 Jin Wang1,2†
Jin Wang1,2† Jiehua Zhang3†
Jiehua Zhang3† Yingze Ye1,2
Yingze Ye1,2 Qingxue Xu1,2Yina Li1,2Shi Feng1,4
Qingxue Xu1,2Yina Li1,2Shi Feng1,4 Xiaoxing Xiong1,4
Xiaoxing Xiong1,4 Zhihong Jian4*
Zhihong Jian4* Lijuan Gu1,2*
Lijuan Gu1,2*- 1Central Laboratory, Renmin Hospital of Wuhan University, Wuhan, China
- 2Department of Anesthesia, Renmin Hospital of Wuhan University, Wuhan, China
- 3Department of Stomatology, Renmin Hospital of Wuhan University, Wuhan, China
- 4Department of Neurosurgery, Renmin Hospital of Wuhan University, Wuhan, China
Stroke is a disease with high incidence, mortality and disability rates. It is also the main cause of adult disability in developed countries. Stroke is often caused by small emboli on the inner wall of the blood vessels supplying the brain, which can lead to arterial embolism, and can also be caused by cerebrovascular or thrombotic bleeding. With the exception of recombinant tissue plasminogen activator (rt-PA), which is a thrombolytic drug used to recanalize the occluded artery, most treatments have been demonstrated to be ineffective. Stroke can also induce peripheral organ damage. Most stroke patients have different degrees of injury to one or more organs, including the lung, heart, kidney, spleen, gastrointestinal tract and so on. In the acute phase of stroke, severe inflammation occurs in the brain, but there is strong immunosuppression in the peripheral organs, which greatly increases the risk of peripheral organ infection and aggravates organ damage. Nonneurological complications of stroke can affect treatment and prognosis, may cause serious short-term and long-term consequences and are associated with prolonged hospitalization and increased mortality. Many of these complications are preventable, and their adverse effects can be effectively mitigated by early detection and appropriate treatment with various medical measures. This article reviews the pathophysiological mechanism, clinical manifestations and treatment of peripheral organ injury after stroke.
1 Introduction
Stroke refers to cerebrovascular damage and focal or widespread brain tissue damage due to a variety of causes, including ischemic stroke and hemorrhagic stroke. Stroke involves brain cell and tissue necrosis and has obvious seasonality, especially during the cold season. Although there are an increasing number of in-depth studies on stroke, few methods can be used to treat stroke (1).
Stroke has the second highest death rate worldwide. Although the mortality rate of stroke has decreased significantly in various countries due to the continuous development of medical technology, the incidence of stroke is on the rise (2). According to incomplete statistics, as of, 2016, there were 67.6 million people suffering from ischemic stroke and 15.3 million people suffering from hemorrhagic stroke. From 2006 to, 2016, the prevalence of ischemic stroke increased by 2.7%, while the prevalence of hemorrhagic stroke decreased by 1.7% (3).
The pathophysiological mechanism of stroke is complex, and brain damage usually affects the normal function of peripheral organs and can even cause serious damage (4). At the same time, injury to peripheral organs after stroke often aggravates brain injury and affects patient recovery (Figure 1). With a focus on pathophysiology, this review discusses the pathophysiological mechanism underlying peripheral organ injury after stroke and closely links the brain with peripheral organs to identify a more effective and comprehensive method for the treatment of stroke.
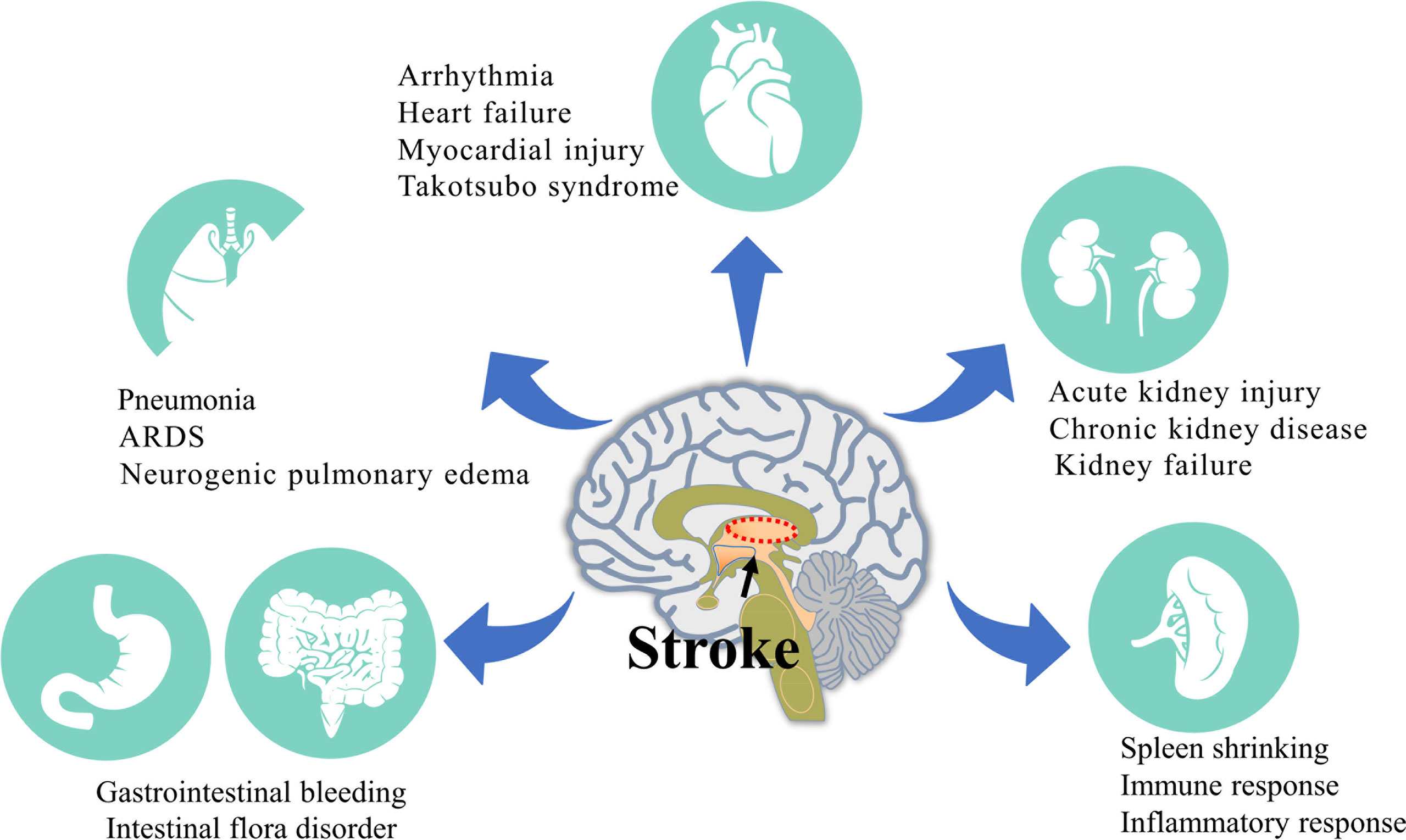
Figure 1 Injury of various peripheral organs after stroke. The activation of sympathetic nerve, hypothalamus-pituitary-adrenal axis and immune system after stroke leads to a series of systemic events and finally leads to the injury of various peripheral organs. The most common peripheral injuries include the Lung, Heart, Gastrointestinal tract, Kidney and Spleen.
1.1 Lung Injury After Stroke
Stroke breaks out a strong inflammatory cascade in the brain, but the peripheral immune system is inhibited by immune regulation of compensatory release of neurotransmitters, this phenomenon is defined as stroke-induced immunosuppression (SIIS) (5). The direct consequence of SIIS is to make stroke patients more vulnerable to infections, of which stroke-associated pneumonia (SAP) is the most common and often fatal (6). It has been proved in clinical trials that stroke-induced immunosuppressive syndrome is an independent risk factor for stroke-associated pneumonia (7). Previous studies have shown that older and more severe nerve damage and dysphagia are important factors for pneumonia after stroke (8–12). About 1/10 of stroke patients develop pneumonia in the acute phase (13). The occurrence of post-stroke pneumonia is related to 30-day and 1-year mortality, longer hospital stays and dependence on discharge (14). In addition, neurogenic pulmonary edema (NPE) is also one of the common pulmonary complications after stroke. The mechanism of NPE may be the damage of alveolar capillary barrier caused by a large number of nervous system discharges after severe craniocerebral injury and the lung volume overload caused by the increase of systemic vascular resistance. Due to the lack of effective and timely treatment, NPE is usually associated with higher mortality, which suggests that NPE may be one of the causes of poor prognosis in patients with stroke (15). In this review, we focus on the pathogenesis and emerging treatments of SAP and NPE, and further speculate on the role of immune factors in it.
1.1.1 Pathophysiological Mechanism of Lung injury After Stroke
There are two main theories about the pathophysiological mechanism of SAP: Aspiration Theory and stroke-induced immunosuppression (SIIS) (16). A small amount of inhaled substances caused by dysphagia in stroke patients is a key factor in SAP, which may be related to the abnormal transmission of dopamine (17). An experiment in guinea pigs has shown that blocking D1 dopamine receptors in guinea pigs can inhibit swallowing reflexes and reduce substance P in terminal organs (18). Due to the decrease of dopamine production in substantia nigra and striatum of stroke patients, the expression of substance P in glossopharyngeal nerve and cervical parasympathetic ganglion decreased, resulting in dysphagia (17). The risk of SAP is related to substance P deficiency. Treatment with angiotensin converting enzyme (ACE) inhibitors in stroke patients can increase the level of substance P and reduce the incidence of pneumonia in patients with dysphagia (19).
The main causes of SIIS are the transformation from Th1 phenotype to Th2 phenotype of lymphocytes, the decrease of lymphocytes and NK cells in blood and spleen, and the impairment of defense mechanism of monocytes and neutrophils (5). One of the causes of SIIS may be that stroke activates the sympathetic system and the hypothalamus-pituitary-adrenal axis (20). An experimental study shows that injection of 200 Streptococcus pneumoniae colony-forming units into the nasal cavity of stroke mice can cause pneumonia and bacteremia, while 200000 colony-forming units are needed in fake animals to induce similar diseases. but it can be prevented by adrenoceptor blockers (21). The immunosuppressive state caused by sympathetic activation after stroke is characterized by Th-mediated lymphopenia and functional inactivation of monocytes and Th1 (22), which is more obvious in mice with large cerebral infarction (23). A series of clinical and experimental evidence shows that damaged brain tissue produces a variety of pro-inflammatory cytokines, which can activate the hypothalamus-pituitary axis system, resulting in increased adrenocorticoid secretion and T lymphocyte apoptosis (20).
Neurogenic pulmonary edema (NPE) after acute stroke is an acute respiratory distress syndrome (ARDS), which is characterized by acute onset, obvious infiltration of pulmonary interstitial fluid and rapid regression (15). The pathophysiological mechanism of NPE is the joint participation of nervous system, circulatory system and respiratory system. The occurrence of NPE may be due to the activation of a specific trigger area of the central nervous system located in the brainstem, resulting in excessive sympathetic nerve activation leading to peripheral vasoconstriction, increased systemic vascular resistance (SVR) and enhanced venous reflux. After these changes, the pulmonary capillary hydrostatic pressure (PCP) increased, the alveolar wall was damaged, and fluid and red blood cells infiltrated into the pulmonary interstitium and alveoli to form a typical NPE (24).
1.1.2 Treatment
Post-stroke pneumonia is usually directly associated with bacterial infections, including aerobic Gram-negative bacilli and Gram-positive cocci (25),the most common bacterial infections are Pseudomonas aeruginosa and Staphylococcus aureus (26). Selecting targeted antibiotics according to the type of bacteria infected is an important step in the treatment of SAP, and ampicillin + sulbactam group is a good choice (27). According to the pathophysiological mechanism of SAP, head raising, oral care and dental treatment for stroke patients can effectively improve swallowing function and cough reflex sensitivity, which is the key to the treatment and prevention of SAP. For elderly stroke patients, angiotensin converting enzyme inhibitors can well improve cough reflex sensitivity, thus reducing the risk of SAP (Table 1).
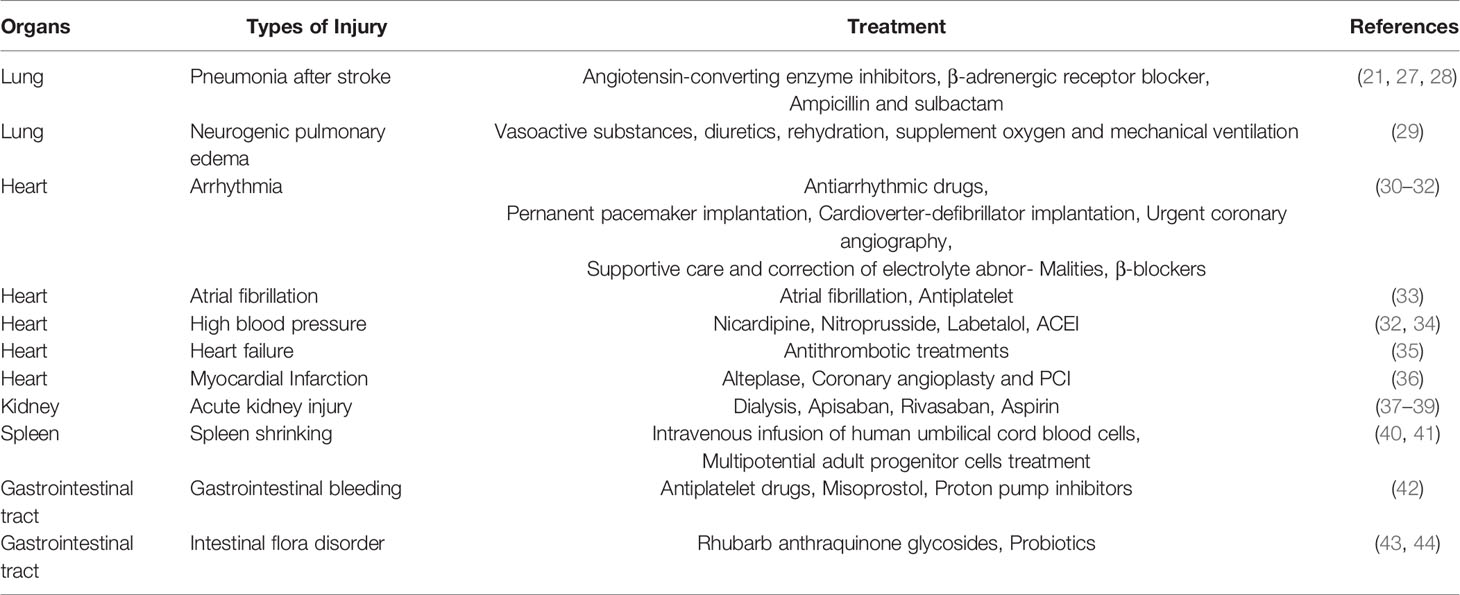
Table 1 Injury types and treatment of different organs after stroke.
The treatment of NPE mainly includes two aspects. on the one hand, it is based on the treatment of primary central nervous system injury, with emphasis on reducing intracranial pressure to prevent sympathetic nerve discharge, which is considered to be the main culprit of lung injury (45). Another important aspect is supportive treatment, including vasoactive substances, diuretics, rehydration, supplement oxygen and mechanical ventilation if necessary (29). Studies have shown that intravenous injection of 25% albumin does not improve the prognosis of patients with ischemic stroke, but increases the risk of pulmonary edema (46, 47). A study on the treatment of 12 cases of aneurysmal SAH with NPE shows that endovascular treatment of severe SAH with NPE is an effective regimen (48). However, due to the lack of sample size, further research is needed to ensure the reliability of the conclusion.
1.2 Heart Injury After Stroke
Adiovascular disease is regarded as the predis-posing risk factor for stroke (49),cardiovascular complications are also the second leading cause of death after stroke (50). In the first few days after stroke, cardiac complications are a common problem, including arrhythmia, heart failure, myocardial injury, non-fatal coronary syndrome, Takotsubo syndrome and other neurocardiogenic syndrome. The study of the interaction between brain and heart has been going on for centuries and has made great progress in the last decade (51), more and more clinical and neuroimaging studies as well as animal studies suggest that this series of cardiac complications may have the same underlying mechanism (52). In this review, we summarize these cardiac complications as stroke-heart syndrome. The main pathophysiological mechanisms include hypothalamus-pituitary-adrenal axis(HPA) (53), the gut dysbiosis (54) and immune and inflammatory response (55), linking the effects of these mechanisms may help us to find new treatments. Some studies have shown that stroke-heart syndrome may originate from the structural or functional changes of central autonomic neural network (CAN) after stroke (52), and affect heart rate and cardiac contractility through sympathetic nervous system and parasympathetic nervous system (56). When brain injury occurs, different areas and degrees of injury will lead to different results. For example, stimulating the orbit of the frontal lobe and anterior cingulate gyrus can affect blood pressure and heart rate, ischemia in the insular cortex may lead to changes in blood pressure and arrhythmias (57). In addition, clinical trials have shown that patients with LDL levels below 70 mg per deciliter have a lower risk of cardiovascular events than patients with LDL levels ranging from 90 mg to 110 mg per deciliter after ischemic stroke. This suggests that LDL may be an independent risk factor for cardiovascular events after stroke (58).
1.2.1 Pathophysiological Mechanism of Heart Injury After Stroke
1.2.1.1 Hypothalamus-Pituitary-Adrenal Axis
The mediator of cortisol is the hypothalamus-pituitary-adrenal axis, and the HPA axis plays a key role in the process of balance in the body (59). Corticotropin-releasing hormone is secreted by the hypothalamic paraventricular nucleus, which stimulates the pituitary gland to release corticotropin, which stimulates the adrenal gland to release the steroid hormone cortisol (60). Elevated cortisol levels increase the mortality of stroke patients (53). In the study of the animal model of middle cerebral artery occlusion (MCAO) in rats, it was found that the activation of paraventricular nucleus was due to the activation of N-methyl-D-aspartate (NMDA) receptor by glutamate, which led to arrhythmia (61). Inhibition of oxidative signals in the paraventricular nucleus can be used as a new method for the treatment of heart failure caused by myocardial infarction (62). Elevated catecholamine levels play an important role in stroke-heart syndrome. Animal studies have shown that plasma catecholamine levels increase after middle cerebral artery occlusion, which may lead to myocardial injury (63). Catecholamine acts on cardiac β-receptors, activating cyclic adenosine monophosphate-protein kinase A signal and increasing intracellular Cyclic adenosine monophosphate (CAMP). CAMP binds to protein kinase A and phosphorylates L-type calcium channels, which makes mitochondrial overload trigger oxidative stress and eventually lead to cardiomyocyte death (50). In vitro and in vivo studies have shown that the intermediates formed during the oxidation of catecholamines are related to cardiotoxicity (64). The consequence of this increase in catecholamine levels is cardiomyocyte necrosis, hypertrophy and fibrosis, and the production of reactive oxygen species, alteration of the calcium-handling proteins, coronary blood flow redistribution (65).
1.2.1.2 Immuno-Inflammatory Response
Immuno-inflammatory response plays a significant role quickly after stroke and is also an important factor in stroke progression (66). In the early stage of stroke, both innate immunity and acquired immunity are involved in local and systemic inflammation (50). The complex interaction involves a variety of mechanisms. In this article, we focus on the immune and inflammatory responses that mediate stroke cardiac syndrome.
In the early stage of acute brain injury, local inflammatory responses in the brain parenchyma (including microglia proliferation, astrocyte proliferation and cytokine/chemokine secretion) maintain endothelial cell activation. During this period of ischemia, a large number of reactive oxygen species (ROS) are produced in the brain and immune cells. Then, reactive oxygen species can activate endothelial cells, cause oxidative stress and destroy the blood-brain barrier (67). Resident macrophages transform into M1 phenotypes, exacerbating inflammation by releasing pro-inflammatory cytokines (68). After ischemia, the brain releases damage-associated molecular patterns (DAMP), stimulates pathogen recognition receptors Toll-like receptors and TLR-4, and increases the production of pro-inflammatory mediators IL-6, IL-1 β, TNF-α, chemokine and their receptors (Figure 2). Through these mediators, the brain recruits peripheral immune cells to the site of brain injury and then crosses the damaged blood-brain barrier (68). Enter the systemic circulation to produce possible secondary heart injury. An animal experimental study reported that abnormal inflammation and apoptotic cells appear in the cerebellum and heart of non-human primate transient global cerebral ischemia (TGI) model (69), suggesting a potential correlation between inflammation and heart injury. Tumor necrosis factor-α leads to ubiquitin and then degradation of troponin I, which eventually leads to a decrease in the contractile function of cardiomyocytes (70). Transforming growth factor-β 1 (TGF-β 1) induces increased expression of IL-6 (71). IL-6 is a cytokine that can regulate the growth, apoptosis and survival of cardiac cells (72). Recent studies have suggested that activation of TLR-4 and elevated levels of IL-1 β are associated with heart failure (73). In patients with ischemic stroke and cerebral hemorrhage, systemic inflammatory response is activated (74). Systemic inflammatory response syndrome is usually characterized by abnormal white blood cell count, increased respiration and heart rate, and abnormal body temperature. This increases the risk of intracranial and systemic complications. The number of CD74+ cells and the expression of CD74mRNA in peripheral blood monocytes increased significantly in patients with ischemic stroke (75). CD74+ cells are mainly distributed on CD4+T cells, monocytes and dendritic cells (75). Monocytes are related to cardiac salvage function damage and poor left ventricular remodeling after acute myocardial infarction (76), and can be used as a new treatment for ischemic brain injury in the future.
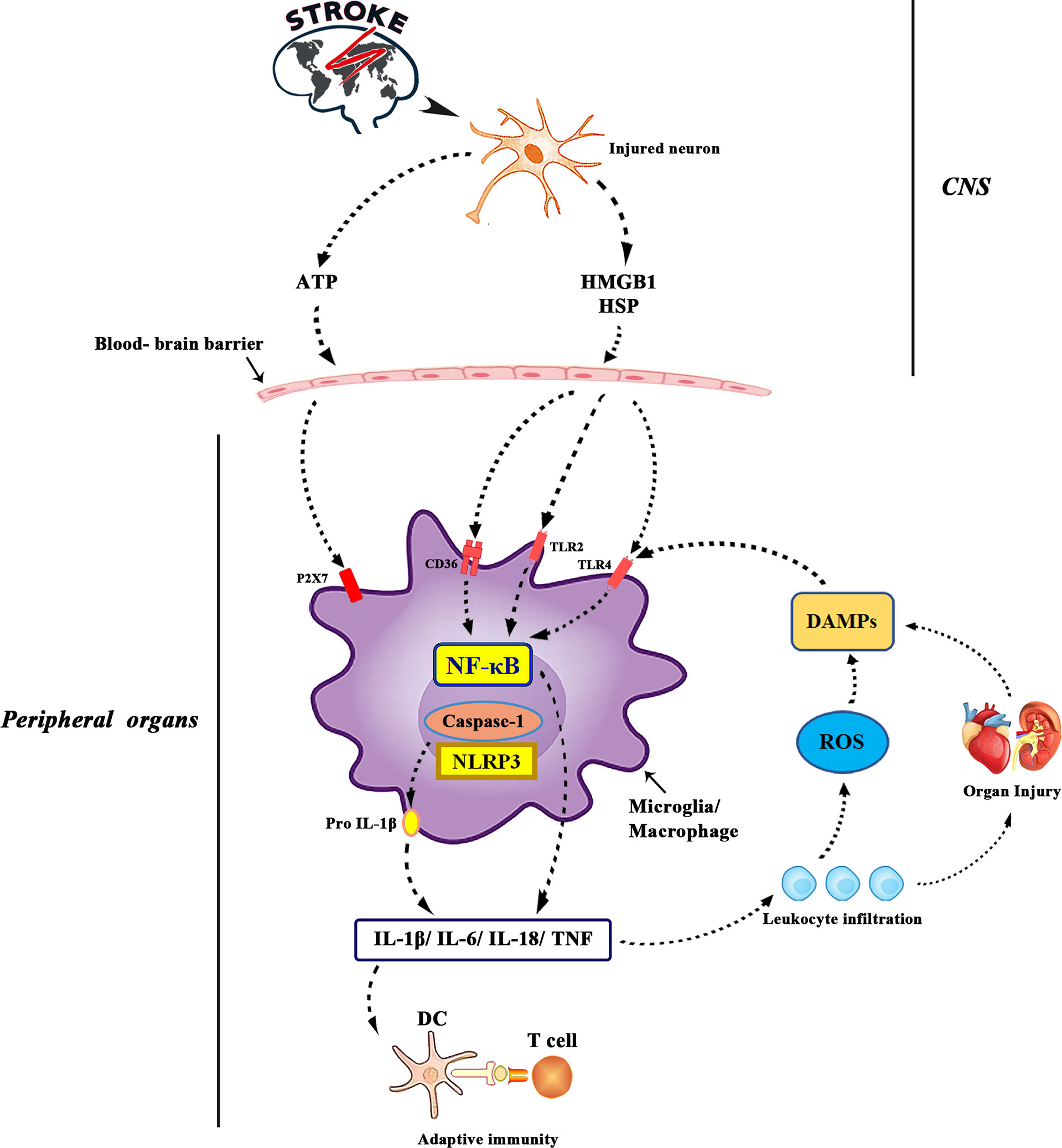
Figure 2 ATP is released from damaged neurons, activating purinergic receptors on microglia and macrophages and leading to the production of pro-inflammatory cytokines. Interleukin-1 converting enzyme (ICE; caspase1) is embedded in a polyprotein complex (NLRP3 or inflammatory body) and activated by P2X7 receptors in microglia. Cell death leads to the formation of DAMPs, which activates TLR, especially TLR2 and TLR4. DAMPs released by ischemia includes high mobility group protein B1, heat shock protein 60 and so on. TLRs binds to scavenger receptors (such as CD36) and up-regulates the expression of inflammatory genes through transcription actor nuclear factor-kB. DAMPs is also produced by matrix decomposition caused by lyases released by dead cells and the effect of reactive oxygen species on lipids. Finally, the production of cytokines and the activation of complement lead to the increase of leukocyte infiltration and tissue damage, which leads to the production of more DAMPs. The antigen revealed by tissue injury was presented to T cells, which laid the foundation for adaptive immunity.
1.2.1.3 Intestinal Biological Disorder
More and more experimental studies have shown that there are interactions between intestinal microbiota and central nervous system as well as heart, which are called brain-gut axis and intestine-heart axis. The main function of the gut-blood barrier is to regulate the absorption of nutrients, electrolytes and water, and to prevent pathogenic microorganisms and toxic substances from entering the bloodstream (54). Stroke can lead to the destruction of the intestinal-blood barrier, thereby increasing intestinal permeability, imbalance of intestinal microflora and transfer to the bloodstream (35).Increased intestinal permeability can promote inflammation (54), while systemic inflammation can aggravate cardiac dysfunction, which has been explained above. Trimethylamine N-oxide (TMAO) is an important gut microbe-dependent metabolite synthesized by dietary choline, betaine and L-carnitine (77). TMAO mediates platelet overactivation and thrombosis, and it also enhances inflammation by acting on dendritic cells, macrophages and platelets (77). Changes in TMAO levels are associated with atherosclerosis, myocardial infarction, thrombosis and heart failure (78, 79).
1.2.2 Treatment
The incidence of stroke-cardiac syndrome after stroke is very high, and the damage of cardiac function affects the prognosis and mortality of patients to a great extent. Almost 80% of AIS patients develop hypertension for various reasons. The side effects of severe hypertension increase the risk of cardiopulmonary complications, cerebral hemorrhage and cytotoxic edema, and are associated with adverse outcomes after AIS (80). Drugs such as nicardipine, nitroprusside and labetalol are recommended for the treatment of hypertension in the acute phase of stroke (systolic blood pressure is greater than 180mmHg) (34). β-blockers can not only inhibit sympathetic activation and inflammation, but also prevent chronic remodeling and treat arrhythmias (81), and may reduce heart injury after stroke. Among them, propranolol has been proved to play a neuroprotective effect by blocking the upregulation of IL-6, thus improving the prognosis after brain injury (82). As recommended in the current clinical guidelines (83), secondary stroke prevention with oral anticoagulants can effectively control the risk factors of cardiovascular disease, but there is no good plan for the treatment and prevention of cardiac complications after stroke. With the in-depth study of the pathophysiology of brain-heart interaction, new and more comprehensive treatments may emerge in the future.
1.3 Renal Injury After Stroke
Acute renal injury (AKI) is one of the common complications after stroke, which can lead to renal failure (84). Studies have shown that acute renal injury after stroke is associated with higher mortality and poor functional prognosis (85). In a systematic retrospective study of 12,325,652 patients with ischemic stroke, the incidence of acute renal injury (AKI) was 9.6% (86). High score of National Institutes of Health Stroke Scale (NIHSS)and hypertension are important indicators for the incidence of AKI in stroke patients on admission (85). In addition, diabetes, elevated plasma osmotic pressure and the use of cyclic diuretics are also risk factors for AKI after stroke (87–89). Chronic kidney disease (CKD) refers to the damage of kidney structure or function caused by various causes for more than 3 months, which is mainly characterized by a decrease in glomerular filtration rate (GFR), which usually develops with the passage of time and increases the risk of stroke (90). CKD is usually diagnosed and staged by measuring glomerular filtration rate (GFR), creatinine clearance and albuminuria, while lower GFR and albuminuria are associated with poor prognosis after stroke (91, 92), which has been demonstrated in other clinical trials (93) (Table 2).
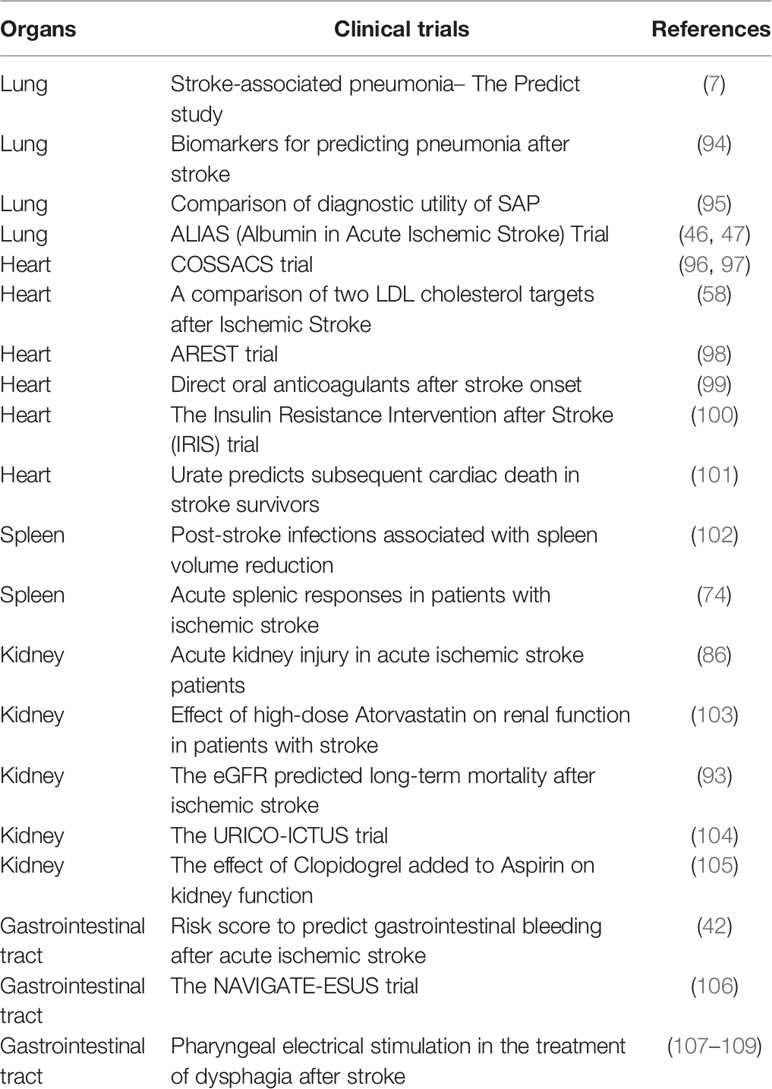
Table 2 Clinical trials targeting the peripheral organs injury after stroke.
There is a strong two-way relationship between stroke and kidney disease, which may be related to their similarities in anatomy, hemodynamics and vascular regulation (110). The glomerular afferent arterioles located in the paramedullary renal artery and cerebral perforating artery in the kidney and brain, respectively, originate directly from the short arterioles and large arteries, and are responsible for maintaining perfusion pressure and blood flow (111). Even if arterial blood pressure is constantly changing, the brain and kidneys can be automatically regulated to maintain a certain range of blood perfusion (84).The pathophysiological interaction between brain and kidney is complex, but many studies have focused on brain dysfunction caused by renal injury. In this review, we focus on the pathophysiological mechanisms of renal injury after stroke, including neuroendocrine system, inflammatory and immune response, extracellular vesicles (EVS) and microRNA(miRNA), as well as related prevention and treatment.
1.3.1 Pathophysiological Mechanism of Renal Injury After Stroke
1.3.1.1 Neuroendocrine System
After stroke, the hypothalamus-pituitary-adrenal axis can be activated to regulate the release of glucocorticoids from the adrenal gland. High levels of glucocorticoids can directly affect the function of glomeruli and renal tubules, resulting in the decrease of GFR (112). At the same time, elevated glucocorticoid levels can lead to vascular and hemodynamic changes, which indirectly affect renal blood flow (RBF) and glomerular function (112) (Figure 3). On the one hand, patients with acute brain injury due to excessive excitation of the sympathetic nerve, resulting in glomerular filtration function decreased, renal blood flow decreased (113). On the other hand, patients with acute cerebral ischemia often present with elevated catecholamines (114), and persistent excitation of the sympathetic nervous system leads to binding of catecholamines and angiotensin II to renal artery receptors, prompting renal artery contraction and renal ischemia (115). It can be seen that the increased levels of glucocorticoid and catecholamine may be related to the damage of renal function after stroke.
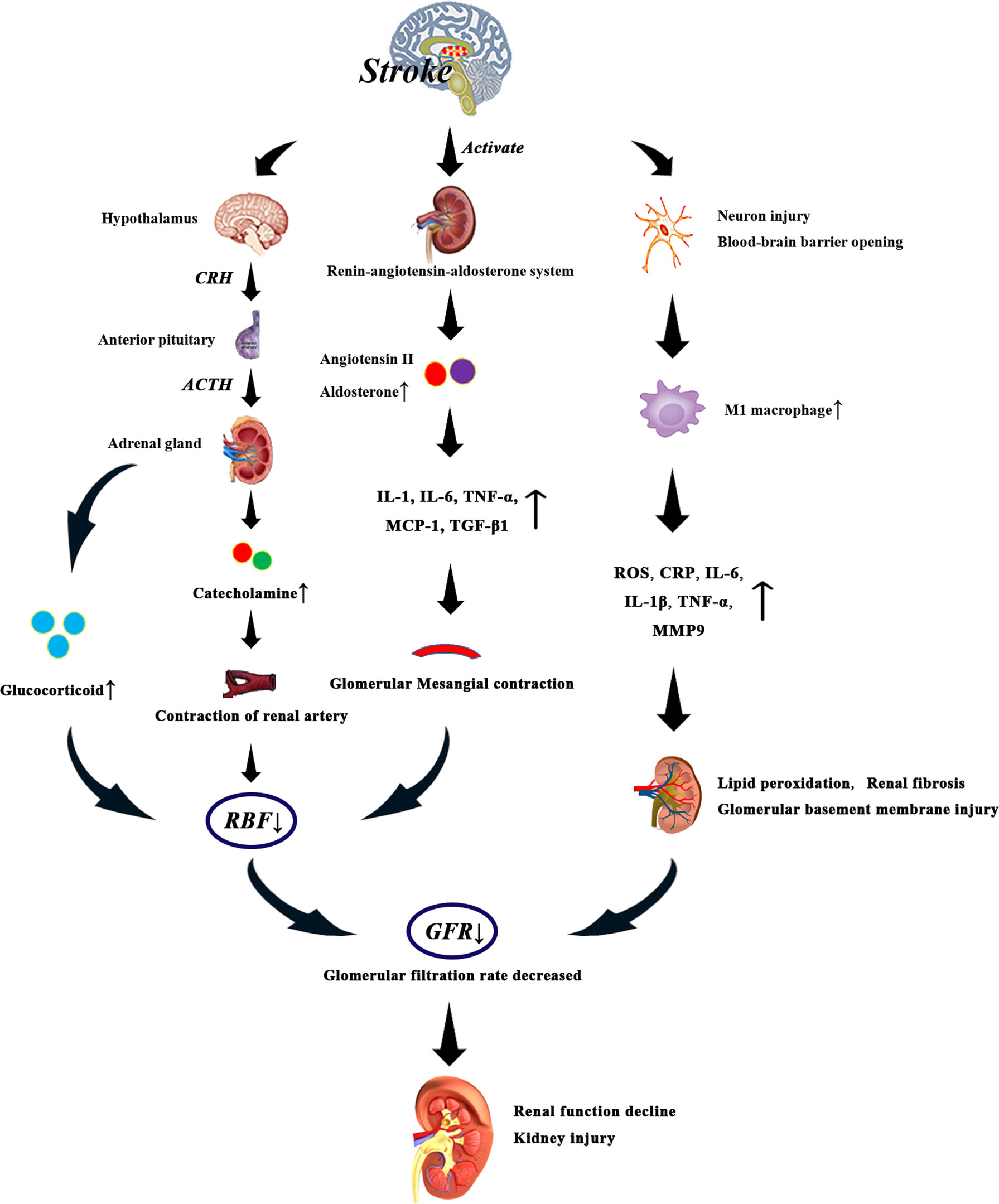
Figure 3 After stroke, the levels of glucocorticoid, catecholamine, angiotensin II and aldosterone increased due to the activation of HPA axis and renin-angiotensin-aldosterone system. Especially when the level of glucocorticoid is too high, the renal blood flow decreases significantly, which leads to the decrease of glomerular filtration rate. Catecholamine and angiotensin II bind to renal artery receptors and promote renal artery contraction and renal ischemia. Activation of renin-angiotensin-aldosterone system can promote systemic and glomerular capillary hypertension, and the direct fibrogenic and pro-inflammatory effects of angiotensin II and aldosterone may also lead to renal injury. In addition, angiotensin II can increase the levels of IL-1, IL-6, tumor necrosis factor-α and monocyte chemoattractant protein-1, thus reducing glomerular blood flow. On the other hand, after stroke, neurons are damaged, the blood-brain barrier is destroyed, and the production of M1 macrophages is increased, which induces the production of inflammatory factors such as C-reactive protein, IL-6, IL-1 β, tumor necrosis factor and matrix metalloproteinase-9, which leads to renal injury. These events eventually lead to decreased glomerular filtration rate, decreased renal function, and irreversible kidney damage.
Activation of the renin-angiotensin-aldosterone system can promote systemic and glomerular capillary hypertension, and the direct fibrogenic and pro-inflammatory effects of angiotensin II and aldosterone may also lead to renal damage (116). Renin-angiotensin system (RAS) is involved in the pathogenesis of ischemic brain injury, and angiotensin II levels are increased in stroke patients (117). Angiotensin II can directly activate the expression of glomerular cytokines, inflammation and fibrosis factors, which has an effect on renal hemodynamics (118). In addition, angiotensin II also stimulates macrophage aggregation in glomeruli and tubular cells, increases the production of cytokines such as IL-1, tumor necrosis factor-α and monocyte chemoattractant protein-1, and acts on glomerular cells to promote the occurrence and development of glomerular injury. Angiotensin II induces the expression of TGF- β 1 and angiotensinogen genes, and induces the proliferation of renal interstitial fibroblasts through AT1 receptors, which may be involved in the pathogenesis of renal fibrosis (119). Another study showed that angiotensin II can induce the production of IL-6 in the kidneys. IL-6 can induce fibrosis gene expression and ET-1 gene expression, which leads to renal injury (120). Excessive activation of the sympathetic nervous system after stroke increases the release of Antidiuretic hormone (ADH), resulting in glomerular Mesangial contraction, glomerular blood flow reduction (121), resulting in renal function damage.
1.3.1.2 Inflammatory and Immune Response
Immune and inflammatory responses play an important role in the progression of stroke and are also important causes of AKI and CKD (84). Macrophages are significant mediators of inflammation and immune regulation, and classical pro-inflammatory M1 macrophages are associated with renal disease (118). Macrophage-derived cytokines (ROS, IL) and inflammatory factors such as C-reactive protein (CRP), IL-6, IL-1 β, TNF- α and matrix metalloproteinase-9 (MMP9) after brain injury are related to renal injury (118). CRP is an acute inflammatory protein, mainly synthesized by hepatocytes, which increases 1000-fold in the site of infection or inflammation (122). CRP levels increased after ischemic stroke (123), and high levels of CRP decreased glomerular filtration rate (124). Reactive oxygen species (ROS) levels increase rapidly in the acute phase of stroke and enter the bloodstream through the blood-brain barrier (BBB) (125). Reactive ROS can cause different types of cell damage, especially lipid peroxidation and membrane damage (126). In the kidney, ROS mainly degrades the glomerular basement membrane and changes the function of glomerular and tubular cells (126). Microglia and macrophages produce and secrete IL-1 β after stroke (127). Il-1β can lead to renal injury and renal fibrosis (128). The expression of MMP-9 is increased during cerebral ischemia, which leads to neuronal injury, apoptosis and opening of BBB (129). It plays a special physiological role in the main cells of renal collecting duct (130). Another experimental study in rats has shown that increased circulating MMP-9 activity is associated with refractory albuminuria, which may lead to the progression of CKD in patients (131).
1.3.1.3 Extracellular Vesicles and microRNA
In the kidney, EVS can come from blood cells, endothelial cells, podocytes or renal tubular epithelial cells (132). EVS may be a biomarker of kidney disease, which is related to inflammation, thrombosis and immunosuppression (132). The production of EVS after brain injury affects the normal physiological function of the kidney. EVS is related to the pathogenesis of acute renal injury and chronic renal disease, including AKI, CKD, renal fibrosis and various glomerular diseases (37). No matter what kind of dialysis treatment is performed, the level of platelet-derived EVS is increased, indicating that dialysis does not clear EVS (38). However, as far as the current research is concerned, the mechanism of EVS causing renal function damage through brain-kidney interaction is not clear. MiRNA is a small non-coding RNA molecule that regulates gene expression and participates in the occurrence and development of tubulointerstitial sclerosis and glomerular lesions (133). The level of miR expression changes after stroke. An experimental study in a mouse model of unilateral ureteral obstruction showed that TGF- β induced up- regulation of miR-21 expression, which was mediated by Smad2 signal, thus promoting renal fibrosis (134). Another in vitro study reported that astragaloside IV (AS-IV) improved renal function and renal fibrosis by inhibiting podocyte dedifferentiation and Mesangial activation induced by overexpression of miR-21 (135). It can be seen that the inhibition of Smad2/miR-21 signal pathway may be used as a new treatment to inhibit renal fibrosis in the future. In addition, miR-29c is the characteristic miRNA in diabetes. By knocking down miR-29c with specific antisense oligonucleotides, the proteinuria and glomerular Mesangial matrix in diabetic mice are significantly decreased (136).
1.3.2 Treatment
When we treat stroke patients with renal injury, we need to evaluate the effectiveness and risk of intravenous thrombolysis and anticoagulation therapy. Studies have shown that stroke patients receiving intravenous injection of rt-PA increase the risk of decreased glomerular filtration rate and cerebral hemorrhage (137). Because the new oral anticoagulants apisaban and rivasaban are excreted mainly or partly by the kidneys, the half-life is prolonged in CKD patients, which not only enhances the antithrombotic effect, but also increases the risk of bleeding (138). There are no significant benefits of aspirin as a traditional antiplatelet drug in the treatment of stroke patients, but studies have shown that aspirin treatment of CKD patients has a greater absolute reduction in major cardiovascular events and mortality, this benefit seems to outweigh the increased risk of massive hemorrhage (139). Many anticoagulants and thrombolytic agents are used clinically to treat stroke, which increases the risk of use when patients are complicated with renal insufficiency. One of the reasons is that we do not know enough about the mechanism of brain-kidney interaction. Further exploration of this interaction will help us to find drugs that have dual protective effects on the brain and kidney.
1.4 Spleen Injury After Stroke
In the human body, the spleen, as one of the most important immune organs, has innate and acquired immune function and plays a vital role after stroke. After stroke, the brain broke out a serious inflammatory cascade reaction. Due to the role of chemokines and cytokines, the brain recruited a large number of spleen-derived immune cells to the brain injury site to combat the inflammatory response (4, 140). Many studies have found that shortly after stroke, the spleen shrinks sharply and the number of cells in the spleen decreases accordingly (74, 140–142). This change may reflect the increase of immune cell outflow from spleen to peripheral circulation and the increase of spleen cell group death (143). In one study, carboxyfluorescein diacetate succinimidyl ester (CFSE) was used to label the migration of splenocytes after cerebral ischemia. It was proved that after cerebral ischemic injury, NK cells, T cells, monocytes and NK cells entered the systemic circulation and migrated to the brain, aggravating brain injury (144). In addition, due to the role of cytokines and chemokines, IL-6, IFN- γ, TNF-α, MCP-1 and other molecules are recruited to the brain injury site and aggravate the brain injury (145, 146). At present, the exact mechanism of spleen activation after stroke is not clear, but the activation of sympathetic nervous system, antigen presentation of central nervous system and the production of chemokines have been proved to be important factors (143).
1.4.1 Pathophysiological Mechanism of Spleen Injury After Stroke
1.4.1.1 Sympathetic Nervous System
Shortly after stroke, activation of the sympathetic nervous system leads to an increase in norepinephrine and epinephrine in the systemic circulation. In rodents, the spleen shrinks after MCAO, which may be due to the expression of α-1 adrenergic receptor in the splenic sac of rats, which leads to splenic contraction after activation (141). Both norepinephrine and epinephrine have been shown to cause significant splenic atrophy (147). Prazosin or carvedilol could prevent the spleen from shrinking, but only carvedilol could significantly reduce the infarct volume (148). These results suggest that α and β adrenergic receptors seem to mediate the response of the spleen to stroke.
1.4.1.2 Antigen Presentation of the Central Nervous System
In the early stage after stroke, the activation of danger-associated molecular patterns (DAMP) causes the damaged brain to secrete various antigens such as ATP, high mobility group protein 1 (HMGB1), heat shock protein (HSP) and nicotinamide adenine dinucleotide (NAD) (149). These antigens interact with antigen-presenting cell receptors to activate innate and acquired immune responses, eventually recruiting immune cells to the injured brain (150).
1.4.1.3 Production of Cytokines and Chemokines
After MCAO, the levels of TNF-α, IFN-γ, IL-6, MCP-1 and IL-2 secreted by mouse splenocytes increased significantly (151). In addition, IFN γ can also activate the expression of chemokine interferon-inducible protein 10, leading to neurodegeneration (40). In cerebral ischemic injury, CCL2 (MCP-1) mediates monocyte and neutrophil infiltration. Inhibition of CCL2/CCR2 axis can reduce brain edema and leukocyte infiltration to improve the results of cerebral reperfusion (41). The treatment of MCAO mice with the antibacterial drug moxifloxacin (MFX) significantly reduced the expression of CCR2 in spleen tissue and brain after ischemia, and reduced the area of cerebral infarction (152). Other cytokines, such as CCL3, CCL5 and CXCR4-CXCL12, have been shown to play a role in splenic response after stroke (153).
1.4.2 Treatment
Some experiments have proved that compared with the rats without splenectomy two weeks before permanent middle cerebral artery occlusion, the area of cerebral infarction was significantly reduced and the number of macrophages, activated microglia and neutrophils was also greatly reduced (154). However, it is obvious that splenectomy can’t be used as a practical clinical stroke treatment, because from a long-term point of view, the risk of infection caused by failure to maintain normal immune function after splenectomy is greatly increased. Therefore, drugs and cell-based therapy for the interaction between the peripheral immune system and the brain may be used as stroke treatment options (155, 156). There is growing evidence that intravenous injection of various types of stem cells can reduce neurological damage caused by stroke to a greater extent than intracerebral administration (157). Intravenous infusion of human umbilical cord blood cells (HUCB) to stroke patients can improve the cerebral ischemic microenvironment and restore neurological function (142, 158). Transplantation of HUCB cells after MCAO increased the production of anti-inflammatory cytokines IL-10, decreased the production of inflammatory cytokines TNF- α and interferon-γ, and inhibited the proliferation of splenic CD8+T cells (157). In addition, multipotential adult progenitor cells (MAPC) treatment can increase the number of Treg cells in the spleen, up-regulate the level of serum IL-10, reduce the release of IL-1 β and IL-6 from splenocytes, and restore the reduction of spleen mass caused by stroke (159). These evidences suggest that spleen is a key target for MAPC to regulate immune response, regulate local cerebral microenvironment and promote rehabilitation after stroke (159).
1.5 Gastrointestinal Bleeding After Stroke
Stroke cuts off the connection between the central nervous system and the gastrointestinal system, resulting in dysphagia, gastrointestinal bleeding, delayed gastrointestinal emptying, etc (160). Dysphagia and gastrointestinal bleeding (GIB) are common complications after stroke and are related to poor prognosis. Since dysphagia has been described in post-stroke pneumonia, we will not repeat it here.
GIB is a common complication in patients with acute stroke and may affect stroke treatment, such as antiplatelet or anticoagulation therapy (161). The incidence of gastrointestinal bleeding after stroke is 1.5%-7.8%, which may be related to the subtype of stroke (162, 163). Patients with gastrointestinal bleeding after stroke were characterized by sudden hematemesis, decreased hemoglobin or orthostatic hypotension. Advanced age, disturbance of consciousness, severe neurological impairment, infection and posterior circulation infarction are independent risk factors for GIB in patients with acute stroke, and GIB is also a high risk factor for death within 1 year after acute stroke (164). At present, clinical trials have demonstrated that AIS-GIB score is an effective clinical grading standard for predicting GIB during hospitalization after acute ischemic stroke, which plays a certain role in helping the incidence of GIB and improving prognosis after AIS (42).
1.5.1 Pathophysiological Mechanism of GIB
At present, the pathogenesis of GIB after stroke is not fully understood. Stress ulcer caused by acute brain injury, increased gastric acid secretion or mucosal ischemia caused by overactivity of vagus nerve may be one of the causes of GIB.
Aspirin is a commonly used antiplatelet drug in patients with stroke, and its low-dose side effects may also cause gastrointestinal mucosal damage, leading to gastroduodenal ulcer (165). A meta-analysis shows that cilostazol is less likely to have gastrointestinal bleeding than aspirin, although other gastrointestinal adverse reactions are more likely to occur (166). In addition to stress ulcer and antiplatelet effect, systemic inflammation and oxidative stress may be the pathophysiological mechanism of gastrointestinal mucosal injury after stroke (167). During ischemic stroke, ulcers can also be formed due to the decrease of gastric mucosal blood flow. Some animal experimental studies have shown that norepinephrine neurons reduce gastric mucosal blood flow through α-adrenergic receptors in rats with cerebral ischemia, resulting in damage to the integrity of gastric mucosa. Vagotomy can eliminate the decrease of gastric mucosal blood flow during cerebral ischemia (168). In addition, malnourished stroke patients are also more likely to develop gastrointestinal bleeding (169).
1.5.2 Treatment
The treatment and prevention of stroke patients with gastrointestinal bleeding should follow routine guidelines. ASA (antiplatelet-statin-antihypertensive) is a drug used for secondary stroke prevention to prevent cerebrovascular thrombosis in patients with cerebrovascular disease or patients at risk of cerebrovascular disease (160). However, antiplatelet drugs should be carefully selected when treating this type of patients, and their anti-platelet and gastrointestinal mucosal damage may increase the risk of bleeding. Therefore, antiplatelet therapy for secondary stroke prevention must be individualized according to the patient’s complications, including the risk of bleeding (170). Studies have shown that antiplatelet drugs in combination with proton pump inhibitors (PPI) or misoprostol can reduce the risk of gastrointestinal injury (171). PPI can effectively inhibit the secretion of basal gastric acid and stimulating gastric acid, and make the PH of gastric juice more than 6.0. therefore, it can induce hemostasis by body fluid and platelet. In addition, PPI can also promote gastric mucosal blood, improve local microcirculation, accelerate mucosal regeneration and repair, and further control bleeding.
2 Conclusion
Peripheral organ injury and dysfunction are very common after stroke, which usually occur within one week after stroke, so measures need to be taken to prevent and treat them in time. The most common complications after stroke include pulmonary infection, heart failure, acute renal injury and gastrointestinal bleeding. Understanding the pathogenesis, high risk factors and onset time of these complications is helpful for early diagnosis and treatment. The interaction between brain and peripheral organs after stroke is almost always carried out through humoral regulation and neuroregulation. The activation of the immune system and inflammatory response are very important for stroke. They affect many peripheral organs and thus affect the outcome of stroke. Here, we also emphasize and discuss the role of immune and inflammatory responses in all the organs under discussion, hoping to clarify the relationship between them and stroke. However, there are still many problems that have not yet been overcome. The injury of peripheral organs after stroke is usually not caused by the interaction of two or several systems, but by the participation of multiple systems throughout the body. There is no experimental model that can summarize all aspects at present. Elucidating the interaction between central and peripheral organs and peripheral organs will help us to develop more effective treatments.
Author Contributions
JW and JZ wrote the initial draft. YY contributed to reviewing the literature. QX and YL prepared the figures and table. SF and XX collected the literature. LG and ZJ designed the manuscript and prepared the final version. The authors read and approved the final manuscript.
Funding
This study was supported by the National Natural Science Foundation of China (Nos. 82071339 and 81771283) and the Natural Science Foundation of Hubei Province (Nos.2020CFB613).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Glossary

References
1. Chen F, Qi Z, Luo Y, Hinchliffe T, Ding G, Xia Y, et al. Non-Pharmaceutical Therapies for Stroke: Mechanisms and Clinical Implications. Prog Neurobiol (2014) 115:246–69. doi: 10.1016/j.pneurobio.2013.12.007
2. Feigin VL, Forouzanfar MH, Krishnamurthi R, Mensah GA, Connor M, Bennett DA, et al. Global and Regional Burden of Stroke During 1990–2010: Findings From the Global Burden of Disease Study 2010. Lancet (2014) 383(9913):245–55. doi: 10.1016/s0140-6736(13)61953-4
3. Benjamin EJ, Muntner P, Alonso A, Bittencourt MS, Callaway CW, Carson AP, et al. Heart Disease and Stroke Statistics-2019 Update: A Report From the American Heart Association. Circulation (2019) 139(10):e56–e528. doi: 10.1161/CIR.0000000000000659
4. Ma S, Zhao H, Ji X, Luo Y. Peripheral to Central: Organ Interactions in Stroke Pathophysiology. Exp Neurol (2015) 272:41–9. doi: 10.1016/j.expneurol.2015.05.014
5. Faura J, Bustamante A, Miro-Mur F, Montaner J. Stroke-Induced Immunosuppression: Implications for the Prevention and Prediction of Post-Stroke Infections. J Neuroinflamm (2021) 18(1):127. doi: 10.1186/s12974-021-02177-0
6. Vermeij JD, Aslami H, Fluiter K, Roelofs JJ, van den Bergh WM, Juffermans NP, et al. Traumatic Brain Injury in Rats Induces Lung Injury and Systemic Immune Suppression. J Neurotrauma (2013) 30(24):2073–9. doi: 10.1089/neu.2013.3060
7. Hoffmann S, Harms H, Ulm L, Nabavi DG, Mackert BM, Schmehl I, et al. Stroke-Induced Immunodepression and Dysphagia Independently Predict Stroke-Associated Pneumonia - The Predict Study. J Cereb Blood Flow Metab (2017) 37(12):3671–82. doi: 10.1177/0271678X16671964
8. Sellars C, Bowie L, Bagg J, Sweeney MP, Miller H, Tilston J, et al. Risk Factors for Chest Infection in Acute Stroke: A Prospective Cohort Study. Stroke (2007) 38(8):2284–91. doi: 10.1161/STROKEAHA.106.478156
9. Ramsey DJ, Smithard DG, Kalra L. Can Pulse Oximetry or a Bedside Swallowing Assessment Be Used to Detect Aspiration After Stroke? Stroke (2006) 37(12):2984–8. doi: 10.1161/01.STR.0000248758.32627.3b
10. Masiero S, Pierobon R, Previato C, Gomiero E. Pneumonia in Stroke Patients With Oropharyngeal Dysphagia: A Six-Month Follow-Up Study. Neurol Sci (2008) 29(3):139–45. doi: 10.1007/s10072-008-0925-2
11. Mann G, Hankey GJ, Cameron D. Swallowing Function After Stroke - Prognosis and Prognostic Factors at 6 Months. Stroke (1999) 30(4):744–8. doi: 10.1161/01.Str.30.4.744
12. Katzan IL, Cebul RD, Husak SH, Dawson NV, Baker DW. The Effect of Pneumonia on Mortality Among Patients Hospitalized for Acute Stroke. Neurology (2003) 60(4):620–5. doi: 10.1212/01.wnl.0000046586.38284.60
13. Badve MS, Zhou Z, van de Beek D, Anderson CS, Hackett ML. Frequency of Post-Stroke Pneumonia: Systematic Review and Meta-Analysis of Observational Studies. Int J Stroke (2019) 14(2):125–36. doi: 10.1177/1747493018806196
14. Saposnik G, Hill MD, O'Donnell M, Fang J, Hachinski V, Kapral MK, et al. Variables Associated With 7-Day, 30-Day, and 1-Year Fatality After Ischemic Stroke. Stroke (2008) 39(8):2318–24. doi: 10.1161/STROKEAHA.107.510362
15. Zhao J, Xuan NX, Cui W, Tian BP. Neurogenic Pulmonary Edema Following Acute Stroke: The Progress and Perspective. BioMed Pharmacother (2020) 130:110478. doi: 10.1016/j.biopha.2020.110478
16. Hannawi Y, Hannawi B, Rao CP, Suarez JI, Bershad EM. Stroke-Associated Pneumonia: Major Advances and Obstacles. Cerebrovasc Dis (2013) 35(5):430–43. doi: 10.1159/000350199
17. Teramoto S. Novel Preventive and Therapeutic Strategy for Post-Stroke Pneumonia. Expert Rev Neurother (2009) 9(8):1187–200. doi: 10.1586/Ern.09.72
18. Jia YX, Sekizawa K, Ohrui T, Nakayama K, Sasaki H. Dopamine D1 Receptor Antagonist Inhibits Swallowing Reflex in Guinea Pigs. Am J Physiol (1998) 274(1):R76–80. doi: 10.1152/ajpregu.1998.274.1.R76
19. Teramoto S, Ouchi Y. Ace Inhibitors and Prevention of Aspiration Pneumonia in Elderly Hypertensives. Lancet (1999) 353(9155). doi: 10.1016/s0140-6736(05)76506-5
20. Dirnagl U, Klehmet J, Braun JS, Harms H, Meisel C, Ziemssen T, et al. Stroke-Induced Immunodepression: Experimental Evidence and Clinical Relevance. Stroke (2007) 38(2 Suppl):770–3. doi: 10.1161/01.STR.0000251441.89665.bc
21. Prass K, Braun JS, Dirnagl U, Meisel C, Meisel A. Stroke Propagates Bacterial Aspiration to Pneumonia in a Model of Cerebral Ischemia. Stroke (2006) 37(10):2607–12. doi: 10.1161/01.STR.0000240409.68739.2b
22. Haeusler KG, Schmidt WU, Fohring F, Meisel C, Helms T, Jungehulsing GJ, et al. Cellular Immunodepression Preceding Infectious Complications After Acute Ischemic Stroke in Humans. Cerebrovasc Dis (2008) 25(1-2):50–8. doi: 10.1159/000111499
23. Liesz A, Hagmann S, Zschoche C, Adamek J, Zhou W, Sun L, et al. The Spectrum of Systemic Immune Alterations After Murine Focal Ischemia: Immunodepression Versus Immunomodulation. Stroke (2009) 40(8):2849–58. doi: 10.1161/STROKEAHA.109.549618
24. Sedy J, Kunes J, Zicha J. Pathogenetic Mechanisms of Neurogenic Pulmonary Edema. J Neurotrauma (2015) 32(15):1135–45. doi: 10.1089/neu.2014.3609
25. Kishore AK, Vail A, Jeans AR, Chamorro A, Di Napoli M, Kalra L, et al. Microbiological Etiologies of Pneumonia Complicating Stroke: A Systematic Review. Stroke (2018) 49(7):1602–9. doi: 10.1161/STROKEAHA.117.020250
26. Hassan A, Khealani BA, Shafqat S, Aslam M, Salahuddin N, Syed NA, et al. Stroke-Associated Pneumonia: Microbiological Data and Outcome. Singapore Med J (2006) 47(3):204–7.
27. Allewelt M, Schuler P, Bolcskei PL, Mauch H, Lode H, Pneumonia SGA. Ampicillin Plus Sulbactam Vs. Clindamycin +/- Cephalosporin for the Treatment of Aspiration Pneumonia and Primary Lung Abscess. Clin Microbiol Infect (2004) 10(2):163–70. doi: 10.1111/j.1469-0691.2004.00774.x
28. Vogelgesang A, Grunwald U, Langner S, Jack R, Broker BM, Kessler C, et al. Analysis of Lymphocyte Subsets in Patients With Stroke and Their Influence on Infection After Stroke. Stroke (2008) 39(1):237–41. doi: 10.1161/STROKEAHA.107.493635
29. Finsterer J. Neurological Perspectives of Neurogenic Pulmonary Edema. Eur Neurol (2019) 81(1-2):94–102. doi: 10.1159/000500139
30. Ruthirago D, Julayanont P, Tantrachoti P, Kim J, Nugent K. Cardiac Arrhythmias and Abnormal Electrocardiograms After Acute Stroke. Am J Med Sci (2016) 351(1):112–8. doi: 10.1016/j.amjms.2015.10.020
31. Kallmunzer B, Breuer L, Kahl N, Bobinger T, Raaz-Schrauder D, Huttner HB, et al. Serious Cardiac Arrhythmias After Stroke: Incidence, Time Course, and Predictors–A Systematic, Prospective Analysis. Stroke (2012) 43(11):2892–7. doi: 10.1161/STROKEAHA.112.664318
32. Ibrahim MS, Samuel B, Mohamed W, Suchdev K. Cardiac Dysfunction in Neurocritical Care: An Autonomic Perspective. Neurocrit Care (2019) 30(3):508–21. doi: 10.1007/s12028-018-0636-3
33. Schnabel RB, Haeusler KG, Healey JS, Freedman B, Boriani G, Brachmann J, et al. Searching for Atrial Fibrillation Poststroke: A White Paper of the Af-Screen International Collaboration. Circulation (2019) 140(22):1834–50. doi: 10.1161/CIRCULATIONAHA.119.040267
34. Powers WJ, Rabinstein AA, Ackerson T, Adeoye OM, Bambakidis NC, Becker K, et al. Guidelines for the Early Management of Patients With Acute Ischemic Stroke: 2019 Update to the 2018 Guidelines for the Early Management of Acute Ischemic Stroke: A Guideline for Healthcare Professionals From the American Heart Association/American Stroke Association. Stroke (2019) 50(12):e344–418. doi: 10.1161/STR.0000000000000211
35. Wen SW, Wong CHY. An Unexplored Brain-Gut Microbiota Axis in Stroke. Gut Microbes (2017) 8(6):601–6. doi: 10.1080/19490976.2017.1344809
36. Boyanpally A, Cutting S, Furie K. Acute Ischemic Stroke Associated With Myocardial Infarction: Challenges and Management. Semin Neurol (2021) 41(4):331–9. doi: 10.1055/s-0041-1726333
37. Zhang W, Zhou X, Zhang H, Yao Q, Liu Y, Dong Z. Extracellular Vesicles in Diagnosis and Therapy of Kidney Diseases. Am J Physiol Renal Physiol (2016) 311(5):F844–F51. doi: 10.1152/ajprenal.00429.2016
38. Daniel L, Fakhouri F, Joly D, Mouthon L, Nusbaum P, Grunfeld JP, et al. Increase of Circulating Neutrophil and Platelet Microparticles During Acute Vasculitis and Hemodialysis. Kidney Int (2006) 69(8):1416–23. doi: 10.1038/sj.ki.5000306
39. Nadkarni GN, Patel AA, Konstantinidis I, Mahajan A, Agarwal SK, Kamat S, et al. Dialysis Requiring Acute Kidney Injury in Acute Cerebrovascular Accident Hospitalizations. Stroke (2015) 46(11):3226–31. doi: 10.1161/STROKEAHA.115.010985
40. Seifert HA, Collier LA, Chapman CB, Benkovic SA, Willing AE, Pennypacker KR. Pro-Inflammatory Interferon Gamma Signaling Is Directly Associated With Stroke Induced Neurodegeneration. J Neuroimmune Pharmacol (2014) 9(5):679–89. doi: 10.1007/s11481-014-9560-2
41. Dimitrijevic OB, Stamatovic SM, Keep RF, Andjelkovic AV. Absence of the Chemokine Receptor Ccr2 Protects Against Cerebral Ischemia/Reperfusion Injury in Mice. Stroke (2007) 38(4):1345–53. doi: 10.1161/01.STR.0000259709.16654.8f
42. Ji R, Shen H, Pan Y, Wang P, Liu G, Wang Y, et al. Risk Score to Predict Gastrointestinal Bleeding After Acute Ischemic Stroke. BMC Gastroenterol (2014) 14:130. doi: 10.1186/1471-230X-14-130
43. Guo Y, Li Q, Yu X, Liang Y. Rhubarb Anthraquinone Glycosides Protect Against Cerebral Ischemia-Reperfusion Injury in Rats by Regulating Brain-Gut Neurotransmitters. BioMed Chromatogr (2021) 35(5):e5058. doi: 10.1002/bmc.5058
44. Holmes A, Finger C, Morales-Scheihing D, Lee J, McCullough LD. Gut Dysbiosis and Age-Related Neurological Diseases; an Innovative Approach for Therapeutic Interventions. Transl Res (2020) 226:39–56. doi: 10.1016/j.trsl.2020.07.012
45. Busl KM, Bleck TP. Neurogenic Pulmonary Edema. Crit Care Med (2015) 43(8):1710–5. doi: 10.1097/CCM.0000000000001101
46. Martin RH, Yeatts SD, Hill MD, Moy CS, Ginsberg MD, Palesch YY, et al. Alias (Albumin in Acute Ischemic Stroke) Trials: Analysis of the Combined Data From Parts 1 and 2. Stroke (2016) 47(9):2355–9. doi: 10.1161/STROKEAHA.116.012825
47. Ginsberg MD, Palesch YY, Hill MD, Martin RH, Moy CS, Barsan WG, et al. High-Dose Albumin Treatment for Acute Ischaemic Stroke (Alias) Part 2: A Randomised, Double-Blind, Phase 3, Placebo-Controlled Trial. Lancet Neurol (2013) 12(11):1049–58. doi: 10.1016/s1474-4422(13)70223-0
48. Meguro T, Tanabe T, Muraoka K, Terada K, Hirotsune N, Nishino S. Endovascular Treatment for Aneurysmal Subarachnoid Hemorrhage With Neurogenic Pulmonary Edema in the Acute Stage. Turk Neurosurg (2016) 26(6):849–53. doi: 10.5137/1019-5149.JTN.13870-15.1
49. Battaglini D, Robba C, Lopes da Silva A, Dos Santos Samary C, Leme Silva P, Dal Pizzol F, et al. Brain-Heart Interaction After Acute Ischemic Stroke. Crit Care (2020) 24(1):163. doi: 10.1186/s13054-020-02885-8
50. Chen Z, Venkat P, Seyfried D, Chopp M, Yan T, Chen J. Brain-Heart Interaction: Cardiac Complications After Stroke. Circ Res (2017) 121(4):451–68. doi: 10.1161/CIRCRESAHA.117.311170
51. Sposato LA, Hilz MJ, Aspberg S, Murthy SB, Bahit MC, Hsieh CY, et al. Post-Stroke Cardiovascular Complications and Neurogenic Cardiac Injury: Jacc State-Of-the-Art Review. J Am Coll Cardiol (2020) 76(23):2768–85. doi: 10.1016/j.jacc.2020.10.009
52. Scheitz JF, Nolte CH, Doehner W, Hachinski V, Endres M. Stroke–Heart Syndrome: Clinical Presentation and Underlying Mechanisms. Lancet Neurol (2018) 17(12):1109–20. doi: 10.1016/s1474-4422(18)30336-3
53. Barugh AJ, Gray P, Shenkin SD, MacLullich AM, Mead GE. Cortisol Levels and the Severity and Outcomes of Acute Stroke: A Systematic Review. J Neurol (2014) 261(3):533–45. doi: 10.1007/s00415-013-7231-5
54. Kelly JR, Kennedy PJ, Cryan JF, Dinan TG, Clarke G, Hyland NP. Breaking Down the Barriers: The Gut Microbiome, Intestinal Permeability and Stress-Related Psychiatric Disorders. Front Cell Neurosci (2015) 9:392. doi: 10.3389/fncel.2015.00392
55. Iadecola C, Anrather J. The Immunology of Stroke: From Mechanisms to Translation. Nat Med (2011) 17(7):796–808. doi: 10.1038/nm.2399
56. Palma JA, Benarroch EE. Neural Control of the Heart: Recent Concepts and Clinical Correlations. Neurology (2014) 83(3):261–71. doi: 10.1212/WNL.0000000000000605
57. Sörös P, Hachinski V. Cardiovascular and Neurological Causes of Sudden Death After Ischaemic Stroke. Lancet Neurol (2012) 11(2):179–88. doi: 10.1016/s1474-4422(11)70291-5
58. Amarenco P, Kim JS, Labreuche J, Charles H, Abtan J, Bejot Y, et al. A Comparison of Two Ldl Cholesterol Targets After Ischemic Stroke. N Engl J Med (2020) 382(1):9. doi: 10.1056/NEJMoa1910355
59. Rosmond R, Bjorntorp P. The Hypothalamic-Pituitary-Adrenal Axis Activity as a Predictor of Cardiovascular Disease, Type 2 Diabetes and Stroke. J Intern Med (2000) 247(2):188–97. doi: 10.1046/j.1365-2796.2000.00603.x
60. Whitnall MH. Regulation of the Hypothalamic Corticotropin-Releasing Hormone Neurosecretory System. Prog Neurobiol (1993) 40(5):573–629. doi: 10.1016/0301-0082(93)90035-q
61. Jia S, Xia Q, Zhang B, Wang L. Involvement of the Paraventricular Nucleus in the Occurrence of Arrhythmias in Middle Cerebral Artery Occlusion Rats. J Stroke Cerebrovasc Dis (2015) 24(4):844–51. doi: 10.1016/j.jstrokecerebrovasdis.2014.11.025
62. Infanger DW, Cao X, Butler SD, Burmeister MA, Zhou Y, Stupinski JA, et al. Silencing Nox4 in the Paraventricular Nucleus Improves Myocardial Infarction-Induced Cardiac Dysfunction by Attenuating Sympathoexcitation and Periinfarct Apoptosis. Circ Res (2010) 106(11):1763–74. doi: 10.1161/CIRCRESAHA.109.213025
63. Hachinski VC, Smith KE, Silver MD, Gibson CJ, Ciriello J. Acute Myocardial and Plasma Catecholamine Changes in Experimental Stroke. Stroke (1986) 17(3):387–90. doi: 10.1161/01.str.17.3.387
64. Costa VM, Carvalho F, Bastos ML, Carvalho RA, Carvalho M, Remiao F. Contribution of Catecholamine Reactive Intermediates and Oxidative Stress to the Pathologic Features of Heart Diseases. Curr Med Chem (2011) 18(15):2272–314. doi: 10.2174/092986711795656081
65. Nef HM, Mollmann H, Hilpert P, Troidl C, Voss S, Rolf A, et al. Activated Cell Survival Cascade Protects Cardiomyocytes From Cell Death in Tako-Tsubo Cardiomyopathy. Eur J Heart Fail (2009) 11(8):758–64. doi: 10.1093/eurjhf/hfp076
66. Fu Y, Liu Q, Anrather J, Shi FD. Immune Interventions in Stroke. Nat Rev Neurol (2015) 11(9):524–35. doi: 10.1038/nrneurol.2015.144
67. Doll DN, Barr TL, Simpkins JW. Cytokines: Their Role in Stroke and Potential Use as Biomarkers and Therapeutic Targets. Aging Dis (2014) 5(5):294–306. doi: 10.14336/AD.2014.0500294
68. Santos Samary C, Pelosi P, Leme Silva P, Rieken Macedo Rocco P. Immunomodulation After Ischemic Stroke: Potential Mechanisms and Implications for Therapy. Crit Care (2016) 20(1):391. doi: 10.1186/s13054-016-1573-1
69. Acosta SA, Mashkouri S, Nwokoye D, Lee JY, Borlongan CV. Chronic Inflammation and Apoptosis Propagate in Ischemic Cerebellum and Heart of Non-Human Primates. Oncotarget (2017) 8(61):102820–34. doi: 10.18632/oncotarget.18330
70. Adams V, Linke A, Wisloff U, Doring C, Erbs S, Krankel N, et al. Myocardial Expression of Murf-1 and Mafbx After Induction of Chronic Heart Failure: Effect on Myocardial Contractility. Cardiovasc Res (2007) 73(1):120–9. doi: 10.1016/j.cardiores.2006.10.026
71. Rustenhoven J, Aalderink M, Scotter EL, Oldfield RL, Bergin PS, Mee EW, et al. Tgf-Beta1 Regulates Human Brain Pericyte Inflammatory Processes Involved in Neurovasculature Function. J Neuroinflamm (2016) 13:37. doi: 10.1186/s12974-016-0503-0
72. Viereck J, Bang C, Foinquinos A, Thum T. Regulatory Rnas and Paracrine Networks in the Heart. Cardiovasc Res (2014) 102(2):290–301. doi: 10.1093/cvr/cvu039
73. Mann DL. Innate Immunity and the Failing Heart: The Cytokine Hypothesis Revisited. Circ Res (2015) 116(7):1254–68. doi: 10.1161/CIRCRESAHA.116.302317
74. Vahidy FS, Parsha KN, Rahbar MH, Lee M, Bui TT, Nguyen C, et al. Acute Splenic Responses in Patients With Ischemic Stroke and Intracerebral Hemorrhage. J Cereb Blood Flow Metab (2016) 36(6):1012–21. doi: 10.1177/0271678X15607880
75. Yang L, Kong Y, Ren H, Li M, Wei CJ, Shi E, et al. Upregulation of Cd74 and Its Potential Association With Disease Severity in Subjects With Ischemic Stroke. Neurochem Int (2017) 107:148–55. doi: 10.1016/j.neuint.2016.11.007
76. Tsujioka H, Imanishi T, Ikejima H, Kuroi A, Takarada S, Tanimoto T, et al. Impact of Heterogeneity of Human Peripheral Blood Monocyte Subsets on Myocardial Salvage in Patients With Primary Acute Myocardial Infarction. J Am Coll Cardiol (2009) 54(2):130–8. doi: 10.1016/j.jacc.2009.04.021
77. Yang S, Li X, Yang F, Zhao R, Pan X, Liang J, et al. Gut Microbiota-Dependent Marker Tmao in Promoting Cardiovascular Disease: Inflammation Mechanism, Clinical Prognostic, and Potential as a Therapeutic Target. Front Pharmacol (2019) 10:1360. doi: 10.3389/fphar.2019.01360
78. Nagatomo Y, Tang WH. Intersections Between Microbiome and Heart Failure: Revisiting the Gut Hypothesis. J Card Fail (2015) 21(12):973–80. doi: 10.1016/j.cardfail.2015.09.017
79. Zhu W, Gregory JC, Org E, Buffa JA, Gupta N, Wang Z, et al. Gut Microbial Metabolite Tmao Enhances Platelet Hyperreactivity and Thrombosis Risk. Cell (2016) 165(1):111–24. doi: 10.1016/j.cell.2016.02.011
80. Smith M, Reddy U, Robba C, Sharma D, Citerio G. Acute Ischaemic Stroke: Challenges for the Intensivist. Intensive Care Med (2019) 45(9):1177–89. doi: 10.1007/s00134-019-05705-y
81. Ibrahim MS, Samuel B, Mohamed W, Suchdev K. Cardiac Dysfunction in Neurocritical Care: An Autonomic Perspective. Neurocrit Care (2018) 30(3):508–21. doi: 10.1007/s12028-018-0636-3
82. Armstead WM, Vavilala MS. Propranolol Protects Cerebral Autoregulation and Reduces Hippocampal Neuronal Cell Death Through Inhibition of Interleukin-6 Upregulation After Traumatic Brain Injury in Pigs. Br J Anaesthesia (2019) 123(5):610–7. doi: 10.1016/j.bja.2019.07.017
83. January CT, Wann LS, Calkins H, Chen LY, Cigarroa JE, Cleveland JC, et al. 2019 Aha/Acc/Hrs Focused Update of the 2014 Aha/Acc/Hrs Guideline for the Management of Patients With Atrial Fibrillation: A Report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines and the Heart Rhythm Society in Collaboration With the Society of Thoracic Surgeons. Circulation (2019) 140(2). doi: 10.1161/cir.0000000000000665
84. Zhao Q, Yan T, Chopp M, Venkat P, Chen J. Brain-Kidney Interaction: Renal Dysfunction Following Ischemic Stroke. J Cereb Blood Flow Metab (2020) 40(2):246–62. doi: 10.1177/0271678X19890931
85. Huang Y, Wan C, Wu G. Acute Kidney Injury After a Stroke: A Prisma-Compliant Meta-Analysis. Brain Behav (2020) 10(9):e01722. doi: 10.1002/brb3.1722
86. Qureshi AI, Aslam H, Zafar W, Huang W, Lobanova I, Naqvi SH, et al. Acute Kidney Injury in Acute Ischemic Stroke Patients in Clinical Trials. Crit Care Med (2020) 48(9):1334–9. doi: 10.1097/CCM.0000000000004464
87. Rodriguez GJ, Cordina SM, Vazquez G, Suri MF, Kirmani JF, Ezzeddine MA, et al. The Hydration Influence on the Risk of Stroke (Thirst) Study. Neurocrit Care (2009) 10(2):187–94. doi: 10.1007/s12028-008-9169-5
88. Jiang F, Su L, Xiang H, Zhang X, Xu D, Zhang Z, et al. Incidence, Risk Factors, and Biomarkers Predicting Ischemic or Hemorrhagic Stroke Associated Acute Kidney Injury and Outcome: A Retrospective Study in a General Intensive Care Unit. Blood Purif (2019) 47(4):317–26. doi: 10.1159/000499029
89. Hu Z, Shang T, Huang R, Li Q, Zheng P, Wang H, et al. Renal Safety of Intra-Arterial Treatment After Acute Ischemic Stroke With Multimodal Ct Imaging Selection. J Stroke Cerebrovasc Dis (2019) 28(7):2031–7. doi: 10.1016/j.jstrokecerebrovasdis.2019.02.027
90. Toyoda K, Ninomiya T. Stroke and Cerebrovascular Diseases in Patients With Chronic Kidney Disease. Lancet Neurol (2014) 13(8):823–33. doi: 10.1016/s1474-4422(14)70026-2
91. Dad T, Weiner DE. Stroke and Chronic Kidney Disease: Epidemiology, Pathogenesis, and Management Across Kidney Disease Stages. Semin Nephrol (2015) 35(4):311–22. doi: 10.1016/j.semnephrol.2015.06.003
92. Hojs Fabjan T, Hojs R. Stroke and Renal Dysfunction. Eur J Intern Med (2014) 25(1):18–24. doi: 10.1016/j.ejim.2013.08.710
93. Putaala J, Haapaniemi E, Gordin D, Liebkind R, Groop PH, Kaste M, et al. Factors Associated With Impaired Kidney Function and Its Impact on Long-Term Outcome in Young Ischemic Stroke. Stroke (2011) 42(9):2459–64. doi: 10.1161/STROKEAHA.110.612721
94. Hotter B, Hoffmann S, Ulm L, Montaner J, Bustamante A, Meisel C, et al. Inflammatory and Stress Markers Predicting Pneumonia, Outcome, and Etiology in Patients With Stroke: Biomarkers for Predicting Pneumonia, Functional Outcome, and Death After Stroke. Neurol Neuroimmunol Neuroinflamm (2020) 7(3). doi: 10.1212/NXI.0000000000000692
95. Kalra L, Hodsoll J, Irshad S, Smithard D, Manawadu D, Investigators S-I. Comparison of the Diagnostic Utility of Physician-Diagnosed With Algorithm-Defined Stroke-Associated Pneumonia. J Neurol Neurosurg Psychiatry (2016) 87(11):1163–8. doi: 10.1136/jnnp-2016-313508
96. Robinson TG, Potter JF, Ford GA, Bulpitt CJ, Chernova J, Jagger C, et al. Effects of Antihypertensive Treatment After Acute Stroke in the Continue or Stop Post-Stroke Antihypertensives Collaborative Study (Cossacs): A Prospective, Randomised, Open, Blinded-Endpoint Trial. Lancet Neurol (2010) 9(8):767–75. doi: 10.1016/s1474-4422(10)70163-0
97. Manning LS, Mistri AK, Potter J, Rothwell PM, Robinson TG. Short-Term Blood Pressure Variability in Acute Stroke: Post Hoc Analysis of the Controlling Hypertension and Hypotension Immediately Post Stroke and Continue or Stop Post-Stroke Antihypertensives Collaborative Study Trials. Stroke (2015) 46(6):1518–24. doi: 10.1161/STROKEAHA.115.009078
98. Labovitz AJ, Rose DZ, Fradley MG, Meriwether JN, Renati S, Martin R, et al. Early Apixaban Use Following Stroke in Patients With Atrial Fibrillation: Results of the Arest Trial. Stroke (2021) 52(4):1164–71. doi: 10.1161/STROKEAHA.120.030042
99. Mizoguchi T, Tanaka K, Toyoda K, Yoshimura S, Itabashi R, Takagi M, et al. Early Initiation of Direct Oral Anticoagulants After Onset of Stroke and Short- and Long-Term Outcomes of Patients With Nonvalvular Atrial Fibrillation. Stroke (2020) 51(3):883–91. doi: 10.1161/STROKEAHA.119.028118
100. Viscoli CM, Brass LM, Carolei A, Conwit R, Ford GA, Furie KL, et al. Pioglitazone for Secondary Prevention After Ischemic Stroke and Transient Ischemic Attack: Rationale and Design of the Insulin Resistance Intervention After Stroke Trial. Am Heart J (2014) 168(6):823–9.e6. doi: 10.1016/j.ahj.2014.07.016
101. Wong KY, MacWalter RS, Fraser HW, Crombie I, Ogston SA, Struthers AD. Urate Predicts Subsequent Cardiac Death in Stroke Survivors. Eur Heart J (2002) 23(10):788–93. doi: 10.1053/euhj.2001.2970
102. Nous A, Peeters I, Nieboer K, Vanbinst AM, De Keyser J, De Raedt S. Post-Stroke Infections Associated With Spleen Volume Reduction: A Pilot Study. PLoS One (2020) 15(5):e0232497. doi: 10.1371/journal.pone.0232497
103. Amarenco P, Callahan A 3rd, Campese VM, Goldstein LB, Hennerici MG, Messig M, et al. Effect of High-Dose Atorvastatin on Renal Function in Subjects With Stroke or Transient Ischemic Attack in the Sparcl Trial. Stroke (2014) 45(10):2974–82. doi: 10.1161/STROKEAHA.114.005832
104. Amaro S, Canovas D, Castellanos M, Gallego J, Marti-Febregas J, Segura T, et al. The Urico-Ictus Study, a Phase 3 Study of Combined Treatment With Uric Acid and Rtpa Administered Intravenously in Acute Ischaemic Stroke Patients Within the First 4.5 H of Onset of Symptoms. Int J Stroke (2010) 5(4):325–8. doi: 10.1111/j.1747-4949.2010.00448.x
105. Ikeme JC, Pergola PE, Scherzer R, Shlipak MG, Benavente OR, Peralta CA. Post Hoc Analyses of Randomized Clinical Trial for the Effect of Clopidogrel Added to Aspirin on Kidney Function. Clin J Am Soc Nephrol (2017) 12(7):1040–7. doi: 10.2215/CJN.00100117
106. Mikulik R, Eckstein J, Pearce LA, Mundl H, Rudilosso S, Olavarria VV, et al. Frequency and Predictors of Major Bleeding in Patients With Embolic Strokes of Undetermined Source: Navigate-Esus Trial. Stroke (2020) 51(7):2139–47. doi: 10.1161/STROKEAHA.119.027995
107. Bath PM, Scutt P, Love J, Clave P, Cohen D, Dziewas R, et al. Pharyngeal Electrical Stimulation for Treatment of Dysphagia in Subacute Stroke: A Randomized Controlled Trial. Stroke (2016) 47(6):1562–70. doi: 10.1161/STROKEAHA.115.012455
108. Dziewas R, Stellato R, van der Tweel I, Walther E, Werner CJ, Braun T, et al. Pharyngeal Electrical Stimulation for Early Decannulation in Tracheotomised Patients With Neurogenic Dysphagia After Stroke (Phast-Trac): A Prospective, Single-Blinded, Randomised Trial. Lancet Neurol (2018) 17(10):849–59. doi: 10.1016/s1474-4422(18)30255-2
109. Vasant DH, Michou E, O'Leary N, Vail A, Mistry S, Hamdy S, et al. Pharyngeal Electrical Stimulation in Dysphagia Poststroke: A Prospective, Randomized Single-Blinded Interventional Study. Neurorehabil Neural Repair (2016) 30(9):866–75. doi: 10.1177/1545968316639129
110. Shah B, Jagtap P, Sarmah D, Datta A, Raut S, Sarkar A, et al. Cerebro-Renal Interaction and Stroke. Eur J Neurosci (2021) 53(4):1279–99. doi: 10.1111/ejn.14983
111. Lau WL, Huisa BN, Fisher M. The Cerebrovascular-Chronic Kidney Disease Connection: Perspectives and Mechanisms. Transl Stroke Res (2017) 8(1):67–76. doi: 10.1007/s12975-016-0499-x
112. Smets P, Meyer E, Maddens B, Daminet S. Cushing's Syndrome, Glucocorticoids and the Kidney. Gen Comp Endocrinol (2010) 169(1):1–10. doi: 10.1016/j.ygcen.2010.07.004
113. Nongnuch A, Panorchan K, Davenport A. Brain-Kidney Crosstalk. Crit Care (2014) 18(3):225. doi: 10.1186/cc13907
114. Meyer JS, Stoica E, Pascu I, Shimazu K, Hartmann A. Catecholamine Concentrations in Csf and Plasma of Patients With Cerebral Infarction and Haemorrhage. Brain (1973) 96(2):277–88. doi: 10.1093/brain/96.2.277
115. Fujii T, Kurata H, Takaoka M, Muraoka T, Fujisawa Y, Shokoji T, et al. The Role of Renal Sympathetic Nervous System in the Pathogenesis of Ischemic Acute Renal Failure. Eur J Pharmacol (2003) 481(2-3):241–8. doi: 10.1016/j.ejphar.2003.09.036
116. Brewster UC, Perazella MA. The Renin-Angiotensin-Aldosterone System and the Kidney: Effects on Kidney Disease. Am J Med (2004) 116(4):263–72. doi: 10.1016/j.amjmed.2003.09.034
117. Mogi M, Kawajiri M, Tsukuda K, Matsumoto S, Yamada T, Horiuchi M. Serum Levels of Renin-Angiotensin System Components in Acute Stroke Patients. Geriatr Gerontol Int (2014) 14(4):793–8. doi: 10.1111/ggi.12167
118. Imig JD, Ryan MJ. Immune and Inflammatory Role in Renal Disease. Compr Physiol (2013) 3(2):957–76. doi: 10.1002/cphy.c120028
119. Ruiz-Ortega M, Egido J. Angiotensin II Modulates Cell Growth-Related Events and Synthesis of Matrix Proteins in Renal Interstitial Fibroblasts. Kidney Int (1997) 52(6):1497–510. doi: 10.1038/ki.1997.480
120. Zhang W, Wang W, Yu H, Zhang Y, Dai Y, Ning C, et al. Interleukin 6 Underlies Angiotensin II-Induced Hypertension and Chronic Renal Damage. Hypertension (2012) 59(1):136–44. doi: 10.1161/HYPERTENSIONAHA.111.173328
121. Davenport A. The Brain and the Kidney–Organ Cross Talk and Interactions. Blood Purif (2008) 26(6):526–36. doi: 10.1159/000167800
122. Sproston NR, Ashworth JJ. Role of C-Reactive Protein at Sites of Inflammation and Infection. Front Immunol (2018) 9:754. doi: 10.3389/fimmu.2018.00754
123. Ladenvall C, Jood K, Blomstrand C, Nilsson S, Jern C, Ladenvall P. Serum C-Reactive Protein Concentration and Genotype in Relation to Ischemic Stroke Subtype. Stroke (2006) 37(8):2018–23. doi: 10.1161/01.STR.0000231872.86071.68
124. Stuveling EM, Hillege HL, Bakker SJL, Gans ROB, de Jong PE, de Zeeuw D. C-Reactive Protein Is Associated With Renal Function Abnormalities in a Non-Diabetic Population. Kidney Int (2003) 63(2):654–61. doi: 10.1046/j.1523-1755.2003.00762.x
125. Chodobski A, Zink BJ, Szmydynger-Chodobska J. Blood-Brain Barrier Pathophysiology in Traumatic Brain Injury. Transl Stroke Res (2011) 2(4):492–516. doi: 10.1007/s12975-011-0125-x
126. Baud L, Ardaillou R. Reactive Oxygen Species: Production and Role in the Kidney. Am J Physiol (1986) 251(5 Pt 2):F765–76. doi: 10.1152/ajprenal.1986.251.5.F765
127. Clausen BH, Lambertsen KL, Meldgaard M, Finsen B. A Quantitative In Situ Hybridization and Polymerase Chain Reaction Study of Microglial-Macrophage Expression of Interleukin-1beta Mrna Following Permanent Middle Cerebral Artery Occlusion in Mice. Neuroscience (2005) 132(4):879–92. doi: 10.1016/j.neuroscience.2005.01.031
128. Faubel S, Lewis EC, Reznikov L, Ljubanovic D, Hoke TS, Somerset H, et al. Cisplatin-Induced Acute Renal Failure Is Associated With an Increase in the Cytokines Interleukin (Il)-1beta, Il-18, Il-6, and Neutrophil Infiltration in the Kidney. J Pharmacol Exp Ther (2007) 322(1):8–15. doi: 10.1124/jpet.107.119792
129. Chaturvedi M, Kaczmarek L. Mmp-9 Inhibition: A Therapeutic Strategy in Ischemic Stroke. Mol Neurobiol (2014) 49(1):563–73. doi: 10.1007/s12035-013-8538-z
130. Piedagnel R, Murphy G, Ronco PM, Lelongt B. Matrix Metalloproteinase 2 (Mmp2) and Mmp9 Are Produced by Kidney Collecting Duct Principal Cells But Are Differentially Regulated by Sv40 Large-T, Arginine Vasopressin, and Epidermal Growth Factor. J Biol Chem (1999) 274(3):1614–20. doi: 10.1074/jbc.274.3.1614
131. Pulido-Olmo H, Garcia-Prieto CF, Alvarez-Llamas G, Barderas MG, Vivanco F, Aranguez I, et al. Role of Matrix Metalloproteinase-9 in Chronic Kidney Disease: A New Biomarker of Resistant Albuminuria. Clin Sci (Lond) (2016) 130(7):525–38. doi: 10.1042/CS20150517
132. Karpman D, Stahl AL, Arvidsson I. Extracellular Vesicles in Renal Disease. Nat Rev Nephrol (2017) 13(9):545–62. doi: 10.1038/nrneph.2017.98
133. Trionfini P, Benigni A, Remuzzi G. Micrornas in Kidney Physiology and Disease. Nat Rev Nephrol (2015) 11(1):23–33. doi: 10.1038/nrneph.2014.202
134. Zhong X, Chung AC, Chen HY, Meng XM, Lan HY. Smad3-Mediated Upregulation of Mir-21 Promotes Renal Fibrosis. J Am Soc Nephrol (2011) 22(9):1668–81. doi: 10.1681/ASN.2010111168
135. Wang X, Gao Y, Tian N, Zou D, Shi Y, Zhang N. Astragaloside Iv Improves Renal Function and Fibrosis Via Inhibition of Mir-21-Induced Podocyte Dedifferentiation and Mesangial Cell Activation in Diabetic Mice. Drug Des Devel Ther (2018) 12:2431–42. doi: 10.2147/DDDT.S170840
136. Long J, Wang Y, Wang W, Chang BH, Danesh FR. Microrna-29c Is a Signature Microrna Under High Glucose Conditions That Targets Sprouty Homolog 1, and Its In Vivo Knockdown Prevents Progression of Diabetic Nephropathy. J Biol Chem (2011) 286(13):11837–48. doi: 10.1074/jbc.M110.194969
137. Naganuma M, Koga M, Shiokawa Y, Nakagawara J, Furui E, Kimura K, et al. Reduced Estimated Glomerular Filtration Rate Is Associated With Stroke Outcome After Intravenous Rt-Pa: The Stroke Acute Management With Urgent Risk-Factor Assessment and Improvement (Samurai) Rt-Pa Registry. Cerebrovasc Dis (2011) 31(2):123–9. doi: 10.1159/000321516
138. Reinecke H, Engelbertz C, Schabitz WR. Preventing Stroke in Patients With Chronic Kidney Disease and Atrial Fibrillation: Benefit and Risks of Old and New Oral Anticoagulants. Stroke (2013) 44(10):2935–41. doi: 10.1161/STROKEAHA.113.001701
139. Jardine MJ, Ninomiya T, Perkovic V, Cass A, Turnbull F, Gallagher MP, et al. Aspirin Is Beneficial in Hypertensive Patients With Chronic Kidney Disease: A Post-Hoc Subgroup Analysis of a Randomized Controlled Trial. J Am Coll Cardiol (2010) 56(12):956–65. doi: 10.1016/j.jacc.2010.02.068
140. Liu ZJ, Chen C, Li FW, Shen JM, Yang YY, Leak RK, et al. Splenic Responses in Ischemic Stroke: New Insights Into Stroke Pathology. CNS Neurosci Ther (2015) 21(4):320–6. doi: 10.1111/cns.12361
141. Seifert HA, Offner H. The Splenic Response to Stroke: From Rodents to Stroke Subjects. J Neuroinflamm (2018) 15(1):195. doi: 10.1186/s12974-018-1239-9
142. Sahota P, Vahidy F, Nguyen C, Bui TT, Yang B, Parsha K, et al. Changes in Spleen Size in Patients With Acute Ischemic Stroke: A Pilot Observational Study. Int J Stroke (2013) 8(2):60–7. doi: 10.1111/ijs.12022
143. Offner H, Subramanian S, Parker SM, Wang C, Afentoulis ME, Lewis A, et al. Splenic Atrophy in Experimental Stroke Is Accompanied by Increased Regulatory T Cells and Circulating Macrophages. J Immunol (2006) 176(11):6523–31. doi: 10.4049/jimmunol.176.11.6523
144. Seifert HA, Hall AA, Chapman CB, Collier LA, Willing AE, Pennypacker KR. A Transient Decrease in Spleen Size Following Stroke Corresponds to Splenocyte Release Into Systemic Circulation. J Neuroimmune Pharmacol (2012) 7(4):1017–24. doi: 10.1007/s11481-012-9406-8
145. Seifert HA, Leonardo CC, Hall AA, Rowe DD, Collier LA, Benkovic SA, et al. The Spleen Contributes to Stroke Induced Neurodegeneration Through Interferon Gamma Signaling. Metab Brain Dis (2012) 27(2):131–41. doi: 10.1007/s11011-012-9283-0
146. Ahmad M, Graham SH. Inflammation After Stroke: Mechanisms and Therapeutic Approaches. Transl Stroke Res (2010) 1(2):74–84. doi: 10.1007/s12975-010-0023-7
147. Mignini F, Streccioni V, Amenta F. Autonomic Innervation of Immune Organs and Neuroimmune Modulation. Auton Autacoid Pharmacol (2003) 23(1):1–25. doi: 10.1046/j.1474-8673.2003.00280.x
148. Ajmo CT Jr., Collier LA, Leonardo CC, Hall AA, Green SM, Womble TA, et al. Blockade of Adrenoreceptors Inhibits the Splenic Response to Stroke. Exp Neurol (2009) 218(1):47–55. doi: 10.1016/j.expneurol.2009.03.044
149. Magnus T, Wiendl H, Kleinschnitz C. Immune Mechanisms of Stroke. Curr Opin Neurol (2012) 25(3):334–40. doi: 10.1097/WCO.0b013e328352ede6
150. Famakin BM. The Immune Response to Acute Focal Cerebral Ischemia and Associated Post-Stroke Immunodepression: A Focused Review. Aging Dis (2014) 5(5):307–26. doi: 10.14336/AD.2014.0500307
151. Offner H, Subramanian S, Parker SM, Afentoulis ME, Vandenbark AA, Hurn PD. Experimental Stroke Induces Massive, Rapid Activation of the Peripheral Immune System. J Cereb Blood Flow Metab (2006) 26(5):654–65. doi: 10.1038/sj.jcbfm.9600217
152. Bao Y, Kim E, Bhosle S, Mehta H, Cho S. A Role for Spleen Monocytes in Post-Ischemic Brain Inflammation and Injury. J Neuroinflamm (2010) 7:92. doi: 10.1186/1742-2094-7-92
153. Wang Z, He D, Zeng YY, Zhu L, Yang C, Lu YJ, et al. The Spleen May Be an Important Target of Stem Cell Therapy for Stroke. J Neuroinflamm (2019) 16(1):20. doi: 10.1186/s12974-019-1400-0
154. Ajmo CT Jr., Vernon DO, Collier L, Hall AA, Garbuzova-Davis S, Willing A, et al. The Spleen Contributes to Stroke-Induced Neurodegeneration. J Neurosci Res (2008) 86(10):2227–34. doi: 10.1002/jnr.21661
155. An C, Shi Y, Li P, Hu X, Gan Y, Stetler RA, et al. Molecular Dialogs Between the Ischemic Brain and the Peripheral Immune System: Dualistic Roles in Injury and Repair. Prog Neurobiol (2014) 115:6–24. doi: 10.1016/j.pneurobio.2013.12.002
156. Pennypacker KR. Targeting the Peripheral Inflammatory Response to Stroke: Role of the Spleen. Transl Stroke Res (2014) 5(6):635–7. doi: 10.1007/s12975-014-0372-8
157. Vendrame M, Gemma C, Pennypacker KR, Bickford PC, Davis Sanberg C, Sanberg PR, et al. Cord Blood Rescues Stroke-Induced Changes in Splenocyte Phenotype and Function. Exp Neurol (2006) 199(1):191–200. doi: 10.1016/j.expneurol.2006.03.017
158. Chen J, Sanberg PR, Li Y, Wang L, Lu M, Willing AE, et al. Intravenous Administration of Human Umbilical Cord Blood Reduces Behavioral Deficits After Stroke in Rats. Stroke (2001) 32(11):2682–8. doi: 10.1161/hs1101.098367
159. Yang B, Hamilton JA, Valenzuela KS, Bogaerts A, Xi X, Aronowski J, et al. Multipotent Adult Progenitor Cells Enhance Recovery After Stroke by Modulating the Immune Response From the Spleen. Stem Cells (2017) 35(5):1290–302. doi: 10.1002/stem.2600
160. Schaller BJ, Graf R, Jacobs AH. Pathophysiological Changes of the Gastrointestinal Tract in Ischemic Stroke. Am J Gastroenterol (2006) 101(7):1655–65. doi: 10.1111/j.1572-0241.2006.00540.x
161. Chou YF, Weng WC, Huang WY. Association Between Gastrointestinal Bleeding and 3-Year Mortality in Patients With Acute, First-Ever Ischemic Stroke. J Clin Neurosci (2017) 44:289–93. doi: 10.1016/j.jocn.2017.06.068
162. Hsu HL, Lin YH, Huang YC, Weng HH, Lee M, Huang WY, et al. Gastrointestinal Hemorrhage After Acute Ischemic Stroke and Its Risk Factors in Asians. Eur Neurol (2009) 62(4):212–8. doi: 10.1159/000229018
163. O'Donnell MJ, Kapral MK, Fang J, Saposnik G, Eikelboom JW, Oczkowski W, et al. Gastrointestinal Bleeding After Acute Ischemic Stroke. Neurology (2008) 71(9):650–5. doi: 10.1212/01.wnl.0000319689.48946.25
164. Fu J. Factors Affecting the Occurrence of Gastrointestinal Bleeding in Acute Ischemic Stroke Patients. Med (Baltimore) (2019) 98(28):e16312. doi: 10.1097/MD.0000000000016312
165. Nema H, Kato M. Investigation of Gastroduodenal Mucosal Injuries Caused by Low-Dose Aspirin Therapy in Patients With Cerebral Infarction. J Gastroenterol Hepatol (2010) 25 Suppl 1:S119–21. doi: 10.1111/j.1440-1746.2010.06229.x
166. Kamal AK, Naqvi I, Husain MR, Khealani BA. Cilostazol Versus Aspirin for Secondary Prevention of Vascular Events After Stroke of Arterial Origin. Cochrane Database Syst Rev (2011) 1)::CD008076. doi: 10.1002/14651858.CD008076.pub2
167. Camara-Lemarroy CR, Ibarra-Yruegas BE, Gongora-Rivera F. Gastrointestinal Complications After Ischemic Stroke. J Neurol Sci (2014) 346(1-2):20–5. doi: 10.1016/j.jns.2014.08.027
168. Kawakubo K, Ibayashi S, Nagao T, Doi K, Aoyagi K, Iida M, et al. Brain Ischemia and Gastric Mucosal Damage in Spontaneously Hypertensive Rats: The Role of Arterial Vagal Adrenoceptors. Dig Dis Sci (1996) 41(12):2383–91. doi: 10.1007/BF02100132
169. Collaboration FT. Poor Nutritional Status on Admission Predicts Poor Outcomes After Stroke: Observational Data From the Food Trial. Stroke (2003) 34(6):1450–6. doi: 10.1161/01.STR.0000074037.49197.8C
170. Malloy RJ, Kanaan AO, Silva MA, Donovan JL. Evaluation of Antiplatelet Agents for Secondary Prevention of Stroke Using Mixed Treatment Comparison Meta-Analysis. Clin Ther (2013) 35(10):1490–500.e7. doi: 10.1016/j.clinthera.2013.09.004
Keywords: stroke, peripheral organ injury, lung, heart, kidney, spleen, gastrointestinal tract
Citation: Wang J, Zhang J, Ye Y, Xu Q, Li Y, Feng S, Xiong X, Jian Z and Gu L (2022) Peripheral Organ Injury After Stroke. Front. Immunol. 13:901209. doi: 10.3389/fimmu.2022.901209
Received: 21 March 2022; Accepted: 21 April 2022;
Published: 01 June 2022.
Edited by:
Junlei Chang, Shenzhen Institutes of Advanced Technology (CAS), ChinaCopyright © 2022 Wang, Zhang, Ye, Xu, Li, Feng, Xiong, Jian and Gu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zhihong Jian, emhpaG9uZ0B3aHUuZWR1LmNu; Lijuan Gu, Z3VsaWp1YW5Ad2h1LmVkdS5jbg==
†These authors have contributed equally to this work