 Shuling Wu1‡
Shuling Wu1‡ Wei Yi2†‡
Wei Yi2†‡ Yuanjiao Gao1‡Wen Deng1
Yuanjiao Gao1‡Wen Deng1 Xiaoyue Bi1
Xiaoyue Bi1 Yanjie Lin3
Yanjie Lin3 Liu Yang1Yao Lu1Ruyu Liu1Min Chang1Ge Shen1Leiping Hu1
Liu Yang1Yao Lu1Ruyu Liu1Min Chang1Ge Shen1Leiping Hu1 Lu Zhang1*
Lu Zhang1* Minghui Li1,3*†
Minghui Li1,3*† Yao Xie1,3*†
Yao Xie1,3*†- 1Department of Hepatology Division 2, Beijing Ditan Hospital, Capital Medical University, Beijing, China
- 2Department of Gynecology and Obstetrics, Beijing Ditan Hospital, Capital Medical University, Beijing, China
- 3Department of Hepatology Division 2, Peking University Ditan Teaching Hospital, Beijing, China
It is considered that chronic hepatitis B patients have obtained functional cure if they get hepatitis B surface antigen (HBsAg) seroclearance after treatment. Serum HBsAg is produced by cccDNA that is extremely difficult to clear and dslDNA that is integrated with host chromosome. High HBsAg serum level leads to failure of host immune system, which makes it unable to produce effective antiviral response required for HBsAg seroclerance. Therefore, it is very difficult to achieve functional cure, and fewer than 1% of chronic hepatitis B patients are cured with antiviral treatment annually. Some chronic hepatitis B patients are coinfected with other chronic viral infections, such as HIV, HCV and HDV, which makes more difficult to cure. However, it is found that the probability of obtaining HBsAg seroclearance in patients with coinfection is higher than that in patients with HBV monoinfection, especially in patients with HBV/HIV coinfection who have an up to 36% of HBsAg 5-year-seroclerance rate. The mechanism of this interesting phenomenon is related to the functional reconstruction of immune system after antiretroviral therapy (ART). The quantity increase and function recovery of HBV specific T cells and B cells, and the higher level of cytokines and chemokines such as IP-10, GM-CSF, promote HBsAg seroclearance. This review summarizes recent studies on the immune factors that have influence on HBsAg seroconversion in the chronic hepatitis B patients with viral coinfection, which might provide new insights for the development of therapeutic approaches to partially restore the specific immune response to HBV and other viruses.
Introduction
There are more than 250 million hepatitis B virus (HBV) carriers in the world, and about 600 000 patients die of HBV-related liver diseases every year (1, 2). The pathogenesis of hepatitis B is considered to be related to the host immune response, but the underlying mechanism is not completely clear at present. In the immune tolerance state, the virus replicates a lot, and the levels of serum hepatitis B surface antigen (HBsAg) and hepatitis B e antigen (HBeAg) are very high. After entering the stage of immune clearance, the virus replication decreases, and the levels of HBsAg and HBeAg decrease as well. HBV DNA can be inhibited by effective antiviral treatment, but it is hard to clear covalently closed circular DNA (cccDNA) and double stranded linear DNA (dslDNA) which integrated with the host chromosome. As a result, it is quite difficult to completely clear serum HBsAg. Clearance of serum HBsAg with or without anti-HBs is defined as hepatitis B functional cure (3, 4). In order to improve the rate of functional cure, some new drugs have been developed and entered the stage of clinical trials. In the long process of chronic hepatitis B virus infection, the human body may also be coinfected with other hepatophilic or non-hepatophilic viruses. These viruses inhibit or activate human immunity, making the immune clearance mechanism of HBsAg more complex.
Three Forms of HBSAG Derived From Two Sources
The total length of HBV genome is about 3.2 kb, containing four partially or completely overlapping open reading frames (ORF) C, S, P and X. HBsAg is encoded by the S ORF, which contains PreS1, PreS2 and S. The production of HBsAg comes mainly from cccDNA. The 2.4 kb and 2.1 kb S mRNA of cccDNA transcripts, that is PreS1/S and PreS2/S respectively, then translates into three sizes of proteins: L-HBs (PreS1+PreS2+S), M-HBs (PreS2+S), and S-HBs (S). The three S proteins differ in their N- terminus but share a common S domain with 4 putative transmembrane (TM) domains on their C-terminus. PreS1 has 108-109 amino acid residues, PreS2 has 55 amino acid residues and S has 226 amino acid residues (see Figure 1).
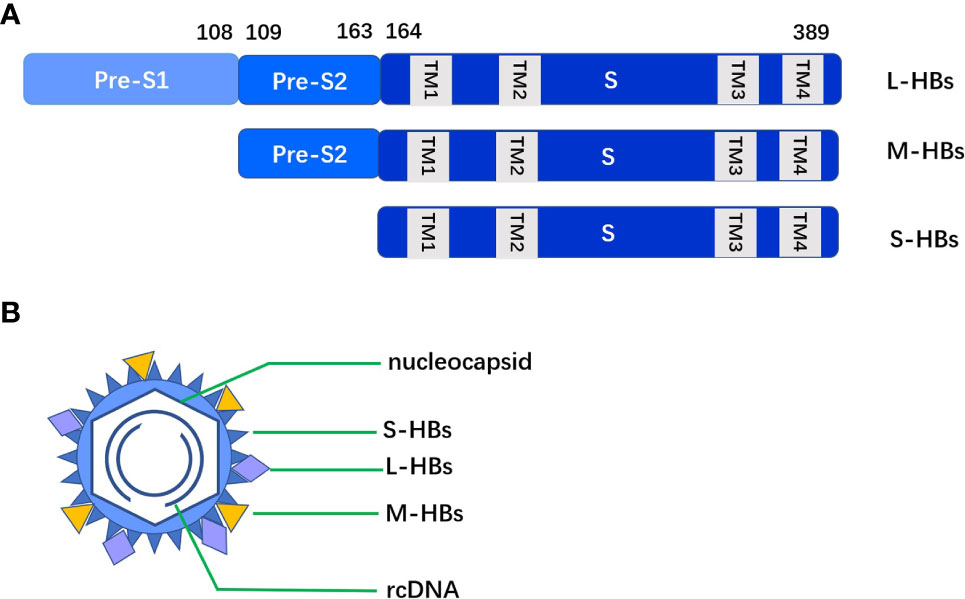
Figure 1 (A) The three S proteins (L-HBs, M-HBs and S-HBs) differ in their N- terminus but share a common S domain with 4 putative TM domains on their C-terminus. (B) Intact HBV particles contain a large amount of S-HBs and the same amount of M-HBs and L-HBs, with a composition ratio of about 4:1:1.
Another source of HBsAg is dslDNA integrated with host genes. The cccDNA transcript pregenomic RNA (pgRNA) is reverse transcribed into negative strand DNA, of which about 90% is synthesized relaxed circular DNA (rcDNA) and about 10% is synthesized dslDNA. After shelling, dslDNA enters the hepatocyte nucleus and integrates in the host gene chromosome. This integration can occur in the early stage of HBV infection, but the integration level in HBeAg positive stage is low, and the integration in HBeAg negative stage is frequent (5). HBsAg production pathways are shown in Figure 2.
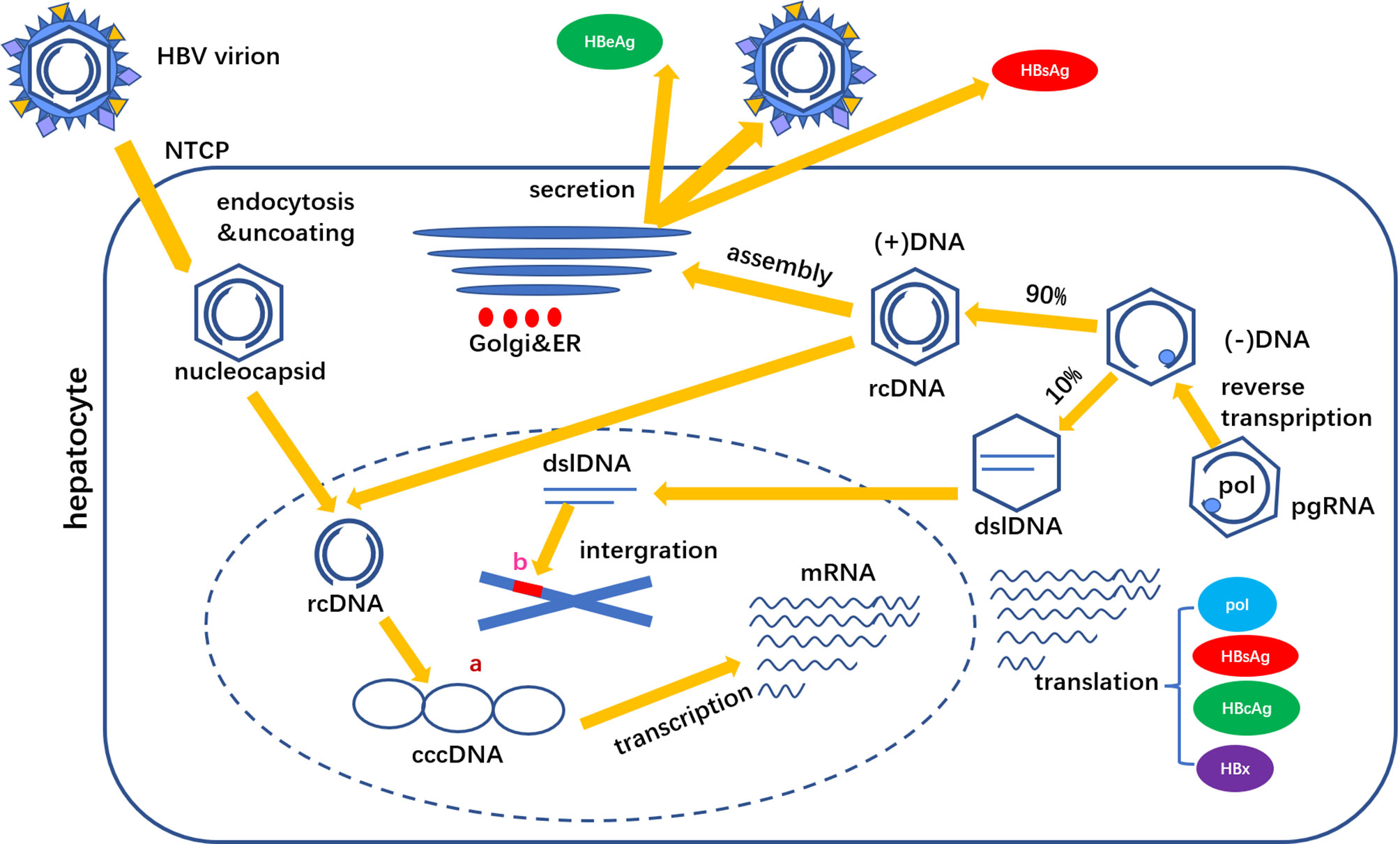
Figure 2 HBsAg is derived from two sources (A) cccDNA and (B) dslDNA. cccDNA is the main source of HBsAg production.
The integrated DNA is no longer involved in the formation of virus particles, but can translate into HBsAg. Due to the deletion of some PreS in dslDNA, the proportion of M-HBs and L-HBs in HBsAg from integrated HBV DNA is low. HBsAg translated through the above two pathways accumulates in the endoplasmic reticulum (ER) and forms agglomerates with different cysteines in the S region through covalent disulfide bonds. Intact HBV particles contain a large amount of S-HBs and the same amount of M-HBs and L-HBs, with a composition ratio of about 4:1:1 (6). HBsAg in virus particles accounts for about 1/3 of the total amount of HBsAg, other HBsAg exists in small spherical subvirus particles and filamentous particles.
A recent study found that the proportion of M-HBs in HBeAg positive patients is the best predictor of early HBsAg clearance before nucleoside analogue (NA) treatment. The median level of M-HBs in patients with HBsAg clearance before treatment is significantly lower than that in patients without HBsAg clearance. The proportion of M-HBs and L-HBs decreases rapidly during treatment, and M-HBs cannot be detected after half a year of treatment. In patients with HBsAg clearance treated with pegylated interferon (PEG-IFN), the proportion of M-HBs and L-HBs also shows similar dynamic changes (7). The mechanisms underlying the change in HBsAg composition prior to HBsAg loss is unknown. It is assumed that the structural arrangement of the integrated dslDNA form does not necessarily affect the expression of S-HBs, but parts of PreS may be missing. HBsAg derived from integrated dslDNA contains a low proportion of M-HBs and L-HBs. M-HBs and L-HBs mainly derived from cccDNA. Consequently, the decrease in M-HBs and L-HBs before HBsAg loss might reflect a progressive shutdown of cccDNA activity, but more research is needed to verify this hypothesis.
HBSAG is Related to HBV Specific Immune Dysfunction
After human body is infected by HBV, the virus is jointly cleared by innate and specific immune responses. However, the simultaneous nonspecific immune response will cause liver inflammation and necrosis, and even occurrence of liver cirrhosis and liver cancer (8). The immune function of patients with chronic hepatitis B (CHB) is impaired. On the one hand, the immune function of HBV specific T cells is low. such as the increase of immune negative regulatory components [regulatory T cells (Treg), myeloid derived suppressor cells (MDSC), programmed death receptor 1 (PD1), transforming growth factor (TGF-β), interleukin (IL)-10, etc], depressed effector function and proliferation ability, imbalance of cytokine network. As a result, the body is unable to eliminate effectively the virus, leading to continuous replication of HBV in the human body.
On the other hand, the immune response of non-HBV specific CD8+ T cells, natural killer (NK) cells and T helper (Th) 17 cells is enhanced, which can cause liver damage. A large number of non-HBV specific CD8+T cells infiltrate in the liver of patients with CHB, and their ability to proliferate and produce IL-2 is significantly reduced, but the function of other proinflammatory factors is not damaged, such as interferon (IFN)-γ and tumor necrosis factor (TNF)- α, resulting in nonspecific inflammatory injury (9, 10).
For those HBeAg positive patients in immune activation stage, there is a significant increase in NK cell activity because of increased expression of IL-12, IL-15 and IL-18 in liver and decreased expression of IL-10, NK cells enhance the killing ability, but the ability of secreting IFN-γ is not enhanced, causing liver damage but not clearance of virus. In addition, NK cells can also mediate hepatocyte apoptosis through the upregulation expression of TNF related apoptosis inducing ligand (TRAIL). A large number of Th17 cells infiltrate in the liver of patients with CHB, and is positively correlated with viral load (HBV DNA), alanine aminotransferase (ALT) level and histological activity. IL-17 secreted by Th17 cells mainly promotes the secretion of IL-1β, IL-6, TNF-α and IL-23 inflammatory factors by myeloid dendritic cells (MDC) and monocytes to mediate liver injury (11, 12).
What role does HBsAg play in immune disorder in patients with chronic hepatitis B? It is found that a large amount of HBsAg is the main factor associated with low anti HBV specific immune function. Although there is no strong evidence to support that HBsAg can directly inhibit HBV specific immune response, some studies suggest that HBsAg is related to HBV specific immune dysfunction (13, 14). High level of HBsAg is associated with the impairment of anti-HBV specific T and B cell immune function. Reducing the HBsAg level should promote the recovery of specific immune function, and in turn promote the clearance of HBsAg. When HBsAg clearance or HBsAg seroconversion is realized, the anti-HBV immune response function of the body nearly returns to normal. The incidence of HBsAg variation increases when there is an excessive immune response, such as slow plus acute liver failure (15). HBsAg can inhibit monocyte activity by binding to specific receptors on monocytes, and HBsAg can cause dysfunction of MDC and plasma cell like dendritic cells (PDC). HBsAg can also increase the response of IL-23/IL-17 axis and mediate liver immune injury (16).
HBsAg and Innate Immunity
Innate immunity is the first defense line against microbial infection, which relies on different pattern recognition receptors (PRRS) to recognize nucleic acids. Hepatitis B virus infection can activate inflammatory factors through two main types of PRR i.e., Toll like receptor (TLR) signal pathway (17–19), and retinoic acid induced gene 1 (RIG-1) signaling pathway (19–21). The expression levels of TLR3, RIG-1 and melanoma differentiation associated gene 5 (MDA5) in peripheral blood of patients with chronic hepatitis B are significantly decreased, which may account for the chronic state of HBV infection. HBsAg can inhibit innate immunity by inhibiting TLR-mediated signaling pathway and inducing IL-10 in Kupffer cells (KCS) and sinusoidal endothelial cells (LSEC) (17). In the presence of HBsAg, the function of myeloid dendritic cells (MDC) is also impaired, which stimulates T cell response (16). In another study, DC isolated from CHB patients is functional, and DC stimulates autologous HBV specific T cell expansion through the cross presentation of circulating HBsAg (22). Most experiments of HBsAg mediated innate immunity are carried out in vitro, which is related to the difference of these results. A recent study shows that HBsAg suppressed the activation of the nuclear factor kappa B (NF-кB) pathway via interaction with the TAK1-TAB2 complex, leading to downregulation of innate immune responses (23).
HBsAg and Cellular Immunity
Dysfunction and failure of HBV specific CD8+ T cell response are markers of chronic HBV infection (24, 25). High levels of HBsAg in circulation and liver may lead to impaired HBsAg specific CD8+ T cell response through continuous antigen stimulation. In addition, HBsAg can inhibit T cell response and enhance regulatory T cell response by promoting the differentiation of monocytes into MDSC (26). In the woodchuck hepatitis virus (WHV) transgenic mouse model, high levels of viral replication and protein expression in male mice induces the expansion of regulatory T cells in the liver, resulting in impaired WHV specific CD8+ T cell response and gender related differences in virus infection results (27). Compared with healthy persons, the proportion of myeloid dendritic cells (MDC) and plasma like dendritic cells (PDC) in patients with chronic hepatitis B is by and large normal, but MDC has the decreased ability of providing costimulatory signals to T cells and secreting cytokines such as TNF-α. The main function of these cytokines is to promote the maturation of DC and proliferation of DC induced T cells (28). In this process, the HBsAg and HBV DNA levels are high, suggesting that the presence of these two viral components may damage the function of MDC. Other studies have shown that in transgenic mouse models, circulating HBsAg clearance does not improve HBV specific CD8+ T cell response in vivo (29).
HBsAg and Humoral Immunity
B cell response may play an important role in controlling HBV infection. For example, the clinical application of rituximab, which consumes B cells, can lead to the reactivation of HBV in controlled patients. This suggests that the response of B cells to HBV is essential for maintaining effective host immune control of HBV (30–32). In chronic hepatitis B patients, the antigen presenting function of HBsAg and the dysfunction of CD4+ T cells due to the high level of DC can affect the secretion of anti-HBs by HBV specific B cells. This may also lead to insufficient affinity or no function of anti-HBs, so it can’t play the role of neutralizing antibody.
HBs-ELISPOT and flow cytometry fluorescence sorting (FACS) technology were used to detect HBsAg specific memory B cells (CD19 cells) in HBV vaccine inoculation staff and CHB patients. These two methods detected a small number of HBsAg specific B cells in HBV vaccine inoculation staff, but none was detected in CHB patients (33–35). Other studies have shown that the frequency of HBsAg specific B cells in blood is similar in patients with acute, chronic and cured HBV infection, and has no relationship with the serum levels of HBsAg, HBV DNA or ALT (36, 37).
HBsAg specific B cells from patients with chronic hepatitis B are atypical B cells, characterized by low expression of CD21 and CD27, but high expression of inhibitory markers such as PD-1 and T-bet. In addition, HBsAg specific B cells from patients with chronic hepatitis B are not able to mature into anti-HBs secreting cells in vitro. However, their function can be partially restored by specific culture conditions, such as PD-1 blocking or adding IL-2, IL-21 and CD40L (38). Le Bert (39) et al. found that in CHB patients, HBcAg specific B cells were more frequent than HBsAg specific B cells. The phenotypic and functional differences between HBsAg and hepatitis B core antigen (HBcAg) specific B cells in a same patient suggest that high levels of HBsAg may lead to programming obstacles of HBsAg specific B cells through continuous stimulation. Follicular helper T cell can improving HBsAg-specific B cell response in chronic hepatitis B patients targeting by TLR8 signaling (40).
In some CHB patients, the presence of anti-HBs and HBsAg coexists. Only the presence of anti-HBs may eliminate HBsAg in peripheral blood, but it will not terminate chronic HBV infection in the liver. Therefore, HBsAg specific B cell response contributes to HBV pathogenesis, clearance and protective immunity, but it is not enough to control HBV infection alone (41).
Immune Mechanisms of Current Drugs and Emerging Therapies Targeting Functional Cure of Hepatitis B
Persistent HBsAg seroclearance after treatment, and with or without anti-HBs serologic conversion, is the ideal end point of antiviral therapy for chronic hepatitis B, which represents sustained virological inhibition and immunological control. Current hepatitis B antiviral treatments include two main classes of drugs, one is the oral NAs and the other is injected IFN/PEG-IFN.
It is difficult to achieve the goal of HBsAg seroclearance with standard antiviral treatments. Functional cure occurs at an average annual rate of 0.22% in CHB patients during first-line oral NAs antiviral treatment (42). PEG-IFN treatment can acquire an average HBsAg clearance annual rate of 3% (43–45), 5-year cumulative rate of 14% and 10-year cumulative rate of 32% (46) in CHB patients. But in inactive HBsAg carriers, the HBsAg clearance rate can reach to 47% after 48 weeks of PEG-IFN treatment (47). New treatment strategies such as combination therapy (initial combination therapy of NAs and IFN/PEG-IFN, continuous combination therapy of NAs and IFN/PEG-IFN) and new therapeutic drugs may help patients improve the negative conversion rate of HBsAg and even the seroconversion rate of HBsAg.
NAs and IFN/PEG-IFN play different roles in host immune response. IFN mainly regulates innate immune response, especially NK cell activity. Micco et al. (48) found that PEG-IFN can induce the production of IL-15 and promote the activation and expansion of CD56bright NK cells, so as to enhance its antiviral activity and promote IFN-γ expression of apoptosis inducing ligand related to soluble TNF. PEG-IFN may lead to the continuous consumption of effector CD8+ T cells, and has limited repair effect on the function of HBV specific CD8+ T cells. NAs cannot resume the antiviral ability of NK cells, but temporarily repair the function of damaged T lymphocytes. In patients with virological inhibition after long-term NAs treatment, the damaged function of HBV specific T lymphocytes is partially restored in vitro (49–55). These studies showed that NAs might promote the recovery of T cell function mainly through inhibiting HBV replication. Besides, increased NK cell function is associated with active hepatitis and HBsAg seroclearance following structured NAs cessation (56). Host immune function repair is a key step to achieve chronic hepatitis B functional cure. The rationality of the combined treatment strategy of NAs and IFN/PEG-IFN lies in that the two kinds of antiviral mechanisms play different roles in innate immunity and adaptive immunity. The inhibition of HBV replication by NAs can enhance the activation of IFN on innate immunity (57, 58).
New drugs targeting at the functional cure of hepatitis B mainly include two main categories: direct antiviral drugs and indirect antiviral drugs. The former directly targets viral diseases and interfere with the replication process of HBV DNA, and the latter targets the host immune system to attack HBV. Direct antiviral drugs include siRNA (ARC-520 and JNJ-3989) (59, 60), HBV entry inhibitor (Bulevirtide, formerly known as Myrcludex B) (61), core protein allosteric regulator (NVR 3-778, JNJ-56136379, RO7049389 and ABI-H0731) (62–65), antisense RNA (IONIS-HBVRx and IONIS-HBVLRx) (66), cccDNA inhibitor (not yet in clinical trial), HBsAg release inhibitor (REP 2139) (67), HBsAg neutralizer (lenvervimab) (68), etc.
Indirect antiviral drugs include Toll like receptor (TLR) agonists (vesatolimod, selgantolimod) (69), immune checkpoint inhibitors (anti-PD-L1) (70), therapeutic vaccines (GS-4774) (71), engineering T cells, etc. Among them, the effect of therapeutic vaccine is disappointing (72). The combination of existing and new antiHBV drugs may improve HBsAg seroclearance rate (73), and the elimination of HBV requires a treatment scheme based on a combination of multiple drugs.
Immune Mechanisms and HBSAG Serum Level Changes in HBV Coinfection Patients
HBV infection here only refers to chronic HBV infection, that is, HBsAg positive lasts for more than half a year. HBV coinfection only includes HBV and other viruses, excluding bacteria, fungi, parasites, protozoa and other infections. According to the tropism of coinfected viruses, they can be divided into coinfected hepatophilic viruses such as hepatitis C virus (HCV), hepatitis D virus (HDV), hepatitis E virus (HEV), hepatitis A virus (HAV), and non-hepatophilic viruses such as human immunodeficiency virus (HIV).
HBV/HAV Coinfection
HAV is often transmitted through fecal-oral route and mostly leads to acute and self-limiting infection. In a few of cases, it can cause severe liver function damage or even liver failure (74). Acute HAV infection used to occur in adolescents who were not vaccinated against hepatitis A. Due to the emergence of hepatitis A vaccine, hepatitis A has become increasingly a disease of adults in many parts of the world. The pathogenesis of acute hepatitis A tends to be dominated by host immune response, and HAV causes a weak interferon response in the liver of infected chimpanzees (75). Compared with CD8+ T cell response, immune control of HAV may be more directly related with CD4+ T cells (76). The frequency of HAV specific CD8+ T cells in blood and liver of patients with jaundice may decrease with the clearance of infection (77).
In Ifnar1-/- transgenic mice, HAV induced hepatocyte apoptosis and inflammatory response are activated by innate immunity (78). Innate cytotoxic cells and Treg cells are transformed into inflammatory phenotypes in symptomatic infected individuals (79, 80). In HBV-infected PXB cells superinfected with HAV, HBV replication was reduced as compared to that in PXB cells infected with HBV alone, which means to a certain extent, HAV infection inhibits HBV replication (81). Earlier study also found that infection with HBV downregulated the expression of the two HBV proteins (HBsAg and PreS2) in PLC/PRF/5 cells (82). The sharp rise in IFN-γ production mediated by the acute HAV infection may be pivotal in the suppression of HBV replication in chronic hepatitis B (83). Fu et al. (84) retrospectively analyzed 211 HBV coinfection patients in a tertiary teaching hospital in China from 2005 to 2014, and 35 patients were coinfected with HAV. Patients with HAV coinfection generally had better outcomes than those with other viruses coinfection. Sagnelli et al. (85) reported that 3 of 9 patients with HBV/HAV coinfection became negative for HBsAg after 6-month follow-up. Beisel et al. (86) reported a 47-year-old patient with HBV-related compensated cirrhosis who had an acute HAV superinfection. The spontaneous HBsAg seroconversion occurred and the non-specific immunity of HAV led to functional cure of hepatitis B. Acute HAV superinfection may trigger sustained clearance of HBsAg in patients with chronic HBV infection.
HBV/HEV Coinfection
HEV is transmitted usually through fecal-oral pathway and occasionally through blood transfusion pathway (87). HEV is also a zoonotic virus in some genotypes. Acute HEV infection generally occurs in adults. Wong et al. (88) found that the seropositive rate of antiHEV-IgG was 19.86% among HBV infected patients by using the data of the National Health and Nutrition Examination Survey from 2011 to 2018. In a cross-sectional study in Vietnam from 2012 to 2013, the seropositive rate of antiHEV-IgM was 11.6% among HBV infected patients (89).
In acute hepatitis E infection patients, the percentage of NK and NKT cells in peripheral blood monocytes decreased significantly, while the ratio of activated NK and NKT cells was higher than that in the uninfected group (90). The expression of activated NK cell markers Granzyme B and CD69 also increased significantly (91). The severe condition of pregnant women infected with HEV is related to the decrease of NK cell activity (92). In pregnant women with HEV infection, the inflammatory cytokine TNF-α, IL-6 and IFN-γ level increased significantly (93).
A large amount of evidence shows that TNF-α and NF-κB signaling pathways play an important role in stimulating inflammatory response in HEV. In cell culture, TNF-α has been shown to moderately inhibit HEV replication. Interestingly, it can cooperate with IFN-α anti HEV effect through NF-κB cascade inducing a subset of IFN-stimulated gene (ISG) (94).
HEV coinfection can accelerate the disease progression of patients with chronic HBV infection and increase the mortality of patients with liver cirrhosis. Acute HEV superinfection was associated with a 1-year mortality rate of 2.4% in non-cirrhotic patients with chronic HBV infection. The 1-year mortality rate increased to 35.7% in patients with compensated liver cirrhosis after HEV superinfection. HEV superinfection increased the long-term risk of cirrhosis, hepatocarcinoma, and liver-related death in patients with chronic HBV infection (95).
Compared with HBV monoinfection, the expression of cytokines related to hepatocyte necrosis such as IL-6, IL-10 and TNF-α increased in HBV/HEV coinfection patients (96). There are scarce and conflicting data regarding the replication of viruses in coinfection patients. The median level of HBV DNA in HBV/HEV coinfection patients is lower than that in HBV monoinfected patients. However, due to the small number of samples, it is not clear whether this difference is statistically significant. In addition, baseline HBV DNA are not available to compare with HBV DNA levels after HEV superinfection (97). The higher HBV DNA level in patients with HBV monoinfection may be explained by that HEV is an RNA virus, which may play a role of ribozyme in HBV DNA replication (96). There is no significant difference in HBV DNA levels between CHB/HAV coinfection patients and CHB/HEV coinfection patients (98). Yeh et al. (99) reported the disappearance of HBsAg in a renal transplant patient with chronic HBV/HEV coinfection. However, we cannot draw a conclusion about the effect of HEV on HBsAg from a single case, and further studies are required to evaluate this hypothesis.
HBV/HCV Coinfection
HCV is a single stranded RNA virus, which mainly leads chronic infection. Innate immune response is very important for HCV infection. It limits virus transmission by inducing apoptosis of infected hepatocytes and stimulates antigen specific adaptive immune response. NK cells destroy infected hepatocytes and cytokine release through cytolysis, which plays a vital role in the innate immune response to acute HCV infection.
IFN produced by NK cells can directly inhibit HCV replication. IFN-γ and TNF-α lead to maturation of dendritic cells, release of IL-12 and differentiation of CD4 and CD8+ T cells. Specific CD8+ T cells destroys HCV infected hepatocytes through human leukocyte antigen (HLA) class I antigen presenting cells and induces cytokines (TNF-α and IFN-γ) secretion. Helper CD4+ T cells support this function through IL-2 to stimulate activation of CD8+ T cells and NK cells (100).
During chronic HCV infection, the production of IL-2 by HCV specific CD4+ T cells decreases, resulting in impaired activation of CD8+ T cells. HCV core protein and PD-1 are also associated with T cell inhibition (101). The strong CD4+ T cell response during acute HCV infection is associated with virus clearance. The lack of strong CD4+ T cell response during acute infection and the decline of CD4+ T cell response after acute infection are related to chronic progression (102). Regulatory T cells such as CD25+ T cells can inhibit CD8+ cells and cytokines (such as IL-10 and transforming growth factor TGF-β) release to inhibit immune response during chronic HCV infection (103, 104).
HBV and HCV share the same transmission mechanism, thus coinfection of HBV and HCV is common, particularly in high endemic areas where individuals have a high risk of parenteral infection. The prevalence of HBV/HCV coinfection is approximately 5%-20% in HBsAg positive patients and 2%-10% in HCV-positive patients (105). A prevalence of overt HBV coinfection in HCV positive patients was reported at 1.4% in the United States (106).
Both HBV and HCV complete their life cycle in hepatocytes, and HCV core protein strongly inhibits HBV replication during HBV/HCV coinfection (107). A recent study found that HCV core protein inhibits HBV replication by downregulating HBx levels via Siah-1-mediated proteasomal degradation during coinfection (108). HCV core gene also inhibits the induction of an immune response to HBsAg. The observed interference effect of the HCV core occurs in the priming stage and is limited to the DNA form of the HBsAg antigen, but not to the protein form (109). HCV plays a dominant role, so high HCV RNA and low HBV DNA levels are observed in most cases with HBV/HCV coinfection. The cure of HCV infection may lead to HBV reactivation, and a meta-analysis showed that the pooled proportion of patients who had HBV reactivation was 24% in patients with chronic HBV infection and 1.4% in those with resolved HBV infection (110). HBV reactivation is the result of the weakening of hepatocyte IFN response after HCV clearance. Higher serum TNF-α at baseline and lower IFN-γ at week 4 were associated with mild clinical reactivation of HBV in HBV/HCV-coinfected patients receiving direct-acting antiviral agents (DAAs) (111). Chemokine ligand CXCL-10 (another name is interferon induced protein-10, IP-10), CCL5 and ALT have predictive value for HBV reactivation after HCV clearance (112). On the other hand, exogenous HBsAg stimulated NKG2D expression on NK cells from CHB patients, which inhibits HCV replication, suggesting that HBsAg may facilitate the clearance of HCV in HBV/HCV-coinfected patients (113).
In HBV/HCV coinfection patients, the HBsAg level is usually lower than that in HBV monoinfection patients, and the decrease of HBsAg production is also related to the increase of CXCL-10 level (114). A 5-year follow-up study in HBV/HCV coinfection patients showed that the cumulative HBsAg seroclearance rate was 30.0%, with 33.1% in the 48-week PEG-IFN plus ribavirin combination therapy group, and 24.3% in the 24-week therapy group (115). DAAs-treated HBV/HCV-coinfected patients had significantly higher rate of HBV seroclearance, particularly among those with low pre-treatment HBsAg titer; on the contrary, those with higher pre-treatment HBsAg titer were at greater risk of HBV reactivation (116).
HBV/HDV Coinfection
HDV is a defective virus, which relies on HBV for packaging, release and transmission. The global total prevalence of hepatitis D varies greatly in various literatures. The rate of HBsAg positive patients complicated with HDV infection ranges from 4.5% to 13.02% (117–121). This difference may be related with the inconsistent diagnostic criteria of hepatitis D. HBV/HDV coinfection can cause the most severe viral hepatitis.
Due to severe inflammation and necrosis of hepatic lobules, liver biopsy showed that the degree of liver injury during coinfection was almost twice that of HBV or HCV monoinfection (122). Due to HDV-induced interferon response (123), pronounced induction of innate immune responses (such as elevated cytokine levels of ISGs, TGF-β, IFN-γ, IP-10, etc.) may lead to a higher degree of liver inflammation compared with HBV monoinfection, resulting in a more severe infection process (124).
In Huh7 and HEK293 cells, large hepatitis D antigen (L-HDAg) can interfere with TNF-α-NF-κB signal transduction axis (125). L-HDAg can enhance TGF-β-c-Jun induced signal cascade, while TGF-β is the main regulator of liver fibrosis and cirrhosis (126). L-HDAg can also induce oxidative stress and activate NF-κB and signal transducer and activator of transcription-3 (STAT-3), leading to liver cirrhosis and cancer (127). HBsAg may increase the translocation of L-HDAg from nucleus to ER, and the translocation is accompanied by an increase in NF-κB activity (128). Compared with HBV monoinfection, the upregulation of antigen processing mechanism leads to higher efficiency of HBV epitope presentation in HBV/HDV coinfected cells, which can promote the recognition of infected cells by T cells (129).
Although HDV has been shown to inhibit HBV replication in many studies, serum HBsAg levels in patients with HBV/HDV coinfection are higher or equal than those in patients with HBV monoinfection (130, 131). HBV/HDV coinfected sequences exhibited certain unique mutations in HBsAg genes. Some of these mutations affected the generation of proteasomal sites, binding of HBsAg epitopes to MHC-I and -II ligands, and subsequent generation of T- cell epitopes. Selective amplification of these mutations at certain strategic locations might not only enable HBV to counteract the inhibitory effects of HDV on HBV replication, but also facilitate its survival by escaping the immune response (132). The percentage of conserved HBsAg-positions was significantly higher in HBV/HDV coinfection than HBV monoinfection. HDV can constrain HBsAg genetic evolution to preserve its fitness (133).
HBV/HIV Coinfection
HIV is a non-hepatophilic virus that mainly invades lymphocytes. The human immune function gradually loses and eventually leads to acquired immune deficiency syndrome (AIDS) by HIV infection. Host and virus jointly determine the disease progression after HIV infection, in which the activation level of innate immunity plays an important role (134).
Evolution during primary HIV infection does not require adaptive immune selection (135). It is found that DC, NK cells, macrophages and NKT cells play an important and irreplaceable role in innate immunity in long-term nonprogressors and elite controllers (that means with HIV-1 infection for many years, long-term asymptomatic, normal CD4+ T cell count and no antiretroviral therapy) of HIV infection. Other innate immune cells are inefficient or even ineffective.
The progression of HIV infection may be related to the number and phenotypic function of DC. In patients with typical progression of HIV-1 infection, the number and phenotype of DC change with the progression of the disease (136), while DC in elite controllers can enhance and expand the ability to stimulate HIV specific CD8+ T cell response by improving the internal immune recognition of HIV infected cells. Type I IFN secreted by DC plays an important role in inducing effective HIV specific CD8+ T cell immunity (137).
The dysfunction of DC after HIV infection contributes to the persistence of the virus (138). NK cell activity is in the normal range in long-term nonprogressors, but decreased in patients with disease progression, indicating that NK cell activity is an important factor in controlling the progression of HIV infection (139). The levels of macrophage inflammatory protein (MIP), IP-10, monocyte chemoattractant protein-1 (MCP-1) and TGF decrease in elite controllers of HIV infection (140). Increased expression of CD224 on NKT cells is associated with HIV disease progression (141), and NKT cells in non-progression patients secrete more IFN-γ, IL-2 and TNF-α than those in progressive patients. These cytokines can significantly reduce HIV viral load and maintain a high number of CD4+ T cells (142).
In addition to the difference of innate immunity, adaptive immunity, such as CD8+ T cell function, has also been enhanced in HIV elite controllers. HIV-1 specific CD8+ T cells of elite controllers can reduce HIV-1 replication in infected CD4+ T cells by 60%-80%, and can also recognize resting infected CD4+ T cells and kill these cells without virus activation (143–146). There are a small amount of HIV-1 specific CD57+ CD4+ T cells in elite controllers, which may play a direct role in killing virus infected cells and supplement the cytotoxic activity of HIV-1 specific CD8+ T cells (147). Elite controls can produce effective anti-HIV-1 antibodies, and the frequency of preserved memory B cells is higher (148, 149).
A meta-analysis showed that the global rate of combined HBV infection in HIV patients was 7.6% (150). HIV coinfection has a negative impact on the progress of HBV infection, which can lead to rapid progression to liver fibrosis and cirrhosis (151). Enhanced production of CXCL10 following coinfection of hepatocytes with both HIV and HBV may contribute to accelerated liver disease in the setting of HIV/HBV coinfection (152). It is known that HBV Pre-S deletion is closely related to HBV-associated terminal liver disease in HBV monoinfection. High-frequency Pre-S quasispecies deletions are predominant in HIV/HBV coinfection patients, providing a reference for the pathogenesis of the accelerated progression of liver disease in HIV/HBV coinfection (153). Even after effective antiretroviral therapy, the chronic immune activation of patients with HIV/HBV coinfection is higher than that of patients with HIV momoinfection. Chronic immune activation may lead to hepatic steatosis and cirrhosis, increase the risk of liver cancer (154). At the same time, the presence of active HBV infection will affect the viral immunological status of patients with HIV/HBV coinfection, which is characterized by the low number of CD4+ T cells at the onset and the slow recovery of CD4+ T cell count after antiretroviral treatment (155, 156).
HBsAg levels and HBsAg production were significantly higher in untreated HIV/HBV coinfection patients compared to HBV monoinfection patients. The highest HBsAg concentrations were observed in patients with more advanced HIV disease (157). Compared with patients with HBV monoinfection, successful long-term tenofovir dipivoxil (TDF) inclusive ART can increase the HBsAg seroclearance rate in HIV/HBV coinfection patients, reaching 3.2%-36% (158–173) (see Table 1). The longer the follow-up time, the higher the HBsAg seroclearance rate. Higher HBsAg seroclearance rate is associated with increased CD4+ T cells. The sudden recovery of adaptive immunity causes immune reconstitution inflammatory syndrome, and then accelerates the production of protective antibodies. Therefore, immune reconstitution under antiretroviral therapy may affect the HBsAg serum conversion rate.
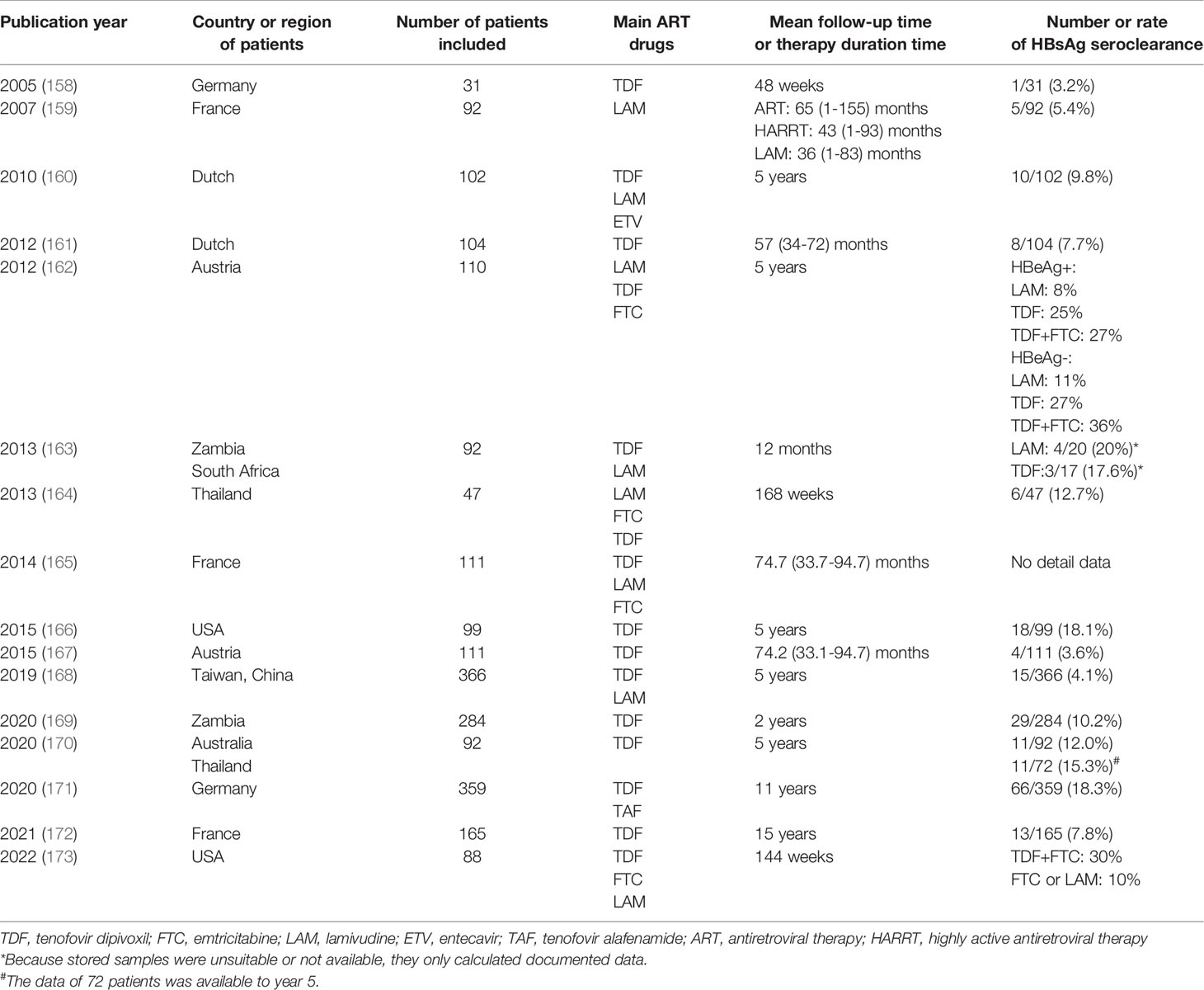
Table 1 HBsAg seroclearance rate in HBV/HIV coinfection patients with TDF inclusive ART.
Conclusions
The pathogenesis of chronic hepatitis B is mainly related with immune mechanisms. The functional cure of hepatitis B with HBsAg seroclearance as the therapeutic target mainly depends on the immune response. When coinfected with HBV and other viruses, the body immune state becomes more complicated. In some cases, coinfection can improve the seroclearance rate of HBsAg. The specific mechanism needs to be further elaborated for better guiding the clinical application.
Author Contributions
ML, LZ, and YX contributed to study concept and design. SW, WY, YG, WD, XB, YJL, LY, YL, RL, MC, GS, and LH collected and sorted out literatures. SW and LZ drew pictures. SW, WY, and YG wrote the first draft. ML and YX edited the English version. YX approved the submitted version after modification. All authors contributed to the article and approved the submitted version.
Funding
This project was supported by National Science and Technology Major Project of China (No. 2017ZX10201201-001-006 and 2017ZX10201201-002-006, and 2018ZX10715-005-003-005), the Beijing Hospitals Authority Clinical medicine Development of Special Funding Support (No. XMLX 201706 and XMLX 202127), the Digestive Medical Coordinated Development Center of Beijing Hospitals Authority (No. XXZ0302 and XXT28), Beijing Science and Technology Commission (No. D161100002716002), Special Public Health Project for Health Development in Capital (2021-1G-4061 and 2022-1-2172), and Beijing Municipal Science and Technology Commission (No. Z151100004015122).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Global Hepatitis Report (2017). Geneva, Switzerland: World Health Organization. Available at: https://www.who.int/hepatitis/publications/global-hepatitis-report2017/en/ (Accessed on 28 August 2019).
2. Polaris Observatory Collaborators. Global Prevalence, Treatment, and Prevention of Hepatitis B Virus Infection in 2016: A Modelling Study. Lancet Gastroenterol Hepatol (2018) 3:383–403. doi: 10.1016/S2468-1253(18)30056-6
3. Terrault NA, Lok ASF, McMahon BJ, Chang KM, Hwang JP, Jonas MM, et al. Update on Prevention, Diagnosis, and Treatment of Chronic Hepatitis B: AASLD 2018 Hepatitis B Guidance. Hepatology (2018) 67:1560–99. doi: 10.1002/hep.29800
4. Cornberg M, Lok AS, Terrault NA, Zoulim F, 2019 EASL-AASLD HBV Treatment Endpoints Conference Faculty. Guidance for Design and Endpoints of Clinical Trials in Chronic Hepatitis B - Report From the 2019 EASL-AASLD HBV Treatment Endpoints Conference‡. J Hepatol (2020) 72:539–57. doi: 10.1016/j.jhep.2019.11.003
5. Bill CA, Summers J. Genomic DNA Double-Strand Breaks are Targets for Hepadnaviral DNA Integration. Proc Natl Acad Sci USA (2004) 101:11135–40. doi: 10.1073/pnas.0403925101
6. Heermann KH, Goldmann U, Schwartz W, Seyffarth T, Baumgarten H, Gerlich WH. Large Surface Proteins of Hepatitis B Virus Containing the Pre-s Sequence. J Virol (1984) 52:396–402. doi: 10.1128/JVI.52.2.396-402.1984
7. Pfefferkorn M, Schott T, Böhm S, Deichsel D, Felkel C, Gerlich WH, et al. Composition of HBsAg Is Predictive of HBsAg Loss During Treatment in Patients With HBeAg-Positive Chronic Hepatitis B. J Hepatol (2021) 74:283–92. doi: 10.1016/j.jhep.2020.08.039
8. Chen Y, Tian Z. HBV-Induced Immune Imbalance in the Development of HCC. Front Immunol (2019) 10:2048. doi: 10.3389/fimmu.2019.02048
9. Thimme R, Wieland S, Steiger C, Ghrayeb J, Reimann KA, Purcell RH, et al. CD8(+) T Cells Mediate Viral Clearance and Disease Pathogenesis During Acute Hepatitis B Virus Infection. J Virol (2003) 77:68–76. doi: 10.1128/jvi.77.1.68-76.2003
10. Maini MK, Boni C, Lee CK, Larrubia JR, Reignat S, Ogg GS, et al. The Role of Virus-Specific CD8(+) Cells in Liver Damage and Viral Control During Persistent Hepatitis B Virus Infection. J Exp Med (2000) 191:1269–80. doi: 10.1084/jem.191.8.1269
11. Gehring AJ, Protzer U. Targeting Innate and Adaptive Immune Responses to Cure Chronic HBV Infection. Gastroenterology (2019) 156:325–37. doi: 10.1053/j.gastro.2018.10.032
12. Kuipery A, Gehring AJ, Isogawa M. Mechanisms of HBV Immune Evasion. Antiviral Res (2020) 179:104816. doi: 10.1016/j.antiviral.2020.104816
13. Kondo Y, Ninomiya M, Kakazu E, Kimura O, Shimosegawa T. Hepatitis B Surface Antigen Could Contribute to the Immunopathogenesis of Hepatitis B Virus Infection. ISRN Gastroenterol (2013) 2013:935295. doi: 10.1155/2013/935295
14. Tsai KN, Kuo CF, Ou JJ. Mechanisms of Hepatitis B Virus Persistence. Trends Microbiol (2018) 26:33–42. doi: 10.1016/j.tim.2017.07.006
15. Gao S, Joshi SS, Osiowy C, Chen Y, Coffin CS, Duan ZP. Chronic Hepatitis B Carriers With Acute on Chronic Liver Failure Show Increased HBV Surface Gene Mutations, Including Immune Escape Variants. Virol J (2017) 14:203. doi: 10.1186/s12985-017-0870-x
16. Op den Brouw ML, Binda RS, van Roosmalen MH, Protzer U, Janssen HL, van der Molen RG, et al. Hepatitis B Virus Surface Antigen Impairs Myeloid Dendritic Cell Function: A Possible Immune Escape Mechanism of Hepatitis B Virus. Immunology (2009) 126:280–9. doi: 10.1111/j.1365-2567.2008.02896.x
17. Wu J, Meng Z, Jiang M, Pei R, Trippler M, Broering R, et al. Hepatitis B Virus Suppresses Toll-Like Receptor-Mediated Innate Immune Responses in Murine Parenchymal and Nonparenchymal Liver Cells. Hepatology (2009) 49:1132–40. doi: 10.1002/hep.22751
18. Jiang M, Broering R, Trippler M, Poggenpohl L, Fiedler M, Gerken G, et al. Toll-Like Receptor-Mediated Immune Responses Are Attenuated in the Presence of High Levels of Hepatitis B Virus Surface Antigen. J Viral Hepat (2014) 21:860–72. doi: 10.1111/jvh.12216
19. Luangsay S, Ait-Goughoulte M, Michelet M, Floriot O, Bonnin M, Gruffaz M, et al. Expression and Functionality of Toll- and RIG-Like Receptors in HepaRG Cells. J Hepatol (2015) 63:1077–85. doi: 10.1016/j.jhep.2015.06.022
20. Sato S, Li K, Kameyama T, Hayashi T, Ishida Y, Murakami S, et al. The RNA Sensor RIG-I Dually Functions as an Innate Sensor and Direct Antiviral Factor for Hepatitis B Virus. Immunity (2015) 42:123–32. doi: 10.1016/j.immuni.2014.12.016
21. Lee S, Goyal A, Perelson AS, Ishida Y, Saito T, Gale M Jr. Suppression of Hepatitis B Virus Through Therapeutic Activation of RIG-I and IRF3 Signaling in Hepatocytes. iScience (2021) 24:101969. doi: 10.1016/j.isci.2020.101969
22. Gehring AJ, Haniffa M, Kennedy PT, Ho ZZ, Boni C, Shin A, et al. Mobilizing Monocytes to Cross-Present Circulating Viral Antigen in Chronic Infection. J Clin Invest (2013) 123:3766–76. doi: 10.1172/JCI66043
23. Deng F, Xu G, Cheng Z, Huang Y, Ma C, Luo C, et al. Hepatitis B Surface Antigen Suppresses the Activation of Nuclear Factor Kappa B Pathway via Interaction With the TAK1-TAB2 Complex. Front Immunol (2021) 12:618196. doi: 10.3389/fimmu.2021.618196
24. Park JJ, Wong DK, Wahed AS, Lee WM, Feld JJ, Terrault N, et al. Hepatitis B Virus–Specific and Global T-Cell Dysfunction in Chronic Hepatitis B. Gastroenterology (2016) 150:684–95.e5. doi: 10.1053/j.gastro.2015.11.050
25. Boni C, Fisicaro P, Valdatta C, Amadei B, Di Vincenzo P, Giuberti T, et al. Characterization of Hepatitis B Virus (HBV)-Specific T-Cell Dysfunction in Chronic HBV Infection. J Virol (2007) 81:4215–25. doi: 10.1128/JVI.02844-06
26. Pal S, Nandi M, Dey D, Chakraborty BC, Shil A, Ghosh S, et al. Myeloid-Derived Suppressor Cells Induce Regulatory T Cells in Chronically HBV Infected Patients With High Levels of Hepatitis B Surface Antigen and Persist After Antiviral Therapy. Aliment Pharmacol Ther (2019) 49:1346–59. doi: 10.1111/apt.15226
27. Kosinska AD, Pishraft-Sabet L, Wu W, Fang Z, Lenart M, Chen J, et al. Low Hepatitis B Virus-Specific T-Cell Response in Males Correlates With High Regulatory T-Cell Numbers in Murine Models. Hepatology (2017) 66:69–83. doi: 10.1002/hep.29155
28. Tavakoli S, Mederacke I, Herzog-Hauff S, Glebe D, Grün S, Strand D, et al. Peripheral Blood Dendritic Cells Are Phenotypically and Functionally Intact in Chronic Hepatitis B Virus (HBV) Infection. Clin Exp Immunol (2008) 151:61–70. doi: 10.1111/j.1365-2249.2007.03547.x
29. Fumagalli V, Di Lucia P, Venzin V, Bono EB, Jordan R, Frey CR, et al. Serum HBsAg Clearance has Minimal Impact on CD8+ T Cell Responses in Mouse Models of HBV Infection. J Exp Med (2020) 217:e20200298. doi: 10.1084/jem.20200298
30. Loomba R, Liang TJ. Hepatitis B Reactivation Associated With Immune Suppressive and Biological Modifier Therapies: Current Concepts, Management Strategies, and Future Directions. Gastroenterology (2017) 152:1297–309. doi: 10.1053/j.gastro.2017.02.009
31. Kusumoto S, Tanaka Y, Ueda R, Mizokami M. Reactivation of Hepatitis B Virus Following Rituximab-Plus-Steroid Combination Chemotherapy. J Gastroenterol (2011) 46:9–16. doi: 10.1007/s00535-010-0331-4
32. Seto WK, Chan TS, Hwang YY, Wong DK, Fung J, Liu KS, et al. Hepatitis B Reactivation in Patients With Previous Hepatitis B Virus Exposure Undergoing Rituximab-Containing Chemotherapy for Lymphoma: A Prospective Study. J Clin Oncol (2014) 32:3736–43. doi: 10.1200/JCO.2014.56.7081
33. Tuaillon E, Tabaa YA, Petitjean G, Huguet MF, Pajeaux G, Fondere JM, et al. Detection of Memory B Lymphocytes Specific to Hepatitis B Virus (HBV) Surface Antigen (HBsAg) From HBsAg-Vaccinated or HBV-Immunized Subjects by ELISPOT Assay. J Immunol Methods (2006) 315:144–52. doi: 10.1016/j.jim.2006.07.016
34. Valats JC, Tuaillon E, Funakoshi N, Hoa D, Brabet MC, Bollore K, et al. Investigation of Memory B Cell Responses to Hepatitis B Surface Antigen in Health Care Workers Considered as Non-Responders to Vaccination. Vaccine (2010) 28:6411–6. doi: 10.1016/j.vaccine.2010.07.058
35. Ward SM, Phalora P, Bradshaw D, Leyendeckers H, Klenerman P. Direct Ex Vivo Evaluation of Long-Lived Protective Antiviral Memory B Cell Responses Against Hepatitis B Virus. J Infect Dis (2008) 198:813–7. doi: 10.1086/591094
36. Burton AR, Pallett LJ, McCoy LE, Suveizdyte K, Amin OE, Swadling L, et al. Circulating and Intrahepatic Antiviral B Cells Are Defective in Hepatitis B. J Clin Invest (2018) 128:4588–603. doi: 10.1172/JCI121960
37. Salimzadeh L, Le Bert N, Dutertre CA, Gill US, Newell EW, Frey C, et al. PD-1 Blockade Partially Recovers Dysfunctional Virus-Specific B Cells in Chronic Hepatitis B Infection. J Clin Invest (2018) 128:4573–87. doi: 10.1172/JCI121957
38. Neumann-Haefelin C, Thimme R. Entering the Spotlight: Hepatitis B Surface Antigen-Specific B Cells. J Clin Invest (2018) 128:4257–9. doi: 10.1172/JCI124098
39. Le Bert N, Salimzadeh L, Gill US, Dutertre CA, Fachetti F, Tan A, et al. Comparative Characterization of B Cells Specific for HBV Nucleocapsid and Envelope Proteins in Patients With Chronic Hepatitis B. J Hepatol (2020) .72:34–44. doi: 10.1016/j.jhep.2019.07.015
40. Ayithan N, Tang L, Tan SK, Chen D, Wallin JJ, Fletcher SP, et al. (TFH) Cell Targeting by TLR8 Signaling for Improving HBsAg-Specific B Cell Response in Chronic Hepatitis B Patients. Front Immunol (2021) 12:735913. doi: 10.3389/fimmu.2021.735913
41. Zhang ZH, Li L, Zhao XP, Glebe D, Bremer CM, Zhang ZM, et al. Elimination of Hepatitis B Virus Surface Antigen and Appearance of Neutralizing Antibodies in Chronically Infected Patients Without Viral Clearance. J Viral Hepat (2011) 18:424–33. doi: 10.1111/j.1365-2893.2010.01322.x
42. Hsu YC, Yeh ML, Wong GL, Chen CH, Peng CY, Buti M, et al. Incidences and Determinants of Functional Cure During Entecavir or Tenofovir Disoproxil Fumarate for Chronic Hepatitis B. J Infect Dis (2021) 224:1890–9. doi: 10.1093/infdis/jiab241
43. Lau GK, Piratvisuth T, Luo KX, Marcellin P, Thongsawat S, Cooksley G, et al. Peginterferon Alfa-2a, Lamivudine, and the Combination for HBeAg-Positive Chronic Hepatitis B. N Engl J Med (2005) 352:2682–95. doi: 10.1056/NEJMoa043470
44. Janssen HL, van Zonneveld M, Senturk H, Zeuzem S, Akarca US, Cakaloglu Y, et al. Pegylated Interferon Alfa-2b Alone or in Combination With Lamivudine for HBeAg-Positive Chronic Hepatitis B: A Randomised Trial. Lancet (2005) 365:123–9. doi: 10.1016/S0140-6736(05)17701-0
45. Li M, Zhang L, Lu Y, Chen Q, Lu H, Sun F, et al. Early Serum HBsAg Kinetics as Predictor of HBsAg Loss in Patients With HBeAg-Negative Chronic Hepatitis B After Treatment With Pegylated Interferonα-2a. Virol Sin (2021) 36:311–20. doi: 10.1007/s12250-020-00290-7
46. Choi HSJ, van Campenhout MJH, van Vuuren AJ, Krassenburg LAP, Sonneveld MJ, de Knegt RJ, et al. Ultra-Long-Term Follow-Up of Interferon Alfa Treatment for HBeAg-Positive Chronic Hepatitis B Virus Infection. Clin Gastroenterol Hepatol (2021) 19:1933–40.e1. doi: 10.1016/j.cgh.2020.09.004
47. Song A, Lin X, Lu J, Ren S, Cao Z, Zheng S, et al. Pegylated Interferon Treatment for the Effective Clearance of Hepatitis B Surface Antigen in Inactive HBsAg Carriers: A Meta-Analysis. Front Immunol (2021) 12:779347. doi: 10.3389/fimmu.2021.779347
48. Micco L, Peppa D, Loggi E, Schurich A, Jefferson L, Cursaro C, et al. Differential Boosting of Innate and Adaptive Antiviral Responses Du Ring Pegylated-Interferon-Alpha Therapy of Chronic Hepatitis B. J Hepatol (2013) 58:225–33. doi: 10.1016/j.jhep.2012.09.029
49. Peppa D, Micco L, Javaid A, Kennedy PT, Schurich A, Dunn C, et al. Blockade of Immunosuppressive Cytokines Restores NK Cell Antiviral Function in Chronic Hepatitis B Virus Infection. PloS Pathog (2010) 6:e1001227. doi: 10.1371/journal.ppat.1001227
50. Boni C, Laccabue D, Lampertico P, Giuberti T, Viganò M, Schivazappa S, et al. Restored Function of HBV-Specific T Cells After Long-Term Effective Therapy With Nucleos(T)Ide Analogues. Gastroenterology (2012) 143:963–73.e9. doi: 10.1053/j.gastro.2012.07.014
51. Boni C, Penna A, Ogg GS, Bertoletti A, Pilli M, Cavallo C, et al. Lamivudine Treatment can Overcome Cytotoxic T-Cell Hyporesponsiveness in Chronic Hepatitis B: New Perspectives for Immune Therapy. Hepatology (2001) 33:963–71. doi: 10.1053/jhep.2001.23045
52. Cooksley H, Chokshi S, Maayan Y, Wedemeyer H, Andreone P, Gilson R, et al. Hepatitis B Virus E Antigen Loss During Adefovir Dipivoxil Therapy Is Associated With Enhanced Virus-Specific CD4+ T-Cell Reactivity. Antimicrob Agents Chemother (2008) 52:312–20. doi: 10.1128/AAC.00467-07
53. Chen Y, Li X, Ye B, Yang X, Wu W, Chen B, et al. Effect of Telbivudine Therapy on the Cellular Immune Response in Chronic Hepatitis B. Antiviral Res (2011) 91:23–31. doi: 10.1016/j.antiviral.2011.04.008
54. Ma L, Cai YJ, Yu L, Feng JY, Wang J, Li C, et al. Treatment With Telbivudine Positively Regulates Antiviral Immune Profiles in Chinese Patients With Chronic Hepatitis B. Antimicrob Agents Chemother (2013) 57:1304–11. doi: 10.1128/AAC.02181-12
55. Jiang Y, Li W, Yu L, Liu J, Xin G, Yan H, et al. Enhancing the Antihepatitis B Virus Immune Response by Adefovir Dipivoxil and Entecavir Therapies. Cell Mol Immunol (2011) 8:75–82. doi: 10.1038/cmi.2010.37
56. Zimmer CL, Rinker F, Höner Zu Siederdissen C, Manns MP, Wedemeyer H, Cornberg M, et al. Increased NK Cell Function After Cessation of Long-Term Nucleos(T)Ide Analogue Treatment in Chronic Hepatitis B Is Associated With Liver Damage and HBsAg Loss. J Infect Dis (2018) 217:1656–66. doi: 10.1093/infdis/jiy097
57. de Niet A, Stelma F, Jansen L, Sinnige MJ, Remmerswaal EB, Takkenberg RB, et al. Restoration of T Cell Function in Chronic Hepatitis B Patients Upon Treatment With Interferon Based Combination Therapy. J Hepatol (2016) 64:539–46. doi: 10.1016/j.jhep.2015.10.013
58. Tan AT, Hoang LT, Chin D, Rasmussen E, Lopatin U, Hart S, et al. Reduction of HBV Replication Prolongs the Early Immunological Response to Ifnα Therapy. J Hepatol (2014) 60:54–61. doi: 10.1016/j.jhep.2013.08.020
59. Yuen MF, Wong DK, Schluep T, Lai CL, Ferrari C, Locarnini S, et al. Long-Term Serological, Virological and Histological Responses to RNA Inhibition by ARC-520 in Chinese Chronic Hepatitis B Patients on Entecavir Treatment. Gut (2021) 12:gutjnl-2020-323445. doi: 10.1136/gutjnl-2020-323445
60. Gane EJ, Locarnini S, Lim TH, Strasser S, Sievert W, Cheng W, et al. First Results With RNA Interference (RNAi) in Chronic Hepatitis B (CHB) Using ARO-HBV. Hepatology (2018) 68:1462A.
61. Wedemeyer H, Schoneweis K, Bogomolov PO, Voronkova N, Chulanov V, Stepanova T, et al. GS-13-Final Results of a Multicenter, Open-Label Phase 2 Clinical Trial (MYR203) to Assess Safety and Efficacy of Myrcludex B in Cwith PEG-Interferon Alpha 2a in Patients With Chronic HBV/HDV Co-Infection. J Hepatol (2019) 70:e81. doi: 10.1016/S0618-8278(19)30141-0
62. Yuen MF, Gane EJ, Kim DJ, Weilert F, Yuen Chan HL, Lalezari J, et al. Antiviral Activity, Safety, and Pharmacokinetics of Capsid Assembly Modulator NVR 3-778 in Patients With Chronic HBV Infection. Gastroenterology (2019) 156:1392–403.e7. doi: 10.1053/j.gastro.2018.12.023
63. Zoulim F, Lenz O, Vandenbossche JJ, Talloen W, Verbinnen T, Moscalu I, et al. JNJ-56136379, an HBV Capsid Assembly Modulator, Is Well-Tolerated and has Antiviral Activity in a Phase 1 Study of Patients With Chronic Infection. Gastroenterology (2020) 159:521–33.e9. doi: 10.1053/j.gastro.2020.04.036
64. Yuen MF, Zhou X, Gane E, Schwabe C, Tanwandee T, Feng S, et al. Safety, Pharmacokinetics, and Antiviral Activity of RO7049389, a Core Protein Allosteric Modulator, in Patients With Chronic Hepatitis B Virus Infection: A Multicenter, Randomized, Placebo-Controlled, Phase 1 Trial. Lancet Gastroenterol Hepatol (2021) 6:723–32. doi: 10.1016/S2468-1253(21)00176-X
65. Yuen MF, Agarwal K, Gane EJ, Schwabe C, Ahn SH, Kim DJ, et al. Safety, Pharmacokinetics, and Antiviral Effects of ABI-H0731, a Hepatitis B Virus Core Inhibitor: A Randomized, Placebo-Controlled Phase 1 Trial. Lancet Gastroenterol Hepatol (2020) 5:52–166. doi: 10.1016/S2468-1253(19)30346-2
66. Han K, Cremer J, Elston R, Oliver S, Baptiste-Brown S, Chen S, et al. A Randomized, Double-Blind, Placebo-Controlled, First-Time-in-Human Study to Assess the Safety, Tolerability, and Pharmacokinetics of Single and Multiple Ascending Doses of GSK3389404 in Healthy Subjects. Clin Pharmacol Drug Dev (2019) 8:790–801. doi: 10.1002/cpdd.670
67. Bazinet M, Pântea V, Cebotarescu V, Cojuhari L, Jimbei P, Albrecht J, et al. Safety and Efficacy of REP 2139 and Pegylated Interferon Alfa-2a for Treatment-Naive Patients With Chronic Hepatitis B Virus and Hepatitis D Virus Co-Infection (REP 301 and REP 301-LTF): A Non-Randomised, Open-Label, Phase 2 Trial. Lancet Gastroenterol Hepatol (2017) 2:877–89. doi: 10.1016/S2468-1253(17)30288-1
68. Lee HW, Park JY, Hong T, Park MS, Ahn SH. Efficacy of Lenvervimab, a Recombinant Human Immunoglobulin, in Treatment of Chronic Hepatitis B Virus Infection. Clin Gastroenterol Hepatol (2020) 18:3043–3045.e1. doi: 10.1016/j.cgh.2019.09.038
69. Janssen HLA, Brunetto MR, Kim YJ, Ferrari C, Massetto B, Nguyen AH, et al. Safety, Efficacy and Pharmacodynamics of Vesatolimod (GS-9620) in Virally Suppressed Patients With Chronic Hepatitis B. J Hepatol (2018) 68:431–40. doi: 10.1111/jvh.12942
70. Gane E, Verdon DJ, Brooks AE, Gaggar A, Nguyen AH, Subramanian GM, et al. Anti-PD-1 Blockade With Nivolumab With and Without Therapeutic Vaccination for Virally Suppressed Chronic Hepatitis B: A Pilot Study. J Hepatol (2019) 71:900–7. doi: 10.1016/j.jhep.2019.06.028
71. Lok AS, Pan CQ, Han SH, Trinh HN, Fessel WJ, Rodell T, et al. Randomized Phase II Study of GS-4774 as a Therapeutic Vaccine in Virally Suppressed Patients With Chronic Hepatitis B. J Hepatol (2016) 65:509–16. doi: 10.1016/j.jhep.2016.05.016
72. Meng Z, Chen Y, Lu M. Advances in Targeting the Innate and Adaptive Immune Systems to Cure Chronic Hepatitis B Virus Infection. Front Immunol (2020) 10:3127. doi: 10.3389/fimmu.2019.03127
73. Yuen MF, Locarnini S, Given B, Schluep T, Hamilton J, Biermer M, et al. First Clinical Experience With RNA Interference-Based Triple Combination Therapy in Chronic Hepatitis B: JNJ-3989, JNJ-6379 and a Nucleos(t) Ide Analogue. Hepatology (2019) 70:1489A.
74. Nakao M, Nakayama N, Uchida Y, Tomiya T, Oketani M, Ido A, et al. Deteriorated Outcome of Recent Patients With Acute Liver Failure and Late-Onset Hepatic Failure Caused by Infection With Hepatitis A Virus: A Subanalysis of Patients Seen Between 1998 and 2015 and Enrolled in Nationwide Surveys in Japan. Hepatol Res (2019) 49:844–52. doi: 10.1111/hepr.13345
75. Lanford RE, Feng Z, Chavez D, Guerra B, Brasky KM, Zhou Y, et al. Acute Hepatitis A Virus Infection is Associated With a Limited Type I Interferon Response and Persistence of Intrahepatic Viral RNA. Proc Natl Acad Sci USA (2011) 108:11223–8. doi: 10.1073/pnas.1101939108
76. Zhou Y, Callendret B, Xu D, Brasky KM, Feng Z, Hensley LL, et al. Dominance of the CD4(+) T Helper Cell Response During Acute Resolving Hepatitis A Virus Infection. J Exp Med (2012) 209:1481–92. doi: 10.1084/jem.20111906
77. Schulte I, Hitziger T, Giugliano S, Timm J, Gold H, Heinemann FM, et al. Characterization of CD8+ T-Cell Response in Acute and Resolved Hepatitis A Virus Infection. J Hepatol (2011) 54:201–8. doi: 10.1016/j.jhep.2010.07.010
78. Hirai-Yuki A, Hensley L, McGivern DR, González-López O, Das A, Feng H, et al. MAVS-Dependent Host Species Range and Pathogenicity of Human Hepatitis A Virus. Science (2016) 353:1541–5. doi: 10.1126/science.aaf8325
79. Choi YS, Jung MK, Lee J, Choi SJ, Choi SH, Lee HW, et al. Tumor Necrosis Factor-Producing T-Regulatory Cells Are Associated With Severe Liver Injury in Patients With Acute Hepatitis a. Gastroenterology (2018) 154:1047–60. doi: 10.1053/j.gastro.2017.11.277
80. Kim J, Chang DY, Lee HW, Lee H, Kim JH, Sung PS, et al. Innate-Like Cytotoxic Function of Bystander-Activated CD8+ T Cells is Associated With Liver Injury in Acute Hepatitis A. Immunity (2018) 48:161–173.e5. doi: 10.1016/j.immuni.2017.11.025
81. Win NN, Kanda T, Ogawa M, Nakamoto S, Haga Y, Sasaki R, et al. Superinfection of Hepatitis A Virus in Hepatocytes Infected With Hepatitis B Virus. Int J Med Sci (2019) 16:1366–70. doi: 10.7150/ijms.32795
82. Berthillon P, Crance JM, Leveque F, Jouan A, Petit MA, Deloince R, et al. Inhibition of the Expression of Hepatitis A and B Viruses (HAV and HBV) Proteins by Interferon in a Human Hepatocarcinoma Cell Line (PLC/PRF/5). J Hepatol (1996) 25:15–9. doi: 10.1016/s0168-8278(96)80322-9
83. van Nunen AB, Pontesilli O, Uytdehaag F, Osterhaus AD, de Man RA. Suppression of Hepatitis B Virus Replication Mediated by Hepatitis A-Induced Cytokine Production. Liver (2001) 21:45–9. doi: 10.1034/j.1600-0676.2001.210107.x
84. Fu J, Guo D, Gao D, Huang W, Li Z, Jia B. Clinical Analysis of Patients Suffering From Chronic Hepatitis B Superinfected With Other Hepadnaviruses. J Med Virol (2016) 88:1003–9. doi: 10.1002/jmv.24417
85. Sagnelli E, Coppola N, Pisaturo M, Pisapia R, Onofrio M, Sagnelli C, et al. Clinical and Virological Improvement of Hepatitis B Virus-Related or Hepatitis C Virus-Related Chronic Hepatitis With Concomitant Hepatitis A Virus Infection. Clin Infect Dis (2006) 42:1536–43. doi: 10.1086/503840
86. Beisel C, Addo MM, Schulze Zur Wiesch J. Seroconversion of HBsAG Coincides With Hepatitis A Super-Infection: A Case Report. World J Clin Cases (2020) 8:1651–5. doi: 10.12998/wjcc.v8.i9.1651
87. Khuroo MS, Kamili S, Yattoo GN. Hepatitis E Virus Infection may be Transmitted Through Blood Transfusions in an Endemic Area. J Gastroenterol Hepatol (2004) 19:778–84. doi: 10.1111/j.1440-1746.2004.03437.x
88. Wong RJ, Cheung R, Gish RG, Chitnis AS. Prevalence of Hepatitis E Infection Among Adults With Concurrent Chronic Liver Disease. J Viral Hepat (2021) 28:1643–55. doi: 10.1111/jvh.13597
89. Hoan NX, Tong HV, Hecht N, Sy BT, Marcinek P, Meyer CG, et al. Hepatitis E Virus Superinfection and Clinical Progression in Hepatitis B Patients. EBioMedicine (2015) 11(2):2080–6. doi: 10.1016/j.ebiom.2015.11.020
90. Prabhu SB, Gupta P, Durgapal H, Rath S, Gupta SD, Acharya SK, et al. Study of Cellular Immune Response Against Hepatitis E Virus (HEV). J Viral Hepat (2011) 18:587–94. doi: 10.1111/j.1365-2893.2010.01338.x
91. Abravanel F, Barragué H, Dörr G, Sauné K, Péron JM, Alric L, et al. Conventional and Innate Lymphocytes Response at the Acute Phase of HEV Infection in Transplanted Patients. J Infect (2016) 72:723–30. doi: 10.1016/j.jinf.2016.02.016
92. Baley JE, Schacter BZ. Mechanisms of Diminished Natural Killer Cell Activity in Pregnant Women and Neonates. J Immunol (1985) 134:3042–8.
93. Kumar A, Devi SG, Kar P, Agarwal S, Husain SA, Gupta RK, et al. Association of Cytokines in Hepatitis E With Pregnancy Outcome. Cytokine (2014) 65:95–104. doi: 10.1016/j.cyto.2013.09.022
94. Wang W, Xu L, Brandsma JH, Wang Y, Hakim MS, Zhou X, et al. Convergent Transcription of Interferon-Stimulated Genes by TNF-α and IFN-α Augments Antiviral Activity Against HCV and HEV. Sci Rep (2016) 6:25482. doi: 10.1038/srep25482
95. Tseng TC, Liu CJ, Chang CT, Su TH, Yang WT, Tsai CH, et al. HEV Superinfection Accelerates Disease Progression in Patients With Chronic HBV Infection and Increases Mortality in Those With Cirrhosis. J Hepatol (2020) 72:1105–11. doi: 10.1016/j.jhep.2020.01.012
96. Kilonzo SB, Wang YL, Jiang QQ, Wu WY, Wang P, Ning Q, et al. Superinfective Hepatitis E Virus Infection Aggravates Hepatocytes Injury in Chronic Hepatitis B. Curr Med Sci (2019) 39:719–26. doi: 10.1007/s11596-019-2097-0
97. Kumar M, Sharma BC, Sarin SK. Hepatitis E Virus as an Etiology of Acute Exacerbation of Previously Unrecognized Asymptomatic Patients With Hepatitis B Virus-Related Chronic Liver Disease. J Gastroenterol Hepatol (2008) 23:883–7. doi: 10.1111/j.1440-1746.2007.05243.x
98. Zhang X, Ke W, Xie J, Zhao Z, Xie D, Gao Z. Comparison of Effects of Hepatitis E or A Viral Superinfection in Patients With Chronic Hepatitis B. Hepatol Int (2010) 4:615–20. doi: 10.1007/s12072-010-9204-4
99. Yeh CT, Yeh CS, Chu YD, Chiang YJ. Seroclearance of Hepatitis B Surface Antigen Following Hepatitis E Exacerbation on Chronic Hepatitis E and B Dual Infection in a Renal Transplant Recipient: A Case Report. J Med Case Rep (2018) 12:50. doi: 10.1186/s13256-018-1586-2
100. Nascimbeni M, Mizukoshi E, Bosmann M, Major ME, Mihalik K, Rice CM, et al. Kinetics of CD4+ and CD8+ Memory T-Cell Responses During Hepatitis C Virus Rechallenge of Previously Recovered Chimpanzees. J Virol (2003) 77:4781–93. doi: 10.1128/jvi.77.8.4781-4793.2003
101. Yao ZQ, Eisen-Vandervelde A, Waggoner SN, Cale EM, Hahn YS. Direct Binding of Hepatitis C Virus Core to Gc1qr on CD4+ and CD8+ T Cells Leads to Impaired Activation of Lck and Akt. J Virol (2004) 78:6409–19. doi: 10.1128/JVI.78.12.6409-6419.2004
102. Wang JH, Layden TJ, Eckels DD. Modulation of the Peripheral T-Cell Response by CD4 Mutants of Hepatitis C Virus: Transition From a Th1 to a Th2 Response. Hum Immunol (2003) 64:662–73. doi: 10.1016/s0198-8859(03)00070-3
103. Barrat FJ, Cua DJ, Boonstra A, Richards DF, Crain C, Savelkoul HF, et al. In Vitro Generation of Interleukin 10-Producing Regulatory CD4(+) T Cells is Induced by Immunosuppressive Drugs and Inhibited by T Helper Type 1 (Th1)- and Th2-Inducing Cytokines. J Exp Med (2002) 195:603–16. doi: 10.1084/jem.20011629
104. Boettler T, Spangenberg HC, Neumann-Haefelin C, Panther E, Urbani S, Ferrari C, et al. T Cells With a CD4+CD25+ Regulatory Phenotype Suppress In Vitro Proliferation of Virus-Specific CD8+ T Cells During Chronic Hepatitis C Virus Infection. J Virol (2005) 79:7860–7. doi: 10.1128/JVI.79.12.7860-7867.2005
105. Caccamo G, Saffioti F, Raimondo G. Hepatitis B Virus and Hepatitis C Virus Dual Infection. World J Gastroenterol (2014) 20:14559–67. doi: 10.3748/wjg.v20.i40.14559
106. Tyson GL, Kramer JR, Duan Z, Davila JA, Richardson PA, El-Serag HB. Prevalence and Predictors of Hepatitis B Virus Coinfection in a United States Cohort of Hepatitis C Virus-Infected Patients. Hepatology (2013) 58:538–45. doi: 10.1002/hep.26400
107. Shih CM, Lo SJ, Miyamura T, Chen SY, Lee YH. Suppression of Hepatitis B Virus Expression and Replication by Hepatitis C Virus Core Protein in HuH-7 Cells. J Virol (1993) 67:5823–32. doi: 10.1128/JVI.67.10.5823-5832.1993
108. Lee S, Yoon H, Han J, Jang KL. Hepatitis C Virus Core Protein Inhibits Hepatitis B Virus Replication by Downregulating HBx Levels via Siah-1-Mediated Proteasomal Degradation During Coinfection. J Gen Virol (2021) 102. doi: 10.1099/jgv.0.001701
109. Zhu W, Wu C, Deng W, Pei R, Wang Y, Cao L, et al. Inhibition of the HCV Core Protein on the Immune Response to HBV Surface Antigen and on HBV Gene Expression and Replication In Vivo. PloS One (2012) 7:e45146. doi: 10.1371/journal.pone.0045146
110. Mücke MM, Backus LI, Mücke VT, Coppola N, Preda CM, Yeh ML, et al. Hepatitis B Virus Reactivation During Direct-Acting Antiviral Therapy for Hepatitis C: A Systematic Review and Meta-Analysis. Lancet Gastroenterol Hepatol (2018) 3:172–80. doi: 10.1016/S2468-1253(18)30002-5
111. Huang SC, Cheng PN, Liu CH, Yang HC, Su TH, Tseng TC, et al. Serum Cytokine/Chemokine Profiles Predict Hepatitis B Reactivation in HBV/HCV Co-Infected Subjects Receiving Direct-Acting Antiviral Agents. J Formos Med Assoc (2022) 121:920–9. doi: 10.1016/j.jfma.2021.09.002
112. Cheng X, Uchida T, Xia Y, Umarova R, Liu CJ, Chen PJ, et al. Diminished Hepatic IFN Response Following HCV Clearance Triggers HBV Reactivation in Coinfection. J Clin Invest (2020) 130:3205–20. doi: 10.1172/JCI135616
113. Wang XX, Pan XB, Han JC, Cong X, Jin Q, Kong XS, et al. HBsAg Stimulates NKG2D Receptor Expression on Natural Killer Cells and Inhibits Hepatitis C Virus Replication. Hepatobi Pancreat Dis Int (2018) 17:233–40. doi: 10.1016/j.hbpd.2018.03.010
114. Wiegand SB, Jaroszewicz J, Potthoff A, Honer Zu Siederdissen C, Maasoumy B, Deterding K, et al. Dominance of Hepatitis C Virus (HCV) Is Associated With Lower Quantitative Hepatitis B Surface Antigen and Higher Serum Interferongamma-Induced Protein 10 Levels in HBV/HCV-Coinfected Patients. Clin Microbiol Infect (2015) 21:710.e1–e9. doi: 10.1016/j.cmi.2015.03.003
115. Yu ML, Lee CM, Chen CL, Chuang WL, Lu SN, Liu CH, et al. Sustained Hepatitis C Virus Clearance and Increased Hepatitis B Surface Antigen Seroclearance in Patients With Dual Chronic Hepatitis C and B During Posttreatment Follow-Up. Hepatology (2013) 57:2135–42. doi: 10.1002/hep.26266
116. Yeh ML, Huang CF, Huang CI, Holmes JA, Hsieh MH, Tsai YS, et al. Hepatitis B-Related Outcomes Following Direct-Acting Antiviral Therapy in Taiwanese Patients With Chronic HBV/HCV Co-Infection. J Hepatol (2020) 73:62–71. doi: 10.1016/j.jhep.2020.01.027
117. Stockdale AJ, Kreuels B, Henrion MYR, Giorgi E, Kyomuhangi I, de Martel C, et al. The Global Prevalence of Hepatitis D Virus Infection: Systematic Review and Meta-Analysis. J Hepatol (2020) 73:523–32. doi: 10.1016/j.jhep.2020.04.008
118. Wranke A, Wedemeyer H. Antiviral Therapy of Hepatitis Delta Virus Infection-Progress and Challenges Towards Cure. Curr Opin Virol (2016) 20:112–8. doi: 10.1016/j.coviro.2016.10.002
119. Lempp FA, Ni Y, Urban S. Hepatitis Delta Virus: Insights Into a Peculiar Pathogen and Novel Treatment Options. Nat Rev Gastroenterol Hepatol (2016) 13:580–9. doi: 10.1038/nrgastro.2016.126
120. Shen DT, Ji DZ, Chen HY, Goyal H, Pan S, Xu HG. Hepatitis D: Not a Rare Disease Anymore: Global Update for 2017-2018. Gut (2020) 69:786–8. doi: 10.1136/gutjnl-2019-318691
121. Miao Z, Zhang S, Ou X, Li S, Ma Z, Wang W, et al. Estimating the Global Prevalence, Disease Progression, and Clinical Outcome of Hepatitis Delta Virus Infection. J Infect Dis (2020) 221:1677–87. doi: 10.1093/infdis/jiz633
122. Kefalakes H, Horgan XJ, Jung MK, Amanakis G, Kapuria D, Bolte FJ, et al. Liver-Resident Bystander CD8+ T Cells Contribute to Liver Disease Pathogenesis in Chronic Hepatitis D Virus Infection. Gastroenterology (2021) 161:1567–83.e1569. doi: 10.1053/j.gastro.2021.07.027
123. Zhang Z, Filzmayer C, Ni Y, Sültmann H, Mutz P, Hiet MS, et al. Hepatitis D Virus Replication Is Sensed by MDA5 and Induces IFN-β/λ Responses in Hepatocytes. J Hepatol (2018) 69:25–35. doi: 10.1016/j.jhep.2018.02.021
124. Giersch K, Allweiss L, Volz T, Helbig M, Bierwolf J, Lohse AW, et al. Hepatitis Delta Co-Infection in Humanized Mice Leads to Pronounced Induction of Innate Immune Responses in Comparison to HBV Mono-Infection. J Hepatol (2015) 63:346–53. doi: 10.1016/j.jhep.2015.03.011
125. Park CY, Oh SH, Kang SM, Lim YS, Hwang SB. Hepatitis Delta Virus Large Antigen Sensitizes to TNF-Alpha-Induced NF-kappaB Signaling. Mol Cells (2009) 28:49–55. doi: 10.1007/s10059-009-0100-5
126. Choi SH, Jeong SH, Hwang SB. Large Hepatitis Delta Antigen Modulates Transforming Growth Factor-Beta Signaling Cascades: Implication of Hepatitis Delta Virus-Induced Liver Fibrosis. Gastroenterology (2007) 132:343–57. doi: 10.1053/j.gastro.2006.10.038
127. Williams V, Brichler S, Khan E, Chami M, Deny D9:24 am 30/04/2022, Kremsdorf M. Large Hepatitis Delta Antigen Activates STAT-3 And NF-KappaB via Oxidative Stress. J Viral Hepat (2012) 19:744–53. doi: 10.1111/j.1365-2893.2012.01597.x
128. Huang IC, Chien CY, Huang CR, Lo SJ. Induction of Hepatitis D Virus Large Antigen Translocation to the Cytoplasm by Hepatitis B Virus Surface Antigens Correlates With Endoplasmic Reticulum Stress and NF-kappaB Activation. J Gen Virol (2006) 87:1715–23. doi: 10.1099/vir.0.81718-0
129. Tham CYL, Kah J, Tan AT, Volz T, Chia A, Giersch K, et al. Hepatitis Delta Virus Acts as an Immunogenic Adjuvant in Hepatitis B Virus-Infected Hepatocytes. Cell Rep Med (2020) 1:100060. doi: 10.1016/j.xcrm.2020.100060
130. Heidrich B, Deterding K, Tillmann HL, Raupach R, Manns MP, Wedemeyer H. Virological and Clinical Characteristics of Delta Hepatitis in Central Europe. J Viral Hepat (2009) 16:883–94. doi: 10.1111/j.1365-2893.2009.01144.x
131. Heidrich B, Serrano BC, Idilman R, Kabacam G, Bremer B, Raupach R, et al. HBeAg-Positive Hepatitis Delta: Virological Patterns and Clinical Long-Term Outcome. Liver Int (2012) 32:1415–25. doi: 10.1111/j.1478-3231.2012.02831.x
132. Sajjad M, Ali S, Baig S, Sharafat S, Khan BA, Khan S, et al. HBV S Antigen Evolution in the Backdrop of HDV Infection Affects Epitope Processing and Presentation. J Med Virol (2021) 93:3714–29. doi: 10.1002/jmv.26711
133. Colagrossi L, Salpini R, Scutari R, Carioti L, Battisti A, Piermatteo L, et al. HDV can Constrain HBV Genetic Evolution in HBsAg: Implications for the Identification of Innovative Pharmacological Targets. Viruses (2018) 10:363. doi: 10.3390/v10070363
134. Zaunders J, van Bockel D. Innate and Adaptive Immunity in Long-Term Non-Progression in HIV Disease. Front Immunol (2013) 4:95. doi: 10.3389/fimmu.2013.00095
135. Swan DA, Rolland M, Herbeck JT, Schiffer JT, Reeves DB. Evolution During Primary HIV Infection Does Not Require Adaptive Immune Selection. Proc Natl Acad Sci USA (2022) 119:e2109172119. doi: 10.1073/pnas.2109172119
136. Poudrier J, Soulas C, Chagnon-Choquet J, Burdo T, Autissier P, Oskar K, et al. High Expression Levels of BLyS/BAFF by Blood Dendritic Cells and Granulocytes are Associated With B-Cell Dysregulation in SIV-Infected Rhesus Macaques. PloS One (2015) 10:e0131513. doi: 10.1371/journal.pone.0131513
137. Martin-Gayo E, Buzon MJ, Ouyang Z, Hickman T, Cronin J, Pimenova D, et al. Potent Cell-Intrinsic Immune Responses in Dendritic Cells Facilitate HIV-1-Specific T Cell Immunity in HIV-1 Elite Controllers. PloS Pathog (2015) 11:e1004930. doi: 10.1371/journal.ppat.1004930
138. Coindre S, Tchitchek N, Alaoui L, Vaslin B, Bourgeois C, Goujard C, et al. Mass Cytometry Analysis Reveals Complex Cell-State Modifications of Blood Myeloid Cells During HIV Infection. Front Immunol (2019) 10:2677. doi: 10.3389/fimmu.2019.02677
139. Mendila M, Heiken H, Becker S, Stoll M, Kemper A, Jacobs R, et al. Immunologic and Virologic Studies in Long-Term Nonprogressors With HIV-1 Infection. Eur J Med Res (1999) 4:417–24.
140. Li JZ, Arnold KB, Lo J, Dugast AS, Plants J, Ribaudo HJ, et al. Differential Levels of Soluble Inflammatory Markers by Human Immunodeficiency Virus Controller Status and Demographics. Open Forum Infect Dis (2015) 2:ofu117. doi: 10.1093/ofid/ofu117
141. Ahmad F, Shankar EM, Yong YK, Tan HY, Ahrenstorf G, Jacobs R, et al. Negative Checkpoint Regulatory Molecule 2B4 (CD244) Upregulation Is Associated With Invariant Natural Killer T Cell Alterations and Human Immunodeficiency Virus Disease Progression. Front Immunol (2017) 8:338. doi: 10.3389/fimmu.2017.00338
142. Singh D, Ghate M, Godbole S, Kulkarni S, Thakar M. Functional Invariant Natural Killer T Cells Secreting Cytokines Are Associated With Non-Progressive Human Immunodeficiency Virus-1 Infection But Not With Suppressive Anti-Retroviral Treatment. Front Immunol (2018) 9:1152. doi: 10.3389/fimmu.2018.01152
143. Betts MR, Nason MC, West SM, De Rosa SC, Migueles SA, Abraham J, et al. HIV Nonprogressors Preferentially Maintain Highly Functional HIV-Specific CD8+ T Cells. Blood (2006) 107:4781–9. doi: 10.1182/blood-2005-12-4818
144. Shasha D, Karel D, Angiuli O, Greenblatt A, Ghebremichael M, Yu X, et al. Elite Controller CD8+ T Cells Exhibit Comparable Viral Inhibition Capacity, But Better Sustained Effector Properties Compared to Chronic Progressors. J Leukoc Biol (2016) 100:1425–33. doi: 10.1189/jlb.4A0915-422R
145. Kwaa AKR, Talana CAG, Blankson JN. Interferon Alpha Enhances NK Cell Function and the Suppressive Capacity of HIV-Specific CD8(+) T Cells. J Virol (2019) 93:01541–18. doi: 10.1128/JVI.01541-18
146. Monel B, McKeon A, Lamothe-Molina P, Jani P, Boucau J, Pacheco Y, et al. HIV Controllers Exhibit Effective CD8+T Cell Recognition of HIV-1-Infected non-Activated CD4+T Cells. Cell Rep (2019) 27:142–53.e4. doi: 10.1016/j.celrep.2019.03.016
147. Phetsouphanh C, Aldridge D, Marchi E, Munier CML, Meyerowitz J, Murray L, et al. Maintenance of Functional CD57+ Cytolytic CD4+ T Cells in HIV+ Elite Controllers. Front Immunol (2019) 10:1844. doi: 10.3389/fimmu.2019.01844
148. Moris A, Pereira M, Chakrabarti L. A Role for Antibodies in Natural HIV Control. Curr Opin HIV AIDS (2019) 14:265–72. doi: 10.1097/COH.0000000000000554
149. Rouers A, Klingler J, Su B, Samri A, Laumond G, Even S, et al. HIV-Specific B Cell Frequency Correlates With Neutralization Breadth in Patients Naturally Controlling HIV-Infection. EBioMedicine (2017) 21:158–69. doi: 10.1016/j.ebiom.2017.05.029
150. Platt L, French CE, McGowan CR, Sabin K, Gower E, Trickey A, et al. Prevalence and Burden of HBV Co-Infection Among People Living With HIV: A Global Systematic Review and Meta-Analysis. J Viral Hepat (2020) 27:294–315. doi: 10.1111/jvh.13217
151. Copeland NK, Eller MA, Chen J, Kim D, Li F, Creegan M, et al. Brief Report: Increased Inflammation And Liver Disease In HIV/HBV-Coinfected Individuals. J Acquir Immune Defic Syndr (2021) 88:310–3. doi: 10.1097/QAI.0000000000002760
152. Singh KP, Zerbato JM, Zhao W, Braat S, Deleage C, Tennakoon GS, et al. Intrahepatic CXCL10 is Strongly Associated With Liver Fibrosis in HIV-Hepatitis B Co-Infection. PloS Pathog (2020) 16:e1008744. doi: 10.1371/journal.ppat.1008744
153. Nie Y, Deng XZ, Lan Y, Li F, Hu FY. Pre-S Deletions are Predominant Quasispecies in HIV/HBV Infection: Quasispecies Perspective. Infect Drug Resist (2020) 8:13. doi: 10.2147/IDR.S255473
154. He Y, Cai W, Zhao W, Chen J, Hu F, Li F, Lin W, et al. Persistent Chronic Immune Activation In HIV/HBV-Coinfected Patients After Antiretroviral Therapy. J Viral Hepat (2021) 28:1355–61. doi: 10.1111/jvh.13559
155. European Pregnancy and Paediatric HIV Cohort Collaboration (EPPICC) Study Group in EuroCoord. Prevalence and Clinical Outcomes of Poor Immune Response Despite Virologically Suppressive Antiretroviral Therapy Among Children and Adolescents With Human Immunodeficiency Virus in Europe and Thailand: Cohort Study. Clin Infect Dis (2020) 70:404–15. doi: 10.1093/cid/ciz253
156. Wandeler G, Gsponer T, Bihl F, Bernasconi E, Cavassini M, Kovari H, et al. Hepatitis B Virus Infection is Associated With Impaired Immunological Recovery During Antiretroviral Therapy in the Swiss HIV Cohort Study. J Infect Dis (2013) 208:1454–8. doi: 10.1093/infdis/jit351
157. Jaroszewicz J, Reiberger T, Meyer-Olson D, Mauss S, Vogel M, Ingiliz P, et al. Hepatitis B Surface Antigen Concentrations in Patients With HIV/HBV Co-Infection. PloS One (2012) 7:e43143. doi: 10.1371/journal.pone.0043143
158. Stephan C, Berger A, Carlebach A, Lutz T, Bickel M, Klauke S, et al. Impact Of Tenofovir-Containing Antiretroviral Therapy On Chronic Hepatitis B In A Cohort Co-Infected With Human Immunodeficiency Virus. J Antimicrob Chemother (2005) 56:1087–93. doi: 10.1093/jac/dki396
159. Miailhes P, Trabaud MA, Pradat P, Lebouché B, Chevallier M, Chevallier P, et al. Impact of Highly Active Antiretroviral Therapy (HAART) on the Natural History of Hepatitis B Virus (HBV) and HIV Coinfection: Relationship Between Prolonged Efficacy of HAART and HBV Surface and Early Antigen Seroconversion. Clin Infect Dis (2007) 45:624–32. doi: 10.1086/520752
160. de Vries-Sluijs TE, Reijnders JG, Hansen BE, Zaaijer HL, Prins JM, Pas SD, et al. Long-Term Therapy With Tenofovir Is Effective for Patients Co-Infected With Human Immunodeficiency Virus and Hepatitis B Virus. Gastroenterology (2010) 139:1934–41. doi: 10.1053/j.gastro.2010.08.045
161. Zoutendijk R, Zaaijer HL, de Vries-Sluijs TE, Reijnders JG, Mulder JW, Kroon FP, et al. Hepatitis B Surface Antigen Declines and Clearance During Long-Term Tenofovir Therapy in Patients Coinfected With HBV and HIV. J Infect Dis (2012) 206:974–80. doi: 10.1093/infdis/jis439
162. Kosi L, Reiberger T, Payer BA, Grabmeier-Pfistershammer K, Strassl R, Rieger A, et al. Five-Year on-Treatment Efficacy of Lamivudine-, Tenofovir- and Tenofovir + Emtricitabine-Based HAART in HBV-HIV-Coinfected Patients. J Viral Hepat (2012) 19:801–10. doi: 10.1111/j.1365-2893.2012.01601.x
163. Hamers RL, Zaaijer HL, Wallis CL, Siwale M, Ive P, Botes ME, et al. HIV-HBV Coinfection in Southern Africa and the Effect of Lamivudine- Versus Tenofovir-Containing cART on HBV Outcomes. J Acquir Immune Defic Syndr (2013) 64:174–82. doi: 10.1097/QAI.0b013e3182a60f7d
164. Matthews GV, Ali RJ, Avihingsanon A, Amin J, Hammond R, Bowden S, et al. Quantitative HBsAg and HBeAg Predict Hepatitis B Seroconversion After Initiation of HAART in HIV-HBV Coinfected Individuals. PloS One (2013) 8:e61297. doi: 10.1371/journal.pone.0061297
165. Boyd A, Gozlan J, Maylin S, Delaugerre C, Peytavin G, Girard PM, et al. Persistent Viremia in Human Immunodeficiency Virus/Hepatitis B Coinfected Patients Undergoing Long-Term Tenofovir: Virological and Clinical Implications. Hepatology (2014) 60:497–507. doi: 10.1002/hep.27182
166. Huang AJ, Núñez M. Outcomes in HIV/HBV-Coinfected Patients in the Tenofovir Era are Greatly Affected by Immune Suppression. J Int Assoc Provid AIDS Care (2015) 14:3608. doi: 10.1177/2325957415586258
167. Boyd A, Maylin S, Gozlan J, Delaugerre C, Simon F, Girard PM, et al. Use of Hepatitis B Surface and "E" Antigen Quantification During Extensive Treatment With Tenofovir in Patients Co-Infected With HIV-HBV. Liver Int (2015) 35:795–804. doi: 10.1111/liv.12521
168. Huang YS, Sun HY, Chang SY, Chuang YC, Cheng A, Huang SH, et al. Long-Term Virological and Serologic Responses of Chronic Hepatitis B Virus Infection to Tenofovir Disoproxil Fumarate-Containing Regimens in Patients With HIV and Hepatitis B Coinfection. Hepatol Int (2019) 13:431–9. doi: 10.1007/s12072-019-09953-4
169. Chihota BV, Wandeler G, Chilengi R, Mulenga L, Chung RT, Bhattacharya D, et al. High Rates of Hepatitis B Virus (HBV) Functional Cure Among Human Immunodeficiency Virus-HBV Coinfected Patients on Antiretroviral Therapy in Zambia. J Infect Dis (2020) 221:218–22. doi: 10.1093/infdis/jiz450
170. Audsley J, Avihingsanon A, Littlejohn M, Bowden S, Matthews GV, Fairley CK, et al. Long-Term TDF-Inclusive ART and Progressive Rates of HBsAg Loss in HIV-HBV Coinfection-Lessons for Functional HBV Cure? J Acquir Immune Defic Syndr (2020) 84:527–33. doi: 10.1097/QAI.0000000000002386
171. van Bremen K, Hoffmann C, Mauss S, Lutz T, Ingiliz P, Spinner CD, et al. Obstacles to HBV Functional Cure: Late Presentation in HIV and its Impact on HBV Seroconversion in HIV/HBV Coinfection. Liver Int (2020) 40:2978–81. doi: 10.1111/liv.14684
172. Dezanet LNC, Miailhes P, Lascoux-Combe C, Chas J, Maylin S, Gabassi A, et al. Persistent HBV Replication and Serological Response During Up to 15 Years of Tenofovir-Based Antiretroviral Therapy in HIV/HBV-Coinfected Patients: A Multicentre Prospective Cohort Study. J Antimicrob Chemother (2021) 76:3009–19. doi: 10.1093/jac/dkab294
Keywords: hepatitis B virus, hepatitis B surface antigen, functional cure, coinfection, immune
Citation: Wu S, Yi W, Gao Y, Deng W, Bi X, Lin Y, Yang L, Lu Y, Liu R, Chang M, Shen G, Hu L, Zhang L, Li M and Xie Y (2022) Immune Mechanisms Underlying Hepatitis B Surface Antigen Seroclearance in Chronic Hepatitis B Patients With Viral Coinfection. Front. Immunol. 13:893512. doi: 10.3389/fimmu.2022.893512
Received: 10 March 2022; Accepted: 11 April 2022;
Published: 11 May 2022.
Edited by:
Yan-Mei Jiao, Fifth Medical Center of the PLA General Hospital, ChinaReviewed by:
Huiying Rao, Peking University People’s Hospital, ChinaBo Feng, Peking University People’s Hospital, China
Copyright © 2022 Wu, Yi, Gao, Deng, Bi, Lin, Yang, Lu, Liu, Chang, Shen, Hu, Zhang, Li and Xie. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yao Xie, eGlleWFvMDAxMjAxODRAc2luYS5jb20=; Minghui Li, d3VobTIwMDBAc2luYS5jb20=; Lu Zhang, emhhbmdsdTEyMThAMTI2LmNvbQ==
†ORCID: Minghui Li, orcid.org/0000-0003-3233-5473
Wei Yi, orcid.org/0000-0003-4241-8205
Yao Xie, orcid.org/0000-0003-4108-7037
‡These authors have contributed equally to this work