 Yu Deng1,2†
Yu Deng1,2† Lupeng Li
Lupeng Li Edward A. Miao
Edward A. Miao Pengda Liu
Pengda Liu- 1Lineberger Comprehensive Cancer Center, The University of North Carolina at Chapel Hill, Chapel Hill, NC, United States
- 2Department of Biochemistry and Biophysics, The University of North Carolina at Chapel Hill, Chapel Hill, NC, United States
- 3Curriculum in Genetics and Molecular Biology, The University of North Carolina at Chapel Hill, Chapel Hill, NC, United States
- 4Department of Immunology and Department of Molecular Genetics and Microbiology, Duke University, Durham, NC, United States
- 5Department of Microbiology and Immunology, The University of North Carolina at Chapel Hill, Chapel Hill, NC, United States
The innate immune response is the first-line host defense against pathogens. Cytosolic nucleic acids, including both DNA and RNA, represent a special type of danger signal to initiate an innate immune response. Activation of cytosolic nucleic acid sensors is tightly controlled in order to achieve the high sensitivity needed to combat infection while simultaneously preventing false activation that leads to pathologic inflammatory diseases. In this review, we focus on post-translational modifications of key cytosolic nucleic acid sensors that can reversibly or irreversibly control these sensor functions. We will describe phosphorylation, ubiquitination, SUMOylation, neddylation, acetylation, methylation, succinylation, glutamylation, amidation, palmitoylation, and oxidation modifications events (including modified residues, modifying enzymes, and modification function). Together, these post-translational regulatory modifications on key cytosolic DNA/RNA sensing pathway members reveal a complicated yet elegantly controlled multilayer regulator network to govern innate immune activation.
Introduction
All cells express a selected subset of innate immune sensors to defend against pathogens. Activation of the innate immune response promotes the production of interferons and proinflammatory cytokines, triggers regulated cell death to clear intracellular pathogens, and promotes adaptive immune responses. Pathogen-associated molecular patterns (PAMPs), which are conserved pathogen-derived molecules (1–3), are recognized by germline-encoded pattern recognition receptors (PRRs) of the innate immune system. Distinct types of PRRs sense a variety of PAMPs. PRRs include Toll-like receptors (TLRs), Nod-like receptors (NLRs), retinoic acid-inducible gene I (RIG-I)-like receptors (RLRs), C-type lectin receptors (CLRs), the absent in melanoma 2 (AIM2)-like receptors (ALRs), and other nucleic acid sensors including cyclic GMP-AMP synthase (cGAS) (4, 5). PAMPs include lipopolysaccharides (LPS), flagellin, lipoteichoic acid, peptidoglycan, and nucleic acid acids. LPS is recognized by TLR4, peptidoglycan is sensed by TLR2 (6), flagellin is recognized by TLR5 and NAIPs, and dsRNA is detected by TLR3 (7). Activation of PRRs by their corresponding PAMPs induces innate immunity and inflammation to clear infection (2, 3, 8–10). This review only focuses on nucleic acid sensing PRRs.
Given that both invasive bacteria/viruses and host cells contain DNA and RNA, how to distinguish self from foreign nucleic acids at first glance seems challenging for host defense. Largely, this is achieved by at least three mechanisms including “availability,” “localization,” and “structure” (11). “Availability” refers to the local concentration, half-life, and whether the nucleic acid is covered by binding partners. “Localization” indicates various cellular compartments where nucleic acids can be detected including the plasma membrane, cytoplasm, and nucleus. “Structure” includes the nucleic acid sequence, secondary structures, and certain modifications occurring in these nucleic acids. Nucleic acid sensors will transduce signals to trigger innate immunity in facilitating transcription of interferon and regulated cell death. Dysregulation of nucleic acid sensing or signal transduction leads to susceptibility to infection and other human diseases, including autoimmune diseases, autoinflammation, and cancer (12). Thus, the activation of nucleic acid sensing is tightly controlled.
The control of nucleic acid sensing is achieved at multiple levels. Upon bacterial or viral infection, type I interferons are synthesized to function through either autocrine or paracrine signaling to activate various STAT pathways to boost transcription of interferon-stimulated gene (13). Host cells can increase the sensitivity of nucleic acid sensors by inactivating sensor inhibitors. Conversely, bacteria or viruses can disable these sensors via post-translational modifications or via inhibitory binding proteins. To date, a plethora of protein post-translational modifications have been reported including phosphorylation (on Ser, Thr, and Tyr residues), ubiquitination (on Lys residues), acetylation (on Lys residues), methylation (on Lys and Arg residues), hydroxylation (on Pro residues), oxidation (on Cys residues), SUMOylation (on Lys and Glu residues), glutamylation (on Glu residues), and amidation (on Gln and Asn residues). These protein modifications control protein localization, stability, activation, and function in a temporal and spatial manner either through direct allosteric conformational changes or through regulating protein binding partners (14). Here, we will summarize major post-translational modifications identified to date on key mammalian nucleic acid sensing pathways, hoping to provide an up-to-date review of the roles of these modifications in fine-tuning innate immune responses, as well as provide novel insights into potentials in targeting certain modifying enzymes in treating human diseases where innate immune sensing is dysregulated.
Cytosolic DNA Sensing
Cytosolic DNA is a danger signal that can be derived from infectious bacteria or viruses. It can also arise from self-DNA, such as from damaged genomic DNA, mitochondrial DNA, or DNA released during apoptosis. Sensing of cytosolic DNA comprises an important component for mammalian innate immunity, and activation of the cytosolic DNA sensors leads to the production of type I IFNs, pro-inflammatory cytokines, and chemokines, as well as regulated cell death for antiviral/antibacterial responses. A large number of candidates have been proposed as cytosolic DNA sensors including members of the ALRs such as AIM2 (15–17), myeloid nuclear differentiation antigen (MNDA) (18), interferon-inducible protein X (IFIX) (19), and interferon-inducible protein 16 (IFI16) (20), as well as non-ALR sensors such as cGAS (21), meiotic recombination 11 homolog A (MRE11) (22), Ku heterodimers (Ku70/Ku80) (23, 24), LRR binding FLII interacting protein 1 (LRRFIP1) (25), DExD/H box helicases (DDX41) (26), Z-DNA binding protein 1 (ZBP1) (27), and RNA polymerase III (28, 29). Notably, activation of different cytosolic DNA sensors leads to distinct downstream signaling. For example, DNA binding and activation of AIM2 in macrophages trigger the formation of inflammasome complexes for caspase 1 activation, leading to pyroptosis (30). Meanwhile, activation of other cytosolic DNA sensors such as cGAS stimulates interferon production. Specifically, DNA binding promotes cGAS dimerization and phase transition to facilitate cGAS activation, which leads to the synthesis of 2′3′-cyclic-GMP-AMP (cGAMP). cGAMP is a second messenger that diffuses throughout the cytosol and binds to a stimulator of interferon genes (STING), an endoplasmic reticulum (ER) transmembrane protein. This dimerizes STING, which then recruits and activates TBK1 to phosphorylate IRF3, promoting IRF3 dimerization and nuclear translocation, thus inducing IRF3-mediated IFN transcription (31). In addition, AIM2 recognizes specific DNA sequences in a cell type-dependent manner (32, 33), while cGAS senses cytosolic DNA in a DNA sequence-independent but DNA length-dependent manner in most cell types. Regardless of different types of cytosolic DNA sensors, hyperactivation of cytosolic DNA sensing and signaling results in autoimmune disease (34), while suppression of cytosolic DNA sensing contributes to evasion of immune destruction during tumorigenesis as well as resistance to cancer immunotherapies (35). Thus, activation of the nucleic acid sensors is tightly controlled under physiological conditions, and dysregulation leads to human pathological conditions. Post-translational modifications occurring on nucleic acid sensing pathway members serve as a critical approach to control and fine-tune pathway activities.
Post-Translational Modifications of Cytosolic DNA Sensing
Phosphorylation
Protein phosphorylation has been widely observed in nature as a reversible modification occurring on Ser, Thr, or Tyr residues to acutely control protein function—a phosphate group is added to target proteins by protein kinases and removed by protein phosphatases (36). Phosphorylation has been observed to recruit binding partners such as well-defined BRCT domains as readers for pSQ/pTQ motifs in DNA damage response (37), regulate protein stability (38), change protein cellular localization (39), or allosterically regulate enzyme activities (40). In this section, we will summarize discoveries associated with phosphorylation-mediated regulations in cytosolic DNA sensing.
The cytosolic DNA sensor cGAS was reported to be phosphorylated on hS305 (human S305 equivalent to mS291: mouse cGAS-S291) by the kinase Akt confirmed by both in vitro kinase assays and mass spectrometry analyses (40) (Table 1 and Figure 1). In addition, the same residue was also reported to be phosphorylated by CDK1 during mitosis, which was validated by both in vitro kinase assays and specific phospho-antibodies (41). Phosphorylation of cGAS on hS305/mS291 in the cGAS enzymatic domain by either kinase suppresses cGAS activity. Considering Akt activation is cell cycle dependent, peaking in S/G2 (101), it is plausible that Akt and CDK1 phosphorylate cGAS at S/G2 and M phases, respectively, to suppress cytosolic cGAS activation. Given that nuclear cGAS has been shown to be suppressed by binding to BAF (102) or tethering to chromatin (103), and during mitosis nuclear DNA is freely accessible to the cytosolic space, cGAS phosphorylation at its N-terminus by mitotic kinases including Aroura kinase B at multiple sites was observed to prevent cGAS sensing chromatin DNA, which tightly keeps cGAS inactive (44). Upon mitosis exit, the phosphatase PP1 dephosphorylates cGAS at hS305/mS291 to restore the ability of cytosolic cGAS in sensing DNA (41). Thus, cGAS phosphorylation in either its enzymatic domain or N-terminus may function in parallel to BAF1 (barrier-to-autointegration factor 1) binding or chromatin tethering in inhibiting cGAS activation during mitosis. Activation of cGAS in mitosis promotes mitotic cell death (104). Given cGAS largely senses cytosolic DNA, retention of cGAS in the cytoplasm at least through BLK (B lymphocyte kinase)-mediated cGAS-Tyr215 phosphorylation (42) facilitates its cytosolic DNA sensing and also evades its nuclear binding to PARP1 (Poly(ADP-Ribose) Polymerase 1) in suppressing homologous recombination. In addition, cGAS phosphorylation was also found to control cGAS activation by modulating cGAS oligomerization. Specifically, through a screen to search for compounds inhibiting VSV infection in THP1 cells in vitro, DNAPK (DNA-dependent protein kinase) inhibitors were found to restrict VSV replication by activating cGAS (43). Moreover, DNAPK was found to phosphorylate hcGAS on T68 and S213, which prevents cGAS oligomerization and activation. This study may provide explanations for why missense mutations of PRKDC, the DNAPK catalytic subunit, are observed in patients with autoimmune diseases (43). At resting states, cGAS is associated with the protein phosphatase PPP6C to retain cGAS in a dephosphorylated state, and upon DNA virus infection, dissociation of PPP6C allowed hcGAS phosphorylation on S435 (mcGAS-S420) residue in the catalytic pocket priming cGAS for activation (45). Thus, depending on the phosphorylation sites, cGAS phosphorylation can either suppress or facilitate cGAS activation.
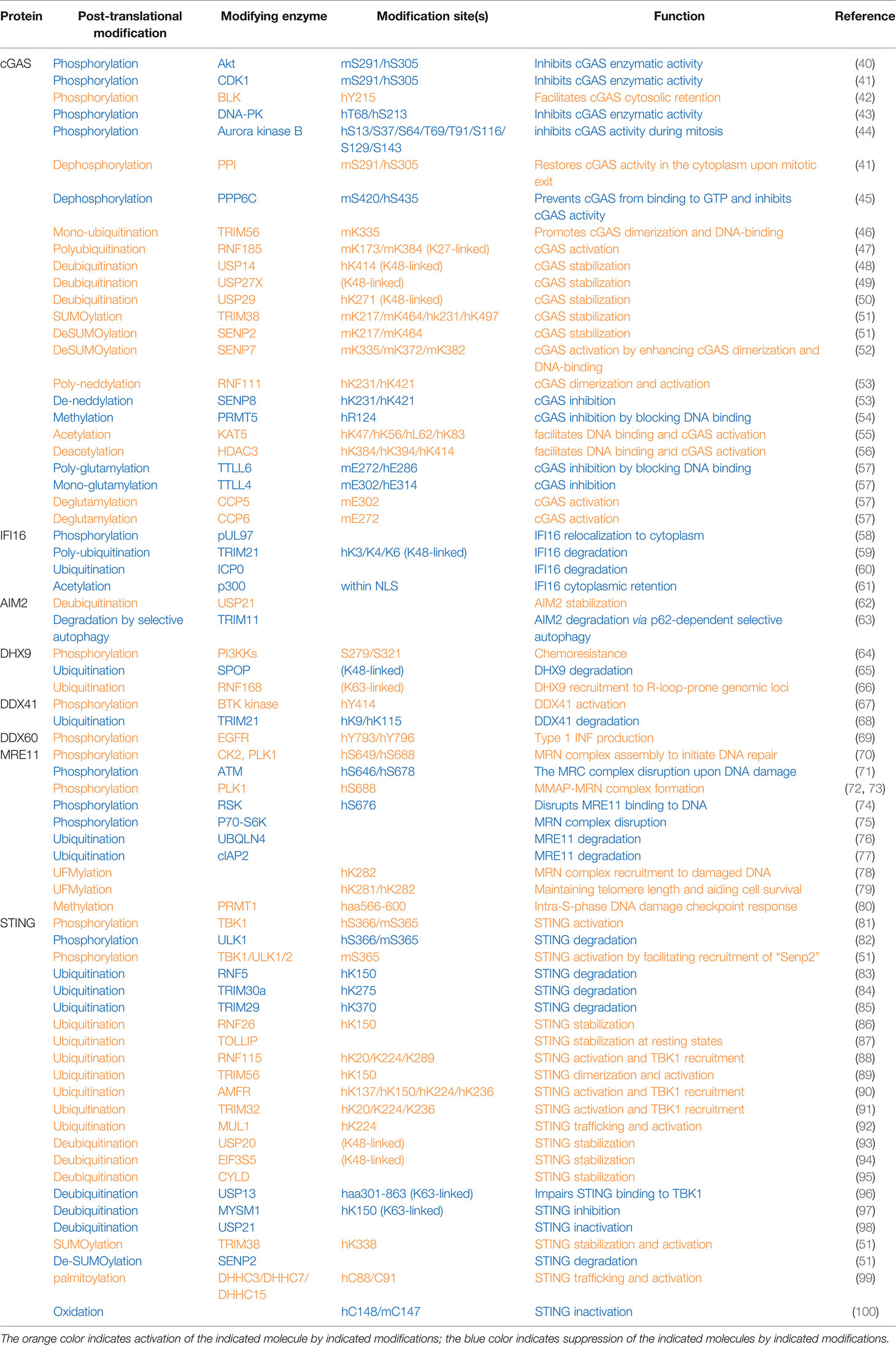
Table 1 Post-translational modifications of proteins in cytosolic DNA sensing signaling pathways.
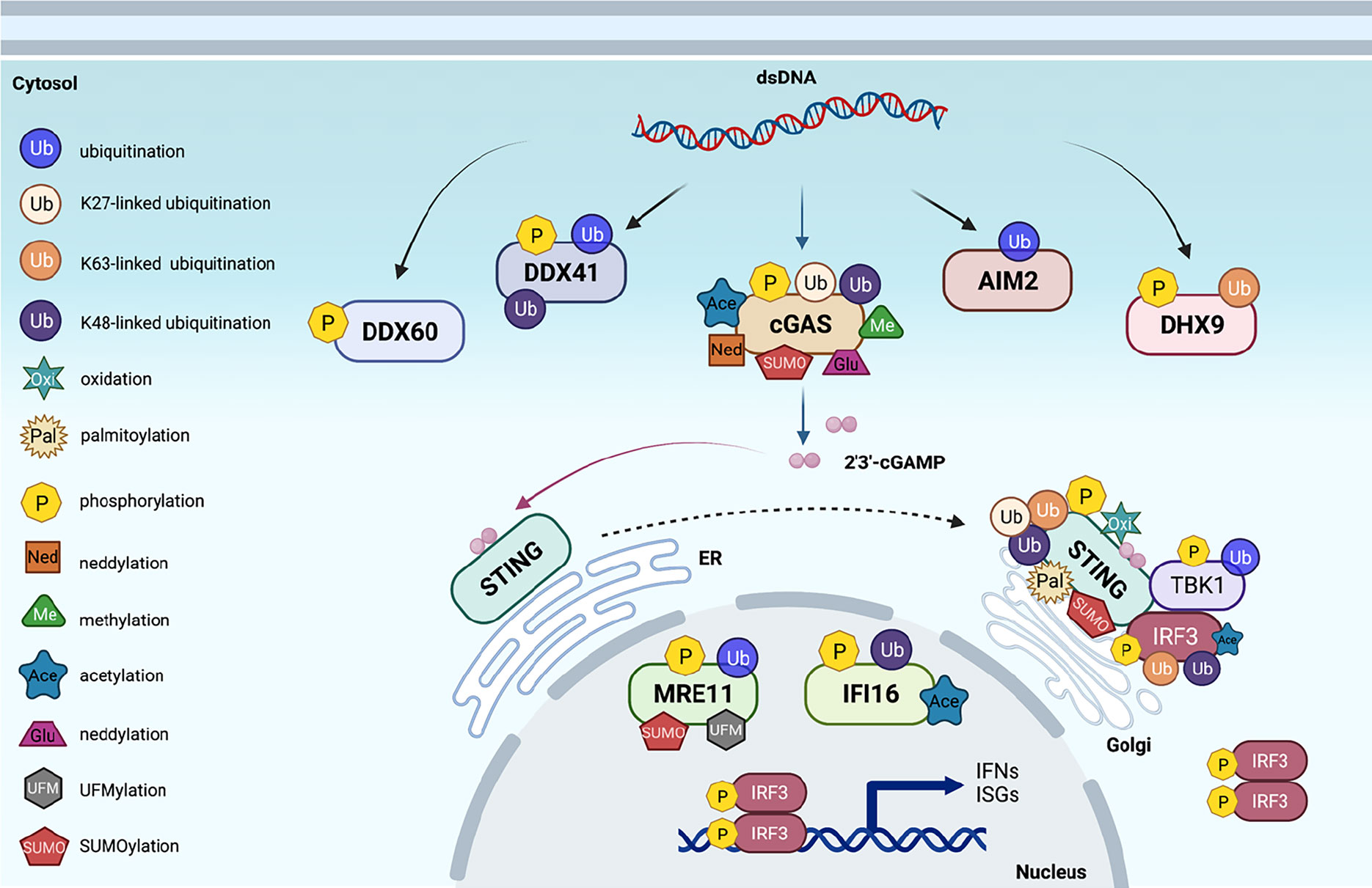
Figure 1 Post-translational modifications of proteins in cytosolic DNA sensing signaling. An overview of cytosolic DNA sensing signaling. Reported post-translational modifications on each DNA sensing signaling pathway member are earmarked by indicated icons. The cartoon illustration is generated by BioRender.
Human cytomegalovirus (HCMV) infection on human embryonic lung fibroblasts induced viral pUL97-mediated phosphorylation on IFNγ-inducible protein 16 (IFI16), which facilitates the mis-localization of IFI16 into the cytosol to disable its viral DNA sensing ability (58) (Table 1). DHX9 phosphorylation close to its substrate-binding domain (may include S239 and S321) by PI3KKs promoted oncogenic circular RNA expression contributing to chemoresistance (64). BTK-mediated DDX41 phosphorylation on Tyr414 was critical for sensing foreign dsDNA and subsequent recruitment of STING for IFN production (67). However, the underlying mechanism(s) for how these phosphorylation events control the function of these sensors remains unclear.
Plk1 phosphorylates MRE11 at S649 and S688 residues, and CK2 phosphorylates MRE11 at S688, both of which promote the assembly of the MRB complex that is necessary to imitate the DNA damage repair (70). In addition, Plk1-mediated MRE11 phosphorylation at S688 also promotes MRN binding to MMAP (C2orf44) to form the MMAP-MRN complex, which further facilitates the repair of damaged DNA (72, 73). In contrast, RSK-mediated MRE11-S676 phosphorylation interferes with MRE11 binding to DNA, leading to impaired homologous recombination (74). Similarly, S6K phosphorylates MRE11-T597 residue, leading to impaired MRN complex formation and subsequent deficient DNA damage repair in colon cancer cells (75). Although it is clear that these various phosphorylation events exert distinct regulatory effects in modulating MRE11 function, whether these phosphorylation events are regulated under viral/bacterial infection during the cytosolic sensing process remains to be determined.
As an ER-localized protein, STING binds di-nucleic acids including 2′3′-cGAMP generated by cGAS upon sensing cytosolic DNA, which facilitates STING dimerization, oligomerization, and trafficking to Golgi, where TBK1 binds STING and phosphorylates STING-S366 (S365 in mice) (81), which is necessary to further recruit IRF3. TBK1 then also phosphorylates IRF3, promoting IRF3 dimerization and nuclear translocation to induce transcription of interferon genes. Afterward, 2′3′-cGAMP also triggers ULK1 activation by releasing its suppression by AMPK to phosphorylate STING-S366, leading to STING degradation, thereby preventing sustained innate immune signaling (82). One possible mechanism to explain how STING-S366 phosphorylation primes STING for degradation might be mediated by STING deSUMOylation, such that STING-S366 phosphorylation promotes SENP2 recognition at the late stage of viral infection that facilitates STING deSUMOylation, allowing STING ubiquitination to occur for STING degradation (51).
Ubiquitination
Protein ubiquitination is an ancient and evolutionarily conserved protein modification in regulating protein function in eukaryotes (105). Ubiquitin is a protein with 76 amino acids containing seven lysine residues that can be conjugated with another ubiquitin molecule to form polyubiquitin chains with distinct lengths. Ubiquitin can also be conjugated in a head-to-toe manner so that overall there are 8 distinct ubiquitin linkages formed including linear (M1, head-to-toe), K6, K11, K27, K29, K33, K48, and K63. To date, K11- and K48-linked ubiquitination has been related to proteasomal protein degradation, and other linkages have been reported to be involved in other biological processes including DNA damage response, protein trafficking, structure, and activity control (106). The ubiquitination process is carried out by a three-step enzymatic cascade including activating ubiquitin by E1, conjugating ubiquitin by E2, and selection of specific substrates for ubiquitin modification by E3 ubiquitin ligases. Given that E3 determines the substrate specificity, there are more than 600 identified E3 ligases in mammals. The poly-ubiquitin chains can be removed by deubiquitinases (DUBs) and largely consist of USPs, OTUs, UCHs, Joshphines, MINDYs, and JAMMs families (107). These DUBs exert ubiquitin chain hydrolysis ability and recognize both ubiquitin chains and substrates. Thus, protein ubiquitination is a dynamic and reversible process governed by E1/E2/E3 and DUBs.
Ubiquitination has been extensively studied in regulating innate immune DNA sensing signaling. Mono-ubiquitination of mcGAS on K335 by the E3 ligase TRIM56 was reported to facilitate cGAS activation upon DNA challenge by enhancing DNA binding and cGAS dimerization (46) (Table 1). RNF185-mediated mcGAS poly-ubiquitination at K173 and K384 residues through a K27 linkage also propagate the cGAS enzymatic activity (47). In contrast, various DUBs have been reported to facilitate cGAS activation largely by stabilizing cGAS proteins—for example, USP14 removes K48-linked polyubiquitin chains on K414 (48), USP29 removes polyubiquitination on K271 (50), and USP27X cleaves K48-linked ubiquitin chains on cGAS (49) to antagonize cGAS degradation. Notably, the E3 ligases governing proteasomal cGAS ubiquitination and degradation remain unclear.
TIRM21-governed IFI16 polyubiquitination on K3/K4/K6 residues earmarks IFI16 for degradation (59). Interestingly, herpesviral nuclear protein ICP0 binds nuclear IFI16 to retain it in the nucleus and additionally facilitates its degradation (60), leading to evasion of innate immune surveillance. Upon DNA virus infection, the E3 ubiquitin ligase TRIM11 binds AIM2, enhancing TRIM11 association with p62 and leading to AIM2 degradation through selective autophagy (63). In contrast, USP21 deubiquitinates AIM2, stabilizing the AIM2 inflammasome and facilitating downstream inflammation signaling (62). The DNA helicase DHX9 has been reported to be ubiquitinated and degraded by the E3 ligase SPOP (65), while RNF168-mediated DHX9 ubiquitination promotes recruitment of DHX9 to genomic loci prone to form R-loops where DHX9 resolves and removes R-loops (66). Whether any of these ubiquitination events occur in cytosolic DNA sensing mediated by DHX9 remains to be further investigated.
The expression of the E3 ligase TRIM21 is induced by interferons, and TRIM21 promotes K48-linked DDX41-K9 and K115 ubiquitination and degradation, serving as a mechanism to restrain the activation of innate immunity upon cytosolic DNA challenges (68). Upon DNA damage, UBQLN4 is recruited to damaged DNA, where UBQLN4 binds ubiquitinated MRE11 to remove it from repairing damaged DNA, leading to degradation of MRE11 to terminate the homologous recombination (76). The E3 ligase cIAP2 binds MRE11 to downregulate MRE11 protein levels by inducing an altered ubiquitination pattern on MRE11 (77).
Ubiquitin modifications on STING have been extensively studied with distinct effects on STING function in innate immunity. The E3 ligase RNF5 has been reported to target STING-K150 for ubiquitination and degradation upon viral infection (83), a process that can be antagonized by RNF26-mediated STING-K150 ubiquitination, presumably through a non-K48 linkage (86). In addition, TRIM29 (85) and TRIM30a (84) have also been reported to ubiquitinate STING-K370 and K275 residues, respectively, to target STING for degradation, serving as mechanisms to restrain innate immune sensing. As a result, Trim30a-deficient mice are more resistant to DNA viral infection (84). Interestingly, TRIM30a expression is induced by HSV-1 infection, suggesting that TRIM30a-mediated STING ubiquitination and degradation may serve as a negative feedback mechanism to shut down interferon signaling to avoid its hyperactivation (84). Moreover, TOLLIP, which usually helps to clear poly-Q-containing protein aggregates, was found in a siRNA-mediated screen as a positive regulator to stabilize STING proteins at the resting state by binding STING to prevent its lysosomal degradation (87). Other than regulating STING protein stability, STING ubiquitination by various E3 ligases has also been shown to be critical for STING dimerization/oligomerization and recruitment of both TBK1 and IRF3. For example, RNF115-mediated STING-K20/K224/K289 ubiquitination facilitates the formation of higher orders of STING structures and TBK1 recruitment (88). TRIM56-dependent STING-K150 (89), AMFR, governed STING-K137/K150/K224/K236 (90), and TRIM32-mediated STING-K20/K224/K236 ubiquitination (91) plays critical roles in STING dimerization and recruitment of TBK1/IRF3 to facilitate IRF3 phosphorylation and interferon production presumably through non-K48-linked ubiquitin chain linkages. In addition, the E3 ligase MUL1 conjugates K63-linked ubiquitin chains to STING-K224, which facilitates proper STING trafficking from ER to Golgi and bridges interactions of TBK1 with IRF3 mediated by STING (92).
DUBs have also been identified to antagonize E3 ligase-induced STING ubiquitination and function. Three DUBs including USP20 (93), EIF30S (94), and CYLD (95), have been reported to largely remove K48-linked polyubiquitin chains on STING, leading to stabilization of STING proteins and sustaining innate immune signaling. Another two DUBs, including USP13 (96) and MYSM1 (97), largely cleave K63-linked ubiquitin chains from STING, leading to impaired STING recruitment of TBK1 and IRF3, resulting in dampened interferon production. USP21 also negatively regulates STING function in promoting interferon production by deubiquitinating STING (98), a process negatively controlled by p38-MAPK (98).
SUMOylation and Neddylation
In addition to ubiquitin, other ubiquitin-like molecules can also be conjugated to target proteins to modulate their function. This includes SUMO (small ubiquitin-related modifier), NEDD8 (neural precursor cell expressed developmentally downregulated protein 8), and UFM1 (ubiquitin-fold modifier 1, ISG15 (ISG15 Ubiquitin-Like Modifier). SUMO is a ~10 KD small protein structurally similar to ubiquitin and can be conjugated to target proteins through lysine residues by an enzyme cascade consisting of E1-activating enzyme, E2-conjugating enzyme, and E3 SUMO ligase (108). SUMOylation regulates target protein stability, cellular location, and function largely through recruiting distinct subsets of downstream binding partners and effectors. NEDD8 is also a ubiquitin-like protein with NEDD8-specific conjugation and deconjugation pathways that can distinguish this modification from other ubiquitin-like modifications (109). The best-characterized proteins regulated by Nedd8 conjugation are cullins, which are scaffold proteins for cullin-ring types of E3 ubiquitin ligases (110), and there are also non-cullin protein targets found with neddylation (109). The UFM1 system is less understood, although it is highly conserved in eukaryotes except for yeast and fungi. Different from other ubiquitin-like molecules, UFM1 is more connected with the function of ER and ER stress controls the UFM1 system (111). Modification of DNA sensing signaling components by SUMOylation, neddylation, or UFMylation has also been observed in controlling innate immune activity.
mcGAS was found to be SUMOylated by TRIM38 at the resting state on K271/K464, which antagonizes ubiquitination-mediated degradation, resulting in cGAS stabilization for acute sensing viral infection (51). At the later stage of infection, the deSUMOylase SENP2 cleaves SUMO conjugates added on cGAS-K217/K464 by TRIM38 to facilitate cGAS ubiquitination and degradation, thus restraining cGAS overactivation (51), while another deSUMOylase, SENP7, through removing SUMO conjugates on mcGAS-K335/K372/K382, facilitates cGAS binding to DNA and cGAS dimer formation and subsequent interferon production (52). Given that TRIM56 mono-ubiquitinates mcGAS on K335, it is plausible that SENP7-mediated cGAS deSUMOylation is necessary for cGAS mono-ubiquitination in order to activate cGAS (46). Similarly, TRIM38 also maintains STING SUMOylation on K338, which prepares STING ready for sensing cytosolic DNA signaling. Upon viral infection, SENP2 deSUMOylates STING, which facilitates STING degradation in terminating this signaling (51). Thus, TRIM38/SENP2 controls both cGAS and STING protein stability in the early and late stages of viral infection to ensure the timely activation and inactivation to fine-tune the pathway responses.
cGAS was also found to be neddylated by UBE2M (E2)/RNF111 (E3) on K231/K421 residues, where neddylated cGAS will be properly positioned to form dimers with the previous cGAS, thus facilitating cGAS activation. In contrast, SENP8 de-neddylates cGAS on these residues and subsequently impairs proper cGAS dimer formation and activation (53).
UFMylation of MRE11-K282 has been observed and reported to be critical for MRN complex formation to ensure a timely location of the MRN complex to damaged DNA (78). In addition, MRE11 UFMylation is also important to recruit the phosphatase PP1 to dephosphorylate NBS1, therefore enhancing MRN complex binding to telomeres to maintain telomere length (79).
Other Protein Modifications
In addition to phosphorylation, ubiquitination, and ubiquitin-like modifications, other post-translational modifications that control activation of cytosolic DNA sensing have also been reported albeit with less attention. cGAS acetylation has been shown to be critical for cGAS binding to DNA. cGAS acetylation on K47/K56/K62/K83 residues in cGAS N-terminus by KAT5 facilitates DNA binding and cGAS activation (55), while deacetylation of cGAS-K384/K394/K414 in the cGAS enzymatic domain by HDAC3 is necessary for cGAS binding DNA (56). This may suggest that although both cGAS N and C domains participate in DNA binding, acetylation is only preferred in the disordered N but not well-ordered C domain for DNA recognition. PRMT5-mediated cGAS-R124 methylation attenuates cGAS-controlled antiviral immune response, largely through disrupting cGAS binding to DNA (54). Considering the R124 residue is also within the N-terminus, it is plausible that R124 methylation destabilizes cGAS conformation, while K47/K56/K62/K83 maintains a suitable cGAS structure for DNA binding, which requires further in-depth investigations. PRMT1 methylates MRE11 (aa566-600) to maintain an intact MRN complex during intra-S-phase DNA damage, which is critical to establish a proper intra-S-phase DNA damage checkpoint (80). In addition, P300 acetylates IFI16 within its nuclear localization signal (NLS) to retain IFI16 in the cytoplasm, disabling its ability to sense nuclear DNA for activation of the innate immune signaling (61).
In addition to S/T targeted phosphorylation and K/R targeted modifications, E (Glu) residues in cGAS have been observed to undergo glutamylation modifications. Specifically, TTLL4-mediated mono-glutamylation of hcGAS-E314 inhibits cGAS enzymatic activity (57), and similarly, TTLL6-governed poly-glutamylation of hcGAS-E272 disrupts cGAS binding to DNA (57). In contrast, CCP5 removes hcGAS-E314 mono-glutamylation, and CCP6 cleaves hcGAS-E272 poly-glutamylation to recover cGAS binding to DNA and activation (57). Both glutamylated and non-glutamylated cGAS species are observed at resting states, while during viral infection, expression of TTLL enzymes is downregulated, leading to increased populations of non-glutamylated cGAS for sensing viral DNA to initiate innate immunity.
Given that the cellular trafficking of STING from ER to Golgi plays a critical role in recruiting TBK1/IRF3 to activate the innate immunity, other modifications on STING than K63-linked ubiquitination (92) have also been observed. C88/C91 palmitoylation of STING by DHHC3/DHHC7/DHHC15 was reported to be necessary to mediate STING leaving ER for activation (99), while the detailed molecular mechanisms remain unclear. In addition to palmitoylation, cysteine residues also undergo oxidation, such that hSTING-C148 oxidation induced by cellular ROS interferes with STING oligomerization and subsequent activation to suppress interferon production (100).
Overview of Cytosolic RNA Sensing
Similar to DNA sensing, infection by RNA viruses that expose viral RNA to host cytoplasm triggers acute nucleic acid sensing via PRRs to initiate signaling pathways leading to the production of type I interferons including IFNα and IFNβ and other cytokines for robust innate immune responses. Depending on the localization, RNA sensors can be divided into endosomal membrane-associated TLRs that function primarily in immune cells, and cytosolic RNA sensors RIG-I (112) and MDA5 (melanoma differentiation-associated gene 5), which are expressed in most cells (2). Both RIG-I and MDA5 are RNA helicases composed of two N-terminal CARDs (caspase recruitment domains), a central DExD/H-box ATPase/helicase domain and a C-terminal regulatory domain that binds RNA (112). RIG-I senses dsRNA, single-strand RNA with 5′-triphosphates (113), or even reversely transcribed 5′-triphosphate RNA from cytosolic viral dsDNA (29). In contrast, MDA5 largely recognizes dsRNA (114). RNA binding stimulates helicase activity in both RIG-I and MDA5 and promotes the formation of prion-like aggregates through oligomerization to expose N-terminal CARDs. These exposed CARDs bind the mitochondrial protein MAVS (also named VISA, IPS-1, CARDIF) (115) to form a signaling platform with the help of the E3 ligase TRAF3 (116) (also other TRAFs (117)) to recruit TBK1 and IKKε to facilitate transcription of interferon genes through IRFs (118) and activate IKKα/β to induce NF-κB-mediated transcription of proinflammatory genes (119). Phosphorylated IRF3 or IRF7 forms homo-dimers and translocates into the nucleus to promote type I interferon transcription.
Post-Translational Modifications of Proteins in Cytosolic RNA Sensing
Phosphorylation
At resting states, both RIG-I and MDA5 are phosphorylated at caspase recruitment domains to keep them inactive. Upon sensing cytosolic RNA, TRIM25 adds K63-linked polyubiquitin on RIG-I at K172 to facilitate RIG-I binding with its downstream effectors MAVS (also known as VISA/IPS-1) (120), thus activating the RNA sensing signaling to induce interferon production (Table 2 and Figure 2). Phosphorylation of RIG-I on S8 and/or T170 impairs its K172 polyubiquitination by PKCα/β via disrupting TRIM25 binding and also disrupts RIG-I interactions with MAVS and subsequent antiviral interferon production (137, 139). In addition, RIG-I phosphorylation by CKII at T770/S854/S855 inhibits RIG-I activation by inhibiting the formation of RIG-I intermolecular interactions and oligomerization (140). Similarly, MDA5 phosphorylation on S88 blocks MDA5 interaction with MAVS at resting states (141). In contrast, an RNAi screen identified PP1α and PP1γ as major phosphatases dephosphorylating RIG-I on S8/T170, which facilitates RIG-I binding to TIRM25 and MAVS (VISA/IPS-1) to promote innate immune activation (141). Therefore, PP1-depleted cells showed a decreased ability to induce interferon, and increased RNA virus replication upon RNA virus infection including influenza virus, paramyxovirus, dengue virus, and picornavirus (141). RNA viruses induced EGFR activation, which led to DDX60-Tyr793/Tyr796 phosphorylation, which attenuated RIG-I signaling and reduced type I IFN production (69).
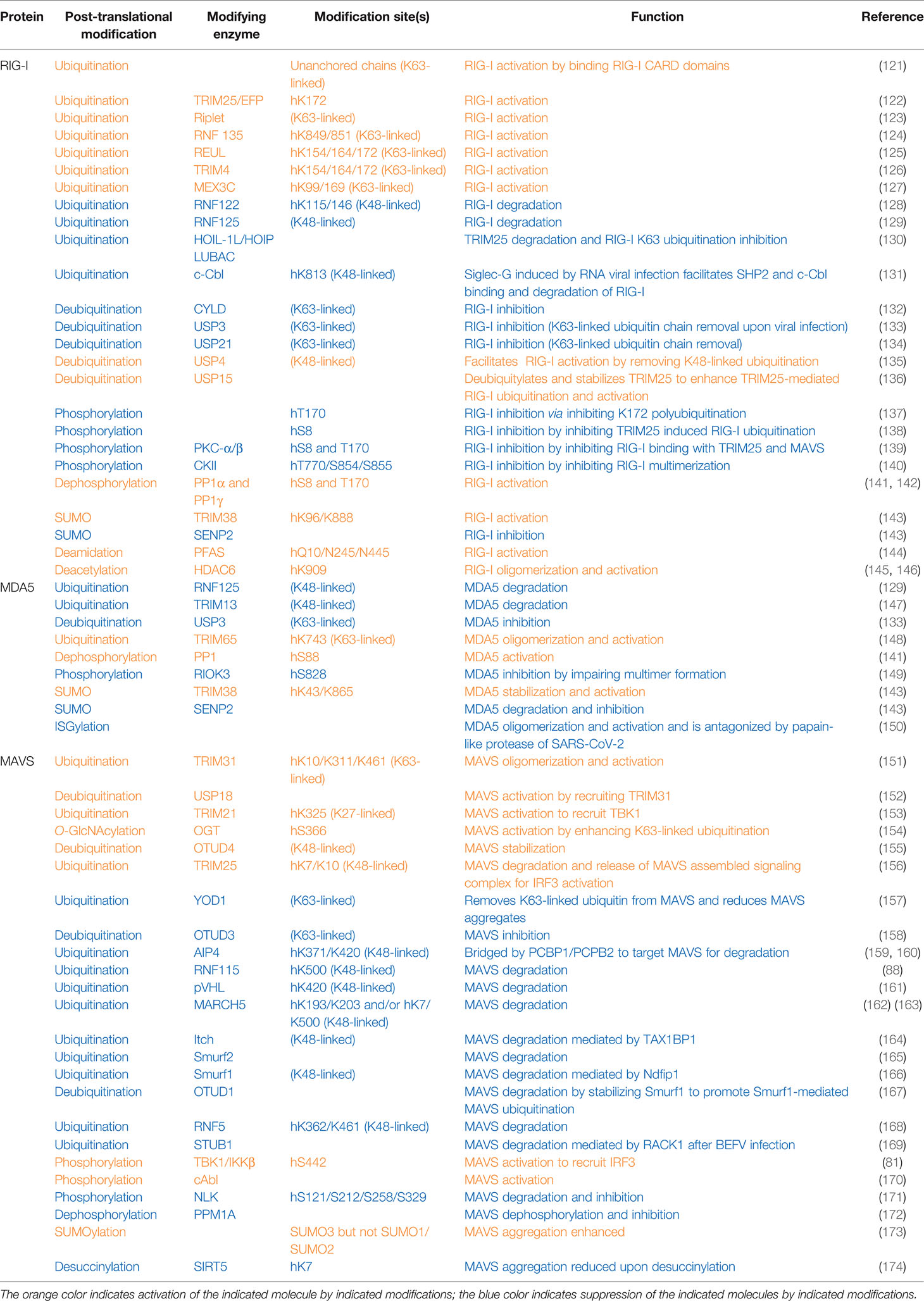
Table 2 Post-translational modifications of proteins in cytosolic RNA sensing signaling pathways.
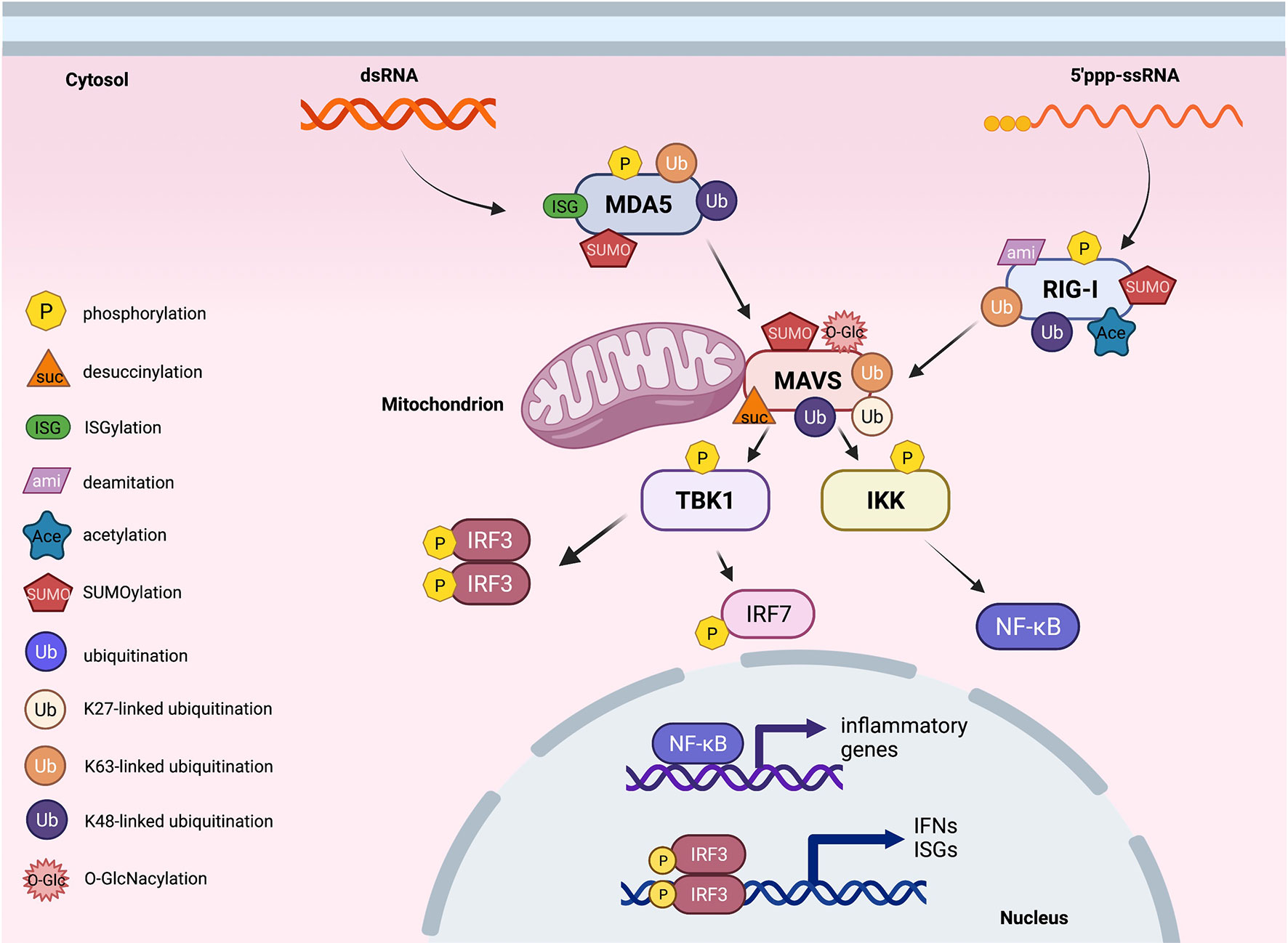
Figure 2 Post-translational modifications of proteins in cytosolic RNA sensing signaling. An overview of cytosolic RNA sensing signaling. Reported post-translational modifications on each RNA sensing signaling pathway member are earmarked by indicated icons. The cartoon illustration is generated by BioRender.
Similar to STING, MAVS undergoes S442 phosphorylation by either TBK1 or IKKβ, and this phosphorylation is essential to recruit IRF3 for its phosphorylation and nuclear translocation in inducing interferon production upon sensing cytosolic RNA (81). Through a yeast two-hybrid screen, the tyrosine kinase c-Abl binds the CARD and TM domains in MAVS to phosphorylate MAVS on Y residues, through unknown mechanisms to facilitate MAVS activation (170). In contrast, NLK-mediated MAVS-S121/S212/S258/S329 phosphorylation upon RNA viral infection promotes MAVS degradation to dampen RNA sensing ability (171). PPM1A (protein phosphatase magnesium-dependent 1A) is complexed with TBK1/IKKε and targets both MAVS and TBK1/IKKε for dephosphorylation, leading to the dissociation of the MAVS/TBK1/IKKε signaling complex and subsequently impaired RNA sensing signaling. As a result, Ppm1a−/− mice are resistant to RNA viral infection (172).
Ubiquitination
Activation of RIG-I requires unanchored K63-linked ubiquitin chains in addition to RNA and ATP. Free K63-linked ubiquitin chains bind to the CARD domains in RIG-I (121), allowing for a transition from a closed inactive conformation to an open active conformation. At the early phase of RNA viral infection, activation of RIG-I requires TRIM25-mediated K63-linked polyubiquitination modification on K172, which serves as a platform to recruit downstream effector binding (122). With help from E2 enzymes UBE2D3 and UBE2N, the E3 ligase Riplet facilitates the conjugation of K63-linked ubiquitination on RIG-I to aid its activation (123). Another study reports the E3 ligase REUL governs RIG-I ubiquitination (presumably through K63 linkage) on K154/K164/K172, also promoting RIG-I-mediated RNA sensing using a yeast two-hybrid assay (125). In addition, through screening human ubiquitin-related enzyme cDNA library, expression of TRIM14 is observed to be able to stimulate IFN-β promoter reporter largely through promoting K63-linked RIG-I ubiquitination on K154/K164/K172 (126). Moreover, MEX3C adds K63-linked ubiquitin chains to RIG-I K99/K169 residues to exert a similar signaling activation function (127). An independent yeast two-hybrid screen identified RNF135 as an additional E3 ligase to promote K63-linked RIG-I ubiquitination on its C-terminal K849/K851 residues, exerting a similar function as TRIM27-mediated RIG-I-K172 ubiquitination to facilitate RIG-I activation (124). With the use of distinct approaches including microarray and DUB cDNA screen, three DUBs including CYLD (132), USP3 (133), and USP21 (134) were reported to remove K63-linked RIG-I ubiquitination to antagonize RIG-I activation. CYLD maintains low RIG-I ubiquitination at the resting state, and during viral infection, CYLD is downregulated, allowing K63-linked RIG-I ubiquitination to occur for RIG-I activation (132). Notably, at the resting state, USP3 does not bind RIG-I, and upon viral infection, an induced USP3 binding to RIG-I leads to the removal of K63-linked ubiquitin chains to restrain or terminate RIG-I signaling (133). Whether USP21 expression or interaction with RIG-I is also regulated by viral infection remains unclear.
In addition to K63-linked ubiquitination that promotes RIG-I signaling complex formation, K48-linked RIG-I ubiquitination has also been observed to control RIG-I protein stability. To this end, RNF122 was observed to co-localize with RIG-I to conjugate K48-linked ubiquitin chains to K115/K146 residues that earmark RIG-I for proteasomal degradation (128). A yeast two-hybrid assay found RNF125 as a RIG-I binding E3 ligase that conjugates K48-linked ubiquitin chains to both RIG-I and MDA5 to promote their destruction (129). Expression of both RNF122 and RNF125 is enhanced by IFN production; thus, RNF122- or RNF125-mediated RIG-I ubiquitination and degradation may serve as a negative feedback mechanism to restrain sustained innate immune activation. Siglec-G induced by RNA viral infection facilitates SHP2 and the E3 ligase c-Cbl binding to RIG-I, where c-Cbl facilitates K48-linked RIG-I-K813 ubiquitination and degradation, serving as a mechanism hijacked by RNA viruses to disable RIG-I-mediated RNA sensing (131). In contrast, the deubiquitinase USP4 is found to remove K48-linked ubiquitin chains from RIG-I, thus stabilizing RIG-I to facilitate its RNA sensing function (135). USP4 expression is attenuated upon RNA viral infection; thus, USP4-mediated RIG-I deubiquitination may serve as a negative regulatory mechanism to restrain RIG-I signaling from overactivation or sustained activation.
Moreover, TRIM25-mediated RIG-I K63-linked ubiquitination and activation can also be antagonized by the linear ubiquitin assembly complex composed of HOIL-1L/HOIP/LUBAC, where HOIL-1L/HOIP targets TRIM25 for degradation, and HOIL-1L also competes with TRIM25 to bind RIG-I (130). These two mechanisms independently lead to the suppression of RIG-I activation when sensing cytosolic RNA. Moreover, the deubiquitinase USP15 removes the ubiquitin moiety from RIG-I to stabilize TRIM25, leading to enhanced RIG-I ability in sensing cytosolic RNA to promote interferon production (136).
Like RIG-I, K63-linked ubiquitination of MDA5 by TRIM65 on K743 residue is critical for MDA5 oligomerization and subsequent activation upon RNA viral infection (148). In addition, viral infection induces USP3 interaction with MDA5 to catalyze the removal of K63-linked ubiquitin chains, thus limiting sustained activation of MDA5 signaling (133). At the later stage of viral infection, expression of either the E3 ligase RNF125 (129) or TRIM13 (147) is induced, leading to conjugation of K48-linked ubiquitin chains to MDA5 for MDA5 degradation, both of which serve as a negative feedback mechanism to terminate MDA5 signaling.
Activation of MAVS can be initiated by either K63-linked or K27-linked ubiquitination events. TRIM31 conjugates K63-linked ubiquitin moieties to MAVS-K10/K311/K461, which is necessary for MAVS oligomerization and subsequent activation (151). Interestingly, a mitochondrion-localized DUB USP18 serves as a scaffold protein to bridge TRIM31 interaction with MAVS, enhancing TIRM31-mediated K63 linkage ubiquitination of MAVS for its activation (152). Expression of TRIM21 is enhanced under viral infection, where TRIM21 catalyzes K27-linked ubiquitination of MAVS-K325, which further recruits TBK1 to transduce innate immune signaling (153), which may serve as a fine-tuning mechanism to enhance innate immunity. In addition, metabolic states also modulate anti-RNA viral infection responses. For example, OGT (O-linked β-N-acetylglucosamine (O-GlcNAc) transferase) adds on O-GlcNAc to MAVS-S366 residue, which promotes K63-linked ubiquitination of MAVS for its activation (154). Another approach to enhance MAVS activation is to stabilize MAVS proteins by removing K48-linked ubiquitin chains from MAVS by OTUD4. Upon viral infection, OTUD4 expression is induced to quickly stabilize MAVS, preparing it for timely response to infection (155). Interestingly, TRIM25 targets MAVS for ubiquitination and degradation after MAVS activation, allowing the release of MAVS assembled signaling complex including TRAF3, NEMO, and TBK1 to translocate to the cytoplasm where TBK1 phosphorylates and activates IRF3 to facilitate interferon production (156).
It seems that at resting states, the levels of MAVS-K63-linked ubiquitination remain low by OTUD3, while upon viral infection, OTUD3 is inactivated by SIRT1-mediated K129 deacetylation, allowing for the buildup of K63-linked ubiquitination of MAVS for its activation (158). MAVS K63-linked ubiquitin moiety added on MAVS for its activation can be removed by YOD1 in the later stage of viral infection to restrain MAVS from overactivation (157). At the resting state, PCBP1 bridges the E3 ligase AIP4 to ubiquitinate MAVS through a K48 linkage to target MAVS for proteasomal degradation, thus maintaining a low level of MAVS expression (159). Upon viral infection, PCBP2 expression is induced and similarly bridges AIP4 to target MAVS-K371/K420 for degradation at later stages of infection, serving as a possible negative feedback mechanism to terminate MAVS signaling (159, 160). Another mechanism to maintain a low level of MAVS under uninfected conditions is achieved by RNF115-mediated K48-linked polyubiquitination on K500 for degradation of MAVS (88).
Ubiquitination of MAVS also plays an important role in preventing sustained activation of the MAVS signaling. To this end, viral infection induces RNF5 binding to MAVS, leading to RNF5-mediated conjugation of K48-linked ubiquitin chains to MAVS-K362/K461 leading to MAVS destruction (168). Similarly, viral infection also induces binding of the mitochondrial E3 ligase MARCH5 to aggregated and active MAVS, where MARCH5 ubiquitinates K193/K203 (162) or K7/K500 (163) through a K48 linkage for MAVS destruction, serving as a negative feedback mechanism to restrain sustained MAVS activation. In addition, viral infection induces expression of TAX1BP1, which recruits the E3 ligase Itch to add on K48-linked ubiquitination to MAVS for MAVS degradation in terminating MAVS signaling (164). Another E3 ligase Smurf2 (Smad ubiquitin regulatory factor 2) also promotes MAVS ubiquitination through a K48 linkage to promote MAVS destruction (165). Similarly, Smurf1 also promotes K48-linked ubiquitination and destruction of MAVS, which depends on Ndfip1 as a recruiter and activator for Smurf1 (166). In addition, RNA viral infection induces OTUD1 expression, which deubiquitinates and stabilizes Smurf1, therefore enhancing Smurf1-mediated MAVS degradation to negatively regulate MAVS function (167). The E3 ligase pVHL also negatively controls MAVS protein stability by adding K48-linked polyubiquitin chains on MAVS-K420 to facilitate its destruction (161).
RNA viruses also hijack MAVS degradation mechanisms to facilitate viral replication and viral infection. For example, RNA viral infection enhances the expression of RACK1 (Receptors for activated C kinase 1), by upregulating the expression of the E3 ligase STUB1 (STIP1 homology and U-box containing protein 1) to target MAVS for ubiquitination and degradation; RACK1 facilitates BEFV (bovine epidemic fever virus) replication (169).
SUMOylation and Neddylation
TIRM38 exerts a protein SUMO E3 ligase activity in governing SUMOylation of RIG-I-K96/K888 and MDA5-K43/K865 in uninfected and early infected cells, respectively, to stabilize both RIG-I and MDA5 by antagonizing K48-linked ubiquitination, leading to an acute and enhanced response to viral infection (143). At the later infection stage, SENP2 removes SUMO conjugates to facilitate RIG-I and MDA5 proteasomal degradation to terminate RNA sensing signaling (143). Recently, ISGylation of MDA5 has also been reported to facilitate MDA5 oligomerization and activation, a process that can be antagonized by the papain-like protease of SARS-CoV-2 (150). In addition, SUMO3, not SUMO2 or SUMO1, addition to MAVS has been reported to enhance MAVS aggregation and activation to stimulate interferon production (173) upon poly(dA:dT) treatments.
Other Protein Modifications
Upon viral infection, HDAC6 binds and deacetylates RIG-I-K909 to enhance its RNA sensing ability by allowing the formation of RIG-I oligomers (145, 146). The viral PFAS (phosphoribosylformylglycinamide synthase), although lacking intrinsic activity, uses host PFAS to deamidate RIG-I on Q10/N245/N445 to activate RIG-I in triggering the host RNA sensing signaling (144). In addition, after viral infection, SIRT5 catalyzes MAVS desuccinylation at residue K7 to reduce the MAVS aggregates to limit MAVS activation and RLR signaling (174).
Conclusions
Given that hyperactivation of cytosolic nucleic acid sensing signaling causes autoimmune diseases while hypoactivation of cytosolic nucleic acid sensing leads to susceptibility to infection and compromised immunotherapeutic effects [summarized in (31)], the timely and concise control of activation of both cytosolic DNA and RNA sensing signaling is tightly controlled through multilayer regulatory mechanisms. Among them, post-translational modifications of key cytosolic nucleic acid sensing pathway members have been extensively studied, and fine-tuning mechanisms have been elucidated. Considering the structural difficulties in distinguishing pathogen DNA or RNA from the host’s nucleic acids, the innate immune system may prefer to enhance sensitivity during infection because the likelihood of a positive is high, and false-negative risk is acceptable for a short period of time.
In echoing this concept, post-translational modifications on key cytosolic nucleic acid sensors have been shown to differently govern sensor activation in different stages of infection. For example, cGAS is found to be unphosphorylated at the resting state to restrain its inappropriate activation by associating with PPP6C, and this interaction is alleviated upon viral infection, allowing for cGAS phosphorylation that primes cGAS activation (45). In contrast, RIG-I and MAVS are phosphorylated at resting states and upon viral infection; removal of phosphorylation on RIG-I and MAVS by PP1α and PP1γ is required for their activation (141, 142). In addition, the expression of a handful of E3 ligases and DUBs is induced by interferons; thus, ubiquitination or deubiquitination of nucleic acid sensors serves as a fine-tuning mechanism to restrain sustained innate immune signaling or terminate nucleic acid sensing signaling. For example, interferon induces expression of TRIM21, which ubiquitinates and degrades IFI16 (59) and DDX41 (68) to restrain DNA sensing, while facilitating K27-linked MAVS (153) to facilitate RNA sensing. HSV-1 infection induces TRIM30a expression, which targets STING for ubiquitination and degradation to shut down interferon signaling (84). Similarly, interferon induces expression of RNF122 (128) and RNF125 (129) to ubiquitinate and degrade RIG-I/MDA5, triggers TRIM13 synthesis to degrade MDA5 (147), or promotes RNF5 (168) and MARCH5 (162, 163) expression to degrade MAVS, all leading to inactivation of RNA sensing signaling after viral infection. Interferon induces downregulation of CYLD to promote RIG-I K63-linked ubiquitination for its activation (132) and reduces OTUD3 expression to allow MAVS to undergo K63-linked ubiquitination and activation (158). Moreover, interferon production also interferes with USP3 binding with both RIG-I (133) and MDA5 (133), allowing for the buildup of K63-linked ubiquitin chains for their activation. Aberrant activation of resting-state nucleic acid sensors might contribute to autoimmune diseases. This can be achieved by modulating nucleic acid sensor-modifying enzymes mentioned above. To this end, PP2A has been reported to confer susceptibility to autoimmune diseases (175). In addition, connections and crosstalks between innate immune responses and tumorigenesis are also observed. For example, TRIM21 facilitates tumorigenesis through ubiquitinating tumor-suppressive (e.g., p53 and p21) substrates (176). Given TRIM21 expression is induced by interferon to terminate DNA sensing (59), whether nucleic acid sensing-related function of TRM21 also contributes to tumorigenesis by evading cytosolic DNA sensing remains to be determined.
Interestingly, K63-linked ubiquitination has been shown to play a critical role in facilitating both cytosolic DNA and RNA sensing. K63-linked ubiquitination of STING is not only important for proper STING conformational changes and proper trafficking (89, 92) but also critical for recruiting its downstream effectors TBK1 and IRF3 (88, 91). Similarly, un-anchored K63-linked ubiquitin chains are necessary to activate RIG-I (121), and K63-linked ubiquitination of RIG-I by TRIM25 is also pivotal for RIG-I aggregate formation and activation (122). Conjugating K63-linked ubiquitin chains to MDA5 by TRIM65 is critical for MDA5 oligomerization and activation (148). MAVS ubiquitination by TIRM31 via a K63 linkage is required for its activation (152). To this end, K27-linked ubiquitination of cGAS has been reported to facilitate cGAS activation (47), while whether K63-linked ubiquitination plays a similar activation function remains to be further determined. In cancer, K63-linked ubiquitination has been observed to promote activation of oncogenic kinases including Akt and TAK1 (177) and various DNA damage response factors including CLASPIN (178), both leading to enhanced tumorigenesis.
It is commonly observed that a given protein can be modified by multiple posttranslational modifications to control distinct functions in a temporal and spatial manner (31). One residue in a given protein can also be regulated by different modifications. For example, TRIM56 mono-ubiquitinates mcGAS on K335 to facilitate its activation (46), while SENP7 deSUMOylates mcGAS-K335 (46). This may suggest that these modifications are mutually exclusive, and it is plausible that deSUMOylation is necessary for cGAS mono-ubiquitination for cGAS activation. In contrast, the same enzyme also controls multiple target functions. For example, TRIM38 functions as a SUMO ligase to differentially control cytosolic nucleic acid sensing. Specifically, TRIM38 SUMOylates cGAS (51) and STING (51) to stabilize both of them by antagonizing degradation-oriented ubiquitination, thus facilitating activation of DNA sensing. Similarly, TRIM38 also SUMOylates RIG-I (143) and MDA5 (143) to stabilize RIG-I and MDA5 for activation. Thus, distinct nucleic acid sensor-modifying enzymes can coordinate or compete by competitively regulating the same residues.
In addition to the timing of the modification (e.g., prior to or post-infection), the location of the modifications most of the time also dictates distinct functions. For example, three DNA binding sites termed site A, site B, and site C are identified in cGAS (179), which span the whole cGAS molecule. Acetylation of the cGAS-N terminus (K47/K56/K62/K83) facilitates DNA binding (55), while deacetylation of the cGAS C-terminus (K384/K394/K414) enhances cGAS binding with DNA (56). Another example is for fine-tuned activation control of RIG-I by multiple ubiquitination events (Figure 3). Specifically, TRIM25-mediated K172 ubiquitination is necessary for RIG-I activation (122). Riplet introduces K63-linked ubiquitination to activate RIG-I (123). Similarly, another two E3 ligases including REUL (125) and TRIM4 (126) add on K63-linked ubiquitin conjugates to K154/K164/K172 for RIG-I activation. In addition, MEX3C aids K63-linked ubiquitination on K99/K169, which also promotes RIG-I activation (127). Even K63-linked ubiquitination on C-terminal K849/K851 by RNF135 facilitates RIG-I activation (124). Why there is a need for multiple E3 ligases to catalyze the same ubiquitination events, why ubiquitination at distinct residues all lead to RIG-I activation, and whether these modifications/expression of enzymes are viral type, infection stage, or tissue-specific remain to be further determined. Nonetheless, these examples reveal multilayers of regulatory mechanisms achieved by the same or crosstalks among different posttranslational modifications. Moreover, whether these modifying enzymes can be targeted for treating patients with either autoimmune diseases or immune deficiency warrants a promising yet understudied direction.
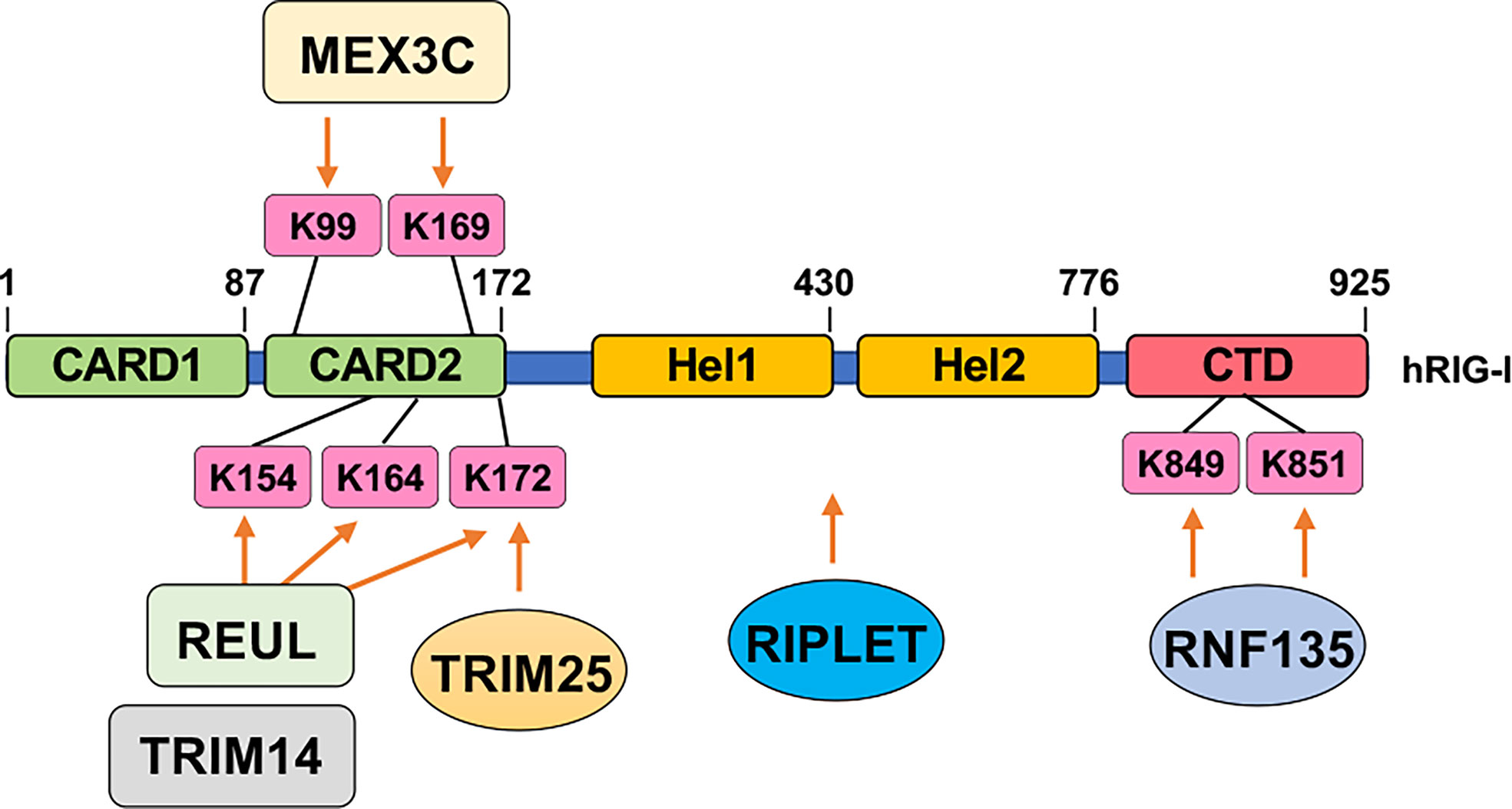
Figure 3 Ubiquitination-mediated RIG-I activation. Indicated E3 ubiquitin ligases add on K63-linked ubiquitin chains to indicated residues in RIG-I to facilitate RIG-I activation upon sensing cytosolic RNA.
Among all the nucleic acid-modifying enzymes, inhibitors targeting kinases have been developed for the treatment of autoimmunity and inflammation, including JAK, IRAK4, RIPK, SYK, BTK, and TPL2 (180). Targeting kinases regulating nucleic acid sensor activities like Akt is also feasible—Akt inhibition suppresses tumor growth not only through intrinsic survival mechanisms but also through releasing Akt-mediated cGAS suppression to facilitate innate immune activation and subsequent increased immune cell infiltrates, which warrants further investigations. In contrast, the application of BLK inhibitors in treating T-cell lymphoma (where BLK was shown as an oncogene (181)) should be used with caution because inhibiting BLK may attenuate cGAS cytosolic retention leading to deficiency in cGAS activation and dampened T-cell recruitment into tumors. In contrast, inhibitors targeting E3 ligases and DUBs are not well developed largely because E3 ligases do not exert enzymatic activity and technical difficulties in developing DUB inhibitors. However, it is plausible that properly targeting nucleic acid sensor-modifying enzymes listed in this review will lead to new therapeutic directions for treating either autoimmune diseases/inflammation or cancer.
Author Contributions
YD and YW: information collection. YD and YW: table/figure construction. YD, YW, and PL: drafting of the manuscript. YD, YW, LL, EM, and PL: revising of the manuscript. All authors listed have made a substantial, direct, and intellectual contribution to the work and approved it for publication.
Funding
This work was supported by an NIH grant (R01CA244825, PL), the Gabrielle’s Angel Foundation Medical Research Award (PL), and the UNC University Cancer Research Fund (PL).
Conflict of Interests
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We sincerely apologize to all colleagues whose important work could not be cited in this review owing to space limitations, especially many prominent and pioneer work in the nucleic acid sensing field.
References
1. Hu MM, Shu HB. Cytoplasmic Mechanisms of Recognition and Defense of Microbial Nucleic Acids. Annu Rev Cell Dev Biol (2018) 34:357–79. doi: 10.1146/annurev-cellbio-100617-062903
2. Akira S, Uematsu S, Takeuchi O. Pathogen Recognition and Innate Immunity. Cell (2006) 124:783–801. doi: 10.1016/j.cell.2006.02.015
3. Atianand MK, Fitzgerald KA. Molecular Basis of DNA Recognition in the Immune System. J Immunol (2013) 190:1911–8. doi: 10.4049/jimmunol.1203162
4. Takeuchi O, Akira S. Pattern Recognition Receptors and Inflammation. Cell (2010) 140:805–20. doi: 10.1016/j.cell.2010.01.022
5. Man SM, Kanneganti TD. Regulation of Inflammasome Activation. Immunol Rev (2015) 265:6–21. doi: 10.1111/imr.12296
6. Dammermann W, Wollenberg L, Bentzien F, Lohse A, Luth S. Toll Like Receptor 2 Agonists Lipoteichoic Acid and Peptidoglycan are Able to Enhance Antigen Specific IFNgamma Release in Whole Blood During Recall Antigen Responses. J Immunol Methods (2013) 396:107–15. doi: 10.1016/j.jim.2013.08.004
7. Mahla RS, Reddy MC, Prasad DV, Kumar H. Sweeten PAMPs: Role of Sugar Complexed PAMPs in Innate Immunity and Vaccine Biology. Front Immunol (2013) 4:248. doi: 10.3389/fimmu.2013.00248
8. Holm CK, Paludan SR, Fitzgerald KA. DNA Recognition in Immunity and Disease. Curr Opin Immunol (2013) 25:13–8. doi: 10.1016/j.coi.2012.12.006
9. Kagan JC. Signaling Organelles of the Innate Immune System. Cell (2012) 151:1168–78. doi: 10.1016/j.cell.2012.11.011
10. Medzhitov R. Recognition of Microorganisms and Activation of the Immune Response. Nature (2007) 449:819–26. doi: 10.1038/nature06246
11. Schlee M, Hartmann G. Discriminating Self From non-Self in Nucleic Acid Sensing. Nat Rev Immunol (2016) 16:566–80. doi: 10.1038/nri.2016.78
12. Kawasaki T, Kawai T, Akira S. Recognition of Nucleic Acids by Pattern-Recognition Receptors and its Relevance in Autoimmunity. Immunol Rev (2011) 243:61–73. doi: 10.1111/j.1600-065X.2011.01048.x
13. Platanias LC. Mechanisms of Type-I- and Type-II-Interferon-Mediated Signalling. Nat Rev Immunol (2005) 5:375–86. doi: 10.1038/nri1604
14. Ramazi S, Zahiri J. Posttranslational Modifications in Proteins: Resources, Tools and Prediction Methods. Database (Oxford) (2021) 2021.
15. Hornung V, Ablasser A, Charrel-Dennis M, Bauernfeind F, Horvath G, Caffrey DR, et al. AIM2 Recognizes Cytosolic dsDNA and Forms a Caspase-1-Activating Inflammasome With ASC. Nature (2009) 458:514–8. doi: 10.1038/nature07725
16. Burckstummer T, Baumann C, Bluml S, Dixit E, Durnberger G, Jahn H, et al. An Orthogonal Proteomic-Genomic Screen Identifies AIM2 as a Cytoplasmic DNA Sensor for the Inflammasome. Nat Immunol (2009) 10:266–72. doi: 10.1038/ni.1702
17. Fernandes-Alnemri T, Yu JW, Datta P, Wu J, Alnemri ES. AIM2 Activates the Inflammasome and Cell Death in Response to Cytoplasmic DNA. Nature (2009) 458:509–13. doi: 10.1038/nature07710
18. Briggs JA, Burrus GR, Stickney BD, Briggs RC. Cloning and Expression of the Human Myeloid Cell Nuclear Differentiation Antigen: Regulation by Interferon Alpha. J Cell Biochem (1992) 49:82–92. doi: 10.1002/jcb.240490114
19. Ding Y, Wang L, Su LK, Frey JA, Shao R, Hunt KK, et al. Antitumor Activity of IFIX, a Novel Interferon-Inducible HIN-200 Gene, in Breast Cancer. Oncogene (2004) 23:4556–66. doi: 10.1038/sj.onc.1207592
20. Unterholzner L, Keating SE, Baran M, Horan KA, Jensen SB, Sharma S, et al. IFI16 is an Innate Immune Sensor for Intracellular DNA. Nat Immunol (2010) 11:997–1004. doi: 10.1038/ni.1932
21. Sun L, Wu J, Du F, Chen X, Chen ZJ. Cyclic GMP-AMP Synthase is a Cytosolic DNA Sensor That Activates the Type I Interferon Pathway. Science (2013) 339:786–91. doi: 10.1126/science.1232458
22. Kondo T, Kobayashi J, Saitoh T, Maruyama K, Ishii KJ, Barber GN, et al. DNA Damage Sensor MRE11 Recognizes Cytosolic Double-Stranded DNA and Induces Type I Interferon by Regulating STING Trafficking. Proc Natl Acad Sci USA (2013) 110:2969–74. doi: 10.1073/pnas.1222694110
23. Zhang X, Brann TW, Zhou M, Yang J, Oguariri RM, Lidie KB, et al. Cutting Edge: Ku70 is a Novel Cytosolic DNA Sensor That Induces Type III Rather Than Type I IFN. J Immunol (2011) 186:4541–5. doi: 10.4049/jimmunol.1003389
24. Sui H, Zhou M, Imamichi H, Jiao X, Sherman BT, Lane HC, et al. STING is an Essential Mediator of the Ku70-Mediated Production of IFN-Lambda1 in Response to Exogenous DNA. Sci Signaling (2017) 10.
25. Yang P, An H, Liu X, Wen M, Zheng Y, Rui Y, et al. The Cytosolic Nucleic Acid Sensor LRRFIP1 Mediates the Production of Type I Interferon via a Beta-Catenin-Dependent Pathway. Nat Immunol (2010) 11:487–94. doi: 10.1038/ni.1876
26. Zhang Z, Yuan B, Bao M, Lu N, Kim T, Liu YJ. The Helicase DDX41 Senses Intracellular DNA Mediated by the Adaptor STING in Dendritic Cells. Nat Immunol (2011) 12:959–65. doi: 10.1038/ni.2091
27. Takaoka A, Wang Z, Choi MK, Yanai H, Negishi H, Ban T, et al. DAI (DLM-1/ZBP1) is a Cytosolic DNA Sensor and an Activator of Innate Immune Response. Nature (2007) 448:501–5. doi: 10.1038/nature06013
28. Chiu YH, Macmillan JB, Chen ZJ. RNA Polymerase III Detects Cytosolic DNA and Induces Type I Interferons Through the RIG-I Pathway. Cell (2009) 138:576–91. doi: 10.1016/j.cell.2009.06.015
29. Ablasser A, Bauernfeind F, Hartmann G, Latz E, Fitzgerald KA, Hornung V. RIG-I-Dependent Sensing of Poly(Da:Dt) Through the Induction of an RNA Polymerase III-Transcribed RNA Intermediate. Nat Immunol (2009) 10:1065–72. doi: 10.1038/ni.1779
30. Schroder K, Tschopp J. The Inflammasomes. Cell (2010) 140:821–32. doi: 10.1016/j.cell.2010.01.040
31. Yu L, Liu P. Cytosolic DNA Sensing by cGAS: Regulation, Function, and Human Diseases. Signal Transduct Target Ther (2021) 6:170. doi: 10.1038/s41392-021-00554-y
32. Rathinam VA, Fitzgerald KA. Innate Immune Sensing of DNA Viruses. Virology (2011) 411:153–62. doi: 10.1016/j.virol.2011.02.003
33. Keating SE, Baran M, Bowie AG. Cytosolic DNA Sensors Regulating Type I Interferon Induction. Trends Immunol (2011) 32:574–81. doi: 10.1016/j.it.2011.08.004
34. Crowl JT, Gray EE, Pestal K, Volkman HE, Stetson DB. Intracellular Nucleic Acid Detection in Autoimmunity. Annu Rev Immunol (2017) 35:313–36. doi: 10.1146/annurev-immunol-051116-052331
35. Iurescia S, Fioretti D, Rinaldi M. Targeting Cytosolic Nucleic Acid-Sensing Pathways for Cancer Immunotherapies. Front Immunol (2018) 9:711. doi: 10.3389/fimmu.2018.00711
36. Ubersax JA, Ferrell JE Jr. Mechanisms of Specificity in Protein Phosphorylation. Nat Rev Mol Cell Biol (2007) 8:530–41. doi: 10.1038/nrm2203
37. Yu X, Chini CC, He M, Mer G, Chen J. The BRCT Domain is a Phospho-Protein Binding Domain. Science (2003) 302:639–42. doi: 10.1126/science.1088753
38. Nishi H, Hashimoto K, Panchenko AR. Phosphorylation in Protein-Protein Binding: Effect on Stability and Function. Structure (2011) 19:1807–15. doi: 10.1016/j.str.2011.09.021
39. Liu P, Gan W, Guo C, Xie A, Gao D, Guo J, et al. Akt-Mediated Phosphorylation of XLF Impairs non-Homologous End-Joining DNA Repair. Mol Cell (2015) 57:648–61. doi: 10.1016/j.molcel.2015.01.005
40. Seo GJ, Yang A, Tan B, Kim S, Liang Q, Choi Y, et al. Akt Kinase-Mediated Checkpoint of cGAS DNA Sensing Pathway. Cell Rep (2015) 13:440–9. doi: 10.1016/j.celrep.2015.09.007
41. Zhong L, Hu M-M, Bian L-J, Liu Y, Chen Q, Shu H-B. Phosphorylation of cGAS by CDK1 Impairs Self-DNA Sensing in Mitosis. Cell Discovery (2020) 6:26. doi: 10.1038/s41421-020-0162-2
42. Liu H, Zhang H, Wu X, Ma D, Wu J, Wang L, et al. Nuclear cGAS Suppresses DNA Repair and Promotes Tumorigenesis. Nature (2018) 563:131–6. doi: 10.1038/s41586-018-0629-6
43. Sun X, Liu T, Zhao J, Xia H, Xie J, Guo Y, et al. DNA-PK Deficiency Potentiates cGAS-Mediated Antiviral Innate Immunity. Nat Commun (2020) 11:6182. doi: 10.1038/s41467-020-19941-0
44. Li T, Huang T, Du M, Chen X, Du F, Ren J, et al. Phosphorylation and Chromatin Tethering Prevent cGAS Activation During Mitosis. Science (2021) 371. doi: 10.1126/science.abc5386
45. Li M, Shu H-B. Dephosphorylation of cGAS by PPP6C Impairs its Substrate Binding Activity and Innate Antiviral Response. Protein Cell (2020) 11:584–99. doi: 10.1007/s13238-020-00729-3
46. Seo GJ, Kim C, Shin W-J, Sklan EH, Eoh H, Jung JU. TRIM56-Mediated Monoubiquitination of cGAS for Cytosolic DNA Sensing. Nat Commun (2018) 9:613. doi: 10.1038/s41467-018-02936-3
47. Wang Q, Huang L, Hong Z, Lv Z, Mao Z, Tang Y, et al. The E3 Ubiquitin Ligase RNF185 Facilitates the cGAS-Mediated Innate Immune Response. PloS Pathog (2017) 13:e1006264. doi: 10.1371/journal.ppat.1006264
48. Chen M, Meng Q, Qin Y, Liang P, Tan P, He L, et al. TRIM14 Inhibits cGAS Degradation Mediated by Selective Autophagy Receptor P62 to Promote Innate Immune Responses. Mol Cell (2016) 64:105–19. doi: 10.1016/j.molcel.2016.08.025
49. Guo Y, Jiang F, Kong L, Li B, Yang Y, Zhang L, et al. Cutting Edge: USP27X Deubiquitinates and Stabilizes the DNA Sensor cGAS to Regulate Cytosolic DNA–Mediated Signaling. J Immunol (2019) 203:2049–54. doi: 10.4049/jimmunol.1900514
50. Zhang Q, Tang Z, An R, Ye L, Zhong B. USP29 Maintains the Stability of cGAS and Promotes Cellular Antiviral Responses and Autoimmunity. Cell Res (2020) 30:914–27. doi: 10.1038/s41422-020-0341-6
51. Hu M-M, Yang Q, Xie X-Q, Liao C-Y, Lin H, Liu T-T, et al. Sumoylation Promotes the Stability of the DNA Sensor cGAS and the Adaptor STING to Regulate the Kinetics of Response to DNA Virus. Immunity (2016) 45:555–69. doi: 10.1016/j.immuni.2016.08.014
52. Cui Y, Yu H, Zheng X, Peng R, Wang Q, Zhou Y, et al. SENP7 Potentiates cGAS Activation by Relieving SUMO-Mediated Inhibition of Cytosolic DNA Sensing. PloS Pathog (2017) 13:e1006156. doi: 10.1371/journal.ppat.1006156
53. Li C, Zhang L, Qian D, Cheng M, Hu H, Hong Z, et al. RNF111-Facilitated Neddylation Potentiates cGAS-Mediated Antiviral Innate Immune Response. PloS Pathog (2021) 17:e1009401. doi: 10.1371/journal.ppat.1009401
54. Ma D, Yang M, Wang Q, Sun C, Shi H, Jing W, et al. Arginine Methyltransferase PRMT5 Negatively Regulates cGAS-Mediated Antiviral Immune Response. Sci Adv (2021) 7. doi: 10.1126/sciadv.abc1834
55. Song Z-M, Lin H, Yi X-M, Guo W, Hu M-M, Shu H-B. KAT5 Acetylates cGAS to Promote Innate Immune Response to DNA Virus. Proc Natl Acad Sci (2020) 117:21568–75. doi: 10.1073/pnas.1922330117
56. Dai J, Huang Y-J, He X, Zhao M, Wang X, Liu Z-S, et al. Acetylation Blocks cGAS Activity and Inhibits Self-DNA-Induced Autoimmunity. Cell (2019) 176:1447–1460.e1414. doi: 10.1016/j.cell.2019.01.016
57. Xia P, Ye B, Wang S, Zhu X, Du Y, Xiong Z, et al. Glutamylation of the DNA Sensor cGAS Regulates its Binding and Synthase Activity in Antiviral Immunity. Nat Immunol (2016) 17:369–78. doi: 10.1038/ni.3356
58. Dell'Oste V, Gatti D, Gugliesi F, Andrea MD, Bawadekar M, Cigno IL, et al. Innate Nuclear Sensor IFI16 Translocates Into the Cytoplasm During the Early Stage of In Vitro Human Cytomegalovirus Infection and Is Entrapped in the Egressing Virions During the Late Stage. J Virol (2014) 88:6970–82. doi: 10.1128/JVI.00384-14
59. Li D, Wu R, Guo W, Xie L, Qiao Z, Chen S, et al. STING-Mediated IFI16 Degradation Negatively Controls Type I Interferon Production. Cell Rep (2019) 29:1249–1260.e1244. doi: 10.1016/j.celrep.2019.09.069
60. Orzalli MH, DeLuca NA, Knipe DM. Nuclear IFI16 Induction of IRF-3 Signaling During Herpesviral Infection and Degradation of IFI16 by the Viral ICP0 Protein. Proc Natl Acad Sci (2012) 109:E3008–17. doi: 10.1073/pnas.1211302109
61. Li T, Diner BA, Chen J, Cristea IM. Acetylation Modulates Cellular Distribution and DNA Sensing Ability of Interferon-Inducible Protein IFI16. Proc Natl Acad Sci USA (2012) 109:10558–63.
62. Hong Y, Lee SO, Oh C, Kang K, Ryoo J, Kim D, et al. USP21 Deubiquitinase Regulates AIM2 Inflammasome Activation. J Immunol (2021) 207:1926–36. doi: 10.4049/jimmunol.2100449
63. Liu T, Tang Q, Liu K, Xie W, Liu X, Wang H, et al. TRIM11 Suppresses AIM2 Inflammasome by Degrading AIM2 via P62-Dependent Selective Autophagy. Cell Rep (2016) 16:1988–2002. doi: 10.1016/j.celrep.2016.07.019
64. Lin Y-C, Yu Y-S, Lin H-H, Hsiao K-Y. Oxaliplatin-Induced DHX9 Phosphorylation Promotes Oncogenic Circular RNA CCDC66 Expression and Development of Chemoresistance. Cancers (Basel) (2020) 12:697. doi: 10.3390/cancers12030697
65. Yuan D, Chen Y, Yang Z, Li G, Wu M, Jiang J, et al. SPOP Attenuates Migration and Invasion of Choriocarcinoma Cells by Promoting DHX9 Degradation. Am J Cancer Res (2020) 10:2428–45.
66. Patel PS, Abraham KJ, Guturi KKN, Halaby M-J, Khan Z, Palomero L, et al. RNF168 Regulates R-Loop Resolution and Genomic Stability in BRCA1/2-Deficient Tumors. J Clin Invest (2021) 131. doi: 10.1172/JCI140105
67. Lee KG, Kim SS, Kui L, Voon DC, Mauduit M, Bist P, et al. Bruton's Tyrosine Kinase Phosphorylates DDX41 and Activates its Binding of dsDNA and STING to Initiate Type 1 Interferon Response. Cell Rep (2015) 10:1055–65. doi: 10.1016/j.celrep.2015.01.039
68. Zhang Z, Bao M, Lu N, Weng L, Yuan B, Liu YJ. The E3 Ubiquitin Ligase TRIM21 Negatively Regulates the Innate Immune Response to Intracellular Double-Stranded DNA. Nat Immunol (2013) 14:172–8. doi: 10.1038/ni.2492
69. Oshiumi H, Miyashita M, Okamoto M, Morioka Y, Okabe M, Matsumoto M, et al. DDX60 Is Involved in RIG-I-Dependent and Independent Antiviral Responses, and Its Function Is Attenuated by Virus-Induced EGFR Activation. Cell Rep (2015) 11:1193–207. doi: 10.1016/j.celrep.2015.04.047
70. Li Z, Li J, Kong Y, Yan S, Ahmad N, Liu X. Plk1 Phosphorylation of Mre11 Antagonizes the DNA Damage Response. Cancer Res (2017) 77:3169–80. doi: 10.1158/0008-5472.CAN-16-2787
71. Bai Y, Wang W, Li S, Zhan J, Li H, Zhao M, et al. C1QBP Promotes Homologous Recombination by Stabilizing MRE11 and Controlling the Assembly and Activation of MRE11/RAD50/NBS1 Complex. Mol Cell (2019) 75:1299–1314.e1296. doi: 10.1016/j.molcel.2019.06.023
72. Rozier L, Guo Y, Peterson S, Sato M, Baer R, Gautier J, et al. The MRN-CtIP Pathway is Required for Metaphase Chromosome Alignment. Mol Cell (2013) 49:1097–107. doi: 10.1016/j.molcel.2013.01.023
73. Xu R, Xu Y, Huo W, Lv Z, Yuan J, Ning S, et al. Mitosis-Specific MRN Complex Promotes a Mitotic Signaling Cascade to Regulate Spindle Dynamics and Chromosome Segregation. Proc Natl Acad Sci USA (2018) 115:E10079–e10088. doi: 10.1073/pnas.1806665115
74. Chen C, Zhang L, Huang N-J, Huang B, Kornbluth S. Suppression of DNA-Damage Checkpoint Signaling by Rsk-Mediated Phosphorylation of Mre11. Proc Natl Acad Sci (2013) 110:20605–10. doi: 10.1073/pnas.1306328110
75. Piscitello D, Varshney D, Lilla S, Vizioli MG, Reid C, Gorbunova V, et al. AKT Overactivation can Suppress DNA Repair via P70s6 Kinase-Dependent Downregulation of MRE11. Oncogene (2018) 37:427–38. doi: 10.1038/onc.2017.340
76. Jachimowicz RD, Beleggia F, Isensee J, Velpula BB, Goergens J, Bustos MA, et al. UBQLN4 Represses Homologous Recombination and Is Overexpressed in Aggressive Tumors. Cell (2019) 176:505–519.e522. doi: 10.1016/j.cell.2018.11.024
77. Nicholson J, Jevons SJ, Groselj B, Ellermann S, Konietzny R, Kerr M, et al. E3 Ligase Ciap2 Mediates Downregulation of MRE11 and Radiosensitization in Response to HDAC Inhibition in Bladder Cancer. Cancer Res (2017) 77:3027–39. doi: 10.1158/0008-5472.CAN-16-3232
78. Wang Z, Gong Y, Peng B, Shi R, Fan D, Zhao H, et al. MRE11 UFMylation Promotes ATM Activation. Nucleic Acids Res (2019) 47:4124–35. doi: 10.1093/nar/gkz110
79. Lee L, Oliva ABP, Martinez-Balsalobre E, Churikov D, Peter J, Rahmouni D, et al. UFMylation of MRE11 is Essential for Telomere Length Maintenance and Hematopoietic Stem Cell Survival. Sci Adv (2021) 7:eabc7371. doi: 10.1126/sciadv.abc7371
80. Boisvert FM, Déry U, Masson JY, Richard S. Arginine Methylation of MRE11 by PRMT1 is Required for DNA Damage Checkpoint Control. Genes Dev (2005) 19:671–6. doi: 10.1101/gad.1279805
81. Liu S, Cai X, Wu J, Cong Q, Chen X, Li T, et al. Phosphorylation of Innate Immune Adaptor Proteins MAVS, STING, and TRIF Induces IRF3 Activation. Science (2015) 347:aaa2630. doi: 10.1126/science.aaa2630
82. Konno H, Konno K, Barber GN. Cyclic Dinucleotides Trigger ULK1 (ATG1) Phosphorylation of STING to Prevent Sustained Innate Immune Signaling. Cell (2013) 155:688–98. doi: 10.1016/j.cell.2013.09.049
83. Zhong B, Zhang L, Lei C, Li Y, Mao AP, Yang Y, et al. The Ubiquitin Ligase RNF5 Regulates Antiviral Responses by Mediating Degradation of the Adaptor Protein MITA. Immunity (2009) 30:397–407. doi: 10.1016/j.immuni.2009.01.008
84. Wang Y, Lian Q, Yang B, Yan S, Zhou H, He L, et al. Trim30α Is a Negative-Feedback Regulator of the Intracellular DNA and DNA Virus-Triggered Response by Targeting STING. PloS Pathog (2015) 11:e1005012.
85. Xing J, Zhang A, Zhang H, Wang J, Li XC, Zeng MS, et al. TRIM29 Promotes DNA Virus Infections by Inhibiting Innate Immune Response. Nat Commun (2017) 8:945. doi: 10.1038/s41467-017-00101-w
86. Qin Y, Zhou MT, Hu MM, Hu YH, Zhang J, Guo L, et al. RNF26 Temporally Regulates Virus-Triggered Type I Interferon Induction by Two Distinct Mechanisms. PloS Pathog (2014) 10:e1004358. doi: 10.1371/journal.ppat.1004358
87. Pokatayev V, Yang K, Tu X, Dobbs N, Wu J, Kalb RG, et al. Homeostatic Regulation of STING Protein at the Resting State by Stabilizer TOLLIP. Nat Immunol (2020) 21:158–67. doi: 10.1038/s41590-019-0569-9
88. Zhang ZD, Xiong TC, Yao SQ, Wei MC, Chen M, Lin D, et al. RNF115 Plays Dual Roles in Innate Antiviral Responses by Catalyzing Distinct Ubiquitination of MAVS and MITA. Nat Commun (2020) 11:5536. doi: 10.1038/s41467-020-19318-3
89. Tsuchida T, Zou J, Saitoh T, Kumar H, Abe T, Matsuura Y, et al. The Ubiquitin Ligase TRIM56 Regulates Innate Immune Responses to Intracellular Double-Stranded DNA. Immunity (2010) 33:765–76. doi: 10.1016/j.immuni.2010.10.013
90. Wang Q, Liu X, Cui Y, Tang Y, Chen W, Li S, et al. The E3 Ubiquitin Ligase AMFR and INSIG1 Bridge the Activation of TBK1 Kinase by Modifying the Adaptor STING. Immunity (2014) 41:919–33. doi: 10.1016/j.immuni.2014.11.011
91. Zhang J, Hu MM, Wang YY, Shu HB. TRIM32 Protein Modulates Type I Interferon Induction and Cellular Antiviral Response by Targeting MITA/STING Protein for K63-Linked Ubiquitination. J Biol Chem (2012) 287:28646–55. doi: 10.1074/jbc.M112.362608
92. Ni G, Konno H, Barber GN. Ubiquitination of STING at Lysine 224 Controls IRF3 Activation. Sci Immunol (2017) 2. doi: 10.1126/sciimmunol.aah7119
93. Zhang M, Zhang MX, Zhang Q, Zhu GF, Yuan L, Zhang DE, et al. USP18 Recruits USP20 to Promote Innate Antiviral Response Through Deubiquitinating STING/MITA. Cell Res (2016) 26:1302–19. doi: 10.1038/cr.2016.125
94. Luo WW, Li S, Li C, Lian H, Yang Q, Zhong B, et al. Irhom2 is Essential for Innate Immunity to DNA Viruses by Mediating Trafficking and Stability of the Adaptor STING. Nat Immunol (2016) 17:1057–66. doi: 10.1038/ni.3510
95. Zhang L, Wei N, Cui Y, Hong Z, Liu X, Wang Q, et al. The Deubiquitinase CYLD is a Specific Checkpoint of the STING Antiviral Signaling Pathway. PloS Pathog (2018) 14:e1007435. doi: 10.1371/journal.ppat.1007435
96. Sun H, Zhang Q, Jing YY, Zhang M, Wang HY, Cai Z, et al. USP13 Negatively Regulates Antiviral Responses by Deubiquitinating STING. Nat Commun (2017) 8:15534. doi: 10.1038/ncomms15534
97. Tian M, Liu W, Zhang Q, Huang Y, Li W, Wang W, et al. MYSM1 Represses Innate Immunity and Autoimmunity Through Suppressing the cGAS-STING Pathway. Cell Rep (2020) 33:108297. doi: 10.1016/j.celrep.2020.108297
98. Chen Y, Wang L, Jin J, Luan Y, Chen C, Li Y, et al. P38 Inhibition Provides Anti-DNA Virus Immunity by Regulation of USP21 Phosphorylation and STING Activation. J Exp Med (2017) 214:991–1010. doi: 10.1084/jem.20161387
99. Mukai K, Konno H, Akiba T, Uemura T, Waguri S, Kobayashi T, et al. Activation of STING Requires Palmitoylation at the Golgi. Nat Commun (2016) 7:11932. doi: 10.1038/ncomms11932
100. Tao L, Lemoff A, Wang G, Zarek C, Lowe A, Yan N, et al. Reactive Oxygen Species Oxidize STING and Suppress Interferon Production. Elife (2020) 9. doi: 10.7554/eLife.57837
101. Liu P, Begley M, Michowski W, Inuzuka H, Ginzberg M, Gao D, et al. Cell-Cycle-Regulated Activation of Akt Kinase by Phosphorylation at its Carboxyl Terminus. Nature (2014) 508:541–5. doi: 10.1038/nature13079
102. Guey B, Wischnewski M, Decout A, Makasheva K, Kaynak M, Sakar MS, et al. BAF Restricts cGAS on Nuclear DNA to Prevent Innate Immune Activation. Science (2020) 369:823–8. doi: 10.1126/science.aaw6421
103. Volkman HE, Cambier S, Gray EE, Stetson DB. Tight Nuclear Tethering of cGAS is Essential for Preventing Autoreactivity. Elife (2019) 8. doi: 10.7554/eLife.47491
104. Zierhut C, Yamaguchi N, Paredes M, Luo JD, Carroll T, Funabiki H. The Cytoplasmic DNA Sensor cGAS Promotes Mitotic Cell Death. Cell (2019) 178:302–315 e323. doi: 10.1016/j.cell.2019.05.035
105. Hochstrasser M. Evolution and Function of Ubiquitin-Like Protein-Conjugation Systems. Nat Cell Biol (2000) 2:E153–157. doi: 10.1038/35019643
106. Komander D, Rape M. The Ubiquitin Code. Annu Rev Biochem (2012) 81:203–29. doi: 10.1146/annurev-biochem-060310-170328
107. Mevissen TET, Komander D. Mechanisms of Deubiquitinase Specificity and Regulation. Annu Rev Biochem (2017) 86:159–92. doi: 10.1146/annurev-biochem-061516-044916
108. Geiss-Friedlander R, Melchior F. Concepts in Sumoylation: A Decade on. Nat Rev Mol Cell Biol (2007) 8:947–56. doi: 10.1038/nrm2293
109. Enchev RI, Schulman BA, Peter M. Protein Neddylation: Beyond Cullin-RING Ligases. Nat Rev Mol Cell Biol (2015) 16:30–44. doi: 10.1038/nrm3919
110. Wang Z, Liu P, Inuzuka H, Wei W. Roles of F-Box Proteins in Cancer. Nat Rev Cancer (2014) 14:233–47. doi: 10.1038/nrc3700
111. Gerakis Y, Quintero M, Li H, Hetz C. The UFMylation System in Proteostasis and Beyond. Trends Cell Biol (2019) 29:974–86. doi: 10.1016/j.tcb.2019.09.005
112. Yoneyama M, Kikuchi M, Natsukawa T, Shinobu N, Imaizumi T, Miyagishi M, et al. The RNA Helicase RIG-I has an Essential Function in Double-Stranded RNA-Induced Innate Antiviral Responses. Nat Immunol (2004) 5:730–7. doi: 10.1038/ni1087
113. Pichlmair A, Schulz O, Tan CP, Naslund TI, Liljestrom P, Weber F, et al. RIG-I-Mediated Antiviral Responses to Single-Stranded RNA Bearing 5'-Phosphates. Science (2006) 314:997–1001. doi: 10.1126/science.1132998
114. Brisse M, Ly H. Comparative Structure and Function Analysis of the RIG-I-Like Receptors: RIG-I and MDA5. Front Immunol (2019) 10:1586. doi: 10.3389/fimmu.2019.01586
115. Seth RB, Sun L, Ea CK, Chen ZJ. Identification and Characterization of MAVS, a Mitochondrial Antiviral Signaling Protein That Activates NF-kappaB and IRF 3. Cell (2005) 122:669–82. doi: 10.1016/j.cell.2005.08.012
116. Paz S, Vilasco M, Werden SJ, Arguello M, Joseph-Pillai D, Zhao T, et al. A Functional C-Terminal TRAF3-Binding Site in MAVS Participates in Positive and Negative Regulation of the IFN Antiviral Response. Cell Res (2011) 21:895–910. doi: 10.1038/cr.2011.2
117. Fang R, Jiang Q, Zhou X, Wang C, Guan Y, Tao J, et al. MAVS Activates TBK1 and IKKepsilon Through TRAFs in NEMO Dependent and Independent Manner. PloS Pathog (2017) 13:e1006720.
118. Paz S, Sun Q, Nakhaei P, Romieu-Mourez R, Goubau D, Julkunen I, et al. Induction of IRF-3 and IRF-7 Phosphorylation Following Activation of the RIG-I Pathway. Cell Mol Biol (Noisy-le-grand) (2006) 52:17–28.
119. tenOever BR, Sharma S, Zou W, Sun Q, Grandvaux N, Julkunen I, et al. Activation of TBK1 and IKKvarepsilon Kinases by Vesicular Stomatitis Virus Infection and the Role of Viral Ribonucleoprotein in the Development of Interferon Antiviral Immunity. J Virol (2004) 78:10636–49. doi: 10.1128/JVI.78.19.10636-10649.2004
120. Gack MU, Kirchhofer A, Shin YC, Inn KS, Liang C, Cui S, et al. Roles of RIG-I N-Terminal Tandem CARD and Splice Variant in TRIM25-Mediated Antiviral Signal Transduction. Proc Natl Acad Sci USA (2008) 105:16743–8.
121. Zeng W, Sun L, Jiang X, Chen X, Hou F, Adhikari A, et al. Reconstitution of the RIG-I Pathway Reveals a Signaling Role of Unanchored Polyubiquitin Chains in Innate Immunity. Cell (2010) 141:315–30. doi: 10.1016/j.cell.2010.03.029
122. Gack MU, Shin YC, Joo C-H, Urano T, Liang C, Sun L, et al. TRIM25 RING-Finger E3 Ubiquitin Ligase is Essential for RIG-I-Mediated Antiviral Activity. Nature (2007) 446:916–20. doi: 10.1038/nature05732
123. Shi Y, Yuan B, Zhu W, Zhang R, Li L, Hao X, et al. Ube2D3 and Ube2N are Essential for RIG-I-Mediated MAVS Aggregation in Antiviral Innate Immunity. Nat Commun (2017) 8:15138. doi: 10.1038/ncomms15138
124. Oshiumi H, Matsumoto M, Hatakeyama S, Seya T. Riplet/RNF135, a RING Finger Protein, Ubiquitinates RIG-I to Promote Interferon-Beta Induction During the Early Phase of Viral Infection. J Biol Chem (2009) 284:807–17. doi: 10.1074/jbc.M804259200
125. Gao D, Yang YK, Wang RP, Zhou X, Diao FC, Li MD, et al. REUL is a Novel E3 Ubiquitin Ligase and Stimulator of Retinoic-Acid-Inducible Gene-I. PloS One (2009) 4:e5760. doi: 10.1371/journal.pone.0005760
126. Yan J, Li Q, Mao AP, Hu MM, Shu HB. TRIM4 Modulates Type I Interferon Induction and Cellular Antiviral Response by Targeting RIG-I for K63-Linked Ubiquitination. J Mol Cell Biol (2014) 6:154–63. doi: 10.1093/jmcb/mju005
127. Kuniyoshi K, Takeuchi O, Pandey S, Satoh T, Iwasaki H, Akira S, et al. Pivotal Role of RNA-Binding E3 Ubiquitin Ligase MEX3C in RIG-I-Mediated Antiviral Innate Immunity. Proc Natl Acad Sci USA (2014) 111:5646–51. doi: 10.1073/pnas.1401674111
128. Wang W, Jiang M, Liu S, Zhang S, Liu W, Ma Y, et al. RNF122 Suppresses Antiviral Type I Interferon Production by Targeting RIG-I CARDs to Mediate RIG-I Degradation. Proc Natl Acad Sci USA (2016) 113:9581–6. doi: 10.1073/pnas.1604277113
129. Arimoto K, Takahashi H, Hishiki T, Konishi H, Fujita T, Shimotohno K. Negative Regulation of the RIG-I Signaling by the Ubiquitin Ligase RNF125. Proc Natl Acad Sci USA (2007) 104:7500–5. doi: 10.1073/pnas.0611551104
130. Inn KS, Gack MU, Tokunaga F, Shi M, Wong LY, Iwai K, et al. Linear Ubiquitin Assembly Complex Negatively Regulates RIG-I- and TRIM25-Mediated Type I Interferon Induction. Mol Cell (2011) 41:354–65. doi: 10.1016/j.molcel.2010.12.029
131. Chen W, Han C, Xie B, Hu X, Yu Q, Shi L, et al. Induction of Siglec-G by RNA Viruses Inhibits the Innate Immune Response by Promoting RIG-I Degradation. Cell (2013) 152:467–78. doi: 10.1016/j.cell.2013.01.011
132. Friedman CS, O'Donnell MA, Legarda-Addison D, Ng A, Cardenas WB, Yount JS, et al. The Tumour Suppressor CYLD is a Negative Regulator of RIG-I-Mediated Antiviral Response. EMBO Rep (2008) 9:930–6. doi: 10.1038/embor.2008.136
133. Cui J, Song Y, Li Y, Zhu Q, Tan P, Qin Y, et al. USP3 Inhibits Type I Interferon Signaling by Deubiquitinating RIG-I-Like Receptors. Cell Res (2014) 24:400–16. doi: 10.1038/cr.2013.170
134. Fan Y, Mao R, Yu Y, Liu S, Shi Z, Cheng J, et al. USP21 Negatively Regulates Antiviral Response by Acting as a RIG-I Deubiquitinase. J Exp Med (2014) 211:313–28. doi: 10.1084/jem.20122844
135. Wang L, Zhao W, Zhang M, Wang P, Zhao K, Zhao X, et al. USP4 Positively Regulates RIG-I-Mediated Antiviral Response Through Deubiquitination and Stabilization of RIG-I. J Virol (2013) 87:4507–15. doi: 10.1128/JVI.00031-13
136. Pauli EK, Chan YK, Davis ME, Gableske S, Wang MK, Feister KF, et al. The Ubiquitin-Specific Protease USP15 Promotes RIG-I-Mediated Antiviral Signaling by Deubiquitylating TRIM25. Sci Signal (2014) 7:ra3. doi: 10.1126/scisignal.2004577
137. Gack MU, Nistal-Villan E, Inn KS, Garcia-Sastre A, Jung JU. Phosphorylation-Mediated Negative Regulation of RIG-I Antiviral Activity. J Virol (2010) 84:3220–9. doi: 10.1128/JVI.02241-09
138. Nistal-Villán E, Gack MU, Martínez-Delgado G, Maharaj NP, Inn K-S, Yang H, et al. Negative Role of RIG-I Serine 8 Phosphorylation in the Regulation of Interferon-β Production*. J Biol Chem (2010) 285:20252–61. doi: 10.1074/jbc.M109.089912
139. Maharaj NP, Wies E, Stoll A, Gack MU. Conventional Protein Kinase C-Alpha (PKC-Alpha) and PKC-Beta Negatively Regulate RIG-I Antiviral Signal Transduction. J Virol (2012) 86:1358–71. doi: 10.1128/JVI.06543-11
140. Sun Z, Ren H, Liu Y, Teeling JL, Gu J. Phosphorylation of RIG-I by Casein Kinase II Inhibits its Antiviral Response. J Virol (2011) 85:1036–47. doi: 10.1128/JVI.01734-10
141. Wies E, Wang MK, Maharaj NP, Chen K, Zhou S, Finberg RW, et al. Dephosphorylation of the RNA Sensors RIG-I and MDA5 by the Phosphatase PP1 is Essential for Innate Immune Signaling. Immunity (2013) 38:437–49. doi: 10.1016/j.immuni.2012.11.018
142. Davis Meredith E, Wang May K, Rennick Linda J, Full F, Gableske S, Mesman Annelies W, et al. Antagonism of the Phosphatase PP1 by the Measles Virus V Protein Is Required for Innate Immune Escape of MDA5. Cell Host Microbe (2014) 16:19–30. doi: 10.1016/j.chom.2014.06.007
143. Hu MM, Liao CY, Yang Q, Xie XQ, Shu HB. Innate Immunity to RNA Virus Is Regulated by Temporal and Reversible Sumoylation of RIG-I and MDA5. J Exp Med (2017) 214:973–89. doi: 10.1084/jem.20161015
144. He S, Zhao J, Song S, He X, Minassian A, Zhou Y, et al. Viral Pseudo-Enzymes Activate RIG-I via Deamidation to Evade Cytokine Production. Mol Cell (2015) 58:134–46. doi: 10.1016/j.molcel.2015.01.036
145. Choi SJ, Lee HC, Kim JH, Park SY, Kim TH, Lee WK, et al. HDAC6 Regulates Cellular Viral RNA Sensing by Deacetylation of RIG-I. EMBO J (2016) 35:429–42. doi: 10.15252/embj.201592586
146. Liu HM, Jiang F, Loo YM, Hsu S, Hsiang TY, Marcotrigiano J, et al. Regulation of Retinoic Acid Inducible Gene-I (RIG-I) Activation by the Histone Deacetylase 6. EBioMedicine (2016) 9:195–206. doi: 10.1016/j.ebiom.2016.06.015
147. Narayan K, Waggoner L, Pham ST, Hendricks GL, Waggoner SN, Conlon J, et al. TRIM13 is a Negative Regulator of MDA5-Mediated Type I Interferon Production. J Virol (2014) 88:10748–57. doi: 10.1128/JVI.02593-13
148. Lang X, Tang T, Jin T, Ding C, Zhou R, Jiang W. TRIM65-Catalized Ubiquitination is Essential for MDA5-Mediated Antiviral Innate Immunity. J Exp Med (2017) 214:459–73. doi: 10.1084/jem.20160592
149. Takashima K, Oshiumi H, Takaki H, Matsumoto M, Seya T. RIOK3-Mediated Phosphorylation of MDA5 Interferes With Its Assembly and Attenuates the Innate Immune Response. Cell Rep (2015) 11:192–200. doi: 10.1016/j.celrep.2015.03.027
150. Liu G, Lee JH, Parker ZM, Acharya D, Chiang JJ, van Gent M, et al. ISG15-Dependent Activation of the Sensor MDA5 is Antagonized by the SARS-CoV-2 Papain-Like Protease to Evade Host Innate Immunity. Nat Microbiol (2021) 6:467–78. doi: 10.1038/s41564-021-00884-1
151. Liu B, Zhang M, Chu H, Zhang H, Wu H, Song G, et al. The Ubiquitin E3 Ligase TRIM31 Promotes Aggregation and Activation of the Signaling Adaptor MAVS Through Lys63-Linked Polyubiquitination. Nat Immunol (2017) 18:214–24. doi: 10.1038/ni.3641
152. Hou J, Han L, Zhao Z, Liu H, Zhang L, Ma C, et al. USP18 Positively Regulates Innate Antiviral Immunity by Promoting K63-Linked Polyubiquitination of MAVS. Nat Commun (2021) 12:2970. doi: 10.1038/s41467-021-23219-4
153. Xue B, Li H, Guo M, Wang J, Xu Y, Zou X, et al. TRIM21 Promotes Innate Immune Response to RNA Viral Infection Through Lys27-Linked Polyubiquitination of MAVS. J Virol (2018) 92. doi: 10.1128/JVI.00321-18
154. Li T, Li X, Attri KS, Liu C, Li L, Herring LE, et al. O-GlcNAc Transferase Links Glucose Metabolism to MAVS-Mediated Antiviral Innate Immunity. Cell Host Microbe (2018) 24:791–803.e796. doi: 10.1016/j.chom.2018.11.001
155. Liuyu T, Yu K, Ye L, Zhang Z, Zhang M, Ren Y, et al. Induction of OTUD4 by Viral Infection Promotes Antiviral Responses Through Deubiquitinating and Stabilizing MAVS. Cell Res (2019) 29:67–79. doi: 10.1038/s41422-018-0107-6
156. Castanier C, Zemirli N, Portier A, Garcin D, Bidere N, Vazquez A, et al. MAVS Ubiquitination by the E3 Ligase TRIM25 and Degradation by the Proteasome is Involved in Type I Interferon Production After Activation of the Antiviral RIG-I-Like Receptors. BMC Biol (2012) 10:44. doi: 10.1186/1741-7007-10-44
157. Liu C, Huang S, Wang X, Wen M, Zheng J, Wang W, et al. The Otubain YOD1 Suppresses Aggregation and Activation of the Signaling Adaptor MAVS Through Lys63-Linked Deubiquitination. J Immunol (2019) 202:2957–70. doi: 10.4049/jimmunol.1800656
158. Zhang Z, Fang X, Wu X, Ling L, Chu F, Li J, et al. Acetylation-Dependent Deubiquitinase OTUD3 Controls MAVS Activation in Innate Antiviral Immunity. Mol Cell (2020) 79:304–319.e307. doi: 10.1016/j.molcel.2020.06.020
159. Zhou X, You F, Chen H, Jiang Z. Poly(C)-Binding Protein 1 (PCBP1) Mediates Housekeeping Degradation of Mitochondrial Antiviral Signaling (MAVS). Cell Res (2012) 22:717–27. doi: 10.1038/cr.2011.184
160. You F, Sun H, Zhou X, Sun W, Liang S, Zhai Z, et al. PCBP2 Mediates Degradation of the Adaptor MAVS via the HECT Ubiquitin Ligase AIP4. Nat Immunol (2009) 10:1300–8. doi: 10.1038/ni.1815
161. Du J, Zhang D, Zhang W, Ouyang G, Wang J, Liu X, et al. pVHL Negatively Regulates Antiviral Signaling by Targeting MAVS for Proteasomal Degradation. J Immunol (2015) 195:1782–90. doi: 10.4049/jimmunol.1500588
162. Park YJ, Oanh NTK, Heo J, Kim SG, Lee HS, Lee H, et al. Dual Targeting of RIG-I and MAVS by MARCH5 Mitochondria Ubiquitin Ligase in Innate Immunity. Cell Signal (2020) 67:109520. doi: 10.1016/j.cellsig.2019.109520
163. Yoo Y-S, Park Y-Y, Kim J-H, Cho H, Kim S-H, Lee H-S, et al. The Mitochondrial Ubiquitin Ligase MARCH5 Resolves MAVS Aggregates During Antiviral Signalling. Nat Commun (2015) 6:7910. doi: 10.1038/ncomms8910
164. Choi YB, Shembade N, Parvatiyar K, Balachandran S, Harhaj EW. TAX1BP1 Restrains Virus-Induced Apoptosis by Facilitating Itch-Mediated Degradation of the Mitochondrial Adaptor MAVS. Mol Cell Biol (2017) 37. doi: 10.1128/MCB.00422-16
165. Pan Y, Li R, Meng JL, Mao HT, Zhang Y, Zhang J. Smurf2 Negatively Modulates RIG-I-Dependent Antiviral Response by Targeting VISA/MAVS for Ubiquitination and Degradation. J Immunol (2014) 192:4758–64. doi: 10.4049/jimmunol.1302632
166. Wang Y, Tong X, Ye X. Ndfip1 Negatively Regulates RIG-I-Dependent Immune Signaling by Enhancing E3 Ligase Smurf1-Mediated MAVS Degradation. J Immunol (2012) 189:5304–13. doi: 10.4049/jimmunol.1201445
167. Zhang L, Liu J, Qian L, Feng Q, Wang X, Yuan Y, et al. Induction of OTUD1 by RNA Viruses Potently Inhibits Innate Immune Responses by Promoting Degradation of the MAVS/TRAF3/TRAF6 Signalosome. PloS Pathog (2018) 14:e1007067. doi: 10.1371/journal.ppat.1007067
168. Zhong B, Zhang Y, Tan B, Liu TT, Wang YY, Shu HB. The E3 Ubiquitin Ligase RNF5 Targets Virus-Induced Signaling Adaptor for Ubiquitination and Degradation. J Immunol (2010) 184:6249–55. doi: 10.4049/jimmunol.0903748
169. Zhang Y, Hou P, He DC, Wang H, He H. RACK1 Degrades MAVS to Promote Bovine Ephemeral Fever Virus Replication via Upregulating E3 Ubiquitin Ligase STUB1. Vet Microbiol (2021) 257:109096. doi: 10.1016/j.vetmic.2021.109096
170. Song T, Wei C, Zheng Z, Xu Y, Cheng X, Yuan Y, et al. C-Abl Tyrosine Kinase Interacts With MAVS and Regulates Innate Immune Response. FEBS Lett (2010) 584:33–8. doi: 10.1016/j.febslet.2009.11.025
171. Li SZ, Shu QP, Song Y, Zhang HH, Liu Y, Jin BX, et al. Phosphorylation of MAVS/VISA by Nemo-Like Kinase (NLK) for Degradation Regulates the Antiviral Innate Immune Response. Nat Commun (2019) 10:3233. doi: 10.1038/s41467-019-11258-x
172. Xiang W, Zhang Q, Lin X, Wu S, Zhou Y, Meng F, et al. PPM1A Silences Cytosolic RNA Sensing and Antiviral Defense Through Direct Dephosphorylation of MAVS and TBK1. Sci Adv (2016) 2:e1501889. doi: 10.1126/sciadv.1501889
173. Choi GW, Lee Y, Yun M, Kang J, Lee SB. Formation of SUMO3-Conjugated Chains of MAVS Induced by Poly(Da:Dt), a Ligand of RIG-I, Enhances the Aggregation of MAVS That Drives the Secretion of Interferon-Beta in Human Keratinocytes. Biochem Biophys Res Commun (2020) 522:939–44. doi: 10.1016/j.bbrc.2019.11.189
174. Liu X, Zhu C, Zha H, Tang J, Rong F, Chen X, et al. SIRT5 Impairs Aggregation and Activation of the Signaling Adaptor MAVS Through Catalyzing Lysine Desuccinylation. EMBO J (2020) 39:e103285. doi: 10.15252/embj.2019103285
175. Crispin JC, Apostolidis SA, Rosetti F, Keszei M, Wang N, Terhorst C, et al. Cutting Edge: Protein Phosphatase 2A Confers Susceptibility to Autoimmune Disease Through an IL-17-Dependent Mechanism. J Immunol (2012) 188:3567–71. doi: 10.4049/jimmunol.1200143
176. Alomari M. TRIM21 - A Potential Novel Therapeutic Target in Cancer. Pharmacol Res (2021) 165:105443. doi: 10.1016/j.phrs.2021.105443
177. Wang G, Long J, Gao Y, Zhang W, Han F, Xu C, et al. K63-Linked Ubiquitination in Kinase Activation and Cancer. Front Oncol (2012) 2:5. doi: 10.3389/fonc.2012.00005
178. Zhu X, Xue J, Jiang X, Gong Y, Gao C, Cao T, et al. TRIM21 Suppresses CHK1 Activation by Preferentially Targeting CLASPIN for K63-Linked Ubiquitination. Nucleic Acids Res (2022) 50:1517–30. doi: 10.1093/nar/gkac011
179. Xie W, Lama L, Adura C, Tomita D, Glickman JF, Tuschl T, et al. Human cGAS Catalytic Domain has an Additional DNA-Binding Interface That Enhances Enzymatic Activity and Liquid-Phase Condensation. Proc Natl Acad Sci USA (2019) 116:11946–55. doi: 10.1073/pnas.1905013116
180. Zarrin AA, Bao K, Lupardus P, Vucic D. Kinase Inhibition in Autoimmunity and Inflammation. Nat Rev Drug Discovery (2021) 20:39–63. doi: 10.1038/s41573-020-0082-8
Keywords: post-translational modifications, DNA sensing, RNA sensing, innate immunity, enzymes
Citation: Deng Y, Wang Y, Li L, Miao EA and Liu P (2022) Post-Translational Modifications of Proteins in Cytosolic Nucleic Acid Sensing Signaling Pathways. Front. Immunol. 13:898724. doi: 10.3389/fimmu.2022.898724
Received: 18 March 2022; Accepted: 17 May 2022;
Published: 20 June 2022.
Edited by:
Brian J. Ferguson, University of Cambridge, United KingdomReviewed by:
Carlos Maluquer De Motes, University of Surrey, United KingdomJianzhong Zhu, Yangzhou University, China
Copyright © 2022 Deng, Wang, Li, Miao and Liu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Pengda Liu, cGVuZ2RhX2xpdUBtZWQudW5jLmVkdQ==
†These authors have contributed equally to this work