Abstract
Cell therapy is a distinguished targeted immunotherapy with great potential to treat solid tumors in the new era of cancer treatment. Cell therapy products include genetically engineered cell products and non-genetically engineered cell products. Several recent cell therapies, especially chimeric antigen receptor (CAR)-T cell therapies, have been approved as novel treatment strategies for cancer. Many clinical trials on cell therapies, in the form of cell therapy alone or in combination with other treatments, in solid tumors, have been conducted or ongoing. However, there are still challenges since adverse events and the limited efficacy of cell therapies have also been observed. Here, we concisely summarize the clinical milestones of the conducted and ongoing clinical trials of cell therapy, introduce the evolution of CARs, discuss the challenges and limitations of these therapeutic modalities taking CAR-T as the main focus, and analyze the disparities in the regulatory policies in different countries.
Introduction to Cell Therapy
While traditional cytotoxic agents still play important roles as one of the critical mainstays in therapeutics for malignant solid tumors, targeted small-molecule drugs, antibodies, and cell therapies are emerging as new directions. Indeed, cell therapies have recently helped transform the treatment blueprint for cancer patients. Cell therapy is generally considered as the transplantation of autologous or allogeneic cellular material into a patient for therapeutic purposes (1). Cell therapy products include genetically engineered cell products and non-genetically engineered cell products. Cell therapy has been regarded as “the third pillar of future medicine” with great potential to treat solid tumors in the new era of cancer treatment (2).
At present, the subcategories of cell therapy products used to treat tumors are as follows: engineered chimeric antigen receptors (CARs), T-cell receptor-engineered T-cell therapy (TCR-T), tumor-infiltrating lymphocytes (TILs), tumor vaccines, stem cells, dendritic cells mixed with cytokine-induced killer (DC-CIK) cells, CIK cells, and natural killer (NK) cells. CARs and TCR-T are parts of specific immune therapy modified by genetic engineering, which have attracted much attention because of their excellent therapeutic effects. Two CAR-T therapies—Kymriah (3) and Yescarta (4)—were approved by the United States (US) Food and Drug Administration (FDA) in 2017, with objective remission rates of 83% (52/63) and 72% (73/101), respectively, for recurrent or refractory B-cell acute leukemia. In 2020, the FDA approved Tecartus (5), the first cell-based gene therapy for relapsed or refractory mantle cell lymphoma, with an objective remission rate of 87% (52/60) and a complete response of 62% (37/60). Most recently, ABECMA (idecabtagene vicleucel) (6) was approved by the FDA for adult patients with relapsed or refractory multiple myeloma after four or more prior lines of therapy in 2021. The National Medical Products Administration (NMPA) approved FKC876, the first cell-based gene therapy for relapsed or refractory indolent non-Hodgkin’s lymphoma in China. These demonstrate the current success of cell therapy, including CAR-T therapy, for blood cancers. However, cell-based therapies are not yet quite effective in treating solid tumors due to heterogeneous antigens in solid tumors, complex microenvironment (7–9), limited targetable antigens (10), difficulties in immune cell migration, and tumor infiltration (11, 12). Despite these difficulties, some cell therapies have already been approved by the FDA for clinical practice in solid tumors. For example, the use of sipuleucel-T, a dendritic cell (DC) vaccine (13), and its related clinical trials have been reported in recent years. Furthermore, the global market value of cell therapy technology has rapidly grown and is expected to exceed $34 billion by 2025 (14). Thus, cell therapies would be a huge leap from traditional therapies.
Cell therapy is closely related to the fields of targeted therapy, gene therapy, and regenerative medicine, making it even more complicated in clinical practice. Meanwhile, cell therapy has become an independent evaluation system different from traditional drugs, especially with regard to the assessment of safety and efficacy. Therefore, the management system of cell therapy varies worldwide (Table 1). Here, we provide a concise overview of cell therapy, especially CAR-T therapy; enumerate the relevant clinical trials initiated in the last decade; and summarize the disparities in regulatory policies in different countries. In addition, we assess the conceptual framework and specific therapeutic strategies as well as the challenges and limitations of CAR-T therapies.
Table 1
| Region | Agency in charge | Government policy | Year of promulgation | Definition | Classification | Reference |
|---|---|---|---|---|---|---|
| European Union | EMA (European Medicines Agency) | Guideline on Human Cell-Based Medicinal Products (Doc. Ref. EMEA/CHMP/410869/2006) | 2008 |
|
| https://www.ema.europa.eu/en/human-cell-based-medicinal-products |
| Japan | PMDA (Pharmaceuticals and Medical Devices Agency) | Act on Securing Quality, Efficacy and Safety of Pharmaceuticals, Medical Devices, Regenerative and Cellular Therapy Products, Gene Therapy Products, and Cosmetics (abbreviated as the PMD Act) | 2014 |
|
| http://www.japaneselawtranslation.go.jp/law/detail_main?re=&vm=2&id=3213 |
| United States | FDA-CBER (The Center for Biologics Evaluation Research) | Considerations for the Design of Early-Phase Clinical Trials of Cellular and Gene Therapy Products Guidance for Industry (FDA-2013-D-0576-0019) | 2015 |
| Cellular therapy products include:
| https://www.fda.gov/vaccines-blood-biologics/cellular-gene-therapy-products |
| China | CFDA (China Food and Drug Administration) | Technical Guidelines for the Research and Evaluation of Cell Therapy Products (Trial version, 2017-NO216) | 2017 |
|
| http://www.nmpa.gov.cn/WS04/CL2138/300457.html |
Government policies regarding cell therapies worldwide.
Milestones of Cell Therapy for Solid Tumors
Important events of cell therapy in solid tumor treatment are summarized in Figure 1. The first adoptive cellular immunotherapy, lymphokine-activated killer (LAK) cells (15), opened the door for cell therapy. The safety of using high-dose interleukin (IL)-2 is still an issue, although it has a very broad spectrum antitumor effect. In 1988, the first reported clinical trial of TILs showed a 60% objective regression rate (16). In 2006, the first study using TCR reported a substantial sustainable regression (17). In 2010, the first DC vaccine was approved by the FDA for metastatic, asymptomatic hormone-refractory prostate cancer (13) with a 3-year survival rate of 34%. In 2013, the first trial of genetically modified MSCs (MSC_apceth_101) was initiated (NCT02008539) (18). Five out of 10 patients treated with MSC_apceth_101 achieved stable disease. The combination of MSC_apceth_101 with ganciclovir was reported to be safe and tolerable in patients with advanced gastrointestinal adenocarcinoma (19, 20). In 2015, the first trial of genetically modified TILs (LN-144) was initiated (NCT02360579). From the current result, LN-144 treatment results in an overall response rate of 36.4% and a median duration of response not reaching 17 months of median follow-up time in metastatic melanoma patients who progressed on multiple prior therapies (21). In 2018, the first clinical trial using CAR-NK was initiated (NCT03415100), which is still ongoing. In 2019, the first TCR-T and CAR-T (CAR-glypican-3) trial in China obtained the CDE license.
Figure 1

Timeline of important events for cell therapy in solid tumors. After the first adoptive cellular immunotherapy, LAK opened the gate of cell therapy in 1985. Cell therapy has rapidly evolved in the last 10 years with a consistent appearance of novel types. The transformation from the non-genetic engineering model to genetic engineering-based products is the most important direction.
Official Government Policies for Cell Therapy Worldwide
The available official regulatory policies around the world regarding cell therapies, including those in the USA, Europe, Japan, and China, were collected, reviewed, and summarized (Table 1).
In September 2008, the guideline on Human Cell-Based Medicinal Products from the Committee for Medicinal Product for Human Use of the Europe Medicines Agency came into effect. It defined cell therapy as cell-based medical products, including self-renewing stem cells, autologous or allogeneic origin cells, and genetically modified cells. The present document applies only to the cellular component of the cell-based medicinal products containing genetically modified cells.
In November 2014, the Pharmaceuticals and Medical Devices Agency (PMDA) of Japan published “Act on Securing Quality, Efficacy and Safety of Pharmaceuticals, Medical Devices, Regenerative and Cellular Therapy Products, Gene Therapy Products, and Cosmetics.” The Act defined cell therapy products as those intended for use in human or animal healthcare, which are obtained after culturing or other processes using human or animal cells, as well as those intended for use in the treatment of disease in humans or animals, which are introduced into the cells of humans or animals and contain genes to be expressed in their bodies.
In June 2015, the Center for Biologics Evaluation and Research (CBER)/Office of Cellular, Tissue, and Gene Therapies of the USFDA issued “Considerations for the Design of Early-Phase Clinical Trials of Cellular and Gene Therapy Products – Guidance for Industry” (FDA-2013-D-0576). This guidance defined cellular therapy products as cellular immunotherapies, cancer vaccines, and other types of both autologous and allogeneic cells for certain therapeutic indications. It also mentioned that the risk and safety problems associated with specific types of cell therapy products should be carefully considered based on limited experiences. Furthermore, it gives specific recommendations and evaluation criteria for a wide range of situations/settings, including manufacturing, preclinical use, clinical trial design, dose determination, and assessment of feasibility and activity of therapy, among others. In 2016, CBER launched a new policy aimed at accelerating the review of cell therapy products for serious or life-threatening diseases, called the “Regenerative Medicine Advanced Therapy Designation,” to guarantee the availability of novel products for patients who most urgently need them. Afterward, the USFDA released “Expedited Programs for Regenerative Medicine Therapies for Serious Conditions” (FDA-2017-D-6159), which further clarifies that products defined as fast track, breakthrough therapy, or regenerative medicine advanced therapy (RMAT) designation are able to go through priority review and accelerated approval. Due to the current coronavirus pandemic, the USFDA (FDA-2020-D-1137) has recently released “Manufacturing Considerations for Licensed and Investigational Cellular and Gene Therapy Products During COVID-19 Public Health Emergency,” highlighting the importance to avoid virus contamination from various sources such as cellular and tissue samples from donors.
In December 2017, China’s FDA [CFDA, now known as the National Medical Product Administration (NMPA)] published the “Technical Guidelines for Research and Evaluation of Cell Therapy Products (Trial version, 2017-NO216).” It describes cell therapy as “products derived from human cells that are used to treat human diseases.” This is a trial version asking for professional inputs, as formal guidance is in urgent need. A “double-track system” is currently used by the Chinese authorities for the oversight of cell therapy products, comprising the drug “registration system” of The Center for Drug Evaluation (CDE) under NMPA and the medical technology “filing system” of the National Health Commission. Many clinical trial studies are carried out through the “filing system,” which has relatively loose requirements, rather than through the “registration system” with relatively strict requirements for investigational new drug (IND) research. This practice has promoted the development of cell therapy products and industry in clinical application. However, more clinical trials of cell therapy were registered on www.clinicaltrials.gov than those registered as IND in CDE. Now, updated guidelines are called, considering the rapid development of innovative pharmaceutical enterprises in China.
To sum up, cell therapy is an interdisciplinary field with complex safety concerns and efficacy standards, which greatly differ from those of traditional drugs, leading to the divergence in definition and administration patterns as summarized in Table 1. It is believed that a refinement of the documents is a pressing need in order to efficiently share, manage, and oversee cell-based products in clinical trials and medical practice. Meanwhile, the rapid research advancement requires a continuously updated management of cell therapy products to ensure both innovation and safety.
Trends of Investigational Products on Cell Therapy
Conducted and Ongoing Clinical Trials During the Past Decade
The clinical trials of cell therapies carried out from January 1, 2011, to January 1, 2021, worldwide were derived and analyzed from the Pharmaprojects database (https://citeline.informa.com/trials/) developed by a leading international research group known as INFORMA (Supplementary Information) to illustrate the status of clinical investigations of cell therapy in solid tumors both worldwide and specifically in mainland China.
A total of 572 clinical trials were recorded, among which 112 trials (19.6%) were CAR-related, 87 (15.2%) were on TCR-T, 17 (3.0%) on stem cells, 188 (32.8%) on cell-based vaccines, and 168 (29.4%) on others (on TILs, DC-CIK, CIK, and NK cells) (Table 2). Most of the trials are phase 1 or phase 1/2 (370/572, 64.7%), and only 24 (4.2%) trials are phase 3. Among those, 198 (34.6%) have been completed; however, only three products investigated in these trials have been approved, namely, sipuleucel-T (22) by the USFDA for treating asymptomatic or minimally symptomatic metastatic castrate-resistant (hormone-refractory) prostate cancer; autologous dendritic cells by the Indian FDA (23) for prostate, ovarian, colorectal, and non-small cell lung carcinomas; and Immuncell-LC in South Korea for liver cancer (24) worldwide.
Table 2
| Items | Type | CAR n (%) | TCR n (%) | Stem n (%) | Vaccine n (%) | Other n (%) | Total n (%) |
|---|---|---|---|---|---|---|---|
| Phase | I | 78 (69.6) | 44 (50.6) | 8 (47.1) | 49 (26.1) | 49 (29.2) | 228 (39.9) |
| I/II | 30 (26.8) | 28 (32.2) | 4 (23.5) | 46 (24.5) | 34 (20.2) | 142 (24.8) | |
| II | 3 (2.7) | 14 (16.1) | 5 (29.4) | 76 (40.4) | 80 (47.6) | 178 (31.1) | |
| III/IV/other | 1 (0.9) | 1 (1.1) | 0 | 17 (9.0) | 5 (3) | 24 (4.2) | |
| Trial status | Open | 71 (63.4) | 37 (42.5) | 11 (64.7) | 38 (20.2) | 58 (34.5) | 215 (37.6) |
| Closed | 18 (16.1) | 26 (29.9) | 4 (23.5) | 57 (30.3) | 54 (32.1) | 159 (27.8) | |
| Completed | 23 (20.5) | 24 (27.6) | 2 (11.8) | 93 (49.5) | 56 (33.3) | 198 (34.6) | |
| Totally (n/572*%) | 112 (19.6) | 87 (15.2) | 17 (3.0) | 188 (32.8) | 168 (29.4) | 572 (100) | |
| Primary endpoint | Safety | 96 (85.7) | 66 (75.9) | 13 (76.5) | 98 (52.1) | 87 (51.8) | 360 (62.9) |
| Efficacy | 16 (14.3) | 21 (24.1) | 4 (23.5) | 90 (47.9) | 81 (48.2) | 212 (37.1) | |
| Patient population | Biomarker selected | 30 (26.8) | 36 (41.4) | 0 | 12 (6.4) | 18 (10.7) | 96 (16.8) |
| Unselected | 82 (73.2) | 51 (58.6) | 17 (100) | 176 (93.6) | 150 (89.3) | 476 (83.2) | |
| Area | China | 60 (53.6) | 20 (23.0) | 2 (11.8) | 6 (3.2) | 52 (31.0) | 140 (24.5) |
| USA | 42 (37.5) | 57 (65.5) | 9 (52.9) | 116 (61.7) | 55 (32.7) | 279 (48.8) | |
| Europe | 6 (5.4) | 2 (2.3) | 3 (17.6) | 39 (20.7) | 21 (12.5) | 71 (12.4) | |
| Other | 4 (3.6) | 8 (9.2) | 3 (17.6) | 27 (14.4) | 40 (23.8) | 82 (14.3) | |
| Therapy method | Neoadjuvant | 3 (2.7) | 2 (2.3) | 0 | 15 (8.0) | 3 (1.8) | 23 (4.0) |
| Adjuvant | 6 (5.4) | 5 (5.7) | 0 | 42 (22.3) | 18 (10.7) | 71 (12.4) |
Classification and characteristics of clinical trials on cell therapy worldwide.
NA, not applicable/available.
In China, 140 trials have been initiated in the last 10 years, consisting of 60 (42.9%) trials on CAR, 20 (14.3%) on TCR-T, 6 (4.3%) on vaccine, 2 (1.4%) on stem cells, and 52 (37.1%) on others, none of which have been approved in China yet. Phase 1 and phase 1/2 trials account for 75.7% (105/140), phase 2 trials account for 20.7% (29/140), and only 5 (3.6%) trials are phase 3 (Table 3).
Table 3
| Items | Classification | CAR n (%) | TCR n (%) | Vaccine n (%) | Stem n (%) | Other n (%) | Total n (%) |
|---|---|---|---|---|---|---|---|
| Phase | I | 36 (60.0) | 16 (80.0) | 1 (16.7) | 0 | 12 (23.1) | 65 (46.4) |
| I/II | 22 (36.7) | 3 (15.0) | 1 (16.7) | 1 (50.0) | 14 (26.9) | 41 (29.3) | |
| II | 2 (3.3) | 1 (5.0) | 3 (50.0) | 1 (50.0) | 22 (42.3) | 29 (20.7) | |
| III/IV/other | 0 | 0 | 1 (16.7) | 0 | 4 (7.7) | 5 (3.6) | |
| Trial status | Open | 40 (66.7) | 13 (65.0) | 2 (33.3) | 1 (50.0) | 18 (34.6) | 74 (52.8) |
| Closed | 6 (10.0) | 1 (5.0) | 0 | 0 | 12 (23.1) | 19 (13.6) | |
| Completed | 14 (23.3) | 6 (30.0) | 4 (66.7) | 1 (50.0) | 22 (42.3) | 47 (33.6) | |
| Totally (n/140*%) | 60 (42.9) | 20 (14.3) | 6 (4.3) | 2 (1.4) | 52 (37.1) | 140 (100) | |
| Tumor types top 5 | Liver | 12 (20.0) | 9 (45.0) | 4 (66.7) | 0 | 12 (23.1) | 37 (26.4) |
| Pancreas | 9 (15.0) | 3 (15.0) | 1 (16.7) | 0 | 17 (32.7) | 30 (21.4) | |
| Lung | 15 (25.0) | 0 | 3 (50.0) | 0 | 9 (17.3) | 27 (19.3) | |
| Gastric | 7 (11.7) | 3 (15.0) | 1 (16.7) | 1 (50.0) | 7 (13.5) | 19 (13.6) | |
| Esophageal | 14 (23.3) | 0 | 0 | 0 | 4 (7.7) | 18 (12.9) | |
| Unspecified solid tumor | 11 (18.3) | 3 (15.0) | 1 (16.7) | 0 | 4 (7.7) | 19 (13.6) | |
| Primary endpoint | Safety | 48 (80.0) | 17 (85.0) | 2 (33.3) | 1 (50.0) | 16 (30.8) | 84 (60.0) |
| Efficacy | 12 (20.0) | 3 (15.0) | 4 (66.7) | 1 (50.0) | 36 (69.2) | 56 (40.0) | |
| Patient population | Biomarker selected | 13 (21.7) | 7 (35.0) | 0 | 0 | 7 (13.5) | 8 (19.3) |
| Unselected | 47 (78.3) | 13 (65.0) | 6 (100) | 2 (100) | 45 (86.5) | 113 (80.7) |
Classification and characteristics of clinical trials on cell therapy in China.
NA, not applicable/available.
Most of the clinical trials on cell therapy are in the early stage of evaluating safety instead of efficacy both worldwide (360, 62.9%) and in China (84, 60.0%). The abundance rate for products with specific targets, such as TCR-T and CAR-T, was 34.8% (199/572) worldwide, which was similar to that in China (57.2%, 80/140). The recruited patient population comprised mostly those with stage III to IV solid tumors in second- or later-line treatment both worldwide and in China. The proportion of biomarker-selected trials on these patients was still low both worldwide and in China (16.8% and 19.3%). The newly initiated clinical trials have increased both in and outside of China (Figures 2A, B).
Figure 2
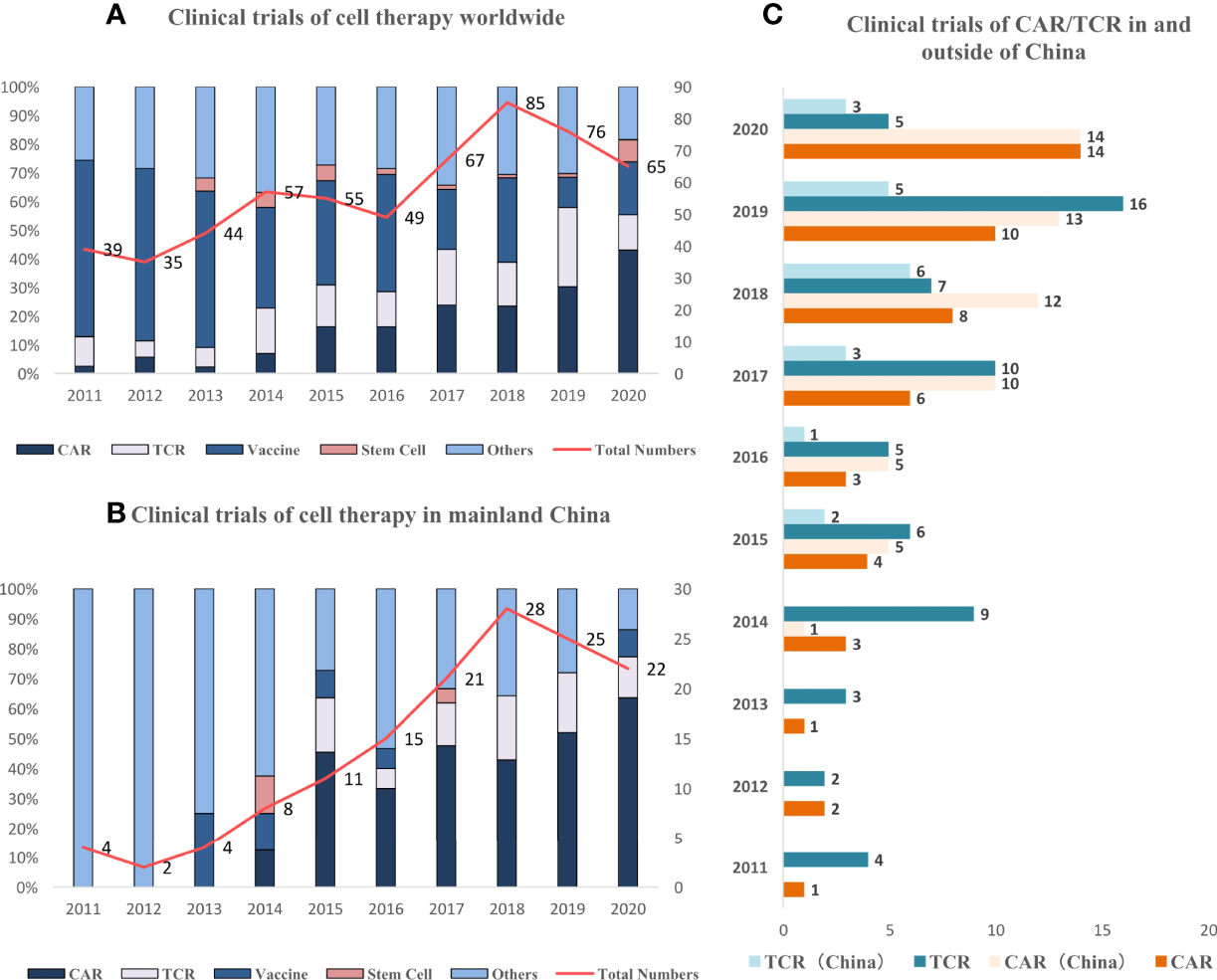
Trend of clinical trials on cell therapy worldwide and in China. (A) Newly initiated active clinical trials on cell therapy have increased especially after 2014, and the dominating type is converting from vaccine to CAR/TCR-T. (B) Newly initiated active clinical trials in China continue to increase since 2015, and the dominating type is CAR/TCR. (C) Newly initiated active clinical trials increase both in and outside of China (since 2015).
The landscape of targets in clinical trials was also different between China and the rest of the world. The top 3 targets of CARs were mesothelin, GPC3, and mucin-1 (MUC1) worldwide and GPC3, MUC1, and Claudin18.2 in China. The top 3 targets of TCR-T were NY-ESO-1, pHLA, and MAGE-4 worldwide. NY-ESO-1, HBV, and HPV were the top 3 targets in China, which was dominated by virus-specific targets. This abundance was much higher compared with that in other regions excluding China, indicating that the incidence of these distinctive virus-specific tumors in China was still high. The clinical trials on CAR-T and TCR-T have increased both inside and outside of China during the last 10 years, especially after 2017 (Figure 2).
The cell therapy of solid tumors has emerged as a novel strategy with promising efficacy data, but it is still under early-phase trial investigations worldwide, including in China.
CAR Therapy for Solid Tumors
More than half of all cell therapy trials in China are CAR-based; hence, the use of the most promising subtype of cell therapies, CAR, in treating solid tumors was discussed hereafter. CAR is a synthetic molecule comprising an extracellular domain, a transmembrane domain, and an intracellular signaling domain (Figure 3) (25). CAR-T immunotherapy has achieved remarkable success in treating hematologic cancers (26). Unfortunately, the clinical findings of CAR in treating solid tumors have been discouraging so far.
Figure 3
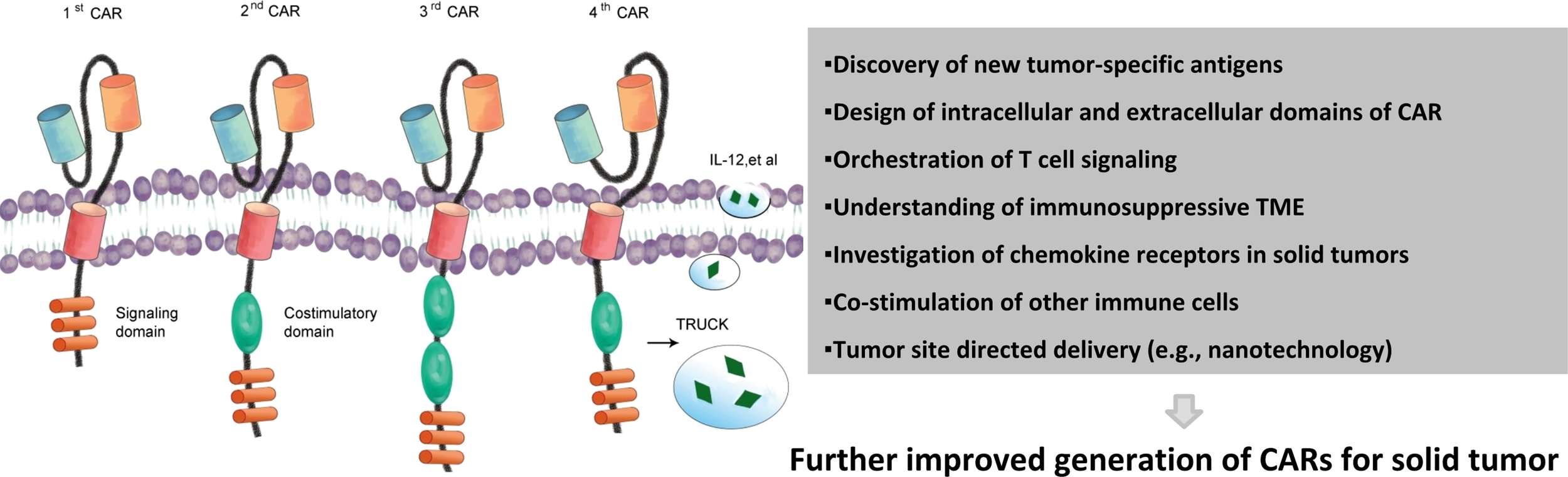
Chimeric antigen receptors and main challenges of treating solid tumors. Evolution of CAR from the first generation to the fourth generation. First-generation CARs. Second-generation CARs embody an additional intracellular signaling domain to the first-generation receptor configuration and provide a co-stimulatory signal. Third-generation receptors incorporate two co-stimulatory domains with the T-cell-activating signaling domain. Fourth-generation CARs or TRUCKS (T cells redirected for universal cytokine killing) carry vectors that encode a CAR and a CAR-responsive promoter as well as respond to the successful signaling of CAR by the transgenic production of cytokines such as IL-12. Upon accumulated knowledge of solid tumors and their surrounding environment, more effective CAR therapeutic products may be generated. CAR, chimeric antigen receptors.
Challenges of CARs for Solid Tumors
Identifying antigens exclusively expressed on tumor cells is the key factor for effective CAR-T exploration in solid tumors, which has been most challenging. A growing number of clinical trials now focus on solid tumors, targeting tumor-associated antigens (TAAs) including disialoganglioside GD2 (e.g., NCT03721068, NCT04637503) (27–30), human epidermal growth factor receptor 2 (HER2) (e.g., NCT04650451, NCT03740256) (31), epidermal growth factor receptor variant III (EGFRvIII) (e.g., NCT02844062, NCT03423992) (31–33), carcinoembryonic antigen (CEA) (e.g., NCT04348643, NCT04513431) (28), interleukin (IL) 13Rα2 (e.g., NCT04003649), prostate-specific membrane antigen (PSMA) (e.g., NCT04227275, NCT04249947) (34, 35), neural cell adhesion molecule L1 (NCAM-L1, CD171) (e.g., NCT02311621) (36), receptor tyrosine kinase-like orphan receptor 1 (ROR1) (e.g., NCT02706392) (37), mesothelin (e.g., NCT03545815, NCT03054298) (38, 39), and B7-H3 (e.g., NCT04185038, NCT04483778, NCT04077866) (40). Another potential immunotherapy target for solid tumors is tumor necrosis factor (TNF)-related apoptosis-inducing ligand (TRAIL) death receptors. The TRAIL ligand receptors, death receptors 4 (DR4)/DR5, are widely expressed in many cancer cells, but not expressed in normal tissues (41, 42). Importantly, on-target/off-tumor toxicities to normal cells attributable to cross-reactivities have not been observed for the aforementioned antigens.
Inadequate antigen expression to trigger optimal CAR-T cell activation remains a significant challenge for the effective clinical application of CAR-T therapeutics. In fact, CAR-T cells directed against MUC1 and PSCA are not able to completely eliminate solid tumors, and tumor cells expressing low densities of the target antigen are the main reason for tumor escape (43). Additionally, solid tumors display substantial heterogeneity of phenotypes and target antigens. In an EGFRvIII-targeted CAR-T first-in-human study, post-therapeutic biopsies of glioblastoma revealed that the levels of EGFRvIII antigen expression reduced after treatment compared with pre-treatment tumors in 71% of the patients (31, 32). The inability of the CAR-T cells to detect and eliminate antigen-low tumor cells may be related to the failure of treatment in this study.
The two additional obstacles in achieving a beneficial therapeutic effect using CAR-T therapy on solid tumors are the poor trafficking to both tumor sites and to the tumor microenvironment (TME) (44, 45). Unlike the situation in blood cancer, CAR-T cells cannot effectively traffic from the blood into the solid tumor mass. The chemokine receptor can be co-expressed in CAR-T cells to increase the trafficking of CAR-T cells to solid tumor sites. Several groups have reported that CAR-T cells, which express the chemokine receptor CCR2b, increase the tumor infiltration and antitumor activity of CAR-T cells (46, 47). Another chemokine receptor combined with CAR-T cells is CXCR2 (48, 49), which has been shown to improve the homing of CAR-T cells to the tumor. CARs expressing IL12β p40 subunit to produce IL-23 upon activation, which activates STAT3 signaling to promote proliferation, showed improved activities in mice (50, 51). The chemokine receptors co-expressed in CAR-T cells need to be selected according to the specific tumor types because different tumor types have different expression patterns of chemokines (52–55). Thus, more chemokine receptors must be found and validated for solid tumors. Alternatively, locoregional delivery of CAR-T cells and repeat treatment may help improve the therapeutic effect of CAR-T therapy. Donovan et al. identified EPHA2 as a tumor antigen for medulloblastoma and designed trivalent CAR-T cells (EPHA2, HER2, IL13Rα2) to perform preclinical studies and validated that intrathecal delivery of the trivalent CAR-T could be an effective treatment for metastatic medulloblastoma (56). Therefore, cancer-type-specific CAR-T delivery and transplantation may also help overcome the obstacle of poor trafficking.
The TME has numerous suppressive immune cells and molecular factors, which impair the cytotoxic function of CAR-T cells (57, 58). Overcoming the immunosuppressive effects of the TME is the main focus of CAR-T therapy research in solid tumors (59). Several strategies have been proposed for CAR-T cells to resist immunosuppression of the TME. Suryadevara et al. reported that the prevention of lymphocyte-specific tyrosine kinase (Lck) binding in the CD28 domain eliminated the secretion of IL-2 from CAR-T cells, and hence, CAR-T cells achieved resistance to regulatory T cells (Tregs) (60). Mohammed et al. showed that CAR-T cells expressing 4/7 inverted cytokine receptors could reverse the immunosuppressive signal from IL-4 to the signal of proliferation (61). Yamamoto et al. found that the apoptosis-inducing ligand FasL (CD95) is overexpressed within the TME of many human cancer types and designed T cells to prevent Fas-induced apoptosis (44). Rafiq and colleagues reported that CAR-T cells expressing PD-1-blocking single-chain variable fragment (scFv) achieved resistance to PD-L1 inhibition (62). However, many inhibitory factors exist in the TME: a strategy to resolve one inhibitory factor may succeed in mouse models, while more efforts may be required to achieve expected therapeutic effects in clinical trials.
CAR products cannot be easily expanded under good manufacturing practice (GMP) conditions which limits their application as “off-the-shelf” products for patients. On the other hand, CAR-modified NK cells may help solve this problem, owing to their tolerable safety profile and low possibility of triggering graft-versus-host disease upon the allogeneic infusion of CAR-modified NK cells without the constraint of autologous cells. An ex-vivo expansion method of NK cells from CD34+ umbilical cord blood is now an option for a clinical-grade protocol for adoptive immunotherapy. Induced pluripotent stem cells (iPSCs) are also sources of functional NK cells. The iPSC-derived NK cells can be expanded 102- to 103-fold with a panel of cytokines, including IL-3, IL-7, IL-15, SCF, and Flt3L, without exogenous stromal cells (63), which is promising for future development. A study comparing the cytotoxicity of NK cells isolated from peripheral blood mononuclear cells and iPSCs for killing ovarian cancer cells (64) indicated that both NK cell populations had a significant effect on the killing of cancer cells. Gene editing of iPSCs with CRISPR/Cas9 could also improve the therapeutic potential of iPSC-derived NK cells (iPSC-NKs). A study by Zhu et al. revealed that knocked-out cytokine-inducible SH2-containing protein (encoded by the gene CISH) developed CISH−/− iPSC-NKs, showing increased expansion and cytotoxic effects on tumor cell lines including ovarian tumor cells (65). The expression of CARs can further boost the antitumor activity of effector NK cells. Another study showed that CAR-iPSC-NK cells inhibited ovarian tumor growth more effectively compared with iPSC-NK cells (66).
Improvements in CAR Design
Currently, CAR-T cells used in preclinical or clinical trials are mostly based on 4-1BB or CD28, but these CAR designs have some downsides, such as cytokine release syndrome and macrophage activation syndrome (67, 68). Guedan et al. reported that CAR-T cells with ICOS and 4-1BB co-stimulation had an optimal antitumor activity and persistence in vivo (67). In contrast to CAR-T, TILs or TCR-engineered T cells that rely on TCR signaling have reported low rates of adverse events (68). Helsen et al. developed a new chimeric receptor termed TAC (T-cell antigen coupler) which transduces signals through the native TCR, and TAC-engineered T cells display both enhanced in-vivo antitumor efficacy and decreased off-tumor toxicity compared to the first- and second-generation CARs (68). Another chimeric receptor utilizing native TCR for transducing signal is TRuC (T-cell receptor fusion construct). TRuC-T cells have shown the capacity for trafficking to tumors and long-term functional persistence in a model (69).
Whether using a typical CAR that is based on co-stimulator 4-1BB/CD28 or the TAC/TRuC that hijacks the endogenous TCR subunits to form a CAR complex, CD3ζ is required for the activation of these artificial receptors. However, in the TME, CD3ζ expression is always downregulated by arginine exhaustion with arginase (70, 71); hence, CARs based on CD3ζ may have a disadvantage in treating solid tumors. Recently, a natural multichain immunoreceptor design showed potential antitumor activity for solid tumors. Wang and colleagues (72) developed a multichain CAR design that is not based on CD3ζ but rather on KIR/DAP12. KIR-CAR/DAP12 expression in T cells is not affected in solid tumors, while CARs based on CD3ζ are not fully expressed in solid tumors. Moreover, Dap12-based CAR-T cells showed resistance to tumor-induced hypofunction and enhanced antitumor activity compared to CD3ζ-based CAR-T cells. Currently, Dap12-based CAR-T immunotherapy is being tested in clinical trials by Wang and colleagues, and the preliminary results show that one ovarian cancer patient achieved SD over 16 months and the ascites of two patients significantly decreased along with effective CAR-T cell expansion both in blood and ascites (data not published). In 2019, another Dap12-based CAR design was reported (73), which was TREM1/Dap12-based and showed faster tumor eradication than BBζ CAR-T cells in mice.
In conclusion, because of the suppression of CD3ζ within TME, Dap12-based multichain CAR showed advantages when applied to solid tumors.
Future Directions of CAR-T Therapy
Current clinical trials show that TAAs for CAR-T therapy in solid tumors are not specific to tumor cells. In fact, it is difficult to find targets highly and uniquely expressed in solid tumors. Thus far, B7-H3 and DR4/DR5 have been established as potential targets for treating solid tumors. In addition to antigen identification, overcoming the dysfunction of CAR-T cells in the TME is the most important challenge for improving therapeutic effects in solid tumors. Some strategies have been proposed to enhance the function of CAR-T in TME, but these strategies need to be clinically validated. As mentioned earlier, mutations in the CD28 domain can improve CAR-T resistance to Treg cells, implying that CAR designs currently in use are not perfect. The bottleneck of CAR-T therapy for solid tumors may be addressed by changing the design of CARs to optimize the T-cell signal transduction pathway (74), especially the endoplasmic domain of CAR.
Most research groups focus on target switching or new antibody generation. However, barriers to CAR-T therapy against solid tumors cannot easily be overcome using this strategy; hence, artificial receptors approaching two destinations, tumor recognition and immune cell activation, need to be designed. Just mimicking TCR may not serve the purpose. Other immune cell-activating receptors may also be mimicked. Single-chain artificial receptors can be substituted by multiple-chain receptors with an architecture closer to that of natural activating receptors. Also, antigen-dependent control of CAR may enhance its effectiveness. For instance, Hernandez-Lopez et al. engineered a two-step positive-feedback circuit attempting to allow cytotoxic T cells to discriminate targets upon the density of TAAs (75). Jan et al. put efforts into generating lenalidomide-controlled reversible ON- and OFF-switch CARs, under which the system CARs are designed to split under the ON-mode and be degraded under the OFF-mode (76). In addition, advanced nanomaterial-based CAR deliveries are also in development (77). With these new designs, the therapeutic effects of CARs against solid tumors may be improved, and TME suppressive effects may also be avoided during CAR-T cell expansion and functionality realization.
CAR-T/immune cell survival and tumor homing have gained increasing attention. Antiregulatory signal antibodies and homing signals have been tested with the so-called “travel trailer style” design to provide more T/immune cell growth factors, which will be validated soon in clinical trials. In addition to the molecular level design, combining CAR-modified immune cell therapies themselves or other treatments, such as immune checkpoint blockade or traditional cancer treatment, are worthy of being tested in the near future (78).
Concluding Remarks
After comparing the regulations from different countries on cell therapy from several sources including engineered cells and products that recruit, mobilize, and activate human cells in treating human malignant tumors, ideally, CAR-T should select the tumor-specific antigens that have no on-target/off-tumor toxicities upon more advanced research. An “off-the-shelf” product for cancer treatment should be well considered for wide application in clinical practice for its ease of availability and large-scale expansion. Most importantly, cell therapy is expected to have a positive impact on the treatment of solid tumors (Figure 1). Thus, supportive and reasonable guidelines from related authorities worldwide may ensure promising, beneficial, and favorable results.
Funding
This work was supported by the Chinese Academy of Medical Sciences Innovation Fund for Medical Sciences (Platform Improvement of Clinical Trial Capability 2020-I2M-2-007) (recipient: NL), Chinese Academy of Medical Sciences (grants 2019XK320068) (recipient: NL), Beijing Municipal Science and Technology Commission (International Pharmaceutical Clinical Research and Development Platform 2015) (recipient: NL), and Corbett Estate Fund for Cancer Research (62285-531021-41800; 62285-531021-51800; 62285-531021-61800; 62285-531021-71800) (recipient: EW).
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Statements
Author contributions
All authors listed have made a substantial, direct, and intellectual contribution to the work and approved it for publication.
Acknowledgments
We thank Mr. Euni Wu for editing the language of the manuscript.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2022.896685/full#supplementary-material
Glossary
| CARs | chimeric antigen receptors, are engineered receptors, which graft an arbitrary specificity onto an immune effector cell (T cell). Typically, these receptors are used to graft the specificity of a monoclonal antibody onto a T cell, with the transfer of their coding sequence facilitated by retroviral vectors. The receptors are called chimeric because they are composed of parts from different sources. CARs are under investigation as a therapy for cancer, using a technique called adoptive cell transfer. T cells are extracted from a patient and modified so that they express receptors specific to the patient’s particular cancer. CARs are synthetic molecules composed of an extracellular domain, a transmembrane domain, and an intracellular signaling domain. The extracellular component is an scFv, which recognizes and binds specific TAAs. The transmembrane domain is typically derived from CD8 molecules, and the intracellular signaling domain consists of CD3ζ and one or two co-stimulatory domains (CD28 and/or 4-1BB). |
| TCR | T-cell receptor, is a protein complex found on the surface of T cells or T lymphocytes that is responsible for recognizing fragments of antigen as peptides bound to major histocompatibility complex (MHC) molecules. The binding between TCR and antigen peptides is of relatively low affinity and is degenerate: that is, many TCRs recognize the same antigen peptide and many antigen peptides are recognized by the same TCR. |
| TILs | tumor-infiltrating lymphocytes, which include B cells, T cells, and natural killer (NK) cells, form an important component in antitumor immune responses. |
| DCs | dendritic cells, are any of the various white blood cells that have long projections from the cell body and function in the immune response by taking in and processing antigens and presenting them to T cells in lymph nodes, thus activating the T cells. Immature dendritic cells are found chiefly in the skin and mucosal surfaces. |
| NK | natural killer cells, are large granular lymphocytes that do not express markers of either T- or B-cell lineage. These cells kill target cells through antibody-dependent cell-mediated cytotoxicity. NK cells can also use perforin to kill cells in the absence of antibody |
| scFv | single-chain variable fragment, consists of a variable heavy (VH) and a variable light (VL) antibody chains linked with a peptide linker. |
| GPCRs | Chemokine receptors are a superfamily of G-protein coupled receptors that control immune cell behavior via the promotion of chemotaxis, cell adhesion, and mediator release. |
| TME | tumor microenvironment, is the environment surrounding a tumor and consists of protean components, such as immune cells, blood or lymphatic vessels, fibroblasts, endothelial cells, pericytes, and extracellular matrix (ECM); the tumor closely interacts with its TME, which contributes to the generation of therapy resistance, metastasis, and immune escape. |
References
1
El-KadiryAERafeiMShammaaR. Cell Therapy: Types, Regulation, and Clinical Benefits. Front Med (Lausanne) (2021) 8:756029. doi: 10.3389/fmed.2021.756029
2
FischbachMABluestoneJALimWA. Cell-Based Therapeutics: The Next Pillar of Medicine. Sci Transl Med (2013) 5:179ps7. doi: 10.1126/scitranslmed.3005568
3
USFDA. KYMRIAH®-HIGHLIGHTS OF PRESCRIBING INFORMATION. FDA (2017).
4
USFDA. YESCARTA®-HIGHLIGHTS OF PRESCRIBING INFORMATION. FDA (2017).
5
StuppRMasonWPvan den BentMJWellerMFisherBTaphoornMJet al. Radiotherapy Plus Concomitant and Adjuvant Temozolomide for Glioblastoma. N Engl J Med (2005) 352:987–96. doi: 10.1056/NEJMoa043330
6
USFDA. ABECMA-HIGHLIGHTS OF PRESCRIBING INFORMATION. FDA (2021).
7
HanahanDCoussensLM. Accessories to the Crime: Functions of Cells Recruited to the Tumor Microenvironment. Cancer Cell (2012) 21:309–22. doi: 10.1016/j.ccr.2012.02.022
8
QiDWuE. Cancer Prognosis: Considering Tumor and its Microenvironment as a Whole. EBioMedicine (2019) 43:28–9. doi: 10.1016/j.ebiom.2019.04.031
9
KlichinskyMRuellaMShestovaOLuXMBestAZeemanMet al. Human Chimeric Antigen Receptor Macrophages for Cancer Immunotherapy. Nat Biotechnol (2020) 38:947–53. doi: 10.1038/s41587-020-0462-y
10
DuperretEKLiuSPaikMTrautzAStoltzRLiuXet al. A Designer Cross-Reactive DNA Immunotherapeutic Vaccine That Targets Multiple MAGE-A Family Members Simultaneously for Cancer Therapy. Clin Cancer Res (2018) 24:6015–27. doi: 10.1158/1078-0432.CCR-18-1013
11
McGrailDJFedericoLLiYDaiHLuYMillsGBet al. Multi-Omics Analysis Reveals Neoantigen-Independent Immune Cell Infiltration in Copy-Number Driven Cancers. Nat Commun (2018) 9:1317. doi: 10.1038/s41467-018-03730-x
12
OzgaAJChowMTLusterAD. Chemokines and the Immune Response to Cancer. Immunity (2021) 54:859–74. doi: 10.1016/j.immuni.2021.01.012
13
HovdenAOAppelS. The First Dendritic Cell-Based Therapeutic Cancer Vaccine is Approved by the FDA. Scand J Immunol (2010) 72:554. doi: 10.1111/j.1365-3083.2010.02464.x
14
Global Cell Therapy Market—Companies to ActionFrost & Sullivan (2018).
15
RosenbergSALotzeMTMuulLMLeitmanSChangAEEttinghausenSEet al. Observations on the Systemic Administration of Autologous Lymphokine-Activated Killer Cells and Recombinant Interleukin-2 to Patients With Metastatic Cancer. N Engl J Med (1985) 313:1485–92. doi: 10.1056/NEJM198512053132327
16
RosenbergSAPackardBSAebersoldPMSolomonDTopalianSLToySTet al. Use of Tumor-Infiltrating Lymphocytes and Interleukin-2 in the Immunotherapy of Patients With Metastatic Melanoma. A preliminary report. N Engl J Med (1988) 319:1676–80. doi: 10.1056/NEJM198812223192527
17
FlotoRAMacAryPABonameJMMienTSKampmannBHairJRet al. Dendritic Cell Stimulation by Mycobacterial Hsp70 Is Mediated Through CCR5. Science (2006) 314:454–8. doi: 10.1126/science.1133515
18
NiessHvon EinemJCThomasMNMichlMAngeleMKHussRet al. Treatment of Advanced Gastrointestinal Tumors With Genetically Modified Autologous Mesenchymal Stromal Cells (TREAT-ME1): Study Protocol of a Phase I/II Clinical Trial. BMC Cancer (2015) 15:237. doi: 10.1186/s12885-015-1241-x
19
von EinemJCGuentherCVolkHDGrutzGHirschDSalatCet al. Treatment of Advanced Gastrointestinal Cancer With Genetically Modified Autologous Mesenchymal Stem Cells: Results From the Phase 1/2 TREAT-ME-1 Trial. Int J Cancer (2019) 145:1538–46. doi: 10.1002/ijc.32230
20
von EinemJCPeterSGuntherCVolkHDGrutzGSalatCet al. Treatment of Advanced Gastrointestinal Cancer With Genetically Modified Autologous Mesenchymal Stem Cells - TREAT-ME-1 - a Phase I, First in Human, First in Class Trial. Oncotarget (2017) 8:80156–66. doi: 10.18632/oncotarget.20964
21
SarnaikAKhushalaniNIChesneyJALewisKDMedinaTMKlugerHMet al. Long-Term Follow Up of Lifileucel (LN-144) Cryopreserved Autologous Tumor Infiltrating Lymphocyte Therapy in Patients With Advanced Melanoma Progressed on Multiple Prior Therapies. J Clin Oncol (2020) 38:10006–6. doi: 10.1200/JCO.2020.38.15_suppl.10006
22
USFDA. PROVENGE (Sipuleucel-T). FDA (2019).
23
APAC Biotech Pvt Ltd. Indian FDA Approves the First Autologous Dendritic Cell-Based Immuno-Oncology Product, APCEDEN®. FDA (2017).
24
LimJ. Current Cellular Therapy in Korea. FDA (2010).
25
DottiGGottschalkSSavoldoBBrennerMK. Design and Development of Therapies Using Chimeric Antigen Receptor-Expressing T Cells. Immunol Rev (2014) 257:107–26. doi: 10.1111/imr.12131
26
ParkJHGeyerMBBrentjensRJ. CD19-Targeted CAR T-Cell Therapeutics for Hematologic Malignancies: Interpreting Clinical Outcomes to Date. Blood (2016) 127:3312–20. doi: 10.1182/blood-2016-02-629063
27
JohnLBDevaudCDuongCPYongCSBeavisPAHaynesNMet al. Anti-PD-1 Antibody Therapy Potently Enhances the Eradication of Established Tumors by Gene-Modified T Cells. Clin Cancer Res (2013) 19:5636–46. doi: 10.1158/1078-0432.CCR-13-0458
28
SridharPPetroccaF. Regional Delivery of Chimeric Antigen Receptor (CAR) T-Cells for Cancer Therapy. Cancers (2017) 9(7). doi: 10.3390/cancers9070092
29
TokarewNOgonekJEndresSvon Bergwelt-BaildonMKoboldS. Teaching an Old Dog New Tricks: Next-Generation CAR T Cells. Br J Cancer (2019) 120:26–37. doi: 10.1038/s41416-018-0325-1
30
LouisCUSavoldoBDottiGPuleMYvonEMyersGDet al. Antitumor Activity and Long-Term Fate of Chimeric Antigen Receptor-Positive T Cells in Patients With Neuroblastoma. Blood (2011) 118:6050–6. doi: 10.1182/blood-2011-05-354449
31
AkhavanDAlizadehDWangDWeistMRShepphirdJKBrownCE. CAR T Cells for Brain Tumors: Lessons Learned and Road Ahead. Immunol Rev (2019) 290:60–84. doi: 10.1111/imr.12773
32
O'RourkeDMNasrallahMPDesaiAMelenhorstJJMansfieldKMorrissetteJJDet al. A Single Dose of Peripherally Infused EGFRvIII-Directed CAR T Cells Mediates Antigen Loss and Induces Adaptive Resistance in Patients With Recurrent Glioblastoma. Sci Transl Med (2017) 9(399). doi: 10.1126/scitranslmed.aaa0984
33
YinYBoesteanuACBinderZAXuCReidRARodriguezJLet al. Checkpoint Blockade Reverses Anergy in IL-13ralpha2 Humanized scFv-Based CAR T Cells to Treat Murine and Canine Gliomas. Mol Ther Oncolytics (2018) 11:20–38. doi: 10.1016/j.omto.2018.08.002
34
CherkasskyLMorelloAVillena-VargasJFengYDimitrovDSJonesDRet al. Human CAR T Cells With Cell-Intrinsic PD-1 Checkpoint Blockade Resist Tumor-Mediated Inhibition. J Clin Invest (2016) 126:3130–44. doi: 10.1172/JCI83092
35
KlossCCLeeJZhangAChenFMelenhorstJJLaceySFet al. Dominant-Negative TGF-Beta Receptor Enhances PSMA-Targeted Human CAR T Cell Proliferation And Augments Prostate Cancer Eradication. Mol Ther (2018) 26:1855–66. doi: 10.1016/j.ymthe.2018.05.003
36
RichardsRMSotilloEMajznerRG. CAR T Cell Therapy for Neuroblastoma. Front Immunol (2018) 9:2380. doi: 10.3389/fimmu.2018.02380
37
WallstabeLGottlichCNelkeLCKuhnemundtJSchwarzTNerreterTet al. ROR1-CAR T Cells are Effective Against Lung and Breast Cancer in Advanced Microphysiologic 3D Tumor Models. JCI Insight (2019) 4(18). doi: 10.1172/jci.insight.126345
38
AdusumilliPSCherkasskyLVillena-VargasJColovosCServaisEPlotkinJet al. Regional Delivery of Mesothelin-Targeted CAR T Cell Therapy Generates Potent and Long-Lasting CD4-Dependent Tumor Immunity. Sci Transl Med (2014) 6:261ra151. doi: 10.1126/scitranslmed.3010162
39
HaasARTanyiJLO'HaraMHGladneyWLLaceySFTorigianDAet al. Phase I Study of Lentiviral-Transduced Chimeric Antigen Receptor-Modified T Cells Recognizing Mesothelin in Advanced Solid Cancers. Mol Ther (2019) 27:1919–29. doi: 10.1016/j.ymthe.2019.07.015
40
TheruvathJSotilloEMountCWGraefCMDelaidelliAHeitzenederSet al. Locoregionally Administered B7-H3-Targeted CAR T Cells for Treatment of Atypical Teratoid/Rhabdoid Tumors. Nat Med (2020) 26:712–9. doi: 10.1038/s41591-020-0821-8
41
PanGNiJWeiYFYuGGentzRDixitVM. An Antagonist Decoy Receptor and a Death Domain-Containing Receptor for TRAIL. Science (1997) 277:815–8. doi: 10.1126/science.277.5327.815
42
LeBlancHNAshkenaziA. Apo2L/TRAIL and its Death and Decoy Receptors. Cell Death Differ (2003) 10:66–75. doi: 10.1038/sj.cdd.4401187
43
YekuOOPurdonTJKoneruMSpriggsDBrentjensRJ. Armored CAR T Cells Enhance Antitumor Efficacy and Overcome the Tumor Microenvironment. Sci Rep (2017) 7:10541. doi: 10.1038/s41598-017-10940-8
44
YamamotoTNLeePHVodnalaSKGurusamyDKishtonRJYuZet al. T Cells Genetically Engineered to Overcome Death Signaling Enhance Adoptive Cancer Immunotherapy. J Clin Invest (2019) 129:1551–65. doi: 10.1172/JCI121491
45
MuradJPTilakawardaneDParkAKLopezLSYoungCGibsonJet al. Pre-Conditioning Modifies the Tumor Microenvironment to Enhance Solid Tumor CAR T Cell Efficacy and Endogenous Protective Immunity. Mol Ther (2021) 29(7):2335–49. doi: 10.1101/2020.11.18.388892
46
MoonEKCarpenitoCSunJWangLCKapoorVPredinaJet al. Expression of a Functional CCR2 Receptor Enhances Tumor Localization and Tumor Eradication by Retargeted Human T Cells Expressing a Mesothelin-Specific Chimeric Antibody Receptor. Clin Cancer Res an Off J Am Assoc Cancer Res (2011) 17:4719–30. doi: 10.1158/1078-0432.CCR-11-0351
47
CraddockJALuABearAPuleMBrennerMKRooneyCMet al. Enhanced Tumor Trafficking of GD2 Chimeric Antigen Receptor T Cells by Expression of the Chemokine Receptor CCR2b. J Immunother (2010) 33:780–8. doi: 10.1097/CJI.0b013e3181ee6675
48
IdornMSkadborgSKKellermannLHalldorsdottirHRHolmen OlofssonGMetOet al. Chemokine Receptor Engineering of T Cells With CXCR2 Improves Homing Towards Subcutaneous Human Melanomas in Xenograft Mouse Model. Oncoimmunology (2018) 7:e1450715. doi: 10.1080/2162402X.2018.1450715
49
WhildingLMHalimLDraperBParente-PereiraACZabinskiTDaviesDMet al. CAR T-Cells Targeting the Integrin Alphavbeta6 and Co-Expressing the Chemokine Receptor CXCR2 Demonstrate Enhanced Homing and Efficacy Against Several Solid Malignancies. Cancers (2019) 11(5). doi: 10.3390/cancers11050674
50
LarsonRCMausMV. Recent Advances and Discoveries in the Mechanisms and Functions of CAR T Cells. Nat Rev Cancer (2021) 21:145–61. doi: 10.1038/s41568-020-00323-z
51
MaXShouPSmithCChenYDuHSunCet al. Interleukin-23 Engineering Improves CAR T Cell Function in Solid Tumors. Nat Biotechnol (2020) 38:448–59. doi: 10.1038/s41587-019-0398-2
52
PayneASCorneliusLA. The Role of Chemokines in Melanoma Tumor Growth and Metastasis. J Invest Dermatol (2002) 118:915–22. doi: 10.1046/j.1523-1747.2002.01725.x
53
RamanDBaugherPJThuYMRichmondA. Role of Chemokines in Tumor Growth. Cancer Lett (2007) 256:137–65. doi: 10.1016/j.canlet.2007.05.013
54
ChowMTLusterAD. Chemokines in Cancer. Cancer Immunol Res (2014) 2:1125–31. doi: 10.1158/2326-6066.CIR-14-0160
55
NagarshethNWichaMSZouW. Chemokines in the Cancer Microenvironment and Their Relevance in Cancer Immunotherapy. Nat Rev Immunol (2017) 17:559–72. doi: 10.1038/nri.2017.49
56
DonovanLKDelaidelliAJosephSKBielamowiczKFousekKHolgadoBLet al. Locoregional Delivery of CAR T Cells to the Cerebrospinal Fluid for Treatment of Metastatic Medulloblastoma and Ependymoma. Nat Med (2020) 26:720–31. doi: 10.1038/s41591-020-0827-2
57
YanLLiuB. Critical Factors in Chimeric Antigen Receptor-Modified T-Cell (CAR-T) Therapy for Solid Tumors. OncoTargets Ther (2019) 12:193–204. doi: 10.2147/OTT.S190336
58
ZhangEGuJXuH. Prospects for Chimeric Antigen Receptor-Modified T Cell Therapy for Solid Tumors. Mol Cancer (2018) 17:7. doi: 10.1186/s12943-018-0759-3
59
HouAJChenLCChenYY. Navigating CAR-T Cells Through the Solid-Tumour Microenvironment. Nat Rev Drug Discovery (2021) 20:531–50. doi: 10.1038/s41573-021-00189-2
60
SuryadevaraCMDesaiRFarberSHChoiBDSwartzAMShenSHet al. Preventing Lck Activation in CAR T Cells Confers Treg Resistance But Requires 4-1bb Signaling for Them to Persist and Treat Solid Tumors in Nonlymphodepleted Hosts. Clin Cancer Res (2019) 25:358–68. doi: 10.1158/1078-0432.CCR-18-1211
61
MohammedSSukumaranSBajgainPWatanabeNHeslopHERooneyCMet al. Improving Chimeric Antigen Receptor-Modified T Cell Function by Reversing the Immunosuppressive Tumor Microenvironment of Pancreatic Cancer. Mol Ther (2017) 25:249–58. doi: 10.1016/j.ymthe.2016.10.016
62
RafiqSYekuOOJacksonHJPurdonTJvan LeeuwenDGDrakesDJet al. Targeted Delivery of a PD-1-Blocking scFv by CAR-T Cells Enhances Anti-Tumor Efficacy In Vivo. Nat Biotechnol (2018) 36:847–56. doi: 10.1038/nbt.4195
63
KnorrDANiZHermansonDHexumMKBendzickLCooperLJet al. Clinical-Scale Derivation of Natural Killer Cells From Human Pluripotent Stem Cells for Cancer Therapy. Stem Cells Trans Med (2013) 2:274–83. doi: 10.5966/sctm.2012-0084
64
ShafferBCLe LuduecJ-BForlenzaCJakubowskiAAPeralesM-AYoungJWet al. Phase II Study of Haploidentical Natural Killer Cell Infusion for Treatment of Relapsed or Persistent Myeloid Malignancies Following Allogeneic Hematopoietic Cell Transplantation. Biol Blood Marrow Transplant (2016) 22:705–9. doi: 10.1016/j.bbmt.2015.12.028
65
ZhuHBlumRHBernareggiDAskEHWuZHoelHJet al. Metabolic Reprograming via Deletion of CISH in Human iPSC-Derived NK Cells Promotes In Vivo Persistence and Enhances Anti-Tumor Activity. Cell Stem Cell (2020) 27:224–237 e6. doi: 10.1016/j.stem.2020.05.008
66
HanK-PZhuXLiuBJengEKongLYovandichJLet al. IL-15: IL-15 Receptor Alpha Superagonist Complex: High-Level Co-Expression in Recombinant Mammalian Cells, Purification and Characterization. Cytokine (2011) 56:804–10. doi: 10.1016/j.cyto.2011.09.028
67
GuedanSPoseyADJr.ShawCWingADaTPatelPRet al. Enhancing CAR T Cell Persistence Through ICOS and 4-1BB Costimulation. JCI Insight (2018) 3(1). doi: 10.1172/jci.insight.96976
68
HelsenCWHammillJALauVWCMwawasiKAAfsahiABezverbnayaKet al. The Chimeric TAC Receptor Co-Opts the T Cell Receptor Yielding Robust Anti-Tumor Activity Without Toxicity. Nat Commun (2018) 9:3049. doi: 10.1038/s41467-018-05395-y
69
Jian DingHHShahSZiebaAKrishnamurphyJAshhurstTMenkAVet al. Preclinical Evaluation of TC-210, a Mesothelin-Specific T Cell Receptor (TCR) Fusion Construct (TRuC™) T Cells for the Treatment of Solid Tumors, in: AACR Annual Meeting, Cancer Research (2019). doi: 10.1158/1538-7445.am2021-ct105
70
RodriguezPCQuicenoDGZabaletaJOrtizBZeaAHPiazueloMBet al. Arginase I Production in the Tumor Microenvironment by Mature Myeloid Cells Inhibits T-Cell Receptor Expression and Antigen-Specific T-Cell Responses. Cancer Res (2004) 64:5839–49. doi: 10.1158/0008-5472.CAN-04-0465
71
RodriguezPCZeaAHDeSalvoJCulottaKSZabaletaJQuicenoDGet al. L-Arginine Consumption by Macrophages Modulates the Expression of CD3 Zeta Chain in T Lymphocytes. J Immunol (2003) 171:1232–9. doi: 10.4049/jimmunol.171.3.1232
72
WangEWangLCTsaiCYBhojVGershensonZMoonEet al. Generation of Potent T-Cell Immunotherapy for Cancer Using DAP12-Based, Multichain, Chimeric Immunoreceptors. Cancer Immunol Res (2015) 3:815–26. doi: 10.1158/2326-6066.CIR-15-0054
73
ChenBZhouMZhangHWangCHuXWangBet al. TREM1/Dap12-Based CAR-T Cells Show Potent Antitumor Activity. Immunotherapy (2019) 11:1043–55. doi: 10.2217/imt-2019-0017
74
WaldmanADFritzJMLenardoMJ. A Guide to Cancer Immunotherapy: From T Cell Basic Science to Clinical Practice. Nat Rev Immunol (2020) 20:651–68. doi: 10.1038/s41577-020-0306-5
75
Hernandez-LopezRAYuWCabralKACreaseyOALopez PazminoMDPTonaiYet al. T Cell Circuits That Sense Antigen Density With an Ultrasensitive Threshold. Science (2021) 371:1166–71. doi: 10.1126/science.abc1855
76
JanMScarfoILarsonRCWalkerASchmidtsAGuirguisAAet al. And OFF-Switch Chimeric Antigen Receptors Controlled by Lenalidomide. Sci Transl Med (2021) 13(575). doi: 10.1126/scitranslmed.abb6295
77
GongNSheppardNCBillingsleyMMJuneCHMitchellMJ. Nanomaterials for T-Cell Cancer Immunotherapy. Nat Nanotechnol (2021) 16:25–36. doi: 10.1038/s41565-020-00822-y
78
WangSSunFHuangHChenKLiQJZhangLet al. The Landscape of Cell and Gene Therapies for Solid Tumors. Cancer Cell (2021) 39:7–8. doi: 10.1016/j.ccell.2020.12.005
Summary
Keywords
cellular immunotherapy, chimeric antigen receptors, genetically engineered, tumor microenvironment, solid tumors
Citation
Chen K, Wang S, Qi D, Ma P, Fang Y, Jiang N, Wu E and Li N (2022) Clinical Investigations of CAR-T Cell Therapy for Solid Tumors. Front. Immunol. 13:896685. doi: 10.3389/fimmu.2022.896685
Received
15 March 2022
Accepted
22 June 2022
Published
18 July 2022
Volume
13 - 2022
Edited by
Katy Rezvani, University of Texas MD Anderson Cancer Center, United States
Reviewed by
Jamshid Hadjati, Tehran University of Medical Sciences, Iran; Youwei Zhang, Case Western Reserve University, United States
Updates
Copyright
© 2022 Chen, Wang, Qi, Ma, Fang, Jiang, Wu and Li.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ning Li, ncctrials@cicams.ac.cn; Erxi Wu, Erxi.Wu@BSWHealth.org
†These authors have contributed equally to this work
This article was submitted to Cancer Immunity and Immunotherapy, a section of the journal Frontiers in Immunology
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.