 Marie Robert
Marie Robert Pierre Miossec
Pierre Miossec Arnaud Hot
Arnaud Hot- 1Department of Clinical Immunology and Rheumatology, and Immunogenomics and Inflammation Research Unit, University of Lyon, Hôpital Edouard Herriot, Lyon, France
- 2Department of Internal Medicine, University of Lyon, Hôpital Edouard Herriot, Lyon, France
The involvement of IL-17A in autoimmune and inflammatory diseases has prompted the development of therapeutic strategies to block the Th17 pathway. Promising results came from their use in psoriasis and in ankylosing spondylitis. IL-17A acts on various cell types and has both local and systemic effects. Considering the premature mortality observed during chronic inflammatory diseases, IL-17A action on vascular cells was studied. Both in vitro and in vivo results suggest that this cytokine favors inflammation, coagulation and thrombosis and promotes the occurrence of cardiovascular events. These observations led to study the role of IL-17A in diseases characterized by vascular inflammation, namely allograft rejection and vasculitis. Increased circulating levels of IL-17A and histological staining reveal that the Th17 pathway is involved in the pathogenesis of these diseases. Vasculitis treatment faces challenges while the use of steroids has many side effects. Regarding results obtained in giant cell arteritis with IL-6 inhibitors, a cytokine involved in Th17 differentiation, the use of anti-IL-17 is a promising strategy. However, lessons from rheumatoid arthritis and multiple sclerosis must be learnt before targeting IL-17 in vasculitis, which may be culprit, consort or both of them.
1 Introduction
Interleukin (IL)-17A is a pro-inflammatory cytokine involved in many autoimmune and inflammatory diseases (1). Its identification in the pathogenesis of these disorders had led to the development of therapeutics with a great success in psoriasis and in ankylosing spondylitis (2, 3). Outside its local effects, IL-17A induces systemic manifestations playing a role in the premature cardiovascular (CV) mortality observed in inflammatory diseases (4–6). IL-17A acts on all cell types that make up the three layers of the vascular wall by promoting inflammation, coagulation and thrombosis (7). These results suggest that IL-17A is involved in vascular inflammation and particularly in allograft rejection and vasculitis.
Vasculitis are defined according to the size and the type of the vessels that are predominantly affected (8, 9). From large to small vasculitis, histopathological lesions are different but evidence hints that T-helper (Th)-17 cells and IL-17A are involved in their pathogenesis (1). In many cases, the treatment relies on steroids which have short and long-term side effects and alternative therapeutics are expected (10–13). Given the role of the Th17 pathway in these diseases, one can expect that the inhibition of IL-17A could be of a great interest. Indirect evidence came from the blockade of IL-6, a cytokine required for Th17 differentiation, that is recommended in selected cases of large vessel vasculitis (LVV) (14, 15). However, disappointing use of IL-17 inhibitors in rheumatoid arthritis (RA) and in multiple sclerosis, while the Th17 pathway is clearly involved in the pathogenesis, must be understood before going further into their application in vasculitis (16, 17).
In this review, the key effects of the Th17 pathway on blood vessels will be detailed, after a brief overview of the Th17 pathway. Then, its involvement in vasculitis will be addressed. Therapeutic implications are finally discussed.
2 Effects of IL-17 on Blood Vessels
2.1 Overview of the Th17 Pathway
2.1.1 IL-17 Cytokines
Six isoforms (IL-17A to IL-17F) compose the IL-17 family (18–23). IL-17A has pleiotropic effects with a key role in host defense against extracellular pathogens, including bacteria and fungi, but also in chronic inflammation and autoimmunity (1, 16). IL-17A and IL-17F bear the greatest homology and are secreted either as homodimer or heterodimer (23, 24). Both IL-17A and IL-17F drive inflammation, IL-17F being less potent than IL-17A (25). In the presence of tumor necrosis factor (TNF)-α, they induce rather similar expression profiles (26, 27).
IL-17E (or IL-25) has the lowest homology with IL-17A and promotes Th2-cell mediated immune responses (28). Infection of the lungs with an IL-25 expressing adenovirus or IL-17E protein induces IL-4, IL-5, IL-13 production and then eosinophil infiltration, mucus secretion and airway hyperreactivity (29). IL-17E axis plays a role in asthma exacerbations and now constitutes an attractive target for the development of new therapies (30). IL-17E also modulates Th17 cell function by acting as a receptor antagonist for IL-17A function (31–33).
2.1.2 IL-17 Receptor Family and Signaling
Five receptors (IL-17RA to IL-17RE) compose the IL-17 receptor (IL-17R) family (32). IL-17RA interacts with other subunits to form receptor complexes. IL-17A, IL-17F or IL-17A/F signal through IL-17RA/RC. IL-17E binds to IL-17RA/RB and IL-17C to IL-17RA/RE (28). IL-17RD is an alternate receptor subunit for IL-17A but not for IL-17F (34).
IL-17 signaling activates nuclear factor kappa B (NFκB), CCAAT/enhancer binding protein (CEBP)-ß/δ and mitogen-activated protein kinase pathways. It activates inflammatory genes encoding cytokines and chemokines (e.g., IL-6 and IL-8). IL-17 can also regulate genes post-transcriptionally and mRNA half-life (34).
IL-17 function is regulated by different mediators. TNFα, IL-1, granulocyte-macrophage colony stimulating factor (GM-CSF) and interferon (IFN) γ regulate positively IL-17 effects whereas IL-17E/IL-25, anti-IL-17 auto-antibodies and soluble IL-17R inhibit its function (17).
2.1.3 IL-17 Producing Cells
Th17 cells undergo differentiation following three steps. The first step corresponds to the initiation of the differentiation and is mediated by transforming growth factor (TGF)-ß and IL-21. Both cytokines induce the transcription of the lineage specific transcription factor receptor-related orphan receptor (RORc). Secondly, IL-6 and IL-1ß allow the amplification of the Th17 lineage. Finally, Th17 cells acquire their pathogenic role thanks to IL-23. Th17 produce many cytokines as IL-17A, IL-17F, IL-21 and IL-22 (15, 35).
A balance exists between Th17 and Treg cells because their developmental pathways are interconnected and reciprocally regulated. TGF-ß is necessary for both Th17 and Treg differentiation. The addition of IL-6 inhibits FoxP3, required for Treg differentiation, and upregulates RORc. This induces a shift toward the Th17 lineage (15). Moreover, mice deficient in exons 2 and 7 of FoxP3 fail to repress RORc, have increased levels of Th1 and Th17 cytokines and exhibit multi-organ inflammation with Treg lacking their suppressive ability (36). In humans, a substantial number of inflammatory diseases are characterized by an increase of Treg lacking exon 2 of FoxP3, with Treg unable to control IL-17+ T cell proliferation. These Δexon 2 FoxP3 Tregs but also some Tregs in inflammatory conditions can also produce IL-17. These results highlight the plasticity between these two cell types (37–39).
Overall, the Th17/Treg balance plays a key role in autoimmune and inflammatory diseases; Treg cells prevent their development while Th17 cells promote them (15). These dynamic changes illustrate the importance of the cytokine environment but are also influenced by local interactions (35).
Other IL-17 sources include immune cells with γδ T cells, invariant natural killer cells, innate lymphoid cells, CD8+ T cells or double-negative T cells (28, 40). Mast cells and neutrophils do not produce IL-17 but engulf it (41, 42).
These elements are summarized in Figure 1.
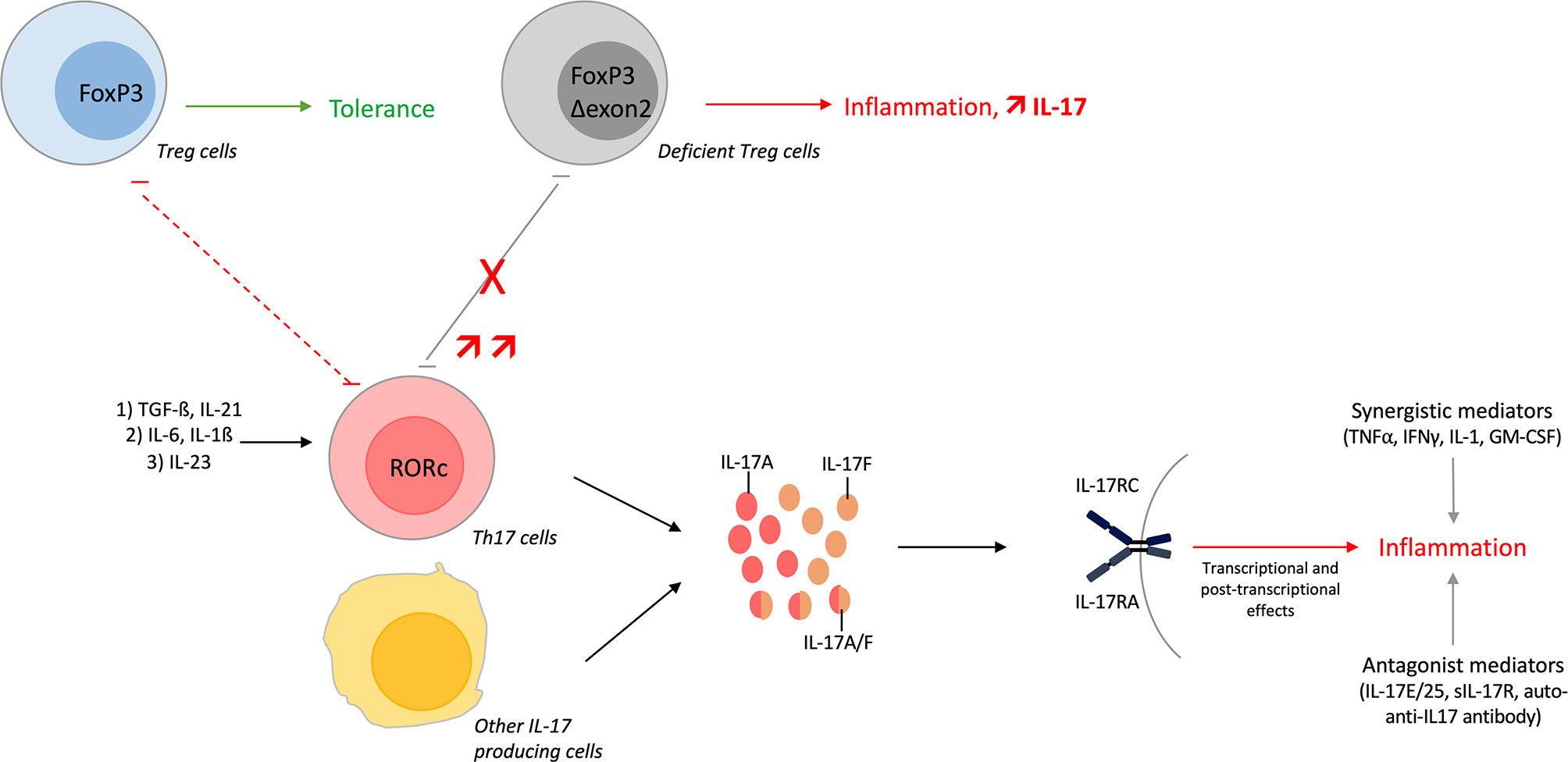
Figure 1 Overview of the Th17 pathway. T-helper (Th) 17 cells undergo differentiation from Th0 cells. The first step involves transforming growth factor (TGF)-ß and interleukin (IL)-21 that initiate the differentiation. Next, IL-6 and IL-1ß amplify the Th17 lineage and finally, IL-23 is required to maintain the lineage and to acquire its pathogenic role. Th17 differentiation can be shifted towards regulatory T (Treg) cells depending on cytokine environment. Developmental pathways of both cells are interconnected and reciprocally inhibited. Treg lacking exon 2 of FoxP3 impairs the Th17/Treg balance with increase amount of IL-17+ T cells. These Δexon 2 FoxP3 Treg cells, and some Tregs in inflammatory conditions, can also produce IL-17. Moreover, IL-17A/F/AF are produced by Th17 cells but also by other immune cells. These cytokines bind the same receptor, activate different pathways and finally induce inflammation. IL-17 function is regulated positively or negatively by different mediators.
2.2 Results on Isolated Cells
The vascular wall is composed of three layers: the intima, the media and the adventitia. Briefly, endothelial cells (EC) are part of the intima, vascular smooth muscle cells (VSMC) of the media and adipocytes, fibroblasts and immune cells of the adventitia. IL-17A alone, and even more when combined with TNFα, acts on these cell types (Figure 2A).
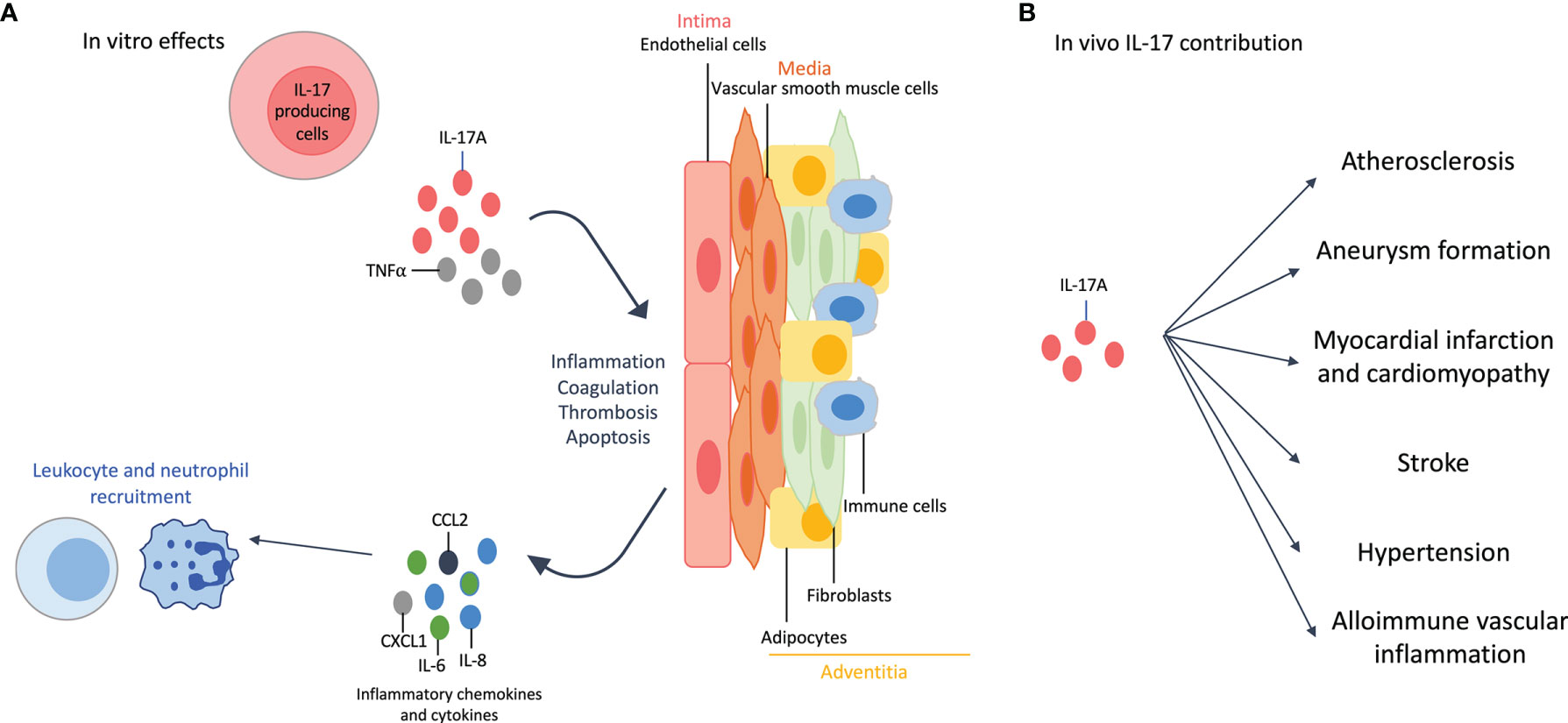
Figure 2 Effects of interleukin (IL)-17A on the cardiovascular system. (A): In vitro effects: IL-17A, and even more when combined with tumor necrosis factor (TNF)-α, affects all cell types of the vascular wall. IL-17A +/- TNFα induces inflammation with the release of pro-inflammatory cytokines (e.g., IL-6) and chemokines (e.g., IL-8, chemokine C-X-C motif ligand 1 CXCL1, C-C Motif Chemokine Ligand 2 CCL2), that in turn enhances neutrophil and leukocyte recruitment. This recruitment is also favored by the increased expression of adhesion molecules. IL-17A induces thrombosis, coagulation and apoptosis. The effects on adipocytes participate to the inflammatory environment. (B): In vivo contribution of IL-17A results in accelerated atherosclerosis, aneurysm formation, myocardial infarction and cardiomyopathy, stroke, hypertension and allo-immune vascular inflammation.
2.2.1 Effects on Intima Cells
IL-17A induces the secretion of pro-inflammatory cytokines (e.g., IL-6) and chemokines (e.g., IL-8, chemokine (C-X-C motif) ligand 1 CXCL1, C-C Motif Chemokine Ligand 2 CCL2) by EC (43). It also increases the level of adhesion molecules, especially when combined with TNFα, and then promotes leukocyte recruitment and invasion of EC (7). IL-17A promotes thrombosis and coagulation by activating tissue factor and reducing anti-coagulation mediators (e.g., CD39 and thrombomodulin) (7, 44). Finally, IL-17A increases EC apoptosis (45).
2.2.2 Effects on Media Cells
VSMC play a key role in atherosclerosis through their ability of proliferation, migration and apoptosis. IL-17A increases the production of pro-inflammatory cytokines and chemokines and the expression of adhesion molecules, plaque destabilizing proteins and tissue factor. VSMC apoptosis is also increased by IL-17A when combined with TNFα and/or IFNγ, that in turn promotes atherosclerosis (45, 46).
2.2.3 Effects on Adventitia Cells
IL-17A triggers inflammation through the production of pro-inflammatory cytokines by the different cell types of the adventitia but also by enhancing adipocyte lipolysis (7, 47).
IL-17 effects on isolated cells are summarized in Figure 2A.
2.3 Results From In Vivo Experiments and Systemic Effects of IL-17A
2.3.1 Effects of IL-17 on the CV System In Vivo
IL-17A, alone or combined with IFNγ, increases lesion size and plaque instability by inducing inflammation and enhancing the recruitment of myeloid cells. Other results show that IL-17A favors aneurysm formation, myocardial infarction, stroke and hypertension. However, some results are contradictory regarding the protective or the deleterious effect of IL-17 on CV outcome. This is particularly true for atherosclerosis where animal models and protocols used are different across experiments (7).
2.3.2 Allo-Immune Vascular Rejection as a Paradigm of IL-17 Vascular Pathology
Blood vessels in allotransplantation remain largely of graft origin and are subject to host allo-immune responses. Vascular pathology can contribute to graft inflammation, ischemia/reperfusion injury and then allograft rejection. Mediators of innate and adaptive immunity are involved in these processes and support both hyper-acute, acute and chronic rejections (48). Among adaptive immune cells, the Th17 subset contributes to allograft rejection (49).
During acute rejection, IL-17 blockade significantly improves cardiac graft survival in a rat model (50). The antagonism of IL-17 decreases mononuclear infiltration and endothelial damage in a murine aortic transplantation model (51). Allograft rejection, partially mediated by IL-17, mainly relies on neutrophil recruitment (49, 52).
Chronic rejection is made up of a parenchymal and a vascular rejection. The latter is caused by a stenosis of vessels due to a progressive immune mediated host response to graft blood vessels. Different mechanisms contribute to stenosis including ischemia/reperfusion, hypertension and immune activation (53). IL-17 and Th17 cells play a role in these phenomenon (17). A model of chronic allograft vasculopathy shows that, in absence of Th1 response, Th17 cells induce severe accelerated allograft rejection and vasculopathy (54). Reducing IL-17 production suppresses chronic allograft rejection and vasculopathy (55). IL-17-/- mice do not develop cardiac fibrosis after mismatched organ transfer, which is considered as secondary to allograft vasculopathy (56). Also, in IL-17 deficient mice, graft coronary artery disease after heterotopic cardiac transplantations is reduced (57).
2.3.3 Circulating Levels of IL-17 in Human Vascular Pathology
In addition to its local effects, IL-17A is circulating and has systemic effects (4). As mentioned above, IL-17A function is regulated by various mediators (17). To counteract these complex interactions, a bioassay was developed to study specifically the bioactive fraction of IL-17A (58). This was particularly well described in rheumatoid patients where bioactive IL-17A is associated with joint destruction and CVE occurrence (5, 59). Similarly, patients with myocardial infarction show a peak of circulating IL-17A at admission (60). IL-17A induces systemic effects by affecting various cell types: cells of the liver, of skeletal and cardiac muscles and of the blood vessels (4).
In vivo contribution of the Th17 lineage is described in Figure 2B.
3 Role of the Th17 Pathway in Vasculitis
Regarding IL-17A effects on the CV system, this cytokine could play a role in vasculitis pathogenesis which is characterized by blood vessel wall inflammation, endothelial injury and tissue damage.
Only noninfectious vasculitis of the 2012 International Chapel Hill Consensus Conference (CHCC2012), partially revised in 2018, are described (8, 9). Vasculitis are classified according to the size and the type of vessels predominantly affected. The effects of IL-17 and Th17 in large vessel vasculitis (LVV), medium vessel vasculitis (MVV), small vasculitis (SVV) and variable vessel vasculitis (VVV) are addressed.
3.1 Large Vessel Vasculitis (LVV)
LVV mainly affect large arteries including the aorta and its major branches. The two major diseases are Takayasu arteritis (TAK) and giant-cell arteritis (GCA) (8). Both disorders occur mainly in females and share histopathologic features with a chronic granulomatous inflammatory reaction (61). They differ by the age of onset: TAK generally occurs before the age of 50 years old whereas GCA after age 50 (62). Chronic inflammation within the vessel wall can lead to aneurysm formation, rupture or dissection where IL-17A and Th17 cells play a role (7, 63). Th17 cells are identifiable both in the peripheral blood and in the vasculitic lesions. It raises the possibility that inflammatory cells recirculate (63). Figure 3A summarizes the results described below.
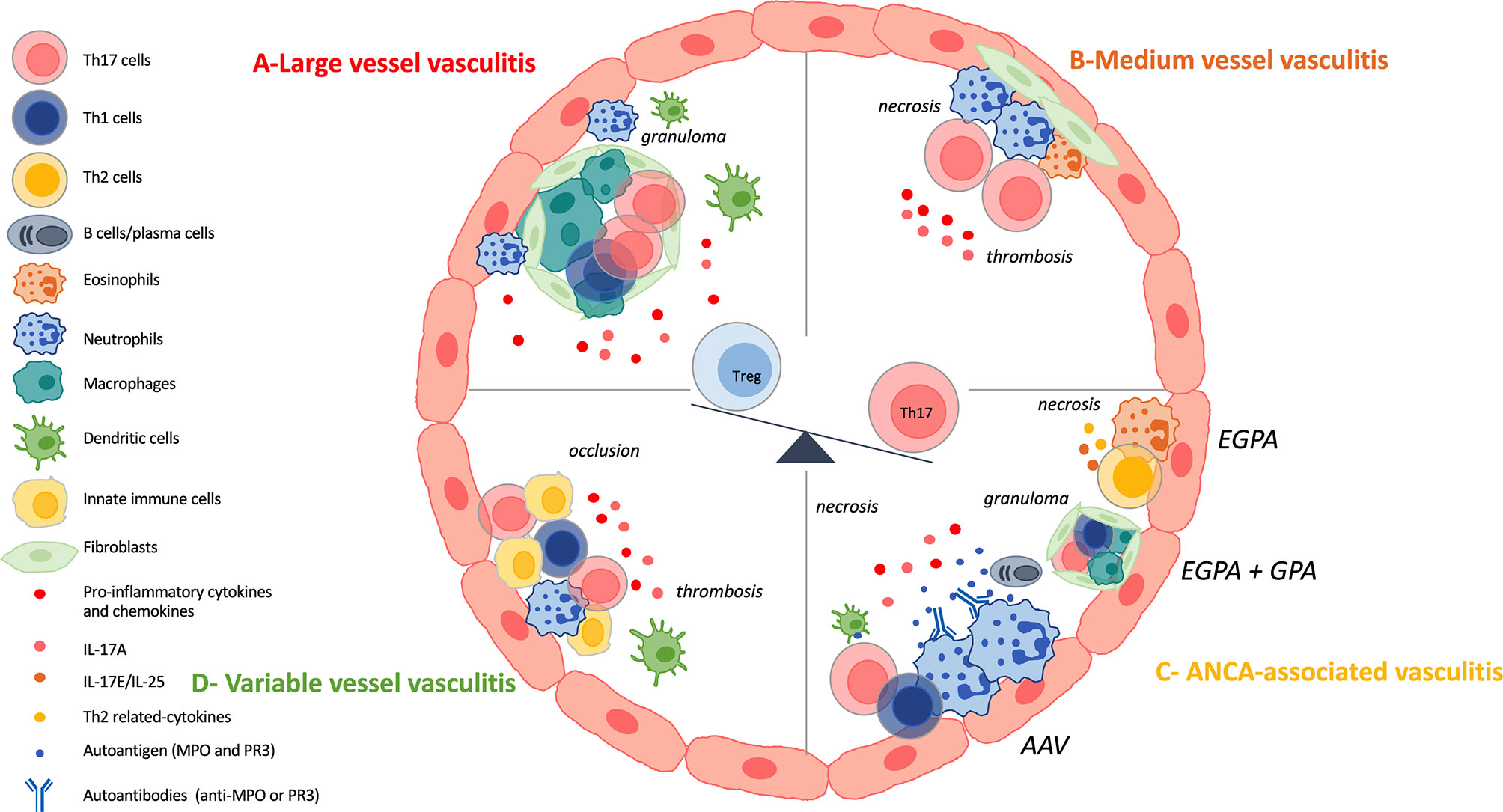
Figure 3 IL-17 and Th17 involvement in vasculitis. (A): Large vessel vasculitis (LVV) include giant cell arteritis (GCA) and Takayasu arteritis (TAK) and are characterized by a chronic granulomatous inflammatory reaction. Vascular adventitial dendritic cells are activated and induce the recruitment, the activation and the polarization of CD4+ T cells. Among CD4+ T cells, T-helper (Th)-17 cells and Th1 cells are involved in the local inflammatory process. Then, CD8+T cells and monocytes are recruited and finally, vascular remodeling occurs. Th17 cells are also circulating and patients harbor increased frequency of them. IL-17A promotes neutrophil recruitment. There is a decrease of regulatory T (Treg) cells at the expense of the Th17 lineage. (B): Medium vessel vasculitis (MVV) comprise polyarteritis nodosa (PAN) and Kawasaki disease (KD). There are few data on Th17 involvement in PAN pathogenesis but patients harbor defective Tregs and increased frequency of Th17 cells. KD is characterized by necrotizing arteritis with neutrophil infiltration. Lymphocytes, eosinophils and myofibroblastic proliferation participate in KD arteriopathy. Th17 cells and their related cytokines are increased in KD and there is a dysregulation of the Th17/Treg balance. (C): Antineutrophil cytoplasmic antibody (ANCA)-associated vasculitis (AAV) are composed of three variants: microscopic polyangiitis (MPA), granulomatosis with polyangiitis (GPA), eosinophilic granulomatosis with polyangiitis (EGPA). They are characterized by fibrinoid necrosis, thrombus formation and immune cell infiltration. GPA and EGPA are associated with granulomatous inflammatory reaction where Th17, Th1 cells and macrophages play a role. EGPA is characterized by the involvement of eosinophils, Th2 cells and their related cytokines. GPA and MPA, and sometimes EGPA, are associated with ANCA. There is a loss of B and T tolerance toward MPO and PR3 antigens, that are found primary in neutrophils. PR3 and MPO-specific Th17 responses occur in the course of the disease. IL-17A also favors the recruitment of neutrophils at the site of injury. There is a dysregulated Th17/Treg cells balance. (D): Variable vessel vasculitis (VVV) can affect any type and any size of vessels. Enhanced innate immune response, early tissue infiltration and late adaptive immunity are the different steps that occurred in Behçet’s disease (BD) pathogenesis. This results in thrombosis, occlusion and aneurysms. Th17 cells are found at the site of tissue injury and interact with other immune cells. There is an hyperactivation of Th17 and Th1 cells at the expense of Treg population.
3.1.1 Takayasu Arteritis (TAK)
TAK is a rare disease that induces acute inflammation, carotidynia, discrepant blood pressure between the arms, absent or asymmetric pulsation, limb claudication and angina (64).
Patients with TAK have significantly increased level of IL-17A and circulating Th17 cells compared with healthy individuals (65, 66). Similar results are obtained when comparing patients with active TAK with those in remission (66–68). After anti-CD3/CD28 stimulation, IL-17A-producing T cell frequency is significantly increased in the presence of serum from TAK patients with active disease compared with those in remission (66). Anti-endothelial protein C receptor (EPCR) antibodies, found in 34.6% of TAK patients, impair Th17 differentiation (69). The imbalance that exists between Th1/Th17 and Treg cells in TAK is driven by type I and II cytokines which signal through the Janus kinase/signal transducers and activators of transcription (JAK/STAT) pathway (70). The differentiation of Th1 and Th17 cells is impaired in TAK patients and mediated by an overactivation of mammalian target of rapamycin complex 1 (mTORC1) (71). Recently, an increase in Th17.1 cells producing both IL-17 and IFNγ was observed in TAK patients compared with healthy controls. Similar results were described for PD1+Th17 cells (72). Finally, IL-17A and Th17-related cytokines trigger neutrophil recruitment and activation that could contribute to vascular lesions (73).
Among Th17-related cytokines (IL-6, IL-21, IL-23, IL-1ß, TGF-ß), the role of IL-6 in TAK pathogenesis has been well described while IL-6 is critically involved in Th17 differentiation (15). Its level is increased in TAK patients compared with controls and linked to disease activity (66, 68). Similar trends are described for IL-23 level in TAK (65, 66).
3.1.2 Giant-Cell Arteritis (GCA)
GCA affects older patients than TAK and typically induces vasculitis of the extracranial branches of the aorta (62, 74). The three layers of the arterial wall are affected by histopathological lesions, especially the internal elastic lamina with multinucleated giant cells, CD4+ T cells and macrophages organized in granuloma (74). Recent understanding of the immunopathological model of GCA has allowed to divide its pathogenesis into four steps. The first corresponds to the activation of vascular adventitial dendritic cells, the second is characterized by the recruitment, the activation, and the polarization of CD4+ T cells, followed by the recruitment of CD8+T cells and monocytes. Finally, vascular remodeling occurs. Th17 cells are mainly involved in the second phase (75, 76). Th17 pathogenic role in vascular remodeling is similar to the one described in myocardial infarction (7).
Frequencies of circulating Th17 cells are increased up to 10-fold in untreated GCA patients compared with healthy controls (77–79). Immunohistochemical analysis of temporal artery biopsy specimens from GCA patients show infiltration of Th17 cells, mainly localized at the junction between the adventitia and the media. Artery-infiltrating Th1 cells may derive from local differentiation of Th17 cells in the presence of IL-12. CD161+CD4+ T cells could be the common precursor that links Th1 and Th17 cells (76, 78). As well as Th1 cells, a population of IL-17/IFNγ double producing cells is expanded in untreated GCA patients to create granuloma and IL-17 regulates macrophage recruitment (63, 77, 78). IFNγ+ CD4+ T cell commitment involves JAK/STAT pathway and type I interferon signature is upregulated in GCA aortas (80, 81). Induction of both T-bet+-CD4+ and RORγt+CD4+ T cells involves the AKT-mTORC1 signaling pathway and there is a constitutive activation of the Notch-AKT-mTORC1 signaling axis in T cells from GCA patients (82).
An imbalance between Th17 and Treg cells is observed in GCA patients compared with healthy subjects. This balance is partially modulated by IL-6 and IL-21, whose levels are correlated with disease activity (78, 79, 83). GCA Tregs may increase vascular inflammation by promoting Th17 polarization (37).
In a model of severe combined-immunodeficiency (SCID) mice engrafted with normal human arteries, treatment with dexamethasone inhibits both mRNA production of IL-17 and Th17 density in the vessel wall (77). Similar results are observed in patients with a decrease of IL-17 producing CD4+ T cells after steroid therapy and the Th17/Treg ratio is significantly reduced (78). However, it does not restore the Treg deficiency observed in GCA and does not affect Th1 response (77, 78). Moreover, in a chimeric mouse model of GCA, the inhibition of JAK1/JAK3 activity reduces RORc expression and IL-17 level, and finally suppresses T-cell invasion and proliferation into the artery (80).
Results described for Th17 cells are confirmed at the cytokine level. IL-17 and Th17-related cytokines (IL-1ß, IL-6, IL-21, IL-23) levels are increased in serum from untreated GCA patients and after PBMCs stimulation with PMA/ionomycin from patients with active disease compared with controls and those in remission (77, 79). After steroid therapy, the level of circulating IL-17 is decreased (77). Similar observations apply for IL-17A, IL-1ß, IL-6 and IL-23 expression in inflamed temporal arteries (77, 79, 84, 85). IL-17 is overexpressed when transmural inflammation and granulomatous reaction occur (84).
Finally, studies of genetic background and epigenetic modifications suggest a role of IL-17 and Th17 pathway in GCA development or pathogenesis (75, 86, 87).
3.2 Medium Vessel Vasculitis (MVV)
Polyarteritis nodosa (PAN) and Kawasaki disease (KD) are the two entities that constitute MVV. Figure 3B gives a brief overview of Th17 involvement in the pathogenesis.
3.2.1 Polyarteritis Nodosa (PAN)
PAN generally occurs in patients of 50 years old and can be primary or secondary to viral infection with the example of hepatitis-B-virus. It induces renal vasculitis with renovascular hypertension, renal infarcts and microaneurysms (88).
Very few studies refer to the effects of IL-17 or Th17 cells in PAN. IL-17 producing CD4+ T cells frequency is higher in PAN patients compared with healthy controls. PAN patients also have defective Tregs in suppressive ability (89). In PAN patients with cutaneous mutations, trends toward a decrease of IL-17 level after treatment are observed without significant difference (90).
3.2.2 Kawasaki Disease (KD)
KD is the leading cause of acquired heart disease in children and primarily involves muscular arteries (91). Three pathological processes have been identified to explain KD arteriopathy: necrotizing arteritis characterized by neutrophilic infiltration, subacute and chronic vasculitis with an inflammatory cell infiltrate composed of lymphocytes, plasma cells and eosinophils and luminal myofibroblastic proliferation. These lesions can induce coronary artery aneurysm, thrombosis, stenosis and myocardial infarction (92). Systemic inflammation goes along with vascular lesions and could be mediated by inflammatory cytokines, as IL-17.
Th17 cells and related cytokines (IL-17A, IL-6, IL-23, IL-21 and IL-22) are markedly increased in KD patients compared with controls. These observations have been made both in the plasma, in the serum and in the supernatants of cultured PBMCs after stimulation with anti-CD3/CD28 (93–95). Some results suggest that IL-17 level is correlated with disease activity (93, 96). Moreover, myofibroblasts expressing IL-17 and IL-6 are observed in the damaged arterial wall of KD autopsies (97). Regarding the Th17/Treg balance, Treg frequency and FoxP3 expression are markedly lower in KD patients compared with controls suggesting a shift towards Th17 differentiation, probably mediated by IL-6 (93, 98).
Cytokine levels and Th17 frequency are decreased after one week of combined therapy including aspirin and intravenous immunoglobulin (IVIG) (94). IVIG-resistant KD have increased plasma levels of IL-17A and IL-6 before treatment compared with sensitive patients. Resistant patients maintain high levels of these cytokines after treatment (93). To counteract this resistance, plasma exchanges have been tested. It induces the removal of IL-17 and IL-6 levels and could participate in therapeutic mechanisms (99).
3.3 Small Vessel Vasculitis (SVV)
Small intraparenchymal arteries, arterioles, capillaries and venules are mainly affected in SVV. Two categories of SVV are described based on paucity or abundance of vessel wall immunoglobulin deposits (8).
3.3.1 Antineutrophil Cytoplasmic Antibody (ANCA)-Associated Vasculitis (AAV)
Results suggest that the Th17 subset is involved in AAV pathogenesis as IL-17 serum level is increased in AAV patients compared with healthy individuals. Similar observations apply for IL-23. However, levels remain elevated in some patients and major relapses occur suggesting that a Th17 memory cell population may persist (100). The proportion CCR6+CD4+RORγt+ T cells is increased in kidney biopsy samples compared to the peripheral blood suggesting recruitment of these cells into the kidney. These results support the role of Th17 in AAV pathogenesis (101, 102). The conversion from Tregs to Th17 effector cells within the inflammatory environment play a role in AAV pathogenesis and the imbalance of Th17/activated Treg cells marks renal involvement (103, 104). Moreover, Treg cells harbor a defect in their suppressive function (39).
Among other IL-17 cytokines, serum IL-17C level is increased in AAV-patients with crescentic glomerulonephritis (GN) compared with controls. Results from mice models confirm that IL-17C promotes tissue renal injury in an IL-17A-dependant manner. IL-17RE promotes Th17 response in crescentic GN and is expressed on Th17 cells. The activation of the IL-17C/IL-17RE axis increases renal expression of IL-17 target genes leading to neutrophil recruitment and then tissue injury (105).
3.3.1.1 Microscopic Polyangiitis (MPA)
MPA belongs to AAV and affects mainly the kidneys with progressive GN and the lungs with alveolar hemorrhage (106). MPA is mainly associated with MPO-ANCA (107). IL-17A level is increased in MPA patients compared with controls (108). Th17 and Th1 cells promote macrophage activation at sites of injury. Macrophages hasten disease progression through their profibrotic properties (109). In murine model of anti-MPO induced GN, IL-17 may enhance antigen deposition in the glomeruli and mediates pathogenic effector functions (110). Toll-like receptor (TLR)-2 ligand promotes Th17-induced MPO autoimmunity (111). MPO-specific Th17 cells are involved earlier in disease while Th1 cells are implicated later (112). CD8+ T cells also cause experimental injury (113).
3.3.1.2 Granulomatosis With Polyangiitis (GPA)
GPA is characterized by a necrotizing granulomatous inflammation and is predominantly associated with PR3-ANCA (107). It usually involves the upper and lower respiratory tract and kidneys. Almost all patients have sinonasal involvement (114). Patients with GPA have an increased percentage of circulating IL-17A+T cells compared with healthy controls (115, 116). Involvement of the eye socket in GPA is rare but cytokine staining for IL-17 and IL-23 are significantly greater in GPA lesions compared with idiopathic inflammatory orbital diseases and sarcoidosis lesions (114, 117). The increased production of IL-17A could in turn enhances neutrophil recruitment and activation that are involved in AAV pathogenesis (118). Stimulation with the PR3 autoantigen increases Th17 cell frequency in ANCA-positive GPA patients compared with ANCA-negative ones and healthy individuals, suggesting the existence of PR3-specific Th17 responses (119, 120). Moreover, sustained remission is characterized by an increase of Treg, Th2 cells and Th2-cytokines levels (116, 121, 122). The increase of Treg cells is not sufficient to confer enhanced suppression (122, 123). Among FoxP3+ cells, there is a marked increase of non Treg cell subtype in GPA-patients in remission compared with healthy donors. These cells produce significantly more pro-inflammatory cytokines (e.g., IL-17) in ANCA-positive patients compared with ANCA-negative ones and healthy controls (124). Finally, recent results suggest an interaction between regulatory B (Breg) cells and Th17 cells. There is a negative correlation between Th17 effector memory cells and Bregs, and in vitro experiments show an expansion of Th17 cells after Breg depletion (125).
3.3.1.3 Eosinophilic Granulomatosis With Polyangiitis (EGPA)
EGPA is associated with asthma, peripheral blood and tissue eosinophilia. This disease is classically considered as a Th2-driven inflammatory response with a key role of IL-5 in eosinophil recruitment (126). Activated eosinophils play a pathogenic role and secrete IL-17E/IL-25 that in turn enhance Th2 cytokine production. Patients with active EGPA have increased levels of IL-25 and tissue staining of nerve tissue specimens reveals the presence of IL-25 and IL-17RB+ T cells (127).
The frequency of circulating IL-17-producing CD4+ T cells is increased in active EGPA compared with inactive disease (128). In a pathologic colonic submucosa from an EGPA patient, there is a positive correlation between eosinophil count, crypt-to-crypt distance, the basement membrane-to-crypt distance with the percentage of Th17 cells (129). Finally, there is an inhibition of Treg differentiation (128, 130).
Results on AAV are summarized in Figure 3C.
3.3.2 Immune-Complex-Mediated Vasculitis
3.3.2.1 Anti-GBM (Anti-Glomerular Basement Membrane) Disease
Anti-GBM disease is an autoimmune disorder characterized by the presence of anti-GBM auto-antibodies bound to basement membrane in glomerular and pulmonary alveolar capillaries. It explains the rapidly progressive GN and alveolar hemorrhage that occur. The main target of auto-antibodies is the non-collagenous domain 1 of the α3 chain of type IV collagen (8, 131).
Many results came from different animal models (102). Mice injected with Th17 cells exhibit a neutrophil signature with increased neutrophil infiltration (132). The Th17 pathway allows the recruitment of destructive neutrophils through the expression of CXCL5 by kidney tubular cells which contribute to renal tissue injury (133, 134). Moreover, the Th17 subset induces GN with crescent formation and antigen-specific Th17 cells are the main contributors to renal tissue injury (135). Additional studies confirmed the pathogenic role of Th17 cells in anti-GBM disease (136–138). The Th17/Th1/Treg balance also plays a role in its pathogenesis (134, 139).
3.3.2.2 Cryoglobulinemic Vasculitis
Cryoglobulinemic vasculitis is characterized by cryoglobulin immune deposits in small vessels that can affect skin, glomeruli and peripheral nerves (8). To our knowledge, there are currently no report on IL-17 involvement in vasculitis due to cryoglobulins. Results from a paper on mixed cryoglobulinemia associated with chronic hepatitis C virus (HCV) show an elevation of IL-17-inducing cytokines in patients with chronic HCV and mixed-cryoglobulinemia compared with HCV patients without vasculitis and healthy controls (140). This may suggest IL-17 involvement in this disease.
3.3.2.3 Immunoglobulin A (IgA) Vasculitis (IgAV)
IgAV is a vasculitis more common in children characterized by IgA1 deposits affecting small vessels. Clinical symptoms include cutaneous purpura, arthralgias and/or arthritis, acute enteritis and glomerulonephritis (8, 141).
Immunostaining of renal biopsies shows IL-17 expression in all specimens studied with IL-17+CD3-CD4- cells in the tubules and glomeruli, and IL-17+CD3+CD4+cells in the interstitium. Compared to control patients, glomerular and tubular grades of IL-17 expression are higher and IL-17 expression is correlated with proteinuria (142). Tubular cells may be an extra-immune source of IL-17 when triggered with injury (143). Moreover, the proportion of circulating Th17 cells and serum IL-17A level are increased in IgAV children compared with healthy individuals. Once again, the Th17/Treg imbalance may play a role in IgAV pathogenesis and is correlated with disease activity (144–146).
3.3.2.4 Hypocomplementemic Urticarial Vasculitis HUV (Anti-C1q Vasculitis)
HUV is a leukocytoclastic immune complex vasculitis accompanied by urticaria and hypocomplementemia with anti-C1q antibodies. This disease can induce GN, arthritis, pulmonary disorder and eye inflammation (147). To our knowledge, no paper reports results on Th17 involvement in HUV.
3.4 Variable Vessel Vasculitis (VVV) – Behçet’s Disease (BD)
VVV can affect vessels of any size and of any type. Behçet’s disease end Cogan’s syndrome are the two entities included in CHCC2012 (8).
BD often refers to the Silk Route disease because of its prevalence in the Middle-East and far-east Asia. Skin and mucosa lesions are the most common clinical manifestations but the prognosis mainly relies on vascular and neurological involvement (148). BD is characterized by thrombosis, aneurysms and occlusions (149).
The frequency of circulating Th17 cells and serum levels of IL-17A are increased in BD compared with healthy controls and are correlated with disease activity. Circulating Treg proportion is decreased in BD and the Th17/Treg ratio is higher in BD patients compared with controls (150–153). The Th17 and Th1 hyperactivation observed in BD is partially mediated by a decreased in B and T lymphocyte attenuator (BTLA) expression (154). Serum amyloid A promotes Th17 differentiation in BD (155). Immunostaining reveals that IL-17+cells infiltrate the erythema nodosum-like eruption in the skin of BD patients. IL-17A producing cells are found in the cerebrospinal fluid, in brain parenchyma inflammatory infiltrates and in intracerebral blood vessels from patients with active disease (156). Finally, IL-17A and IFNγ production are associated with enhanced innate immune response, early neutrophil tissue infiltration and late adaptive immunity (149, 157, 158). Results concerning BD pathogenesis are presented in Figure 3D.
4 Targeting IL-17 and Th17 Cells in Vascular Inflammation
Almost all studies described above only show an increased level of IL-17 and/or of Th17 cells but results on their real pathogenic roles are limited. IL-17A acts as a primer, or a consort, in a complex network of cytokines and is looking for synergy, with the typical example of TNFα. Lessons from RA and multiple sclerosis, where IL-17 inhibitors do not work as expected, must be learnt when considering new therapeutic strategies for vasculitis (17, 159). The targeting of IL-17 alone may not be sufficient to control these diseases and combined inhibition should be considered. To potentiate an eventual benefit from targeting this cytokine, the identification of patients with bioactive IL-17A would be of interest (17). Nonetheless, targeting IL-17 pathway could be part of new strategies to control vasculitis and methods to target it are firstly described. Then, approved biologics and on-going clinical trials are presented. Finally, other treatments to inhibit IL-17 are detailed.
4.1 Tools to Target the IL-17 Pathway
4.1.1 Direct Modulation of the IL-17 Pathway
Two antibodies are now available for targeting directly IL-17A (sekukinumab and ixekizumab) and constitute the more straightforward option. Recently, bi-specific antibody directed against both IL-17A and IL-17F (bimekizumab) was tested in psoriasis and may be more effective than secukinumab, which only inhibits IL-17A (160). These results were expected as IL-17A and IL-17F act synergistically (27). In the same vein, bispecific antibodies that block TNFα and IL-17A are currently developing. Finally, the targeting of IL-17RA with brodalumab and the inhibition of RORc constitute alternative strategies (6, 35).
4.1.2 Indirect Inhibition of the IL-17 Pathway
Th17 differentiation is a multi-step and a dynamic process. Cytokines required to Th17 differentiation include IL-1ß, IL-6 and IL-23. In addition to these cytokines, low dose of IL-2 allows a shift toward Treg cells at the expense of Th17 population (15, 161). Conversely, targeting of IL-6 receptor in GCA increases Treg population and reverts their pathogenic phenotype observed during active disease (38). Regarding these elements, the inhibition of a cytokine involved in these processes is a way to interfere with the Th17 pathway. Many biologics are available to target these cytokines but only the ones tested in vasculitis are described here (Figure 4). Statins and metformin, that are widely used, are also described, as in vitro inhibitors of the IL-17 pathway.
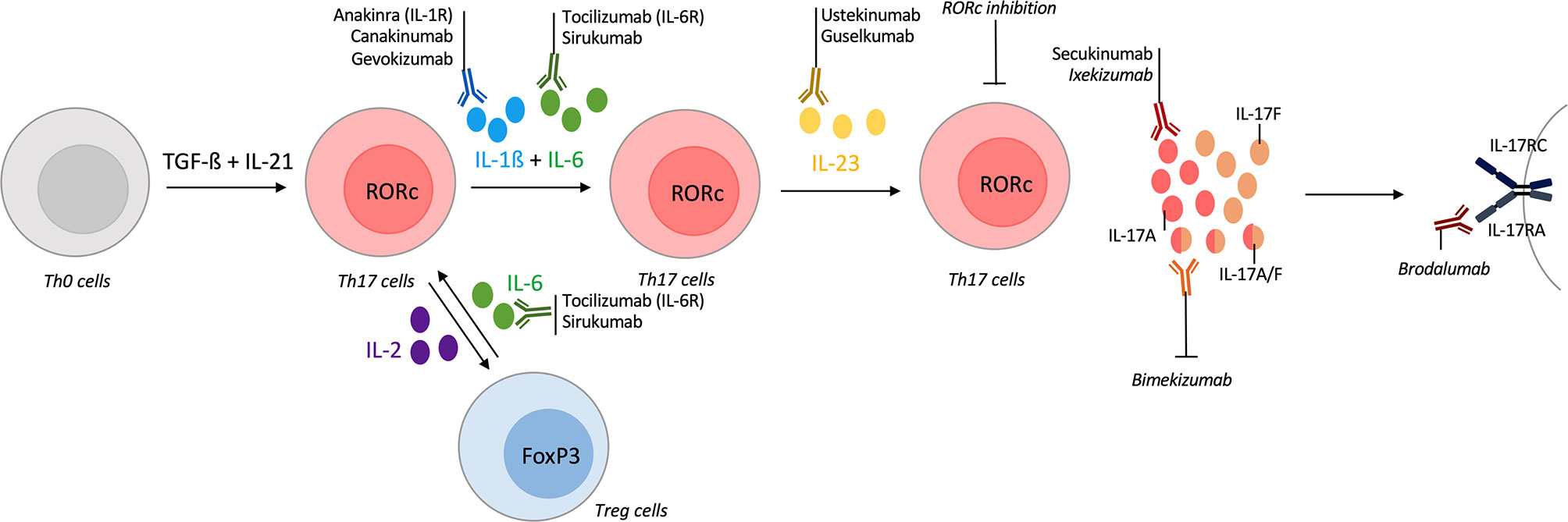
Figure 4 Tools for targeting the IL-17/Th17 pathway in vasculitis. T-helper (Th) 17 differentiation is a multi-step process with different cytokines involved. Their inhibitions could act on Th17 differentiation and modify the course of vasculitis. The first step of Th17 differentiation is mediated by interleukin (IL)-1ß and IL-6. Inhibitors of IL-1ß (canakinumab, gevokizumab) or IL-1 receptor antagonist (anakinra) are available and some were tested in vasculitis. Inhibitors of IL-6 (sirukumab) or of its receptor (tocilizumab) were approved in selected giant-cell arteritis (GCA) patients. IL-6 inhibition also acts on the Th17/regulatory T cells (Treg) dysregulation that occurs in vasculitis. Another way to modulate the Th17/Treg balance is to use low-dose of IL-2. Receptor-related orphan receptor (RORc) inhibition also suppresses Th17 lineage. Then, IL-23 is required for Th17 differentiation and ustekinumab is an inhibitor of the IL-12/IL-23 axis, guselkumab specifically targets IL-23. Promising results in Behçet disease (BD) are described. After their differentiation, Th17 cells produce IL-17A which could be inhibited by secukinumab or ixekizumab. Bispecific antibodies against IL-17A/IL-17F are currently developing with bimekizumab. brodalumab is an IL-17 receptor antagonist. All biologics targeting directly the IL-17 pathway are represented but secukinumab was the only one tested in vasculitis.
4.2 Approved Biologics in Vasculitis and On-Going Clinical Trials
Biologics that have been tested in vasculitis are described with the idea that the inhibition of the Th17 pathway could modulate disease’s activity. However, the cytokine itself that is inhibited could also play a role in the pathogenesis. The discussion below only provides some elements for thought.
4.2.1 Inhibition of IL-17
To date, very few trials have been conducted in vasculitis. A trial is on hold with secukinumab in patients naïve to biologics and with newly diagnosed or relapsing GCA (162).
Some results suggest that secukinumab could be efficient in BD as promising results come from patients with non-infectious uveitis (163). Trials to modify the Th17/Treg balance have been performed with low dose IL-2 administration and even more are on-going (NCT01988506, NCT04065672, NCT04387942) (161).
4.2.2 Inhibition of IL-6
The inhibition of IL-6 in vasculitis could have a therapeutic effect in different manner, notably in an IL-17 dependent fashion. First, IL-6 is required for Th17 differentiation. This cytokine also plays a role in the Th17/Treg imbalance and IL-17A up-regulates IL-6 expression through NFκB pathway (1, 15, 16).
Tocilizumab is an IL-6 receptor inhibitor that was firstly used in RA (164). As IL-6 pathway is absolutely central in GCA pathogenesis, its targeting through tocilizumab was firstly tested (75). Tolicizumab was shown to restore a better Treg function than glucocorticoids (37). Trials were conducted in GCA and allowed for reductions in steroid doses and maintain remission (165, 166). Recent EULAR guidelines recommend the use of Tocilizumab as adjunctive therapy in selected GCA patients (14). Sirukumab efficacy, an IL-6 production inhibitor, is currently testing in GCA (167).
Tocilizumab can be considered in relapsing or refractory TAK when conventional disease modifying anti-rheumatic drugs (DMARDs) or TNF blockers are not sufficient to control disease activity (14, 168–170). Apart from LVV, Tocilizumab was also tested in BD with promising results and clinical trials are on-going (NCT03554161) (171, 172). In AAV, some case reports have been published but larger clinical trial are required (173).
4.2.3 Inhibition of IL-1ß
IL-1ß is required for Th17 differentiation and enhances Th17 cell differentiation, as shown in BD (174). Different tools are available to block this pathway, either with IL-1 receptor antagonist (anakinra) or by targeting directly the cytokine itself (canakinumab, gevokizumab) (175). Clinical trials are on-going to test anakinra in addition to corticosteroids in GCA (NCT02902731). Anakinra is currently testing in KD (NCT02179853, NCT02390596). In BD, canakinumab (NCT02756650), anakinra and gevokizumab were tested and results suggest a mixed effect depending on clinical manifestations (176–181). Canakinumab was also tried in HUV and results are expected (NCT01170936).
4.2.4 Inhibition of IL-23
As mentioned above, IL-23 is required for Th17 differentiation (15). Different biologics were developed to target this cytokine with the example of ustekinumab, that targets p40 subunit and thus potentially inhibits both Th1 and Th17 pathways (182). Precisely, ustekinumab was tested in a GCA patient and inhibited Th1 and Th17 polarization (183). A recent trial showed disappointing results but they must be analyzed carefully for several reasons (uncontrolled trial, time of analysis) (184, 185). Other trials displayed promising results and this treatment is currently testing in relapse or refractory GCA (NCT03711448) (186). Among LVV, ustekinumab was also tested in three TAK patients with promising results (187–189). Recently, guselkumab, that specifically binds to the p19 subunit of IL-23, is currently tested in GCA (NCT04633447). Concerning VVV, ustekinumab was tried in BD; at week 12, 70% of patients fulfilled criteria for complete response. As compared to baseline values, IL-17A levels were significantly decreased at week 12 (190). These observations were subsequently confirmed but results from other trials are expected (NCT02648581) (191).
4.3 Other Treatments to Target the IL-17 Pathway
Outside biologics, some treatments currently used in cardiovascular prevention and in diabetes can target the IL-17 pathway. For instance, statins are shown to reduce the pro-inflammatory and pro-thrombotic effects of IL-17A and TNFα on EC (192).
Indirect evidence of metformin’s effect on the IL-17 pathway comes from cancer. Metformin is an agonist of Sirtuin-1 whose activation reduces Th17 frequency in patients (193). A Chinese trial tested metformin in thirty BD patients. In terms of clinical manifestations, the overall favorable response rate was almost 90% and partial remission was obtained in the rest of patients. Inflammatory parameters were lowered by the treatment. FoxP3 and TGF-ß protein levels were increased while IL-17 expression was lowered suggesting that metformin mediates the Th17/Treg imbalance (194).
Other treatments used in RA or in chronic inflammatory diseases were shown to decrease Th17 response, as glucocorticoids suppress Th17 responses in GCA patients (77). JAK inhibitors were tested in TAK patients with a response in two of three patients treated with a decrease in Th17 cells and an increase of Treg ones (70). Recently, tofacitinib (TOF), which preferentially inhibits JAK1 and JAK3, was compared with methotrexate (MTX) in TAK. TOF and MTX were associated with glucocorticoids, the study included 53 patients (26 in MTX group, 27 in TOF group) and showed the advantages of TOF regarding complete remission induction, the prevention of relapse and the tapering of steroid dose compared with MTX. No serious side effects were observed in the TOF group during 12 months of treatment but results must be confirmed in larger cohort with longer follow-up (195). Similarly, baricitinib, which inhibits JAK1/JAK2, has been tested in GCA. Fifteen relapsing GCA patients were enrolled in an open-label pilot study and preliminary results demonstrated evidence of both efficacy and safety. Discontinuation of glucocorticoids was allowed in the majority of patients with relapsing GCA but larger trials are needed to confirm these results (196). Finally, the inhibition of the complement pathway with avacopan (anti-C5a receptor) in AAV showed promising results but its effects on the Th17 pathway have to be characterized (197).
Overall, more clinical data are needed to conclude on the potential benefit to specifically target IL-17 in these diseases.
5 Conclusion
It is now well established that IL-17A is involved in autoimmunity and in chronic inflammation. This demonstration came from both in vitro and in vivo experiments and was confirmed by the efficacy of IL-17 inhibitors in various articular and cutaneous diseases. The effects of IL-17A on the CV system were described locally and then at a systemic level. Interacting in a complex network of cytokines, IL-17A promotes inflammation, thrombosis and coagulation. Regarding these effects, results suggest that this cytokine is involved in various diseases ranging from atherosclerosis to vasculitis. Vasculitis treatment faces challenges as steroids constitute the cornerstone of care. The indirect modulation of the Th17 pathway through different biologics has shown efficacy, particularly in LVV with tocilizumab which is now recommended in selected cases. As an extension, IL-17A could constitute an attractive target in inflammatory CV diseases if the cytokine is considered in its complex network. Disappointing expectations from the use of IL-17 inhibitors in RA and in multiple sclerosis must be considered before going further into their application in vasculitis where the Th17 pathway may act as a culprit and a consort. Given that, the thoughtful use of these drugs could be of great benefit to patients.
Author Contributions
MR: writing and figures. PM and AH: concept and proof reading. All authors contributed to the article and approved the submitted version.
Funding
MR is supported by the Ecole de l’Inserm Liliane Bettencourt Programme. PM is a senior member of the Institut Universitaire de France. His laboratory is supported in part by the IHU OPERA.
Conflict of Interest
PM holds a patent on the determination of bioactive IL-17A.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Miossec P, Kolls JK. Targeting IL-17 and TH17 Cells in Chronic Inflammation. Nat Rev Drug Discov (2012) 11:763–76. doi: 10.1038/nrd3794
2. Bilal J, Berlinberg A, Bhattacharjee S, Trost J, Riaz IB, Kurtzman DJB. A Systematic Review and Meta-Analysis of the Efficacy and Safety of the Interleukin (IL)-12/23 and IL-17 Inhibitors Ustekinumab, Secukinumab, Ixekizumab, Brodalumab, Guselkumab and Tildrakizumab for the Treatment of Moderate to Severe Plaque Psoriasis. J Dermatol Treat (2018) 29:569–78. doi: 10.1080/09546634.2017.1422591
3. Dubash S, Bridgewood C, McGonagle D, Marzo-Ortega H. The Advent of IL-17A Blockade in Ankylosing Spondylitis: Secukinumab, Ixekizumab and Beyond. Expert Rev Clin Immunol (2019) 15:123–34. doi: 10.1080/1744666x.2019.1561281
4. Beringer A, Miossec P. Systemic Effects of IL-17 in Inflammatory Arthritis. Nat Rev Rheumatol (2019) 15:491–501. doi: 10.1038/s41584-019-0243-5
5. Robert M, Hot A, Mifsud F, Ndongo-Thiam N, Miossec P. Joint Destruction Is Associated With All Types of Cardiovascular Events in French Rheumatoid Patients: A Real-Life Study With Very Long Follow-Up. Front Med (Lausanne) (2020) 7:556086. doi: 10.3389/fmed.2020.556086
6. Miossec P. Local and Systemic Effects of IL-17 in Joint Inflammation: A Historical Perspective From Discovery to Targeting. Cell Mol Immunol (2021) 18:860–5. doi: 10.1038/s41423-021-00644-5
7. Robert M, Miossec P. Effects of Interleukin 17 on the Cardiovascular System. Autoimmun Rev (2017) 16:984–91. doi: 10.1016/j.autrev.2017.07.009
8. Jennette JC, Falk RJ, Bacon PA, Basu N, Cid MC, Ferrario F, et al. 2012 Revised International Chapel Hill Consensus Conference Nomenclature of Vasculitides. Arthritis Rheumatol (2013) 65:1–11. doi: 10.1002/art.37715
9. Sunderkotter CH, Zelger B, Chen KR, Requena L, Piette W, Carlson JA, et al. Nomenclature of Cutaneous Vasculitis: Dermatologic Addendum to the 2012 Revised International Chapel Hill Consensus Conference Nomenclature of Vasculitides. Arthritis Rheumatol (2018) 70:171–84. doi: 10.1002/art.40375
10. Caplan A, Fett N, Rosenbach M, Werth VP, Micheletti RG. Prevention and Management of Glucocorticoid-Induced Side Effects: A Comprehensive Review: Ocular, Cardiovascular, Muscular, and Psychiatric Side Effects and Issues Unique to Pediatric Patients. J Am Acad Dermatol (2017) 76:201–7. doi: 10.1016/j.jaad.2016.02.1241
11. Caplan A, Fett N, Rosenbach M, Werth VP, Micheletti RG. Prevention and Management of Glucocorticoid-Induced Side Effects: A Comprehensive Review: Infectious Complications and Vaccination Recommendations. J Am Acad Dermatol (2017) 76:191–8. doi: 10.1016/j.jaad.2016.02.1240
12. Caplan A, Fett N, Rosenbach M, Werth VP, Micheletti RG. Prevention and Management of Glucocorticoid-Induced Side Effects: A Comprehensive Review: Gastrointestinal and Endocrinologic Side Effects. J Am Acad Dermatol (2017) 76:11–6. doi: 10.1016/j.jaad.2016.02.1239
13. Caplan A, Fett N, Rosenbach M, Werth VP, Micheletti RG. Prevention and Management of Glucocorticoid-Induced Side Effects: A Comprehensive Review: A Review of Glucocorticoid Pharmacology and Bone Health. J Am Acad Dermatol (2017) 76:1–9. doi: 10.1016/j.jaad.2016.01.062
14. Hellmich B, Agueda A, Monti S, Buttgereit F, de Boysson H, Brouwer E, et al. Update of the EULAR Recommendations for the Management of Large Vessel Vasculitis. Ann Rheum Dis (2018) 79:19–30. doi: 10.1136/annrheumdis-2019-215672
15. Noack M, Miossec P. Th17 and Regulatory T Cell Balance in Autoimmune and Inflammatory Diseases. Autoimmun Rev (2014) 13:668–77. doi: 10.1016/j.autrev.2013.12.004
16. Miossec P, Korn T, Kuchroo VK. Interleukin-17 and Type 17 Helper T Cells. N Engl J Med (2009) 361:888–98. doi: 10.1056/NEJMra0707449
17. Robert M, Miossec P. IL-17 in Rheumatoid Arthritis and Precision Medicine: From Synovitis Expression to Circulating Bioactive Levels. Front Med (Lausanne) (2018) 5:364. doi: 10.3389/fmed.2018.00364
18. Rouvier E, Luciani MF, Mattei MG, Denizot F, Golstein P. CTLA-8, Cloned From an Activated T Cell, Bearing AU-Rich Messenger RNA Instability Sequences, and Homologous to a Herpesvirus Saimiri Gene. J Immunol (1993) 150:5445–56.
19. Yao Z, Painter SL, Fanslow WC, Ulrich D, Macduff BM, Spriggs MK, et al. Human IL-17: A Novel Cytokine Derived From T Cells. J Immunol (1995) 155:5483–6.
20. Li H, Chen J, Huang A, Stinson J, Heldens S, Foster J, et al. Cloning and Characterization of IL-17B and IL-17C, Two New Members of the IL-17 Cytokine Family. Proc Natl Acad Sci USA (2000) 97:773–8. doi: 10.1073/pnas.97.2.773
21. Starnes T, Broxmeyer HE, Robertson MJ, Hromas R. Cutting Edge: IL-17D, a Novel Member of the IL-17 Family, Stimulates Cytokine Production and Inhibits Hemopoiesis. J Immunol (2002) 169:642–6. doi: 10.4049/jimmunol.169.2.642
22. Lee J, Ho WH, Maruoka M, Corpuz RT, Baldwin DT, Foster JS, et al. IL-17E, a Novel Proinflammatory Ligand for the IL-17 Receptor Homolog IL-17rh1. J Biol Chem (2001) 276:1660–4. doi: 10.1074/jbc.M008289200
23. Hymowitz SG, Filvaroff EH, Yin JP, Lee J, Cai L, Risser P, et al. IL-17s Adopt a Cystine Knot Fold: Structure and Activity of a Novel Cytokine, IL-17F, and Implications for Receptor Binding. EMBO J (2001) 20:5332–41. doi: 10.1093/emboj/20.19.5332
24. Wright JF, Guo Y, Quazi A, Luxenberg DP, Bennett F, Ross JF, et al. Identification of an Interleukin 17F/17A Heterodimer in Activated Human CD4+ T Cells. J Biol Chem (2007) 282:13447–55. doi: 10.1074/jbc.M700499200
25. Li X, Bechara R, Zhao J, McGeachy MJ, Gaffen SL. IL-17 Receptor-Based Signaling and Implications for Disease. Nat Immunol (2019) 20:1594–602. doi: 10.1038/s41590-019-0514-y
26. Zrioual S, Ecochard R, Tournadre A, Lenief V, Cazalis MA, Miossec P. Genome-Wide Comparison Between IL-17A- and IL-17F-Induced Effects in Human Rheumatoid Arthritis Synoviocytes. J Immunol (2009) 182:3112–20. doi: 10.4049/jimmunol.0801967
27. Hot A, Miossec P. Effects of Interleukin (IL)-17A and IL-17F in Human Rheumatoid Arthritis Synoviocytes. Ann Rheum Dis (2011) 70:727–32. doi: 10.1136/ard.2010.143768
28. Beringer A, Noack M, Miossec P. IL-17 in Chronic Inflammation: From Discovery to Targeting. Trends Mol Med (2016) 22:230–41. doi: 10.1016/j.molmed.2016.01.001
29. Hurst SD, Muchamuel T, Gorman DM, Gilbert JM, Clifford T, Kwan S, et al. New IL-17 Family Members Promote Th1 or Th2 Responses in the Lung: In Vivo Function of the Novel Cytokine IL-25. J Immunol (2002) 169:443–53. doi: 10.4049/jimmunol.169.1.443
30. Beale J, Jayaraman A, Jackson DJ, Macintyre JDR, Edwards MR, Walton RP, et al. Rhinovirus-Induced IL-25 in Asthma Exacerbation Drives Type 2 Immunity and Allergic Pulmonary Inflammation. Sci Transl Med (2014) 6:256ra134. doi: 10.1126/scitranslmed.3009124
31. Kleinschek MA, Owyang AM, Joyce-Shaikh B, Langrish CL, Chen Y, Gorman DM, et al. IL-25 Regulates Th17 Function in Autoimmune Inflammation. J Exp Med (2007) 204:161–70. doi: 10.1084/jem.20061738
32. Gaffen SL. Structure and Signalling in the IL-17 Receptor Family. Nat Rev Immunol (2009) 9:556–67. doi: 10.1038/nri2586
33. Lavocat F, Ndongo-Thiam N, Miossec P. Interleukin-25 Produced by Synoviocytes Has Anti-Inflammatory Effects by Acting As a Receptor Antagonist for Interleukin-17a Function. Front Immunol (2017) 8:647. doi: 10.3389/fimmu.2017.00647
34. Su Y, Huang J, Zhao X, Lu H, Wang W, Yang XO, et al. Interleukin-17 Receptor D Constitutes an Alternative Receptor for Interleukin-17A Important in Psoriasis-Like Skin Inflammation. Sci Immunol (2019) 4. doi: 10.1126/sciimmunol.aau9657
35. Robert M, Miossec P. Interleukin-17 and Lupus: Enough to be a Target? For Which Patients? Lupus (2020) 29:6–14. doi: 10.1177/0961203319891243
36. Joly AL, Liu S, Dahlberg CI, Mailer RK, Westerberg LS, Andersson J. Foxp3 Lacking Exons 2 and 7 Is Unable to Confer Suppressive Ability to Regulatory T Cells In Vivo. J Autoimmun (2015) 63:23–30. doi: 10.1016/j.jaut.2015.06.009
37. Samson M, Greigert H, Ciudad M, Gerard C, Ghesquière T, Trad M, et al. Improvement of Treg Immune Response After Treatment With Tocilizumab in Giant Cell Arteritis. Clin Transl Immunol (2021) 10:e1332. doi: 10.1002/cti2.1332
38. Miyabe C, Miyabe Y, Strle K, Kim ND, Stone JH, Luster AD, et al. An Expanded Population of Pathogenic Regulatory T Cells in Giant Cell Arteritis Is Abrogated by IL-6 Blockade Therapy. Ann Rheum Dis (2017) 76:898–905. doi: 10.1136/annrheumdis-2016-210070
39. Free ME, Bunch DO, McGregor JA, Jones BE, Berg EA, Hogan SL, et al. Patients With Antineutrophil Cytoplasmic Antibody-Associated Vasculitis Have Defective Treg Cell Function Exacerbated by the Presence of a Suppression-Resistant Effector Cell Population. Arthritis Rheumatol (2013) 65:1922–33. doi: 10.1002/art.37959
40. Crispin JC, Oukka M, Bayliss G, Cohen RA, Van Beek CA, Stillman IE, et al. Expanded Double Negative T Cells in Patients With Systemic Lupus Erythematosus Produce IL-17 and Infiltrate the Kidneys. J Immunol (2008) 181:8761–6. doi: 10.4049/jimmunol.181.12.8761
41. Lin AM, Rubin CJ, Khandpur R, Wang JY, Riblett M, Yalavarthi S, et al. Mast Cells and Neutrophils Release IL-17 Through Extracellular Trap Formation in Psoriasis. J Immunol (2011) 187:490–500. doi: 10.4049/jimmunol.1100123
42. Noordenbos T, Blijdorp I, Chen S, Stap J, Mul E, Canete JD, et al. Human Mast Cells Capture, Store, and Release Bioactive, Exogenous IL-17a. J Leukoc Biol (2016) 100:453–62. doi: 10.1189/jlb.3HI1215-542R
43. Fossiez F, Djossou O, Chomarat P, Flores-Romo L, Ait-Yahia S, Maat C, et al. T Cell Interleukin-17 Induces Stromal Cells to Produce Proinflammatory and Hematopoietic Cytokines. J Exp Med (1996) 183:2593–603. doi: 10.1084/jem.183.6.2593
44. Hot A, Lenief V, Miossec P. Combination of IL-17 and TNFalpha Induces a Pro-Inflammatory, Pro-Coagulant and Pro-Thrombotic Phenotype in Human Endothelial Cells. Ann Rheum Dis (2012) 71:768–76. doi: 10.1136/annrheumdis-2011-200468
45. Zhu F, Wang Q, Guo C, Wang X, Cao X, Shi Y, et al. IL-17 Induces Apoptosis of Vascular Endothelial Cells: A Potential Mechanism for Human Acute Coronary Syndrome. Clin Immunol (2011) 141:152–60. doi: 10.1016/j.clim.2011.07.003
46. Erbel C, Chen L, Bea F, Wangler S, Celik S, Lasitschka F, et al. Inhibition of IL-17A Attenuates Atherosclerotic Lesion Development in apoE-Deficient Mice. J Immunol (2009) 183:8167–75. doi: 10.4049/jimmunol.0901126
47. Shin JH, Shin DW, Noh M. Interleukin-17A Inhibits Adipocyte Differentiation in Human Mesenchymal Stem Cells and Regulates Pro-Inflammatory Responses in Adipocytes. Biochem Pharmacol (2009) 77:1835–44. doi: 10.1016/j.bcp.2009.03.008
48. Abrahimi P, Liu R, Pober JS. Blood Vessels in Allotransplantation. Am J Transplant (2015) 15:1748–54. doi: 10.1111/ajt.13242
49. Chadha R, Heidt S, Jones ND, Wood KJ. Th17: Contributors to Allograft Rejection and a Barrier to the Induction of Transplantation Tolerance? Transplantation (2011) 91:939–45. doi: 10.1097/TP.0b013e3182126eeb
50. Antonysamy MA, Fanslow WC, Fu F, Li W, Qian S, Troutt AB, et al. Evidence for a Role of IL-17 in Organ Allograft Rejection: IL-17 Promotes the Functional Differentiation of Dendritic Cell Progenitors. J Immunol (1999) 162:577–84.
51. Tang JL, Subbotin VM, Antonysamy MA, Troutt AB, Rao AS, Thomson AW. Interleukin-17 Antagonism Inhibits Acute But Not Chronic Vascular Rejection. Transplantation (2001) 72:348–50. doi: 10.1097/00007890-200107270-00035
52. Liu Z, Fan H, Jiang S. CD4(+) T-Cell Subsets in Transplantation. Immunol Rev (2013) 252:183–91. doi: 10.1111/imr.12038
53. Libby P, Pober JS. Chronic Rejection. Immunity (2001) 14:387–97. doi: 10.1016/s1074-7613(01)00119-4
54. Yuan X, Paez-Cortez J, Schmitt-Knosalla I, D'Addio F, Mfarrej B, Donnarumma M, et al. A Novel Role of CD4 Th17 Cells in Mediating Cardiac Allograft Rejection and Vasculopathy. J Exp Med (2008) 205:3133–44. doi: 10.1084/jem.20081937
55. Shi X, Zhang M, Liu F, Wang Z, Zhang L, Cheng H, et al. Tim-1-Fc Suppresses Chronic Cardiac Allograft Rejection and Vasculopathy by Reducing IL-17 Production. Int J Clin Exp Pathol (2014) 7:509–20.
56. Faust SM, Lu G, Marini BL, Zou W, Gordon D, Iwakura Y, et al. Role of T Cell TGFbeta Signaling and IL-17 in Allograft Acceptance and Fibrosis Associated With Chronic Rejection. J Immunol (2009) 183:7297–306. doi: 10.4049/jimmunol.0902446
57. Itoh S, Nakae S, Axtell RC, Velotta JB, Kimura N, Kajiwara N, et al. IL-17 Contributes to the Development of Chronic Rejection in a Murine Heart Transplant Model. J Clin Immunol (2010) 30:235–40. doi: 10.1007/s10875-009-9366-9
58. Ndongo-Thiam N, Miossec P. A Cell-Based Bioassay for Circulating Bioactive IL-17: Application to Destruction in Rheumatoid Arthritis. Ann Rheum Dis (2015) 74:1629–31. doi: 10.1136/annrheumdis-2014-207110
59. Robert M, Hot A, Mifsud F, Ndongo-Thiam N, Miossec P. Synergistic Interaction Between High Bioactive IL-17A and Joint Destruction for the Occurrence of Cardiovascular Events in Rheumatoid Arthritis. Front Immunol (2020) 11:1998. doi: 10.3389/fimmu.2020.01998
60. Bochaton T, Mewton N, Thiam N, Lavocat F, Baetz D, Dufay N, et al. Early Kinetics of Serum Interleukine-17A and Infarct Size in Patients With Reperfused Acute ST-Elevated Myocardial Infarction. PloS One (2017) 12:e0188202. doi: 10.1371/journal.pone.0188202
61. Harky A, Fok M, Balmforth D, Bashir M. Pathogenesis of Large Vessel Vasculitis: Implications for Disease Classification and Future Therapies. Vasc Med (2019) 24:79–88. doi: 10.1177/1358863X18802989
62. Weyand CM, Goronzy JJ. Medium- and Large-Vessel Vasculitis. N Engl J Med (2003) 349:160–9. doi: 10.1056/NEJMra022694
63. Weyand CM, Goronzy JJ. Immune Mechanisms in Medium and Large-Vessel Vasculitis. Nat Rev Rheumatol (2013) 9:731–40. doi: 10.1038/nrrheum.2013.161
64. Mason JC. Takayasu Arteritis–Advances in Diagnosis and Management. Nat Rev Rheumatol (2010) 6:406–15. doi: 10.1038/nrrheum.2010.82
65. Misra DP, Chaurasia S, Misra R. Increased Circulating Th17 Cells, Serum IL-17A, and IL-23 in Takayasu Arteritis. Autoimmune Dis (2016) 2016:7841718. doi: 10.1155/2016/7841718
66. Saadoun D, Garrido M, Comarmond C, Desbois AC, Domont F, Savey L, et al. Th1 and Th17 Cytokines Drive Inflammation in Takayasu Arteritis. Arthritis Rheumatol (2015) 67:1353–60. doi: 10.1002/art.39037
67. Matsumoto K, Suzuki K, Yoshimoto K, Seki N, Tsujimoto H, Chiba K, et al. Significant Association Between Clinical Characteristics and Changes in Peripheral Immuno-Phenotype in Large Vessel Vasculitis. Arthritis Res Ther (2019) 21:304. doi: 10.1186/s13075-019-2068-7
68. Savioli B, Abdulahad WH, Brouwer E, Kallenberg CGM, de Souza AWS. Are Cytokines and Chemokines Suitable Biomarkers for Takayasu Arteritis? Autoimmun Rev (2017) 16:1071–8. doi: 10.1016/j.autrev.2017.07.023
69. Mutoh T, Shirai T, Ishii T, Shirota Y, Fujishima F, Takahashi F, et al. Identification of Two Major Autoantigens Negatively Regulating Endothelial Activation in Takayasu Arteritis. Nat Commun (2020) 11:1253. doi: 10.1038/s41467-020-15088-0
70. Regnier P, Le Joncour A, Maciejewski-Duval A, Desbois AC, Comarmond C, Rosenzwajg M, et al. Targeting JAK/STAT Pathway in Takayasu's Arteritis. Ann Rheum Dis (2020) 79:951–9. doi: 10.1136/annrheumdis-2019-216900
71. Zhang J, Zhao L, Wang J, Cheng Z, Sun M, Zhao J, et al. Targeting Mechanistic Target of Rapamycin Complex 1 Restricts Proinflammatory T Cell Differentiation and Ameliorates Takayasu Arteritis. Arthritis Rheumatol (2020) 72:303–15. doi: 10.1002/art.41084
72. Singh K, Rathore U, Rai MK, Behera MR, Jain N, Ora M, et al. Novel Th17 Lymphocyte Populations, Th17.1 and PD1+Th17, Are Increased in Takayasu Arteritis, and Both Th17 and Th17.1 Sub-Populations Associate With Active Disease. J Inflamm Res (2022) 15:1521–41. doi: 10.2147/jir.S355881
73. Arnaud L, Haroche J, Mathian A, Gorochov G, Amoura Z. Pathogenesis of Takayasu's Arteritis: A 2011 Update. Autoimmun Rev (2011) 11:61–7. doi: 10.1016/j.autrev.2011.08.001
74. Ly KH, Regent A, Tamby MC, Mouthon L. Pathogenesis of Giant Cell Arteritis: More Than Just an Inflammatory Condition? Autoimmun Rev (2010) 9:635–45. doi: 10.1016/j.autrev.2010.05.002
75. Samson M, Corbera-Bellalta M, Audia S, Planas-Rigol E, Martin L, Cid MC, et al. Recent Advances in Our Understanding of Giant Cell Arteritis Pathogenesis. Autoimmun Rev (2017) 16:833–44. doi: 10.1016/j.autrev.2017.05.014
76. Samson M, Audia S, Martin L, Janikashvili N, Bonnotte B. Pathogenesis of Giant Cell Arteritis: New Insight Into the Implication of CD161+ T Cells. Clin Exp Rheumatol (2013) 31:S65–73.
77. Deng J, Younge BR, Olshen RA, Goronzy JJ, Weyand CM. Th17 and Th1 T-Cell Responses in Giant Cell Arteritis. Circulation (2010) 121:906–15. doi: 10.1161/CIRCULATIONAHA.109.872903
78. Samson M, Audia S, Fraszczak J, Trad M, Ornetti P, Lakomy D, et al. Th1 and Th17 Lymphocytes Expressing CD161 Are Implicated in Giant Cell Arteritis and Polymyalgia Rheumatica Pathogenesis. Arthritis Rheumatol (2012) 64:3788–98. doi: 10.1002/art.34647
79. Terrier B, Geri G, Chaara W, Allenbach Y, Rosenzwajg M, Costedoat-Chalumeau N, et al. Interleukin-21 Modulates Th1 and Th17 Responses in Giant Cell Arteritis. Arthritis Rheumatol (2012) 64:2001–11. doi: 10.1002/art.34327
80. Zhang H, Watanabe R, Berry GJ, Tian L, Goronzy JJ, Weyand CM. Inhibition of JAK-STAT Signaling Suppresses Pathogenic Immune Responses in Medium and Large Vessel Vasculitis. Circulation (2018) 137:1934–48. doi: 10.1161/circulationaha.117.030423
81. Vieira M, Régnier P, Maciejewski-Duval A, Le Joncour A, Darasse-Jèze G, Rosenzwajg M, et al. Interferon Signature in Giant Cell Arteritis Aortitis. J Autoimmun (2022) 127:102796. doi: 10.1016/j.jaut.2022.102796
82. Wen Z, Shen Y, Berry G, Shahram F, Li Y, Watanabe R, et al. The Microvascular Niche Instructs T Cells in Large Vessel Vasculitis via the VEGF-Jagged1-Notch Pathway. Sci Transl Med (2017) 9. doi: 10.1126/scitranslmed.aal3322
83. Roche NE, Fulbright JW, Wagner AD, Hunder GG, Goronzy JJ, Weyand CM. Correlation of Interleukin-6 Production and Disease Activity in Polymyalgia Rheumatica and Giant Cell Arteritis. Arthritis Rheumatol (1993) 36:1286–94. doi: 10.1002/art.1780360913
84. Ciccia F, Rizzo A, Guggino G, Cavazza A, Alessandro R, Maugeri R, et al. Difference in the Expression of IL-9 and IL-17 Correlates With Different Histological Pattern of Vascular Wall Injury in Giant Cell Arteritis. Rheumatol (Oxford) (2015) 54:1596–604. doi: 10.1093/rheumatology/kev102
85. Espigol-Frigole G, Corbera-Bellalta M, Planas-Rigol E, Lozano E, Segarra M, Garcia-Martinez A, et al. Increased IL-17A Expression in Temporal Artery Lesions Is a Predictor of Sustained Response to Glucocorticoid Treatment in Patients With Giant-Cell Arteritis. Ann Rheum Dis (2013) 72:1481–7. doi: 10.1136/annrheumdis-2012-201836
86. Marquez A, Hernandez-Rodriguez J, Cid MC, Solans R, Castaneda S, Fernandez-Contreras ME, et al. Influence of the IL17A Locus in Giant Cell Arteritis Susceptibility. Ann Rheum Dis (2014) 73:1742–5. doi: 10.1136/annrheumdis-2014-205261
87. Coit P, De Lott LB, Nan B, Elner VM, Sawalha AH. DNA Methylation Analysis of the Temporal Artery Microenvironment in Giant Cell Arteritis. Ann Rheum Dis (2016) 75:1196–202. doi: 10.1136/annrheumdis-2014-207116
88. Guillevin L, Lhote F. Polyarteritis Nodosa and Microscopic Polyangiitis. Clin Exp Immunol (1995) 101(Suppl 1):22–3. doi: 10.1111/j.1365-2249.1995.tb06157.x
89. Shimojima Y, Ishii W, Kishida D, Fukushima K, Ikeda SI. Imbalanced Expression of Dysfunctional Regulatory T Cells and T-Helper Cells Relates to Immunopathogenesis in Polyarteritis Nodosa. Mod Rheumatol (2017) 27:102–9. doi: 10.3109/14397595.2016.1172999
90. Okano T, Takeuchi S, Soma Y, Suzuki K, Tsukita S, Ishizu A, et al. Presence of Anti-Phosphatidylserine-Prothrombin Complex Antibodies and Anti-Moesin Antibodies in Patients With Polyarteritis Nodosa. J Dermatol (2017) 44:18–22. doi: 10.1111/1346-8138.13491
91. McCrindle BW, Rowley AH, Newburger JW, Burns JC, Bolger AF, Gewitz M, et al. Diagnosis, Treatment, and Long-Term Management of Kawasaki Disease: A Scientific Statement for Health Professionals From the American Heart Association. Circulation (2017) 135:e927–99. doi: 10.1161/CIR.0000000000000484
92. Shulman ST, Rowley AH. Kawasaki Disease: Insights Into Pathogenesis and Approaches to Treatment. Nat Rev Rheumatol (2015) 11:475–82. doi: 10.1038/nrrheum.2015.54
93. Jia S, Li C, Wang G, Yang J, Zu Y. The T Helper Type 17/Regulatory T Cell Imbalance in Patients With Acute Kawasaki Disease. Clin Exp Immunol (2010) 162:131–7. doi: 10.1111/j.1365-2249.2010.04236.x
94. Rasouli M, Heidari B, Kalani M. Downregulation of Th17 Cells and the Related Cytokines With Treatment in Kawasaki Disease. Immunol Lett (2014) 162:269–75. doi: 10.1016/j.imlet.2014.09.017
95. Guo MM, Tseng WN, Ko CH, Pan HM, Hsieh KS, Kuo HC. Th17- and Treg-Related Cytokine and mRNA Expression Are Associated With Acute and Resolving Kawasaki Disease. Allergy (2015) 70:310–8. doi: 10.1111/all.12558
96. Sohn MH, Noh SY, Chang W, Shin KM, Kim DS. Circulating Interleukin 17 Is Increased in the Acute Stage of Kawasaki Disease. Scand J Rheumatol (2003) 32:364–6. doi: 10.1080/03009740410005034
97. Shimizu C, Oharaseki T, Takahashi K, Kottek A, Franco A, Burns JC. The Role of TGF-Beta and Myofibroblasts in the Arteritis of Kawasaki Disease. Hum Pathol (2013) 44:189–98. doi: 10.1016/j.humpath.2012.05.004
98. Furuno K, Yuge T, Kusuhara K, Takada H, Nishio H, Khajoee V, et al. CD25+CD4+ Regulatory T Cells in Patients With Kawasaki Disease. J Pediatr (2004) 145:385–90. doi: 10.1016/j.jpeds.2004.05.048
99. Fujimaru T, Ito S, Masuda H, Oana S, Kamei K, Ishiguro A, et al. Decreased Levels of Inflammatory Cytokines in Immunoglobulin-Resistant Kawasaki Disease After Plasma Exchange. Cytokine (2014) 70:156–60. doi: 10.1016/j.cyto.2014.07.003
100. Nogueira E, Hamour S, Sawant D, Henderson S, Mansfield N, Chavele KM, et al. Serum IL-17 and IL-23 Levels and Autoantigen-Specific Th17 Cells Are Elevated in Patients With ANCA-Associated Vasculitis. Nephrol Dial Transplant (2010) 25:2209–17. doi: 10.1093/ndt/gfp783
101. Krebs CF, Paust HJ, Krohn S, Koyro T, Brix SR, Riedel JH, et al. Autoimmune Renal Disease Is Exacerbated by S1P-Receptor-1-Dependent Intestinal Th17 Cell Migration to the Kidney. Immunity (2016) 45:1078–92. doi: 10.1016/j.immuni.2016.10.020
102. Krebs CF, Schmidt T, Riedel JH, Panzer U. T Helper Type 17 Cells in Immune-Mediated Glomerular Disease. Nat Rev Nephrol (2017) 13:647–59. doi: 10.1038/nrneph.2017.112
103. Abdulahad WH, Lamprecht P, Kallenberg CG. T-Helper Cells as New Players in ANCA-Associated Vasculitides. Arthritis Res Ther (2011) 13:236. doi: 10.1186/ar3362
104. Wang Y, Zhang S, Zhang N, Feng M, Liang Z, Zhao X, et al. Reduced Activated Regulatory T Cells and Imbalance of Th17/activated Treg Cells Marks Renal Involvement in ANCA-Associated Vasculitis. Mol Immunol (2020) 118:19–29. doi: 10.1016/j.molimm.2019.11.010
105. Krohn S, Nies JF, Kapffer S, Schmidt T, Riedel JH, Kaffke A, et al. IL-17c/IL-17 Receptor E Signaling in CD4(+) T Cells Promotes TH17 Cell-Driven Glomerular Inflammation. J Am Soc Nephrol (2018) 29:1210–22. doi: 10.1681/ASN.2017090949
106. Villiger PM, Guillevin L. Microscopic Polyangiitis: Clinical Presentation. Autoimmun Rev (2010) 9:812–9. doi: 10.1016/j.autrev.2010.07.009
107. Kallenberg CG. Key Advances in the Clinical Approach to ANCA-Associated Vasculitis. Nat Rev Rheumatol (2014) 10:484–93. doi: 10.1038/nrrheum.2014.104
108. Maruyama H, Hirayama K, Nagai M, Ebihara I, Shimohata H, Kobayashi M. Serum Decoy Receptor 3 Levels Are Associated With the Disease Activity of MPO-ANCA-Associated Renal Vasculitis. Clin Rheumatol (2016) 35:2469–76. doi: 10.1007/s10067-016-3321-y
109. O'Sullivan KM, Lo CY, Summers SA, Elgass KD, McMillan PJ, Longano A, et al. Renal Participation of Myeloperoxidase in Antineutrophil Cytoplasmic Antibody (ANCA)-Associated Glomerulonephritis. Kidney Int (2015) 88:1030–46. doi: 10.1038/ki.2015.202
110. Gan PY, Steinmetz OM, Tan DS, O'Sullivan KM, Ooi JD, Iwakura Y, et al. Th17 Cells Promote Autoimmune Anti-Myeloperoxidase Glomerulonephritis. J Am Soc Nephrol (2010) 21:925–31. doi: 10.1681/ASN.2009070763
111. Summers SA, Steinmetz OM, Gan PY, Ooi JD, Odobasic D, Kitching AR, et al. Toll-Like Receptor 2 Induces Th17 Myeloperoxidase Autoimmunity While Toll-Like Receptor 9 Drives Th1 Autoimmunity in Murine Vasculitis. Arthritis Rheumatol (2011) 63:1124–35. doi: 10.1002/art.30208
112. Gan PY, Chan A, Ooi JD, Dick J, Nagai K, O'Sullivan KM, et al. Biologicals Targeting T Helper Cell Subset Differentiating Cytokines Are Effective in the Treatment of Murine Anti-Myeloperoxidase Glomerulonephritis. Kidney Int (2019) 96:1121–33. doi: 10.1016/j.kint.2019.05.012
113. Chang J, Eggenhuizen P, O'Sullivan KM, Alikhan MA, Holdsworth SR, Ooi JD, et al. CD8+ T Cells Effect Glomerular Injury in Experimental Anti-Myeloperoxidase Gn. J Am Soc Nephrol (2017) 28:47–55. doi: 10.1681/ASN.2015121356
114. Comarmond C, Cacoub P. Granulomatosis With Polyangiitis (Wegener): Clinical Aspects and Treatment. Autoimmun Rev (2014) 13:1121–5. doi: 10.1016/j.autrev.2014.08.017
115. Wilde B, Thewissen M, Damoiseaux J, Hilhorst M, van Paassen P, Witzke O, et al. Th17 Expansion in Granulomatosis With Polyangiitis (Wegener's): The Role of Disease Activity, Immune Regulation and Therapy. Arthritis Res Ther (2012) 14:R227. doi: 10.1186/ar4066
116. Szczeklik W, Jakiela B, Wawrzycka-Adamczyk K, Sanak M, Hubalewska-Mazgaj M, Padjas A, et al. Skewing Toward Treg and Th2 Responses Is a Characteristic Feature of Sustained Remission in ANCA-Positive Granulomatosis With Polyangiitis. Eur J Immunol (2017) 47:724–33. doi: 10.1002/eji.201646810
117. Isa H, Luthert P, Rose G, Verity D, Pusey C, Tomkins-Netzer O, et al. Tissue Interleukin-17 and Interleukin-23 as Biomarkers for Orbital Granulomatosis With Polyangiitis. Ophthalmology (2015) 122:2140–2. doi: 10.1016/j.ophtha.2015.04.016
118. Schreiber A, Choi M. The Role of Neutrophils in Causing Antineutrophil Cytoplasmic Autoantibody-Associated Vasculitis. Curr Opin Hematol (2015) 22:60–6. doi: 10.1097/MOH.0000000000000098
119. Abdulahad WH, Stegeman CA, Limburg PC, Kallenberg CG. Skewed Distribution of Th17 Lymphocytes in Patients With Wegener's Granulomatosis in Remission. Arthritis Rheumatol (2008) 58:2196–205. doi: 10.1002/art.23557
120. Rani L, Minz RW, Sharma A, Anand S, Gupta D, Panda NK, et al. Predominance of PR3 Specific Immune Response and Skewed TH17 vs. T-Regulatory Milieu in Active Granulomatosis With Polyangiitis. Cytokine (2015) 71:261–7. doi: 10.1016/j.cyto.2014.10.005
121. Cosmi L. Th17 and Treg Lymphocytes as Cellular Biomarkers of Disease Activity in Granulomatosis With Polyangiitis. Eur J Immunol (2017) 47:633–6. doi: 10.1002/eji.201746986
122. Abdulahad WH, Stegeman CA, van der Geld YM, Doornbos-van der Meer B, Limburg PC, Kallenberg CG. Functional Defect of Circulating Regulatory CD4+ T Cells in Patients With Wegener's Granulomatosis in Remission. Arthritis Rheumatol (2007) 56:2080–91. doi: 10.1002/art.22692
123. Morgan MD, Day CJ, Piper KP, Khan N, Harper L, Moss PA, et al. Patients With Wegener's Granulomatosis Demonstrate a Relative Deficiency and Functional Impairment of T-Regulatory Cells. Immunology (2010) 130:64–73. doi: 10.1111/j.1365-2567.2009.03213.x
124. Reijnders TDY, Stegeman CA, Huitema MG, Rutgers A, Heeringa P, Abdulahad WH. Unraveling the Identity of FoxP3+ Regulatory T Cells in Granulomatosis With Polyangiitis Patients. Sci Rep (2019) 9:8273. doi: 10.1038/s41598-019-44636-y
125. von Borstel A, Lintermans LL, Heeringa P, Rutgers A, Stegeman CA, Sanders JS, et al. Circulating CD24hiCD38hi Regulatory B Cells Correlate Inversely With the ThEM17 Cell Frequency in Granulomatosis With Polyangiitis Patients. Rheumatol (Oxford) (2019). doi: 10.1093/rheumatology/key412
126. Wu EY, Hernandez ML, Jennette JC, Falk RJ. Eosinophilic Granulomatosis With Polyangiitis: Clinical Pathology Conference and Review. J Allergy Clin Immunol Pract (2018) 6:1496–504. doi: 10.1016/j.jaip.2018.07.001
127. Terrier B, Bieche I, Maisonobe T, Laurendeau I, Rosenzwajg M, Kahn JE, et al. Interleukin-25: A Cytokine Linking Eosinophils and Adaptive Immunity in Churg-Strauss Syndrome. Blood (2010) 116:4523–31. doi: 10.1182/blood-2010-02-267542
128. Saito H, Tsurikisawa N, Tsuburai T, Oshikata C, Akiyama K. Cytokine Production Profile of CD4+ T Cells From Patients With Active Churg-Strauss Syndrome Tends Toward Th17. Int Arch Allergy Immunol (2009) 149 Suppl 1:61–5. doi: 10.1159/000210656
129. Tsurikisawa N, Oshikata C, Tsuburai T, Sugano S, Nakamura Y, Shimoda T, et al. Th17 Cells Reflect Colon Submucosal Pathologic Changes in Active Eosinophilic Granulomatosis With Polyangiitis. BMC Immunol (2015) 16:75. doi: 10.1186/s12865-015-0138-4
130. Saito H, Tsurikisawa N, Tsuburai T, Akiyama K. Involvement of Regulatory T Cells in the Pathogenesis of Churg-Strauss Syndrome. Int Arch Allergy Immunol (2008) 146 Suppl 1:73–6. doi: 10.1159/000126065
131. Cui Z, Zhao MH. Advances in Human Antiglomerular Basement Membrane Disease. Nat Rev Nephrol (2011) 7:697–705. doi: 10.1038/nrneph.2011.89
132. Summers SA, Steinmetz OM, Li M, Kausman JY, Semple T, Edgtton KL, et al. Th1 and Th17 Cells Induce Proliferative Glomerulonephritis. J Am Soc Nephrol (2009) 20:2518–24. doi: 10.1681/ASN.2009030337
133. Disteldorf EM, Krebs CF, Paust HJ, Turner JE, Nouailles G, Tittel A, et al. CXCL5 Drives Neutrophil Recruitment in TH17-Mediated GN. J Am Soc Nephrol (2015) 26:55–66. doi: 10.1681/ASN.2013101061
134. Krebs CF, Panzer U. Plasticity and Heterogeneity of Th17 in Immune-Mediated Kidney Diseases. J Autoimmun (2018) 87:61–8. doi: 10.1016/j.jaut.2017.12.005
135. Tulone C, Giorgini A, Freeley S, Coughlan A, Robson MG. Transferred Antigen-Specific T(H)17 But Not T(H)1 Cells Induce Crescentic Glomerulonephritis in Mice. Am J Pathol (2011) 179:2683–90. doi: 10.1016/j.ajpath.2011.08.017
136. Hunemorder S, Treder J, Ahrens S, Schumacher V, Paust HJ, Menter T, et al. TH1 and TH17 Cells Promote Crescent Formation in Experimental Autoimmune Glomerulonephritis. J Pathol (2015) 237:62–71. doi: 10.1002/path.4559
137. Lee H, Lee JW, Yoo KD, Yoo JY, Lee JP, Kim DK, et al. Cln 3-Requiring 9 Is a Negative Regulator of Th17 Pathway-Driven Inflammation in Anti-Glomerular Basement Membrane Glomerulonephritis. Am J Physiol Renal Physiol (2016) 311:F505–19. doi: 10.1152/ajprenal.00533.2015
138. Zhang Q, Luan H, Wang L, He F, Zhou H, Xu X, et al. Galectin-9 Ameliorates Anti-GBM Glomerulonephritis by Inhibiting Th1 and Th17 Immune Responses in Mice. Am J Physiol Renal Physiol (2014) 306:F822–32. doi: 10.1152/ajprenal.00294.2013
139. Yang C, Huang XR, Fung E, Liu HF, Lan HY. The Regulatory T-Cell Transcription Factor Foxp3 Protects Against Crescentic Glomerulonephritis. Sci Rep (2017) 7:1481. doi: 10.1038/s41598-017-01515-8
140. Antonelli A, Ferri C, Ferrari SM, Ghiri E, Goglia F, Pampana A, et al. Serum Levels of Proinflammatory Cytokines Interleukin-1beta, Interleukin-6, and Tumor Necrosis Factor Alpha in Mixed Cryoglobulinemia. Arthritis Rheumatol (2009) 60:3841–7. doi: 10.1002/art.25003
141. Audemard-Verger A, Pillebout E, Guillevin L, Thervet E, Terrier B. IgA Vasculitis (Henoch-Shonlein Purpura) in Adults: Diagnostic and Therapeutic Aspects. Autoimmun Rev (2015) 14:579–85. doi: 10.1016/j.autrev.2015.02.003
142. Gulhan B, Orhan D, Kale G, Besbas N, Ozen S. Studying Cytokines of T Helper Cells in the Kidney Disease of IgA Vasculitis (Henoch-Schonlein Purpura). Pediatr Nephrol (2015) 30:1269–77. doi: 10.1007/s00467-015-3051-4
143. Lin FJ, Jiang GR, Shan JP, Zhu C, Zou J, Wu XR. Imbalance of Regulatory T Cells to Th17 Cells in IgA Nephropathy. Scand J Clin Lab Invest (2012) 72:221–9. doi: 10.3109/00365513.2011.652158
144. Li YY, Li CR, Wang GB, Yang J, Zu Y. Investigation of the Change in CD4(+) T Cell Subset in Children With Henoch-Schonlein Purpura. Rheumatol Int (2012) 32:3785–92. doi: 10.1007/s00296-011-2266-3
145. Chen O, Zhu XB, Ren H, Wang YB, Sun R. The Imbalance of Th17/Treg in Chinese Children With Henoch-Schonlein Purpura. Int Immunopharmacol (2013) 16:67–71. doi: 10.1016/j.intimp.2013.03.027
146. Li B, Ren Q, Ling J, Tao Z, Yang X, Li Y. The Change of Th17/Treg Cells and IL-10/IL-17 in Chinese Children With Henoch-Schonlein Purpura: A PRISMA-Compliant Meta-Analysis. Med (Baltimore) (2019) 98:e13991. doi: 10.1097/MD.0000000000013991
147. Wisnieski JJ, Baer AN, Christensen J, Cupps TR, Flagg DN, Jones JV, et al. Hypocomplementemic Urticarial Vasculitis Syndrome. Clinical and Serologic Findings in 18 Patients. Med (Baltimore) (1995) 74:24–41. doi: 10.1097/00005792-199501000-00003
148. Yazici H, Seyahi E, Hatemi G, Yazici Y. Behcet Syndrome: A Contemporary View. Nat Rev Rheumatol (2018) 14:119. doi: 10.1038/nrrheum.2018.3
149. Tong B, Liu X, Xiao J, Su G. Immunopathogenesis of Behcet's Disease. Front Immunol (2019) 10:665. doi: 10.3389/fimmu.2019.00665
150. Ahmadi M, Yousefi M, Abbaspour-Aghdam S, Dolati S, Aghebati-Maleki L, Eghbal-Fard S, et al. Disturbed Th17/Treg Balance, Cytokines, and miRNAs in Peripheral Blood of Patients With Behcet's Disease. J Cell Physiol (2019) 234:3985–94. doi: 10.1002/jcp.27207
151. Sonmez C, Yucel AA, Yesil TH, Kucuk H, Sezgin B, Mercan R, et al. Correlation Between IL-17a/F, IL-23, IL-35 and IL-12/-23 (P40) Levels in Peripheral Blood Lymphocyte Cultures and Disease Activity in Behcet's Patients. Clin Rheumatol (2018) 37:2797–804. doi: 10.1007/s10067-018-4049-7
152. Touzot M, Cacoub P, Bodaghi B, Soumelis V, Saadoun D. IFN-Alpha Induces IL-10 Production and Tilt the Balance Between Th1 and Th17 in Behcet Disease. Autoimmun Rev (2015) 14:370–5. doi: 10.1016/j.autrev.2014.12.009
153. Filleron A, Tran TA, Hubert A, Letierce A, Churlaud G, Koné-Paut I, et al. Regulatory T Cell/Th17 Balance in the Pathogenesis of Pediatric Behçet Disease. Rheumatol (Oxford) (2021) 61:422–9. doi: 10.1093/rheumatology/keab253
154. Ye Z, Deng B, Wang C, Zhang D, Kijlstra A, Yang P. Decreased B and T Lymphocyte Attenuator in Behcet's Disease may Trigger Abnormal Th17 and Th1 Immune Responses. Sci Rep (2016) 6:20401. doi: 10.1038/srep20401
155. Lucherini OM, Lopalco G, Cantarini L, Emmi G, Lopalco A, Venerito V, et al. Critical Regulation of Th17 Cell Differentiation by Serum Amyloid-A Signalling in Behcet's Disease. Immunol Lett (2018) 201:38–44. doi: 10.1016/j.imlet.2018.10.013
156. Nanke Y, Yago T, Kotake S. The Role of Th17 Cells in the Pathogenesis of Behcet's Disease. J Clin Med (2017) 6. doi: 10.3390/jcm6070074
157. Deniz R, Tulunay-Virlan A, Ture Ozdemir F, Unal AU, Ozen G, Alibaz-Oner F, et al. Th17-Inducing Conditions Lead to In Vitro Activation of Both Th17 and Th1 Responses in Behcet's Disease. Immunol Invest (2017) 46:518–25. doi: 10.1080/08820139.2017.1306865
158. Leccese P, Alpsoy E. Behcet's Disease: An Overview of Etiopathogenesis. Front Immunol (2019) 10:1067. doi: 10.3389/fimmu.2019.01067
159. Ruiz de Morales JMG, Puig L, Dauden E, Canete JD, Pablos JL, Martin AO, et al. Critical Role of Interleukin (IL)-17 in Inflammatory and Immune Disorders: An Updated Review of the Evidence Focusing in Controversies. Autoimmun Rev (2020) 19:102429. doi: 10.1016/j.autrev.2019.102429
160. Reich K, Warren RB, Lebwohl M, Gooderham M, Strober B, Langley RG, et al. Bimekizumab Versus Secukinumab in Plaque Psoriasis. N Engl J Med (2021) 385:142–52. doi: 10.1056/NEJMoa2102383
161. Rosenzwajg M, Lorenzon R, Cacoub P, Pham HP, Pitoiset F, El Soufi K, et al. Immunological and Clinical Effects of Low-Dose Interleukin-2 Across 11 Autoimmune Diseases in a Single, Open Clinical Trial. Ann Rheum Dis (2019) 78:209–17. doi: 10.1136/annrheumdis-2018-214229
162. Venhoff N, Schmidt WA, Lamprecht P, Tony HP, App C, Sieder C, et al. Efficacy and Safety of Secukinumab in Patients With Giant Cell Arteritis: Study Protocol for a Randomized, Parallel Group, Double-Blind, Placebo-Controlled Phase II Trial. Trials (2021) 22:543. doi: 10.1186/s13063-021-05520-1
163. Letko E, Yeh S, Foster CS, Pleyer U, Brigell M, Grosskreutz CL, et al. Efficacy and Safety of Intravenous Secukinumab in Noninfectious Uveitis Requiring Steroid-Sparing Immunosuppressive Therapy. Ophthalmology (2015) 122:939–48. doi: 10.1016/j.ophtha.2014.12.033
164. Noack M, Miossec P. Selected Cytokine Pathways in Rheumatoid Arthritis. Semin Immunopathol (2017) 39:365–83. doi: 10.1007/s00281-017-0619-z
165. Stone JH, Tuckwell K, Dimonaco S, Klearman M, Aringer M, Blockmans D, et al. Trial of Tocilizumab in Giant-Cell Arteritis. N Engl J Med (2017) 377:317–28. doi: 10.1056/NEJMoa1613849
166. Villiger PM, Adler S, Kuchen S, Wermelinger F, Dan D, Fiege V, et al. Tocilizumab for Induction and Maintenance of Remission in Giant Cell Arteritis: A Phase 2, Randomised, Double-Blind, Placebo-Controlled Trial. Lancet (2016) 387:1921–7. doi: 10.1016/S0140-6736(16)00560-2
167. Schmidt WA, Dasgupta B, Luqmani R, Unizony SH, Blockmans D, Lai Z, et al. A Multicentre, Randomised, Double-Blind, Placebo-Controlled, Parallel-Group Study to Evaluate the Efficacy and Safety of Sirukumab in the Treatment of Giant Cell Arteritis. Rheumatol Ther (2020) 7:793–810. doi: 10.1007/s40744-020-00227-2
168. Mekinian A, Comarmond C, Resche-Rigon M, Mirault T, Kahn JE, Lambert M, et al. Efficacy of Biological-Targeted Treatments in Takayasu Arteritis: Multicenter, Retrospective Study of 49 Patients. Circulation (2015) 132:1693–700. doi: 10.1161/CIRCULATIONAHA.114.014321
169. Nakaoka Y, Isobe M, Takei S, Tanaka Y, Ishii T, Yokota S, et al. Efficacy and Safety of Tocilizumab in Patients With Refractory Takayasu Arteritis: Results From a Randomised, Double-Blind, Placebo-Controlled, Phase 3 Trial in Japan (the TAKT Study). Ann Rheum Dis (2018) 77:348–54. doi: 10.1136/annrheumdis-2017-211878
170. Mekinian A, Saadoun D, Vicaut E, Thietart S, Lioger B, Jego P, et al. Tocilizumab in Treatment-Naive Patients With Takayasu Arteritis: TOCITAKA French Prospective Multicenter Open-Labeled Trial. Arthritis Res Ther (2020) 22:218. doi: 10.1186/s13075-020-02311-y
171. Atienza-Mateo B, Calvo-Rio V, Beltran E, Martinez-Costa L, Valls-Pascual E, Hernandez-Garfella M, et al. Anti-Interleukin 6 Receptor Tocilizumab in Refractory Uveitis Associated With Behcet's Disease: Multicentre Retrospective Study. Rheumatol (Oxford) (2018) 57:856–64. doi: 10.1093/rheumatology/kex480
172. Ding Y, Li C, Liu J, Yu X, Wang Y, Shi J, et al. Tocilizumab in the Treatment of Severe and/or Refractory Vasculo-Behcet's Disease: A Single-Centre Experience in China. Rheumatol (Oxford) (2018) 57:2057–9. doi: 10.1093/rheumatology/key230
173. Sakai R, Kondo T, Kikuchi J, Shibata A, Chino K, Okuyama A, et al. Corticosteroid-Free Treatment of Tocilizumab Monotherapy for Microscopic Polyangiitis: A Single-Arm, Single-Center, Clinical Trial. Mod Rheumatol (2016) 26:900–7. doi: 10.3109/14397595.2016.1160968
174. Shimizu J, Takai K, Takada E, Fujiwara N, Arimitsu N, Ueda Y, et al. Possible Association of Proinflammatory Cytokines Including IL1beta and TNFalpha With Enhanced Th17 Cell Differentiation in Patients With Behcet's Disease. Clin Rheumatol (2016) 35:1857–63. doi: 10.1007/s10067-015-2966-2
175. Gabay C, Lamacchia C, Palmer G. IL-1 Pathways in Inflammation and Human Diseases. Nat Rev Rheumatol (2010) 6:232–41. doi: 10.1038/nrrheum.2010.4
176. Grayson PC, Yazici Y, Merideth M, Sen HN, Davis M, Novakovich E, et al. Treatment of Mucocutaneous Manifestations in Behcet's Disease With Anakinra: A Pilot Open-Label Study. Arthritis Res Ther (2017) 19:69. doi: 10.1186/s13075-017-1222-3
177. Gul A, Tugal-Tutkun I, Dinarello CA, Reznikov L, Esen BA, Mirza A, et al. Interleukin-1beta-Regulating Antibody XOMA 052 (Gevokizumab) in the Treatment of Acute Exacerbations of Resistant Uveitis of Behcet's Disease: An Open-Label Pilot Study. Ann Rheum Dis (2012) 71:563–6. doi: 10.1136/annrheumdis-2011-155143
178. Cantarini L, Lopalco G, Caso F, Costa L, Iannone F, Lapadula G, et al. Effectiveness and Tuberculosis-Related Safety Profile of Interleukin-1 Blocking Agents in the Management of Behcet's Disease. Autoimmun Rev (2015) 14:1–9. doi: 10.1016/j.autrev.2014.08.008
179. Emmi G, Talarico R, Lopalco G, Cimaz R, Cantini F, Viapiana O, et al. Efficacy and Safety Profile of Anti-Interleukin-1 Treatment in Behcet's Disease: A Multicenter Retrospective Study. Clin Rheumatol (2016) 35:1281–6. doi: 10.1007/s10067-015-3004-0
180. Cantarini L, Vitale A, Scalini P, Dinarello CA, Rigante D, Franceschini R, et al. Anakinra Treatment in Drug-Resistant Behcet's Disease: A Case Series. Clin Rheumatol (2015) 34:1293–301. doi: 10.1007/s10067-013-2443-8
181. Vitale A, Rigante D, Caso F, Brizi MG, Galeazzi M, Costa L, et al. Inhibition of Interleukin-1 by Canakinumab as a Successful Mono-Drug Strategy for the Treatment of Refractory Behcet's Disease: A Case Series. Dermatology (2014) 228:211–4. doi: 10.1159/000358125
182. Reddy M, Davis C, Wong J, Marsters P, Pendley C, Prabhakar U. Modulation of CLA, IL-12r, CD40L, and IL-2Ralpha Expression and Inhibition of IL-12- and IL-23-Induced Cytokine Secretion by CNTO 1275. Cell Immunol (2007) 247:1–11. doi: 10.1016/j.cellimm.2007.06.006
183. Samson M, Ghesquiere T, Berthier S, Bonnotte B. Ustekinumab Inhibits Th1 and Th17 Polarisation in a Patient With Giant Cell Arteritis. Ann Rheum Dis (2018) 77:e6. doi: 10.1136/annrheumdis-2017-211622
184. Matza MA, Fernandes AD, Stone JH, Unizony SH. Ustekinumab for the Treatment of Giant Cell Arteritis. Arthritis Care Res (Hoboken) (2021) 73:893–7. doi: 10.1002/acr.24200
185. Samson M, Bonnotte B. Ustekinumab for the Treatment of Giant Cell Arteritis: Comment on the Paper of Martza Et al. Arthritis Care Res (Hoboken) (2021) 73:1058–9. doi: 10.1002/acr.24376
186. Conway R, O'Neill L, Gallagher P, McCarthy GM, Murphy CC, Veale DJ, et al. Ustekinumab for Refractory Giant Cell Arteritis: A Prospective 52-Week Trial. Semin Arthritis Rheumatol (2018) 48:523–8. doi: 10.1016/j.semarthrit.2018.04.004
187. Terao C, Yoshifuji H, Nakajima T, Yukawa N, Matsuda F, Mimori T. Ustekinumab as a Therapeutic Option for Takayasu Arteritis: From Genetic Findings to Clinical Application. Scand J Rheumatol (2016) 45:80–2. doi: 10.3109/03009742.2015.1060521
188. Yachoui R, Kreidy M, Siorek M, Sehgal R. Successful Treatment With Ustekinumab for Corticosteroid- and Immunosuppressant-Resistant Takayasu's Arteritis. Scand J Rheumatol (2018) 47:246–7. doi: 10.1080/03009742.2017.1278788
189. Gon Y, Yoshifuji H, Nakajima T, Murakami K, Nakashima R, Ohmura K, et al. Long-Term Outcomes of Refractory Takayasu Arteritis Patients Treated With Biologics Including Ustekinumab. Mod Rheumatol (2020) 31:1–6. doi: 10.1080/14397595.2020.1800560
190. Mirouse A, Barete S, Monfort JB, Resche-Rigon M, Bouyer AS, Comarmond C, et al. Ustekinumab for Behcet's Disease. J Autoimmun (2017) 82:41–6. doi: 10.1016/j.jaut.2017.05.002
191. Mirouse A, Barete S, Desbois AC, Comarmond C, Sene D, Domont F, et al. Long-Term Outcome of Ustekinumab Therapy for Behcet's Disease. Arthritis Rheumatol (2019) 71:1727–32. doi: 10.1002/art.40912
192. Hot A, Lavocat F, Lenief V, Miossec P. Simvastatin Inhibits the Pro-Inflammatory and Pro-Thrombotic Effects of IL-17 and TNF-Alpha on Endothelial Cells. Ann Rheum Dis (2013) 72:754–60. doi: 10.1136/annrheumdis-2012-201887
193. Limagne E, Thibaudin M, Euvrard R, Berger H, Chalons P, Vegan F, et al. Sirtuin-1 Activation Controls Tumor Growth by Impeding Th17 Differentiation via STAT3 Deacetylation. Cell Rep (2017) 19:746–59. doi: 10.1016/j.celrep.2017.04.004
194. Yong C, Dan L, Chenhong L, Yan S, Jianfei C, Jianlong G. Efficacy and Safety of Metformin for Behcet's Disease and Its Effect on Treg/Th17 Balance: A Single-Blinded, Before-After Study. Nan Fang Yi Ke Da Xue Xue Bao (2019) 39:127–33. doi: 10.12122/j.issn.1673-4254.2019.02.01
195. Kong X, Sun Y, Dai X, Wang L, Ji Z, Chen H, et al. Treatment Efficacy and Safety of Tofacitinib Versus Methotrexate in Takayasu Arteritis: A Prospective Observational Study. Ann Rheum Dis (2022) 81:117–23. doi: 10.1136/annrheumdis-2021-220832
196. Koster MJ, Crowson CS, Giblon RE, Jaquith JM, Duarte-Garcia A, Matteson EL, et al. Baricitinib for Relapsing Giant Cell Arteritis: A Prospective Open-Label 52-Week Pilot Study. Ann Rheum Dis (2022). doi: 10.1136/annrheumdis-2021-221961
Keywords: interleukin-17, Th17 cells, cardiovascular system, allo-immune vascular inflammation, vasculitis, IL-17 inhibitors
Citation: Robert M, Miossec P and Hot A (2022) The Th17 Pathway in Vascular Inflammation: Culprit or Consort? Front. Immunol. 13:888763. doi: 10.3389/fimmu.2022.888763
Received: 03 March 2022; Accepted: 22 March 2022;
Published: 11 April 2022.
Edited by:
Michael V. Volin, Midwestern University, United StatesReviewed by:
Maxime Samson, Centre Hospitalier Regional Universitaire De Dijon, FranceRyu Watanabe, Osaka City University, Japan
Copyright © 2022 Robert, Miossec and Hot. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Marie Robert, bWFyaWUucm9iZXJ0MDdAb3V0bG9vay5jb20=