 Tracoyia Roach
Tracoyia Roach Laurence Morel
Laurence Morel- Department of Pathology, Immunology, and Laboratory Medicine, University of Florida, Gainesville, FL, United States
Immune homeostasis is a constant balancing act between effector T cells and regulatory T cells defined by Foxp3 expression, the transcription factor that drives their differentiation and immunosuppressive activity. Immune homeostasis is altered when Treg cells are not generated or maintained in sufficient numbers. Treg cells rendered unstable by loss of Foxp3 expression, known as ex-Treg cells, gain pro-inflammatory functions. Treg cells may also become dysfunctional and lose their suppressive capabilities. These alterations can cause an imbalance between effector and regulatory subsets, which may ultimately lead to autoimmunity. This review discusses recent studies that identified genetic factors that maintain Treg cell stability as well as preserve their suppressive function. We focus on studies associated with systemic lupus erythematosus and highlight their findings in the context of potential therapeutic gene targeting in Treg cells to reverse the phenotypic changes and functional dysregulation inducing autoimmunity.
Introduction
Regulatory T cells maintain immune homeostasis and prevent autoimmune diseases by limiting the responses of proinflammatory and autoimmune T cells. Several subsets of Treg cells have been characterized, among which the classical Foxp3+ CD4+ T cells, referred to here as Treg cells, play an essential role. The mechanisms by which these cells maintain immune homeostasis involve inhibitory cytokines, cytolysis, and metabolic disruption of effector T (Teff) cells (1). Treg cells are defined by the stable expression of Foxp3, a forkhead/winged helix transcription factor, and high levels of the high affinity interleukin-2 receptor (IL-2R) α chain (CD25) on their surface (2), which are the main genes required for Treg cell development, maintenance, and function (3). There are two major types of Treg cells: thymus Tregs (tTregs) that develop in the thymus, and peripheral Treg (pTregs) cells that are generated in peripheral sites. In addition, studies have been conducted on induced Treg (iTregs) cells that are induced in vitro by T cell receptor (TCR) activation in the presence of TGFβ (4). Treg cell stability, i.e. the maintenance of their transcriptional program, is indispensable for the preservation of their function. Furthermore, unstable or “ex-Treg” cells induce inflammation not just by a lack of suppression but also in a direct manner by secreting inflammatory cytokines (5).
Defective Number or Frequency of Tregs Lead to Autoimmune Diseases
Numerical and/or functional Treg anomalies contribute to autoimmune diseases such as type 1 diabetes (6), rheumatoid arthritis (7), and systemic lupus erythematosus (SLE) (8). The absolute number of circulating Treg cells is decreased in SLE patients with active disease as compared to healthy controls (9–13). The number of Treg cells was shown to have a strong inverse correlation with SLEDAI (Systemic Lupus Erythematosus Disease Activity Index) scores, showing the lower numbers of Treg cells corresponding to increased disease severity (11, 12). Controversially, other studies have reported increased (14–16) or similar (17, 18) Treg cell numbers in SLE patients as compared to healthy controls. The discrepancy between these studies has been attributed to different definitions and gating strategies for Treg cells, including the fact that expression of Foxp3 alone is not a reliable marker for human Treg cells, further complicating analyses of their function and stability (12, 19). Variations in the treatment regimen with immunosuppressive drugs may also contribute to the large variations in relative Treg cell frequencies in SLE patients. Dysfunctional Treg cells have also been reported in SLE patients (8), including the expansion of a Treg population with a low CD25 expression (20).
This review discusses recent studies that have identified intrinsic genetic factors maintaining Treg cell stability as well as preserving their suppressive function. We focus on studies associated with systemic lupus erythematosus pathogenesis, or with a lupus-like phenotype, and we highlight their findings in the context of potential therapeutic gene targeting in Treg cells to reverse the phenotypic changes and functional dysregulation inducing systemic autoimmunity. The many studies that have reported gene targeting affecting Treg cells in other autoimmune diseases such as arthritis, uveitis or experimental autoimmune encephalomyelitis are not included in this review. Additionally, other studies that have reported deletions of specific genes in other cell types, such as dendritic cells, that affect Treg cell development and function are also not mentioned in this review.
Single Gene Determinants of Treg Cell Homeostasis
Scrufy mice do not produce Treg cells due to a mutation in Foxp3, causing them to develop a severe inflammatory disease with autoimmune components, including lupus-like manifestations (21). A large number of studies have now defined Foxp3 as the master regulator of Treg cell differentiation and functions (3). A recent study has shown that Foxp3 sustained expression is also necessary to maintain Treg functions once they have differentiated (22). Reverse genetic approaches have identified several genes that control Treg cell number, stability and/or functions through Foxp3 expression, and whose deficiency or overexpression lead to autoimmunity or lupus-like manifestations.
Negative Regulators of Foxp3 Expression
The AP1 transcription complex is comprised of a network of heterodimers formed by proteins of the Jun, Fos, ATF, and MAF families. Fos/Jun dimers promote the expression of Foxp3 through direct binding to its promoter in response to TCR signaling (23). Within this transcription complex, Fos-like 2 (Fosl2) inhibits Treg development in a cell-intrinsic manner (24). Fosl2 transgenic mice develop spontaneous autoimmunity and systemic inflammation with disease phenotypes resembling that of Treg-deficient IPEX patients and scurfy mice. On the other hand, mice lacking Fosl2 in CD4+ T cells display less severe disease phenotypes. Mechanistically, Fosl2 interrupts Treg development by repressing the expression of Foxp3 as well as that of other genes involved in Treg differentiation or function (24).
NFIL3 (Nuclear factor Interleukin 3 regulated, also known as E4 binding protein 4, E4BP4) represses numerous genes and regulates diverse biological processes (25, 26). In the immune system, NFIL3/E4BP4 has a vital role for many cell types including Th1, Th2, NKT and Treg cells by regulating the plasticity of cytokine production (27, 28). Treg cells are the T cell subset with the lowest Nfil3 expression, and its overexpression attenuated the suppressive ability and stability of these cells (29). Not only does NFIL3 binds directly to the Foxp3 promoter reducing Foxp3 expression, but it also downregulates the promoter activity of Treg hallmark genes such as Icos, Tnfrsf18, Ctla4, and Il2ra, in both Foxp3-independent and dependent pathways (29). Accordingly, Nfl3-deficiency in T cells increased Foxp3 expression, but decreased the frequency of Foxp3-expressing follicular regulatory T (Tfr) cells, resulting in an expansion of follicular helper T (Tfh) cells and the production of autoantibodies (30). Tfr cells are a specialized subsets of tissue Treg cells that work to constrain the activity of Tfh cells and germinal center (GC) B cells with whom they share the Bcl6 transcription factor (31). A decreased relative frequency of Tfr cells has been correlated with disease activity in SLE patients (32). NFIL3 expression was increased and its phosphorylation was decreased in CD4+ T cells from patients with SLE with a positive correlation to disease activity (30). These alterations were associated with the characteristic expansion of Tfh cells in SLE. It would be of great interest to follow up this study with an analysis of the impact that NFIL3 increased expression and decreased phosphorylation has on Treg and Tfr cell numbers and functions in SLE.
Positive Regulators of Foxp3 Expression
NF-kβ is one of the multi-molecular complexes that interacts with Foxp3 to control Treg cell transcriptional programs and biology. c-Rel, one of its subunits activated by TCR signaling, supports tTreg development and Foxp3 expression by binding to its promoter and one of its regulatory non-coding sequences (CNS3) (33). NF-kβ maintains the stability of mature Treg cells by preventing them from converting into effector-like T cells through mechanisms involving IKKα and IKKβ kinases, which are upstream activators of the NF-kβ pathway (34, 35). Foxp3 forms a complex with Rel-A, one of the most abundant NF-kβ subunits in conventional T cells, and with other transcription factors including Helios and p300, leading to its full functionality as a transcriptional activator (36). Foxp3-Cre mediated depletion of Rel-A in established Treg cells resulted in defective effector Treg cells that led to the development of an autoimmune syndrome characterized by a massive T cell activation, immune infiltrations of several tissues, as well as the production of inflammatory cytokines, and autoantibodies (36, 37). Furthermore, Rel-A deficient Treg cells were unstable and lost Foxp3 expression becoming ex-Tregs expressing high amounts of proinflammatory cytokines IFNγ and TNFα (36).
Bcl10 is a gene in the Carma1-Bcl10-Malt1 (CBM) signaling complex that controls NF-kB and MAPK activation in T cells following TCR activation (38). Bcl10 is necessary for the development of Treg cells and their suppressive function. Bcl10-deficient Treg cells converted in proinflammatory effector T cells secreting IFNγ, leading to a fatal systemic autoimmunity (39). This indicated that Bcl10-mediated NF-kB activation is required for Treg cell development and function. Previous studies have reported that HIF1-α directly binds to the IFNγ promoter in VHL-deficient Treg cells, a model described later in the text, provoking an increased IFNγ production and impairing Treg cell function (40). This phenotype is also displayed in Bcl10-deficient Treg cells (39).
Sclerostin domain-containing protein 1 (SOSTDC1) is selectively expressed in Tfh cells (41), which secretes this factor once they have lost the ability to help GC B cells (42). SOSTDC1 deficiency greatly reduced the generation Tfr cells, which in turn enhanced humoral immunity against viruses (42). Mechanistically, SOSTDC1 inhibits the canonical WNT-β-catenin pathway (43), which in turn inhibits Treg cell differentiation (44, 45). It should be noted that an autoimmune phenotype was not reported in these mice. This implies that although the differentiation of tTreg cells into Tfr cells was impaired, Treg cells themselves were functional and the effect of SOSTDC1 secreted by Tfh cells is confined to the GCs. Inhibition of Tfr cell differentiation in SOSTDC1-deficient mice was mediated by the stabilization of β-catenin (42). As a negative feedback loop, late-stage Tfh cells secrete SOSTDC1, which commits Treg cells in the GC to the Tfr fate by blocking WNT stimuli. Uncontrolled WNT-β-catenin signaling plays a role in autoimmune diseases (46), which may be due, at least in part, to defective Treg and Tfr cell differentiation.
Genes Regulating Treg Cell Function and Stability Through Their Metabolism
Mammalian target of rapamycin corresponds to two kinase complexes, mTORC1 and mTORC2, which function as a central metabolic checkpoint. The functional links between metabolism and effector functions has been dissected in T cells, in which the integration by mTOR of the stimulatory signals and the energy status of the cells plays a critical role (47). Treg cells display diminished activity of the mTOR pathway as compared to Teff cells (46, 47), and increased mTOR activity negatively affects the generation and function of Treg cells (48–51). However, mTORC1 deficiency profoundly impairs Treg development and function (52). Mechanistically, mTORC1 enables cholesterol synthesis and lipid metabolism that are triggered by IL-2 signaling, both for which being required for Treg cell proliferation and the upregulation of suppressive molecules. mTOR signaling is required for the generation and function of both tTregs and pTregs, and its Foxp3-driven deletion impairs mitochondrial metabolism and oxidative phosphorylation, which is the main source of energy in Treg cells (53). Accordingly, Treg-specific deletion of the mitochondrial transcription factor Tfam severely impaired Treg suppressive functions (53). A recent genome-wide CRISPR/Cas9 screen combined with in silico analyses of protein-protein interaction networks identified novel regulatory modules that mediate mTORC1 signaling in Treg cells (54). The requirement for the expression of Sec31a and Ccdc101, two key genes in these modules, was validated when their deficiency in Treg cells impaired their suppressive functions and led to inflammatory phenotypes. SEC31A promotes mTORC1 activation by interacting with the GATOR2 component SEC13 to protect it from SKP1-dependent proteasomal degradation. Therefore, SEC31A expression is necessary to maintain mTORC1 activation in Treg cells. On the other hand, CCDC101 is a member of the SAGA complex, a potent inhibitor of mTORC1. Therefore, CCDC101 limits the expression of glucose and amino acid transporters and maintains a relative metabolic quiescence that characterizes Treg cells. Ccdc101-deficiency impairs Treg cells by unleashing an overreactive mTORC1. Additionally, Lamtor1, a lysosomal scaffold protein for mTORC1 is also important for Treg cell survival. Mice with Lamtor1-deficient Treg cells develop severe autoimmunity showing that Lamtor1 is a vital intrinsic factor for Treg suppressive functions, but not for their development and survival (55).
PP2A is a serine-threonine phosphatase composed of a catalytic C subunit PP2Ac, a scaffold A subunit PP2AA and a regulatory B subunit PP2AB (56). PP2A is highly expressed in Treg cells, and mice with a Treg-specific deletion of a member of the PP2AA subunit developed multi-organ autoimmunity with similarities to the scurfy phenotype (57). This indicated that PP2A activity is required to maintain Treg cells. PP2AA-deficiency increased mTORC1 activity in Treg cells, resulting in enhanced glycolysis and oxidative phosphorylation (57), a phenotype that was reversed by a treatment with mTOR inhibitor rapamycin. Therefore, PP2A activity is necessary to prevent mTORc1 overactivation, a process essential for suppressive function of Treg cells. In addition, PP2Ac is required for Treg cell to function by preventing the loss of expression of the IL-2Rβ chain, enabling IL-2 signaling (58). PPP2R2D is a regulatory subunit of PP2A whose expression is increased in T cells from patients with SLE. Mice with PPP2R2D-deficient T cells developed a reduced systemic autoimmunity in response to TLR7 activation (59). Furthermore, PPP2R2D-deficiency enhanced the suppressive function of Treg cells, which was supported by an increased IL-2 transcription in conventional T cells, a process that is negatively regulated by PPP2R2D (59). Therefore, PPP2R2D regulates Treg cells through PP2A in a cell-extrinsic manner (IL-2 secretion from conventional T cells), as opposed to PP2A controlling Treg function through mTORC1 in a cell-intrinsic manner.
HIF-1α and HIF-2α are two master transcription factors responsible for the physiological responses to hypoxia (60). Under normoxic conditions, prolyl hydroxylase domain proteins (PHD2/PHD3) hydroxylate HIF-1α and HIF-2α allowing for their recognition by von-Hippel Lindau tumor suppressor (VHL)-containing E3 complex, ubiquinating the transcription factors for proteasomal degradation. This process is interrupted under hypoxic conditions, allowing the accumulation of HIF-1α and HIF-2α (61). In immune cells under normoxic conditions, the expression of HIF-1α can also be increased by mTOR activation (62) and induce glycolysis (63). Germline Hif1a-deficiency promoted the differentiation of Treg cells over Th17 cells (64, 65). Mechanistically, HIF-1α promotes Foxp3 degradation by the proteasome (64). Germline Hif1a deficiency also inhibited glycolysis in favor of mitochondrial metabolism, which promoted Treg cell differentiation (65). Interestingly, Hif1a-deficiency in established Treg cells (through Foxp3-Cre mediated deletion) did not impair Treg cell function (66). This indicated that HIF-1α regulates Treg cell differentiation but not their maintenance and function. Hif2a-deficiency in established Treg cells impaired their suppressive activity despite normal Foxp3 expression (66). Moreover, Hif2a-deficient Treg cells showed an enhanced secretion of IL-17 (66). Importantly, patients with SLE and associated lupus nephritis have increased numbers of IL-17-producing Treg cells in their peripheral blood (67). These studies demonstrate a complex crosstalk between HIF-1α and HIF-2α in Treg cells in which HIF-1α prevents their differentiation and HIF-2α stabilizes their function.
VHL-deficiency in Treg cells impaired their suppressive activity and stability leading to massive inflammation (40). VHL-deletion induced a HIF-1α-mediated expression of glycolytic enzymes in Treg cells that promoted Th1 differentiation. Moreover, HIF-1α directly activates the Ifng promoter. These results contrast with the lack of phenotype resulting from direct deletion of Hif1a in Treg cells (59), and suggest that HIF-2α constitutive expression in VHL-deficient Treg cells is likely to play a role.
Serine/Arginine-rich splicing factor 1 (SRSF1) is the prototype member of the highly conserved serine 1 arginine (SR) family of RNA-binding proteins (68). SRSF1 expression was decreased in the T cells of SLE patients with severe disease showing an overactive T cell phenotype (69). Deletion of SRSF1 in T cells led to systemic autoimmunity and lupus nephritis that was associated with mTOR activation in T cells (70). Treg-specific deletion of SRSF1 also led to systemic autoimmunity with Treg cells losing their suppressive function and producing proinflammatory cytokines (70). As with pan-T cell deletion, SRSF1-deficient Tregs displayed a highly glycolytic metabolism and mTOR activation.
Treg Cell Regulation in Spontaneous Mouse Models of Lupus
Many studies have documented alterations in Treg numbers and functions in spontaneous mouse models of lupus (71). Multiple mechanisms are responsible for these phenotypes, with a major contribution of the inflammatory milieu created by cytokines such as Type 1 IFN and IL-6. Whether the genetic susceptibility that drives lupus pathogenesis in these models affects intrinsically Treg cells, at least in part, is less understood. The frequency of Treg cells varies across a wide range in mice and humans without pathogenic consequences (72). NZW mice do not develop autoimmunity, but their genome contains lupus susceptibility genes that are revealed when combined with other genomes such as NZB or BXSB (73). NZW mice present a low frequency of Treg cells, which was found to be cell-intrinsic and due to a low Foxp3 expression leading to a poor stability of the Treg program (72). Although NZW Treg cells express a distinctive transcriptional profile, it could not be attributed to a single genetic defect. Therefore, the NZW Treg phenotype is likely to be supported by a complex polygenic inheritance, similar to lupus susceptibility as a whole in NZW-derived strains (74). However, we propose that these intrinsically defective NZW Treg cells become pathogenic when combined with other immune defects induced by alleles from lupus-prone strains.
The (NZB x NZW) F1-derived NZM2410 strain is a model of lupus in which an analysis of genetic susceptibility has been conducted, and genes regulating T cell function have been identified (74). NZM2410 mice carry three major susceptibility loci associated with lupus nephritis, Sle1, Sle2, and Sle3 (75). Congenic strains carrying separately each of these loci on a non-autoimmune C57BL/6 (B6) background present distinct autoimmune endophenotypes that correspond in combination to the lupus phenotype of the parental strain (76). Sle1 had the strongest linkage to lupus nephritis and its expression is necessary for the development of autoimmunity in NZM2410 mice (77). Sle1 regulates the function of T cells (78) in a cell-intrinsic manner (79), and it decreases the number and function of Treg cells (78). Sle1 corresponds to at least three sub-loci, Sle1a, Sle1b, and Sle1c (80). Within Sle1a, genetic linkage analysis identified an interacting locus Sle1a1 responsible for expanding the number of activated CD4+ T cells while reducing the frequency of pTreg cells (81). Sle1a1 only contains one functional gene, Pbx1 (81), a transcription factor required for mammalian organogenesis (82). Pbx1 is required for the development of B cells and the function of hematopoietic stem cells (83, 84), but its function in T cells had not been characterized. Sle1a1 corresponds to the overexpression of the truncated splice isoform Pbx1-d over Pbx1-b, the normal isoform, in T cells (85). Pbx1-d lacks both the DNA-binding and HOX-binding domains and functions as a dominant negative (86). The mouse and human PBX1 proteins share complete homology, and PBX1-D was found more frequently in the CD4+ T cells from SLE patients than healthy controls (85). Furthermore, PBX1-D expression in human CD4+ T cells is associated with defective Treg cells (87). Mice overexpressing Pbx1-d in T cells replicated the phenotypes of B6.Sle1a1 congenic mice as previously mentioned (88). Pbx1-d transgenic overexpression in T cells impaired iTreg differentiation as well as the induction or maintenance of pTreg cells in a cell-intrinsic manner (88). On the other hand, Pbx1-d overexpression in CD4+ T cells expanded Tfh cell differentiation (88). These results suggest that Pbx1 regulates the balance between Treg and Tfh cells, and that Pbx1-d contributes to autoimmunity by tilting the balance in favor of Tfh over Treg cells. This impaired Pbx1-d-mediated T cell homeostasis has consequences on lupus associated atherosclerosis, with chimeric atherosclerosis-prone mice carrying Pbx1-d expressing T cells developing more severe lesions than mice carrying Pbx1-b expressing T cells (89). Furthermore, there is evidence that dyslipidemia and Pbx1-d expression synergized to impair Treg cell functions. The mechanism by which Pbx1 and its dominant negative Pbx1-d isoform regulate T cell function has not been established yet. Interestingly, Pbx1 directly upregulates NFIL3 expression (90), and NFIL3 regulates the expression of Foxp3 and other Treg-associated genes (29). A disruption of the Pbx1/NFIL3 axis is therefore a potential mechanism by which Pbx1-d may alter the Treg/Tfh cell balance in favor of autoimmunity.
Within the Sle1c locus (91), recombinant congenic analysis mapped an activated CD4+ T cell phenotype to the Sle1c2 sub-locus and the estrogen-related receptor gamma (Esrrg) gene it contains (91). Esrrg is essential in maintaining mitochondrial metabolism through activation of oxidative phosphorylation, the electron transport chain and ATP production in multiple cell types (91), but its function in T cells was unknown. Esrrg expression is reduced in the CD4+ T cells of B6.Sle1c2 congenic mice, in association with altered mitochondrial functions and a decreased mitochondrial mass (91). This phenotype is consistent with that of CD4+ T cells of SLE patients in which mitochondrial defects have been described (92). Esrrg deletion in Treg cells altered the expression of genes involved in mitochondrial and Treg programs (93). This led to impaired suppressive function as well as differentiation into Tfr cells, which allowed for greater Tfh cell and humoral responses. These results suggest that the hypomorph Esrrg lupus susceptibility allele contributes to autoimmune pathogenesis by reducing the metabolic fitness of Treg cells.
Conclusion
In summary, several genes have been identified as being responsible for sustaining the differentiation, function, and stability of Treg cells. The most common approach has been reverse genetics. Only a few Treg-specific studies have been conducted, but continued analyses of selective gene knockouts or overexpression models could advance our knowledge of novel genes that negatively or positively control Treg cells. However, CRISPR/Cas9 screens such as the one recently performed for mTORC1 activation in Treg cells (54) are likely to accelerate the speed of discovery and uncover novel genetic pathways through a less biased evaluation than classical reverse genetic approaches. The dissection of genetic susceptibility in a spontaneous mouse model of lupus has identified two genes that directly impact Treg cell homeostasis. So far, genetic loci associated with human lupus susceptibility, or susceptibility to other autoimmune diseases, have not been linked with Treg phenotypes. It is therefore unknown if allelic variations directly impacting Treg phenotypes confer autoimmune susceptibility in human populations.
The majority of genes that have been identified to regulate Treg cells either directly control Foxp3 expression or their cellular metabolism (Figure 1). Treg cells are highly sensitive to mTOR activation, requiring “just the right amount” for optimal differentiation and suppressive function. Several genes have been identified in mice to maintain this “Goldilocks” homeostasis. The maintenance of mitochondrial metabolism or glycolysis, which is partially under mTORC1 control, is also required by Treg cells. It is predicted that other metabolic genes are also involved, and in silico analyses of protein-protein networks may be useful in pinpointing critical nodes in these networks.
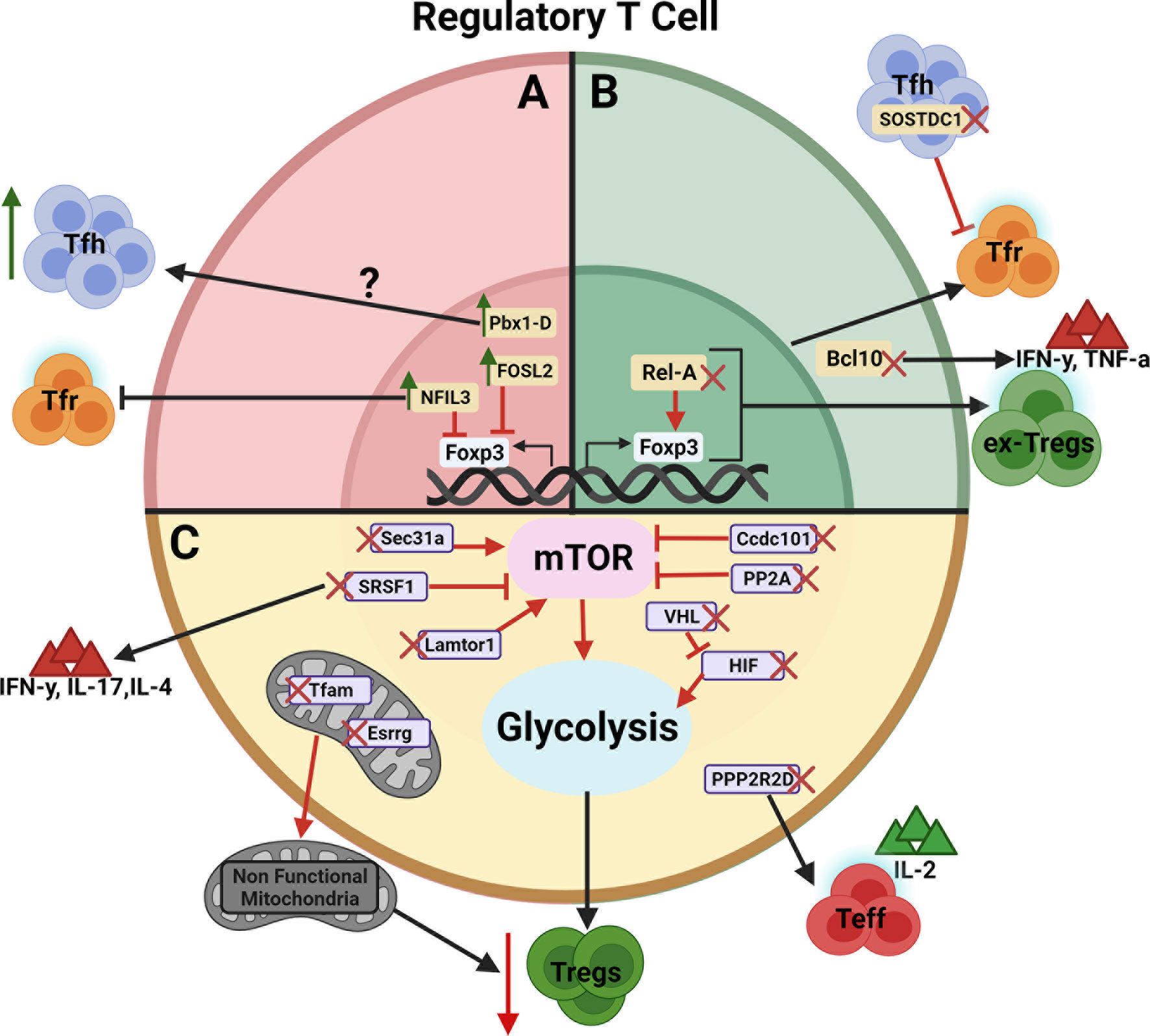
Figure 1 Schematic view of genes regulating Treg cell development, function, and/or stability. The genes are presented according to their effect on Treg cells. (A) Negative regulators whose over expression leads to an expansion of Tfh cells or inhibition of Tfr cells (Green arrows indicates gene overexpression).? indicates that the Pbx1-d direct target is unknown. (B) Positive regulators whose deletion leads to the inhibition of Tfr cells or the generation of ex-Treg cells producing IFNγ and TNFα (Red X indicates gene deletion). (C) Genes regulating Treg cells through their metabolism by way of mTOR, glycolysis and/or mitochondria metabolism leading to decreased immunosuppressive activity. Red arrows between genes and their target indicate an enhancing effect with expression of the target being decreased by the gene deletion. Red blocked arrows indicate an inhibitory effect with expression of the target being increased by the gene overexpression. Figure created with BioRender.com.
Adoptive Treg cell therapies are being evaluated in clinical trials for autoimmune diseases and transplantation (94). The identification of regulatory networks that ensure their stability hand functions has great translational potentials to maximize these approaches. This knowledge could also benefit efforts to deactivate Treg cells in the tumor microenvironment to potentiate immunotherapies. This will require a comprehensive validation of these genetic pathways in human Treg cells, although the restraints of the read-out to in vitro suppression greatly limit the scope and the interpretation of these translation studies.
Author Contributions
TR and LM wrote the review. All authors contributed to the article and approved the submitted version.
Funding
This publication is supported by a grant from the NIH R01 AI045050 to LM
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Vignali DAA, Collison LW, Workman CJ. How Regulatory T Cells Work. Nat Rev Immunol (2008) 8:523–32. doi: 10.1038/NRI2343
2. Plitas G, Rudensky AY. Regulatory T Cells: Differentiation and Function. Cancer Immunol Res (2016) 4:721. doi: 10.1158/2326-6066.CIR-16-0193
3. Sakaguchi S, Mikami N, Wing JB, Tanaka A, Ichiyama K, Ohkura N. Regulatory T Cells and Human Disease. Ann Rev Immunol (2020) 38:541–66. doi: 10.1146/ANNUREV-IMMUNOL-042718-041717
4. Shevach EM, Thornton AM. Ttregs, Ptregs, and Itregs: Similarities and Differences. Immunol Rev (2014) 259:88. doi: 10.1111/IMR.12160
5. Pandiyan P, Zhu J. Origin and Functions of Pro-Inflammatory Cytokine Producing Foxp3+ Regulatory T Cells. Cytokine (2015) 76:13–24. doi: 10.1016/J.CYTO.2015.07.005
6. Brusko TM, Wasserfall CH, Clare-Salzler MJ, Schatz DA, Atkinson MA. Functional Defects and the Influence of Age on the Frequency of CD4+ CD25+ T-Cells in Type 1 Diabetes. Diabetes (2005) 54:1407–14. doi: 10.2337/DIABETES.54.5.1407
7. van Roon JAG, Hartgring SAY, van der Wurff-Jacobs KMG, Bijlsma JWJ, Lafeber FPJG. Numbers of CD25+Foxp3+ T Cells That Lack the IL-7 Receptor are Increased Intra-Articularly and Have Impaired Suppressive Function in RA Patients. Rheumatol (2010) 49:2084–9. doi: 10.1093/RHEUMATOLOGY/KEQ237
8. Scheinecker C, Göschl L, Bonelli M. Treg Cells in Health and Autoimmune Diseases: New Insights From Single Cell Analysis. J Autoimmun (2020) 110:102376. doi: 10.1016/J.JAUT.2019.102376
9. Crispin JC, Martínez A, Alcocer-Varela J. Quantification of Regulatory T Cells in Patients With Systemic Lupus Erythematosus. J Autoimmun (2003) 21:273–6. doi: 10.1016/S0896-8411(03)00121-5
10. Miyara M, Amoura Z, Parizot C, Badoual C, Dorgham K, Trad S, et al. Global Natural Regulatory T Cell Depletion in Active Systemic Lupus Erythematosus. J Immunol (2005) 175:8392–400. doi: 10.4049/JIMMUNOL.175.12.8392
11. Tselios K, Sarantopoulos A, Gkougkourelas I, Boura P. The Influence of Therapy on CD4+CD25highFOXP3+ Regulatory T Cells in Systemic Lupus Erythematosus Patients: A Prospective Study. Scand J Rheumatol (2015) 44:29–35. doi: 10.3109/03009742.2014.922214
12. Żabińska M, Krajewska M, Kościelska-Kasprzak K, Jakuszko K, Bartoszek D, Myszka M, et al. CD4+CD25+CD127– and CD4+CD25+Foxp3+ Regulatory T Cell Subsets in Mediating Autoimmune Reactivity in Systemic Lupus Erythematosus Patients. Arch Immunol Ther Exper (2016) 64:399. doi: 10.1007/S00005-016-0399-5
13. Li W, Deng C, Yang H, Wang G. The Regulatory T Cell in Active Systemic Lupus Erythematosus Patients: A Systemic Review and Meta-Analysis. Front Immunol (2019) 10:159. doi: 10.3389/FIMMU.2019.00159
14. Lin SC, Chen KH, Lin CH, Kuo CC, Ling QD, Chan CH. The Quantitative Analysis of Peripheral Blood FOXP3-Expressing T Cells in Systemic Lupus Erythematosus and Rheumatoid Arthritis Patients. Eur J Clin Invest (2007) 37:987–96. doi: 10.1111/J.1365-2362.2007.01882.X
15. Yan B, Ye S, Chen G, Kuang M, Shen N, Chen S. Dysfunctional CD4+,CD25+ Regulatory T Cells in Untreated Active Systemic Lupus Erythematosus Secondary to Interferon-Alpha-Producing Antigen-Presenting Cells. Arthritis Rheum (2008) 58:801–12. doi: 10.1002/ART.23268
16. Bonelli M, Savitskaya A, Steiner C-W, Rath E, Smolen JS, Scheinecker C. Phenotypic and Functional Analysis of CD4+ CD25- Foxp3+ T Cells in Patients With Systemic Lupus Erythematosus. J Immunol (2009) 182:1689–95. doi: 10.4049/JIMMUNOL.182.3.1689
17. Venigalla RKC, Tretter T, Krienke S, Max R, Eckstein V, Blank N, et al. Reduced CD4+,CD25- T Cell Sensitivity to the Suppressive Function of CD4+,CD25high,CD127 -/Low Regulatory T Cells in Patients With Active Systemic Lupus Erythematosus. Arthritis Rheum (2008) 58:2120–30. doi: 10.1002/ART.23556
18. Vargas-Rojas MI, Crispín JC, Richaud-Patin Y, Alcocer-Varela J. Quantitative and Qualitative Normal Regulatory T Cells Are Not Capable of Inducing Suppression in SLE Patients Due to T-Cell Resistance. Lupus (2008) 17:289–94. doi: 10.1177/0961203307088307
19. Long SA, Buckner JH. CD4+FOXP3+ T Regulatory Cells in Human Autoimmunity: More Than a Numbers Game. J Immunol (2011) 187:2061–6. doi: 10.4049/JIMMUNOL.1003224
20. Horwitz DA. Identity of Mysterious CD4+CD25-Foxp3+ Cells in SLE. Arthritis Res Ther (2010) 12:101. doi: 10.1186/AR2894
21. Hadaschik EN, Wei X, Leiss H, Heckmann B, Niederreiter B, Steiner G, et al. Regulatory T Cell-Deficient Scurfy Mice Develop Systemic Autoimmune Features Resembling Lupus-Like Disease. Arthritis Res Ther (2015) 17:35. doi: 10.1186/S13075-015-0538-0
22. Hu W, Wang ZM, Feng Y, Schizas M, Hoyos BE, van der Veeken J, et al. Regulatory T Cells Function in Established Systemic Inflammation and Reverse Fatal Autoimmunity. Nat Immunol (2021) 22:1163–74. doi: 10.1038/S41590-021-01001-4
23. Ogawa C, Tone Y, Tsuda M, Peter C, Waldmann H, Tone M. TGF-β-Mediated Foxp3 Gene Expression is Cooperatively Regulated by Stat5, Creb, and AP-1 Through CNS2. J Immunol (2014) 192:475–83. doi: 10.4049/JIMMUNOL.1301892
24. Renoux F, Stellato M, Haftmann C, Vogetseder A, Huang R, Subramaniam A, et al. The AP1 Transcription Factor Fosl2 Promotes Systemic Autoimmunity and Inflammation by Repressing Treg Development. Cell Rep (2020) 31:107826. doi: 10.1016/j.celrep.2020.107826
25. Keniry M, Pires MM, Mense S, Lefebvre C, Gan B, Justiano K, et al. Survival Factor NFIL3 Restricts FOXO-Induced Gene Expression in Cancer. Genes Dev (2013) 27:916. doi: 10.1101/GAD.214049.113
26. Keniry M, Dearth RK, Persans M, Parsons R. New Frontiers for the NFIL3 bZIP Transcription Factor in Cancer, Metabolism and Beyond. Discoveries (2014) 2:e15. doi: 10.15190/D.2014.7
27. Motomura Y, Kitamura H, Hijikata A, Matsunaga Y, Matsumoto K, Inoue H, et al. The Transcription Factor E4BP4 Regulates the Production of IL-10 and IL-13 in CD4+ T Cells. Nat Immunol (2011) 12:450–9. doi: 10.1038/ni.2020
28. Kashiwada M, Cassel SL, Colgan JD, Rothman PB. NFIL3/E4BP4 Controls Type 2 T Helper Cell Cytokine Expression. EMBO J (2011) 30:2071–82. doi: 10.1038/EMBOJ.2011.111
29. Kim HS, Sohn H, Jang SW, Lee GR. The Transcription Factor NFIL3 Controls Regulatory T-Cell Function and Stability. Exp Mol Med (2019) 51:1–15. doi: 10.1038/s12276-019-0280-9
30. Wang Z, Zhao M, Yin J, Liu L, Hu L, Huang Y, et al. E4BP4-Mediated Inhibition of T Follicular Helper Cell Differentiation is Compromised in Autoimmune Diseases. J Clin Invest (2020) 130:3717. doi: 10.1172/JCI129018
31. Sage PT, Sharpe AH. The Multifaceted Functions of Follicular Regulatory T Cells. Curr Opin Immunol (2020) 67:68–74. doi: 10.1016/J.COI.2020.10.009
32. Fu W, Liu X, Lin X, Feng H, Sun L, Li S, et al. Deficiency in T Follicular Regulatory Cells Promotes Autoimmunity. J Exp Med (2018) 215:815. doi: 10.1084/JEM.20170901
33. Isomura I, Palmer S, Grumont RJ, Bunting K, Hoyne G, Wilkinson N, et al. C-Rel is Required for the Development of Thymic Foxp3+ CD4 Regulatory T Cells. J Exp Med (2009) 206:3001–14. doi: 10.1084/JEM.20091411
34. Chang JH, Xiao Y, Hu H, Jin J, Yu J, Zhou X, et al. Ubc13 Maintains the Suppressive Function of Regulatory T Cells and Prevents Their Conversion Into Effector-Like T Cells. Nat Immunol (2012) 13:481–90. doi: 10.1038/NI.2267
35. Heuser C, Gotot J, Piotrowski EC, Philipp MS, Courrèges CJF, Otte MS, et al. Prolonged Ikkβ Inhibition Improves Ongoing CTL Antitumor Responses by Incapacitating Regulatory T Cells. Cell Rep (2017) 21:578–86. doi: 10.1016/J.CELREP.2017.09.082
36. Ronin E, di Ricco ML, Vallion R, Divoux J, Kwon HK, Grégoire S, et al. The Nf-κb Rela Transcription Factor is Critical for Regulatory T Cell Activation and Stability. Front Immunol (2019) 10:2487/FULL. doi: 10.3389/FIMMU.2019.02487/FULL
37. Messina N, Fulford T, O’Reilly L, Loh WX, Motyer JM, Ellis D, et al. The NF-κb Transcription Factor RelA is Required for the Tolerogenic Function of Foxp3(+) Regulatory T Cells. J Autoimm (2016) 70:52–62. doi: 10.1016/J.JAUT.2016.03.017
38. Lin X, Wang D. The Roles of CARMA1, Bcl10, and MALT1 in Antigen Receptor Signaling. Semin Immunol (2004) 16:429–35. doi: 10.1016/J.SMIM.2004.08.022
39. Yang D, Zhao X, Lin X. Bcl10 is Required for the Development and Suppressive Function of Foxp3+ Regulatory T Cells. Cell Mol Immunol (2021) 18:206–18. doi: 10.1038/s41423-019-0297-y
40. Lee JH, Elly C, Park Y, Liu YC. E3 Ubiquitin Ligase VHL Regulates Hypoxia-Inducible Factor-1α to Maintain Regulatory T Cell Stability and Suppressive Capacity. Immunity (2015) 42:1062–74. doi: 10.1016/J.IMMUNI.2015.05.016
41. Liu X, Yan X, Zhong B, Nurieva RI, Wang A, Wang X, et al. Bcl6 Expression Specifies the T Follicular Helper Cell Program In Vivo. J Exp Med (2012) 209:1841–52. doi: 10.1084/JEM.20120219
42. Wu X, Wang Y, Huang R, Gai Q, Liu H, Shi M, et al. SOSTDC1-Producing Follicular Helper T Cells Promote Regulatory Follicular T Cell Differentiation. Science (2020) 369:984–8. doi: 10.1126/science.aba6652
43. Cho SW, Kwak S, Woolley TE, Lee MJ, Kim EJ, Baker RE, et al. Interactions Between Shh, Sostdc1 and Wnt Signaling and a New Feedback Loop for Spatial Patterning of the Teeth. Development (2011) 138:1807–16. doi: 10.1242/DEV.056051
44. vanLoosdregt J, Fleskens V, Tiemessen MM, Mokry M, vanBoxtel R, Meerding J, et al. Canonical Wnt Signaling Negatively Modulates Regulatory T Cell Function. Immunity (2013) 39:298–310. doi: 10.1016/J.IMMUNI.2013.07.019
45. Sumida T, Lincoln MR, Ukeje CM, Rodriguez DM, Akazawa H, Noda T, et al. Activated β-Catenin in Foxp3+ Regulatory T Cells Links Inflammatory Environments to Autoimmunity. Nat Immunol 2018 19:12 (2018) 19:1391–402. doi: 10.1038/s41590-018-0236-6
46. Shi J, Chi S, Xue J, Yang J, Li F, Liu X. Emerging Role and Therapeutic Implication of Wnt Signaling Pathways in Autoimmune Diseases. J Immunol Res (2016) 2016:9392132. doi: 10.1155/2016/9392132
47. Chi H. Regulation and Function of mTOR Signalling in T Cell Fate Decision. Nat Rev Immunol (2012) 12:325. doi: 10.1038/NRI3198
48. Delgoffe GM, Pollizzi KN, Waickman AT, Heikamp E, Meyers DJ, Horton MR, et al. The Kinase mTOR Regulates the Differentiation of Helper T Cells Through the Selective Activation of Signaling by Mtorc1 and Mtorc2. Nat Immunol (2011) 12:295–304. doi: 10.1038/NI.2005
49. Shrestha S, Yang K, Guy C, Vogel P, Neale G, Chi H. Regulatory T Cells Require the Phosphatase PTEN to Restrain Type 1 and Follicular Helper T-Cell Responses. Nat Immunol (2015) 16:178. doi: 10.1038/NI.3076
50. Liu G, Yang K, Burns S, Shrestha S, Chi H. S1P1-mTOR Axis Directs the Reciprocal Differentiation of TH1 and Regulatory T Cells. Nat Immunol (2010) 11:1047. doi: 10.1038/NI.1939
51. Battaglia M, Stabilini A, Roncarolo MG. Rapamycin Selectively Expands CD4+CD25+FoxP3+ Regulatory T Cells. Blood (2005) 105:4743–8. doi: 10.1182/BLOOD-2004-10-3932
52. Zeng H, Yang K, Cloer C, Neale G, Vogel P, Chi H. Mtorc1 Couples Immune Signals and Metabolic Programming to Establish T(reg)-Cell Function. Nature (2013) 499:485–90. doi: 10.1038/NATURE12297
53. Chapman NM, Zeng H, Nguyen TLM, Wang Y, Vogel P, Dhungana Y, et al. mTOR Coordinates Transcriptional Programs and Mitochondrial Metabolism of Activated Treg Subsets to Protect Tissue Homeostasis. Nat Comm (2018) 9:2095. doi: 10.1038/S41467-018-04392-5
54. Long L, Wei J, Lim SA, Raynor JL, Shi H, Connelly JP, et al. CRISPR Screens Unveil Signal Hubs for Nutrient Licensing of T Cell Immunity. Nature (2021) 600:308–13. doi: 10.1038/S41586-021-04109-7
55. Hosokawa T, Kimura T, Nada S, Okuno T, Ito D, Kang S, et al. Lamtor1 is Critically Required for CD4 + T Cell Proliferation and Regulatory T Cell Suppressive Function. J Immunol (2017) 199:2008–19. doi: 10.4049/JIMMUNOL.1700157
56. Shi Y. Serine/threonine Phosphatases: Mechanism Through Structure. Cell (2009) 139:468–84. doi: 10.1016/J.CELL.2009.10.006
57. Apostolidis SA, Rodríguez-Rodríguez N, Suárez-Fueyo A, Dioufa N, Ozcan E, Crispín JC, et al. Phosphatase PP2A is Requisite for the Function of Regulatory T Cells. Nat Immunol (2016) 17:556–64. doi: 10.1038/NI.3390
58. Sharabi A, Li H, Kasper IR, Pan W, Meidan E, Tsokos MG, et al. PP2A Enables IL-2 Signaling by Preserving IL-2rβ Chain Expression During Treg Development. JCI Insight (2019) 5(9):e126294. doi: 10.1172/JCI.INSIGHT.126294
59. Pan W, Sharabi A, Ferretti A, Zhang Y, Burbano C, Yoshida N, et al. PPP2R2D Suppresses IL-2 Production and Treg Function. JCI Insight (2020) 5(19):e138215. doi: 10.1172/jci.insight.138215
60. Eltzschig HK, Bratton DL, Colgan SP. Targeting Hypoxia Signalling for the Treatment of Ischaemic and Inflammatory Diseases. Nat Rev Drug Discovery (2014) 13:852. doi: 10.1038/NRD4422
61. Gossage L, Eisen T, Maher ER. VHL, the Story of a Tumour Suppressor Gene. Nat Rev Cancer (2015) 15:55–64. doi: 10.1038/NRC3844
62. Pollizzi KN, Powell JD. Integrating Canonical and Metabolic Signaling Programmes in the Regulation of T Cell Responses. Nat Rev Immunol (2014) 14:435–46. doi: 10.1038/NRI3701
63. Corcoran SE, O’Neill LAJ. Hif1α and Metabolic Reprogramming in Inflammation. J Clin Invest (2016) 126:3699–707. doi: 10.1172/JCI84431
64. Dang E v., Barbi J, Yang HY, Jinasena D, Yu H, Zheng Y, et al. Control of TH17/Treg Balance by Hypoxia-Inducible Factor 1. Cell (2011) 146:772. doi: 10.1016/J.CELL.2011.07.033
65. Shi LZ, Wang R, Huang G, Vogel P, Neale G, Green DR, et al. HIF1alpha-Dependent Glycolytic Pathway Orchestrates a Metabolic Checkpoint for the Differentiation of TH17 and Treg Cells. J Exp Med (2011) 208:1367–76. doi: 10.1084/JEM.20110278
66. Hsu TS, Lin YL, Wang YA, Mo ST, Chi PY, Lai ACY, et al. HIF-2α is Indispensable for Regulatory T Cell Function. Nat Comm (2020) 11:5005. doi: 10.1038/S41467-020-18731-Y
67. Jiang C, Wang H, Xue M, Lin L, Wang J, Cai G, et al. Reprograming of Peripheral Foxp3 + Regulatory T Cell Towards Th17-Like Cell in Patients With Active Systemic Lupus Erythematosus. Clin Immunol (2019) 209:108276. doi: 10.1016/J.CLIM.2019.108267
68. Das S, Krainer AR. Emerging Functions of SRSF1, Splicing Factor and Oncoprotein, in RNA Metabolism and Cancer. Mol Cancer Res (2014) 12:1195–204. doi: 10.1158/1541-7786.MCR-14-0131
69. Kono M, Kurita T, Yasuda S, Kono M, Fujieda Y, Bohgaki T, et al. Decreased Expression of Serine/Arginine-Rich Splicing Factor 1 in T Cells From Patients With Active Systemic Lupus Erythematosus Accounts for Reduced Expression of RasGRP1 and DNA Methyltransferase 1. Arthritis Rheum (2018) 70:2046–56. doi: 10.1002/ART.40585/
70. Katsuyama T, Li H, Comte D, Tsokos GC, Moulton VR. Splicing Factor SRSF1 Controls T Cell Hyperactivity and Systemic Autoimmunity. J Clin Invest (2019) 129:5411. doi: 10.1172/JCI127949
71. Ohl K, Tenbrock K. Regulatory T Cells in Systemic Lupus Erythematosus. Eur J Immunol (2015) 45:344–55. doi: 10.1002/EJI.201344280
72. Dépis F, Kwon H-K, Mathis D, Benoist C. Unstable FoxP3+ T Regulatory Cells in NZW Mice. Proc Natl Acad Sci USA (2016) 113:1345–50. doi: 10.1073/PNAS.1524660113
73. Li W, Titov AA, Morel L. An Update on Lupus Animal Models. Curr Opin Rheumatol (2017) 29:434–41. doi: 10.1097/BOR.0000000000000412
74. Morel L. Mapping Lupus Susceptibility Genes in the NZM2410 Mouse Model. Adv Immunol (2012) 115:113–39. doi: 10.1016/B978-0-12-394299-9.00004-7
75. Morel L, Rudofsky UH, Longmate JA, Schiffenbauer J, Wakeland EK. Polygenic Control of Susceptibility to Murine Systemic Lupus Erythematosus. Immunity (1994) 1:219–29. doi: 10.1016/1074-7613(94)90100-7
76. Morel L Mohan C Yu Y, Croker BP, Tian N, Deng A, Wakeland EK. Functional Dissection of Systemic Lupus Erythematosus Using Congenic Mouse Strains. J Immunol (1997) 158:6019–28.
77. Morel L, Tian X-H, Croker BP, Wakeland EK. Epistatic Modifiers of Autoimmunity in a Murine Model of Lupus Nephritis Disease. Immunity (1999) 11:131–9. doi: 10.1016/S1074-7613(00)80088-6
78. Chen Y, Perry D, Boackle SA, Sobel ES, Molina H, Croker BP, et al. Genetic Determination of T Cell Help in Loss of Tolerance to Nuclear Antigens. J Immunol (2005) 174:7692–702. doi: 10.4049/JIMMUNOL.174.12.7692
79. Sobel ES, Satoh M, Chen Y, Wakeland EK, Morel L. The Major Murine Systemic Lupus Erythematosus Susceptibility Locus Sle1 Results in Abnormal Functions of Both B and T Cells. J Immunol (2002) 169:2694–700. doi: 10.4049/JIMMUNOL.169.5.2694
80. Morel L, Blenman KR, Croke BP, Wakeland EK. The Major Murine Systemic Lupus Erythematosus Susceptibility Locus, Sle1, is a Cluster of Functionally Related Genes. Proc Natl Acad Sci USA (2001) 98:1787–92. doi: 10.1073/pnas.031336098
81. Cuda CM, Wan S, Sobel ES, Croker BP, Morel L. Murine Lupus Susceptibility Locus Sle1a Controls Regulatory T Cell Number and Function Through Multiple Mechanisms. J Immunol (2007) 179:7439–47. doi: 10.4049/JIMMUNOL.179.11.7439
82. Cuda CM, Li S, Liang S, Yin Y, Potula HHSK, Xu Z, et al. Pre-B Cell Leukemia Homeobox 1 is Associated With Lupus Susceptibility in Mice and Humans. J Immunol (2012) 188:604–14. doi: 10.4049/JIMMUNOL.1002362
83. Schnabel CA, Selleri L, Jacobs Y, Warnke R, Cleary ML. Expression of Pbx1b During Mammalian Organogenesis. Mech Dev (2001) 100:131–5. doi: 10.1016/s0925-4773(00)00516-5
84. Sanyal M, Tung JW, Karsunky H, Zeng H, Selleri L, Weissman IL, et al. B-Cell Development Fails in the Absence of the Pbx1 Proto-Oncogene. Blood (2007) 109:4191. doi: 10.1182/BLOOD-2006-10-054213
85. Sengupta M, Liang S, Potula HHS, Chang LJ, Morel L. The SLE-Associated Pbx1-D Isoform Acts as a Dominant-Negative Transcriptional Regulator. Genes Immun 2012 (2012) 13:653–7. doi: 10.1038/gene.2012.43
86. Ficara F, Murphy MJ, Lin M, Cleary ML. Pbx1 Regulates Self-Renewal of Long-Term Hematopoietic Stem Cells by Maintaining Their Quiescence. Cell Stem Cell (2008) 2:484–96. doi: 10.1016/J.STEM.2008.03.004
87. Sobel ES, Brusko TM, Butfiloski EJ, Hou W, Li S, Cuda CM, et al. Defective Response of CD4+T Cells to Retinoic Acid and Tgfβ in Systemic Lupus Erythematosus. Arthritis Res Ther (2011) 13:1–15. doi: 10.1186/AR3387/FIGURES/7
88. Choi SC, Hutchinson TE, Titov AA, Seay HR, Li S, Brusko TM, et al. The Lupus Susceptibility Gene Pbx1 Regulates the Balance Between Follicular Helper T Cell and Regulatory T Cell Differentiation. J Immunol (2016) 197:458–69. doi: 10.4049/jimmunol.1502283
89. Li W, Elshikha AS, Cornaby C, Teng X, Abboud G, Brown J, et al. T Cells Expressing the Lupus Susceptibility Allele Pbx1d Enhance Autoimmunity and Atherosclerosis in Dyslipidemic Mice. JCI Insight (2020) 5(11):e138274. doi: 10.1172/JCI.INSIGHT.138274
90. Xu X, Zhou Y, Fu B, Zhang J, Dong Z, Zhang X, et al. PBX1 Promotes Development of Natural Killer Cells by Binding Directly to the Nfil3 Promoter. FASEB J (2020) 34:6479–92. doi: 10.1096/FJ.202000121R
91. Perry DJ, Yin Y, Telarico T, Baker H v., Dozmorov I, Perl A, et al. Murine Lupus Susceptibility Locus Sle1c2 Mediates CD4+ T Cell Activation and Maps to Estrogen-Related Receptor γ. J Immunol (2012) 189:793–803. doi: 10.4049/JIMMUNOL.1200411
92. Perl A. Review: Metabolic Control of Immune System Activation in Rheumatic Diseases. Arthritis Rheumatol (2017) 69:2259–70. doi: 10.1002/ART.40223
93. Li W, Gong M, Park YP, Elshikha AS, Choi SC, Brown J, et al. Lupus Susceptibility Gene Esrrg Modulates Regulatory T Cells Through Mitochondrial Metabolism. JCI Insight (2021) 6(14):e143540. doi: 10.1172/JCI.INSIGHT.143540
Keywords: regulatory T cells, Foxp3, autoimmunity, lupus, genetics
Citation: Roach T and Morel L (2022) Genetic Variations Controlling Regulatory T Cell Development and Activity in Mouse Models of Lupus-Like Autoimmunity. Front. Immunol. 13:887489. doi: 10.3389/fimmu.2022.887489
Received: 01 March 2022; Accepted: 22 April 2022;
Published: 26 May 2022.
Edited by:
José Carlos Crispín, Instituto Nacional de Ciencias Médicas y Nutrición Salvador Zubirán (INCMNSZ), MexicoReviewed by:
Florencia Rosetti, Instituto Nacional de Ciencias Médicas y Nutrición Salvador Zubirán (INCMNSZ), MexicoKlaus Tenbrock, RWTH Aachen University, Germany
Copyright © 2022 Roach and Morel. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Laurence Morel, bW9yZWxAdWZsLmVkdQ==