
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Immunol. , 26 May 2022
Sec. Microbial Immunology
Volume 13 - 2022 | https://doi.org/10.3389/fimmu.2022.894069
This article is part of the Research Topic Immune Interactions with Pathogenic and Commensal Fungi View all 17 articles
 Jingyun Zhang1,2
Jingyun Zhang1,2 Jingwen Peng1,2
Jingwen Peng1,2 Dongmei Li3
Dongmei Li3 Huan Mei1,2Yu Yu1,2Xiaofang Li1,2
Huan Mei1,2Yu Yu1,2Xiaofang Li1,2 Xiaodong She1,2*
Xiaodong She1,2* Weida Liu1,2,4*
Weida Liu1,2,4*Vulvovaginal candidiasis (VVC) is characterized by symptomatic inflammatory responses in the vagina caused by Candida albicans and non-albicans Candida (NAC) species. The epidermal growth factor receptor (EGFR) -mitogen-activated protein kinase (MAPK) signaling pathway has been linked to immune responses of oral mucosa after C. albicans exposure, but whether this pathway plays a similar response in vaginal epithelial cells is not known. Here, we observed that phosphorylation of EGFR and p38 was continuously activated in vaginal epithelial cells by C. albicans strain SC5314. This differs markedly from oral epithelial cells, which respond in a biphasic manner in order to properly discriminate the morphology of C. albicans. When compared with SC5314, a highly azole-resistant C. albicans isolate 1052 can induce a stronger phosphorylated signal of EGFR and p38, while clinically-isolated NAC strains including C. tropicalis, C. glabrata, C. parapsilosis and C. auris trigger higher levels of phosphorylated ERK1/2 and c-Fos than C. albicans. Inhibition of EGFR significantly reduces inflammatory response and epithelial damage induced by C. albicans both in vitro and in vivo, while inhibition of p38 leads to significant repair of epithelial damage triggered by both C. albicans and NAC species. These results confirm the importance of the EGFR-MAPK signaling in VVC pathogenesis and highlight the remarkable immunogenic differences between C. albicans and NAC species in host-microbe interactions.
Vulvovaginal candidiasis (VVC) is the most common mucosal infection, characterized by symptomatic inflammation of the vagina (1). VVC has been reported to affect 70–75% of healthy women during their reproductive years, and about 5–8% of them evolve into recurrent VVC (RVVC) (2, 3). For Candida infection in the oral mucosa, both early innate immune responses and T cell-mediated adaptive immunity are critical and protective against the invasion of Candida pathogen (4–8). On the contrary, human challenge studies indicate that adaptive immune response is not defective and provides no protection upon Candida stimulation in both VVC and RVVC patients (9–11). Moreover, while an aggressive innate immune response is the primary determinant of the pathological process of VVC, fungal factors such as the presence of pseudo hyphae/hyphae are required but not sufficient to induce the pathological process of VVC (12). It seems that the hyperactive inflammatory response due to strong neutrophil recruitments and mucosal damage is responsible for symptoms of VVC (1). However, elevated neutrophil infiltration was demonstrated to be ineffective at reducing fungal burden and invasion during VVC (13–15), which appears to contradict the protective role of neutrophil recruitment in oral cavity or gastrointestinal tract for Candida clearance at the site of infection (6). Therefore, host mediated-epithelial damage and inflammatory response appear to be more important during the pathogenesis of VVC.
The mucosal epithelium not only functions as a physical barrier restraining commensal microbes, but also plays a critical role in orchestrating immune responses to activate myeloid cells in the submucosal layers to clear the invading pathogens (16, 17). The mechanism of how oral epithelial cells respond to candidal pathogens is well characterized (8, 18, 19) and epidermal growth factor receptor (EGFR) has also been shown to facilitate epithelial cell invasion via endocytosis of C. albicans while simultaneously promoting protective innate immune responses against C. albicans (20, 21). EGFR is a membrane-bound tyrosine kinase receptor. After binding to the fungal ligands, EGFR signaling will activate inflammatory response via several pathways, including MAPK and nuclear factor kappa light chain enhancer of activated B cells (NF-ĸB) (20, 22, 23). MAP kinase phosphatase 1(MKP1) and c-Fos in the MAPK signaling pathway are both activated by the filamentous form of C. albicans in a biphasic manner in oral and vaginal epithelial infection in vitro (24, 25). However, the function of EGFR and its link to the main MAPK proteins (ERK1/2, JNK and p38) in VVC have never been explored.
The significance of pathogenic variation between different strains of C. albicans has been highlighted by a number of comparative studies. From the standpoint of fungus factors, genetic variation at the ECE1 locus, encoding a critical toxin of C. albicans (candidalysin), has recently been shown to influence the pathogenicity of C. albicans strains, SC5314 and 529L (26). Clinical C. albicans vaginal isolates and the standard strain SC5314 also differ during interaction with macrophages (27). By the same token, the immune responses of vaginal epithelial cells to clinical C. albicans strain and SC5314 could also be differently initiated in the context of VVC. Given the increasing population share of antifungal-resistant Candida spp. among clinical isolates over the past several decades (28), particular to azoles (29, 30), understanding how immune system responds to drug-resistant Candida also help us to develop more effective immunotherapeutic agents against drug-susceptible and-resistant fungi.
Despite the fact that C. albicans remains the principal etiologic agent of VVC, results from others also reveal an increasing prevalence of non-albicans Candida (NAC) species globally, affecting 10– 30% of VVC cases (31). Most of these cases are due to C. glabrata, followed by C. parapsilosis, C. tropicalis, C. krusei and C. dubliniensis (32, 33). Compared with C. albicans, NAC species are generally more resistant to antifungal drugs (34), making treatment for VVC more complicated. Although less commonly reported, the highly antifungal-resistant emerging pathogen C. auris was described in one case of VVC (35). In a study with VVC murine model, although single isolate of each NAC species was tested, the preliminary data indicated that most NAC species are incapable of forming hyphae and do not elicit robust immunopathology in vivo (36), but NAC do actively contribute to symptomatic VVC (37, 38). While significant attention has been given to C. albicans-induced host immune response, little is known regarding the interaction of vaginal epithelial cells and clinical NAC species.
To elucidate the function of EGFR and MAPK pathways in the pathogenesis of vulvovaginal candidiasis, we analyzed the immune activation of vaginal epithelial cells infected by both C. albicans and some NAC species with respect to the two signaling pathways. Our results reveal that EGFR is significantly activated by C. albicans in human vaginal epithelial cells both in vitro and in vivo, which governs the activation of MAPK proteins (p38 and ERK1/2). While the highly azole-resistant C. albicans induced stronger phosphorylation of EGFR and p38 than the drug-susceptible SC5314, the clinical NAC isolates and C. auris induced stronger phosphorylation of ERK1/2 and c-Fos in human vaginal epithelial cells than C. albicans strains. Furthermore, EGFR inhibitor greatly suppressed inflammatory cytokines production in vitro and also significantly decreased vaginal inflammation and fungal burden in vivo during C. albicans infection. Consistently, p38 and ERK1/2 inhibitor strongly suppressed inflammatory cytokines production but only the p38 inhibitor significantly improved cell survival of vaginal epithelium infected by both C. albicans and NAC species.
Clinical C. albicans isolates (1052 and C1-14) and NAC species (Candida glabrata, Candida parapsilosis, Candida tropicalis) were selected from our previous multicenter epidemiology study on VVC in China (38). Two C. auris isolates (10913 and 14918) were obtained from Wester Dijk Fungal Biodiversity Institute, and the standard strains (SC5314 and ATCC 90028) were reserved in the Institute of Dermatology, Chinese Academy of Medical Sciences. The susceptibility of two clinical C. albicans isolates (1052 and C1-14) to antifungal drugs was determined as described previously (39) and the minimum inhibitory concentration (MIC) values showed both isolates were highly azole-resistant (Fluconazole MIC >256). All cultures were streaked on yeast extract peptone dextrose (YPD) agar plates, and single colony was picked and grown in liquid YPD broth in an orbital shaker for 12h at 28°C (C. albicans, C. tropicalis, C. glabrata and C. parapsilosis) or 35°C (C. auris). Yeast cells were then pelleted by centrifugation and washed twice with PBS. The cells were suspended and counted via a hemacytometer. For Hyphae form of SC5314, the above yeast cells were grown in fresh 1640 medium and shaking at 37°C for 3-5 h.
The human vaginal epithelial cell line VK2/E6E7 cells (ATCC #CRL-2616, VA) were cultured in keratinocyte-serum free medium (KSFM) (Gibco, USA) supplemented with 50 μg/ml of bovine pituitary extract (BPE) and 0.1 ng/ml of epidermal growth factor (EGF) at 37°C with 5% CO2. Experiments were carried out in KSFM without supplements and performed independently at least 3 times.
After 6h stimulation with C. albicans, VK2/E6E7 cells were rinsed with cold PBS and the total RNA was extracted using Tri reagent solution (Ambion). RNA-seq libraries were constructed according to the technical protocol and single-end 50-base reads were produced on a BGISEQ-500 platform at BGI-Shenzhen following the manufacturer’s procedures. Genes were considered significantly differentially expressed according to the criteria of a fold change ≥2 and Q value (adjusted P value) ≤0.05.
The VK2/E6E7 vaginal epithelial cells were seeded onto six-well tissue culture plates and incubated in supplement-free KSFM for 12 h and then infected with Candida cells with a multiplicity of infection (MOI) of 5. EGFR inhibitors were added to the host cells 2h before the fungal stimulation. At various time points, the epithelial cells were rinsed with cold PBS and lysed using a modified RIPA lysis buffer containing protease (Cell Signaling Technology) and phosphatase (Sigma-Aldrich) inhibitors, left on ice for 30 min. The cells were collected by centrifugation and supernatants were assayed for total protein. 20 ug of the protein was separated by SDS-PAGE and the proteins were detected by immunoblotting with specific antibodies, including anti-phospho-EGFR Tyr1068 (#3777), anti-phospho-c-Fos Ser32 (Cell signaling; #5348), anti-phospho-p65 Ser536 (Cell signaling; #3033), anti-phospho-JNK Thr183/Tyr185 (Cell signaling; #9255),anti-phospho-Erk1/2 Thr202/Thr204 (Cell signaling; #4370), anti-phospho-p38 Thr180/Tyr182 (Cell signaling; #4511). For the extraction of total protein from vaginal tissue, half of the dissected vagina was firstly grinded and then lysed using Minute™ Total Protein Extraction Kit for Animal Cultured Cells/Tissues (SD-001/SN-002, invent biotechnologies, America) at 4°C. After quantitation, the tissue protein was separated by SDS-PAGE and detected as above.
VK2/E6E7 cells were plated into 6-well tissue culture plates at 6.0 × 105 cells per well and then transfected with the lentivirus (Hanbio Biotechnology, Shanghai, China) containing LV-EGFR-sh2 for 24h according to the manufacturer’s protocol. The overexpression efficiencies of EGFR were identified using Western blotting.
The xCELLigence Real-Time Cell Analyzer (RTCA) system (Acea Biosciences, San Diego, CA, USA) was adopted to measure cell damage induced by Candida pathogens. VK2/E6E7 cells were counted and then seeded (5×104) into the E-plate 96 wells, incubating at 37°C for at least 20 h prior to treatment. When the cell growth curve reached a plateau, inhibitors or DMSO (0.8%) were added to the host cells 2h prior to the fungal stimulation and the vaginal epithelial cells were infected by different Candida strains at MOI of 10. EGFR inhibitor AG1478(Absin, abs810610) was used at 4.5μM, ERK1/2 inhibitor SD5978(Biyotime) was used at 5μM and p38 inhibitor SB203580 (selleckchem) was used at 5uM. The cell index was automatically recorded every 15 min during the incubation period. This experiment was performed in duplicates and run for 24h postinfection.
The VK2/E6E7 cells were grown in 6-well tissue culture plates for 48h and then infected with Candida yeast cells at MOI of 0.01 for 24 h. The culture supernatants were collected and clarified by centrifugation. The levels of CCL20, GM-CSF, G-CSF, IL-1β, IL-6 and IL-17A in the supernatants were determined using the Luminex multipex assay (R&D Systems; number LXSAHM-08).
Our previously described murine model of vulvovaginal candidiasis was modified for investigating early vaginal infection events (40). The estrogen (β-estradiol 17-valerate; Sigma) was firstly dissolved in DMSO (20mg/mL) and then in sesame oil (2mg/mL). The female mice were injected subcutaneously with 0.2 mg of estrogen every other day to a total of three times. The mice were then anesthetized with 1 mg/kg urethane and were inoculated into the vaginal lumen with 10 µL of 2×108 cfu/ml C. albicans yeast in sterile saline. After 1 day, mice were sacrificed, the vagina was harvested and divided by two. One-half was weighed and homogenized for Immunoblotting. The other one-half was processed for histopathology and immunofluorescence. For EGFR inhibition studies, the mice were administrated with 30 mg/kg AG1478 at day -2 and day -1 with respect to C. albicans inoculation. Mice were infected with C. albicans spores and processed as above.
The vaginal tissue was transferred to 4% paraformaldehyde solution, embedded in paraffin and sectioned at an optimal cutting temperature. 5-µm sections were selected and stained with Periodic Acid-Schiff (PAS), hematoxylin-eosin (H&E) or antibody. Stained sections were scanned and evaluated for severity of inflammation and fungal burden. For immunofluorescence, slides were probed with indicated antibodies at 4°C overnight and washed in PBS three times the next day, followed by fluorescence-conjugated secondary antibodies. They were subjected to DAPI counter-staining before being air-dried and mounted. Immunofluorescent images were collected by LSM 510 Laser Scanning Microscope and analyzed by ImageJ software.
Continuous variables are expressed as the means ± standard deviation(SD), and categorical data are presented as frequencies or percentages. we performed the student’s t-test (comparing the two groups) and one-way ANOVA (comparing three or more groups), followed by Tukey’s (comparing all groups) or Dunnett’s (comparing treatments vs. control) post-tests to assess the significance of changes relative to controls. 2-sided tests were used in all statistical analyses and a P value<0.05 was considered significant. All tests were performed with the GraphPad Prism 9.0.
To determine whether EGFR and possible downstream signaling pathways in vaginal epithelial cells are involved in VVC inflammation, we first performed RNA–seq analysis on VK2/E6E7 cells at 6 h post incubation with C. albicans standard strain SC5314. We found that ErbB, MAPK, NF-κB and IL-17 signaling pathways were significantly more up-regulated in stimulated VK2/E6E7 cells than phosphate-buffered saline (PBS)-treated cells (Figure 1A). The heat map shows most up-regulated genes including CSF2, CSF3, FOS, IL-1, GADD45 (Figure 1A). Since numerous studies have shown that phosphorylation of EGFR and MAPK signaling are critical for the activation of oral epithelial cells in response to C. albicans (20, 25, 41, 42), we next investigated the phosphorylation of EGFR, MAPK (p38, ERK1/2, JNK), c-Fos and NF-κB(p65) in VK2/E6E7 vaginal epithelial cells at different time points post co-culture with SC5314 at 5 of multiplicity of infection (MOI). The phosphorylation level of EGFR on tyrosine residue 1068 immediately increased after1 h post infection and sharply increased at 4-6 h and then remained high levels until 10 h post infection (Figure 1B). The trend of phosphorylated p38 elevations was similar to phosphorylated EGFR. The phosphorylation of ERK1/2 or c-Fos was also induced by the pathogen at 1 h post-infection and sustained until 10 h. However, phosphorylated ERK1/2 gradually reached its highest point at 6 h, accompanied with a sharp increase of phosphorylated c-Fos at 4 h (2 h earlier than p38) (Figure 1B). In conjunction with the different trajectory of these MAPKs, the phosphorylation of NF-κB quickly elevated during the first 2 h and subsided afterwards and phosphorylated JNK remained unchanged at each time points (Figure 1B). In terms of an earlier NF-κB response, our data suggest that EGFR signaling and its downstream MAPKs (p38, ERK1/2) may be more critical for Candida- induced earlier inflammatory response in vaginal epithelial cells. Among these MAPK(s), the time course of ERK1/2 activation is better lined up with c-Fos phosphorylation. Unchanged phosphorylation of JNK, opposite the responses of oral epithelial cells to the same C. albicans strain (25), argues roles of JNK in vaginal immune responses to Candida spp.
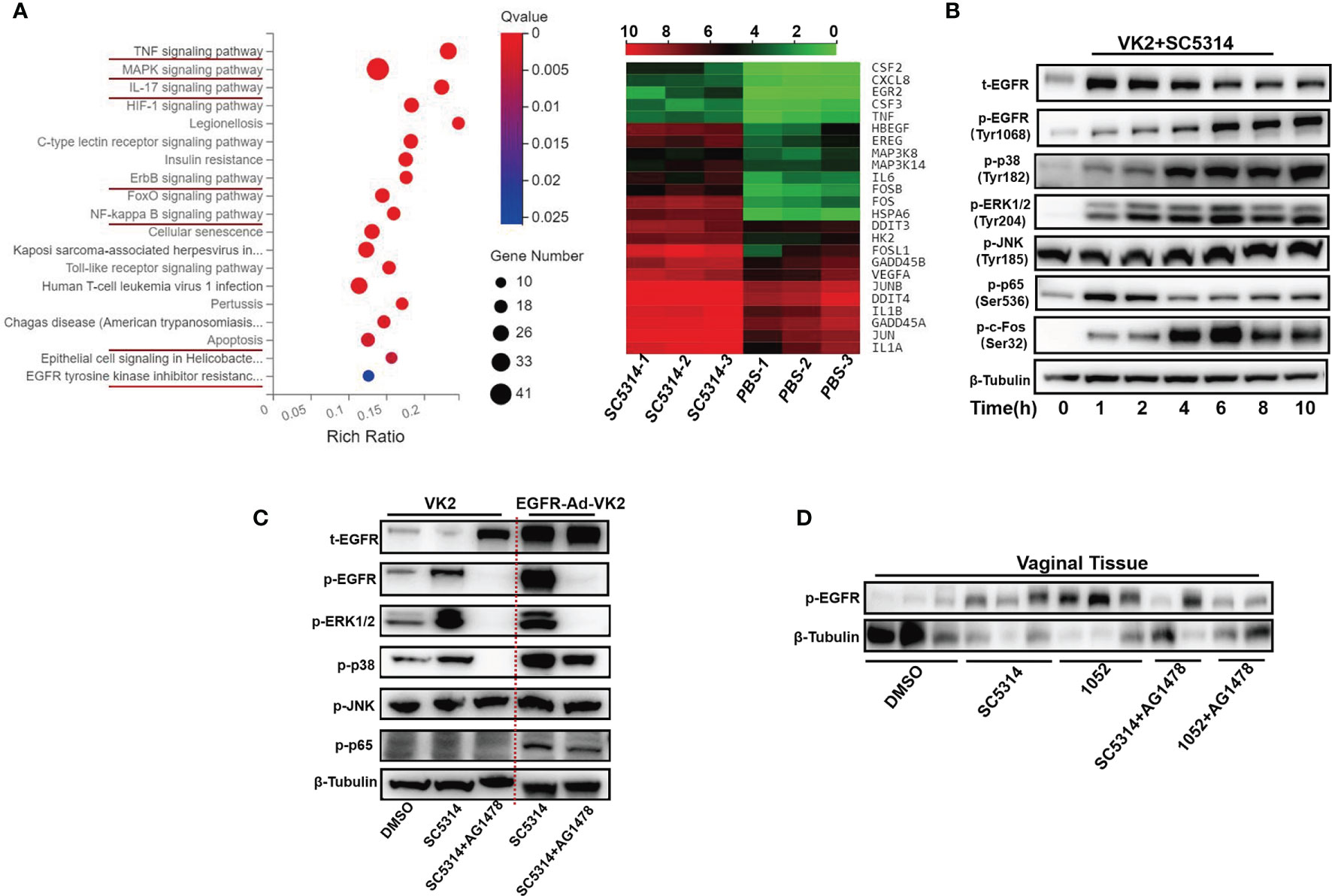
Figure 1 C. albicans activates EGFR, NF-κB, MAPK and c-Fos signaling in vaginal epithelial cells. Pathway enrichment analysis and heatmaps of VK2/E6E7 vaginal epithelial cells stimulating with SC5314 and PBS. Using the phyper function in the R software to perform enrichment analysis and calculate the P value. Then performing FDR correction on the P value to obtain the Q value. A Q value < 0.05 is regarded as a significant enrichment. Red underlines highlight the upregulated pathways related to EGFR and MAPK. Heat map displaying the upregulated(red) or downregulated(blue) genes. Each column represents an individual sample(n=3) (A). VK2/E6E7 cells were stimulated with SC5314 (MOI=5) for the indicated time. Cell lysates were analyzed by immunoblotting for the indicated proteins. The phosphorylated forms of proteins are preceded by “p” (B). SC5314 elicited significantly stronger phosphorylation of p38 in EGFR-Ad-VK2, VK2/E6E7 cells overexpressing EGFR, when compared to VK2/E6E7 cells. EGFR inhibitor (AG1478) suppressed SC5314-induced phosphorylation of ERK1/2 and p38 in EGFR-Ad-VK2 and VK2/E6E7 cells (C). Female ICR mice (n=3 or 2/each group) were infected with SC5314 or clinical isolate1052 after pre-treatment with DMSO or AG1478 for 2d and vaginal tissues were harvested 1 d after infection for detection of EGFR phosphorylation by immunoblotting analysis (D). Data are representative of three independent experiments and bands are shown relative to β-tubulin loading control.
In oral epithelial cells, EGFR inhibitor can significantly suppress C. albicans-induced pEGFR, c-Fos and pMKP1 (20). To verify the EGFR roles in activating downstream target MAPK proteins, phosphorylated levels of three MAPKs (p38, ERK1/2, JNK) in C. albicans infected vaginal epithelial cells were assessed using an EGFR antagonist AG1478 and EGFR overexpressed epithelial cells (EGFR-Ad-VK2). We first treated VK2/E6E7 epithelial cells with AG1478 2 h prior to C. albicans challenge. As expected, the phosphorylations of EGFR, p38 and ERK1/2 at 6 h post-infection were completely inhibited by AG1478, while phosphorylated JNK remained the same level (Figure 1C). By contrast, Candida-induced EGFR and p38 phosphorylation levels, but not ERK1/2 phosphorylation, were increased in EGFR-Ad-VK2 cells that again were effectively reversed in the presence of AG1478 (Figure 1C). EGFR, p38 and ERK1/2 inhibitors did not affect C. albicans hyphal growth (Figure S1).
To verify EGFR signaling responses in vivo, we used a mouse VVC model to analyze activation of EGFR during VVC. We found that infections with standard C. albicans SC5314 and drug resistant strain 1052 can induce high level of phosphorylated EGFR in collected vaginal tissues, especially in the case of 1052 infection (Figure 1D). Treatment of mice with EGFR antagonist AG1478 greatly reduced phosphorylation of EGFR in both C. albicans infected mice, especially in 1052 infected vaginal tissues (Figure 1D). Taken together, in vivo results agree with in vitro data to show that EGFR signaling is necessary for activating C. albicans-induced vaginal inflammation.
The in vitro data above suggest that EGFR signaling via p38 and ERK1/2 pathway activates C. albicans-induced inflammatory response in vaginal epithelial cells. To understand the impact of EGFR signaling activation for the downstream inflammatory response during vaginal inflammatory response, antagonists AG1478 (EGFR), SD5978 (ERK1/2) and SB203580 (p38) were used to treat VK2/E6E7 vaginal epithelial cells 2 h prior to infection with SC5314. We found that inhibition of phosphorylation of EGFR, ERK1/2 or p38 respectively resulted in significant reduction in the levels of CCL20 (75.70%, 79.73%, 80.94%), G-CSF (93.61%, 93.75%, 95.53%), GM-CSF (93.33%, 94.15%, 95.38%), IL-1β (81.04%, 69.96%, 93.46%), IL-6 (75.32%, 51.58%, 93.26%) and IL-17A (57.94%, 57.94%, 59.63%) (Figure 2A). Among the three inhibitors, SB203580 showed the strongest inhibitory effect on all the cytokines induced by SC5314, particularly on G-CSF, IL-1β and IL-6.
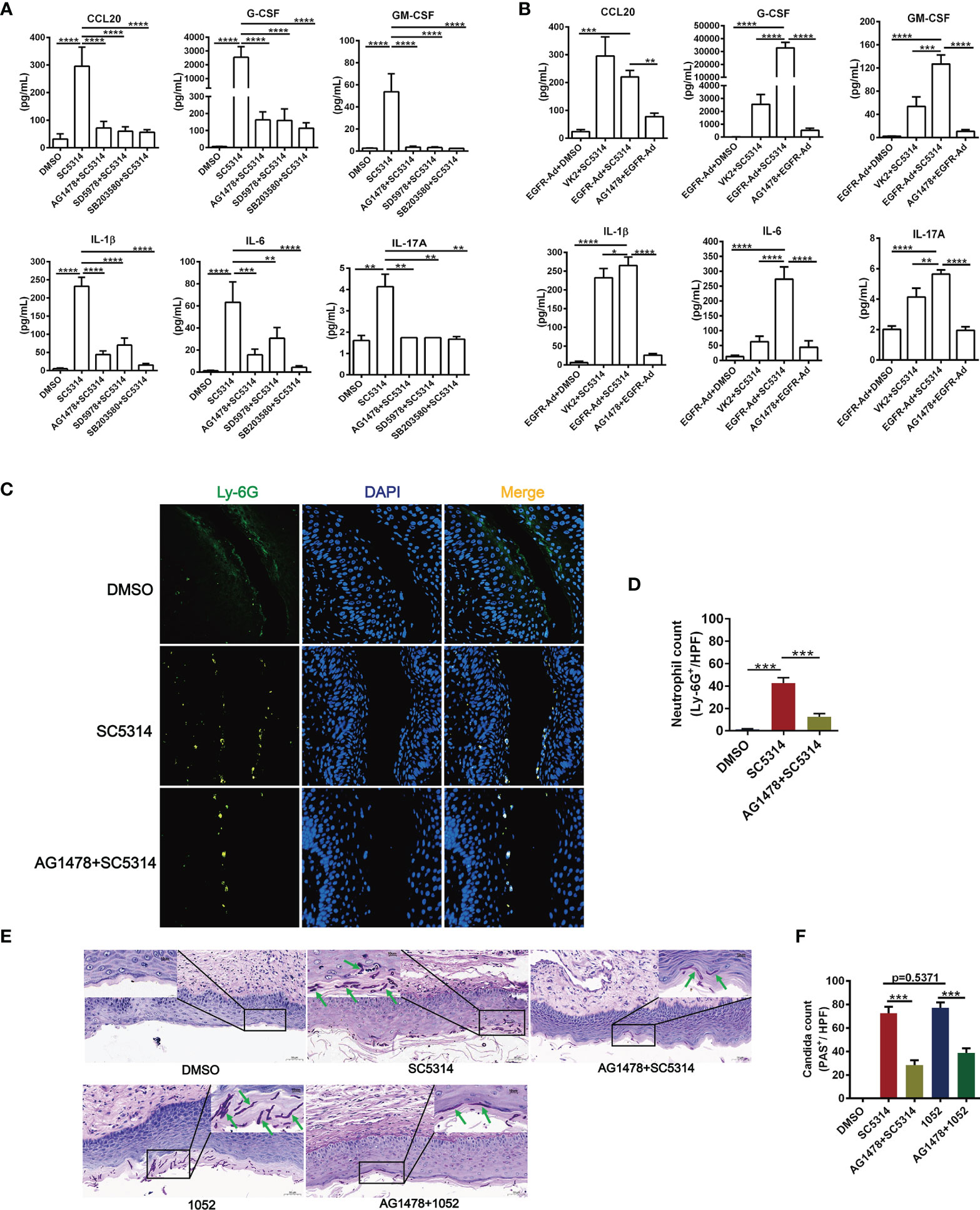
Figure 2 Functional role of EGFR and MAPK signaling in regulating vaginal epithelial inflammatory responses. Quantification of cytokines were assessed by multiplex microbead assay (luminex), secreted from VK2/E6E7 vaginal epithelial cells stimulating with SC5314(MOI=0.01) for 24 h. EGFR inhibitor AG1478, p38 inhibitor SB203580 or ERK1/2 inhibitor SD5978 was added to the cells 2 h prior to fungal stimulation. All these inhibitors suppressed SC5314-induced secretion of cytokines (CCL20, G-CSF, GM-CSF, IL-1β, IL-6, IL-17A) (A). EGFR-Ad-VK2 released higher levels of cytokines (G-CSF, GM-CSF, IL-1β, IL-6, IL-17A) induced by SC5314 when compared to VK2/E6E7 cells, which can be suppressed by AG1478 (B). VVC mice were treated as previously (Figure 1D). Vaginal sections from C. albicans–infected mice were assayed for immunofluorescence staining of Ly-6G (neutrophil marker, green) and nuclei were counterstained with DAPI (blue) (C). Numbers in graph (D) provide the mean neutrophils count per group (n=3/each group). Fungal load in vaginal tissues was assessed by periodic acid-Schiff (PAS). Insets show regions of vaginal fungal burden and higher-magnification images of the organisms indicated by the arrows (E). Numbers in graph (F) provide the mean C. albicans count per group (n=3/each group). Data are representative of three independent experiments. Error bars represent SD, *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
Compared with SC5314-infected VK2/E6E7 cells, EGFR-Ad-VK2 cells secreted much higher levels of cytokines such as G-CSF, GM-CSF, IL-1β, IL-6 and IL-17A – except CCL20 upon SC5314 stimulation (Figure 2B). Although the CCL20 level in SC5314-infected EGFR-Ad-VK2 was more highly produced than in the DMSO control, the quantity was less than for SC5314 infected VK2/E6E7 cells. Again, the upregulation of all the cytokines – including CCL20 in EGFR-Ad-VK2 cells – can be suppressed by EGFR, p38 and ERK1/2 inhibitors as effectively as those in infected VK2/E6E7 cells (Figure 2B). Moreover, in the murine model of VVC, treatment of AG1478 greatly ameliorated vaginal inflammation caused by SC5314. The effects of AG1478 were confirmed by histopathologic and immunofluorescent analysis, which revealed that the vaginas of AG1478-treated mice had low levels of neutrophil recruitment (Figures 2C, D) and fungal burden (Figures 2E, F). In all, these data imply that EGFR-p38/ERK1/2 pathways are required for vaginal epithelial cells to mount inflammatory responses to C. albicans and inhibition of EGFR signaling would reduce vaginal neutrophil recruitment and fungal burden during VVC.
The above experiments were mostly performed with the standard C. albicans strain SC5314, an original clinical isolate that has been widely used to evaluate fungal and host interaction. Pathogenic variability has been noted in C. albicans isolates (6). To discriminate the possible difference in host responses to clinical drug-sensitive and drug-resistant C. albicans, the EGFR responses in VK2/E6E7 cells infected by SC5314 (drug-sensitive) and a clinical azole-resistant C. albicans 1052 (38) were compared. Total proteins of VK2/E6E7 cells were extracted at 1, 2, 4, 6, 8 and 10 h post fungal infection for measurement of the activation of EGFR-MAPK pathway. We found that EGFR and p38 phosphorylation were significantly higher in 1052 infected epithelial cells, especially in the 4 to 10 h period (Figure 3A). We find it intriguing that ERK1/2 phosphorylation induced by 1052 appeared at early stages (1- 4 h) and gradually subsided at later stages, which was different from an overall increased ERK1/2 response in SC5314 infection especially (Figure 3A). In accord with the early ERK1/2 response in both strain infections, p65(NF-κB) phosphorylation levels were higher in SC5314 and 1052 infected epithelial cells during 1- 4 h. The second difference between two types of infections was that the levels of c-Fos phosphorylation induced by 1052 were significantly lower than the levels induced by SC5314 at both early and later stage (Figure 3A).
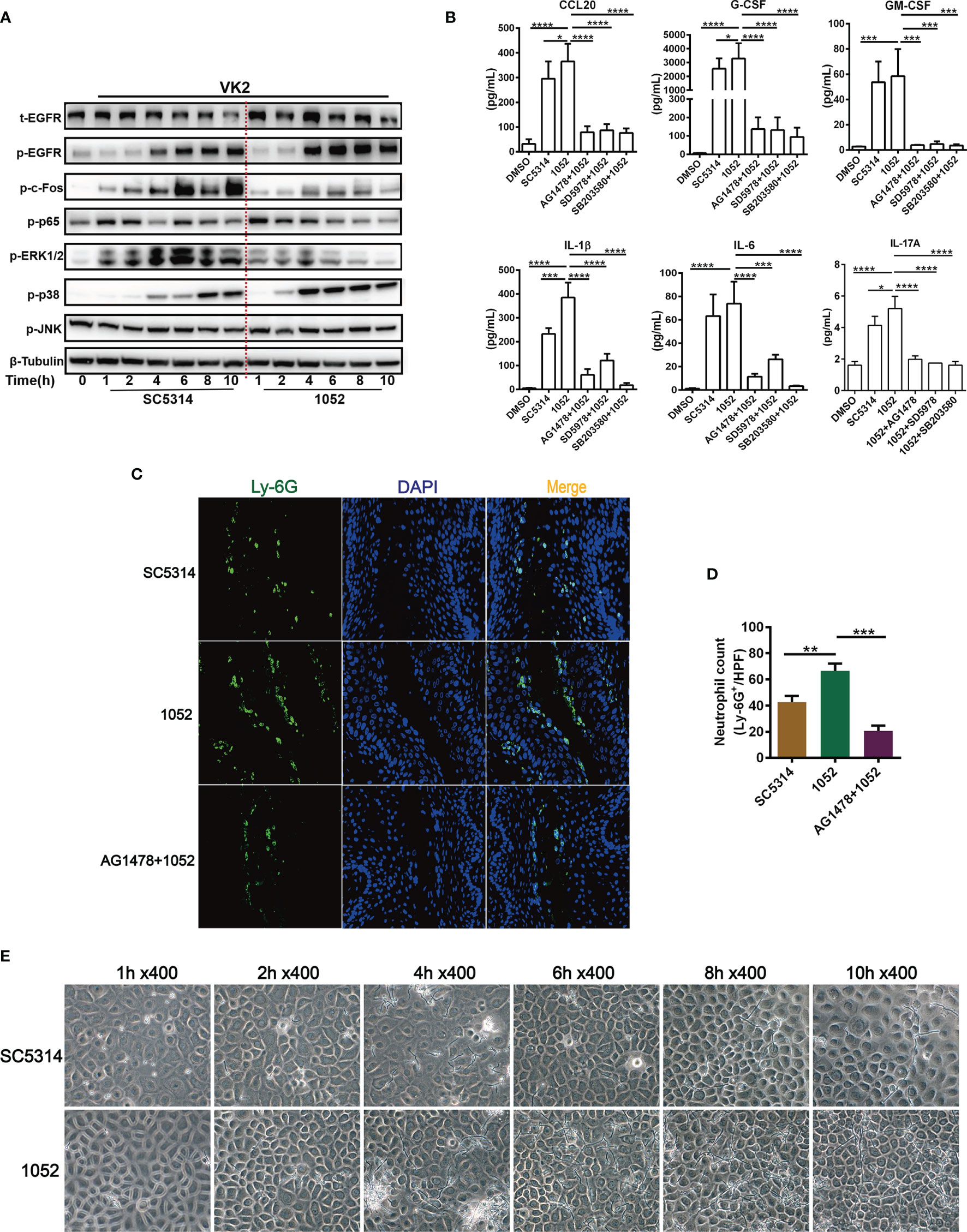
Figure 3 Differential activation of EGFR and MAPK and cytokine induction by clinical isolate 1052 and WT strain SC5314. VK2/E6E7 cells were stimulated with SC5314 and clinical isolate 1052 (MOI=5) for the indicated time respectively. Phosphorylation of EGFR, MAPK proteins, NF-κB (p65) and c-Fos was detected by immunoblotting of cell lysates (A). 1052 induced higher levels of cytokines secretion (CCL20, G-CSF, GM-CSF, IL-1β, IL-6, IL-17A) than SC5314. All inhibitors (EGFR, p38 and ERK1/2 inhibitor) suppressed 1052-induced secretion of CCL20, G-CSF, GM-CSF, IL-1β, IL-6 and IL-17A (B). Infection with 1052 in the vaginal mucosa of mice induced significantly increased neutrophil recruitment at the infection foci when compared with SC5314 and pre-treatment of AG1478 resulted in significantly decreased neutrophil recruitment (C). Numbers in graph (D) provide the mean neutrophils count per group (n=3/each group). Morphology of SC5314 and clinical isolate 1052 adhering to VK2/E6E7 cells at 0, 1, 2, 4, 6, 8, 10 h post infection (E), gently washed with PBS for at least 3 times. Data are representative of three independent experiments. Error bars represent SD, *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
To better understand the meanings of these divergent host responses in fungal infection, we collected micrographs of Candida morphology adhering to vaginal epithelial cells at different time points. We found that the azole-resistant isolate was more filamentous than SC5314. The hyphal lengths of 1052 adhering to VK2/E6E7 cells were longer and the quantities of hyphae were greater than SC5314 at each time point after 2 h (Figure 3E). In accord with these in vitro results, the hyphal lengths of 1052 adhering to vaginal mucosa were also longer than SC5314 in the mouse model of VVC at 24 h post infection (Figure 2E). The different MAPK responses in vaginas infected by drug-sensitive and drug-resistant C. albicans certainly require a further investigation with more strains. However, the extent of hyphal formation could be a convenient explanation for such a difference in MAPK responses. Nevertheless, EGFR responses are better lined up with p38 response in both strain infections.
The inflammatory cytokines in supernatants of VK2/E6E7 cells infected by SC5314 and 1052 were also measured and compared. The results showed that levels of all cytokines (CCL20, G-CSF, GM-CSF, IL-1β, IL-6 and IL-17A) in 1052 infected epithelial cells were higher than in SC5314 infected cells and each highly-produced cytokine in the 1052-stimulated epithelial cells can be suppressed by EGFR, p38 and ERK1/2 inhibitors (Figure 3B). In particular, the inhibition of p38 resulted in a much greater reduction of 1052-triggered G-CSF, IL-1β and IL-6 than the other two inhibitors (Figure 3B). Along with stronger inflammatory cytokine production, levels of neutrophil infiltrates in the vaginal tissue infected by 1052 were significantly higher than SC5314 (Figures 3C, D). Apparently, inflammatory cytokines and neutrophil infiltrate levels were dependent on EGFR since both phenotypes in SC5314 and 1052 infection can be reversed by AG1478(Figures 3C, D).
Vulvovaginal candidiasis (VVC) caused by non-albicans Candida (NAC) species are increasingly reported today. Given that more drug-resistant strains have been found in NAC species, we next analyze the EGFR-MAPK responses during the NAC species infections by using 3 clinical NAC isolates of C. tropicalis, C. glabrata, C. parapsilosis previously isolated from VVC patients (38) and 2 strains of C. auris (CBS10913 and CBS14918). The results were compared with 4 strains of C. albicans (SC5314, ATCC90028, 1052 and C1-14). To examine the extent of EGFR/MAPK pathway activation in vaginal epithelial cells infected by each of these strains, proteins of infected epithelial cells 6 h post infection at an MOI of 5 were extracted for analysis. We found that 4 C. albicans strains induced significantly higher levels of EGFR and p38 phosphorylation in VK2/E6E7 cells than all NAC strains (Figure 4A). When compared with the C. albicans strains, NAC strains consistently triggered much stronger activation of ERK1/2 and c-Fos phosphorylation in VK2/E6E7 cells (Figure 4A). In general, there were no significant difference in phosphorylation of EGFR, p38 or ERK1/2 among these NAC infected epithelial cells except that a slightly decreased ERK1/2 phosphorylation and a slightly increased p38 phosphorylation were seen in C. tropicalis (Figure 4A). When one consideres that C. tropicalis is phylogenically closer to C. albicans than other species, the ERK1/2 and p38 responses observed in C. tropicalis are not surprising. Nevertheless, activation of ERK1/2 and c-Fos phosphorylation seems to mediate the immune responses of vaginal epithelial cells by NAC species.
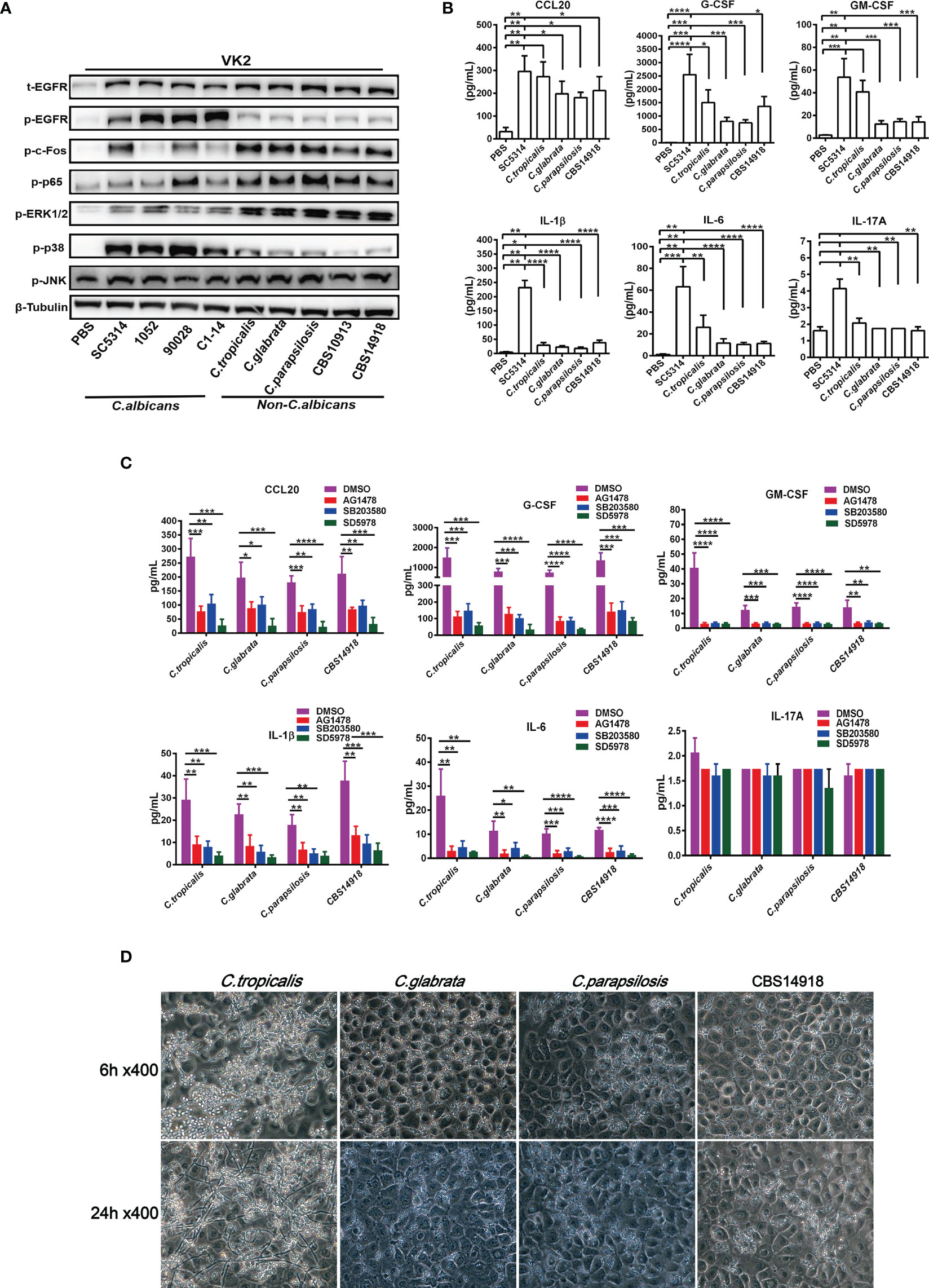
Figure 4 Divergent activation of EGFR and MAPK and cytokine induction by C. albicans and non-albicans Candida species. VK2/E6E7 cells were stimulated with C. albicans (SC5314, 1052, ATCC90028 and C1-14) and non-albicans Candida (NAC) isolates C. tropicalis, C. glabrata, C. parapsilosis, CBS10913 and CBS14918) (MOI=5) respectively. Protein lysates were taken at 6 h post-infection for western blotting analysis (A). Cytokines induced by C. albicans and NAC isolates (MOI=0.01) were assessed at 24h post-infection via Luminex (B). All inhibitors suppressed NAC isolates-induced cytokines production (C). Morphology of C. tropicalis, C. glabrata, C. parapsilosis and CBS14918 strains on VK2/E6E7 cells at 6 and 24 h post infection (D). Data are representative of three independent experiments. Error bars represent SD, *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
Typically, MAPK signaling in oral epithelial cells is correlated with hyphal formation of C. albicans (41). However, clinical NAC species used in this study that were not able to form the true hyphae in epithelial cells (Figure 4D) also effectively activated ERK1/2 and the protein c-Fos (Figure 4A). We found that only a small population of C. tropicalis switched to pseudohyphae, while C. glabrata, C. parapsiolosis and C. auris grew only in the yeast morphology until 24 h (Figure 4D). These results suggested that ERK1/2 phosphorylation may be more yeast-specific while EGFR and p38 phosphorylation are more hyphae-specific in vaginal epithelial cells, which is also supported by the higher EGFR and p38 responses induced by drug resistant 1052 with high filamentation in Figures 3A, E.
Regarding the downstream cytokine levels, 4 NAC species generally triggered less cytokine production in VK2/E6E7 cells when compared to SC5314 (Figure 4B). Inhibition of EGFR, p38 and ERK1/2 led to further reductions of all cytokines by 4 NAC species (Figure 4C) except IL-17A. Particularly, ERK1/2 inhibitor showed the strongest inhibitory effects on NAC-induced CCL20, G-CSF, GM-CSF, IL-1β, and IL-6, which is consistent with the strong activation of ERK1/2 phosphorylation induced by NAC species (Figure 4A). This highlights the importance of ERK1/2 in mediating the inflammatory responses in NAC species-induced vaginal infection.
In addition to vaginal inflammation, epithelial damage also contributes to the symptoms of VVC (1, 43). Since inhibition of EGFR and p38/ERK1/2 can effectively reduce epithelial inflammatory responses to SC5314, we then wonder whether blocking these pathways would lessen epithelial damage during the VVC infection. The real-time damage of vaginal epithelial cells during Candida infection was monitored using an xCELLigence (real time cell analyzer) RTCA Instrument. For SC5314 infection, normalized cell index of VK2/E6E7 cells increased sharply in the first 2 hours and then subsided. However, EGFR and p38 inhibitors, but not ERK1/2 inhibitor, significantly improved cell survival of the infected VK2/E6E7 cells at 24 h post infection (Figures 5A, D). Conversely, EGFR-Ad-VK2 cells resulted in a significant increase in cell death when infected by SC5314 (Figures 5B, E). EGFR inhibitor in vivo also greatly reduced the apoptosis of vaginal epithelium (Figures 5G, H). Taken together, these data clearly demonstrated that the EGFR/p38 pathway is critical in driving the epithelial damage caused by C. albicans during VVC.
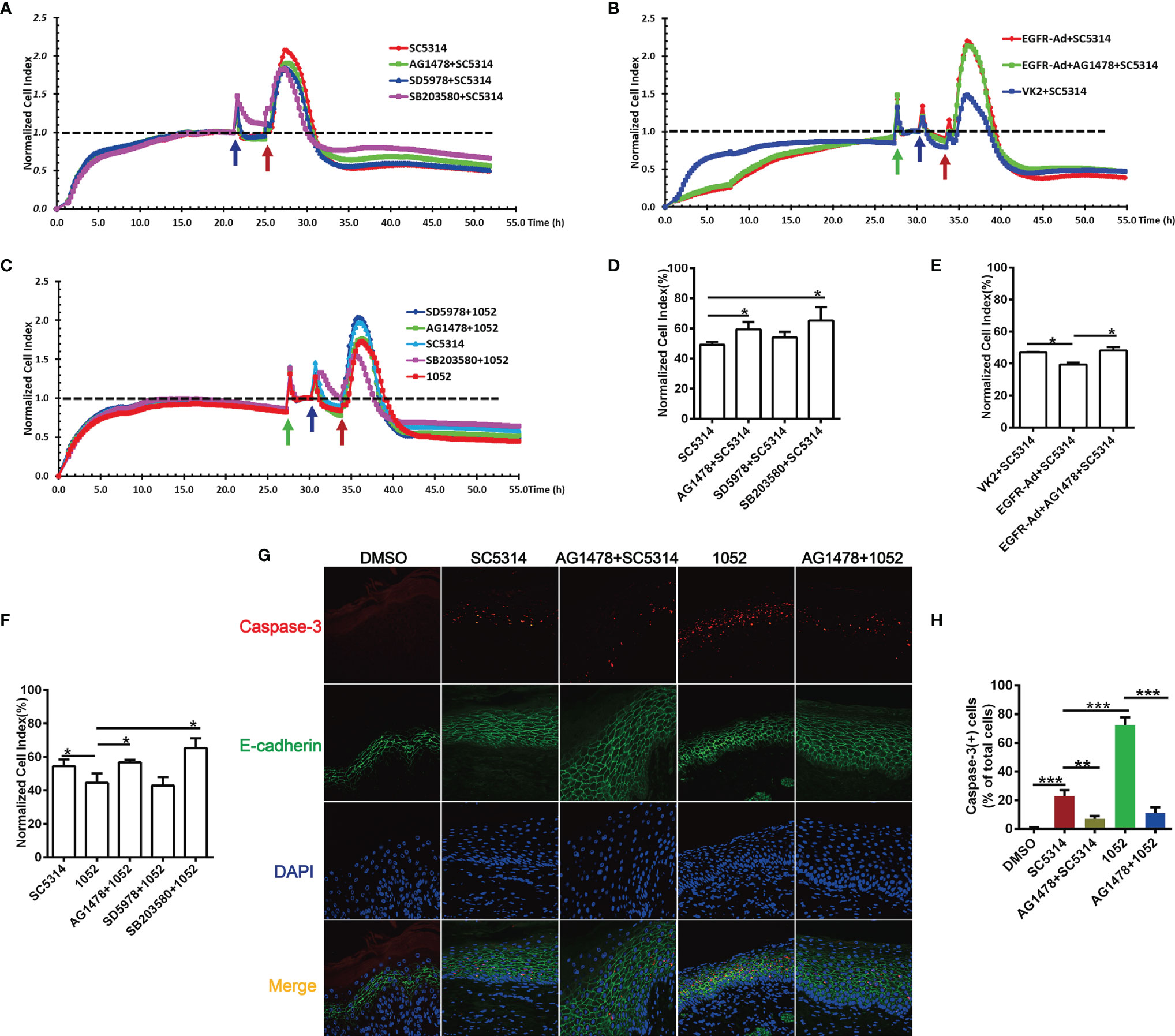
Figure 5 EGFR and MAPK signaling regulate the viability of vaginal epithelial cells after infection with C. albicans. Cell damage was monitored continuously by xCELLigence Real-Time Cellular Analysis (RTCA) system. Normalized Cell Index (NCI) demonstrated cell viability. 1052-infected VK2/E6E7 cells exhibited a significant decrease in viability when compared to SC5314 infections and pre-treatment of the EGFR and p38 inhibitors protected cells against SC5314 or 1052-killing (A, C). SC5314-infected EGFR-Ad cells exhibited increased damage when compared to the infected VK2/E6E7 cells (B). NCI at 24 h post infection in graph (D–F) provides the mean cell viability. Green arrow represents the time points on which the medium was changed. Blue and red arrows represent the time point at which inhibitors and Candida strains were added into the well respectively. VVC mice were treated as previously (Figure 1D). Immunofluorescent staining of E-cadherin (green), caspase-3(red) and DAPI (blue) demonstrated apoptotic vaginal epithelial cells in vaginal tissues sections (G). Numbers in graph (H) provide the mean apoptotic vaginal epithelial cells count per group (n=3/each group). Data are representative of three independent experiments. Error bars represent SD, *p < 0.05, Error bars represent SD, *p < 0.05, **p < 0.01, ***p < 0.001.
Considering the fact that 1052 activated higher levels of EGFR and p38 phosphorylation, we hypothesized that the azole-resistant isolate may cause more damage to vaginal epithelial cells than SC5314. As expected, the viability of VK2/E6E7 cells stimulated with 1052 was reduced compared to the viability of SC5314-infected cells (Figures 5C, F). Similarly, blocking EGFR and p38 resulted in a significant increase in cell survival, but ERK1/2 inhibitor had no effect on cell viability (Figures 5C, F). In the murine model of VVC, levels of cell apoptosis in 1052-infected vaginal tissue were significantly higher than in SC5314-infected vaginal tissue and blocking EGFR also decreased cell apoptosis induced by 1052 (Figures 5G, H).
Despite lower levels of phosphorylation of EGFR and p38 in NAC infections than C. albicans infection (Figure 4A), epithelial damage caused by C. tropicalis and C. glabrata was as great as that caused by C. albicans; cell damage by C. parapsilosis and C. auris was even more extensive than C. albicans (Figures 6A, F). For all the NAS strains, the p38 inhibitor significantly improved the epithelial cell survival during the infection, but this was not the case for the EGFR or ERK1/2 inhibitor (Figures 6B–E, G), which highlights that p38 signaling regulates NAC-induced epithelial damage. While both EGFR and p38 are required for C. albicans-induced epithelial damage (Figure 5D), the absence of EGFR response in NAC induced epithelial damage (Figure 6G) suggests a likely different upstream signal for p38-mediated epithelial responses in NAC infections.
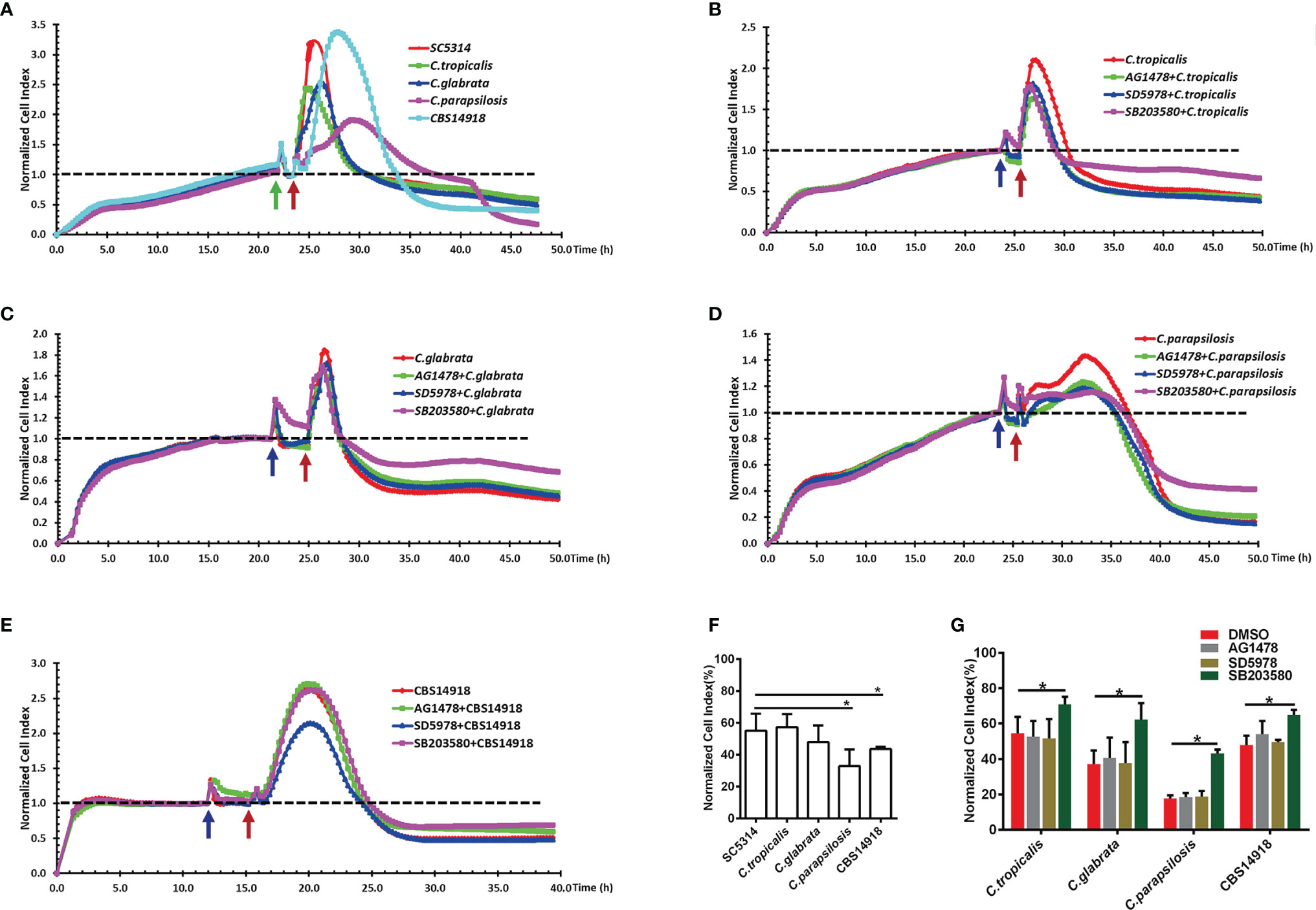
Figure 6 EGFR and MAPK signaling regulate the viability of vaginal epithelial cells after infection with non-albicans Candida species. Cells damage caused by SC5314 and non-albicans Candida (NAC) isolates were monitored continuously for 24 h (A). Only pre-treatment of p38 inhibitor (SB203580) exhibited a significant increased protective capacity against NAC isolates compared to EGFR and ERK1/2 inhibitor (B–E). NCI at 24 h post infection in graph (F, G) provides the mean cell viability. Green arrow represents the time points at which the medium was changed. Blue and red arrows represent the time point on which inhibitors and Candida strains were added into the well respectively. Data are representative of three independent experiments. Error bars represent SD, *p < 0.05.
The pathological process of VVC is largely driven by local innate immune response, where vaginal epithelial cells have been shown to play a central role (1, 12). In this study, the function of the EGFR signaling pathway and the possible key players are investigated in mouse VVC infected tissue and Candida stimulated vaginal epithelial cells in vitro. Through divergent patterns of EGFR and respective downstream MAPK proteins activated by C. albicans, drug resistant C. albicans and non-albicans Candida species, our in vivo and in vitro data confirm that the EGFR/MAPK pathway in vaginal epithelial cells is sustained by Candida stimulation with a time course that is different from the biphasic manner of oral epithelial cells based on yeast to hyphal conversion (17). We see that C. albicans prefers to induce an EGFR-p38 mediated inflammatory response and epithelial damage, while NAC species elevate phosphorylated levels of ERK1/2 and c-Fos for downstream NK-ĸB activation and cytokine productions, however, p38 is still required for the promotion of epithelial damage (summarized in Figure 7).
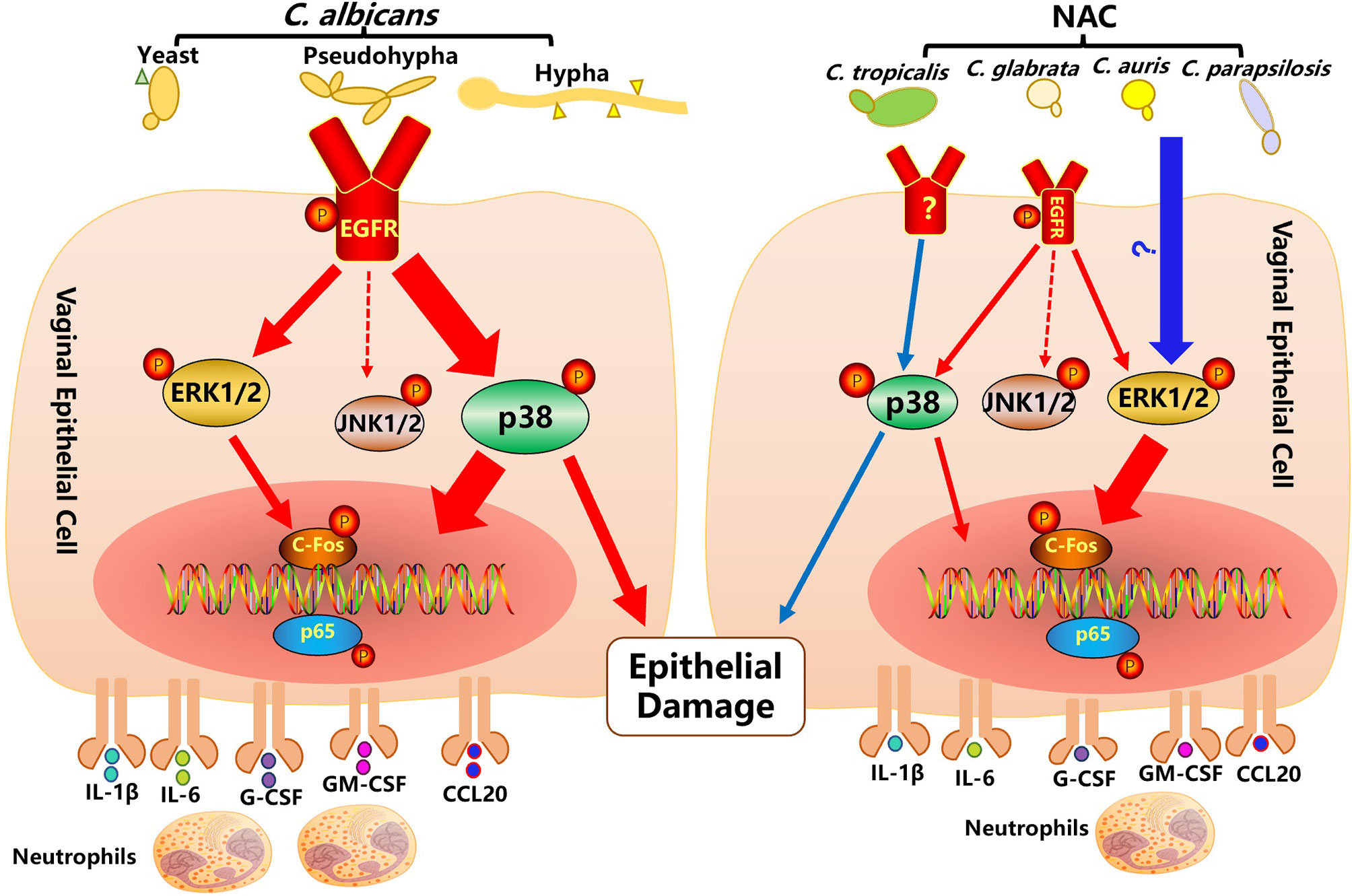
Figure 7 Activation of the EGFR-MAPK signaling pathway in vaginal epithelial cells by Candida pathogens. C. albicans infection on vaginal epithelial cells manifests by a modest yeast-to-hyphal transition within the first 2 hours, when the EGFR-ERK1/2 signaling pathway (small red arrow) is moderately activated. At a later stage of stimulation (4 to 10 hours), after the formation of a greater number of hyphae, the EGFR-p38 signaling pathway (thick red arrow) is strongly activated. Activated EGFR-p38 and-ERK1/2 signaling pathways lead to an increased phosphorylation of c-Fos and p65 and an increased secretion of cytokines (IL-1, IL-6, G-CSF, GM-CSF, CCL20). In particular, the EGFR-p38 signaling pathway also moderates epithelial damage induced by C. albicans. While non-albicans Candida (NAC) spp. infections of vaginal epithelial cells rarely produce hyphae, they activate a strong phosphorylation of ERK1/2, c-fos and p65 and a subsequent secretion of cytokines. Although not strongly activated, the p38 pathway still has some effect in the moderation of epithelial damage.
In Candida-infected oral epithelial cells, activation of EGFR and downstream MAPK target proteins (c-Fos and MKP1) are key for candidalysin-induced immune responses (20, 44). Activation of this c-Fos/MKP1 signaling is also associated with a biphasic immune response pattern that enable oral epithelial cells to discriminate the morphology of C. albicans (25). During this biphasic response, phosphorylation of p38 and JNK induced by C. albicans reach their highest levels at 2 h post-infection and then decrease in oral epithelial cells. However, in Candida-infected vaginal epithelial cells in this study, phosphorylation of p38 increased over time and peaked at a later stage. In addition, phosphorylated JNK remains normal during at the time points. Our data support the notion that the variations in mucosal responses against C. albicans are niche-specific (6). As the over expression of EGFR in VK2/E6E7 cells resulted in a significant increase of p38 phosphorylation, but not of ERK1/2 phosphorylation, we believe that the primary downstream target protein of an activating EGFR signaling for inflammatory cytokines during VVC infection is p38 MAPK.
Symptomatic VVC is associated with massive neutrophil migration to the vagina and the subsequent uncontrolled inflammation triggered by inflammatory mediators secreted from infected vaginal epithelial cells and resident immune cells (1, 45). In this study, the phosphorylation of EGFR and neutrophil recruitment appeared in vaginal mucosa in mouse VVC model and blocking EGFR or p38 signaling significantly reduced vaginal neutrophil recruitment, fungal burden, and inflammatory cytokines production induced by C. albicans. These results are contradictory with an observation with oral epithelial cells, in which EGFR-mediated immune responses and neutrophil recruitments protect the host against oral C. albicans infection (20). Nevertheless, in terms of the effects of p38 inhibition, our finding is consistent with the results from myeloid cells, in which p38γ/p38δ deletion protects against C. albicans infection and decreased host damage in vivo (46). The data again confirm that activation of the EGFR/p38 pathway is the key event to causing those deleterious inflammatory episodes and neutrophil recruitment during VVC.
Compared to drug-susceptible C. albicans infection, drug-resistant strain showed stronger phosphorylation of EGFR and p38 and lower ERK1/2 and c-Fos. The mechanism could be due to the differently-secreted or surface-mounted fungal ligands between two fungal strains for activating EGFR, for example hyphal-derived ligands, which is supported by better developing hyphae in drug-resistant strain in this study. The impact of p38 activation on vaginal epithelial cells is confirmed by p38 inhibitor in this study. Together with a strong positive correlation between hyphae and p38 phosphorylation in symptomatic VVC patients and lower levels of phosphorylated p38 in most yeast form of NAC infected cells (12), it is tempting to expect that EGFR- p38 is more specific for filamentous fungi. However, when we stimulated VK2/E6E7 cells with true hyphae of SC5314, we found that only phosphorylation of p38 was increased steadily, not EGFR or other MAPK protein (Figure S2). Obviously, other non-EGFR signaling could be also responsible for p38 activation in vaginal epithelial cells. Since phosphorylation levels of ERK1/2 and c-Fos occur at early stages of yeast form growth and subside at a later stage, ERK1/2 and c-Fos phosphorylation may be yeast-specific in VVC.
Both pathogen-mediated damage and host-mediated damage contribute to the outcome of an infectious disease (43, 47, 48). During VVC, epithelial damage is mediated by the host innate immune responses, and triggered by virulence factors of Candida species (47, 49). Our results have shown that EGFR, p38 and ERK1/2 inhibitor effectively suppress the inflammatory responses in vaginal epithelial cells to both clinical C. albicans and NAC species. A representative panel of NAC species has been shown to induce less damage and neutrophil recruitment than C. albicans (36). In this study, clinical NAC species including C. tropicalis, C. glabrata, C. parapsilosis and C. auris can trigger a stronger phosphorylation of ERK1/2 or c-Fos than C. albicans strains at 6 h post challenge. All our tested NAC strains were unable to form hyphae in vaginal tissue except C. tropicalis, which showed the strongest p38 and the least phosphorylation of ERK1/2 among the NAC species. This further supports that ERK1/2 may be yeast specific and p38 may be hyphae specific during VVC infection. In accord with the MAPK responses and hyphae formation, yeast-formed NAC strains induced less inflammatory cytokine production and filamentous fungal cells induced stronger cytokines. We note that C. auris, an emergent but rare pathogen of VVC (35) can induce higher levels of G-CSF and IL-1β than C. glabrata and C. parapsilosis although all of three species obstinately remain as yeast form. The precise identity of C. auris in immunophathology needs to be unraveled, but both G-CSF and IL-1β could lead to stronger inflammatory response. In addition, C. auris and C. parapsilosis triggered more damage to vaginal epithelial cells than C. albicans after 24 h post infection, indicating that such a strong virulence from each of these two NAC species was not contributed by hypha formation. Damage-driven responses in Candida-infected vaginal epithelial cells persisted at later stages of infection (50) and the survival curve lines of the infected VK2/E6E7 vaginal epithelial cells also indicated a species-specific damage-response to Candida pathogens.
In summary, the EGFR/MAPK pathway is differently activated in vaginal epithelial cells by clinical C. albicans and NAC species. Inhibition of EGFR markedly decreased vaginal inflammatory responses and epithelial damage induced by C. albicans in vitro and in vivo, while blocking p38 significantly reduced inflammatory cytokine production and cell damage induced by clinical C. albicans and NAC species. It is also worthy of note that ERK1/2 phosphorylation may be specific to the yeast morphology of Candida in VVC. These results indicate that targeting EGFR/MAPK pathway or the downstream signaling could be an effective therapeutic strategy against VVC caused by drug-susceptible C. albicans, NAC species and even drug-resistant Candida spp.
The datasets presented in this study can be found in online repositories. The name of the repository and accession number can be found below: NCBI Sequence Read Archive (PRJNA830650).
The animal study was reviewed and approved by the Animal Study Committee of the Institute of Dermatology at CAMS.
JZ designed the experiments, analyzed data and wrote the manuscript. JZ, JP, HM, YY and XL undertook experiments. DL were involved in reviewing and revising the paper. WL and XS funded and supervised the project and provided scientific guidance. All authors have approved the final version.
This study was financially supported by the National Natural Science Foundation of China (No. 81972949) to WL, the National Natural Science Foundation of Jiangsu Province, China (BK20191137) to XS.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2022.894069/full#supplementary-material
1. Ardizzoni A, Wheeler RT, Pericolini E. It Takes Two to Tango: How a Dysregulation of the Innate Immunity, Coupled With Candida Virulence, Triggers VVC Onset. Front Microbiol (2021) 12:692491. doi: 10.3389/fmicb.2021.692491
2. Sobel JD, Faro S, Force RW, Foxman B, Ledger WJ, Nyirjesy PR, et al. Vulvovaginal Candidiasis: Epidemiologic, Diagnostic, and Therapeutic Considerations. Am J Obstet Gynecol (1998) 178:203–11. doi: 10.1016/S0002-9378(98)80001-X
3. Cassone A. Vulvovaginal Candida Albicans Infections: Pathogenesis, Immunity and Vaccine Prospects. BJOG (2015) 122:785–94. doi: 10.1111/1471-0528.12994
4. Farah CS, Elahi S, Drysdale K, Pang G, Gotjamanos T, Seymour GJ, et al. Primary Role for CD4(+) T Lymphocytes in Recovery From Oropharyngeal Candidiasis. Infect Immun (2002) 70:724–31. doi: 10.1128/IAI.70.2.724-731.2002
5. Verma AH, Zafar H, Ponde NO, Hepworth OW, Sihra D, Aggor F, et al. IL-36 and IL-1/IL-17 Drive Immunity to Oral Candidiasis via Parallel Mechanisms. J Immunol (2018) 201:627–34. doi: 10.4049/jimmunol.1800515
6. D'Enfert C, Kaune AK, Alaban LR, Chakraborty S, Cole N, Delavy M, et al. The Impact of the Fungus-Host-Microbiota Interplay Upon Candida Albicans Infections: Current Knowledge and New Perspectives. FEMS Microbiol Rev (2021) 45(3):fuaa060. doi: 10.1093/femsre/fuaa060
7. Verma AH, Richardson JP, Zhou C, Coleman BM, Moyes DL, Ho J, et al. Oral Epithelial Cells Orchestrate Innate Type 17 Responses to Candida Albicans Through the Virulence Factor Candidalysin. Sci Immunol (2017) 2(17):eaam8834. doi: 10.1126/sciimmunol.aam8834
8. Break TJ, Oikonomou V, Dutzan N, Desai JV, Swidergall M, Freiwald T, et al. Aberrant Type 1 Immunity Drives Susceptibility to Mucosal Fungal Infections. Science (2021) 371(6526):eaay5731. doi: 10.1126/science.aay5731
9. Rosati D, Bruno M, Jaeger M, Kullberg BJ, van de Veerdonk F, Netea MG, et al. An Exaggerated Monocyte-Derived Cytokine Response to Candida Hyphae in Patients With Recurrent Vulvovaginal Candidiasis. J Infect Dis (2020) jiaa444. doi: 10.1093/infdis/jiaa444
10. Wormley FJ, Cutright J, Fidel PJ. Multiple Experimental Designs to Evaluate the Role of T-Cell-Mediated Immunity Against Experimental Vaginal Candida Albicans Infection. Med Mycol (2003) 41:401–9. doi: 10.1080/3693780310001597683
11. Wozniak KL, Wormley FJ, Fidel PJ. Candida-Specific Antibodies During Experimental Vaginal Candidiasis in Mice. Infect Immun (2002) 70:5790–9. doi: 10.1128/IAI.70.10.5790-5799.2002
12. Roselletti E, Perito S, Sabbatini S, Monari C, Vecchiarelli A. Vaginal Epithelial Cells Discriminate Between Yeast and Hyphae of Candida Albicans in Women Who Are Colonized or Have Vaginal Candidiasis. J Infect Dis (2019) 220:1645–54. doi: 10.1093/infdis/jiz365
13. Fidel PJ, Barousse M, Espinosa T, Ficarra M, Sturtevant J, Martin DH, et al. An Intravaginal Live Candida Challenge in Humans Leads to New Hypotheses for the Immunopathogenesis of Vulvovaginal Candidiasis. Infect Immun (2004) 72:2939–46. doi: 10.1128/IAI.72.5.2939-2946.2004
14. Yano J, Noverr MC, Fidel PJ. Vaginal Heparan Sulfate Linked to Neutrophil Dysfunction in the Acute Inflammatory Response Associated With Experimental Vulvovaginal Candidiasis. MBIO (2017) 8(2):e00211–17. doi: 10.1128/mBio.00211-17
15. Yano J, Peters BM, Noverr MC, Fidel PJ. Novel Mechanism Behind the Immunopathogenesis of Vulvovaginal Candidiasis: "Neutrophil Anergy". Infect Immun (2018) 86(3):e00684–17. doi: 10.1128/IAI.00684-17
16. Weindl G, Naglik JR, Kaesler S, Biedermann T, Hube B, Korting HC, et al. Human Epithelial Cells Establish Direct Antifungal Defense Through TLR4-Mediated Signaling. J Clin Invest (2007) 117:3664–72. doi: 10.1172/JCI28115
17. Netea MG, Kullberg BJ. Epithelial Sensing of Fungal Invasion. Cell Host Microbe (2010) 8:219–20. doi: 10.1016/j.chom.2010.08.008
18. Swidergall M, Solis NV, Millet N, Huang MY, Lin J, Phan QT, et al. Activation of EphA2-EGFR Signaling in Oral Epithelial Cells by Candida Albicans Virulence Factors. PloS Pathog (2021) 17:e1009221. doi: 10.1371/journal.ppat.1009221
19. Swidergall M, Solis NV, Lionakis MS, Filler SG. EphA2 is an Epithelial Cell Pattern Recognition Receptor for Fungal β-Glucans. Nat Microbiol (2018) 3:53–61. doi: 10.1038/s41564-017-0059-5
20. Ho J, Yang X, Nikou SA, Kichik N, Donkin A, Ponde NO, et al. Candidalysin Activates Innate Epithelial Immune Responses via Epidermal Growth Factor Receptor. Nat Commun (2019) 10:2297. doi: 10.1038/s41467-019-09915-2
21. Zhu W, Phan QT, Boontheung P, Solis NV, Loo JA, Filler SG. EGFR and HER2 Receptor Kinase Signaling Mediate Epithelial Cell Invasion by Candida Albicans During Oropharyngeal Infection. Proc Natl Acad Sci USA (2012) 109:14194–9. doi: 10.1073/pnas.1117676109
22. Hardbower DM, Singh K, Asim M, Verriere TG, Olivares-Villagómez D, Barry DP, et al. EGFR Regulates Macrophage Activation and Function in Bacterial Infection. J Clin Invest (2016) 126:3296–312. doi: 10.1172/JCI83585
23. Shostak K, Zhang X, Hubert P, Göktuna SI, Jiang Z, Klevernic I, et al. NF-κb-Induced KIAA1199 Promotes Survival Through EGFR Signalling. Nat Commun (2014) 5:5232. doi: 10.1038/ncomms6232
24. Moyes DL, Murciano C, Runglall M, Islam A, Thavaraj S, Naglik JR. Candida Albicans Yeast and Hyphae are Discriminated by MAPK Signaling in Vaginal Epithelial Cells. PloS One (2011) 6:e26580. doi: 10.1371/journal.pone.0026580
25. Moyes DL, Runglall M, Murciano C, Shen C, Nayar D, Thavaraj S, et al. A Biphasic Innate Immune MAPK Response Discriminates Between the Yeast and Hyphal Forms of Candida Albicans in Epithelial Cells. Cell Host Microbe (2010) 8:225–35. doi: 10.1016/j.chom.2010.08.002
26. Liu J, Willems H, Sansevere EA, Allert S, Barker KS, Lowes DJ, et al. A Variant ECE1 Allele Contributes to Reduced Pathogenicity of Candida Albicans During Vulvovaginal Candidiasis. PloS Pathog (2021) 17:e1009884. doi: 10.1371/journal.ppat.1009884
27. Gerwien F, Dunker C, Brandt P, Garbe E, Jacobsen ID, Vylkova S. Clinical Candida Albicans Vaginal Isolates and a Laboratory Strain Show Divergent Behaviors During Macrophage Interactions. Msphere (2020) 5(4):e00393–20. doi: 10.1128/mSphere.00393-20
28. Pfaller MA, Diekema DJ, Gibbs DL, Newell VA, Ellis D, Tullio V, et al. Results From the ARTEMIS DISK Global Antifungal Surveillance Study, 1997 to 2007: A 10.5-Year Analysis of Susceptibilities of Candida Species to Fluconazole and Voriconazole as Determined by CLSI Standardized Disk Diffusion. J Clin Microbiol (2010) 48:1366–77. doi: 10.1128/JCM.02117-09
29. Chen J, Hu N, Xu H, Liu Q, Yu X, Zhang Y, et al. Molecular Epidemiology, Antifungal Susceptibility, and Virulence Evaluation of Candida Isolates Causing Invasive Infection in a Tertiary Care Teaching Hospital. Front Cell Infect Microbiol (2021) 11:721439. doi: 10.3389/fcimb.2021.721439
30. Luna-Tapia A, Willems H, Parker JE, Tournu H, Barker KS, Nishimoto AT, et al. Loss of Upc2p-Inducible ERG3 Transcription Is Sufficient To Confer Niche-Specific Azole Resistance Without Compromising Candida Albicans Pathogenicity. MBIO (2018) 9(3):e00225–18. doi: 10.1128/mBio.00225-18
31. Brandolt TM, Klafke GB, Gonçalves CV, Bitencourt LR, Martinez AM, Mendes JF, et al. Prevalence of Candida Spp. In Cervical-Vaginal Samples and the In Vitro Susceptibility of Isolates. Braz J Microbiol (2017) 48:145–50. doi: 10.1016/j.bjm.2016.09.006
32. Parazzini F, Di Cintio E, Chiantera V, Guaschino S. Determinants of Different Candida Species Infections of the Genital Tract in Women. Sporachrom Study Geoup. Eur J Obstet Gynecol Reprod Biol (2000) 93:141–5. doi: 10.1016/S0301-2115(00)00289-X
33. Makanjuola O, Bongomin F, Fayemiwo SA. An Update on the Roles of Non-Albicans Candida Species in Vulvovaginitis. J Fungi (Basel) (2018) 4(4):121. doi: 10.3390/jof4040121
34. Bitew A, Abebaw Y. Vulvovaginal Candidiasis: Species Distribution of Candida and Their Antifungal Susceptibility Pattern. BMC WOMENS Health (2018) 18:94. doi: 10.1186/s12905-018-0607-z
35. Kumar D, Banerjee T, Pratap CB, Tilak R. Itraconazole-Resistant Candida Auris With Phospholipase, Proteinase and Hemolysin Activity From a Case of Vulvovaginitis. J Infect Dev Ctries (2015) 9:435–7. doi: 10.3855/jidc.4582
36. Willems H, Lowes DJ, Barker KS, Palmer GE, Peters BM. Comparative Analysis of the Capacity of the Candida Species To Elicit Vaginal Immunopathology. Infect Immun (2018) 86(12):e00527–18. doi: 10.1128/IAI.00527-18
37. Hamad M, Kazandji N, Awadallah S, Allam H. Prevalence and Epidemiological Characteristics of Vaginal Candidiasis in the UAE. MYCOSES (2014) 57:184–90. doi: 10.1111/myc.12141
38. Song N, Kan S, Pang Q, Mei H, Zheng H, Li D, et al. A Prospective Study on Vulvovaginal Candidiasis: Multicentre Molecular Epidemiology of Pathogenic Yeasts in China. J Eur Acad Dermatol Venereol (2021) 36(4):566–72. doi: 10.1111/jdv.17874
39. Dong J, Liang G, Zheng H, Kan S, Song N, Zhang M, et al. In Vitro Activity of Ravuconazole Against Candida Auris and Vaginal Candida Isolates. MYCOSES (2021) 64:651–5. doi: 10.1111/myc.13260
40. Wang L, Wang C, Mei H, Shen Y, Lv G, Zeng R, et al. Combination of Estrogen and Immunosuppressive Agents to Establish a Mouse Model of Candidiasis With Concurrent Oral and Vaginal Mucosal Infection. Mycopathologia (2016) 181:29–39. doi: 10.1007/s11046-015-9947-5
41. Moyes DL, Murciano C, Runglall M, Kohli A, Islam A, Naglik JR. Activation of MAPK/c-Fos Induced Responses in Oral Epithelial Cells is Specific to Candida Albicans and Candida Dubliniensis Hyphae. Med Microbiol Immunol (2012) 201:93–101. doi: 10.1007/s00430-011-0209-y
42. Hanaoka M, Domae E. IL-1α Released From Oral Epithelial Cells Upon Candidalysin Exposure Initiates an Early Innate Epithelial Response. Int Immunol (2021) 33:161–70. doi: 10.1093/intimm/dxaa070
43. Fidel PJ, Yano J, Esher SK, Noverr MC. Applying the Host-Microbe Damage Response Framework to Candida Pathogenesis: Current and Prospective Strategies to Reduce Damage. J Fungi (Basel) (2020) 6(1):35. doi: 10.3390/jof6010035
44. Moyes DL, Wilson D, Richardson JP, Mogavero S, Tang SX, Wernecke J, et al. Candidalysin is a Fungal Peptide Toxin Critical for Mucosal Infection. Nature (2016) 532:64–8. doi: 10.1038/nature17625
45. Kolaczkowska E, Kubes P. Neutrophil Recruitment and Function in Health and Inflammation. Nat Rev Immunol (2013) 13:159–75. doi: 10.1038/nri3399
46. Alsina-Beauchamp D, Escós A, Fajardo P, González-Romero D, Díaz-Mora E, Risco A, et al. Myeloid Cell Deficiency of P38γ/P38δ Protects Against Candidiasis and Regulates Antifungal Immunity. EMBO Mol Med (2018) 10(5):e8485. doi: 10.15252/emmm.201708485
47. Jabra-Rizk MA, Kong EF, Tsui C, Nguyen MH, Clancy CJ, Fidel PJ, et al. Candida Albicans Pathogenesis: Fitting Within the Host-Microbe Damage Response Framework. Infect Immun (2016) 84:2724–39. doi: 10.1128/IAI.00469-16
48. Casadevall A, Pirofski LA. The Damage-Response Framework of Microbial Pathogenesis. Nat Rev Microbiol (2003) 1:17–24. doi: 10.1038/nrmicro732
49. Richardson JP, Willems H, Moyes DL, Shoaie S, Barker KS, Tan SL, et al. Candidalysin Drives Epithelial Signaling, Neutrophil Recruitment, and Immunopathology at the Vaginal Mucosa. Infect Immun (2018) 86(2):e00645–17. doi: 10.1128/IAI.00645-17
50. Pekmezovic M, Hovhannisyan H, Gresnigt MS, Iracane E, Oliveira-Pacheco J, Siscar-Lewin S, et al. Candida Pathogens Induce Protective Mitochondria-Associated Type I Interferon Signalling and a Damage-Driven Response in Vaginal Epithelial Cells. Nat Microbiol (2021) 6:643–57. doi: 10.1038/s41564-021-00875-2
Keywords: Candida albicans, non-albicans Candida species, vaginal epithelial cells, EGFR, MAPK, vulvovaginal candidiasis (VVC)
Citation: Zhang J, Peng J, Li D, Mei H, Yu Y, Li X, She X and Liu W (2022) Divergent EGFR/MAPK-Mediated Immune Responses to Clinical Candida Pathogens in Vulvovaginal Candidiasis. Front. Immunol. 13:894069. doi: 10.3389/fimmu.2022.894069
Received: 11 March 2022; Accepted: 25 April 2022;
Published: 26 May 2022.
Edited by:
Wanqing Liao, Shanghai Changzheng Hospital, ChinaCopyright © 2022 Zhang, Peng, Li, Mei, Yu, Li, She and Liu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xiaodong She, c2hleGQxOTc5QDE2My5jb20=; Weida Liu, bGl1d2RAcHVtY2Rlcm0uY2Ftcy5jbg==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.