
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Immunol. , 17 May 2022
Sec. Microbial Immunology
Volume 13 - 2022 | https://doi.org/10.3389/fimmu.2022.893921
This article is part of the Research Topic Pore-Forming Toxin Effects on Immune Cells View all 4 articles
 Xiaobing Han1,2
Xiaobing Han1,2 Roger Ortines3
Roger Ortines3 Ipsita Mukherjee3
Ipsita Mukherjee3 Tulasikumari Kanipakala3Thomas Kort3
Tulasikumari Kanipakala3Thomas Kort3 Shardulendra P. Sherchand3Grant Liao3
Shardulendra P. Sherchand3Grant Liao3 Mark Mednikov3Agnes L. Chenine3
Mark Mednikov3Agnes L. Chenine3 M. Javad Aman3
M. Javad Aman3 Cory L. Nykiforuk1*
Cory L. Nykiforuk1* Rajan P. Adhikari3*
Rajan P. Adhikari3*Staphylococcus aureus has been acquiring multiple drug resistance and has evolved into superbugs such as Methicillin/Vancomycin-resistant S. aureus (MRSA/VRSA) and, consequently, is a major cause of nosocomial and community infections associated with high morbidity and mortality for which no FDA-approved vaccines or biotherapeutics are available. Previous efforts targeting the surface-associated antigens have failed in clinical testing. Here, we generated hyperimmune products from sera in rabbits against six major S. aureus toxins targeted by an experimental vaccine (IBT-V02) and demonstrated significant efficacy for an anti-virulence passive immunization strategy. Extensive in vitro binding and neutralizing titers were analyzed against six extracellular toxins from individual animal sera. All IBT-V02 immunized animals elicited the maximum immune response upon the first boost dose against all pore-forming vaccine components, while for superantigen (SAgs) components of the vaccine, second and third doses of a boost were needed to reach a plateau in binding and toxin neutralizing titers. Importantly, both anti-staphylococcus hyperimmune products consisting of full-length IgG (IBT-V02-IgG) purified from the pooled sera and de-speciated F(ab’)2 (IBT-V02-F(ab’)2) retained the binding and neutralizing titers against IBT-V02 target toxins. F(ab’)2 also exhibited cross-neutralization titers against three leukotoxins (HlgAB, HlgCB, and LukED) and four SAgs (SEC1, SED, SEK, and SEQ) which were not part of IBT-V02. F(ab’)2 also neutralized toxins in bacterial culture supernatant from major clinical strains of S. aureus. In vivo efficacy data generated in bacteremia and pneumonia models using USA300 S. aureus strain demonstrated dose-dependent protection by F(ab’)2. These efficacy data confirmed the staphylococcal toxins as viable targets and support the further development effort of hyperimmune products as a potential adjunctive therapy for emergency uses against life-threatening S. aureus infections.
Staphylococcus aureus (S. aureus) is a Gram-positive bacterium that is responsible for a variety of infections in humans ranging from mild skin infection to invasive diseases like sepsis, endocarditis, and pneumonia, which cause severe morbidity and mortality and pose a major challenge to public health. Annually, S. aureus is estimated to cause up to 20,000 deaths and cost approximately $15 billion to the health system in the U.S.A (1, 2). In addition, the widespread antimicrobial resistance among S. aureus clinical isolates results in severely restricted treatment options using existing antibiotics (3, 4). Alternative approaches to prevent and treat resistant Staphylococcal infections are urgently needed. Currently, there are no licensed vaccines or biotherapeutics targeting invasive Staphylococcal diseases.
S. aureus has a broad arsenal of virulence factors that enhance bacterial in vivo fitness by exploiting host nutrients, enabling host colonization, or evading host immune responses (5). S. aureus cell surface-associated antigens and toxins are two major groups of targets that have been intensively studied for vaccine and antibody development (6–8). Considerable effort has been undertaken to develop a vaccine or immunotherapeutic targeted at S. aureus cell surface-associated antigens including clumping factor A (ClfA) [Pfizer SA4Ag, Veronate], iron-regulated surface determinant protein B (IsdB) [Merck V710], lipoteichoic acid (LTA) [Biosynexus pagibaximab], capsular polysaccharide (CP3 and CP8) [Pfizer SA4-Ag, Nabi StaphVax], and manganese transporter protein C (MntC) [Pfizer SA4-Ag], and yet none has imparted protective immunity in controlled clinical trials (9). Indeed, in one case, the vaccination actually worsened the clinical outcome (10, 11). Collectively, these studies suggest that vaccination of surface antigens might induce pathological immune effects mediated by deleterious CD4 T-cell-dependent interferon-gamma (IFNγ) response (12). Corroborating this evidence, an animal study also demonstrated the role of vaccination against cell-surface antigens in enhancing disease severity in an infective endocarditis model (13). For the second major group of targets, seroepidemiology studies suggested a correlation between higher serum levels of toxin-specific antibodies and favorable clinical outcomes (14, 15). The observation that colonized patients present milder disease outcomes as compared to non-colonized patients demonstrated that pre-existing immunity plays some role in protecting humans against severe staphylococcal infections (16). The studies of anti-staphylococcal antibody repertoire in the general population and convalescent patients had highlighted the importance of antibody responses to leukocidins and superantigens (17, 18). These studies suggest multiple staphylococcal toxins may play a major role in the pathogenesis and impact disease outcome. Therefore, passive immunization with antibodies against major virulence factors, like toxins, should have a beneficial effect for patients having low endogenous levels of antibodies or having difficulties mounting an effective humoral response.
Staphylococcal toxins, in particular the cytolytic and superantigenic toxins, mediate tissue destruction and immune evasion (19, 20), target both innate and adaptive immune systems, and impair a wide range of immune cell functions (21). Alpha-hemolysin (Hla, or alpha-toxin), a beta-barrel pore-forming toxin that damages epithelial and endothelial cells as well as keratinocytes, plays a central role in Staphylococcal pathogenesis (22, 23). Bicomponent leukocidins kill immune cells, including phagocytes, natural killer cells, dendritic cells and T lymphocytes, lyse red blood cells to extract iron, and thus represent major virulent factors for immune disruption (24). Superantigens (SAgs) can cause massive polyclonal activation of T cells leading to cytokine storm and toxic shock, induce intoxication and dysregulation of a variety of specialized T cell subsets, and induce anergy and lymphocyte apoptosis (25). Therapeutic antibodies targeting these toxins could rescue the immune cells, preserve the immune system architecture, and ensure an effective immune response elicited against infection while not interfering with the administration of antibiotics. The anti-virulence approach could also reduce the risk of bacteria’s acquisition of resistance, due to the absence of selective pressure that directly affects bacterial survival.
Several monoclonal antibodies (mAbs) are currently being evaluated in human trials. Tosatoxumab (AR-301, an anti-Hla human IgG1), developed by Aridis, is recruiting for a phase III trial in patients (study #NCT03816956) with S. aureus ventilator-associated pneumonia (VAP) in addition to standard of care treatment. Suvratoxumab (AR-320, another anti-Hla human IgG), developed by AstraZeneca and recently licensed by Aridis, is engineered for extended half-life, and has displayed some efficacy in reducing the development of S. aureus VAP (phase II trial) (26). X-Biotech has developed 514G3, a mAb against Protein A (SpA), completed a combined phase I/II clinical trial (ClinicalTrials.gov Identifier: NCT02357966) on bacteremia patients, and reported a reduction in S. aureus-related adverse effects, but statistical significance was not reached. Overall, mAb immunotherapeutics for S. aureus prophylaxis and treatment showed promising clinical outcomes but require further validation in larger controlled clinical studies (27). Given the complexity of S. aureus pathogenesis and the diversity of virulent mechanisms, a polyclonal approach can simultaneously neutralize multiple virulent factors and is highly resistant to escape. In fact, intravenous immunoglobulin (IVIG), which contains pooled human polyclonal antibodies, can neutralize Hla and PVL produced by methicillin-resistant S. aureus (MRSA) and confer protection in a rabbit model of necrotizing pneumonia (28). However, affinity-purified anti-LukS/F-PV and anti-Hla antibodies from IVIG at 1 mg/kg provided similar protective efficacy as IVIG at 200 mg/kg, which indicated that a hyperimmune approach may be needed for providing higher titer polyclonal antibodies at lower administration volume to achieve the necessary benefits. De-speciated equine polyclonal immune globulin (EIG) is an attractive alternative for IVIG, due to its high antibody yield, ease of management of antibody response, low risk of human virus or adventitious agent contamination, and removal of the Fc region could avoid enhancement effects (mediated through Fc effector function) or aggravate inflammation observed in vaccine clinical trials. The new generation of EIG products containing highly purified F(ab’)2 fragments were demonstrated to be safe and well-tolerated in clinical use (29, 30). However, the effect of removal of Fc fragment on Ab-mediated toxin neutralization is inconclusive (31, 32), and the impact of reduced half-life of EIG on toxin clearance needs to be evaluated before proceeding to large-scale EIG manufacture.
In the present study, we immunized rabbits using a multicomponent vaccine IBT-V02, comprised of toxoids for Hla (HlaH35LH48L), subunits of PVL (LukSmut9, LukFmut1), LukAB (LukABmut50), and a fusion of three SAgs (toxoid of TSST-1, SEB, and SEA; TBA225) (8). As a surrogate to equine hyperimmune products, we generated rabbit F(ab’)2 and performed proof-of-concept studies, which demonstrated that the multivalent antibody approach targeting extracellular toxins provides significant protection against MRSA infection, the anti-Staph F(ab’)2 (IBT-V02-F(ab’)2) fragment provided equivalent in vitro Ab-mediated toxin neutralization as the full-length IgG (IBT-V02-IgG), and by increasing the bioavailability, IBT-V02-F(ab’)2 is as effective as the full-length IBT-V02-IgG in protecting animals in stringent MRSA infection models.
S. aureus strains: USA300 NRS384, USA100, MN8, Newman, and MNHOCH were received from Network on Antimicrobial Resistance in S. aureus (NARSA) (currently BEI, beiresources.org). Previously, these strains had been well characterized for their toxin profile by western blots as well as culture supernatant toxicity in rabbit blood (33, 34). Bacterial strains were further confirmed by the hemolytic pattern on sheep blood agar. Bacterial culture supernatants were prepared in 7 ml BHI media in 50 ml Falcon tubes incubating overnight at 37°C in a shaking incubator (230 rpm). The next day, OD600 was measured and normalized to 6.0 using BHI media. Culture supernatants were prepared by centrifuging the cultures followed by sterile filtration passing through 0.2 μm filters. Culture supernatants were aliquoted and stored at -80°C.
For challenge dose preparations, fresh BHI plates were inoculated by a glycerol stock generated from a single colony USA300 NRS384. The next day, colonies were transferred into 7 ml BHI broth and grown overnight to prepare the inoculum. On the 3rd day, 1 liter of BHI broth was inoculated with inoculum and grown for 6 hours at 37°C shaker flask (230 rpm). Culture suspension was then centrifuged, washed in PBS, and frozen stock aliquots were prepared and stored at -80°C.
S. aureus toxins (Hla, PVL, LukAB, LukED, HlgAB, and HlgCB) and superantigens (SEA, SEB, SED, TSST-1, SEK, SEQ, and SEC1) were provided by IBT Bioservices (ibtbioservices.com) with > 98% purity. The toxins were reconstituted in PBS and stored at -80°C until use.
The HL-60 cells, from ATCC (atcc.org), Manassas, VA were cultured in RPMI media containing 82 U/ml of penicillin and streptomycin with 16% FBS. Cells were differentiated in media with 1.5% dimethyl sulfoxide (DMSO) as described previously (6, 8, 33). PBMCs from healthy volunteers were obtained from StemExpress (stemexpress.com, Cat #LE010F).
IBT-V02 vaccine consists of five rationally designed toxoid proteins (8) including Hla toxoid, PVL toxoid consisting of mutated S and F subunit proteins, and a dimeric LukAB toxoid. To generate the inactivated toxoids of SEA, SEB, and TSST-1, three-point mutations were introduced in each corresponding wild type toxin (SEAL48R/D70R/Y92A, SEBL45R/Y89A/Y94A, and TSST-1L30R/D27A/I46A), which are the critical amino acid residues in the MHC binding sites of these toxins. These mutants had previously been tested in different animal models (35). For SEAL48R/D70R/Y92A, we introduced another extra mutation H225A to generate SEAL48R/D70R/Y92A/H225A, based on the published literature on the role of this amino acid to activate the T-cell response (36, 37). Individual toxoids were linked together by a flexible linker (three GGGGS repeats (3xG4S)) in order of TSST-1L30R/D27A/I46A, SEBL45R/Y89A/Y94A, and SEAL48R/D70R/Y92A/H225A. Hence, the name TBA225. This construct was expressed in E. coli, and the fusion protein was purified by column chromatography as described below as well as in our previously published paper (7).
The individual toxoid proteins were cloned in the vector pET24a+ and expressed in BL21 DE3 E. coli. IPTG (0.3 mM) was used to induce the expression of proteins. Cells were lysed by lysis buffer with lysozyme (1 mg/ml) followed by sonication. The supernatant containing the proteins was precipitated with ammonium sulfate. The pellet was then resuspended and desalted into capture column equilibration buffer and loaded over a Poros 50 HS cation exchange column. Fractions were collected and tested to identify the eluted fractions containing the toxoids by running and staining SDS gel against standards. Fractions were then collected and dialyzed. Each protein was thoroughly tested and stored at -80°C in storage buffer. A detailed purification method for different components of IBT-V02 has been published elsewhere (6, 7, 38). The finished products have a high level of purity, very low level of endotoxin, and expected molecular weight as characterized by SEC-HPLC, SDS-PAGE, and other QC methods described in our previous publication (8).
For rabbit immunizations, five toxoid proteins were blended, based on equal weight ratios, and used as a pentavalent immunogen. Immunogen was emulsified with Complete Freund’s Adjuvant (CFA) for prime and incomplete Freund’s Adjuvant (IFA) for boosts in a 1:1 ratio (v/v) before subcutaneous (s.c.) injection. The immunization dose was 200 µg/animal of total protein (40 µg of each toxoid) for the prime and final push, and 100 µg total protein (20 µg of each toxoid) for boosts. A total of 15 New Zealand White (NZW) specific pathogen-free (SPF) rabbits were immunized 5 times before final bleed to collect serum at Rockland under an IACUC approved protocol. Test bleeds were collected one week after each immunization. The immunization schedule is illustrated in Figure 1.

Figure 1 Rabbit Immunization Schedule. The immunization of rabbits is depicted along the top of the timeline by orange arrows at 0, 22, 36, 50, and 71 days representing the primary and subsequent boosts, respectively. The pre-immunization bleed (yellow arrow) was conducted at day zero with subsequent test bleeds performed along the timeline as denoted by the green arrows at 29, 43, and 57 days, with a terminal bleed at day 78.
After the terminal bleed, the serum from each rabbit was pooled and prepared for Low Endotoxin Unit (EU) purification (performed by Rockland Immunochemicals SOP 073A) using liquid chromatography (Protein A Sepharose 4 Fast Flow; performed by Rockland Immunochemicals SOP 073A). Purified rabbit IgG, meeting specification for purity and low endotoxin, was shipped to Emergent BioSolutions for final preparation of the test articles, IBT-V02-IgG and IBT-V02-F(ab’)2 (EBSI protocol PRO047450).
In general, the purified antibody was dialyzed in PBS buffers and transferred aseptically to the final container. The purified IgG was analyzed by HPLC using a Tosoh TSKgel UP‐SW3000 (4.6 mm x 30 cm) Column on an Agilent 1100, and by SDS‐PAGE under reduced and non‐reduced conditions. Part of purified IgG (9.82 mg/ml) was formulated into 10% (w/w) maltose, 0.03% (w/w) polysorbate 80 (PS80) as final formulation to provide an intact IgG hyperimmune control (IBT-V02-IgG). Another part of purified IgG was digested with pepsin to produce F(ab’)2 plus F(ab’)2-related immune globulin fragments. The digested material was further purified through diafiltration and anion exchange chromatography. The purified F(ab’)2 product was diafiltrated and formulated into 10% maltose and 0.03% PS80 for the F(ab’)2 test articles (IBT-V02-F(ab’)2). Sera collected from naïve rabbits were used as a negative control, the same purification process and formulation were used to purify control Rabbit F(ab’)2 (RF(ab’)2). For experiments where indicated, purified intact control Rabbit IgG (R-IgG) was purchased from Rockland. Part of the IBT-V02-F(ab’)2 was further polished using EndoTrap® HD resin and columns to meet established specifications for endotoxin levels. Two different lots of final products: lot#1 (Lot#740_POC_17_001_004) and lot #2 (Lot# 510063-20-001) were characterized using standard QC methods and the results are summarized in Table 1.
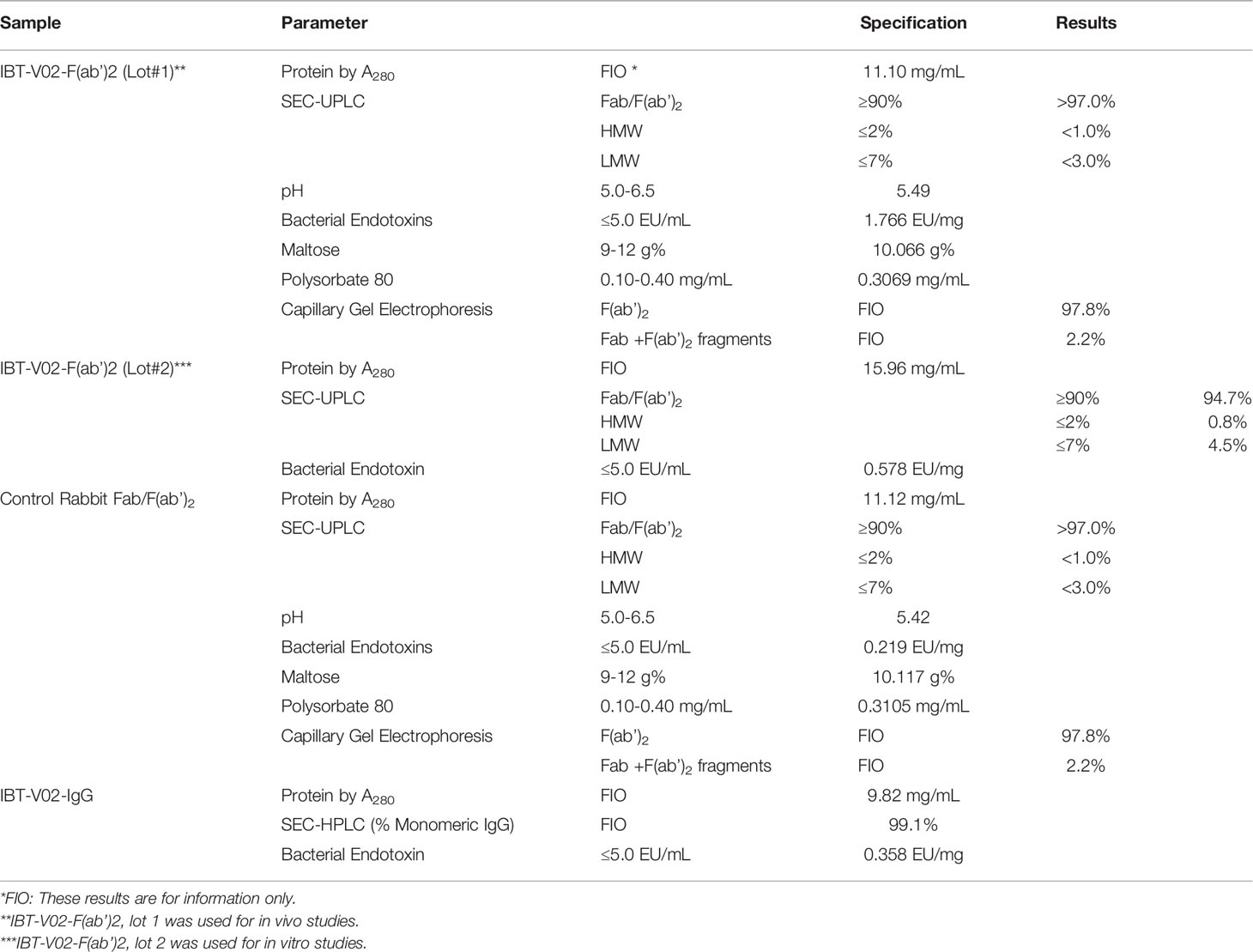
Table 1 Test Articles and Control Articles with Characterization Parameters. All test articles and controls met specifications for in vitro and in vivo testing.
A multiplex assay to detect serum IgG titers to cytolytic toxins and SAgs has been previously developed at IBT using the Luminex© xMAP technology. Briefly, IBT-V02 target antigens, Hla, LukS-PV, LukF-PV, LukAB, SEA, SEB, and TSST-1, were coupled to carboxylated MagPlex microsphere beads with distinct spectral regions via a carbodiimide reaction. Individual bead stocks (bead regions) were mixed in equal ratios to generate a multiplex master mix. The master mix was co-incubated with serum samples in a 96-well plate for 2 hours at room temperature with shaking, washed three times on a magnetic plate washer and co-incubated with a secondary PE-labeled anti-rabbit IgG for detection. Plates were washed, and samples were acquired on a Luminex 200 instrument. Data were analyzed using a 4-parameter logistic (4PL) curve fit in XLFit (IDBS, Microsoft). IgG titers were expressed as the effective dilution at the point of the 4PL curve where 50% (ED50) of antigen binding was detected by toxin-specific antibodies present in the serum sample.
Hla TNAs were performed as previously described (8). In brief, 8% rabbit red blood cells (RRBCs) were co-incubated with wildtype Hla ± serially diluted serum samples at 37°C. Cells were centrifuged after 30 min and absorbance determined at OD416nm. PVL and LukAB TNAs were performed with human promyelocytic leukemia (HL-60) cells as previously described (25). In brief, differentiated HL-60 cells were incubated with either PVL or LukAB ± serially diluted serum samples at 37°C with 5% CO2 for 3 hours, and CellTiter Glo was added to the culture to measure cell viability.
For superantigen TNAs, serum or purified antibodies were serially diluted in RPMI 1640 media (Gibco A10491), mixed with EC98 concentration of superantigens, and incubated at room temperature for 1 hour in a 96-well plate (BD Falcon BD353376). PBMCs (StemExpress #LE010F) from healthy volunteers were added such that the final density was 1 × 105 cells per well; plates were incubated at 37°C with 5% CO2 for 48 hours. The supernatants were collected and IFN-γ was measured by ELISA (R&D Systems #DY285B), to calculate neutralization as described in our previous publications (7, 8). A 4-parameter logistic (4PL) curve fit in XLfit (IDBS, Microsoft) was used to analyze the data. Based on the 4PL plots, ND50 (50% neutralizing dilution for serum) or NC50 (50% neutralizing concentration for purified antibody) were calculated.
Female BALB/c mice were purchased from Charles River Laboratories. The age of mice for each experiment was 6-8 weeks. Mice were challenged intraperitoneally (IP) with a lethal dose of USA300 NRS384 in 200 µl volume and monitored for health, weight, and survival for 14 days post-challenge. For the full length (IBT-V02-IgG) and F(ab’)2 (IBT-V02-F(ab’)2) comparative study, mice were administered IP (150 mg/kg) -2 hours prior to bacterial challenge once for the IBT-V02-IgG group, whereas for F(ab’)2 control (RF(ab’)2) and IBT-V02-F(ab’)2, mice were dosed at -2, +24, 48, 72, and 96 hours post challenge in 500 µl volume. Two different challenge doses 1xLD90 and 2xLD90 were used. For IBT-V02-F(ab’)2 dose-response study, we treated mice twice a day in two different doses: (75 mg/kg per dose; 150 mg/kg/day) or (50 mg/kg per dose; 100 mg/kg/day) in two different challenge doses 1xLD90 or 1.5xLD90. Mice were maintained under pathogen-free conditions and fed laboratory chow and water ad libitum.
For the pneumonia model, mice (6-8 wks.) were anesthetized with isoflurane and were intranasally (IN) challenged with a lethal dose of S. aureus (USA300 NRS384) in 20 or 30 µl DPBS. F(ab’)2 was administered two times per day (50 mg/kg, 75 mg/kg, or 100 mg/kg per dose) through IP route. Mice were dosed at days: D0, D1, D2, D3, and D4 post challenge in 250 µl volume per dose. Four different challenge doses were used: 5x108, 4x108, 3.5x108, and 3x108 CFU/mouse. Animals were monitored for survival, health scores, and body weight two times a day (AM and PM) within the first 4 days of challenge and then once a day until termination of the study (day 14) as described previously (39).
Data were analyzed using GraphPad PRISM v8.4.3. Statistical analysis was performed using an unpaired t-test for the comparison of area under the curve (AUC) for supernatant-based antibody neutralization assays with the controls. An overall significance level of 5% was used and the probability of a type 1 error for each test was adjusted for multiple comparisons. Kaplan-Meier curves along with log-rank (Mantel-Cox tests) were used to compare the survival rates for each treatment group to the negative control group.
IBT-V02 vaccine consists of five rationally designed proteins, seven fully attenuated toxoids, and can elicit immune responses against over 15 Staphylococcal toxins by homological cross-neutralization (8). Hla toxoid harbors two mutations (H35L and H48L), PVL toxoid consists of mutated S and F subunit proteins (LukSmut9 and LukFmut1), a dimeric toxoid LukABmut50, and a fusion protein TBA225, which represents three superantigen toxoids fused together via a flexible linker (TSST-1, SEB and SEA). Each of these toxoids is fully attenuated, and the safety and immunogenicity had been extensively studied previously (40). Using five toxoid proteins blended at equal weight ratios as immunogen to immunize rabbits, strong antibody responses were elicited. Sera from test bleeds were collected one week after each boost (Figure 1) and antibody titers to seven IBT-V02 targeted antigens (Hla, LukS-PV, LukF-PV, LukAB, SEA, SEB, and TSST-1) as determined by a multiplex Luminex assay for each rabbit are presented as the effective dilution of the sera that produced 50% maximal response (ED50) (Figure 2). IBT-V02 induced robust antibody responses after the first boost to all the antigens except SEA. The average ED50 titer for Hla is 136,673, LukS-PV is 72,458, LukF-PV is 32,083, LukAB is 77,572, SEB is 14,369, and TSST-1 is 47,006. The magnitude of the responses to each antigen was comparable to the titers obtained from previous studies, except for SEA (8). However, the second boost did induce antibody response to SEA, and the titers steadily increased until the last terminal bleed, reaching an average ED50 titer of 50,385. The antibody titers to other antigens were also gradually increased upon subsequent boosts. The terminal bleed titers against all target antigens were significantly higher than the assay internal control, which consisted of antibodies obtained by immunizing rabbits with IBT-V02 two times (IBT generated rabbit polyclonal sera). Individual rabbit responses were variable, but in close range against each antigen. There were no “non-responders” observed in the group, indicating that IBT-V02 is highly immunogenic and can successfully induce universal responsiveness with a multivalent approach.
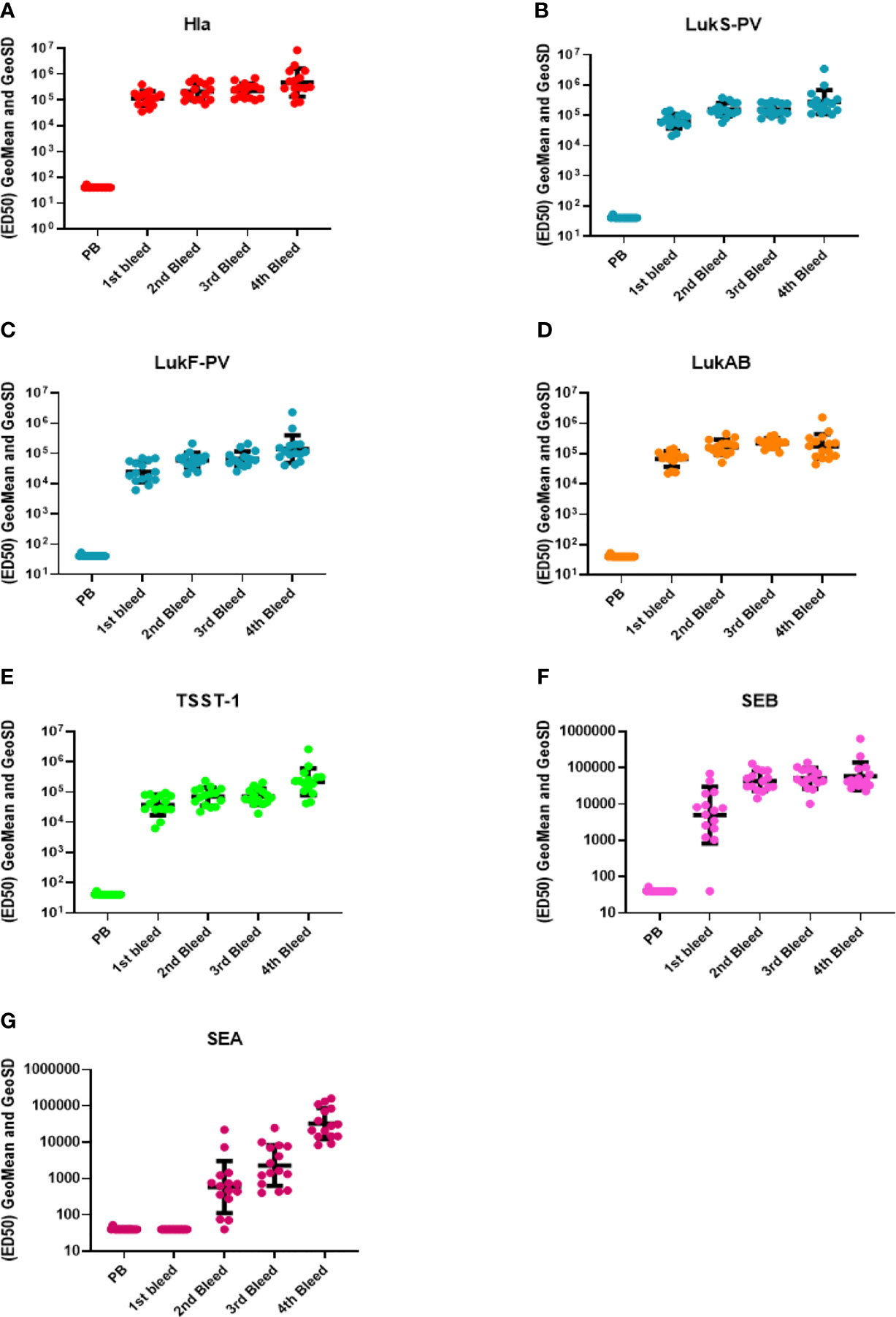
Figure 2 Antibody titers during rabbit immunization. The 50% effective dilution (ED50) for the antigen binding determined by the Luminex assay whereby 50% of antigen was detected by toxin-specific antibodies present in the serum sample, was measured in the pre-bleed (PB) and three test bleeds along with the fourth and final terminal bleed (refer to Figure 1). Toxin components are (A) Hla, (B) LukS-PV, (C) LukF-PV, (D) LukAB, (E) TSST-1, (F) SEB, and (G) SEA. Geomean values (black lines) were plotted along with GeoSD.
Three types of toxin neutralization assays (TNA) were used to further examine the function of antibodies elicited by IBT-V02 immunization. The potency of antibodies against Hla was measured by the inhibition of alpha-toxin dependent rabbit red blood cell lysis. The potency against PVL and LukAB was determined by the inhibition of leukocidin-induced cytotoxicity in PMN-like HL-60 cells. Human PBMC based superantigen TNAs were carried out to study the antibody potency against SEA, SEB, and TSST-1. As observed in the Luminex titers (Figure 2), the first boost elicited strong neutralizing antibodies to Hla, PVL, LukAB, SEA, SEB, and TSST-1, with few exceptions where neutralizing antibodies were not generated against SEA. The second boost increased the antibody response to all the antigens except SEA (Figure 3). The response to Hla, PVL, LukAB, and TSST-1 plateaued after the second boost. The response to SEB increased by subsequent boost, and upon the last boost, the neutralization titer was significantly increased for SEA. Taken together, the binding and neutralization studies showed that highly potent antibodies against all vaccine components were generated by IBT-V02 immunization.
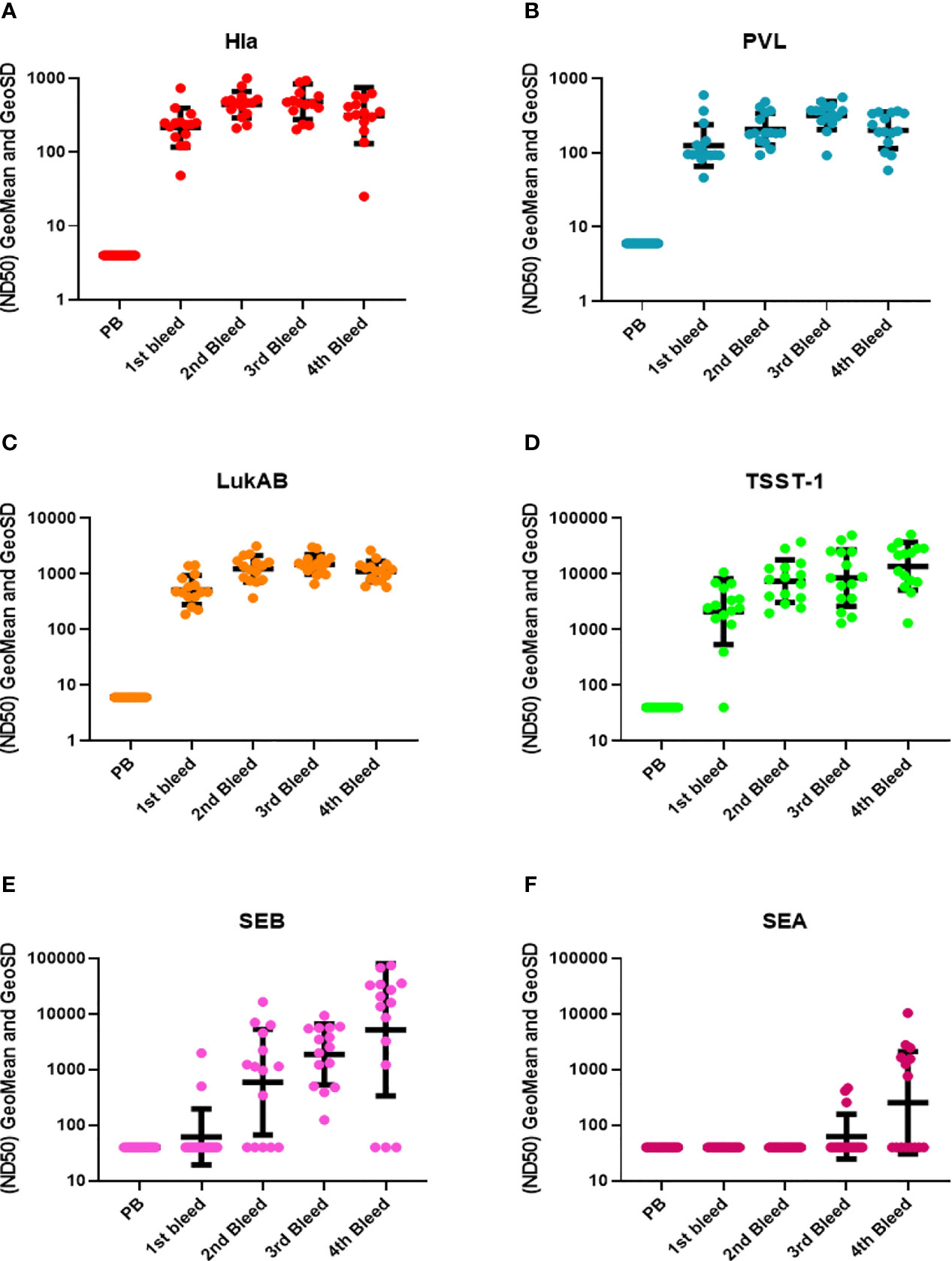
Figure 3 TNA titers during rabbit immunization. The 50% neutralizing dilution (ND50) for six different toxins were measured as described in materials and method sections. Neutralizing antibody titers (ND50s) were measured in the pre-bleed (PB) and three test bleeds along with the 4th and final terminal bleed (refer to Figure 1). Toxins included were (A) Hla, (B) PVL (LukS+LukF), (C) LukAB, (D) TSST-1, (E) SEB, and (F) SEA. Geomean values (black lines) were plotted along with GeoSD.
Next, we sought to characterize the anti-Staph hyperimmune products and compare the profiles of full-length IgG and de-speciated F(ab’)2 derived from the pooled sera of all 15 immunized rabbits. In this series of experiments, IBT-V02-IgG and IBT-V02-F(ab’)2 antibody products were tested for their binding and neutralization potencies. The Luminex EC50 for antibodies against all antigens were in hundreds of ng/mL (Supplementary Table S1) range, significantly lower than the control rabbit F(ab’)2 (RF(ab’)2) generated from naïve animals. Purified antibody products also exhibited strong neutralization against toxins. The TNA NC50s were in the single or double µg/mL range (Supplementary Table S2). When the binding and neutralization potencies were compared among the two IBT-V02-F(ab’)2 lots and one full-length IgG (IBT-V02-IgG) lot, there was no significant difference among different antibody products, indicating that the removal of Fc region did not affect IBT-V02-F(ab’)2 in vitro potency (Figure 4).
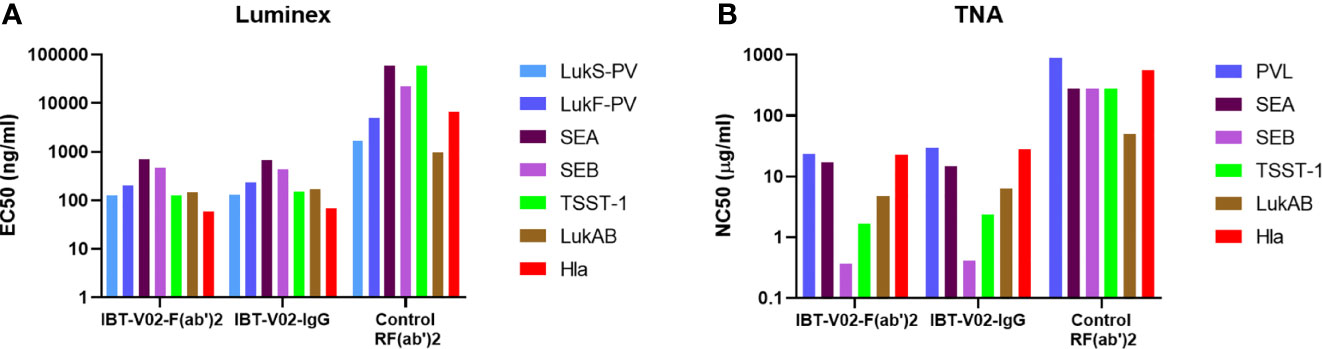
Figure 4 Anti-Staph IBT-V02-F(ab’)2 and IBT-V02-IgG binding and neutralization potency were compared with the control RF(ab’)2. (A) Luminex binding titers were compared based on 50% effective concentration (EC50) for seven antigens (components of IBT-V02), Hla, LukS-PV, LukF-PV, LukAB, SEA, SEB, and TSST-1. (B) Toxin neutralizing titers were compared based on 50% neutralizing concentration (NC50) for six toxins, Hla, PVL (LukS-PV + LukF-PV), LukAB, SEA, SEB, and TSST-1. EC50 and NC50 values were determined using a 4PL curve fit in XLFit (IDBS, Microsoft). Since no significant difference was noted among the tested two lots, data from one lot were plotted as a representation.
The bicomponent leukocidins produced by S. aureus include PVL, gamma hemolysins (HlgAB and HlgCB), LukED, and LukAB. The S and F subunits of bicomponent leukocidin have a high degree of homology respectively across the family except for LukAB. S. aureus also produces more than 20 types of SAgs, which have limited primary sequence identity but exhibit considerable structural homology. IBT-V02 vaccine contains toxoids for SEA, SEB, and TSST-1, which represent three divergent subgroups of SAgs. We tested the cross-neutralization potential first in terminal bleed samples from individual animals against leukotoxins (Figure 5) and then in purified antibodies (Figure 6). To further evaluate the toxin neutralization ability of newly purified anti-Staph hyperimmune product, we examined the cross-neutralization of IBT-V02-F(ab’)2 against leukocidins and superantigens that were not part of the vaccine components. Results showed that IBT-V02-F(ab’)2 exhibited neutralization against HlgCB and LukED when compared to control RF(ab’)2, (Figure 6) with the NC50 values in the μg/ml range, so the potency was around 10 times lower than that against PVL (Supplementary Tables S2, S3). The superantigens SEC1, SED, SEK, and SEQ were used to investigate the cross-neutralization ability of IBT-V02-F(ab’)2 against superantigens, and results indicated that IBT-V02-F(ab’)2 can totally neutralize SEC1 at two concentrations tested, and neutralize SED and SEK in higher concentrations, but to a lesser extent against SEQ (Figure 7). These data demonstrated that the cross-neutralizing activity of anti-Staph hyperimmune product can be achieved by a rationally designed multivalent vaccine with specific strategies targeting homology and specificity.
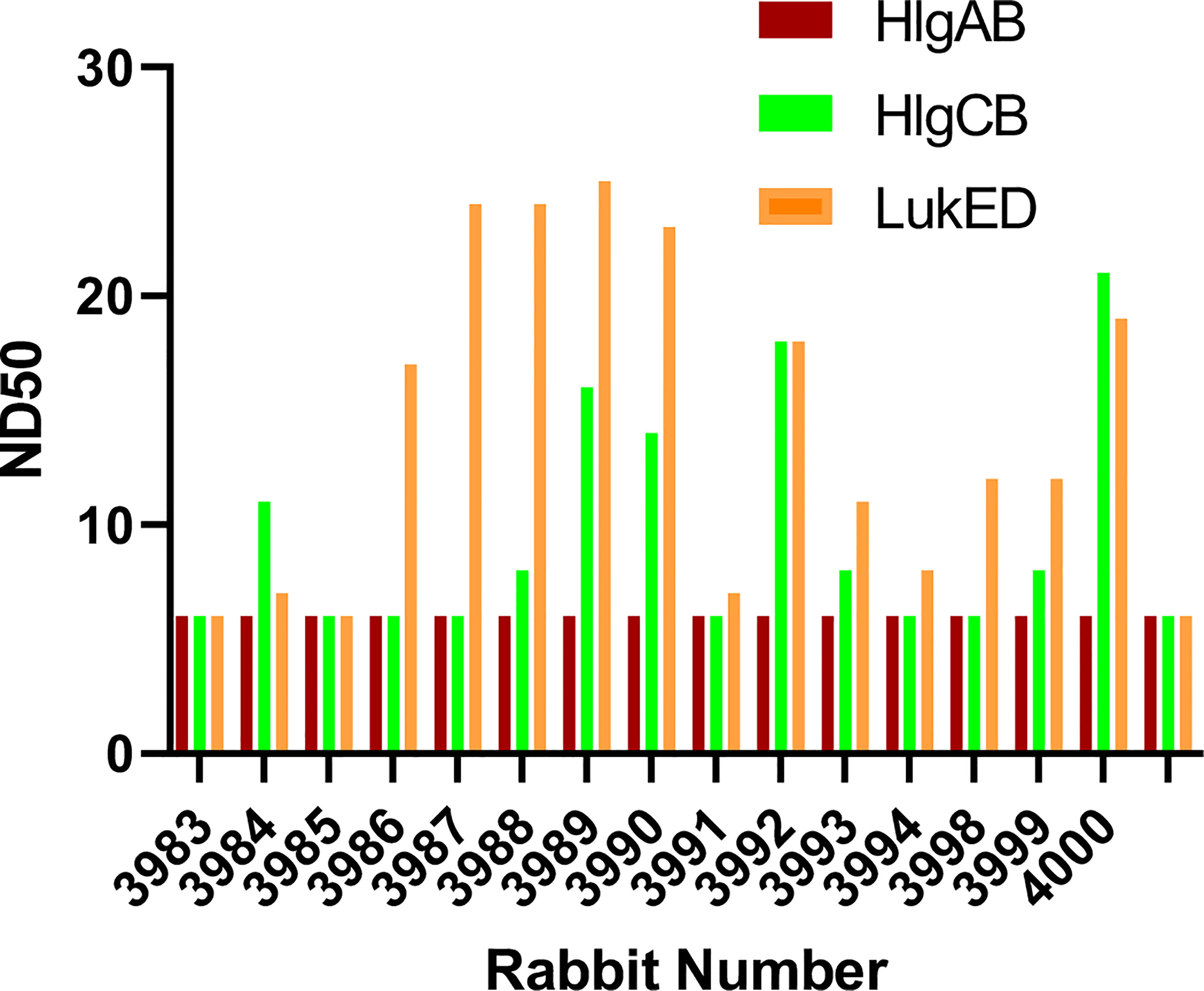
Figure 5 The cross-neutralization of terminal bleeds against other leukocidins. All 15 rabbit samples from terminal bleed were run in full curve and 50% neutralizing dilutions (ND50) were determined for three leukotoxins: HlgAB, HlgCB, and LukED.
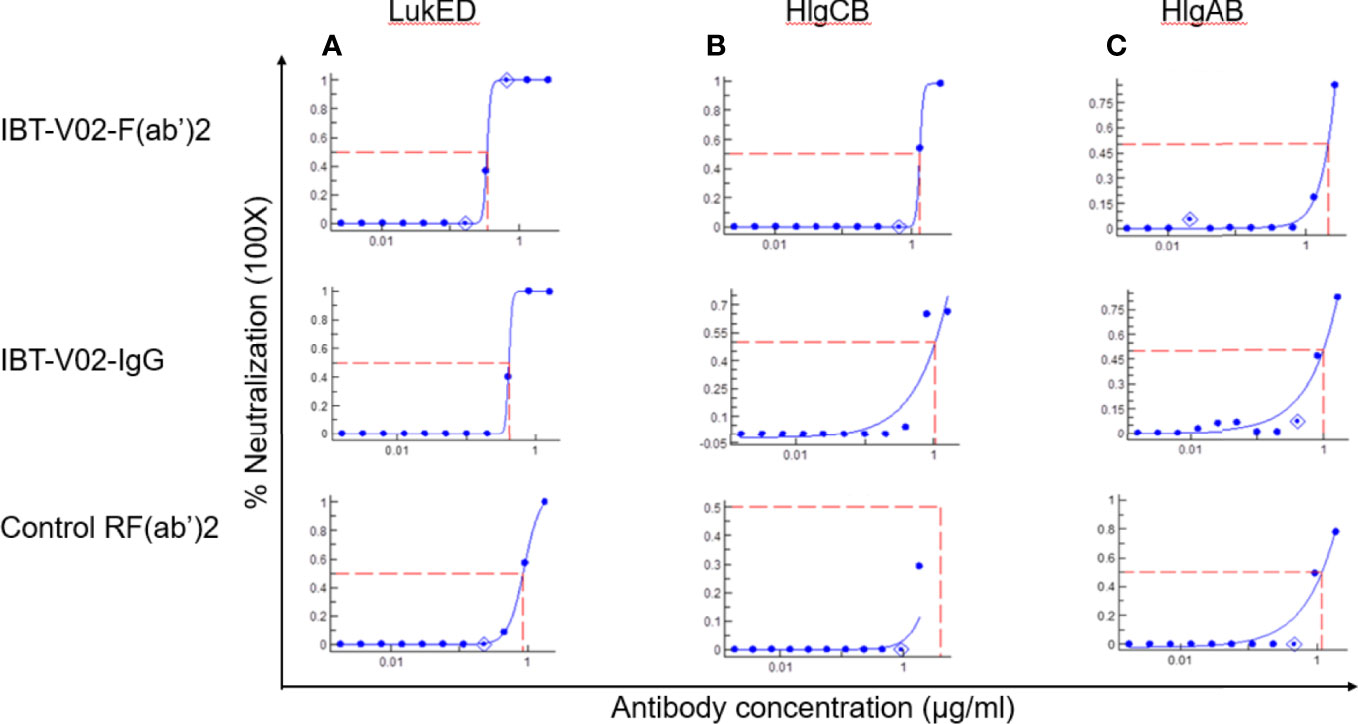
Figure 6 Cross neutralization data against 90% effective concentration (EC90) of (A) LukED, (B) HlgCB, and (C) HlgAB by IBT-V02-F(ab’)2, IBT-V02-IgG and negative control RF(ab’)2 tested in full curve and analyzed by a curve fit in XLFit (IDBS, Microsoft).
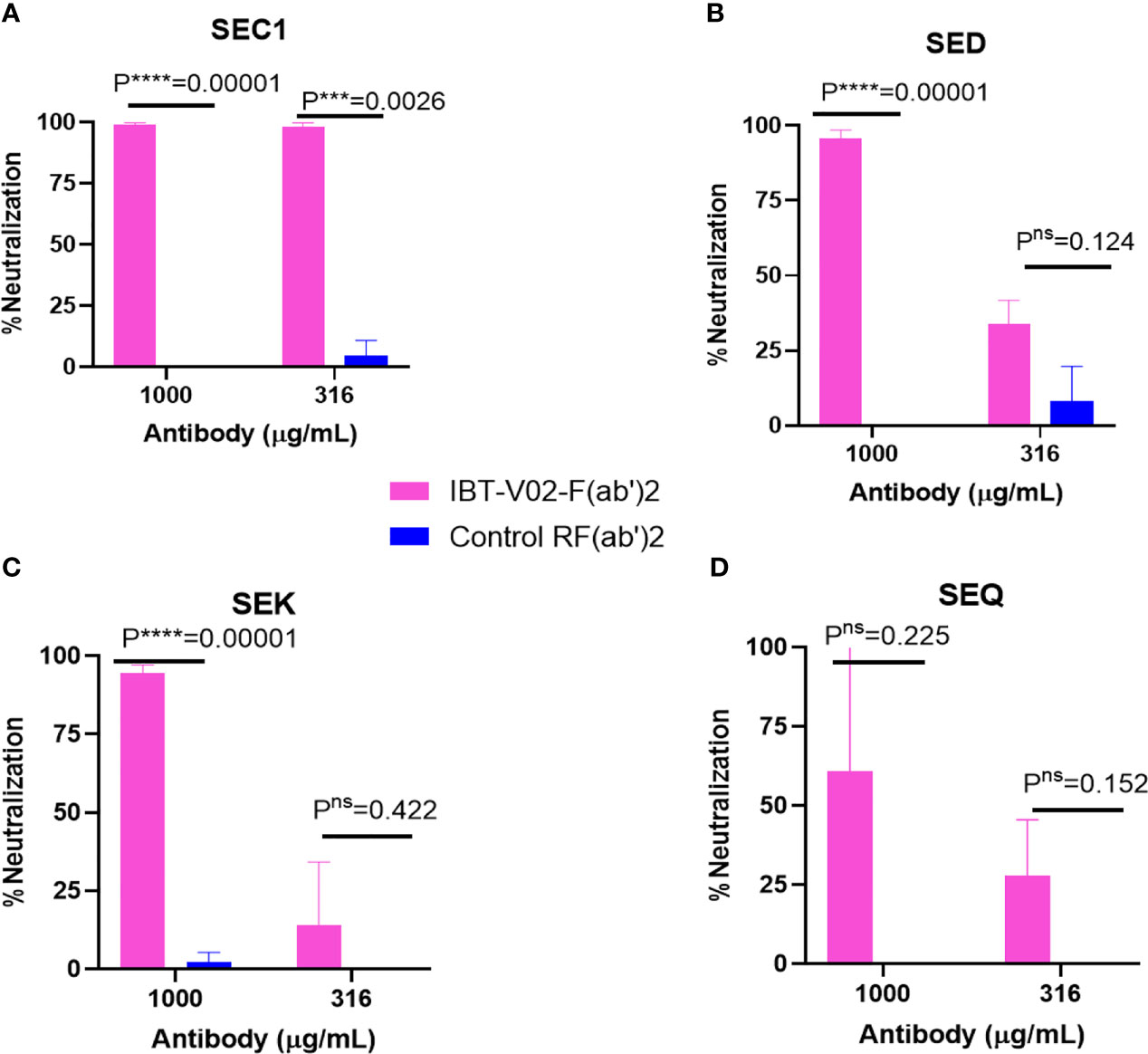
Figure 7 Cross neutralization data against 90% effective concentration (EC90) of (A) SEC1, (B) SED, (C) SEK, and (D) SEQ by IBT-V02-F(ab’)2 and control RF(ab’)2 tested in two different concentrations (1,000 and 316 µg/ml) of antibodies. The data were analyzed by unpaired multiple t-tests.p- values are represented as: ns, P > 0.05; ***, P ≤ 0.001; ****, P ≤ 0.0001.
S. aureus strains in different clonal complexes display considerable variability in toxin profiles. The neutralizing potentials of IBT-V02-F(ab’)2 were compared in bacterial culture supernatant from different clinical strains with control RF(ab’)2 and PBS alone. Among the selected strains were the USA300 LAC (JE2), USA100, and S. aureus strains which are positive for SEB, SEA, and TSST-1 superantigens. As shown in Figure 8, supernatant toxicity (PBS group) varies widely among the strains, and as expected, USA300 LAC exhibited the highest toxicity (rabbit blood-based hemolysis assay) followed by SEA positive Newman strain. We observed ED50s of 400; 150; 64; 50; and 40 for JE2 (USA300, LAC), Newman, MNHOCH, MN8, and USA100, respectively. IBT-V02-F(ab’)2 neutralizing activities were most prominent in USA300 LAC and USA100 strains when compared with MN8, Newman, and MNHOCH in presence of 40 µg/ml when compared with Control-RF(ab’)2 (Figure 8). AUC showed a significant reduction in toxicity in (IBT-V02-F(ab’)2) when compared either with PBS alone or with control RF(ab’)2 (Figure 8 AUC bar diagrams inset figures next to each plot). For supernatant neutralization of superantigens, PBMC based assays demonstrated that 100 µg/ml of IBT-V02-F(ab’)2 significantly decreased the amount of IFNγ production (readout of superantigenicity) when compared with either media alone or with control RF(ab’)2 (Supplementary Figure S1) in all three tested strains: MN8 (TSST-1); Newman (SEA); and MNHOCH (SEB). Similarly, as proof of concept, we selected two bacterial culture supernatants, JE2 and Newman, to measure the neutralizing potency of IBT-V02-F(ab’)2 against leucotoxins. As shown in Supplementary Figure S2, IBT-V02-F(ab’)2 significantly decreased the % lysis in HL-60 based assays. Nevertheless, cross-neutralizing activities of specific IBT-V02-F(ab’)2 were clearly demonstrated in all clinical isolates tested.
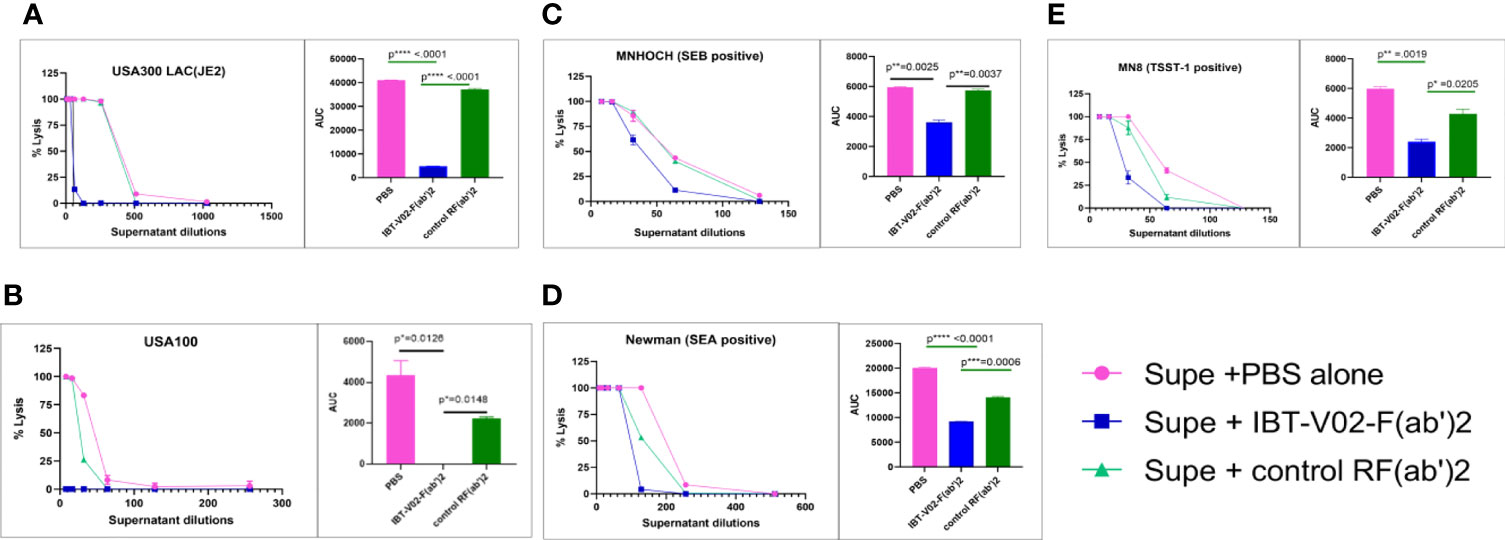
Figure 8 The bacterial culture supernatant neutralization by IBT-V02-F(ab’2) in RRBC hemolytic assay. (A) USA300 LAC JE2, (B) USA100, (C) MNHOCH, (D) Newman, and (E) MN8 neutralization by F(ab’)2. In each figure, the AUC is shown as an inset. The data were analyzed by unpaired t-test. p- values are represented as: ns, P > 0.05; *, P ≤ 0.05; **, P ≤ 0.01; ***, P ≤ 0.001; ****, P ≤ 0.0001
Given the equivalent in vitro potency observed for IBT-V02-F(ab’)2 and IBT-V02-IgG, we investigated whether these antibody products protected in a murine model of MRSA-induced bacteremia and could demonstrate equivalent in vivo potency. The bacteremia was established by intraperitoneal injection of USA300 strain NRS384. Upon 1 x 108 CFU/mouse inoculation, high bacterial loads could be recovered from the liver, kidneys, spleen, and lungs 2 days after infection, with bacteria disseminated to blood by day 4 and could still be recovered from internal organs 6 days after infection (data not included). The LD90 was determined as 2 x 108 CFU/mouse, which caused 90% mortality among the five inoculation doses tested. A single dose of 150 mg/kg of IBT-V02-IgG at 2 hours before infection and five consecutive doses of IBT-V02-F(ab’)2 at 2 hours before, and 1 day, 2 days, 3 days, and 4 days post-infection (dpi) were given to animals infected with 1xLD90 or 2xLD90 USA300 NRS384 along with the control. Upon 2xLD90 infection, all control group animals died within 24 hours, and only 10% of survival was observed for each treated group. When animals were challenged with 1xLD90 of USA300 NRS384, 90% of control group animals were dead within 48 hours; IBT-V02-IgG provided 60% survival, while IBT-V02-F(ab’)2 protected 50% of animals at the end of the 14-day study. Compared to the gradually decreased survival in the RF(ab’)2 treated group, IBT-V02-IgG treated group maintained 90% for 7 days (Figure 9A), which is about the half-life of the rabbit antibody in the murine model (41). When we closely examined the health scores, we found that the animals in the IBT-V02-F(ab’)2 group had lower health scores (i.e., healthier) in the afternoon after treatment compared to their conditions in the morning before treatment, when most mortality occurred. Based on previous studies, we have found that the half-life of other F(ab’)2 hyperimmunes in mice is about 5 hours (unpublished data). In comparison, full-length or intact IgG hyperimmunes typically have a half-life of ~3-4 days in mice (unpublished data). Taken together, we hypothesized that the protection could be improved by increasing the bioavailability of IBT-V02-F(ab’)2. To test this hypothesis, animals were treated with IBT-V02-F(ab’)2 or RF(ab’)2 control twice per day, 8 hours apart for 5 consecutive days after 1xLD90 or 1.5xLD90 infection, at two treatment doses, 75 mg/kg and 50 mg/kg. Interestingly, even the lower dose at shorter treatment intervals significantly protected the animals, with 100% and 90% of survival observed against the 1xLD90 challenge (Figure 9B). The treatment only provided 20% survival upon 1.5xLD90 infection, but significantly increased the mean time to death (MTD) of treated groups compared to control group (Log-Rank, Mantel-Cox test). The dose and bioavailability dependent protection provided by IBT-V02-F(ab’)2 treatment strongly confirmed the in vivo potency of de-speciated anti-toxin antibodies against MRSA bacteremia. The survival data were also reflected in the health score and change in weight data (Supplemental Figures: S3A, B).
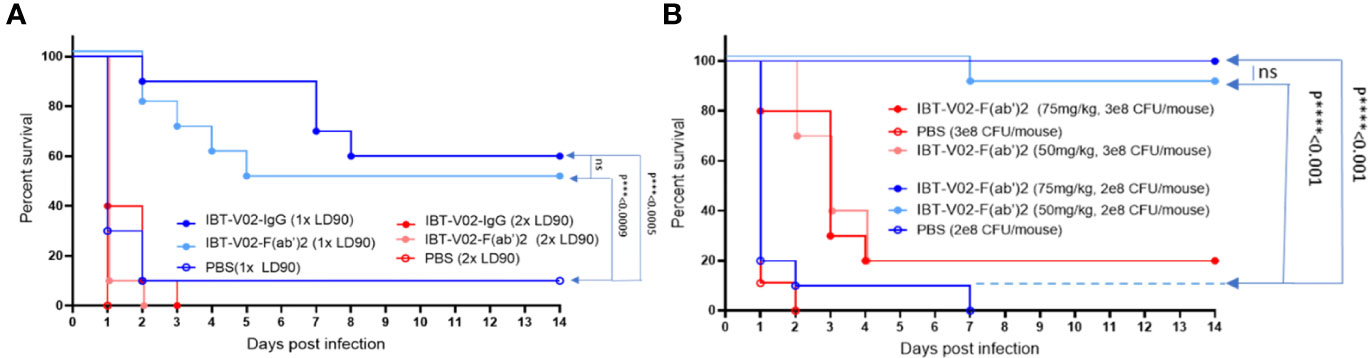
Figure 9 Efficacy studies in BALB/c mice challenged with USA300 (NRS384) in bacteremia model. (A) Comparison of IBT-V02-IgG and IBT-V02-F(ab’)2 either at 1xLD90 or at 2xLD90 with different antibody treatment doses. (B) Different treatment doses of IBT-V02-F(ab’)2 are compared with two different challenge doses. The data were analyzed by log-rank (Mantel-Cox) test. ns, not significant; ***, P < 0.001; ****, P < 0.0001.
To further evaluate the in vivo potency of anti-toxin hyperimmune products, we investigated the protective efficacy of IBT-V02-F(ab’)2 in a mouse pneumonia model. LD90 for this model was not accurately determined due to the observation of either 100% mortality upon higher inoculation or 100% survival upon lower inoculation. Therefore, we tested the efficacy using four different interpolated doses of USA300 strain NRS384 via intranasal challenge (Figures 10A, B). Upon 5x108 CFU/mouse challenge, only 10% of 75 mg/kg IBT-V02-F(ab’)2 treated animals survived, no significant protection was observed. Upon 4x108 CFU/mouse challenge, 75 mg/kg of treatment significantly extended the MTD but didn’t significantly improve the survival rate (Figure 10A). When animals were infected with 3x108 CFU/mouse USA300 NRS384, 100 mg/kg of IBT-V02-F(ab’)2 treatment provided an 80% survival rate, significantly higher than the control group. With the challenge dose increased to 3.5x108 CFU/mouse, the 40% survival was not significant compared to control, but the treatment significantly extended the MTD (Figure 10B). The survival data also correlate with the health score and change in weight data (Supplemental Figures: S4A, B). These results further confirmed the function of IBT-V02-F(ab’)2 in stringent pneumonia models and validated the efficacy of an anti-toxin passive immunization strategy against S. aureus MRSA infection.
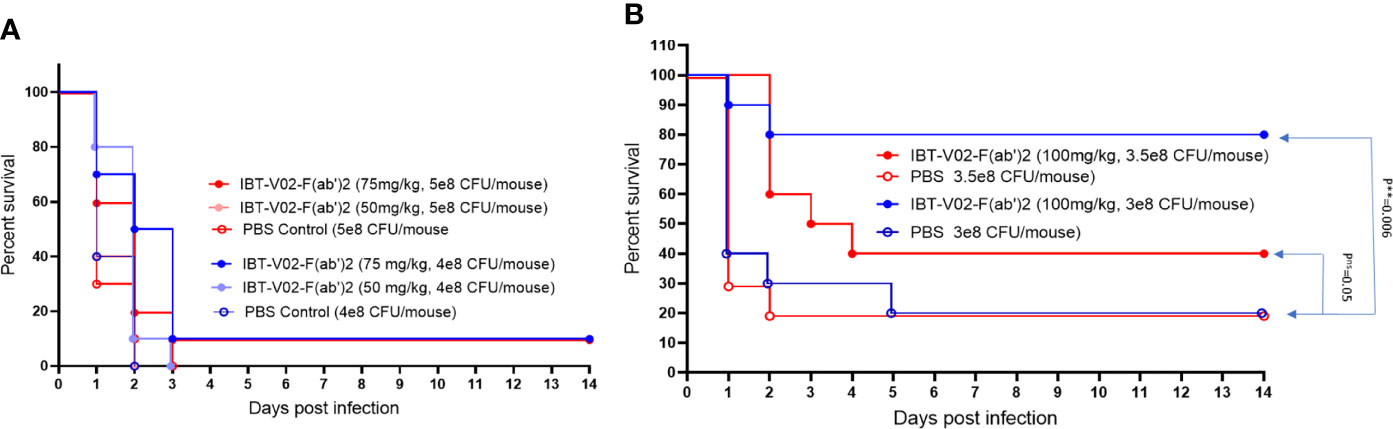
Figure 10 The dose-dependent efficacy studies in BALB/c mice challenged with USA300 (NRS384) in pneumonia model. (A) High challenged dose (5 x 108 CFU/mouse and 4 x 108 CFU/mouse) (B) Titrated challenge dose (3.5 x 108 CFU/mouse and 3 x 108 CFU/mouse). The data were analyzed by log-rank (Mantel-Cox) test. ns, P > 0.05; **, P ≤ 0.01.
In this study, we have presented for the first time that an F(ab’)2 hyperimmune generated against multivalent Staphylococcal toxoid vaccine provided significant protection against USA300 MRSA infection in both bacteremia and pneumonia mouse models. The de-speciated rabbit product (IBT-V02-F(ab’)2) retained the in vitro binding and neutralizing efficacy against IBT-V02 target toxins compared with full-length IgG (IBT-V02-IgG) and provided similar or even better protection against in vivo infection when the product’s bioavailability was improved.
De-speciated equine hyperimmune products instead of full-length IgG have been developed as therapeutics against bacterial toxins (Botulism antitoxin heptavalent types A-G, Emergent BioSolutions). In addition, antibody fragments instead of full-length IgG have been developed to enhance pharmacokinetic properties and/or the efficiency of penetration into tissues or tumor masses (caplacizumab, ranibizumab, abciximab, certolizumab pegol) (42). When the hyperimmune products were generated from heterologous hosts, the IgG Fc fragment was also removed to reduce the incidence of hypersensitivity reactions (43). Currently, there are six anti-toxin F(ab’)2 hyperimmune products that are licensed by the FDA, including Botulism antitoxin heptavalent generated from horses, and five antivenin hyperimmunes generated from horse or sheep (44). However, even with the clinical efficacy in these approved products, antibody Fc-mediated clearance is thought to play an important role in toxin neutralization, owing to both effective immune complex (IC) clearance mediated by Fcγ-receptor (FcγR) (45, 46) and IgG stabilization in the bloodstream contributed by the neonatal FcR (FcRn). In BoNT toxicity studies, the antitoxin agents that do not bear Fc had a much shorter half-life and exhibited reduced efficacy due to rapid clearance from the bloodstream (32, 47). Nevertheless, Mazuet et al. (31) showed one mAb (F(ab’)2 displayed a much higher affinity and retained its neutralization efficacy compared with intact IgG mAb (31). This evidence suggested that the high affinity of F(ab’)2 may compensate for the lack of Fc-mediated mechanisms and may induce neutralization solely via toxin blocking. In our study, the anti-staphylococcal toxins F(ab’)2 generated by immunization with IBT-V02, have a high binding affinity (Figure 4A and Supplementary Table S1) and high anti-toxin potency (Figure 4B), as indicated by in vitro characterization results (Figures 4A, B and Supplementary Tables S1, S2). In addition, when the daily dose of F(ab’)2 was split into two administrations to maintain the F(ab’)2 steady-state concentration, it improved the bioavailability, hence full protection was observed by IBT-V02-F(ab’)2. Whereas only 60% of protection was observed in the full-length IgG in the MRSA bacteremia model (Figures 9A, B). Our data successfully demonstrated that the efficacy of F(ab’)2 against toxins could be achieved by improving potency and bioavailability.
The pathogenesis of S. aureus is mediated by a vast array of surface-associated proteins, carbohydrate structures, and secreted virulence factors. The complexity of diseases and multiplicity of components pose a significant challenge for the selection of viable targets for active and passive immunization. Previous passive immunization approaches had evaluated a number of surface-associated antigens as targets, including type 5 and type 8 capsular polysaccharide (CPS) (Altastaph), fibrinogen-binding proteins clumping factor A (ClfA) (Veronate and tefibazumab), lipoteichoic acid (LTA) (Pagibaximab), ATP-binding cassette (ABC) transporter GrfA (Aurograb), poly-N-acetylated glucosamine (PNAG) (SAR279356). Unfortunately, these approaches have all failed in clinical trials. Additionally, no clinical success was obtained to date when cell surface-associated targets were developed as vaccine candidates. In fact, in the Phase III clinical trial of V710, a vaccine that targets iron-regulated surface determinant B (IsdB), vaccine recipients were five times more likely to die than the unvaccinated patients when they developed postoperative S. aureus infection (11). When cell surface-associated antigens were targeted by antibody-based strategies, phagocytic uptake and/or opsonophagocytic killing were used as indicators to demonstrate the antibody efficacy, despite the fact that no clinical studies have established such outcome as correlate of immunity (48). In the current study, we have taken an alternative approach, which is to target important extracellular secreted toxins to mitigate host tissue damage and prevent toxin-induced immune dysregulation, combined with the removal of the Fc region to avoid enhancement effects observed in clinical trials aimed at inducing direct bacterial killing.
In our study, two major types of virulence factors, pore-forming toxins (PFTs) and superantigens (SAgs), were selected as immunogens to produce the hyperimmune products with the desired function. The multivalent toxoid vaccine IBT-V02 targets Hla, PVL, LukAB, and three superantigens (SEA, SEB, and TSST-1). Hla has been recognized as a key virulence factor of S. aureus skin and soft tissue infections (SSTI), bacteremia/sepsis and pneumonia (49, 50) and is currently being evaluated as a therapeutic target in clinical development (27). PVL and LukAB are two of the five leukocidins produced by S. aureus isolates that infected humans, the other three are γ-hemolysins AB (HlgAB) and CB (HlgCB) and leukocidin ED (LukED). Leukocidins target an array of human leukocytes that are critical for innate immune defenses (21) and adaptive immunity and are associated with invasive infections such as necrotizing pneumonia (51, 52) and sepsis. Leukocidins have two subunits that are classified as the host cell targeting S component and the polymerization F component. Different from LukAB, which is pre-assembled as a heterodimer, the other S and F components are secreted as monomers before the receptor binding, assembly, and polymerization, and display 68 to 80% amino acid sequence homology. LukA and LukB share only 30 and 40% homology with the other S- and F-components respectively (53–55). Hence, we used toxoid proteins of LukF-PV and LukS-PV components separately and in combination with LukAB as immunogens, with the expectation of generating antibodies with cross-neutralization activities to other PFTs. In fact, we found that the IBT-V02-F(ab’)2 has neutralization activities against HlgAB, HlgCB and LukED (Supplementary Table 3), indicating that the anti-Staph hyperimmune generated from the IBT-V02 vaccine comprised of antibodies recognizing all five bicomponent PFTs.
There are more than 29 types of S. aureus superantigens (SAgs) in various strains, including staphylococcal enterotoxins (SEs), staphylococcal enterotoxin-like toxins (SEls), and toxic shock syndrome toxin-1 (TSST-1). Most virulent S. aureus isolates produce at least one or more SAgs (56). SAgs are not only important for toxic shock syndrome but also play a role in sepsis and possibly other S. aureus invasive diseases (57, 58). According to the homology of nucleotide and amino acid sequences, SE and SEI toxins can be classified into several groups, such as the SEA group, SEB group, and SEI group (59). Based on the genetic diversity and the clinical prevalence, the IBT-V02 vaccine includes toxoids of three SAgs (TSST-1, SEB, and SEA) expressed as a fusion protein. When IBT-V02 was used to immunize the rabbits, we found that antibodies raised against SEA and SEB were delayed in comparison to the other vaccine components. That is interesting since high titer antibodies against SEA and SEB were generated upon the first immunization in NHP (40). As immunogenicity is dependent upon the nature of the antigen and species-specific processing and presentation with the host’s MHC molecule on the surface of Antigen Presenting Cells (APCs), we suspect that SAgs are more immunogenic to NHPs, since they are closely related to humans, who are very susceptible to SAgs (60, 61). Nonetheless, after hyperimmunization, high titer antibodies were generated against SEB and SEA, and the potency (NC50) of the final product IBT-V02-F(ab’)2 is in the single or double nanomolar range, comparable with some of the anti-SAg mAbs (62, 63). Despite differences in amino acid sequence homology among SAg subgroups, these toxins share high similarity of secondary and tertiary structures (7). The combination of the three most prevalent SAgs also results in an extended cross-neutralization activity (as shown by Figure 7) and may provide broad protection against the effects of major SAgs, such as SEC1, SED, SEK and SEQ. These cross-neutralization activities may not be extended to each completed group because of the high molecular diversity of SAgs, but are more relevant for the USA300 epidemic strains where SEQ and SEK SAgs are more prevalent (64).
The multivalent approach we used in this study is not only for neutralizing multiple toxins, but also to provide broad strain coverage. S. aureus has a myriad of virulence factors and many of the genes are located on mobile genetic elements (MGE), which can be easily transferred horizontally between strains, causing diverse toxin profiles of different strains (65). On the other hand, many of the virulence factors have some degree of functional redundancy, and the expression is regulated by a complex regulation network that allows bacterial adaptation and survival in the host (66, 67). Various studies have linked specific S. aureus clonal groups with particular infections or specific virulence factors (68), but the toxin profiles of S. aureus strains are quite heterogeneous, varying remarkably even within each clonal group (69, 70). Given the genotypic and phenotypic diversity of the S. aureus strains, we tested the neutralization ability of IBT-V02-F(ab’)2 directly against the toxins present in the bacterial culture supernatants and found that IBT-V02-F(ab’)2 can neutralize the toxins produced by diverse clinical isolates. Furthermore, significant efficacy against USA300 strain was observed (Figure 8A and Supplementary Figure S2) in rabbit RBC based as well as HL-60 PMN based toxin neutralization assays. The tested USA300 clinical isolate belongs to clonal complex 8 (CC8) group and is the current epidemic lineage and predominant type of MRSA in the United States (71). The strains from this group often produce high levels of cytotoxins and are responsible for severe infections and invasive diseases. Therefore, we used a strain of USA300 MRSA in the animal studies, and results clearly indicate that IBT-V02-F(ab’)2 can provide significant protection against virulent MRSA infection in stringent infection models (Figures 9, 10).
Vaccines could play a pivotal role in the prevention of disease in populations at high-risk for S. aureus infection. IBT-V02 has been developed for preventing recurrent SSTI and has shown excellent preclinical efficacy (8). In the current study, we studied whether passive immunization with antibody products could also be applied in acute care treatment settings when there is not enough time for developing an effective immunological response, such as in bacteremia and pneumonia infections, or for vulnerable segments of the population recalcitrant to vaccine response, immunosuppressed or immunocompromised. In the treatment of S. aureus infections, we envision that the IBT-V02-F(ab’)2 protects cell and tissue integrity by neutralizing Hla, rescues immune cell function by disabling PFTs, and ensures the balanced response by obstructing SAg-induced over inflammation. IBT-V02-F(ab’)2 could be developed as adjunctive therapy to the standard of care (SOC) antibiotics to improve the clinical efficacy. Therefore, in future studies, we would test combining IBT-V02-F(ab’)2 with SOC antibiotics and examine the synergy and efficacy. Furthermore, the ability to test the combination of treatment and co-vaccination could be explored to prevent recurrence.
In summary, we generated anti-staphylococcus hyperimmune using multivalent IBT-V02 as an immunogen, demonstrating that the de-speciated F(ab’)2 (IBT-V02-F(ab’)2) retained the binding and neutralizing efficacy compared to full-length IgG, and also exhibited cross-neutralization titers against other homological toxins. IBT-V02-F(ab’)2 had broad strain coverage against toxins produced by different clinical isolates. The in vivo efficacy data generated in bacteremia and pneumonia models using USA300 S. aureus strain demonstrated a dose-dependent protection. These efficacy data confirmed that staphylococcal toxins are viable targets and support the further development effort of hyperimmune products as a potential adjunctive therapy for emergency uses against life-threatening S. aureus infections.
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding authors.
The animal study was reviewed and approved by IACUC.
All authors were directly involved in designing the experiments, data analysis, and drafting the manuscript. RO, IM, ThK, TuK, SS, GL, MM, and AC performed experiments. XH, MA, CN, and RA were involved in supervision, data analysis, and writing manuscript. All authors contributed to the article and approved the submitted version.
Part of this research was funded by grant from National Institute of Allergy and Infectious Diseases (NIAID) (R43AI136143) to RA.
RA and MA have stocks in Integrated Biotherapeutics Inc. RO, ThK, TuK, SS, GL, AC, MA, and RA are the currently employed by IBT. IM and MM were IBT employees when the study was conducted. Similarly, CN is currently employed by Emergent, and XH was employed by Emergent when the data was generated. Both have stock options in Emergent BioSolutions.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We thank Jean Moir and members of Process Development team, EBCI, for the technical assistance in IBT-V02-F(ab’)2 production. The quality control testing was performed by the EBCI QC team. We would like to thank Dr. Frederick W. Holtsberg for supervising the Luminex assay development team at IBT.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2022.893921/full#supplementary-material
1. Kourtis AP, Hatfield K, Baggs J, Mu Y, See I, Epson E, et al. Vital Signs: Epidemiology and Recent Trends in Methicillin-Resistant and in Methicillin-Susceptible Staphylococcus Aureus Bloodstream Infections - United States. MMWR Morb Mortal Wkly Rep (2019) 68(9):214–9. doi: 10.15585/mmwr.mm6809e1
2. Suaya JA, Mera RM, Cassidy A, O'Hara P, Amrine-Madsen H, Burstin S, et al. Incidence and Cost of Hospitalizations Associated With Staphylococcus Aureus Skin and Soft Tissue Infections in the United States From 2001 Through 2009. BMC Infect Dis (2014) 14:296. doi: 10.1186/1471-2334-14-296
3. Spellberg B, Guidos R, Gilbert D, Bradley J, Boucher HW, Scheld WM, et al. The Epidemic of Antibiotic-Resistant Infections: A Call to Action for the Medical Community From the Infectious Diseases Society of America. Clin Infect Dis (2008) 46(2):155–64. doi: 10.1086/524891
4. Park JW, Lee H, Kim JW, Kim B. Characterization of Infections With Vancomycin-Intermediate Staphylococcus Aureus (VISA) and Staphylococcus Aureus With Reduced Vancomycin Susceptibility in South Korea. Sci Rep (2019) 9(1):6236. doi: 10.1038/s41598-019-42307-6
5. Shettigar K, Murali TS. Virulence Factors and Clonal Diversity of Staphylococcus Aureus in Colonization and Wound Infection With Emphasis on Diabetic Foot Infection. Eur J Clin Microbiol Infect Dis (2020) 39(12):2235–46. doi: 10.1007/s10096-020-03984-8
6. Kailasan S, Kort T, Mukherjee I, Liao GC, Kanipakala T, Williston N, et al. Rational Design of Toxoid Vaccine Candidates for Staphylococcus Aureus Leukocidin AB (LukAB). Toxins (Basel) (2019) 11(6). doi: 10.3390/toxins11060339
7. Venkatasubramaniam A, Adhikari RP, Kort T, Liao GC, Conley S, Abaandou L, et al. TBA225, a Fusion Toxoid Vaccine for Protection and Broad Neutralization of Staphylococcal Superantigens. Sci Rep (2019) 9(1):3279. doi: 10.1038/s41598-019-39890-z
8. Karauzum H, Venkatasubramaniam A, Adhikari RP, Kort T, Holtsberg FW, Mukherjee I, et al. IBT-V02: A Multicomponent Toxoid Vaccine Protects Against Primary and Secondary Skin Infections Caused by Staphylococcus Aureus. Front Immunol (2021) 12:624310. doi: 10.3389/fimmu.2021.624310
9. Giersing BK, Dastgheyb SS, Modjarrad K, Moorthy V. Status of Vaccine Research and Development of Vaccines for Staphylococcus Aureus. Vaccine (2016) 34(26):2962–6. doi: 10.1016/j.vaccine.2016.03.110
10. Fowler VG, Allen KB, Moreira ED, Moustafa M, Isgro F, Boucher HW, et al. Effect of an Investigational Vaccine for Preventing Staphylococcus Aureus Infections After Cardiothoracic Surgery: A Randomized Trial. JAMA (2013) 309(13):1368–78. doi: 10.1001/jama.2013.3010
11. McNeely TB, Shah NA, Fridman A, Joshi A, Hartzel JS, Keshari RS, et al. Mortality Among Recipients of the Merck V710 Staphylococcus Aureus Vaccine After Postoperative S. Aureus Infections: An Analysis of Possible Contributing Host Factors. Hum Vaccin Immunother (2014) 10(12):3513–6. doi: 10.4161/hv.34407
12. Karauzum H, Chen G, Abaandou L, Mahmoudieh M, Boroun AR, Shulenin S, et al. Synthetic Human Monoclonal Antibodies Toward Staphylococcal Enterotoxin B (SEB) Protective Against Toxic Shock Syndrome. J Biol Chem (2012) 287(30):25203–15. doi: 10.1074/jbc.M112.364075
13. Spaulding AR, Salgado-Pabon W, Merriman JA, Stach CS, Ji Y, Gillman AN, et al. Vaccination Against Staphylococcus Aureus Pneumonia. J Infect Dis (2014) 209(12):1955–62. doi: 10.1093/infdis/jit823
14. Adhikari RP, Ajao AO, Aman MJ, Karauzum H, Sarwar J, Lydecker AD, et al. Lower Antibody Levels to Staphylococcus Aureus Exotoxins Are Associated With Sepsis in Hospitalized Adults With Invasive S. Aureus Infections. J Infect Dis (2012) 206(6):915–23. doi: 10.1093/infdis/jis462
15. Fritz SA, Tiemann KM, Hogan PG, Epplin EK, Rodriguez M, Al-Zubeidi DN, et al. A Serologic Correlate of Protective Immunity Against Community-Onset Staphylococcus Aureus Infection. Clin Infect Dis (2013) 56(11):1554–61. doi: 10.1093/cid/cit123
16. Wertheim HF, Vos MC, Ott A, van Belkum A, Voss A, Kluytmans JA, et al. Risk and Outcome of Nosocomial Staphylococcus Aureus Bacteraemia in Nasal Carriers Versus Non-Carriers. Lancet (2004) 364(9435):703–5. doi: 10.1016/S0140-6736(04)16897-9
17. Radke EE, Brown SM, Pelzek AJ, Fulmer Y, Hernandez DN, Torres VJ, et al. Hierarchy of Human IgG Recognition Within the Staphylococcus Aureus Immunome. Sci Rep (2018) 8(1):13296. doi: 10.1038/s41598-018-31424-3
18. Meyer TC, Michalik S, Holtfreter S, Weiss S, Friedrich N, Volzke H, et al. A Comprehensive View on the Human Antibody Repertoire Against Staphylococcus Aureus Antigens in the General Population. Front Immunol (2021) 12:651619. doi: 10.3389/fimmu.2021.651619
19. Kong C, Neoh HM, Nathan S. Targeting Staphylococcus Aureus Toxins: A Potential Form of Anti-Virulence Therapy. Toxins (Basel) (2016) 8(3). doi: 10.3390/toxins8030072
20. Otto M. Staphylococcus Aureus Toxins. Curr Opin Microbiol (2014) 17:32–7. doi: 10.1016/j.mib.2013.11.004
21. Scherr TD, Hanke ML, Huang O, James DB, Horswill AR, Bayles KW, et al. Staphylococcus Aureus Biofilms Induce Macrophage Dysfunction Through Leukocidin AB and Alpha-Toxin. mBio (2015) 6(4). doi: 10.1128/mBio.01021-15
22. Inoshima I, Inoshima N, Wilke GA, Powers ME, Frank KM, Wang Y, et al. A Staphylococcus Aureus Pore-Forming Toxin Subverts the Activity of ADAM10 to Cause Lethal Infection in Mice. Nat Med (2011) 17(10):1310–4. doi: 10.1038/nm.2451
23. Berube BJ, Bubeck Wardenburg J. Staphylococcus Aureus Alpha-Toxin: Nearly a Century of Intrigue. Toxins (Basel) (2013) 5(6):1140–66. doi: 10.3390/toxins5061140
24. Spaan AN, van Strijp JAG, Torres VJ. Leukocidins: Staphylococcal Bi-Component Pore-Forming Toxins Find Their Receptors. Nat Rev Microbiol (2017) 15(7):435–47. doi: 10.1038/nrmicro.2017.27
25. Oliveira D, Borges A, Simoes M. Staphylococcus Aureus Toxins and Their Molecular Activity in Infectious Diseases. Toxins (Basel) (2018) 10(6). doi: 10.3390/toxins10060252
26. Francois B, Mercier E, Gonzalez C, Asehnoune K, Nseir S, Fiancette M, et al. Safety and Tolerability of a Single Administration of AR-301, a Human Monoclonal Antibody, in ICU Patients With Severe Pneumonia Caused by Staphylococcus Aureus: First-in-Human Trial. Intensive Care Med (2018) 44(11):1787–96. doi: 10.1007/s00134-018-5229-2
27. Clegg J, Soldaini E, McLoughlin RM, Rittenhouse S, Bagnoli F, Phogat S. Staphylococcus Aureus Vaccine Research and Development: The Past, Present and Future, Including Novel Therapeutic Strategies. Front Immunol (2021) 12:705360. doi: 10.3389/fimmu.2021.705360
28. Diep BA, Le VT, Visram ZC, Rouha H, Stulik L, Dip EC, et al. Improved Protection in a Rabbit Model of Community-Associated Methicillin-Resistant Staphylococcus Aureus Necrotizing Pneumonia Upon Neutralization of Leukocidins in Addition to Alpha-Hemolysin. Antimicrob Agents Chemother (2016) 60(10):6333–40. doi: 10.1128/AAC.01213-16
29. Kodihalli S, Emanuel A, Takla T, Hua Y, Hobbs C, LeClaire R, et al. Therapeutic Efficacy of Equine Botulism Antitoxin in Rhesus Macaques. PloS One (2017) 12(11):e0186892. doi: 10.1371/journal.pone.0186892
30. Both L, White J, Mandal S, Efstratiou A. Access to Diphtheria Antitoxin for Therapy and Diagnostics. Euro Surveill (2014) 19(24). doi: 10.2807/1560-7917.ES2014.19.24.20830
31. Mazuet C, Dano J, Popoff MR, Creminon C, Volland H. Characterization of Botulinum Neurotoxin Type A Neutralizing Monoclonal Antibodies and Influence of Their Half-Lives on Therapeutic Activity. PloS One (2010) 5(8):e12416. doi: 10.1371/journal.pone.0012416
32. Torgeman A, Ozeri E, Ben David A, Diamant E, Rosen O, Schwartz A, et al. Role of Homologous Fc Fragment in the Potency and Efficacy of Anti-Botulinum Antibody Preparations. Toxins (Basel) (2017) 9(6). doi: 10.3390/toxins9060180
33. Adhikari RP, Kort T, Shulenin S, Kanipakala T, Ganjbaksh N, Roghmann MC, et al. Antibodies to S. Aureus LukS-PV Attenuated Subunit Vaccine Neutralize a Broad Spectrum of Canonical and Non-Canonical Bicomponent Leukotoxin Pairs. PloS One (2015) 10(9):e0137874. doi: 10.1371/journal.pone.0137874
34. Venkatasubramaniam A, Kanipakala T, Ganjbaksh N, Mehr R, Mukherjee I, Krishnan S, et al. A Critical Role for HlgA in Staphylococcus Aureus Pathogenesis Revealed by A Switch in the SaeRS Two-Component Regulatory System. Toxins (Basel) (2018) 10(9). doi: 10.3390/toxins10090377
35. Ulrich RG, Olson MA, Bavari S. Development of Engineered Vaccines Effective Against Structurally Related Bacterial Superantigens. Vaccine (1998) 16(19):1857–64. doi: 10.1016/S0264-410X(98)00176-5
36. Hudson KR, Tiedemann RE, Urban RG, Lowe SC, Strominger JL, Fraser JD. Staphylococcal Enterotoxin A Has Two Cooperative Binding Sites on Major Histocompatibility Complex Class II. J Exp Med (1995) 182(3):711–20. doi: 10.1084/jem.182.3.711
37. Kozono H, Parker D, White J, Marrack P, Kappler J. Multiple Binding Sites for Bacterial Superantigens on Soluble Class II MHC Molecules. Immunity (1995) 3(2):187–96. doi: 10.1016/1074-7613(95)90088-8
38. Karauzum H, Adhikari RP, Sarwar J, Devi VS, Abaandou L, Haudenschild C, et al. Structurally Designed Attenuated Subunit Vaccines for S. Aureus LukS-PV and LukF-PV Confer Protection in a Mouse Bacteremia Model. PloS One (2013) 8(6):e65384. doi: 10.1371/journal.pone.0065384
39. Adhikari RP, Karauzum H, Sarwar J, Abaandou L, Mahmoudieh M, Boroun AR, et al. Novel Structurally Designed Vaccine for S. Aureus Alpha-Hemolysin: Protection Against Bacteremia and Pneumonia. PloS One (2012) 7(6):e38567. doi: 10.1371/journal.pone.0038567
40. Venkatasubramaniam A, Liao G, Cho E, Adhikari RP, Kort T, Holtsberg FW, et al. Safety and Immunogenicity of a 4-Component Toxoid-Based Staphylococcus Aureus Vaccine in Rhesus Macaques. Front Immunol (2021) 12:621754. doi: 10.3389/fimmu.2021.621754
41. Catunda Lemos AP, Cervenak J, Bender B, Hoffmann OI, Baranyi M, Kerekes A, et al. Characterization of the Rabbit Neonatal Fc Receptor (FcRn) and Analyzing the Immunophenotype of the Transgenic Rabbits That Overexpresses FcRn. PloS One (2012) 7(1):e28869. doi 10.1371/journal.pone.0028869
42. Bates A, Power CA. David vs. Goliath: The Structure, Function, and Clinical Prospects of Antibody Fragments. Antibodies (Basel) (2019) 8(2). doi: 10.3390/antib8020028
43. Tharmalingam T, Han X, Wozniak A, Saward L. Polyclonal Hyper Immunoglobulin: A Proven Treatment and Prophylaxis Platform for Passive Immunization to Address Existing and Emerging Diseases. Hum Vaccin Immunother (2022) 18(2):1886560. doi: 10.1080/21645515.2021.1886560
44. Crowe JE Jr. Human Antibodies for Viral Infections. Annu Rev Immunol (2022) 40:349–386. doi: 10.1146/annurev-immunol-042718-041309
45. Abboud N, Chow SK, Saylor C, Janda A, Ravetch JV, Scharff MD, et al. A Requirement for FcgammaR in Antibody-Mediated Bacterial Toxin Neutralization. J Exp Med (2010) 207(11):2395–405. doi: 10.1084/jem.20100995
46. Varshney AK, Wang X, Aguilar JL, Scharff MD, Fries BC. Isotype Switching Increases Efficacy of Antibody Protection Against Staphylococcal Enterotoxin B-Induced Lethal Shock and Staphylococcus Aureus Sepsis in Mice. mBio (2014) 5(3):e01007-14. doi: 10.1128/mBio.01007-14
47. Nowakowski A, Wang C, Powers DB, Amersdorfer P, Smith TJ, Montgomery VA, et al. Potent Neutralization of Botulinum Neurotoxin by Recombinant Oligoclonal Antibody. Proc Natl Acad Sci USA (2002) 99(17):11346–50. doi: 10.1073/pnas.172229899
48. Miller LS, Fowler VG, Shukla SK, Rose WE, Proctor RA. Development of a Vaccine Against Staphylococcus Aureus Invasive Infections: Evidence Based on Human Immunity, Genetics and Bacterial Evasion Mechanisms. FEMS Microbiol Rev (2020) 44(1):123–53. doi: 10.1093/femsre/fuz030
49. Inoshima N, Wang Y, Bubeck Wardenburg J. Genetic Requirement for ADAM10 in Severe Staphylococcus Aureus Skin Infection. J Invest Dermatol (2012) 132(5):1513–6. doi: 10.1038/jid.2011.462
50. Powers ME, Kim HK, Wang Y, Bubeck Wardenburg J. ADAM10 Mediates Vascular Injury Induced by Staphylococcus Aureus Alpha-Hemolysin. J Infect Dis (2012) 206(3):352–6. doi: 10.1093/infdis/jis192
51. Jung N, Lehmann C, Hellmann M, Seifert H, Valter MM, Hallek M, et al. Necrotizing Pneumonia Caused by Panton-Valentine Leucocidin-Producing Staphylococcus Aureus Originating From a Bartholin's Abscess. Infect Dis Obstet Gynecol (2008) 2008:491401. doi: 10.1155/2008/491401
52. Rajova J, Pantucek R, Petras P, Varbanovova I, Maslanova I, Benes J. Necrotizing Pneumonia Due to Clonally Diverse Staphylococcus Aureus Strains Producing Panton-Valentine Leukocidin: The Czech Experience. Epidemiol Infect (2016) 144(3):507–15. doi: 10.1017/S0950268815001521
53. Aman MJ, Adhikari RP. Staphylococcal Bicomponent Pore-Forming Toxins: Targets for Prophylaxis and Immunotherapy. Toxins (Basel) (2014) 6(3):950–72. doi: 10.3390/toxins6030950
54. Alonzo F 3rd, Torres VJ. The Bicomponent Pore-Forming Leucocidins of Staphylococcus Aureus. Microbiol Mol Biol Rev (2014) 78(2):199–230. doi: 10.1128/MMBR.00055-13
55. Badarau A, Rouha H, Malafa S, Battles MB, Walker L, Nielson N, et al. Context Matters: The Importance of Dimerization-Induced Conformation of the LukGH Leukocidin of Staphylococcus Aureus for the Generation of Neutralizing Antibodies. MAbs (2016) 8(7):1347–60. doi: 10.1080/19420862.2016.1215791
56. Hu D-L, Li S, Fang R, Ono HK. Update on Molecular Diversity and Multipathogenicity of Staphylococcal Superantigen Toxins. Anim Diseases (2021) 1(1):7. doi: 10.1186/s44149-021-00007-7
57. Spaulding AR, Salgado-Pabon W, Kohler PL, Horswill AR, Leung DY, Schlievert PM. Staphylococcal and Streptococcal Superantigen Exotoxins. Clin Microbiol Rev (2013) 26(3):422–47. doi: 10.1128/CMR.00104-12
58. Strandberg KL, Rotschafer JH, Vetter SM, Buonpane RA, Kranz DM, Schlievert PM. Staphylococcal Superantigens Cause Lethal Pulmonary Disease in Rabbits. J Infect Dis (2010) 202(11):1690–7. doi: 10.1086/657156
59. Suzuki Y, Ono HK, Shimojima Y, Kubota H, Kato R, Kakuda T, et al. A Novel Staphylococcal Enterotoxin SE02 Involved in a Staphylococcal Food Poisoning Outbreak That Occurred in Tokyo in 2004. Food Microbiol (2020) 92:103588. doi: 10.1016/j.fm.2020.103588
60. Krakauer T. Staphylococcal Superantigens: Pyrogenic Toxins Induce Toxic Shock. Toxins (Basel) (2019) 11(3). doi: 10.3390/toxins11030178
61. Krakauer T, Stephens J, Buckley M, Tate M. Superantigen-Induced Cytokine Release From Whole-Blood Cell Culture as a Functional Measure of Drug Efficacy After Oral Dosing in Nonhuman Primates. Res Vet Sci (2007) 83(2):182–7. doi: 10.1016/j.rvsc.2006.12.004
62. Larkin EA, Stiles BG, Ulrich RG. Inhibition of Toxic Shock by Human Monoclonal Antibodies Against Staphylococcal Enterotoxin B. PloS One (2010) 5(10):e13253. doi: 10.1371/journal.pone.0013253
63. Kum WW, Chow AW. Inhibition of Staphylococcal Enterotoxin A-Induced Superantigenic and Lethal Activities by a Monoclonal Antibody to Toxic Shock Syndrome Toxin-1. J Infect Dis (2001) 183(12):1739–48. doi: 10.1086/320732
64. Glaser P, Martins-Simoes P, Villain A, Barbier M, Tristan A, Bouchier C, et al. Demography and Intercontinental Spread of the USA300 Community-Acquired Methicillin-Resistant Staphylococcus Aureus Lineage. mBio (2016) 7(1):e02183-15. doi: 10.1128/mBio.02183-15
65. Lindsay JA, Holden MT. Understanding the Rise of the Superbug: Investigation of the Evolution and Genomic Variation of Staphylococcus Aureus. Funct Integr Genomics (2006) 6(3):186–201. doi: 10.1007/s10142-005-0019-7
66. Bronner S, Monteil H, Prevost G. Regulation of Virulence Determinants in Staphylococcus Aureus: Complexity and Applications. FEMS Microbiol Rev (2004) 28(2):183–200. doi: 10.1016/j.femsre.2003.09.003
67. Novick RP. Autoinduction and Signal Transduction in the Regulation of Staphylococcal Virulence. Mol Microbiol (2003) 48(6):1429–49. doi: 10.1046/j.1365-2958.2003.03526.x
68. Tong SY, Lilliebridge RA, Bishop EJ, Cheng AC, Holt DC, McDonald MI, et al. Clinical Correlates of Panton-Valentine Leukocidin (PVL), PVL Isoforms, and Clonal Complex in the Staphylococcus Aureus Population of Northern Australia. J Infect Dis (2010) 202(5):760–9. doi: 10.1086/655396
69. King JM, Kulhankova K, Stach CS, Vu BG, Salgado-Pabon W. Phenotypes and Virulence Among Staphylococcus Aureus USA100, USA200, USA300, USA400, and USA600 Clonal Lineages. mSphere (2016) 1(3). doi: 10.1128/mSphere.00071-16
70. Thurlow LR, Joshi GS, Richardson AR. Virulence Strategies of the Dominant USA300 Lineage of Community-Associated Methicillin-Resistant Staphylococcus Aureus (CA-MRSA). FEMS Immunol Med Microbiol (2012) 65(1):5–22. doi: 10.1111/j.1574-695X.2012.00937.x
Keywords: S. aureus, hyperimmune, IBT-V02-F(ab’)2, bacteremia model, pneumonia model, IBT-V02 vaccine
Citation: Han X, Ortines R, Mukherjee I, Kanipakala T, Kort T, Sherchand SP, Liao G, Mednikov M, Chenine AL, Aman MJ, Nykiforuk CL and Adhikari RP (2022) Hyperimmune Targeting Staphylococcal Toxins Effectively Protect Against USA 300 MRSA Infection in Mouse Bacteremia and Pneumonia Models. Front. Immunol. 13:893921. doi: 10.3389/fimmu.2022.893921
Received: 11 March 2022; Accepted: 07 April 2022;
Published: 17 May 2022.
Edited by:
Sandra Sousa, Universidade do Porto, PortugalReviewed by:
Eduard Babiychuk, University of Bern, SwitzerlandCopyright © 2022 Han, Ortines, Mukherjee, Kanipakala, Kort, Sherchand, Liao, Mednikov, Chenine, Aman, Nykiforuk and Adhikari. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Cory L. Nykiforuk, Y255a2lmb3J1a0BlYnNpLmNvbQ==; Rajan P. Adhikari, cmFqYW5ASW50ZWdyYXRlZEJpb3RoZXJhcGV1dGljcy5jb20=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.