
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Immunol., 28 July 2022
Sec. Viral Immunology
Volume 13 - 2022 | https://doi.org/10.3389/fimmu.2022.889372
This article is part of the Research TopicHost-microbe Interaction in SARS-CoV-2 Infection: Mechanism and InterventionView all 14 articles
 Bruce D. Wines1,2,3,4
Bruce D. Wines1,2,3,4 Liriye Kurtovic2,3
Liriye Kurtovic2,3 Halina M. Trist1Sandra Esparon1
Halina M. Trist1Sandra Esparon1 Ester Lopez5
Ester Lopez5 Klasina Chappin1Li-Jin Chan6,7
Klasina Chappin1Li-Jin Chan6,7 Francesca L. Mordant5
Francesca L. Mordant5 Wen Shi Lee5
Wen Shi Lee5 Nicholas A. Gherardin5
Nicholas A. Gherardin5 Sheila K. Patel8Gemma E. Hartley3Phillip Pymm6,7James P. Cooney6,7James G. Beeson2,3,9,10
Sheila K. Patel8Gemma E. Hartley3Phillip Pymm6,7James P. Cooney6,7James G. Beeson2,3,9,10 Dale I. Godfrey5
Dale I. Godfrey5 Louise M. Burrell8
Louise M. Burrell8 Menno C. van Zelm3,11
Menno C. van Zelm3,11 Adam K. Wheatley5,12
Adam K. Wheatley5,12 Amy W. Chung5
Amy W. Chung5 Wai-Hong Tham6,7Kanta Subbarao5,13
Wai-Hong Tham6,7Kanta Subbarao5,13 Stephen J. Kent5,12,14
Stephen J. Kent5,12,14 P. Mark Hogarth1,2,3*
P. Mark Hogarth1,2,3*Joining a function-enhanced Fc-portion of human IgG to the SARS-CoV-2 entry receptor ACE2 produces an antiviral decoy with strain transcending virus neutralizing activity. SARS-CoV-2 neutralization and Fc-effector functions of ACE2-Fc decoy proteins, formatted with or without the ACE2 collectrin domain, were optimized by Fc-modification. The different Fc-modifications resulted in distinct effects on neutralization and effector functions. H429Y, a point mutation outside the binding sites for FcγRs or complement caused non-covalent oligomerization of the ACE2-Fc decoy proteins, abrogated FcγR interaction and enhanced SARS-CoV-2 neutralization. Another Fc mutation, H429F did not improve virus neutralization but resulted in increased C5b-C9 fixation and transformed ACE2-Fc to a potent mediator of complement-dependent cytotoxicity (CDC) against SARS-CoV-2 spike (S) expressing cells. Furthermore, modification of the Fc-glycan enhanced cell activation via FcγRIIIa. These different immune profiles demonstrate the capacity of Fc-based agents to be engineered to optimize different mechanisms of protection for SARS-CoV-2 and potentially other viral pathogens.
Recent history has seen regular deadly zoonotic coronavirus spillover events with the emergence of severe acute respiratory syndrome coronavirus (SARS-CoV) in 2002 (1), Middle East respiratory syndrome (MERS) coronavirus in 2012 (2) and SARS-CoV-2 in December 2019 (3). SARS related coronaviruses are found in bats throughout Southeast Asia (4) and the serology of people living in proximity to a Rhinolophus spp bat colony suggests these zoonotic infections are not uncommon (5). Since the publication of the SARS-CoV-2 genome in January 2020 (3) there has been rapid development and deployment of vaccines for SARS-CoV-2 (6) and the clinical development of multiple SARS-CoV-2 spike specific neutralizing monoclonal antibodies (mAbs) from convalescent patients or animals, reviewed in (7).
Evolution of the SARS-COV-2 spike protein has selected for increased transmissibility, for example by increased affinity for host cells (8), with the emergence and then dominance of many new variants of concern (VOC) including Alpha, B.1.1.7; Beta, B.1.351; Gamma, P.1; Delta, B.1.617.2 (9) and most recently, Omicron, B.1.1.529 (WHO) that impact the neutralization efficacy of antibodies generated against the spike antigen of earlier strains (10). This includes profound escape from neutralization by some mAbs (11–15) and significant loss of neutralization activity of convalescent sera (13, 16, 17) and of humoral responses to first generation vaccines (12, 18, 19) reviewed in (10). Reinfection by neutralization-escape variants (20, 21) and break-through infection in vaccinees is now a feature of the pandemic (13, 19). Furthermore, protective antibody responses in humans are largely restricted to specific coronavirus species since few Abs to SARS-CoV-2 receptor binding domain (RBD) cross-neutralize SARS-CoV or MERS-CoV (22) but the recent identification of spike-specific broadly neutralizing mAbs may be a key to future pan-beta-coronavirus pandemic preparedness (23). Overall, despite increased surveillance and biosecurity (24) and the development of SARS-CoV-2 vaccines and mAbs, a critical vulnerability to variants of concern (VOC) and future pandemic novel coronaviruses persists. There is a need for prophylactic and therapeutic approaches that are more broadly effective against Sarbecoviruses.
Decoy proteins, based on the host entry receptor, inhibit viral entry, and achieve cross neutralization of multiple virus species or strains (25–31). Angiotensin-converting enzyme 2 (ACE2), is the principal entry receptor for the major human pathogenic coronaviruses, SARS-CoV and SARS-CoV-2 (32, 33), as well as the human endemic coronavirus NL63 (34). ACE2 is a transmembrane carboxypeptidase (35) with the ectodomain comprised of a catalytic domain, as well as a collectrin domain likely involved in dimerization (36). It normally plays a role in cardiovascular homeostasis by cleaving angiotensin II, the key agonist of the renin–angiotensin–aldosterone system (RAAS) that regulates blood pressure and electrolytes (37, 38). SARS-CoV-2 entry into host cells is blocked by the recombinant soluble ACE2 catalytic ectodomain in its native form (39, 40) or when engineered for higher avidity (31, 41, 42) or affinity (28, 29, 31, 41–45) for the ancestral spike, which has thus far been retained against later VOC (28, 29, 31, 41). Enzyme inactive forms of antiviral ACE2 decoy proteins have also been developed (26, 46, 47). However, ACE2 enzymatic activity, by cleaving angiotensin II, is protective in lung injury models and may therefore be beneficial to retain in an ACE2-based biological for COVID19 (38, 39, 48, 49).
To improve virus neutralization potency or pharmacokinetic properties of SARS-CoV-2 decoys, the ACE2 ectodomain, with or without the collectrin domain, has been fused to the Fc portion of IgG (26, 28, 29, 42–47, 50–55), which results in increased neutralisation potency by bivalency and increased serum half-life (56). In mAb studies using in vivo SARS-CoV-2 challenge models, Fc-dependent immune effector functions, which include antibody-dependent cellular cytotoxicity (ADCC), phagocytosis and clearance of viruses, bolstered protection against infection and pathology above that provided by neutralization alone (57–61). Similarly, ACE-Fc decoys have demonstrated protective activity in human ACE2 transgenic mouse models (29, 49, 62) and in hamsters (28) challenged with SARS-CoV-2.
We report the development of multifunctional antiviral proteins by applying novel mutations of the Fc and glycan modification to manipulate the Fc component of ACE2-Fc which resulted variously in increased virus neutralization, complement directed killing and activation of FcγRIIIa.
-trACE2 and trACE2-Fc Truncated ACE2 (trACE2) comprised the catalytic portion of the ACE2 ectodomain and a sequence encoding trACE2 (aa 19-615, Accession BAB40370) in pHLsec (63) was a gift from Merlin Thomas (64). This trACE2 sequence was fused to a synthetic DNA for human IgG1 Fc (Accession AXN93652.1) in pcDNA3.4 (ThermoFisher) with an encoded linker sequence D615-GSGSGSG-T223, where D615 is the last residue of ACE2 and T223 (Eu numbering) is the fusion point to IgG1-Fc on the amino terminal side of the Fc core hinge containing the inter-heavy chain disulfides (for full amino acid sequences see Supplementary Text). -flACE2-Fc comprised the full-length ACE2 ectodomain fused to human IgG1-Fc. The incorporation of a synthetic DNA encoding the collectrin domain (GeneArt, ThermoFisher) formed a full length ACE2 ectodomain encoding sequence (aa 19-740) fused to the human IgG1-Fc via a linker with the sequence S740-GGGGS-T223, where S740 is the last residue of ACE2 and T223 is the fusion point to IgG1-Fc. EflACE2-Fc. EflACE2-Fc was equivalent to the flACE2-Fc, except it incorporated the three mutations, T27Y, L79T, N330Y reported as sACE2.v2.4 and having enhanced affinity for SARS-CoV-2 spike RBD (43). The EflACE2-Fc construct was a synthetic DNA (GeneArt) in pcDNA3.4 (ThermoFisher). The mutations H429F, H429Y and E430G in the Fc were introduced using cleavage at a unique Afe I (New England Biolabs) site within codons for E430-L432 and the insertion of appropriate mutagenic oligos with NEBuilder according to the manufacturer’s instructions (NEB).
ACE2-Fc protein expression used transient transfection of Expi293 cells (Thermo Fisher Scientific). The supernatant of Expi293 transiently transfected for the expression of ACE2-Fc was extensively dialysed against 10mM TrisHCl pH 8 and applied to a High-Q column (BioRad Laboratories). Bound proteins were eluted with the indicated gradient to buffer A with 0.4 M NaCl and washed with 1 M NaCl. Fractions were examined by SDS-PAGE, fractions containing ACE2-Fc were pooled and concentrated using a 30 kDa cut-off filtration device (Merck) and separated by SEC using a Superose 6 column (GE Lifesciences). Lamelli native PAGE (150V, 2.5 h, 4°C), was performed according to (65).
Recombinant Spike receptor binding domain (RBD; aa328-514, GenBank: MN908947.3) of SARS-CoV-2 Wuhan strain was produced with the N-terminal Fel d 1 leader sequence and C-terminal biotin ligase (BirA) AviTag and a hexahistidine affinity tag (Hartley et al., 2020). Specific mutations were introduced in this construct to generate SARS-CoV-2 variant RBD proteins, representing those from three lineages of concern: B.1.351 (beta; N501Y, E484K, K417N), P.1 (gamma; N501Y, E4848K, K417T) and B.1.167.2 (delta; T478K, L452R). The DNA constructs were codon-optimized for H. sapiens and cloned into a pCR3 expression vector. Plasmid DNA was purified from E. coli by Maxiprep (Zymo Research, Irvine, CA), and 30 μg DNA was transfected into Expi 293F cells using the Expi293 Expression system (Thermo Fisher, Waltham, MA). Supernatants from 25 ml cell cultures were collected 5 days post-transfection and purified by application to a Talon NTA-cobalt affinity column (Takara Bio, Kusatsu, Shiga, Japan) with elution in 200 mM Imidazole. Eluted proteins were then dialyzed against 10 mM Tris for 48 hours at 4°C.
Antiviral activity was determined using SARS-CoV-2 (CoV/Australia/VIC01/2020) in a microneutralization assay where cytopathic effect was titred to limiting dilution on Vero cells as described previously (64, 66).
Measurements of the affinity of ACE2 proteins for S protein RBD (64) were performed on the Octet RED96e (FortéBio). All assays were performed at 25°C using anti-human IgG Fc capture (AHC) biosensor tips (FortéBio) in kinetics buffer (PBS pH 7.4 supplemented with 0.1% (w/v) BSA and 0.05% (v/v) Tween-20). After a 60 second (60s) biosensor baseline step, ACE2-Fc recombinant proteins (20 μg/mL) were loaded onto the AHC sensors by submerging sensor tips for 200s and then washing in kinetics buffer for 60s. For most ACE2-Fc recombinant proteins, association measurements were performed by dipping into a two-fold dilution series of SARS-CoV-2 spike RBD (64) from 16–250 or 500nM for 180s and dissociation was measured in kinetics buffer for 180s. For EflACE2-Fc WT a two-fold dilution series of 2 – 31 or 63nM was used. Sensor tips were regenerated five times using a cycle of 5s in 10 mM glycine pH 1.5 and 5 s in kinetics buffer. Baseline drift was corrected by subtracting the average shift of an ACE2-Fc-loaded sensor not incubated with SARS-CoV-2 spike RBD, and an unloaded sensor incubated with SARS-CoV-2 spike RBD. Curve fitting analysis was performed with Octet Data Analysis 10.0 software using a global fit 1:1 model to determine KD values and kinetic parameters. Curves that could not be fitted were excluded from the analyses.
ELISA plates were coated with 5μg/ml CoV-2 receptor binding domain fused to mIgGFc (RBD-Ig, RBD aa residues 334-527) and blocked with phosphate buffered saline (PBS) containing 0.05% (w/v) Tween-20 and 2% (w/v) bovine serum albumin (BSA). RBD-Ig was reacted with ACE2-Fc proteins diluted in ½ log titrations (1 hour, 25°C) followed by washing 5 times with PBS, 0.05% Tween-20. Bound ACE2-Fc was detected with sequential incubation with mouse anti-human IgG1-biotin (Thermo MH1515, clone HP6070, at 1μg/ml for 1 hour, 25°C), high sensitivity streptavidin-HRP (1/10,000 dil, 1 hour, 25°C, Pierce, Thermo Scientific) and TMB substrate.
The ACE2-Fc proteins or the anti-CD20 mAb, Rituximab, at 5µg/ml, or the indicated concentrations were incubated with Ramos cells expressing transfected spike proteins (Ramos-S cells) (67) at 5x106 cells/ml in 25µl in fluorescence activated cell sorting (FACS) buffer- PBS containing 0.5% (w/v) BSA, 1mM glucose (PBS/BSA/G), for 30 min on ice. Cells were washed twice with FACS-buffer, incubated with APC conjugated anti-human IgG-Fc for thirty minutes on ice, washed again and resuspended in 25 µl of FACS-buffer.
Evaluation of the binding of dimeric rsFcγR was performed as described in (68). ACE2-Fc opsonized Ramos-S cells were resuspended in 0.5µg/ml of dimeric rsFcγRIIIa (V158 form) or FACS-buffer and incubated for 30 min on ice followed by 1/500 streptavidin-APC (or anti-hIgG-Fc labelled with fluorescein isothiocyanate for confirmation of ACE2-Fc opsonization) for 20 min on ice. The cells were washed, resuspended in FACS buffer and analyzed on a Canto II flow cytometer (Becton Dickinson).
Ninety-six well flat-bottom MaxiSorp Nunc plates (ThermoFischer Scientific) were coated with 5 μg/ml Avidin in PBS overnight, blocked, and then incubated with either two-fold dilution of biotinylated RBD (69) or 2.5 μg/ml in 0.1% casein for 1 hour at RT. The ACE2-Fc proteins were then added over the indicated concentration range. In experiments to measure C5b-C9 fixation, the plates were incubated with 10% fresh human serum for 30 minutes at RT followed by 1/2000 dilution of rabbit anti-C5b-C9 (Millipore) for 1 hour at RT, washed and then incubated with goat anti-rabbit IgG conjugated to HRP (Millipore) at 1/2000 dilution for 1 hour at RT, followed by TMB substrate for 15-20 minutes at RT (70). Reactivity was stopped using 1 M sulfuric acid and absorbance was measured at 450 nm. Test samples and reagents were prepared in PBS 0.1% (w/v) casein and plates washed thrice between each step using PBS, 0.05% (v/v) Tween 20. Samples were tested in duplicate and corrected for background reactivity using negative control wells from which ACE2-Fc proteins were omitted. The mean and SEM from independent experiments are shown.
CDC was measured by opsonizing Ramos-S cells as above (5x106 cells/ml in 25µl in PBS/BSA/G for 30 min on ice) before resuspending in 1/3 diluted normal human serum for 30 min at 37°C. Cells were washed twice with PBS and the dead cells were enumerated by staining with 1/500 Zombie green (BioLegend) before fixing with 2% paraformaldehyde in PBS and analysis on a Canto II flow cytometer.
This assay used IIA1.6/FcR-γ/FcγRIIIa V158 cells expressing a NF-κB response element driven nanoluciferase (NanoLuc, pNL3.2.NF-κB-RE[NlucP/NF-κB-RE/Hygro], Promega N111) and was performed essentially as described previously (67). Briefly, Ramos cells expressing the Spike-IRES-orange2 were used as target cells and were incubated with agonists and the FcγRIIIa/NF-κB-RE reporter cells for 5h before measurement of induced nanoluciferase with Nano-Glo substrate (Promega).
A custom coronavirus multiplex array (71) was performed using SARS-S1 subunit (S1N-S52H5, Acrobiosystems), SARS-CoV-2 S1 (40591-V08B1) and HCoV NL63 S1 and S2 subunits (40604-V08B, Sino Biological), NL63 S trimer [100788, bpsbioscience], and hexahistidine tagged RBD WT (SARS CoV-2, isolate Wuhan-Hu-1, NCBI Reference Sequence: YP_009724390.1, aa residues 319-541 (72),) and 24 variants identified from the GISAID RBD surveillance repository (71). TrACE2-Fc was biotinylated using EZ-Link® Sulfo-NHS-LC-Biotin (ThermoFisher Scientific) according to the manufacturer’s instructions. Biotinylated trACE2-Fc (70 nM) was incubated with a concentration series, eight two-fold dilutions from 282 nM, of unlabelled trACE2-Fc, flACE2-Fc, EflACE2-Fc fusion proteins or the inhibitory human mAb S35 (AcroBiosystems) and binding to RBD or S proteins coupled to beads was determined using first Streptavidin, R-Phycoerythrin Conjugate (SAPE) (Thermo Fisher) at 4µg/ml (1 h), followed by 10µg/ml of R-Phycoerythrin, Biotin-XX Conjugate (Thermo Fisher) (1 h) and multiplex analysis. Apparent IC50 (nM) values are indicated from curve fits.
Alphafold v2.2 (73, 74) was run on the EflACE2-Fc sequence using five models and specifying two homo-oligomers. The output of this recapitulated the observed structure of the ACE2 homodimer (PDB ID: 6M17, ACE2 residues 19-729) with an RMSD of 1.378 Å, however, the IgG1-Fc domains did not pair. The IgG1-Fc plus the G4S linker and collectrin domain of ACE2 (residues 615-729) was therefore run on Alphafold v2.2 specifying two homo-oligomers and an output of five models. Of these, one model showed correctly paired IgG1 Fc domains and a collectrin domain folded as in the full-length ACE2 structure (PDB ID: 6M17) with an RMSD of 0.718 Å. Superimposition of the collectrin domains of the model with the ACE2 homodimer and that with the paired IgG1-Fc allowed reconstruction for the complete EflACE2-Fc sequence. Positioning of the linkers was manually modelled based on the human B12 IgG crystal structure (PDB ID: 1HZH) to allow the correct pairing of the Fc-hinge disulphide residues at positions 749 and 752 (Figures 1A, B). The trACE2-Fc structure was modelled manually on the EflACE2-Fc model, maintaining the relative position of the ACE2 catalytic domains as in the full-length homodimer. Coordinate files are available from the authors on request.
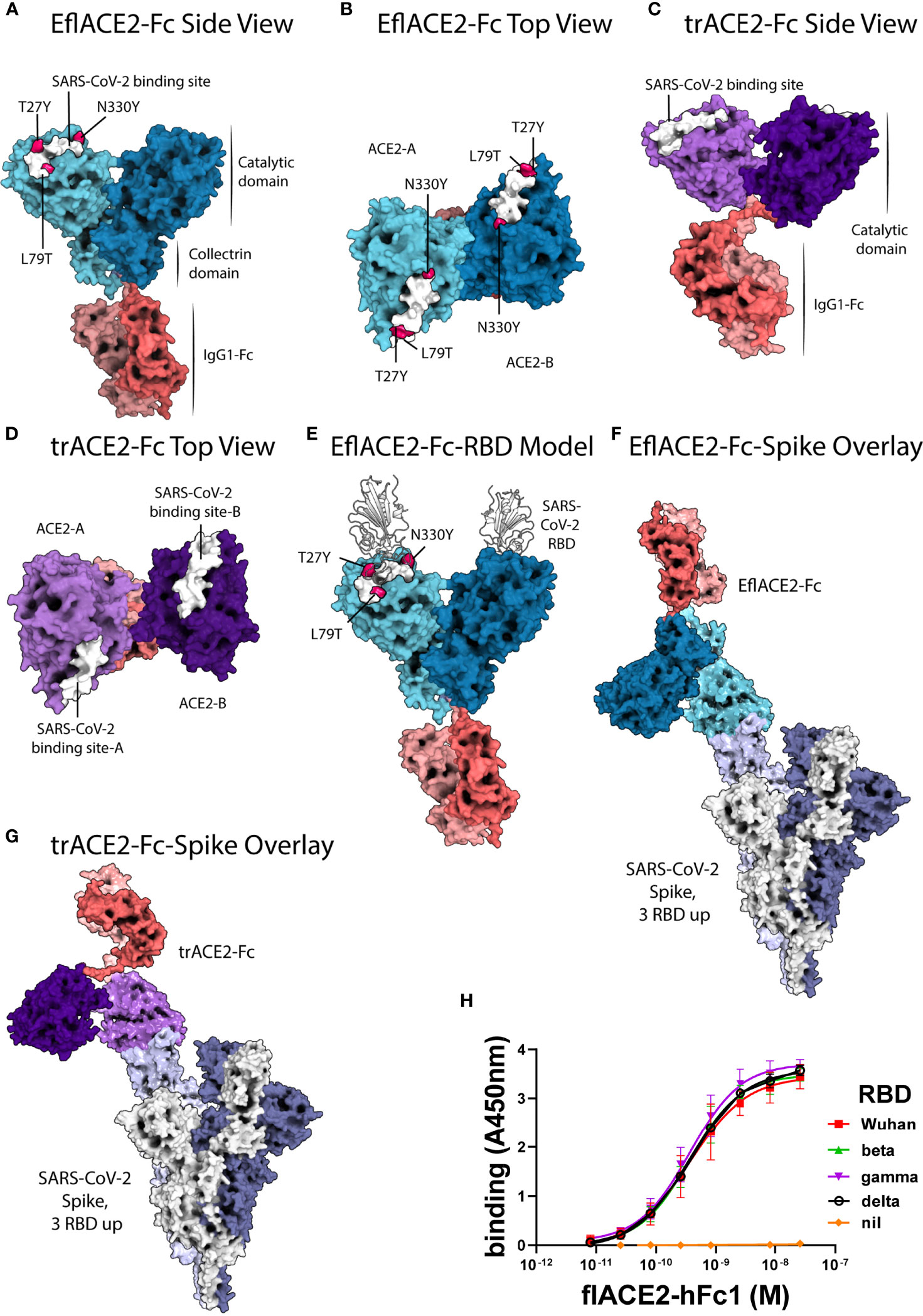
Figure 1 ACE2-Fc protein modelling and the interaction with SARS-CoV-2. (A-E) SARS-CoV-2 spike RBD binding footprint in white. The flACE2-Fc comprised a full-length ACE2 ectodomain (aa 19-740) fused to human IgG1-Fc. (A, B) EflACE2-Fc, having improved RBD binding is a variant of flACE2-Fc wherein three-point mutations, T27Y, L79T and N330Y, have been incorporated into ACE2 component (43) to enhance binding affinity to SARS-CoV-2 spike RBD. (C, D) The truncated ACE2 ectodomain (aa 19-615) was fused to human IgG1 Fc generating the trACE2-Fc fusion protein. (E) HADDOCK model of SARS-CoV-2 spike RBD binding to EflACE2 SARS-CoV-2 spike RBD shown in cartoon representation in white. (F) EflACE2-Fc shown overlayed on, and aligned by, ACE2 residues 19-614 of the 7VXM cryo-EM complex of SARS-CoV-2 spike and ACE2. (G) trACE2-Fc shown overlayed on, and aligned by, ACE2 residues 19-614 of the 7VXM cryo-EM complex of SARS-CoV-2 Spike and ACE2. In (F, G), Positioning of the ACE2 dimer and Fc disulfides respectively indicate the ACE2-Fc constructs are unlikely to bind multiple RBD on a single spike trimer. (H) ACE2-Fc binding to variant SARS-CoV-2 spike RBD. The flACE2-Fc-WT fusion protein binds to the ancestral Wuhan RBD, and the beta, gamma and delta VOC RBDs with equivalent EC50 values. Plotted values are mean ± SD, n = 3, except for delta RBD n = 2. Agonist versus response curve fitting EC50 ranged from 0.31 to 0.40 nM.
Docking of the SARS-CoV-2 spike RBD to the EflACE2-Fc construct was modelled using HADDOCK v2.4 (75, 76) and the best model from the top scoring cluster was taken, having a HADDOCK score of -151 ± 4.2 and an RMSD from the overall lowest energy structure of 0.7 Å ± 0.5 (Figure 1E). The SARS-CoV-2 spike RBD and ACE2 catalytic domain (residues 19-614) had an overall RMSD of 2.733 Å from the observed SARS-CoV-2 binding to native ACE2 (PDB ID: 6M0J), with the SARS-CoV-2 and ACE2 chains aligning more closely with RMSDs of 0.400 Å and 1.400 Å respectively. A HADDOCK SARS-CoV-2 spike RBD docking model generated using an Alphafold prediction of the native ACE2 structure aligned similarly with the observed structure (PDB ID: 6M0J) with an RMSD of 2.940 Å, and overlayed the EflACE2-Fc structure with an RMSD of 0.573 Å.
Data and Statistical analysis used the Prism software package (GraphPad Software 9.0.2, San Diego, CA). Curve fitting to agonist(inhibitor) response curves for EC50 (IC50) determination and ANOVA with multiple comparisons tests were used as indicated in the Figure legends.
A series of ACE2-Fc fusion proteins (Table 1) were produced and analyzed for improved capacity to neutralize SARS-CoV-2 infection and to enhance or transform Fc-dependent effector functions attributed normally to the mechanisms of action of antibodies. Three versions of the ACE2 ectodomain were fused to the human IgG1 Fc portion. The first ACE2 fusion comprised the full length ACE2 ectodomain (flACE2-Fc, aa 19-740), including both the catalytic and collectrin domains (Figure 1A) and the second, an enhanced full length ACE2 ectodomain, EflACE2-Fc, with enhanced binding to SARS-CoV-2 S protein-RBD resulting from three amino acid mutations in the RBD binding site of the ACE2 protein (T27Y, L79T and N330Y) (43) and the third comprised a truncated ectodomain (trACE2-Fc, aa 19-615) containing the ACE2 catalytic domain but lacking the collectrin domain. Models of the trACE2-Fc (Figures 1C, D) and EflACE2-Fc (Figures 1A, B) decoy proteins were generated using Alphafold 2 (73, 74) and EflACE2-Fc was docked to SARS-CoV-2 spike RBD (Figure 1E). A comparison of the RBD docked to the Alphafold prediction of the EflACE2 and native ACE2 structures aligned similarly with the observed structure (PDB ID: 6M0J) with an RMSD of 2.733 Å and 2.940 Å respectively, and overlayed the RBD-EflACE2-Fc docked structure with an RMSD of 0.573 Å. This indicates that the affinity enhancing mutations do not impact the docking position of the SARS-CoV-2 spike RBD using this modelling approach. To evaluate the interaction with trimeric spike and assess the relative distance between ACE2 catalytic domains and adjacent RBD, the EflACE2-Fc-SARS-CoV-2 spike RBD or the trACE2-Fc-SARS-CoV-2 spike RBD model was overlayed on the RBD of chain A of the observed Spike-ACE2 complex structure (PDB ID: 7VXM) (Figures 1F, G). This showed that the ACE2 dimer in the EflACE2-Fc construct is not able to bind adjacent RBD on a single spike trimer due to distance restraints. Though the two ACE2 catalytic domains in the trACE2-Fc construct are likely not dimeric, through the lack of a collectrin domain (36), restraints imposed by disulphide bonding at the N-terminus of the Fc similarly act to restrict the distance between the ACE2 domains and likely also prevent binding to adjacent RBD for this construct (Figure 1G).
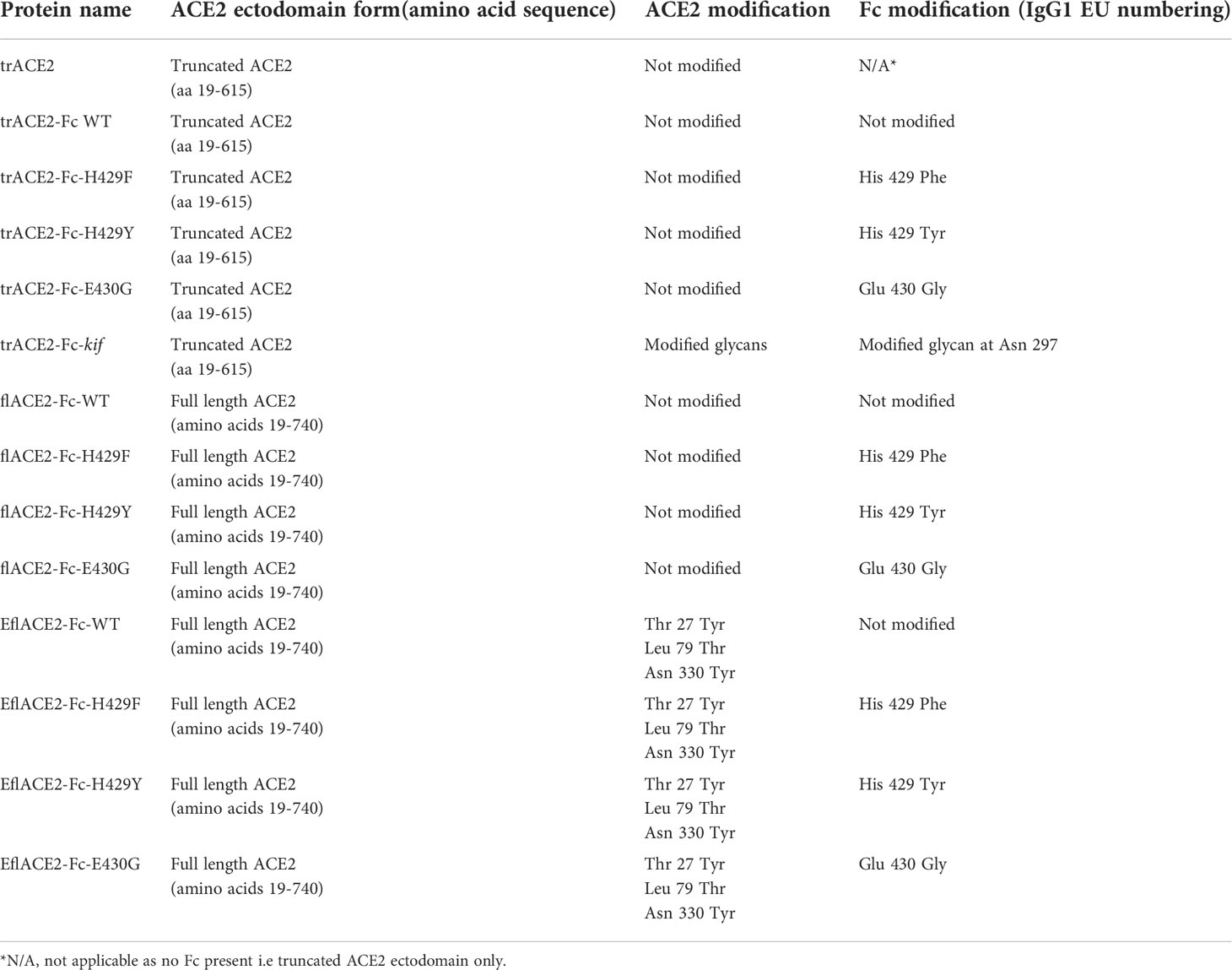
Table 1 ACE2 proteins used in this study.
The key rationale for the development of a ACE2 decoy antiviral protein as a biosecurity agent against a future pandemic is the presumption it will similarly inhibit SARS-CoV-2 variants and ACE2 tropic coronaviruses generally. Indeed, using an ELISA flACE2-Fc bound near equally to both the ancestral RBD and RBD from the beta, gamma and delta VOCs (Figure 1H).
The activity of ACE2-Fc against SARS-CoV-2 variants was further addressed using a bead array. The inhibition of binding of biotinylated trACE2-Fc to an established array of 24 SARS-CoV-2 spike RBD variants (71) by unlabeled ACE2-Fc decoys was examined. Inhibition of binding to RBD-WT followed the hierarchy trACE2-Fc-WT < flACE2-Fc-WT < EflACE2-Fc-WT (IC50 = 114, 80, 10 nM respectively) with effective inhibition of binding to all the individual RBD variants reached, with IC50 values within two-fold of that observed with the ancestral RBD (Figure 2). Thus, across the array of RBD variants the average IC50 values (110 ± 4; 86 ± 4 nM; 9.5 ± 0.9 nM) simply replicated this hierarchy of increasing neutralization potency over trACE2-Fc-WT as variants with increased affinity for ACE2 have equivalent increased susceptibility to inhibition by ACE2, including the N439K, S477N and E484K RBDs and other variants associated with escape from neutralizing antibodies (11, 54, 77). This contrasted sharply with the neutralizing mAb S35 where binding to the L455F and A475V RBD variants was abrogated. Furthermore, the decoy proteins were also effective inhibitors of binding to the spike proteins of the SARS and NL63 beta-coronaviruses (Figure 2). This illustrates the intrinsic resistance of ACE2 based antiviral decoys to escape by spike mutation and their applicability to other viruses that also use ACE2 for entry.
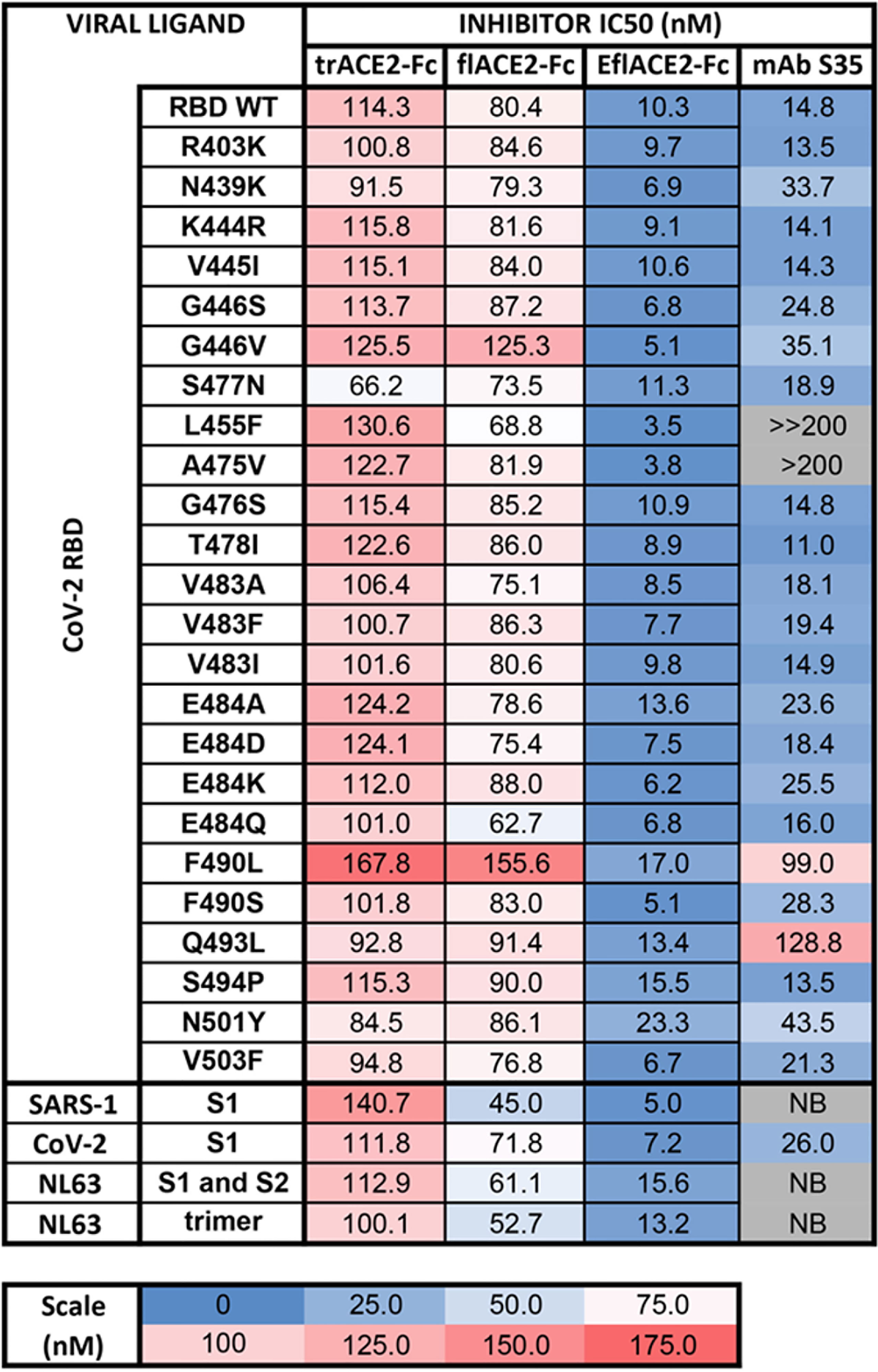
Figure 2 Human ACE2-Fc decoy proteins broadly inhibit binding to RBD variants and S from variants and related Sarbecoviruses. Biotinylated trACE2-Fc was incubated with a concentration series of unlabelled trACE2-Fc, flACE2-Fc, EflACE2-Fc fusion proteins or the inhibitory human mAb S35. Binding to RBD or S proteins coupled to beads was determined. Apparent IC50 (nM) values are indicated. NB, no binding.
In addition to fusion to wild-type (WT) IgG1-Fc, these ACE2 formats were also fused to a Fc carrying novel substitutions of histidine 429 (Eu numbering) with phenylalanine (H429F) or tyrosine (H429Y), or in the adjacent residue, a known IgG hexamerising mutation E430G (78, 79). A glycan-modified form of trACE2-Fc was also produced in the presence of the mannosidase inhibitor kifunensine (trACE2-Fc-kif). The recombinant ACE2-Fc fusion proteins were purified first by anion exchange followed by size exclusion chromatography (SEC) and comprised largely a single species by SEC (Figure 3A) except the H429Y mutant Fc proteins which in all formats were resolved by SEC as oligomeric and monomeric species (Figure 3B).
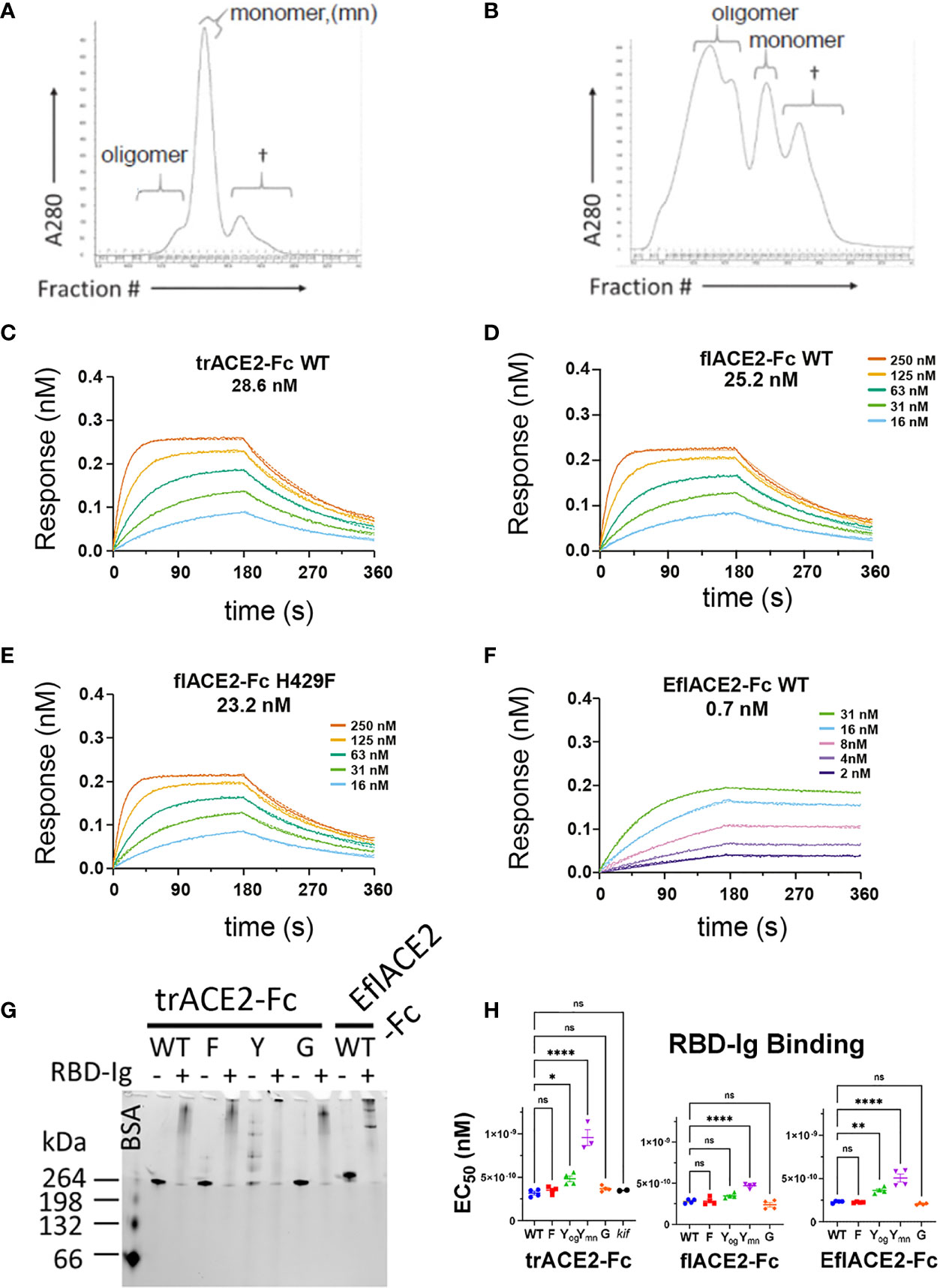
Figure 3 Characterization of engineered human flACE2-Fc and trACE2–Fc fusion proteins. (A) Size-exclusion chromatography (SEC) of IEX fractions containing flACE2-Fc-WT using a Superose 6 column, with oligomeric, monomeric forms and low mw impurities (†) indicated; and (B) SEC of IEX fractions containing flACE2-Fc H429Y, showing the high proportion of oligomeric species. (C–F) Biolayer interferometry (BLI) analysis of ACE2-Fc proteins which were immobilized on anti-human Fc (BLI) sensors and reacted with the indicated concentrations of RBD. The dissociation constants, KD (nM), are derived from global fitting of the association and dissociation curves to a Langmuir binding model. The ACE2-Fc proteins were, (C) trACE2-Fc WT (D) flACE2-Fc WT, (E) flACE2-Fc H429F and (F) the RBD binding-enhanced triple mutant of ACE2 fused to Fc; EflACE2-Fc WT (representative of n = 2 independent experiments). (G) Native Gel-shift analysis of ACE2-Fc proteins (1 mg, ~ 5 pmol) alone or combined with SARS-CoV-2 spike RBD-Ig (0.5 mg, ~ 5 pmol) and analyzed by native PAGE. The resulting shift in size of the proteins in the mixtures demonstrated the formation of ACE2-Fc: Cov2-RBD complexes. (H) Binding of different formats of ACE2-Fc-WT, and their Fc variants to immobilized RBD-Ig was determined by ELISA. EC50 (nM) values are from agonist versus response curve fits,mean ± SD, n is indicated by individual symbols for each independent experiment. One-way ANOVA with Dunnett’s multiple comparisons test, p > 0.05 (ns), ≤ 0.05 (*), ≤0.01 (**), ≤ 0.0001 (****).
Fc modification did not affect the intrinsic affinity for the SARS-CoV-2 spike RBD (e.g. trACE2-Fc-WT, KD = 28.6nM; flACE2-Fc-WT, 25.2 nM; flACE2-Fc-H429F, KD = 23.2 nM, Figures 3C-E) which was comparable with that of the reported affinity 22nM for the flACE2 (43). As expected, the EflACE2-Fc WT protein with the enhanced RBD-binding mutant ACE2 domain showed a ~30-fold increase in affinity to KD = 0.7 nM (Figure 3F) (43) compared to the flACE2-Fc.
Native PAGE (N-PAGE) analysis showed that ACE2-Fc WT fusion proteins migrate as a single species, at ~ 260 kDa for trACE2-Fc and at > 260 kDa for the flACE2-Fc and EflACE2-Fc fusion proteins, reflecting the additional presence of the collectrin domain (Figure 3G). Notably the ACE2-Fc-H429Y variants (e.g. trACE2-Fc-H429Y Fc, Figure 3G, 5th trACE2-Fc lane “Y”) migrated in N-PAGE as several distinct higher molecular weight oligomer species, that were not apparent in denaturing SDS-PAGE, i.e. these comprise non-covalent oligomers. N-PAGE shift analysis showed that the normal and enhanced ACE2 (e.g. trACE2-Fc and EflACE2-Fc) proteins, and the Fc mutants, had high-specific binding activity for SARS-CoV-2 spike RBD, visualized by their shift to high molecular weight complexes following interaction with SARS-CoV-2 spike RBD-Ig (RBD-Ig “+” lanes, Figure 3G). When quantified by ELISA the ACE2-Fc proteins bound the bivalent ligand RBD-Ig with subnanomolar avidity and were unaffected by mutation of the Fc, excepting the oligomer forming H429Y Fc mutants which exhibited weaker binding (Figure 3H).
The antiviral activities of the ACE2-Fc fusion proteins were determined in a microneutralization assay using SARS-CoV-2 infection of Vero cells (64) where the EC50 endpoint corresponds to neutralization of ~99% of the inoculum virions (66). The SARS-CoV-2 neutralization endpoint (EC50 2.70 µM) of the unfused truncated ectodomain (trACE2 alone) was improved ~10-fold by its fusion with the unmodified wildtype Fc region of IgG1 (trACE2-Fc-WT, EC50 283 nM), consistent with its improved binding avidity (Figure 4A). In accord with its increased intrinsic affinity for the RBD (Figure 3F), the EflACE2-Fc-WT (EC50 11 nM) was a further ~11 and 25-fold more inhibitory than the unmodified flACE2-Fc-WT (EC50 124 nM) and trACE2-Fc-WT respectively (Figures 4A–C). Thus overall, EflACE2-Fc-WT (Figure 4C) was ~240-fold more active in virus neutralization than trACE2 alone (Figure 4A).
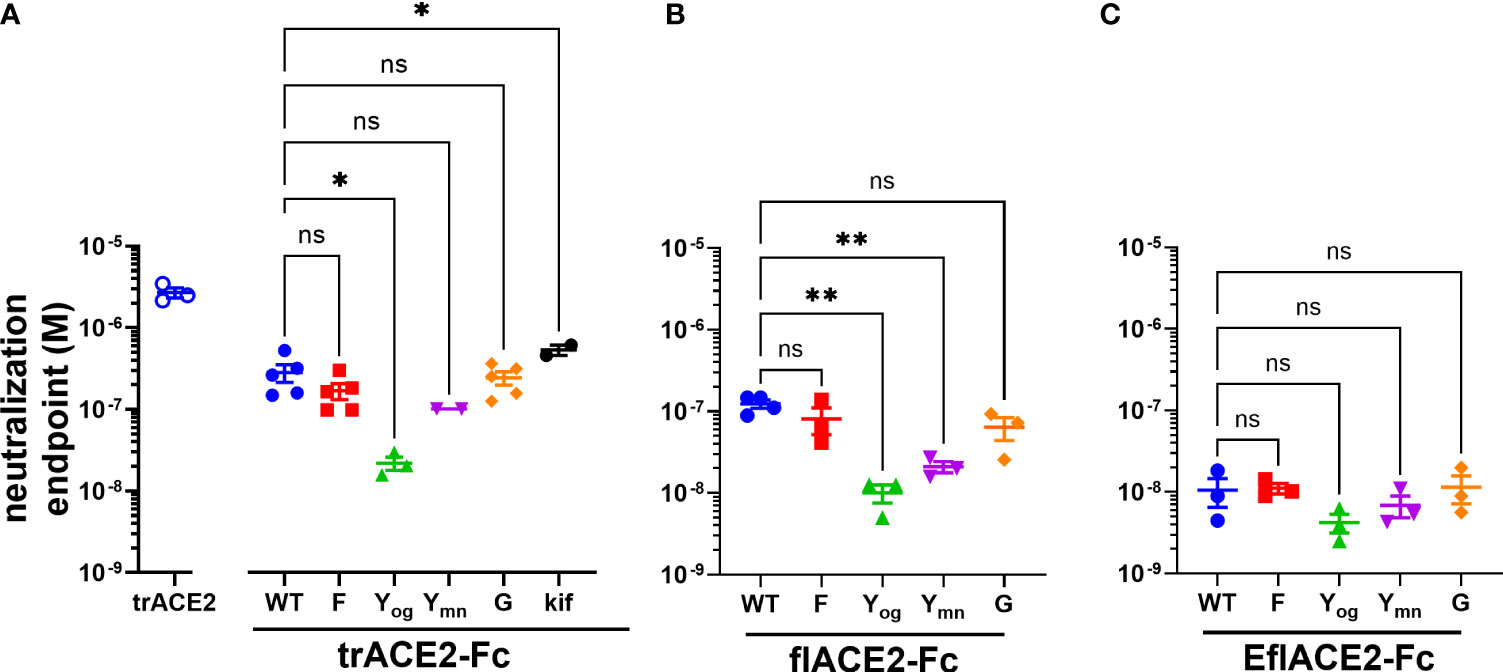
Figure 4 (A–C) SARS-CoV-2 neutralization potency of ACE2-Fc fusion proteins is increased by both the ACE2 scaffold and the H429Y Fc mutation. Neutralization potencies of the ACE2 enzymatic ectodomain polypeptide (trACE2) and the three formats of ACE2-Fc-WT fusion and variant proteins were determined by titration of the cytopathic effect to endpoint in a micro-neutralization assay. The fusion proteins were (A) trACE2-Fc-WT, (B) flACE2-Fc-WT and (C) EflACE2-Fc-WT, incorporating triple mutation of ACE2 engineered (43) for enhanced affinity to RBD and their Fc variants (Eu numbering), E430G, G; H429F, F; H429Y oligomers on SEC, Yog; and H429Y monomers on SEC, Ymn. A further variant trACE2-Fc fusion protein is the glycan-modified trACE2-Fc-kif produced in the presence of kifunensine. Mean ± SEM, one-way ANOVA with Dunnett’s multiple comparisons test, p < 0.05 (ns), ≤ 0.05 (*), ≤ 0.01 (**), independent experiments (n) are indicated as individual symbols.
Of the five Fc modifications, the oligomeric (og) form of the H429Y Fc mutants fused with any ACE2 format, consistently displayed superior neutralization activity within its ACE2 format class. Thus, the oligomeric trACE2-Fc-H429Yog, isolated by SEC, had a neutralization activity (EC50 21.9 nM) that was 13-fold improved over the monomeric trACE2-Fc-WT (EC50 283nM, Figure 4A). Similarly, flACE2-Fc-H429Yog (EC50 10.0 nM) showed greater potency than flACE2-Fc-WT (EC50 124 nM) (Figure 4B). Indeed, it was equivalent in neutralization activity to the EflACE2-Fc WT neutralization (EC50 10.6 nM). Finally, the most potent inhibitor, EflACE2-Fc-H429Yog (EC50 4.23 nM) (Figure 4C), was ~ 600-fold more active than the monovalent trACE2 (Figure 4A). This improved neutralization by the H429Y decoy contrasted with the H429F and the E430G modifications which did not significantly alter SARS-CoV-2 neutralization activity in any ACE2-Fc format (Figures 4A-C). As a comparator the laboratory equivalent of the therapeutic mAb REGN 10933 (casirivimab) had an EC50 of 3.6 nM, (n = 2).
The Fc receptors of leukocytes and serum complement provide the two major effector systems harnessed normally by the Fc portion of antibodies. FcγR functions, which may include ADCC, phagocytosis and clearance of opsonized viruses are important antiviral effector mechanisms and are increasingly found to play a protective role during SARS-CoV-2 infection (57, 58, 60, 61, 80, 81). The interaction of FcγRIIIa with the ACE2-Fc fusion proteins was evaluated by flow cytometry using Ramos cells expressing SARS-CoV-2 spike protein (Ramos-S cells) opsonized with the different formats of ACE2-Fc. Dimeric recombinant soluble FcγRIIIa (68) bound the Fc-WT and H429F and E430G mutant fusion proteins within each ACE2 format class near equivalently. However, the H429Y mutation of the Fc largely ablated FcγR binding in all the ACE2-Fc formats (Supplementary Figure S1A). The loss of FcγRIIIa binding was not due to lack of opsonization of the Ramos-S cells by the H429Y variants as all ACE2-Fc proteins showed similar binding of the SARS-CoV-2 spike protein on the cell surface (Table 2).
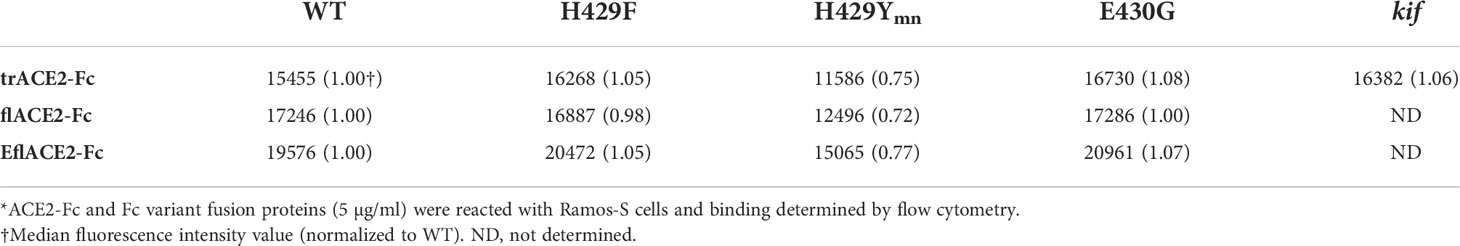
Table 2 Flow cytometric analysis of Ramos-S cells by opsonized ACE2-Fc proteins*.
Next, FcγRIIIa activation by ACE2-Fc fusion proteins was evaluated as a validated surrogate of ADCC (67). FcγRIIIa was activated by Ramos-S cells opsonized with a Fc-WT fusion of any ACE2 format (Figure 5A). The flACE2-Fc-WT induced FcγRIIIa-mediated activation of the reporter cell at 2.7-fold lower concentration than the trACE2-Fc-WT (EC50 1.7 nM) (Figure 4A, p < 0.0001, Supplementary Figures S1B, C), indicating that inclusion of the ACE2 collectrin domain, improved FcγRIIIa activation. Notably, the increased affinity of the EflACE2-Fc WT for SARS-CoV-2 spike protein, did not increase FcγRIIIa activation above that of flACE2-Fc WT (Figure 5A).
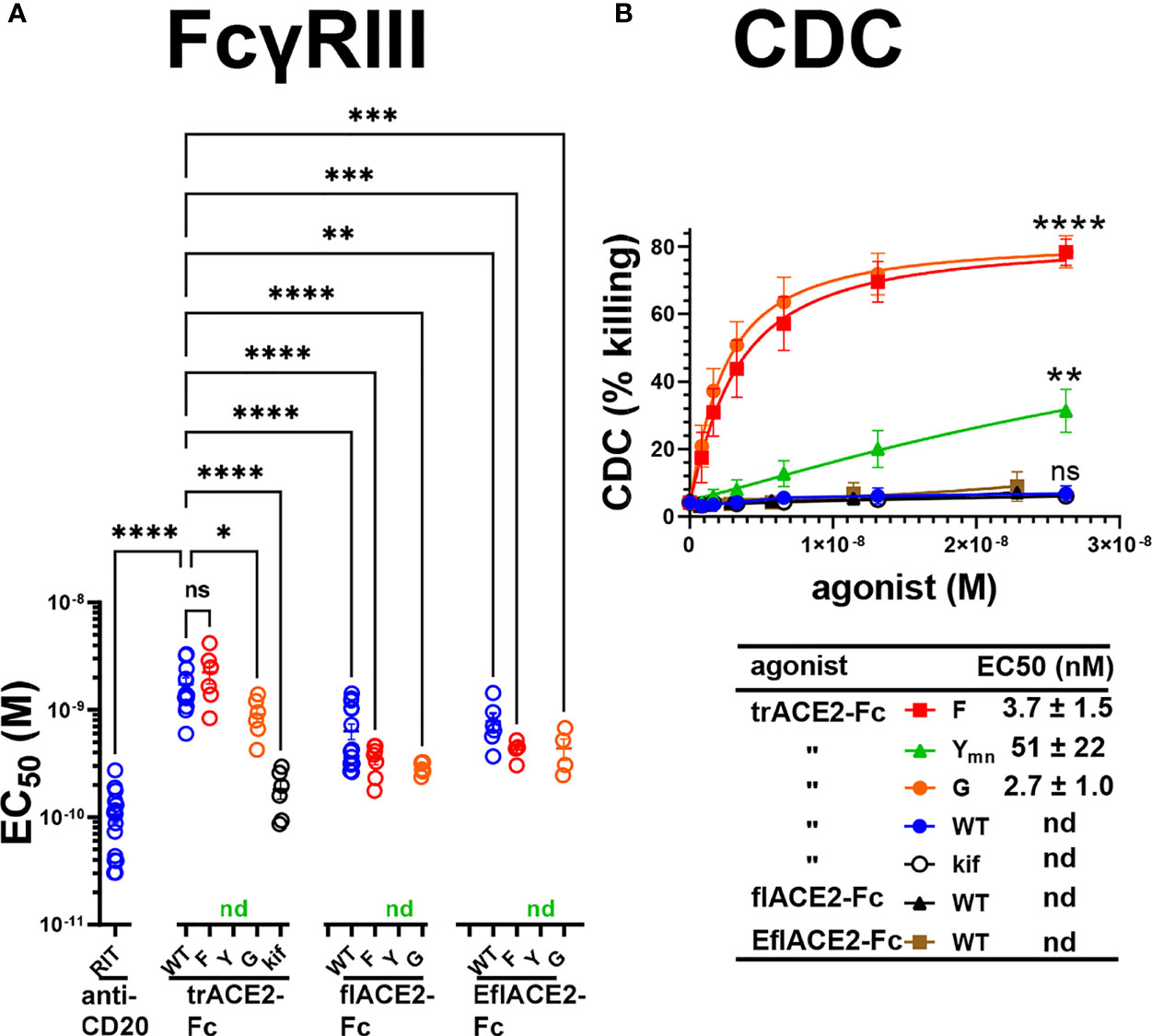
Figure 5 FcγR and complement dependent effector functions of the ACE2-Fc decoy proteins. (A) Activation of FcγRIIIa by ACE2-Fc proteins. ACE2-Fc proteins activated FcγRIIIa, except for the Fc H429Y mutants which failed to stimulate in any ACE2 format either as oligomeric or monomeric forms. Ramos-S target cells were opsonized with trACE2-Fc, flACE2-Fc and EflACE2-Fc, WT and Fc variants, including H429F, F; H429Y, Y; E430G, G or trACE2-Fc-kif, produced from trACE2-Fc WT in 293Expi cells in the presence of the mannosidase inhibitor kifunensine. In some experiments Ramos-S target cells were separately opsonized with Rituximab, RIT. These opsonized targets were incubated with FcγRIIIa/NF-κB-RE nanoluciferase reporter cells and FcγRIIIa activation measured by the induction of nanoluciferase (RLU). Activation data (Supplementary Figures S1B, C) were fitted to agonist response curves to estimate EC50(nM); nd, not determined as there was insufficient activity for the data to be fitted. EC50 values from the curve fits are shown. Mean ± SEM, n is indicated by individual symbols for each independent experiment, one-way ANOVA with Dunnett’s multiple comparisons test, comparing to trACE2-Fc WT. p > 0.05 (ns), ≤ 0.05 (*), ≤ 0.01 (**), ≤ 0.001 (***), ≤ 0.0001 (****). (B) H429F, and E430G Fc mutant ACE2-Fc proteins are potent mediators of complement lysis of SARS-CoV-2 S expressing cells. Flow cytometric analysis of complement-dependent cytotoxicity (CDC) of opsonized Ramos-S cells was determined in the presence of a 1/3 dilution of a pool of normal human serum (from >5 individuals) as a source of complement. Plots are mean ± SEM, n = 3 independent experiments. Two-way ANOVA with Dunnett’s multiple comparisons test comparing to trACE2-Fc-WT for main column effect, p > 0.05 (ns), ≤ 0.01 (**), ≤ 0.0001 (****). EC50 (nM) values are mean ± SEM each from 3 curve fits.
However, the most potent FcγRIIIa activation was achieved following glycan-modification by kifunensine (82) during the production of the trACE2-Fc. Thus, despite the lower activity of the trACE2-Fc format, FcγRIIIa activation by trACE2-Fc-kif exceeded that of the flACE2-Fc and EflACE2-Fc and approached that of the therapeutic anti-CD20 mAb rituximab used as a comparator on the CD20+ Ramos-S cells (Figure 5A; Supplementary Figure S1B). Thus, the hierarchy of FcγRIII activation by the proteins was trACE2-Fc-kif > EflACE2-Fc WT ~ flACE2-Fc- WT > trACE2-Fc-WT.
In accord with the FcγRIIIa binding data (Supplementary Figure S1A), modification of ACE2-Fc decoys by the H429F or E430G mutation had only modest effects on FcγRIIIa activation (Figure 5A; Supplementary Figures S1B, C). In contrast, the H429Y mutation in all ACE2-Fc formats ablated FcγRIIIa activation of cells which is consistent with their abrogated binding to FcγRIIIa (Supplementary Figure S1A). Thus, while enhancing virus neutralization, the H429Y modified Fc in trACE2-Fc, flACE2-Fc and EflACE2-Fc formats were largely inactive in FcγR binding and consequently unable to activate cells via FcγRIIIa (Figure 5A).
The second major Fc-dependent effector system is the classical complement pathway. The activation of complement by the ACE2-Fc proteins was tested initially by ELISA for the capacity to fix complement components C5b-9 and then to mediate complement-dependent killing of cells expressing SARS-CoV-2 spike protein. The fixing of C5b-9 (Supplementary Figure S1D) which forms the membrane attack complex, was achieved by all Fc fusions but was enhanced by both the H429F Fc mutation and the hexamerising E430G mutation of trACE2-Fc compared to unmodified Fc-WT. Despite the H429Y mutated Fc preforming oligomers, which might be anticipated to confer superior complement fixation, this was not apparent and the trACE2-Fc-H429Y oligomer form, showed similar C5b-9 fixation as the trACE2-Fc-WT and glycomodifed trACE2-Fc-kif, (Supplementary Figure S1D).
Despite the ELISA showing that the trACE2-Fc-WT fusions with an unmodified Fc fix C5b-9, analysis of cell killing showed that the unmodified Fc-WT fusion proteins of any ACE2 format failed to mediate significant CDC (Figure 5B). In stark contrast to this CDC inactivity, both the H429F and the hexamerising E430G Fc mutants of trACE2-Fc fusion proteins were remarkably active in mediating complement lysis of Ramos-S cells (Figure 5B). The monomeric form of trACE2-Fc-H429Ymn was active, although substantially less potent than the H429F mutant. H429 in the Fc is thus a site for modification that remarkably potentiates the Fc’s capacity for stimulating complement-mediated target lysis.
In this study we examined different antiviral functions of ACE2-Fc virus decoy proteins. As in IgG antibodies, the Fc-region drives the effector responses mediated by the IgG-Fc fusion proteins. In vivo SARS-CoV-2 challenge models (57–61) have found Fc immune functions of antibodies decreased virus load, spread from nasal tissue to major organ systems, cytokine storm and inflammation, and mortality. In contrast, ablating Fc function resulted in increased disease severity or mortality (58–60, 83). Recently a non-neutralizing human mAb with Fc-enhanced ADCC activity conferred partial protection in a SARS-CoV-2 infection model and contributed to complete protection in combination with a neutralizing mAb (80). Indeed, ADCC potency is an indicator of humoral responses that protect against severe disease in humans (81). While complement activation features in the pathophysiology of severe COVID-19 it is likely to be initially protective (84) and is an identified function of anti-SARS-CoV-2 therapeutic antibodies (85). The Fc portion is thus an important element to optimize for the development of ACE2-Fc as an anti-SARS-CoV-2 antiviral molecule and for viral entry receptors fused to Fc more generally.
Hence, we have manipulated three major antiviral activities of ACE2-Fc by modifying its Fc portion to enhance the existing decoy (neutralization) action of the ACE2 component, complement mediated killing and activation of FcγR. Firstly, the H429Y mutation, in the Fc CH3 domain outside the Fcγ receptor or complement contact sites of the CH2 domain, resulted in the formation of oligomers of the decoy protein which resulted in improved neutralization potency. The improved neutralization activity and oligomeric nature of the H429Y Fc mutant decoys mimic the polymeric antibody classes, IgA (86) and IgM (87) where avidity contributes to the efficacy of SARS-CoV-2 neutralization. Fc : Fc interactions are a recognized property of IgG antibodies (88) and their stabilization by mutation can lead to the formation of in solution oligomers (89). In contrast, the E430G modification of IgG is known to promote “on-target” oligomerization (hexamerization) of IgG (79), but did not significantly alter SARS-CoV-2 neutralization activity in any ACE2-Fc format. In contrast, the H429Y Fc mutation enhanced neutralization potency of all formats of the ACE2-Fc decoy proteins. H429Y Fc mutation in combination with the inclusion of the collectrin domain and the triple ACE2 mutations enhancing affinity for S (43), (i.e. EflACE2-Fc-H429Y) resulted in an overall 600-fold increased SARS-CoV-2 neutralization potency over that of the monomeric truncated ACE2 domain. The neutralization potency of EflACE2-Fc-H429Y (4.2 nM) was comparable to that of the laboratory equivalent of the therapeutic mAb REGN 10933 (casirivimab, 3.6 nM). A feature of the H429Y mutation was the loss of binding by FcγRIIIa. Mutations at the CH2/CH3 interface can affect low affinity FcγR binding to the Fc (90), suggesting these sites, though distant, can affect each other (91).
Secondly, the phenylalanine substitution of histidine 429 (H429F) of the ACE2-Fc proteins did not enhance neutralization but did transform CDC against S expressing targets. This improved CDC activity was like that of E430G mutated ACE2-Fc, a known “on-target” Fc-hexamerising mutation, a format optimal for C1 binding and activation (78, 79). Lastly, FcγR potency of trACE2-Fc was improved by modifying the Fc glycan (82) to enhance FcγRIII binding (92). It is likely that similar treatment of flACE2-Fc WT and EflACE2-Fc WT, or alternatively, amino acid substitution to increase affinity for FcγRIIIa (93), would similarly further improve their FcγRIII activating potency. Notably, FcγRIII activation was a little reduced for the decoy lacking the collection domain, indicating the formatting of Fc-fusion proteins can impact Fc-mediated activity.
We have demonstrated ACE2-Fc to be a potent agent against SARS-CoV-2, not only for neutralization but also for the harnessing of Fc-mediated effector functions. The exemplar Fc modifications demonstrated herein illustrate the potential for the tuning of Fc function to optimize virus neutralization, FcγR interaction and complement activation. This selection of desired functional profiles could aid the deployment of broadly effective ACE2-Fc, mAbs and other Fc therapeutics. There has been a rapid progression of multiple different SARS-CoV-2 mAbs to clinical use that is likely to herald increased deployment of mAbs clinically for infectious diseases. The optimization of Fc functions will make a significant difference to their clinical success. Furthermore, the world remains susceptible to new pandemics and vaccine escape variants. Thus, an antiviral decoy comprising optimized Fc fusion to a viral entry receptor such as ACE2-Fc, is an important option for deploying a rapid first line of defense to contain new zoonotic viral threats while vaccines, mAbs and antiviral drugs are being developed.
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation. PDB coordinate files are available from the authors on request.
BW and PMH conceived and planned the experiments. BW, LK, HT, SE, EL, KC, L-JC, FM, WL, NG, SP, GH, PP, and JC performed the experiments and analyzed the data. JB, DG, LB, MvZ, AW, AC, W-HT, KS, SK, and PMH provided supervision and analyzed the data. BW and PMH wrote the manuscript with input from all other authors. All authors contributed to the article and approved the submitted version.
The Medical Research Future Fund (MRFF 2002073) and Victorian State Government COVID research funding supported the research of AW, DG, W-HT, SK, PH, and SP, LB (1175865) with contribution from the Victorian Operational Infrastructure Support Program and Australian Government NHMRC Independent Research Institutes Infrastructure Support Scheme. NHMRC project grants supported PH, BW. (1145303). Investigator Grants are held by JB (1173046), KS (1177174) and DG (2008913) and program grants by SK (1149990), LB (1055214). DG (1117766), W-HT, SK, MZ (1117687) and AW receive National Health and Medical Research Council (NHMRC) fellowships. DG is supported by the Australian Research Council (ARC; CE140100011). NG was supported by an ARC DECRA Fellowship (DE210100705). SP and LB funded by a Heart Foundation Vanguard Grant (105798). The Melbourne WHO Collaborating Centre for Reference and Research on Influenza is supported by the Australian Government Department of Health. W-HT is a Howard Hughes Medical Institute–Wellcome Trust International Research Scholar (208693/Z/17/Z).
Authors PMH and BW are inventors on a provisional patent filing by the Burnet Institute.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We thank Dr Gaoqian Feng for pooled human serum. Thanks to Reema Bajaj for reading the manuscript.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2022.889372/full#supplementary-material
Supplementary Figure 1 | Human ACE2-Fc proteins activate FcγRIIIa and Complement. (A) FcγRIIIa binding. The ACE2-Fc WT fusion proteins and their variants (5 µg/ml) were reacted with Ramos-S cells (Ramos cells expressing spike protein) and Fc receptor binding evaluated by flow cytometry using biotinylated dimeric rsFcγRIIIa, followed by streptavidin-APC. (mean of 3 replicates). (B, C) Activation of FcγRIIIa. ACE2-Fc proteins are potent activators of FcγRIIIa apart from the Fc H429Y mutants which fail to stimulate FcγRIIIa in any ACE2 format. Ramos-S target cells were opsonized with (B) trACE2-Fc and (C) flACE2-Fc, WT and separately with Fc variants, including H429F, F; H429Y unfractionated, Y; H429Y oligomers, Yog; H429Y monomer, Ymn; E430G, G or trACE2-Fc kif produced from trACE2-Fc WT in 293Expi cells in the presence of the mannosidase inhibitor kifunensine. Ramos-S target cells were separately opsonized with Rituximab, RIT. These opsonized targets were incubated with FcγRIIIa-NF-κB-RE nanoluciferase reporter cells and FcγRIIIa activation measured by the induction of nanoluciferase (RLU). Representative activation data showing fitting to agonist response curves to determine each EC50 (nM) data point shown in Figure 5A. (D) ACE2-Fc fusion proteins comprising Fc regions with either of the H429F and E430G mutations, strongly fix complement C5b-9. In ELISA analysis the indicated concentration series of trACE2-Fc or its Fc variants, was bound to SARS-CoV-2 spike RBD-biotin (2.5 μg/ml) captured by plate bound avidin (2 μg/ml). Following incubation with human serum the formation of C5b-9 was determined, (mean ± SD); two independent experiments.
1. Zhong NS, Zheng BJ, Li YM, Poon ZH, Chan KH, Li PH, et al. Guan: Epidemiology and cause of severe acute respiratory syndrome (SARS) in guangdong, people’s republic of China, in February, 2003. Lancet (2003) 362(9393):1353–8. doi: 10.1016/s0140-6736(03)14630-2
2. Zaki AM, van Boheemen S, Bestebroer TM, Osterhaus AD, Fouchier RA. Isolation of a novel coronavirus from a man with pneumonia in Saudi Arabia. N Engl J Med (2012) 367(19):1814–20. doi: 10.1056/NEJMoa1211721
3. Zhu N, Zhang D, Wang W, Li X, Yang B, Song J, et al. A novel coronavirus from patients with pneumonia in China, 2019. N Engl J Med (2020) 382(8):727–33. doi: 10.1056/NEJMoa2001017
4. Wacharapluesadee S, Tan CW, Maneeorn P, Duengkae P, Zhu F, Joyjinda Y, et al. Evidence for SARS-CoV-2 related coronaviruses circulating in bats and pangolins in southeast Asia. Nat Commun (2021) 12(1):972. doi: 10.1038/s41467-021-21240-1
5. Wang N, Li SY, Yang XL, Huang HM, Zhang YJ, Guo H, et al. Serological evidence of bat SARS-related coronavirus infection in humans, China. Virol Sin (2018) 33(1):104–7. doi: 10.1007/s12250-018-0012-7
6. Heinz FX, Stiasny K. Distinguishing features of current COVID-19 vaccines: knowns and unknowns of antigen presentation and modes of action. NPJ Vaccines (2021) 6(1):104. doi: 10.1038/s41541-021-00369-6
7. Corti D, Purcell LA, Snell G, Veesler D. Tackling COVID-19 with neutralizing monoclonal antibodies. Cell (2021) 184(12):3086–108. doi: 10.1016/j.cell.2021.05.005
8. Tragni V, Preziusi F, Laera L, Onofrio A, Mercurio I, Todisco S, et al. Modeling SARS-CoV-2 spike/ACE2 protein-protein interactions for predicting the binding affinity of new spike variants for ACE2, and novel ACE2 structurally related human protein targets, for COVID-19 handling in the 3PM context. EPMA J (2022) 13(1):149–75. doi: 10.1007/s13167-021-00267-w
9. Tegally H, Wilkinson E, Giovanetti M, Iranzadeh A, Fonseca V, Giandhari J, et al. Detection of a SARS-CoV-2 variant of concern in south Africa. Nature (2021) 592(7854):438–43. doi: 10.1038/s41586-021-03402-9
10. Harvey WT, Carabelli AM, Jackson B, Gupta RK, Thomson EC, Harrison EM, et al. SARS-CoV-2 variants, spike mutations and immune escape. Nat Rev Microbiol (2021) 19(7):409–24. doi: 10.1038/s41579-021-00573-0
11. Yuan M, Huang D, Lee CD, Wu NC, Jackson AM, Zhu X, et al. Structural and functional ramifications of antigenic drift in recent SARS-CoV-2 variants. Science (2021) 373(6556):818–23. doi: 10.1126/science.abh1139
12. Collier DA, De Marco A, Ferreira I, Meng B, Datir RP, Walls AC, et al. Sensitivity of SARS-CoV-2 B.1.1.7 to mRNA vaccine-elicited antibodies. Nature (2021) 593(7857):136–41. doi: 10.1038/s41586-021-03412-7
13. Wang P, Nair MS, Liu L, Iketani S, Luo Y, Guo Y, et al. Antibody resistance of SARS-CoV-2 variants B.1.351 and B.1.1.7. Nature (2021) 593(7857):130–5. doi: 10.1038/s41586-021-03398-2
14. Chen RE, Winkler ES, Case JB, Aziati ID, Bricker TL, Joshi A, et al. In vivo monoclonal antibody efficacy against SARS-CoV-2 variant strains. Nature (2021) 596(7870):103–8. doi: 10.1038/s41586-021-03720-y
15. Liu H, Wei P, Zhang Q, Chen Z, Aviszus K, Downing W, et al. 501Y.V2 and 501Y.V3 variants of SARS-CoV-2 lose binding to bamlanivimab in vitro. MAbs (2021) 13(1):1919285. doi: 10.1080/19420862.2021.1919285
16. Cele S, Gazy I, Jackson L, Hwa SH, Tegally H, Lustig G, et al. Escape of SARS-CoV-2 501Y.V2 from neutralization by convalescent plasma. Nature (2021) 593(7857):142–6. doi: 10.1038/s41586-021-03471-w
17. Wibmer CK, Ayres F, Hermanus T, Madzivhandila M, Kgagudi P, Oosthuysen B, et al. SARS-CoV-2 501Y.V2 escapes neutralization by south African COVID-19 donor plasma. Nat Med (2021) 27(4):622–5. doi: 10.1038/s41591-021-01285-x
18. Wang Z, Schmidt F, Weisblum Y, Muecksch F, Barnes CO, Finkin S, et al. mRNA vaccine-elicited antibodies to SARS-CoV-2 and circulating variants. Nature (2021) 592(7855):616–22. doi: 10.1038/s41586-021-03324-6
19. Kustin T, Harel N, Finkel U, Perchik S, Harari S, Tahor M, et al. Evidence for increased breakthrough rates of SARS-CoV-2 variants of concern in BNT162b2-mRNA-vaccinated individuals. Nat Med (2021) 27(8):1379–84. doi: 10.1038/s41591-021-01413-7
20. Sabino EC, Buss LF, Carvalho MPS, Prete CA Jr., Crispim MAE, Fraiji NA, et al. Resurgence of COVID-19 in manaus, Brazil, despite high seroprevalence. Lancet (2021) 397(10273):452–5. doi: 10.1016/S0140-6736(21)00183-5
21. Pulliam JRC, van Schalkwyk C, Govender N, von Gottberg A, Cohen C, Groome MJ, et al. Increased risk of SARS-CoV-2 reinfection associated with emergence of Omicron in South Africa. Science (2022) 76(6593):eabn4947. doi: 10.1126/science.abn4947
22. Ju B, Zhang Q, Ge J, Wang R, Sun J, Ge X, et al. Human neutralizing antibodies elicited by SARS-CoV-2 infection. Nature (2020) 584(7819):115–9. doi: 10.1038/s41586-020-2380-z
23. Zhou P Fau - Yuan M, Yuan G, Auid M, Song G, Beutler N, Shaabani N, et al. A human antibody reveals a conserved site on beta-coronavirus spike proteins and confers protection against SARS-CoV-2 infection. Sci Transl Med (2022) 14(637):eabi9215. doi: 10.1126/scitranslmed.abi9215
24. Daszak P, Olival KJ, Li H. A strategy to prevent future epidemics similar to the 2019-nCoV outbreak. Biosaf Health (2020) 2(1):6–8. doi: 10.1016/j.bsheal.2020.01.003
25. Stegmann C, Hochdorfer D, Lieber D, Subramanian N, Stohr D, Laib Sampaio K, et al. A derivative of platelet-derived growth factor receptor alpha binds to the trimer of human cytomegalovirus and inhibits entry into fibroblasts and endothelial cells. PloS Pathog (2017) 13(4):e1006273. doi: 10.1371/journal.ppat.1006273
26. Zhang Z, Zeng E, Zhang L, Wang W, Jin Y, Sun J, et al. Potent prophylactic and therapeutic efficacy of recombinant human ACE2-Fc against SARS-CoV-2 infection in vivo. Cell Discovery (2021) 7(1):65. doi: 10.1038/s41421-021-00302-0
27. Zhang L, Narayanan KK, Cooper L, Chan KK, Devlin CA, Aguhob A, et al. An engineered ACE2 decoy receptor can be administered by inhalation and potently targets the BA.1 and BA.2 omicron variants of SARS-CoV-2. bioRxiv (2022) 2022:3.28.486075. doi: 10.1101/2022.03.28.486075
28. Higuchi Y, Suzuki T, Arimori T, Ikemura N, Mihara E, Kirita Y, et al. Engineered ACE2 receptor therapy overcomes mutational escape of SARS-CoV-2. Nat Commun (2021) 12(1):3802. doi: 10.1038/s41467-021-24013-y
29. Sims JJ, Greig JA, Michalson KT, Lian S, Martino RA, Meggersee R, et al. Intranasal gene therapy to prevent infection by SARS-CoV-2 variants. PloS Pathog (2021) 17(7):e1009544. doi: 10.1371/journal.ppat.1009544
30. Ikemura N, Taminishi S, Inaba T, Arimori T, Motooka D, Katoh K, et al. Engineered ACE2 counteracts vaccine-evading SARS-CoV-2 omicron variant. bioRxiv (2022) 2021:12.22.473804. doi: 10.1101/2021.12.22.473804
31. Tanaka S, Nelson G, Olson CA, Buzko O, Higashide W, Shin A, et al. An ACE2 triple decoy that neutralizes SARS-CoV-2 shows enhanced affinity for virus variants. Sci Rep (2021) 11(1):12740. doi: 10.1038/s41598-021-91809-9
32. Walls AC, Park YJ, Tortorici MA, Wall A, McGuire AT, Veesler D. Structure, function, and antigenicity of the SARS-CoV-2 spike glycoprotein. Cell (2020) 183(6):1735. doi: 10.1016/j.cell.2020.11.032
33. Zhou P, Yang XL, Wang XG, Hu B, Zhang L, Zhang W, et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature (2020) 579(7798):270–3. doi: 10.1038/s41586-020-2012-7
34. Devarakonda CKV, Meredith E, Ghosh M, Shapiro LH. Coronavirus receptors as immune modulators. J Immunol (2021) 206(5):923–9. doi: 10.4049/jimmunol.2001062
35. Obukhov AG, Stevens BR, Prasad R, Li Calzi S, Boulton ME, Raizada MK, et al. SARS-CoV-2 infections and ACE2: Clinical outcomes linked with increased morbidity and mortality in individuals with diabetes. Diabetes (2020) 69(9):1875–86. doi: 10.2337/dbi20-0019
36. Yan R, Zhang Y, Li Y, Xia L, Guo Y, Zhou Q. Structural basis for the recognition of SARS-CoV-2 by full-length human ACE2. Science (2020) 367(6485):1444–8. doi: 10.1126/science.abb2762
37. Bourgonje AR, Abdulle AE, Timens W, Hillebrands JL, Navis GJ, Gordijn SJ, et al. Angiotensin-converting enzyme 2 (ACE2), SARS-CoV-2 and the pathophysiology of coronavirus disease 2019 (COVID-19). J Pathol (2020) 251(3):228–48. doi: 10.1002/path.5471
38. Liu P, Wysocki J, Souma T, Ye M, Ramirez V, Zhou B, et al. Novel ACE2-Fc chimeric fusion provides long-lasting hypertension control and organ protection in mouse models of systemic renin angiotensin system activation. Kidney Int (2018) 94(1):114–25. doi: 10.1016/j.kint.2018.01.029
39. Zoufaly A, Poglitsch M, Aberle JH, Hoepler W, Seitz T, Traugott M, et al. Human recombinant soluble ACE2 in severe COVID-19. Lancet Respir Med (2020) 8(11):1154–8. doi: 10.1016/S2213-2600(20)30418-5
40. Monteil V, Kwon H, Prado P, Hagelkruys A, Wimmer RA, Stahl M, et al. Inhibition of SARS-CoV-2 infections in engineered human tissues using clinical-grade soluble human ACE2. Cell (2020) 181(4):905–913 e7. doi: 10.1016/j.cell.2020.04.004
41. Xiao T, Lu J, Zhang J, Johnson RI, McKay LGA, Storm N, et al. A trimeric human angiotensin-converting enzyme 2 as an anti-SARS-CoV-2 agent. Nat Struct Mol Biol (2021) 28(2):202–9. doi: 10.1038/s41594-020-00549-3
42. Li Y, Wang H, Tang X, Fang S, Ma D, Du C, et al. SARS-CoV-2 and three related coronaviruses utilize multiple ACE2 orthologs and are potently blocked by an improved ACE2-ig. J Virol (2020) 94(22):e01283–20. doi: 10.1128/JVI.01283-20
43. Chan KK, Dorosky D, Sharma P, Abbasi SA, Dye JM, Kranz DM, et al. Engineering human ACE2 to optimize binding to the spike protein of SARS coronavirus 2. Science (2020) 369(6508):1261–5. doi: 10.1126/science.abc0870
44. Glasgow A, Glasgow J, Limonta D, Solomon P, Lui I, Zhang Y, et al. Engineered ACE2 receptor traps potently neutralize SARS-CoV-2. Proc Natl Acad Sci U.S.A. (2020) 117(45):28046–55. doi: 10.1073/pnas.2016093117
45. Cohen-Dvashi H, Weinstein J, Katz M, Eilon M, Mor Y, Shimon A, et al. Coronacept – a potent immunoadhesin against SARS-CoV-2. bioRxiv (2020) 2020:8.12.247940. doi: 10.1101/2020.08.12.247940
46. Lei C, Qian K, Li T, Zhang S, Fu W, Ding M, et al. Neutralization of SARS-CoV-2 spike pseudotyped virus by recombinant ACE2-ig. Nat Commun (2020) 11(1):2070. doi: 10.1038/s41467-020-16048-4
47. Liu P, Xie X, Gao L, Jin J. Designed variants of ACE2-Fc that decouple anti-SARS-CoV-2 activities from unwanted cardiovascular effects. Int J Biol Macromol (2020) 165(Pt B):1626–33. doi: 10.1016/j.ijbiomac.2020.10.120
48. Abd El-Aziz TM, Al-Sabi A, Stockand JD. Human recombinant soluble ACE2 (hrsACE2) shows promise for treating severe COVID-19. Signal Transduct Target Ther (2020) 5(1):258. doi: 10.1038/s41392-020-00374-6
49. Zhang L, Dutta S, Xiong S, Chan M, Chan KK, Fan TM, et al. Engineered ACE2 decoy mitigates lung injury and death induced by SARS-CoV-2 variants. Nat Chem Biol (2022) 18(3):342–51. doi: 10.1038/s41589-021-00965-6
50. Iwanaga N, Cooper L, Rong L, Maness NJ, Beddingfield B, Qin Z, et al. ACE2-IgG1 fusions with improved in vitro and in vivo activity against SARS-CoV-2. iScience (2022) 25(1):103670. doi: 10.1016/j.isci.2021.103670
51. Tada T, Fan C, Chen JS, Kaur R, Stapleford KA, Gristick H, et al. An ACE2 microbody containing a single immunoglobulin Fc domain is a potent inhibitor of SARS-CoV-2. Cell Rep (2020) 33(12):108528. doi: 10.1016/j.celrep.2020.108528
52. Huang KY, Lin MS, Kuo TC, Chen CL, Lin CC, Chou YC, et al. Humanized COVID-19 decoy antibody effectively blocks viral entry and prevents SARS-CoV-2 infection. EMBO Mol Med (2020) 13(1):e12828. doi: 10.15252/emmm.202012828
53. Svilenov HL, Sacherl J, Reiter A, Wolff LS, Cheng CC, Stern M, et al. Picomolar inhibition of SARS-CoV-2 variants of concern by an engineered ACE2-IgG4-Fc fusion protein. Antiviral Res (2021) 196:105197. doi: 10.1016/j.antiviral.2021.105197
54. Liu Z, VanBlargan LA, Bloyet LM, Rothlauf PW, Chen RE, Stumpf S, et al. Identification of SARS-CoV-2 spike mutations that attenuate monoclonal and serum antibody neutralization. Cell Host Microbe (2021) 29(3):477–488 e4. doi: 10.1016/j.chom.2021.01.014
55. Jing W, Procko E. ACE2-based decoy receptors for SARS coronavirus 2. Proteins (2021) 89(9):1065–78. doi: 10.1002/prot.26140
56. Stapleton NM, Einarsdottir HK, Stemerding AM, Vidarsson G. The multiple facets of FcRn in immunity. Immunol Rev (2015) 268(1):253–68. doi: 10.1111/imr.12331
57. Schafer A, Muecksch F, Lorenzi JCC, Leist SR, Cipolla M, Bournazos S, et al. Antibody potency, effector function, and combinations in protection and therapy for SARS-CoV-2 infection in vivo. J Exp Med (2021) 218(3):e20201993. doi: 10.1084/jem.20201993
58. Ullah I, Prevost J, Ladinsky MS, Stone H, Lu M, Anand SP, et al. Live imaging of SARS-CoV-2 infection in mice reveals that neutralizing antibodies require Fc function for optimal efficacy. Immunity (2021) 54(9):2143–2158 e15. doi: 10.1016/j.immuni.2021.08.015
59. Suryadevara N, Shrihari S, Gilchuk P, VanBlargan LA, Binshtein E, Zost SJ, et al. Neutralizing and protective human monoclonal antibodies recognizing the n-terminal domain of the SARS-CoV-2 spike protein. Cell (2021) 184(9):2316–2331 e15. doi: 10.1016/j.cell.2021.03.029
60. Yamin R, Jones AT, Hoffmann HH, Schafer A, Kao KS, Francis RL, et al. Fc-engineered antibody therapeutics with improved anti-SARS-CoV-2 efficacy. Nature (2021) 599(7885):465–70. doi: 10.1038/s41586-021-04017-w
61. Li D, Edwards RJ, Manne K, Martinez DR, Schafer A, Alam SM, et al. In vitro and in vivo functions of SARS-CoV-2 infection-enhancing and neutralizing antibodies. Cell (2021) 184(16):4203–4219 e32. doi: 10.1016/j.cell.2021.06.021
62. Chen Y, Sun L, Ullah I, Beaudoin-Bussières G, Anand SP, Hederman AP, et al. Engineered ACE2-Fc counters murine lethal SARS-CoV-2 infection through direct neutralization and Fc-effector activities. bioRxiv (2021) 2021:11.24.469776. doi: 10.1101/2021.11.24.469776
63. Aricescu AR, Lu W, Jones EY. A time- and cost-efficient system for high-level protein production in mammalian cells. Acta Crystallogr D Biol Crystallogr (2006) 62(Pt 10):1243–50. doi: 10.1107/S0907444906029799
64. Juno JA, Tan HX, Lee WS, Reynaldi A, Kelly HG, Wragg K, et al. Humoral and circulating follicular helper T cell responses in recovered patients with COVID-19. Nat Med (2020) 26(9):1428–34. doi: 10.1038/s41591-020-0995-0
65. Wines BD, Hulett MD, Jamieson GP, Trist HM, Spratt JM, Hogarth PM. Identification of residues in the first domain of human Fc alpha receptor essential for interaction with IgA. J Immunol (1999) 162(4):2146–53.
66. Khoury DS, Wheatley AK, Ramuta MD, Reynaldi A, Cromer D, Subbarao K, et al. Measuring immunity to SARS-CoV-2 infection: comparing assays and animal models. Nat Rev Immunol (2020) 20(12):727–38. doi: 10.1038/s41577-020-00471-1
67. Lee WS, Selva KJ, Davis SK, Wines BD, Reynaldi A, Esterbauer R, et al. Decay of Fc-dependent antibody functions after mild to moderate COVID-19. Cell Rep Med (2021) 2(6):100296. doi: 10.1016/j.xcrm.2021.100296
68. Wines BD, Vanderven HA, Esparon SE, Kristensen AB, Kent SJ, Hogarth PM. Dimeric FcgammaR ectodomains as probes of the Fc receptor function of anti-influenza virus IgG. J Immunol (2016) 197(4):1507–16. doi: 10.4049/jimmunol.1502551
69. Hartley GE, Edwards ESJ, Aui PM, Varese N, Stojanovic S, McMahon J, et al. Rapid generation of durable b cell memory to SARS-CoV-2 spike and nucleocapsid proteins in COVID-19 and convalescence. Sci Immunol (2020) 5(54):eabf8891. doi: 10.1126/sciimmunol.abf8891
70. Kurtovic L, Agius PA, Feng G, Drew DR, Ubillos I, Sacarlal J, et al. Induction and decay of functional complement-fixing antibodies by the RTS,S malaria vaccine in children, and a negative impact of malaria exposure. BMC Med (2019) 17(1):45. doi: 10.1186/s12916-019-1277-x
71. Lopez E, Haycroft ER, Adair A, Mordant FL, O’Neill MT, Pymm P, et al. Simultaneous evaluation of antibodies that inhibit SARS-CoV-2 variants via multiplex assay. JCI Insight (2021) 6(16):e150012. doi: 10.1172/jci.insight.150012
72. Amanat F, Stadlbauer D, Strohmeier S, Nguyen THO, Chromikova V, McMahon M, et al. A serological assay to detect SARS-CoV-2 seroconversion in humans. Nat Med (2020) 26(7):1033–6. doi: 10.1038/s41591-020-0913-5
73. Jumper J, Evans R, Pritzel A, Green T, Figurnov M, Ronneberger O, et al. Highly accurate protein structure prediction with AlphaFold. Nature (2021) 596(7873):583–9. doi: 10.1038/s41586-021-03819-2
74. Varadi M, Anyango S, Deshpande M, Nair S, Natassia C, Yordanova G, et al. AlphaFold protein structure database: massively expanding the structural coverage of protein-sequence space with high-accuracy models. Nucleic Acids Res (2022) 50(D1):D439–44. doi: 10.1093/nar/gkab1061
75. Honorato RV, Koukos PI, Jimenez-Garcia B, Tsaregorodtsev A, Verlato M, Giachetti A, et al. Structural biology in the clouds: The WeNMR-EOSC ecosystem. Front Mol Biosci (2021) 8:729513. doi: 10.3389/fmolb.2021.729513
76. van Zundert GCP, Rodrigues J, Trellet M, Schmitz C, Kastritis PL, Karaca E, et al. The HADDOCK2.2 web server: User-friendly integrative modeling of biomolecular complexes. J Mol Biol (2016) 428(4):720–5. doi: 10.1016/j.jmb.2015.09.014
77. Prevost J, Finzi A. The great escape? SARS-CoV-2 variants evading neutralizing responses. Cell Host Microbe (2021) 29(3):322–4. doi: 10.1016/j.chom.2021.02.010
78. Diebolder CA, Beurskens FJ, de Jong RN, Koning RI, Strumane K, Lindorfer MA, et al. Complement is activated by IgG hexamers assembled at the cell surface. Science (2014) 343(6176):1260–3. doi: 10.1126/science.1248943
79. Strasser J, de Jong RN, Beurskens FJ, Wang G, Heck AJR, Schuurman J, et al. Unraveling the macromolecular pathways of IgG oligomerization and complement activation on antigenic surfaces. Nano Lett (2019) 19(7):4787–96. doi: 10.1021/acs.nanolett.9b02220
80. Beaudoin-Bussières G, Chen Y, Ullah I, Prévost J, Tolbert WD, Symmes K, et al. A Fc-enhanced NTD-binding non-neutralizing antibody delays virus spread and synergizes with a nAb to protect mice from lethal SARS-CoV-2 infection. Cell Rep (2022) 38(7):110368. doi: 10.1016/j.celrep.2022.110368
81. Zohar T, Loos C, Fischinger S, Atyeo C, Wang C, Slein MD, et al. Compromised humoral functional evolution tracks with SARS-CoV-2 mortality. Cell (2020) 183(6):1508–1519 e12. doi: 10.1016/j.cell.2020.10.052
82. van Berkel PH, Gerritsen J, van Voskuilen E, Perdok G, Vink T, van de Winkel JG, et al. Rapid production of recombinant human IgG with improved ADCC effector function in a transient expression system. Biotechnol Bioeng (2010) 105(2):350–7. doi: 10.1002/bit.22535
83. Winkler ES, Gilchuk P, Yu J, Bailey AL, Chen RE, Chong Z, et al. Human neutralizing antibodies against SARS-CoV-2 require intact Fc effector functions for optimal therapeutic protection. Cell (2021) 184(7):1804–1820 e16. doi: 10.1016/j.cell.2021.02.026
84. Afzali B, Noris M, Lambrecht BN, Kemper C. The state of complement in COVID-19. Nat Rev Immunol (2022) 22(2):77–84. doi: 10.1038/s41577-021-00665-1
85. Yu J, Tostanoski LH, Peter L, Mercado NB, McMahan K, Mahrokhian SH, et al. DNA Vaccine protection against SARS-CoV-2 in rhesus macaques. Science (2020) 369(6505):806–11. doi: 10.1126/science.abc6284
86. Wang Z, Lorenzi JCC, Muecksch F, Finkin S, Viant C, Gaebler C, et al. Enhanced SARS-CoV-2 neutralization by dimeric IgA. Sci Transl Med (2021) 13(577):eabf1555. doi: 10.1126/scitranslmed.abf1555
87. Gasser R, Cloutier M, Prevost J, Fink C, Ducas E, Ding S, et al. Major role of IgM in the neutralizing activity of convalescent plasma against SARS-CoV-2. Cell Rep (2021) 34(9):108790. doi: 10.1016/j.celrep.2021.108790
88. Moller NP. Fc-mediated immune precipitation. i. a new role of the Fc-portion of IgG. Immunology (1979) 38(3):631–40.
89. de Jong RN, Beurskens FJ, Verploegen S, Strumane K, van Kampen MD, Voorhorst M, et al. A novel platform for the potentiation of therapeutic antibodies based on antigen-dependent formation of IgG hexamers at the cell surface. PloS Biol (2016) 14(1):e1002344. doi: 10.1371/journal.pbio.1002344
90. Shields RL, Namenuk AK, Hong K, Meng YG, Rae J, Briggs J, et al. High resolution mapping of the binding site on human IgG1 for Fc gamma RI, Fc gamma RII, Fc gamma RIII, and FcRn and design of IgG1 variants with improved binding to the Fc gamma R. J Biol Chem (2001) 276(9):6591–604. doi: 10.1074/jbc.M009483200
91. Orlandi C, Deredge D, Ray K, Gohain N, Tolbert W, DeVico AL, et al. Antigen-induced allosteric changes in a human IgG1 Fc increase low-affinity Fcgamma receptor binding. Structure (2020) 28(5):516–527 e5. doi: 10.1016/j.str.2020.03.001
92. Shinkawa T, Nakamura K, Yamane N, Shoji-Hosaka E, Kanda Y, Sakurada M, et al. The absence of fucose but not the presence of galactose or bisecting n-acetylglucosamine of human IgG1 complex-type oligosaccharides shows the critical role of enhancing antibody-dependent cellular cytotoxicity. J Biol Chem (2003) 278(5):3466–73. doi: 10.1074/jbc.M210665200
Keywords: coronavirus, SARS-CoV-2, COVID-19, ACE2-Fc, neutralization, antibody effector function, ADCC, complement
Citation: Wines BD, Kurtovic L, Trist HM, Esparon S, Lopez E, Chappin K, Chan L-J, Mordant FL, Lee WS, Gherardin NA, Patel SK, Hartley GE, Pymm P, Cooney JP, Beeson JG, Godfrey DI, Burrell LM, van Zelm MC, Wheatley AK, Chung AW, Tham W-H, Subbarao K, Kent SJ and Hogarth PM (2022) Fc engineered ACE2-Fc is a potent multifunctional agent targeting SARS-CoV2. Front. Immunol. 13:889372. doi: 10.3389/fimmu.2022.889372
Received: 04 March 2022; Accepted: 27 June 2022;
Published: 28 July 2022.
Edited by:
Penghua Wang, University of Connecticut Health Center, United StatesReviewed by:
Ciro Leonardo Pierri, University of Bari Aldo Moro, ItalyCopyright © 2022 Wines, Kurtovic, Trist, Esparon, Lopez, Chappin, Chan, Mordant, Lee, Gherardin, Patel, Hartley, Pymm, Cooney, Beeson, Godfrey, Burrell, van Zelm, Wheatley, Chung, Tham, Subbarao, Kent and Hogarth. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: P. Mark Hogarth, bWFyay5ob2dhcnRoQGJ1cm5ldC5lZHUuYXU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.