 Julien C. Marie
Julien C. Marie Edith Bonnelye
Edith Bonnelye
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Immunol. , 23 May 2022
Sec. Cancer Immunity and Immunotherapy
Volume 13 - 2022 | https://doi.org/10.3389/fimmu.2022.899104
This article is part of the Research Topic The Bone/Bone Marrow Microenvironment: A Hub for Immune Regulation of the Tumor Cells Fate View all 6 articles
Bone loss associated with estrogen deficiency indicates a fundamental role of these hormones in skeletal growth and bone remodeling. In the last decades, growing recent evidence demonstrated that estrogens can also affect the immune compartment of the bone. In this review, we summarize the impacts of estrogens on bone immune cells and their consequences on bone homeostasis, metastasis settlement into the bone and tumor progression. We also addressed the role of an orphan nuclear receptor ERRalpha (“Estrogen-receptor Related Receptor alpha”) on macrophages and T lymphocytes, and as an immunomodulator in bone metastases. Hence, this review links estrogens to bone immune cells in osteo-oncology.
Estrogens family of steroid hormones composed of estradiol, estrone, estriol and estetrol. Estradiol (E2) is the most prevalent and the most potent estrogen. Though ovaries are the main producers of estrogens, fat tissues, testes, the adrenal cortex, and the liver also contribute to their production (1). Estrogens are major players in both skeletal growth, particularly during puberty, and skeletal maintenance, including normal bone mineral density and trabecular bone mass, during adult life. Reduced estrogen levels at menopause for women and later in life for men lead to decreased bone density and microarchitecture deterioration resulting in a high risk of fracture (1).
Estrogens exert their actions through genotropic and non-genotropic signaling pathways. They can bind to two nuclear receptor isoforms, estrogen receptor-α (ERα) and ERβ which, once their ligand pocket is occupied, migrate to the nucleus and fulfil their roles as transcription factors by binding to dedicated DNA sequences (2). ERα can also activate kinases (MEK, ERK, JNK) and modulates several transcription factor activities (c-jun, Elk-1) (3). More recently, a G Protein-Coupled Estrogen Receptor (GPER1) has been discovered. Unlike ERα/β, GPER1 binds estrogens with lower affinity (4). In addition, based on a strong similarity with ERα DNA binding domain (68%) but a moderate similarity (36%) with the ligand pocket of ER, precluding estrogen binding, three orphan nuclear receptors referred to as Estrogen receptor-Related Receptors (ERRs) ERRα, ERRβ and ERRγ have been described (5). Despite the identification of putative ligands, many data support the fact that ERRα impinges on the estrogen signaling pathway in numerous tissues including the bone (6–8).
Skeletal metastases are frequent complications of many cancers of which prostate (PCa) and breast (BCa) cancers are the most frequent with a 73% and 68% incidence of bone metastases (BMet) (9). BMet development requires first cancer cell extravasation and homing to the bone marrow through interactions with endothelial cells and with osteoblasts (bone-forming cells). Once in the bone, cancer cells can then disrupt the osteoblast/osteoclast balance; in favor of osteoclasts (bone-resorbing cells) or osteoblasts inducing osteolysis or osteoblastic/mixed lesions. Aside from bone cells, immune cells in the bone marrow (BM) also strongly influence BMet (10). In pre-clinical models, estrogen deficiency (OVX) has been shown to fuel BMet, since the declining production of the sex steroid by the ovaries and inflammatory tone associated with estrogen deficiency modified the bone microenvironment, mainly the osteoclasts, in such a way that cancer cell anchorage, survival and osteolytic phenotype were stimulated (11). Conversely in clinical studies, incidence of BM disseminated tumor cells (DTC) was reported to be slightly higher in pre-menopausal women (32.7%) versus postmenopausal (29.5%), suggesting that the BM of post-menopausal women is less attractive to metastasis (12). Of note, all immune cells present in the bone express the ERs (13), and growing evidence suggests a role for estrogens in BMet through their action on bone immune cells. Moreover, regarding basic science, it is important to keep in mind that a large part of the cultures of hematopoietic cells are carried out with media containing phenol red, known to have estrogenic activity. This review aims at presenting our current knowledge and our own thoughts on the links between estrogen signaling in bone immune cells and their impact on metastatic cell homing and progression within the bone/BM microenvironment.
Aside from erythrocytes, myeloid cells, including mainly neutrophils, monocytes/macrophages and osteoclasts are the most abundant hematopoietic cells in the bone (14). All of these myeloid cells, the differentiation of which occurs in partially or completely in the BM, express not only ERα/β but also GPER1 (15). We will see that estrogens affect both their differentiation and function in the bone with consequences on BMet niche, cancer cell homing and progression in the bone.
Neutrophils largely outnumber the rest of the myeloid cells in the bone (16), where their numbers are influenced by estrogens. In the 80’s, experiments based on the injection of estrogens to male mice revealed a profound neutropenia in the BM (17, 18). Since then, it has been reported that estrogens impact both neutrophil differentiation and functions. Neutrophil differentiation is largely promoted by the Granulocyte-Colony-Stimulating Factor (GM-CSF). In the absence of estrogen, B cells present in the BM secrete more G-CSF, which can contribute to the neutrophilia observed in estrogen-deficient patients (19). Neutropenia in the bone associated with estrogens can also be explained by the ability of estrogens to down-regulate the production of CXC chemokine ligand (CXCL)12 by osteoblasts and BM stromal cells (20). Indeed, if a large portion of mature neutrophils leave the BM, the fraction expressing CXCR4, receptor of CXCL12, stays within the bone (20). These BM-resident neutrophils secrete Proteinase 3 (PR3), a serine protease which through its interaction with Receptor for Advanced Glycation Endproducts (RAGE) at the surface of metastatic PCa could enhance their homing to the bone (21). Though only the depletion of neutrophils in vivo will firmly validate this conclusion, this observation strongly suggests that a large number of neutrophils in the bone could contribute to BMet incidence from PCa cells by encouraging PCa cell anchorage in the BM. One feature of neutrophils is that they form NETs (Neutrophil Extracellular Traps), which not only prevents pathogen progression but also, as recently demonstrated, contributes to the sequestring of liver and lung metastatic cells in the bone promoting their implantation (22). Inhibition of ERα and ERβ signaling in neutrophils by using selective antagonist raloxifene was associated with inhibition of NETs suggesting that estrogen promote NETs formation into the bone (23). It is noteworthy that NET fails to attract primary cancer cells from breast to bone. This difference depending on metastases origin was associated with the ability of metastasis to express or not CCD25, which recognizes DNA chromatin filaments composing the NET (22). Once settled in the bone, BMets originating from PCa DTC stimulate oxidative bursts in neutrophils, increasing NET formation. In turn, neutrophils induce apoptosis of PCa cells by inhibiting STAT5 signaling in cancer cells in a manner that remains to be understood (24). Interestingly, this cytotoxic property of neutrophils decreases with time and in late-stage bone tumors. High levels of E2 also lead to a decrease in degranulation of β-glucuronidase and lysozyme, as well as in the intracellular concentration of reactive oxygen species (ROS) in neutrophils known to stimulate osteoclasts and osteoblast apoptosis (25–27). Hence, by controlling both neutrophil homeostasis and functions in the bone, estrogens repress the trapping of disseminated cells in the bone, but diminish the anti-tumor activity at an early stage and decrease the osteogenic feature associated with neutrophils once the tumor is settled within the bone (Figure 1).
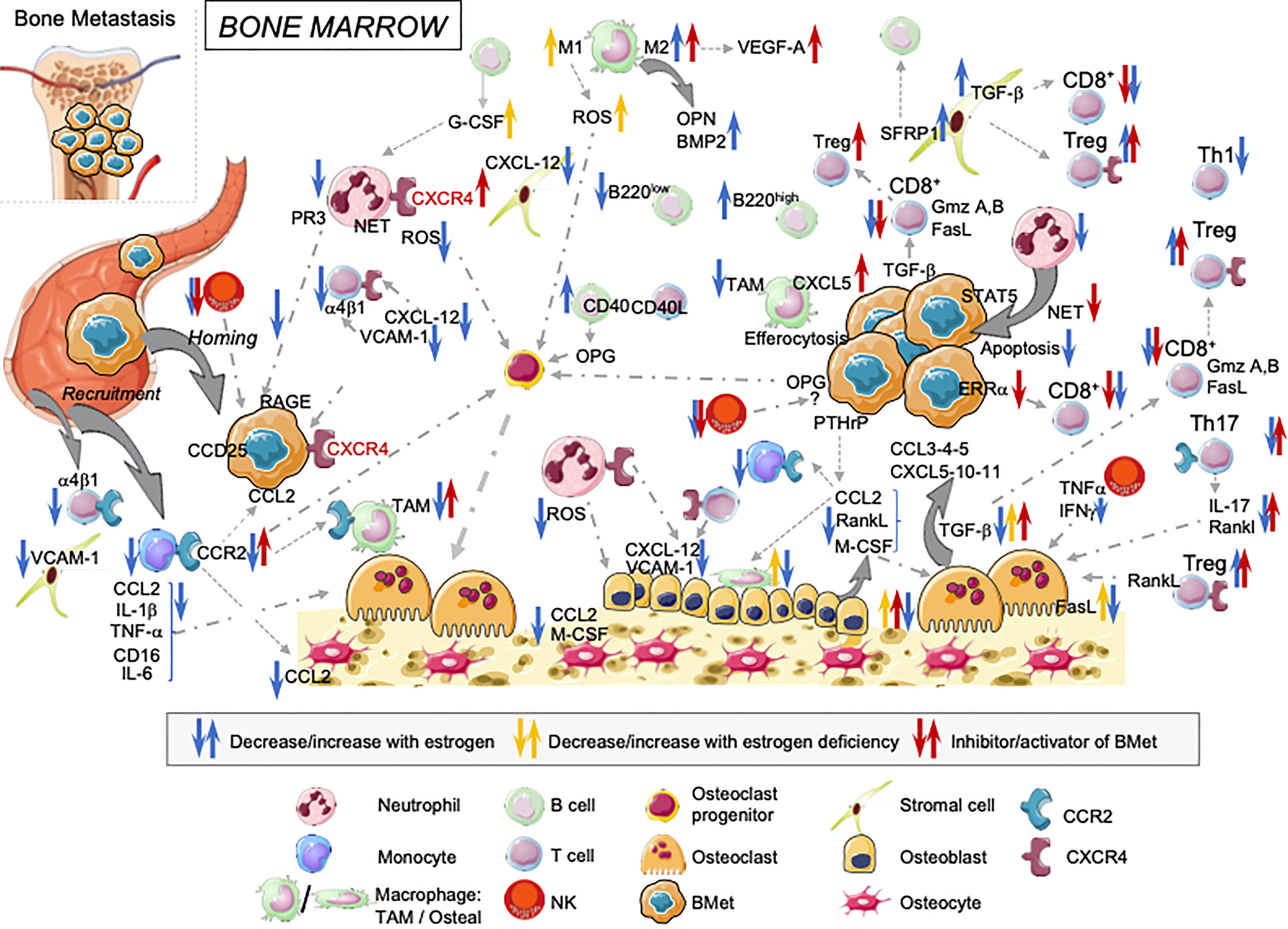
Figure 1 Overview of estrogens regulating immune landscape on the bone marrow cells: consequences on cancer cells anchorage and tumor progression in bone. Estrogens influence BM cells at different steps. They modulate both their development and functions with direct consequences on bone osteogenesis, cancer cell implantation and tumor growth in the bone.
In addition to neutropenia, estrogens also repress the differentiation of mononuclear phagocytic cells. Indeed, estrogens reduce the levels of Macrophage-Colony-Stimulating Factors (M-CSF), mainly produced by osteocytes, osteoblasts, and osteoblast precursors as well as Granulocyte-Macrophage Colony-Stimulating Factor (GM-CSF), this latter being secreted by multiple cell types in the bone mainly in response to danger stimuli (28–31). Bone macrophages, mostly arise through the differentiation of monocytes returning to the bone. In addition to repressing monocyte/macrophage lineage differentiation, estrogens also reduce concentrations of CCL2 (C-C Motif Chemokine Ligand 2) produced by osteoblasts and osteocytes, and thus the recruitment to the bone of monocytes and macrophage-precursors which expressed CCR2 (32). Estrogens also directly inhibit the production of CCL2 by monocyte/macrophage and thus the positive loop of macrophage recruitment to the bone (31). Interestingly, depending on the ER engaged, estrogens can have different direct effects on monocyte/macrophage functions. In monocytes/macrophages, E2/ERα inhibits the production of pro-inflammatory cytokines, such as IL-1β, TNF-α, IL-6, documented to reduce osteoclast differentiation (31–33). Conversely, E2/ERβ represses CD16 surface expression and thus ADCC (antigen-dependent cellular cytotoxicity) (34). Whether different stages of macrophage maturation are more strongly associated with the expression of one type of ER than another remains to be addressed. ERRα, whose expression is up-regulated by estrogens in several tissues including the bone (8, 35, 36), is involved in macrophage functions. More precisely, it can regulate macrophage response to TLR4 and IFNγ as well as ROS production (37–39). Through their potent phagocytic activity, macrophages represent powerful cells to eliminate metastatic cells reaching the bone, albeit they can also influence BMet through other mechanisms that can be targeted by estrogens. “Indeed, estrogens strongly influence macrophage polarization by promoting M2 (pro-osteoblastic) and suppressing M1 (pro-osteoclastic) (34, 40). Therefore, by promoting M1 macrophages estrogen deficiency inhibits BMP2 production by M2 and stimulate pro-osteoclastic molecules production ROS, nitric oxide, and pro-inflammatory cytokines by M1, promoting bone resorption (41). If this balance in favor of M2 contributes to preventing the establishment of the metastatic niche in bone, once metastasis is implanted, M2-like macrophages promote metastasis angiogenesis and tumor progression through their production of VEGF-A (10). In the bone, a peculiar subset of macrophages named osteal macrophages, or osteomacs, has been described (42). Osteal macrophages (TRAP- F4/80+, CD68+, Mac3+) are present at the bone surface in the close vicinity of mature osteoblasts where they support osteoblastic function and bone anabolism (43). Data have connected estrogen levels and osteal macrophage functions, since the number of osteal macrophages increased on both trabecular and endocortical bone post-ovariectomy. Interestingly, in this condition, osteal macrophages contain TRAP+ intracellular vesicles, attesting to their “clean up” role of exocytosed post-resorption vesicles released by osteoclast (TRAP+) activity (44). PCa-derived BMet increases osteoblast functions through osteoblastic production of CCL2, which favors osteal macrophage positioning (45). Tumor-associated macrophages (TAM) have been proposed potentiate BCa and PCa BMet. Increased numbers of CD206+ M2-like macrophages have also been found in PCa BMet (46, 47). CCL2 expression by cancer cells promotes the recruitment of TAM expressing CCR2 to facilitate cancer cell anchorage in the bone (33). In the same line, the expression of Parathyroid Hormone-Related Protein (PTHRP) by BCa and PCa cells up-regulates the production of CCL2 in osteoblasts that contribute to macrophage recruitment to the bone, bone remodeling and BMet progression (48, 49). Moreover, the clearance of tumor apoptotic cells by efferocytosis promotes CXCL5 production by macrophages and an inflammatory bone microenvironment supporting BMet development (50). Thus, estrogens repress the number of monocytes/macrophages in the bone including osteal macrophages and affect their functions. Estrogens enhance their non-inflammatory cytokine production and repress bone remodeling, preventing formation of a fertile soil for BMet anchorage. However, once BMet manage to settle in the bone, estrogens provide through their action on macrophages a microenvironment facilitating tumor progression (Figure 1).
Osteoclasts are also derived from the monocyte/macrophage lineage. However, on inflammatory conditions, osteoclasts can arise from either dendritic cells or erythromyeloid progenitors (51–54). In addition to the indirect effects described above of estrogens on osteoclast lineage differentiation, selective ablation of ERα in osteoclasts was reported to induce trabecular bone loss in female mice due to decreased FasL expression and autocrine regulation, leading to the inhibition of osteoclast apoptosis (55). As for macrophages/monocytes, osteoclasts are also antigen-presenting cells able to activate CD4+T cells and CD8+ T cells, and are endowed with a unique ability to induce Foxp3+ regulatory T cells (Treg) (56, 57). Moreover, osteoclasts produce a number of chemokines (Cc13, Cc14, Ccl5, Cxcl5, Cxl10, Cxcl11) attracting multiple immune cells e.g. monocytes-macrophages, T-cells, NK and DC cells (58). PCa-derived BMet increase osteoblast functions and facilitate their growth in bone by activating osteoclastogenesis through osteoblastic production of the Receptor Activator of Nuclear factor Kappa-B ligand (RANKL), a key factor for osteoclast differentiation (45). ERRα is also a strong regulator of osteoclast differentiation (59–61). Interestingly, depending on the metastasis origin, overexpression of ERRα in cancer cells either inhibits or stimulates osteoclasts by stimulating the expression of either the decoy receptor of RANKL, OPG (osteoprotegerin), a major inhibitor of osteoclast activity in BCa cells, or VEGF-A and WNT5a in PCa cells (62, 63). Now the impact of estrogens on ERRα in oncology and their role on BMet modulation remains to be determined. Through their action on osteoclasts, estrogens repress bone resorption but also modify the ability of osteoclasts to interact and recruit other immune cells to the bone with direct consequences on the development of a pro-inflammatory microenvironment in favor of BMet (Figure 1).
T lymphocytes present in the BM account for less than 5% of CD45+ cells and have a reduced CD4/CD8 ratio compared to the blood. The BM T cell compartment is almost exclusively composed of α,β T lymphocytes (64). A large part of the T cells present in the BM are memory cells, which either circulate or permanently inhabit the BM, suggesting that in the context of BMet, BM T cells could compose a pool of T cells with specificity against antigens bared by cancer cells (65). The entry of T cells into the BM is largely supported by CXCL12 produced by osteoblasts and stromal cells, and down-regulated by estrogens (20). In the absence of intravital/in situ labeling data, there is so far no direct evidence that estrogens affect T cell retention within the bone. However, this phenomenon is largely dictated by the interaction between α4β1 integrin, present at the surface of T cells, and VCAM-1 expressed by stromal cells and endothelial cells and depicted to be down-regulated by estrogens (66, 67). One of the main roles of estrogens on BM T cells is to repress their osteoclastogenic ability. Evidence of this function emerged more than 20 years ago, after the observation that in contrast to wild type mice, nude mice are protected from trabecular bone loss induced by ovariectomy (68, 69). Later, CD4 T cells producing IL-17 and RANK-L in the bone were identified as potent stimulators of osteoclastogenesis (70). More recently a Th17 osteoclastogenic T cell subset expressing high amounts of TNF-α, and established in the gut, was proposed to reach the bone in both mice and humans (71, 72), in a CCL2-dependent manner (73). Thanks to their osteoclastogenic ability Th17 cells have been suggested to establish a bone pre-metastatic niche, which facilitates cancer cell implantation in the bone (74) (75). In addition, Th17 cells were suggested to preclude BMet control anti-PD1 treatment (76). Estrogens directly control the Th17 cell pool since ERα binds the promoter of RORc inhibiting Rorgt expression (77, 78).
In addition to Th17 cell differentiation, estrogens repress the Th1 cell program (79). In the BM, estrogen deficiency impairs the production of TGF-β by stromal cells, a cytokine that plays a key role in the repression of T-Bet and IFN-γ/TNF-α expression in T cells (80) and thus in osteoclastogenesis (81). Given the potent role of TGF-β in repressing the expression of granzyme-A,-B, and FasL in CD8+ T cells (82), the ability of estrogens to sustain TGF-β levels in the BM impairs CD8+ T cell cytotoxic functions, and promotes BMet progression (83). However, the control by estrogens/estrogen signaling of TGF-β levels in bone and in BMet is more complex. Indeed, bone contains a large source of TGF-β stored in its mineralized matrix, which is released and activated by osteoclast activity. In addition, BMet can also contribute to increasing the levels of TGF-β (84). ERRα is also clearly involved in effector T cell activation (85, 86). In BCa BMet, ERRα expression restrains TGF-β production by cancer cells, leading to exacerbated cytotoxic features in CD8+ T cells in the bone and efficient BMet control (87). TGF-β has been associated with an increase in the generation and stability of Tregs (88). In the BM, the proportion of Tregs among CD4+ T cells is much higher than in the lymph nodes, likely due to high levels of CXCR4 expression on Tregs (89) largely promoted by estrogens (90). Once the cancer cell is anchored, Tregs produce high amounts of RANKL promoting osteolysis associated with a feedback loop of TGF-β release in BCa BMet (91).
Hence, in the bone, the effects of estrogens on T cells are dual: by contributing to an immune-suppressive environment associated with reduced activity of CD8+ T cells and increased number of Tregs which can favor tumor growth and by limiting osteolysis due to inhibition of RANKL production by Th1 and Th17 cells (Figure 1).
BM is the main site of B lymphopoiesis and maturation. In mice, estrogen treatment was linked to a decrease in the numbers of pro/pre- (B220low/IgM−) and immature (B220low/IgM+) B subpopulations, whereas the mature (B220high/IgM+) subpopulation increased in the BM (92). This modification of B cell precursor homeostasis does not seems to be associated with a loss of survival of pro/pre- and immature B cells since estrogens induce an increase in the anti-apoptotic factor Bcl2 in these cells (93). The exact mechanism of action of estrogens on B lymphopoiesis in vivo remains unknown. However, co-culture approaches suggested that estrogens modify the ability of BM stromal cells to support efficient B lymphopoiesis (94). Estrogens promote the production of soluble Frizzled-related protein 1 by BM stromal cells, which in turn stabilizes β-catenin and blocks early B lymphoid progression (95). Of note, B cell interactions with other immune cells in the BM conditions bone homeostasis. Through CD40/CD40L interaction, B cells and T cells cooperate to sustain normal bone mass and mineral density. Signals delivered by CD40 engagement stimulates in B cells the production of OPG and consequently reduce osteoclast differentiation. Interestingly, in post-menopausal women, a switch from OPG to RANK-L production is observed in B cells emphasizing bone loss (96, 97). The role of B cells in the direct control of BMet seems elusive, since in mice with BMet, as in humans, mature B cells are scattered over the BM close to osteoclasts rather than metastasis (98). However, by controlling B lymphopoiesis, and modifying mature B cell profile in the BM, estrogen levels modify bone homeostasis by reducing osteolysis and thus may limit cancer cell anchorage and progression in the bone.
In the adult, the BM is regarded as the main site for ILC-poiesis. Several multipotent ILC precursors have been defined in mice with different degrees of pluripotency that are still under debate (99). Among ILCs, ILC-1/Natural killer (NK) cells express both ERα/β and are prevalent in the BM. In contrast, outside the uterus ILC-2 fails to express ERs, and ILC-3 are endowed with a clear tropism for the mucosa (100). If the effects of estrogen on NK cell ontogeny in the BM are not well documented, there is more evidence of a role, either direct or indirect, for estrogens on NK functions in BM. Interestingly, both development and activation of BM NK cells are dependent on IL-15, a cytokine also important for osteoclast development (101) and abundantly produced by a fraction of mesenchymal cells named CAR cells (CXCL12-abundant reticular) (102, 103), the functions of which are orchestrated by estrogens. Once NK cells have matured, the down-regulation of CXCR4 and up-regulation of S1P5 drive their egress from the CXCL12 enriched BM microenvironment (104). A large population of resident CXCR6+CD69+ NK cells is observed but seems to be endowed with a weak cytotoxic ability against cancer cells, whereas the major anti-tumor activity of NK is supported by recirculating mature NK cells (105). Estrogens reduce the cytotoxic activity (production of granzymes and FasL) of mature NK cells, likely through the down-regulation of their activation receptors (NKp46, NKG2D/L) and the increased expression of the inhibiting receptor CD94 (106). However in the bone, no effect of NK on BMet progression has clearly been documented (107). Hence, the reduced cytotoxic function of NK cells in response to estrogens likely increases the number of disseminated cells in the blood that could reach the bone, but do not contribute to the metastasis immune escape once settled in the bone. However activated NK cells can either promote or inhibit osteoclastogenesis depending on the release of TNF-α or IFN-γ, respectively and this regulation is influenced by estrogens which repress IFNγ in NK cells (106, 108, 109). Activated NK cells can also directly lyse mature osteoclasts (110). Conversely, zoledronate treatment, the most potent bisphosphonate for the treatment of BMet, protects osteoclasts from NK cell cytotoxicity (110). Thus, estrogens induce a large repression of NK cell functions in the bone, which in the light of the ability of NK cells to regulate bone resorbing cells could impact bone remodeling and BMet progression (Figure 1).
The last decades of works on estrogens and BM brought to light the fact that estrogens affect the immune compartment of the bone and possibly the global system. Therefore, this observation may explain why adult females mount stronger immune responses than males, and strongly questions the consequences of modifications in estrogen levels on innate and adaptive immune responses during aging and gender reassignment. Moreover, the emergence of the immunomodulatory role of estrogens suggests that anti-estrogenic drugs, or estrogen administration, by injection or topical applications, may influence the BM. Subsequently this treatment may have an indirect anti-tumor effects by modulating the immune system and may therefore participate to the modulation of pre-metastatic niches and/or modulate the tumor immune environment in the bone. Anti-estrogenic/and estrogenic therapies may also modify the ability of patients to respond to immunotherapy by affecting BM immune cell development and function. This area needs to be particularly explored given the increasing number of people either treated with anti-estrogenic drugs or receiving estrogen.
JM and EB contributed equally to this work. All authors contributed to the article and approved the submitted version.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
This work was supported by the National Center for Scientific Research (CNRS) to EB, the National Institute of Health and Medical Research (INSERM) to JM, by the Labex DevWECan ANR-10-LABX-6 (JM). The SIRIC LYriCAN INCa-DGOS-inserm_12563 is acknowledged.
We thank Brigitte Manship for the editing.
1. Almeida M, Laurent MR, Dubois V, Claessens F, O’Brien CA, Bouillon R, et al. Estrogens and Androgens in Skeletal Physiology and Pathophysiology. Physiol Rev (2017) 97(1):135–87. doi: 10.1152/physrev.00033.2015
2. Evans RM, Mangelsdorf DJ. Nuclear Receptors, RXR, and the Big Bang. Cell (2014) 157(1):255–66. doi: 10.1016/j.cell.2014.03.012
3. Hammes SR, Levin ER. Extranuclear Steroid Receptors: Nature and Actions. Endocrine Rev (2007) 28(7):726–41. doi: 10.1210/er.2007-0022
4. Zimmerman MA, Budish RA, Kashyap S, Lindsey SH. GPER–Novel Membrane Oestrogen Receptor. Clin Sci (2016) 130(12):1005–16. doi: 10.1042/CS20160114
5. Audet-Walsh E, Giguére V. The Multiple Universes of Estrogen-Related Receptor α and γ in Metabolic Control and Related Diseases. Acta Pharmacol Sin (2015) 36(1):51–61. doi: 10.1038/aps.2014.121
6. Giguère V. To ERR in the Estrogen Pathway. Trends Endocrinol Metab (2002) 13(5):220–5. doi: 10.1016/S1043-2760(02)00592-1
7. Bonnelye E, Aubin JE. An Energetic Orphan in an Endocrine Tissue: A Revised Perspective of the Function of Estrogen Receptor-Related Receptor Alpha in Bone and Cartilage. J Bone Miner Res (2013) 28(2):225–33. doi: 10.1002/jbmr.1836
8. Fujimoto J, Nakagawa Y, Toyoki H, Sakaguchi H, Sato E, Tamaya T. Estrogen-Related Receptor in Reproductive Organs. Reprod Med Biol (2005) 4(2):129–31. doi: 10.1111/j.1447-0578.2005.00100.x
9. Clézardin P, Coleman R, Puppo M, Ottewell P, Bonnelye E, Paycha F, et al. Bone Metastasis: Mechanisms, Therapies, and Biomarkers. Physiol Rev (2021) 101(3):797–855. doi: 10.1152/physrev.00012.2019
10. Mendoza-Reinoso V, McCauley LK, Fournier PGJ. Contribution of Macrophages and T Cells in Skeletal Metastasis. Cancers (2020) 12:1014. doi: 10.3390/cancers12041014
11. Wright LE, Guise TA. The Microenvironment Matters: Estrogen Deficiency Fuels Cancer Bone Metastases. Clin Cancer Res (2014) 20(11):2817–9. doi: 10.1158/1078-0432.CCR-14-0576
12. Braun S, Vogl FD, Naume B, Janni W, Osborne MP, Coombes RC, et al. A Pooled Analysis of Bone Marrow Micrometastasis in Breast Cancer. N Engl J Med (2005) 353(8):793–802. doi: 10.1056/NEJMoa050434
13. Kovats S. Estrogen Receptors Regulate Innate Immune Cells and Signaling Pathways. Cell Immunol (2015) 294(2):63–9. doi: 10.1016/j.cellimm.2015.01.018
14. Zhao E, Xu H, Wang L, Kryczek I, Wu K, Hu Y, et al. Bone Marrow and the Control of Immunity. Cell Mol Immunol (2012) 9(1):11–9. doi: 10.1038/cmi.2011.47
15. Notas G, Kampa M, Castanas E. G Protein-Coupled Estrogen Receptor in Immune Cells and Its Role in Immune-Related Diseases. Front Endocrinol (2020) 11:579420. doi: 10.3389/fendo.2020.579420
16. von Vietinghoff S, Ley K. Homeostatic Regulation of Blood Neutrophil Counts. J Immunol (2008) 181(8):5183–8. doi: 10.4049/jimmunol.181.8.5183
17. Crandall TL, Joyce RA, Boggs DR. Estrogens and Hematopoiesis: Characterization and Studies on the Mechanism of Neutropenia. J Lab Clin Med (1980) 95(6):857–67. doi: 10.5555/uri:pii:002221438090387X
18. Gaunt SD, Pierce KR. Myelopoiesis and Marrow Adherent Cells in Estradiol-Treated Mice. Vet Pathol (1985) 22(4):403–8. doi: 10.1177/030098588502200416
19. Nordqvist J, Bernardi A, Islander U, Carlsten H. Effects of a Tissue-Selective Estrogen Complex on B Lymphopoiesis and B Cell Function. Immunobiology (2017) 222(8–9):918–23. doi: 10.1016/j.imbio.2017.05.013
20. Ucer S, Iyer S, Kim H-N, Han L, Rutlen C, Allison K, et al. The Effects of Aging and Sex Steroid Deficiency on the Murine Skeleton Are Independent and Mechanistically Distinct. J Bone Miner Res (2017) 32(3):560–74. doi: 10.1002/jbmr.3014
21. Kolonin MG, Sergeeva A, Staquicini DI, Smith TL, Tarleton CA, Molldrem JJ, et al. Interaction Between Tumor Cell Surface Receptor RAGE and Proteinase 3 Mediates Prostate Cancer Metastasis to Bone. Cancer Res (2017) 77(12):3144–50. doi: 10.1158/0008-5472.CAN-16-0708
22. Yang L, Liu Q, Zhang X, Liu X, Zhou B, Chen J, et al. DNA of Neutrophil Extracellular Traps Promotes Cancer Metastasis Via CCDC25. Nature (2020) 583(7814):133–8. doi: 10.1038/s41586-020-2394-6
23. Flores R, Döhrmann S, Schaal C, Hakkim A, Nizet V, Corriden R. The Selective Estrogen Receptor Modulator Raloxifene Inhibits Neutrophil Extracellular Trap Formation. Front Immunol (2016) 7:566. doi: 10.3389/fimmu.2016.00566
24. Costanzo-Garvey DL, Keeley T, Case AJ, Watson GF, Alsamraae M, Yu Y, et al. Neutrophils Are Mediators of Metastatic Prostate Cancer Progression in Bone. Cancer Immunol Immunother (2020) 69(6):1113–30. doi: 10.1007/s00262-020-02527-6
25. Nowak K, Jabłońska E, Ratajczak-Wrona W. Neutrophils Life Under Estrogenic and Xenoestrogenic Control. J Steroid Biochem Mol Biol (2019) 186:203–11. doi: 10.1016/j.jsbmb.2018.10.015
26. Singh P, Hu P, Hoggatt J, Moh A, Pelus LM. Expansion of Bone Marrow Neutrophils Following G-CSF Administration in Mice Results in Osteolineage Cell Apoptosis and Mobilization of Hematopoietic Stem and Progenitor Cells. Leukemia (2012) 26(11):2375–83. doi: 10.1038/leu.2012.117
27. Agidigbi TS, Kim C. Reactive Oxygen Species in Osteoclast Differentiation and Possible Pharmaceutical Targets of ROS-Mediated Osteoclast Diseases. Int J Mol Sci (2019) 20(14):E3576. doi: 10.3390/ijms20143576
28. Srivastava S, Weitzmann MN, Kimble RB, Rizzo M, Zahner M, Milbrandt J, et al. Estrogen Blocks M-CSF Gene Expression and Osteoclast Formation by Regulating Phosphorylation of Egr-1 and Its Interaction With Sp-1. J Clin Invest (1998) 102(10):1850–9. doi: 10.1172/JCI4561
29. Simfia I, Schiavi J, McNamara LM. Alterations in Osteocyte Mediated Osteoclastogenesis During Estrogen Deficiency and Under ROCK-II Inhibition: An In Vitro Study Using a Novel Postmenopausal Multicellular Niche Model. Exp Cell Res (2020) 392(1):112005. doi: 10.1016/j.yexcr.2020.112005
30. Hamilton JA, Anderson GP. Gm-CSF Biology. Growth Factors (2004) 22(4):225–31. doi: 10.1080/08977190412331279881
31. Riggs BL. The Mechanisms of Estrogen Regulation of Bone Resorption. J Clin Invest (2000) 106(10):1203–4. doi: 10.1172/JCI11468
32. Yang X-W, Wang X-S, Cheng F-B, Wang F, Wan L, Wang F, et al. Elevated CCL2/MCP-1 Levels Are Related to Disease Severity in Postmenopausal Osteoporotic Patients. Clin Lab (2016) 62(11):2173–81. doi: 10.7754/Clin.Lab.2016.160408
33. Buenrostro D, Park SI, Sterling JA. Dissecting the Role of Bone Marrow Stromal Cells on Bone Metastases. BioMed Res Int (2014) 2014:875305. doi: 10.1155/2014/875305
34. Trenti A, Tedesco S, Boscaro C, Trevisi L, Bolego C, Cignarella A. Estrogen, Angiogenesis, Immunity and Cell Metabolism: Solving the Puzzle. Int J Mol Sci (2018) 19(3):E859. doi: 10.3390/ijms19030859
35. Liu D, Zhang Z, Gladwell W, Teng CT. Estrogen Stimulates Estrogen-Related Receptor Alpha Gene Expression Through Conserved Hormone Response Elements. Endocrinology (2003) 144(11):4894–904. doi: 10.1210/en.2003-0432
36. Bonnelye E, Kung V, Laplace C, Galson DL, Aubin JE. Estrogen Receptor-Related Receptor Alpha Impinges on the Estrogen Axis in Bone: Potential Function in Osteoporosis. Endocrinology (2002) 143(9):3658–70. doi: 10.1210/en.2002-220095
37. Yuk J, Kim S, Lee H, Han J, Dufour C, Kim J, et al. Orphan Nuclear Receptor Errα Controls Macrophage Metabolic Signaling and A20 Expression to Negatively Regulate TLR-Induced Inflammation. Immunity (2015) 43(1):80–91. doi: 10.1016/j.immuni.2015.07.003
38. Sonoda J, Laganiere J, Mehl IR, Barish GD, Chong LW, Li X, et al. Nuclear Receptor ERR Alpha and Coactivator PGC-1 Beta Are Effectors of IFN-gamma-induced Host Defense. Genes Dev (2007) 21(15):1909–20. doi: 10.1101/gad.1553007
39. O’Neill L, Pearce E. Immunometabolism Governs Dendritic Cell and Macrophage Function. J Exp Med (2016) 213(1):15–23. doi: 10.1084/jem.20151570
40. Dou C, Ding N, Zhao C, Hou T, Kang F, Cao Z, et al. Estrogen Deficiency-Mediated M2 Macrophage Osteoclastogenesis Contributes to M1/M2 Ratio Alteration in Ovariectomized Osteoporotic Mice. J Bone Miner Res (2018) 33(5):899–908. doi: 10.1002/jbmr.3364
41. Chen K, Jiao Y, Liu L, Huang M, He C, He W, et al. Communications Between Bone Marrow Macrophages and Bone Cells in Bone Remodeling. Front Cell Dev Biol (2020) 8:598263. doi: 10.3389/fcell.2020.598263
42. Michalski MN, McCauley LK. Macrophages and Skeletal Health. Pharmacol Ther (2017) 174:43–54. doi: 10.1016/j.pharmthera.2017.02.017
43. Cho SW, Soki FN, Koh AJ, Eber MR, Entezami P, Park SI, et al. Osteal Macrophages Support Physiologic Skeletal Remodeling and Anabolic Actions of Parathyroid Hormone in Bone. Proc Natl Acad Sci USA (2014) 111(4):1545–50. doi: 10.1073/pnas.1315153111
44. Batoon L, Millard SM, Raggatt LJ, Wu AC, Kaur S, Sun LWH, et al. Osteal Macrophages Support Osteoclast-Mediated Resorption and Contribute to Bone Pathology in a Postmenopausal Osteoporosis Mouse Model. J Bone Miner Res (2021) 36(11):2214–28. doi: 10.1002/jbmr.4413
45. Siddiqui JA, Seshacharyulu P, Muniyan S, Pothuraju R, Khan P, Vengoji R, et al. GDF15 Promotes Prostate Cancer Bone Metastasis and Colonization Through Osteoblastic CCL2 and RANKL Activation. Bone Res (2022) 10(1):6. doi: 10.1038/s41413-021-00178-6
46. Lo CH, Lynch CC. Multifaceted Roles for Macrophages in Prostate Cancer Skeletal Metastasis. Front Endocrinol (Lausanne) (2018) 9:247. doi: 10.3389/fendo.2018.00247
47. Soki FN, Cho SW, Kim YW, Jones JD, Park SI, Koh AJ, et al. Bone Marrow Macrophages Support Prostate Cancer Growth in Bone. Oncotarget (2015) 6(34):35782–96. doi: 10.18632/oncotarget.6042
48. Mulholland BS, Forwood MR, Morrison NA. Monocyte Chemoattractant Protein-1 (Mcp-1/Ccl2) Drives Activation of Bone Remodelling and Skeletal Metastasis. Curr Osteoporos Rep (2019) 17(6):538–47. doi: 10.1007/s11914-019-00545-7
49. Li X, Loberg R, Liao J, Ying C, Snyder LA, Pienta KJ, et al. A Destructive Cascade Mediated by CCL2 Facilitates Prostate Cancer Growth in Bone. Cancer Res (2009) 69(4):1685–92. doi: 10.1158/0008-5472.CAN-08-2164
50. Roca H, Jones JD, Purica MC, Weidner S, Koh AJ, Kuo R, et al. Apoptosis-Induced CXCL5 Accelerates Inflammation and Growth of Prostate Tumor Metastases in Bone. J Clin Invest (2018) 128(1):248–66. doi: 10.1172/JCI92466
51. Jacome-Galarza CE, Percin GI, Muller JT, Mass E, Lazarov T, Eitler J, et al. Developmental Origin, Functional Maintenance and Genetic Rescue of Osteoclasts. Nature (2019) 568(7753):541–5. doi: 10.1038/s41586-019-1105-7
52. Hasegawa T, Kikuta J, Sudo T, Matsuura Y, Matsui T, Simmons S, et al. Identification of a Novel Arthritis-Associated Osteoclast Precursor Macrophage Regulated by Foxm1. Nat Immunol (2019) 20(12):1631–43. doi: 10.1038/s41590-019-0526-7
53. Rivollier A, Mazzorana M, Tebib J, Piperno M, Aitsiselmi T, Rabourdin-Combe C, et al. Immature Dendritic Cell Transdifferentiation Into Osteoclasts: A Novel Pathway Sustained by the Rheumatoid Arthritis Microenvironment. Blood (2004) 104(13):4029–37. doi: 10.1182/blood-2004-01-0041
54. Yahara Y, Barrientos T, Tang YJ, Puviindran V, Nadesan P, Zhang H, et al. Erythromyeloid Progenitors Give Rise to a Population of Osteoclasts That Contribute to Bone Homeostasis and Repair. Nat Cell Biol (2020) 22(1):49–59. doi: 10.1038/s41556-019-0437-8
55. Nakamura T, Imai Y, Matsumoto T, Sato S, Takeuchi K, Igarashi K, et al. Estrogen Prevents Bone Loss Via Estrogen Receptor Alpha and Induction of Fas Ligand in Osteoclasts. Cell (2007) 130(5):811–23. doi: 10.1016/j.cell.2007.07.025
56. Li H, Hong S, Qian J, Zheng Y, Yang J, Yi Q. Cross Talk Between the Bone and Immune Systems: Osteoclasts Function as Antigen-Presenting Cells and Activate CD4+ and CD8+ T Cells. Blood (2010) 116(2):210–7. doi: 10.1182/blood-2009-11-255026
57. Kiesel J, Buchwald Z, Aurora R. Cross-Presentation by Osteoclasts Induces FoxP3 in CD8+ T Cells. J Immunol (2009) 182(9):5477–87. doi: 10.4049/jimmunol.0803897
58. Kiesel J, Miller C, Abu-Amer Y, Aurora R. Systems Level Analysis of Osteoclastogenesis Reveals Intrinsic and Extrinsic Regulatory Interactions. Dev Dyn (2007) 236(8):2181–97. doi: 10.1002/dvdy.21206
59. Bae S, Lee M, Mun S, Giannopoulou E, Yong-Gonzalez V, Cross J, et al. MYC-Dependent Oxidative Metabolism Regulates Osteoclastogenesis Via Nuclear Receptor Errα. J Clin Invest (2017) 127(7):2555–68. doi: 10.1172/JCI89935
60. Bonnelye E, Saltel F, Chabadel A, Zirngibl RA, Aubin JE, Jurdic P. Involvement of the Orphan Nuclear Estrogen Receptor-Related Receptor Alpha in Osteoclast Adhesion and Transmigration. J Mol Endocrinol (2010) 45(6):365–77. doi: 10.1677/JME-10-0024
61. Wei W, Schwaid A, Wang X, Wang X, Chen S, Chu Q, et al. Ligand Activation of Errα by Cholesterol Mediates Statin and Bisphosphonate Effects. Cell Metab (2016) 23(3):479–91. doi: 10.1016/j.cmet.2015.12.010
62. Fradet A, Sorel H, Bouazza L, Goehrig D, Depalle B, Bellahcene A, et al. Dual Function of ERRalpha in Breast Cancer and Bone Metastasis Formation: Implication of VEGF and Osteoprotegerin. Cancer Res (2011) 71(17):5728–38. doi: 10.1158/0008-5472.CAN-11-1431
63. Fradet A, Bouchet M, Delliaux C, Gervais M, Kan C, Benetollo C, et al. Estrogen Related Receptor Alpha in Castration-Resistant Prostate Cancer Cells Promotes Tumor Progression in Bone. Oncotarget (2016) 7(47):77071–86. doi: 10.18632/oncotarget.12787
64. Hillhouse EE, Delisle J-S, Lesage S. Immunoregulatory CD4(-)CD8(-) T Cells as a Potential Therapeutic Tool for Transplantation, Autoimmunity, and Cancer. Front Immunol (2013) 4:6. doi: 10.3389/fimmu.2013.00006
65. Beckhove P, Feuerer M, Dolenc M, Schuetz F, Choi C, Sommerfeldt N, et al. Specifically Activated Memory T Cell Subsets From Cancer Patients Recognize and Reject Xenotransplanted Autologous Tumors. J Clin Invest (2004) 114(1):67–76. doi: 10.1172/JCI200420278
66. Hou X, Pei F. Estradiol Inhibits Cytokine-Induced Expression of VCAM-1 and ICAM-1 in Cultured Human Endothelial Cells Via Ampk/Pparα Activation. Cell Biochem Biophys (2015) 72(3):709–17. doi: 10.1007/s12013-015-0522-y
67. Ulyanova T, Scott LM, Priestley GV, Jiang Y, Nakamoto B, Koni PA, et al. VCAM-1 Expression in Adult Hematopoietic and Nonhematopoietic Cells Is Controlled by Tissue-Inductive Signals and Reflects Their Developmental Origin. Blood (2005) 106(1):86–94. doi: 10.1182/blood-2004-09-3417
68. Roggia C, Gao Y, Cenci S, Weitzmann MN, Toraldo G, Isaia G, et al. Up-Regulation of TNF-Producing T Cells in the Bone Marrow: A Key Mechanism by Which Estrogen Deficiency Induces Bone Loss In Vivo. Proc Natl Acad Sci USA (2001) 98(24):13960–5. doi: 10.1073/pnas.251534698
69. Cenci S, Weitzmann MN, Roggia C, Namba N, Novack D, Woodring J, et al. Estrogen Deficiency Induces Bone Loss by Enhancing T-cell Production of TNF-Alpha. J Clin Invest (2000) 106(10):1229–37. doi: 10.1172/JCI11066
70. Okamoto K, Nakashima T, Shinohara M, Negishi-Koga T, Komatsu N, Terashima A, et al. Osteoimmunology: The Conceptual Framework Unifying the Immune and Skeletal Systems. Physiol Rev (2017) 97(4):1295–349. doi: 10.1152/physrev.00036.2016
71. Yu M, Pal S, Paterson CW, Li J-Y, Tyagi AM, Adams J, et al. Ovariectomy Induces Bone Loss Via Microbial-Dependent Trafficking of Intestinal TNF+ T Cells and Th17 Cells. J Clin Invest (2021) 131(4):143137. doi: 10.1172/JCI143137
72. Ciucci T, Ibáñez L, Boucoiran A, Birgy-Barelli E, Pène J, Abou-Ezzi G, et al. Bone Marrow Th17 Tnfα Cells Induce Osteoclast Differentiation, and Link Bone Destruction to IBD. Gut (2015) 64(7):1072–81. doi: 10.1136/gutjnl-2014-306947
73. Uehara IA, Soldi LR, Silva MJB. Current Perspectives of Osteoclastogenesis Through Estrogen Modulated Immune Cell Cytokines. Life Sci (2020) 256:117921. doi: 10.1016/j.lfs.2020.117921
74. Monteiro AC, Leal AC, Gonçalves-Silva T, Mercadante ACT, Kestelman F, Chaves SB, et al. T Cells Induce Pre-Metastatic Osteolytic Disease and Help Bone Metastases Establishment in a Mouse Model of Metastatic Breast Cancer. PloS One (2013) 8(7):e68171. doi: 10.1371/journal.pone.0068171
75. Monteiro AC, Bonomo A. Cd8+ T Cells From Experimental in Situ Breast Carcinoma Interfere With Bone Homeostasis. Bone (2021) 150:116014. doi: 10.1016/j.bone.2021.116014
76. Jiao S, Subudhi SK, Aparicio A, Ge Z, Guan B, Miura Y, et al. Differences in Tumor Microenvironment Dictate T Helper Lineage Polarization and Response to Immune Checkpoint Therapy. Cell (2019) 179(5):1177–1190.e13. doi: 10.1016/j.cell.2019.10.029
77. Chen R-Y, Fan Y-M, Zhang Q, Liu S, Li Q, Ke G-L, et al. Estradiol Inhibits Th17 Cell Differentiation Through Inhibition of Rorγt Transcription by Recruiting the Erα/REA Complex to Estrogen Response Elements of the Rorγt Promoter. J Immunol (2015) 194(8):4019–28. doi: 10.4049/jimmunol.1400806
78. Tyagi AM, Srivastava K, Mansoori MN, Trivedi R, Chattopadhyay N, Singh D. Estrogen Deficiency Induces the Differentiation of IL-17 Secreting Th17 Cells: A New Candidate in the Pathogenesis of Osteoporosis. PloS One (2012) 7(9):e44552. doi: 10.1371/journal.pone.0044552
79. Cenci S, Toraldo G, Weitzmann MN, Roggia C, Gao Y, Qian WP, et al. Estrogen Deficiency Induces Bone Loss by Increasing T Cell Proliferation and Lifespan Through IFN-γ-Induced Class II Transactivator. Proc Natl Acad Sci USA (2003) 100(18):10405–10. doi: 10.1073/pnas.1533207100
80. Marie J, Liggitt D, Rudensky A. Cellular Mechanisms of Fatal Early-Onset Autoimmunity in Mice With the T Cell-Specific Targeting of Transforming Growth Factor-Beta Receptor. Immunity (2006) 25(3):441–54. doi: 10.1016/j.immuni.2006.07.012
81. Gao Y, Qian W-P, Dark K, Toraldo G, Lin ASP, Guldberg RE, et al. Estrogen Prevents Bone Loss Through Transforming Growth Factor Beta Signaling in T Cells. Proc Natl Acad Sci USA (2004) 101(47):16618–23. doi: 10.1073/pnas.0404888101
82. Thomas DA, Massagué J. TGF-Beta Directly Targets Cytotoxic T Cell Functions During Tumor Evasion of Immune Surveillance. Cancer Cell (2005) 8(5):369–80. doi: 10.1016/j.ccr.2005.10.012
83. Janssen LME, Ramsay EE, Logsdon CD, Overwijk WW. The Immune System in Cancer Metastasis: Friend or Foe? J Immunother Cancer (2017) 5(1):79. doi: 10.1186/s40425-017-0283-9
84. Trivedi T, Pagnotti GM, Guise TA, Mohammad KS. The Role of TGF-β in Bone Metastases. Biomolecules (2021) 11(11):1643. doi: 10.3390/biom11111643
85. Michalek RD, Gerriets VA, Nichols AG, Inoue M, Kazmin D, Chang CY, et al. Estrogen-Related Receptor-Alpha Is a Metabolic Regulator of Effector T-cell Activation and Differentiation. Proc Natl Acad Sci USA (2011) 108(45):18348–53. doi: 10.1073/pnas.1108856108
86. Liebmann M, Hucke S, Koch K, Eschborn M, Ghelman J, Chasan AI, et al. Nur77 Serves as a Molecular Brake of the Metabolic Switch During T Cell Activation to Restrict Autoimmunity. Proc Natl Acad Sci USA (2018) 115(34):E8017–26. doi: 10.1073/pnas.1721049115
87. Bouchet M, Lainé A, Boyault C, Proponnet-Guerault M, Meugnier E, Bouazza L, et al. Errα Expression in Bone Metastases Leads to an Exacerbated Anti-Tumor Immune Response. Cancer Res (2020) 80(13):2914–26. doi: 10.1158/0008-5472.CAN-19-3584
88. Marie JC, Letterio JJ, Gavin M, Rudensky AY. TGF-Beta1 Maintains Suppressor Function and Foxp3 Expression in CD4+CD25+ Regulatory T Cells. J Exp Med (2005) 201(7):1061–7. doi: 10.1084/jem.20042276
89. Hirata Y, Furuhashi K, Ishii H, Li HW, Pinho S, Ding L, et al. Cd150high Bone Marrow Tregs Maintain Hematopoietic Stem Cell Quiescence and Immune Privilege Via Adenosine. Cell Stem Cell (2018) 22(3):445–53.e5. doi: 10.1016/j.stem.2018.01.017
90. Fan X-L, Duan X-B, Chen Z-H, Li M, Xu J-S, Ding G-M. Lack of Estrogen Down-Regulates CXCR4 Expression on Treg Cells and Reduces Treg Cell Population in Bone Marrow in OVX Mice. Cell Mol Biol (Noisy-le-grand) (2015) 61(2):13–7. doi: 10.14715/cmb/2015.61.2.3
91. Tan W, Zhang W, Strasner A, Grivennikov S, Cheng JQ, Hoffman RM, et al. Tumour-Infiltrating Regulatory T Cells Stimulate Mammary Cancer Metastasis Through RANKL-RANK Signalling. Nature (2011) 470(7335):548–53. doi: 10.1038/nature09707
92. Thurmond TS, Murante FG, Staples JE, Silverstone AE, Korach KS, Gasiewicz TA. Role of Estrogen Receptor Alpha in Hematopoietic Stem Cell Development and B Lymphocyte Maturation in the Male Mouse. Endocrinology (2000) 141(7):2309–18. doi: 10.1210/endo.141.7.7560
93. Grimaldi CM, Cleary J, Dagtas AS, Moussai D, Diamond B. Estrogen Alters Thresholds for B Cell Apoptosis and Activation. J Clin Invest (2002) 109(12):1625–33. doi: 10.1172/JCI0214873
94. Masuzawa T, Miyaura C, Onoe Y, Kusano K, Ohta H, Nozawa S, et al. Estrogen Deficiency Stimulates B Lymphopoiesis in Mouse Bone Marrow. J Clin Invest (1994) 94(3):1090–7. doi: 10.1172/JCI117424
95. Yokota T, Oritani K, Garrett KP, Kouro T, Nishida M, Takahashi I, et al. Soluble Frizzled-Related Protein 1 Is Estrogen Inducible in Bone Marrow Stromal Cells and Suppresses the Earliest Events in Lymphopoiesis. J Immunol (2008) 181(9):6061–72. doi: 10.4049/jimmunol.181.9.6061
96. Fujiwara Y, Piemontese M, Liu Y, Thostenson JD, Xiong J, O’Brien CA. Rankl (Receptor Activator of Nfκb Ligand) Produced by Osteocytes Is Required for the Increase in B Cells and Bone Loss Caused by Estrogen Deficiency in Mice. J Biol Chem (2016) 291(48):24838–50. doi: 10.1074/jbc.M116.742452
97. Eghbali-Fatourechi G, Khosla S, Sanyal A, Boyle WJ, Lacey DL, Riggs BL. Role of RANK Ligand in Mediating Increased Bone Resorption in Early Postmenopausal Women. J Clin Invest (2003) 111(8):1221–30. doi: 10.1172/JCI200317215
98. Li M, Sasaki T, Ono K, de Freitas PHL, Sobhan U, Kojima T, et al. Distribution of Macrophages, Osteoclasts and the B-lymphocyte Lineage in Osteolytic Metastasis of Mouse Mammary Carcinoma. BioMed Res (2007) 28(3):127–37. doi: 10.2220/biomedres.28.127
99. Diefenbach A, Colonna M, Koyasu S. Development, Differentiation, and Diversity of Innate Lymphoid Cells. Immunity (2014) 41(3):354–65. doi: 10.1016/j.immuni.2014.09.005
100. Blanquart E, Laffont S, Guéry J-C. Sex Hormone Regulation of Innate Lymphoid Cells. BioMed J (2021) 44(2):144–56. doi: 10.1016/j.bj.2020.11.007
101. Amarasekara DS, Yun H, Kim S, Lee N, Kim H, Rho J. Regulation of Osteoclast Differentiation by Cytokine Networks. Immune Netw (2018) 18(1):e8. doi: 10.4110/in.2018.18.e8
102. Sugiyama T, Kohara H, Noda M, Nagasawa T. Maintenance of the Hematopoietic Stem Cell Pool by CXCL12-CXCR4 Chemokine Signaling in Bone Marrow Stromal Cell Niches. Immunity (2006) 25(6):977–88. doi: 10.1016/j.immuni.2006.10.016
103. Noda M, Omatsu Y, Sugiyama T, Oishi S, Fujii N, Nagasawa T. Cxcl12-CXCR4 Chemokine Signaling Is Essential for NK-Cell Development in Adult Mice. Blood (2011) 117(2):451–8. doi: 10.1182/blood-2010-04-277897
104. Bernardini G, Sciumè G, Santoni A. Differential Chemotactic Receptor Requirements for NK Cell Subset Trafficking Into Bone Marrow. Front Immunol (2013) 4:12. doi: 10.3389/fimmu.2013.00012
105. Melsen JE, Lugthart G, Vervat C, Kielbasa SM, van der Zeeuw SAJ, Buermans HPJ, et al. Human Bone Marrow-Resident Natural Killer Cells Have a Unique Transcriptional Profile and Resemble Resident Memory Cd8+ T Cells. Front Immunol (2018) 9:1829. doi: 10.3389/fimmu.2018.01829
106. Hao S, Zhao J, Zhou J, Zhao S, Hu Y, Hou Y. Modulation of 17beta-Estradiol on the Number and Cytotoxicity of NK Cells In Vivo Related to MCM and Activating Receptors. Int Immunopharmacol (2007) 7(13):1765–75. doi: 10.1016/j.intimp.2007.09.017
107. Liu S, Song A, Wu Y, Yao S, Wang M, Niu T, et al. Analysis of Genomics and Immune Infiltration Patterns of Epithelial-Mesenchymal Transition Related to Metastatic Breast Cancer to Bone. Transl Oncol (2021) 14(2):100993. doi: 10.1016/j.tranon.2020.100993
108. Takayanagi H, Ogasawara K, Hida S, Chiba T, Murata S, Sato K, et al. T-Cell-Mediated Regulation of Osteoclastogenesis by Signalling Cross-Talk Between RANKL and IFN-Gamma. Nature (2000) 408(6812):600–5. doi: 10.1038/35046102
109. Lam J, Takeshita S, Barker JE, Kanagawa O, Ross FP, Teitelbaum SL. TNF-Alpha Induces Osteoclastogenesis by Direct Stimulation of Macrophages Exposed to Permissive Levels of RANK Ligand. J Clin Invest (2000) 106(12):1481–8. doi: 10.1172/JCI11176
Keywords: estrogen, bone marrow, immune cells, bone metastasis, estrogen receptor
Citation: Marie JC and Bonnelye E (2022) Effects of Estrogens on Osteoimmunology: A Role in Bone Metastasis. Front. Immunol. 13:899104. doi: 10.3389/fimmu.2022.899104
Received: 18 March 2022; Accepted: 26 April 2022;
Published: 23 May 2022.
Edited by:
Antonio Maurizi, University of L’Aquila, ItalyReviewed by:
Teun J. De Vries, VU Amsterdam, NetherlandsCopyright © 2022 Marie and Bonnelye. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Edith Bonnelye, ZWRpdGguYm9ubmVseWVAaW5zZXJtLmZy; Julien C. Marie, anVsaWVuLm1hcmllQGluc2VybS5mcg==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.