 Jianan Zhao1,2,3
Jianan Zhao1,2,3 Kai Wei1,2,3
Kai Wei1,2,3 Ping Jiang1,2,3
Ping Jiang1,2,3 Cen Chang1,2,3
Cen Chang1,2,3 Lingxia Xu1,2,3
Lingxia Xu1,2,3 Linshuai Xu1,2,3
Linshuai Xu1,2,3 Yiming Shi1,2,3
Yiming Shi1,2,3 Shicheng Guo4,5*
Shicheng Guo4,5* Yu Xue6*
Yu Xue6* Dongyi He1,2,3,7*
Dongyi He1,2,3,7*- 1Guanghua Clinical Medical College, Shanghai University of Traditional Chinese Medicine, Shanghai, China
- 2Department of Rheumatology, Shanghai Guanghua Hospital, Shanghai University of Traditional Chinese Medicine, Shanghai, China
- 3Institute of Arthritis Research in Integrative Medicine, Shanghai Academy of Traditional Chinese Medicine, Shanghai, China
- 4Computation and Informatics in Biology and Medicine, University of Wisconsin-Madison, Madison, WI, United States
- 5Department of Medical Genetics, School of Medicine and Public Health, University of Wisconsin-Madison, Madison, WI, United States
- 6Department of Rheumatology, Huashan Hospital, Institute of Rheumatology, Immunology and Allergy, Fudan University, Shanghai, China
- 7Arthritis Institute of Integrated Traditional and Western Medicine, Shanghai Chinese Medicine Research Institute, Shanghai, China
Gout, a chronic inflammatory arthritis disease, is characterized by hyperuricemia and caused by interactions between genetic, epigenetic, and metabolic factors. Acute gout symptoms are triggered by the inflammatory response to monosodium urate crystals, which is mediated by the innate immune system and immune cells (e.g., macrophages and neutrophils), the NACHT, LRR, and PYD domains-containing protein 3 (NLRP3) inflammasome activation, and pro-inflammatory cytokine (e.g., IL-1β) release. Recent studies have indicated that the multiple programmed cell death pathways involved in the inflammatory response include pyroptosis, NETosis, necroptosis, and apoptosis, which initiate inflammatory reactions. In this review, we explore the correlation and interactions among these factors and their roles in the pathogenesis of gout to provide future research directions and possibilities for identifying potential novel therapeutic targets and enhancing our understanding of gout pathogenesis.
Introduction
Gout is a type of arthritis associated with inflammation and is primarily caused by the deposition of monosodium urate (MSU) crystals in joints because of increased serum uric acid levels. Elevated serum uric acid levels gradually develop into hyperuricemia and lead to MSU formation, which is stimulated by various factors and leads to joint inflammation. The risk factors for gout include genetic, metabolic, and multiple comorbidities, including metabolic syndrome, cardiovascular disease, and renal disease (1). The global healthcare burden of gout has increased. Worldwide prevalence and incidence vary by study region, population, and assessment methods, ranging from <1% to 6.8%, with an incidence of 0.58–2.89 per 1,000 person-years (1). Patients with gout are primarily treated with uric acid-lowering and anti-inflammatory therapies (1, 2). Uric acid-lowering drugs such as allopurinol, febuxostat, probenecid sulfinpyrazone, and benzbromarone—typically used in combination—increase the metabolic burden on the kidney, the risk of kidney stone disease, and uric acid excretion and primarily reduce purine catabolism (3). Additionally, some patients do not accept long-term uric acid-lowering therapies due to lack compliance and, therefore, exhibit a poor treatment response. Anti-inflammatory treatments include nonsteroidal anti-inflammatory agents, colchicine, and corticosteroids. However, these drugs may induce multiple side effects; the development of new drugs has been slow (3). Additionally, currently used clinical drugs do not cover all clinical stages of gout.
Cell death commonly occurs in various physiological and pathological processes in the human body. In 2018, the Nomenclature Committee on Cell Death classified cell death as either accidental or regulated based on functional differences, which includes 11 primary forms: apoptosis, necroptosis, pyroptosis, ferroptosis, parthanatos, entotic cell death, NETotic cell death, autophagy-dependent cell death, alkaliptosis, oxeiptosis, and lysosome-dependent cell death (4). Numerous studies have characterized these types of cell death and their underlying molecular mechanisms. However, the role of cell death in certain diseases remains unclear. Different cell death patterns may be associated with the different clinical stages of gout. MSUs in gout can function as pattern recognition receptors and thus act as danger signals to activate multiple immune cells and multiple cell death pathways, possibly through mutual crosstalk (5). For example, tumor necrosis factor (TNF)-α and interleukin (IL)-1β are important molecules or products of apoptosis, necroptosis, and pyroptosis. These molecules can be secreted by monocytes and promote inflammatory cell infiltration in patients with gout (6–9). In this review, we provide insights that may be useful for developing new drugs for gout and for determining the associated pathological mechanisms by describing the multiple forms of cell death, including pyroptosis, NETosis, necroptosis, and apoptosis, in gout and their roles.
Pyroptosis
Pyroptosis is a form of pro-inflammatory cell death (10). The various key molecules involved in pyroptosis include the inflammasome [apoptosis-associated speck-like protein containing a C-terminal caspase activation and recruitment domain (ASC), NLRP3, and pro-caspase1], members of the gasdermin family, caspase1, and caspases4/5/11, which mediate the non-classical pathway (11). The activation of the NLRP3 inflammasome hydrolyzes IL-1β precursor (pro-IL-1β), IL-18 precursor (pro-IL-18) and gasdermin D via caspase-1, resulting in the maturation and release of IL-18 and IL-1β and N-terminal fragments of gasdermin D. It results in gasdermin-mediated pore formation and membrane rupture, the release of cellular contents to mediate inflammation (11). The activation of the NLRP3 inflammasome and release of IL-1β are thought to be important in the progression of hyperuricemia to gout. Both soluble uric acid and MSU in gout act as damage-associated molecular patterns (12). Soluble uric acid level and MSU accumulation depend on mitochondrial reactive oxygen species (ROS) production and the Toll-like receptor (TLR)/myeloid differentiation primary response (MyD)88-nuclear factor kappa B (NF-κB) signaling pathway to activate the NLRP3 inflammasome and pro-IL-1β transcription, respectively. When inflammasomes are activated, pro-IL-1β is cleaved by caspase1, which promotes maturation and pyroptosis to cause the release of biologically active IL-1β (12). In the following section, we focus on the potential link between pyroptosis, which is a potential therapeutic target, and gout.
NLRP3 Inflammasome Activation and IL-1β Release Promote Inflammation in Gout
In gout, high uric acid levels typically lead to hyperuricemia, inflammation, and other complications. Uric acid precipitates into MSU and triggers the assembly of multiple cells with activated NLRP3 inflammasome and the maturation and release of IL-1β (13, 14). Numerous biological mediators contribute to this process via various mechanisms. Experimental mice lacking critical components of pyroptosis (NLRP3, ASC, or caspase1) showed decreased knee neutrophil infiltration in response to MSU stimulation. Macrophages of these mice were defective in the MSU-induced IL-1β release process compared to those in the control group (15). Moreover, for monocyte/macrophage subsets in gout patients, granulocyte-macrophage colony-stimulating factor, G protein receptors (GPR43 and P2Y14), and microbial populations assist in inducing the expression of key molecules of MSU-promoted pyroptosis through different mechanisms, such as inhibition of cyclic adenosine monophosphate synthesis, production of metabolites, or inhibition of histone deacetylases activity (16–18). Cold-inducible RNA-binding protein, an endogenous damage-associated molecular pattern, promotes MSU-stimulated neutrophil infiltration, a CXC-motif receptor 2-dependent process associated with the NLRP3/ASC/caspase1/IL-1β/MyD88 pathway (19, 20).
We further explored the mechanisms by which MSU activates the NLRP3 inflammasome, which can be described as follows: (Figure 1) (i) MSU is endocytosed by phagocytes and causes the release of histone B by disrupting lysosomes (21). It also releases intracellular ATP through the pannexin/connexin channels. Extracellular ATP is degraded by different extracellular ATPases to diphosphate and monophosphate and further degraded to adenosine or metabolites that interact with P2X/P2Y, which further increases ATP release and activates the NLRP3 inflammasome by activating purinergic receptors (P2X/P2Y) on the cell surface or in adjacent cells (22). Interestingly, histones B and S can directly cleave receptor interacting serine/threonine kinase 1 (RIPK1), a key molecule involved in necroptosis and apoptosis, thereby inhibiting macrophages from undergoing necroptosis or apoptosis and tending toward pyroptosis (23). In addition, Toll/IL-1 receptor-domain-containing adapter-inducing interferon-β and the apoptosis protein inhibitor IAP modulate the ubiquitination of receptor interacting serine/threonine kinase 3 (RIPK3) and mixed lineage kinase domain-like protein (MLKL) in lipopolysaccharide (LPS)-induced necroptosis in vitro. RIPK3 in IAP-deficient mice promotes autoantibody-mediated arthritis by promoting NLRP3 inflammasome formation and IL-1β release, which may be a consequence of RIPK3 inhibition of necroptosis by shifting the form of cell death to pyroptosis (24). The crosstalk between multiple cell death mechanisms requires further elucidation. (ii) MSU activates the NLRP3 inflammasome by regulating cell surface ion channel activity and intracellular and extracellular potassium ion concentrations upon contact with the macrophage surface independent of histone proteinase B (25). In addition, phagocytosis of MSU decreases intracellular K+ concentrations via a mechanism that may involve the low-pH environment of lysosomes, causing the release of abundant sodium, increased intracellular osmotic pressure, cell swelling, and decreased potassium ion concentration (26). We also found that the mechanosensitive transient receptor potential vanilloid 4 (TRPV4) channel expression level is upregulated in MSU-stimulated synovial macrophages and human peripheral blood mononuclear cells. Both genetic ablation and repression of TRPV4 attenuated MSU inflammation by inhibiting NLRP3 inflammasome and IL-1β production, suggesting that TRPV4 is required for MSU-mediated activation of the NLRP3 inflammasome cascade (27). TRPV4 is an osmotic cation channel. MSU-stimulated reduction in intracellular ATP levels leads to mitochondrial membrane depolarization, activating NLRP3 and causing IL-1β release through the regulation of K+- and Ca2+-mediated mitochondrial dysfunction (28). (iii) After MSU engulfment by the plasma membrane of phagocytes, MSU forms electrostatic bonds and lipid raft aggregations with cholesterol in the plasma membrane of dendritic cells, leading to further activation of the Syk/phosphatidylinositol-3-kinase (PI3K) signaling pathway by an intracellular immunoreceptor tyrosine-based activation motif (29). Activation of Syk signaling can lead to the production of ROS and promote the release of cytokines and chemokines that induce inflammation, possibly accompanied by NLRP3 activation (30, 31). Clec12a, a C-type lectin receptor widely expressed on the surface of innate immune cells, negatively regulates the Syk signaling pathway through an intracellular immunoreceptor tyrosine-based activation motif (32) and may inhibit MSU-induced cell death through a mechanism involving physical sensing of monosodium urate or recognition of a protein ligand of dead cells (33). (iv) The complement system also contributes to NLRP3 inflammasome activation. MSU can activate the complement component C5a to enhance ROS production, triggering NLRP3 inflammasome assembly and IL-1β release (34, 35). In response to C5a, neutrophils infiltrating the peritoneum release phosphatidylserine-positive neutrophil-derived microvesicles, thereby inhibiting the C5a response (34). Microvesicles also inhibit inflammation by releasing transforming growth factor-β through a phosphatidylserine/MerTK receptor-independent pathway (34). Intracellular C5a and C5a receptor 2 (C5aR2) interactions amplify double-stranded DNA-dependent protein kinase R expression via the mitogen-activated protein kinase/extracellular signal-regulated kinase pathway and type I interferon signaling. A deficiency in C5aR2 inhibits NLRP3 inflammasome activation and high-mobility group box 1 release from mouse macrophages (36). The function of C5aR2 varies among cells and can negatively regulate C5aR1 expression in T cells (37). Intracellular C5 and C5a receptor 1 (C5aR1) stimulate NLRP3 assembly and initiate caspase1-dependent IL-1β secretion, IFN-γ production, and Th1 differentiation in human CD4+ T cells (37). C-reactive protein was found to be the second most abundant protein on the surface of the MSU crystals. Both erythrocyte sedimentation rate and C-reactive protein levels are significantly elevated in elderly patients with gout during acute attacks (38). C-reactive protein recruits complement component C1 and mannose-binding lectin pathway protease MASP1 on the surface of MSU and enhances binding of C3 and terminal complement complexes, suggesting that the activation of the complement cascade pathway contributes to the subsequent cell death pathway; however, further studies are needed to verify this hypothesis (39). (v). MSU promoted NLRP3 inflammasome activation and IL-1β release by affecting mitochondrial function and oxidative stress. Mitochondria are important organelles that maintain intracellular homeostasis (40). Analysis of the effect of mitochondrial genetic variation and copy number variants on gout susceptibility showed that a reduced mtDNA copy number significantly increased the risk of gout. Mitochondria are important for the colocalization of NLRP3 and ASC and release of IL-1β (41). Thus, mitochondrial dysfunction may lead to changes in the mitochondrial membrane potential, release of mitochondrial ROS, and other mechanisms that activate the NLRP3 inflammasome and contribute to the release of IL-1β to promote gout progression. For example, a small heterodimer partner can inhibit NLRP3 activation by competing for NLRP3 binding to an apoptosis-associated speck-like protein containing CARD (ASC). Deletion of a small heterodimer partner caused an excessive NLRP3 response in a mouse model of gout via a mechanism involving severe damage to and accumulation of mitochondria and the sustained action of NLRP3 and ASC (42). Similarly, Raf kinase inhibitor protein negatively regulates inflammasome activation by competing for NLRP1, NLRP3, and NLPC4 inflammasome binding to ASC. The depletion of Raf kinase inhibitor protein also exacerbates gouty arthritis (43). Heat shock protein 60 (HSP60) expression is upregulated in the peripheral blood mononuclear cells and serum of patients with acute gout. MSU crystals can also induce macrophage HSP60 expression, thereby promoting the collapse of mitochondrial membrane potential and mitochondrial ROS production, leading to mitochondrial dysfunction and activation of the NLRP3 inflammasome via a mechanism involving the TLR4/MyD88/NF-κB signaling pathway. Knockdown or overexpression of HSP60 affects TLR4 and MyD88 expression, degradation of Ikβα, and the nuclear localization of NF-κB (44).
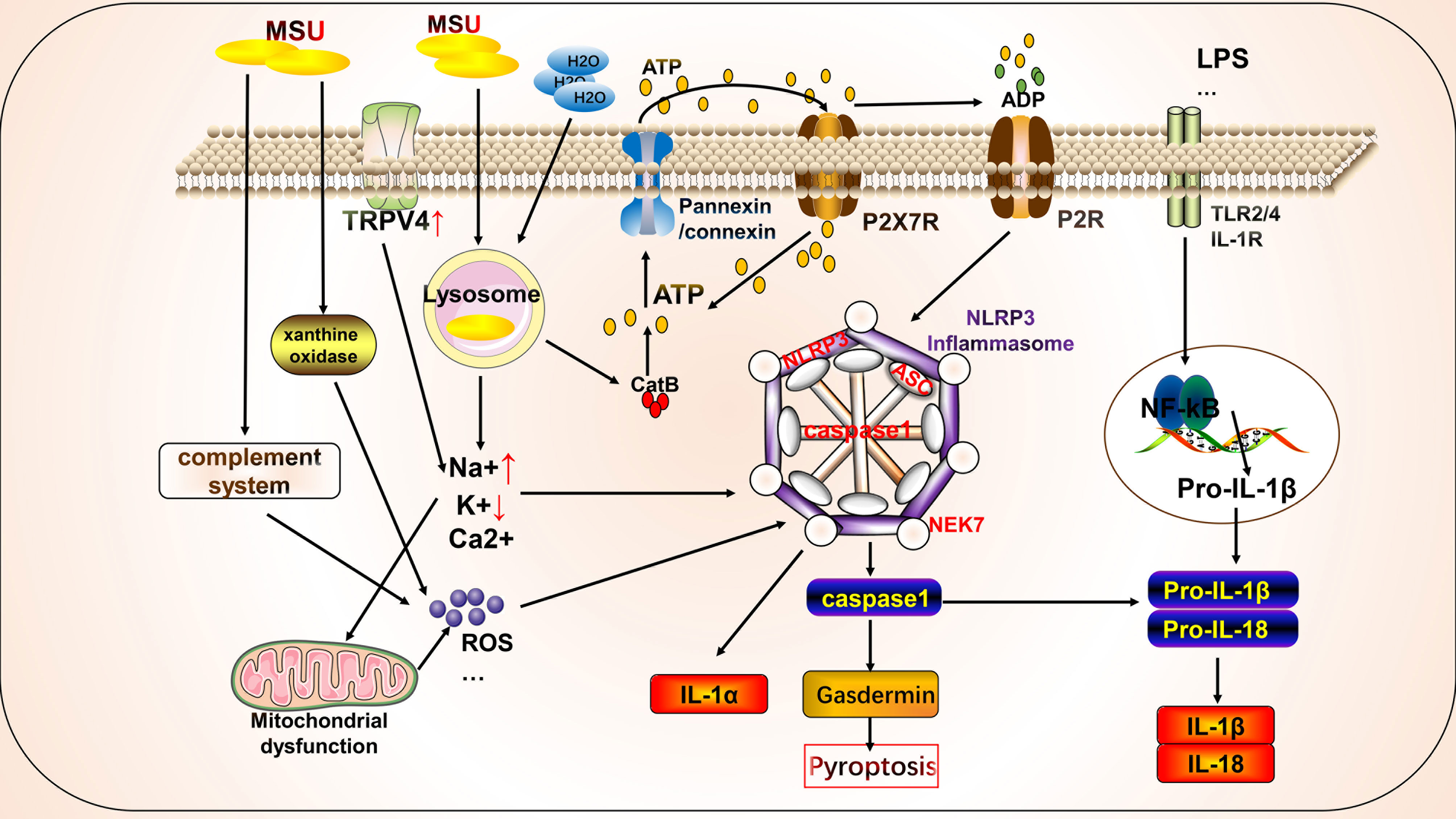
Figure 1 Mechanism of activation of key molecules for pyroptosis by monosodium urate (MSU) in gout. MSU can activate key molecules involved in pyroptosis, such as NLRP3 inflammasome, and promote the release of IL-1β through various mechanisms. MSU-stimulated destabilization of lysosomes leads to the release of cathepsin, which activates the NLRP3 inflammasome by regulating ATP metabolism through the cell surface pannexin/connexin channels and purinergic receptors. In addition, MSU can regulate intracellular ion concentrations and mitochondrial function, leading to the release and production of substances, such as ROS, to activate the NLRP3 inflammasome. The complement system, particularly the lectin system, is also partially involved in the activation of the NLRP3 inflammasome.
Therapeutic Targets and Molecules Associated With Pyroptosis Improve Gout
Pyroptosis plays a vital role in the progression of gout and has thus been targeted because of its clinical treatment potential. Organisms have several mechanisms that negatively regulate pyroptosis. For example, an MSU-induced increase in macrophage CD44 increases caspase1 activity and IL-1β production by decreasing phosphatase 2A activity (45). Proteoglycan-4, a glycoprotein produced by synovial fibroblasts, inhibits MSU-induced inflammatory responses by interacting with CD44 receptors on macrophages, thereby reducing NF-κB nuclear translocation, NLRP3 expression, caspase1 activation, and mature IL-1β production (46). The mechanism underlying the negative regulation of pyroptosis appears insufficient to suppress the inflammatory response. Therefore, drugs that inhibit pyroptosis, either directly or indirectly, should be developed to treat gout (Table 1).
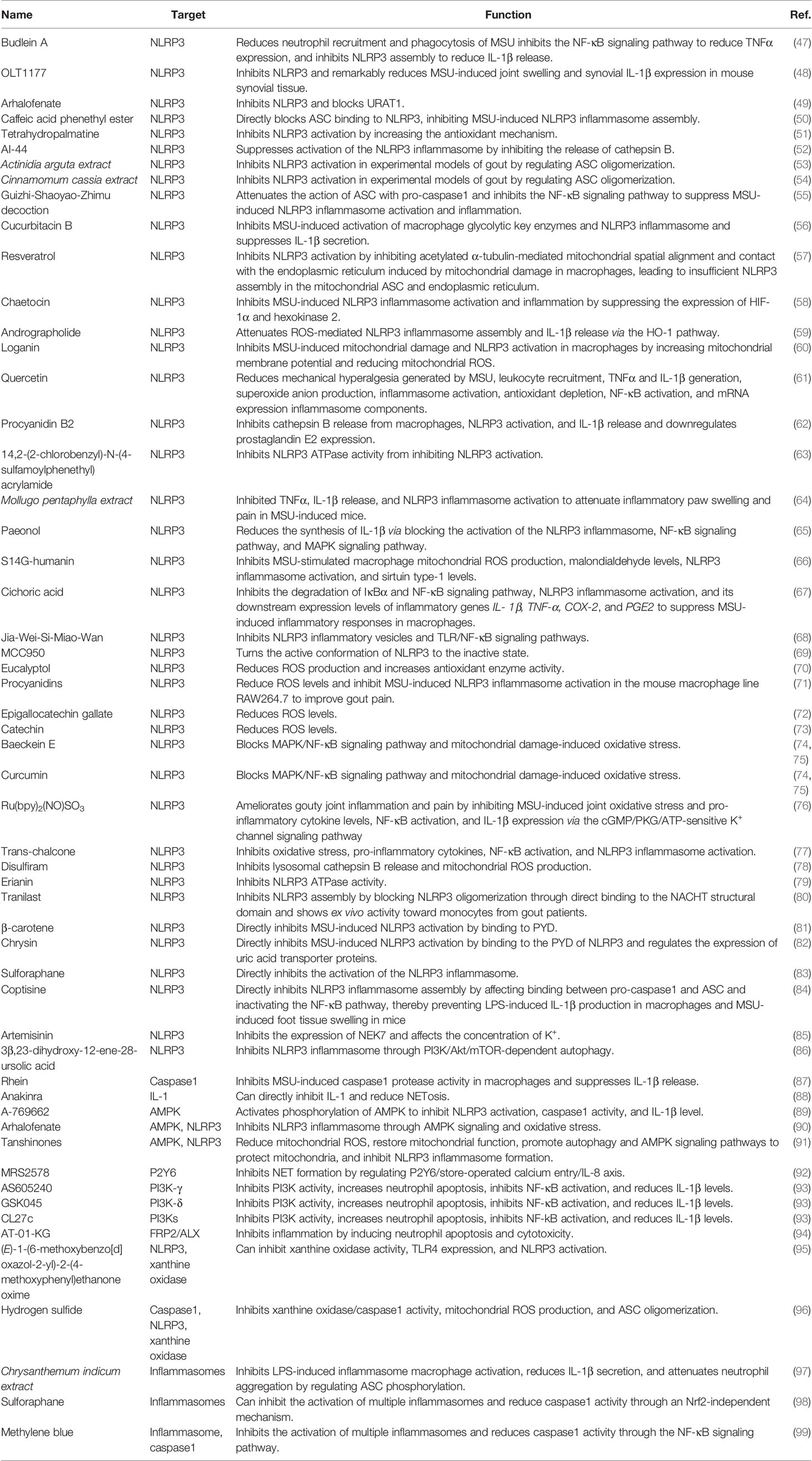
Table 1 Multiple potential compounds targeting cell death for the treatment of gout.
Several existing natural ingredients and artificial pharmacological inhibitors improve gout by directly inhibiting the assembly or activation of the NLRP3 inflammasome. For example, erianin showed ex vivo activity in synoviocytes and monocytes of patients with gout. Erianin inhibits the assembly of NLRP3 by directly interacting with it and is associated with the Walker A motif in the NACHT structural domain, inhibiting NLRP3 ATPase activity (79). Gavage of sulforaphane in a mouse model of acute gout revealed that sulforaphane reduced MSU-induced tissue swelling and inflammatory cell infiltration in the mouse foot by directly inhibiting NLRP3 inflammasome activation independent of ROS (83). The mRNA expression of NIMA-related kinase 7 (NEK7) and NLRP3 is upregulated in gout patients (85). NEK7 is an essential component of NLRP3 inflammasome (100). Artemisinin inhibits LPS- and MSU-induced NEK7 and NLRP3 expression levels in macrophages and attenuates K+ efflux from macrophages to reduce foot and ankle swelling in mice with arthritis (85).
In addition, some mechanisms indirectly inhibit MSU activation of the NLRP3 inflammasome and thus prevent its activation by restoring mitochondrial function and reducing ROS production. Many antioxidants derived from natural sources are responsible for this effect. Eucalyptol inhibited inflammatory cell infiltration, upregulation of TRPV1 expression, activation of the NLRP3 inflammasome, and production of pro-inflammatory cytokines induced by MSU injection in mouse ankle joints to suppress gout and joint inflammation primarily through antioxidant mechanisms. The underlying mechanisms included reducing ROS production and increasing antioxidant enzyme activity (70). Epigallocatechin gallate and catechin are tea-like bioactive polyphenols. Epigallocatechin gallate can inhibit MSU-induced neutrophil infiltration, NLRP3 expression, and pro-inflammatory factor secretion to ameliorate inflammation (101). Epigallocatechin gallate inhibits de novo mitochondrial DNA synthesis and ROS production in mouse macrophages and the NLRP3 inflammasome and prevents gouty inflammation (72). Catechin inhibits NLRP3 inflammasome activation by upregulating the mitochondrial survival protein BCL2 level in monocytes, restoring MSU-induced impairment of the mitochondrial transmembrane potential, and reducing intracellular ROS and Ca2+ levels (73). In addition to natural antioxidants, other substances have similar effects. For example, soluble decoy receptor 3 (DcR3) promotes the differentiation of anti-inflammatory M2 macrophages and inhibits NLRP3 activation by reducing MSU-induced mitochondrial dysfunction and lysosomal rupture to decrease ROS production and cathepsin levels and to improve gout (102). Disulfiram inhibits NLRP3 activation by inhibiting the release of lysosomal histone B into the cytoplasmic lysate and inhibiting mitochondrial ROS production and has significant efficacy against MSU-induced gout inflammation (78). Interestingly, another study tested the degree of NLRP3 activation in MSU-stimulated mouse macrophages at different temperatures (25, 33, 37, 39, and 42°C) and found that 37°C was the optimal temperature. Greater NLRP3 activation was observed at lower temperatures, indicating that low temperatures contribute to the MSU-induced activation of the NLRP3 inflammasome. Therefore, thermotherapy may be an effective form of physiotherapy for gout (103).
IL-1 inhibitors, such as anakinra, have also been shown to ameliorate gout. A pilot, open-label study (trial registration number ISRCTN10862635) evaluated the efficacy of anakinra in 10 patients with gout who were intolerant or had failed other conventional therapies and found that all patients responded well to anakinra without demonstrating any adverse effects (88). Another clinical trial on the efficacy and safety of anakinra in 40 patients with gout revealed that most responded well to anakinra, suggesting that it can be used as an alternative to other therapies or for short-term use. However, long-term administration is associated with complications, such as infection (104). MSU-activated NLRP3 inflammasomes induce IL-1β and IL-1α release. NLRP3 inflammasomes are required to activate calpain and the intracellular cleavage of IL-1α, thereby aiding IL-1α release (105, 106). Given the efficacy of IL-1 receptor antagonists in gout, neutralizing antibodies against IL-1α may also be beneficial. Notably, targeted inhibition of the pyroptosis effector protein gasdermin D may not be as necessary for MSU-induced IL-1β release as gasdermin D mediated membrane destruction. MSU may also disrupt cells through other mechanisms, leading to IL-1β release (107). Therefore, studies on the link between pyroptosis and gout should focus on upstream mechanisms, such as the NLRP3 inflammasome.
Effect of Crosstalk Between Autophagy and Pyroptosis on Gout
Autophagy is a conserved cellular pathway that controls the degradation of proteins and organelles and plays a vital role in maintaining homeostasis and disease progression (108). Its molecular mechanisms have been extensively reviewed, primarily phosphorylation involving the mammalian target of rapamycin (mTOR), autophagy initiation recognition, formation of autophagy-induced complexes, and formation of final degradation products of autophagic lysosomes (11). Autophagy contributes to the pathogenesis of gout through crosstalk with pyroptosis (Figure 2). Osteoblasts phagocytose MSU, causing upregulation of NLRP3 activation to promote autophagy and autophagosome formation by reducing mTOR levels and promoting LC3-II cleavage, but not IL-1β release. This phagocytosis may prevent the harmful effects of MSU by maintaining it inside the autophagosome (109). In addition, Cyclocarya paliurus ameliorates hyperuricemia and gout via a mechanism that may involve 3β,23-dihydroxy-12-ene-28-ursolic acid, a potent substance derived from C. paliurus that inhibits NLRP3 inflammasome formation via PI3K/Akt/mTOR-dependent autophagy (86). Similarly, resveratrol improves gout inflammation by upregulating sirtuin1 expression, inhibiting the mRNA expression of NLRP3 and NF-κB, and promoting autophagy (110).
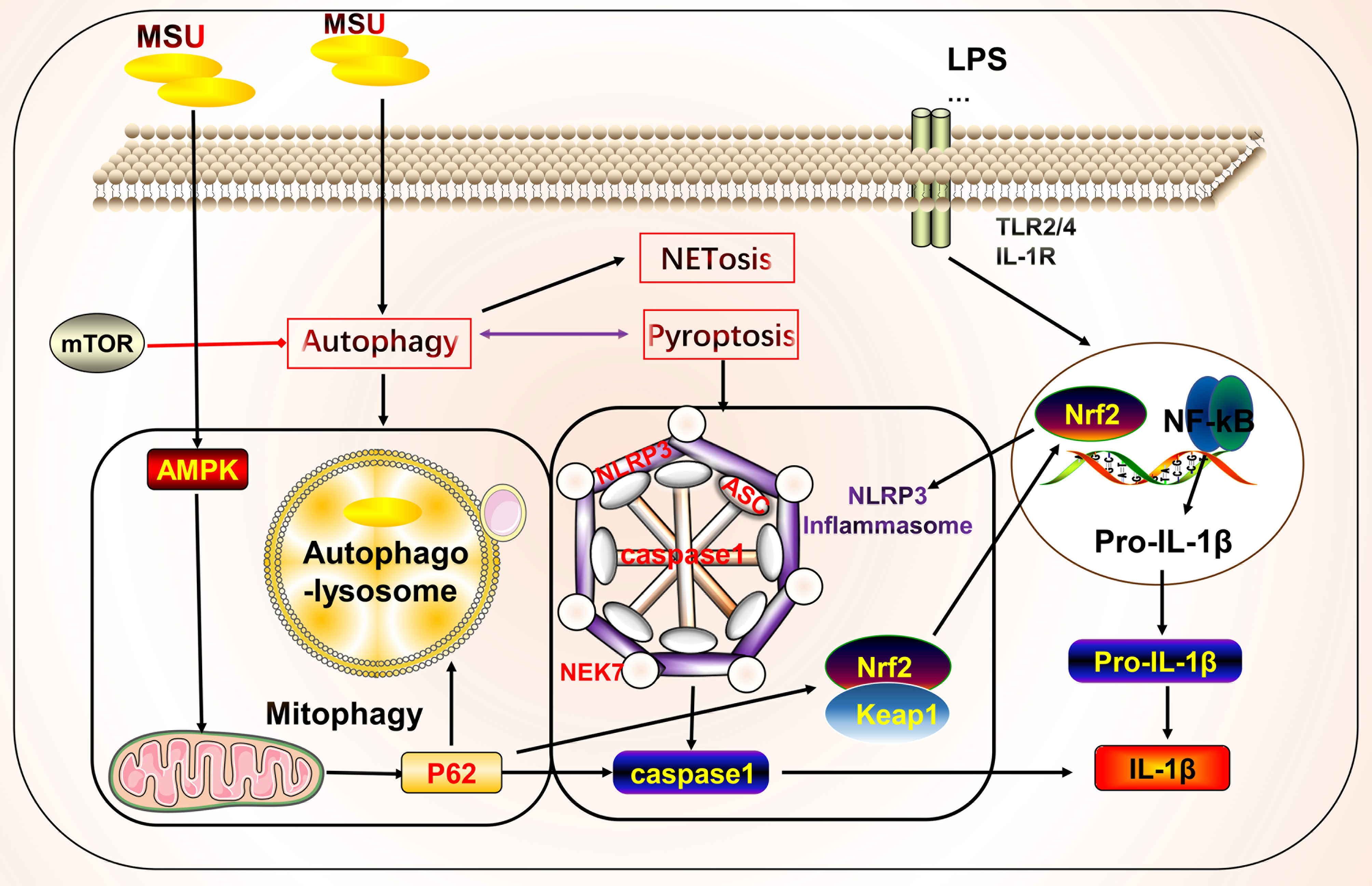
Figure 2 Crosstalk between autophagy and pyroptosis on gout. Autophagy is primarily associated with pyroptosis in gout. Autophagy induced by monosodium urate (MSU) leads to the formation of autophagolysosomes that engulf MSU to isolate further inflammatory responses. AMPK is involved in autophagy regulation by regulating mitochondrial function. The autophagy protein P62 plays a dual role, participating in mitochondrial autophagy with anti-inflammatory effects and in crosstalk with pyroptosis to promote inflammation. Autophagy also cross-talks with other cell death pathways, such as NETosis.
We focused on the roles of AMP-activated protein kinase (AMPK) and P62 in the crosstalk between autophagy and pyroptosis. Protein kinase AMP-activated non-catalytic subunit gamma 2 (PRKAG2) (cg09817217 and cg07012178), encoding the γ2 chain of AMPK, showed a gene body hypomethylation pattern in peripheral blood mononuclear cells of gouty arthritis patients (111). MSU inhibits AMPKα phosphorylation in macrophages in vitro. The AMPK activator, A-769662, inhibits the inflammatory response to MSU by promoting AMPK-dependent polarization of the M2 anti-inflammatory macrophage phenotype and AMPK kinase phosphorylation, inhibiting NLRP3 expression, and suppressing caspase1 and IL-1β activation. In vitro intervention with 10 nM colchicine resulted in effects similar to those of A-769662 (89). Colchicine can also inhibit NLRP3 inflammasome signaling by irreversibly inhibiting tubulin polymerization and microtubules (112). Arhalofenate enhances AMPK activity in macrophages to regulate mitochondrial function and oxidative stress-related AMPK downstream gene expression, inhibits MSU-induced NLRP3 inflammasome activation and IL-1β release, and promotes autophagic flux to suppress inflammation (90). Tanshinones attenuate LPS-induced mitochondrial ROS production, restore mitochondrial function, promote autophagy and AMPK signaling pathways to protect the mitochondria, and inhibit NLRP3 inflammasome formation in macrophages (91). P62 is a selective autophagy receptor that plays a dual role in gout. NF-κB induces the expression of pro-IL-1β and NLRP3 to initiate the assembly of NLRP3 inflammasomes. NF-κB also induces a specific mechanism for the delayed accumulation of P62/SQSTM1 during inflammation. When NLRP3 is activated and the mitochondria are damaged, the damaged mitochondria undergo parkin-dependent ubiquitin-binding and are thus recognized by P62, which then undergoes cell-selective autophagy and mitochondrial autophagy, thereby clearing damaged mitochondria to prevent the release of mitochondrial ROS and other substances to suppress inflammation (113). By contrast, MSU stimulates autophagosome formation, leading to impaired proteasomal degradation and P62 accumulation. Accumulation of P62 leads to IL-1β and caspase1 expression through activation of extracellular signal-regulated kinase and JNK signaling. IL-1β, in turn, induces the P62 protein and creates a vicious cycle; additionally, the silencing of Atg16L1, a protein essential for autophagosome formation, leads to P62 accumulation and enhances inflammation (114). In addition, P62 promotes its nuclear translocation by binding to Kelch-like ECH-associated protein 1 and releasing nuclear factor erythroid 2-related factor 2, which induces the transcription of heme oxygenase-1 and superoxide dismutase, thereby activating the NLRP3 inflammasome (115, 116). Thus, when mitochondrial autophagy functions effectively, P62 can inhibit NLRP3 activation by suppressing inflammation. However, when mitochondrial autophagy is inadequate or impaired, P62 is produced in large amounts and promotes inflammation.
NETosis
Neutrophils can participate in gout inflammation and gout stones by inducing the formation of neutrophil extracellular traps (NETs), which release a range of mediators including histones, ROS, and a variety of proteases (117, 118). NETs are extracellular meshworks containing chromatin and granule proteins and were initially considered a means of targeting microorganisms. These features include disassembly of nuclear and granule membranes and hypercitrullination of histones by peptidylarginine deiminase 4, which mediates chromatin decondensation, membrane rupture, and concomitant ROS production by NADPH oxidase (119). ROS trigger the release of neutrophil elastase into the cytoplasm and activate neutrophil elastase enzymatic activity in a myeloperoxidase-dependent manner, contributing to its degradation of F-actin and translocation to the nucleus for continued degradation of core histones, such as H1 and H4, subsequently promoting chromatin depolymerization and NETosis (118, 120–122) (Figure 3).
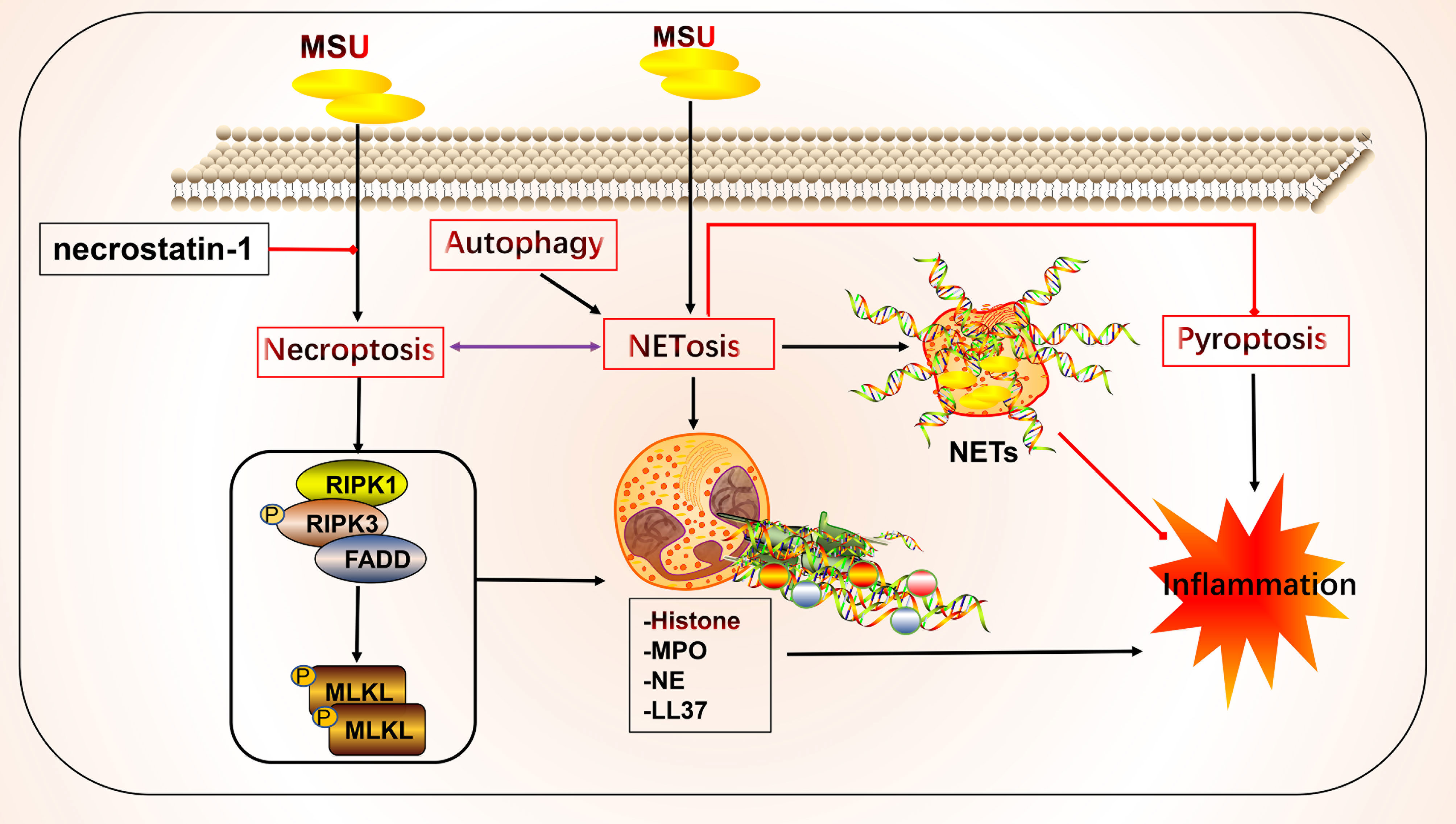
Figure 3 Crosstalk of NETosis with other forms of cell death and implications for gout. Monosodium urate (MSU) -stimulated NETs require key molecules involved in necroptosis. Neutrophils can promote cytokine degradation by forming NETs, phagocytosing MSU, and physically isolating them to suppress inflammation. Histones released during NETosis may have cytotoxic and immunostimulatory effects on cells. In addition, crosstalk with other forms of cell death, including pyroptosis and autophagy, may occur.
NETs and NETosis may play both anti-inflammatory and pro-inflammatory roles in gout. Histones released during NETosis may be important in the progression of gout and its related clinical symptoms; however, histones exert cytotoxic and immunostimulatory effects on glomerular cells (123), have cytotoxic effects, and increase lethality in endothelial cells and mouse models of sepsis in vitro (124). By contrast, MSU can trigger NET formation and NETosis. In standard synovial fluid from MSU-induced arthritis, non-refractive fibrous material, cells, and crystals were observed that primarily consisted of fibrin and collagen fibers that formed NETs (125). MSU and low-grade inflammation are often detected in the synovial fluid of joints of patients with asymptomatic gout, possibly in a state of clinical sub-inflammation (126, 127). Although MSU is thought to be an essential driver of inflammation, in practice it may not lead to a robust inflammatory response, which is consistent with the disease profile of clinically asymptomatic patients with gout. The robust inflammatory response also requires stimulation by other factors that lead to intense inflammation, such as the NLRP3 inflammasome (128–131). In this process, NETs may act as a physical barrier by phagocytosing MSU, thus forming a large reticulation of NETs to prevent the spread of harmful substances and influence disease progression (125). Studies have shown that neutrophils that phagocytose MSU form NETs by extruding DNA, packing crystals, and degrading cytokines to suppress excessive inflammatory responses (132). Additionally, NETs limit the inflammatory response by degrading cytokines and chemokines and inhibiting neutrophil recruitment and activation; however, the onset of NET formation leads to inflammation (133, 134).
NETs and NETosis may be linked to necroptosis (135). The critical molecules involved in necroptosis include RIPK1, RIPK3, and MLKL. RIPK1 and RIPK3 form a complex and activate MLKL, which leads to membrane pore formation, membrane leakage, and cell lysis, resulting in the release of cellular components. The detailed molecular mechanisms underlying these effects have been previously reviewed (11, 136, 137). Mulay et al. found that crystals of calcium oxalate, MSU, calcium pyrophosphate dihydrate, and cystine induce cysteinase-independent cell death in renal tubular epithelial cells, which is blocked by the RIPK1 inhibitor necrostatin-1, suggesting that crystal-induced cell death is associated with necroptosis (138). Garcia-Gonzalez et al. evaluated NETs in the synovial fluid of patients during a gout attack. Neutrophils in the synovial fluid release NETs in response to MSU, depending on the number of crystals, but not on cell density, which is accompanied by the activation of MLKL, a key molecule of necroptosis (125). Similarly, MSU-induced NETs were accompanied by RIPK3 expression and MLKL phosphorylation, and the inhibition of RIPK1, RIPK3, and MLKL inhibited NET formation in human and mouse neutrophils (139). The bridge linking the two forms of death may be ROS. Preferential activation of the three critical molecules of necroptosis leads to membrane rupture in neutrophils and ROS production, stimulating NET formation and NETosis (139). Thus, in addition to the association between necroptosis and NETs, pyroptosis of macrophages and NETs in neutrophils are interlinked. The latter may be a compensatory measure to suppress inflammation via NET formation (140). Sil et al. found that macrophage-derived IL-1β enhances the formation of MSU-stimulated NETs. Anakinra reduced NETosis in macrophages. In addition, caspase11-deficient experimental mice with gout and their source macrophages responded to MSU stimulation by significantly reducing the production of cytokines, such as IL-1β, TNFα, and IL-6. Caspase11-deficient neutrophils also failed to produce NETs, the mechanism of which may involve the effect of caspase11 on the regulation of macrophage pyroptosis in gout and NET formation (141). The P2Y6 receptor in endothelial cells in vitro activates LPS-induced transendothelial migration of neutrophils via Rho kinase (142). The P2Y6 receptor antagonist MRS2578 inhibits MSU-induced NETosis via a mechanism that may involve the P2Y6/store-operated calcium entry/IL-8 axis (92). In addition to the aforementioned link between NETosis, necroptosis, and pyroptosis, mutual crosstalk with autophagy may occur. Nuclear factor, interleukin 3 regulated (NFIL3), which is highly expressed in neutrophils of patients with gout, promotes its transcription by binding to the REDD1 promoter, which enhances autophagy and NET formation by inhibiting the mTOR pathway, to promote inflammatory responses (143). Inhibition of autophagy to prevent intracellular chromatin depletion leads to apoptosis, because intracellular chromatin depletion is necessary for NET formation and NETosis (144). Neutrophils from patients with gout form NETs in an autophagy-dependent manner, and inhibition of phagolysosome and PI3K signaling pathways prevents MSU-induced NETs and NETosis (145).
Apoptosis
Apoptosis is a controlled form of cell death that primarily involves the exogenous death receptor and endogenous mitochondrial pathways. Its key molecules include multiple death receptors, caspase enzymes, and mitochondrial proteins that mediate apoptosis (11). The morphological features of apoptosis include cytoplasmic concentration, breakage of DNA, and formation of apoptotic vesicles. This process generally does not result in the release of cellular contents. However, many apoptotic cells lead to inefficient clearance, causing an inflammatory response (11). However, few studies have directly examined the relationship between gout and apoptosis (Figure 4). Studies have shown that new macrophages are constantly recruited within the gout tophi and that these macrophages produce TNF-α and matrix metalloproteinase-2/9 to induce degradation of the stroma. However, apoptosis occurs in CD68+ macrophages, which limits inflammation (146). MSU stimulation can affect apoptotic processes in various cells and gout through different mechanisms, with most studies focusing on apoptosis in neutrophils and endothelial cells. MSU stimulates the release of ROS and reactive nitrogen species from fibroblast-like synoviocytes, leading to endoplasmic reticulum oxidative stress and possibly mitochondrial dysfunction, thereby promoting fibroblast-like synoviocyte apoptosis (147). The adhesion between neutrophils and endothelial cells is key to the development of acute gout (148). MSU is also gradually deposited on the vessel wall to induce a vascular inflammatory response and damage the patient’s vascular endothelial cells, thereby causing gout complications. During inflammation regression, neutrophils migrating to the inflammatory site first undergo apoptosis and are phagocytosed by phagocytic cells (149). Some apoptosis-related mechanisms in the body improve inflammation in order to cope with the inflammatory response in gout. Promoting the apoptosis of neutrophils and inhibiting the apoptosis of endothelial cells are beneficial for suppressing the inflammatory response to improve gout. CD300a is an inhibitory receptor whose expression increased in neutrophils from the knees of mice after MSU stimulation, which promotes apoptosis of neutrophils by increasing the cleavage of caspase8 of the exogenous death receptor pathway (150). Similarly, annexin A1, a glucocorticoid regulatory protein, reduced neutrophil infiltration and IL-1β release by inducing apoptosis in neutrophils in a MSU-induced gouty arthritis mouse model (151). Although multiple anti-inflammatory mechanisms exist, their actions may be insufficient to inhibit inflammation. Therefore, promoting endogenous anti-inflammatory substances or inhibiting the production of pro-inflammatory molecules—and thus promoting inflammatory cell apoptosis or inhibiting beneficial cell apoptosis through artificial agonists or inhibitors—is a potential future therapeutic direction. For example, inhibition of Rho-associated kinase, a serine/threonine kinase, attenuates neutrophil accumulation, IL-1β levels, and hypernociception in experimental models of gout through mechanisms involving reduced MYPT phosphorylation, decreased Iκβα activity, and increased caspase-dependent apoptosis of neutrophils (152). Similarly, PI3K inhibitors AS605240, GSK045, and CL27c significantly reduced MSU-induced neutrophil infiltration by increasing apoptosis and reducing myeloperoxidase activity, NF-κB activation, and IL-1β levels, thus improving inflammation (93). Simiaowan reduced neutrophil infiltration, inhibited endothelial cell apoptosis in rats with MSU-induced gout, and improved the inflammatory response by attenuating the expression of ICAM-1 (148). AT-01-KG, an artificial agonist of FRP2/ALX, inhibits inflammation by inducing neutrophil apoptosis and cytarabine in a mouse model of gouty arthritis (94). Natural traditional medicines have also been shown to inhibit apoptosis of chondrocytes in gouty arthritis, as summarized in a previous review (153). Anemonoides nemorosa and Tecomella undulata are used in traditional medicines to treat gout. Studies showed that Aqueous extracts of A. nemorosa can induce cell cycle arrest and apoptosis by regulating mitochondrial function (154). Tecomella undulata induces apoptosis in tumor cells by promoting Fas, Fas-associated death domain, caspase3/7/8, and DNA fragmentation (155). In addition, some clinical medications for gout are effective in modulating apoptosis. Uromodulin gene mutations cause premature hyperuricemia, gout, and kidney damage (156). Uromodulin gene mutations affect the processing of uromodulin/tammhorsfall glycoprotein (156). Colchicine promotes the secretion of tammhorsfall glycoprotein from the endoplasmic reticulum, which significantly reduces apoptosis, whereas intracellular endoplasmic reticulum accumulation of tammhorsfall glycoprotein promotes apoptosis (156). Allopurinol reduces apoptosis and increases osteoblast viability in an animal model of hyperuricemia and reduces the risk of vascular calcification by decreasing Wnt3a, Runx2, Sp7, Bglap, Col1a1, SM22a, and Acta2 expression in vascular smooth muscle cells (157).
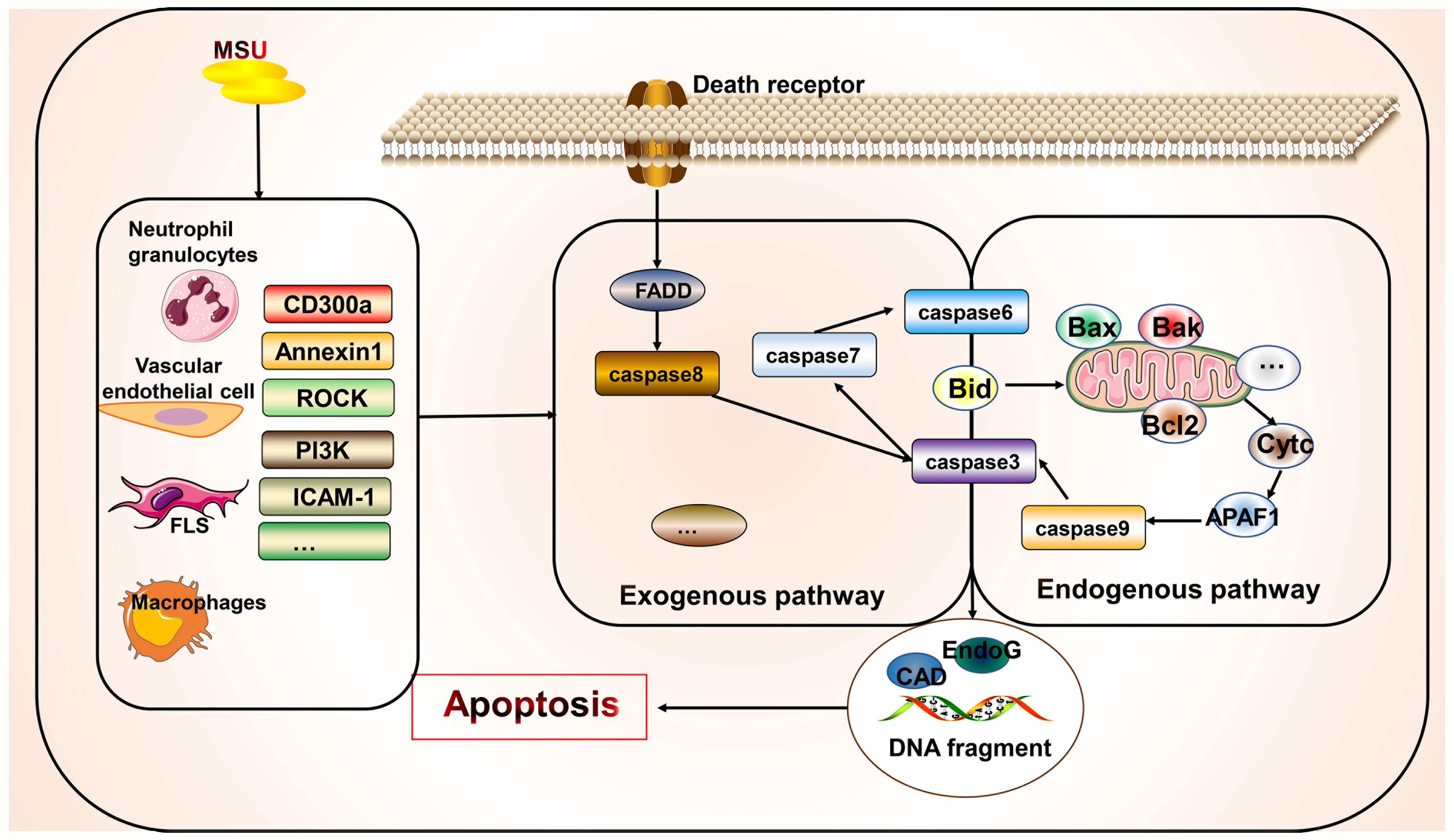
Figure 4 Association of apoptosis with gout. Apoptosis occurs primarily via endogenous and exogenous pathways. Apoptosis exists in various cells in gout but has not been widely examined. However, the managing gout involves inhibiting the apoptosis of beneficial cells and promoting the apoptosis of harmful cells. Some apoptosis-related molecular targets such as CD300a have been identified and require further investigation.
Conclusion
Cell survival and death are fundamental processes. The molecular mechanisms and morphological characteristics of various types of cell death have been revealed to be closely related to multiple diseases. First, we explored the role of pyroptosis in gout, in which the assembly of NLRP3, a key molecule in pyroptosis, is considered a critical step in the progression from hyperuricemia to gout. In response to MSU stimulation, NLRP3 inflammasomes undergo assembly and release IL-1β, which promotes inflammation. Second, we discussed the crosstalk between NETosis and other forms of cell death and its relationship with gout. NETosis appears to require the assistance of crucial molecules involved in necroptosis for activation. The occurrence of NETs and NETosis in neutrophils appears to be a compensatory mechanism that suppresses inflammation. Finally, we described the roles of these two forms of cell death in gout, autophagy, and apoptosis, which have not been widely evaluated and require further investigation. Studies should focus on the impact of crosstalk between different forms of cell death on gout pathogenesis. In addition, using artificial agonists and inhibitors to target cell death or biological mediators that regulate cell death is a promising therapeutic approach. Whether the combined targeting of multiple pathways and molecules is beneficial for intercellular crosstalk should be examined. Forms of cell death other than those covered in this study may exist and be relevant to gout pathogenesis; however, current research seems to be limited. Thus, unexplored forms of cell death will remain a potential direction of future research. Although translating preclinical results into clinical practice remains challenging, there is great potential to target the precise mechanisms of cell death in gout.
Author Contributions
JZ is responsible for the collection, collation and writing of the original manuscript. KW, PJ, CC, LXX, LSX and YS is responsible for the collection of the original manuscript. SG, YX, and DH are responsible for the revision and review of the manuscript. All authors reviewed and accepted with the final version.
Funding
This work was funded by the National Natural Science Funds of China (82074234 and 82071756), Shanghai Chinese Medicine Development Office, National Administration of Traditional Chinese Medicine, Regional Chinese Medicine (Specialist) Diagnosis and Treatment Center Construction Project-Rheumatology, State Administration of Traditional Chinese Medicine, National TCM Evidence-Based Medicine Research and Construction Project, Basic TCM Evidence-Based Capacity Development Program, Shanghai Municipal Health Commission, and East China Region-based Chinese and Western Medicine Joint Disease Specialist Alliance.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abbreviations
NLRP3, the NACHT, LRR, and PYD domains-containing protein 3; MSU, monosodium urate; TNF, tumor necrosis factor; IL, interleukin; ASC, apoptosis-associated speck-like protein containing a C-terminal caspase activation and recruitment domain; ROS, reactive oxygen species; TLR, Toll-like receptor; MyD, myeloid differentiation primary response; NF-κB, nuclear factor kappa B; RIPK1, receptor interacting serine/threonine kinase 1; RIPK3, receptor interacting serine/threonine kinase 3; MLKL, mixed lineage kinase domain-like protein; LPS, lipopolysaccharide; TRPV4, transient receptor potential vanilloid 4; PI3K, phosphatidylinositol-3-kinase; C5aR2, C5a receptor 2; C5aR1, C5a receptor 1; HSP60, heat shock protein 60; NEK7, NIMA-related kinase 7; DcR3, decoy receptor 3; mTOR, the mammalian target of rapamycin; AMPK, AMP-activated protein kinase; PRKAG2, protein kinase AMP-activated non-catalytic subunit gamma 2; NETs, neutrophil extracellular traps; NFIL3, nuclear factor, interleukin 3 regulated.
References
1. Dehlin M, Jacobsson L, Roddy E. Global Epidemiology of Gout: Prevalence, Incidence, Treatment Patterns and Risk Factors. Nat Rev Rheumatol (2020) 16(7):380–90. doi: 10.1038/s41584-020-0441-1
2. Dalbeth N, Gosling AL, Gaffo A, Abhishek A. Gout. Lancet (2021) 397(10287):1843–55. doi: 10.1016/S0140-6736(21)00569-9
3. Singh JA. Quality of Life and Quality of Care for Patients With Gout. Curr Rheumatol Rep (2009) 11(2):154–60. doi: 10.1007/s11926-009-0022-4
4. Tang D, Kang R, Berghe TV, Vandenabeele P, Kroemer G. The Molecular Machinery of Regulated Cell Death. Cell Res (2019) 29(5):347–64. doi: 10.1038/s41422-019-0164-5
5. Chen J, Wu M, Yang J, Wang J, Qiao Y, Li X. The Immunological Basis in the Pathogenesis of Gout. Iran J Immunol (2017) 14(2):90–8.
6. Yokose K, Sato S, Asano T, Yashiro M, Kobayashi H, Watanabe H, et al. TNF-α Potentiates Uric Acid-Induced Interleukin-1β (IL-1β) Secretion in Human Neutrophils. Mod Rheumatol (2018) 28(3):513–7. doi: 10.1080/14397595.2017.1369924
7. Temmoku J, Fujita Y, Matsuoka N, Urano T, Furuya MY, Asano T, et al. Uric Acid-Mediated Inflammasome Activation in IL-6 Primed Innate Immune Cells Is Regulated by Baricitinib. Mod Rheumatol (2021) 31(1):270–5. doi: 10.1080/14397595.2020.1740410
8. Jeong JH, Hong S, Kwon OC, Ghang B, Hwang I, Kim YG, et al. CD14(+) Cells With the Phenotype of Infiltrated Monocytes Consist of Distinct Populations Characterized by Anti-Inflammatory as Well as Pro-Inflammatory Activity in Gouty Arthritis. Front Immunol (2017) 8:1260. doi: 10.3389/fimmu.2017.01260
9. Amaral FA, Bastos LF, Oliveira TH, Dias AC, Oliveira VL, Tavares LD, et al. Transmembrane TNF-α Is Sufficient for Articular Inflammation and Hypernociception in a Mouse Model of Gout. Eur J Immunol (2016) 46(1):204–11. doi: 10.1002/eji.201545798
10. Kovacs SB, Miao EA. Gasdermins: Effectors of Pyroptosis. Trends Cell Biol (2017) 27(9):673–84. doi: 10.1016/j.tcb.2017.05.005
11. Zhao J, Hu Y, Peng J. Targeting Programmed Cell Death in Metabolic Dysfunction-Associated Fatty Liver Disease (MAFLD): A Promising New Therapy. Cell Mol Biol Lett (2021) 26(1):17. doi: 10.1186/s11658-021-00254-z
12. Cabău G, Crișan TO, Klück V, Popp RA, Joosten LAB. Urate-Induced Immune Programming: Consequences for Gouty Arthritis and Hyperuricemia. Immunol Rev (2020) 294(1):92–105. doi: 10.1111/imr.12833
13. Reber LL, Marichal T, Sokolove J, Starkl P, Gaudenzio N, Iwakura Y, et al. Contribution of Mast Cell-Derived Interleukin-1β to Uric Acid Crystal-Induced Acute Arthritis in Mice. Arthritis Rheumatol (2014) 66(10):2881–91. doi: 10.1002/art.38747
14. Nakamura Y, Franchi L, Kambe N, Meng G, Strober W, Núñez G. Critical Role for Mast Cells in Interleukin-1β-Driven Skin Inflammation Associated With an Activating Mutation in the Nlrp3 Protein. Immunity (2012) 37(1):85–95. doi: 10.1016/j.immuni.2012.04.013
15. Martinon F, Pétrilli V, Mayor A, Tardivel A, Tschopp J. Gout-Associated Uric Acid Crystals Activate the NALP3 Inflammasome. Nature (2006) 440(7081):237–41. doi: 10.1038/nature04516
16. Vieira AT, Macia L, Galvão I, Martins FS, Canesso MC, Amaral FA, et al. A Role for Gut Microbiota and the Metabolite-Sensing Receptor GPR43 in a Murine Model of Gout. Arthritis Rheumatol (2015) 67(6):1646–56. doi: 10.1002/art.39107
17. Cleophas MC, Crişan TO, Lemmers H, Toenhake-Dijkstra H, Fossati G, Jansen TL, et al. Suppression of Monosodium Urate Crystal-Induced Cytokine Production by Butyrate Is Mediated by the Inhibition of Class I Histone Deacetylases. Ann Rheum Dis (2016) 75(3):593–600. doi: 10.1136/annrheumdis-2014-206258
18. Li H, Jiang W, Ye S, Zhou M, Liu C, Yang X, et al. P2Y(14) Receptor has a Critical Role in Acute Gouty Arthritis by Regulating Pyroptosis of Macrophages. Cell Death Dis (2020) 11(5):394. doi: 10.1038/s41419-020-2609-7
19. Amaral FA, Costa VV, Tavares LD, Sachs D, Coelho FM, Fagundes CT, et al. NLRP3 Inflammasome-Mediated Neutrophil Recruitment and Hypernociception Depend on Leukotriene B(4) in a Murine Model of Gout. Arthritis Rheumatol (2012) 64(2):474–84. doi: 10.1002/art.33355
20. Fujita Y, Yago T, Matsumoto H, Asano T, Matsuoka N, Temmoku J, et al. Cold-Inducible RNA-Binding Protein (CIRP) Potentiates Uric Acid-Induced IL-1β Production. Arthritis Res Ther (2021) 23(1):128. doi: 10.1186/s13075-021-02508-9
21. Orlowski GM, Colbert JD, Sharma S, Bogyo M, Robertson SA, Rock KL. Multiple Cathepsins Promote Pro-IL-1β Synthesis and NLRP3-Mediated IL-1β Activation. J Immunol (2015) 195(4):1685–97. doi: 10.4049/jimmunol.1500509
22. Riteau N, Baron L, Villeret B, Guillou N, Savigny F, Ryffel B, et al. ATP Release and Purinergic Signaling: A Common Pathway for Particle-Mediated Inflammasome Activation. Cell Death Dis (2012) 3(10):e403. doi: 10.1038/cddis.2012.144
23. McComb S, Shutinoski B, Thurston S, Cessford E, Kumar K, Sad S. Cathepsins Limit Macrophage Necroptosis Through Cleavage of Rip1 Kinase. J Immunol (2014) 192(12):5671–8. doi: 10.4049/jimmunol.1303380
24. Lawlor KE, Khan N, Mildenhall A, Gerlic M, Croker BA, D'Cruz AA, et al. RIPK3 Promotes Cell Death and NLRP3 Inflammasome Activation in the Absence of MLKL. Nat Commun (2015) 6:6282. doi: 10.1038/ncomms7282
25. Hari A, Zhang Y, Tu Z, Detampel P, Stenner M, Ganguly A, et al. Activation of NLRP3 Inflammasome by Crystalline Structures via Cell Surface Contact. Sci Rep (2014) 4:7281. doi: 10.1038/srep07281
26. Schorn C, Frey B, Lauber K, Janko C, Strysio M, Keppeler H, et al. Sodium Overload and Water Influx Activate the NALP3 Inflammasome. J Biol Chem (2011) 286(1):35–41. doi: 10.1074/jbc.M110.139048
27. Lan Z, Chen L, Feng J, Xie Z, Liu Z, Wang F, et al. Mechanosensitive TRPV4 Is Required for Crystal-Induced Inflammation. Ann Rheum Dis (2021) 80(12):1604–14. doi: 10.1136/annrheumdis-2021-220295
28. Nomura J, So A, Tamura M, Busso N. Intracellular ATP Decrease Mediates NLRP3 Inflammasome Activation Upon Nigericin and Crystal Stimulation. J Immunol (2015) 195(12):5718–24. doi: 10.4049/jimmunol.1402512
29. Ng G, Sharma K, Ward SM, Desrosiers MD, Stephens LA, Schoel WM, et al. Receptor-Independent, Direct Membrane Binding Leads to Cell-Surface Lipid Sorting and Syk Kinase Activation in Dendritic Cells. Immunity (2008) 29(5):807–18. doi: 10.1016/j.immuni.2008.09.013
30. Sancho D, Reis e Sousa C. Sensing of Cell Death by Myeloid C-Type Lectin Receptors. Curr Opin Immunol (2013) 25(1):46–52. doi: 10.1016/j.coi.2012.12.007
31. Sancho D, Reis e Sousa C. Signaling by Myeloid C-Type Lectin Receptors in Immunity and Homeostasis. Annu Rev Immunol (2012) 30:491–529. doi: 10.1146/annurev-immunol-031210-101352
32. Marshall AS, Willment JA, Lin HH, Williams DL, Gordon S, Brown GD. Identification and Characterization of a Novel Human Myeloid Inhibitory C-Type Lectin-Like Receptor (MICL) That Is Predominantly Expressed on Granulocytes and Monocytes. J Biol Chem (2004) 279(15):14792–802. doi: 10.1074/jbc.M313127200
33. Neumann K, Castiñeiras-Vilariño M, Höckendorf U, Hannesschläger N, Lemeer S, Kupka D, et al. Clec12a Is an Inhibitory Receptor for Uric Acid Crystals That Regulates Inflammation in Response to Cell Death. Immunity (2014) 40(3):389–99. doi: 10.1016/j.immuni.2013.12.015
34. Cumpelik A, Ankli B, Zecher D, Schifferli JA. Neutrophil Microvesicles Resolve Gout by Inhibiting C5a-Mediated Priming of the Inflammasome. Ann Rheum Dis (2016) 75(6):1236–45. doi: 10.1136/annrheumdis-2015-207338
35. Khameneh HJ, Ho AW, Laudisi F, Derks H, Kandasamy M, Sivasankar B, et al. C5a Regulates IL-1β Production and Leukocyte Recruitment in a Murine Model of Monosodium Urate Crystal-Induced Peritonitis. Front Pharmacol (2017) 8:10. doi: 10.3389/fphar.2017.00010
36. Yu S, Wang D, Huang L, Zhang Y, Luo R, Adah D, et al. The Complement Receptor C5aR2 Promotes Protein Kinase R Expression and Contributes to NLRP3 Inflammasome Activation and HMGB1 Release From Macrophages. J Biol Chem (2019) 294(21):8384–94. doi: 10.1074/jbc.RA118.006508
37. Arbore G, West EE, Spolski R, Robertson AAB, Klos A, Rheinheimer C, et al. T Helper 1 Immunity Requires Complement-Driven NLRP3 Inflammasome Activity in CD4+ T Cells. Science (2016) 352(6292):aad1210. doi: 10.1126/science.aad1210
38. Lee JH, Yang JA, Shin K, Lee GH, Lee WW, Lee EY, et al. Elderly Patients Exhibit Stronger Inflammatory Responses During Gout Attacks. J Korean Med Sci (2017) 32(12):1967–73. doi: 10.3346/jkms.2017.32.12.1967
39. Alberts A, Klingberg A, Wessig AK, Combes C, Witte T, Brand K, et al. C-Reactive Protein (CRP) Recognizes Uric Acid Crystals and Recruits Proteases C1 and MASP1. Sci Rep (2020) 10(1):6391. doi: 10.1038/s41598-020-63318-8
40. Annesley SJ, Fisher PR. Mitochondria in Health and Disease. Cells (2019) 8(7):680. doi: 10.3390/cells8070680
41. Gosling AL, Boocock J, Dalbeth N, Harré Hindmarsh J, Stamp LK, Stahl EA, et al. Mitochondrial Genetic Variation and Gout in Māori and Pacific People Living in Aotearoa New Zealand. Ann Rheum Dis (2018) 77(4):571–8. doi: 10.1136/annrheumdis-2017-212416
42. Yang CS, Kim JJ, Kim TS, Lee PY, Kim SY, Lee HM, et al. Small Heterodimer Partner Interacts With NLRP3 and Negatively Regulates Activation of the NLRP3 Inflammasome. Nat Commun (2015) 6:6115. doi: 10.1038/ncomms7115
43. Qin Q, Liu H, Shou J, Jiang Y, Yu H, Wang X. The Inhibitor Effect of RKIP on Inflammasome Activation and Inflammasome-Dependent Diseases. Cell Mol Immunol (2021) 18(4):992–1004. doi: 10.1038/s41423-020-00525-3
44. Huang Q, Gao W, Mu H, Qin T, Long F, Ren L, et al. HSP60 Regulates Monosodium Urate Crystal-Induced Inflammation by Activating the TLR4-NF-κb-MyD88 Signaling Pathway and Disrupting Mitochondrial Function. Oxid Med Cell Longev (2020) 2020:8706898. doi: 10.1155/2020/8706898
45. Bousoik E, Qadri M, Elsaid KA. CD44 Receptor Mediates Urate Crystal Phagocytosis by Macrophages and Regulates Inflammation in A Murine Peritoneal Model of Acute Gout. Sci Rep (2020) 10(1):5748. doi: 10.1038/s41598-020-62727-z
46. Qadri M, Jay GD, Zhang LX, Wong W, Reginato AM, Sun C, et al. Recombinant Human Proteoglycan-4 Reduces Phagocytosis of Urate Crystals and Downstream Nuclear Factor Kappa B and Inflammasome Activation and Production of Cytokines and Chemokines in Human and Murine Macrophages. Arthritis Res Ther (2018) 20(1):192. doi: 10.1186/s13075-018-1693-x
47. Fattori V, Zarpelon AC, Staurengo-Ferrari L, Borghi SM, Zaninelli TH, Da Costa FB, et al. Budlein A, a Sesquiterpene Lactone From Viguiera Robusta, Alleviates Pain and Inflammation in a Model of Acute Gout Arthritis in Mice. Front Pharmacol (2018) 9:1076. doi: 10.3389/fphar.2018.01076
48. Marchetti C, Swartzwelter B, Koenders MI, Azam T, Tengesdal IW, Powers N, et al. NLRP3 Inflammasome Inhibitor OLT1177 Suppresses Joint Inflammation in Murine Models of Acute Arthritis. Arthritis Res Ther (2018) 20(1):169. doi: 10.1186/s13075-018-1664-2
49. Abhishek A. New Urate-Lowing Therapies. Curr Opin Rheumatol (2018) 30(2):177–82. doi: 10.1097/BOR.0000000000000476
50. Lee HE, Yang G, Kim ND, Jeong S, Jung Y, Choi JY, et al. Targeting ASC in NLRP3 Inflammasome by Caffeic Acid Phenethyl Ester: A Novel Strategy to Treat Acute Gout. Sci Rep (2016) 6:38622. doi: 10.1038/srep38622
51. Wang Y, Zhu W, Lu D, Zhang C, Wang Y. Tetrahydropalmatine Attenuates MSU Crystal-Induced Gouty Arthritis by Inhibiting ROS-Mediated NLRP3 Inflammasome Activation. Int Immunopharmacol. (2021) 100:108107. doi: 10.1016/j.intimp.2021.108107
52. Gu Y, Zhu Y, Deng G, Liu S, Sun Y, Lv W. Curcumin Analogue AI-44 Alleviates MSU-Induced Gouty Arthritis in Mice via Inhibiting Cathepsin B-Mediated NLRP3 Inflammasome Activation. Int Immunopharmacol. (2021) 93:107375. doi: 10.1016/j.intimp.2021.107375
53. Heo KH, Sun X, Shim DW, Kim MK, Koppula S, Yu SH, et al. Actinidia Arguta Extract Attenuates Inflammasome Activation: Potential Involvement in NLRP3 Ubiquitination. J Ethnopharmacol (2018) 213:159–65. doi: 10.1016/j.jep.2017.11.023
54. Shin WY, Shim DW, Kim MK, Sun X, Koppula S, Yu SH, et al. Protective Effects of Cinnamomum Cassia (Lamaceae) Against Gout and Septic Responses via Attenuation of Inflammasome Activation in Experimental Models. J Ethnopharmacol (2017) 205:173–7. doi: 10.1016/j.jep.2017.03.043
55. Zhou GQ, Chen G, Yang J, Qin WY, Ping J. Guizhi-Shaoyao-Zhimu Decoction Attenuates Monosodium Urate Crystal-Induced Inflammation Through Inactivation of NF-κb and NLRP3 Inflammasome. J Ethnopharmacol (2022) 283:114707. doi: 10.1016/j.jep.2021.114707
56. Xue Y, Li R, Fang P, Ye ZQ, Zhao Y, Zhou Y, et al. NLRP3 Inflammasome Inhibitor Cucurbitacin B Suppresses Gout Arthritis in Mice. J Mol Endocrinol (2021) 67(2):27–40. doi: 10.1530/JME-20-0305
57. Misawa T, Saitoh T, Kozaki T, Park S, Takahama M, Akira S. Resveratrol Inhibits the Acetylated α-Tubulin-Mediated Assembly of the NLRP3-Inflammasome. Int Immunol (2015) 27(9):425–34. doi: 10.1093/intimm/dxv018
58. Mian W, Zhang M, Ma Y, Liu F, Chen S, Lu J, et al. Chaetocin Attenuates Gout in Mice Through Inhibiting HIF-1α and NLRP3 Inflammasome-Dependent IL-1β Secretion in Macrophages. Arch Biochem Biophys (2019) 670:94–103. doi: 10.1016/j.abb.2019.06.010
59. Lo CW, Lii CK, Hong JJ, Chuang WT, Yang YC, Huang CS, et al. Andrographolide Inhibits IL-1β Release in Bone Marrow-Derived Macrophages and Monocyte Infiltration in Mouse Knee Joints Induced by Monosodium Urate. Toxicol Appl Pharmacol (2021) 410:115341. doi: 10.1016/j.taap.2020.115341
60. Choi N, Yang G, Jang JH, Kang HC, Cho YY, Lee HS, et al. Loganin Alleviates Gout Inflammation by Suppressing NLRP3 Inflammasome Activation and Mitochondrial Damage. Molecules (2021) 26(4):1071. doi: 10.3390/molecules26041071
61. Ruiz-Miyazawa KW, Staurengo-Ferrari L, Mizokami SS, Domiciano TP, Vicentini F, Camilios-Neto D, et al. Quercetin Inhibits Gout Arthritis in Mice: Induction of an Opioid-Dependent Regulation of Inflammasome. Inflammopharmacology (2017). doi: 10.1007/s10787-017-0356-x
62. Qiao CY, Li Y, Shang Y, Jiang M, Liu J, Zhan ZY, et al. Management of Gout-Associated MSU Crystals-Induced NLRP3 Inflammasome Activation by Procyanidin B2: Targeting IL-1β and Cathepsin B in Macrophages. Inflammopharmacology (2020) 28(6):1481–93. doi: 10.1007/s10787-020-00758-8
63. Cocco M, Miglio G, Giorgis M, Garella D, Marini E, Costale A, et al. Design, Synthesis, and Evaluation of Acrylamide Derivatives as Direct NLRP3 Inflammasome Inhibitors. ChemMedChem (2016) 11(16):1790–803. doi: 10.1002/cmdc.201600055
64. Lee YM, Shon EJ, Kim OS, Kim DS. Effects of Mollugo Pentaphylla Extract on Monosodium Urate Crystal-Induced Gouty Arthritis in Mice. BMC Complement Altern Med (2017) 17(1):447. doi: 10.1186/s12906-017-1955-1
65. Chen G, Guo T, Yang L. Paeonol Reduces IL-β Production by Inhibiting the Activation of Nucleotide Oligomerization Domain-Like Receptor Protein-3 Inflammasome and Nuclear Factor-κb in Macrophages. Biochem Cell Biol (2022) 100(1):28–36. doi: 10.1139/bcb-2021-0255
66. Zhang J, Lei H, Li X. The Protective Effects of S14G-Humanin (HNG) Against Mono-Sodium Urate (MSU) Crystals- Induced Gouty Arthritis. Bioengineered (2022) 13(1):345–56. doi: 10.1080/21655979.2021.2001911
67. Wang Q, Lin B, Li Z, Su J, Feng Y. Cichoric Acid Ameliorates Monosodium Urate-Induced Inflammatory Response by Reducing NLRP3 Inflammasome Activation via Inhibition of NF-kB Signaling Pathway. Evid Based Complement Alternat Med (2021) 2021:8868527. doi: 10.1155/2021/8868527
68. Yuan X, Fan YS, Xu L, Xie GQ, Feng XH, Qian K. Jia-Wei-Si-Miao-Wan Alleviates Acute Gouty Arthritis by Targeting NLRP3 Inflammasome. J Biol Regul Homeost Agents. (2019) 33(1):63–71.
69. Tapia-Abellán A, Angosto-Bazarra D, Martínez-Banaclocha H, de Torre-Minguela C, Cerón-Carrasco JP, Pérez-Sánchez H, et al. MCC950 Closes the Active Conformation of NLRP3 to an Inactive State. Nat Chem Biol (2019) 15(6):560–4. doi: 10.1038/s41589-019-0278-6
70. Yin C, Liu B, Wang P, Li X, Li Y, Zheng X, et al. Eucalyptol Alleviates Inflammation and Pain Responses in a Mouse Model of Gout Arthritis. Br J Pharmacol (2020) 177(9):2042–57. doi: 10.1111/bph.14967
71. Liu HJ, Pan XX, Liu BQ, Gui X, Hu L, Jiang CY, et al. Grape Seed-Derived Procyanidins Alleviate Gout Pain via NLRP3 Inflammasome Suppression. J Neuroinflammation. (2017) 14(1):74. doi: 10.1186/s12974-017-0849-y
72. Lee HE, Yang G, Park YB, Kang HC, Cho YY, Lee HS, et al. Epigallocatechin-3-Gallate Prevents Acute Gout by Suppressing NLRP3 Inflammasome Activation and Mitochondrial DNA Synthesis. Molecules (2019) 24(11):2138. doi: 10.3390/molecules24112138
73. Jhang JJ, Lu CC, Ho CY, Cheng YT, Yen GC. Protective Effects of Catechin Against Monosodium Urate-Induced Inflammation Through the Modulation of NLRP3 Inflammasome Activation. J Agric Food Chem (2015) 63(33):7343–52. doi: 10.1021/acs.jafc.5b02605
74. Lin X, Wang H, An X, Zhang J, Kuang J, Hou J, et al. Baeckein E Suppressed NLRP3 Inflammasome Activation Through Inhibiting Both the Priming and Assembly Procedure: Implications for Gout Therapy. Phytomedicine (2021) 84:153521. doi: 10.1016/j.phymed.2021.153521
75. Chen B, Li H, Ou G, Ren L, Yang X, Zeng M. Curcumin Attenuates MSU Crystal-Induced Inflammation by Inhibiting the Degradation of Iκbα and Blocking Mitochondrial Damage. Arthritis Res Ther (2019) 21(1):193. doi: 10.1186/s13075-019-1974-z
76. Rossaneis AC, Longhi-Balbinot DT, Bertozzi MM, Fattori V, Segato-Vendrameto CZ, Badaro-Garcia S, et al. [Ru(bpy)(2)(NO)SO(3)](PF(6)), a Nitric Oxide Donating Ruthenium Complex, Reduces Gout Arthritis in Mice. Front Pharmacol (2019) 10:229. doi: 10.3389/fphar.2019.00229
77. Staurengo-Ferrari L, Ruiz-Miyazawa KW, Pinho-Ribeiro FA, Fattori V, Zaninelli TH, Badaro-Garcia S, et al. Trans-Chalcone Attenuates Pain and Inflammation in Experimental Acute Gout Arthritis in Mice. Front Pharmacol (2018) 9:1123. doi: 10.3389/fphar.2018.01123
78. Deng W, Yang Z, Yue H, Ou Y, Hu W, Sun P. Disulfiram Suppresses NLRP3 Inflammasome Activation to Treat Peritoneal and Gouty Inflammation. Free Radic Biol Med (2020) 152:8–17. doi: 10.1016/j.freeradbiomed.2020.03.007
79. Zhang X, Hu L, Xu S, Ye C, Chen A. Erianin: A Direct NLRP3 Inhibitor With Remarkable Anti-Inflammatory Activity. Front Immunol (2021) 12:739953. doi: 10.3389/fimmu.2021.739953
80. Huang Y, Jiang H, Chen Y, Wang X, Yang Y, Tao J, et al. Tranilast Directly Targets NLRP3 to Treat Inflammasome-Driven Diseases. EMBO Mol Med (2018) 10(4):e8689. doi: 10.15252/emmm.201708689
81. Yang G, Lee HE, Moon SJ, Ko KM, Koh JH, Seok JK, et al. Direct Binding to NLRP3 Pyrin Domain as a Novel Strategy to Prevent NLRP3-Driven Inflammation and Gouty Arthritis. Arthritis Rheumatol (2020) 72(7):1192–202. doi: 10.1002/art.41245
82. Chang YH, Chiang YF, Chen HY, Huang YJ, Wang KL, Hong YH, et al. Anti-Inflammatory and Anti-Hyperuricemic Effects of Chrysin on a High Fructose Corn Syrup-Induced Hyperuricemia Rat ModelVia the Amelioration of Urate Transporters and Inhibition of NLRP3 Inflammasome Signaling Pathway. Antioxidants (Basel) (2021) 10(4):564. doi: 10.3390/antiox10040564
83. Yang G, Yeon SH, Lee HE, Kang HC, Cho YY, Lee HS, et al. Suppression of NLRP3 Inflammasome by Oral Treatment With Sulforaphane Alleviates Acute Gouty Inflammation. Rheumatol (Oxford) (2018) 57(4):727–36. doi: 10.1093/rheumatology/kex499
84. Wu J, Luo Y, Jiang Q, Li S, Huang W, Xiang L, et al. Coptisine From Coptis Chinensis Blocks NLRP3 Inflammasome Activation by Inhibiting Caspase-1. Pharmacol Res (2019) 147:104348. doi: 10.1016/j.phrs.2019.104348
85. Kim SK, Choe JY, Park KY. Anti-Inflammatory Effect of Artemisinin on Uric Acid-Induced NLRP3 Inflammasome Activation Through Blocking Interaction Between NLRP3 and NEK7. Biochem Biophys Res Commun (2019) 517(2):338–45. doi: 10.1016/j.bbrc.2019.07.087
86. Lou D, Zhang X, Jiang C, Zhang F, Xu C, Fang S, et al. 3β,23-Dihydroxy-12-Ene-28-Ursolic Acid Isolated From Cyclocarya Paliurus Alleviates NLRP3 Inflammasome-Mediated Gout via PI3K-AKT-mTOR-Dependent Autophagy. Evid Based Complement Alternat Med (2022) 2022:5541232. doi: 10.1155/2022/5541232
87. Chang WC, Chu MT, Hsu CY, Wu YJ, Lee JY, Chen TJ, et al. Rhein, An Anthraquinone Drug, Suppresses the NLRP3 Inflammasome and Macrophage Activation in Urate Crystal-Induced Gouty Inflammation. Am J Chin Med (2019) 47(1):135–51. doi: 10.1142/S0192415X19500071
88. So A, De Smedt T, Revaz S, Tschopp J. A Pilot Study of IL-1 Inhibition by Anakinra in Acute Gout. Arthritis Res Ther (2007) 9(2):R28. doi: 10.1186/ar2143
89. Wang Y, Viollet B, Terkeltaub R, Liu-Bryan R. AMP-Activated Protein Kinase Suppresses Urate Crystal-Induced Inflammation and Transduces Colchicine Effects in Macrophages. Ann Rheum Dis (2016) 75(1):286–94. doi: 10.1136/annrheumdis-2014-206074
90. McWherter C, Choi YJ, Serrano RL, Mahata SK, Terkeltaub R, Liu-Bryan R. Arhalofenate Acid Inhibits Monosodium Urate Crystal-Induced Inflammatory Responses Through Activation of AMP-Activated Protein Kinase (AMPK) Signaling. Arthritis Res Ther (2018) 20(1):204. doi: 10.1186/s13075-018-1699-4
91. Yue H, Yang Z, Ou Y, Liang S, Deng W, Chen H, et al. Tanshinones Inhibit NLRP3 Inflammasome Activation by Alleviating Mitochondrial Damage to Protect Against Septic and Gouty Inflammation. Int Immunopharmacol. (2021) 97:107819. doi: 10.1016/j.intimp.2021.107819
92. Sil P, Hayes CP, Reaves BJ, Breen P, Quinn S, Sokolove J, et al. P2Y6 Receptor Antagonist MRS2578 Inhibits Neutrophil Activation and Aggregated Neutrophil Extracellular Trap Formation Induced by Gout-Associated Monosodium Urate Crystals. J Immunol (2017) 198(1):428–42. doi: 10.4049/jimmunol.1600766
93. Galvão I, Queiroz-Junior CM, de Oliveira VLS, Pinho V, Hirsch E, Teixeira MM. The Inhibition of Phosphoinositide-3 Kinases Induce Resolution of Inflammation in a Gout Model. Front Pharmacol (2018) 9:1505. doi: 10.3389/fphar.2018.01505
94. Galvão I, Melo EM, de Oliveira VLS, Vago JP, Queiroz-Junior C, de Gaetano M, et al. Therapeutic Potential of the FPR2/ALX Agonist AT-01-KG in the Resolution of Articular Inflammation. Pharmacol Res (2021) 165:105445. doi: 10.1016/j.phrs.2021.105445
95. Huang J, Zhou Z, Zhou M, Miao M, Li H, Hu Q. Development of Benzoxazole Deoxybenzoin Oxime and Acyloxylamine Derivatives Targeting Innate Immune Sensors and Xanthine Oxidase for Treatment of Gout. Bioorg Med Chem (2018) 26(8):1653–64. doi: 10.1016/j.bmc.2018.02.013
96. Castelblanco M, Lugrin J, Ehirchiou D, Nasi S, Ishii I, So A, et al. Hydrogen Sulfide Inhibits NLRP3 Inflammasome Activation and Reduces Cytokine Production Both In Vitro and in a Mouse Model of Inflammation. J Biol Chem (2018) 293(7):2546–57. doi: 10.1074/jbc.M117.806869
97. Yu SH, Sun X, Kim MK, Akther M, Han JH, Kim TY, et al. Chrysanthemum Indicum Extract Inhibits NLRP3 and AIM2 Inflammasome Activation via Regulating ASC Phosphorylation. J Ethnopharmacol (2019) 239:111917. doi: 10.1016/j.jep.2019.111917
98. Greaney AJ, Maier NK, Leppla SH, Moayeri M. Sulforaphane Inhibits Multiple Inflammasomes Through an Nrf2-Independent Mechanism. J Leukoc Biol (2016) 99(1):189–99. doi: 10.1189/jlb.3A0415-155RR
99. Ahn H, Kang SG, Yoon SI, Ko HJ, Kim PH, Hong EJ, et al. Methylene Blue Inhibits NLRP3, NLRC4, AIM2, and Non-Canonical Inflammasome Activation. Sci Rep (2017) 7(1):12409. doi: 10.1038/s41598-017-12635-6
100. Schmid-Burgk JL, Chauhan D, Schmidt T, Ebert TS, Reinhardt J, Endl E, et al. A Genome-Wide CRISPR (Clustered Regularly Interspaced Short Palindromic Repeats) Screen Identifies NEK7 as an Essential Component of NLRP3 Inflammasome Activation. J Biol Chem (2016) 291(1):103–9. doi: 10.1074/jbc.C115.700492
101. Jhang JJ, Lu CC, Yen GC. Epigallocatechin Gallate Inhibits Urate Crystals-Induced Peritoneal Inflammation in C57BL/6 Mice. Mol Nutr Food Res (2016) 60(10):2297–303. doi: 10.1002/mnfr.201600106
102. Pan YG, Huang MT, Sekar P, Huang DY, Lin WW, Hsieh SL. Decoy Receptor 3 Inhibits Monosodium Urate-Induced NLRP3 Inflammasome Activation via Reduction of Reactive Oxygen Species Production and Lysosomal Rupture. Front Immunol (2021) 12:638676. doi: 10.3389/fimmu.2021.638676
103. Ahn H, Lee G, Lee GS. Lower Temperatures Exacerbate NLRP3 Inflammasome Activation by Promoting Monosodium Urate Crystallization, Causing Gout. Cells (2021) 10(8):1919. doi: 10.3390/cells10081919
104. Ottaviani S, Moltó A, Ea HK, Neveu S, Gill G, Brunier L, et al. Efficacy of Anakinra in Gouty Arthritis: A Retrospective Study of 40 Cases. Arthritis Res Ther (2013) `15(5):R123. doi: 10.1186/ar4303
105. Gross O, Yazdi AS, Thomas CJ, Masin M, Heinz LX, Guarda G, et al. Inflammasome Activators Induce Interleukin-1α Secretion via Distinct Pathways With Differential Requirement for the Protease Function of Caspase-1. Immunity (2012) 36(3):388–400. doi: 10.1016/j.immuni.2012.01.018
106. Yazdi AS, Drexler SK. Regulation of Interleukin 1α Secretion by Inflammasomes. Ann Rheum Dis (2013) 72 Suppl 2:ii96–9. doi: 10.1136/annrheumdis-2012-202252
107. Rashidi M, Simpson DS, Hempel A, Frank D, Petrie E, Vince A, et al. The Pyroptotic Cell Death Effector Gasdermin D Is Activated by Gout-Associated Uric Acid Crystals But Is Dispensable for Cell Death and IL-1β Release. J Immunol (2019) 203(3):736–48. doi: 10.4049/jimmunol.1900228
108. Klionsky DJ, Petroni G, Amaravadi RK, Baehrecke EH, Ballabio A, Boya P, et al. Autophagy in Major Human Diseases. EMBO J (2021) 40(19):e108863. doi: 10.15252/embj.2021108863
109. Allaeys I, Marceau F, Poubelle PE. NLRP3 Promotes Autophagy of Urate Crystals Phagocytized by Human Osteoblasts. Arthritis Res Ther (2013) 15(6):R176. doi: 10.1186/ar4365
110. Yang QB, He YL, Zhong XW, Xie WG, Zhou JG. Resveratrol Ameliorates Gouty Inflammation via Upregulation of Sirtuin 1 to Promote Autophagy in Gout Patients. Inflammopharmacology (2019) 27(1):47–56. doi: 10.1007/s10787-018-00555-4
111. Wang Z, Zhao Y, Phipps-Green A, Liu-Bryan R, Ceponis A, Boyle DL, et al. Differential DNA Methylation of Networked Signaling, Transcriptional, Innate and Adaptive Immunity, and Osteoclastogenesis Genes and Pathways in Gout. Arthritis Rheumatol (2020) 72(5):802–14. doi: 10.1002/art.41173
112. Nolasco S, Bellido J, Serna M, Carmona B, Soares H, Zabala JC. Colchicine Blocks Tubulin Heterodimer Recycling by Tubulin Cofactors TBCA, TBCB, and TBCE. Front Cell Dev Biol (2021) 9:656273. doi: 10.3389/fcell.2021.656273
113. Zhong Z, Umemura A, Sanchez-Lopez E, Liang S, Shalapour S, Wong J, et al. NF-κb Restricts Inflammasome Activation via Elimination of Damaged Mitochondria. Cell (2016) 164(5):896–910. doi: 10.1016/j.cell.2015.12.057
114. Choe JY, Jung HY, Park KY, Kim SK. Enhanced P62 Expression Through Impaired Proteasomal Degradation is Involved in Caspase-1 Activation in Monosodium Urate Crystal-Induced Interleukin-1b Expression. Rheumatol (Oxford) (2014) 53(6):1043–53. doi: 10.1093/rheumatology/ket474
115. Jhang JJ, Cheng YT, Ho CY, Yen GC. Monosodium Urate Crystals Trigger Nrf2- and Heme Oxygenase-1-Dependent Inflammation in THP-1 Cells. Cell Mol Immunol (2015) 12(4):424–34. doi: 10.1038/cmi.2014.65
116. Cleophas MC, Crişan TO, Joosten LA. Factors Modulating the Inflammatory Response in Acute Gouty Arthritis. Curr Opin Rheumatol (2017) 29(2):163–70. doi: 10.1097/BOR.0000000000000366
117. Papayannopoulos V. Neutrophil Extracellular Traps in Immunity and Disease. Nat Rev Immunol (2018) 18(2):134–47. doi: 10.1038/nri.2017.105
118. Rada B. Neutrophil Extracellular Traps. Methods Mol Biol (2019) 1982:517–28. doi: 10.1007/978-1-4939-9424-3_31
119. Fuchs TA, Abed U, Goosmann C, Hurwitz R, Schulze I, Wahn V, et al. Novel Cell Death Program Leads to Neutrophil Extracellular Traps. J Cell Biol (2007) 176(2):231–41. doi: 10.1083/jcb.200606027
120. Metzler KD, Goosmann C, Lubojemska A, Zychlinsky A, Papayannopoulos V. A Myeloperoxidase-Containing Complex Regulates Neutrophil Elastase Release and Actin Dynamics During NETosis. Cell Rep (2014) 8(3):883–96. doi: 10.1016/j.celrep.2014.06.044
121. Papayannopoulos V, Metzler KD, Hakkim A, Zychlinsky A. Neutrophil Elastase and Myeloperoxidase Regulate the Formation of Neutrophil Extracellular Traps. J Cell Biol (2010) 191(3):677–91. doi: 10.1083/jcb.201006052
122. Desai J, Mulay SR, Nakazawa D, Anders HJ. Matters of Life and Death. How Neutrophils Die or Survive Along NET Release and Is "NETosis" = Necroptosis? Cell Mol Life Sci (2016) 73(11-12):2211–9. doi: 10.1007/s00018-016-2195-0
123. Kumar SV, Kulkarni OP, Mulay SR, Darisipudi MN, Romoli S, Thomasova D, et al. Neutrophil Extracellular Trap-Related Extracellular Histones Cause Vascular Necrosis in Severe Gn. J Am Soc Nephrol (2015) 26(10):2399–413. doi: 10.1681/ASN.2014070673
124. Xu J, Zhang X, Pelayo R, Monestier M, Ammollo CT, Semeraro F, et al. Extracellular Histones are Major Mediators of Death in Sepsis. Nat Med (2009) 15(11):1318–21. doi: 10.1038/nm.2053
125. Garcia-Gonzalez E, Gamberucci A, Lucherini OM, Alì A, Simpatico A, Lorenzini S, et al. Neutrophil Extracellular Traps Release in Gout and Pseudogout Depends on the Number of Crystals Regardless of Leukocyte Count. Rheumatol (Oxford) (2021) 60(10):4920–8. doi: 10.1093/rheumatology/keab087
126. Vedder D, Gerritsen M, Nurmohamed MT, van Vollenhoven RF, Lood C. A Neutrophil Signature Is Strongly Associated With Increased Cardiovascular Risk in Gout. Rheumatol (Oxford) (2021) 60(6):2783–90. doi: 10.1093/rheumatology/keaa712
127. Pascual E. Persistence of Monosodium Urate Crystals and Low-Grade Inflammation in the Synovial Fluid of Patients With Untreated Gout. Arthritis Rheum (1991) 34(2):141–5. doi: 10.1002/art.1780340203
128. Oliviero F, Scanu A, Punzi L. Metabolism of Crystals Within the Joint. Reumatismo (2012) 63(4):221–9. doi: 10.4081/reumatismo.2011.221
129. De Miguel E, Puig JG, Castillo C, Peiteado D, Torres RJ, Martín-Mola E. Diagnosis of Gout in Patients With Asymptomatic Hyperuricaemia: A Pilot Ultrasound Study. Ann Rheum Dis (2012) 71(1):157–8. doi: 10.1136/ard.2011.154997
130. Dalbeth N, House ME, Aati O, Tan P, Franklin C, Horne A, et al. Urate Crystal Deposition in Asymptomatic Hyperuricaemia and Symptomatic Gout: A Dual Energy CT Study. Ann Rheum Dis (2015) 74(5):908–11. doi: 10.1136/annrheumdis-2014-206397
131. Andrés M, Quintanilla MA, Sivera F, Sánchez-Payá J, Pascual E, Vela P, et al. Silent Monosodium Urate Crystal Deposits Are Associated With Severe Coronary Calcification in Asymptomatic Hyperuricemia: An Exploratory Study. Arthritis Rheumatol (2016) 68(6):1531–9. doi: 10.1002/art.39581
132. Schett G, Schauer C, Hoffmann M, Herrmann M. Why Does the Gout Attack Stop? A Roadmap for the Immune Pathogenesis of Gout. RMD Open (2015) 1(Suppl 1):e000046. doi: 10.1136/rmdopen-2015-000046
133. Schorn C, Janko C, Latzko M, Chaurio R, Schett G, Herrmann M. Monosodium Urate Crystals Induce Extracellular DNA Traps in Neutrophils, Eosinophils, and Basophils But Not in Mononuclear Cells. Front Immunol (2012) 3:277. doi: 10.3389/fimmu.2012.00277
134. Schauer C, Janko C, Munoz LE, Zhao Y, Kienhöfer D, Frey B, et al. Aggregated Neutrophil Extracellular Traps Limit Inflammation by Degrading Cytokines and Chemokines. Nat Med (2014) 20(5):511–7. doi: 10.1038/nm.3547
135. Desai J, Foresto-Neto O, Honarpisheh M, Steiger S, Nakazawa D, Popper B, et al. Particles of Different Sizes and Shapes Induce Neutrophil Necroptosis Followed by the Release of Neutrophil Extracellular Trap-Like Chromatin. Sci Rep (2017) 7(1):15003. doi: 10.1038/s41598-017-15106-0
136. Zhang J, Yang Y, He W, Sun L. Necrosome Core Machinery: MLKL. Cell Mol Life Sci (2016) 73(11-12):2153–63. doi: 10.1007/s00018-016-2190-5
137. Zhao J, Jiang P, Guo S, Schrodi SJ, He D. Apoptosis, Autophagy, NETosis, Necroptosis, and Pyroptosis Mediated Programmed Cell Death as Targets for Innovative Therapy in Rheumatoid Arthritis. Front Immunol (2021) 12:809806. doi: 10.3389/fimmu.2021.809806
138. Mulay SR, Desai J, Kumar SV, Eberhard JN, Thomasova D, Romoli S, et al. Cytotoxicity of Crystals Involves RIPK3-MLKL-Mediated Necroptosis. Nat Commun (2016) 7:10274. doi: 10.1038/ncomms10274
139. Desai J, Kumar SV, Mulay SR, Konrad L, Romoli S, Schauer C, et al. PMA and Crystal-Induced Neutrophil Extracellular Trap Formation Involves RIPK1-RIPK3-MLKL Signaling. Eur J Immunol (2016) 46(1):223–9. doi: 10.1002/eji.201545605
140. Sil P, Wicklum H, Surell C, Rada B. Macrophage-Derived IL-1β Enhances Monosodium Urate Crystal-Triggered NET Formation. Inflammation Res (2017) 66(3):227–37. doi: 10.1007/s00011-016-1008-0
141. Caution K, Young N, Robledo-Avila F, Krause K, Abu Khweek A, Hamilton K, et al. Caspase-11 Mediates Neutrophil Chemotaxis and Extracellular Trap Formation During Acute Gouty Arthritis Through Alteration of Cofilin Phosphorylation. Front Immunol (2019) 10:2519. doi: 10.3389/fimmu.2019.02519
142. Kukulski F, Ben Yebdri F, Bahrami F, Fausther M, Tremblay A, Sévigny J. Endothelial P2Y2 Receptor Regulates LPS-Induced Neutrophil Transendothelial Migration In Vitro. Mol Immunol (2010) 47(5):991–9. doi: 10.1016/j.molimm.2009.11.020
143. Tang H, Tan C, Cao X, Liu Y, Zhao H, Liu Y, et al. NFIL3 Facilitates Neutrophil Autophagy, Neutrophil Extracellular Trap Formation and Inflammation During Gout via REDD1-Dependent mTOR Inactivation. Front Med (Lausanne) (2021) 8:692781. doi: 10.3389/fmed.2021.692781
144. Remijsen Q, Vanden Berghe T, Wirawan E, Asselbergh B, Parthoens E, De Rycke R, et al. Neutrophil Extracellular Trap Cell Death Requires Both Autophagy and Superoxide Generation. Cell Res (2011) 21(2):290–304. doi: 10.1038/cr.2010.150
145. Mitroulis I, Kambas K, Chrysanthopoulou A, Skendros P, Apostolidou E, Kourtzelis I, et al. Neutrophil Extracellular Trap Formation is Associated With IL-1β and Autophagy-Related Signaling in Gout. PloS One (2011) 6(12):e29318. doi: 10.1371/journal.pone.0029318
146. Schweyer S, Hemmerlein B, Radzun HJ, Fayyazi A. Continuous Recruitment, Co-Expression of Tumour Necrosis Factor-Alpha and Matrix Metalloproteinases, and Apoptosis of Macrophages in Gout Tophi. Virchows Arch (2000) 437(5):534–9. doi: 10.1007/s004280000282
147. Zamudio-Cuevas Y, Martínez-Flores K, Fernández-Torres J, Loissell-Baltazar YA, Medina-Luna D, López-Macay A, et al. Monosodium Urate Crystals Induce Oxidative Stress in Human Synoviocytes. Arthritis Res Ther (2016) 18(1):117. doi: 10.1186/s13075-016-1012-3
148. Shi L, Xu L, Yang Y, Song H, Pan H, Yin L. Suppressive Effect of Modified Simiaowan on Experimental Gouty Arthritis: An In Vivo and In Vitro Study. J Ethnopharmacol (2013) 150(3):1038–44. doi: 10.1016/j.jep.2013.10.023
149. El Kebir D, Filep JG. Targeting Neutrophil Apoptosis for Enhancing the Resolution of Inflammation. Cells (2013) 2(2):330–48. doi: 10.3390/cells2020330
150. Valiate BVS, Queiroz-Junior CM, Levi-Schaffer F, Galvão I, Teixeira MM. CD300a Contributes to the Resolution of Articular Inflammation Triggered by MSU Crystals by Controlling Neutrophil Apoptosis. Immunology (2021) 164(2):305–17. doi: 10.1111/imm.13371
151. Galvão I, Vago JP, Barroso LC, Tavares LP, Queiroz-Junior CM, Costa VV, et al. Annexin A1 Promotes Timely Resolution of Inflammation in Murine Gout. Eur J Immunol (2017) 47(3):585–96. doi: 10.1002/eji.201646551
152. Galvão I, Athayde RM, Perez DA, Reis AC, Rezende L, de Oliveira VLS, et al. ROCK Inhibition Drives Resolution of Acute Inflammation by Enhancing Neutrophil Apoptosis. Cells (2019) 8(9):964. doi: 10.3390/cells8090964
153. Liang H, Deng P, Ma YF, Wu Y, Ma ZH, Zhang W, et al. Advances in Experimental and Clinical Research of the Gouty Arthritis Treatment With Traditional Chinese Medicine. Evid Based Complement Alternat Med (2021) 2021:8698232. doi: 10.1155/2021/8698232
154. Swanepoel B, Venables L, Olaru OT, Nitulescu GM, van de Venter M. In Vitro Anti-Proliferative Activity and Mechanism of Action of Anemone Nemorosa. Int J Mol Sci (2019) 20(5):1217. doi: 10.3390/ijms20051217
155. Ravi A, Mallika A, Sama V, Begum AS, Khan RS, Reddy BM. Antiproliferative Activity and Standardization of Tecomella Undulata Bark Extract on K562 Cells. J Ethnopharmacol (2011) 137(3):1353–9. doi: 10.1016/j.jep.2011.07.067
156. Choi SW, Ryu OH, Choi SJ, Song IS, Bleyer AJ, Hart TC. Mutant Tamm-Horsfall Glycoprotein Accumulation in Endoplasmic Reticulum Induces Apoptosis Reversed by Colchicine and Sodium 4-Phenylbutyrate. J Am Soc Nephrol (2005) 16(10):3006–14. doi: 10.1681/ASN.2005050461
157. Yan B, Liu D, Zhu J, Pang X. The Effects of Hyperuricemia on the Differentiation and Proliferation of Osteoblasts and Vascular Smooth Muscle Cells are Implicated in the Elevated Risk of Osteopenia and Vascular Calcification in Gout: An In Vivo and In Vitro Analysis. J Cell Biochem (2019) 120(12):19660–72. doi: 10.1002/jcb.29272
Keywords: gout, inflammation, programmed cell death, pyroptosis, NETosis, necroptosis, apoptosis
Citation: Zhao J, Wei K, Jiang P, Chang C, Xu L, Xu L, Shi Y, Guo S, Xue Y and He D (2022) Inflammatory Response to Regulated Cell Death in Gout and Its Functional Implications. Front. Immunol. 13:888306. doi: 10.3389/fimmu.2022.888306
Received: 02 March 2022; Accepted: 17 March 2022;
Published: 06 April 2022.
Edited by:
Haitao Wang, National Cancer Institute (NIH), United StatesReviewed by:
Heng Sun, University of Macau, ChinaXinwei Wu, National Institutes of Health (NIH), United States
Wenjie Wang, National Institutes of Health (NIH), United States
Weiqian Tian, Affiliated Hospital of Nanjing University of Chinese Medicine, China
Copyright © 2022 Zhao, Wei, Jiang, Chang, Xu, Xu, Shi, Guo, Xue and He. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yu Xue, eXh1ZUB1bmlyaGV1bWEub3Jn; Shicheng Guo, U2hpY2hlbmcuR3VvQHdpc2MuZWR1; Dongyi He, ZG9uZ3lpaGVAbWVkbWFpbC5jb20uY24=