 Christoph Tabeling1,2,3
Christoph Tabeling1,2,3 Carla R. González Calera1Jasmin Lienau1
Carla R. González Calera1Jasmin Lienau1 Jakob Höppner4
Jakob Höppner4 Thomas Tschernig5
Thomas Tschernig5 Olivia Kershaw6Birgitt Gutbier1Jan Naujoks2Julia Herbert1
Olivia Kershaw6Birgitt Gutbier1Jan Naujoks2Julia Herbert1 Bastian Opitz2Achim D. Gruber6
Bastian Opitz2Achim D. Gruber6 Berthold Hocher7,8,9
Berthold Hocher7,8,9 Norbert Suttorp2,10
Norbert Suttorp2,10 Harald Heidecke11
Harald Heidecke11 Gerd-R. Burmester4
Gerd-R. Burmester4 Gabriela Riemekasten12
Gabriela Riemekasten12 Elise Siegert3,4
Elise Siegert3,4 Wolfgang M. Kuebler10,13,14,15,16
Wolfgang M. Kuebler10,13,14,15,16 Martin Witzenrath1,2,10*
Martin Witzenrath1,2,10*- 1Division of Pulmonary Inflammation, Charité - Universitätsmedizin Berlin, Corporate Member of Freie Universität Berlin and Humboldt-Universität zu Berlin, Berlin, Germany
- 2Department of Infectious Diseases and Respiratory Medicine, Charité - Universitätsmedizin Berlin, Corporate Member of Freie Universität Berlin and Humboldt-Universität zu Berlin, Berlin, Germany
- 3Berlin Institute of Health at Charité - Universitätsmedizin Berlin, Berlin, Germany
- 4Department of Rheumatology and Clinical Immunology, Charité - Universitätsmedizin Berlin, Corporate Member of Freie Universität Berlin and Humboldt-Universität zu Berlin, Berlin, Germany
- 5Institute of Anatomy and Cell Biology, University of Saarland, Homburg, Germany
- 6Department of Veterinary Pathology, Freie Universität Berlin, Berlin, Germany
- 7Fifth Department of Medicine (Nephrology/Endocrinology/Rheumatology), University of Heidelberg, University Medical Centre Mannheim, Heidelberg, Germany
- 8Key Laboratory of Study and Discovery of Small Targeted Molecules of Hunan Province, School of Medicine, Hunan Normal University, Changsha, China
- 9Reproductive and Genetic Hospital of CITIC-Xiangya, Changsha, China
- 10German Center for Lung Research (DZL), Partner Site Charité, Berlin, Germany
- 11CellTrend GmbH, Luckenwalde, Germany
- 12Department of Rheumatology, University of Lübeck, Lübeck, Germany
- 13Institute of Physiology, Charité - Universitätsmedizin Berlin, Corporate Member of Freie Universität Berlin and Humboldt-Universität zu Berlin, Berlin, Germany
- 14German Center for Cardiovascular Research (DZHK), Partner Site, Berlin, Germany
- 15St. Michael’s Hospital, Keenan Research Centre for Biomedical Science, Toronto, ON, Canada
- 16Departments of Physiology and Surgery, University of Toronto, Toronto, ON, Canada
Introduction: Inflammation is a major pathological feature of pulmonary arterial hypertension (PAH), particularly in the context of inflammatory conditions such as systemic sclerosis (SSc). The endothelin system and anti-endothelin A receptor (ETA) autoantibodies have been implicated in the pathogenesis of PAH, and endothelin receptor antagonists are routinely used treatments for PAH. However, immunological functions of the endothelin B receptor (ETB) remain obscure.
Methods: Serum levels of anti-ETB receptor autoantibodies were quantified in healthy donors and SSc patients with or without PAH. Age-dependent effects of overexpression of prepro-endothelin-1 or ETB deficiency on pulmonary inflammation and the cardiovascular system were studied in mice. Rescued ETB-deficient mice (ETB-/-) were used to prevent congenital Hirschsprung disease. The effects of pulmonary T-helper type 2 (Th2) inflammation on PAH-associated pathologies were analyzed in ETB-/- mice. Pulmonary vascular hemodynamics were investigated in isolated perfused mouse lungs. Hearts were assessed for right ventricular hypertrophy. Pulmonary inflammation and collagen deposition were assessed via lung microscopy and bronchoalveolar lavage fluid analyses.
Results: Anti-ETB autoantibody levels were elevated in patients with PAH secondary to SSc. Both overexpression of prepro-endothelin-1 and rescued ETB deficiency led to pulmonary hypertension, pulmonary vascular hyperresponsiveness, and right ventricular hypertrophy with accompanying lymphocytic alveolitis. Marked perivascular lymphocytic infiltrates were exclusively found in ETB-/- mice. Following induction of pulmonary Th2 inflammation, PAH-associated pathologies and perivascular collagen deposition were aggravated in ETB-/- mice.
Conclusion: This study provides evidence for an anti-inflammatory role of ETB. ETB seems to have protective effects on Th2-evoked pathologies of the cardiovascular system. Anti-ETB autoantibodies may modulate ETB-mediated immune homeostasis.
Introduction
Despite modern therapy, pulmonary arterial hypertension (PAH) remains a fatal condition. PAH is characterized by construction and remodelling of pulmonary arteries leading to increased pulmonary vascular resistance and right heart failure (1).
An increasing body of evidence points to inflammation as a central pathogenic factor in idiopathic PAH (iPAH) as well as PAH secondary to other conditions (2–8). Perivascular inflammation and lymphoid tissue are found in lungs of PAH patients, and concordantly in murine models of pulmonary hypertension (2, 9–12). Elevated numbers of mast cells and T-helper type 2 (Th2) lymphocytes as well as increased expression of Th2 cytokines were repeatedly found in patients with pulmonary hypertension (2, 8, 13–16). Analogously, preclinical studies indicate an important underlying role of Th2-mediated immune signaling in the induction of morphological and functional changes found in PAH (2, 16–23).
The concept that the endothelium-derived peptide endothelin-1 (ET-1) serves as a major driver of PAH pathobiology is broadly accepted (1, 24–26). Prepro-endothelin-1 is the precursor of big-ET-1, which is converted to mature, bioactive ET-1 (24, 26, 27). Patients with pulmonary hypertension show elevated pulmonary vascular expression of ET-1 as well as increased levels of circulating ET-1 (25, 28).
ET-1 is a potent vasoconstrictor in the pulmonary circulation through activation of the G protein-coupled receptors endothelin A receptor (ETA) and endothelin B receptor (ETB) expressed on pulmonary arterial smooth muscle cells (PASMCs) (26, 27). The vasoconstrictive response induced by ET-1 involves thromboxane A2 (TXA2) release and consecutive TXA2 receptor activation (29, 30). Downstream signalling of ET-1-evoked vasoconstriction critically depends on protein kinase C isozyme alpha (PKCα) (31). Contrariwise, activation of ETB located on endothelial cells induces vasodilation via release of nitric oxide (NO) and prostaglandins (26, 27), partially dampening the vasoconstrictive effects of ET-1.
Besides its vasomotor actions, ET-1 promotes immune cell trafficking (32), such as via release of tumor necrosis factor alpha (TNF-α), interleukin (IL)-1β, and IL-6 from monocytes and macrophages (33), or via ETA-dependent release of IL-6 from vascular smooth muscle cells (34).
Endothelin receptor antagonists (ERAs) are routinely used for the treatment of PAH. However, whether ETA-selective targeting or dual inhibition of ETA/ETB is superior to the other is controversially discussed.
In systemic sclerosis (SSc), PAH is a common vascular complication, and an important driver of mortality (24). The prognosis of PAH secondary to SSc (SSc-PAH) has been critically linked to the presence of anti-ETA autoantibodies (AAb) (35). Autoimmunity is believed to play a significant role in PAH pathobiology (2, 5, 6, 35–40), and blood plasmablast levels were shown to be elevated in PAH patients (36). However, the potential involvement of anti-ETB AAb in PAH is currently unknown and the immunomodulatory role of ETB in the context of pulmonary hypertension remains largely elusive.
In this study, anti-ETB AAb levels were assessed in PAH patients for the first time. To better characterize the immunomodulatory functions of ETB, we additionally studied the effects of rescued ETB deficiency in mice on pulmonary vascular disease, independent of and dependent on pulmonary Th2 inflammation.
Circulating ET-1 is cleared from the blood via ET-1/ETB complex internalization (27, 41–43) and ETB deficiency results in increased ET-1 plasma levels (44–46). Thus, effects on PAH-associated cardiovascular pathologies were studied in parallel in mice overexpressing prepro-ET-1 (preETtg) to allow differentiation between ET-1- and ETB-mediated effects.
We hypothesized that ETB plays an anti-inflammatory role, alleviating Th2-evoked pathologies of the cardiovascular system.
Materials and Methods
Patients and Clinical Manifestations
Serum samples from 177 SSc patients referred to the Department of Rheumatology and Clinical Immunology at Charité - Universitätsmedizin Berlin were collected. Patients with SSc met the American College of Rheumatology/European League Against Rheumatism 2013 classification (47). SSc patients were classified as having either limited cutaneous SSc or diffuse cutaneous SSc or SSc sine scleroderma, according to the LeRoy criteria, depending on the distribution and history of skin sclerosis at the study visit. The first non-Raynaud symptom was considered as disease onset.
Under clinical routine conditions, patients were screened for PAH at least in 1-year intervals by assessment of World Health Organization functional class, echocardiography, lung function including single-breath diffusion capacity for carbon monoxide (DLCOSB), and, during the last few years, also by the detection of N-terminal pro-brain natriuretic peptide (NT-proBNP) levels. In all SSc patients in which PAH was suspected, diagnosis was confirmed by right heart catheterization. Interstitial lung disease was identified on the basis of a high-resolution computed tomographic scan, as confirmed by an expert radiologist. Additional serum samples were obtained from 10 iPAH patients, confirmed by right heart catheterization. Control serum samples were obtained from 26 healthy subjects.
The epidemiologic data of patients and healthy donors are shown in Supplementary Table 1. The study protocol was approved by the ethics committee (Charité - Universitätsmedizin Berlin; EA1/179/17). A written informed consent was obtained from each patient. The study was conducted in accordance with the principles of the Declaration of Helsinki.
Detection of Anti-ETB AAb
Prior to analysis, serum samples were stored at −80°C. Serum ETB antibody levels were measured in duplicate by ELISA (CellTrend GmbH, Luckenwalde, Germany) in a blinded manner as described (35, 48). In brief, microtiter plates were coated with extracts from Chinese hamster ovary cells overexpressing human ETB. Calcium chloride (1 mmol/L) was administered to each buffer for maintenance of conformational epitopes. Diluted serum samples were incubated (1:100, 4°C, 2 h). For detection, plates were washed and incubated for 1 h with horseradish peroxidase-labeled goat anti-human immunoglobulin G (IgG; 1:20,000; Jackson, West Grove, Pennsylvania, USA), followed by enzymatic substrate reaction. Optical densities were converted into concentrations (U/ml) by comparison to a standard curve. Concentrations below the limit of detection (LOD) were depicted as LOD/√2.
Mice
All animal procedures were approved by institutional authorities of the Charité - Universitätsmedizin Berlin and the Local State Office of Health and Social Affairs Berlin (LAGeSo; Berlin, Germany). Transgenic mice overexpressing human prepro-ET-1 (preETtg) on a mixed NMRI/C57BL/6 background rescued endothelin B receptor-deficient mice (ETB-/-) on a mixed C57BL/6/129 background and the respective corresponding wild-type mice were housed under specific pathogen-free conditions. The generation of preETtg mice (49) and rescued ETB-deficient mice (50) has been described elsewhere. Rescued ETB-deficient mice hold a dopamine-β-hydroxylase ETB transgene to prevent fatal congenital Hirschsprung disease (50).
Isolated Perfused Mouse Lung
Here, we used the isolated perfused mouse lung preparation to evaluate pulmonary hemodynamics ex vivo. While this approach does not allow determining whether a specific model fulfills the clinical criteria of pulmonary hypertension in vivo (which is, however, obscured anyway by the fact that invasive hemodynamic measurements in mice are commonly restricted to recordings of right ventricular systolic pressure rather than mean Ppa), constant perfusion rates and defined left atrial pressures allow for a sensitive assessment of differences in pulmonary vascular resistance independent of right and left ventricular function.
Anesthetized mice were prepared, lungs were isolated, and pulmonary artery and left atrium were cannulated as described (51–53). Lungs were perfused constantly (1 mL/min, nonrecirculating, left atrial pressure 2.2 cmH2O) with 37°C sterile Krebs-Henseleit hydroxyethyl amylopectin buffer (Serag-Wiessner, Naila, Germany). Negative pressure ventilation was performed (Pexp −4.5, Pins −9.0 cmH2O, 90 breaths/min). Pulmonary arterial pressure (Ppa) and dynamic lung compliance (Cdyn) were measured and recorded via Pulmodyn software (17). ET-1, thromboxane analog U46619, or serotonin (all Merck KGaA, Darmstadt, Germany) was administered to the perfusion buffer for 10, 3, or 0.5 min, respectively (31). Doses were increased following intervals of 24 (ET-1), 12 (U46619), or 8 min (serotonin). Vasopressor responses were calculated (maximal difference in Ppa, Δ Ppa). To determine the role of ETA, ETA inhibitor BQ-123 (8 μmol/L; Merck KGaA) or solvent (aqua dest.) was added to the perfusion buffer 10 min prior to ET-1 application. Lungs with signs of edema, atelectasis, or hemostasis were excluded from further analyses.
Fulton Index
Hearts were excised. Right ventricle and left ventricle plus adjacent septum were microscopically dissected and weighed. Fulton index [quotient of right ventricle (RV) and left ventricle (LV) including septum (S)] was calculated.
Pulmonary Th2 Inflammation
After systemic sensitization via i.p. injections of 20 μg of ovalbumin (OVA; grade V; Merck KGaA) dissolved in 100 µL of aluminum hydroxide suspension (1.3%; SERVA Electrophoresis GmbH, Heidelberg, Germany) and 10 µL of phosphate-buffered saline (PBS) on days 0 and 14, mice were repeatedly exposed to aerosolized ovalbumin (1%) in PBS on days 28–30 for 20 min/day (53, 54). On the respective days, sham-treated mice received i.p. injections of 100 µL of aluminum hydroxide suspension and 10 µL of PBS, followed by exposure to aerosolized PBS. Effects of pulmonary Th2 inflammation were analyzed on day 32.
Bronchoalveolar Lavage
Bronchoalveolar lavage (BAL) of the right lung was performed twice with 650 µL of ice-cold PBS containing protease inhibitor (cOmplete™ mini; Merck KGaA) (55, 56). Total cell numbers and leukocyte differentiation were determined by microscopic analysis blinded to the study groups. Cytokines from the BAL fluid supernatant of the first lavage were quantified according to the manufacturer´s instructions using a cytokine multiplex assay (Bioplex®; Bio-Rad Laboratories GmbH, Feldkirchen, Germany).
Lung Histopathology
Lungs were removed and immersion fixed for 24 h with 4% buffered formaldehyde solution pH 7.0 (Merck KGaA). After embedding in paraffin, tissue sections were cut with a microtome, mounted onto glass slides, and stained.
For histopathological analyses of naïve mice, 3-µm sections were either stained with hematoxylin and eosin (H&E) or immunostained for CD45R/B220 (B cells, monoclonal, 1:1,000, clone RA3-6B2, 553086; BD Biosciences, Heidelberg, Germany) or CD3 (T cells, polyclonal, 1:800; reference A0452; Dako, Santa Clara, CA, USA). Positive immunostaining was visualized using diaminobenzidine, and slides were counterstained with hematoxylin. For analyses of the effects of pulmonary Th2 inflammation, 5-µm tissue sections were either stained with H&E or Masson–Goldner trichrome.
Microscopic analyses were performed (Axiophot; Carl Zeiss Microscopy GmbH, Jena, Germany) in a blinded fashion by a board-certified veterinary pathologist or an anatomist and images were digitized (Color View II camera, CellSens software; Olympus Europa SE Co. KG, Hamburg, Germany).
Real-Time Quantitative RT-PCR
Gene expression was analyzed as described (31, 57). Lung tissue was homogenized in Trizol (Thermo Fisher Scientific, Dreieich, Germany), and RNA was extracted. Reverse transcription (RT) of total RNA was performed (high-capacity reverse transcription kit; Thermo Fisher Scientific). For quantitative RT-PCR (ABI 7300 instrument; Thermo Fisher Scientific), TaqMan assays (Life Technologies) were applied for the target genes ETA, TXA2 receptor, and ET-1. TaqMan assay IDs were Mm01243722_m1 (ETA), Mm00436917_m1 (TXA2 receptor), and Mm00438656_m1 (ET-1). Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) served as internal reference. GAPDH primer sequences were TGTGTCCGTCGTGGATCTGA (forward, 5’ to 3’), CCTGCTTCACCACCTTCTTGA (reverse, 5’ to 3’), and CCGCCTGGAGAAACCTGCCAAGTATG (probe, 5’-FAM to 3’-TAMRA) (57). The relative expression (relative quantity, RQ) of each target gene was quantified using the comparative Ct method, with relative expression set to 1 in PBS-treated WT mice (31, 57).
TXB2 and VIP Quantification
Thromboxane B2 (TXB2) perfusate levels and vasoactive intestinal peptide (VIP) plasma levels were quantified via enzyme immunoassay (EIA) according to the respective manufacturer’s guide (TXB2 EIA; Cayman Chemical, MI, USA; detection limit 7.8 pg/mL; VIP EIA; Phoenix Europe GmbH, Karlsruhe, Germany; detection limit 0.05 ng/mL).
Statistical Analysis
For comparison of autoantibody levels, data were analyzed by one-way ANOVA followed by Dunnett’s multiple comparisons test. Dose–response curves were compared using two-way repeated measures ANOVA. Mann–Whitney U test was performed for comparison between two groups. *p < 0.05, **p < 0.01, ***p < 0.001.
Results
Elevated Anti-ETB Autoantibody Serum Levels in Patients With PAH Secondary to Systemic Sclerosis
Autoantibodies against ETA were shown to be elevated in SSc-PAH patients (35). Analogously, in this study, anti-ETB autoantibody serum levels were quantified in SSc patients with or without PAH as well as in iPAH patients and healthy donors. Compared to healthy donors, SSc-PAH patients showed increased levels of anti-ETB AAb (Figure 1). In SSc patients without PAH as well as in iPAH patients, anti-ETB AAb serum levels were increased by trend when compared to healthy donors (Figure 1). However, the relatively small number of iPAH serum samples needs to be considered. Detailed patient characteristics are found in Supplementary Table 1.
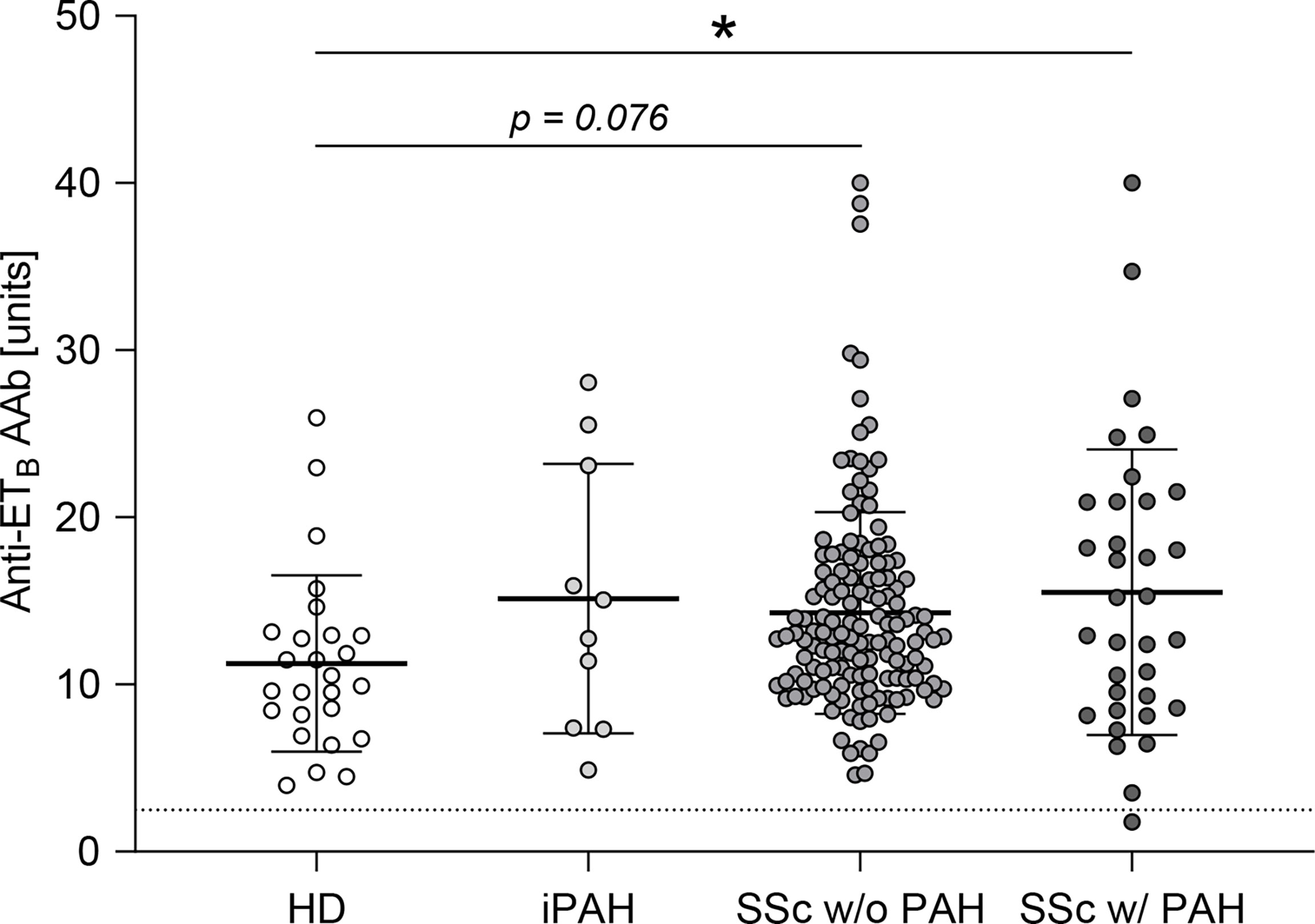
Figure 1 Anti-ETB autoantibody serum levels were elevated in patients with PAH secondary to SSc. Serum levels of anti-endothelin B receptor (ETB) autoantibodies (AAb) were quantified in healthy donors (HD), patients with idiopathic pulmonary arterial hypertension (iPAH), and systemic sclerosis (SSc) patients (+/- interstitial lung disease) with or without PAH. Data are expressed as single values with mean ± SD; N = 26 (HD) or N = 10 (iPAH) or N = 143 (SSc w/o PAH) or N = 34 (SSc w/PAH). The dotted line indicates the lower detection limit of the ELISA. *p < 0.05 (one-way ANOVA and Dunnett’s multiple comparisons test).
Pulmonary Hypertension, Right Ventricular Hypertrophy, and Lymphocytic Alveolitis in preETtg and ETB-/- Mice
To confirm the dual vasomotor role of ET-1 via ETA and ETB receptors in the pulmonary vasculature, functional analyses in isolated perfused mouse lungs were performed. In isolated WT lungs, application of the ETA inhibitor BQ-123 resulted in an almost complete reduction of the pulmonary vascular pressure response to ET-1 compared with the solvent control (Supplementary Figure 1A), while rescued ETB deficiency resulted in an elevated pulmonary vascular pressure response to ET-1 or the thromboxane receptor agonist U46619 compared to WT controls (Supplementary Figures 1B, C).
To dissect ET-1-specific as well as ETB-specific effects on pulmonary inflammation and PAH-associated cardiovascular pathologies independent of additional inflammatory stimuli, we first investigated age-dependent effects in two transgenic mouse models of (i) prepro-ET-1 overexpression (preETtg) and (ii) rescued ETB deficiency (ETB-/-). In line with the literature (58), young to mature-adult (2–6 months old) preETtg mice did not show pulmonary hypertension. However, in highly aged (16–18 months old) mice, prepro-ET-1 overexpression resulted in a significant increase in pulmonary arterial pressure compared to corresponding WT controls (Figure 2A). Furthermore, 2- to 6-month-old preETtg mice demonstrated a moderate increase in pulmonary vascular responsiveness to thromboxane receptor agonist U46619 compared to corresponding WT mice, whereas pulmonary vascular responsiveness of 16- to 18-month old mice was comparable within both groups (preETtg vs. WT) (Figure 2B). Cardiac analysis using the Fulton index (right ventricular weight/weight of left ventricle including septum) revealed right ventricular hypertrophy associated with chronic prepro-ET-1 overexpression in 16- to 18-month-old but not in 2- to 6-month-old mice (Figure 2C). Long-term overexpression of prepro-ET-1 was also associated with an increase in lymphocyte numbers in the BAL (Figure 2D). Independent of prepro-ET-1 overexpression, less BAL macrophages were found in 16- to 18-month-old vs. 2- to 6-month-old mice (Figure 2D).
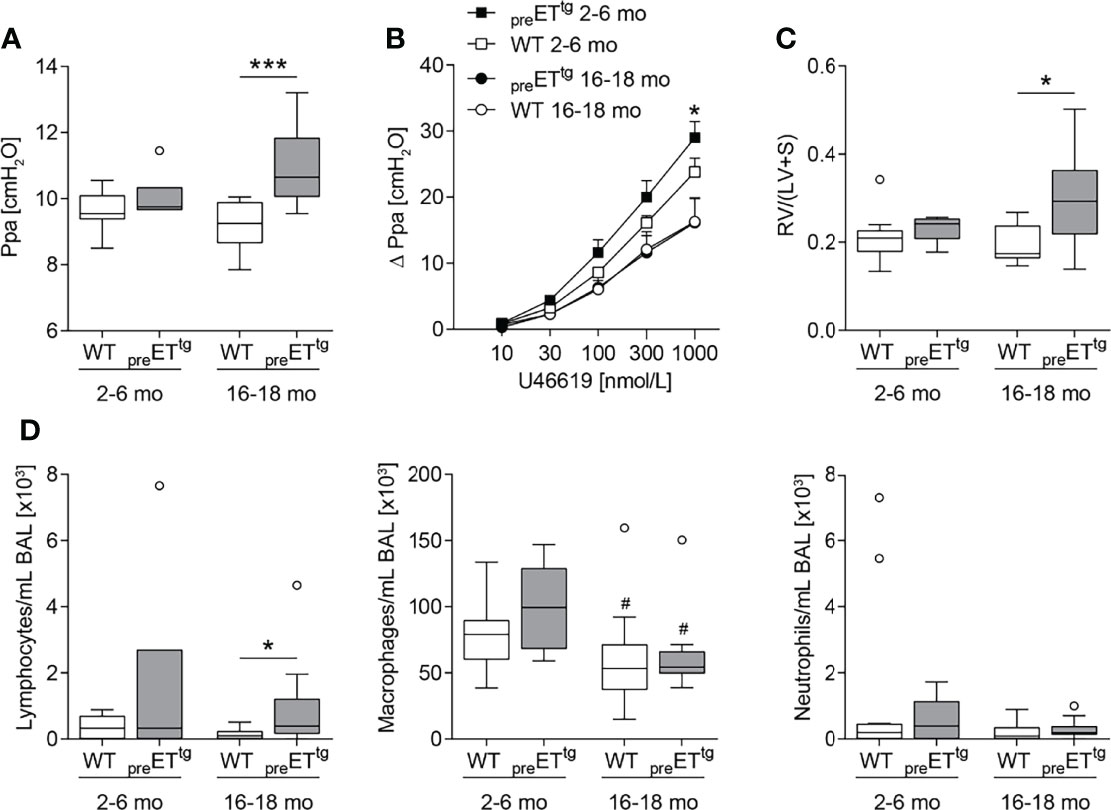
Figure 2 Prepro-endothelin-1 overexpression was age-dependently associated with increased pulmonary arterial pressure, vascular hyperresponsiveness, right ventricular hypertrophy, and increased number of lymphocytes in bronchoalveolar lavage. Lungs and hearts of 2- to 6-month (mo)-old or 16- to 18-mo-old prepro-endothelin-1 overexpressing (preETtg) mice and corresponding wild-type (WT) mice were prepared, or bronchoalveolar lavage (BAL) was performed. (A) In isolated perfused and ventilated lungs, under basal conditions, 16- to 18-mo-old preETtg showed a higher pulmonary arterial pressure (Ppa) compared to WT mice of the same age. (B) Pulmonary vascular responsiveness to intravascular application of the thromboxane receptor agonist U46619 was increased in 2- to 6-mo-old preETtg mice compared to WT controls of the same age. Data (Δ Ppa) represent the difference between the highest pressure response to U46619 and the basal Ppa. (C) Fulton index [quotient of right ventricle (RV) and left ventricle (LV) including septum (S)] determined after weighing the cardiac compartments was higher in 16- to 18-mo-old preETtg compared to WT mice of the same age. (D) Analysis of differentially quantified leukocytes in BAL showed increased number of lymphocytes in 16- to 18-mo-old preETtg compared to WT mice of the same age, whereas macrophages decreased with age, independent of prepro-ET-1 overexpression. In (A, C, D), data are represented as box plots depicting median, quartiles, and ranges excluding outliers (open circles), and analyzed using Mann–Whitney U test. # indicates significant difference between 16- to 18-mo-old vs. 2- to 6-mo-old groups, * indicates significant difference between preETtg vs. corresponding WT group (as indicated). In (B), values are given as mean and SEM, and analyzed using two-way repeated measures ANOVA, followed by a single Mann–Whitney U test between values of preETtg and WT mice of the same age at the highest dose of U46619 (*). In (A–C), N = 5–12; in (D), N = 7–17. */#p < 0.05, ***p < 0.001.
In mature-adult (6 months old) ETB-/- mice, basal pulmonary arterial pressure was significantly increased compared to WT mice of the same age (Figure 3A). Weight analysis of cardiac compartments revealed that ETB-/- mice of both age groups exhibited right ventricular hypertrophy compared with their respective WT counterparts (Figure 3B). Furthermore, splenomegaly was present in ETB-/- compared to WT mice and was progressive with age (Figure 3C) whereas relative liver weight was comparable in all groups (Supplementary Figure 2A). Cellular analysis of BAL fluid showed increased numbers of lymphocytes and macrophages in young ETB-/- compared to WT mice (Figure 3D).
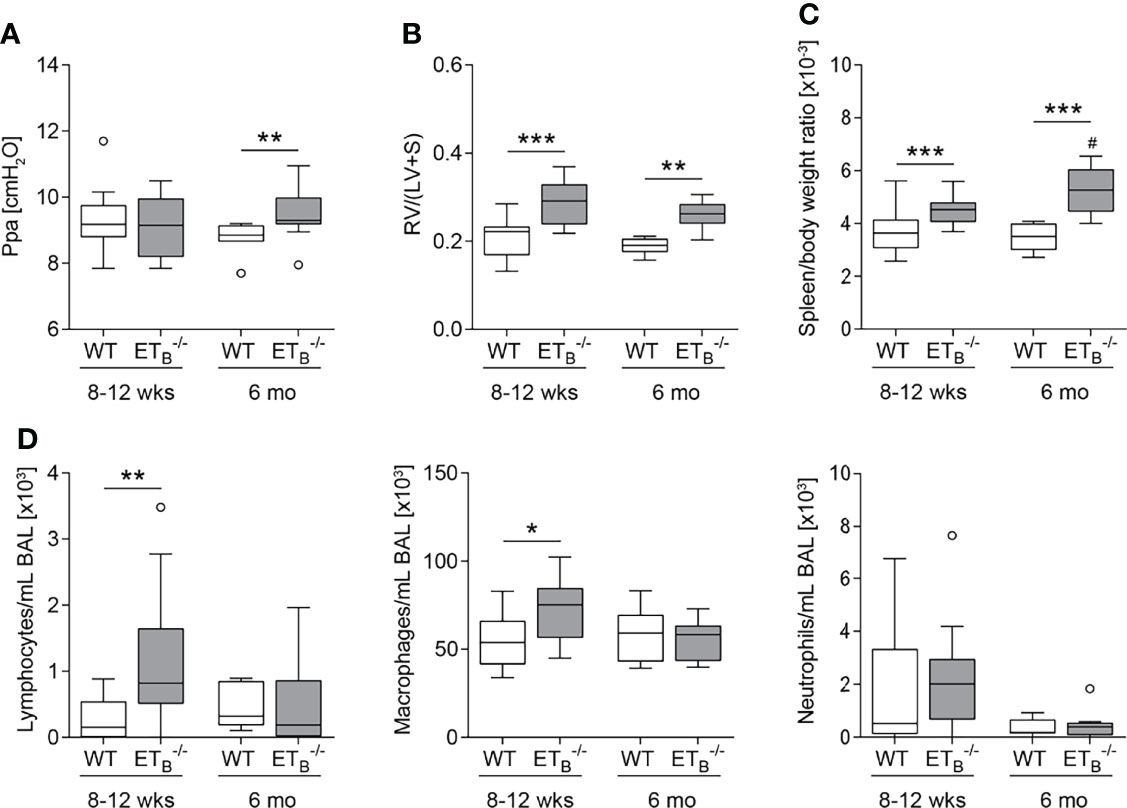
Figure 3 ETB deficiency was age-dependently associated with increased pulmonary arterial pressure, right ventricular hypertrophy, splenomegaly, and increased number of lymphocytes in bronchoalveolar lavage. Lungs, hearts, and spleens of 8- to 12-week (wk)-old and 6-mo-old rescued endothelin B receptor-deficient (ETB-/-) and corresponding wild-type (WT) mice were removed, or bronchoalveolar lavage (BAL) was performed. (A) In isolated perfused and ventilated lungs, under basal conditions, pulmonary arterial pressure (Ppa) was increased in 6-mo-old ETB-/- compared to WT mice of the same age. (B) Fulton index [ratio of right ventricle (RV) and left ventricle (LV) including septum (S)] determined after weighing the cardiac compartments was higher in ETB-/- compared to WT mice. (C) Determination of spleen weight related to body weight revealed splenomegaly in ETB-/- mice. (D) Analysis of differentially quantified leukocytes in BAL revealed increased number of lymphocytes and macrophages in BAL from 8- to 12-wk-old ETB-/- vs. WT mice of the same age. Data are represented as box plots depicting median, quartiles, and ranges excluding outliers (open circles). In (A–C), N = 7–28; in (D), N = 7–17. # indicates significant difference in the 6-mo-old vs. the corresponding 8- to 12-wk-old group, * indicates significant difference between ETB-/- vs. the corresponding WT group. */#p < 0.05, **p < 0.01, ***p < 0.001 (Mann–Whitney U test).
Basal dynamic lung compliance was found to be reduced in ETB-/- compared to corresponding WT mice in both age groups (Supplementary Figure 2B), in contrast to preETtg mice, which showed unaltered dynamic lung compliance (Supplementary Figure 3).
Perivascular Lymphoid Infiltrates in ETB-/- Lungs
Compared to WT controls, ETB-/- mice developed marked perivascular lymphocytic infiltrates (Supplementary Figure 4). In ETB-/- mice, perivascular lymphocytic infiltrates were particularly observed in the peripheral lung tissue, which were absent from WT lungs (Figure 4A). These infiltrates mostly consisted of B cells (Figure 4B) and T cells (Figure 4C). Both the prevalence and number of these cell clusters increased with age, and infiltrates were present in almost all (14/15) >16 months old ETB-/- mice (Figure 4D). Such perivascular cell clusters adjacent to small pulmonary arteries were absent in preETtg mice (Supplementary Figure 5) pointing to an immunomodulatory role of ETB in the lungs in the context of pulmonary hypertension.
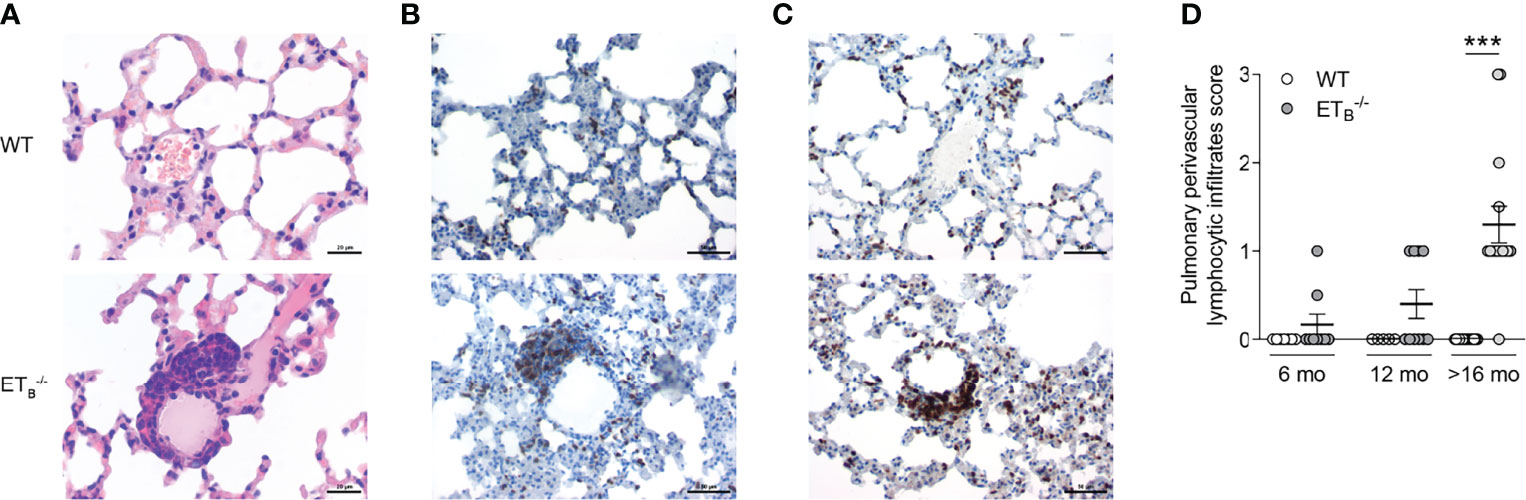
Figure 4 ETB deficiency was associated with peripheral perivascular lymphocytic infiltrates in the lung. Lungs of 6-, 12-, and >16-mo-old rescued endothelin B receptor-deficient (ETB-/-) and the corresponding wild-type (WT) mice were assessed histologically following hematoxylin and eosin (H&E) stain (A) or immunohistochemical stains for CD45R/B220 (B cells; B) or CD3 (T cells; C). The scale bars represent 20 µm (A) or 50 µm (B, C). Representative images of ≥12-mo-old mice are shown; N = 20–25 per group (A) or N = 3–5 per group (B, C). (D) H&E-stained lung sections were analyzed and scored (0, no peripheral perivascular infiltrates; 1, mild; 2 moderate; 3, pronounced) by an independent board-certified pathologist, blinded to the study groups. Data are expressed as single values with mean ± SEM; N = 7–9 (6-mo-old group), N = 5–10 (12-mo-old group), or N = 15 (>16-mo-old group). ***p < 0.001 (Mann–Whitney U test).
Th2 Inflammation Aggravates PAH-Associated Pathologies in ETB-/- Lungs
Next, we studied the effects of pulmonary Th2 inflammation as a second inflammatory hit in ETB-/- mice. Pulmonary Th2 inflammation was induced via systemic ovalbumin sensitization and ovalbumin airway exposure (OVA/OVA).
Pulmonary arterial pressure was elevated in ETB-/- compared to WT mice as described before, independent of pulmonary Th2 inflammation (Figure 5A). Following OVA/OVA treatment, pulmonary vascular hyperresponsiveness to ET-1 and U46619 was highly increased in ETB-/- mice compared to WT mice (Figure 5A). Pulmonary vascular hyperresponsiveness to serotonin, however, was not increased in ETB-/- as compared to WT mice (Supplementary Figure 6A).
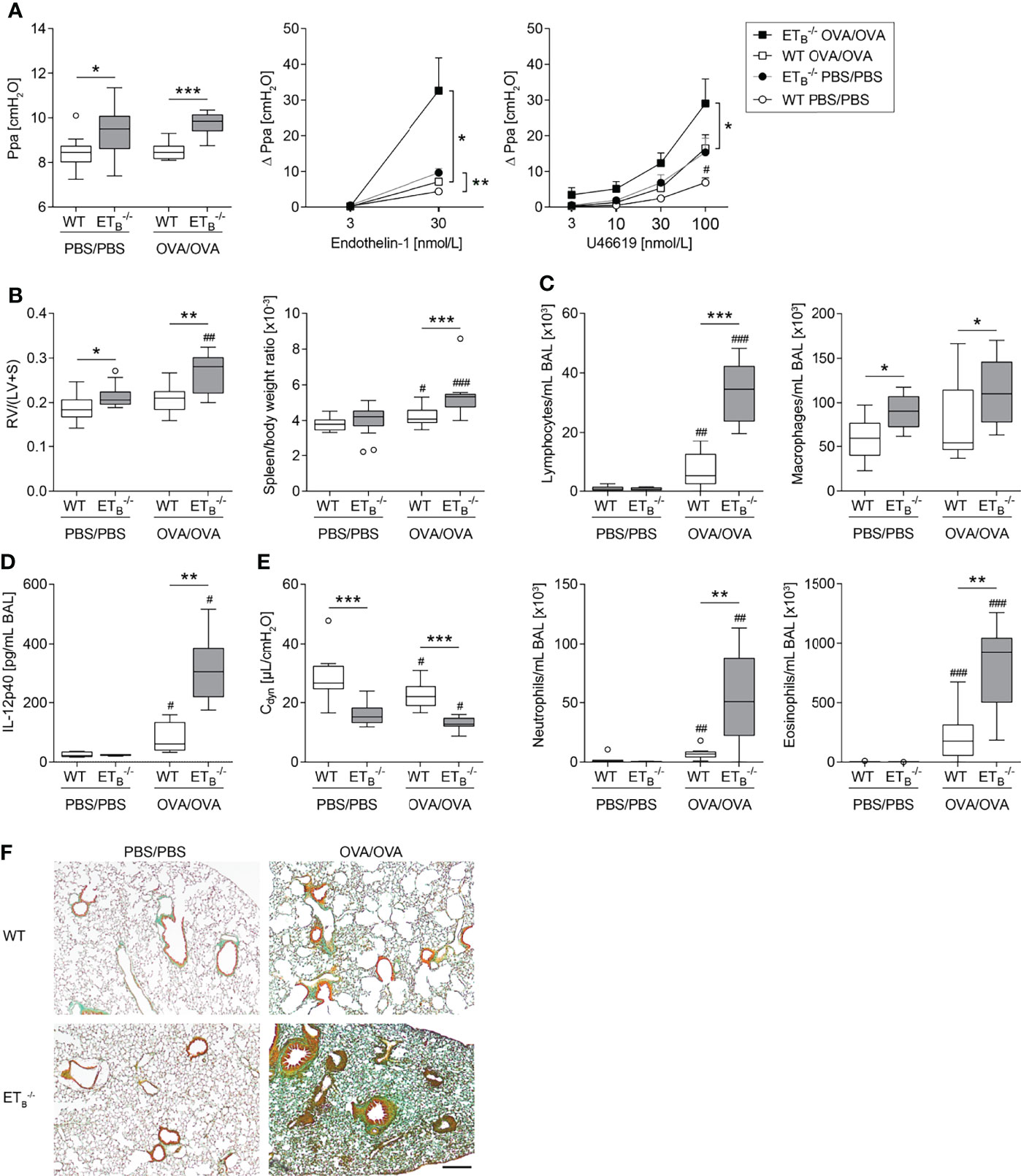
Figure 5 ETB deficiency aggravated Th2-mediated vascular pathologies and inflammation in the lung. Rescued endothelin B receptor-deficient (ETB-/-) and corresponding wild-type (WT) mice were systemically sensitized with ovalbumin (OVA) (or PBS as control) and repeatedly exposed to aerosolized OVA (OVA/OVA) or PBS (PBS/PBS). Forty-eight hours after the last challenge, lungs, hearts, and spleens of 12-wk-old mice were harvested, or bronchoalveolar lavage (BAL) was performed. (A) In isolated perfused and ventilated lungs, pulmonary arterial pressure (Ppa) was measured under basal conditions, and pulmonary vascular responsiveness to increasing concentrations of endothelin-1 or thromboxane receptor agonist U46619 was determined. Data (Δ Ppa) represent the difference between the highest pressure response to the respective stimulus and the basal Ppa. (B) Fulton index [quotient of right ventricle (RV) and left ventricle (LV) including septum (S)] was determined after weighing the cardiac compartments (left) and spleen weight was determined and related to body weight (right). (C) Leukocytes were differentially quantified in BAL. (D) IL-12p40 was determined in BAL (lower detection limit was 0.54 pg/mL). (E) In isolated perfused and ventilated mouse lungs, dynamic lung compliance (Cdyn) was measured. (F) Lung tissue sections were stained with Masson–Goldner trichrome that revealed more pronounced pulmonary collagen deposition in ETB-/- than WT mice after OVA/OVA treatment. The scale bar represents 100 µm and is valid for all photomicrographs. Representative images (N = 7 per group) are shown. In (A left, B–E), data are represented as box plots depicting median, quartiles, and ranges excluding outliers (open circles), and analyzed using Mann–Whitney U test. # indicates significant difference between OVA/OVA vs. the corresponding PBS/PBS group, * indicates significant difference between ETB-/- vs. the corresponding WT group. In (A middle-right), values are given as mean and SEM, and analyzed using two-way repeated measures ANOVA (*). In (A right), additional Mann–Whitney U test was performed comparing values of ETB-/- and WT mice treated with PBS at the highest dose of U46619 (#). N = 5–14 (A–C, E) or N = 3–6 (D). */#p < 0.05, **/##p < 0.01, ***/###p < 0.001.
Importantly, pulmonary Th2 inflammation aggravated right ventricular hypertrophy as well as splenomegaly in ETB-/- mice compared to WT controls (Figure 5B). Liver weight in relation to body weight was comparable in all groups (Supplementary Figure 6B).
The Th2-mediated inflammatory cell influx into the lung was increased in ETB-/- mice as reflected by elevated BAL cell numbers including lymphocytes, neutrophils, and eosinophils (Figure 5C) and as revealed by more pronounced perivascular leukocyte infiltrates in ETB-/- lungs (Supplementary Figure 7). While BAL Th2 cytokines IL-4, IL-5, and IL-13 in OVA/OVA-treated ETB-/- mice were comparable with the respective WT mice (Supplementary Table 2), IL-12 subunit p40 (IL-12p40) levels were greatly increased in BAL of ETB-/- mice (Figure 5D).
ETB Deficiency Aggravates Th2-Mediated Collagen Deposition in the Lung
Th2 immune responses have been associated with pulmonary collagen deposition (59) and IL-12p40 is believed to possess profibrotic properties in the lung (60). Dynamic lung compliance was lowest in ETB-/- mice after induction of pulmonary Th2 inflammation (Figure 5E). Accordingly, histological analyses of Masson–Goldner trichrome-stained lung slices revealed more pronounced collagen deposition in ETB-/- lungs than in WT lungs following OVA/OVA treatment (Figure 5F).
ETB Mediates Thromboxane Release Evoked by ET-1
To mechanistically dissect the pronounced increase in pulmonary vascular responsiveness to ET-1 in ETB-/- mice (Figure 5A), we indirectly assessed pulmonary vascular TXA2 release in isolated mouse lungs via quantification of stable TXB2 in the perfusate before and after intravascular application of ET-1. Indeed, vascular thromboxane release following ET-1 application was highly elevated in ETB-/- lungs (Figure 6A).
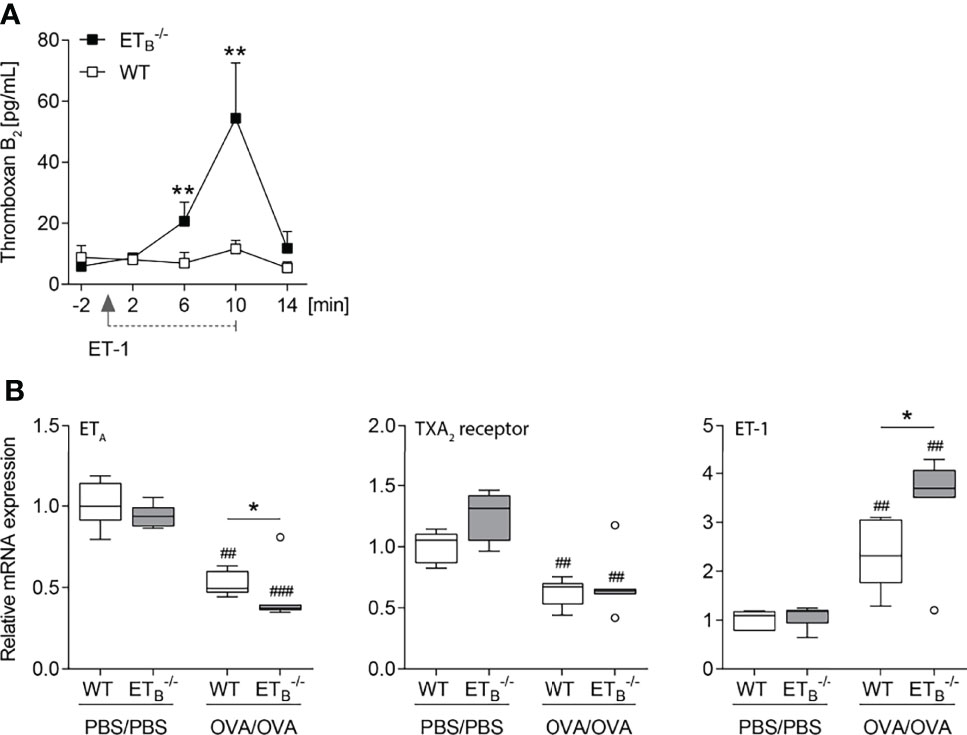
Figure 6 ET-1-mediated thromboxane release is increased in ETB-/- mice. (A) In isolated perfused lungs of 10- to 12-wk-old rescued endothelin B receptor-deficient (ETB-/-) and corresponding wild-type (WT) mice, perfusate samples were collected 2 min before application of 100 nmol/L ET-1 (for 10 min), and 2, 6, 10, and 14 min after the start of ET-1 application, and TXB2 levels were determined. The detection limit was 7.8 pg/ml. Values are given as mean and SEM (N = 7), and analyzed using Mann–Whitney U tests at each time point comparing both groups. (B) ETB-/- and corresponding WT mice were systemically sensitized with OVA (or PBS as control) and repeatedly exposed to aerosolized OVA (OVA/OVA) or PBS (PBS/PBS). Forty-eight hours after the last inhalative OVA challenge, lungs of 15- to 17-wk-old mice were isolated for mRNA expression analyses by quantitative PCR. Relative quantification of mRNA was performed using the comparative Ct method. Data are represented as box plots depicting median, quartiles, and ranges excluding outliers (open circles), and analyzed using Mann–Whitney U test; N = 6–7 per group. # indicates significant difference between OVA/OVA vs. the corresponding PBS/PBS group, * indicates significant difference between ETB-/- vs. the corresponding WT group. *p < 0.05, **/##p < 0.01, ###p < 0.001.
Increased vascular hyperresponsiveness secondary to ETA and/or TXA2 receptor upregulation, however, was ruled out via mRNA expression analyses. In ETB-/- lungs, ETA mRNA expression was downregulated while TXA2 receptor mRNA expression was comparable (Figure 6B). Of note, rescued ETB deficiency led to increased pulmonary ET-1 expression following induction of pulmonary Th2 inflammation (Figure 6B).
To investigate a potential pathomechanistic link between the endothelin system in pulmonary Th2 inflammation and the VIP, VIP plasma levels were quantified. Neither rescued ETB deficiency nor OVA/OVA treatment had an effect on VIP plasma levels (Supplementary Figure 8).
Discussion
In our study, we evaluated anti-ETB AAb in PAH patients for the first time and found increased levels in SSc-PAH patients. Furthermore, we characterized the immunomodulatory role of ETB in the context of PAH using a mouse model of PAH due to rescued ETB deficiency. Our data point to an important role of ETB in immune homeostasis, with functional ETB deficiency unleashing PAH development under inflammatory conditions.
ETB deficiency is associated with defective ET-1 clearance (27, 41–43), and increased levels of plasma ET-1 (44–46). In order to distinguish ET-1- and ETB-dependent effects in ETB-/- mice, we studied a second transgenic mouse model in parallel, namely, prepro-endothelin-1 overexpressing (preETtg) mice.
Here, we show that pulmonary hypertension, pulmonary vascular hyperresponsiveness, and right ventricular hypertrophy were present in preETtg as well as in ETB-/- mice, arguing for ET-1-specific effects. In preETtg mice, both pulmonary hypertension and right ventricular hypertrophy, however, were exclusively detected in highly aged (≥16 months old) mice, possibly as a result of decreasing NO-mediated compensatory effects with increasing age (61). In contrast, in ETB-/- mice, PAH-associated alterations were generally observed at a younger age than in preETtg mice, which may be the result of synergistic unfavorable effects of ETB deficiency and consecutive defective clearance of ET-1. Yet, as opposed to the characteristic findings in PAH patients, relevant pulmonary arterial remodeling was absent in both transgenic mouse lines, suggesting that the observed pulmonary hypertensive phenotypes are primarily driven by an increased vascular tone due to an imbalance of vasoconstrictive and vasodilatory mechanisms, rather than by extensive vascular remodeling in the pulmonary circulation.
Increased numbers of lymphocytes were found in BAL of both transgenic mouse models, again pointing to ET-1 as causative trigger. This finding is in line with previously described chronic lymphocytic lung inflammation as a result of prepro-ET-1 overexpression (58).
Of note, pronounced peripheral perivascular cluster of lymphoid infiltrates were exclusively found in ETB-/- lungs, arguing for a dysregulation of the immune system due to the loss of ETB. The perivascular space is a unique lung compartment, which might have been underestimated (62). Capillaries as well as lymphatic vessels from the periphery are found in the perivascular space, predominantly around the pulmonary arteries. This compartment is rather inactive in healthy lungs, but gains major significance in many types of lung inflammation. It is hypothesized that in certain conditions, inflammatory mediators induce extravasation of fluid and leukocytes from the periarterial capillaries, leading to thick cellular cuffs around the pulmonary arteries (63, 64). This defense mechanism may contribute to secondary lesions or processes such as the development of tertiary lymphoid tissue.
The data presented here give a strong hint that ETB is involved in the regulation of perivascular infiltration of pulmonary arteries. This finding is of specific interest as perivascular infiltrates and lymphoid tissue are commonly found in lungs of PAH patients and in preclinical models of pulmonary hypertension (2, 9–12).
As previously shown by us and others, induction of pulmonary Th2 inflammation in mice induces relevant PAH-associated features such as perivascular inflammation, hyperresponsiveness of the pulmonary vasculature to vasoconstrictive stimuli, complex pulmonary arterial remodeling, and increase in right ventricular systolic pressure (2, 16–23). Importantly, a key role of Th2 inflammation in the pathogenesis of pulmonary hypertension has been demonstrated in lung-specific IL-13-overexpressing mice, which develop spontaneous pulmonary hypertension, pulmonary arterial remodeling, and right ventricular hypertrophy (22). Interestingly, also in Fra-2 transgenic mice, a well-described model of SSc-PAH and interstitial lung disease, a strong underlying Th2 phenotype is present (56, 65).
Here, we analyzed the effects of pulmonary Th2 inflammation as a second hit in ETB-/- mice. Notably, both pulmonary vascular hyperresponsiveness and right ventricular hypertrophy were aggravated secondary to Th2 inflammation in ETB-/- mice. Moreover, in ETB-/- lungs, pulmonary perivascular inflammation and collagen deposition were increased.
On the cytokine level, IL-12 subunit p40 (IL-12p40) levels were largely increased in BAL of ETB-/- mice, whereas Th2 cytokines were similar in ETB-/- and WT mice. Increased IL-12p40 levels are notable with respect to the exaggerated PAH phenotype in ETB-/- mice as well as the increased pulmonary collagen deposition, since PAH patients show elevated levels of circulating IL-12p40 (12). Analogously, IL-12p40 serum levels are increased in mice deficient for chemokine receptor CCR7, which develop pulmonary hypertension, pulmonary arterial remodeling, and perivascular lymphoid infiltrates in the lung (12). Moreover, IL-12p40 has been identified as a central profibrotic mediator in murine lung fibrosis (60).
Pulmonary vascular hyperresponsiveness to the stimuli ET-1 and TXA2 analog U46619 was shown to be aggravated in ETB-/- lungs compared to the WT lungs. Interestingly, pulmonary vascular hyperresponsiveness to serotonin was unchanged in ETB-/- lungs, arguing for stimulus-specific alterations of the here assessed vasomotor responses. Mechanistically, ET-1-evoked hyperresponsiveness was most likely based on increased TXA2 release following vascular ET-1 application in ETB-/- mice, as indicated here by the elevated TXB2 perfusate levels.
Increased responsiveness as a result of ETA and/or TXA2 receptor upregulation, however, was ruled out in this study. In fact, ETA was downregulated in ETB-/- lungs following induction of Th2 inflammation, possibly as a counter-response to the increase in local ET-1 expression in ETB-/- lungs. Taken together, both the upregulation of ET-1 expression and the increased release of IL-12p40 may have contributed to the here observed exaggeration of the PAH phenotype in ETB-/- mice following induction of pulmonary Th2 inflammation.
It can be assumed that the anti-inflammatory effects of ETB signaling described in this study are primarily mediated via ETB receptor activation on vascular and inflammatory cells. In contrast, secondary immunomodulatory effects in response to ETB-regulated sodium and water reabsorption in the kidney are unlikely to underlie the detected anti-inflammatory properties of ETB in the lung. Specifically, ETB exerts natriuretic functions (66), and collecting duct-specific deficiency of ETB accordingly causes systemic hypertension with decreased urinary aldosterone excretion and plasma renin activity (67). Reduced activation of the renin–angiotensin–aldosterone system is, however, characteristically associated with mitigated inflammation (68). The same holds true for natriuretic peptides, which are abundantly released upon volume expansion (69) and have protective immunomodulatory properties (70). These anti-inflammatory effects of renal ETB deficiency stand in contrast to the pro-inflammatory effects in the lung detected in our study. Therefore, the anti-inflammatory role of ETB in the lung seems unrelated to its natriuretic function.
Our finding that Th2 inflammation as a second hit augments hallmarks of PAH is in line with previous findings in mice expressing a hypomorphic bone morphogenetic protein receptor type 2 (BMPR2) transgene, which showed an increase in right ventricular systolic pressure following induction of a Th2 immune response (23). As Th2-mediated aggravation of PAH phenotypes has been repeatedly shown, it is tempting to speculate that mediators of Th2 signaling may serve as potential targets in PAH. This needs to be evaluated in further studies.
PAH may occur in SSc patients with or without accompanying interstitial lung disease and/or digital ulcers (71, 72). The endothelin system is believed to play a central role in SSc-PAH, and SSc-PAH patients benefit from ERA treatment (24, 71, 72). ERAs are further indicated to treat SSc-related digital ulcers (72). Additional SSc-associated complications seem to involve the endothelin system. Alveolitis is frequently observed in SSc patients (73, 74). Intriguingly, alveolitis was also experimentally induced in mice treated with anti-ETA AAb and anti-angiotensin II type 1 receptor (AT1R) AAb-positive IgG derived from SSc patients (75). Moreover, expression of type I collagen in fibroblasts following treatment with IgG from SSc patients correlated with anti-ETA AAb levels, suggesting an underlying role of the endothelin system in SSc-associated fibrosis (75), as also discussed elsewhere (76).
The immunomodulatory actions of ETB shown here may be of relevance for the early phase of PAH, in which inflammation, endothelial dysfunction, and hyperresponsiveness of the pulmonary vasculature are believed to play relevant mechanistic roles. In the chronic disease state, reflected by profound remodeling of the pulmonary arteries, ETB, however, may play a less prominent role, as indicated by the fact that ETA-selective blockers do not appear to be superior to dual ETA/ETB therapy in established PAH (77, 78). Prospective clinical trials comparing selective ERA (inhibition of ETA) head-to-head against dual ERA (combined inhibition of ETA/ETB) therapy at early disease time points may be required to identify a potential advantage of selective ERA therapy in PAH.
In conclusion, our data show an anti-inflammatory role of ETB. ETB deficiency as a single hit is associated with spontaneous formation of marked lymphoid infiltrates in the perivascular space of the lung, in addition to pulmonary hypertension, pulmonary vascular hyperresponsiveness, and right ventricular hypertrophy. Th2 inflammation as a second hit aggravates PAH-associated pathologies in ETB-/- mice. The pathogenic role of anti-ETB AAb in SSc-PAH needs to be evaluated in further studies.
Data Availability Statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Ethics Statement
The studies involving human participants were reviewed and approved by the ethics committee (Charité - Universitätsmedizin Berlin, Germany; EA1/179/17). The patients/participants provided their written informed consent to participate in this study. The animal study was reviewed and approved by institutional authorities of the Charité – Universitätsmedizin Berlin, Germany, and the Local State Office of Health and Social Affairs Berlin (LAGeSo; Berlin, Germany).
Author Contributions
CT and MW conceived and designed the research. CT, CG, TT, OK, BG, JN, and JHe performed experiments. CT, CG, JL, JHö, TT, OK, BG, JN, BO, AG, HH, ES, and MW analyzed data. All authors interpreted the results of the experiments. CT, CG, JL, JHö, TT, OK, and ES prepared figures. CT and JL drafted the manuscript. All authors edited and revised the manuscript and approved the final version of the manuscript.
Funding
This study received funding from the German Research Foundation (SFB-TR84, C6/C9 to MW and Z01b to AG) and Actelion Pharmaceuticals Germany GmbH. The funders were not involved in the study design, collection, analysis, interpretation of data, the writing of this article or the decision to submit it for publication.
Conflict of Interest
CT received funding for research from Deutsche Gesellschaft für Pneumologie, Bayer HealthCare, Boehringer Ingelheim, and for lectures from Actelion Pharmaceuticals, Boehringer Ingelheim. HH is CEO of CellTrend GmbH, Luckenwalde, Germany. MW received funding for research from Deutsche Forschungsgemeinschaft, Bundesministerium für Bildung und Forschung, Deutsche Gesellschaft für Pneumologie, European Respiratory Society, Marie Curie Foundation, Else Kröner Fresenius Stiftung, CAPNETZ STIFTUNG, International Max Planck Research School, Actelion, Bayer Health Care, Biotest AG, Boehringer Ingelheim, NOXXON Pharma, Pantherna, Quark Pharma, Silence Therapeutics, Vaxxilon, and for lectures and advisory from Actelion, Alexion, Aptarion, Astra Zeneca, Bayer Health Care, Berlin Chemie, Biotest, Boehringer Ingelheim, Chiesi, Glaxo Smith Kline, Insmed, Novartis, Teva and Vaxxilon.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
The authors would like to thank Udo Schneider, Daniel Grund, and Vincent Casteleyn for their support on patient recruitment. The technical assistance of Katharina Krause-Relle is greatly appreciated. The present study is part of the doctoral thesis of CG. CT and ES are participants in the BIH-Charité Clinician Scientist Program funded by the Charité - Universitätsmedizin Berlin and the Berlin Institute of Health.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2022.895501/full#supplementary-material
Abbreviations
AT1R, angiotensin II type 1 receptor; AAb, autoantibody; BAL, bronchoalveolar lavage; BMPR2, bone morphogenetic protein receptor type 2; Cdyn, dynamic lung compliance; ET-1, endothelin-1; ETA, endothelin A receptor; ETB, endothelin B receptor; ERA, endothelin receptor antagonist; H&E, hematoxylin and eosin; IgG, immunoglobulin G; IL, interleukin; IL-12p40, IL-12 subunit p40; NO, nitric oxide; OVA, ovalbumin; OVA/OVA, OVA-sensitized and OVA-challenged; PAH, pulmonary arterial hypertension; PBS, phosphate buffered saline; PBS/PBS, non-sensitized and non-challenged (PBS-treated); PASMCs, pulmonary arterial smooth muscle cells; Ppa, pulmonary arterial pressure; SSc, systemic sclerosis; Th2, T-helper type 2; TNF-α, tumor necrosis factor alpha; TXA2, thromboxane A2; TXB2, thromboxane B2; VIP, vasoactive intestinal peptide.
References
2. Price LC, Wort SJ, Perros F, Dorfmüller P, Huertas A, Montani D, et al. Inflammation in Pulmonary Arterial Hypertension. Chest (2012) 141(1):210–21. doi: 10.1378/chest.11-0793
3. Hassoun PM, Mouthon L, Barberà JA, Eddahibi S, Flores SC, Grimminger F, et al. Inflammation, Growth Factors, and Pulmonary Vascular Remodeling. J Am Coll Cardiol (2009) 54(1 Suppl):S10–9. doi: 10.1016/j.jacc.2009.04.006
4. Huertas A, Tu L, Humbert M, Guignabert C. Chronic Inflammation Within the Vascular Wall in Pulmonary Arterial Hypertension: More Than a Spectator. Cardiovasc Res (2020) 116(5):885–93. doi: 10.1093/cvr/cvz308
5. Kherbeck N, Tamby MC, Bussone G, Dib H, Perros F, Humbert M, et al. The Role of Inflammation and Autoimmunity in the Pathophysiology of Pulmonary Arterial Hypertension. Clin Rev Allergy Immunol (2013) 44(1):31–8. doi: 10.1007/s12016-011-8265-z
6. Le Pavec J, Humbert M, Mouthon L, Hassoun PM. Systemic Sclerosis-Associated Pulmonary Arterial Hypertension. Am J Respir Crit Care Med (2010) 181(12):1285–93. doi: 10.1164/rccm.200909-1331PP
7. Dorfmüller P, Perros F, Balabanian K, Humbert M. Inflammation in Pulmonary Arterial Hypertension. Eur Respir J (2003) 22(2):358–63. doi: 10.1183/09031936.03.00038903
8. Soon E, Holmes AM, Treacy CM, Doughty NJ, Southgate L, Machado RD, et al. Elevated Levels of Inflammatory Cytokines Predict Survival in Idiopathic and Familial Pulmonary Arterial Hypertension. Circulation (2010) 122(9):920–7. doi: 10.1161/CIRCULATIONAHA.109.933762
9. Hu Y, Chi L, Kuebler WM, Goldenberg NM. Perivascular Inflammation in Pulmonary Arterial Hypertension. Cells (2020) 9(11):2338. doi: 10.3390/cells9112338
10. Colvin KL, Cripe PJ, Ivy DD, Stenmark KR, Yeager ME. Bronchus-Associated Lymphoid Tissue in Pulmonary Hypertension Produces Pathologic Autoantibodies. Am J Respir Crit Care Med (2013) 188(9):1126–36. doi: 10.1164/rccm.201302-0403OC
11. Perros F, Dorfmüller P, Montani D, Hammad H, Waelput W, Girerd B, et al. Pulmonary Lymphoid Neogenesis in Idiopathic Pulmonary Arterial Hypertension. Am J Respir Crit Care Med (2012) 185(3):311–21. doi: 10.1164/rccm.201105-0927OC
12. Larsen KO, Yndestad A, Sjaastad I, Løberg EM, Goverud IL, Halvorsen B, et al. Lack of CCR7 Induces Pulmonary Hypertension Involving Perivascular Leukocyte Infiltration and Inflammation. Am J Physiol Lung Cell Mol Physiol (2011) 301(1):L50–9. doi: 10.1152/ajplung.00048.2010
13. Harbaum L, Renk E, Yousef S, Glatzel A, Lüneburg N, Hennigs JK, et al. Acute Effects of Exercise on the Inflammatory State in Patients With Idiopathic Pulmonary Arterial Hypertension. BMC Pulm Med (2016) 16(1):145. doi: 10.1186/s12890-016-0301-6
14. Heath D, Yacoub M. Lung Mast Cells in Plexogenic Pulmonary Arteriopathy. J Clin Pathol (1991) 44(12):1003–6. doi: 10.1136/jcp.44.12.1003
15. Hamada H, Terai M, Kimura H, Hirano K, Oana S, Niimi H. Increased Expression of Mast Cell Chymase in the Lungs of Patients With Congenital Heart Disease Associated With Early Pulmonary Vascular Disease. Am J Respir Crit Care Med (1999) 160(4):1303–8. doi: 10.1164/ajrccm.160.4.9810058
16. Kumar R, Mickael C, Chabon J, Gebreab L, Rutebemberwa A, Garcia AR, et al. The Causal Role of Il-4 and Il-13 in Schistosoma Mansoni Pulmonary Hypertension. Am J Respir Crit Care Med (2015) 192(8):998–1008. doi: 10.1164/rccm.201410-1820OC
17. Haberberger RV, Tabeling C, Runciman S, Gutbier B, König P, Andratsch M, et al. Role of Sphingosine Kinase 1 in Allergen-Induced Pulmonary Vascular Remodeling and Hyperresponsiveness. J Allergy Clin Immunol (2009) 124(5):933–41.e1-9. doi: 10.1016/j.jaci.2009.06.034
18. Witzenrath M, Ahrens B, Kube SM, Hocke AC, Rosseau S, Hamelmann E, et al. Allergic Lung Inflammation Induces Pulmonary Vascular Hyperresponsiveness. EurRespirJ (2006) 28(2):370–7. doi: 10.1183/09031936.06.00080105
19. Daley E, Emson C, Guignabert C, de Waal MR, Louten J, Kurup VP, et al. Pulmonary Arterial Remodeling Induced by a Th2 Immune Response. J Exp Med (2008) 205(2):361–72. doi: 10.1084/jem.20071008
20. Gomez-Arroyo J, Saleem SJ, Mizuno S, Syed AA, Bogaard HJ, Abbate A, et al. A Brief Overview of Mouse Models of Pulmonary Arterial Hypertension: Problems and Prospects. Am J Physiol Lung Cell Mol Physiol (2012) 302(10):L977–91. doi: 10.1152/ajplung.00362.2011
21. Mushaben EM, Hershey GK, Pauciulo MW, Nichols WC, Le Cras TD. Chronic Allergic Inflammation Causes Vascular Remodeling and Pulmonary Hypertension in BMPR2 Hypomorph and Wild-Type Mice. PloS One (2012) 7(3):e32468. doi: 10.1371/journal.pone.0032468
22. Cho WK, Lee CM, Kang MJ, Huang Y, Giordano FJ, Lee PJ, et al. Il-13 Receptor α2-Arginase 2 Pathway Mediates Il-13-Induced Pulmonary Hypertension. Am J Physiol Lung Cell Mol Physiol (2013) 304(2):L112–24. doi: 10.1152/ajplung.00101.2012
23. Park SH, Chen WC, Hoffman C, Marsh LM, West J, Grunig G. Modification of Hemodynamic and Immune Responses to Exposure With a Weak Antigen by the Expression of a Hypomorphic BMPR2 Gene. PloS One (2013) 8(1):e55180. doi: 10.1371/journal.pone.0055180
24. McLaughlin V, Humbert M, Coghlan G, Nash P, Steen V. Pulmonary Arterial Hypertension: The Most Devastating Vascular Complication of Systemic Sclerosis. Rheumatol (Oxford) (2009) 48(Suppl 3):iii25–31. doi: 10.1093/rheumatology/kep107
25. Giaid A, Yanagisawa M, Langleben D, Michel RP, Levy R, Shennib H, et al. Expression of Endothelin-1 in the Lungs of Patients With Pulmonary Hypertension. N Engl J Med (1993) 328(24):1732–9. doi: 10.1056/NEJM199306173282402
26. Dhaun N, Webb DJ. Endothelins in Cardiovascular Biology and Therapeutics. Nat Rev Cardiol (2019) 16(8):491–502. doi: 10.1038/s41569-019-0176-3
27. Davenport AP, Hyndman KA, Dhaun N, Southan C, Kohan DE, Pollock JS, et al. Endothelin. Pharmacol Rev (2016) 68(2):357–418. doi: 10.1124/pr.115.011833
28. Stewart DJ, Levy RD, Cernacek P, Langleben D. Increased Plasma Endothelin-1 in Pulmonary Hypertension: Marker or Mediator of Disease? Ann Intern Med (1991) 114(6):464–9. doi: 10.7326/0003-4819-114-6-464
29. de Nucci G, Thomas R, D'Orleans-Juste P, Antunes E, Walder C, Warner TD, et al. Pressor Effects of Circulating Endothelin Are Limited by Its Removal in the Pulmonary Circulation and by the Release of Prostacyclin and Endothelium-Derived Relaxing Factor. Proc Natl Acad Sci USA (1988) 85(24):9797–800. doi: 10.1073/pnas.85.24.9797
30. Horgan MJ, Pinheiro JM, Malik AB. Mechanism of Endothelin-1-Induced Pulmonary Vasoconstriction. Circ Res (1991) 69(1):157–64. doi: 10.1161/01.res.69.1.157
31. Tabeling C, Noe E, Naujoks J, Doehn JM, Hippenstiel S, Opitz B, et al. PKCα Deficiency in Mice Is Associated With Pulmonary Vascular Hyperresponsiveness to Thromboxane A2 and Increased Thromboxane Receptor Expression. J Vasc Res (2015) 52(4):279–88. doi: 10.1159/000443402
32. Cabral-Marques O, Riemekasten G. Functional Autoantibodies Targeting G Protein-Coupled Receptors in Rheumatic Diseases. Nat Rev Rheumatol (2017) 13(11):648–56. doi: 10.1038/nrrheum.2017.134
33. Helset E, Sildnes T, Seljelid R, Konopski ZS. Endothelin-1 Stimulates Human Monocytes in Vitro to Release TNF-alpha, Il-1beta and Il-6. Mediat Inflamm (1993) 2(6):417–22. doi: 10.1155/S0962935193000596
34. Browatzki M, Schmidt J, Kübler W, Kranzhöfer R. Endothelin-1 Induces Interleukin-6 Release Via Activation of the Transcription Factor NF-kappaB in Human Vascular Smooth Muscle Cells. Basic Res Cardiol (2000) 95(2):98–105. doi: 10.1007/s003950050170
35. Becker MO, Kill A, Kutsche M, Guenther J, Rose A, Tabeling C, et al. Vascular Receptor Autoantibodies in Pulmonary Arterial Hypertension Associated With Systemic Sclerosis. Am J Respir Crit Care Med (2014) 190(7):808–17. doi: 10.1164/rccm.201403-0442OC
36. Blum LK, Cao RRL, Sweatt AJ, Bill M, Lahey LJ, Hsi AC, et al. Circulating Plasmablasts Are Elevated and Produce Pathogenic Anti-Endothelial Cell Autoantibodies in Idiopathic Pulmonary Arterial Hypertension. Eur J Immunol (2018) 48(5):874–84. doi: 10.1002/eji.201747460
37. Tamby MC, Humbert M, Guilpain P, Servettaz A, Dupin N, Christner JJ, et al. Antibodies to Fibroblasts in Idiopathic and Scleroderma-Associated Pulmonary Hypertension. Eur Respir J (2006) 28(4):799–807. doi: 10.1183/09031936.06.00152705
38. Tamby MC, Chanseaud Y, Humbert M, Fermanian J, Guilpain P, Garcia-de-la-Peña-Lefebvre P, et al. Anti-Endothelial Cell Antibodies in Idiopathic and Systemic Sclerosis Associated Pulmonary Arterial Hypertension. Thorax (2005) 60(9):765–72. doi: 10.1136/thx.2004.029082
39. Arends SJ, Damoiseaux JG, Duijvestijn AM, Debrus-Palmans L, Boomars KA, Brunner-La Rocca HP, et al. Functional Implications of IgG Anti-Endothelial Cell Antibodies in Pulmonary Arterial Hypertension. Autoimmunity (2013) 46(7):463–70. doi: 10.3109/08916934.2013.812080
40. Arends SJ, Damoiseaux J, Duijvestijn A, Debrus-Palmans L, Boomars K, Broers B, et al. Prevalence of Anti-Endothelial Cell Antibodies in Idiopathic Pulmonary Arterial Hypertension. Eur Respir J (2010) 35(4):923–5. doi: 10.1183/09031936.00164209
41. Kelland NF, Kuc RE, McLean DL, Azfer A, Bagnall AJ, Gray GA, et al. Endothelial Cell-Specific ETB Receptor Knockout: Autoradiographic and Histological Characterisation and Crucial Role in the Clearance of Endothelin-1. Can J Physiol Pharmacol (2010) 88(6):644–51. doi: 10.1139/Y10-041
42. Fukuroda T, Fujikawa T, Ozaki S, Ishikawa K, Yano M, Nishikibe M. Clearance of Circulating Endothelin-1 by ETB Receptors in Rats. Biochem Biophys Res Commun (1994) 199(3):1461–5. doi: 10.1006/bbrc.1994.1395
43. Dupuis J, Goresky CA, Fournier A. Pulmonary Clearance of Circulating Endothelin-1 in Dogs in Vivo: Exclusive Role of ETB Receptors. J Appl Physiol (1985) (1996) 81(4):1510–5. doi: 10.1152/jappl.1996.81.4.1510
44. Ivy DD, Yanagisawa M, Gariepy CE, Gebb SA, Colvin KL, McMurtry IF. Exaggerated Hypoxic Pulmonary Hypertension in Endothelin B Receptor-Deficient Rats. Am J Physiol Lung Cell Mol Physiol (2002) 282(4):L703–12. doi: 10.1152/ajplung.00272.2001
45. Gariepy CE, Ohuchi T, Williams SC, Richardson JA, Yanagisawa M. Salt-Sensitive Hypertension in Endothelin-B Receptor-Deficient Rats. J Clin Invest (2000) 105(7):925–33. doi: 10.1172/JCI8609
46. Bagnall AJ, Kelland NF, Gulliver-Sloan F, Davenport AP, Gray GA, Yanagisawa M, et al. Deletion of Endothelial Cell Endothelin B Receptors Does Not Affect Blood Pressure or Sensitivity to Salt. Hypertension (2006) 48(2):286–93. doi: 10.1161/01.HYP.0000229907.58470.4c
47. van den Hoogen F, Khanna D, Fransen J, Johnson SR, Baron M, Tyndall A, et al. Classification Criteria for Systemic Sclerosis: An American College of Rheumatology/European League Against Rheumatism Collaborative Initiative. Ann Rheum Dis (2013) 72(11):1747–55. doi: 10.1136/annrheumdis-2013-204424
48. Riemekasten G, Philippe A, Näther M, Slowinski T, Müller DN, Heidecke H, et al. Involvement of Functional Autoantibodies Against Vascular Receptors in Systemic Sclerosis. Ann Rheum Dis (2011) 70(3):530–6. doi: 10.1136/ard.2010.135772
49. Hocher B, Thöne-Reineke C, Rohmeiss P, Schmager F, Slowinski T, Burst V, et al. Endothelin-1 Transgenic Mice Develop Glomerulosclerosis, Interstitial Fibrosis, and Renal Cysts But Not Hypertension. J Clin Invest (1997) 99(6):1380–9. doi: 10.1172/JCI119297
50. Quaschning T, Rebhan B, Wunderlich C, Wanner C, Richter CM, Pfab T, et al. Endothelin B Receptor-Deficient Mice Develop Endothelial Dysfunction Independently of Salt Loading. J Hypertens (2005) 23(5):979–85. doi: 10.1097/01.hjh.0000166838.55688.7e
51. von Bethmann AN, Brasch F, Nüsing R, Vogt K, Volk HD, Müller KM, et al. Hyperventilation Induces Release of Cytokines From Perfused Mouse Lung. Am J Respir Crit Care Med (1998) 157(1):263–72. doi: 10.1164/ajrccm.157.1.9608052
52. Tabeling C, Yu H, Wang L, Ranke H, Goldenberg NM, Zabini D, et al. CFTR and Sphingolipids Mediate Hypoxic Pulmonary Vasoconstriction. Proc Natl Acad Sci USA (2015) 112(13):E1614–23. doi: 10.1073/pnas.1421190112
53. Tabeling C, Scheer H, Schönrock SM, Runge F, Gutbier B, Lienau J, et al. Nucleotide Oligomerization Domain 1 Ligation Suppressed Murine Allergen-Specific T-Cell Proliferation and Airway Hyperresponsiveness. Am J Respir Cell Mol Biol (2014) 50(5):903–11. doi: 10.1165/rcmb.2013-0333OC
54. Schulze T, Golfier S, Tabeling C, Räbel K, Gräler MH, Witzenrath M, et al. Sphingosine-1-Phospate Receptor 4 (S1P4) Deficiency Profoundly Affects Dendritic Cell Function and Th17-Cell Differentiation in a Murine Model. FASEB J (2011) 25(11):4024–36. doi: 10.1096/fj.10-179028
55. Heine G, Tabeling C, Hartmann B, González Calera CR, Kühl AA, Lindner J, et al. 25-Hydroxvitamin D3 Promotes the Long-Term Effect of Specific Immunotherapy in a Murine Allergy Model. J Immunol (2014) 193(3):1017–23. doi: 10.4049/jimmunol.1301656
56. Tabeling C, Wienhold SM, Birnhuber A, Brack MC, Nouailles G, Kershaw O, et al. Pulmonary Fibrosis in Fra-2 Transgenic Mice Is Associated With Decreased Numbers of Alveolar Macrophages and Increased Susceptibility to Pneumococcal Pneumonia. Am J Physiol Lung Cell Mol Physiol (2021) 320(5):L916–L25. doi: 10.1152/ajplung.00505.2020
57. Naujoks J, Tabeling C, Dill BD, Hoffmann C, Brown AS, Kunze M, et al. IFNs Modify the Proteome of Legionella-Containing Vacuoles and Restrict Infection Via IRG1-Derived Itaconic Acid. PloS Pathog (2016) 12(2):e1005408. doi: 10.1371/journal.ppat.1005408
58. Hocher B, Schwarz A, Fagan KA, Thöne-Reineke C, El Hag K, Kusserow H, et al. Pulmonary Fibrosis and Chronic Lung Inflammation in ET-1 Transgenic Mice. Am J Respir Cell Mol Biol (2000) 23(1):19–26. doi: 10.1165/ajrcmb.23.1.4030
59. Tabeling C, Herbert J, Hocke AC, Lamb DJ, Wollin SL, Erb KJ, et al. Spleen Tyrosine Kinase Inhibition Blocks Airway Constriction and Protects From Th2-Induced Airway Inflammation and Remodeling. Allergy (2017) 72(7):1061–72. doi: 10.1111/all.13101
60. Huaux F, Arras M, Tomasi D, Barbarin V, Delos M, Coutelier JP, et al. A Profibrotic Function of Il-12p40 in Experimental Pulmonary Fibrosis. J Immunol (2002) 169(5):2653–61. doi: 10.4049/jimmunol.169.5.2653
61. Kalk P, Mach A, Thone-Reineke C, Godes M, Heiden S, Sharkovska Y, et al. Pulmonary Fibrosis in L-NAME-Treated Mice Is Dependent on an Activated Endothelin System. Can J Physiol Pharmacol (2008) 86(8):541–5. doi: 10.1139/Y08-047
62. Pabst R, Tschernig T. Perivascular Capillaries in the Lung: An Important But Neglected Vascular Bed in Immune Reactions? J Allergy Clin Immunol (2002) 110(2):209–14. doi: 10.1067/mai.2002.126836
63. Lindsey AS, Sullivan LM, Housley NA, Koloteva A, King JA, Audia JP, et al. Analysis of Pulmonary Vascular Injury and Repair During Pseudomonas Aeruginosa Infection-Induced Pneumonia and Acute Respiratory Distress Syndrome. Pulm Circ (2019) 9(1):2045894019826941. doi: 10.1177/2045894019826941
64. Pabst R, Tschernig T. Periarterial Leukocyte Accumulation in Allergic Airway Inflammation. Am J Respir Crit Care Med (2006) 174(7):840. doi: 10.1164/ajrccm.174.7.840
65. Birnhuber A, Biasin V, Schnoegl D, Marsh LM, Kwapiszewska G. Transcription Factor Fra-2 and Its Emerging Role in Matrix Deposition, Proliferation and Inflammation in Chronic Lung Diseases. Cell Signal (2019) 64:109408. doi: 10.1016/j.cellsig.2019.109408
66. Zhang Y, Jose PA, Zeng C. Regulation of Sodium Transport in the Proximal Tubule by Endothelin. Contrib Nephrol (2011) 172:63–75. doi: 10.1159/000328684
67. Ge Y, Bagnall A, Stricklett PK, Strait K, Webb DJ, Kotelevtsev Y, et al. Collecting Duct-Specific Knockout of the Endothelin B Receptor Causes Hypertension and Sodium Retention. Am J Physiol Renal Physiol (2006) 291(6):F1274–80. doi: 10.1152/ajprenal.00190.2006
68. Pacurari M, Kafoury R, Tchounwou PB, Ndebele K. The Renin-Angiotensin-Aldosterone System in Vascular Inflammation and Remodeling. Int J Inflam (2014) 2014:689360. doi: 10.1155/2014/689360
69. McFarlane SI, Winer N, Sowers JR. Role of the Natriuretic Peptide System in Cardiorenal Protection. Arch Intern Med (2003) 163(22):2696–704. doi: 10.1001/archinte.163.22.2696
70. Ramakrishnan V, Burnett JC Jr. Natriuretic Peptides, Inflammation, and Sounding the Alarm. Circ Heart Fail (2020) 13(7):e007208. doi: 10.1161/CIRCHEARTFAILURE.120.007208
71. Naranjo M, Hassoun PM. Systemic Sclerosis-Associated Pulmonary Hypertension: Spectrum and Impact. Diagnostics (Basel) (2021) 11(5):911. doi: 10.3390/diagnostics11050911
72. Kowal-Bielecka O, Fransen J, Avouac J, Becker M, Kulak A, Allanore Y, et al. Update of EULAR Recommendations for the Treatment of Systemic Sclerosis. Ann Rheum Dis (2017) 76(8):1327–39. doi: 10.1136/annrheumdis-2016-209909
73. Manganelli P, Salaffi F, Pesci A. Clinical and Subclinical Alveolitis in Connective Tissue Diseases Assessed by Bronchoalveolar Lavage. Semin Arthritis Rheum (1997) 26(5):740–54. doi: 10.1016/s0049-0172(97)80042-x
74. Schmidt K, Martinez-Gamboa L, Meier S, Witt C, Meisel C, Hanitsch LG, et al. Bronchoalveoloar Lavage Fluid Cytokines and Chemokines as Markers and Predictors for the Outcome of Interstitial Lung Disease in Systemic Sclerosis Patients. Arthritis Res Ther (2009) 11(4):R111. doi: 10.1186/ar2766
75. Kill A, Tabeling C, Undeutsch R, Kühl AA, Gunther J, Radic M, et al. Autoantibodies to Angiotensin and Endothelin Receptors in Systemic Sclerosis Induce Cellular and Systemic Events Associated With Disease Pathogenesis. Arthritis Res Ther (2014) 16(1):R29. doi: 10.1186/ar4457
76. Grune J, Kuebler WM. Is There a Role for Endothelin-1 Receptor Antagonists in the Treatment of Lung Fibrosis Associated With Pulmonary Hypertension? Eur Respir J (2018) 52(2):1801287. doi: 10.1183/13993003.01287-2018
77. Vizza CD, Fedele F, Pezzuto B, Rubin LJ. Safety and Efficacy Evaluation of Ambrisentan in Pulmonary Hypertension. Expert Opin Drug Saf (2012) 11(6):1003–11. doi: 10.1517/14740338.2012.714770
Keywords: endothelin B receptor, autoantibody, Th2 inflammation, pulmonary vascular hyperresponsiveness, pulmonary arterial hypertension, systemic sclerosis
Citation: Tabeling C, González Calera CR, Lienau J, Höppner J, Tschernig T, Kershaw O, Gutbier B, Naujoks J, Herbert J, Opitz B, Gruber AD, Hocher B, Suttorp N, Heidecke H, Burmester GR, Riemekasten G, Siegert E, Kuebler WM and Witzenrath M (2022) Endothelin B Receptor Immunodynamics in Pulmonary Arterial Hypertension. Front. Immunol. 13:895501. doi: 10.3389/fimmu.2022.895501
Received: 13 March 2022; Accepted: 09 May 2022;
Published: 09 June 2022.
Edited by:
Rudolf Lucas, Augusta University, United StatesReviewed by:
Dustin Fraidenburg, University of Illinois at Chicago, United StatesRobert Naeije, Université libre de Bruxelles, Belgium
Copyright © 2022 Tabeling, González Calera, Lienau, Höppner, Tschernig, Kershaw, Gutbier, Naujoks, Herbert, Opitz, Gruber, Hocher, Suttorp, Heidecke, Burmester, Riemekasten, Siegert, Kuebler and Witzenrath. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Martin Witzenrath, martin.witzenrath@charite.de