
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 17 May 2022
Sec. Molecular Innate Immunity
Volume 13 - 2022 | https://doi.org/10.3389/fimmu.2022.888897
This article is part of the Research Topic Insights in Molecular Innate Immunity: 2021 View all 13 articles
 Mahdi Eskandarian Boroujeni1
Mahdi Eskandarian Boroujeni1 Agata Sekrecka1Aleksandra Antonczyk1Sanaz Hassani1
Agata Sekrecka1Aleksandra Antonczyk1Sanaz Hassani1 Michal Sekrecki1Hanna Nowicka1Natalia Lopacinska1Arta Olya1
Michal Sekrecki1Hanna Nowicka1Natalia Lopacinska1Arta Olya1 Katarzyna Kluzek1Joanna Wesoly2
Katarzyna Kluzek1Joanna Wesoly2 Hans A. R. Bluyssen1*
Hans A. R. Bluyssen1*A disease outbreak in December 2019, caused by a novel coronavirus SARS-CoV-2, was named COVID-19. SARS-CoV-2 infects cells from the upper and lower respiratory tract system and is transmitted by inhalation or contact with infected droplets. Common clinical symptoms include fatigue, fever, and cough, but also shortness of breath and lung abnormalities. Still, some 5% of SARS-CoV-2 infections progress to severe pneumonia and acute respiratory distress syndrome (ARDS), with pulmonary edema, acute kidney injury, and/or multiple organ failure as important consequences, which can lead to death. The innate immune system recognizes viral RNAs and triggers the expression of interferons (IFN). IFNs activate anti-viral effectors and components of the adaptive immune system by activating members of the STAT and IRF families that induce the expression of IFN-stimulated genes (ISG)s. Among other coronaviruses, such as Middle East respiratory syndrome coronavirus (MERS-CoV) and SARS-CoV, common strategies have been identified to antagonize IFN signaling. This typically coincides with hyperactive inflammatory host responses known as the “cytokine storm” that mediate severe lung damage. Likewise, SARS-CoV-2 infection combines a dysregulated IFN response with excessive production of inflammatory cytokines in the lungs. This excessive inflammatory response in the lungs is associated with the local recruitment of immune cells that create a pathogenic inflammatory loop. Together, it causes severe lung pathology, including ARDS, as well as damage to other vulnerable organs, like the heart, spleen, lymph nodes, and kidney, as well as the brain. This can rapidly progress to multiple organ exhaustion and correlates with a poor prognosis in COVID-19 patients. In this review, we focus on the crucial role of different types of IFN that underlies the progression of SARS-CoV-2 infection and leads to immune cell hyper-activation in the lungs, exuberant systemic inflammation, and multiple organ damage. Consequently, to protect from systemic inflammation, it will be critical to interfere with signaling cascades activated by IFNs and other inflammatory cytokines. Targeting members of the STAT family could therefore be proposed as a novel therapeutic strategy in patients with severe COVID-19.
Since the end of 2019, a novel coronavirus (SARS-CoV-2) caused a respiratory disease outbreak in Wuhan, that quickly spread in China and subsequently worldwide. Soon after that, the WHO declared SARS-CoV-2 infection a global health threat and named it coronavirus disease-19 (COVID-19). Clinically, COVID-19 varied from mild to severe symptoms, including acute respiratory distress syndrome (ARDS), as observed after SARS-CoV infection. As of February 2022, more than 350 million confirmed cases, including 5.6 million deaths have been reported.
SARS-CoV-2 is a SARS-CoV-like β-lineage coronavirus originating from bats, that initially infects cells from the upper and lower respiratory tract system through inhalation or contact with infected droplets (1, 2). Common clinical symptoms include fatigue, fever, and cough, but also shortness of breath and lung abnormalities. Still, some 5% of SARS-CoV-2 infections progress to severe pneumonia and ARDS, with pulmonary edema, acute kidney injury, and/or multiple organ failure as important consequences, which can lead to death (3–6).
The innate immune system recognizes viral RNAs via endosomal and cytosolic pattern recognition receptors, including TLRs or RIG-1 and MDA5. This triggers the expression of interferons (IFN), which are the mediators of cellular homeostatic responses to virus infection. IFNs comprise a family of antiviral cytokines, classified as type I, II, and III. Through binding to their cognate cellular receptors, they activate transcription factors of the STAT and IRF family and expression of IFN-stimulated genes (ISG)s, which turn on the anti-viral state and render cells more resistant to virus infection and thereby limit virus spread (7, 8). Among other coronaviruses, such as Middle East respiratory syndrome coronavirus (MERS-CoV) and SARS-CoV, common strategies have been identified to antagonize IFN signaling. This typically coincides with hyperactive inflammatory host responses known as the “cytokine storm” that mediate severe lung damage.
Based on recent COVID-19 patient and animal studies it can be concluded that early during SARS-CoV-2 infection of the upper airways a dysregulated IFN response, mediated by antagonizing IFN induced signaling pathways, is a hallmark of SARS-CoV-2 infections. Later in the lungs, this is accompanied by the production of inflammatory cytokines, together with excessive IFN production, known as the “cytokine storm”. The excessive inflammatory response in the lungs is associated with the local recruitment of immune cells, including macrophages, effector T-cells, dendritic cells, and neutrophils that create a pathogenic inflammatory loop (9, 10). Together, it causes severe lung pathology, including ARDS, as well as damage to other vulnerable organs, like the heart, spleen, lymph nodes, and kidney, as well as the brain. This can rapidly progress to multiple organ exhaustion and correlates with a poor prognosis in COVID-19 patients (6, 9, 10).
So far, several effective vaccines have been generated to prevent infection and transmission. On the other hand, efforts to increase our understanding of the biology of SARS-CoV-2 infection and the host immune response are urgently pursued to design therapeutic strategies that reduce the risk of severe COVID-19 and the overall mortality rate.
SARS-CoV-2 infection starts with its entrance into human cells. Like SARS-CoV, SARS-CoV-2 engages angiotensin-converting enzyme-2 (ACE2) as a cellular entry receptor. In addition, it uses the cellular serine protease TMPRSS2 for S protein priming, whereas Neuropilin1 (NRP1) serves as a co-factor in cell entry (11–14). Thus, effective SARS-CoV-2 transmission requires high co-expression of ACE2 and TMPRSS2 in target cells, including nasal epithelial cells and type II alveolar cells (AT2) of the lungs (15). Viral-Track was developed as a new computational pipeline, to detect viral RNA in single-cell transcriptome data of bronchoalveolar lavage fluid (BALF) from COVID-19 patients. Detecting viral RNA only in severe disease (16), particularly in ciliated and epithelial progenitors and in a subset of macrophages (SPP1+ macrophages), suggested differential progression of infection in the lung (16, 17).
In addition to the nose and lung, the extrapulmonary spread of SARS-CoV-2 was observed (13). Interestingly, Zou et al. recently analyzed single-cell RNA sequencing datasets combined with ACE2 expression and identified potential risk organs, including the lung, heart, kidney, ileum, esophagus, and bladder (18). At the same time, specific cell types could be located (i.e., ileum and esophagus epithelial cells, bladder urothelial cells, AT2, myocardial cells, and proximal tubule cells of the kidney), being sensitive to SARS-CoV-2 infection (19). In fact, ACE2 distribution in the host organism importantly correlated with organ injury. Likewise, expression differences and/or ACE2 polymorphisms determine SARS-CoV-2 infection susceptibility and clinical manifestations. As part of the renin-angiotensin-aldosterone system (RAAS), ACE2 converts angiotensin II (Ang II) to Ang (1–7). Therefore, ACE2 has a dual role in both SARS-CoV and SARS-CoV-2 infection. On the one hand, it acts as the host cell virus entry receptor, but on the other by protecting the lungs and other organs from Ang II-mediated vasoconstrictive, pro-inflammatory, and pro-fibrotic injury on pulmonary vascular and epithelial cells (6, 20).
Common clinical symptoms of SARS-CoV-2 infection include fatigue, fever, and cough, but also shortness of breath and lung abnormalities. A mere 20% of COVID-19 patients require hospitalization and about 1 in 4 of these progress to severe pneumonia and ARDS, with pulmonary edema, acute kidney injury, and/or multiple organ failure as important consequences, that require invasive mechanical ventilation (3–5). In common with SARS-CoV and MERS-CoV infections, ARDS causes similar immunopathogenic features and is the main cause of death in COVID-19 disease (4). Advanced age and the presence of certain comorbidities, such as coronary heart disease, hypertension, chronic obstructive pulmonary disease, and diabetes mellitus, are among the risk factors for the development of severe COVID-19 and poor outcome (3, 21–25).
An important hallmark of ARDS is the cytokine storm - an excessive inflammatory response of immune effector cells combined with the release of pro-inflammatory cytokines and chemokines (4). Indeed, SARS-CoV-2 infected individuals with severe symptoms display increased serum concentrations of many pro-inflammatory mediators, including interferon (IFN)-g-induced protein 10 (IP-10), monocyte chemoattractant protein 1 (MCP1), macrophage inflammatory protein (MIP)-1a, platelet-derived growth factor (PDGF), TNF-a, vascular endothelial growth factor (VEGF), granulocyte colony-stimulating factor (G-CFS), IL-1b, IL-2, IL-6, IL-7, IL-8 (4, 26–29). Also, recent single-cell expression profiling studies in bronchoalveolar cells, PBMCs, and airway epithelium from COVID-19 patients, have shown that upregulation of pro-inflammatory pathways correlates with severe symptoms (17, 30). Therefore, an excessive inflammatory response, associated with high levels of circulating pro-inflammatory cytokines and chemokines, profound lymphopenia, and substantial infiltration of hyper-active mononuclear cells in the lungs, is a major cause of disease severity and death in patients with COVID-19. In addition, it has also become clear that SARS-CoV-2 infected individuals can rapidly progress to a multiple organ dysfunction syndrome (MODS). In this respect, increased fibrin degradation products (identified as D- dimers) and coagulation abnormalities (i.e, apparent thrombotic manifestations and prolonged prothrombin time) as well as lower platelets counts, promote vascular leakage and disseminated intravascular coagulation. These vascular abnormalities could lead to organ failure and death and are a clear link with poor prognosis in patients with severe COVID-19 (3–5, 31, 32).
The innate immune system recognizes viral RNAs via endosomal and cytosolic pattern recognition receptors, including Toll-like receptor (TLR) 3 and TLR7 or retinoic acid–inducible gene I (RIG-I) and melanoma differentiation-associated protein 5 (MDA5) (33, 34). After infection of the target cells, they trigger the activation of the downstream transcription factors, namely interferon regulatory factor (IRF)3 and IRF7 and nuclear factor-kappa B (NF-κB). These factors collectively induce the production of IFNs, the main mediators of cellular homeostatic responses to virus infection (7, 8).
IFNs consist of three sub-types, including IFN-I, IFN-II, and IFN-III. IFN-II, also known as IFNγ, mediates broad immune responses to pathogens by binding to the IFNγ receptor (IFNGR) complex. In response to foreign antigens or mitogens, IFN-II is mainly produced by T lymphocytes and Natural Killer (NK) cells, to modulate adaptive immunity as well as inflammation. Although IFN-II exhibits anti-viral activity, IFN-I is the main anti-viral IFN subtype, predominantly consisting of IFNα and IFNβ subtypes. IFN-I can be produced by many cell types and collectively bind to the IFNα receptor (IFNAR) to potently inhibit viral infection as part of a robust innate immune response. IFN-III (consisting of IFNΛ1 to -4 subtypes) is largely produced by epithelial cells, hepatocytes, and dendritic cells, in a tissue-specific manner. All IFNΛ subtypes bind the heterodimeric IFN-Λ receptor (IFNLR) (35), which is restricted to certain cell types and tissues, including macrophages, conventional dendritic cells (DCs), and plasmacytoid dendritic cells (pDCs), neutrophils, and respiratory epithelial cells. IFN-Λ responses, therefore, confer more localized antiviral protection at the site of infection. In addition to antiviral and pro-inflammatory activity, IFNs exert anti-proliferative and pro-apoptotic functions (7, 8).
As potent IFN-producing cells also plasmacytoid (p)DCs are important cellular mediators of the antiviral response by producing massive amounts of IFN-I and IFN-III. In addition, they produce other pro-inflammatory cytokines, which modulate the function of innate and adaptive immune cells (35).
Coronaviruses, such as SARS-CoV and MERS-CoV, have developed strategies to antagonize IFN responses. This typically coincides with hyperactive inflammatory host responses that lead to severe lung damage. Using mice infected with SARS-CoV, it was shown that robust virus replication accompanied by delayed IFN-I signaling triggers hyperinflammatory and severe immune pathology of the lungs as well as reduced survival. This dysregulated IFN-I signaling promoted the infiltration of pathogenic inflammatory monocyte macrophages (IMM), leading to increased lung cytokine/chemokine levels, and resulting in vascular leakage and impaired virus-specific T cell responses (30). Likewise, robust and persistent expression of IFN and IFN-stimulated genes (ISGs) in association with impaired T cell and antibody responses was observed in fatal SARS in humans (36). Thus, during SARS-CoV and MERS-CoV infection, an ineffective IFN-I response, accompanied by excessive production of inflammatory cytokines, correlates with disease severity and poor prognosis and appears to be a hallmark of coronavirus infections (37–39).
As stated by Zhang et al: “Combined immunodeficiencies, immune dysregulation disorders and defects of innate immunity impairing type I interferon responses show a higher rate of severe disease and higher mortality. In addition, inborn errors of type I interferon responses and inborn errors of immunity in which patients harbor autoantibodies against these cytokines, confer a high risk for critical COVID-19” (40). Together, this reflects the key role of IFN-I in the host defense against SARS-CoV-2 (Figure 1). Using peripheral blood mononuclear cells (PBMCs), Lee et al. (30) performed single-cell RNA-seq to identify driving factors of COVID-19 progression. By comparing healthy donors, patients with mild or severe COVID-19, and patients with severe influenza, they observed that all PBMC cell types of COVID-19 displayed hyper-inflammatory signatures, marked by a TNF/IL-1β-mediated inflammatory response, as compared to severe influenza. Moreover, classical monocytes from severe COVID-19 patients displayed a type I IFN response together with the TNF/IL-1β-mediated inflammation, being absent in mild COVID-19 patients. Remarkably, patients with severe influenza also presented these type I IFN-dependent inflammatory features. Consequently, it was proposed that the type I IFN response plays an important role in the excessive inflammation seen in severe COVID-19 patients (30) (Figure 1).
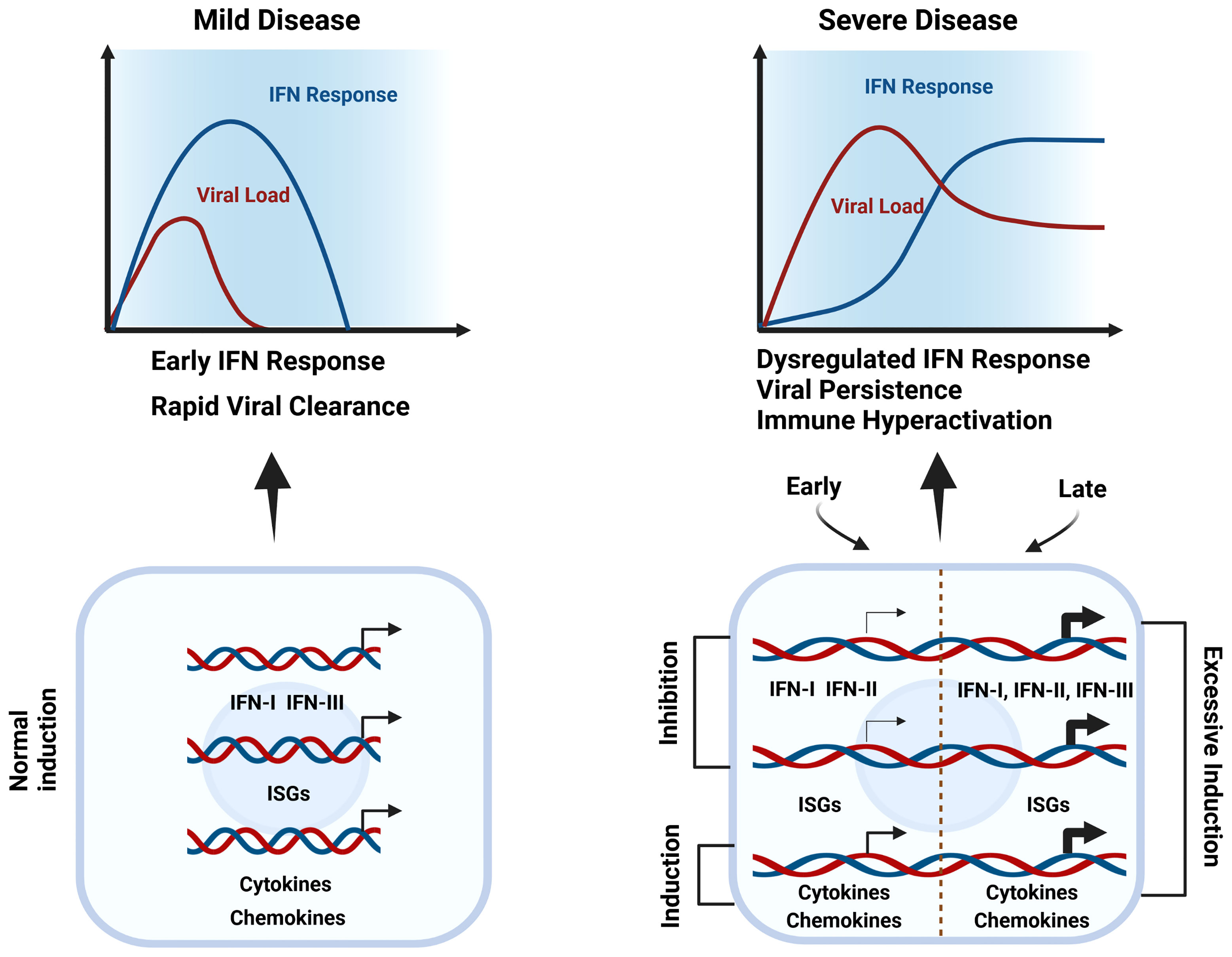
Figure 1 Dysregulated IFN responses & immune hyper-activation in severe COVID-19. During mild disease, the initial viral burden is low and IFNs, together with cytokines, chemokines, and other ISGs are induced early and rapidly to mediate effective viral clearance (left). In severe COVID-19 (right), in the upper respiratory tract interferon production and ISG expression are inhibited by SARS-CoV-2, diminishing the immune response and promoting virus survival (Early). But by the time the virus enters the lower respiratory tract, an exuberant immune response is activated, including excessive upregulation of interferons, cytokines, chemokines, and other ISGs that at this point are harmful (Late). Thus, both early-stage deficiencies in type I IFN responses and late-stage IFN hyper-responsiveness are clear characteristics of severe COVID-19.
Conversely, by studying peripheral blood responses of COVID-19 patients, Hadjadj et al. (41) observed reduced IFN responses in critically ill patients paired with a pro-inflammatory response. By comparing the transcriptional responses of several respiratory viruses, including SARS-CoV-2, Blanco-Melo et al. demonstrated in vitro that the host response to SARS-CoV-2 is unable to activate a robust IFN-I and -III response whereas high levels of chemokines were presented for the recruitment of immune cells (42). Along the same lines, Broggi and colleagues evaluated the ability of SARS-CoV-2 to induce IFNs in the upper or lower airways of COVID-19 patients. Importantly, they observed that IFN levels in the upper respiratory tract of COVID-19 patients did not significantly differ from healthy individuals. But bronchoalveolar lavage fluid from these patients exhibited increased levels of inflammatory cytokines, IFN-I, and IFN-III. This indicated that in the upper airways interferon production is inhibited by SARS-CoV-2, diminishing the immune response and promoting virus survival (Figure 1). But by the time the virus enters the lower respiratory tract, an excessive immune response is activated, together with upregulation of interferons that at this point is harmful. In conclusion, both early-stage deficiencies in type I IFN responses and late stage IFN hyper-responsiveness are a characteristic of severe COVID-19 (43, 44). Indeed, an early transient response of IFNa and IFNΛ was observed in mild-to-moderate COVID-19, whereas in severe patients, levels further enhanced towards the second week (45). The longitudinal dynamic changes of IFN-I production in severe patients correlate with experiments in a mouse SARS-CoV-2 model, further proving in vivo that IFN-I is unable to control SARS-CoV-2 replication but importantly drives hyperactive immune responses (46).
Therefore, it is tempting to propose that during early SARS-CoV-2 infection of the upper respiratory tract, intervention with recombinant interferons and other antivirals offer important solutions. However, during hyper-inflammation in the lower respiratory tract, it will be critical to interfere with signaling cascades activated by IFNs and other inflammatory cytokines, probably with anti-inflammatory drugs.
As mentioned above, after infection of target cells, RNA viruses trigger the activation of downstream IRF3 and IRF7 and NF-κB transcription factors and the production of IFNs. Subsequently, through binding to their cognate cellular receptors and Janus kinase (JAK)-dependent phosphorylation, IFNs activate transcription factors of the Signal Transducer and Activator of Transcription (STAT) family and IFN-stimulated gene (ISG) expression, which turn on the anti-viral state and render cells more resistant to virus infection and thereby limits virus spread. Thus, IFN-I, IFN-II, and IFN-III induce ISG expression by phosphorylating STAT1 and/or STAT2. IFN-II-induced STAT1 homodimers regulate gene expression of IFN-II activation site (GAS) containing genes. The association of IRF9 with STAT1/2 heterodimers (known as ISGF3), expands the range of enhancer elements targeted by IFN-I and IFN-III to IFN-stimulated response element (ISRE) (Figure 2). Similarly, IRF family members, including IRF1, IRF7 and IRF8, can regulate ISG expression in response to all three types of IFN by binding the ISRE. Together, this leads to the transcriptional activation of hundreds of ISGs, such as MX1 and 2, ISG15, IFIT1, IFIT2 and IFIT3, and OAS1, OAS2, and OAS3 that importantly participate in the antiviral response (7, 8).
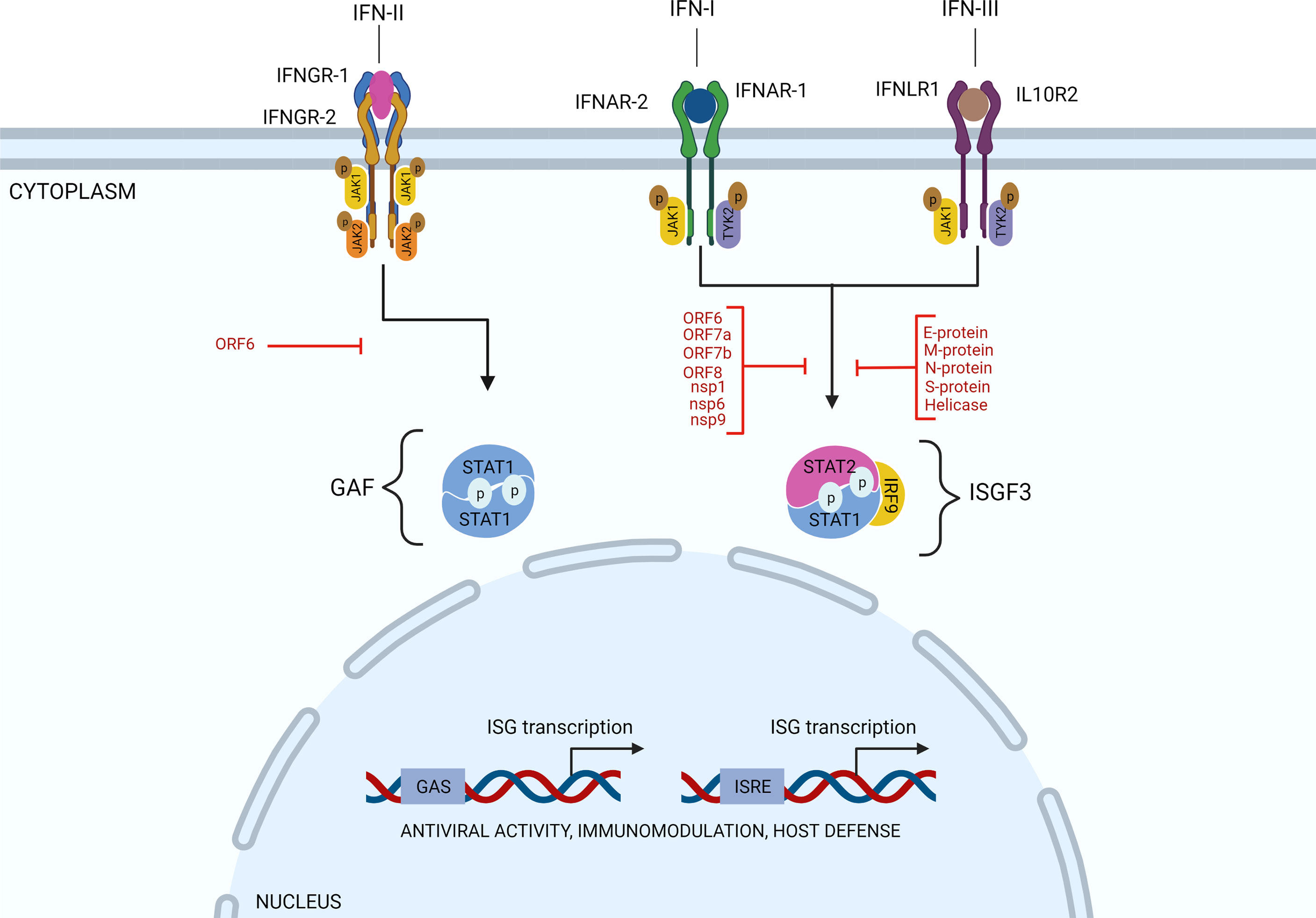
Figure 2 SARS-CoV-2 evolved strategies to target IFN signaling. Through binding to their cognate cellular receptors and Janus kinase (JAK)-dependent phosphorylation, IFNs activate transcription factors of the Signal Transducer and Activator of Transcription (STAT) family and expression of IFN-stimulated genes (ISG)s. Thus, IFN-I, IFN-II, and IFN-III induce ISG expression by phosphorylating STAT1 and/or STAT2. IFN-II-induced STAT1 homodimers regulate gene expression of IFN-II activation site (GAS) containing genes. The association of IRF9 with STAT1/2 heterodimers (known as ISGF3), expands the range of enhancer elements targeted by IFN-I and IFN-III to IFN-stimulated response element (ISRE). Various SARS-CoV-2 proteins, as indicated (in red), have been identified to inhibit the IFN response. Particularly, by interfering with STAT1 and/or STAT2 activation and ISG expression, as explained in the text.
Among coronaviruses, such as MERS-CoV and SARS-CoV, common strategies have been identified to antagonize IFN production and inhibit IFN signaling (39, 47, 48). For example, SARS-CoV was shown to antagonize the IFN response using nsp1, papain-like protease (PLpro), nsp7, nsp15, ORF3b, M, ORF6, and N proteins (49, 50). Thus, several antagonizing mechanisms have been identified, including inhibition of IRF3 and IRF7 activation, blocking expression of STAT1, and interaction with ISG15.
Concerning SARS-CoV-2 infection and dysregulated IFN-I responses, likewise, nsp1, nsp12, nsp13, nsp14, and, nsp15 and orf6 and the M protein (51–53) were among potent IFN-I inhibitors. In addition, Li et al. observed that ORF6, ORF8, and nucleocapsid proteins significantly inhibited IFN-I production, and the IFN-I-mediated innate immune response (53). The same was true for ORF3b (54). A more detailed functional analysis of these proteins in relation to COVID-19 progression was performed by Xia et al. Importantly, they found that nsp6 suppressed IRF3 phosphorylation by binding TANK binding kinase 1 (TBK1), nsp13 blocked and bound TBK1 phosphorylation, and ORF6 inhibited IRF3 nuclear translocation by binding importin Karyopherin a 2 (KPNA2). They also identified nsp1 and nsp6 to block IFN-I signaling by interfering with the phosphorylation of STAT1/STAT2 or their nuclear translocation (Figure 2). Interestingly, nsp1 and nsp6 proteins of SARS-CoV-2 antagonized IFN-I signaling more efficiently than the counterparts of SARS-CoV and MERS-CoV suggesting that SARS-CoV and SARS-CoV-2 use different mechanisms to affect disease onset and progression (55). Along the same lines, Zhang et al. observed that the SARS-CoV-2 M protein did not significantly affect IRF3 phosphorylation, but blocked its nuclear translocation. Likewise, the S protein was found to suppress STAT1 phosphorylation and nuclear translocation by intervening with JAK1-STAT1 interaction (56). On the other hand, without directly influencing STAT1 and STAT2 activation, nsp7 and ORF7a clearly inhibited ISRE promoter activity (56). Nsp9 and ORF8 exhibited inhibition of STAT1 activation, whereas Helicase and E protein intervened with STAT1 and STAT2 activation (56). The same was shown by Shi et al. with the M protein significantly blocking STAT1 phosphorylation and ISG expression (55). This study by Shi et al. also provided evidence to suggest that ORF7a inhibited STAT2 phosphorylation, whereas ORF7b inhibited phosphorylation of both STAT1 and STAT2 (55). In a separate study, Mu et al. found that SARS-CoV-2 replication was efficiently enhanced upon N protein overexpression in infected human cells, by interacting with STAT1 and STAT2 and inhibiting their phosphorylation and nuclear translocation (57). (Figure 2). Finally, the S protein also interfered with IFN-I signaling, however with a currently unknown mechanism (51, 57).
Analysis of gene expression profiles from COVID-19 patient airway epithelial cells together with epithelial cell lines infected with SARS-CoV-2 revealed that also IFN-II signaling is antagonized in a SARS-CoV-2 dependent manner (58). Particularly, it was shown that the SARS-CoV-2 ORF6 protein suppressed NLRC5, an MHC class I trans-activator, both transcriptionally and functionally (Figure 2). This was mediated by blocking type II interferon-mediated STAT1 signaling, and subsequent NLRC5 and IRF1 gene expression. At the same time, ORF6 was shown to inhibit NLRC5 nuclear import, uncovering a novel immune evasion mechanism toward modulation of the MHC class I pathway.
Finally, it was assessed how JAK/STAT signaling was altered in immune cells in COVID-19 patients and if an imbalance in JAK/STAT signaling contributed to disease severity. Rincon-Arevalo et al. reported increased expression in mild and severe COVID-19 patients of STAT1 and IRF9, in correlation with peripheral monocytes exhibiting IFN signatures [marked by Siglec-1 [CD169] expression (59)]. Remarkably, CD14+ monocytes and plasmablasts in severe patients displayed reduced expression of STAT1 and Siglec-1 in comparison to mild patients. In contrast, STAT1 phosphorylation was increased in severe COVID-19 cases, pointing to an imbalanced JAK/STAT signaling and lack of ISG induced transcription. This defect was still present in PBMCs isolated from severe COVID-19 patients stimulated with IFN-α and IFN-γ. This strongly implies that impaired JAK/STAT signaling could serve as a potential predictive biomarker of severe COVID-19 in combination with patient stratification and IFN response targeted therapy.
Together a picture emerges in which multiple components of the type I, II and III IFN systems are targeted by SARS-CoV-2, including members of the STAT family (Figure 2) and also IRFs. This affects important aspects of the innate and adaptive immune system and leads to a dysregulated immune response.
As with SARS-CoV and MERS-CoV infected patients, acute pneumonia is the most frequent and critical clinical manifestation presented among severe COVID-19 patients (2–6). Severe lung pathology after MERS-CoV and SARS-CoV infection is a consequence of dysregulated IFN-I responses and rapid virus replication, which promote the accumulation of pathogenic inflammatory monocyte macrophages (IMM) and IFN-producing pDCs, and the release of elevated lung cytokine/chemokine levels. Subsequently increased influx of IMM resulted in exuberant inflammation. In addition, T cell responses are reduced through IFN-I-mediated T cell apoptosis. Together, this causes severe lung pathology that correlated with poor outcome of patients infected with MERS-CoV and SARS-CoV (60–63).
Likewise, SARS-CoV-2 infection combines a dysregulated IFN response with excessive production of inflammatory cytokines in the lungs. In COVID-19 patients, this also has proven to be a main cause of disease severity and death and is associated with high levels of circulating pro-inflammatory cytokines and chemokines and the local recruitment of immune cells (6, 10, 64, 65). A current model suggests that upon upper airway entry, SARS-CoV-2 impregnates the lower lungs, where it infects a number of target cells, including the alveolar type 2 cells (AT2), alveolar macrophages, and vascular endothelial cells (Figure 3). Virus cell entry recognition via endosomal and cytosolic pattern recognition receptors (as TLR3 and TLR7) triggers the activation of downstream IRF3 and IRF7 and NF-κB transcription factors and the production of IFNs and proinflammatory mediators. Severe COVID-19 also displays increased CD4+ and CD8+ T cell activation. Yet, with the observed lymphopenia in these patients it is anticipated that in response to the virus, effector T cells are recruited to the lung. In this respect, lymphopenia has been recognized as a hallmark of severe COVID-19 (4, 5, 10), whereas an elevated neutrophil/lymphocyte ratio has been shown to serve as a biomarker of disease severity (65–68). Interestingly, plasmacytoid DC (pDCs), the main source of IFN-I and IFN-III, were also reduced in abundance in the circulation (65, 69).
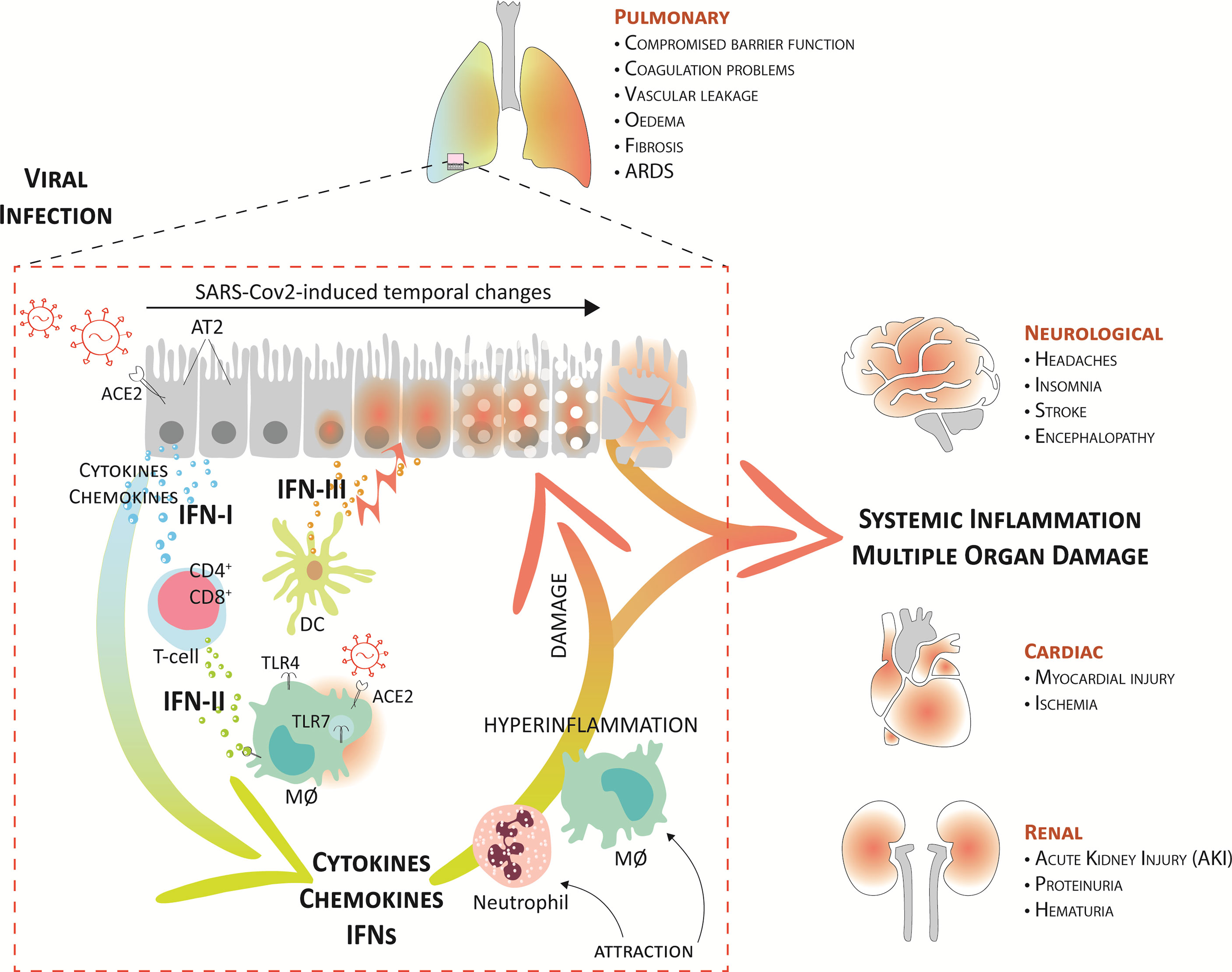
Figure 3 Systemic inflammation and multiple organ damage in severe COVID-19. A current model suggests that upon upper airway entry, SARS-CoV-2 impregnates the lower lungs via ACE2, where it infects several target cells, including the alveolar type 2 cells (AT2), alveolar macrophages (MØ), and vascular endothelial cells. Virus cell entry recognition via endosomal and cytosolic pattern recognition receptors (as TLR3 and TLR7) trigger the production of IFNs and proinflammatory mediators. Together, the local recruitment of effector T-cells (CD4+ and CD8+) and dendritic cells (DCs), further perpetuates higher proinflammatory cytokines, IFNs, and chemokines in the lung fluid. Consequently, this further augments damage to the lung tissue, mediated by endothelial dysfunction and vasodilation, and leads to the attraction of more inflammatory neutrophils and macrophages that create a pathogenic inflammatory loop in the lung. On the one hand, this leads to respiratory/organ failure and on the other hand, it also results in a “systemic cytokine storm” that causes widespread inflammation and damage to other vulnerable organs, like the heart, kidney, and brain. This can rapidly progress to multiple organ exhaustion, which correlates with a poor prognosis in COVID-19 patients.
Together, the local recruitment of immune cells further perpetuates proinflammatory cytokine and chemokine expression in BALF (CCL2, CCL3, CCL4, and CXCL10) but also in the circulation (IL-1, IFN-I, IFN-II, and IFN-III, IL-17, TNF, IP-10, MCP-1, G-CSF, GM-CSF, IL-1RA, CCL2, CCL3, CCL5, CCL8, CXCL2, CXCL8, CXCL9, and CXCL16) (4–6, 41, 42, 64, 70–72). Consequently, this further augments damage to the lung tissue, mediated by endothelial dysfunction and vasodilation, and leads to the attraction of more inflammatory neutrophils and macrophages that create a pathogenic inflammatory loop in the lung (Figure 3). These systemic cytokine profiles show overlap with cytokine release syndromes, and this highly suggests that COVID-19-associated hyper-inflammation is mediated by an over-activated mononuclear phagocyte (MNP) compartment (73, 74).
Interestingly, excessive IFN production, especially IFN-III, has shown to be an important factor impairing lung epithelial regeneration. IFN-III, unlike other IFNs, activates antiviral response and at the same time limits tissue-damaging functions of neutrophils. Major et al. (75) and Broggi et al. (43) showed that both IFN-I and IFN-III, produced by lung-resident dendritic cells through TLR-3 stimulation, disrupt lung epithelial cell repair during influenza recovery in mice in lower airways. IFN-λ is prevalent compared to IFN-I in virus-infected lungs. Specifically, IFN-λ directly affects epithelial cell proliferation and viability to inhibit tissue repair. With the presence of IFN-I and IFN-III in the lower, but not upper, airways of patients with COVID-19, it is therefore proposed that also during SARS-CoV-2 infection, especially IFN-III produced by activated DCs, acts on lung epithelial cells to compromise lung barrier function (76, 77) (Figure 3).
Recently, the cyclic GMP-AMP synthase (cGAS)–stimulator of interferon genes (STING) pathway was implicated in the pathological type I IFN responses in COVID-19. Indeed, a STING-dependent type I IFN signature was observed primarily in macrophages adjacent to areas of endothelial cell damage of COVID-19 skin manifestations. Under these conditions, SARS-CoV-2 infection activated cGAS–STING signaling in endothelial cells through mitochondrial DNA release, resulting in cell death and type I IFN production. Likewise, in lung samples from patients with COVID-19 with prominent tissue destruction, cGAS–STING activity was associated with type I IFN responses. Finally, in SARS-CoV-2 infected epithelial cell lines, such as Calu-3 and A549-ACE2, cGAS–STING activation was also linked to the production of pro-inflammatory cytokines. Together, this potentially identified the cGAS–STING pathway, activated by damaged mitochondrial DNA and not SARS-CoV-2, as a major driver of pathological type I IFN and pro-inflammatory cytokine responses in severe COVID-19 (78–80).
The excessive infiltration of inflammatory immune cells and the amplified production of cytokines, chemokines, and IFNs, observed in severe COVID-19, result in vascular leakage, coagulation abnormalities, and diminished lung barrier function, which stimulate endotheliitis and lung edema. This limits oxygen exchange and promotes hypoxia, leading to lung failure (81, 82). On the other hand, these excessive inflammatory conditions also result in a “systemic cytokine storm” that causes widespread inflammation and damage to other vulnerable organs, like the heart, spleen, lymph nodes, and kidney, as well as the brain. This can rapidly progress to multiple organ exhaustion, which correlates with a poor prognosis in COVID-19 patients (6, 9, 10) (Figure 3).
It is important to realize that under these COVID-19 associated excessive inflammatory conditions, immune cell activation and cytokine and IFN release is tightly regulated by IFN and TLR signaling. Accordingly, maximum expression of many of these pro-inflammatory cytokines and IFN-stimulated genes has shown to depend on combinatorial actions of transcription factors of the STAT, IRF, and NF-kB families. This identifies STATs as potential therapeutic targets in severe COVID-19 (83–88) (Figure 4).
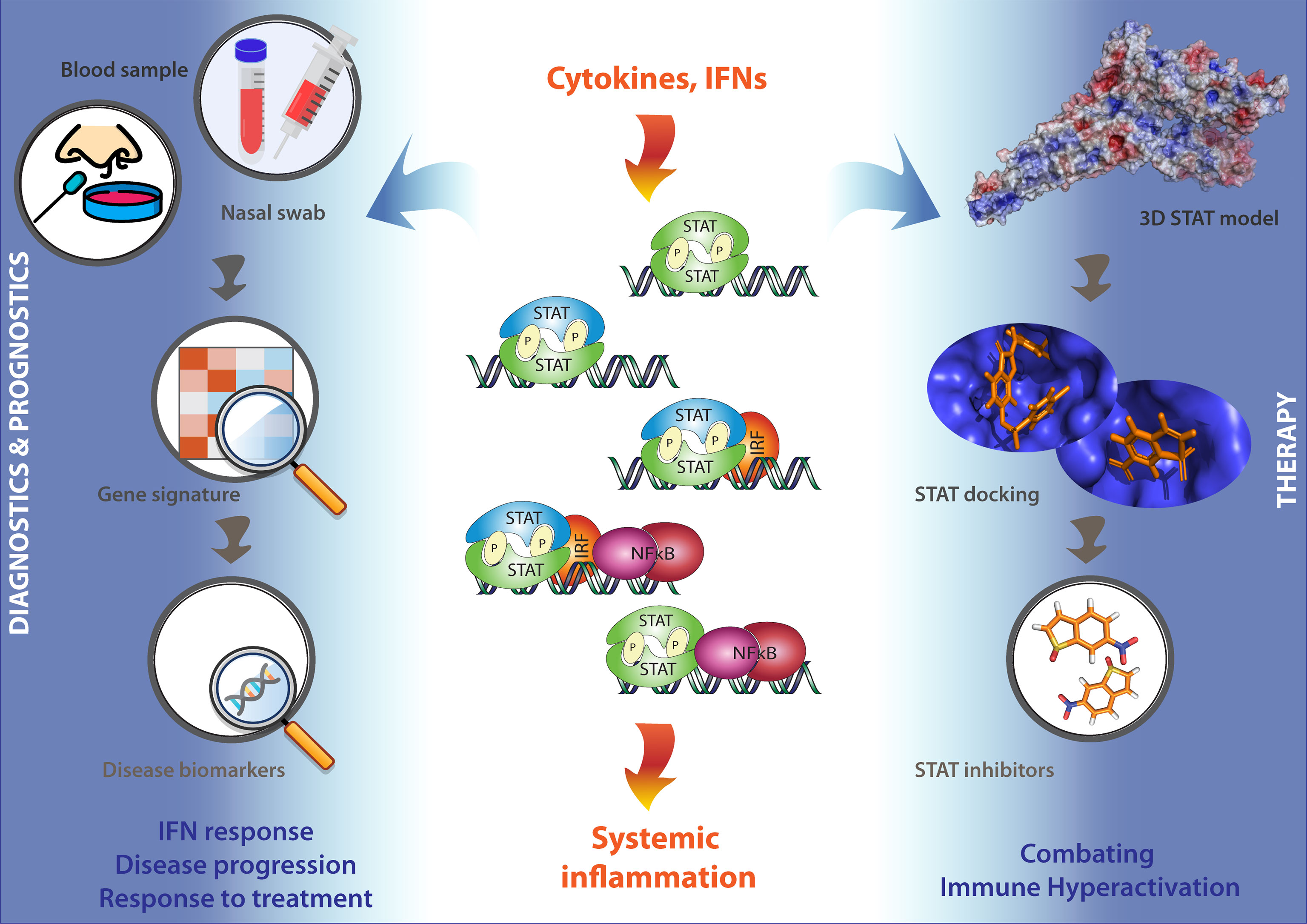
Figure 4 STATs as potential therapeutic targets in COVID-19 patients. Under conditions of excessive inflammation, maximum expression of many pro-inflammatory cytokine and IFN-stimulated genes has shown to depend on combinatorial actions of transcription factors of the STAT, IRF, and NF-kB families. This identifies STATs as potential therapeutic targets in severe COVID-19. Thus, a STAT-dependent pro-inflammatory gene signature could serve as a novel diagnostic tool to monitor and diagnose IFN responses, disease progression, and responses to treatment using blood or nasal swabs from COVID-19 patients (Left). On the other hand, the further testing and optimizing of already available Statinhibs, together with the identification of new Statinhibs using in silico 3D STAT modeling, virtual docking and screening, and in vitro validation, may offer new clinical benefits in combating immune hyper-activation during severe COVID-19 (Right).
Further evidence for this was proposed in one publication where hyperactivation of STAT3 was linked to COVID-19-associated coagulopathy and lung damage. Specifically, plasminogen activator inhibitor-1 (PAI-1) upregulation, as an important STAT3 target, was linked to coagulopathy. TLR4 bound PAI-1 on macrophages could also induce the local production of proinflammatory cytokines and chemokines. This leads to the influx of active innate immune cells to the SARS-CoV-2 infected lung and drives the destruction of lung architecture. Based on this, STAT3 inhibition was proposed as a treatment strategy for COVID- 19 (89). In another study, in Syrian hamsters, which are highly sensitive to SARS-CoV-2 infection and virus-induced lung pathology (90), a STAT2-mediated dysregulated immune response was identified as the driving force. In particular STAT2-dependent type I and III IFN signaling was shown to restrict infection and systemic dissemination of SARS-CoV-2. This was in agreement with the effect of STAT1 in a mouse SARS-CoV infection model (91). Surprisingly, the severe lung damage presented in SARS-CoV-2 infected WT hamsters was absent in STAT2 KO hamsters, contrary to general observations of viral infections in STAT2−/− mice (92) or STAT2−/− hamsters (93–95). Indeed, pneumonia was absent in SARS-CoV-2 infected STAT2−/− hamsters. Accordingly, this points to an important role of STAT2 in systemic inflammation and severe COVID-19 and identifies it as a novel therapeutic target. Moreover, hamsters could serve as a preferred infection model for the preclinical assessment of vaccines or antiviral therapies, and of anti-inflammatory strategies controlling the hyperactive immune response in severe COVID-19 (96, 97).
As shown above, SARS-CoV-2 mainly infects the upper and lower respiratory system but also targets extrapulmonary organs. An important cause of death in COVID-19 disease is ARDS, which is characterized by the “cytokine storm”. This results from the release of pro-inflammatory cytokines and chemokines and IFNs by immune effector cells, which mediates widespread inflammation and damage to other vulnerable organs. This can rapidly progress to multiple organ damage, which correlates with a poor prognosis in COVID-19 patients. So far several molecular mechanisms have been described that cause widespread inflammation and damage to vulnerable organs, including cardiovascular, renal, and neurological systems, and contribute to multiple organ exhaustion and death in COVID-19.
With the growing number of COVID-19 infected cases and accumulating clinical data, COVID-19 related cardiovascular diseases have attracted a great deal of concern over an associated mortality rise among infected patients. Likewise, the strong correlation of risk factors such as hypertension and cardiovascular disease with the severity of COVID-19 outcome has already been well established (98). In this aspect, the cardiovascular manifestations in patients with COVID-19 encompass a wide range of complications including myocardial infarction, arrhythmias, heart failure, and hypercoagulation (99). It is well known that ACE2 is abundant in the heart, especially in pericytes, coronary endothelial cells, cardiomyocytes, and cardiac fibroblasts (100). Following SARS-CoV-2 infection and downregulation of ACE2, RAAS is over-activated and may contribute to thrombotic events and inflammatory responses and ultimately leading to microvascular dysfunction and injury (101). Coagulation abnormalities are evidently seen in nearly 20%–55% of hospitalized patients with COVID-19 (102). For instance, in a cohort study of confirmed COVID-19 patients, the elevated levels of D-dimer and fibrin degradation product (FDP) along with prolonged prothrombin time and activated partial thromboplastin time were frequently seen in COVID-19 non-survivors (103). This observation indicates the high incidence of thromboembolic events in COVID-19 cases which might impact vascular homeostasis (104).
In a recent systemic review investigating the potential contribution of hyper-inflammation in the induction of cardiac injuries in the COVID-19 context, the authors concluded that systematic hyper-inflammatory response may trigger cardiac injury in COVID-19 patients including children and adolescents without having pre-existing cardiovascular abnormalities (105). For the hyperinflammation assessment, the markers C-reactive protein(CRP) and/or ferritin and/or IL-6 were employed in their investigation, as both ferritin and IL-6 have been previously shown increases in severe COVID-19 cases in contrast to survivors (106). A retrospective study that explored the relationship between SARS-CoV-2-induced hyperinflammatory response in 317 patients with COVID-19, reported that 14 out of 39 patients with a myocardial injury who showed immune dysregulation, developed myocardial injury nine days after admission (107). This finding may imply that cardiac injury is elicited by the development of inflammatory storms in severe COVID-19 cases. In other words, cytokine storms may contribute to apoptosis or necrosis of the myocardial cells, compromising the hemodynamics of coronary circulation, destabilizing coronary plaque, and instigating vascular microthrombosis (108).
Kidney injury in a quarter of hospitalized patients with COVID-19 has been described in several studies. Indeed, COVID-19 associated acute kidney injury (AKI) is reported with a high mortality rate, particularly in critically ill patients with comorbidities such as hypertension and diabetes (109). Urine analysis of COVID-19 patients displayed alterations in urine sediments including proteinuria and hematuria which is linked with elevated mortality in hospitalized COVID-19 patients (110). Moreover, adequate evidence for the presence of SARS-CoV-2 RNA in the urine of COVID-19-positive patients does not seem robust to accurately predict renal dysfunction (111).
The pathophysiological mechanisms underlying COVID-19 associated AKI remains to be clarified. However, there are COVID-19-specific mechanisms that might play a role in renal dysfunction. Among all, the relationship between ACE2 and the renin-angiotensin-aldosterone system (RAAS) which is disturbed by COVID-19 infection is regarded as a major mechanism engaged in kidney injury (112). In fact, ACE2 as the main receptor for SARS-CoV-2 and highly expressed in kidney (113), is able to convert angiotensin II to angiotensin 1–7, down-regulating RAAS-mediated platelet and endothelial activation, vasoconstriction and release of proinflammatory mediators. However, upon binding of SARS-CoV-2 to ACE2, followed by accumulation of angiotensin II and overactivation of RAAS, leading subsequently to hypercoagulability, endothelial injury, and inflammatory response (114). Based on this understanding, drugs that inhibit RAAS have been suggested as a potential treatment for COVID-19 (115), but it still demands more tailor-made prospective randomized controlled studies to confirm the efficacy of these drugs.
Cytokine storm syndrome which is portrayed as a hyperactive state of the immune system through extensive recruitment of immune cells, is believed to substantially contribute to the severity of COVID-19 infection (116). Likewise, upregulation of cytokines namely IL-6 which is a key element in multi-organ damage including kidney injury, has been frequently recorded in COVID-19 patients (117). It was shown that IFN-α and IFN-β display mutual and differential effects on podocytes and parietal epithelial cells, which collectively advance glomerulosclerosis by inducing podocyte loss and also restricting podocyte regeneration from local progenitors (118). This is of considerable importance since inflammation-induced proteinuria is mediated by podocyte injury and aberrant type I interferon response can further aggravate proteinuric kidney disease (119). Moreover, a growing body of evidence showed the renal levels of C5b-9 which engaged in the complement membrane attack complex, were elevated in severe COVID-19 patients compared with healthy controls. Similarly, C5a triggers abnormal epigenetic changes in renal tubular epithelial cells causing tubular senescence. In addition, C5b–9 is involved in endotheliopathy and thromboinflammation (117, 120). In light of these findings, complement activation might drive inflammation associated with coagulation and thrombosis in the context of COVID-19, exacerbating kidney dysfunction towards to progression of AKI.
There is also a growing body of evidence pointing to complications of the nervous system in patients with COVID-19. These neurological and neuropsychiatric symptoms range from smell and taste alterations to infectious encephalitis. The underlying mechanisms involved in these neurological manifestations are not entirely understood. However, it seems systemic inflammation and deregulated immune activity might cast new light on the causes of SARS‐CoV‐2-associated neuropathogenesis.
There are conflicting reports about the direct invasion of SARS‐CoV‐2 to the brain (121–123). Indeed, the varied expression levels of SARS-CoV-2 entry receptors in the central nervous system propose potential routes including the olfactory system and blood-brain barrier (BBB) for direct viral infection. For instance, it was shown that ACE2 and TMPRSS2 are expressed in the cells located in the olfactory epithelium, except for olfactory sensory neurons (124). Likewise, the expression of ACE2 is detected in pericytes which is a part of the neurovascular unit in BBB (125).
According to a meta-analysis of postmortem examinations in COVID-19 patients with neurological manifestations, the main neuropathological features that were documented among over 190 cases were gliosis including astrogliosis and microgliosis, hypoxic alterations, infiltration of inflammatory cells such as macrophages and T- lymphocytes, cerebral microthrombosis, ischaemic brain lesions. In light of these observations, it seems that systemic inflammation and dysregulated coagulation substantially drive neuropathological outcomes including hemorrhages and microthrombi in the context of COVID-19 infection, compared with the injuries caused directly by SARS-CoV-2 infection (126). The contribution of gliosis is further supported by the neuropathological examinations of 43 patients with COVID-19, in which the interplay between activated immune response and nervous system was of considerable note in all examined brain regions where reactive astrogliosis was detectable at varying levels of intensity (122). In fact, reactive astrocytes are regarded to be an indispensable part of the neuroimmune system by promoting neuroprotection in infectious diseases. However, following SARS-CoV-2 infection, the expression levels of glial fibrillary acidic protein (GFAP), as an astrogliosis marker, are highly induced in patients with moderate to severe COVID-19 (127, 128), suggesting the possible role of reactive astrocytes along with aberrant microglial functions in shaping COVID-19-induced cytokine storm, leading ultimately to multiple-organ failure.
Current research on dysregulated immune activity in COVID-19 shows the upregulation of circulating cytokines such as TNF-α, IL-6, and IL-10 (4, 129), suggesting peripheral inflammation induced by COVID-19 might be disseminated to brain parenchyma through compromised BBB and severely affect neurons and glial cells. In this regard, we recently showed the increased levels of neuroinflammatory genes along with gliosis and neuronal death in the cerebral cortex of COVID-19 patients (127), as also reported in (130). Additionally in our study, upon the close histopathologic examination of the post-mortem cerebral cortex of patients with COVID-19, inflammation of blood vessels (vasculitis) was detected, which might pave the way for lymphocytic infiltration and neuronal damage, as already documented in a case report of COVID-19 (131). Thus, disrupted BBB undermines the regulation of BBB permeability and makes the brain vulnerable to the invasion of pathogens and entry of pro-inflammatory mediators, leading to severe systemic disease.
Implications for recombinant IFN therapy have strictly been proposed in the early stages of COVID-19 disease, especially during upper respiratory tract infection of SARS-CoV-2. In this respect, the kinetics of IFN-I secretion versus the kinetics of viral replication should help in identifying the window of therapeutic opportunity.
On the other hand, significant progress has been made on therapeutic approaches that can manage the systemic inflammation and hyperactive immune response in severe COVID-19 patients (29, 132–134). Currently, at clinicaltrials.gov over one hundred clinical trials are registered connected to inflammatory cytokine blocking medications as potential treatment strategy in COVID-19 patients.
As the JAK/STAT pathway modulates important inflammatory and immunological pathways, JAKs and STATs serve as potential therapeutic targets in severe COVID-19. Anti-IL6 antibodies (Tocilizumab), BTK inhibition with ibrutinib and acalabrutinib, as well as the known JAK inhibitors- Ruxolitinib, Fedratinib, and Baricitinib are part of potential treatment strategies against COVID-19 that combine antiviral and anti-inflammatory agents (135–139). Ruxolitinib entered phase III clinical trials (NCT04120090, NCT03533790) and Fedratinib phase II, both were used in connection with pneumonia-associated COVID-19 disease.
Strategies to inhibit the JAK-STAT pathway identified numerous JAK inhibitors (Jakinibs), which mostly are known as pan-JAK inhibitors. For example, the FDA approved Tofacitinib, Ruxolitinib, Fedratinib, and Baricitinib, which inhibit multiple JAKs. Likewise, a high number of small molecules have been identified that target the pTyr-SH2 interaction area of STAT3, and in analogy to Jakinhibs indicated as Statinibs (140, 141). They were recently compiled by our group in SINBAD, a curated open-access database of more than 175 Statinhibs which have been published and described in scientific articles providing proof of their inhibitory properties. It is a tool allowing user-friendly analysis of experimental conditions and provides detailed information about known STAT inhibitory compounds (142); datasets are available at http://sinbad.amu.edu.pl as well as through the public repository https://doi.org/10.6084/m9.figshare.14975136.v1 (142).
Among the first reported Statinhibs was STA-21, which was shown to inhibit STAT3 dimerization, DNA binding, and STAT3-dependent transcription in breast cancer cells (143). Similarly, the known Statinhibs STATTIC and STX-0119 inhibited phosphorylation, dimerization and nuclear translocation of STAT3, leading to increased apoptosis of cancer cells [reviewed in (87, 144, 145)].
As part of a novel strategy to characterize and understand STAT-inhibition, we combined comparative in silico docking and virtual screening of multiple STAT-SH2 models with in vitro validation of STAT inhibition (146, 147). This led to the identification of a novel class of multi-STAT or pan-STAT inhibitor, C01L_F03 that commonly and with equal affinity targets the SH2 domain of STAT1, 2, and 3. Interestingly, STATTIC and STX-0119 displayed similar STAT cross-binding characteristics. During anti-microbial and inflammatory responses, macrophage activation, and cytokine release is tightly regulated by IFN and TLR signaling. Accordingly, maximum expression of important pro-inflammatory and chemokine genes depends on cross-talk between IFNs and TLR signaling through collaborative actions of STATs, IRFs, and NF-kB. Recently, we further proved that in macrophages, dendritic cells and vascular smooth muscle cells, under conditions of IFN and TLR cross-talk, the above-mentioned novel pan-STAT inhibitors (with STATTIC being the most potent one) managed genome-wide inhibition of multiple cytokines, chemokines and anti-viral products. Accordingly, a novel STAT-inhibitory strategy was developed for the inhibition of pro-inflammatory cytokine-dependent migration of endothelial cells, adhesion of leukocytes to endothelial cells, and loss of contractility of mesenteric arteries (148).
Promising results for several FDA-approved inhibitors [Tofacitinib: pan-JAK inhibition; Ruxolitinib: JAK1/2-inhibition; Tocilizumab: IL6 receptor antibody); or (pre)Clinical Trial tested [sb1578: JAK2-inhibition; WP1066: JAK2-inhibition; STA-21: STAT3-SH2 inhibition; STATTIC: STAT3-SH2 inhibition], promises entry of Statinhibs to the clinic shortly (87). So far, Statinhibs have not been implicated in connection to COVID-19. However, the further testing and optimizing of already available Statinhibs, together with the identification of new Statinhibs, may offer new clinical benefits in combating immune hyperactivation during severe COVID-19. In this context, according to Toubiana et al., “care must be taken to apply STAT targeting therapeutic strategies in a stage-appropriate manner, because some approaches that may help in the earlier stages of the disease could be detrimental if used in its later stages. With the potential use for COVID-19 treatment of existing drugs that enhance STAT1 or inhibit STAT3 functions, problems could arise. For example, chronic mucocutaneous candidiasis (CMC) was present in 98% of patients with gain of function (GOF) mutations in STAT1 in one study of 274 individuals, and the few that did not have CMC, had invasive fungal or bacterial infections. Generally, the immune profile was relatively normal, but 82% had decreased Th17 cells” (149). Conversely, a STAT-dependent pro-inflammatory gene signature could serve as a novel diagnostic tool to monitor and diagnose IFN responses, disease progression, and responses to treatment using blood or nasal swabs from COVID-19 patients (Figure 4).
MEB was involved in concept development and writing and editing of the manuscript. AS, AA and SH designed figures. MS, HN, NL, AO, KK, and JW participated in the development of the concept and critically evaluated and edited the manuscript. HB developed the concept and was involved in writing and editing the manuscript and coordinated input from all co-authors. All authors contributed to the article and approved the submitted version.
This publication was supported by grant UMO 2016/23/B/NZ2/00623 (HAR.B.) from the National Science Centre Poland and grant 7/2020 (HAR.B.) from the Adam Mickiewicz University in Poznan.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Wu F, Zhao S, Yu B, Chen Y-M, Wang W, Song Z-G, et al. A New Coronavirus Associated With Human Respiratory Disease in China. Nature (2020) 579(7798):265–9. doi: 10.1038/s41586-020-2008-3
2. Zhou P, Yang X-L, Wang X-G, Hu B, Zhang L, Zhang W, et al. A Pneumonia Outbreak Associated With a New Coronavirus of Probable Bat Origin. Nature (2020) 579(7798):270–3. doi: 10.1038/s41586-020-2012-7
3. Guan W-J, Ni Z-Y, Hu Y, Liang W-H, Ou C-Q, He J-X, et al. Clinical Characteristics of Coronavirus Disease 2019 in China. N Engl J Med (2020) 382(18):1708–20. doi: 10.1056/NEJMoa2002032
4. Huang C, Wang Y, Li X, Ren L, Zhao J, Hu Y, et al. Clinical Features of Patients Infected With 2019 Novel Coronavirus in Wuhan, China. Lancet (2020) 395(10223):497–506. doi: 10.1016/S0140-6736(20)30183-5
5. Wang D, Hu B, Hu C, Zhu F, Liu X, Zhang J, et al. Clinical Characteristics of 138 Hospitalized Patients With 2019 Novel Coronavirus–Infected Pneumonia in Wuhan, China. Jama (2020) 323(11):1061–9. doi: 10.1001/jama.2020.1585
6. Lopes-Pacheco M, Silva PL, Cruz FF, Battaglini D, Robba C, Pelosi P, et al. Pathogenesis of Multiple Organ Injury in Covid-19 and Potential Therapeutic Strategies. Front Physiol (2021) 29. doi: 10.3389/fphys.2021.593223
7. Antonczyk A, Krist B, Sajek M, Michalska A, Piaszyk-Borychowska A, Plens-Galaska M, et al. Direct Inhibition of Irf-Dependent Transcriptional Regulatory Mechanisms Associated With Disease. Front Immunol (2019) 10:1176. doi: 10.3389/fimmu.2019.01176
8. Michalska A, Blaszczyk K, Wesoly J, Bluyssen HA. A Positive Feedback Amplifier Circuit That Regulates Interferon (Ifn)-Stimulated Gene Expression and Controls Type I and Type Ii Ifn Responses. Front Immunol (2018) 9:1135. doi: 10.3389/fimmu.2018.01135
9. Park A, Iwasaki A. Type I and Type Iii Interferons–Induction, Signaling, Evasion, and Application to Combat Covid-19. Cell Host Microbe (2020) 27(6):870–8. doi: 10.1016/j.chom.2020.05.008
10. Schultze JL, Aschenbrenner AC. Covid-19 and the Human Innate Immune System. Cell (2021) 184(7):1671–92. doi: 10.1016/j.cell.2021.02.029
11. Li W, Moore MJ, Vasilieva N, Sui J, Wong SK, Berne MA, et al. Angiotensin-Converting Enzyme 2 Is a Functional Receptor for the Sars Coronavirus. Nature (2003) 426(6965):450–4. doi: 10.1038/nature02145
12. Wan Y, Shang J, Graham R, Baric RS, Li F. Receptor Recognition by the Novel Coronavirus From Wuhan: An Analysis Based on Decade-Long Structural Studies of Sars Coronavirus. J Virol (2020) 94(7):e00127–20. doi: 10.1128/JVI.00127-20
13. Hoffmann M, Kleine-Weber H, Schroeder S, Krüger N, Herrler T, Erichsen S, et al. Sars-Cov-2 Cell Entry Depends on Ace2 and Tmprss2 and Is Blocked by a Clinically Proven Protease Inhibitor. cell (2020) 181(2):271–80. doi: 10.1016/j.cell.2020.02.052
14. Matsuyama S, Nagata N, Shirato K, Kawase M, Takeda M, Taguchi F. Efficient Activation of the Severe Acute Respiratory Syndrome Coronavirus Spike Protein by the Transmembrane Protease Tmprss2. J Virol (2010) 84(24):12658–64. doi: 10.1128/JVI.01542-10
15. Cantuti-Castelvetri L, Ojha R, Pedro LD, Djannatian M, Franz J, Kuivanen S, et al. Neuropilin-1 Facilitates Sars-Cov-2 Cell Entry and Infectivity. Science (2020) 370(6518):856–60. doi: 10.1126/science.abd2985
16. Bost P, Giladi A, Liu Y, Bendjelal Y, Xu G, David E, et al. Host-Viral Infection Maps Reveal Signatures of Severe Covid-19 Patients. Cell (2020) 181(7):1475–88. doi: 10.1016/j.cell.2020.05.006
17. Chua RL, Lukassen S, Trump S, Hennig BP, Wendisch D, Pott F, et al. Covid-19 Severity Correlates With Airway Epithelium–Immune Cell Interactions Identified by Single-Cell Analysis. Nat Biotechnol (2020) 38(8):970–9. doi: 10.1038/s41587-020-0602-4
18. Zou X, Chen K, Zou J, Han P, Hao J, Han Z. Single-Cell Rna-Seq Data Analysis on the Receptor Ace2 Expression Reveals the Potential Risk of Different Human Organs Vulnerable to 2019-Ncov Infection. Front Med (2020) 14(2):185–92. doi: 10.1007/s11684-020-0754-0
19. Barbry P, Muus C, Luecken M, Eraslan G, Waghray A, Heimberg G, et al. Integrated Analyses of Single-Cell Atlases Reveal Age, Gender, and Smoking Status Associations With Cell Type-Specific Expression of Mediators of Sars-Cov-2 Viral Entry and Highlights Inflammatory Programs in Putative Target Cells. bioRxiv (2020) 2020.04.19.049254. doi: 10.1101/2020.04.19.049254
20. Gheblawi M, Wang K, Viveiros A, Nguyen Q, Zhong J-C, Turner AJ, et al. Angiotensin-Converting Enzyme 2: Sars-Cov-2 Receptor and Regulator of the Renin-Angiotensin System: Celebrating the 20th Anniversary of the Discovery of Ace2. Circ Res (2020) 126(10):1456–74. doi: 10.1161/CIRCRESAHA.120.317015
21. Leung JM, Yang CX, Tam A, Shaipanich T, Hackett T-L, Singhera GK, et al. Ace-2 Expression in the Small Airway Epithelia of Smokers and Copd Patients: Implications for Covid-19. Eur Respir J (2020) 55(5):2000688. doi: 10.1183/13993003.00688-2020
22. Iaccarino G, Grassi G, Borghi C, Ferri C, Salvetti M, Volpe M. Age and Multimorbidity Predict Death Among Covid-19 Patients: Results of the Sars-Ras Study of the Italian Society of Hypertension. Hypertension (2020) 76(2):366–72. doi: 10.1161/HYPERTENSIONAHA.120.15324
23. Williamson EJ, Walker AJ, Bhaskaran K, Bacon S, Bates C, Morton CE, et al. Opensafely: Factors Associated With Covid-19 Death in 17 Million Patients. Nature 584:430–36 (2020). doi: 10.1038/s41586-020-2521-4
24. Wu Z, McGoogan JM. Characteristics of and Important Lessons From the Coronavirus Disease 2019 (Covid-19) Outbreak in China: Summary of a Report of 72 314 Cases From the Chinese Center for Disease Control and Prevention. JAMA (2020) 323(13):1239–42. doi: 10.1001/jama.2020.2648
25. Zhou F, Yu T, Du R, Fan G, Liu Y, Liu Z, et al. Clinical Course and Risk Factors for Mortality of Adult Inpatients With Covid-19 in Wuhan, China: A Retrospective Cohort Study. Lancet (2020) 395(10229):1054–62. doi: 10.1016/S0140-6736(20)30566-3
26. Ackermann M, Verleden SE, Kuehnel M, Haverich A, Welte T, Laenger F, et al. Pulmonary Vascular Endothelialitis, Thrombosis, and Angiogenesis in Covid-19. N Engl J Med (2020) 383(2):120–8. doi: 10.1056/NEJMoa2015432
27. Chen N, Zhou M, Dong X, Qu J, Gong F, Han Y, et al. Epidemiological and Clinical Characteristics of 99 Cases of 2019 Novel Coronavirus Pneumonia in Wuhan, China: A Descriptive Study. Lancet (2020) 395(10223):507–13. doi: 10.1016/S0140-6736(20)30211-7
28. Liu J, Li S, Liu J, Liang B, Wang X, Wang H, et al. Longitudinal Characteristics of Lymphocyte Responses and Cytokine Profiles in the Peripheral Blood of Sars-Cov-2 Infected Patients. EBioMedicine (2020) 55:102763. doi: 10.1016/j.ebiom.2020.102763
29. Zhang C, Wu Z, Li J-W, Zhao H, Wang G-Q. Cytokine Release Syndrome in Severe Covid-19: Interleukin-6 Receptor Antagonist Tocilizumab May Be the Key to Reduce Mortality. Int J Antimicrob Agents (2020) 55(5):105954. doi: 10.1016/j.ijantimicag.2020.105954
30. Lee JS, Park S, Jeong HW, Ahn JY, Choi SJ, Lee H, et al. Immunophenotyping of Covid-19 and Influenza Highlights the Role of Type I Interferons in Development of Severe Covid-19. Sci Immunol (2020) 5(49):eabd1554. doi: 10.1126/sciimmunol.abd1554
31. Helms J, Tacquard C, Severac F, Leonard-Lorant I, Ohana M, Delabranche X, et al. High Risk of Thrombosis in Patients With Severe Sars-Cov-2 Infection: A Multicenter Prospective Cohort Study. Intensive Care Med (2020) 46(6):1089–98. doi: 10.1007/s00134-020-06062-x
32. Tang N, Bai H, Chen X, Gong J, Li D, Sun Z. Anticoagulant Treatment Is Associated With Decreased Mortality in Severe Coronavirus Disease 2019 Patients With Coagulopathy. J Thromb Haemostasis (2020) 18(5):1094–9. doi: 10.1111/jth.14817
33. Mazaleuskaya L, Veltrop R, Ikpeze N, Martin-Garcia J, Navas-Martin S. Protective Role of Toll-Like Receptor 3-Induced Type I Interferon in Murine Coronavirus Infection of Macrophages. Viruses (2012) 4(5):901–23. doi: 10.3390/v4050901
34. Sa Ribero M, Jouvenet N, Dreux M, Nisole S. Interplay Between Sars-Cov-2 and the Type I Interferon Response. PloS Pathog (2020) 16(7):e1008737. doi: 10.1371/journal.ppat.1008737
35. Gilliet M, Cao W, Liu Y-J. Plasmacytoid Dendritic Cells: Sensing Nucleic Acids in Viral Infection and Autoimmune Diseases. Nat Rev Immunol (2008) 8(8):594–606. doi: 10.1038/nri2358
36. Cameron MJ, Bermejo-Martin JF, Danesh A, Muller MP, Kelvin DJ. Human Immunopathogenesis of Severe Acute Respiratory Syndrome (Sars). Virus Res (2008) 133(1):13–9. doi: 10.1016/j.virusres.2007.02.014
37. Lim YX, Ng YL, Tam JP, Liu DX. Human Coronaviruses: A Review of Virus–Host Interactions. Diseases (2016) 4(3):26. doi: 10.3390/diseases4030026
38. Nelemans T, Kikkert M. Viral Innate Immune Evasion and the Pathogenesis of Emerging Rna Virus Infections. Viruses (2019) 11(10):961. doi: 10.3390/v11100961
39. Totura AL, Baric RS. Sars Coronavirus Pathogenesis: Host Innate Immune Responses and Viral Antagonism of Interferon. Curr Opin Virol (2012) 2(3):264–75. doi: 10.1016/j.coviro.2012.04.004
40. Zhang Q, Bastard P, Cobat A, Casanova J-L. Human Genetic and Immunological Determinants of Critical Covid-19 Pneumonia. Nature (2022) 603:587–98. doi: 10.1038/s41586-022-04447-0
41. Hadjadj J, Yatim N, Barnabei L, Corneau A, Boussier J, Smith N, et al. Impaired Type I Interferon Activity and Inflammatory Responses in Severe Covid-19 Patients. Science (2020) 369(6504):718–24. doi: 10.1126/science.abc6027
42. Blanco-Melo D, Nilsson-Payant BE, Liu W-C, Uhl S, Hoagland D, Møller R, et al. Imbalanced Host Response to Sars-Cov-2 Drives Development of Covid-19. Cell (2020) 181(5):1036–45. doi: 10.1016/j.cell.2020.04.026
43. Broggi A, Ghosh S, Sposito B, Spreafico R, Balzarini F, Lo Cascio A, et al. Type Iii Interferons Disrupt the Lung Epithelial Barrier Upon Viral Recognition. Science (2020) 369(6504):706–12. doi: 10.1126/science.abc3545
44. King C, Sprent J. Dual Nature of Type I Interferons in Sars-Cov-2-Induced Inflammation. Trends Immunol (2021) 42(4):312–22. doi: 10.1016/j.it.2021.02.003
45. Lucas C, Wong P, Klein J, Castro TB, Silva J, Sundaram M, et al. Longitudinal Analyses Reveal Immunological Misfiring in Severe Covid-19. Nature (2020) 584(7821):463–9. doi: 10.1038/s41586-020-2588-y
46. Israelow B, Song E, Mao T, Lu P, Meir A, Liu F, et al. Mouse Model of Sars-Cov-2 Reveals Inflammatory Role of Type I Interferon Signaling. J Exp Med (2020) 217(12):e20201241. doi: 10.1084/jem.20201241
47. Channappanavar R, Perlman S eds. Pathogenic Human Coronavirus Infections: Causes and Consequences of Cytokine Storm and Immunopathology. Semin Immunopathol (2017) 39:529–39. doi: 10.1007/s00281-017-0629-x
48. Kindler E, Thiel V, Weber F. Interaction of Sars and Mers Coronaviruses With the Antiviral Interferon Response. Adv Virus Res (2016) 96:219–43. doi: 10.1016/bs.aivir.2016.08.006
49. Frieman M, Ratia K, Johnston RE, Mesecar AD, Baric RS. Severe Acute Respiratory Syndrome Coronavirus Papain-Like Protease Ubiquitin-Like Domain and Catalytic Domain Regulate Antagonism of Irf3 and Nf-Kb Signaling. J Virol (2009) 83(13):6689–705. doi: 10.1128/JVI.02220-08
50. Niemeyer D, Mösbauer K, Klein EM, Sieberg A, Mettelman RC, Mielech AM, et al. The Papain-Like Protease Determines a Virulence Trait That Varies Among Members of the Sars-Coronavirus Species. PloS Pathog (2018) 14(9):e1007296. doi: 10.1371/journal.ppat.1007296
51. Lei X, Dong X, Ma R, Wang W, Xiao X, Tian Z, et al. Activation and Evasion of Type I Interferon Responses by Sars-Cov-2. Nat Commun (2020) 11(1):1–12. doi: 10.1038/s41467-020-17665-9
52. Yuen C-K, Lam J-Y, Wong W-M, Mak L-F, Wang X, Chu H, et al. Sars-Cov-2 Nsp13, Nsp14, Nsp15 and Orf6 Function as Potent Interferon Antagonists. Emerging Microbes Infect (2020) 9(1):1418–28. doi: 10.1080/22221751.2020.1780953
53. Li G, Fan Y, Lai Y, Han T, Li Z, Zhou P, et al. Coronavirus Infections and Immune Responses. J Med Virol (2020) 92(4):424–32. doi: 10.1002/jmv.25685
54. Konno Y, Kimura I, Uriu K, Fukushi M, Irie T, Koyanagi Y, et al. Sars-Cov-2 Orf3b Is a Potent Interferon Antagonist Whose Activity Is Increased by a Naturally Occurring Elongation Variant. Cell Rep (2020) 32(12):108185. doi: 10.1016/j.celrep.2020.108185
55. Xia H, Cao Z, Xie X, Zhang X, Chen JY-C, Wang H, et al. Evasion of Type I Interferon by Sars-Cov-2. Cell Rep (2020) 33(1):108234. doi: 10.1016/j.celrep.2020.108234
56. Zhang Q, Chen Z, Huang C, Sun J, Xue M, Feng T, et al. Severe Acute Respiratory Syndrome Coronavirus 2 (Sars-Cov-2) Membrane (M) and Spike (S) Proteins Antagonize Host Type I Interferon Response. Front Cell Infect Microbiol (2021) 1242. doi: 10.3389/fcimb.2021.766922
57. Mu J, Fang Y, Yang Q, Shu T, Wang A, Huang M, et al. Sars-Cov-2 N Protein Antagonizes Type I Interferon Signaling by Suppressing Phosphorylation and Nuclear Translocation of Stat1 and Stat2. Cell Discov (2020) 6(1):1–4. doi: 10.1038/s41421-020-00208-3
58. Yoo J-S, Sasaki M, Cho SX, Kasuga Y, Zhu B, Ouda R, et al. Sars-Cov-2 Inhibits Induction of the Mhc Class I Pathway by Targeting the Stat1-Irf1-Nlrc5 Axis. Nat Commun (2021) 12(1):1–17. doi: 10.1038/s41467-021-26910-8
59. Rincon-Arevalo H, Aue A, Ritter J, Szelinski F, Khadzhynov D, Zickler D, et al. Altered Increase in Stat1 Expression and Phosphorylation in Severe Covid-19. Eur J Immunol (2022) 52(1):138–48. doi: 10.1002/eji.202149575
60. De Wit E, Van Doremalen N, Falzarano D, Munster VJ. Sars and Mers: Recent Insights Into Emerging Coronaviruses. Nat Rev Microbiol (2016) 14(8):523–34. doi: 10.1038/nrmicro.2016.81
61. Fehr AR, Channappanavar R, Perlman S. Middle East Respiratory Syndrome: Emergence of a Pathogenic Human Coronavirus. Annu Rev Med (2017) 68:387–99. doi: 10.1146/annurev-med-051215-031152
62. Newton AH, Cardani A, Braciale TJ. The Host Immune Response in Respiratory Virus Infection: Balancing Virus Clearance and Immunopathology. Semin Immunopathol 38(4):471–82 (2016). doi: 10.1007/s00281-016-0558-0
63. Channappanavar R, Fehr AR, Vijay R, Mack M, Zhao J, Meyerholz DK, et al. Dysregulated Type I Interferon and Inflammatory Monocyte-Macrophage Responses Cause Lethal Pneumonia in Sars-Cov-Infected Mice. Cell Host Microbe (2016) 19(2):181–93. doi: 10.1016/j.chom.2016.01.007
64. Harrison AG, Lin T, Wang P. Mechanisms of Sars-Cov-2 Transmission and Pathogenesis. Trends Immunol (2020) 41(12):1100–15. doi: 10.1016/j.it.2020.10.004
65. Kuri-Cervantes L, Pampena MB, Meng W, Rosenfeld AM, Ittner CA, Weisman AR, et al. Comprehensive Mapping of Immune Perturbations Associated With Severe Covid-19. Sci Immunol (2020) 5(49):eabd7114. doi: 10.1126/sciimmunol.abd7114
66. Lagunas-Rangel FA. Neutrophil-To-Lymphocyte Ratio and Lymphocyte-To-C-Reactive Protein Ratio in Patients With Severe Coronavirus Disease 2019 (Covid-19): A Meta-Analysis. J Med Virol 92(10):1733–34. (2020). doi: 10.1002/jmv.25819
67. Liu Y, Du X, Chen J, Jin Y, Peng L, Wang HH, et al. Neutrophil-To-Lymphocyte Ratio as an Independent Risk Factor for Mortality in Hospitalized Patients With Covid-19. J Infect (2020) 81(1):e6–e12. doi: 10.1016/j.jinf.2020.04.002
68. Qin C, Zhou L, Hu Z, Zhang S, Yang S, Tao Y, et al. Dysregulation of Immune Response in Patients With Coronavirus 2019 (Covid-19) in Wuhan, China. Clin Infect Dis (2020) 71(15):762–8. doi: 10.1093/cid/ciaa248
69. Arunachalam PS, Wimmers F, Mok CKP, Perera RA, Scott M, Hagan T, et al. Systems Biological Assessment of Immunity to Mild Versus Severe Covid-19 Infection in Humans. Science (2020) 369(6508):1210–20. doi: 10.1126/science.abc6261
70. Xiong Y, Liu Y, Cao L, Wang D, Guo M, Jiang A, et al. Transcriptomic Characteristics of Bronchoalveolar Lavage Fluid and Peripheral Blood Mononuclear Cells in Covid-19 Patients. Emerging Microbes Infect (2020) 9(1):761–70. doi: 10.1080/22221751.2020.1747363
71. Stephenson E, Reynolds G, Botting RA, Calero-Nieto FJ, Morgan MD, Tuong ZK, et al. Single-Cell Multi-Omics Analysis of the Immune Response in Covid-19. Nat Med (2021) 27(5):904–16. doi: 10.1038/s41591-021-01329-2
72. Merad M, Martin JC. Pathological Inflammation in Patients With Covid-19: A Key Role for Monocytes and Macrophages. Nat Rev Immunol (2020) 20(6):355–62. doi: 10.1038/s41577-020-0331-4
73. Mehta P, McAuley DF, Brown M, Sanchez E, Tattersall RS, Manson JJ. Covid-19: Consider Cytokine Storm Syndromes and Immunosuppression. Lancet (2020) 395(10229):1033–4. doi: 10.1016/S0140-6736(20)30628-0
74. Schulert GS, Grom AA. Pathogenesis of Macrophage Activation Syndrome and Potential for Cytokine-Directed Therapies. Annu Rev Med (2015) 66:145–59. doi: 10.1146/annurev-med-061813-012806
75. Major J, Crotta S, Llorian M, McCabe TM, Gad HH, Priestnall SL, et al. Type I and Iii Interferons Disrupt Lung Epithelial Repair During Recovery From Viral Infection. Science (2020) 369(6504):712–7. doi: 10.1126/science.abc2061
76. Broggi A, Granucci F, Zanoni I. Type Iii Interferons: Balancing Tissue Tolerance and Resistance to Pathogen Invasion. J Exp Med (2020) 217(1):e20190295. doi: 10.1084/jem.20190295
77. Prokunina-Olsson L, Alphonse N, Dickenson RE, Durbin JE, Glenn JS, Hartmann R, et al. Covid-19 and Emerging Viral Infections: The Case for Interferon Lambda. J Exp Med (2020) 217(5):e20200653. doi: 10.1084/jem.20200653
78. Di Domizio J, Gulen MF, Saidoune F, Thacker VV, Yatim A, Sharma K, et al. The Cgas-Sting Pathway Drives Type I Ifn Immunopathology in Covid-19. Nature (2022) 603:145–51. doi: 10.1038/s41586-022-04421-w
79. Neufeldt CJ, Cerikan B, Cortese M, Frankish J, Lee J-Y, Plociennikowska A, et al. Sars-Cov-2 Infection Induces a Pro-Inflammatory Cytokine Response Through Cgas-Sting and Nf-Kb. Commun Biol (2022) 5:45. doi: 10.1038/s42003-021-02983-5
80. Andreakos E. Stinging Type I Ifn-Mediated Immunopathology in Covid-19. Nat Immunol (2022) 23:478–80. doi: 10.1038/s41590-022-01174-6
81. Berlin DA, Gulick RM, Martinez FJ. Severe Covid-19. N Engl J Med (2020) 383(25):2451–60. doi: 10.1056/NEJMcp2009575
82. Gandhi RT, Lynch JB, Del Rio C. Mild or Moderate Covid-19. N Engl J Med (2020) 383(18):1757–66. doi: 10.1056/NEJMcp2009249
83. Schroder K, Sweet MJ, Hume DA. Signal Integration Between Ifnγ and Tlr Signalling Pathways in Macrophages. Immunobiology (2006) 211(6-8):511–24. doi: 10.1016/j.imbio.2006.05.007
84. Hu X, Chen J, Wang L, Ivashkiv LB. Crosstalk Among Jak-Stat, Toll-Like Receptor, and Itam-Dependent Pathways in Macrophage Activation. J Leukocyte Biol (2007) 82(2):237–43. doi: 10.1189/jlb.1206763
85. Hu X, Chakravarty SD, Ivashkiv LB. Regulation of Interferon and Toll-Like Receptor Signaling During Macrophage Activation by Opposing Feedforward and Feedback Inhibition Mechanisms. Immunol Rev (2008) 226(1):41–56. doi: 10.1111/j.1600-065X.2008.00707.x
86. Hu X, Ivashkiv LB. Cross-Regulation of Signaling Pathways by Interferon-Γ: Implications for Immune Responses and Autoimmune Diseases. Immunity (2009) 31(4):539–50. doi: 10.1016/j.immuni.2009.09.002
87. Szelag M, Piaszyk-Borychowska A, Plens-Galaska M, Wesoly J, Bluyssen HA. Targeted Inhibition of Stats and Irfs as a Potential Treatment Strategy in Cardiovascular Disease. Oncotarget (2016) 7(30):48788. doi: 10.18632/oncotarget.9195
88. Piaszyk-Borychowska A, Széles L, Csermely A, Chiang H-C, Wesoły J, Lee C-K, et al. Signal Integration of Ifn-I and Ifn-Ii With Tlr4 Involves Sequential Recruitment of Stat1-Complexes and Nfκb to Enhance Pro-Inflammatory Transcription. Front Immunol (2019) 10:1253. doi: 10.3389/fimmu.2019.01253
89. Matsuyama T, Kubli SP, Yoshinaga SK, Pfeffer K, Mak TW. An Aberrant Stat Pathway Is Central to Covid-19. Cell Death Differ (2020) 27(12):3209–25. doi: 10.1038/s41418-020-00633-7
90. Boudewijns R, Thibaut HJ, Kaptein SJ, Li R, Vergote V, Seldeslachts L, et al. Stat2 Signaling Restricts Viral Dissemination But Drives Severe Pneumonia in Sars-Cov-2 Infected Hamsters. Nat Commun (2020) 11(1):1–10. doi: 10.1038/s41467-020-19684-y
91. Frieman MB, Chen J, Morrison TE, Whitmore A, Funkhouser W, Ward JM, et al. Sars-Cov Pathogenesis Is Regulated by a Stat1 Dependent But a Type I, Ii and Iii Interferon Receptor Independent Mechanism. PloS Pathog (2010) 6(4):e1000849. doi: 10.1371/journal.ppat.1000849
92. Park C, Li S, Cha E, Schindler C. Immune Response in Stat2 Knockout Mice. Immunity (2000) 13(6):795–804. doi: 10.1016/S1074-7613(00)00077-7
93. Toth K, Lee SR, Ying B, Spencer JF, Tollefson AE, Sagartz JE, et al. Stat2 Knockout Syrian Hamsters Support Enhanced Replication and Pathogenicity of Human Adenovirus, Revealing an Important Role of Type I Interferon Response in Viral Control. PloS Pathog (2015) 11(8):e1005084. doi: 10.1371/journal.ppat.1005084
94. Gowen BB, Westover JB, Miao J, Van Wettere AJ, Rigas JD, Hickerson BT, et al. Modeling Severe Fever With Thrombocytopenia Syndrome Virus Infection in Golden Syrian Hamsters: Importance of Stat2 in Preventing Disease and Effective Treatment With Favipiravir. J Virol (2017) 91(3):e01942–16. doi: 10.1128/JVI.01942-16
95. Siddharthan V, Van Wettere AJ, Li R, Miao J, Wang Z, Morrey JD, et al. Zika Virus Infection of Adult and Fetal Stat2 Knock-Out Hamsters. Virology (2017) 507:89–95. doi: 10.1016/j.virol.2017.04.013
96. Zumla A, Hui DS, Azhar EI, Memish ZA, Maeurer M. Reducing Mortality From 2019-Ncov: Host-Directed Therapies Should Be an Option. Lancet (2020) 395(10224):e35–e6. doi: 10.1016/S0140-6736(20)30305-6
97. Liu Q, Zhou Y-h, Yang Z-q. The Cytokine Storm of Severe Influenza and Development of Immunomodulatory Therapy. Cell Mol Immunol (2016) 13(1):3–10. doi: 10.1038/cmi.2015.74
98. Asokan I, Rabadia SV, Yang EH. The Covid-19 Pandemic and Its Impact on the Cardio-Oncology Population. Curr Oncol Rep (2020) 22(6):60. doi: 10.1007/s11912-020-00945-4
99. Kwenandar F, Japar KV, Damay V, Hariyanto TI, Tanaka M, Lugito NPH, et al. Coronavirus Disease 2019 and Cardiovascular System: A Narrative Review. IJC Heart Vasculature (2020) 29:100557. doi: 10.1016/j.ijcha.2020.100557
100. Guo J, Huang Z, Lin L, Lv J. Coronavirus Disease 2019 (Covid-19) and Cardiovascular Disease: A Viewpoint on the Potential Influence of Angiotensin-Converting Enzyme Inhibitors/Angiotensin Receptor Blockers on Onset and Severity of Severe Acute Respiratory Syndrome Coronavirus 2 Infection. J Am Heart Assoc (2020) 9(7):e016219. doi: 10.1161/JAHA.120.016219
101. Silhol F, Sarlon G, Deharo J-C, Vaïsse B. Downregulation of Ace2 Induces Overstimulation of the Renin–Angiotensin System in Covid-19: Should We Block the Renin–Angiotensin System? Hypertension Res (2020) 43(8):854–6. doi: 10.1038/s41440-020-0476-3
102. Lee SG, Fralick M, Sholzberg M. Coagulopathy Associated With Covid-19. CMAJ (2020) 192(21):E583–E. doi: 10.1503/cmaj.200685
103. Tang N, Li D, Wang X, Sun Z. Abnormal Coagulation Parameters Are Associated With Poor Prognosis in Patients With Novel Coronavirus Pneumonia. J Thromb Haemostasis (2020) 18(4):844–7. doi: 10.1111/jth.14768
104. Robba C, Battaglini D, Ball L, Valbusa A, Porto I, Della Bona R, et al. Coagulative Disorders in Critically Ill Covid-19 Patients With Acute Distress Respiratory Syndrome: A Critical Review. J Clin Med (2021) 10(1):140. doi: 10.3390/jcm10010140
105. Aldien AS, Ganesan GS, Wahbeh F, Al-Nassr N, Altarawneh H, Al Theyab L, et al. Systemic Inflammation May Induce Cardiac Injury in Covid-19 Patients Including Children and Adolescents Without Underlying Cardiovascular Diseases: A Systematic Review. Cardiovasc Revasc Med (2021) 35:169–78. doi: 10.1016/j.carrev.2021.04.007
106. Henry BM, De Oliveira MHS, Benoit S, Plebani M, Lippi G. Hematologic, Biochemical and Immune Biomarker Abnormalities Associated With Severe Illness and Mortality in Coronavirus Disease 2019 (Covid-19): A Meta-Analysis. Clin Chem Lab Med (CCLM) (2020) 58(7):1021–8. doi: 10.1515/cclm-2020-0369
107. Li D, Chen Y, Jia Y, Tong L, Tong J, Wang W, et al. Sars-Cov-2–Induced Immune Dysregulation and Myocardial Injury Risk in China: Insights From the Ers-Covid-19 Study. Circ Res (2020) 127(3):397–9. doi: 10.1161/CIRCRESAHA.120.317070
108. Guo T, Fan Y, Chen M, Wu X, Zhang L, He T, et al. Cardiovascular Implications of Fatal Outcomes of Patients With Coronavirus Disease 2019 (Covid-19). JAMA Cardiol (2020) 5(7):811–8. doi: 10.1001/jamacardio.2020.1017
109. Gabarre P, Dumas G, Dupont T, Darmon M, Azoulay E, Zafrani L. Acute Kidney Injury in Critically Ill Patients With Covid-19. Intensive Care Med (2020) 46(7):1339–48. doi: 10.1007/s00134-020-06153-9
110. Cheng Y, Luo R, Wang K, Zhang M, Wang Z, Dong L, et al. Kidney Disease Is Associated With in-Hospital Death of Patients With Covid-19. Kidney Int (2020) 97(5):829–38. doi: 10.1016/j.kint.2020.03.005
111. Frithiof R, Bergqvist A, Järhult JD, Lipcsey M, Hultström M. Presence of Sars-Cov-2 in Urine Is Rare and Not Associated With Acute Kidney Injury in Critically Ill Covid-19 Patients. Crit Care (2020) 24(1):1–3. doi: 10.1186/s13054-020-03302-w
112. Cohen JB, Hanff TC, Bress AP, South AM. Relationship Between Ace2 and Other Components of the Renin-Angiotensin System. Curr Hypertension Rep (2020) 22(7):1–5. doi: 10.1007/s11906-020-01048-y
113. Li Z, Wu M, Yao J, Guo J, Liao X, Song S, et al. Caution on Kidney Dysfunctions of Covid-19 Patients. (2020). doi: 10.1101/2020.02.08.20021212
114. Ekholm M, Kahan T. The Impact of the Renin-Angiotensin-Aldosterone System on Inflammation, Coagulation, and Atherothrombotic Complications, and to Aggravated Covid-19. Front Pharmacol (2021) 12:1534. doi: 10.3389/fphar.2021.640185
115. Vaduganathan M, Vardeny O, Michel T, McMurray JJ, Pfeffer MA, Solomon SD. Renin–Angiotensin–Aldosterone System Inhibitors in Patients With Covid-19. N Engl J Med (2020) 382(17):1653–9. doi: 10.1056/NEJMsr2005760
116. Fajgenbaum DC, June CH. Cytokine Storm. N Engl J Med (2020) 383(23):2255–73. doi: 10.1056/NEJMra2026131
117. Legrand M, Bell S, Forni L, Joannidis M, Koyner JL, Liu K, et al. Pathophysiology of Covid-19-Associated Acute Kidney Injury. Nat Rev Nephrol (2021) 17(11):751–64. doi: 10.1038/s41581-021-00452-0
118. Migliorini A, Angelotti ML, Mulay SR, Kulkarni OO, Demleitner J, Dietrich A, et al. The Antiviral Cytokines Ifn-A and Ifn-B Modulate Parietal Epithelial Cells and Promote Podocyte Loss: Implications for Ifn Toxicity, Viral Glomerulonephritis, and Glomerular Regeneration. Am J Pathol (2013) 183(2):431–40. doi: 10.1016/j.ajpath.2013.04.017
119. Gurkan S, Cabinian A, Lopez V, Bhaumik M, Chang JM, Rabson AB, et al. Inhibition of Type I Interferon Signalling Prevents Tlr Ligand-Mediated Proteinuria. J Pathol (2013) 231(2):248–56. doi: 10.1002/path.4235
120. Castellano G, Franzin R, Sallustio F, Stasi A, Banelli B, Romani M, et al. Complement Component C5a Induces Aberrant Epigenetic Modifications in Renal Tubular Epithelial Cells Accelerating Senescence by Wnt4/Bcatenin Signaling After Ischemia/Reperfusion Injury. Aging (Albany NY) (2019) 11(13):4382–406. doi: 10.18632/aging.102059
121. Yang AC, Kern F, Losada PM, Agam MR, Maat CA, Schmartz GP, et al. Dysregulation of Brain and Choroid Plexus Cell Types in Severe Covid-19. Nature (2021) 595(7868):565–71. doi: 10.1038/s41586-021-03710-0
122. Matschke J, Lütgehetmann M, Hagel C, Sperhake JP, Schröder AS, Edler C, et al. Neuropathology of Patients With Covid-19 in Germany: A Post-Mortem Case Series. Lancet Neurol (2020) 19(11):919–29. doi: 10.1016/S1474-4422(20)30308-2
123. Song E, Zhang C, Israelow B, Lu-Culligan A, Prado AV, Skriabine S, et al. Neuroinvasion of Sars-Cov-2 in Human and Mouse Brain. J Exp Med (2021) 218(3):e20202135. doi: 10.1084/jem.20202135
124. Brann DH, Tsukahara T, Weinreb C, Lipovsek M, Van den Berge K, Gong B, et al. Non-Neuronal Expression of Sars-Cov-2 Entry Genes in the Olfactory System Suggests Mechanisms Underlying Covid-19-Associated Anosmia. Sci Adv (2020) 6(31):eabc5801. doi: 10.1126/sciadv.abc5801
125. He L, Mäe MA, Muhl L, Sun Y, Pietilä R, Nahar K, et al. Pericyte-Specific Vascular Expression of Sars-Cov-2 Receptor Ace2–Implications for Microvascular Inflammation and Hypercoagulopathy in Covid-19. medRxiv (2020) 2020.05.11.088500. doi: 10.1101/2020.05.11.088500
126. Maiese A, Manetti AC, Bosetti C, Del Duca F, La Russa R, Frati P, et al. Sars-Cov-2 and the Brain: A Review of the Current Knowledge on Neuropathology in Covid-19. Brain Pathol (2021) 31(6):e13013. doi: 10.1111/bpa.13013
127. Boroujeni ME, Simani L, Bluyssen HA, Samadikhah HR, Zamanlui Benisi S, Hassani S, et al. Inflammatory Response Leads to Neuronal Death in Human Post-Mortem Cerebral Cortex in Patients With Covid-19. ACS Chem Neurosci (2021) 12(12):2143–50. doi: 10.1021/acschemneuro.1c00111
128. Kanberg N, Ashton NJ, Andersson L-M, Yilmaz A, Lindh M, Nilsson S, et al. Neurochemical Evidence of Astrocytic and Neuronal Injury Commonly Found in Covid-19. Neurology (2020) 95(12):e1754–e9. doi: 10.1212/WNL.0000000000010111
129. Diao B, Wang C, Tan Y, Chen X, Liu Y, Ning L, et al. Reduction and Functional Exhaustion of T Cells in Patients With Coronavirus Disease 2019 (Covid-19). Front Immunol (2020) 827. doi: 10.3389/fimmu.2020.00827
130. Deigendesch N, Sironi L, Kutza M, Wischnewski S, Fuchs V, Hench J, et al. Correlates of Critical Illness-Related Encephalopathy Predominate Postmortem Covid-19 Neuropathology. Acta Neuropathol (2020) 140(4):583–6. doi: 10.1007/s00401-020-02213-y
131. Efe IE, Aydin OU, Alabulut A, Celik O, Aydin K. Covid-19– Associated Encephalitis Mimicking Glial Tumor. World Neurosurg (2020) 140:46–8. doi: 10.1016/j.wneu.2020.05.194
132. Gupta A, Madhavan MV, Sehgal K, Nair N, Mahajan S, Sehrawat TS, et al. Extrapulmonary Manifestations of Covid-19. Nat Med (2020) 26(7):1017–32. doi: 10.1038/s41591-020-0968-3
133. Li H, Chen C, Hu F, Wang J, Zhao Q, Gale RP, et al. Impact of Corticosteroid Therapy on Outcomes of Persons With Sars-Cov-2, Sars-Cov, or Mers-Cov Infection: A Systematic Review and Meta-Analysis. Leukemia (2020) 34(6):1503–11. doi: 10.1038/s41375-020-0848-3
134. Robba C, Battaglini D, Ball L, Loconte M, Brunetti I, Vena A, et al. Distinct Phenotypes Require Distinct Respiratory Management Strategies in Severe Covid-19. Respir Physiol Neurobiol (2020) 279:103455. doi: 10.1016/j.resp.2020.103455
135. Seif F, Aazami H, Khoshmirsafa M, Kamali M, Mohsenzadegan M, Pornour M, et al. Jak Inhibition as a New Treatment Strategy for Patients With Covid-19. Int Arch Allergy Immunol (2020) 181(6):467–75. doi: 10.1159/000508247
136. Wu D, Yang XO. Th17 Responses in Cytokine Storm of Covid-19: An Emerging Target of Jak2 Inhibitor Fedratinib. J Microbiol Immunol Infect (2020) 53(3):368–70. doi: 10.1016/j.jmii.2020.03.005
137. Venugopal S, Bar-Natan M, Mascarenhas JO. Jaks to Stats: A Tantalizing Therapeutic Target in Acute Myeloid Leukemia. Blood Rev (2020) 40:100634. doi: 10.1016/j.blre.2019.100634
138. Wang A, Singh K, Ibrahim W, King B, Damsky W. Focus: Skin: The Promise of Jak Inhibitors for Treatment of Sarcoidosis and Other Inflammatory Disorders With Macrophage Activation: A Review of the Literature. Yale J Biol Med (2020) 93(1):187–95.
139. Bagca BG, Avci CB. The Potential of Jak/Stat Pathway Inhibition by Ruxolitinib in the Treatment of Covid-19. Cytokine Growth Factor Rev (2020) 54:51–61. doi: 10.1016/j.cytogfr.2020.06.013
140. Furqan M, Akinleye A, Mukhi N, Mittal V, Chen Y, Liu D. Stat Inhibitors for Cancer Therapy. J Hematol Oncol (2013) 6(1):1–11. doi: 10.1186/1756-8722-6-90
141. Miklossy G, Hilliard TS, Turkson J. Therapeutic Modulators of Stat Signalling for Human Diseases. Nat Rev Drug Discov (2013) 12(8):611–29. doi: 10.1038/nrd4088
142. Plens-Gałąska M, Woźniak T, Wesoły J, Bluyssen HA, Sinbad. Structural, Experimental and Clinical Characterization of Stat Inhibitors and Their Potential Applications. Sci Data (2022) 9:139. doi: 10.1038/s41597-022-01243-3
143. Song H, Wang R, Wang S, Lin J. A Low-Molecular-Weight Compound Discovered Through Virtual Database Screening Inhibits Stat3 Function in Breast Cancer Cells. Proc Natl Acad Sci (2005) 102(13):4700–5. doi: 10.1073/pnas.0409894102
144. Schust J, Sperl B, Hollis A, Mayer TU, Berg T. Stattic: A Small-Molecule Inhibitor of Stat3 Activation and Dimerization. Chem Biol (2006) 13(11):1235–42. doi: 10.1016/j.chembiol.2006.09.018
145. Ashizawa T, Miyata H, Ishii H, Oshita C, Matsuno K, Masuda Y, et al. Antitumor Activity of a Novel Small Molecule Stat3 Inhibitor Against a Human Lymphoma Cell Line With High Stat3 Activation. Int J Oncol (2011) 38(5):1245–52. doi: 10.3892/ijo.2011.957
146. Czerwoniec A, Szelag M, Juszczak K, Wesoly J, Bluyssen HA. Cavs—Novel In Silico Selection Strategy of Specific Stat Inhibitory Compounds. J Comput Sci (2015) 10:186–94. doi: 10.1016/j.jocs.2015.03.001
147. Szelag M, Czerwoniec A, Wesoly J, Bluyssen HA. Identification of Stat1 and Stat3 Specific Inhibitors Using Comparative Virtual Screening and Docking Validation. PloS One (2015) 10(2):e0116688. doi: 10.1371/journal.pone.0116688
148. Plens-Galaska M, Szelag M, Collado A, Marques P, Vallejo S, Ramos-González M, et al. Genome-Wide Inhibition of Pro-Atherogenic Gene Expression by Multi-Stat Targeting Compounds as a Novel Treatment Strategy of Cvds. Front Immunol (2018) 9:2141. doi: 10.3389/fimmu.2018.02141
Keywords: COVID – 19, interferon response, immune hyperactivation, multiple organ damage, STAT signaling, Statinhib, therapy
Citation: Eskandarian Boroujeni M, Sekrecka A, Antonczyk A, Hassani S, Sekrecki M, Nowicka H, Lopacinska N, Olya A, Kluzek K, Wesoly J and Bluyssen HAR (2022) Dysregulated Interferon Response and Immune Hyperactivation in Severe COVID-19: Targeting STATs as a Novel Therapeutic Strategy. Front. Immunol. 13:888897. doi: 10.3389/fimmu.2022.888897
Received: 03 March 2022; Accepted: 13 April 2022;
Published: 17 May 2022.
Edited by:
Uday Kishore, Brunel University London, United KingdomReviewed by:
Béatrice Nal, INSERM U1104 Centre d’immunologie de Marseille-Luminy (CIML), FranceCopyright © 2022 Eskandarian Boroujeni, Sekrecka, Antonczyk, Hassani, Sekrecki, Nowicka, Lopacinska, Olya, Kluzek, Wesoly and Bluyssen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hans A. R. Bluyssen, aC5ibHV5c3NAYW11LmVkdS5wbA==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.