 Rilan Bai
Rilan Bai Jiuwei Cui
Jiuwei Cui
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 12 May 2022
Sec. Cancer Immunity and Immunotherapy
Volume 13 - 2022 | https://doi.org/10.3389/fimmu.2022.886931
This article is part of the Research Topic NK cell modifications to advance their anti-tumor activities View all 13 articles
Antibodies targeting programmed death receptor-1 (PD-1)/programmed death ligand-1 (PD-L1) have been considered breakthrough therapies for a variety of solid and hematological malignancies. Although cytotoxic T cells play an important antitumor role during checkpoint blockade, they still show a potential killing effect on tumor types showing loss of/low major histocompatibility complex (MHC) expression and/or low neoantigen load; this knowledge has shifted the focus of researchers toward mechanisms of action other than T cell-driven immune responses. Evidence suggests that the blockade of the PD-1/PD-L1 axis may also improve natural killer (NK)-cell function and activity through direct or indirect mechanisms, which enhances antitumor cytotoxic effects; although important, this topic has been neglected in previous studies. Recently, some studies have reported evidence of PD-1 and PD-L1 expression in human NK cells, performed exploration of the intrinsic mechanism by which PD-1/PD-L1 blockade enhances NK-cell responses, and made some progress. This article summarizes the recent advances regarding the expression of PD-1 and PD-L1 molecules on the surface of NK cells as well as the interaction between anti-PD-1/PD-L1 drugs and NK cells and associated molecular mechanisms in the tumor microenvironment.
Antibodies targeting programmed death receptor-1 (PD-1) and programmed death ligand 1 (PD-L1) have been approved for the treatment of a variety of solid and hematologic malignancies; patients with various malignancies and even those with a very advanced disease showed durable responses to this treatment (1–3). However, only 10–20% of patients with different tumor types respond well to PD-1/PD-L1 blocking therapy (3, 4). In addition, treatment with anti-PD-1/PD-L1 monoclonal antibodies (mAbs) can lead to unexplained clinical responses of tumors with no or low expression of major histocompatibility complex (MHC) and/or PD-L1 (2, 4, 5). Therefore, a better understanding of the mechanisms of action of anti-PD-L1 (or anti-PD-1) mAb therapy and the impact of this therapy on each component of the tumor immune microenvironment will help improve the precision of cancer immunotherapeutics in the future. Most believe that antibodies targeting PD-1 and PD-L1 are largely only beneficial for eliciting T cell-driven responses, but accumulating evidence suggests that blocking the PD-1/PD-L1 axis may also improve natural killer (NK)-cell function and activity and enhance antitumor cytotoxicity through direct/indirect but crucial mechanisms. For example, Hodgkin lymphoma cells with defective MHC class I expression responds well to anti-PD-1 mAb therapy, which suggests the presence of immune responses that are independent of cytotoxic CD8+T cells, inhibited by PD-1, and rescued by anti-PD-1 therapy (5–7). NK cells show MHC-independent antitumor cytotoxicity, which allows them to exhibit killing effects on many tumor types with absent or low MHC expression and/or low neoantigen burden. Recently, some studies have reported evidence of PD-1 and PD-L1 expression in human NK cells (8, 9). In vivo mechanistic studies on whether and how PD-1 or PD-L1 plays a role in NK-cell response to tumors and whether and how PD-1/PD-L1 blockade mobilizes NK cell response remain scarce; however, with the development of multi-omics and high-throughput sequencing technologies, the understanding of this field has gradually deepened and progressed. Based on this knowledge, in this article, we comprehensively review the expression of PD-1 and PD-L1 molecules on the surface of NK cells and discuss the interactions between anti-PD-1/PD-L1 drugs and NK cells in the tumor microenvironment (TME) as well as the associated molecular mechanisms.
NK cells are mainly involved in killing microbes and malignantly transformed allogeneic and autologous cells without prior sensitization and exhibit non-MHC-restricted antitumor cytotoxicity (10). Activated NK cells exert a strong cytotoxic effect by inducing the secretion of cytotoxic mediators and the production of inflammatory cytokines and chemokines through integration of adhesion molecules and receptor signaling activation (11). According to the CD56 density on the cell surface, NK cells can be divided into CD56dim (~90%) and CD56dim (~10%) cells (12). CD56bright mainly performs immunoregulatory function by secreting cytokines, while CD56dim mainly performs cytolytic function by secreting granzyme B and perforin, which enhance the expression of immunoglobulin-like receptors and Fcγ receptor III (FcγRIII)/CD16 (13, 14). Induction and regulation of NK cell function is mediated by a range of activating or inhibitory surface receptors. In humans, the main activating receptors involved in target-cell killing include natural cytotoxic receptors (NCRs) (including NKp46, NKp30, and NKp44) and NKG2D (15). FcγRIII is also an activating receptor that is mainly expressed by CD56dimNK cells and is essential for antibody-dependent cell-mediated cytotoxicity (ADCC) against IgG-coated target cells (16). Conversely, MHC class I antigen-specific inhibitory receptors on the surface of NK cells, namely, killer cell immunoglobulin-like receptors (KIRs), leukocyte immunoglobulin-like receptors (LIRs), and natural killer group 2 A (NKG2A), tightly regulate the cytotoxicity mediated by these cells and the production of lymphokines, by recognizing self MHC-I antigens (17, 18). In addition, several non-MHC-specific inhibitory NK receptors have been identified, including classical cytotoxic T-lymphocyte-associated protein 4 (CTLA-4), PD-1, and T cell immunoreceptor with Ig and immunoreceptor tyrosine-based inhibitory motif (ITIM) domains (TIGIT), CD96, lymphocyte-activation gene 3 (LAG-3), and T cell immunoglobulin-3 (TIM-3) (19). Sheffer M et al. (20) systematically defined the molecular signature of human tumor cells that determines their sensitivity to human allogeneic NK cells and found that the transcriptional signature of NK cell-sensitive tumor cells correlates with immune checkpoint inhibitor (ICI) resistance in clinical samples. The study has also applied genome-scale CRISPR-based gene editing screens in several solid tumor cell lines to functionally interrogate which genes in tumor cells regulate responses to NK cells. Tumor cells escape immune responses by regulating the expression of inhibitory receptors on NK cells, and in this context, PD-1/PD-L1 axis has been studied extensively; understanding the expression of NK cell surface receptors and their role in the functional activity of NK cells is essential for the development of effective immunotherapies.
PD-1 is a member of the immunoglobulin superfamily. It can be expressed on various immune cells, including T (CD4+ and CD8+) cells, B cells, bone marrow cells, NK cells, and other innate lymphocytes (ILCs) (21–23). When bound to ligands (PD-L1 and PD-L2) that may be expressed on tumor cells, PD-1 may play a role in impairing antitumor effects and facilitating tumor immune escape (21–23). In recent years, researchers have shown interest in targeting PD-1 to increase NK activity. However, PD-1 expression on NK cells is diverse and difficult to clarify. High expression of PD-1 on NK cells can be detected in the peripheral blood of approximately one-quarter of healthy individuals (22). PD-1, which is usually not expressed on CD56bright NK cells, is confined to fully mature NK cells of NKG2A−KIR+CD57+CD56dim phenotype (24). In the TMEs of various cancers, such as ovarian cancer (ascites), Kaposi’s sarcoma (peripheral blood), renal cell carcinoma, and multiple myeloma, the proportion of PD-1+ NK cells and the expression of PD-1 on NK cells increase (25–28). PD-1 expression on peripheral and tumor infiltrating NK cells from patients with digestive cancers was increased (29). In addition, chronic infections such as those caused by human immunodeficiency virus (HIV), hepatitis C virus (HCV), and human cytomegalovirus (HCMV) have also been shown to enhance PD-1 expression on NK cells (30, 31).
PD-1, an important checkpoint of NK activation, is more abundantly expressed in activated NK cells than in inactivated NK cells (9). Functional and phenotypic assays showed that PD-1+ NK cells had the highest functional activity when stimulated and that most of these cells expressed activation markers (CD69 and Sca-1) (32). A higher frequency of circulating PD-1+ NK cells (mean > 9%) was associated with a better overall survival (OS) in patients with head and neck cancers (HNCs) (33), which indicates that PD-1+ NK cells are not necessarily inactive in PD-L1+ tumors but may only be inhibited from killing tumor cells. The interaction of PD-1 with its ligands provides a negative signal for the activation of NK cells, which causes the NK cells to express a dysfunctional, exhausted phenotype (26) and an inhibited antitumor ability (29). Several murine tumor models have shown that PD-1+ NK cells present at the site of PD-L1+ tumors exhibit an exhaustive phenotype and that PD-1/PD-L1 interaction strongly inhibits NK cell-mediated antitumor immunity (32). Overall, PD-1+ NK cells exhibit enhanced apoptotic sensitivity, reduced cytolytic activity, impaired cytotoxic and cytokine production efficiencies, and reduced proliferative capacity in PD-L1+ tumors (24, 29). In addition, immune cells in TME can also affect NK cells by expressing PD-L1. For example, tumor-associated neutrophils (TANs) can impair the cytotoxicity and infiltration capacity of NK cells, and downregulation of CCR1 leads to diminished infiltration capacity of NK cells and reduced responsiveness of the NK-activating receptors NKp46 and NKG2D (34); CD56dimPD-1+ NK cells expressed in patients with Hodgkin lymphoma are efficiently inhibited by PD-L1-expressing myeloid cells (35).
The molecular mechanisms regulating PD-1 expression in human NK cells have not yet been elucidated. Signaling by cells and/or soluble factors in the TME may play a major role, and cytokines may mediate crosstalk between different immune checkpoints. For example, it has been demonstrated that tumor-derived interleukin (IL)-18 increases the immunosuppressive CD56dimCD16dim/- NK cell fraction in patients with triple-negative breast cancer (TNBC) and induces the expression of PD-1 in these cells (36). The G-CSF/STAT3 pathway and IL-18 are responsible for upregulating PD-L1 expression on TANs and NK cells, respectively; transforming growth factor-β (TGF-β) and interferon-γ (IFN-γ) impair NK cell cytotoxicity by upregulating the expressions of PD-L1 and PD-1 on tumor cells and NK cells, respectively (37). In addition, chemotherapeutic agents can upregulate PD-1 expression on NK cells and PD-L1 expression on tumor cells through nuclear factor kappa B (NF-κB) (38). In future studies, it is important to identify the mechanisms that lead to the expression of PD-1 on NK cells in the TME and their importance in NK cell-based immunotherapy.
IFN-γ secreted by activated T cells can stimulate the upregulation of PD-L1 expression on the surface of tumor cells and transmit inhibitory signals to T cells after PD-L1–PD-1 binding, which results in T cell dysfunction and tumor immune escape (39). PD-L1 is reported to be expressed on tumor cells as well as immune cells within the TME, including antigen presenting cells (APCs) (mainly macrophages and dendritic cells [DCs]), activated/depleted T and B lymphocytes, regulatory T cells (Tregs), and NK cells (8, 40). According to a comprehensive review by Sun et al. (41), the regulation of PD-L1 expression and function occurs at different levels. Several inflammatory mediators, including TNF-α, IFN-γ, IL-10, IL-17, and C5a, are inducers of PD-L1 expression (42–44). The JAK/STAT, RAS/MAPK, and PTEN-PI3K/AKT pathways are involved in the control of PD-L1 gene expression through different downstream transcription factors, such as STAT1, STAT3, IRF1, IRF3, HIF-1α, MYC, JUN, BRD4, and NF-κB (45). Corresponding DNA-binding elements other than IRF3 have been described on the PD-L1 gene promoter (46–48). Other regulatory mechanisms include microRNA (e.g., miR-513, miR-34a, miR-200, and miR-570)-mediated post-transcriptional repression and the presence of soluble PD-L1 (sPD-L1) in the blood, which may compete with membrane-bound PD-L1 for binding to PD-1 to regulate cell surface PD-L1 expression (41, 49).
IFN-γ is one of the most studied inducers of PD-L1 expression in tumors, and NF-κB (a major transcription factor of inflammation and immunity) is involved in NK cell activation to regulate IFN-γ production (50). Researchers have proposed that NF-κB directly induces PD-L1 gene transcription by binding to the latter’s promoter and that it post-transcriptionally regulates PD-L1 through an indirect pathway; thus, NF-κB may be a key positive regulator of PD-L1 expression in tumors (51). In addition, the PD-L1 promoter contains a hypoxia-inducible factor 1-alpha (HIF-1α) response element (52, 53), which drives the IKK-β gene transcription through a hypoxia response element present in the promoter while supporting NF-κB pathway activation by directly inducing p65 (54, 55); NF-κB can also induce HIF-1α transcription by directly binding to the HIF-1α promoter (56). Thus, both HIF-1α and NF-κB pathways can initiate and maintain PD-L1 expression and reinforce each other through positive feedback. Thus, the possibility that these signaling pathways can regulate PD-L1 expression in NK cells must be explored. A recent study confirmed that some myeloid leukemia cell lines and acute myeloid leukemia (AML) blasts from patients can induce PD-L1 signaling in NK cells through the PI3K/AKT/NF-κB pathway (8). Notably, the expression levels of two activating antigens, CD69 and CD25, were significantly higher in PD-L1+ NK cells than in PD-L1- NK cells, which indicates that PD-L1+ NK cells show an increase in antitumor cytotoxic effector function and IFN-γ-mediated CD69 expression. The study revealed that the higher the sensitivity of the target cells to NK cell cytotoxicity (due to its negative correlation with MHC-I molecule expression), the greater the directness of the cell-cell contact between NK cells and target cells, the higher the expression of PD-L1, and the stronger the activation of CD69 NK cells (8). Therefore, PD-L1 expression on NK cells may serve as an in vivo biomarker for sensitivity to NK cell lysis in patients with tumors. Nonetheless, the expression and function of PD-L1 on NK cells and the involvement of PD-L1+ NK cells in anti-PD-L1 mAb therapy has not been comprehensively explored. An improvement in the understanding of PD-L1 expression on NK cells and signaling regulatory mechanisms is essential to understand tumor and immune cell biological characteristics and to develop NK cell-based antitumor immunotherapy.
Existing literature has shown the potential benefits of anti-PD-1/PD-L1 therapy in rescuing and/or improving NK cell function and improving antitumor immune responses (28, 29, 33, 57). Researchers have found that disrupting the PD-1/PD-L1 interaction can enhance the killing effect of NK cells on tumor cells of mice with several cancers, and in some models, PD-1/PD-L1 blockers are completely ineffective when mouse NK cells are depleted (32). In humanized (Hu) NOD.Cg-PrkdcscidIl2rgtm1Wjl/SzJ (NSG) mice xenografted with dedifferentiated liposarcoma (DDLPS), abundance of hCD8+ T subsets, such as hCD8+ IFN-γ+, hCD8+PD-1+, and hCD8+Ki-67+ cells, and hNK subsets, such as hCD56+IFN-γ+, hCD56+PD-1+, and hCD56+Ki-67+ cells, is functionally associated with anti-PD-1 effects (58). In addition, a significant increase in the number of activated hCD56+NKp46+NKG2D+ NK cells was also detected, which indicates that NK cells play a pivotal role in the antitumor effect of anti-PD-1 therapy (58).
PD-1/PD-L1 blocking may rescue multiple aspects of NK cell-mediated antitumor immune activity (Figure 1). Firstly, most studies have shown that PD-1 expression on the surface of NK cells is upregulated in the TME, and this expression plays a negative immunoregulatory role after binding to the corresponding ligands and is associated with poor tumor prognosis (29, 32, 33). PD-1/PD-L1 blockade can activate NK cells by preventing inhibitory signals between PD-1+ NK cells and PD-L1+ target cells. Secondly, the activity of NK cells is determined by a series of activation and inhibition signals, and PD-1 blockade via antibodies activates some positive regulatory signaling pathways or prevents other intracellular inhibitory signals (8); furthermore, the unique ADCC effect of NK cells may be activated and enhanced by some PD-1/PD-L1 blockers, and this effect triggers strong antitumor activity (59). In addition, anti-PD-1/PD-L1 therapy may indirectly affect NK cell function through other immune cells in the TME (60, 61). NK cells may, in turn, enhance tumor immune responsiveness to PD-1 antibodies by affecting other immune factors in the TME. These mechanisms are reviewed in detail below.
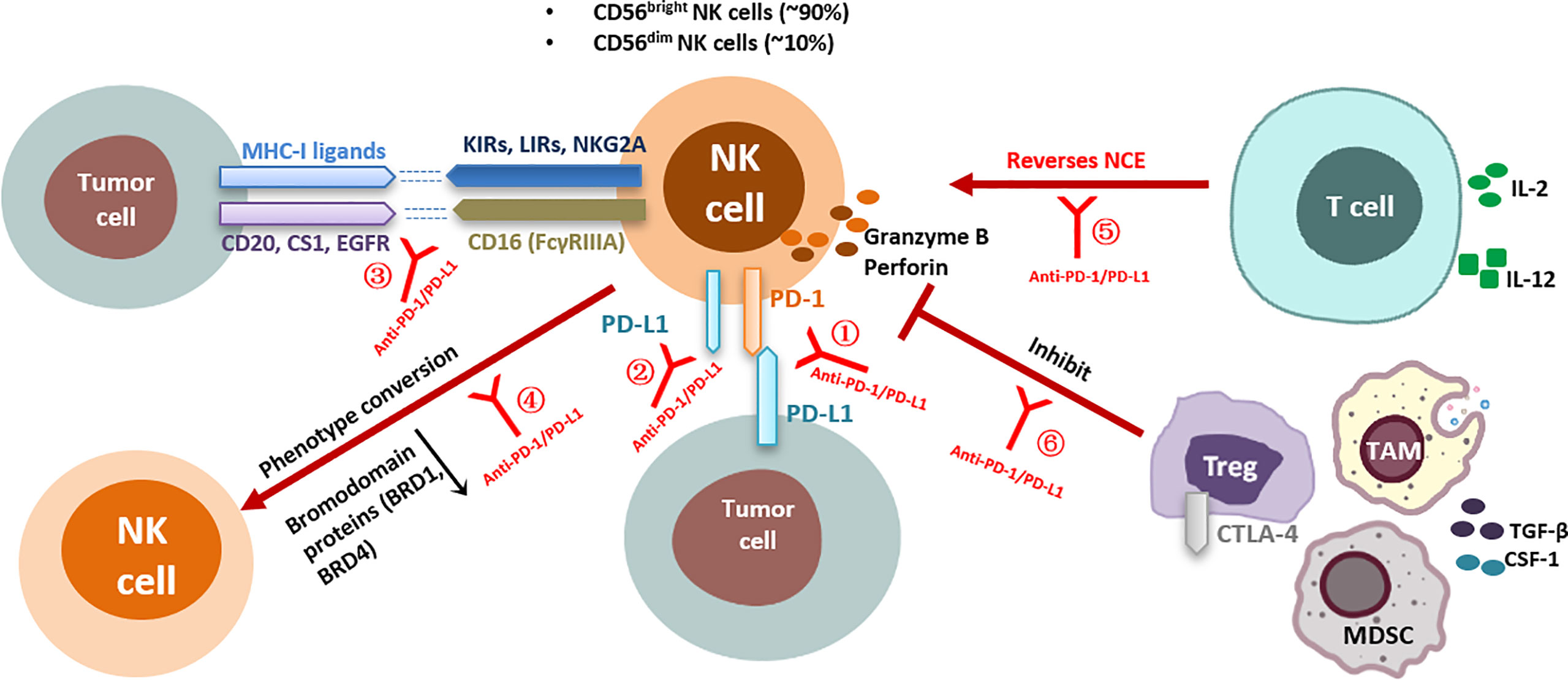
Figure 1 Effects of anti-PD-1/PD-L1 antibodies on NK cell function and corresponding regulatory mechanisms. Direct interaction of anti-PD-1/PD-L1 antibodies with NK cells: ①Anti-PD-1/PD-L1 antibodies block PD-1/PD-L1 inhibitory signaling in NK cells; ②Anti-PD-1/PD-L1 antibodies enhance PD-L1+ NK-cell antitumor activity; ③Anti-PD-1/PD-L1 antibodies enhance the ADCC effect of NK cells; ④Anti-PD-1/PD-L1 bispecific antibodies induce phenotypic transformation of NK cells; Indirect interaction of anti-PD-1/PD-L1 antibodies with NK cells: ⑤Anti-PD-1/PD-L1 antibodies indirectly reverse NK cell exhaustion by affecting CD8+ T cell activity; ⑥Anti-PD-1/PD-L1 antibodies relieve the inhibition of NK cell function by affecting Tregs. KIRs, killer cell immunoglobulin-like receptors; LIRs, leukocyte immunoglobulin-like receptors; NKG2A, natural killer group 2 A; CTLA-4, cytotoxicT-lymphocyte-associated protein 4; IL, interleukin; NCE, NK cell exhaustion; PD-1, programmed death receptor-1; PD-L1, programmed death-ligand 1; MHC, major histocompatibility complex; TGF-β, transformation growth factor-β; IFN-γ, interferon-γ; Tregs, regulatory T cells; CSF-1, colony-stimulating factor 1; EGFR, epidermal growth factor receptor; NK cell, natural killer cell; TAMs, tumor-associated macrophages; MDSCs, myeloid-derived suppressor cells.
As discussed above, the binding of PD-L1 expressed by tumor cell surface to PD-1 expressed by activated NK cells potentially inhibits NK cell-dependent immune surveillance and mediates antitumor immune responses. However, studies on the mechanism by which PD-1 inhibits NK cell response against tumors and the possibility of inducing NK cell responses via PD-1/PD-L1 blockade are still scarce. In one study, CD16- or ILCs-activated NK cells showed upregulated expression of PD-1, as well as CD69, CD107a, IFN-γ, and granzyme B; however, the expression of these molecules was downregulated and that of CD16 surface density was significantly downregulated after the binding of PD-1 to PD-L1 (33). PD-1 blockade increased NK cell activation and cytotoxicity, but only in patients with HNCs showing high PD-L1 expression (33). Another study used anti-PD-1/PD-L1 antibodies to block the interaction between PD-1and PD-L1 and found that this strategy could help reverse PD-1 inhibition and the dysfunctional state of PD-1+ NK cells. This blockade leads to a significant increase in NK cell cytotoxicity and cytokine production, inhibition of tumor growth in vivo, and obvious improvement in NK cell-based antitumor response (32). Similarly, a previous study found that blocking PD-1/PD-L1 signaling on the surface of NK cells significantly enhanced IFN-γ production, CD107a expression, and cell degranulation, and inhibited NK cell apoptosis in vitro. More importantly, PD-1 blocking antibody was found to significantly inhibit the growth of xenograft tumors, and NK depletion completely abolished this inhibition of tumor growth (29). However, inhibition of tumor growth could be completely abolished by NK depletion. Furthermore, PD-1 may exert its inhibitory effect on NK cells by interfering with AKT activation, and PD-1/PD-L1 blockade activates NK cells by enhancing the PI3K/AKT signaling pathway in them (29). In addition, anti-PD-1/PD-L1 antibodies can enhance the secretion of tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) by inhibiting PD-1 in NK cells, effectively improving the antitumor activity of these cells (62). IFN-β has been shown to activate NK cells and induce cytotoxicity against tumor cells by upregulating NK cell-surface membrane-bound and soluble TRAIL expression, which leads to subsequent activation of the TRAIL receptor signaling pathway and apoptosis in nasopharyngeal carcinoma (NPC) cells (62). Subsequently, blocking the PD-1/PD-L1 checkpoint in the presence of IFN-β further increased the killing activity of NK cells against NPC cells, and this suggests that blocking PD-1 in activated NK cells may increase the secretion of soluble TRAIL and contribute to the killing of TRAIL NPC cells (62). This study revealed a new mechanism by which IFN-β and anti-PD-1 antibodies enhance the antitumor effect of NK cells and provide ideas for the therapeutic strategy of the combination of IFN-β and anti-PD-1.
Recently, researchers have found that repetitive irradiation can increase PD-L1 levels in non-small cell lung carcinoma (NSCLC) cells while reducing NKG2D ligand levels, which may reduce the sensitivity of lung tumor cells to the cytotoxic effect of NK cells and the possibility that tumor cells escape immune responses (63). Mechanistic studies revealed that IL-6-MEK/ERK signaling contributes most significantly to the upregulation of PD-L1 or downregulation of NKG2D ligand in radioresistant cells (63), while in another similar study, IL‐6‐JAK/STAT3 signaling was shown to contribute significantly to this process (64). Subsequently, researchers examined PD‐1 levels in NK cells and found that PD‐1 expression could not be detected in primary NK cells, but this expression increased when NK cells were exposed to tumor cells; in other words, there was a PD‐L1/PD‐1 interaction between tumor cells and NK cells, which inhibited the activity of NK cells (63). When neutralizing antibodies against PD-L1 were added to radioresistant cell/NK cell co-cultures, this resistance was reduced and the susceptibility of tumor cells to NK cell cytotoxicity increased, presumably because PD‐L1/PD‐1 interaction was blocked (63). In addition, the added PD-L1 antibodies effectively reversed NK cell activity by releasing PD-1 from the PD-L1–PD-1 complex; these antibodies also effectively reversed the expression of NKG2D ligand on tumor cells, which further enhanced the killing ability of NK cells against tumor cells (63). In summary, the PD-1/PD-L1 axis is an inhibitory/regulatory signal for the interaction between tumor cells and NK cells, and blocking of PD-1/PD-L1 interaction may be an effective antitumor immunotherapeutic strategy that is based on the reversal of NK cell dysfunction.
Anti-PD-1/L1 mAbs may act on NK cells through other non-PD-1 dependent pathways in addition to the PD-1/PD-L1 pathway. Upon encountering and being activated by NK-susceptible tumor cells, NK cells not only secrete cytokines and cytolytic granules, but also upregulate the expression of PD-L1 on their own surface through the PI3K/AKT/NF-κB pathway (8). This population of PD-L1+ NK cells is essential for the antitumor activity of PD-L1 mAbs. Upon application of atezolizumab, an mAb medication against PD-L1, PD-L1+ NK cells express significantly increased levels of granzyme B, IFN-γ, and CD107a, which results in a significant increase in antitumor activity and ultimately a significant decrease in tumor burden in mice (8). Furthermore, it was confirmed that anti-PD-L1 mAbs directly activate PD-L1+ NK cells through the p38/NF-κB pathway in PD-L1− tumors (8). Upregulation of PD-L1 by NK cells, as well as the direct effect of atezolizumab on NK cells, leads to NF-κB activation, which creates a positive feedback loop in the presence of excessive immunotherapeutic agents to consistently induce PD-L1 expression and further activate NK cells (8). In the loop, the binding of anti-PD-L1 mAbs to PD-L1 upregulates PD-L1 expression on the surface of NK cells, thus, increasing the number of binding sites for the drug; this, in turn, leads to sustained activation of p38, which further transmits strong activation signals to NK cells to maintain their cytotoxic and cytokine secretion characteristics (8). The study suggested that anti-PD-L1 mAb therapy has a unique therapeutic effect against PD-L1-negative tumors (8). Based on the fact that PD-L1+ NK cells act through a PD-1-independent pathway, the discovery of new antitumor mechanisms provides insights into the activation of NK cells and a potential explanation for the response to anti-PD-L1 mAb therapy in some patients who lack PD-L1 expression.
Most PD-1/PD-L1 inhibitors (including nivolumab, pembrolizumab, etc.) have human IgG4 which has low fragment crystallizable (Fc) effector activity. These would be expected to have low ADCC. Atezolizumab is aglycosylated and would be expected to have no ADCC. These all prevent potent ADCC against non-tumor cells expressing PD-L1. However, avelumab, a fully human IgG1 anti-PD-L1 mAb containing a wild-type Fc that induces ADCC (65), has shown toxicity and efficacy similar to those of Fc-modified anti-PD-1/PD-L1 mAbs in several phase I and II clinical trials (66). FcγRIIIA expressed on NK cells can activate NK cell-induced cytotoxicity by recognizing the Fc portion of tumor-bound antibodies and releasing cytotoxic factors and cytokines that recruit and activate other immune cells with specific antitumor activity in the presence of tumor antigen-targeting antibodies (16). Investigators have demonstrated that avelumab triggers and enhances NK cell-mediated ADCC against TNBC cells expressing a certain level of PD-L1, which results in a significant increase in tumor cell lysis. Park et al. (59) also demonstrated that when NK cells were co-cultured with wild-type Fc anti-PD-L1 mAbs, which can induce ADCC effects on NK cells, the cytotoxicity against PD-L1-positive tumor cell lines was significantly enhanced. Therefore, some anti-PD-L1 mAbs may act as both tumor antigen-targeting antibodies and anti-PD-L1 inhibitors, which leads to the activation of NK cell and CD8+ T cell and improvement in therapeutic efficacy.
The magnitude of the ADCC effect of NK cells evoked by anti-PD-L1 mAbs may be modulated by other factors. First, polymorphisms in FcγRIIIA may affect NK cell-mediated interindividual variability in the ADCC effect (67). Although this association was identified in studies involving different tumor antigen-targeting mAbs of the IgG1 isotype (rituximab, trastuzumab, and cetuximab), it may not be generalizable to different settings (68). Preliminary in vitro results showed that avelumab-mediated ADCC was more effective in patients with NK cells expressing the high-affinity CD16 valine (V) allele than in those with NK cells expressing the low-affinity phenylalanine (F) allele and F/F genotype (65); however, this difference in effectiveness needs to be verified in clinical studies. Second, the release of some cytokines, such as IL-2 and IL-15, as well as subsequently stimulated IFN-γ, can stimulate the enhancement of avelumab-triggered cytokine production and degranulation in NK cells while increasing lytic activity against tumor cells (67). In addition, in a previous study, investigators enhanced the ADCC of avelumab against many types of cancer cells through epigenetic priming of NK cells and tumors (69). This evidence suggests that therapeutic strategies that block PD-1/PD-L1 while inducing the ADCC action of NK cells may enhance existing immunotherapeutic efficacy. Considering that the magnitude of NK cell-based ADCC effects induced by anti-PD-L1 mAbs can be regulated by some factors such as immunomodulators (IL-15 or IL-2), combining drugs targeting these factors (67) may improve the effectiveness of immunotherapeutic strategy.
Bromodomain proteins, such as BRD1 and BRD4, play a role in the development of immune and hematological cells and in the regulation of tumor inflammation (70–72). In a previous study on high-grade serous ovarian cancer (HGSC), BRD1 expression was found to be low in tumor cells and high in immune cells, which is associated with significant downregulation of T cell- and NK cell-surface activity markers (GZMA, GZMB, IFNG, and NKG7) and upregulation of the naïve T cell marker TCF7 (73). This study performed immune function and single-cell RNA-seq transcriptional profiling of novel HGSC organoid/immune cell co-cultures treated with unique bispecific anti-PD-1/PD-L1 antibodies versus monospecific anti-PD-1 or anti-PD-L1 antibodies (control) (73). It revealed that bispecific antibodies uniquely induced a transition from inert to more active and cytotoxic NK cell phenotypes and a transition from naïve to more active and cytotoxic progenitor-exhausted phenotypes of CD8+ T cells after treatment. It was further found that these superior cell state changes were driven in part by the downregulation of BRD1 expression induced by bispecific antibodies in immune cells (73). The inhibitory effect of the small molecule inhibitor, BAY-299, on BRD1 partially leads to increased NK cell maturation, activation, and tumor cell killing via alteration of the chromatin pathway of key immune transcription factors (e.g., GATA3, TBX21, and TBXT), induces similar state transitions in immune cells in vitro and in vivo and demonstrates the in vivo efficacy of bispecific antibodies (73). Therefore, changes in the activity and cytotoxic status of NK cells and T cells may be the key to driving the induction of effective antitumor immune responses using bispecific antibodies, by partially eliminating some TME-driven dysfunction through epigenetic changes (BRD1 downregulation or inhibition).
Regulative effects of NK and CD8+ T cells on each other have been reported in many infection models (74, 75) and antigen-independent IL-2 models (76), and PD-1/PD-L1 inhibitors may indirectly alter NK cell function by affecting CD8+ T cell activity. NK cell exhaustion (NCE) has been identified as a self-regulatory mechanism responsible for inducing dysfunctional phenotypes to prevent exacerbated immune responses under conditions of chronic stimulation (77), and it is a crucial mechanism involved in tumor or viral evasion of immune responses. IL-2 is a pleiotropic cytokine that activates T cells, NK cells, and dendritic cells. IL-2 binds to IL-2 receptors (IL-2Rs), including CD32 (IL-2Rγc), CD122 (IL-2Rβ), and CD25 (IL-2Rα). IL-2Rs showed different affinities for cytokines, with CD25 (IL-2Rα) having the highest IL-2 affinity. CD25 is constitutively expressed on Tregs; therefore, IL-2 treatment can lead to the expansion of these cells, thus mediating NK and CD8+ T cell suppression through multiple mechanisms (76, 78, 79). A recent study evaluated the role of the PD-1/PD-L1 pathway in NK cell activation, function, and depletion in a mouse model of IL-2-dependent depletion (80) and found that anti-PD-1 therapy provides an activating advantage to CD8+ T cells in the competition for IL-2 by promoting CD8+ T cell expansion, activation, and functional phenotype; consequently, the quantity of stimulatory cytokines available to NK cells becomes limited, which results in a delayed NCE, improved NK cell activation, higher proliferative capacity, and enhanced granzyme B production. This evidence suggests that the phenotypic and functional benefits observed after the chronic stimulation of NK cells by anti-PD-1 therapy are indirectly mediated by the effect of anti-PD-1 antibodies on CD8+ T cells, rather than by the direct effect of anti-PD-1 drugs on NK cells; this can be demonstrated by the reversal of the benefit of anti-PD-1 therapy on NK cells by the depletion of CD8+ T cells. Disruption of the balance between NK and CD8+ T cells affects the homeostatic response to each other’s stimuli, and this resource competition (i.e., IL-2) delays the onset of NCE. Thus, there is a delicate balance between CD8+ T cells, Tregs, and NK cells that is partly regulated by their ability to respond to cytokines. Achieving a balance between these immune cells may be important for achieving long-term efficacy of immunotherapy. Although dual regulation between CD8+ T cells and NK cells is caused by competition for space and resources, direct or indirect lysis, and functional inhibition, these two populations can also work together to mount a stronger response. Another study found that dual blockade of PD-1 and IL-10 enhanced cytokine secretion, NK degranulation, and killing target cell function of NK cells by restoring HIV-specific CD4+ T cell function, thus establishing a previously unappreciated relationship between CD4+ T cell injury and NK cell depletion in HIV infection (81). This important evidence also fully suggests that PD-1 blockade may enhance immunotherapy by improving collaboration between CD4+ T cells and NK cells.
Immunosuppressive cells such as tumor-associated macrophages (TAMs), myeloid-derived suppressor cells (MDSCs), and Tregs in the TME inhibit NK cell proliferation, infiltration, and activation by secreting immunosuppressive cytokines (TGF-β and IL-10) or by interfering with NK cell receptor expression and activation; thus, these cells facilitate tumor immune escape and promote progression and metastasis (82, 83). It can, therefore, be hypothesized that PD-1/PD-L1 blockade prevents the induction of immunosuppression and improves NK cell efficacy to increase the survival of a tumor-bearing animal. A previous study showed that anti-PD-L1 therapy has no direct effect on the cytotoxicity or cytokine secretion of PD-1-negative PM21 particle-expanded NK cells in response to PD-L1+ targets in vitro; however, secretion of a large quantity of IFN-γ by NK cells significantly improved antitumor efficacy, and long-lasting retention of their cytotoxic phenotype by NK cells can be observed in vivo (84). Thus, the investigators continued to explore the specific mechanism by which anti-PD-L1 therapy enhances the antitumor efficacy of NK cells. They found that PM21-NK cells are highly cytotoxic to tumor cells and secrete IFN-γ upon stimulation, which leads to the induction of PD-L1 expression in tumors and consequently to the induction and in situ proliferation of Tregs in the TME (84). Subsequently, blockade of PD-L1 mitigated the induction of Treg expansion and associated immunosuppressive responses, which in turn improved the NK cell phenotype and cytotoxic activity, as well as survival in treated animals (84). The number of CD57+ NK cells has increased, a population with high cytotoxic capacity and responsiveness via CD16 binding, and the presence of these cells is correlated with better outcomes in patients with squamous cell lung cancer in other reports (85, 86). The expansion of Tregs may occur in the TME with PD-L1 expression, and Tregs have been reported to suppress the survival and cytotoxic function of NK cells through several mechanisms, including TGF-β surface presentation (87–89). Thus, anti-PD-L1 therapy may improve NK cell antitumor efficacy by reducing in situ Treg generation and preventing Treg induction.
NK cells and other immune cells in the TME mutually regulate each other; for example, PD-L1+ liver-resident NK (LrNK) cells have an inhibitory interaction with PD-1+ T cells (90). NK cells can indirectly affect the antitumor immune function by acting on these immune cells, thereby enhancing the response of tumors to anti-PD-1/PD-L1 antibodies. The large number of beneficial effects of tumor-infiltrating NK cells can be mediated by modulating the TME and promoting T cell recruitment and activation. An earlier study that used mixed lymphocyte cultures showed that NK cells are required for the differentiation and activation of CD8+ T cells with cytotoxic functions (91). Another study that used in vitro co-culture and in vivo xenograft adoptive transfer experiments demonstrated that high-quality NK cells (iNK) derived from induced pluripotent stem cells (iPSCs) can recruit and activate T cells, allow them to respond to PD-1 blockade, and enhance inflammatory cytokine production and tumor elimination (92). NK cells in tumor-infiltrating and draining lymph nodes were reported to show upregulation of the inhibitory molecule PD-L1, which transmits an inhibitory signal by interacting with PD-1 expressed on DCs to limit the activation of these cells; this in turn leads to a decrease in the priming ability and memory response of tumor-specific CD8+ T cells (61). When blocking the interaction between NK cells and DCs, a significantly higher frequency of CD8+ T cells, level of IFN-γ production, and capacity for cytotoxicity were observed (61). In this model, tumor cells induced the modulation of DC activation via PD-L1hiNK cells, which reduced the priming capacity of CD8+ T cells. Conversely, PD-1/PD-L1 inhibitors may reverse the inhibitory effect of PD-1-PD-L1 interaction on CD8+ T cells by disrupting the direct interaction pathway between NK cells and DC cells; this presumably activates NK cell activity and needs to be explored further in future studies. In addition to interacting with DCs through the PD-1/PD-L1 axis, a study has shown in human melanoma that NK cells stably form conjugates with stimulatory dendritic cells (SDCs) in mouse TME and positively regulate the abundance of SDCs in tumors by producing FLT3LG, the cDC1 formative cytokine. SDCs are important in stimulating cytotoxic T cells and driving anticancer immune responses (60). Although anti-PD-1 immunotherapy for cancer primarily targets T cells, NK cell frequencies correlate with protective SDCs in human cancers, with patient responsiveness to anti-PD-1 immunotherapy, and with prolonged overall survival (60).
In summary, blocking PD-1/PD-L1 may activate the systemic immune response by reversing the inhibitory effect of NK cells on other immune cells or activating the antitumor capability of other immune cells by NK cells, which is an effective antitumor immunotherapy strategy. Therefore, a combination therapy based on NK cells and anti-PD-1/PD-L1 drugs can be established.
Considering NK cell-mediated cytotoxicity does not require MHC class I and the unique effects of anti-PD-1/PD-L1 mAb therapy on NK cell function described above, adoptive transfer of autologous or allogeneic NK cells together with anti-PD-1/PD-L1 antibodies may potentially enhance the outcomes of patients receiving cancer immunotherapy. A previous study explored the effects of a combination of NK cells with PD-L1 blockers, regardless of PD-1 expression on NK cells or the initial PD-L1 status of the tumor cells (84). The OS of animals treated with a combination of anti-PD-L1 antibodies and PM21-NK cells was twice as higher than that of those treated with anti-PD-L1 antibodies alone (48 days vs. 24 days, P = 0.0001) (84). The first clinical study on the combination of anti-PD-1 antibodies (pembrolizumab) and allogeneic NK cells in patients with advanced NSCLC in China was recently published (93), and it reported the doubling of the number of NK cells, significant increases in the levels of cytokines such as IL-2, TNF-β, and IFN-γ, and significant decreases in the levels of multiple tumor markers such as circulating tumor cells (CTCs) after treatment in the combination therapy group. Patients in the combined therapy group had a significantly better overall response rate (36.5% vs. 18.5%) and a significantly better survival outcome (OS: 15.5 months vs. 13.3 months; PFS: 6.5 months vs. 4.3 months; all P < 0.05) than did those in the anti-PD-1 antibody alone group; moreover, the benefits were more significant in the combination therapy group [tumor proportion score [TPS] ≥ 50%] than in the latter group. In addition, patients who received multiple courses of NK cell infusion showed a better OS than those who received a single course of NK cell infusion (18.5 months vs. 13.5 months) (93). The treatment was well tolerated throughout the treatment. All adverse events were below grade 4, with grade 2 events comprising the majority of events. All symptoms were relieved after symptomatic treatment. Therefore, a combination therapy of anti-PD-L1 antibodies and NK cells can significantly enhance the antitumor effect, improve the survival benefit, and serve as new treatment regimen for previously treated patients with advanced PD-L1+ NSCLC. In addition, the combination of anti-PD-1/PD-L1 antibody therapy and drugs that enhance the antitumor effects of NK cells through other mechanisms, including a combination of anti-NKG2A mAb (monalizumab) (94), KIR blockade (95), cytokine therapy (96), and therapies targeting other checkpoint receptors such as CTLA-4, LAG-3, CD96, and TIGIT, may have a synergistic effect (97–99); therefore, this strategy may be beneficial for improving the efficacy of immunotherapy and overcoming drug resistance.
The use of anti-PD-1/PD-L1 antibodies could be a promising option for the successful treatment of malignancies as they help overcome T-cell depletion by blocking the PD-1/PD-L1 signaling pathway in the TME. However, most patients and tumor types have shown low/no response to these therapies, or some patients with predicted no or low response to treatment have shown significant benefit; therefore, in-depth exploration of the mechanism of action and efficacy of ICIs is necessary to effectively screen for suitable patients to expand the benefits to them. Accumulating evidence suggests that immune cells other than T cells, such as NK cells, are also involved and play an important role in the PD-1/PD-L1 blocking process; these non-T cells have become an effective complement to T cell immune responses because of their unique advantages, especially against MHC-deficient tumors that show low antigenicity or are resistant to T cell recognition and cytolysis. At present, findings of studies on the expression of PD-1 and PD-L1 on NK cells and their functions are not completely consistent, and further evidence derived from human NK cell-based research is necessary. According to our review, PD-1/PD-L1 antibodies can rescue NK cell from multiple aspects of dysfunction caused by TME, revitalize the cytotoxic activity of these cells against tumors, and further initiate and enhance T cell-mediated adaptive antitumor immunity; NK cells can also indirectly enhance the efficacy of PD-1/PD-L1 blockade by affecting other immune cells in TME. On the basis of these findings, multiple combination strategies, such as the use of a combination of PD-1/PD-L1 inhibitors with drugs that promote NK cell infiltration, persistence, and activation in tumors (e.g.,cytokines, stimulator of IFN gene [STING] agonists) and resistance to inhibitory TMEs (e.g., anti-TGF-β mAbs), are under investigation in basis research studies or clinical trials; these strategies aim to further increase the antitumor activity of NK cells and improve the tumor response to immunotherapy. At present, our understanding of the role of NK cells during PD-1/PD-L1 blockade is still insufficient and needs to be further explored and validated using basic mechanistic studies. A comprehensive and in-depth understanding of the response mechanism of PD-1/PD-L1-based immunotherapy and the interaction mechanism between each immune cell in TME with another or with tumors can lay a foundation for optimizing the efficacy of existing treatments, developing new immunotherapies based on NK cells, and developing combination therapy strategies.
All authors listed have made a substantial, direct, and intellectual contribution to the work and approved it for publication.
This work was supported by grants from Jilin Provincial Department of Science and Technology Project (20190303146SF); Jilin Provincial Department of Finance Project (JLSWSRCZX2020-0023); Jilin Province Biotherapeutic Science and Technology Innovation Center Project (20200602032ZP).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Robert C, Long GV, Brady B, Dutriaux C, Maio M, Mortier L, et al. Nivolumab in Previously Untreated Melanoma Without BRAF Mutation. N Engl J Med (2015) 372(4):320–30. doi: 10.1056/NEJMoa1412082
2. Powles T, Eder JP, Fine GD, Braiteh FS, Loriot Y, Cruz C, et al. MPDL3280A (Anti-PD-L1) Treatment Leads to Clinical Activity in Metastatic Bladder Cancer. Nature (2014) 515(7528):558–62. doi: 10.1038/nature13904
3. Tumeh PC, Harview CL, Yearley JH, Shintaku IP, Taylor EJ, Robert L, et al. PD-1 Blockade Induces Responses by Inhibiting Adaptive Immune Resistance. Nature (2014) 515(7528):568–71. doi: 10.1038/nature13954
4. Herbst RS, Soria JC, Kowanetz M, Fine GD, Hamid O, Gordon MS, et al. Predictive Correlates of Response to the Anti-PD-L1 Antibody MPDL3280A in Cancer Patients. Nature (2014) 515(7528):563–7. doi: 10.1038/nature14011
5. Ansell SM, Lesokhin AM, Borrello I, Halwani A, Scott EC, Gutierrez M, et al. PD-1 Blockade With Nivolumab in Relapsed or Refractory Hodgkin's Lymphoma. N Engl J Med (2015) 372(4):311–9. doi: 10.1056/NEJMoa1411087
6. Ansell SM. Hodgkin Lymphoma: MOPP Chemotherapy to PD-1 Blockade and Beyond. Am J Hematol (2016) 91(1):109–12. doi: 10.1002/ajh.24226
7. Cader FZ, Schackmann RCJ, Hu X, Wienand K, Redd R, Chapuy B, et al. Mass Cytometry of Hodgkin Lymphoma Reveals a CD4(+) Regulatory T-Cell-Rich and Exhausted T-Effector Microenvironment. Blood (2018) 132(8):825–36. doi: 10.1182/blood-2018-04-843714
8. Dong W, Wu X, Ma S, Wang Y, Nalin AP, Zhu Z, et al. The Mechanism of Anti-PD-L1 Antibody Efficacy Against PD-L1-Negative Tumors Identifies NK Cells Expressing PD-L1 as a Cytolytic Effector. Cancer Discov (2019) 9(10):1422–37. doi: 10.1158/2159-8290.Cd-18-1259
9. Leiberman NAP, Degolier K, Haberthur K, Chinn H, Moyes KW, Bouchlaka MN, et al. An Uncoupling of Canonical Phenotypic Markers and Functional Potency of Ex Vivo-Expanded Natural Killer Cells. Front Immunol (2018) 9:150. doi: 10.3389/fimmu.2018.00150
10. Kärre K, Ljunggren HG, Piontek G, Kiessling R. Selective Rejection of H-2-Deficient Lymphoma Variants Suggests Alternative Immune Defence Strategy. Nature (1986) 319(6055):675–8. doi: 10.1038/319675a0
11. Bryceson YT, Ljunggren HG, Long EO. Minimal Requirement for Induction of Natural Cytotoxicity and Intersection of Activation Signals by Inhibitory Receptors. Blood (2009) 114(13):2657–66. doi: 10.1182/blood-2009-01-201632
12. Li H, Zhai N, Wang Z, Song H, Yang Y, Cui A, et al. Regulatory NK Cells Mediated Between Immunosuppressive Monocytes and Dysfunctional T Cells in Chronic HBV Infection. Gut (2018) 67(11):2035–44. doi: 10.1136/gutjnl-2017-314098
13. Koirala P, Roth ME, Gill J, Piperdi S, Chinai JM, Geller DS, et al. Immune Infiltration and PD-L1 Expression in the Tumor Microenvironment are Prognostic in Osteosarcoma. Sci Rep (2016) 6:30093. doi: 10.1038/srep30093
14. Fu B, Tian Z, Wei H. Subsets of Human Natural Killer Cells and Their Regulatory Effects. Immunology (2014) 141(4):483–9. doi: 10.1111/imm.12224
15. Moretta L, Montaldo E, Vacca P, Del Zotto G, Moretta F, Merli P, et al. Human Natural Killer Cells: Origin, Receptors, Function, and Clinical Applications. Int Arch Allergy Immunol (2014) 164(4):253–64. doi: 10.1159/000365632
16. Wang W, Erbe AK, Hank JA, Morris ZS, Sondel PM. NK Cell-Mediated Antibody-Dependent Cellular Cytotoxicity in Cancer Immunotherapy. Front Immunol (2015) 6:368. doi: 10.3389/fimmu.2015.00368
17. Wagtmann N, Rajagopalan S, Winter CC, Peruzzi M, Long EO. Killer Cell Inhibitory Receptors Specific for HLA-C and HLA-B Identified by Direct Binding and by Functional Transfer. Immunity (1995) 3(6):801–9. doi: 10.1016/1074-7613(95)90069-1
18. Godal R, Bachanova V, Gleason M, Mccullar V, Yun GH, Cooley S, et al. Natural Killer Cell Killing of Acute Myelogenous Leukemia and Acute Lymphoblastic Leukemia Blasts by Killer Cell Immunoglobulin-Like Receptor-Negative Natural Killer Cells After NKG2A and LIR-1 Blockade. Biol Blood Marrow Transplant (2010) 16(5):612–21. doi: 10.1016/j.bbmt.2010.01.019
19. Sivori S, Vacca P, Del Zotto G, Munari E, Mingari MC, Moretta L. Human NK Cells: Surface Receptors, Inhibitory Checkpoints, and Translational Applications. Cell Mol Immunol (2019) 16(5):430–41. doi: 10.1038/s41423-019-0206-4
20. Sheffer M, Lowry E, Beelen N, Borah M, Amara SN, Mader CC, et al. Genome-Scale Screens Identify Factors Regulating Tumor Cell Responses to Natural Killer Cells. Nat Genet (2021) 53(8):1196–206. doi: 10.1038/s41588-021-00889-w
21. Buchbinder EI, Desai A. CTLA-4 and PD-1 Pathways: Similarities, Differences, and Implications of Their Inhibition. Am J Clin Oncol (2016) 39(1):98–106. doi: 10.1097/coc.0000000000000239
22. Taylor S, Huang Y, Mallett G, Stathopoulou C, Felizardo TC, Sun MA, et al. PD-1 Regulates KLRG1(+) Group 2 Innate Lymphoid Cells. J Exp Med (2017) 214(6):1663–78. doi: 10.1084/jem.20161653
23. Brunner-Weinzierl MC, Rudd CE. CTLA-4 and PD-1 Control of T-Cell Motility and Migration: Implications for Tumor Immunotherapy. Front Immunol (2018) 9:2737. doi: 10.3389/fimmu.2018.02737
24. Pesce S, Greppi M, Tabellini G, Rampinelli F, Parolini S, Olive D, et al. Identification of a Subset of Human Natural Killer Cells Expressing High Levels of Programmed Death 1: A Phenotypic and Functional Characterization. J Allergy Clin Immunol (2017) 139(1):335–46.e3. doi: 10.1016/j.jaci.2016.04.025
25. Mariotti FR, Petrini S, Ingegnere T, Tumino N, Besi F, Scordamaglia F, et al. PD-1 in Human NK Cells: Evidence of Cytoplasmic mRNA and Protein Expression. Oncoimmunology (2019) 8(3):1557030. doi: 10.1080/2162402x.2018.1557030
26. Beldi-Ferchiou A, Lambert M, Dogniaux S, Vély F, Vivier E, Olive D, et al. PD-1 Mediates Functional Exhaustion of Activated NK Cells in Patients With Kaposi Sarcoma. Oncotarget (2016) 7(45):72961–77. doi: 10.18632/oncotarget.12150
27. Macfarlane AWT, Jillab M, Plimack ER, Hudes GR, Uzzo RG, Litwin S, et al. PD-1 Expression on Peripheral Blood Cells Increases With Stage in Renal Cell Carcinoma Patients and is Rapidly Reduced After Surgical Tumor Resection. Cancer Immunol Res (2014) 2(4):320–31. doi: 10.1158/2326-6066.Cir-13-0133
28. Giuliani M, Janji B, Berchem G. Activation of NK Cells and Disruption of PD-L1/PD-1 Axis: Two Different Ways for Lenalidomide to Block Myeloma Progression. Oncotarget (2017) 8(14):24031–44. doi: 10.18632/oncotarget.15234
29. Liu Y, Cheng Y, Xu Y, Wang Z, Du X, Li C, et al. Increased Expression of Programmed Cell Death Protein 1 on NK Cells Inhibits NK-Cell-Mediated Anti-Tumor Function and Indicates Poor Prognosis in Digestive Cancers. Oncogene (2017) 36(44):6143–53. doi: 10.1038/onc.2017.209
30. Alvarez IB, Pasquinelli V, Jurado JO, Abbate E, Musella RM, de la Barrera SS, et al. Role Played by the Programmed Death-1-Programmed Death Ligand Pathway During Innate Immunity Against Mycobacterium Tuberculosis. J Infect Dis (2010) 202(4):524–32. doi: 10.1086/654932
31. Norris S, Coleman A, Kuri-Cervantes L, Bower M, Nelson M, Goodier MR. PD-1 Expression on Natural Killer Cells and CD8(+) T Cells During Chronic HIV-1 Infection. Viral Immunol (2012) 25(4):329–32. doi: 10.1089/vim.2011.0096
32. Hsu J, Hodgins JJ, Marathe M, Nicolai CJ, Bourgeois-Daigneault MC, Trevino TN, et al. Contribution of NK Cells to Immunotherapy Mediated by PD-1/PD-L1 Blockade. J Clin Invest (2018) 128(10):4654–68. doi: 10.1172/jci99317
33. Concha-Benavente F, Kansy B, Moskovitz J, Moy J, Chandran U, Ferris RL. PD-L1 Mediates Dysfunction in Activated PD-1(+) NK Cells in Head and Neck Cancer Patients. Cancer Immunol Res (2018) 6(12):1548–60. doi: 10.1158/2326-6066.Cir-18-0062
34. Sun R, Xiong Y, Liu H, Gao C, Su L, Weng J, et al. Tumor-Associated Neutrophils Suppress Antitumor Immunity of NK Cells Through the PD-L1/PD-1 Axis. Transl Oncol (2020) 13(10):100825. doi: 10.1016/j.tranon.2020.100825
35. Vari F, Arpon D, Keane C, Hertzberg MS, Talaulikar D, Jain S, et al. Immune Evasion via PD-1/PD-L1 on NK Cells and Monocyte/Macrophages is More Prominent in Hodgkin Lymphoma Than DLBCL. Blood (2018) 131(16):1809–19. doi: 10.1182/blood-2017-07-796342
36. Park IH, Yang HN, Lee KJ, Kim TS, Lee ES, Jung SY, et al. Tumor-Derived IL-18 Induces PD-1 Expression on Immunosuppressive NK Cells in Triple-Negative Breast Cancer. Oncotarget (2017) 8(20):32722–30. doi: 10.18632/oncotarget.16281
37. Zheng G, Guo Z, Li W, Xi W, Zuo B, Zhang R, et al. Interaction Between HLA-G and NK Cell Receptor KIR2DL4 Orchestrates HER2-Positive Breast Cancer Resistance to Trastuzumab. Signal Transduct Target Ther (2021) 6(1):236. doi: 10.1038/s41392-021-00629-w
38. Makowska A, Meier S, Shen L, Busson P, Baloche V, Kontny U. Anti-PD-1 Antibody Increases NK Cell Cytotoxicity Towards Nasopharyngeal Carcinoma Cells in the Context of Chemotherapy-Induced Upregulation of PD-1 and PD-L1. Cancer Immunol Immunother (2021) 70(2):323–36. doi: 10.1007/s00262-020-02681-x
39. Mandai M, Hamanishi J, Abiko K, Matsumura N, Baba T, Konishi I. Dual Faces of Ifnγ in Cancer Progression: A Role of PD-L1 Induction in the Determination of Pro- and Antitumor Immunity. Clin Cancer Res (2016) 22(10):2329–34. doi: 10.1158/1078-0432.Ccr-16-0224
40. Diskin B, Adam S, Cassini MF, Sanchez G, Liria M, Aykut B, et al. PD-L1 Engagement on T Cells Promotes Self-Tolerance and Suppression of Neighboring Macrophages and Effector T Cells in Cancer. Nat Immunol (2020) 21(4):442–54. doi: 10.1038/s41590-020-0620-x
41. Sun C, Mezzadra R, Schumacher TN. Regulation and Function of the PD-L1 Checkpoint. Immunity (2018) 48(3):434–52. doi: 10.1016/j.immuni.2018.03.014
42. An LL, Gorman JV, Stephens G, Swerdlow B, Warrener P, Bonnell J, et al. Complement C5a Induces PD-L1 Expression and Acts in Synergy With LPS Through Erk1/2 and JNK Signaling Pathways. Sci Rep (2016) 6:33346. doi: 10.1038/srep33346
43. Wang X, Ni S, Chen Q, Ma L, Jiao Z, Wang C, et al. Bladder Cancer Cells Induce Immunosuppression of T Cells by Supporting PD-L1 Expression in Tumour Macrophages Partially Through Interleukin 10. Cell Biol Int (2017) 41(2):177–86. doi: 10.1002/cbin.10716
44. Wang X, Yang L, Huang F, Zhang Q, Liu S, Ma L, et al. Inflammatory Cytokines IL-17 and TNF-α Up-Regulate PD-L1 Expression in Human Prostate and Colon Cancer Cells. Immunol Lett (2017) 184:7–14. doi: 10.1016/j.imlet.2017.02.006
45. Ritprajak P, Azuma M. Intrinsic and Extrinsic Control of Expression of the Immunoregulatory Molecule PD-L1 in Epithelial Cells and Squamous Cell Carcinoma. Oral Oncol (2015) 51(3):221–8. doi: 10.1016/j.oraloncology.2014.11.014
46. Barsoum IB, Smallwood CA, Siemens DR, Graham CH. A Mechanism of Hypoxia-Mediated Escape From Adaptive Immunity in Cancer Cells. Cancer Res (2014) 74(3):665–74. doi: 10.1158/0008-5472.Can-13-0992
47. Casey SC, Tong L, Li Y, Do R, Walz S, Fitzgerald KN, et al. MYC Regulates the Antitumor Immune Response Through CD47 and PD-L1. Science (2016) 352(6282):227–31. doi: 10.1126/science.aac9935
48. Zhu H, Bengsch F, Svoronos N, Rutkowski MR, Bitler BG, Allegrezza MJ, et al. BET Bromodomain Inhibition Promotes Anti-Tumor Immunity by Suppressing PD-L1 Expression. Cell Rep (2016) 16(11):2829–37. doi: 10.1016/j.celrep.2016.08.032
49. Chen Y, Wang Q, Shi B, Xu P, Hu Z, Bai L, et al. Development of a Sandwich ELISA for Evaluating Soluble PD-L1 (CD274) in Human Sera of Different Ages as Well as Supernatants of PD-L1+ Cell Lines. Cytokine (2011) 56(2):231–8. doi: 10.1016/j.cyto.2011.06.004
50. Tato CM, Mason N, Artis D, Shapira S, Caamano JC, Bream JH, et al. Opposing Roles of NF-kappaB Family Members in the Regulation of NK Cell Proliferation and Production of IFN-Gamma. Int Immunol (2006) 18(4):505–13. doi: 10.1093/intimm/dxh391
51. Antonangeli F, Natalini A, Garassino MC, Sica A, Santoni A, Di Rosa F. Regulation of PD-L1 Expression by NF-κb in Cancer. Front Immunol (2020) 11:584626. doi: 10.3389/fimmu.2020.584626
52. Jiang BH, Jiang G, Zheng JZ, Lu Z, Hunter T, Vogt PK. Phosphatidylinositol 3-Kinase Signaling Controls Levels of Hypoxia-Inducible Factor 1. Cell Growth Differ (2001) 12(7):363–9. doi: 10.1073/pnas.040560897
53. Noman MZ, Desantis G, Janji B, Hasmim M, Karray S, Dessen P, et al. PD-L1 Is a Novel Direct Target of HIF-1α, and its Blockade Under Hypoxia Enhanced MDSC-Mediated T Cell Activation. J Exp Med (2014) 211(5):781–90. doi: 10.1084/jem.20131916
54. Walmsley SR, Print C, Farahi N, Peyssonnaux C, Johnson RS, Cramer T, et al. Hypoxia-Induced Neutrophil Survival is Mediated by HIF-1alpha-Dependent NF-kappaB Activity. J Exp Med (2005) 201(1):105–15. doi: 10.1084/jem.20040624
55. Mak P, Li J, Samanta S, Mercurio AM. Erβ Regulation of NF-kB Activation in Prostate Cancer Is Mediated by HIF-1[J]. Oncotarget (2015) 6(37):40247–54. doi: 10.18632/oncotarget.5377
56. Yao G, Zhang Q, Doeppner TR, Niu F, Li Q, Yang Y, et al. LDL Suppresses Angiogenesis Through Disruption of the HIF Pathway via NF-κb Inhibition Which is Reversed by the Proteasome Inhibitor Bsc2118. Oncotarget (2015) 6(30):30251–62. doi: 10.18632/oncotarget.4943
57. Bellucci R, Martin A, Bommarito D, Wang K, Hansen SH, Freeman GJ, et al. Interferon-γ-Induced Activation of JAK1 and JAK2 Suppresses Tumor Cell Susceptibility to NK Cells Through Upregulation of PD-L1 Expression. Oncoimmunology (2015) 4(6):e1008824. doi: 10.1080/2162402x.2015.1008824
58. Choi B, Lee JS, Kim SJ, Hong D, Park JB, Lee KY. Anti-Tumor Effects of Anti-PD-1 Antibody, Pembrolizumab, in Humanized NSG PDX Mice Xenografted With Dedifferentiated Liposarcoma. Cancer Lett (2020) 478:56–69. doi: 10.1016/j.canlet.2020.02.042
59. Park JE, Kim SE, Keam B, Park HR, Kim S, Kim M, et al. Anti-Tumor Effects of NK Cells and Anti-PD-L1 Antibody With Antibody-Dependent Cellular Cytotoxicity in PD-L1-Positive Cancer Cell Lines. J Immunother Cancer (2020) 8(2):e000873. doi: 10.1136/jitc-2020-000873
60. Barry KC, Hsu J, Broz ML, Cueto FJ, Binnewies M, Combes AJ, et al. A Natural Killer-Dendritic Cell Axis Defines Checkpoint Therapy-Responsive Tumor Microenvironments. Nat Med (2018) 24(8):1178–91. doi: 10.1038/s41591-018-0085-8
61. Iraolagoitia XL, Spallanzani RG, Torres NI, Araya RE, Ziblat A, Domaica CI, et al. NK Cells Restrain Spontaneous Antitumor CD8+ T Cell Priming Through PD-1/PD-L1 Interactions With Dendritic Cells. J Immunol (2016) 197(3):953–61. doi: 10.4049/jimmunol.1502291
62. Makowska A, Braunschweig T, Denecke B, Shen L, Baloche V, Busson P, et al. Interferon β and Anti-PD-1/PD-L1 Checkpoint Blockade Cooperate in NK Cell-Mediated Killing of Nasopharyngeal Carcinoma Cells. Transl Oncol (2019) 12(9):1237–56. doi: 10.1016/j.tranon.2019.04.017
63. Shen MJ, Xu LJ, Yang L, Tsai Y, Keng PC, Chen Y, et al. Radiation Alters PD-L1/NKG2D Ligand Levels in Lung Cancer Cells and Leads to Immune Escape From NK Cell Cytotoxicity via IL-6-MEK/Erk Signaling Pathway. Oncotarget (2017) 8(46):80506–20. doi: 10.18632/oncotarget.19193
64. Xu L, Chen X, Shen M, Yang DR, Fang L, Weng G, et al. Inhibition of IL-6-JAK/Stat3 Signaling in Castration-Resistant Prostate Cancer Cells Enhances the NK Cell-Mediated Cytotoxicity via Alteration of PD-L1/NKG2D Ligand Levels. Mol Oncol (2018) 12(3):269–86. doi: 10.1002/1878-0261.12135
65. Boyerinas B, Jochems C, Fantini M, Heery CR, Gulley JL, Tsang KY, et al. Antibody-Dependent Cellular Cytotoxicity Activity of a Novel Anti-PD-L1 Antibody Avelumab (MSB0010718C) on Human Tumor Cells. Cancer Immunol Res (2015) 3(10):1148–57. doi: 10.1158/2326-6066.Cir-15-0059
66. Hassan R, Thomas A, Patel MR, Nemunaitis JJ, Bennouna J, Powderly JD, et al. Avelumab (MSB0010718C; Anti-PD-L1) in Patients With Advanced Unresectable Mesothelioma From the JAVELIN Solid Tumor Phase Ib Trial: Safety, Clinical Activity, and PD-L1 Expression. J Clin Oncol (2016) 34(15_suppl):8503. doi: 10.1200/JCO.2016.34.15_suppl.8503
67. Juliá EP, Amante A, Pampena MB, Mordoh J, Levy EM. Avelumab, an IgG1 Anti-PD-L1 Immune Checkpoint Inhibitor, Triggers NK Cell-Mediated Cytotoxicity and Cytokine Production Against Triple Negative Breast Cancer Cells. Front Immunol (2018) 9:2140. doi: 10.3389/fimmu.2018.02140
68. Mimura K, Teh JL, Okayama H, Shiraishi K, Kua LF, Koh V, et al. PD-L1 Expression is Mainly Regulated by Interferon Gamma Associated With JAK-STAT Pathway in Gastric Cancer. Cancer Sci (2018) 109(1):43–53. doi: 10.1111/cas.13424
69. Hicks KC, Fantini M, Donahue RN, Schwab A, Knudson KM, Tritsch SR, et al. Epigenetic Priming of Both Tumor and NK Cells Augments Antibody-Dependent Cellular Cytotoxicity Elicited by the Anti-PD-L1 Antibody Avelumab Against Multiple Carcinoma Cell Types. Oncoimmunology (2018) 7(11):e1466018. doi: 10.1080/2162402x.2018.1466018
70. Mishima Y, Miyagi S, Saraya A, Negishi M, Endoh M, Endo TA, et al. The Hbo1-Brd1/Brpf2 Complex Is Responsible for Global Acetylation of H3K14 and Required for Fetal Liver Erythropoiesis. Blood (2011) 118(9):2443–53. doi: 10.1182/blood-2011-01-331892
71. Mishima Y, Wang C, Miyagi S, Saraya A, Hosokawa H, Mochizuki-Kashio M, et al. Histone Acetylation Mediated by Brd1 Is Crucial for Cd8 Gene Activation During Early Thymocyte Development. Nat Commun (2014) 5:5872. doi: 10.1038/ncomms6872
72. White ME, Fenger JM, Carson WE 3rd. Emerging Roles of and Therapeutic Strategies Targeting BRD4 in Cancer. Cell Immunol (2019) 337:48–53. doi: 10.1016/j.cellimm.2019.02.001
73. Wan C, Keany MP, Dong H, Al-Alem LF, Pandya UM, Lazo S, et al. Enhanced Efficacy of Simultaneous PD-1 and PD-L1 Immune Checkpoint Blockade in High-Grade Serous Ovarian Cancer. Cancer Res (2021) 81(1):158–73. doi: 10.1158/0008-5472.Can-20-1674
74. Waggoner SN, Daniels KA, Welsh RM. Therapeutic Depletion of Natural Killer Cells Controls Persistent Infection. J Virol (2014) 88(4):1953–60. doi: 10.1128/jvi.03002-13
75. Xu HC, Huang J, Pandyra AA, Lang E, Zhuang Y, Thöns C, et al. Lymphocytes Negatively Regulate NK Cell Activity via Qa-1b Following Viral Infection. Cell Rep (2017) 21(9):2528–40. doi: 10.1016/j.celrep.2017.11.001
76. Alvarez M, Bouchlaka MN, Sckisel GD, Sungur CM, Chen M, Murphy WJ. Increased Antitumor Effects Using IL-2 With Anti-TGF-β Reveals Competition Between Mouse NK and CD8 T Cells. J Immunol (2014) 193(4):1709–16. doi: 10.4049/jimmunol.1400034
77. Alvarez M, Simonetta F, Baker J, Pierini A, Wenokur AS, Morrison AR, et al. Regulation of Murine NK Cell Exhaustion Through the Activation of the DNA Damage Repair Pathway. JCI Insight (2019) 5(14):e127729. doi: 10.1172/jci.insight.127729
78. Marcoe JP, Lim JR, Schaubert KL, Fodil-Cornu N, Matka M, Mccubbrey AL, et al. TGF-β is Responsible for NK Cell Immaturity During Ontogeny and Increased Susceptibility to Infection During Mouse Infancy. Nat Immunol (2012) 13(9):843–50. doi: 10.1038/ni.2388
79. Trzonkowski P, Szmit E, Myśliwska J, Dobyszuk A, Myśliwski A. CD4+CD25+ T Regulatory Cells Inhibit Cytotoxic Activity of T CD8+ and NK Lymphocytes in the Direct Cell-to-Cell Interaction. Clin Immunol (2004) 112(3):258–67. doi: 10.1016/j.clim.2004.04.003
80. Alvarez M, Simonetta F, Baker J, Morrison AR, Wenokur AS, Pierini A, et al. Indirect Impact of PD-1/PD-L1 Blockade on a Murine Model of NK Cell Exhaustion. Front Immunol (2020) 11:7. doi: 10.3389/fimmu.2020.00007
81. Porichis F, Hart MG, Massa A, Everett HL, Morou A, Richard J, et al. Immune Checkpoint Blockade Restores HIV-Specific CD4 T Cell Help for NK Cells. J Immunol (2018) 201(3):971–81. doi: 10.4049/jimmunol.1701551
82. Zou W. Mechanistic Insights Into Cancer Immunity and Immunotherapy. Cell Mol Immunol (2018) 15(5):419–20. doi: 10.1038/s41423-018-0011-5
83. Zhai L, Ladomersky E, Lenzen A, Nguyen B, Patel R, Lauing KL, et al. IDO1 in Cancer: A Gemini of Immune Checkpoints. Cell Mol Immunol (2018) 15(5):447–57. doi: 10.1038/cmi.2017.143
84. Oyer JL, Gitto SB, Altomare DA, Copik AJ. PD-L1 Blockade Enhances Anti-Tumor Efficacy of NK Cells. Oncoimmunology (2018) 7(11):e1509819. doi: 10.1080/2162402x.2018.1509819
85. Lopez-Vergès S, Milush JM, Pandey S, York VA, Arakawa-Hoyt J, Pircher H, et al. CD57 Defines a Functionally Distinct Population of Mature NK Cells in the Human CD56dimCD16+ NK-Cell Subset. Blood (2010) 116(19):3865–74. doi: 10.1182/blood-2010-04-282301
86. Villegas FR, Coca S, Villarrubia VG, Jiménez R, Chillón MJ, Jareño J, et al. Prognostic Significance of Tumor Infiltrating Natural Killer Cells Subset CD57 in Patients With Squamous Cell Lung Cancer. Lung Cancer (2002) 35(1):23–8. doi: 10.1016/s0169-5002(01)00292-6
87. Sitrin J, Ring A, Garcia KC, Benoist C, Mathis D. Regulatory T Cells Control NK Cells in an Insulitic Lesion by Depriving Them of IL-2. J Exp Med (2013) 210(6):1153–65. doi: 10.1084/jem.20122248
88. Keskin DB, Allan DS, Rybalov B, Andzelm MM, Stern JN, Kopcow HD, et al. TGFbeta Promotes Conversion of CD16+ Peripheral Blood NK Cells Into CD16- NK Cells With Similarities to Decidual NK Cells. Proc Natl Acad Sci USA (2007) 104(9):3378–83. doi: 10.1073/pnas.0611098104
89. Ghiringhelli F, Ménard C, Terme M, Flament C, Taieb J, Chaput N, et al. CD4+CD25+ Regulatory T Cells Inhibit Natural Killer Cell Functions in a Transforming Growth Factor-Beta-Dependent Manner. J Exp Med (2005) 202(8):1075–85. doi: 10.1084/jem.20051511
90. Zhou J, Peng H, Li K, Qu K, Wang B, Wu Y, et al. Liver-Resident NK Cells Control Antiviral Activity of Hepatic T Cells via the PD-1-PD-L1 Axis. Immunity (2019) 50(2):403–17.e4. doi: 10.1016/j.immuni.2018.12.024
91. Kos FJ, Engleman EG. Requirement for Natural Killer Cells in the Induction of Cytotoxic T Cells. J Immunol (1995) 155(2):578–84. doi: 10.1016/0022-1759(95)00097-T
92. Cichocki F, Bjordahl R, Gaidarova S, Mahmood S, Abujarour R, Wang H, et al. iPSC-Derived NK Cells Maintain High Cytotoxicity and Enhance In Vivo Tumor Control in Concert With T Cells and Anti-PD-1 Therapy. Sci Transl Med (2020) 12(568):eaaz5618. doi: 10.1126/scitranslmed.aaz5618
93. Lin M, Luo H, Liang S, Chen J, Liu A, Niu L, et al. Pembrolizumab Plus Allogeneic NK Cells in Advanced non-Small Cell Lung Cancer Patients. J Clin Invest (2020) 130(5):2560–9. doi: 10.1172/jci132712
94. André P, Denis C, Soulas C, Bourbon-Caillet C, Lopez J, Arnoux T, et al. Anti-NKG2A mAb Is a Checkpoint Inhibitor That Promotes Anti-Tumor Immunity by Unleashing Both T and NK Cells. Cell (2018) 175(7):1731–43.e13. doi: 10.1016/j.cell.2018.10.014
95. Romagné F, André P, Spee P, Zahn S, Anfossi N, Gauthier L, et al. Preclinical Characterization of 1-7F9, a Novel Human Anti-KIR Receptor Therapeutic Antibody That Augments Natural Killer-Mediated Killing of Tumor Cells. Blood (2009) 114(13):2667–77. doi: 10.1182/blood-2009-02-206532
96. Ardolino M, Azimi CS, Iannello A, Trevino TN, Horan L, Zhang L, et al. Cytokine Therapy Reverses NK Cell Anergy in MHC-Deficient Tumors. J Clin Invest (2014) 124(11):4781–94. doi: 10.1172/jci74337
97. Stojanovic A, Fiegler N, Brunner-Weinzierl M, Cerwenka A. CTLA-4 is Expressed by Activated Mouse NK Cells and Inhibits NK Cell IFN-γ Production in Response to Mature Dendritic Cells. J Immunol (2014) 192(9):4184–91. doi: 10.4049/jimmunol.1302091
98. Triebel F, Jitsukawa S, Baixeras E, Roman-Roman S, Genevee C, Viegas-Pequignot E, et al. LAG-3, a Novel Lymphocyte Activation Gene Closely Related to CD4. J Exp Med (1990) 171(5):1393–405. doi: 10.1084/jem.171.5.1393
Keywords: tumor, immune checkpoint inhibitor, natural killer cell, programmed death receptor-1, programmed death-ligand 1
Citation: Bai R and Cui J (2022) Burgeoning Exploration of the Role of Natural Killer Cells in Anti-PD-1/PD-L1 Therapy. Front. Immunol. 13:886931. doi: 10.3389/fimmu.2022.886931
Received: 01 March 2022; Accepted: 19 April 2022;
Published: 12 May 2022.
Edited by:
Alessandro Poggi, San Martino Hospital (IRCCS), Italy Reviewed by: Gordon Freeman, Dana–Farber Cancer Institute, United StatesCopyright © 2022 Bai and Cui. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jiuwei Cui, Y3VpandAamx1LmVkdS5jbg==; orcid.org/0000-0001-6496-7550
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.