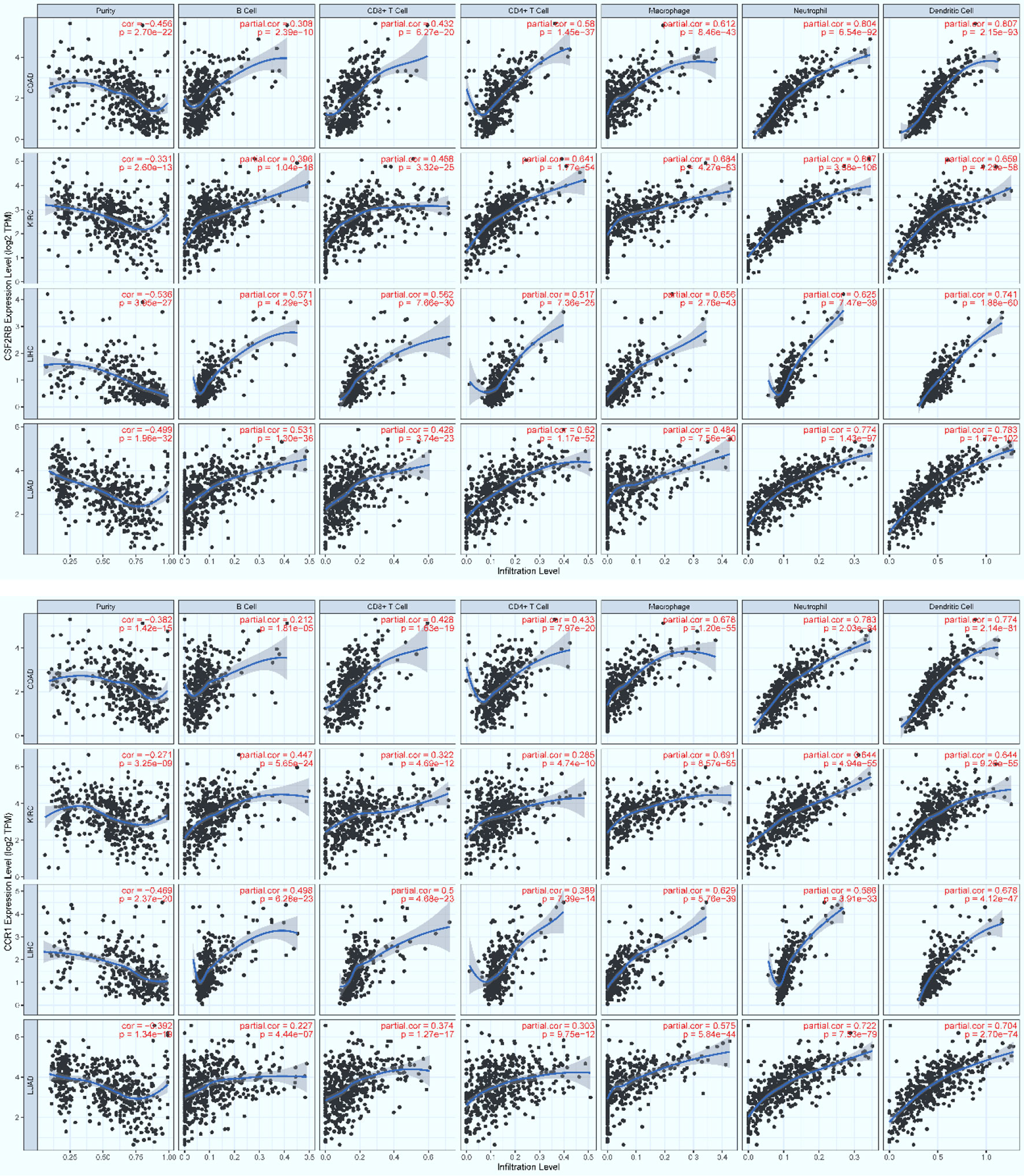
Figure 9 The positive, significant correlation between the infiltration level of core immune cells (X-axis) and the expression level of key regulators (Y-axis), Csf2rb (top panel) and Ccr1 (bottom panel). COAD: colon, KIRC: kidney, LIHC: liver, and LUAD: lung. p < 0.001***.
This study aimed to elucidate the systemic mechanisms underlying the standard CASP-model sepsis in the body through top-down data driven analysis on high-throughput functional genomics data. The success is attributed to the excellent GSE24327 GEO data with three experimental treatments (24): (a). septic KO MyD88 vs. septic WT (GSE24327_A); (b). septic KO MyD88 vs. untreated WT (GSE24327_B); and (c). septic WT vs. untreated WT (GSE24327_C). These data were generated from samples recovered from spleen at 12 hours after the CASP-surgery in mice, which were used for mRNAs profiling on microarray (24). From this, we have observed novel facts that have not been addressed thus far. Our data suggest that the leakage of intestinal contents effectively induced polymicrobial sepsis through provoking excessive innate immune responses at the systems-level (Figures 3–7; Supplementary Figures S1–S16) in wildtype mice, which usually caused post-operative failure of multi-organs (including colon, kidney, liver and lung). Moreover, the complex phenotypic syndromes, born by the identified and characterized 21 (KO MyD88-associated) target signaling pathways, contributed to the rapid progression of severe sepsis and even possible emerging death from complicated syndromes in post-operative mice, much beyond what has been perceived in literature (24). Our results update a significant advance in understanding the CASP-model sepsis mechanisms that have been explored for decades (4, 8, 24, 27, 31, 52–56).
Based on the 21 KO MyD88-associated target signaling pathways, we conceptualize the systemic mechanisms underlying the innate immune responses triggered by the CASP-model sepsis (Figure 10). The core regulators intersected across all data (Table 2) are coordinately, significantly, and differentially expressed at the systems-level, in both wildtype and deficient mice, suggesting unknown mechanisms that have not been addressed thus far. Such core regulators (e.g., CCR1, CSF2RB and IL2RA) would recruit the infiltration of core immune cells (CICs) (including B, CD4+T, CD8+T, macrophage, neutrophil, and dendritic cells) of the immune system (Figures 8, 9; Supplementary Figures S17–S20), likely initiated in spleen and transferred into remote multi-organs (progressing via the axis of colon–heart–spleen–liver–kidney–lung, etc.) through blood circulation in the body. The recruitment of infiltrating CICs fighting pathogens leaked from gut contents would result in severe sepsis progression across remote multi-organs after the CASP-surgery, contributing to the newly observed phenomena: “systemic syndrome” in (C) treatment, “cytokine storm” in (B) treatment, and “KO MyD88-attenuation” in (A) treatment in the current study, which are discussed as follows.
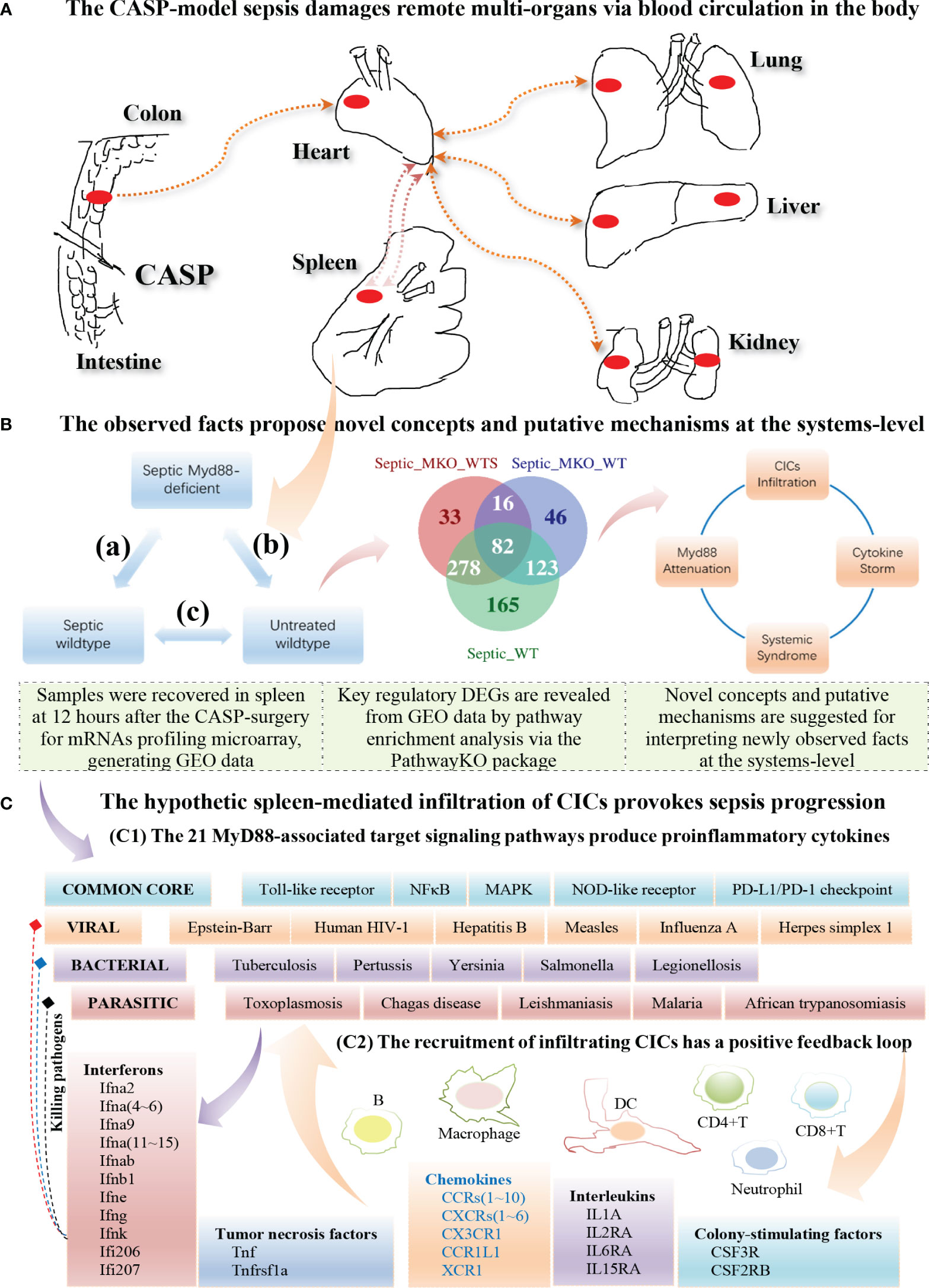
Figure 10 Hypothetic mechanisms underlying the innate immune responses at the systems-level triggered by the CASP-model sepsis at 12 hours after the CASP-surgery. (A) The CASP-model sepsis damages remote multi-organs through blood circulation in the body (top panel). (B) The observed facts propose novel concepts and putative mechanisms at the systems-level (middle panel). (C) The hypothetic spleen-mediated infiltration of core immune cells (CICs) (including B, CD8+T, CD4+T, Macrophage, Neutrophil and Dendritic cells) recruited by key cytokines (including CCR1, CSF2RB and IL2RA) likely drives the rapid progression of sepsis (bottom panel).
“Systemic inflammatory response syndrome (SIRS)” has been perceived to be an inflammatory state affecting the whole body, responding to an infectious or noninfectious insult. It should have both pro- and anti-inflammatory components although its definition is simply referred to an “inflammatory” response (57). The (C) treatment (compared septic wildtype mice with untreated wildtype mice) displays SIRS because of the strongest innate immune responses (Tables 1, 2; Figures 3–7; Supplementary Figures S1–S16), which suggest possible emerging death from systemic syndromes. We term this newly observed fact in wildtype mice as “systemic syndrome” (Figure 10). Our findings endorse the previous report that septic wildtype mice had the highest mortality rate (24). Moreover, the identified and characterized 21 significant (pGFdr <0.0001) signaling pathways (Table 1), much beyond the one determined by previous bench-experiments (24), provide a broader vision at the systems-level of the innate immune responses triggered by the CASP-surgery as early as 12 hours after the surgery causing severe sepsis (Table 1; Figure 1). However, we remind that these target pathways would not necessarily mean any co-existing at the same time in a single event of CASP-surgery or across all patients simultaneously. Rather, they would be due to signaling-crosstalk at the systems-level, sharing a similar set of true key regulators across the entirety of target pathways (Tables 1, 2), as marked on these target signaling pathways (Figures 3–7; Supplementary Figures S1–S16), which may result in an emerging death from systemic syndromes (at high risk for the same or undistinguishable cause) in clinical practices. These data also suggest that it is unlikely that such systemic syndromes can be precisely attributed to a specific type (viral, bacterial or parasitic) of infections, thus increasing the difficulty of early diagnosis and prevention of severe sepsis. Our findings provide novel insights into mechanisms underlying complicated syndromes in a broader vision, which warrant validations at the systems-level in the future.
“Cytokine storm” is a fatal immune reaction, consisting of a positive feedback loop between cytokines and white blood cells (58–61), which significantly elevates the levels of various cytokines (including chemokines, interleukins, interferons, tumor necrosis factors, and transforming growth factors). No consensus definition is accepted due to disagreement on the distinction between the cytokine storm and a physiologic inflammatory response (62). Cytokine storm is also known as cytokine cascade or hypercytokinemia, which defines the immune disorders with systemic inflammation, hyperinflammation, and multiple organ failure (58). The increased production of cytokines controls infections and the sustained excessive elevation in the levels of certain cytokines, cause negative systemic effects such as organ damage. In the present study, we obtained core regulators (see Table 2) once overlapped the DEGs from the (A), (B) and (C) treatments (Supplementary Tables S1–S3), which are involved in immune-cell growth, activation and function. These core regulators include 1) ligands, receptors and adaptors (MyD88 and Lepr); 2) signal transducers (Jak1~Jak3, Mapk1~Mapk3, Mapk8~Mapk14, Stat3, Rela, Jun, Fos, Mmp9, Osmr and Pdgfra); 3) transcriptional factors (Nfkb1, Hif1a, Bcl3 and Eif3e); and 4) downstream effectors, such as interleukins (Il1a, Il2ra, Il6ra and Il15ra), chemokines (Ccr1~Ccr10, Cxcr1~Cxcr6, Cx3cr1, Ccr1l1 and Xcr1), and colony-stimulating factors (Csf3r and Csf2rb). Our data highlight these core regulators intersected across all data (see AandBandC in Table 2) in both wildtype and deficient mice. Intriguingly, interferons (Ifna2, Ifna4~Ifna6, Ifna9, Ifna11~Ifna15, Ifnab, Ifnb1, Ifne, Ifng, Ifnk, Ifi206 and Ifi207) and tumor necrosis factors (Tnf and Tnfrsf1a) only belong to the (B) treatment (see Bonly in Table 2), unlike interleukins (Il1a, Il2ra Il6ra and Il15ra), chemokines (Ccr1~Ccr10, Cxcr1~Cxcr6, Ccl3, Cx3cr1, Ccr1l1 and Xcr1) and colony-stimulating factors (Csf2rb and Csf3r) that are intersected across the (A), (B) and (C) treatments. We term such newly observed facts as “cytokine-storm” (Figure 10) in the (B) treatment that compared septic KO MyD88 mice with untreated wildtype mice (Table 2), similar to that in viral infections (13, 58, 62, 63). The majority of these pro-inflammatory cytokines (Table 2) are newly characterized in the present study although some of them coincide with literature evidence, e.g., Ifng (interferon-γ) in the CASP-model sepsis (24, 27); CCRs and Il2ra (soluble interleukin-2 receptor alpha is a marker of T-cell activation) in Covid-19 disease (58). We recommend that such enriched cytokines warrant bench-experimental validations at the systems-level in the future.
The (C) treatment compared septic wildtype mice with untreated wildtype mice, which produced 648 DEGs (Supplementary Table S3), provoking the strongest innate immune responses. However, the (A) treatment that compared septic KO MyD88 mice with septic wildtype mice produced 408 DEGs (Supplementary Table S1), and the (B) treatment that compared septic KO MyD88 mice with untreated wildtype mice produced 266 DEGs (Supplementary Table S2), both yielding less number of DEGs and provoking relatively weaker innate immune responses. Obviously, the wildtype mice in the (C) treatment produced much more DEGs than the KO MyD88 deficient mice in the (A) and (B) treatment, respectively. We term these newly observed facts as “MyD88-deficiency attenuation” (Figure 10). Our results suggest that the absence of MyD88 balances the innate immune responses in a favorable manner by attenuating deleterious responses, thus preventing excessive cytokine release while maintaining intact protective IFNs responsible for anti-viral, anti-bacterial, and/or anti-parasitic activities in severe septic peritonitis (see Table 2; Figure 10) and as such, the post-operative survival rates of mice were increased (24). We conclude that it is the excessive innate immune response causing excessive inflammation that leads to the rapid progression of sepsis at the systems-level, which results in the fatal failure of remote multi-organs and possible death from systemic syndromes in post-operative mice. In contrast, the KO MyD88-deficiency has attenuated the excessive inflammation by directly reducing the production of cytokines through having impaired the 21 MyD88-associated target signaling pathways (Table 1; Figures 3–7; Supplementary Figures S1–S16; Figure 10).
The majority of core regulators intersected across all data (see AandBandC in Table 2) are chemokines. Chemokines are conceived to recruit core immune cells (CICs) of the immune system (including B, CD4+T, CD8+T, macrophage, neutrophil, and dendritic cells) to the on-site of infection (64) or sterile injury (including tumor progression) (51). Although no databases are available for testing our perspectives, we have attempted on utilizing the database TIMER2.0 (51) of multiple cancer cohorts since local injury caused by tumor-progression promotes distant infiltration of CICs. Therefore, we have observed a positive, significant (p<0.001) correlation (Figures 8, 9; Supplementary Figures S17–S20) between the expression level of key regulators (e.g., MyD88, Nfkb1, Ccr1, Csf2rb, Jak3, Il2ra, Csf3r and Ccr7) and the infiltration level of CICs in multiple cancer cohorts (e.g., COAD for colon, KIRC for kidney, LIHC for liver, and LUAD for lung). Our findings tentatively suggest that the infiltration of CICs likely plays a critical role in promoting the progression of the CASP-model sepsis at the systems-level as early as at 12 hours after the CASP-surgery.
The spleen is the largest lymphatic tissue in the body and is a center for the production and storage of lymphocytes (B lymphocytes, T lymphocytes and NK cells) of the immune system (65). The structure of the spleen enables it to remove older erythrocytes from the circulation and leads to the efficient removal of blood-borne microorganisms and cellular debris because macrophages, neutrophils, and dendritic cells engulf and ingest such toxic agents (65). The red pulp area of the spleen breaks down toxic agents; the white pulp area of the spleen surrounding the splenic arteries is lymphatic tissue, providing a storehouse for lymphocytes (65). We infer that once the polymicrobial agents leaked from gut contents during the CASP-surgery, they were transferred into the spleen, and the degraded debris and the provoked downstream immune effectors were, thereby, transferred into remote multi-organs through circulation of the blood filtrated from the spleen. In fact, all phenomena we have observed in the present study had factually occurred in spleen, rather than in intestine where the CASP-surgery was performed, since GSE24327 GEO data were generated from samples recovered from the spleen 12 hours after the CASP-surgery (24). Taken together, we hypothesize that it is the spleen-mediated infiltration of CICs via blood circulation across remote multi-organs in the body that drives the rapid progression of the CASP-model sepsis across remote multi-organs at the systems-level as early as at 12 hours after the CASP-surgery in the body (Figure 10). In other words, infiltrating CICs across remote multi-organs (including colon, heart, spleen, kidney, liver and lung) would be achieved by enhancing the expression levels of those key regulators at the systems-level (Table 2; Figures 8, 9). Such that, the infiltration of CICs would be recruited by those true key regulators (including chemokines) and would result in the rapid progression of CASP-model sepsis in the body (Table 2; Figures 8–10). Most importantly, there is likely a positive feedback loop among such consequent events of sepsis progression (Figure 10), thus causing a high mortality rate in wildtype mice (C). Such a positive feedback loop is also initiated in MyD88-deficient mice (B) at the early stage of sepsis progression (Table 2; Figures 8–10), but later attenuated owing to the impaired MyD88-assoicated signaling pathways, which keeps a balance between the moderate innate immunity responses and the living status of deficient mice (B), leading to a low mortality rate (24). We recommend these hypotheses be tested by bench-experiments at the systems-level in the future.
This study aimed to elucidate systemic mechanisms underlying the standard CASP-model sepsis in the body through top-down data driven analysis on high-throughput functional genomics data. The novelty of this article roots in six aspects. 1) The in-house PathwayKO package is effective in conducting cutting-edge knockout pathway enrichment analysis in order to identify and assess the entirety of knockout signaling pathways at the systems-level. 2) The real-world GEO data (GSE24327_A, B and C) generated from the spleen 12 hours after the CASP-surgery safeguard unbiased choice of samples for the CASP-model sepsis in mice (both MyD88-deficient and wildtype). Therefore, we can discuss the full range of polymicrobial (viral, bacterial and parasitic) sepsis in provoking severe innate immune responses, resulting in failure of distant multi-organs (including colon, liver, kidney and lung) at the systems-level. 3) The 21 target KO MyD88-associated signaling pathways create a broader vision, providing novel insights into mechanisms underlying the CASP-model sepsis. 4) The true key regulators (ligands, receptors, adaptors, transducers, transcriptional factors, and cytokines) that were coordinately, significantly, and differentially expressed at the systems-level, as marked on those true target signaling pathways, provide massive potential biomarkers that warrant experimental validations in the future. 5) Based on the newly observed facts in the CASP-model sepsis, we discuss “systemic syndrome”, “cytokine storm”, and “KO MyD88 attenuation”, as well as the proposed hypothesis of “spleen-mediated immune-cell infiltration” at the systems-level. 6) Our results provide novel angles in a broader vision towards a better understanding of the CASP-model sepsis in mice, which may serve as a model for humans, to ultimately guide formulating the research paradigms and composite strategies for the early diagnosis and prevention of severe sepsis.
Publicly available datasets were analyzed in this study. This data can be found here: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE24327.
HA and YA designed the project. HA conducted computations and analyzed data. HA, BL, FM, and YA interpreted results, wrote manuscript and approved the final manuscript.
This work was supported by a grant-in-aid from National Science and Technology Major Programs of China (2014ZX0801105B-002) and Supercomputing Program of National Natural Science Foundation of China (No. U1501501-534) to YA.
Author HA was employed by SINOMACH-IT.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The authors sincerely acknowledge TH-2 supercomputing resources in National Supercomputing Center (Guangzhou) at Sun Yat-sen University. We sincerely thank numerous colleagues at Sun Yat-sen University and Guangzhou Medical University for their kind consultations on clinical cases under study.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2022.907646/full#supplementary-material
GEO, gene expression omnibus; KEGG, Kyoto encyclopedia of genes and genome; DEGs, differentially expressed genes; ROC, receiver operating characteristic; AUC, area under the ROC curve; pAUC_SP, partial AUC focusing on the specificity; pAUC_SE, partial AUC focusing on the sensitivity; HES, high-edge-scores; WT, wildtype; KO, knockout; TLR, toll-like receptor; MyD88, myeloid differentiating factor 88 (encoded by Myd88).
1. Liu V, Escobar GJ, Greene JD, Soule J, Whippy A, Angus DC, et al. Hospital Deaths in Patients With Sepsis From 2 Independent Cohorts. JAMA (2014) 312:90–2. doi: 10.1001/jama.2014.5804
2. Sakr Y, Jaschinski U, Wittebole X, Szakmany T, Lipman J, Ñamendys-Silva SA, et al. Sepsis in Intensive Care Unit Patients: Worldwide Data From the Intensive Care Over Nations Audit. Open Forum Infect Dis (2018) 5:ofy313. doi: 10.1093/ofid/ofy313
3. Huang CY, Daniels R, Lembo A, Hartog C, O’Brien J, Heymann T, et al. Life After Sepsis: An International Survey of Survivors to Understand the Post-Sepsis Syndrome. Int J Qual Health Care (2019) 31:191–8. doi: 10.1093/intqhc/mzy137
4. Walker WE. Sepsis: Methods and Protocols. New York. USA: Humana Press (2021). doi: 10.1007/978-1-0716-1488-4
5. Sonego F, Castanheira FVS, Czaikoski PG, Kanashiro A, Souto FO, Franc¸ RO, et al. MyD88-, But Not Nod1- and/or Nod2-Deficient Mice, Show Increased Susceptibility to Polymicrobial Sepsis Due to Impaired Local Inflammatory Response. PloS One (2014) 9:e103734. doi: 10.1371/journal.pone.0103734
6. Gong T, Liu L, Jiang W, Zhou R. DAMP-Sensing Receptors in Sterile Inflammation and Inflammatory Diseases. Nat Rev Immunol (2020) 20:95–112. doi: 10.1038/s41577-019-0215-7
7. Sewnath ME, Olszyna DP, Birjmohun R, ten Kate FJ, Gouma DJ, van der Poll T. IL-10-Deficient Mice Demonstrate Multiple Organ Failure and Increased Mortality During Escherichia Coli Peritonitis Despite an Accelerated Bacterial Clearance. Immunol J (2001) 166:6323–31. doi: 10.4049/jimmunol.166.10.6323
8. Schick MA, Baar W, Flemming S, Schlegel N, Wollborn J, Held C, et al. Sepsis-Induced Acute Kidney Injury by Standardized Colon Ascendens Stent Peritonitis in Rats - A Simple, Reproducible Animal Model. Intensive Care Med Exp (2014) 2:34. doi: 10.1186/s40635-014-0034-x
9. Hotson AN, Gopinath S, Nicolau M, Khasanova A, Finck R, Monack D, et al. Coordinate Actions of Innate Immune Responses Oppose Those of the Adaptive Immune System During Salmonella Infection of Mice. Sci Signal (2016) 9:ra4. doi: 10.1126/scisignal.aaa9303
10. Sauter IP, Madrid KG, de Assis JB, Sá-Nunes A, Torrecilhas AC, Staquicini DI, et al. TLR9/MyD88/TRIF Signaling Activates Host Immune Inhibitory CD200 in Leishmania Infection. JCI Insight (2019) 4:e126207. doi: 10.1172/jci.insight.126207
11. Yang D, Liu N, Zuo C, Lei S, Wu X, Zhou F, et al. Innate Host Response in Primary Human Hepatocytes With Hepatitis C Virus Infection. PloS One (2011) 6:e27552. doi: 10.1371/journal.pone.0027552
12. Hsua JC-C, Laurent-Rollea M, Pawlak JB, Wilen CB, Cresswella P. Translational Shutdown and Evasion of the Innate Immune Response by SARS-CoV-2 NSP14 Protein. Proc Natl Acad Sci USA (2021) 118:e2101161118. doi: 10.1073/pnas.2101161118
13. Xu R, Liu C, Xu X, Hu Y, Zhu B, Yang C. Role of Cytokine Storm in Coronavirus Infections: Culprit or Accomplice? Front Biosci (Landmark Ed) (2022) 27:102. doi: 10.31083/j.fbl2703102
14. Weighardt H, Jusek G, Mages J, Lang R, Hoebe K, Beutler B, et al. Identification of a TLR4- and TRIF-Dependent Activation Program of Dendritic Cells. Eur J Immunol (2004) 34:558–64. doi: 10.1002/eji.200324714
15. Gais P, Reim D, Jusek G, Rossmann-Bloeck T, Weighardt H, Pfeffer K, et al. Cutting Edge: Divergent Cell-Specific Functions of MyD88 for Inflammatory Responses and Organ Injury in Septic Peritonitis. J Immunol (2012) 188:5833–7. doi: 10.4049/jimmunol.1200038
16. Feng Y, Zou L, Zhang M, Li Y, Chen C, Chao W. MyD88 and Trif Signaling Play Distinct Roles in Cardiac Dysfunction and Mortality During Endotoxin Shock and Polymicrobial Sepsis. Anesthesiology (2011) 115:555–67. doi: 10.1097/ALN.0b013e31822a22f7
17. Chen Z, Shao Z, Mei S, Yan Z, Ding X, Billiar T, et al. Sepsis Upregulates CD14 Expression in a MyD88-Dependent and TRIF-Independent Pathway. Shock (2018) 49:82–9. doi: 10.1097/SHK.0000000000000913
18. Dyevoich AM, Disher NS, Haro MA, Karen M, Haas KM. A TLR4-TRIF-Dependent Signaling Pathway Is Required for Protective Natural Tumor-Reactive IgM Production by B1 Cells. Cancer Immunol Immunother (2020) 69:2113–24. doi: 10.1007/s00262-020-02607-7
19. Saikh KU. MyD88 and Beyond: A Perspective on MyD88−Targeted Therapeutic Approach for Modulation of Host Immunity. Immunol Res (2021) 69:117–28. doi: 10.1007/s12026-021-09188-2
20. Gupta S, Kaplan MJ. Bite of the Wolf: Innate Immune Responses Propagate Autoimmunity in Lupus. J Clin Invest (2021) 131:e144918. doi: 10.1172/JCI144918
21. Laitano O, Robinson GP, Murray KO, Garcia CK, Mattingly AJ, Morse D, et al. Skeletal Muscle Fibers Play a Functional Role in Host Defense During Sepsis in Mice. Sci Rep (2021) 11:7316. doi: 10.1038/s41598-021-86585-5
22. Liu Y, Hu J-N, Luo N, Zhao J, Liu S-C, Ma T, et al. The Essential Involvement of the Omentum in the Peritoneal Defensive Mechanisms During Intra-Abdominal Sepsis. Front Immunol (2021) 12:631609. doi: 10.3389/fimmu.2021.631609
23. Menz J, Hundt L, Schulze T, Schmoeckel K, Menges P, Domanska G. Increased Mortality and Altered Local Immune Response in Secondary Peritonitis After Previous Visceral Operations in Mice. Sci Rep (2021) 11:16175. doi: 10.1038/s41598-021-95592-5
24. Reim D, Rossmann-Bloeck T, Jusek G, da Costa OP, Holzmann B. Improved Host Defense Against Septic Peritonitis in Mice Lacking MyD88 and TRIF Is Linked to a Normal Interferon Response. J Leukoc Biol (2011) 90:613–20. doi: 10.1189/jlb.1110602
25. Stelmasiak M, Slotwinski R. Infection-Induced Innate Antimicrobial Response Disorders: From Signaling Pathways and Their Modulation to Selected Biomarkers. Cent Eur J Immunol (2020) 45:104–16. doi: 10.5114/ceji.2020.94712
26. Hyland EM, Webb AE, Kennedy KF, Ince ZNG, Loscher CE, O’Connell MJ. Adaptive Evolution in TRIF Leads to Discordance Between Human and Mouse Innate Immune Signaling. Genome Biol Evol (2021) 13:evab268. doi: 10.1093/gbe/evab268
27. Zantl N, Uebe AE, Neumann B, Wagner H, Siewert J-R, Holzmann B, et al. Essential Role of Gamma Interferon in Survival of Colon Ascendens Stent Peritonitis, a Novel Murine Model of Abdominal Sepsis. Infect Immun (1998) 66:2300–9. doi: 10.1128/IAI.66.5.2300-2309.1998
28. Weighardt H, Kaiser-Moore S, Schlautkotter S, Rossmann-Bloeck T, Schleicher U, Bogdan C, et al. Type I IFN Modulates Host Defense and Late Hyperinflammation in Septic Peritonitis. J Immunol (2006) 177:5623–30. doi: 10.4049/jimmunol.177.8.5623
29. Weighardt H, Mages J, Jusek G, Kaiser-Moore S, Lang R, Holzmann B. Organ-Specific Role of MyD88 for Gene Regulation During Polymicrobial Peritonitis. Infect Immun (2006) 74:3618–32. doi: 10.1128/IAI.01681-05
30. Tan W, Pruett SB. Effects of Sodium Methyldithiocarbamate on Selected Parameters of Innate Immunity and Clearance of Bacteria in a Mouse Model of Sepsis. Life Sci (2015) 15:1–7. doi: 10.1016/j.lfs.2015.08.001
31. Beyer K, Stollhof L, Poetschke C, von Bernstorff W, Partecke LI, Diedrich S, et al. TNF-Related Apoptosis-Inducing Ligand Deficiency Enhances Survival in Murine Colon Ascendens Stent Peritonitis. J Inflamm Res (2016) 9:103–13. doi: 10.2147/jir.s99887
32. Utiger JM, Glas M, Levis A, Prazak J, Haenggi M. Description of a Rat Model of Polymicrobial Abdominal Sepsis Mimicking Human Colon Perforation. BMC Res Notes (2021) 14:14. doi: 10.1186/s13104-020-05438-y
33. Bucciol G, Moens L, Corveleyn A, Dreesman A, Meyts I. A Novel Kindred With MyD88 Deficiency. J Clin Immunol (2022) 14:1–4. doi: 10.1007/s10875-022-01240-6
34. Luo W, Brouwer C. Pathview: An R/Bioconductor Package for Pathway-Based Data Integration and Visualization. Bioinformatics (2013) 29:1830–1. doi: 10.1093/bioinformatics/btt285
35. Zhang JD, Wiemann S. KEGGgraph: A Graph Approach to KEGG Pathway in R and Bioconductor. Bioinformatics (2009) 25:1470–1. doi: 10.1093/bioinformatics/btp167
36. Kanehisa M, Sato Y, Kawashima M, Furumichi M, Tanabe M. KEGG as a Reference Resource for Gene and Protein Annotation. Nucleic Acids Res (2016) 44:D475–62. doi: 10.1093/nar/gkv1070
37. Barry WT, Nobel AB, Wright FA. Significance Analysis of Functional Categories in Gene Expression Studies: A Structured Permutation Approach. Bioinformatics (2005) 21:1943–9. doi: 10.1093/bioinformatics/bti260
38. Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, et al. Gene Set Enrichment Analysis: A Knowledge-Based Approach for Interpreting Genome-Wide Expression Profiles. Proc Natl Acad Sci USA (2005) 102:15545–50. doi: 10.1073/pnas.0506580102
39. Efron B, Tibshirani R. On Testing the Significance of Sets of Genes. Ann Appl Stat (2007) 1:107–29. doi: 10.1214/07-AOAS101
40. Tarca AL, Draghici S, Bhatti G, Romero R. Down-Weighting Overlapping Genes Improves Gene Set Analysis. BMC Bioinform (2012) 13:136. doi: 10.1186/1471-2105-13-136
41. Draghici S, Khatri P, Tarca AL, Amin K, Done A, Voichita C, et al. A Systems Biology Approach for Pathway Level Analysis. Genome Res (2007) 17:1537–45. doi: 10.1101/gr.6202607
42. Nguyen T, Shafi A, Nguyen T, Draghici S. Identifying Significantly Impacted Pathways: A Comprehensive Review and Assessment. Genome Biol (2019) 20:203. doi: 10.1186/s13059-019-1790-4
43. Tarca AL, Draghici S, Khatri P, Hassan SS, Mitta P, Kim J, et al. A Novel Signaling Pathway Impact Analysis. Bioinformatics (2009) 25:75–82. doi: 10.1093/bioinformatics/btn577
44. Killick R, Eckley IA. Changepoint: An R Package for Changepoint Analysis. J Stat Softw (2014) 58:1–19. doi: 10.18637/jss.v058.i03
45. Robin X, Turck N, Hainard A, Tiberti N, Lisacek F, Sanchez J-C, et al. pROC: An Open-Source Package for R and S+ to Analyze and Compare ROC Curves. BMC Bioinform (2011) 12:77. doi: 10.1186/1471-2105-12-77
47. McClish DK. Analyzing a Portion of the ROC Curve. Med Decis Making (1989) 9:190–5. doi: 10.1177/0272989X8900900307
48. Benjamini Y, Hochberg Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J R Stat Soc B (1995) 57:289–300. doi: 10.1111/j.2517-6161.1995.tb02031.x
49. Benjamini Y, Yekutieli D. The Control of the False Discovery Rate in Multiple Testing Under Dependency. Ann Stat (2001) 29:1165–88. doi: 10.1214/aos/1013699998
50. Hanoudi S, Donato M, Draghici S. Identifying Biologically Relevant Putative Mechanisms in a Given Phenotype Comparison. PloS One (2017) 12:e0176950. doi: 10.1371/journal.pone.0176950
51. Li T, Fan J, Wang B, Traugh N, Chen Q, Liu J, et al. TIMER: A Web Server for Comprehensive Analysis of Tumor-Infiltrating Immune Cells. Cancer Res (2017) 77:e108–10. doi: 10.1158/0008-5472.CAN-17-0307
52. Maier S, Traeger T, Entleutner M, Westerholt A, Kleist B, Hüser N, et al. Cecal Ligation and Puncture Versus Colon Ascendens Stent Peritonitis: Two Distinct Animal Models for Polymicrobial Sepsis. Shock (2004) 21:505–11. doi: 10.1097/01.shk.0000126906.52367.dd
53. Lustig MK, Bac VH, Pavlovic D, Maier S, Grundling M, Grisk O, et al. Colon Ascendens Stent Peritonitis–A Model of Sepsis Adopted to the Rat: Physiological, Microcirculatory and Laboratory Changes. Shock (2007) 28:59–64. doi: 10.1097/SHK.0b013e31802e454f
54. Beyer K, Poetschke C, Partecke LI, von Bernstorff W, Maier S, Broeker BM, et al. TRAIL Induces Neutrophil Apoptosis and Dampens Sepsis-Induced Organ Injury in Murine Colon Ascendens Stent Peritonitis. PloS One (2014) 9:e97451. doi: 10.1371/journal.pone.0097451
55. Flemming S, Schlege N, Wunder C, Meir M, Baar W, Wollborn J, et al. Phosphodiesterase 4 Inhibition Dose Dependently Stabilizes Microvascular Barrier Functions and Microcirculation in a Rodent Model of Polymicrobial Sepsis. Shock (2014) 41:537–45. doi: 10.1097/SHK.0000000000000152
56. Flemming S, Burkard N, Meir M, Schick MA, Germer C-T, Schlegel N. Sphingosine-1-Phosphate Receptor-1 Agonist SEW2871 Causes Severe Cardiac Side Effects and Does Not Improve Microvascular Barrier Breakdown in Sepsis. Shock (2018) 49:71–81. doi: 10.1097/SHK.0000000000000908
57. Balk RA. Systemic Inflammatory Response Syndrome (SIRS): Where Did It Come From and Is It Still Relevant Today? Virulence (2014) 5:20–6. doi: 10.4161/viru.27135
58. Fajgenbaum DC, June CH. Cytokine Storm. N Engl J Med (2020) 383:2255–73. doi: 10.1056/NEJMra2026131
59. Kang S, Kishimoto T. Interplay Between Interleukin-6 Signaling and the Vascular Endothelium in Cytokine Storms. Exp Mol Med (2021) 53:1116–23. doi: 10.1038/s12276-021-00649-0
60. Lee DW, Santomasso BD, Locke FL, Ghobadi A, Turtle CJ, Brudno JN, et al. ASTCT Consensus Grading for Cytokine Release Syndrome and Neurologic Toxicity Associated With Immune Effector Cells. Biol Blood Marrow Transplant (2019) 25:625–38. doi: 10.1016/j.bbmt.2018.12.758
61. Ramani T, Auletta CS, Weinstock D, Mounho-Zamora B, Ryan PC, Salcedo TW, et al. Cytokines: The Good, the Bad, and the Deadly. Int J Toxicol (2015) 34:355–65. doi: 10.1177/1091581815584918
62. Sinha P, Matthay MA, Calfee CS. Is a “Cytokine Storm” Relevant to COVID-19? JAMA Intern Med (2020) 180:1152–4. doi: 10.1001/jamainternmed.2020.3313
63. Greenfield KG, Badovinac VP, Griffith TS, Knoop KA. Sepsis, Cytokine Storms, and Immunopathology: The Divide Between Neonates and Adults. Immunohorizons (2021) 28:512–22. doi: 10.4049/immunohorizons.2000104
64. McGuigan AJ, Coleman HG, McCain RS, Kelly RJ, Johnston DI, Taylor MA, et al. Immune Cell Infiltrates as Prognostic Biomarkers in Pancreatic Ductal Adenocarcinoma: A Systematic Review and Meta-Analysis. J Pathol Clin Res (2021) 7:99–112. doi: 10.1002/cjp2.192
Keywords: microbial immunology, colon ascendens stent peritonitis (CASP), signaling pathways, innate immune responses, transcriptional regulation network, innate immunity, systems biology, bioinformatics
Citation: Ai H, Li B, Meng F and Ai Y (2022) CASP-Model Sepsis Triggers Systemic Innate Immune Responses Revealed by the Systems-Level Signaling Pathways. Front. Immunol. 13:907646. doi: 10.3389/fimmu.2022.907646
Received: 30 March 2022; Accepted: 28 April 2022;
Published: 14 June 2022.
Edited by:
Ian Marriott, University of North Carolina at Charlotte, United StatesReviewed by:
Sven Flemming, University Hospital of Wuerzburg, GermanyCopyright © 2022 Ai, Li, Meng and Ai. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hannan Ai, QWxsZW5haTk0QGljbG91ZC5jb20=; Yuncan Ai, THNzYXljQG1haWwuc3lzdS5lZHUuY24=
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.