 Araz Kouyoumdjian
Araz Kouyoumdjian Jean Tchervenkov
Jean Tchervenkov Steven Paraskevas
Steven Paraskevas- 1Division of Experimental Surgery, Faculty of Medicine and Health Sciences, McGill University, Montreal, QC, Canada
- 2Division of General Surgery, Department of Surgery, McGill University, Montreal, QC, Canada
Tumor necrosis factor receptor 2 (TNFR2) has been shown to play a crucial role in CD4+ T regulatory cells (CD4+Tregs) expansion and suppressive function. Increasing evidence has also demonstrated its role in a variety of immune regulatory cell subtypes such as CD8+ T regulatory cells (CD8+ Tregs), B regulatory cells (Bregs), and myeloid-derived suppressor cells (MDSCs). In solid organ transplantation, regulatory immune cells have been associated with decreased ischemia-reperfusion injury (IRI), improved graft survival, and improved overall outcomes. However, despite TNFR2 being studied in the context of autoimmune diseases, cancer, and hematopoietic stem cell transplantation, there remains paucity of data in the context of solid organ transplantation and islet cell transplantation. Interestingly, TNFR2 signaling has found a clinical application in islet transplantation which could guide its wider use. This article reviews the current literature on TNFR2 expression in immune modulatory cells as well as IRI, cell, and solid organ transplantation. Our results highlighted the positive impact of TNFR2 signaling especially in kidney and islet transplantation. However, further investigation of TNFR2 in all types of solid organ transplantation are required as well as dedicated studies on its therapeutic use during induction therapy or treatment of rejection.
Introduction
Tumor necrosis factor (TNF) signaling is central to many aspects of ischemia-reperfusion injury (IRI) in solid organ transplantation and has a central role in regulating acute and chronic anti-donor immune responses which can determine or limit the functional life of the transplanted organ. TNFα is mainly produced by macrophages and neutrophils during innate immune activation after recognition of either pathogen-associated molecular patterns (PAMPs) or danger-associated molecular patterns (DAMPs) via pattern recognition receptors (1). It can also be produced by CD4+ effector T cells and natural killer cells during certain conditions (2). It exerts its actions through binding with its receptors TNF receptor type 1 (TNFR1) and TNF receptor type 2 (TNFR2) as either a transmembrane protein (mTNF) or soluble protein (sTNF) (3, 4). With pertinence to the organ transplant context, TNFR2 is expressed in lymphocytes as well as endothelial cells (5), the latter being the primary donor-recipient interface from the immunological perspective. On the other hand, TNFR1 is ubiquitously expressed on most cell types (6).
The inflammatory phenotypes instigated by TNFα have been studied extensively in the context of innate and adaptive immune cell activation (7–13). Indeed, TNFα signaling through both TNFR1 and TNFR2 has been found to have a central role in many pathological conditions such as autoimmunity, allergy, and malignancy (14–16). Despite TNFα’s key role in mediating and perpetuating pro-inflammatory signals during all stages of the immune activation, increasing evidence has highlighted a strong counterbalancing and immune regulatory role for TNFR2 when compared to TNFR1.
While TNFR2 has been studied extensively in the context of cancer and autoimmunity, there remains paucity of data in the transplant field. The first and most significant immunologic event in the transplant process is ischemia reperfusion injury (IRI). During the ‘reperfusion’ phase of IRI, products of cell death are released into the circulation, stimulating the innate and adaptive immune responses (17). In view of the dichotomous role of TNFα signaling, it is important to better illustrate the role that TNFα-TNFR2 holds in all aspects of solid organ transplantation, beginning with IRI and extending to chronic alloimmune processes throughout the life of the organ.
In this review article, we will aim to summarize the evidence regarding TNFα-TNFR2 signaling in the context of regulatory immune cells as well as summarize the role of TNFα-TNFR2 signaling during IRI, solid organ transplantation, and rejection.
TNFα Signaling Pathway
TNFα is a type II transmembrane protein which has a characteristic homotrimeric configuration and exists in two forms, membrane-bound and soluble (4, 18, 19). mTNF can be cleaved in its stalk region by the matrix metalloproteinase TNF-alpha-converting enzyme to form sTNF (20). After its release, sTNF can circulate in the body and thus can exert its effects away from its initial site of production. mTNF and sTNF have been involved in differential pathway signaling. mTNF and sTNF both bind to TNFR1, however, mTNF preferentially binds TNFR2 (21). This suggests that activation of TNFR2 associated pathways are mostly stimulated by neighboring cells in a paracrine fashion while activation of TNFR1 via sTNF functions in an endocrine manner.
Upon binding of TNFα to TNFR1, signaling via the TNFR1-associated death domain is initiated which can in turn activate one of two distinct cellular responses (22). Through its death domain, TNFR1 signaling leads to activation of the downstream canonical NF-κB, mitogen-activated protein kinase (MAPK), and c-Jun N-terminal kinase (JNK) signaling pathways (23). These three signaling pathways promote inflammation and proliferation. On the other hand, TNFR1 can also induce cell death through activation of the caspase cascade (24).
Upon binding of TNFα to TNFR2, the classical and alternative NF-κB signaling pathway will be activated (25). TNFR2 has also been shown to recruit the TNF receptor associated factor 2 (TRAF2) and the single cellular inhibitor of apoptosis 1 or 2 molecules (cIAP1/2) complex more efficiently than TNFR1 (25, 26). This preferential binding to TNFR2 leads to TRAF2-cIAP1/2 complex depletion and subsequent inhibition of downstream TNFR1 inflammatory signaling (26). On the other hand, as TNFR2 lacks a death domain, it has been shown to recruit and activate the AKT pathway which promotes cell survival, migration, proliferation, CD4+Treg function, and an overall cytoprotective phenotype (27).
TNFR2 in Immune Regulatory Cells
TNFR2 in CD4+ T Regulatory Cells
The dichotomous role of TNFα in pro-inflammatory versus regulatory pathways has largely been studied in the context of CD4+ T cells. While initial exposure to TNFα will prompt CD4+ effector functions, prolonged exposure will trigger habituation, downregulation, and exhaustion of CD4+ effector T cell signaling (28). CD4+ T regulatory cells (CD4+Tregs), classically defined as CD4+CD25+CD127- and forehead box P3+ (FoxP3) cells, are a subset of CD4+ T cells which play a crucial role in immune suppression, homeostasis, and self-tolerance through inhibition of CD4+ T effector cell proliferation as well as production of inhibitory cytokines (29, 30). Their dysregulation has been associated with a variety of autoimmune diseases, cancers, as well as solid organ rejection (31–34). Notably, the expression of TNFR2 has been shown to preferentially associate with FoxP3+CD4+Tregs as well as identify a highly suppressive subset of CD4+Tregs in mice as well as humans (35–37). This was shown in experimental models of autoimmune encephalomyelitis, TNFR2 signaling on Tregs has been shown to maintain Treg suppressive activity and protect against disease progression (38). Another functional role which has been found has been CD4+Treg population expansion and proliferation via TNFR2 co-stimulation (39). These in vitro findings have also been observed in vivo murine models of graft-versus-host disease (GvHD). Upon treatment with a TNFR2 agonist, CD4+Treg population expansion and activation was observed (40). Other novel TNFR2 agonists are also being studied as potential therapeutic targets for treatment of autoimmunity and other inflammatory disorders and have been shown to expand highly suppressive CD4+Tregs capable of CD8+ T cell repression in vitro (41). CD4+Tregs have also been found to respond to proinflammatory cytokines. Indeed, IL-6 and TNFα have been observed to drive human CD4+Tregs proliferation by increasing TNFR2 expression (42). Recent investigations have shown that binding to TNFR2 on conventional CD4+ T cells increases their interleukin (IL)-2 production which in turn leads to signal transducer and activator of transcription (STAT) 5 phosphorylation and proliferation of neighboring CD4+Tregs (43–45). These findings were confirmed in studies where blocking TNFR2 signaling was shown to increase T helper 17 cell differentiation via STAT3 activity and retinoid acid-receptor-related orphan receptor-γt induction (22, 46). TNFR2 has also been studied in the context of thymic CD4+Tregs, or tTregs (formerly called natural Tregs versus peripheral CD4+Tregs, or pTregs (formerly called induced Tregs. In the past, it was believed that TNFR2 signaling was required for optimal tTregs suppressive function under inflammatory conditions alone (47). However, more recent data has put this into question and has rather observed a role for TNFα-TNFR2 signaling in the differentiation and function of pTregs in autoimmunity models (48). On the other hand, there have also been reports that anti-TNFα therapy such as infliximab increase pTreg frequency in human models (49). Together, these data suggest an important role for TNFR2 in CD4+Treg function, however, the exact mechanisms remain elusive, and the discrepancies could be explained by different functionality of pTregs and tTregs in various pathological conditions (Figure 1).
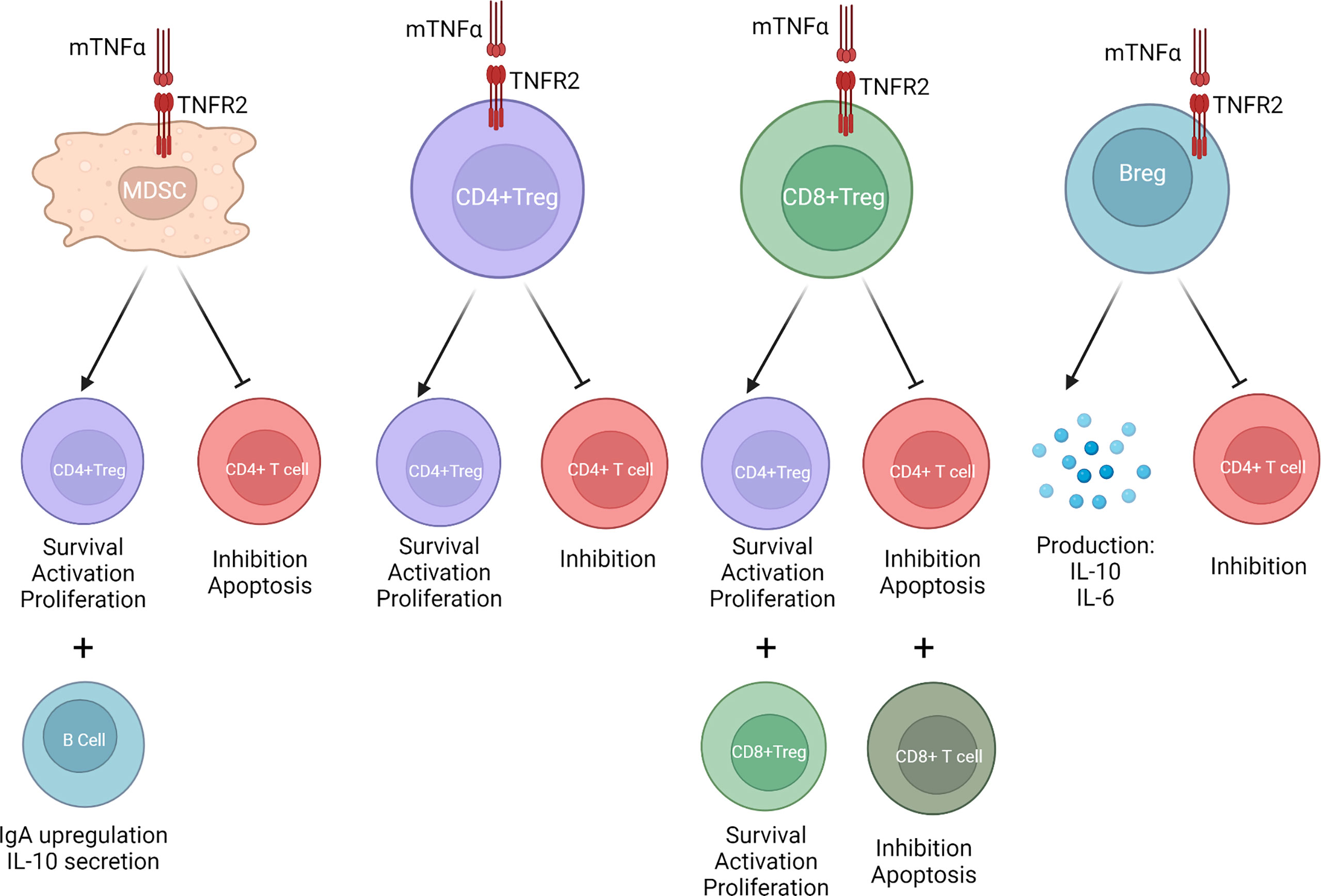
Figure 1 When mTNFα binds to TNFR2, MDSCs, CD4+Tregs, CD8+Tregs, and Bregs are activated. This increases CD4+Treg and CD8+Treg stability and expansion. It stimulates production of anti-inflammatory cytokines such as IL-10 and TGF-β. It inhibits CD4+ T effector cell function. It promotes upregulation of immunoglobulin A (IgA) B cells.
TNFR2 in CD8+ T Regulatory Cells
CD8+FoxP3+ T regulatory cells (CD8+Tregs) are another important class of adaptive regulatory immune cell (50, 51). They are often characterized as CD8+CD25+ Tregs and have similar functions as CD4+CD25+ Tregs (52). They have also been shown to induce tolerogenic antigen-presenting cells as well as induce T effector cell killing in addition to T effector cell function suppression (53). In the context of solid organ transplantation, their capacity to induce tolerance and their role as a potential therapeutic avenue is being increasingly studied (53, 54). In addition, TNFR2 has been found to be a more reliable surface marker than CD25 for characterization of functional CD8+Tregs (51). The induction of CD8+Foxp3+TNFR2+ T cells has also been observed after treatment with anti-CD3 monoclonal antibody treatment in patients with Type 1 diabetes mellitus patients (55). In this study, combined expression of CD8+ with CD25+ identified a highly suppressive CD8+Treg subset which were able to suppress CD4+T cell activity the most. Research has also been performed in the context of hematopoietic stem cell transplantation. In these in vivo mice studies, it was suggested that TNFR2+CD8+Tregs can preferentially target allogeneic T cells and thus effectively treat GvHD and rejection (56, 57). However, as in CD4+ T cells, TNFα-TNFR2 expression and signaling within the CD8+ population is varied. TNFR2 is expressed on CD8+ T effector cells and has been shown to be a marker of their proliferative and cytotoxic effects during early phases of the immune response as well as a signal for CD8+ T effector cell apoptosis and activity termination during the later phases of the immune response (51). These diverse functions highlight the complexity required to maintain homeostasis as well as the care that must be taken when studying these cells and developing targeted therapies.
TNFR2 in B Regulatory Cells
B regulatory cells (Bregs) are a highly diverse suppressive subset of B cells which can arise at any stage in B cell development and have strong suppressive ability, particularly in response to inflammation (58, 59). Bregs have a hallmark production of cytokines such as IL-10. IL-10 release by B cells has been shown to reduce T cell proliferation (60). Bregs have also shown to take part in tolerance induction in solid organ transplantation (61–63). Previously, no common surface marker for Bregs were known. However, TNFR2 expression on B cells has been recently shown to characterize Bregs (64). In this same study, administration of TNFR2 agonist to cell cultures enhanced both production of IL-10 and IL-6 by these B cells. TNFR2 expression has also been related to human memory B cells with suppressive function and increasing evidence suggests that expression of TNFR2 on B cells is characteristic of a suppressive B cell subset (59).
TNFR2 in Myeloid-Derived Suppressor Cells
Myeloid-derived suppressor cells (MDSCs) are important negative immune regulators and are characterized by a heterogenous population of both immature and progenitor myeloid cells (65). In the context of lung transplantation, granulocyte derived MDSCs have been shown to be correlated with a stable patient phenotype while lower levels have been associated with post-transplant complications (66). The expression of TNFR2 on MDSCs has also been revealed (Figure 1). Monocyte-derived MDSCs (Mo-MDSC), which are a subset of MDSC, have been shown to be highly effective in suppressing proliferation of CD4+ T effector cells as well as expanding CD4+Tregs. TNFR2 expression on these Mo-MDSCs has been associated with their generation and function (67, 68). In addition, mTNF has been shown to promote MDSC suppressive function and this activity appears to be mediated via TNFR2 expression (68, 69). In cancer mice models, TNFR2 expression on MDSCs has been found to crosstalk with B cells in germinal centers. Interestingly, this interaction has been shown to mediate immunosuppression by upregulating immunoglobulin A (IgA) responses and promoting IL-10 production (70). TNFR2 expression on MDSCs has also been studied in the context of tuberculosis and has been associated as a strong driver of anti-inflammatory function (71).
TNFR2 Expression and Ischemia Reperfusion Injury
Ischemia-reperfusion injury (IRI) is an inevitable component of organ transplantation and strongly contributes to short and long-term graft outcomes. Increasing evidence now links IRI with activation of the innate and adaptive immune systems (72). During the ischemia phase, adenosine triphosphate (ATP) and glycogen depletion occur, which then leads to mitochondrial dysfunction and ultimately cell death of both endothelial cells and tubular epithelial cells. During reperfusion, there is release of reactive oxygen species, chemokines, cytokines, as well as the products of cell death into the circulation (17). This ultimately stimulates the innate immune response which then enhances activation of the adaptive immune responses and promotes graft allorecognition.
TNFα has been found to be a key cytokine in inducing apoptosis during progression of IRI in many contexts including acute kidney injury (AKI) models (5, 73, 74). In these studies, rat experimental models of AKI were studied and anti-TNFα therapy was found to improve renal IRI recovery. However, these studies lack information on the differential contributions that TNFR1 and TNFR2 hold during IRI.
The differential role of TNFα signaling via TNFR1 and TNFR2 has been studied in IRI in the context of arteriogenesis and angiogenesis (75, 76). In these studies, TNFR2 expression on vascular endothelial cells was found to induce endothelial cell angiogenesis, proliferation, survival, and migration while TNFR1 caused endothelial cell apoptosis and inhibition of migration.
Other in vivo models of the role of TNFR2 in IRI have also been studied. In mouse hind-limb ischemia models, TNFR2 deletion has been shown to be associated with an increase in inflammatory response as well as in a decrease in post hind limb ischemia recovery (77). This same group has shown that TNFR2 is required in ischemia-induced neovascularization and is protective in human adult myocardial infarctions (78, 79). There have also been studies of IRI in cardiomyocyte-induced ischemia (80).. In these settings, TNFR2 expression on cardiomyocytes was associated with cardioprotection while TNFR1 expression on cardiomyocytes was associated with cardiac dysfunction, fibrosis, and cell death.
Etanercept is a fusion protein consisting of TNFR2 combined with the immunoglobulin G1 fragment crystallizable region, developed for use in autoimmune disorders (81). It is the first therapeutic agent to specifically target TNFR2 signaling, by simulating TNFR2 shedding. While data is limited in the transplant context, etanercept use has been shown to be advantageous in limiting sequelae of IRI in various contexts, including kidney (82), heart (83, 84), brain (85, 86), and intestine (87, 88).
Therapeutic Use in Islet Transplantation
Islet transplantation is a treatment of choice in many jurisdictions, particularly for diabetes patients with severe hypoglycemic unawareness, thanks to improvements in insulin independence rates among recipients. Infusion-related cytokine release and neutrophil chemotaxis has long been appreciated as a cause of islet graft loss, leading to a syndrome known as the immediate, blood-mediated inflammatory response (IBMIR) (89). Furthermore, studies of islet cell apoptosis following isolation suggested that TNFα production by the cell preparation was associated with higher levels of cell death (90). In experimental models, etanercept use was associated with improved function and lower rates of beta-cell apoptosis, particularly when used in conjunction with an anti-IL-1β agent (91). Early adoption in the clinical setting suggested that etanercept use was associated with improved graft function and a higher degree of success following a single infusion of donor islets (92, 93). This was confirmed in larger analyses as greater experience was gained with multiple immunosuppressive protocols (94), with administration of etanercept being associated with preservation of insulin independence at 5 years post-transplant, compared to protocols using similar induction therapy, but without etanercept (Figure 2). More robust evidence of the utility of this approach was seen in multivariable statistical analysis of international registry data, in which etanercept use was associated with a significant reduction in risk of graft failure (95). Similar conclusions were drawn in a systematic analysis of reported clinical data (96). These results cemented etanercept use as a mainstay in peri-transplant immunotherapy in this context (97). Early clinical experience with combination therapy targeting TNFα and IL-1β signaling has also been shown to be safe (98). As newer, stem-cell derived products become available to treat type 1 diabetes mellitus, their use may still be complicated by IBMIR and the effects of IRI on the graft during pre-transplant culture and infusion. While etanercept use may provide an advantage during this period, some evidence suggests a narrow therapeutic window beyond which toxicity may be seen (99).
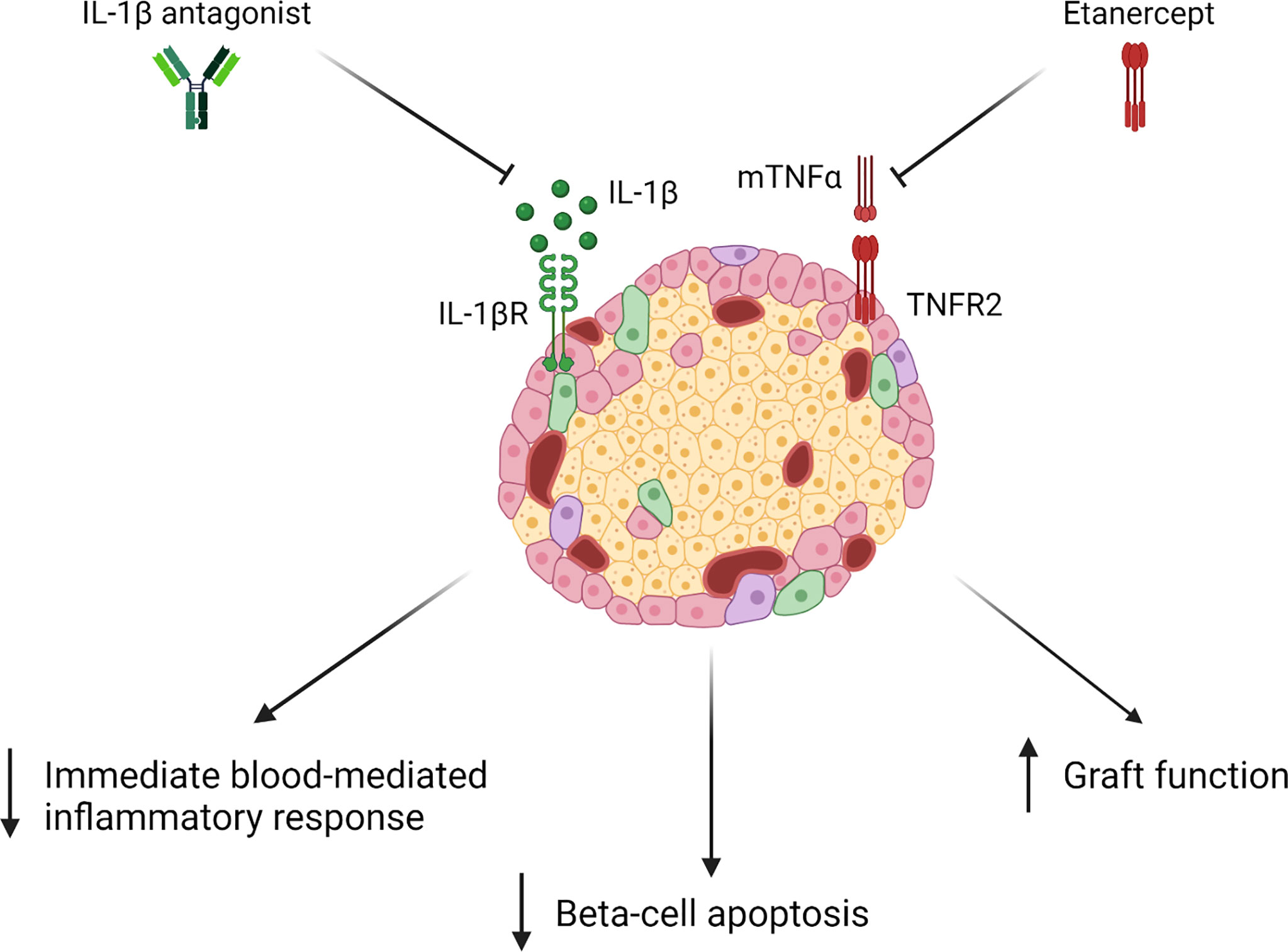
Figure 2 Upon administration of Etanercept with an IL-1β antagonist, studies have shown a decrease in immediate blood-mediated inflammatory response (IBMIR), beta-cell apoptosis, and an improvement in graft function (84–86).
TNFR2 in Solid Organ Transplantation and Acute Rejection
Early graft function is crucial to early patient survival and to long term outcomes in many organ transplant contexts, and is closely related to degree of IRI. In kidney transplantation, IRI manifests as delayed graft function (DGF) (100). DGF, which is defined as the need for dialysis after kidney transplantation, affects between 25 and 40% of grafts from deceased donors, and has long been associated with a higher lifetime risk of acute rejection episodes and poorer long term allograft survival (101). In the context of liver transplantation, IRI is linked to increased liver enzymes, higher rates of biliary strictures, increase risk of acute and chronic rejection, and ultimately poorer long-term graft outcomes (102–104). In the context of pancreas transplantation, IRI is associated with pancreatitis, graft thrombosis, as well as graft loss (105). For lung transplantation, IRI increases chance of alveolar lung damage, lung edema, hypoxemia, and graft failure (106). Finally, for heart transplantation, IRI is associated with early graft dysfunction, primary graft dysfunction, and cardiac allograft vasculopathy (107).
As described earlier, TNFα-TNFR2 interactions have been shown to dampen IRI response in a variety of non-transplant contexts. While the role of TNFR2 expression in renal IRI after transplantation hasn’t yet been studied, its role as a marker of graft function has been increasingly explored. De novo TNFR2 expression on human tubular epithelial cells was first observed during renal transplant biopsies of acutely rejecting allografts (108). It was also discovered that amount of TNFR2 expression correlated with severity of rejection episode. These findings were confirmed in both human and rat studies where biopsies of renal allografts with acute rejection were taken and showed significantly higher staining for TNFR2 observed on podocytes and renal tubular epithelial cells (109). However, despite TNFR2 expression on tubular epithelial cells correlating with acute rejection episode and severity, no long-term data was available for human patients in studying if TNFR2 was also related to faster resolution or improved return to baseline creatinine. Also, no mechanistic studies were performed during these models and thus these studies could not differentiate if this increase in TNFR2 expression was an instigator of rejection or a response to rejection for its resolution. Finally, TNFR2 expression in peripheral blood sample of the patients was not studied and could have been an interesting addition to assess if any changes in TNFR2 frequency on immune regulatory versus pro-inflammatory cells was observed. An increase in serum TNFR2 expression in patients with acute rejection post kidney transplant has also been observed (110, 111). However, these studies were lacking correlation with long-term graft and patient outcomes as well as was characterization of the cell type which had an increase TNFR2 frequency. Together, these studies suggest a role for TNFR2 measurement in conjunction with serum creatinine as a diagnostic marker of acute rejection however, they lack the mechanistic link between TNFR2 and acute rejection and lack long-term graft and patient outcomes data to further clarify this link.
The mechanistic links and signaling pathways involved in TNFR2 expressing tubular epithelial cells in the context of acute rejection have since been studied (112, 113). TNFR1 signaling was shown to colocalize with apoptosis signal-regulating kinase-1 (ASK1) on glomerular cells and peritubular capillary endothelial cells while TNFR2 colocalized with endothelial/epithelial tyrosine kinase (EKT) on tubular epithelial cells and glomerular cells. While TNFR1-ASK1 were shown to induce proapoptotic pathways, TNFR2-EKT signaling was shown to induce tubular epithelial cell adhesion, migration, proliferation, and survival during acute rejection episodes. The same group revealed an associated upregulation as well as colocalization of TNFα converting enzyme (TACE) with TNFR2 on tubular epithelial cell during acute rejection episode. This suggests a role for TACE to promote TNFR2 shedding and limit proinflammatory TNFα effects during acute rejection episodes (114). More recently, the study (115) reported that TNFR2 signaling induces regeneration of tubular epithelial cells during acute cell-mediated kidney rejection. In this study, a mechanistic link between TNFR2 signaling and induction of tubular epithelial stem cell properties during acute rejection was observed.
While these studies observed an association between TNFR2 expression on tubular epithelial cells and acute rejection, there is paucity of data looking at TNFR2 expression on immune cells during solid organ transplantation. Our group has previously studied the role of the frequency of recipient pretransplant TNFR2+CD4+Tregs as a predictor of post kidney transplant DGF and short to medium-term outcomes (116). Increase pretransplant circulating peripheral CD4+Treg TNFR2 frequency was associated with decreased post-transplant DGF rates and was hypothesized to be linked to its role in inducing and maintaining higher CD4+Treg suppressive function. Also, TNFR2 expression on Mo-MDSCs has been associated with increased CD4+Tregs frequency in human renal transplantation (117).
TNFR2 expression has also been briefly explored in heart transplantation. In cardiac transplant mice models an increase in TNFR2 expression was observed on cardiomyocytes and was associated with an increase in cell cycle entry and proliferation of these cardiomyocytes during acute rejection episodes (118). On the other hand, both TNFR1 and TNFR2 have been associated with graft arterial disease in cardiac allografts (119). Also, in rat cardiac allograft models, treatment with TNFR2 recombinant protein decreased TNFα expression as well as decreased cardiac remodeling (120).
These paradoxical results can be related to differential expression of TNF receptors on distinct cell types which can thus lead to contradictory phenotypes (Figure 3). In addition, while some data is available on TNFR2 expression on immune regulatory cells in the context of solid organ transplantation, there remains many unstudied avenues to pursue. Finally, as a link between TNFR2 signaling and stem cell property induction in tubular epithelial cells has been demonstrated, research exploring therapeutic potentials for TNFR2 targeting in transplant rejection and modulation of ischemia reperfusion injury can be considered.
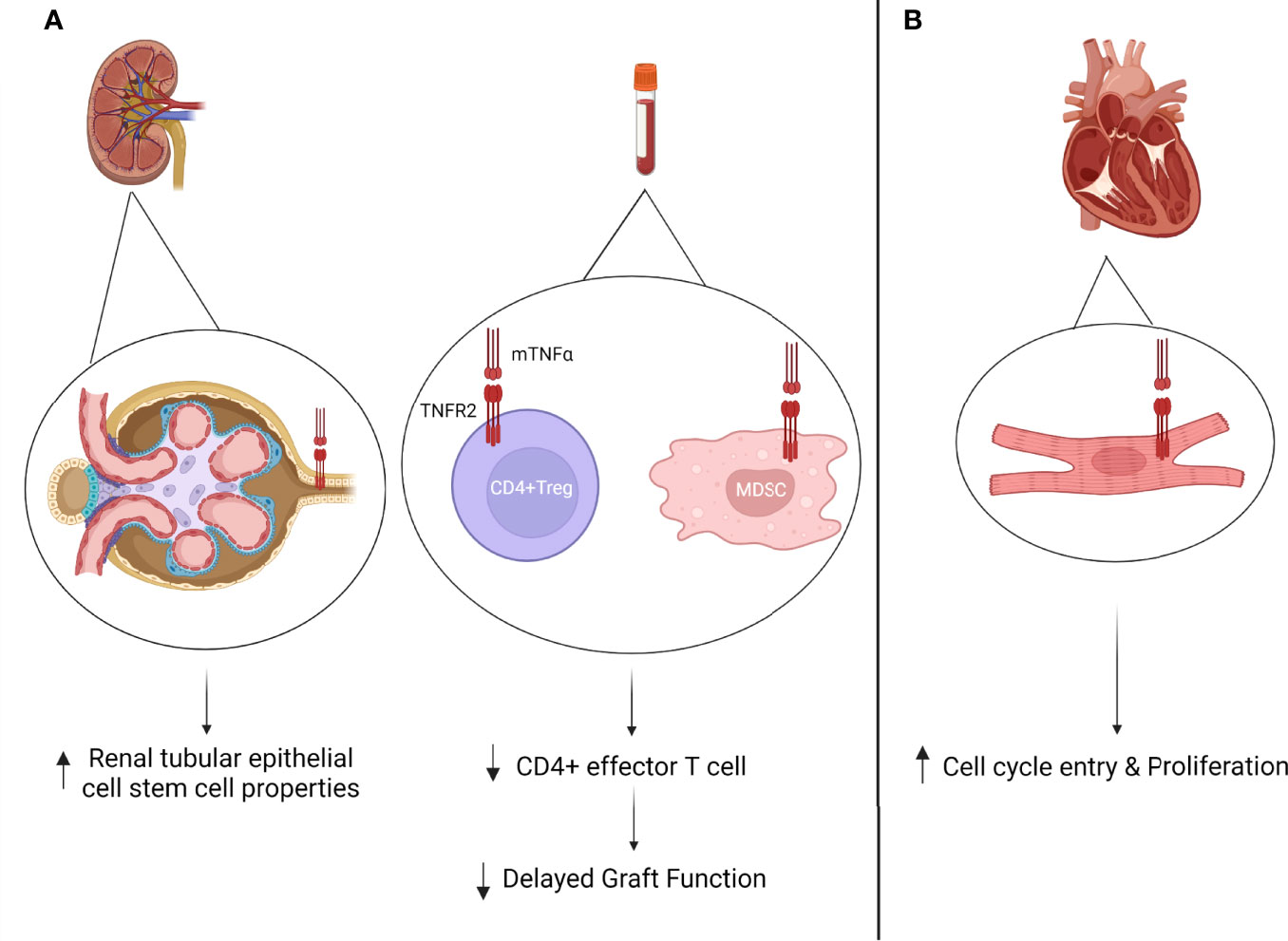
Figure 3 (A) TNFR2 expression in kidney transplantation. Increased TNFR2 expression on tubular epithelial cells during acute rejection episode has been associated with renal tubular regeneration (108). Circulating pre-transplant TNFR2 expression on CD4+Tregs has been associated with decreased delayed graft function (DGF) rates (109). Circulating post-transplant MDSCs has been associated with increased CD4+Treg frequency (105). (B) Increased TNFR2 expression on cardiomyocyte post-transplantation has been associated with increased cell cycle entry and proliferation during acute rejection episodes (111).
Therapeutic Potential in Solid Organ Transplantation
The therapeutic potential of TNFR2 agonists and antagonists has been progressively more studied in the context of CD4+Treg expansion or depletion in both autoimmunity, GvHD, and cancer (121, 122). However, there remains a lack of studies in solid organ transplantation. In this section, we will discuss potential therapeutic avenues with regard to the emerging evidence available on TNFR2 pharmacological agents.
First, selective TNFR2 targeting has been increasingly study in the setting of GvHD (40, 123–126). Indeed, TNFR2 agonist treatment was shown to promote CD4+Treg expansion and improve resolution of rejection while TNFR2 antagonists abrogated these results. As CD4+Treg TNFR2 frequency has been associated with improved short term kidney transplantation outcomes, this could be an exciting avenue to follow. By doing so, both improved graft survival in solid organ transplantation as well as improved treatments against rejection could be possible.
Finally, as TNFR2 has been shown to induce stem cell properties of tubular epithelial cells and their subsequent regeneration (115). TNFR2 expression in mesenchymal stem cells has also been shown to maintain their regenerative functions and induce CD4+Tregs while TNFR2 inhibition has been shown to decrease expression of the mesenchymal stem cell characteristics (127). These novel findings could also allow for TNFR2 targeting in transplantation to induce epithelial regeneration. However, this will have to be done with care as TNFR2 expression varies greatly in different cell types and its effects can be quite dichotomous.
Conclusion
The expression of TNFR2 on CD4+Tregs, CD8+Tregs, MDSCs, and Bregs has been associated with an immune regulatory phenotype. In solid organ transplantation, TNFR2+CD4+Tregs and TNFR2+MDSCs have been infrequently studied and have been associated with improved short-term outcomes after renal transplantation as well as increased CD4+Treg frequency and suppressive function. TNFR2 has also been observed on tubular epithelial cells as well as cardiomyocytes of renal and cardiac allografts respectively. In the context of tubular epithelial cells, recent evidence has shown a correlation between TNFR2 expression and stem cell phenotype induction while in the context of TNFR2 expression on cardiomyocytes, contradictory results have been observed.
This review highlights the positive impact that TNFα-TNFR2 signaling appears to have in solid transplantation. However, the scarcity of data requires further investigation in the role of TNFR2 in all types of solid organ transplantation as well as dedicated studies on the therapeutic use of TNFR2 agents during induction, maintenance therapy, or in the treatment of rejection.
Author Contributions
AK, JT, and SP reviewed the literature. AK drafted the manuscript. All authors contributed to the editing of the manuscript. All authors contributed to the article and approved the submitted version.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
AK is supported by the Clinician-Investigator Program (Faculty of Medicine, McGill University, Montreal, Quebec, Canada). Figures 1–3 were created with Biorender.com.
References
1. Grivennikov SI, Tumanov AV, Liepinsh DJ, Kruglov AA, Marakusha BI, Shakhov AN, et al. Distinct and Nonredundant in Vivo Functions of Tnf Produced by T Cells and Macrophages/Neutrophils: Protective and Deleterious Effects. Immunity (2005) 22(1):93–104. doi: 10.1016/j.immuni.2004.11.016
2. Aste-Amezaga M, D'Andrea A, Kubin M, Trinchieri G. Cooperation of Natural Killer Cell Stimulatory Factor/Interleukin-12 With Other Stimuli in the Induction of Cytokines and Cytotoxic Cell-Associated Molecules in Human T and Nk Cells. Cell Immunol (1994) 156(2):480–92. doi: 10.1006/cimm.1994.1192
3. Aggarwal BB. Signalling Pathways of the Tnf Superfamily: A Double-Edged Sword. Nat Rev Immunol (2003) 3(9):745–56. doi: 10.1038/nri1184
4. Wajant H, Pfizenmaier K, Scheurich P. Tumor Necrosis Factor Signaling. Cell Death Differ (2003) 10(1):45–65. doi: 10.1038/sj.cdd.4401189
5. Al-Lamki RS, Mayadas TN. Tnf Receptors: Signaling Pathways and Contribution to Renal Dysfunction. Kidney Int (2015) 87(2):281–96. doi: 10.1038/ki.2014.285
6. Faustman D, Davis M. Tnf Receptor 2 Pathway: Drug Target for Autoimmune Diseases. Nat Rev Drug Discov (2010) 9(6):482–93. doi: 10.1038/nrd3030
7. Rossol M, Meusch U, Pierer M, Kaltenhäuser S, Häntzschel H, Hauschildt S, et al. Interaction Between Transmembrane Tnf and Tnfr1/2 Mediates the Activation of Monocytes by Contact With T Cells. J Immunol (2007) 179(6):4239–48. doi: 10.4049/jimmunol.179.6.4239
8. Wajant H, Siegmund D. Tnfr1 and Tnfr2 in the Control of the Life and Death Balance of Macrophages. Front Cell Dev Biol (2019) 7:91. doi: 10.3389/fcell.2019.00091
9. Maney NJ, Reynolds G, Krippner-Heidenreich A, Hilkens CMU. Dendritic Cell Maturation and Survival Are Differentially Regulated by Tnfr1 and Tnfr2. J Immunol (2014) 193(10):4914–23. doi: 10.4049/jimmunol.1302929
10. Salamone G, Giordano M, Trevani AS, Gamberale R, Vermeulen M, Schettinni J, et al. Promotion of Neutrophil Apoptosis by Tnf-Alpha. J Immunol (2001) 166(5):3476–83. doi: 10.4049/jimmunol.166.5.3476
11. Michlewska S, Dransfield I, Megson IL, Rossi AG. Macrophage Phagocytosis of Apoptotic Neutrophils Is Critically Regulated by the Opposing Actions of Pro-Inflammatory and Anti-Inflammatory Agents: Key Role for Tnf-Alpha. FASEB J (2009) 23(3):844–54. doi: 10.1096/fj.08-121228
12. Croft M. The Tnf Family in T Cell Differentiation and Function–Unanswered Questions and Future Directions. Semin Immunol (2014) 26(3):183–90. doi: 10.1016/j.smim.2014.02.005
13. Zimmerer JM, Horne PH, Fiessinger LA, Fisher MG, Pham TA, Saklayen SL, et al. Cytotoxic Effector Function of Cd4-Independent, Cd8(+) T Cells Is Mediated by Tnf-α/Tnfr. Transplantation (2012) 94(11):1103–10. doi: 10.1097/TP.0b013e318270f3c0
14. Ahmad S, Azid NA, Boer JC, Lim J, Chen X, Plebanski M, et al. The Key Role of Tnf-Tnfr2 Interactions in the Modulation of Allergic Inflammation: A Review. Front Immunol (2018) 9:2572. doi: 10.3389/fimmu.2018.02572
15. Wu Y, Zhou BP. Tnf-Alpha/Nf-Kappab/Snail Pathway in Cancer Cell Migration and Invasion. Br J Cancer (2010) 102(4):639–44. doi: 10.1038/sj.bjc.6605530
16. Bystrom J, Clanchy FI, Taher TE, Mangat P, Jawad AS, Williams RO, et al. Tnfα in the Regulation of Treg and Th17 Cells in Rheumatoid Arthritis and Other Autoimmune Inflammatory Diseases. Cytokine (2018) 101:4–13. doi: 10.1016/j.cyto.2016.09.001
17. Kalogeris T, Baines CP, Krenz M, Korthuis RJ. Ischemia/Reperfusion. Compr Physiol (2016) 7(1):113–70. doi: 10.1002/cphy.c160006
18. Horiuchi T, Mitoma H, Harashima S, Tsukamoto H, Shimoda T. Transmembrane Tnf-Alpha: Structure, Function and Interaction With Anti-Tnf Agents. Rheumatol (Oxford) (2010) 49(7):1215–28. doi: 10.1093/rheumatology/keq031
19. Boschert V, Krippner-Heidenreich A, Branschädel M, Tepperink J, Aird A, Scheurich P. Single Chain Tnf Derivatives With Individually Mutated Receptor Binding Sites Reveal Differential Stoichiometry of Ligand Receptor Complex Formation for Tnfr1 and Tnfr2. Cell Signal (2010) 22(7):1088–96. doi: 10.1016/j.cellsig.2010.02.011
20. Kriegler M, Perez C, DeFay K, Albert I, Lu SD. A Novel Form of Tnf/Cachectin Is a Cell Surface Cytotoxic Transmembrane Protein: Ramifications for the Complex Physiology of Tnf. Cell (1988) 53(1):45–53. doi: 10.1016/0092-8674(88)90486-2
21. Grell M, Douni E, Wajant H, Löhden M, Clauss M, Maxeiner B, et al. The Transmembrane Form of Tumor Necrosis Factor Is the Prime Activating Ligand of the 80 Kda Tumor Necrosis Factor Receptor. Cell (1995) 83(5):793–802. doi: 10.1016/0092-8674(95)90192-2
22. Yang S, Wang J, Brand DD, Zheng SG. Role of Tnf-Tnf Receptor 2 Signal in Regulatory T Cells and Its Therapeutic Implications. Front Immunol (2018) 9:784. doi: 10.3389/fimmu.2018.00784
23. Wajant H, Scheurich P. Tnfr1-Induced Activation of the Classical Nf-κb Pathway. FEBS J (2011) 278(6):862–76. doi: 10.1111/j.1742-4658.2011.08015.x
24. Sedger LM, McDermott MF. Tnf and Tnf-Receptors: From Mediators of Cell Death and Inflammation to Therapeutic Giants - Past, Present and Future. Cytokine Growth Factor Rev (2014) 25(4):453–72. doi: 10.1016/j.cytogfr.2014.07.016
25. Medler J, Wajant H. Tumor Necrosis Factor Receptor-2 (Tnfr2): An Overview of an Emerging Drug Target. Expert Opin Ther Targets (2019) 23(4):295–307. doi: 10.1080/14728222.2019.1586886
26. Fotin-Mleczek M, Henkler F, Samel D, Reichwein M, Hausser A, Parmryd I, et al. Apoptotic Crosstalk of Tnf Receptors: Tnf-R2-Induces Depletion of Traf2 and Iap Proteins and Accelerates Tnf-R1-Dependent Activation of Caspase-8. J Cell Sci (2002) 115(Pt 13):2757–70. doi: 10.1242/jcs.115.13.2757
27. Pegoretti V, Baron W, Laman JD, Eisel ULM. Selective Modulation of Tnf-Tnfrs Signaling: Insights for Multiple Sclerosis Treatment. Front Immunol (2018) 9:925. doi: 10.3389/fimmu.2018.00925
28. Salomon BL. Insights Into the Biology and Therapeutic Implications of Tnf and Regulatory T Cells. Nat Rev Rheumatol (2021) 17(8):487–504. doi: 10.1038/s41584-021-00639-6
29. Sakaguchi S, Sakaguchi N, Asano M, Itoh M, Toda M. Immunologic Self-Tolerance Maintained by Activated T Cells Expressing Il-2 Receptor Alpha-Chains (Cd25). Breakdown of a Single Mechanism of Self-Tolerance Causes Various Autoimmune Diseases. J Immunol (1995) 155(3):1151–64.
30. Fontenot JD, Gavin MA, Rudensky AY. Foxp3 Programs the Development and Function of Cd4+Cd25+ Regulatory T Cells. Nat Immunol (2003) 4(4):330–6. doi: 10.1038/ni904
31. Sakaguchi S, Wing K, Onishi Y, Prieto-Martin P, Yamaguchi T. Regulatory T Cells: How Do They Suppress Immune Responses? Int Immunol (2009) 21(10):1105–11. doi: 10.1093/intimm/dxp095
32. Attias M, Al-Aubodah T, Piccirillo CA. Mechanisms of Human Foxp3(+) T(Reg) Cell Development and Function in Health and Disease. Clin Exp Immunol (2019) 197(1):36–51. doi: 10.1111/cei.13290
33. Hall BM. Cd4+Cd25+ T Regulatory Cells in Transplantation Tolerance: 25 Years on. Transplantation (2016) 100(12):2533–47. doi: 10.1097/tp.0000000000001436
34. Hori S, Nomura T, Sakaguchi S. Pillars Article: Control of Regulatory T Cell Development by the Transcription Factor Foxp3. Science (2003) 299:1057–61. doi: 10.1126/science.1079490
35. Chen X, Subleski JJ, Kopf H, Howard OM, Männel DN, Oppenheim JJ. Cutting Edge: Expression of Tnfr2 Defines a Maximally Suppressive Subset of Mouse Cd4+Cd25+Foxp3+ T Regulatory Cells: Applicability to Tumor-Infiltrating T Regulatory Cells. J Immunol (2008) 180(10):6467–71. doi: 10.4049/jimmunol.180.10.6467
36. Chen X, Subleski JJ, Hamano R, Howard OM, Wiltrout RH, Oppenheim JJ. Co-Expression of Tnfr2 and Cd25 Identifies More of the Functional Cd4+Foxp3+ Regulatory T Cells in Human Peripheral Blood. Eur J Immunol (2010) 40(4):1099–106. doi: 10.1002/eji.200940022
37. Chen X, Oppenheim JJ. The Phenotypic and Functional Consequences of Tumour Necrosis Factor Receptor Type 2 Expression on Cd4(+) Foxp3(+) Regulatory T Cells. Immunology (2011) 133(4):426–33. doi: 10.1111/j.1365-2567.2011.03460.x
38. Ronin E, Pouchy C, Khosravi M, Hilaire M, Grégoire S, Casrouge A, et al. Tissue-Restricted Control of Established Central Nervous System Autoimmunity by Tnf Receptor 2-Expressing Treg Cells. Proc Natl Acad Sci USA (2021) 118(13):e2014043118. doi: 10.1073/pnas.2014043118
39. Chen X, Bäumel M, Männel DN, Howard OM, Oppenheim JJ. Interaction of Tnf With Tnf Receptor Type 2 Promotes Expansion and Function of Mouse Cd4+Cd25+ T Regulatory Cells. J Immunol (2007) 179(1):154–61. doi: 10.4049/jimmunol.179.1.154
40. Chopra M, Biehl M, Steinfatt T, Brandl A, Kums J, Amich J, et al. Exogenous Tnfr2 Activation Protects From Acute Gvhd Via Host T Reg Cell Expansion. J Exp Med (2016) 213(9):1881–900. doi: 10.1084/jem.20151563
41. Torrey H, Kühtreiber WM, Okubo Y, Tran L, Case K, Zheng H, et al. A Novel Tnfr2 Agonist Antibody Expands Highly Potent Regulatory T Cells. Sci Signal (2020) 13(661):eaba9600. doi: 10.1126/scisignal.aba9600
42. Skartsis N, Peng Y, Ferreira LMR, Nguyen V, Ronin E, Muller YD, et al. Il-6 and Tnfα Drive Extensive Proliferation of Human Tregs Without Compromising Their Lineage Stability or Function. Front Immunol (2021) 12:783282. doi: 10.3389/fimmu.2021.783282
43. Mahmud SA, Manlove LS, Schmitz HM, Xing Y, Wang Y, Owen DL, et al. Costimulation Via the Tumor-Necrosis Factor Receptor Superfamily Couples Tcr Signal Strength to the Thymic Differentiation of Regulatory T Cells. Nat Immunol (2014) 15(5):473–81. doi: 10.1038/ni.2849
44. Nguyen DX, Ehrenstein MR. Anti-Tnf Drives Regulatory T Cell Expansion by Paradoxically Promoting Membrane Tnf-Tnf-Rii Binding in Rheumatoid Arthritis. J Exp Med (2016) 213(7):1241–53. doi: 10.1084/jem.20151255
45. Zorn E, Nelson EA, Mohseni M, Porcheray F, Kim H, Litsa D, et al. Il-2 Regulates Foxp3 Expression in Human Cd4+Cd25+ Regulatory T Cells Through a Stat-Dependent Mechanism and Induces the Expansion of These Cells in Vivo. Blood (2006) 108(5):1571–9. doi: 10.1182/blood-2006-02-004747
46. Miller PG, Bonn MB, McKarns SC. Transmembrane Tnf-Tnfr2 Impairs Th17 Differentiation by Promoting Il2 Expression. J Immunol (2015) 195(6):2633–47. doi: 10.4049/jimmunol.1500286
47. Housley WJ, Adams CO, Nichols FC, Puddington L, Lingenheld EG, Zhu L, et al. Natural But Not Inducible Regulatory T Cells Require Tnf-Alpha Signaling for in Vivo Function. J Immunol (2011) 186(12):6779–87. doi: 10.4049/jimmunol.1003868
48. Yang S, Xie C, Chen Y, Wang J, Chen X, Lu Z, et al. Differential Roles of Tnfα-Tnfr1 and Tnfα-Tnfr2 in the Differentiation and Function of Cd4(+)Foxp3(+) Induced Treg Cells in Vitro and in Vivo Periphery in Autoimmune Diseases. Cell Death Dis (2019) 10(1):27. doi: 10.1038/s41419-018-1266-6
49. Nadkarni S, Mauri C, Ehrenstein MR. Anti-Tnf-Alpha Therapy Induces a Distinct Regulatory T Cell Population in Patients With Rheumatoid Arthritis Via Tgf-Beta. J Exp Med (2007) 204(1):33–9. doi: 10.1084/jem.20061531
50. Maggi E, Cosmi L, Liotta F, Romagnani P, Romagnani S, Annunziato F. Thymic Regulatory T Cells. Autoimmun Rev (2005) 4(8):579–86. doi: 10.1016/j.autrev.2005.04.010
51. Ye LL, Wei XS, Zhang M, Niu YR, Zhou Q. The Significance of Tumor Necrosis Factor Receptor Type Ii in Cd8(+) Regulatory T Cells and Cd8(+) Effector T Cells. Front Immunol (2018) 9:583. doi: 10.3389/fimmu.2018.00583
52. Cosmi L, Liotta F, Lazzeri E, Francalanci M, Angeli R, Mazzinghi B, et al. Human Cd8+Cd25+ Thymocytes Share Phenotypic and Functional Features With Cd4+Cd25+ Regulatory Thymocytes. Blood (2003) 102(12):4107–14. doi: 10.1182/blood-2003-04-1320
53. Bézie S, Anegon I, Guillonneau C. Advances on Cd8+ Treg Cells and Their Potential in Transplantation. Transplantation (2018) 102(9):1467–78. doi: 10.1097/tp.0000000000002258
54. Picarda E, Bézie S, Venturi V, Echasserieau K, Mérieau E, Delhumeau A, et al. Mhc-Derived Allopeptide Activates Tcr-Biased Cd8+ Tregs and Suppresses Organ Rejection. J Clin Invest (2014) 124(6):2497–512. doi: 10.1172/jci71533
55. Ablamunits V, Bisikirska B, Herold KC. Acquisition of Regulatory Function by Human Cd8(+) T Cells Treated With Anti-Cd3 Antibody Requires Tnf. Eur J Immunol (2010) 40(10):2891–901. doi: 10.1002/eji.201040485
56. Horwitz DA, Pan S, Ou JN, Wang J, Chen M, Gray JD, et al. Therapeutic Polyclonal Human Cd8+ Cd25+ Fox3+ Tnfr2+ Pd-L1+ Regulatory Cells Induced Ex-Vivo. Clin Immunol (2013) 149(3):450–63. doi: 10.1016/j.clim.2013.08.007
57. Wang W, Hong T, Wang X, Wang R, Du Y, Gao Q, et al. Newly Found Peacekeeper: Potential of Cd8+ Tregs for Graft-Versus-Host Disease. Front Immunol (2021) 12:764786. doi: 10.3389/fimmu.2021.764786
58. Rosser EC, Mauri C. Regulatory B Cells: Origin, Phenotype, and Function. Immunity (2015) 42(4):607–12. doi: 10.1016/j.immuni.2015.04.005
59. Ticha O, Slanina P, Moos L, Stichova J, Vlkova M, Bekeredjian-Ding I. Tnfr2 Expression Is a Hallmark of Human Memory B Cells With Suppressive Function. Eur J Immunol (2021) 51(5):1195–205. doi: 10.1002/eji.202048988
60. Catalán D, Mansilla MA, Ferrier A, Soto L, Oleinika K, Aguillón JC, et al. Immunosuppressive Mechanisms of Regulatory B Cells. Front Immunol (2021) 12:611795. doi: 10.3389/fimmu.2021.611795
61. Durand J, Chiffoleau E. B Cells With Regulatory Properties in Transplantation Tolerance. World J Transplant (2015) 5(4):196–208. doi: 10.5500/wjt.v5.i4.196
62. Peng B, Ming Y, Yang C. Regulatory B Cells: The Cutting Edge of Immune Tolerance in Kidney Transplantation. Cell Death Dis (2018) 9(2):109. doi: 10.1038/s41419-017-0152-y
63. Alhabbab RY, Nova-Lamperti E, Aravena O, Burton HM, Lechler RI, Dorling A, et al. Regulatory B Cells: Development, Phenotypes, Functions, and Role in Transplantation. Immunol Rev (2019) 292(1):164–79. doi: 10.1111/imr.12800
64. Ticha O, Moos L, Wajant H, Bekeredjian-Ding I. Expression of Tumor Necrosis Factor Receptor 2 Characterizes Tlr9-Driven Formation of Interleukin-10-Producing B Cells. Front Immunol (2017) 8:1951. doi: 10.3389/fimmu.2017.01951
65. Ochando J, Conde P, Utrero-Rico A, Paz-Artal E. Tolerogenic Role of Myeloid Suppressor Cells in Organ Transplantation. Front Immunol (2019) 10:374. doi: 10.3389/fimmu.2019.00374
66. Heigl T, Singh A, Saez-Gimenez B, Kaes J, Van Herck A, Sacreas A, et al. Myeloid-Derived Suppressor Cells in Lung Transplantation. Front Immunol (2019) 10:900. doi: 10.3389/fimmu.2019.00900
67. Zhao X, Rong L, Zhao X, Li X, Liu X, Deng J, et al. Tnf Signaling Drives Myeloid-Derived Suppressor Cell Accumulation. J Clin Invest (2012) 122(11):4094–104. doi: 10.1172/jci64115
68. Polz J, Remke A, Weber S, Schmidt D, Weber-Steffens D, Pietryga-Krieger A, et al. Myeloid Suppressor Cells Require Membrane Tnfr2 Expression for Suppressive Activity. Immun Inflamm Dis (2014) 2(2):121–30. doi: 10.1002/iid3.19
69. Hu X, Li B, Li X, Zhao X, Wan L, Lin G, et al. Transmembrane Tnf-α Promotes Suppressive Activities of Myeloid-Derived Suppressor Cells Via Tnfr2. J Immunol (2014) 192(3):1320–31. doi: 10.4049/jimmunol.1203195
70. Xu X, Meng Q, Erben U, Wang P, Glauben R, Kühl AA, et al. Myeloid-Derived Suppressor Cells Promote B-Cell Production of Iga in a Tnfr2-Dependent Manner. Cell Mol Immunol (2017) 14(7):597–606. doi: 10.1038/cmi.2015.103
71. Chavez-Galan L, Vesin D, Uysal H, Blaser G, Benkhoucha M, Ryffel B, et al. Transmembrane Tumor Necrosis Factor Controls Myeloid-Derived Suppressor Cell Activity Via Tnf Receptor 2 and Protects From Excessive Inflammation During Bcg-Induced Pleurisy. Front Immunol (2017) 8:999. doi: 10.3389/fimmu.2017.00999
72. Slegtenhorst BR, Dor FJ, Rodriguez H, Voskuil FJ, Tullius SG. Ischemia/Reperfusion Injury and Its Consequences on Immunity and Inflammation. Curr Transplant Rep (2014) 1(3):147–54. doi: 10.1007/s40472-014-0017-6
73. Jang HR, Rabb H. The Innate Immune Response in Ischemic Acute Kidney Injury. Clin Immunol (2009) 130(1):41–50. doi: 10.1016/j.clim.2008.08.016
74. Nagata Y, Fujimoto M, Nakamura K, Isoyama N, Matsumura M, Fujikawa K, et al. Anti-Tnf-α Agent Infliximab and Splenectomy Are Protective Against Renal Ischemia-Reperfusion Injury. Transplantation (2016) 100(8):1675–82. doi: 10.1097/tp.0000000000001222
75. Luo D, Luo Y, He Y, Zhang H, Zhang R, Li X, et al. Differential Functions of Tumor Necrosis Factor Receptor 1 and 2 Signaling in Ischemia-Mediated Arteriogenesis and Angiogenesis. Am J Pathol (2006) 169(5):1886–98. doi: 10.2353/ajpath.2006.060603
76. Luo Y, Xu Z, Wan T, He Y, Jones D, Zhang H, et al. Endothelial-Specific Transgenesis of Tnfr2 Promotes Adaptive Arteriogenesis and Angiogenesis. Arterioscler Thromb Vasc Biol (2010) 30(7):1307–14. doi: 10.1161/atvbaha.110.204222
77. Sasi SP, Rahimi L, Yan X, Silver M, Qin G, Losordo DW, et al. Genetic Deletion of Tnfr2 Augments Inflammatory Response and Blunts Satellite-Cell-Mediated Recovery Response in a Hind Limb Ischemia Model. FASEB J (2015) 29(4):1208–19. doi: 10.1096/fj.14-249813
78. Kishore R, Tkebuchava T, Sasi SP, Silver M, Gilbert HY, Yoon YS, et al. Tumor Necrosis Factor-α Signaling Via Tnfr1/P55 Is Deleterious Whereas Tnfr2/P75 Signaling Is Protective in Adult Infarct Myocardium. Adv Exp Med Biol (2011) 691:433–48. doi: 10.1007/978-1-4419-6612-4_45
79. Goukassian DA, Qin G, Dolan C, Murayama T, Silver M, Curry C, et al. Tumor Necrosis Factor-Alpha Receptor P75 Is Required in Ischemia-Induced Neovascularization. Circulation (2007) 115(6):752–62. doi: 10.1161/circulationaha.106.647255
80. Kleinbongard P, Schulz R, Heusch G. Tnfα in Myocardial Ischemia/Reperfusion, Remodeling and Heart Failure. Heart Fail Rev (2011) 16(1):49–69. doi: 10.1007/s10741-010-9180-8
81. Peppel K, Crawford D, Beutler B. A Tumor Necrosis Factor (Tnf) Receptor-Igg Heavy Chain Chimeric Protein as a Bivalent Antagonist of Tnf Activity. J Exp Med (1991) 174(6):1483–9. doi: 10.1084/jem.174.6.1483
82. Choi DE, Jeong JY, Lim BJ, Na KR, Shin YT, Lee KW. Pretreatment With the Tumor Nerosis Factor-Alpha Blocker Etanercept Attenuated Ischemia-Reperfusion Renal Injury. Transplant Proc (2009) 41(9):3590–6. doi: 10.1016/j.transproceed.2009.05.042
83. Yang M, Chen J, Zhao J, Meng M. Etanercept Attenuates Myocardial Ischemia/Reperfusion Injury by Decreasing Inflammation and Oxidative Stress. PLoS One (2014) 9(9):e108024. doi: 10.1371/journal.pone.0108024
84. Pei H, Song X, Peng C, Tan Y, Li Y, Li X, et al. Tnf-α Inhibitor Protects Against Myocardial Ischemia/Reperfusion Injury Via Notch1-Mediated Suppression of Oxidative/Nitrative Stress. Free Radic Biol Med (2015) 82:114–21. doi: 10.1016/j.freeradbiomed.2015.02.002
85. Arango-Dávila CA, Vera A, Londoño AC, Echeverri AF, Cañas F, Cardozo CF, et al. Soluble or Soluble/Membrane Tnf-α Inhibitors Protect the Brain From Focal Ischemic Injury in Rats. Int J Neurosci (2015) 125(12):936–40. doi: 10.3109/00207454.2014.980906
86. Iwata N, Takayama H, Xuan M, Kamiuchi S, Matsuzaki H, Okazaki M, et al. Effects of Etanercept Against Transient Cerebral Ischemia in Diabetic Rats. BioMed Res Int (2015) 2015:189292. doi: 10.1155/2015/189292
87. Gerlach UA, Atanasov G, Wallenta L, Polenz D, Reutzel-Selke A, Kloepfel M, et al. Short-Term Tnf-Alpha Inhibition Reduces Short-Term and Long-Term Inflammatory Changes Post-Ischemia/Reperfusion in Rat Intestinal Transplantation. Transplantation (2014) 97(7):732–9. doi: 10.1097/tp.0000000000000032
88. Ozis SE, Akhayeva T, Guner S, Kilicoglu SS, Pampal A. Etanercept Restores Vasocontractile Sensitivity Affected by Mesenteric Ischemia Reperfusion. J Surg Res (2018) 226:8–14. doi: 10.1016/j.jss.2018.01.005
89. Bennet W, Sundberg B, Groth CG, Brendel MD, Brandhorst D, Brandhorst H, et al. Incompatibility Between Human Blood and Isolated Islets of Langerhans: A Finding With Implications for Clinical Intraportal Islet Transplantation? Diabetes (1999) 48(10):1907–14. doi: 10.2337/diabetes.48.10.1907
90. Hanley S, Liu S, Lipsett M, Castellarin M, Rosenberg L, Tchervenkov J, et al. Tumor Necrosis Factor-Alpha Production by Human Islets Leads to Postisolation Cell Death. Transplantation (2006) 82(6):813–8. doi: 10.1097/01.tp.0000234787.05789.23
91. McCall M, Pawlick R, Kin T, Shapiro AM. Anakinra Potentiates the Protective Effects of Etanercept in Transplantation of Marginal Mass Human Islets in Immunodeficient Mice. Am J Transplant (2012) 12(2):322–9. doi: 10.1111/j.1600-6143.2011.03796.x
92. Hering BJ, Kandaswamy R, Ansite JD, Eckman PM, Nakano M, Sawada T, et al. Single-Donor, Marginal-Dose Islet Transplantation in Patients With Type 1 Diabetes. JAMA (2005) 293(7):830–5. doi: 10.1001/jama.293.7.830
93. Gangemi A, Salehi P, Hatipoglu B, Martellotto J, Barbaro B, Kuechle JB, et al. Islet Transplantation for Brittle Type 1 Diabetes: The Uic Protocol. Am J Transplant (2008) 8(6):1250–61. doi: 10.1111/j.1600-6143.2008.02234.x
94. Bellin MD, Barton FB, Heitman A, Harmon JV, Kandaswamy R, Balamurugan AN, et al. Potent Induction Immunotherapy Promotes Long-Term Insulin Independence After Islet Transplantation in Type 1 Diabetes. Am J Transplant (2012) 12(6):1576–83. doi: 10.1111/j.1600-6143.2011.03977.x
95. Alejandro R, Barton FB, Hering BJ, Wease S. 2008 Update From the Collaborative Islet Transplant Registry. Transplantation (2008) 86(12):1783–8. doi: 10.1097/TP.0b013e3181913f6a
96. Szempruch KR, Banerjee O, McCall RC, Desai CS. Use of Anti-Inflammatory Agents in Clinical Islet Cell Transplants: A Qualitative Systematic Analysis. Islets (2019) 11(3):65–75. doi: 10.1080/19382014.2019.1601543
97. Hering BJ, Clarke WR, Bridges ND, Eggerman TL, Alejandro R, Bellin MD, et al. Phase 3 Trial of Transplantation of Human Islets in Type 1 Diabetes Complicated by Severe Hypoglycemia. Diabetes Care (2016) 39(7):1230–40. doi: 10.2337/dc15-1988
98. Onaca N, Takita M, Levy MF, Naziruddin B. Anti-Inflammatory Approach With Early Double Cytokine Blockade (Il-1β and Tnf-α) Is Safe and Facilitates Engraftment in Islet Allotransplantation. Transplant Direct (2020) 6(3):e530. doi: 10.1097/txd.0000000000000977
99. Brandhorst D, Brandhorst H, Acreman S, Abraham A, Johnson PRV. High Concentrations of Etanercept Reduce Human Islet Function and Integrity. J Inflamm Res (2021) 14:599–610. doi: 10.2147/jir.S294663
100. Ponticelli C. Ischaemia-Reperfusion Injury: A Major Protagonist in Kidney Transplantation. Nephrol Dial Transplant (2014) 29(6):1134–40. doi: 10.1093/ndt/gft488
101. Wu WK, Famure O, Li Y, Kim SJ. Delayed Graft Function and the Risk of Acute Rejection in the Modern Era of Kidney Transplantation. Kidney Int (2015) 88(4):851–8. doi: 10.1038/ki.2015.190
102. Henderson JM. Liver Transplantation and Rejection: An Overview. Hepatogastroenterology (1999) 46 Suppl 2:1482–4.
103. Nakano R, Tran LM, Geller DA, Macedo C, Metes DM, Thomson AW. Dendritic Cell-Mediated Regulation of Liver Ischemia-Reperfusion Injury and Liver Transplant Rejection. Front Immunol (2021) 12:705465. doi: 10.3389/fimmu.2021.705465
104. Cursio R, Gugenheim J. Ischemia-Reperfusion Injury and Ischemic-Type Biliary Lesions Following Liver Transplantation. J Transplant (2012) 2012:164329. doi: 10.1155/2012/164329
105. Troppmann C. Complications After Pancreas Transplantation. Curr Opin Organ Transplant (2010) 15(1):112–8. doi: 10.1097/MOT.0b013e3283355349
106. Chen-Yoshikawa TF. Ischemia-Reperfusion Injury in Lung Transplantation. Cells (2021) 10(6):1333. doi: 10.3390/cells10061333
107. Subramaniam K. Early Graft Failure After Heart Transplantation: Prevention and Treatment. Int Anesthesiol Clin (2012) 50(3):202–27. doi: 10.1097/AIA.0b013e3182603ead
108. Al-Lamki RS, Wang J, Skepper JN, Thiru S, Pober JS, Bradley JR. Expression of Tumor Necrosis Factor Receptors in Normal Kidney and Rejecting Renal Transplants. Lab Invest (2001) 81(11):1503–15. doi: 10.1038/labinvest.3780364
109. Hoffmann U, Bergler T, Rihm M, Pace C, Krüger B, Rümmele P, et al. Upregulation of Tnf Receptor Type 2 in Human and Experimental Renal Allograft Rejection. Am J Transplant (2009) 9(4):675–86. doi: 10.1111/j.1600-6143.2008.02536.x
110. Budak D, Yilmaz VT, Akbas H, Suleymanlar G, Yucel G. Association Between Graft Function and Serum Tnf-α, Tnfr1 and Tnfr2 Levels in Patients With Kidney Transplantation. Renal Failure (2015) 37(5):871–6. doi: 10.3109/0886022X.2015.1015425
111. Diller R, Winde G, Kötting S, Senninger N, Dietl KH, Spiegel HU. Stnf-Rii: Is It Useful for the Early Diagnosis of Rejection and for Prognosis After Renal Transplantation? Transpl Int (2002) 15(7):335–40. doi: 10.1007/s00147-002-0421-1
112. Al-Lamki RS, Wang J, Vandenabeele P, Bradley JA, Thiru S, Luo D, et al. Tnfr1- and Tnfr2-Mediated Signaling Pathways in Human Kidney Are Cell Type-Specific and Differentially Contribute to Renal Injury. FASEB J (2005) 19(12):1637–45. doi: 10.1096/fj.05-3841com
113. Wang J, Al-Lamki RS. Tumor Necrosis Factor Receptor 2: Its Contribution to Acute Cellular Rejection and Clear Cell Renal Carcinoma. BioMed Res Int (2013) 2013:821310. doi: 10.1155/2013/821310
114. Wang J, Li Z, Al-Lamki R, Wang J, Zuo N, Bradley JR, et al. The Role of Tumor Necrosis Factor-α Converting Enzyme in Renal Transplant Rejection. Am J Nephrol (2010) 32(4):362–8. doi: 10.1159/000320467
115. Bradley JR, Wang J, Bardsley V, Broecker V, Thiru S, Pober JS, et al. Signaling Through Tumor Necrosis Receptor 2 Induces Stem Cell Marker in Cd133(+) Regenerating Tubular Epithelial Cells in Acute Cell-Mediated Rejection of Human Renal Allografts. Am J Transplant (2020) 20(9):2380–91. doi: 10.1111/ajt.15846
116. Nguyen M-TJP, Fryml E, Sahakian SK, Liu S, Cantarovich M, Lipman M, et al. Pretransplant Recipient Circulating Cd4+Cd127lo/– Tumor Necrosis Factor Receptor 2+ Regulatory T Cells: A Surrogate of Regulatory T Cell–Suppressive Function and Predictor of Delayed and Slow Graft Function After Kidney Transplantation. Transplantation (2016) 100(2):314–24. doi: 10.1097/tp.0000000000000942
117. Luan Y, Mosheir E, Menon MC, Wilson D, Woytovich C, Ochando J, et al. Monocytic Myeloid-Derived Suppressor Cells Accumulate in Renal Transplant Patients and Mediate Cd4(+) Foxp3(+) Treg Expansion. Am J Transplant (2013) 13(12):3123–31. doi: 10.1111/ajt.12461
118. Al-Lamki RS, Brookes AP, Wang J, Reid MJ, Parameshwar J, Goddard MJ, et al. Tnf Receptors Differentially Signal and Are Differentially Expressed and Regulated in the Human Heart. Am J Transplant (2009) 9(12):2679–96. doi: 10.1111/j.1600-6143.2009.02831.x
119. Suzuki J, Cole SE, Batirel S, Kosuge H, Shimizu K, Isobe M, et al. Tumor Necrosis Factor Receptor -1 and -2 Double Deficiency Reduces Graft Arterial Disease in Murine Cardiac Allografts. Am J Transplant (2003) 3(8):968–76. doi: 10.1034/j.1600-6143.2003.00164.x
120. Sihvola RK, Koskinen PK, Pulkkinen VP, Tikkanen JM, Lemström KB. Inhibition of Tumor Necrosis Factor-Alpha Attenuates Myocardial Remodeling in Rat Cardiac Allografts. J Heart Lung Transplant (2006) 25(5):569–78. doi: 10.1016/j.healun.2006.01.002
121. Zou H, Li R, Hu H, Hu Y, Chen X. Modulation of Regulatory T Cell Activity by Tnf Receptor Type Ii-Targeting Pharmacological Agents. Front Immunol (2018) 9:594. doi: 10.3389/fimmu.2018.00594
122. Wajant H, Beilhack A. Targeting Regulatory T Cells by Addressing Tumor Necrosis Factor and Its Receptors in Allogeneic Hematopoietic Cell Transplantation and Cancer. Front Immunol (2019) 10:2040. doi: 10.3389/fimmu.2019.02040
123. Mancusi A, Piccinelli S, Velardi A, Pierini A. The Effect of Tnf-α on Regulatory T Cell Function in Graft-Versus-Host Disease. Front Immunol (2018) 9:356. doi: 10.3389/fimmu.2018.00356
124. Leclerc M, Naserian S, Pilon C, Thiolat A, Martin GH, Pouchy C, et al. Control of Gvhd by Regulatory T Cells Depends on Tnf Produced by T Cells and Tnfr2 Expressed by Regulatory T Cells. Blood (2016) 128(12):1651–9. doi: 10.1182/blood-2016-02-700849
125. Rodriguez-Barbosa JI, Schneider P, Graca L, Bühler L, Perez-Simon JA, Del Rio ML. The Role of Tnfr2 and Dr3 in the in Vivo Expansion of Tregs in T Cell Depleting Transplantation Regimens. Int J Mol Sci (2020) 21(9):3347. doi: 10.3390/ijms21093347
126. Pierini A, Strober W, Moffett C, Baker J, Nishikii H, Alvarez M, et al. Tnf-α Priming Enhances Cd4+Foxp3+ Regulatory T-Cell Suppressive Function in Murine Gvhd Prevention and Treatment. Blood (2016) 128(6):866–71. doi: 10.1182/blood-2016-04-711275
Keywords: TNFR2, immune regulation, transplantation, rejection, ischemia reperfusion injury (IRI)
Citation: Kouyoumdjian A, Tchervenkov J and Paraskevas S (2022) TFNR2 in Ischemia-Reperfusion Injury, Rejection, and Tolerance in Transplantation. Front. Immunol. 13:903913. doi: 10.3389/fimmu.2022.903913
Received: 24 March 2022; Accepted: 10 June 2022;
Published: 07 July 2022.
Edited by:
Magdalena Plebanski, RMIT University, AustraliaReviewed by:
Leonardo M.R. Ferreira, Medical University of South Carolina, United StatesMin Hu, The University of Sydney, Australia
Copyright © 2022 Kouyoumdjian, Tchervenkov and Paraskevas. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Araz Kouyoumdjian, YXJhei5rb3V5b3VtZGppYW5AbWFpbC5tY2dpbGwuY2E=