 Luke B. Roberts
Luke B. Roberts Graham M. Lord
Graham M. Lord Jane K. Howard
Jane K. Howard- 1School of Immunology and Microbial Sciences, King’s College London, London, United Kingdom
- 2Faculty of Biology, Medicine and Health, University of Manchester, Manchester, United Kingdom
- 3School of Cardiovascular and Metabolic Medicine & Sciences, King’s College London, London, United Kingdom
Cardiovascular diseases (CVDs) are responsible for most pre-mature deaths worldwide, contributing significantly to the global burden of disease and its associated costs to individuals and healthcare systems. Obesity and associated metabolic inflammation underlie development of several major health conditions which act as direct risk factors for development of CVDs. Immune system responses contribute greatly to CVD development and progression, as well as disease resolution. Innate lymphoid cells (ILCs) are a family of helper-like and cytotoxic lymphocytes, typically enriched at barrier sites such as the skin, lung, and gastrointestinal tract. However, recent studies indicate that most solid organs and tissues are home to resident populations of ILCs - including those of the cardiovascular system. Despite their relative rarity, ILCs contribute to many important biological effects during health, whilst promoting inflammatory responses during tissue damage and disease. This mini review will discuss the evidence for pathological and protective roles of ILCs in CVD, and its associated risk factor, obesity.
Introduction
Cardiovascular diseases (CVDs) are the major global cause of premature human death. Obesity and associated metabolic inflammation underlie the development of several major health issues including type 2 diabetes (T2DM), insulin resistance, hypertension, dyslipidaemia, and increased expression of inflammatory mediators - all of which are risk factors for CVDs. Tackling obesity therefore remains a key goal the effort to reduce the increasing burden of CVDs worldwide.
Five major subsets of Innate lymphoid cells (ILCs) are acknowledged: cytotoxic natural killer cells (NKs), lymphoid tissue inducers (LTi) and helper-like ILC1s, ILC2s and ILC3s (1). ILC1s depend on the transcription factor (TF) T-bet for their development (2) and peripheral maintenance (3). They produce IFNγ, tumor necrosis factor (TNF) (4, 5), and TGF-β under some contexts (6). ILC2s are regulated by a suite of TFs including GATA3 and RORα, express type 2 cytokines including interleukin (IL)-4, IL-5, IL-9 and IL-13, and tissue repair factors including amphiregulin. LTi and helper-like ILC3s express TF RORγt and produce IL-17, IL-22, and TNF-superfamily members, including lymphotoxins. ILCs are activated by environmental signals including cytokines, tissue-derived danger signals, metabolites, neurotransmitters, and neuropeptides (1, 7).
While NKs are predominantly circulatory, helper-like ILCs reside in tissues and are enriched at barrier mucosal sites such as the lung and intestinal tract. However, since their discovery, almost all organs have been found to play host to ILC subsets, including tissues of the cardiovascular system (8). Cardiovascular-associated ILCs (cILCs) reside in the pericardium and pericardial fluid (9), in the adventitia of arteries including the aorta (10), and in fat-associated lymphoid clusters (FALC) (11) of perivascular (12) and pericardial adipose tissue (13). The majority of cILCs are ILC2s or ILC2-commited precursors, with minimal presence of ILC1s and ILC3s (14, 15). As observed in other tissues, cILC2 populations are dependent on IL-33 signalling for their development as well as their activation and function. cILC2s may even be more responsive to IL-33 signalling than counterparts at barrier sites such as the lungs, due to unique phenotypic attributes including greater expression of GATA3 (15). Cardiac fibroblasts are a main source of IL-33 in the heart, responsible for ILC2 homeostasis and activity (9), while adventitial stromal cells provide IL-33 and thymic stromal lymphopoietin (TSLP) to ILC2s in arteries and FALC, providing a tissue supportive niche for their development and activation (16). Developmental, adoptive transfer, and parabiosis studies in mice suggest that, similarly to ILCs from barrier sites (17–19), cILC populations are tissue-resident cells, seeded during early embryonic development and shortly after birth, sustaining themselves through local renewal, even during cardiac inflammation (14, 15).
This mini review will summarise the known protective and pathological actions of ILCs in the context of CVDs. As a major cardiometabolic risk factor, obesity, and its relatedness to ILC activity will also be discussed. Figure 1 summarises the information presented. Table 1 provides a list of the abbreviations used throughout.
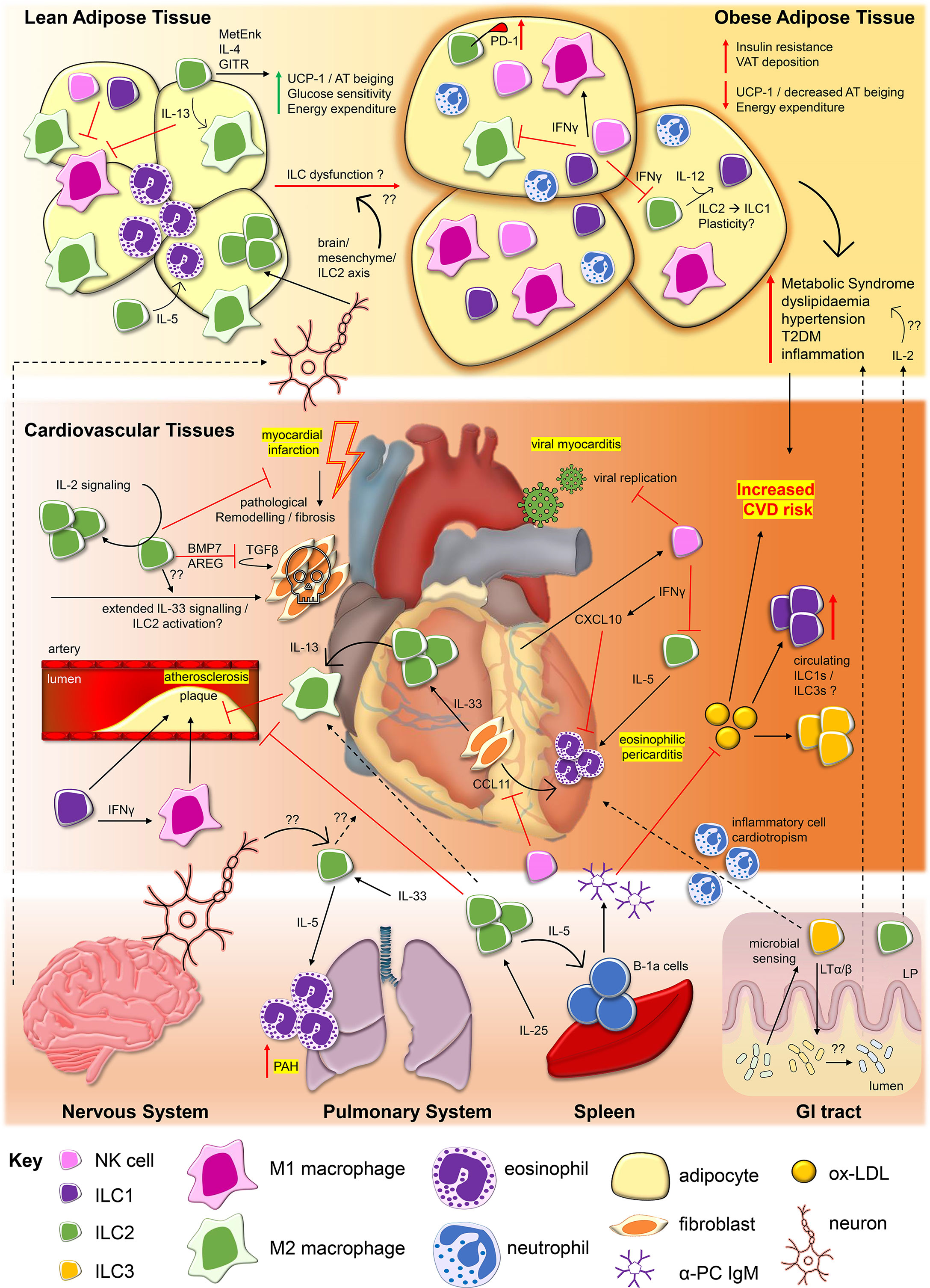
Figure 1 Innate lymphoid cells contribute to protective and pathological processes in cardiovascular diseases and obesity. During viral myocarditis caused by agents such as Coxsackie virus B3 (CV-B3), NKs restrict viral replication (20) and are recruited to cardiac tissues by cardiomyocyte production of C-X-C motif chemokine ligand 10 (CXCL10), itself promoted by NK-derived interferon gamma (IFNγ) (21). IFNγ may also restrict ILC2 activity, influencing ILC2 capacity to drive inflammation associated with eosinophilic pericarditis. Interleukin (IL)-33 derived from cardiac fibroblasts drives cardiac ILC2 proliferation and along with fibroblast production of eotaxins (i.e. eotaxin-1/CCL11), ILC2 production of IL-5 may facilitate recruitment of eosinophils to the pericardium (9). ILC2 production of IL-13 can also promote polarisation of M2 macrophages, which may serve protective roles in atherosclerosis (12, 22). Conversely, ILC1s and promotion of classically activated proinflammatory M1 macrophages may promote atherosclerotic plaque formation (23–25). ILC2s, regulated by IL-2 signalling, may also serve protective roles for cardiac tissue repair following major adverse events such as myocardial infarction, via the production of amphiregulin (AREG) and bone morphogenic protein 7 (BMP7), inhibiting pathological tissue remodelling and fibrosis (26). The impact of prolonged activation of ILC2s by factors such as IL-33 on the outcome of ILC2 repair responses in this context require further study (27). In the circulation, increased frequencies of ILC1s and ILC3s are associated with major cardiovascular and cerebrovascular events including ST-elevated myocardial infarction (STEMI) and acute cerebrovascular infarction (ACI/Stroke) (28, 29). This may be related to increased circulating levels of oxidised low-density lipoproteins (ox-LDL) in patients, but the impact of these alterations to ILC subset frequencies inpatients remain to be elucidated. Obesity, and its associated metabolic inflammation is associated with major risk factors for the development of CVDs, including type 2 diabetes mellitus (T2DM), dyslipidaemia, hypertension, and inflammation. As observed in cardiovascular tissues, ILC2s are the dominant ILC subtype found within lean adipose tissue. ILC2s promote alternatively activated M2 macrophage phenotypes and eosinophils within adipose tissue via their production of IL-5 and IL-13, contributing to lean adipose tissue homeostasis (30–33). ILC2 expression of methionine enkephalin (MetEnk) (31) and IL-4 (34), in addition to signaling via Glucocorticoid-induced TNFR-related protein (GITR) (32) can protect from obesity by promoting beiging of white adipose tissue, linked to upregulation of mitochondrial uncoupling protein 1 (UCP-1), increasing glucose sensitivity and energy expenditure. In obese adipose tissue ILC2 numbers and functions are impaired, contributing to increased visceral adipose tissue (VAT) depots, insulin resistance and decreased beiging. Upregulation of inhibitory receptor PD-1 by ILC2s (35), negative regulation by IFNγ derived from ILC1s and natural killer cells (NKs) (36, 37), and ILC2 to ILC1 conversion (38) may be among the mechanisms which result in ILC2 dysfunction in obesity. NKs and ILC1s numbers are also altered in obesity, potentially impacting upon the ratio between inflammatory M1 and anti-inflammatory M2 macrophages (33, 39). Influences from physiological signals and ILCs present in other tissues may also impact upon CVDs and obesity pathogenesis. The nervous system can regulate ILC2 activity within lean adipose tissue via a brain/mesenchymal/ILC2 axis (40). Dysregulation of this axis may contribute to development of obesity, however, broader effects of neuronal signalling on ILCs in the context of CVDs requires further study. In the induction of pulmonary arterial hypertension (PAH), IL-5 derived from pulmonary system ILC2s are responsible for tissue eosinophilia which may drive arterial damage (41). IL-25 drives ILC2 proliferation in the spleen, promoting atheroprotective effects, IL-5-dependent B-1a expansion and production of anti- phosphorylcholine (PC) Immunoglobulin M (IgM) which targets ox-LDL (42, 43). In the gastrointestinal (GI) tract, sensing of microbial composition by ILC3s may influence inflammatory cell cardiotropism in the context of myocarditis (44), while ILC2s and ILC3s within the intestinal lamina propria (LP) may also contribute to processes driving obesity, through factors such as production of IL-2 (45).

Table 1 Meanings of abbreviations used in this article.
Protective and Reparative Roles of ILCs in CVD
NK Cells
Viral myocarditis is predominantly caused by infection with Coxsackievirus B3. NKs limit viral replication primarily through their potent cytotoxicity, among other protective mechanisms (20). NKs are recruited to the infected heart by cardiomyocyte upregulation of CXCL10, a chemokine ligand of the NK-expressed receptor CXCR3 (21). Expression of CXCL10 is promoted by NK-derived IFNγ - a feed-forward mechanism, driven by NKs for further expansion of the population with in the tissue (21).
Observational studies linking decreased NK numbers and activities, with low grade inflammation and increased disease severity, implicate a protective role for NK cells in atherosclerosis (AS) and coronary artery disease (CAD) (46–48). These effects may be related to NK apoptosis (49) and increased expression of inhibitory molecules (50). However, results from murine models complicate interpretation of these findings, suggesting either pro-atherogenic roles for NKs (51), or conversely, no impact on AS development (52).
NKs may also serve cardioprotective functions during eosinophilic myocarditis, restricting eosinophil influx and survival. Anti-asialo GM1-mediated NK depletion in an anti-MHC (myosin heavy chain) immunisation model of experimental autoimmune myocarditis (EAM) resulted in worsened disease outcome and enhanced eosinophilic influx, accompanied by greater cardiac tissue fibrosis (53). NKs may defend against eosinophilic inflammation directly, by promoting eosinophil apoptosis (36, 53, 54), and indirectly by suppressing fibroblast production of the eosinophil recruiting chemokine eotaxin-1 (CCL11). Furthermore, CXCL10 is an inhibitor of eosinophil recruitment via antagonism of the eosinophil trafficking receptor CCR3 (53, 55). IFNγ also restricts ILC2 cytokine expression and limits the active niche in which ILC2s can exert their effects (56, 57). As will be discussed, ILC2s may promote pathological recruitment of eosinophils in the setting of cardiac inflammation (9). IFNγ production by NKs may therefore also act to inhibit cardiac ILC2 activity.
ILC2s
Apolipoprotein E deficient mice (Apoe-/-) display defective lipoprotein clearance and accrue abnormal levels of low-density lipids, making them prone to development of AS. IL-25 administration limits initiation and progression of AS in high fat diet (HFD)-fed Apoe-/- mice. This is concomitant with expansion of splenic ILC2 populations and enhanced IL-5 production (42, 43). Furthermore, IL-25 treatment increases levels of circulating anti-phosphorylcholine (PC) IgM, dependent on intact IL-5 expression, indicative of an atheroprotective effect of ILC2 activation by IL-25. The PC epitope is a component of oxidised low-density lipoprotein (ox-LDL), strongly associated with AS development. Anti-PC IgM is produced by B-1a cells – an atheroprotective, innate-like B cell subtype. B-1a cells expand following IL-25 treatment (43), and depend on IL-5 for their survival and maturation to produce natural IgM antibodies (58, 59). Transfer of IL-25-expanded, wild type ILC2s to Apoe-/- mice reduces the lipid content of AS lesions and augments B-1a cells and anti-PC IgM, suggestive of a therapeutic avenue to tackle AS (42). Of note, the ILC2/IL-5/B-1a axis is critically dependent on ILC2 expression of the helix loop-helix TF, ID3 (60). As an ID3 single nucleotide polymorphism (rs11574) is associated with increased carotid intimal media thickness, this may link ILC2 dysfunction in humans with enhanced AS risk (61).
HFD feeding also reduces peripheral ILC2 numbers and alters their functional cytokine responses in AS-prone mouse strains - linking defective ILC2 activity with disease (12). Low-density lipoprotein receptor deficient mice (Ldlr-/-) share a similar functional defect in lipid clearance as Apoe-/- mice and are similarly AS-prone. Ldlr-/- mice reconstituted with bone marrow from ILC2 deficient mice (Staggerer/RoraFlox x Il7raCre) displayed accelerated AS plaque lesion development. This was associated with reduced collagen deposition found in larger plaques (indicative of increased plaque destabilisation) and altered plaque immune cell composition, dependent on loss of ILC2-derived IL-13. Plaque macrophage phenotypes were disrupted by ILC2 loss, with reduced arginase-1 expression suggestive of a restricted capacity for tissue repair. This supports work identifying IL-13 as an atheroprotective cytokine, acting via induction of alternatively activated (M2) macrophages (22), potentially supporting the augmentation of ILC2 responses as a novel therapeutic strategy for AS. Indeed, expansion of CD25+ ILC2s by IL-2/anti-IL-2 complexes reduces circulating ox-LDL and AS lesion development in HFD-fed lymphopenic Rag-/-Ldlr-/- mice (10), indicating that IL-2 therapy may be a novel treatment option in some forms of CVD. Supportive of this, genetic ablation of ILC2s in a murine model of myocardial infarction (MI) delayed recovery of cardiac function. MI-protective functions of ILC2s were associated with an upstream regulatory IL-2 signalling axis (13). Furthermore, analysis of peripheral blood from patients recruited to a placebo-controlled, double-blind trial designed to assess the safety of Low-dose InterLeukin-2 treatment in patients with stable ischaemic heart disease and Acute Coronary Syndromes [LILACS trial (62)], revealed a short-term increase in circulating ILC2s, serum IL-5, and eosinophil counts (13). This indicates that IL-2 therapy activates human ILC2s in vivo. However, putative protective roles of ILC2s in this context remain to be fully investigated. Post-cardiac injury, augmentation of ILC2 repair responses may also be an attractive therapeutic option, inhibiting TGF-β1-driven pathological fibrosis and remodelling by cardiac fibroblasts which can lead to terminal heart failure (26).
ILC3s
ILC3 associated cytokines IL-17A and IL-22 have been linked to CVDs, but may be protective under certain contexts (63–65). ILC3s might also be protective during hepatic (66) and intestinal (67) ischemia-reperfusion-injury (IRI), suggestive, potentially, of similar roles during reperfusion following MI. However, in both the liver and the intestinal tract, ILC3s are far more populous than in cardiovascular tissues, where Th17 cells are also more likely to be the major source of ILC3-associated effector cytokines. Whether ILC3s serve protective roles in CVDs requires further investigation.
ILCs and CVD Pathogenesis
ILC1s
ILC1s constitute only a minor population of the ILCs identified within murine cardiovascular tissues (14, 15). Despite this, Ldlr-/- and Apoe-/- models combining genetic knockout of T-bet or IL-12, implicate roles for ILC1s in development of HFD-induced AS (23, 24). However, these models also deplete other cells important in AS pathogenesis, including Th1 and NKs (68). ILC1s have been more specifically linked to AS plaque development using adaptive lymphocyte deficient Rag-/-Apoe-/- models. Anti-NK1.1 or anti-IL-15R antibodies were used to deplete ILC1s and/or NK cells respectively, followed by adoptive transfer of splenic ILC1s, identifying a possible role for ILC1s in aggravation of AS (25).
Involvement of ILC1s in the pathology of major cardiovascular and cerebrovascular adverse events has been proposed, based on correlations between disease severity and circulating frequency of these cells. Total number and proportions of ILCs among circulating leukocytes were increased in acute ST-segment elevation myocardial infarction (STEMI) patients, compared to healthy controls (28). This was accounted for by a specific increase in ILC1s, while numbers of ILC2s and ILC3s remained unaltered, indicating an expansion of ILC1s rather than ILC subset plasticity towards the ILC1 phenotype (69, 70). Similar observations were made in acute cerebral infarction (ACI) patients (29). A significantly increased proportion of ILC1s among total circulating ILCs, profiled at the time of admission, was observed compared to healthy controls. Concomitant with this was a decrease in the proportion of ILC2s and no impact on circulating ILC3s. However, as total number of ILCs among leukocytes was not reported, whether alterations to subset prevalence among total ILCs represents phenotypic plasticity between ILC2s to ILC1s (38, 71–73) in the context of ACI, or reflects a specific expansion of ILC1s, remains unclear.
ILC2s
ILC2s may drive the recruitment of eosinophils to the pericardium via production of IL-5 and promotion of cardiac fibroblast-derived eotaxins (9). Choi et al., utilised repeated IL-33 administration to induce eosinophilic pericarditis in mice, finding that pericardial IL-33R+ (ST2+) ILC2s greatly expanded in response to treatment (9). IL-5 deficiency prevented pericardial eosinophil infiltration, however Il13-/- mice displayed a similar level of pericarditis to WT mice. Furthermore, total ILCs were enriched in the pericardial fluid of pericarditis patients, relative to patients with other heart diseases or healthy controls. However, deeper analysis of ILC subsets was not reported, impeding specific association of human pericarditis with ILC2s. ILC2-derived IL-5 also promotes eosinophil accumulation around pulmonary arteries, following chronic activation by IL-33 (41). This accumulation resulted in severe arterial occlusion and pulmonary arterial hypertrophy (PAH). This was ablated in IL-5 deficient and eosinophil deficient mice, but not in Rag-/- mice, supporting an ILC2/IL-5/eosinophil dependent axis in the aetiology of PAH. Furthermore, PAH has previously been associated with vascular remodelling driven by IL-33 (74). Despite eosinophilia and other abnormalities in IL-5 overexpressing mice, pulmonary arteries in these animals do not appear to be affected (75). Therefore, while the ILC2/eosinophil axis plays a role, other factors also contribute to PAH development. Prolonged IL-33/ST2 signalling may also contribute to poorer cardiac remodelling and promote heart failure following MI (27), despite augmenting ILC2-driven type 2 immune activity, which may be protective or reparative of ventricular function in the acute phase post-MI (57). ILC2s therefore act as a ‘‘double-edged sword’’ (37), the activity of which must be precisely balanced to provide protection, while limiting pathological outcomes.
ILC3s
Few studies have investigated ILC3s in CVD pathogenesis. During EAM, differential disease susceptibility in mice from different sources has been associated with intestinal ILC3s and specific microbiome profiles (44). Anti-CD90 antibody mediated depletion of ILCs restricted inflammatory leukocyte trafficking to the heart during EAM, indicating that microbial sensing via ILC3s plays a role in development of inflammatory heart diseases via cardiotropic immune cell chemotaxis. Additionally, a study investigating ILC3s in axial spondyloarthritis (axSpA) indirectly suggests ILC3 differentiation is promoted by risk factors driving CVD (76). Patients with inflammatory joint conditions, including axSpA, have an increased risk of developing CVD (77). Therefore, clinical management of CVD risk factors, including dyslipidaemia, is important. Circulating IL-22+ ILC3s were increased in axSPa patients with dyslipidaemia compared to patients without dyslipidaemia or healthy controls. Furthermore, ox-LDL promoted IL-22+ and IL-17+ ILC3 differentiation from axSPa patient peripheral blood mononuclear cells (PBMCs) (76) and ILC3s expressed the ox-LDL receptor CD36, blockade of which prevented ox-LDL-mediated differentiation of the cells (76). This indicates that ox-LDL affects ILC3 generation, similar to observations made for ILC1 differentiation from ACI patient PBMCs (29). Although in that study, no effect of ox-LDL stimulation on ILC3s was observed, suggestive of other disease-specific effects of ox-LDL signalling on ILCs. ILC3s are regulated by hypoxia, acting via hypoxia inducible factor (HIF)-1α (78). Tissue hypoxia can be induced by, and contribute to CVDs (79). Hypoxia-driven ILC3 responses might therefore also play a role in some forms of CVD.
ILCs, Adipose Tissue and Obesity
Lean adipose tissue (AT) contains populations of immune cells important for maintaining metabolic homeostasis, including regulatory T cells (TREGs), eosinophils and M2 macrophages. These cells are regulated by ILC2s resident in AT-depots in mice and humans (30–33) and these functions are reliant on IL-33 signalling (30, 33). Disruption of IL-33 signalling in mice reduces ILC2s in white AT (WAT), increasing visceral fat depots, impairing glucose homeostasis, and disrupting energy metabolism by prevention of WAT beiging (33). Beiging converts white adipocytes, or adipogenic precursors within WAT, to brown-like AT and is associated with the upregulation of mitochondrial uncoupling protein 1 (UCP1). This increases thermogenesis - raising body heat and calorific expenditure, protecting from obesity, at least in mice. Mechanistically, IL-33-mediated beiging is dependent on the ILC2-expressed opioid peptide methionine enkephalin, which acts on WAT to induce UCP1 expression (31), similarly to the actions of other ILC2-associated molecules like IL-4 (34) and activation of Glucocorticoid-induced TNFR-related protein (GITR), expressed by both human and murine AT ILC2s (32).
During obesity in humans and in murine diet-induced obesity (DIO) models, AT ILC2 frequencies are reduced, and their functions become dysregulated. This may negatively impact the homeostatic, immunometabolic roles of ILC2s, such as restricting differentiation of obesity-protective M2 AT macrophages (ATM) (30). The mechanisms responsible for reduction of ILC2 responses during obesity are likely multifaceted and remain under study. Recent developments suggest they include the upregulation and activation of inhibitory co-stimulatory receptor PD-1 on ILC2s during obesity (35), and dysregulation of brain-AT circuits, which modulate ILC2 activation indirectly via neuro-mesenchymal interactions (40). Furthermore, as human and murine AT is also populated by group 1 ILCs (AT1-ILCs), including mature and immature NKs, and non-NK ILC1s, AT ILC2 activities may be affected by aberrant IFNγ signaling, which restrains ILC2 responses (30, 56). An increase in AT ILC1s during DIO has been reported by some studies. This might drive ATM M1 polarization via IFNγ production, promoting insulin resistance and metabolic dysfunction in a manner dependent on IL-12 and downstream STAT4 signalling (80). IL-12-mediated plasticity of AT ILC2s to ILC1-like cells may further contribute to these effects (38). ATM M1 responses may also be promoted by increased WAT expression of CD36 in the context of defective ILC2 functions, resulting in enhanced absorption of saturated fatty acids by adipocytes, driving M1 polarisation (33). Conversely, Boulenouar and colleagues report that obesity is associated with a decrease in the number and cytotoxicity of AT1-ILCs. This may contribute to ATM accumulation and alterations to the ratio of proinflammatory M1 to anti-inflammatory M2, thereby impacting insulin resistance and metabolic dysfunction (39). The contribution of AT1-ILC responses and their roles in AT homeostasis and during obesity is an area that requires further investigation.
While AT ILC2s have emerged as important metabolic regulators which promote AT homeostasis, ILC2s from other tissues may also act to promote obesity. In the setting of lymphopenic DIO murine models, adoptive transfer of small intestinal ILC2s, but not WAT ILC2s, promotes obesity via an axis dependent on their production of IL-2 (45) – supportive of a role for this cytokine in obesity induction (81). Intestinal ILC3s also moderately contributed to DIO (45). Currently, few studies have investigated the roles of ILC3s in obesity. However, increased frequency of circulating ILC3s in children who are overweight and asthmatic (compared to children who are asthmatic but not-overweight) suggests that obesity may be an independent risk-factor for promotion of ILC3 differentiation (82). Furthermore, the lymphotoxin pathway may promote DIO by driving a pro-obesity intestinal microbiota (83). As LTi and helper-ILC3s produce lymphotoxins, this potentially implicates these cells in obesity pathogenesis.
Conclusions and Future Perspectives
CVD and obesity are major global health concerns. ILCs are emerging as cells capable of both driving or protecting from CVD pathology. As the field expands, it will shed light on current gaps in our knowledge – for instance, whether ILC1s or ILC3s serve any CVD protective functions. Studies which investigate whether and how cILCs are regulated by factors known to control ILC activity in other tissues and disease states are also required. This may include investigation of cILC regulation by neurotransmitters and neuropeptides (1, 84), post-transcriptional regulation by non-coding RNAs (85–87), and effects of metabolic dysfunction, such as lactic acidosis (88) - a known indicator of cardiac stress. Further studies which compare functions and transcriptional profiles of cILCs with more widely studied barrier ILC populations would also be useful. This would facilitate understanding of similarities and unique differences between these populations, related to the pressures and requirements exerted by the disparate tissue environments these cells function within. Such work may inform new, tissue-specific therapeutic strategies. Encouragingly, there are now several studies which demonstrate how modification of ILC responses might show promise for therapeutic intervention of CVD and obesity in humans. However, most of our mechanistic knowledge about ILC activity is still derived from murine models. As these models are often lymphopenic in nature, the potential translational meaning of these observations to immunocompetent human disease settings is somewhat confounded. Future research should continue to explore and unravel cardiovascular-associated ILC responses, particularly focusing on their roles in the context of human disease.
Author Contributions
LR conceptualised the manuscript’s theme. LR researched and wrote the manuscript. LR designed and created the figures. GL and JH advised on content and edited the manuscript. All authors contributed to the article and approved the submitted version.
Funding
LR is supported by the National Institute for Health Research (NIHR) Biomedical Research Centre based at Guy’s and St Thomas’ NHS Foundation Trust and King’s College London. JH is supported by the Medical Research Council (grant MR/K002996/1). GL is supported by the Wellcome Trust (grant number 091009), the British Heart Foundation (award number PG/12/36/29444), the MRC (grant number MR/M003493/1), and the NIHR at Guy’s & St Thomas’ NHS Foundation Trust in partnership with King’s College London and King’s College Hospital NHS Foundation Trust.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
The authors thank Joana F. Neves (King’s College London) for critically reading the manuscript and providing useful feedback, and Andy Forest for the creation of heart and organ graphics used in Figure 1.
References
1. Vivier E, Artis D, Colonna M, Diefenbach A, Di Santo JP, Eberl G, et al. Innate Lymphoid Cells: 10 Years on. Cell (2018) 174:1054–66. doi: 10.1016/j.cell.2018.07.017
2. Klose CSN, Flach M, Möhle L, Rogell L, Hoyler T, Ebert K, et al. Differentiation of Type 1 ILCs From a Common Progenitor to All Helper-Like Innate Lymphoid Cell Lineages. Cell (2014) 157:340–56. doi: 10.1016/j.cell.2014.03.030
3. Schroeder JH, Roberts LB, Meissl K, Lo JW, Hromadová D, Hayes K, et al. Sustained Post-Developmental T-Bet Expression Is Critical for the Maintenance of Type One Innate Lymphoid Cells In Vivo. Front Immunol (2021) 12:760198. doi: 10.3389/fimmu.2021.760198
4. Daussy C, Faure F, Mayol K, Viel S, Gasteiger G, Charrier E, et al. T-Bet and Eomes Instruct the Development of Two Distinct Natural Killer Cell Lineages in the Liver and in the Bone Marrow. J Exp Med (2014) 211:563–77. doi: 10.1084/jem.20131560
5. Fuchs A, Vermi W, Lee JS, Lonardi S, Gilfillan S, Newberry RD, et al. Intraepithelial Type 1 Innate Lymphoid Cells Are a Unique Subset of IL-12- and IL-15-Responsive IFN-γ-Producing Cells. Immunity (2013) 38:769–81. doi: 10.1016/j.immuni.2013.02.010
6. Jowett GM, Norman MDA, Yu TTL, Rosell Arévalo P, Hoogland D, Lust ST, et al. ILC1 Drive Intestinal Epithelial and Matrix Remodelling. Nat Mater (2020) 20:250–9. doi: 10.1038/s41563-020-0783-8
7. McGinty JW, von Moltke J. A Three Course Menu for ILC and Bystander T Cell Activation. Curr Opin Immunol (2020) 62:15–21. doi: 10.1016/j.coi.2019.11.005
8. Murphy JM, Ngai L, Mortha A, Crome SQ. Tissue-Dependent Adaptations and Functions of Innate Lymphoid Cells. Front Immunol (2022) 13. doi: 10.3389/fimmu.2022.836999
9. Choi HS, Won T, Hou X, Chen G, Bracamonte-Baran W, Talor MV, et al. Innate Lymphoid Cells Play a Pathogenic Role in Pericarditis. Cell Rep (2020) 30:2989–3003.e6. doi: 10.1016/j.celrep.2020.02.040
10. Engelbertsen D, Foks AC, Alberts-Grill N, Kuperwaser F, Chen T, Lederer JA, et al. Expansion of CD25+ Innate Lymphoid Cells Reduces Atherosclerosis. Arterioscler Thromb Vasc Biol (2015) 35:2526–35. doi: 10.1161/ATVBAHA.115.306048
11. Moro K, Yamada T, Tanabe M, Takeuchi T, Ikawa T, Kawamoto H, et al. Innate Production of T(H)2 Cytokines by Adipose Tissue-Associated C-Kit(+)Sca-1(+) Lymphoid Cells. Nature (2010) 463:540–4. doi: 10.1038/nature08636
12. Newland SA, Mohanta S, Clément M, Taleb S, Walker JA, Nus M, et al. Type-2 Innate Lymphoid Cells Control the Development of Atherosclerosis in Mice. Nat Commun (2017) 8:1–11. doi: 10.1038/ncomms15781
13. Yu X, Newland SA, Zhao TX, Lu Y, Sage AS, Sun Y, et al. Innate Lymphoid Cells Promote Recovery of Ventricular Function After Myocardial Infarction. J Am Coll Cardiol (2021) 78:1127–42. doi: 10.1016/j.jacc.2021.07.018
14. Bracamonte-Baran W, Chen G, Hou X, Talor MV, Choi HS, Davogustto G, et al. Non-Cytotoxic Cardiac Innate Lymphoid Cells Are a Resident and Quiescent Type 2-Commited Population. Front Immunol (2019) 10:634. doi: 10.3389/fimmu.2019.00634
15. Deng Y, Wu S, Yang Y, Meng M, Chen X, Chen S, et al. Unique Phenotypes of Heart Resident Type 2 Innate Lymphoid Cells. Front Immunol (2020) 11:802. doi: 10.3389/fimmu.2020.00802
16. Dahlgren MW, Jones SW, Cautivo KM, Dubinin A, Ortiz-Carpena JF, Farhat S, et al. Adventitial Stromal Cells Define Group 2 Innate Lymphoid Cell Tissue Niches. Immunity (2019) 50:707–722.e6. doi: 10.1016/j.immuni.2019.02.002
17. Bando JK, Liang H-E, Locksley RM. Identification and Distribution of Developing Innate Lymphoid Cells in the Fetal Mouse Intestine. Nat Immunol (2015) 16:153–60. doi: 10.1038/ni.3057
18. Zeis P, Lian M, Fan X, Herman JS, Hernandez DC, Gentek R, et al. In Situ Maturation and Tissue Adaptation of Type 2 Innate Lymphoid Cell Progenitors. Immunity (2020) 53:775–792.e9. doi: 10.1016/j.immuni.2020.09.002
19. Ghaedi M, Shen ZY, Orangi M, Martinez-Gonzalez I, Wei L, Lu X, et al. Single-Cell Analysis of Rorα Tracer Mouse Lung Reveals ILC Progenitors and Effector ILC2 Subsets. J Exp Med (2020) 217:1–19. doi: 10.1084/jem.20182293
20. Ong S, Rose NR, Čiháková D. Natural Killer Cells in Inflammatory Heart Disease. Clin Immunol (2017) 175:26–33. doi: 10.1016/j.clim.2016.11.010
21. Yuan J, Liu Z, Lim T, Zhang H, He J, Walker E, et al. CXCL10 Inhibits Viral Replication Through Recruitment of Natural Killer Cells in Coxsackievirus B3-Induced Myocarditis. Circ Res (2009) 104:628–38. doi: 10.1161/CIRCRESAHA.108.192179
22. Cardilo-Reis L, Gruber S, Schreier SM, Drechsler M, Papac-Milicevic N, Weber C, et al. Interleukin-13 Protects From Atherosclerosis and Modulates Plaque Composition by Skewing the Macrophage Phenotype. EMBO Mol Med (2012) 4:1072–86. doi: 10.1002/emmm.201201374
23. Buono C, Binder CJ, Stavrakis G, Witztum JL, Glimcher LH, Lichtman AH. T-Bet Deficiency Reduces Atherosclerosis and Alters Plaque Antigen-Specific Immune Responses. Proc Natl Acad Sci USA (2005) 102:1596–601. doi: 10.1073/pnas.0409015102
24. Davenport P, Tipping PG. The Role of Interleukin-4 and Interleukin-12 in the Progression of Atherosclerosis in Apolipoprotein E-Deficient Mice. Am J Pathol (2003) 163:1117–25. doi: 10.1016/S0002-9440(10)63471-2
25. Wu C, He S, Liu J, Wang B, Lin J, Duan Y, et al. Type 1 Innate Lymphoid Cell Aggravation of Atherosclerosis Is Mediated Through TLR4. Scand J Immunol (2018) 87:1–14. doi: 10.1111/sji.12661
26. Chen WY, Wu YH, Tsai TH, Li RF, Lai ACY, Li LC, et al. Group 2 Innate Lymphoid Cells Contribute to IL-33-Mediated Alleviation of Cardiac Fibrosis. Theranostics (2021) 11:2594–611. doi: 10.7150/THNO.51648
27. Ghali R, Habeichi NJ, Kaplan A, Tannous C, Abidi E, Bekdash A, et al. IL-33 Induces Type-2-Cytokine Phenotype But Exacerbates Cardiac Remodeling Post-Myocardial Infarction With Eosinophil Recruitment, Worsened Systolic Dysfunction, and Ventricular Wall Rupture. Clin Sci (2020) 134:1191–218. doi: 10.1042/CS20200402
28. Li J, Wu J, Zhang M, Zheng Y. Dynamic Changes of Innate Lymphoid Cells in Acute ST-Segment Elevation Myocardial Infarction and Its Association With Clinical Outcomes. Sci Rep (2020) 10:1–12. doi: 10.1038/s41598-020-61903-5
29. Li Q, Liu M, Fu R, Cao Q, Wang Y, Han S, et al. Alteration of Circulating Innate Lymphoid Cells in Patients With Atherosclerotic Cerebral Infarction. Am J Transl Res (2018) 10:4322–30.
30. Molofsky AB, Nussbaum JC, Liang HE, Dyken SJV, Cheng LE, Mohapatra A, et al. Innate Lymphoid Type 2 Cells Sustain Visceral Adipose Tissue Eosinophils and Alternatively Activated Macrophages. J Exp Med (2013) 210:535–49. doi: 10.1084/jem.20121964
31. Brestoff JR, Kim BS, Saenz SA, Stine RR, Monticelli LA, Sonnenberg GF, et al. Group 2 Innate Lymphoid Cells Promote Beiging of White Adipose Tissue and Limit Obesity. Nature (2015) 519:242–6. doi: 10.1038/nature14115
32. Galle-Treger L, Sankaranarayanan I, Hurrell BP, Howard E, Lo R, Maazi H, et al. Costimulation of Type-2 Innate Lymphoid Cells by GITR Promotes Effector Function and Ameliorates Type 2 Diabetes. Nat Commun (2019) 10:1–14. doi: 10.1038/s41467-019-08449-x
33. Okamura T, Hashimoto Y, Mori J, Yamaguchi M, Majima S, Senmaru T, et al. ILC2s Improve Glucose Metabolism Through the Control of Saturated Fatty Acid Absorption Within Visceral Fat. Front Immunol (2021) 12:669629. doi: 10.3389/fimmu.2021.669629
34. Qiu Y, Nguyen KD, Odegaard JI, Cui X, Tian X, Locksley RM, et al. Eosinophils and Type 2 Cytokine Signaling in Macrophages Orchestrate Development of Functional Beige Fat. Cell (2014) 157:1292–308. doi: 10.1016/j.cell.2014.03.066
35. Oldenhove G, Boucquey E, Taquin A, Acolty V, Bonetti L, Ryffel B, et al. PD-1 Is Involved in the Dysregulation of Type 2 Innate Lymphoid Cells in a Murine Model of Obesity. Cell Rep (2018) 25:2053–2060.e4. doi: 10.1016/j.celrep.2018.10.091
36. Awad A, Yassine H, Barrier M, Vorng H, Marquillies P, Tsicopoulos A, et al. Natural Killer Cells Induce Eosinophil Activation and Apoptosis. PloS One (2014) 9:1–9. doi: 10.1371/journal.pone.0094492
37. Duan Z, Liu M, Yuan L, Du X, Wu M, Yang Y, et al. Innate Lymphoid Cells Are Double-Edged Swords Under the Mucosal Barrier. J Cell Mol Med (2021) 25:8579–87. doi: 10.1111/jcmm.16856
38. Lim AI, Menegatti S, Bustamante J, Le Bourhis L, Allez M, Rogge L, et al. IL-12 Drives Functional Plasticity of Human Group 2 Innate Lymphoid Cells. J Exp Med (2016) 213:569–83. doi: 10.1084/jem.20151750
39. Boulenouar S, Michelet X, Duquette D, Alvarez D, Hogan AE, Dold C, et al. Adipose Type One Innate Lymphoid Cells Regulate Macrophage Homeostasis Through Targeted Cytotoxicity. Immunity (2017) 46:273–86. doi: 10.1016/j.immuni.2017.01.008
40. Cardoso F, Klein Wolterink RGJ, Godinho-Silva C, Domingues RG, Ribeiro H, da Silva JA, et al. Neuro-Mesenchymal Units Control ILC2 and Obesity via a Brain–Adipose Circuit. Nature (2021) 597:410–4. doi: 10.1038/s41586-021-03830-7
41. Ikutani M, Tsuneyama K, Kawaguchi M, Fukuoka J, Kudo F, Nakae S, et al. Prolonged Activation of IL-5-Producing ILC2 Causes Pulmonary Arterial Hypertrophy. JCI Insight (2017) 2:e90721. doi: 10.1172/jci.insight.90721
42. Mantani PT, Dunér P, Ljungcrantz I, Nilsson J, Björkbacka H, Fredrikson GN. ILC2 Transfers to Apolipoprotein E Deficient Mice Reduce the Lipid Content of Atherosclerotic Lesions. BMC Immunol (2019) 20:1–13. doi: 10.1186/s12865-019-0330-z
43. Mantani PT, Dunér P, Bengtsson E, Alm R, Ljungcrantz I, Söderberg I, et al. IL-25 Inhibits Atherosclerosis Development in Apolipoprotein E Deficient Mice. PloS One (2015) 10:1–18. doi: 10.1371/journal.pone.0117255
44. Barin JG, Talor MV, Diny NL, Ong SF, Schaub JA, Gebremariam E, et al. Regulation of Autoimmune Myocarditis by Host Responses to the Microbiome. Exp Mol Pathol (2017) 103:141–52. doi: 10.1016/j.yexmp.2017.08.003
45. Sasaki T, Moro K, Kubota T, Kubota N, Kato T, Ohno H, et al. Innate Lymphoid Cells in the Induction of Obesity. Cell Rep (2019) 28:202–217.e7. doi: 10.1016/j.celrep.2019.06.016
46. Hak Ł, Mysłiwska J, Wieckiewicz J, Szyndler K, Trzonkowski P, Siebert J, et al. NK Cell Compartment in Patients With Coronary Heart Disease. Immun Ageing (2007) 4:1–8. doi: 10.1186/1742-4933-4-3
47. Jonasson L, Backteman K, Ernerudh J. Loss of Natural Killer Cell Activity in Patients With Coronary Artery Disease. Atherosclerosis (2005) 183:316–21. doi: 10.1016/j.atherosclerosis.2005.03.011
48. Backteman K, Ernerudh J, Jonasson L. Natural Killer (NK) Cell Deficit in Coronary Artery Disease: No Aberrations in Phenotype But Sustained Reduction of NK Cells Is Associated With Low-Grade Inflammation. Clin Exp Immunol (2014) 175:104–12. doi: 10.1111/cei.12210
49. Szymanowski A, Li W, Lundberg A, Evaldsson C, Nilsson L, Backteman K, et al. Soluble Fas Ligand Is Associated With Natural Killer Cell Dynamics in Coronary Artery Disease. Atherosclerosis (2014) 233:616–22. doi: 10.1016/j.atherosclerosis.2014.01.030
50. Hou N, Zhao D, Liu Y, Gao L, Liang X, Liu X, et al. Increased Expression of T Cell Immunoglobulin- and Mucin Domain-Containing Molecule-3 on Natural Killer Cells in Atherogenesis. Atherosclerosis (2012) 222:67–73. doi: 10.1016/j.atherosclerosis.2012.02.009
51. Selathurai A, Deswaerte V, Kanellakis P, Tipping P, Toh BH, Bobik A, et al. Natural Killer (NK) Cells Augment Atherosclerosis by Cytotoxic-Dependent Mechanisms. Cardiovasc Res (2014) 102:128–37. doi: 10.1093/cvr/cvu016
52. Nour-Eldine W, Joffre J, Zibara K, Esposito B, Giraud A, Zeboudj L, et al. Genetic Depletion or Hyperresponsiveness of Natural Killer Cells Do Not Affect Atherosclerosis Development. Circ Res (2018) 122:47–57. doi: 10.1161/CIRCRESAHA.117.311743
53. Ong S, Ligons DL, Barin JG, Wu L, Talor MV, Diny N, et al. Natural Killer Cells Limit Cardiac Inflammation and Fibrosis by Halting Eosinophil Infiltration. Am J Pathol (2015) 185:847–61. doi: 10.1016/j.ajpath.2014.11.023
54. Barnig C, Cernadas M, Dutile S, Liu X, Perrella MA, Kazani S, et al. Lipoxin A4 Regulates Natural Killer Cell and Type 2 Innate Lymphoid Cell Activation in Asthma. Sci Transl Med (2013) 5:174ra26. doi: 10.1126/scitranslmed.3004812
55. Xanthou G, Duchesnes CE, Williams TJ, Pease JE. CCR3 Functional Responses Are Regulated by Both CXCR3 and Its Ligands CXCL9, CXCL10 and CXCL11. Eur J Immunol (2003) 33:2241–50. doi: 10.1002/eji.200323787
56. Molofsky AB, Van Gool F, Liang H-E, Van Dyken SJ, Nussbaum JC, Lee J, et al. Interleukin-33 and Interferon-γ Counter-Regulate Group 2 Innate Lymphoid Cell Activation During Immune Perturbation. Immunity (2015) 43:161–74. doi: 10.1016/j.immuni.2015.05.019
57. Cautivo KM, Matatia PR, Lizama CO, Mroz NM, Dahlgren MW, Yu X, et al. Interferon Gamma Constrains Type 2 Lymphocyte Niche Boundaries During Mixed Inflammation. Immunity (2022) 55:254–271.e7. doi: 10.1016/j.immuni.2021.12.014
58. Moon B, Takaki S, Miyake K, Takatsu K. The Role of IL-5 for Mature B-1 Cells in Homeostatic Proliferation, Cell Survival, and Ig Production. J Immunol (2004) 172:6020–9. doi: 10.4049/jimmunol.172.10.6020
59. Baumgarth N. The Double Life of a B-1 Cell: Self-Reactivity Selects for Protective Effector Functions. Nat Rev Immunol (2011) 11:34–46. doi: 10.1038/nri2901
60. Perry HM, Oldham SN, Fahl SP, Que X, Gonen A, Harmon DB, et al. Helix-Loop-Helix Factor Inhibitor of Differentiation 3 Regulates Interleukin-5 Expression and B-1a B Cell Proliferation. Arterioscler Thromb Vasc Biol (2013) 33:2771–9. doi: 10.1161/ATVBAHA.113.302571
61. Doran AC, Lehtinen AB, Meller N, Lipinski MJ, Slayton RP, Oldham SN, et al. Id3 Is a Novel Atheroprotective Factor Containing a Functionally Significant Single-Nucleotide Polymorphism Associated With Intima-Media Thickness in Humans. Circ Res (2010) 106:1303–11. doi: 10.1161/CIRCRESAHA.109.210294
62. Zhao TX, Kostapanos M, Griffiths C, Arbon EL, Hubsch A, Kaloyirou F, et al. Low-Dose Interleukin-2 in Patients With Stable Ischaemic Heart Disease and Acute Coronary Syndromes (LILACS): Protocol and Study Rationale for a Randomised, Double-Blind, Placebo-Controlled, Phase I/II Clinical Trial. BMJ Open (2018) 8. doi: 10.1136/bmjopen-2018-022452
63. Robert M, Miossec P. Effects of Interleukin 17 on the Cardiovascular System. Autoimmun Rev (2017) 16:984–91. doi: 10.1016/j.autrev.2017.07.009
64. Luo JW, Hu Y, Liu J, Yang H, Huang P. Interleukin-22: A Potential Therapeutic Target in Atherosclerosis. Mol Med (2021) 27. doi: 10.1186/s10020-021-00353-9
65. Che Y, Su Z, Xia L. Effects of IL-22 on Cardiovascular Diseases. Int Immunopharmacol (2020) 81:106277. doi: 10.1016/j.intimp.2020.106277
66. Eggenhofer E, Sabet-Rashedi M, Lantow M, Renner P, Rovira J, Koehl GE, et al. Rorγt+ IL-22-Producing NKp46+ Cells Protect From Hepatic Ischemia Reperfusion Injury in Mice. J Hepatol (2016) 64:128–34. doi: 10.1016/j.jhep.2015.08.023
67. Geha M, Tsokos MG, Bosse RE, Sannikova T, Iwakura Y, Dalle Lucca JJ, et al. IL-17a Produced by Innate Lymphoid Cells Is Essential for Intestinal Ischemia-Reperfusion Injury. J Immunol (2017) 199:2921–9. doi: 10.4049/jimmunol.1700655
68. Mallat Z, Taleb S, Ait-Oufella H, Tedgui A. The Role of Adaptive T Cell Immunity in Atherosclerosis. J Lipid Res (2009) 50:364–9. doi: 10.1194/jlr.R800092-JLR200
69. Colonna M. Innate Lymphoid Cells: Diversity, Plasticity, and Unique Functions in Immunity. Immunity (2018) 48:1104–17. doi: 10.1016/j.immuni.2018.05.013
70. Bal SM, Golebski K, Spits H. Plasticity of Innate Lymphoid Cell Subsets. Nat Rev Immunol (2020) 20:552–65. doi: 10.1038/s41577-020-0282-9
71. Ohne Y, Silver JS, Thompson-Snipes LA, Collet MA, Blanck JP, Cantarel BL, et al. IL-1 Is a Critical Regulator of Group 2 Innate Lymphoid Cell Function and Plasticity. Nat Immunol (2016) 17:646–55. doi: 10.1038/ni.3447
72. Silver JS, Kearley J, Copenhaver AM, Sanden C, Mori M, Yu L, et al. Inflammatory Triggers Associated With Exacerbations of COPD Orchestrate Plasticity of Group 2 Innate Lymphoid Cells in the Lungs. Nat Immunol (2016) 17:626–35. doi: 10.1038/ni.3443
73. Bal SM, Bernink JH, Nagasawa M, Groot J, Shikhagaie MM, Golebski K, et al. IL-1β, IL-4 and IL-12 Control the Fate of Group 2 Innate Lymphoid Cells in Human Airway Inflammation in the Lungs. Nat Immunol (2016) 17:636–45. doi: 10.1038/ni.3444
74. Kumar R, Graham B. IL-33-Hif1α Axis in Hypoxic Pulmonary Hypertension. EBioMedicine (2018) 33:8–9. doi: 10.1016/j.ebiom.2018.07.004
75. Lee JJ, McGarry MP, Farmer SC, Denzler KL, Larson KA, Carrigan PE, et al. Interleukin-5 Expression in the Lung Epithelium of Transgenic Mice Leads to Pulmonary Changes Pathognomonic of Asthma. Pneumologie (1998) 52(6). doi: 10.1084/jem.185.12.2143
76. Min HK, Moon J, Lee S, Lee AR, Lee CR, Lee J, et al. Expanded IL-22 + Group 3 Innate Lymphoid Cells and Role of Oxidized LDL-C in the Pathogenesis of Axial Spondyloarthritis With Dyslipidaemia. Immune Netw (2021) 21:e43. doi: 10.4110/in.2021.21.e43
77. Agca R, Heslinga SC, Rollefstad S, Heslinga M, McInnes IB, Peters MJL, et al. EULAR Recommendations for Cardiovascular Disease Risk Management in Patients With Rheumatoid Arthritis and Other Forms of Inflammatory Joint Disorders: 2015/2016 Update. Ann Rheum Dis (2016) 76:17–28. doi: 10.1136/annrheumdis-2016-209775
78. Fachi JL, Pral LP, dos Santos JAC, Codo AC, de Oliveira S, Felipe JS, et al. Hypoxia Enhances ILC3 Responses Through HIF-1α-Dependent Mechanism. Mucosal Immunol (2021) 14:828–41. doi: 10.1038/s41385-020-00371-6
79. Liu M, Galli G, Wang Y, Fan Q, Wang Z, Wang X, et al. Novel Therapeutic Targets for Hypoxia-Related Cardiovascular Diseases: The Role of HIF-1. Front Physiol (2020) 11:774. doi: 10.3389/fphys.2020.00774
80. O’Sullivan TE, Rapp M, Fan X, Weizman O-E, Bhardwaj P, Adams NM, et al. Adipose-Resident Group 1 Innate Lymphoid Cells Promote Obesity-Associated Insulin Resistance. Immunity (2016) 45:428–41. doi: 10.1016/j.immuni.2016.06.016
81. Kochumon S, Al Madhoun A, Al-Rashed F, Thomas R, Sindhu S, Al-Ozairi E, et al. Elevated Adipose Tissue Associated IL-2 Expression in Obesity Correlates With Metabolic Inflammation and Insulin Resistance. Sci Rep (2020) 10:1–13. doi: 10.1038/s41598-020-73347-y
82. Wu Y, Yue J, Wu J, Zhou W, Li D, Ding K, et al. Obesity may Provide Pro-ILC3 Development Inflammatory Environment in Asthmatic Children. J Immunol Res (2018) 2018. doi: 10.1155/2018/1628620
83. Upadhyay V, Poroyko V, Kim T, Devkota S, Fu S, Liu D, et al. Lymphotoxin Regulates Commensal Responses to Enable Diet-Induced Obesity. Nat Immunol (2012) 13:947–53. doi: 10.1038/ni.2403
84. Roberts LB, Schnoeller C, Berkachy R, Darby M, Pillaye J, Oudhoff MJ, et al. Acetylcholine Production by Group 2 Innate Lymphoid Cells Promotes Mucosal Immunity to Helminths. Sci Immunol (2021) 6(57). doi: 10.1126/sciimmunol.abd0359
85. Berrien-Elliott MM, Sun Y, Neal C, Ireland A, Trissal MC, Sullivan RP, et al. MicroRNA-142 Is Critical for the Homeostasis and Function of Type 1 Innate Lymphoid Cells. Immunity (2019) 51:479–490.e6. doi: 10.1016/j.immuni.2019.06.016
86. Roberts LB, Jowett GM, Read E, Zabinski T, Berkachy R, Selkirk ME, et al. MicroRNA-142 Critically Regulates Group 2 Innate Lymphoid Cell Homeostasis and Function. J Immunol (2021) 206(11):2725–39. doi: 10.4049/jimmunol.2000647
87. Grimaldi A, Pietropaolo G, Stabile H, Kosta A, Capuano C, Gismondi A, et al. The Regulatory Activity of Noncoding Rnas in Ilcs. Cells (2021) 10:1–15. doi: 10.3390/cells10102742
Keywords: ILCs, innate lymphoid cells, cardiovascular disease, obesity, heart
Citation: Roberts LB, Lord GM and Howard JK (2022) Heartbreakers or Healers? Innate Lymphoid Cells in Cardiovascular Disease and Obesity. Front. Immunol. 13:903678. doi: 10.3389/fimmu.2022.903678
Received: 24 March 2022; Accepted: 19 April 2022;
Published: 11 May 2022.
Edited by:
Carolina Jancic, Consejo Nacional de Investigaciones Científicas y Técnicas (CONICET), ArgentinaReviewed by:
Marek Wagner, University of Bergen, NorwayYi Ding, National Cancer Institute (NIH), United States
Copyright © 2022 Roberts, Lord and Howard. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Luke B. Roberts, bHVrZS5yb2JlcnRzQGtjbC5hYy51aw==