
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 10 June 2022
Sec. T Cell Biology
Volume 13 - 2022 | https://doi.org/10.3389/fimmu.2022.902318
This article is part of the Research Topic The role of regulatory T cells in controlling inflammatory responses View all 12 articles
 Jing Zhang1†
Jing Zhang1† Yuan Zou1†
Yuan Zou1† Longmin Chen2
Longmin Chen2 Qianqian Xu1
Qianqian Xu1 Yi Wang1
Yi Wang1 Min Xie1,3
Min Xie1,3 Xiansheng Liu1,3
Xiansheng Liu1,3 Jianping Zhao1*
Jianping Zhao1* Cong-Yi Wang1*
Cong-Yi Wang1*Asthma is a multifactorial disorder characterized by the airway chronic inflammation, hyper-responsiveness (AHR), remodeling, and reversible obstruction. Although asthma is known as a heterogeneous group of diseases with various clinical manifestations, recent studies suggest that more than half of the clinical cases are ‘‘T helper type 2 (Th2)-high’’ type, whose pathogenesis is driven by Th2 responses to an inhaled allergen from the environmental exposures. The intensity and duration of inflammatory responses to inhaled allergens largely depend on the balance between effector and regulatory cells, but many questions regarding the mechanisms by which the relative magnitudes of these opposing forces are remained unanswered. Regulatory T cells (Tregs), which comprise diverse subtypes with suppressive function, have long been attracted extensive attention owing to their capability to limit the development and progression of allergic diseases. In this review we seek to update the recent advances that support an essential role for Tregs in the induction of allergen tolerance and attenuation of asthma progression once allergic airway inflammation established. We also discuss the current concepts about Treg induction and Treg-expressed mediators relevant to controlling asthma, and the therapies designed based on these novel insights against asthma in clinical settings.
Asthma is a chronic airway inflammatory disease that affects more than 350 million individuals worldwide (1). The etiology underlying asthma includes both genetic predisposition and environmental exposures (2). In general, the airway inflammation in asthmatic setting arises from the reaction in response to allergens and/or other environmental factors, thereby leading to an aberrant airway Th2-type immune response (3). Although a great effort of studies had advanced the understanding of pathologic features of asthma, its mechanisms underlying the regulation of allergic airway inflammation are yet to be fully elucidated. As a result, the development of novel therapeutic strategies against asthma is confronted with formidable challenges.
There is strong evidence in animals that regulatory T cells (Tregs) act as a key regulator of allergic diseases and are essential to limit antigen-specific immune responses. For example, mice deficient in CD4+CD25+ Tregs resulted from loss-of-function mutations in the Foxp3 gene are featured by the development of spontaneous autoimmunity, lymphoproliferation, allergic airway inflammation, hyper IgE syndrome, and eosinophilia (4). Similarly, adoptive transfer of ovalbumin (OVA) peptide-specific CD4+CD25+ Tregs into the OVA-sensitized mice attenuated airway hyper-responsiveness (AHR) along with reduced number of eosinophils and production of Th2 cytokines in the lung following allergen challenge (5). Foxp3+ Tregs also suppress chronic allergic inflammation to establish allergen-tolerance in the respiratory mucosa (6). Furthermore, manipulation of steroid responsiveness in Tregs could represent a novel strategy to treat steroid refractory asthma, as their responsiveness determines steroid sensitivity during allergic airway inflammation (7). Collectively, these studies underscore the significance of Tregs in the regulation of allergic airway inflammation in mouse models.
Unlike the impact observed in animal models, the role of Tregs in asthmatic patients is yet to be well defined. Studies revealed that adult asthmatic patients with either stable or exacerbated symptoms displayed lower percentage of Tregs along with impaired suppressive function in the blood and airway (8). Similarly, decreased pulmonary Treg number coupled with lower capability to suppress pulmonary Th2 responses were observed in asthmatic children (9). In sharp contrast, some studies also demonstrated that the amount of airway Tregs was increased in adult patients with moderate to severe asthma as compared to both mild asthmatic patients and healthy subjects (10), especially in response to bronchial allergen provocation (11). The discrepancy between these findings could be caused by the differences of study cohorts and the approaches for Treg analysis. Nevertheless, a consistent conclusion could probably be reached for the impaired Treg function in asthmatic patients, particularly for their capability to suppress Th2 responses. A recent study further suggested that the numerical and functional defects of Tregs may render the children and younger adults more susceptible to asthma, while the relationship between Tregs and asthma risk or severity in older patients is relatively weak (12). Although the contribution of Tregs in asthma is not fully addressed, clinical improvement following allergen immunotherapy (AIT) for asthma suggested an association with the induction of IL-10-, IL-35- and TGF-β-producing Tregs and Foxp3+ Tregs (13). Therefore, in this review we seek to summarize the immunological features of allergic asthma, and then update the recent advances that support the role of Tregs in allergen tolerance induction and in limiting disease severity once allergic airway inflammation established. We also discuss the current concepts about Treg induction and Treg-expressed mediators relevant to controlling asthma, and the therapies designed based on these novel insights against asthma in clinical settings.
Type 2 immunity has now been well recognized to be a critical feature relevant to a complex network of immunologic mechanisms in allergic asthma (14). Type 2 immune response involves an ever-expanding repertoire of immune cells, including Th2 cells, B cells, natural killer (NK) cells, NKT cells, basophils, eosinophils, mast cells, and group 2 innate lymphoid cells (ILC2s) and their associated cytokines (15). IL-4, IL-5, IL-9, and IL-13 are predominantly produced by immunocytes, while IL-25, IL-31, IL-33, and thymic stromal lymphopoietin (TSLP) are released from tissue cells, particularly epithelial cells (16).
The immunological mechanisms underlying allergic response can be classified into two main phases: (1) the sensitization and memory phase, and (2) the effector phase. The later can be subdivided into the immediate and late-phase reactions (17, 18). During the sensitization and memory phase, the differentiated and clonal expanded allergen-specific Th2 cells produce copious amount of IL-4 and IL-13 to drive the class switching of antibody isotypes to the ε heavy chain. The allergen-specific IgE then binds to the high-affinity FcϵRI on the surface of mast cells and basophils, thereby contributing to the IgE sensitization of individuals against allergens. In this phase, a memory pool of allergen-specific Th2 and B cells is also generated, which is ready to act upon allergen encounters. The immediate reaction of allergic response is also termed as type 1 hypersensitive reaction. Upon the challenge from same allergens, crosslinking of the IgE-FcϵRI complexes on the sensitized basophils and mast cells leads to the release of anaphylactogenic mediators (such as vasoactive amines, prostaglandin D, platelet-activating factor, leukotriene, chemokines, and other cytokines) responsible for the classical immediate symptoms of allergic disease. The late-stage reaction generally occurs following 4-6 hours of allergen stimulation and lasts for more than a few days, and is featured by the localized inflammatory responses mediated by the infiltrated eosinophils, neutrophils, macrophages, Th2 cells and basophils. The ongoing inflammation results in more severe clinical manifestations of allergy, such as chronic persistent asthma, allergic rhinitis, and in extreme cases, systemic anaphylactic reactions (18).
Recent studies also suggested the involvement of epithelial cells in allergic pathology. Barrier epithelial cells not only represent the very first line of defense against environmental insults, but also produce cytokines (e.g., IL-25, IL-31, IL-33, and TSLP) and alarmins (e.g., uric acid, ATP, HMGB1, and S100 proteins) following allergen exposures (19). These signals constitute important factors in the early phase of asthma and promote Th2 differentiation through their effect on tissue dendritic cells and ILC2s (20). In particular, there is evidence that a neutralizing mAb against IL-25 results in a significantly reduced production of IL-5 and IL-13 along with attenuated eosinophil infiltration, goblet cell hyperplasia, and serum IgE secretion, by which it prevents AHR following OVA-induced allergic airway inflammation in mice (21). More excitingly, blocking antibodies against either TSLP or IL-33/ST2 signaling are currently at different stages of clinical trials, which could be promising candidates for asthma treatment in clinical settings (22, 23).
ILCs are defined as ILC type1, 2, and 3 cells based on their transcription factors and cytokine production patterns, and among which, ILC2s play a substantial role in the initiation, progression, and steroid resistance of allergic airway inflammation (20). It was noted that IL-33 targets ILC2 to produce IL-5 and IL-13, thereby enhancing eosinophil recruitment, goblet cell hyperplasia, macrophage M2 polarization, dendritic cell (DC) activation and Th2 differentiation (24–27). Studies revealed that the number of total and type 2 cytokine-expressing ILC2s is significantly higher in the peripheral blood and airway of patients with systemic steroid-dependent severe eosinophilic asthma than those of patients with mild asthma (28). Given that the intracellular cytokine expression by Th2 cells within the airways did not differ between the above two groups of patients, The observations support that uncontrolled ILC2s rather than Th2 cells, represent a steroid-insensitive population of cells to exacerbate the development of airway inflammation in patients with severe prednisone-dependent eosinophilic asthma (28). Other distinct types of effector T cells (Teffs) may also get involved in continuous allergic inflammation as well. For example, although Th1 and IFN-γ secreting NKT cells induce epithelial apoptosis through cell-mediated cytotoxicity, they also exert an inhibitory role in Th2 cells and suppress IgE isotype switching (29). While IL-17 producing Th17 cells mediate neutrophilic type inflammation other than exacerbating Th2-related allergic inflammation (30). Moreover, Th9 cells employ multiple mechanisms to orchestrate allergic inflammation, and particularly, their interaction with diverse cell types including mast cells, ILCs, and DCs, to promote coordinated regulation of allergic airway inflammation (31, 32). Other than secretion of their signature cytokine IL-9, Th9 cells from mice and humans also secret other cytokines such as IL-10, IL-17, IL-21, and IL-22, to facilitate immune responses in the setting of allergic asthma (33, 34). Furthermore, studies on atopic dermatitis demonstrated feasible evidence supporting that the expansion of Th2/Tc2 and Th22/Tc22 may also exert an important role in allergic inflammation (14, 35).
Tregs are one of the main bastions against inappropriate or over-exuberant inflammatory responses, and play an indispensable role in the maintenance of immune tolerance in asthma (36). However, subsets of CD8+ T cells, CD4-CD8- T cells, γδ T cells, regulatory B cells (Bregs), IL-10-producing DCs, IL-10-producing NK cells, and macrophage subsets with suppressive properties also contribute to the suppressive and regulatory events (37). Currently, two main subsets of Tregs have been defined: the thymus-derived naturally occurring CD4+CD25hi Foxp3+ Tregs, also called tTregs, and the peripherally induced adaptive Tregs (pTregs) (38, 39). pTregs are further divided into Foxp3+ pTregs, Foxp3− IL-10-producing Tr1 cells, and Foxp3− TGF-β-expressing Th3 cells. Studies in animals suggest the Foxp3+ pTregs and IL-10-producing Tr1 cells may contribute to the differences of asthma susceptibility associated with different genetic background (40). Generally, Foxp3 induction in tTregs can occur at the double positive (DP) stage or preferentially at the CD4 single positive (SP) stage or during the transition to this stage in the thymus (41). Interaction with antigen presented by either cortical or medullary thymic epithelial cells is sufficient to induce Foxp3 expression, thereby committing T-cell precursor to Treg lineage (42). pTregs are differentiated in the secondary lymphoid organs and tissues, and they are particularly enriched in the intestinal mucosa and lung during chronic inflammation, with specificities directed against food antigens, gut microflora, and environmental allergens (43). The induction of pTregs at the gastrointestinal tract is facilitated by the CD103+CD11c+ DCs in a TGF-β and retinoic acid-dependent manner (44–46), while lung tissue-resident macrophages constitutively coexpressing TGF-β and retinal dehydrogenases (RALDH1 and RALDH2) are the main subset of cells driving pTreg generation from naïve CD4+ T cells (47). It is worthy of note that the classification of Tregs could vary based on the specific markers employed. For example, Tregs can be also classified into nTregs, iTregs, ICOS+ Tregs, Tr1, CD8+ Tregs and IL-17-producing Tregs (48); however, some of these Treg subsets could be functionally overlapping or synergizing each other.
tTregs and pTregs are phenotypically distinct, and display different specificities and complementary functions in vivo (49). Generally, TCR on tTregs primarily recognizes self-antigens, which is crucial for establishing self-tolerance and preventing autoimmunity, while pTregs are thought to predominantly govern tolerogenic responses against non-self-antigens, such as allergens, food, and the commensal microbiota (50, 51). In a mouse model of chronic asthma, passive transfer of pTregs efficiently suppressed all aspects of asthmatic phenotype, whereas equal amount of tTregs only manifested a modest impact in this model, indicating that pTreg are substantially more tolerogenic in this setting (52). Although both tTregs and pTregs attenuate the development of asthma in WT recipients, those cells, however, enhance lung allergic responses in CD8-/- recipients (53). The reprogramming pathways and enhancement appear to be distinct and cytokine specific, in which IL-13 production in nTreg depends on the GITR signaling, while IL-17 production in pTregs is induced by IL-6 signaling (53). There is evidence that tTregs stability in allergic settings is maintained by the epigenetic mechanisms and metabolites generated by themselves such as cyclic adenosine monophosphate (cAMP) (50). In contrast, the instability of Foxp3 expression and loss of suppressive function in pTreg are closely related to the methylation state of the Treg-specific demethylated region (TSDR) (50). Therefore, the regulatory mechanisms underlying the maintenance of Treg stability and functionality are essential to the development of effective strategies against allergic airway diseases.
The suppressive functions of Tregs in allergic inflammation are mediated by an ever-growing list of mechanisms (Figure 1). Tregs can suppress antigen presentation cells (APCs) to activate Teffs, while enhancing the function of tolerogenic DCs. Tregs inhibit the migration and functionality of Teffs including Th1, Th2, and Th17 cells. Tregs also repress the secretion of allergen-specific IgE from plasma cells and induce IgG4-secreting B cells and IL-10-producing Bregs. Other than Teffs, Tregs are able to suppress the activation of eosinophils, basophils, mast cells, NKT cells, and ILC2s (14, 54). The above-mentioned suppressive functions from Tregs are introduced by a number of soluble and membrane-bound molecules such as cytokines with inhibitory effect (e.g., IL-10, TGF-β and IL-35), enzymes or proteins relevant to cytolysis (e.g., granzymes A, B and K, and perforin), membrane proteins and signaling molecules relevant to metabolic homeostasis (e.g., CD25, CD39, CD73, cAMP, LAG3, adenosine receptor 2, and histamine receptor 2), and surface molecules (e.g., PD-1 and CTLA-4) for targeting DCs (38, 55–57).
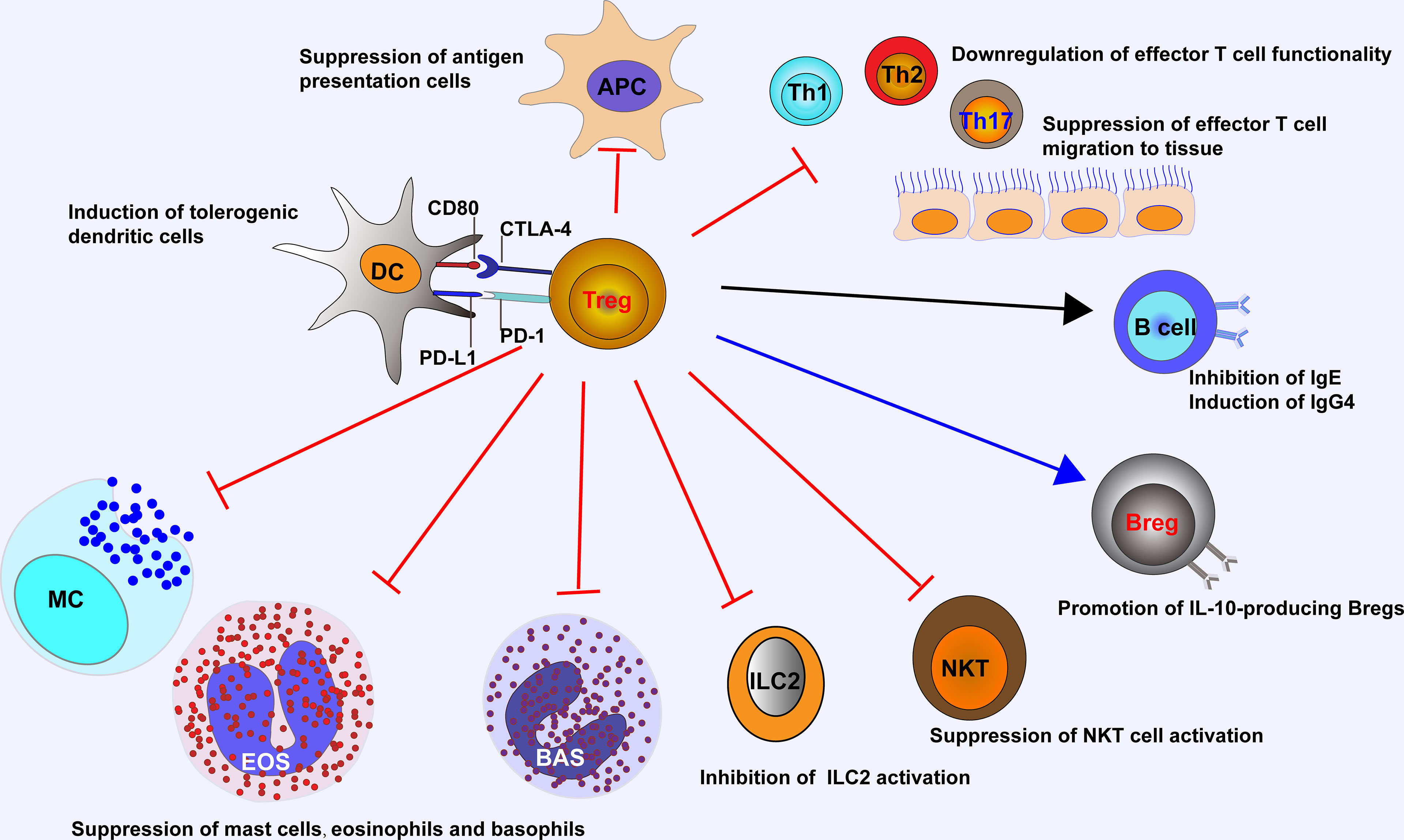
Figure 1 Tregs control ongoing inflammation by acting on major cells that drive allergic reaction, including antigen presentation cells, Teffs, NKT cells, ILC2s, eosinophils, basophils, and mast cells. Tregs suppress IgE-producing B cells, while induce IgG4-producing B cells and IL-10-producing Bregs, and promote the generation of tolerogenic dendritic cells. APC, antigen presentation cell; EOS, eosinophil; BAS, basophil; MC, mast cell.
The key role played by IL-10 and TGF-β in the context of allergic asthma is now well established (summarized in Table 1). Apart from Tregs, IL-10 is also released by Bregs, monocytes, a small fraction of NK cells, macrophages, DCs and ILCs (15). IL-10 exerts its effect on both innate and adaptive immune responses, thereby inducing immune tolerance and dampening tissue inflammation (90). For example, transfer of OVA peptide-specific Tregs to OVA-sensitized mice repressed AHR, eosinophil recruitment, and Th2 cytokine expression in the lung following allergen challenge (5), which was reversed by the application of IL-10R blocking antibody (5). It is worthy of note that the IL-10-producing Tr1 cells also represent an essential mechanism in immune tolerance to a high dose of allergens in nonallergic individuals, such as high dose bee venom exposure in beekeepers by natural bee stings (55).
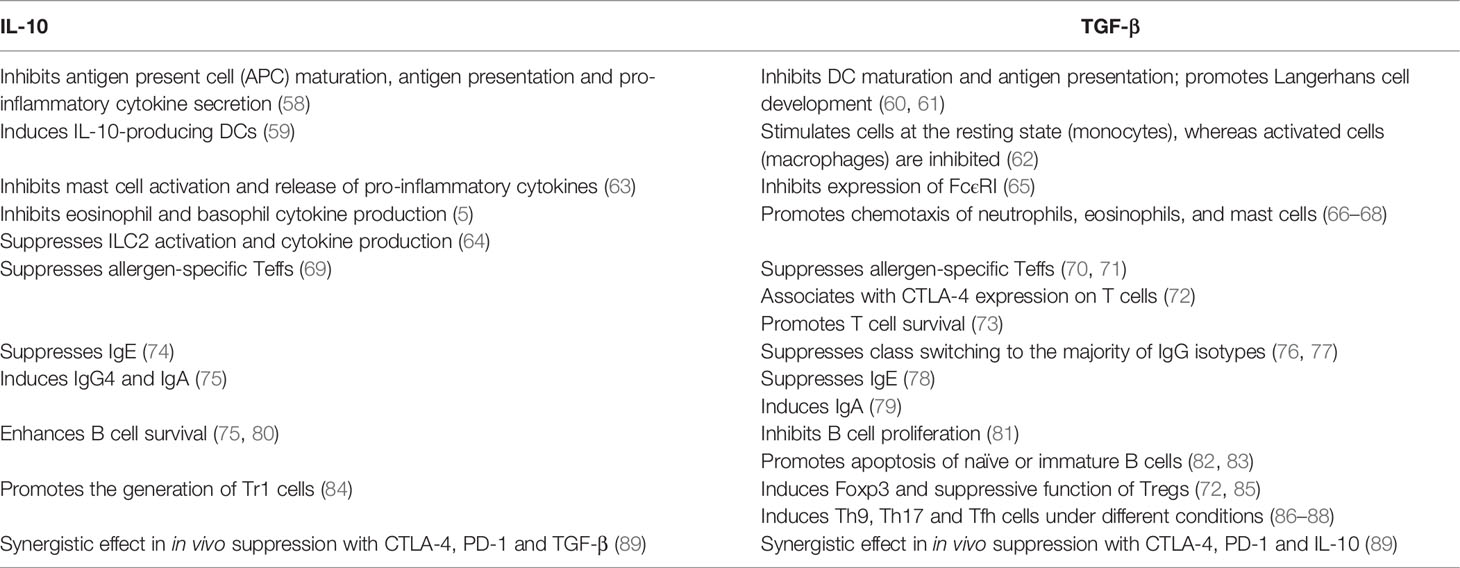
Table 1 Summarized functions of IL-10 and TGF-β in allergic asthma.
TGF-β is a pleiotropic cytokine and Tregs are the major source of its secretion. The implication of TGF-β in allergic asthma is complicated, and confronting effects are observed (38). It has been recognized that TGF-β produced by Tregs is indispensable for the in vivo pTreg expansion and immunomodulatory functionality in an autocrine manner, which is crucial for the induction of immune tolerance and design of alternative mucosal vaccination strategies. However, TGF-β has also been noted to increase mucus production, promote airway remodeling and fibrosis in asthmatic settings, which could be prevented by the anti-TGF-β therapy in allergen challenged animals (91). Therefore, further studies are required to elucidate the paradoxical role of TGF-β in the regulation of allergic asthma.
Airway remodeling refers to the pathological restructuring of the small and large airways in asthma, including neovascularization, subepithelial fibrosis, loss of epithelial integrity, goblet cell and mucus gland enlargement, and increased airway smooth muscle (ASM) mass (92). These pathophysiological changes result in alterations in the composition and structural organization of molecular and cellular components that constitute the airway. As a consequence, the asthmatic patients manifest the presence of airway narrowing and edema, AHR, and mucus hypersecretion, which is relevant to the poor clinical outcomes in asthmatic patients (93).
Airway remodeling is induced by factors from both inflammatory cells and structural cells. In general, the change of structural cells is under the influence of inflammatory cells (94). A variety of inflammatory cells presented in the asthmatic airways is able to produce mediators that have the potential to induce airway remodeling, such as cysteinyl leukotrienes (CysLTs), IL-13, endothelins, TGF-β, and epidermal growth factor (EGF). Vascular endothelial growth factor (VEGF) is an angiogenic factor, which has also been shown to induce airway remodeling and enhance Th2-mediated lung inflammation (95). Secretion of VEGF by cultured ASM cells is upregulated in response to the proinflammatory cytokines, TNF-α and IL-1β (96), or the Th2 cytokines, IL-13 and IL-4 (97). Therefore, the infiltrated immune cells are considered as “amplifiers” of airway remodeling (98). Indeed, sustained immune responses are key drivers in exacerbating the development of airway remodeling. For example, cytokines produced by the infiltrated Th2 cells, such as IL-4 and IL-13 enhance subepithelial fibrosis, mucous hyperplasia, and collagen deposition to promote airway remodeling (99–101). Similarly, alveolar macrophages contribute to airway remodeling through the release of matrix metalloproteinases to alter the extracellular matrix (ECM) and airway structure (102). Although the role of Th17 cells in airway remodeling remains controversial, a synergistic effect of DCs together with Th17 cytokines to promote the accumulation of fibrotic matrix components that correlate with TGF-β expression had been observed (103). It is noteworthy that all of above mentioned immune responses are relevant to the suppressive function of Tregs, and therefore, the role of Tregs in airway remodeling has recently been highly appreciated.
Indeed, data collected from animal studies characterized signaling molecules and transcription factors implicated in airway remodeling, which are closely related to Treg function (94, 104, 105). Specifically, transfer of CD4+CD25+ Tregs at the peak of acute inflammation before the onset of airway remodeling reversed established airway inflammation and prevented the development of airway remodeling (106), supporting a role of Tregs in the prevention of airway remodeling. Similarly, adoptive transfer of tTregs caused a substantial reduction in bronchoalveolar lavage eosinophil composition and suppressed airway remodeling and T cell migration into the lung of STAT6 and RAG2 double knockout mice, confirming the role of Tregs in repressing allergic airway inflammation and remodeling (107). There is also feasible evidence that Th17 responses in chronic allergic airway inflammation abrogate Treg-mediated tolerance, and thereby contributing to airway remodeling (108). More interestingly, adoptive transfer of Tregs into OVA-induced asthmatic mice at the chronic stage selectively reduced the vessel numbers in both peritracheal and peribronchial regions and the lung parenchyma (109), which indicate a potential role of Tregs in the regulation of structural cells such as endothelial cells, smooth muscle cells and mesenchymal cells during the development of airway remodeling in asthmatic settings other than immune cells. Therefore, the exact impact of Tregs on airway remodeling in asthmatic setting is worthy of further investigations.
Asthmatic patients generally manifest different levels of chronic airway inflammation with airway epithelial damage that occurs even in mild, early and nonfatal asthma (110, 111). Damage and shedding of airway epithelial cells are important pathological features of asthma, and altered epithelium in the airway is more susceptible to injury and apoptosis than those from non-asthmatic individuals (112). Specifically, epithelial cells derived from asthmatic patients collected by bronchial brushing seem to be more hyperreactive and less viable (113), which likely results from inflammatory damage. Furthermore, the airway of asthmatic patients is characterized by the dysregulation of airway epithelial repair, leading to a chronic cycle of wound repair coupled with bronchial remodeling (110).
Except for the immunosuppressive function and capacity to limit the intensity and sustained time of immune responses, Tregs also participate in non-immunological processes such as tissues repair during extensive inflammation. The presence of Tregs has been documented in several non-lymphoid tissues, including lung, skin, placenta, intestinal mucosa, adipose tissue, and atherosclerotic plaques (114). Tregs rapidly accumulate in the acutely injured skeletal muscle of mice. Ablation of Tregs impairs muscle repair due to decreased amphiregulin, an epidermal growth factor family member known to promote healing and tissue regeneration (115). Another study demonstrates that amphiregulin deficiency in Tregs results in severe acute lung damage and a rapid decline in lung function during influenza virus infection. In addition, anti-viral immune responses and suppressive function of Tregs are unaltered, suggesting these two functions are invoked by separable cues (116). Nevertheless, implication of Tregs in airway epithelial repair in the context of asthma has not yet been reported, which is necessary for further investigations.
Increasing clinical evidence supports that dysregulated Tregs play an important role in the pathogenesis and chronicity of allergic asthma. In patients with asthma and other allergic diseases, the expression of FOXP3 is reduced as compared to that of healthy controls (117). In atopic children, tTreg maturation is significantly delayed as compared to that of age-matched nonatopic children (118). Additionally, Tregs in patients with allergic asthma exhibit impaired suppressive function compared to those Tregs from healthy controls (11, 119).
Several subsets of dysfunctional Tregs are relevant to allergic asthma. Chemoattractant receptor-homologous molecule expressed on Th2 cells (CRTH2) is one of the functional prostaglandin D2 (PGD2) receptors, and regarded as a potent inducer of type 2 cytokine secretion (48). The allergic asthma patients have more CRTH2+ Tregs in the peripheral blood than healthy controls (120). These CRTH2+ Tregs produce greater amounts of IL-4 and show less suppressive function than that of CRTH2− Tregs in the PGD2-stimulated cultures (120). Another dysregulated Treg subpopulation is the ILT3 (also known as gp49B or CD85k)-expressing Tregs. Flow cytometry detected a substantially elevated percentage of ILT3+ Tregs in mice with massive asthma-like airway pathologies, which promoted the maturation of IRF4+PD-L2+ DCs to activate Th2 cells (121). Simultaneously, ILT3+ Tregs displayed a compromised suppressive function owing to low expression of FOXP3 and Helios (122). In mice, the expression of IL-33 receptor ST2 has been identified in Foxp3+ Tregs in the lung (123). In the presence of IL-33, Tregs display upregulated expression of canonical Th2 transcription factor GATA3 and IL-33 receptor ST2 along with enhanced secretion of type 2 cytokines (122). Furthermore, Tregs lose their ability to suppress Teffs once exposed to IL-33 (122). However, in vitro studies suggest that ST2+ Tregs are highly activated and superior to ST2- Tregs in suppressing CD4+ T cell proliferation through IL-10 and TGF-β release (124). Although those in vitro data may not mimic the in vivo situation, further investigations would be necessary to fully address this question. Pro-inflammatory cytokine-secreting Tregs such as IFN-γ-producing FOXP3+ cells, IL-4-producing FOXP3+ cells, and IL-17-producing FOXP3+ cells are also noticed in asthmatic patients, which are strongly correlated with the severity of asthma and might be insensitive to corticosteroids (125, 126).
It is generally accepted that Tregs are a promising candidate for developing effective therapies to treat immune disorders such as allergic asthma. Current preclinical studies and clinical trials of Treg-based therapies are mainly on the basis of two approaches: one of which is to directly boost Treg number and functionality in vivo, and the other is re-administration of purified, ex vivo modified, GMP (good manufacturing practice)-compliant Tregs back to patients (127).
Several approaches have been employed to promote the in vivo expansion of Tregs or depletion of Teffs, leading to an increased Treg/Teff ratio. These therapies include the administration of IL-2 or IL-2/anti-IL-2 mAb complex, anti-CD3 mAbs, mTOR inhibitors, and dietary or microbe-derived pro-tolerogenic stimulators (127). Although high dose of IL-2 has been used for immunotherapy against metastatic cancer (128), low-dose of IL-2, however, preferentially stimulates Tregs and has shown a great potential of success in Treg-based immunosuppressive strategies against autoimmune and inflammatory diseases (129). Advances in the knowledge of the functional, biophysical and structural characteristics of IL-2 have promoted the generation of IL-2 formulations, such as IL-2/anti-IL-2 mAb complexes (130). Depending on the clone of the anti-IL-2 mAb, IL-2/anti-IL-2 mAb complex exerts differential effect on the expansion of T cell subsets (131). Studies in mice revealed that IL-2/anti-IL-2 mAb complexed with S4B6 clone induces the preferential proliferation of CD8+ T cells, while the IL-2/JES6-1 complex preferentially induces the expansion of Tregs by blocking the interaction of IL-2 with IL-2Rβ (CD122) and IL-2Rγ (CD132), and promoting the interaction of IL-2 with IL-2Rα (CD25) (131, 132). Particularly, the IL-2/JES6-1 complexes have already manifested exciting results in terms of suppressing organ transplant rejection (133), autoimmune and inflammatory diseases in mice such as type 1 diabetes (134), dextran sodium sulfate-induced colitis (132), experimental myasthenia (135), collagen-induced arthritis (136), experimental autoimmune encephalomyelitis (133), and allergic airway disease (137). In the model of established airway allergy, treatment with IL-2/JES6-1 complex dampens eosinophilia and airway inflammation, and inhibits the production of eotaxin-1 and IL-5 (137). Mucus production, AHR to methacholine, and parenchymal tissue inflammation are also dramatically reduced following IL-2/anti-IL-2 mAb complex administration, which is dependent on Treg-derived IL-10 (137). Interestingly, administration of IL-2/JES6-1 complex also improves some manifestations of metabolic diseases, such as obesity related chronic inflammation and insulin resistance, which are characterized by the inflammatory infiltration of immune cells in the adipose tissues that are amenable to Treg modulation (138).
In neoplastic diseases, adoptive cell therapies (ACT) use T cells engineered to express either Ag-specific TCRs or chimeric Ag receptors (CARs) targeting specific tumor antigens to selectively eliminate target cells, which have been approved for the treatment of acute lymphoblastic leukemia and advanced lymphomas (127). In addition to killing cancerous cells, ACT can also be used to regain appropriate Treg function in the inflammatory context. Polyclonal expansion of Tregs via TCR represents the initial strategy for ACT. Unlike other type of Tregs, antigen-specific Tregs are more potent in controlling local inflammation and inhibiting T cell priming in secondary lymphoid tissues (139). More recently, a number of publications demonstrate the utility of CARs in Tregs (140). In this case, Tregs are reinfused after engineering with chimeric TCR of different types. CAR-Tregs have several characteristics versus TCR-Tregs: (1) non-MHC-restriction and less dependent on IL-2; (2) the hinge region provides flexibility, which enables CARs binding to antigen in various orientations; (3) higher antigen affinity than TCRs; and (4) more precise control of the type of antigen-stimulated response (141).
Current preclinical studies and clinical trials for Treg ACTs in inflammatory disorders have indicated the efficacy and technical feasibility of these methods (142, 143). In experimentally induced allergic asthma, CAR-redirected Tregs suppressed allergic airway inflammation, prevented excessive pulmonary mucus production, and attenuated the increase of allergen-specific IgE and Th2 cytokine levels (144). Over the past few decades, autoimmune involvement in the pathogenesis of asthma has been proposed due to the presence of circulating autoantibodies against diverse self-antigens/structures (145). Some patients with severe asthma have autoantibodies against eosinophil peroxidase (EPX) and autologous cellular components in the sputum, which may necessitate an increase for the maintenance of corticosteroids (146). These findings raise the potential of utilizing CAR-Treg ACT in severe and therapy-refractory asthmatics. However, many important issues such as managing the stability and plasticity of Tregs, directing their homing to the desired sites, and safety concerns are still waiting to be worked out.
Allergic asthma involves complex innate and adaptive immune responses to environmental allergens, resulting in airway inflammation predominately mediated by Th2-type cells and allergen-specific IgE (147). Both human and animal studies show that Tregs are essential for the maintenance of self-tolerance and immune homeostasis, and therefore, Tregs defects are observed in asthmatic individuals as compared to healthy controls in terms of their functionality. These discoveries promoted the development of technologies with Treg-based therapies, such as Treg expansion and CAR-Tregs, which may represent a viable approach for curative therapy of allergic diseases. Despite the current achievements, some critical issues, such as how to improve the safety of Tregs, increase the stability of Tregs, and direct their homing to the desired sites, are yet to be elucidated. As a result, additional in-depth studies are necessary to improve current therapeutic approaches against asthma in clinical settings.
JZ, YZ, and LC wrote the manuscript and prepared the figure. QX collected and analyzed the information. YW, MX, and XL reviewed the manuscript. JPZ and C-YW supervised the conception and writing of the manuscript. All authors contributed to the article and approved the submitted version.
Our study was supported by the National Natural Science Foundation of China (82130023, 81920108009, 82100892, 82070808, 81873656, 82100823, 82100931, 91749207, 81770823 and 81800068), Department of Science and Technology of Hubei Province Program project (2020DCD014), the Postdoctoral Science Foundation of China (54000-0106540081 and 54000-0106540080), Hubei Health Committee Program (WJ2021ZH0002), the Integrated Innovative Team for Major Human Diseases Program of Tongji Medical College, Huazhong University of Science and Technology, and the Innovative Funding for Translational Research from Tongji Hospital.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Komlosi ZI, van de Veen W, Kovacs N, Szucs G, Sokolowska M, O'Mahony L, et al. Cellular and Molecular Mechanisms of Allergic Asthma. Mol Aspects Med (2022) 85:100995. doi: 10.1016/j.mam.2021.100995
2. Chabra R, Gupta M. Allergic And Environmental Induced Asthma. Treasure Island (Fl: StatPearls (2022).
3. Tuazon JA, Kilburg-Basnyat B, Oldfield LM, Wiscovitch-Russo R, Dunigan-Russell K, Fedulov AV, et al. Emerging Insights Into the Impact of Air Pollution on Immune-Mediated Asthma Pathogenesis. Curr Allergy Asthma Rep (2022). doi: 10.1007/s11882-022-01034-1
4. Lin W, Truong N, Grossman WJ, Haribhai D, Williams CB, Wang J, et al. Allergic Dysregulation and Hyperimmunoglobulinemia E in Foxp3 Mutant Mice. J Allergy Clin Immunol (2005) 116:1106–15. doi: 10.1016/j.jaci.2005.08.046
5. Kearley J, Barker JE, Robinson DS, Lloyd CM. Resolution of Airway Inflammation and Hyperreactivity After In Vivo Transfer of CD4+CD25+ Regulatory T Cells is Interleukin 10 Dependent. J Exp Med (2005) 202:1539–47. doi: 10.1084/jem.20051166
6. Chiang CY, Chang JH, Chuang HC, Fan CK, Hou TY, Lin CL, et al. Schisandrin B Promotes Foxp3(+) Regulatory T Cell Expansion by Activating Heme Oxygenase-1 in Dendritic Cells and Exhibits Immunomodulatory Effects in Th2-Mediated Allergic Asthma. Eur J Pharmacol (2022) 918:174775. doi: 10.1016/j.ejphar.2022.174775
7. Nguyen QT, Kim D, Iamsawat S, Le HT, Kim S, Qiu KT, et al. Cutting Edge: Steroid Responsiveness in Foxp3(+) Regulatory T Cells Determines Steroid Sensitivity During Allergic Airway Inflammation in Mice. J Immunol (2021) 207:765–70. doi: 10.4049/jimmunol.2100251
8. Mamessier E, Nieves A, Lorec AM, Dupuy P, Pinot D, Pinet C, et al. T-Cell Activation During Exacerbations: A Longitudinal Study in Refractory Asthma. Allergy (2008) 63:1202–10. doi: 10.1111/j.1398-9995.2008.01687.x
9. Hartl D, Koller B, Mehlhorn AT, Reinhardt D, Nicolai T, Schendel DJ, et al. Quantitative and Functional Impairment of Pulmonary CD4+CD25hi Regulatory T Cells in Pediatric Asthma. J Allergy Clin Immunol (2007) 119:1258–66. doi: 10.1016/j.jaci.2007.02.023
10. Smyth LJ, Eustace A, Kolsum U, Blaikely J, Singh D. Increased Airway T Regulatory Cells in Asthmatic Subjects. Chest (2010) 138:905–12. doi: 10.1378/chest.09-3079
11. Thunberg S, Gafvelin G, Nord M, Gronneberg R, Grunewald J, Eklund A, et al. Allergen Provocation Increases TH2-Cytokines and FOXP3 Expression in the Asthmatic Lung. Allergy (2010) 65:311–8. doi: 10.1111/j.1398-9995.2009.02218.x
12. Birmingham JM, Chesnova B, Wisnivesky JP, Calatroni A, Federman J, Bunyavanich S, et al. The Effect of Age on T-Regulatory Cell Number and Function in Patients With Asthma. Allergy Asthma Immunol Res (2021) 13:646–54. doi: 10.4168/aair.2021.13.4.646
13. Kappen JH, Durham SR, Veen HI, Shamji MH. Applications and Mechanisms of Immunotherapy in Allergic Rhinitis and Asthma. Ther Adv Respir Dis (2017) 11:73–86. doi: 10.1177/1753465816669662
14. Palomares O, Akdis M, Martin-Fontecha M, Akdis CA. Mechanisms of Immune Regulation in Allergic Diseases: The Role of Regulatory T and B Cells. Immunol Rev (2017) 278:219–36. doi: 10.1111/imr.12555
15. Akdis M, Aab A, Altunbulakli C, Azkur K, Costa RA, Crameri R, et al. Interleukins (From IL-1 to IL-38), Interferons, Transforming Growth Factor Beta, and TNF-Alpha: Receptors, Functions, and Roles in Diseases. J Allergy Clin Immunol (2016) 138:984–1010. doi: 10.1016/j.jaci.2016.06.033
16. Agache I, Akdis CA. Endotypes of Allergic Diseases and Asthma: An Important Step in Building Blocks for the Future of Precision Medicine. Allergol Int (2016) 65:243–52. doi: 10.1016/j.alit.2016.04.011
17. Larche M, Akdis CA, Valenta R. Immunological Mechanisms of Allergen-Specific Immunotherapy. Nat Rev Immunol (2006) 6:761–71. doi: 10.1038/nri1934
18. Palomares O, Yaman G, Azkur AK, Akkoc T, Akdis M, Akdis CA. Role of Treg in Immune Regulation of Allergic Diseases. Eur J Immunol (2010) 40:1232–40. doi: 10.1002/eji.200940045
19. Hammad H, Lambrecht BN. Barrier Epithelial Cells and the Control of Type 2 Immunity. Immunity (2015) 43:29–40. doi: 10.1016/j.immuni.2015.07.007
20. Morita H, Moro K, Koyasu S. Innate Lymphoid Cells in Allergic and Nonallergic Inflammation. J Allergy Clin Immunol (2016) 138:1253–64. doi: 10.1016/j.jaci.2016.09.011
21. Ballantyne SJ, Barlow JL, Jolin HE, Nath P, Williams AS, Chung KF, et al. Blocking IL-25 Prevents Airway Hyperresponsiveness in Allergic Asthma. J Allergy Clin Immunol (2007) 120:1324–31. doi: 10.1016/j.jaci.2007.07.051
22. Guilleminault L, Conde E, Reber LL. Pharmacological Approaches to Target Type 2 Cytokines in Asthma. Pharmacol Ther (2022) 237:108167. doi: 10.1016/j.pharmthera.2022.108167
23. Braun H, Afonina IS, Mueller C, Beyaert R. Dichotomous Function of IL-33 in Health and Disease: From Biology to Clinical Implications. Biochem Pharmacol (2018) 148:238–52. doi: 10.1016/j.bcp.2018.01.010
24. Halim TY, Steer CA, Matha L, Gold MJ, Martinez-Gonzalez I, McNagny KM, et al. Group 2 Innate Lymphoid Cells are Critical for the Initiation of Adaptive T Helper 2 Cell-Mediated Allergic Lung Inflammation. Immunity (2014) 40:425–35. doi: 10.1016/j.immuni.2014.01.011
25. Kondo Y, Yoshimoto T, Yasuda K, Futatsugi-Yumikura S, Morimoto M, Hayashi N, et al. Administration of IL-33 Induces Airway Hyperresponsiveness and Goblet Cell Hyperplasia in the Lungs in the Absence of Adaptive Immune System. Int Immunol (2008) 20:791–800. doi: 10.1093/intimm/dxn037
26. Kurowska-Stolarska M, Stolarski B, Kewin P, Murphy G, Corrigan CJ, Ying S, et al. IL-33 Amplifies the Polarization of Alternatively Activated Macrophages That Contribute to Airway Inflammation. J Immunol (2009) 183:6469–77. doi: 10.4049/jimmunol.0901575
27. Besnard AG, Togbe D, Guillou N, Erard F, Quesniaux V, Ryffel B. IL-33-Activated Dendritic Cells are Critical for Allergic Airway Inflammation. Eur J Immunol (2011) 41:1675–86. doi: 10.1002/eji.201041033
28. Smith SG, Chen R, Kjarsgaard M, Huang C, Oliveria JP, O'Byrne PM, et al. Increased Numbers of Activated Group 2 Innate Lymphoid Cells in the Airways of Patients With Severe Asthma and Persistent Airway Eosinophilia. J Allergy Clin Immunol (2016) 137:75–86 e78. doi: 10.1016/j.jaci.2015.05.037
29. Khan MA. Regulatory T Cells Mediated Immunomodulation During Asthma: A Therapeutic Standpoint. J Transl Med (2020) 18:456. doi: 10.1186/s12967-020-02632-1
30. Jin W, Dong C. IL-17 Cytokines in Immunity and Inflammation. Emerg Microbes Infect (2013) 2:e60. doi: 10.1038/emi.2013.58
31. Moretti S, Renga G, Oikonomou V, Galosi C, Pariano M, Iannitti RG, et al. A Mast Cell-ILC2-Th9 Pathway Promotes Lung Inflammation in Cystic Fibrosis. Nat Commun (2017) 8:14017. doi: 10.1038/ncomms14017
32. Sehra S, Yao W, Nguyen ET, Glosson-Byers NL, Akhtar N, Zhou B, et al. TH9 Cells are Required for Tissue Mast Cell Accumulation During Allergic Inflammation. J Allergy Clin Immunol (2015) 136:433–40 e431. doi: 10.1016/j.jaci.2015.01.021
33. Huang M, Dong J. Critical Roles of Balanced T Helper 9 Cells and Regulatory T Cells in Allergic Airway Inflammation and Tumor Immunity. J Immunol Res (2021) 2021:8816055. doi: 10.1155/2021/8816055
34. Leyva-Castillo JM, Yoon J, Geha RS. IL-22 Promotes Allergic Airway Inflammation in Epicutaneously Sensitized Mice. J Allergy Clin Immunol (2019) 143:619–30.e617. doi: 10.1016/j.jaci.2018.05.032
35. Czarnowicki T, Gonzalez J, Shemer A, Malajian D, Xu H, Zheng X, et al. Severe Atopic Dermatitis is Characterized by Selective Expansion of Circulating TH2/TC2 and TH22/TC22, But Not TH17/TC17, Cells Within the Skin-Homing T-Cell Population. J Allergy Clin Immunol (2015) 136:104–15.e107. doi: 10.1016/j.jaci.2015.01.020
36. Li J, Sha J, Sun L, Zhu D, Meng C. Contribution of Regulatory T Cell Methylation Modifications to the Pathogenesis of Allergic Airway Diseases. J Immunol Res (2021) 2021:5590217. doi: 10.1155/2021/5590217
37. Akdis CA, Akdis M. Mechanisms and Treatment of Allergic Disease in the Big Picture of Regulatory T Cells. J Allergy Clin Immunol (2009) 123:735–46. doi: 10.1016/j.jaci.2009.02.030
38. Palomares O, Martin-Fontecha M, Lauener R, Traidl-Hoffmann C, Cavkaytar O, Akdis M, et al. Regulatory T Cells and Immune Regulation of Allergic Diseases: Roles of IL-10 and TGF-Beta. Genes Immun (2014) 15:511–20. doi: 10.1038/gene.2014.45
39. Ray A, Khare A, Krishnamoorthy N, Qi Z, Ray P. Regulatory T Cells in Many Flavors Control Asthma. Mucosal Immunol (2010) 3:216–29. doi: 10.1038/mi.2010.4
40. Azevedo CT, Cotias AC, Arantes ACS, Ferreira TPT, Martins MA, Olsen PC. Assessment of Allergen-Responsive Regulatory T Cells in Experimental Asthma Induced in Different Mouse Strains. Mediators Inflamm (2021) 2021:7584483. doi: 10.1155/2021/7584483
41. Fontenot JD, Dooley JL, Farr AG, Rudensky AY. Developmental Regulation of Foxp3 Expression During Ontogeny. J Exp Med (2005) 202:901–6. doi: 10.1084/jem.20050784
42. Zhang J, Chen L, Xiong F, Zhang S, Huang K, Zhang Z, et al. Autophagy in Regulatory T Cells: A Double-Edged Sword in Disease Settings. Mol Immunol (2019) 109:43–50. doi: 10.1016/j.molimm.2019.02.004
43. Noval Rivas M, Chatila TA. Regulatory T Cells in Allergic Diseases. J Allergy Clin Immunol (2016) 138:639–52. doi: 10.1016/j.jaci.2016.06.003
44. Sun CM, Hall JA, Blank RB, Bouladoux N, Oukka M, Mora JR, et al. Small Intestine Lamina Propria Dendritic Cells Promote De Novo Generation of Foxp3 T Reg Cells via Retinoic Acid. J Exp Med (2007) 204:1775–85. doi: 10.1084/jem.20070602
45. Siddiqui KR, Powrie F. CD103+ GALT DCs Promote Foxp3+ Regulatory T Cells. Mucosal Immunol (2008) 1 (Suppl 1):S34–38. doi: 10.1038/mi.2008.43
46. Lathrop SK, Bloom SM, Rao SM, Nutsch K, Lio CW, Santacruz N, et al. Peripheral Education of the Immune System by Colonic Commensal Microbiota. Nature (2011) 478:250–4. doi: 10.1038/nature10434
47. Soroosh P, Doherty TA, Duan W, Mehta AK, Choi H, Adams YF, et al. Lung-Resident Tissue Macrophages Generate Foxp3+ Regulatory T Cells and Promote Airway Tolerance. J Exp Med (2013) 210:775–88. doi: 10.1084/jem.20121849
48. Boonpiyathad T, Sozener ZC, Akdis M, Akdis CA. The Role of Treg Cell Subsets in Allergic Disease. Asian Pac J Allergy Immunol (2020) 38:139–49. doi: 10.12932/AP-030220-0754
49. Bilate AM, Lafaille JJ. Induced CD4+Foxp3+ Regulatory T Cells in Immune Tolerance. Annu Rev Immunol (2012) 30:733–58. doi: 10.1146/annurev-immunol-020711-075043
50. Lan F, Zhang N, Bachert C, Zhang L. Stability of Regulatory T Cells in T Helper 2-Biased Allergic Airway Diseases. Allergy (2020) 75:1918–26. doi: 10.1111/all.14257
51. Josefowicz SZ, Lu LF, Rudensky AY. Regulatory T Cells: Mechanisms of Differentiation and Function. Annu Rev Immunol (2012) 30:531–64. doi: 10.1146/annurev.immunol.25.022106.141623
52. Huang H, Ma Y, Dawicki W, Zhang X, Gordon JR. Comparison of Induced Versus Natural Regulatory T Cells of the Same TCR Specificity for Induction of Tolerance to an Environmental Antigen. J Immunol (2013) 191:1136–43. doi: 10.4049/jimmunol.1201899
53. Joetham A, Schedel M, O'Connor BP, Kim S, Takeda K, Abbott J, et al. Inducible and Naturally Occurring Regulatory T Cells Enhance Lung Allergic Responses Through Divergent Transcriptional Pathways. J Allergy Clin Immunol (2017) 139:1331–42. doi: 10.1016/j.jaci.2016.06.051
54. Boonpiyathad T, Satitsuksanoa P, Akdis M, Akdis CA. Il-10 Producing T and B Cells in Allergy. Semin Immunol (2019) 44:101326. doi: 10.1016/j.smim.2019.101326
55. Meiler F, Zumkehr J, Klunker S, Ruckert B, Akdis CA, Akdis M. In Vivo Switch to IL-10-Secreting T Regulatory Cells in High Dose Allergen Exposure. J Exp Med (2008) 205:2887–98. doi: 10.1084/jem.20080193
56. Siegmund K, Ruckert B, Ouaked N, Burgler S, Speiser A, Akdis CA, et al. Unique Phenotype of Human Tonsillar and In Vitro-Induced FOXP3+CD8+ T Cells. J Immunol (2009) 182:2124–30. doi: 10.4049/jimmunol.0802271
57. Novak N, Mete N, Bussmann C, Maintz L, Bieber T, Akdis M, et al. Early Suppression of Basophil Activation During Allergen-Specific Immunotherapy by Histamine Receptor 2. J Allergy Clin Immunol (2012) 130:1153–58.e1152. doi: 10.1016/j.jaci.2012.04.039
58. Moore KW, de Waal Malefyt R, Coffman RL, O'Garra A. Interleukin-10 and the Interleukin-10 Receptor. Annu Rev Immunol (2001) 19:683–765. doi: 10.1146/annurev.immunol.19.1.683
59. Morante-Palacios O, Fondelli F, Ballestar E, Martinez-Caceres EM. Tolerogenic Dendritic Cells in Autoimmunity and Inflammatory Diseases. Trends Immunol (2021) 42:59–75. doi: 10.1016/j.it.2020.11.001
60. Strobl H, Knapp W. TGF-Beta1 Regulation of Dendritic Cells. Microbes Infect (1999) 1:1283–90. doi: 10.1016/s1286-4579(99)00256-7
61. Borkowski TA, Letterio JJ, Farr AG, Udey MC. A Role for Endogenous Transforming Growth Factor Beta 1 in Langerhans Cell Biology: The Skin of Transforming Growth Factor Beta 1 Null Mice is Devoid of Epidermal Langerhans Cells. J Exp Med (1996) 184:2417–22. doi: 10.1084/jem.184.6.2417
62. Ashcroft GS. Bidirectional Regulation of Macrophage Function by TGF-Beta. Microbes Infect (1999) 1:1275–82. doi: 10.1016/s1286-4579(99)00257-9
63. Nagata K, Nishiyama C. IL-10 in Mast Cell-Mediated Immune Responses: Anti-Inflammatory and Proinflammatory Roles. Int J Mol Sci (2021) 22(9):4972. doi: 10.3390/ijms22094972
64. Bonne-Annee S, Bush MC, Nutman TB. Differential Modulation of Human Innate Lymphoid Cell (ILC) Subsets by IL-10 and TGF-Beta. Sci Rep (2019) 9:14305. doi: 10.1038/s41598-019-50308-8
65. Gomez G, Ramirez CD, Rivera J, Patel M, Norozian F, Wright HV, et al. TGF-Beta 1 Inhibits Mast Cell Fc Epsilon RI Expression. J Immunol (2005) 174:5987–93. doi: 10.4049/jimmunol.174.10.5987
66. Brandes ME, Mai UE, Ohura K, Wahl SM. Type I Transforming Growth Factor-Beta Receptors on Neutrophils Mediate Chemotaxis to Transforming Growth Factor-Beta. J Immunol (1991) 147:1600–6.
67. Luttmann W, Franz P, Matthys H, Virchow JC Jr. Effects of TGF-Beta on Eosinophil Chemotaxis. Scand J Immunol (1998) 47:127–30. doi: 10.1046/j.1365-3083.1998.00298.x
68. Olsson N, Piek E, ten Dijke P, Nilsson G. Human Mast Cell Migration in Response to Members of the Transforming Growth Factor-Beta Family. J Leukoc Biol (2000) 67:350–6. doi: 10.1002/jlb.67.3.350
69. Coomes SM, Kannan Y, Pelly VS, Entwistle LJ, Guidi R, Perez-Lloret J, et al. CD4(+) Th2 Cells are Directly Regulated by IL-10 During Allergic Airway Inflammation. Mucosal Immunol (2017) 10:150–61. doi: 10.1038/mi.2016.47
70. Gorelik L, Fields PE, Flavell RA. Cutting Edge: TGF-Beta Inhibits Th Type 2 Development Through Inhibition of GATA-3 Expression. J Immunol (2000) 165:4773–7. doi: 10.4049/jimmunol.165.9.4773
71. Heath VL, Murphy EE, Crain C, Tomlinson MG, O'Garra A. TGF-Beta1 Down-Regulates Th2 Development and Results in Decreased IL-4-Induced STAT6 Activation and GATA-3 Expression. Eur J Immunol (2000) 30:2639–49. doi: 10.1002/1521-4141(200009)30:9<2639::AID-IMMU2639>3.0.CO;2-7
72. Chen W, Jin W, Hardegen N, Lei KJ, Li L, Marinos N, et al. Conversion of Peripheral CD4+CD25- Naive T Cells to CD4+CD25+ Regulatory T Cells by TGF-Beta Induction of Transcription Factor Foxp3. J Exp Med (2003) 198:1875–86. doi: 10.1084/jem.20030152
73. Ouyang W, Oh SA, Ma Q, Bivona MR, Zhu J, Li MO. TGF-Beta Cytokine Signaling Promotes CD8+ T Cell Development and Low-Affinity CD4+ T Cell Homeostasis by Regulation of Interleukin-7 Receptor Alpha Expression. Immunity (2013) 39:335–46. doi: 10.1016/j.immuni.2013.07.016
74. Jeannin P, Lecoanet S, Delneste Y, Gauchat JF, Bonnefoy JY. IgE Versus IgG4 Production can be Differentially Regulated by IL-10. J Immunol (1998) 160:3555–61.
75. Rousset F, Garcia E, Defrance T, Peronne C, Vezzio N, Hsu DH, et al. Interleukin 10 is a Potent Growth and Differentiation Factor for Activated Human B Lymphocytes. Proc Natl Acad Sci U S A (1992) 89:1890–3. doi: 10.1073/pnas.89.5.1890
76. Kehrl JH, Roberts AB, Wakefield LM, Jakowlew S, Sporn MB, Fauci AS. Transforming Growth Factor Beta is an Important Immunomodulatory Protein for Human B Lymphocytes. J Immunol (1986) 137:3855–60.
77. Kehrl JH, Thevenin C, Rieckmann P, Fauci AS. Transforming Growth Factor-Beta Suppresses Human B Lymphocyte Ig Production by Inhibiting Synthesis and the Switch From the Membrane Form to the Secreted Form of Ig mRNA. J Immunol (1991) 146:4016–23.
78. Snapper CM, Waegell W, Beernink H, Dasch JR. Transforming Growth Factor-Beta 1 is Required for Secretion of IgG of All Subclasses by LPS-Activated Murine B Cells In Vitro. J Immunol (1993) 151:4625–36.
79. Cazac BB, Roes J. TGF-Beta Receptor Controls B Cell Responsiveness and Induction of IgA In Vivo. Immunity (2000) 13:443–51. doi: 10.1016/s1074-7613(00)00044-3
80. Chung F. Anti-Inflammatory Cytokines in Asthma and Allergy: Interleukin-10, Interleukin-12, Interferon-Gamma. Mediators Inflamm (2001) 10:51–9. doi: 10.1080/09629350120054518
81. Petit-Koskas E, Genot E, Lawrence D, Kolb JP. Inhibition of the Proliferative Response of Human B Lymphocytes to B Cell Growth Factor by Transforming Growth Factor-Beta. Eur J Immunol (1988) 18:111–6. doi: 10.1002/eji.1830180117
82. Kee BL, Rivera RR, Murre C. Id3 Inhibits B Lymphocyte Progenitor Growth and Survival in Response to TGF-Beta. Nat Immunol (2001) 2:242–7. doi: 10.1038/85303
83. Warner GL, Ludlow JW, Nelson DA, Gaur A, Scott DW. Anti-Immunoglobulin Treatment of Murine B-Cell Lymphomas Induces Active Transforming Growth Factor Beta But pRB Hypophosphorylation is Transforming Growth Factor Beta Independent. Cell Growth Differ (1992) 3:175–81.
84. Gregori S, Tomasoni D, Pacciani V, Scirpoli M, Battaglia M, Magnani CF, et al. Differentiation of Type 1 T Regulatory Cells (Tr1) by Tolerogenic DC-10 Requires the IL-10-Dependent ILT4/HLA-G Pathway. Blood (2010) 116:935–44. doi: 10.1182/blood-2009-07-234872
85. Ouyang W, Beckett O, Ma Q, Li MO. Transforming Growth Factor-Beta Signaling Curbs Thymic Negative Selection Promoting Regulatory T Cell Development. Immunity (2010) 32:642–53. doi: 10.1016/j.immuni.2010.04.012
86. Veldhoen M, Hocking RJ, Atkins CJ, Locksley RM, Stockinger B. TGFbeta in the Context of an Inflammatory Cytokine Milieu Supports De Novo Differentiation of IL-17-Producing T Cells. Immunity (2006) 24:179–89. doi: 10.1016/j.immuni.2006.01.001
87. Dardalhon V, Awasthi A, Kwon H, Galileos G, Gao W, Sobel RA, et al. IL-4 Inhibits TGF-Beta-Induced Foxp3+ T Cells and, Together With TGF-Beta, Generates IL-9+ IL-10+ Foxp3(-) Effector T Cells. Nat Immunol (2008) 9:1347–55. doi: 10.1038/ni.1677
88. Schmitt N, Liu Y, Bentebibel SE, Munagala I, Bourdery L, Venuprasad K, et al. The Cytokine TGF-Beta Co-Opts Signaling via STAT3-STAT4 to Promote the Differentiation of Human TFH Cells. Nat Immunol (2014) 15:856–65. doi: 10.1038/ni.2947
89. Akdis CA, Akdis M. Mechanisms of Immune Tolerance to Allergens: Role of IL-10 and Tregs. J Clin Invest (2014) 124:4678–80. doi: 10.1172/JCI78891
90. Ouyang W, O'Garra A. IL-10 Family Cytokines IL-10 and IL-22: From Basic Science to Clinical Translation. Immunity (2019) 50:871–91. doi: 10.1016/j.immuni.2019.03.020
91. McMillan SJ, Xanthou G, Lloyd CM. Manipulation of Allergen-Induced Airway Remodeling by Treatment With Anti-TGF-Beta Antibody: Effect on the Smad Signaling Pathway. J Immunol (2005) 174:5774–80. doi: 10.4049/jimmunol.174.9.5774
92. Bergeron C, Tulic MK, Hamid Q. Airway Remodelling in Asthma: From Benchside to Clinical Practice. Can Respir J (2010) 17:e85–93. doi: 10.1155/2010/318029
93. Al Heialy S, Ramakrishnan RK, Hamid Q. Recent Advances in the Immunopathogenesis of Severe Asthma. J Allergy Clin Immunol (2022) 149:455–65. doi: 10.1016/j.jaci.2021.12.765
94. Fixman ED, Stewart A, Martin JG. Basic Mechanisms of Development of Airway Structural Changes in Asthma. Eur Respir J (2007) 29:379–89. doi: 10.1183/09031936.00053506
95. Lee CG, Link H, Baluk P, Homer RJ, Chapoval S, Bhandari V, et al. Vascular Endothelial Growth Factor (VEGF) Induces Remodeling and Enhances TH2-Mediated Sensitization and Inflammation in the Lung. Nat Med (2004) 10:1095–103. doi: 10.1038/nm1105
96. Alagappan VK, McKay S, Widyastuti A, Garrelds IM, Bogers AJ, Hoogsteden HC, et al. Proinflammatory Cytokines Upregulate mRNA Expression and Secretion of Vascular Endothelial Growth Factor in Cultured Human Airway Smooth Muscle Cells. Cell Biochem Biophys (2005) 43:119–29. doi: 10.1385/CBB:43:1:119
97. Faffe DS, Flynt L, Bourgeois K, Panettieri RA Jr, Shore SA. Interleukin-13 and Interleukin-4 Induce Vascular Endothelial Growth Factor Release From Airway Smooth Muscle Cells: Role of Vascular Endothelial Growth Factor Genotype. Am J Respir Cell Mol Biol (2006) 34:213–8. doi: 10.1165/rcmb.2005-0147OC
98. Hough KP, Curtiss ML, Blain TJ, Liu RM, Trevor J, Deshane JS, et al. Airway Remodeling in Asthma. Front Med (Lausanne) (2020) 7:191. doi: 10.3389/fmed.2020.00191
99. Wen FQ, Liu XD, Terasaki Y, Fang QH, Kobayashi T, Abe S, et al. Interferon-Gamma Reduces Interleukin-4- and Interleukin-13-Augmented Transforming Growth Factor-Beta2 Production in Human Bronchial Epithelial Cells by Targeting Smads. Chest (2003) 123:372S–3S. doi: 10.1016/S0012-3692(15)35217-X
100. Oeser K, Schwartz C, Voehringer D. Conditional IL-4/IL-13-Deficient Mice Reveal a Critical Role of Innate Immune Cells for Protective Immunity Against Gastrointestinal Helminths. Mucosal Immunol (2015) 8:672–82. doi: 10.1038/mi.2014.101
101. Tukler Henriksson J, Coursey TG, Corry DB, De Paiva CS, Pflugfelder SC. IL-13 Stimulates Proliferation and Expression of Mucin and Immunomodulatory Genes in Cultured Conjunctival Goblet Cells. Invest Ophthalmol Vis Sci (2015) 56:4186–97. doi: 10.1167/iovs.14-15496
102. Balhara J, Gounni AS. The Alveolar Macrophages in Asthma: A Double-Edged Sword. Mucosal Immunol (2012) 5:605–9. doi: 10.1038/mi.2012.74
103. Peters M, Kohler-Bachmann S, Lenz-Habijan T, Bufe A. Influence of an Allergen-Specific Th17 Response on Remodeling of the Airways. Am J Respir Cell Mol Biol (2016) 54:350–8. doi: 10.1165/rcmb.2014-0429OC
104. Balkrishna A, Solleti SK, Singh H, Verma S, Sharma N, Nain P, et al. Herbal Decoction Divya-Swasari-Kwath Attenuates Airway Inflammation and Remodeling Through Nrf-2 Mediated Antioxidant Lung Defence in Mouse Model of Allergic Asthma. Phytomedicine (2020) 78:153295. doi: 10.1016/j.phymed.2020.153295
105. Koopmans T, Crutzen S, Menzen MH, Halayko AJ, Hackett TL, Knight DA, et al. Selective Targeting of CREB-Binding Protein/Beta-Catenin Inhibits Growth of and Extracellular Matrix Remodelling by Airway Smooth Muscle. Br J Pharmacol (2016) 173:3327–41. doi: 10.1111/bph.13620
106. Kearley J, Robinson DS, Lloyd CM. CD4+CD25+ Regulatory T Cells Reverse Established Allergic Airway Inflammation and Prevent Airway Remodeling. J Allergy Clin Immunol (2008) 122:617–24.e616. doi: 10.1016/j.jaci.2008.05.048
107. Dorsey NJ, Chapoval SP, Smith EP, Skupsky J, Scott DW, Keegan AD. STAT6 Controls the Number of Regulatory T Cells In Vivo, Thereby Regulating Allergic Lung Inflammation. J Immunol (2013) 191:1517–28. doi: 10.4049/jimmunol.1300486
108. Zhao J, Lloyd CM, Noble A. Th17 Responses in Chronic Allergic Airway Inflammation Abrogate Regulatory T-Cell-Mediated Tolerance and Contribute to Airway Remodeling. Mucosal Immunol (2013) 6:335–46. doi: 10.1038/mi.2012.76
109. Huang MT, Dai YS, Chou YB, Juan YH, Wang CC, Chiang BL. Regulatory T Cells Negatively Regulate Neovasculature of Airway Remodeling via DLL4-Notch Signaling. J Immunol (2009) 183:4745–54. doi: 10.4049/jimmunol.0804371
110. Yang Y, Jia M, Ou Y, Adcock IM, Yao X. Mechanisms and Biomarkers of Airway Epithelial Cell Damage in Asthma: A Review. Clin Respir J (2021) 15:1027–45. doi: 10.1111/crj.13407
111. Faul JL, Tormey VJ, Leonard C, Burke CM, Farmer J, Horne SJ, et al. Lung Immunopathology in Cases of Sudden Asthma Death. Eur Respir J (1997) 10:301–7. doi: 10.1183/09031936.97.10020301
112. Davies DE, Wicks J, Powell RM, Puddicombe SM, Holgate ST. Airway Remodeling in Asthma: New Insights. J Allergy Clin Immunol (2003) 111:215–25. doi: 10.1067/mai.2003.128
113. Campbell A, Vignola A, Chanez P, Couret I, Michel FB, Bousquet J, et al. Functional Assessment of Viability of Epithelial Cells. Comparison of Viability and Mediator Release in Healthy Subjects and Asthmatics. Chest (1992) 101:25S–7S. doi: 10.1378/chest.101.3_Supplement.25S
114. Burzyn D, Benoist C, Mathis D. Regulatory T Cells in Nonlymphoid Tissues. Nat Immunol (2013) 14:1007–13. doi: 10.1038/ni.2683
115. Burzyn D, Kuswanto W, Kolodin D, Shadrach JL, Cerletti M, Jang Y, et al. A Special Population of Regulatory T Cells Potentiates Muscle Repair. Cell (2013) 155:1282–95. doi: 10.1016/j.cell.2013.10.054
116. Arpaia N, Green JA, Moltedo B, Arvey A, Hemmers S, Yuan S, et al. A Distinct Function of Regulatory T Cells in Tissue Protection. Cell (2015) 162:1078–89. doi: 10.1016/j.cell.2015.08.021
117. Marques CR, Costa RS, Costa GNO, da Silva TM, Teixeira TO, de Andrade EMM, et al. Genetic and Epigenetic Studies of FOXP3 in Asthma and Allergy. Asthma Res Pract (2015) 1:10. doi: 10.1186/s40733-015-0012-4
118. Tulic MK, Andrews D, Crook ML, Charles A, Tourigny MR, Moqbel R, et al. Changes in Thymic Regulatory T-Cell Maturation From Birth to Puberty: Differences in Atopic Children. J Allergy Clin Immunol (2012) 129:199–206.e191-194. doi: 10.1016/j.jaci.2011.10.016
119. Lin YL, Shieh CC, Wang JY. The Functional Insufficiency of Human CD4+CD25 High T-Regulatory Cells in Allergic Asthma is Subjected to TNF-Alpha Modulation. Allergy (2008) 63:67–74. doi: 10.1111/j.1398-9995.2007.01526.x
120. Boonpiyathad T, Capova G, Duchna HW, Croxford AL, Farine H, Dreher A, et al. Impact of High-Altitude Therapy on Type-2 Immune Responses in Asthma Patients. Allergy (2020) 75:84–94. doi: 10.1111/all.13967
121. Ulges A, Klein M, Reuter S, Gerlitzki B, Hoffmann M, Grebe N, et al. Protein Kinase CK2 Enables Regulatory T Cells to Suppress Excessive TH2 Responses In Vivo. Nat Immunol (2015) 16:267–75. doi: 10.1038/ni.3083
122. Boonpiyathad T, Sokolowska M, Morita H, Ruckert B, Kast JI, Wawrzyniak M, et al. Der P 1-Specific Regulatory T-Cell Response During House Dust Mite Allergen Immunotherapy. Allergy (2019) 74:976–85. doi: 10.1111/all.13684
123. Chen CC, Kobayashi T, Iijima K, Hsu FC, Kita H. IL-33 Dysregulates Regulatory T Cells and Impairs Established Immunologic Tolerance in the Lungs. J Allergy Clin Immunol (2017) 140:1351–63 e1357. doi: 10.1016/j.jaci.2017.01.015
124. Siede J, Frohlich A, Datsi A, Hegazy AN, Varga DV, Holecska V, et al. IL-33 Receptor-Expressing Regulatory T Cells Are Highly Activated, Th2 Biased and Suppress CD4 T Cell Proliferation Through IL-10 and TGFbeta Release. PLoS One (2016) 11:e0161507. doi: 10.1371/journal.pone.0161507
125. Xin L, Gao J, Ge X, Tian C, Ma W, Tian Z, et al. Increased Pro-Inflammatory Cytokine-Secreting Regulatory T Cells are Correlated With the Plasticity of T Helper Cell Differentiation and Reflect Disease Status in Asthma. Respir Med (2018) 143:129–38. doi: 10.1016/j.rmed.2018.09.007
126. Chen T, Hou X, Ni Y, Du W, Han H, Yu Y, et al. The Imbalance of FOXP3/GATA3 in Regulatory T Cells From the Peripheral Blood of Asthmatic Patients. J Immunol Res (2018) 2018:3096183. doi: 10.1155/2018/3096183
127. Roth-Walter F, Adcock IM, Benito-Villalvilla C, Bianchini R, Bjermer L, Boyman O, et al. Immune Modulation via T Regulatory Cell Enhancement: Disease-Modifying Therapies for Autoimmunity and Their Potential for Chronic Allergic and Inflammatory Diseases-An EAACI Position Paper of the Task Force on Immunopharmacology (TIPCO). Allergy (2021) 76:90–113. doi: 10.1111/all.14478
128. Rosenberg SA. IL-2: The First Effective Immunotherapy for Human Cancer. J Immunol (2014) 192:5451–8. doi: 10.4049/jimmunol.1490019
129. Klatzmann D, Abbas AK. The Promise of Low-Dose Interleukin-2 Therapy for Autoimmune and Inflammatory Diseases. Nat Rev Immunol (2015) 15:283–94. doi: 10.1038/nri3823
130. Arenas-Ramirez N, Woytschak J, Boyman O. Interleukin-2: Biology, Design and Application. Trends Immunol (2015) 36:763–77. doi: 10.1016/j.it.2015.10.003
131. Hong SW, O E, Lee JY, Yi J, Cho K, Kim J, et al. Interleukin-2/Antibody Complex Expanding Foxp3() Regulatory T Cells Exacerbates Th2-Mediated Allergic Airway Inflammation. BMB Rep (2019) 52:283–8. doi: 10.5483/BMBRep.2019.52.4.271
132. Spangler JB, Tomala J, Luca VC, Jude KM, Dong S, Ring AM, et al. Antibodies to Interleukin-2 Elicit Selective T Cell Subset Potentiation Through Distinct Conformational Mechanisms. Immunity (2015) 42:815–25. doi: 10.1016/j.immuni.2015.04.015
133. Webster KE, Walters S, Kohler RE, Mrkvan T, Boyman O, Surh CD, et al. In Vivo Expansion of T Reg Cells With IL-2-mAb Complexes: Induction of Resistance to EAE and Long-Term Acceptance of Islet Allografts Without Immunosuppression. J Exp Med (2009) 206:751–60. doi: 10.1084/jem.20082824
134. Tang Q, Adams JY, Penaranda C, Melli K, Piaggio E, Sgouroudis E, et al. Central Role of Defective Interleukin-2 Production in the Triggering of Islet Autoimmune Destruction. Immunity (2008) 28:687–97. doi: 10.1016/j.immuni.2008.03.016
135. Liu R, Zhou Q, La Cava A, Campagnolo DI, Van Kaer L, Shi FD. Expansion of Regulatory T Cells via IL-2/Anti-IL-2 mAb Complexes Suppresses Experimental Myasthenia. Eur J Immunol (2010) 40:1577–89. doi: 10.1002/eji.200939792
136. Lee SY, Cho ML, Oh HJ, Ryu JG, Park MJ, Jhun JY, et al. Interleukin-2/Anti-Interleukin-2 Monoclonal Antibody Immune Complex Suppresses Collagen-Induced Arthritis in Mice by Fortifying Interleukin-2/STAT5 Signalling Pathways. Immunology (2012) 137:305–16. doi: 10.1111/imm.12008
137. Wilson MS, Pesce JT, Ramalingam TR, Thompson RW, Cheever A, Wynn TA. Suppression of Murine Allergic Airway Disease by IL-2:Anti-IL-2 Monoclonal Antibody-Induced Regulatory T Cells. J Immunol (2008) 181:6942–54. doi: 10.4049/jimmunol.181.10.6942
138. Feuerer M, Herrero L, Cipolletta D, Naaz A, Wong J, Nayer A, et al. Lean, But Not Obese, Fat is Enriched for a Unique Population of Regulatory T Cells That Affect Metabolic Parameters. Nat Med (2009) 15:930–9. doi: 10.1038/nm.2002
139. Tang Q, Henriksen KJ, Bi M, Finger EB, Szot G, Ye J, et al. In Vitro-Expanded Antigen-Specific Regulatory T Cells Suppress Autoimmune Diabetes. J Exp Med (2004) 199:1455–65. doi: 10.1084/jem.20040139
140. Dawson NAJ, Levings MK. Antigen-Specific Regulatory T Cells: Are Police CARs the Answer? Transl Res (2017) 187:53–8. doi: 10.1016/j.trsl.2017.06.009
141. Dawson NAJ, Vent-Schmidt J, Levings MK. Engineered Tolerance: Tailoring Development, Function, and Antigen-Specificity of Regulatory T Cells. Front Immunol (2017) 8:1460. doi: 10.3389/fimmu.2017.01460
142. Biswas M, Kumar SRP, Terhorst C, Herzog RW. Gene Therapy With Regulatory T Cells: A Beneficial Alliance. Front Immunol (2018) 9:554. doi: 10.3389/fimmu.2018.00554
143. Ferreira LMR, Muller YD, Bluestone JA, Tang Q. Next-Generation Regulatory T Cell Therapy. Nat Rev Drug Discov (2019) 18:749–69. doi: 10.1038/s41573-019-0041-4
144. Skuljec J, Chmielewski M, Happle C, Habener A, Busse M, Abken H, et al. Chimeric Antigen Receptor-Redirected Regulatory T Cells Suppress Experimental Allergic Airway Inflammation, a Model of Asthma. Front Immunol (2017) 8:1125. doi: 10.3389/fimmu.2017.01125
145. Mukherjee M, Nair P. Autoimmune Responses in Severe Asthma. Allergy Asthma Immunol Res (2018) 10:428–47. doi: 10.4168/aair.2018.10.5.428
146. Mukherjee M, Bulir DC, Radford K, Kjarsgaard M, Huang CM, Jacobsen EA, et al. Sputum Autoantibodies in Patients With Severe Eosinophilic Asthma. J Allergy Clin Immunol (2018) 141:1269–79. doi: 10.1016/j.jaci.2017.06.033
Keywords: regulatory T cells, allergic airway inflammation, asthma, airway epithelial repair, therapeutic strategies
Citation: Zhang J, Zou Y, Chen L, Xu Q, Wang Y, Xie M, Liu X, Zhao J and Wang C-Y (2022) Regulatory T Cells, a Viable Target Against Airway Allergic Inflammatory Responses in Asthma. Front. Immunol. 13:902318. doi: 10.3389/fimmu.2022.902318
Received: 23 March 2022; Accepted: 13 May 2022;
Published: 10 June 2022.
Edited by:
Marco Romano, King’s College London, United KingdomReviewed by:
Jelena Skuljec, Essen University Hospital, GermanyCopyright © 2022 Zhang, Zou, Chen, Xu, Wang, Xie, Liu, Zhao and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Cong-Yi Wang, d2FuZ2N5QHRqaC50am11LmVkdS5jbg==; Jianping Zhao, emhhb2pwODhAMTI2LmNvbQ==
†These authors contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.