 Desalegn Tadesse Egu
Desalegn Tadesse Egu Thomas Schmitt
Thomas Schmitt Jens Waschke
Jens Waschke- Chair of Vegetative Anatomy, Institute of Anatomy, Faculty of Medicine, Ludwig-Maximilians-Universität Munich, Munich, Germany
Pemphigus vulgaris (PV) is an autoimmune bullous skin disease caused primarily by autoantibodies (PV-IgG) against the desmosomal adhesion proteins desmoglein (Dsg)1 and Dsg3. PV patient lesions are characterized by flaccid blisters and ultrastructurally by defined hallmarks including a reduction in desmosome number and size, formation of split desmosomes, as well as uncoupling of keratin filaments from desmosomes. The pathophysiology underlying the disease is known to involve several intracellular signaling pathways downstream of PV-IgG binding. Here, we summarize our studies in which we used transmission electron microscopy to characterize the roles of signaling pathways in the pathogenic effects of PV-IgG on desmosome ultrastructure in a human ex vivo skin model. Blister scores revealed inhibition of p38MAPK, ERK and PLC/Ca2+ to be protective in human epidermis. In contrast, inhibition of Src and PKC, which were shown to be protective in cell cultures and murine models, was not effective for human skin explants. The ultrastructural analysis revealed that for preventing skin blistering at least desmosome number (as modulated by ERK) or keratin filament insertion (as modulated by PLC/Ca2+) need to be ameliorated. Other pathways such as p38MAPK regulate desmosome number, size, and keratin insertion indicating that they control desmosome assembly and disassembly on different levels. Taken together, studies in human skin delineate target mechanisms for the treatment of pemphigus patients. In addition, ultrastructural analysis supports defining the specific role of a given signaling molecule in desmosome turnover at ultrastructural level.
Introduction
Epithelial cells are tethered to one another by different types of intercellular adhesion complexes. Desmosomes form the core of these junctional complexes and provide resilience to tissues that constantly encounter mechanical forces (1, 2). They consist of members of three protein families, the cadherin superfamily which comprises two subclasses of Ca2+- binding transmembrane proteins, the desmogleins (Dsg) and desmocollins (Dsc), each with distinct isoforms, Dsg1-4 and Dsc1-3, respectively (3); armadillo protein family including the plakoglobin and plakophilins 1-3 (Pg and Pkp 1-3); as well as the plakin family member desmoplakin (Dp) also are among the core components of desmosomes (4). Besides, plectin, which also is a member of the plakin family, is involved in desmosome organization by crosslinking the peripheral intermediate filament and actin cytoskeleton (5).
The essential function of desmosomes is compromised under diseased conditions such as pemphigus. Pemphigus is a rare group of autoimmune diseases affecting the skin and oral mucosa but less frequently involves mucous membranes of other organs such as the eyes and genitals (6). Based on immunological and histological characteristics, three major phenotypes of pemphigus are recognized; pemphigus vulgaris (PV), pemphigus foliaceus (PF), and paraneoplastic pemphigus (PNP) (7). PV is caused by autoantibodies which primarily target Dsg1 and Dsg3 (8–10). It is characterized by suprabasal splitting in the epidermis and/or oral epithelia. PF lesions are confined to the epidermis and are triggered by anti-Dsg1 autoantibodies which results in erosions and flaccid blisters in the superficial epidermis, mainly in the granular layer (11). PF is most frequent in some countries in South America and North Africa due to the presence of an endemic form of the disease affecting mainly young adults (12). PNP is characterized by mucocutaneous lesions with diverse clinical presentations including suprabasal blisters and interface dermatitis (13, 14). The presence of neoplasms associated with tissue lesions is the main distinguishing feature of PNP from PV and PF (15). PNP is caused by autoantibodies directed against a variety of autoantigens including Dsg1, Dsg3, and also Dsc1and Dsc3 (16) as well as plakin family proteins (17). Other very rare variants of pemphigus include pemphigus vegetans, pemphigus erythematosus, and pemphigus herpetiformis (7).
Available treatment options mainly focus on modulation of the immune system such as depletion of autoantibody-producing B cells as well as non-specific immunosuppressive agents including corticosteroids and others with associated side effects emanating from long-term administration (18). Besides, chemical inhibitors such as rilzabrutinib, a potent inhibitor of Bruton tyrosine kinase (BTK) (19), has been reported as a promising therapeutic strategy at phase II clinical trial (20). Because of an unmet medical need to treat patients until autoantibody formation can be suppressed, current research focuses on devising novel therapeutic approaches including suppressing specific signaling pathways involved in pemphigus pathogenesis (21). Therefore, in this mini-review we will discuss the role of signaling pathways, which have been delineated to ameliorate acantholysis in several models of PV in vitro, in vivo and ex vivo (22), for the regulation of desmosome ultrastructure as revealed by transmission electron microscopy. We will highlight the significance of a human skin organ model as a useful tool to understand the underlying pathophysiology of pemphigus diseases by providing a physiological relevant near-to-patient situation.
Desmosomes
Desmosomes are recognized in electron micrographs by spatial distribution of electron dense plaques of varying densities identified as outer dense plaques (ODP), inner dense plaques (IDP), and extracellular core (EC) (23, 24) (Figure 1A). The components of these plaques were identified using immunoelectron microscopy (25). A more precise localization of the terminal domains of the main desmosomal proteins has been achieved using the quantitative immunogold method (23). Accordingly, the intracellular core of Dsg and Dsc, Pg and Pkp, as well as the N terminus of Dp constitute the ODP, whereas the N domain of Dp forms the IDP (23) and anchors the desmosomal plaques to the intermediate filament cytoskeleton (26).
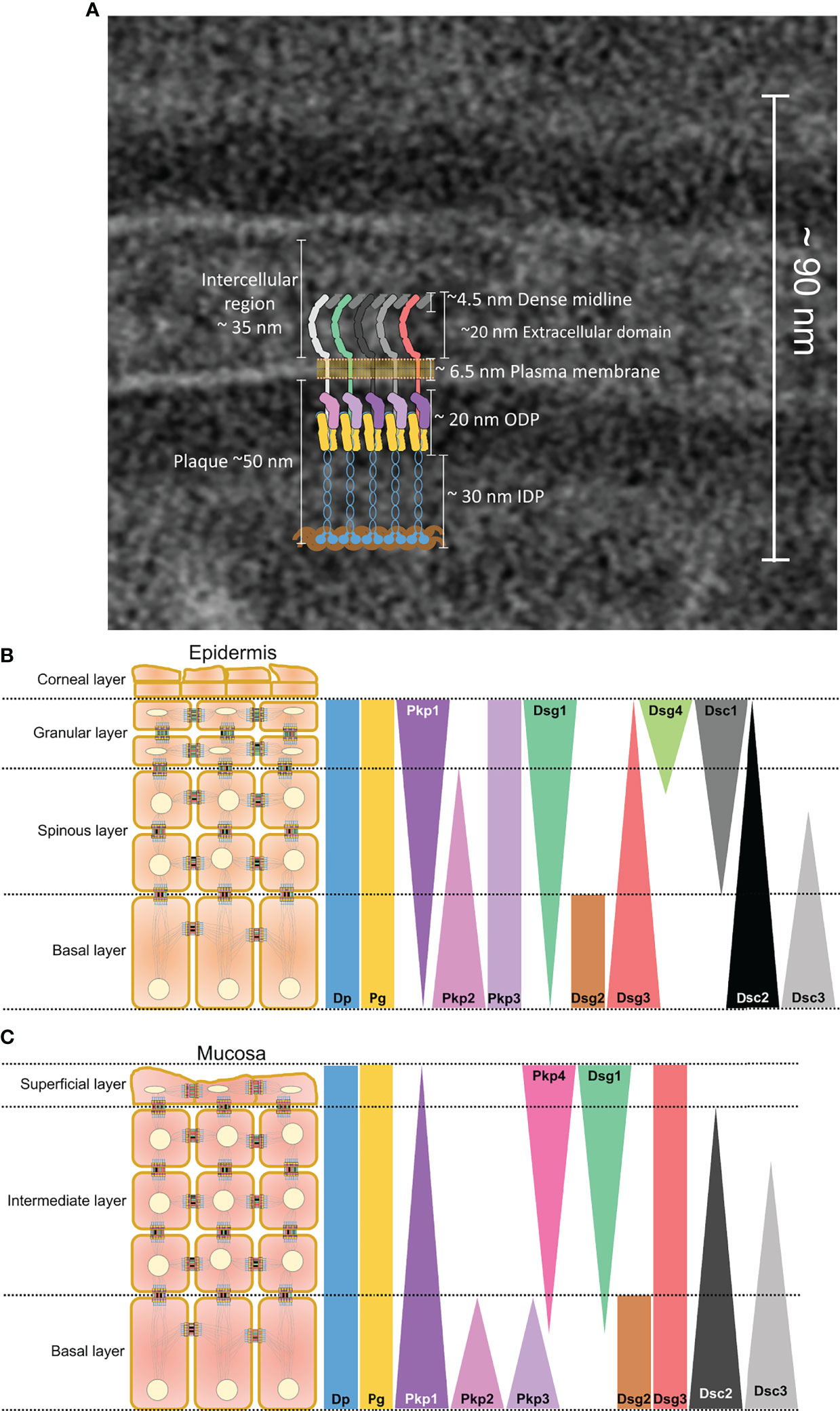
Figure 1 (A) Electron micrograph showing single desmosome in a healthy skin with a superimposed schematic representation depicting molecular structure of a desmosome. Colour coding of each single molecule is the same as those represented in (B, C). A schematic representation of the distribution of desmosomal proteins along the different layers of (B) epidermis and (C) mucosa. Small schematic desmosomes are colour-coded the same as the bars representing the distribution.
The different desmosome proteins exhibit different distribution patterns across the different layers of epithelia as a function of tissue type and differentiation status (27–29) (Figures 1B, C). In the epidermis, Dsg2 and Dsg3 are predominantly expressed in basal keratinocytes, whereas Dsg1 and Dsg4 are localised to the differentiated suprabasal layers (27). Dsg1together with Dsc1 shows an inverse distribution gradient with Dsg3 and Dsc3 across the suprabasal compartments in which Dsg1 and Dsc1 are strongly expressed as the cells differentiate and stratify. Dsg2 and Dsc2 are ubiquitously present in all desmosomes bearing tissues including the heart and simple epithelia (30). Dsg2 is present in the basal cell layer of oral mucosa (31, 32) and neonatal epidermis but restricted to hair follicles in adult human epidermis (33). Moreover, Dsg3 is the dominant desmoglein present in mucosa, whereas Dsg1 is distributed in all layers except the proliferating basal layer but at low levels (27, 32, 34, 35).
Through their extracellular N- domains, desmosomal cadherins are known to make cis and trans interactions with their counterparts on the same or adjacent cells, respectively, to form knot-like structures with desmosomes (36, 37). Although cis interactions are thought to be weaker, both mechanisms synergistically contribute to the formation of a stable adhesion complex (38). Interaction between similar cadherins (homophilic) as well as between different cadherin subclasses (heterophilic) has been reported (4, 39–41). Recent studies using bead assay identified heterophilic trans-interactions between Dsg1/Dsc1 and Dsg3/Dsc3 as the strongest and dominant form of adhesion in desmosomes (42, 43). In line with this, in atomic force microscopy (AFM) experiments Dsg2/Dsc2 formed a more stable dimer with a prolonged bond lifetime (44). In addition, homophilic interaction of Dsg1, Dsg3, and Dsc3 have been demonstrated as well (45–49). For Dsg2, it was reported recently that the interaction modes with desmosomal cadherins Dsg2 and Dsc2 differ from binding events with classical cadherins E-cadherin and N-cadherin (50). Moreover, heterophilic interaction of Dsg2, which is up-regulated in PV, with Dsg3 was proposed as compensatory mechanism because it was found to be resistant to autoantibody-induced steric hindrance (51). Taken together, both homo- and heterophilic interactions of desmosomal cadherins contribute to tissue integrity in epithelia (52, 53).
Other Desmosome-Related Diseases
Desmosomes have been studied extensively in recent years in consequence of diverse disease phenotypes including genetic and acquired diseases as well as autoimmune or microbial-mediated diseases which weaken intercellular adhesion (54).
Gene mutations involving obligate desmosomal proteins such as Dsg, Dsc, or Dp may bring about a wide spectrum of genetic diseases of the skin and other tissues in which the proteins are strongly expressed (55–57). In the skin, mutations of Dsg1 have been shown to cause severe dermatitis, multiple allergies, and metabolic wasting (SAM syndrome), in which loss of Dsg1 besides epidermal splitting causes a profound alteration of the epidermal barrier as well as the immune system, as shown in Dsg1-deficient animal models (58–61). In humans, mutations in desmosomal components are also associated with heart diseases. Mutations in Dsg2 and Dsc2 are known to cause arrhythmogenic right ventricular cardiomyopathy whereas those in Pg and Dp cause cardiomyopathy as well as various cardio-cutaneous syndromes (62–65). Some aspects in the pathophysiology of arrhythmogenic right ventricular cardiomyopathy may be similar to pemphigus due to common aspects in the structure and regulation of desmosomal contacts in cardiac intercalated discs and desmosomes of the epidermis (66). Moreover, mutations in desmosomal proteins have been shown to affect skin appendages. For example, homozygous and heterozygous mutations of Dsg4 have been reported to be associated with a spectrum of disease phenotypes such as hypotrichosis and monilethrix (67–69). Similarly, genetic alteration in Dsc3 was identified as the underlying cause for hair loss and vesicle formation in skin (70). Mutations in Dsg3, although not known in humans so far, caused diverse clinical features, including severe skin and mucosal lesions and hair loss as in squeaking (sqk) phenotype mice (71).
Microbial and viral agents are among the extrinsic factors that alter expression of desmosomal proteins (72). Exfoliative toxin A produced by staphylococcus bacteria is known to target Dsg1 and proteolyzes its adhesive ectodomain resulting in a PF-like lesion as in a bullous impetigo or staphylococcal-scalded skin syndrome (73) An adenovirus known for affecting the epithelial lining of the respiratory and urinary tracts was identified to bind to Dsg2 destabilizing cell-cell attachment (74). Studies in other acquired diseases such as cancer have revealed dysregulation of desmosomal proteins in tumor cells. For example, Dsg1 is downregulated in squamous cell carcinoma whereas Dsg3 is upregulated in head and neck carcinoma (57). Moreover, some molecular mechanisms underlying desmosome dysregulation in cancer cells have been reported (75, 76). Finally, alterations in desmosome ultrastructure have been detected in patients suffering from inflammatory bowel disease (77, 78). Because animal models deficient for Dsg2, Dsc2, and Dp have been shown to have intestinal epithelial barrier defects and disturbed wound healing and are prone to colitis (79–82), several lines of evidence indicate that disturbed desmosomal adhesion contributes to the pathogenesis of inflammatory bowel diseases (83). In this respect, a new function of desmosomes has been elucidated as they control tight junction structure and function (84, 85).
Autoantibody Profiles and Their Roles in PV
Autoantibody profiles in pemphigus patients’ serum dictate the specific disease phenotype manifested (22, 52, 86). Titers of Dsg-specific autoantibodies in pemphigus indicate disease activity as well as progression (87–89). It is known that these autoantibodies consist of both pathogenic and non-pathogenic forms (90, 91) which possess distinct epitopes they preferentially bind to (92, 93). It has been identified that the pathogenic autoantibodies target the EC subdomains of Dsg3 (EC1-3) and cause cell-cell detachment, whereas nonpathogenic antibodies bind to membrane-proximal domains without affecting cell adhesion (94, 95). Skin biopsies from pemphigus patients were examined to identify the tissue- and layer-specific binding of IgG from various pemphigus phenotypes (96, 97). The tissue as well as layer-specific distribution of lesions has been attributed to differential expression of desmosomal proteins among various tissues and across the layers of stratified epithelia (86). According to this hypothesis, anti-Dsg3 reactive antibodies cause suprabasal blistering owing to low expression of Dsg1 in deep layers so that it cannot compensate for Dsg3 as the case in PV. Similarly, anti-Dsg1 autoantibodies cause depletion of Dsg1 in the superficial layers where Dsg3 expression is very low as observed in PF (34, 96, 97). However, this theory has been challenged because it cannot explain why blister formation in PV is restricted to the basal-suprabasal interface. Besides, involvement of other non-Dsg autoantibodies in the disease in some cases or lack of a strong correlation between anti-Dsg titers and disease manifestation in some patients was reported (98–100). In light of this, several non-Dsg antigenic targets, which exhibit a strong autoreactivity to PV sera and with a potential to cause acantholysis, have been identified. These target antigens include cholinergic receptors (101), mitochondrial proteins (102), as well as other desmosomal proteins, such as desmocollins (16, 103–105) and Pkp3 (105), adherens junction protein E-cadherin (106), and others (100). However, the fact that Dsg-specific autoantibodies cause altered distribution followed by internalization of desmosomal proteins and are sufficient to cause skin blistering (107–112) underscores the role of anti-Dsg autoantibodies as a major pathogenic factor in pemphigus. In line with this, immunoadsorption of pathologic autoantibodies from PV sera by the entire EC domains of Dsg1 and Dsg3 abolished the blister-inducing ability of IgG fractions (113, 114).
Models for Studying Pemphigus
Several experimental models have been established to explore the underlying pathomechanisms of pemphigus. These diverse setups have allowed the characterization of the immunological and molecular mechanisms involved in autoimmune blistering diseases by reproducing some features of the disease as manifested in patients. Moreover, it enabled testing the efficacy of some therapeutic agents in a physiological setup which otherwise would be difficult to undertake in humans (115). The models include cell culture, organotypic tissue culture, animal models mainly mice, and ex vivo human skin model.
Cell and tissue cultures
Isolated cell lines from humans and murine sources have been utilized to investigate the pathogenic effects of pemphigus autoantibodies in vitro. The most extensively used epidermal cells are immortalized human keratinocyte (HaCaT cells: Human adult high Calcium low Temperature) (116) and primary human or mouse keratinocytes. Both cells were utilized mainly as monocultures but were also used to construct a three- dimensional (3D) organ culture model representing a stratified and differentiated epidermis (117). Both cell types have pros and cons in terms of originality, reproducibility, cost, and so on (118). The models are vital to assess the level of pathogenicity of autoantibodies derived from PV patients through various functional dissociation assays as well as to determine the effects of different pharmacological mediators in response to the autoantibodies (119–121).
Mouse model
Animal models are used extensively in medical research. Several mouse models have been developed which rendered great insights into PV pathogenesis (122). Based on the method in which the disease is induced and persistence of clinical features, the models can be identified as active or passive. Passive animal model refers to administration of pemphigus autoantibodies into a healthy animal which results in production of a transient disease phenotype (123, 124). The active models represent the generation of animals which manifest the disease through genetic modification as in Dsg3 or Dsg1 knockout mice (9, 125) or immunization in which the mice actively produce antibodies against Dsg3 (90, 126). In the latter, splenocytes from Dsg3 knockout mice, following immunization with recombinant Dsg3, were transferred into Rag2 knockout mice expressing Dsg3 (126–128). This model is more relevant as it involves active production of anti-Dsg3 autoantibodies similar to the situation in PV patients, whereas Dsg3 knockout mice produce a disease phenotype which is an ultrastructural correlate of only the acute phase of the disease (129).
Ex vivo model
Organ culture has been an important experimental model for studying pemphigus since the pioneer work done by Schiltz and Michel (130) in which they placed a skin biopsy on a lens paper floating on a liquid medium containing unpurified PV sera. This model is of paramount importance because of the viability of the tissues at optimal time point, 24 h, as well as the potential of the method to reproduce all the major histological and clinical features of the disease. This model stands out to be better than animal models since it enables overcoming the genetic and immunological differences which otherwise would elicit respective species-specific autoreactions (120). Furthermore, it favors the assessment of the pathogenicity of a given autoantibody and helps to correlate to disease activity in patients to develop more specific therapeutic strategies (120, 121). Although organ culture models are not best suited for biochemical studies, they provide a physiologically relevant setup to investigate mechanisms causing altered expression of desmosomal proteins and the resulting acantholysis (131, 132) which closely reflects the human in vivo situation (115). Thus, the model is ideal to conduct experiments under controlled conditions with sizeable samples which otherwise are not feasible to apply to human subjects. Therefore, we employed a human skin organ culture model as well as a novel mucosa ex vivo model to investigate the role of various signaling molecules in PV pathogenesis (32, 59, 133–136). Large blisters with associated ultrastructural changes in desmosomes including reduction in desmosome density and size as well as formation of split desmosomes and keratin filament uncoupling from desmosomes was observed in samples treated with PV-IgG (Figures 2A–C) (135) which shows that the skin model reflects the ultrastructural hallmarks known from pemphigus patients’ lesions (137, 138). Interestingly, the outer and inner plaque could not be differentiated after treatment with PV-IgG, an observation which requires further attention, especially because it has recently been shown that reorganization of the desmosomal plaque occurs during desmosome maturation and it is conceivable that these events may be reverted in pemphigus pathogenesis (139). Also, it must be noted that PV-IgG containing both autoantibodies against Dsg1 and Dsg3 were required for acantholysis whereas mucosal-dominant PV-IgG with autoantibodies against Dsg3 but not against Dsg1 similar to AK23, which is specific for Dsg3, were not sufficient (135). Taken together, the model better reflects the situation in patients compared to mice where high concentrations of anti-Dsg3-specific IgG are sufficient to cause skin blisters (114, 140).
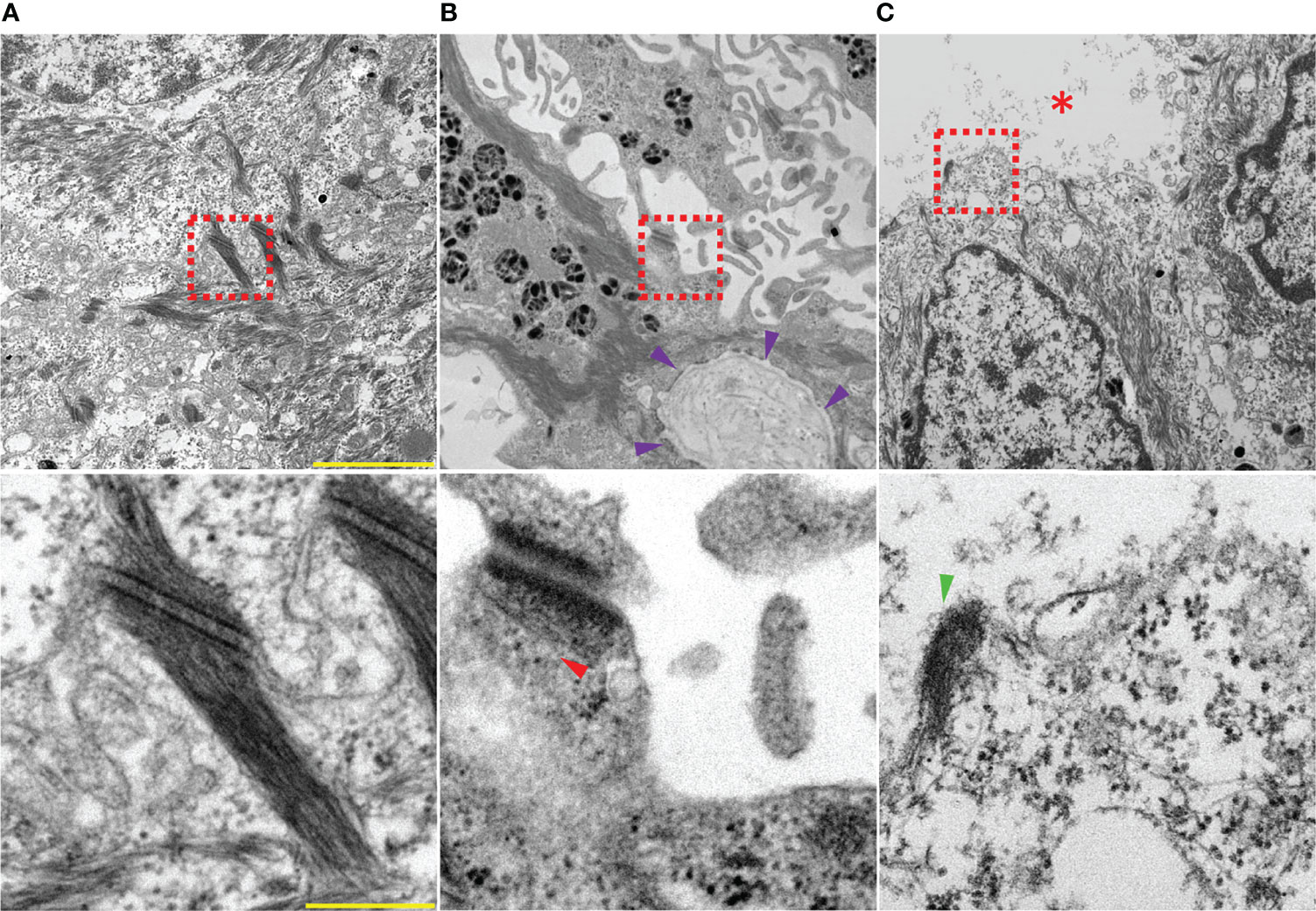
Figure 2 Electron micrographs showing an overview (top) and zoomed in to a single desmosome (bottom) of (A) healthy skin injected with IgG from healthy volunteers. (B, C) skin injected with PV-IgG showing suprabasal blistering, interdesmosomal widening and altered desmosomes: (B) reduced keratin insertion (red arrow head) into a damaged plaque. Note that the distinction between the outer and inner desmosomal plaque is lost after incubation with PV-IgG; violet arrow heads indicate the basement membrane, (C) a split desmosome with half plaque (green arrow head), red asterisk indicates blister cavity. Scale bars: 2 µm (top) and 250 nm (bottom).
Mechanisms Causing Acantholysis
Although not fully unraveled, steric hindrance and signaling have been proposed as the major overarching pathomechanisms that drive loss of intercellular contacts downstream of PV-IgG binding to keratinocytes (52, 141). Both mechanisms are believed to be involved, but not strictly independent of each other (57), and the exact chronology of events as well as contribution of each remains unknown.
Steric Hindrance
Passive transfer of IgG from PV patients or anti-Dsg3 monoclonal antibodies to a healthy neonatal mouse has been shown to induce epidermal blisters (90, 94) which has been reproduced in human skin organ culture, as well (142). These pathogenic immunoglobulins were shown to preferentially target the EC1 and EC2 adhesive regions of Dsg3 (90, 92, 94, 143). The latter has been indicated to predominatly contain epitopes recognised by PV autoantibodies (92, 144, 145). As a result, it has been suggested that PV-IgG autoantibodies directly interfere with the adhesive interaction of Dsg, thereby triggering the initial events leading to acantholysis (90, 127). AFM studies on cell-free surfaces (45, 46, 146) or on living keratinocyte cell surfaces, combined with bead assays, demonstrated direct inhibition of Dsg interaction for Dsg3 but not for Dsg1 (45, 53, 146), but suggested that direct inhibition is not sufficient to cause complete loss of keratinocyte adhesion. In addition, a recent study using bead assays under cell-free conditions showed that PV-IgG and PF-IgG blocked the heterophilic interaction between Dsg3/Dsc3 and Dsc1/Dsc1, respectively (43, 106).
Role of Signaling
The first signaling pathway to be triggered by pemphigus autoantibodies was PLC-mediated influx of Ca2+ (147). Along the years, several studies have shown that steric hindrance alone does not adequately account for acantholysis (45, 111, 146, 148–150), which indicated the involvement of other mechanisms underlying the pathogenic effect of PV-IgG. Phosphorylation and activation of signaling cascades downstream of PV-IgG binding to target antigens in keratinocyte both in vitro and in vivo has highlighted the crucial role of signaling in PV pathogenesis (151–154). As a result, a great number of signaling molecules that are implicated in PV have been identified and characterized (22, 155). These include mitogen-activated kinases (MAPKs) such as p38MAPK, protein kinase C (PKC), extracellular signal-regulated kinases (ERK1/2), Rous sarcoma-related kinases (Src), phospholipase C (PLC), Epidermal growth factor receptor (EGFR), and other cellular responses that alter adhesive interactions (22, 48, 59, 110, 112, 131, 147, 151, 156–162). For several years, the plethora of signaling mechanisms appeared to be triggered without recognizable hirarchy upon binding of autoantibodies. However, it was shown that signaling molecules such as p38MAPK, PI4K, PLC, and PKC directly bind to desmogleins and that Dsg1 and Dsg3, together with Pg, organize overlapping yet distinct signaling hubs (136, 140, 163). These findings help to explain why autoantibodies against Dsg3 and Dsg1 were observed to cause different sets of signaling responses (48) and led us to propose that the different clinical phenotypes of pemphigus with respect to mucosal and skin involvement, as well as suprabasal versus superficial epidermal blistering, may at least in part be caused by the different signaling profiles observed in PV and PF (22).
p38MAPK Regulates Autoantibody-Mediated Ultrastructural Alteration of Desmosomes
p38MAPK has been thoroughly characterized due to its essential role in pemphigus pathophysiology. The different isoforms (α, β, γ, δ) display a species-specific expression pattern (164–166) among which the α subtype is the most commonly expressed isoform in adult tissues (165, 167). Activation of p38MAPK was detected in keratinocyte cell cultures treated with PV-IgG and in mouse skin (151). Moreover, p38MAPK was phosphorylated in perilesional skin of PV patients (153, 168) as well as in the skin of Dsg3-deficient mice (169). This signaling molecule has also been shown to be associated with Dsg3 (33, 112, 136, 140), Dsc3 (169), and Dsg1 (136). p38MAPK-mediated Dsg3 internalization followed by depletion from endosomes was also detected in keratinocyte cultures and patient skin (108, 112, 170). Interstingly, previous studies have shown that pharmacologic inhibition of p38MAPK was sufficient to avert cell dissociation in vitro (33, 48, 151, 171), rescue membrane-bound as well as cytoskeletal fractions of Dsg3 (112, 172, 173), and reorganize keratin cytoskeleton (49, 140, 151). Moreover, it sufficiently abolished blister formation after passive transfer of PV-IgG (152, 171) or PF-IgG (174, 175) in mice.
There are strata of protein kinase cascades functionally subordinate to p38MAPK (176). Mitogen-activated protein kinase 2 (MK2) regulates several cellular activities such as actin remodeling (177), an event which can be correlated to PV pathogenesis. Phosphorylation of MK2 has been detected upon p38MAPK activation by PV-IgG (173), the inhibition of which was protective both in vitro and in vivo. Rho A is crucial to maintain a strong keratin association with desmosomes, enhance cortical actin filaments, stabilize cytoskeletal bound Dsg1 and Dsg3, and was found to be inactivated following PV-IgG and PF-IgG in p38MAPK-dependent manner (131). Besides, toxin-mediated inhibition of Rho GTPases recapitulated the PV-IgG-induced suprabasal blistering in human skin (132).
We employed human organ culture models to assess the role of p38MAPK in mediating the ultrastructural changes of desmosomes in PV pathogenesis and found that inhibition of p38MAPK abolished blister formation in epidermis but not in mucosa, indicating that p38MAPK is crucial for the mechanisms causing epidermal blisters but not mucosal erosions (32, 135). Interestingly, loss of desmosomes as well as all ultrastructural alterations of desmosomes, including reduction in size, splitting, and keratin filament dissociation, were averted by inhibition of p38MAPK (Figures 3A–D).
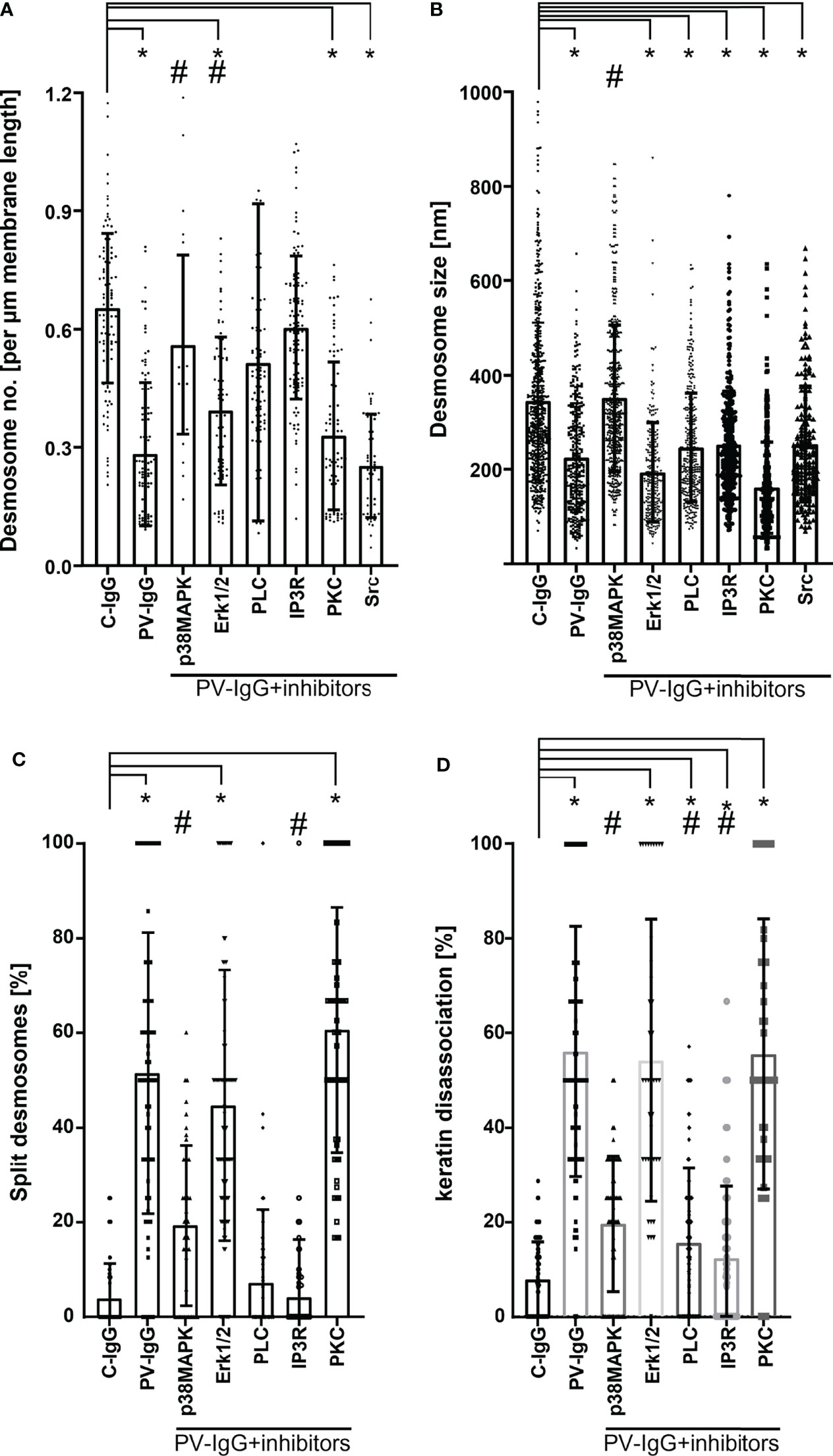
Figure 3 Ultrastructural quantification of desmosomes. (A) Desmosome density expressed in number of desmosomes per µm membrane length. Only desmosomes along clearly delineated cell borders of basal cells were considered. (B) Desmosome size measured along the linear length of the plaques and expressed in nm. (C) Percentage of split desmosomes both in acantholytic and non acantholytic areas. (D) Percentage of keratin dissociation from desmosomal plaques. Each data point represents individual desmosomes for (B), and the average per electron micrograph for (A, C and D) (n= 3-5 for each pathway. *p < 0.05 vs. control, #p < 0.05 vs. PV-IgG). Inhibitors used: SB202190 – p38MAPK inhibitor, Pp2 – Src inhibitor, Bim-X – PKC inhibitor, UO126 MEK (upstream of Erk1/2) inhibitor, U-73122 – PLC inhibitor, Xest (xestospongin) – IP3R inhibitor.
These results suggest that modulation of this signaling pathway would be effective in treating pemphigus patients. However, a clinical trial using a p38MAPK inhibitor was terminated because of dose-limiting hepatotoxicity and did not show therapeutic benefits (178). This observation is important because it reveals that not all signaling pathways that are sufficient to stabilize desmosomal adhesion are druggable in patients.
ERK But Not PKC or Src Partially Regulates Desmosome Ultrastructure
The MAP kinase ERK, a downstream signal effector of EGFR, was phosphorylated upon activation by PV-IgG and specific inhibition of which prevented acantholysis in cell cultures (48, 158, 161). Interestingly, ERK was activated in the presence of Dsg1 autoantibodies only (48, 162) and its specific inhibition was protective against PV-IgG or PF-IgG in cultured keratinocytes (48). Dsg1 is required to suppress ERK1/2 signaling to promote keratinocyte differentiation in the suprabasal epidermis where it is strongly expressed (159) by interacting with ErbB2 binding protein (Erbin2) (179). Mek1 inhibition, an upstream target of ERK, was sufficient to avert epidermal blistering in human skin ex vivo (21, 134) and reduce in number of desmosomes primarily along the basal-suprabasal interface but did not prevent desmosome splitting and keratin detachment from desmosomes (Figures 3A–D) (134).
On the other hand, for PKC and Src, the human ex vivo skin model yielded results different from what was found in cultured keratinocytes in vitro or for in vivo mouse models. PKC has been shown to be involved in signaling, Dsg3 depletion, loss of cell adhesion, and blister formation in cultured keratinocytes as well as in mice (48, 110, 147, 156, 180). Nevertheless, PKC inhibition did not ameliorate skin blistering in the human skin model and failed to modulate ultrastructural alterations (181) (Figures 3A–D). This discrepancy may be caused by the fact that several PKC isoforms exist which might be involved differently in both desmosome assembly and disassembly (22, 64). Src regulates desmosome assembly via interaction of Dsg3 with E-cadherin (169, 182). Reduced phosphorylation of Src was detected along with decreased Dsg3 expression in basal keratinocytes surrounding blister cavities, linking this pathway to PV pathogenesis (182). Inhibition of Src prevented cell dissociation (48, 59, 183, 184) and abrogated manifestation of skin lesions in mice but not in human skin culture where it was also not sufficient to modulate ultrastructural alterations of desmosomes (Figures 3A–D) (59). Since Src, similar to PKC, appears both to participate in desmosome assembly, a process which may require cortactin (59, 162), and to cause loss of desmosome adhesion in response to PV-IgG, this may explain the discrepancy between in vivo data in mice and ex vivo studies in human skin. Alternatively, the role of Src in PV pathogenesis may depend on the autoantibody profile of patients because the PV-IgG fraction used in human skin included higher levels of antibodies targeting Dsg1. It was shown that Src-mediated EGFR activation is associated with anti-Dsg3 autoantibody-mediated signaling rather than with signaling caused by autoantibodies against Dsg1 or Dsc3 (162, 185). Therefore, it is possible that Src inhibition may be effective in treating some PV patients but not others.
However, for all studies using electron microscopy evaluation of human epidermis, the limitation is that they are feasible by using a very limited number of patients’ IgG fractions only. Thus, no preliminary dose-response characterization is possible, and it cannot be ruled out that, with autoantibody samples from other patients or using higher concentrations of pharmacological inhibitors, a protective effect may be found for other signaling molecules as well. Therefore, negative data must be taken with extreme care. On the other hand, if a protective effect under all conditions and in all models, including human skin, is observed for modulation of a specific signaling pathway, this is a strong indication for a potentially interesting treatment paradigm.
PLC Mediates Desmosomal Adhesion by Maintaining Keratin Anchorage to Desmosomes
PV-IgG augments intracellular Ca2+ and inositol 1,4,5-trisphosphate (IP3) levels (186) via activation of Phosphoinositid-Phospholipase C (PLC) (147) leading to cell dissociation. IP3, via activation of IP3 receptor (IP3R), causes Ca2+ release, which in turn activates PKC (187). Inhibition of PLC was protective against the dyscohesive effect of PV-IgG in cell culture (147), in vivo (157), and in human skin explant (133, 136). Interestingly, specific inhibition of PLC and Ca2+ signaling significantly ameliorated keratin dissociation and desmosome splitting (Figures 3A–D) (133).
Signaling Pathways Regulate Desmosome Turnover
It is well established that pemphigus is a desmosome turn-over disease (188) because the signaling pathways involved interfere with different steps of desmosome assembly and maturation on one hand, and desmosome internalization and disassembly on the other. The interesting question is whether the ultrastructural analyses as outlined above allow to allocate signaling pathways to specific steps of desmosome turnover. Loss of desmosomes, which was caused by mucocutaneous PV-IgG but not by mucosal dominant PV-IgG and AK23, is the ultimate consequence of a dysbalance between assembly and disassembly. Thus, depletion of extradesmosomal Dsg molecules, which serve as a pool for incorporation into existing desmosomes, may account for this phenomenon (109, 189). Similarly, it was shown that following depletion of Triton-soluble extradesmosomal Dsg3, Triton insoluble Dsg3 was reduced as well (109). This brings up the important question of whether the latter Dsg3 molecules were derived from desmosomes, which would be characterized best as desmosome disassembly, or whether this is just the consequence of impaired assembly. Experiments showing that Dsg3 internalization following autoantibody binding is a coordinated process involving endocytosis are compatible with both interpretations (111, 170, 171, 190–193).
Recently, it was reported in Madin–Darby canine kidney (MDCK) cells that hepatocyte growth factor (HGF) induces internalization of intact desmosomes with no alteration in desmosome size and composition, which was interpreted as desmosome internalization (194). As outlined above, the situation in pemphigus is different and more complex. PV-IgG cause significant shrinkage of remaining desmosomes (Figure 3B). In addition, transmission electron microscopy and structured illumination microscopy investigations have revealed that desmosomes in neonatal mice were split prior to internalization (124) and were present at cell surfaces surrounding blister cavities in patients’ lesions (129, 138, 195–198) and human ex vivo skin (Figure 3C) (181). These data show that desmosome splitting occurs when desmosomes are weakened due to an imbalance of desmosome assembly and disassembly which is facilitated by mechanical stress (198). In parallel to desmosomes with reduced size, double-membrane structures together with desmosomes which were uncoupled from keratin filaments were also observed (135), indicating that desmosome internalization in skin occurs similar to MDCK cells, which represent a single-layer epithelium. However, internalized desmosomes were rare and thus could not be evaluated in quantitative terms.
These results show that desmosome disassembly and internalization may be dependent on both cell type and stimulus. For both events, uncoupling of Dsg molecules from the cytoskeleton was shown (Figure 3D) (32, 133, 181, 194). It remains unclear to which extent mechanical destabilization of desmosomes resulting in desmosome splitting under force results from desmosome shrinkage or keratin filament dissociation. The data from ultrastructural evaluation of desmosomes indicate that inhibition of p38MAPK was the only pharmacologic intervention to rescue desmosome size (Figure 3B). On the other hand, both p38MAPK and PLC/Ca2+ were effective to modulate keratin dissociation and splitting of desmosomes, which can be interpreted that cytoskeletal anchorage is sufficient to prevent desmosome splitting. The data are in line with experiments showing that phosphorylation of Dp by PKC, which is activated downstream of PLC/Ca2+, in PKP1-dependent manner regulates intermediate filament anchorage, and thereby causes desmosome hyperadhesion by trapping of desmosome components (199–201). In response to PV-IgG, PKC-mediated Dp phosphorylation reverts the hyperadhesive state and causes loss of desmosome adhesion (180, 202). Another conclusion is that steric hindrance alone cannot explain these ultrastructural alterations observed in patient lesions and ex vivo skin model because this mechanism would cause split desmosomes with intact size and keratin filament anchorage.
Taken together, p38MAPK and PLC/Ca2+/PKC were shown to be involved in depletion of Dsg1 and Dsg3 (110, 112, 136), as well as in keratin filament dissociation (133, 135), indicating that these signaling pathways are important for disturbed desmosome assembly as well as for desmosome disassembly and internalization in pemphigus pathogenesis. For Src and ERK, it remains less clear which mechanisms are involved in loss of keratinocyte adhesion. It is likely that they contribute to the same mechanisms impairing desmosome turnover like p38MAPK and PLC-mediated signaling but may be less central for these events.
Concluding Remarks
The ultrastructural analysis in human epidermis revealed that for all signaling pathways where pharmacologic modulation was protective, an ultrastructural correlate in desmosomes was found (Table 1). Based on this, we conclude that investigations on desmosome composition are helpful to advance our understanding of the regulation of desmosome turnover and to ultimately decipher a signaling pathway which may be druggable in patients as an additional line of therapy until depletion of pathogenic autoantibodies is effective and to reduce potential side effects.
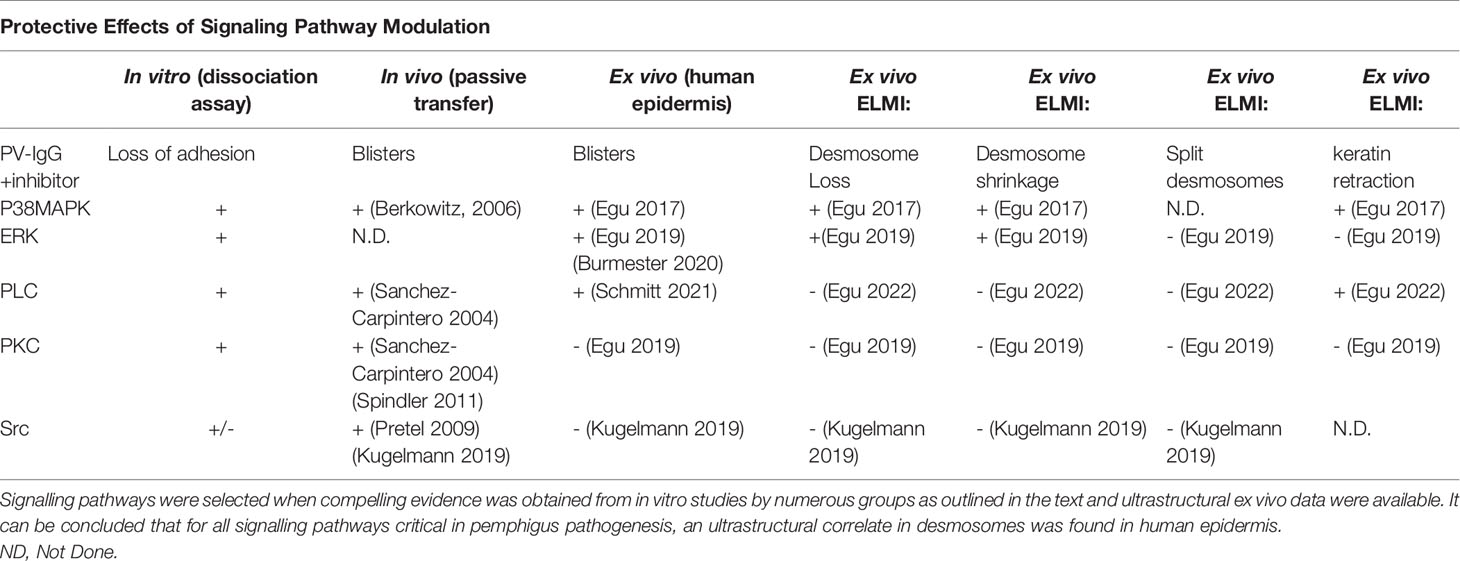
Table 1 Protective effects of signalling pathway modulation.
Electron microscopy is a gold standard method to define ultrastructural alterations of desmosomes in pemphigus patient lesions (138) and to investigate the underlying mechanisms of a given signaling molecule in human skin, the role of which has been suggested by studies in cultured keratinocytes or mouse models. By this approach, mechanisms occurring in cell culture and mouse skin, but not in human epidermis, such as apoptotic cell death, also can be evaluated (138, 203). In the long run, super-resolution microscopy will be applied to study the pathogenesis of pemphigus in more detail as has been started already (170).
Author Contributions
DE: concept, original draft, and revision. JW: concept and revision. TS: figure design, schematic drawing. DE and JW equally contributed to the article. All authors read and approved the final manuscript.
Funding
This work was supported by the DFG FOR2497 (PEGASUS) grant to JW.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Garrod D, Chidgey M. Desmosome Structure, Composition and Function. Biochim Biophys Acta (2008) 1778(3):572–87. doi: 10.1016/j.bbamem.2007.07.014
2. Price AJ, Cost AL, Ungewiss H, Waschke J, Dunn AR, Grashoff C. Mechanical Loading of Desmosomes Depends on the Magnitude and Orientation of External Stress. Nat Commun (2018) 9(1):5284. doi: 10.1038/s41467-018-07523-0
3. Garrod DR, Merritt AJ, Nie Z. Desmosomal Cadherins. Curr Opin Cell Biol (2002) 14(5):537–45. doi: 10.1016/S0955-0674(02)00366-6
4. Green KJ, Simpson CL. Desmosomes: New Perspectives on a Classic. J Invest Dermatol (2007) 127(11):2499–515. doi: 10.1038/sj.jid.5701015
5. Prechova M, Adamova Z, Schweizer AL, Maninova M, Bauer A, Kah D, et al. Plectin-Mediated Cytoskeletal Crosstalk Controls Cell Tension and Cohesion in Epithelial Sheets. J Cell Biol (2022) 221(3). doi: 10.1083/jcb.202105146
6. Schmidt E, Kasperkiewicz M, Joly P. Pemphigus. Lancet (2019) 394(10201):882–94. doi: 10.1016/S0140-6736(19)31778-7
7. Kasperkiewicz M, Ellebrecht CT, Takahashi H, Yamagami J, Zillikens D, Payne AS, et al. Pemphigus. Nat Rev Dis Primers (2017) 3:17026. doi: 10.1038/nrdp.2017.26
8. Amagai M, Klaus-Kovtun V, Stanley JR. Autoantibodies Against a Novel Epithelial Cadherin in Pemphigus Vulgaris, a Disease of Cell Adhesion. Cell (1991) 67(5):869–77. doi: 10.1016/0092-8674(91)90360-B
9. Koch PJ, Mahoney MG, Ishikawa H, Pulkkinen L, Uitto J, Shultz L, et al. Targeted Disruption of the Pemphigus Vulgaris Antigen (Desmoglein 3) Gene in Mice Causes Loss of Keratinocyte Cell Adhesion With a Phenotype Similar to Pemphigus Vulgaris. J Cell Biol (1997) 137(5):1091–102. doi: 10.1083/jcb.137.5.1091
10. Amagai M, Hashimoto T, Green KJ, Shimizu N, Nishikawa T. Antigen-Specific Immunoadsorption of Pathogenic Autoantibodies in Pemphigus Foliaceus. J Invest Dermatol (1995) 104(6):895–901. doi: 10.1111/1523-1747.ep12606168
11. Pollmann R, Schmidt T, Eming R, Hertl M. Pemphigus: A Comprehensive Review on Pathogenesis, Clinical Presentation and Novel Therapeutic Approaches. Clin Rev Allergy Immunol (2018) 54(1):1–25. doi: 10.1007/s12016-017-8662-z
12. Culton DA, Qian Y, Li N, Rubenstein D, Aoki V, Filhio GH, et al. Advances in Pemphigus and its Endemic Pemphigus Foliaceus (Fogo Selvagem) Phenotype: A Paradigm of Human Autoimmunity. J Autoimmun (2008) 31(4):311–24. doi: 10.1016/j.jaut.2008.08.003
13. Joly P, Richard C, Gilbert D, Courville P, Chosidow O, Roujeau JC, et al. Sensitivity and Specificity of Clinical, Histologic, and Immunologic Features in the Diagnosis of Paraneoplastic Pemphigus. J Am Acad Dermatol (2000) 43(4):619–26. doi: 10.1067/mjd.2000.107488
14. Ohzono A, Sogame R, Li X, Teye K, Tsuchisaka A, Numata S, et al. Clinical and Immunological Findings in 104 Cases of Paraneoplastic Pemphigus. Br J Dermatol (2015) 173(6):1447–52. doi: 10.1111/bjd.14162
15. Paolino G, Didona D, Magliulo G, Iannella G, Didona B, Mercuri SR, et al. Paraneoplastic Pemphigus: Insight Into the Autoimmune Pathogenesis, Clinical Features and Therapy. Int J Mol Sci (2017) 18(12):2532. doi: 10.3390/ijms18122532
16. Ishii N, Teye K, Fukuda S, Uehara R, Hachiya T, Koga H, et al. Anti-Desmocollin Autoantibodies in Nonclassical Pemphigus. Br J Dermatol (2015) 173(1):59–68. doi: 10.1111/bjd.13711
17. Bouameur JE, Favre B, Borradori L. Plakins, A Versatile Family of Cytolinkers: Roles in Skin Integrity and in Human Diseases. J Invest Dermatol (2014) 134(4):885–94. doi: 10.1038/jid.2013.498
18. Schmidt E, Spindler V, Eming R, Amagai M, Antonicelli F, Baines JF, et al. Meeting Report of the Pathogenesis of Pemphigus and Pemphigoid Meeting in Munich, September 2016. J Invest Dermatol (2017) 137(6):1199–203. doi: 10.1016/j.jid.2017.01.028
19. Bradshaw JM, McFarland JM, Paavilainen VO, Bisconte A, Tam D, Phan VT, et al. Prolonged and Tunable Residence Time Using Reversible Covalent Kinase Inhibitors. Nat Chem Biol (2015) 11(7):525–31. doi: 10.1038/nchembio.1817
20. Murrell DF, Patsatsi A, Stavropoulos P, Baum S, Zeeli T, Kern JS, et al. Proof of Concept for the Clinical Effects of Oral Rilzabrutinib, the First Bruton Tyrosine Kinase Inhibitor for Pemphigus Vulgaris: The Phase II BELIEVE Study. Br J Dermatol (2021) 185(4):745–55. doi: 10.1111/bjd.20431
21. Burmester IAK, Flaswinkel S, Thies CS, Kasprick A, Kamaguchi M, Bumiller-Bini V, et al. Identification of Novel Therapeutic Targets for Blocking Acantholysis in Pemphigus. Br J Pharmacol (2020) 177(22):5114–30. doi: 10.1111/bph.15233
22. Schmitt T, Waschke J. Autoantibody-Specific Signalling in Pemphigus. Front Med (2021) 8(1238). doi: 10.3389/fmed.2021.701809
23. North AJ, Bardsley WG, Hyam J, Bornslaeger EA, Cordingley HC, Trinnaman B, et al. Molecular Map of the Desmosomal Plaque. J Cell Sci (1999) 112(Pt 23):4325–36. doi: 10.1242/jcs.112.23.4325
24. Steinberg MS, Shida H, Giudice GJ, Shida M, Patel NH, Blaschuk OW. On the Molecular Organization, Diversity and Functions of Desmosomal Proteins. Ciba Found Symp (1987) 125:3–25. doi: 10.1002/9780470513408.ch2
25. Miller K, Mattey D, Measures H, Hopkins C, Garrod D. Localisation of the Protein and Glycoprotein Components of Bovine Nasal Epithelial Desmosomes by Immunoelectron Microscopy. EMBO J (1987) 6(4):885–9. doi: 10.1002/j.1460-2075.1987.tb04834.x
26. Kowalczyk AP, Bornslaeger EA, Borgwardt JE, Palka HL, Dhaliwal AS, Corcoran CM, et al. The Amino-Terminal Domain of Desmoplakin Binds to Plakoglobin and Clusters Desmosomal Cadherin-Plakoglobin Complexes. J Cell Biol (1997) 139(3):773–84. doi: 10.1083/jcb.139.3.773
27. Mahoney MG, Hu Y, Brennan D, Bazzi H, Christiano AM, Wahl JK 3rd. Delineation of Diversified Desmoglein Distribution in Stratified Squamous Epithelia: Implications in Diseases. Exp Dermatol (2006) 15(2):101–9. doi: 10.1111/j.1600-0625.2006.00391.x
28. Arnemann J, Sullivan KH, Magee AI, King IA, Buxton RS. Stratification-Related Expression of Isoforms of the Desmosomal Cadherins in Human Epidermis. J Cell Sci (1993) 104(Pt 3):741–50. doi: 10.1242/jcs.104.3.741
29. Johnson JL, Najor NA, Green KJ. Desmosomes: Regulators of Cellular Signaling and Adhesion in Epidermal Health and Disease. Cold Spring Harb Perspect Med (2014) 4(11):a015297. doi: 10.1101/cshperspect.a015297
30. Lowndes M, Rakshit S, Shafraz O, Borghi N, Harmon RM, Green KJ, et al. Different Roles of Cadherins in the Assembly and Structural Integrity of the Desmosome Complex. J Cell Sci (2014) 127(Pt 10):2339–50. doi: 10.1242/jcs.146316
31. Teh MT, Parkinson EK, Thurlow JK, Liu F, Fortune F, Wan H. A Molecular Study of Desmosomes Identifies a Desmoglein Isoform Switch in Head and Neck Squamous Cell Carcinoma. J Oral Pathol Med (2011) 40(1):67–76. doi: 10.1111/j.1600-0714.2010.00951.x
32. Egu DT, Sigmund AM, Schmidt E, Spindler V, Walter E, Waschke J. A New Ex Vivo Human Oral Mucosa Model Reveals That p38MAPK Inhibition Is Not Effective in Preventing Autoantibody-Induced Mucosal Blistering in Pemphigus. Br J Dermatol (2020) 182(4):987–94. doi: 10.1111/bjd.18237
33. Hartlieb E, Rötzer V, Radeva M, Spindler V, Waschke J. Desmoglein 2 Compensates for Desmoglein 3 But Does Not Control Cell Adhesion Via Regulation of p38 Mitogen-Activated Protein Kinase in Keratinocytes. J Biol Chem (2014) 289(24):17043–53. doi: 10.1074/jbc.M113.489336
34. Shirakata Y, Amagai M, Hanakawa Y, Nishikawa T, Hashimoto K. Lack of Mucosal Involvement in Pemphigus Foliaceus may be Due to Low Expression of Desmoglein 1. J Invest Dermatol (1998) 110(1):76–8. doi: 10.1046/j.1523-1747.1998.00085.x
35. Donetti E, Bedoni M, Boschini E, Dellavia C, Barajon I, Gagliano N. Desmocollin 1 and Desmoglein 1 Expression in Human Epidermis and Keratinizing Oral Mucosa: A Comparative Immunohistochemical and Molecular Study. Arch Dermatol Res (2005) 297(1):31–8. doi: 10.1007/s00403-005-0573-9
36. Al-Amoudi A, Diez DC, Betts MJ, Frangakis AS. The Molecular Architecture of Cadherins in Native Epidermal Desmosomes. Nature (2007) 450(7171):832–7. doi: 10.1038/nature05994
37. Sikora M, Ermel UH, Seybold A, Kunz M, Calloni G, Reitz J, et al. Desmosome Architecture Derived From Molecular Dynamics Simulations and Cryo-Electron Tomography. Proc Natl Acad Sci USA (2020) 117(44):27132–40. doi: 10.1073/pnas.2004563117
38. Harrison OJ, Jin X, Hong S, Bahna F, Ahlsen G, Brasch J, et al. The Extracellular Architecture of Adherens Junctions Revealed by Crystal Structures of Type I Cadherins. Structure (2011) 19(2):244–56. doi: 10.1016/j.str.2010.11.016
39. Thomason HA, Scothern A, McHarg S, Garrod DR. Desmosomes: Adhesive Strength and Signalling in Health and Disease. Biochem J (2010) 429(3):419–33. doi: 10.1042/BJ20100567
40. Chitaev NA, Troyanovsky SM. Direct Ca2+-dependent Heterophilic Interaction Between Desmosomal Cadherins, Desmoglein and Desmocollin, Contributes to Cell-Cell Adhesion. J Cell Biol (1997) 138(1):193–201. doi: 10.1083/jcb.138.1.193
41. Nie Z, Merritt A, Rouhi-Parkouhi M, Tabernero L, Garrod D. Membrane-Impermeable Cross-Linking Provides Evidence for Homophilic, Isoform-Specific Binding of Desmosomal Cadherins in Epithelial Cells. J Biol Chem (2011) 286(3):2143–54. doi: 10.1074/jbc.M110.192245
42. Harrison OJ, Brasch J, Lasso G, Katsamba PS, Ahlsen G, Honig B, et al. Structural Basis of Adhesive Binding by Desmocollins and Desmogleins. Proc Natl Acad Sci USA (2016) 113(26):7160–5. doi: 10.1073/pnas.1606272113
43. Ishii K, Yoshida K, Stanley JR, Yamagami J, Amagai M, Ishiko A. Pemphigus Vulgaris and Foliaceus Igg Autoantibodies Directly Block Heterophilic Transinteraction Between Desmoglein and Desmocollin. J Invest Dermatol (2020) 140(10):1919–26.e7. doi: 10.1016/j.jid.2020.02.010
44. Shafraz O, Rübsam M, Stahley SN, Caldara AL, Kowalczyk AP, Niessen CM, et al. E-Cadherin Binds to Desmoglein to Facilitate Desmosome Assembly. Elife (2018) 7:e37629. doi: 10.7554/eLife.37629
45. Waschke J, Bruggeman P, Baumgartner W, Zillikens D, Drenckhahn D. Pemphigus Foliaceus IgG Causes Dissociation of Desmoglein 1-Containing Junctions Without Blocking Desmoglein 1 Transinteraction. J Clin Invest (2005) 115(11):3157–65. doi: 10.1172/JCI23475
46. Heupel WM, Zillikens D, Drenckhahn D, Waschke J. Pemphigus Vulgaris IgG Directly Inhibit Desmoglein 3-Mediated Transinteraction. J Immunol (2008) 181(3):1825–34. doi: 10.4049/jimmunol.181.3.1825
47. Spindler V, Heupel WM, Efthymiadis A, Schmidt E, Eming R, Rankl C, et al. Desmocollin 3-Mediated Binding is Crucial for Keratinocyte Cohesion and is Impaired in Pemphigus. J Biol Chem (2009) 284(44):30556–64. doi: 10.1074/jbc.M109.024810
48. Walter E, Vielmuth F, Rotkopf L, Sardy M, Horvath ON, Goebeler M, et al. Different Signaling Patterns Contribute to Loss of Keratinocyte Cohesion Dependent on Autoantibody Profile in Pemphigus. Sci Rep (2017) 7(1):3579. doi: 10.1038/s41598-017-03697-7
49. Vielmuth F, Wanuske MT, Radeva MY, Hiermaier M, Kugelmann D, Walter E, et al. Keratins Regulate the Adhesive Properties of Desmosomal Cadherins Through Signaling. J Invest Dermatol (2018) 138(1):121–31. doi: 10.1016/j.jid.2017.08.033
50. Fuchs M, Kugelmann D, Schlegel N, Vielmuth F, Waschke J. Desmoglein 2 can Undergo Ca(2+)-dependent Interactions With Both Desmosomal and Classical Cadherins Including E-cadherin and N-Cadherin. Biophys J (2022) 121(7):1322–35. doi: 10.1016/j.bpj.2022.02.023
51. Sigmund AM, Steinert LS, Egu DT, Bayerbach FC, Waschke J, Vielmuth F. Dsg2 Upregulation as a Rescue Mechanism in Pemphigus. Front Immunol (2020) 11:581370. doi: 10.3389/fimmu.2020.581370
52. Spindler V, Waschke J. Pemphigus-a Disease of Desmosome Dysfunction Caused by Multiple Mechanisms. Front Immunol (2018) 9:136. doi: 10.3389/fimmu.2018.00136
53. Vielmuth F, Spindler V, Waschke J. Atomic Force Microscopy Provides New Mechanistic Insights Into the Pathogenesis of Pemphigus. Front Immunol (2018) 9:485. doi: 10.3389/fimmu.2018.00485
54. Broussard JA, Getsios S, Green KJ. Desmosome Regulation and Signaling in Disease. Cell Tissue Res (2015) 360(3):501–12. doi: 10.1007/s00441-015-2136-5
55. Al-Jassar C, Bikker H, Overduin M, Chidgey M. Mechanistic Basis of Desmosome-Targeted Diseases. J Mol Biol (2013) 425(21):4006–22. doi: 10.1016/j.jmb.2013.07.035
56. Ishida-Yamamoto A, Igawa S. Genetic Skin Diseases Related to Desmosomes and Corneodesmosomes. J Dermatol Sci (2014) 74(2):99–105. doi: 10.1016/j.jdermsci.2014.02.005
57. Stahley SN, Kowalczyk AP. Desmosomes in Acquired Disease. Cell Tissue Res (2015) 360(3):439–56. doi: 10.1007/s00441-015-2155-2
58. Samuelov L, Sarig O, Harmon RM, Rapaport D, Ishida-Yamamoto A, Isakov O, et al. Desmoglein 1 Deficiency Results in Severe Dermatitis, Multiple Allergies and Metabolic Wasting. Nat Genet (2013) 45(10):1244–8. doi: 10.1038/ng.2739
59. Kugelmann D, Rotzer V, Walter E, Egu DT, Fuchs MT, Vielmuth F, et al. Role of Src and Cortactin in Pemphigus Skin Blistering. Front Immunol (2019) 10:626. doi: 10.3389/fimmu.2019.00626
60. Godsel LM, Roth-Carter QR, Koetsier JL, Tsoi LC, Huffine AL, Broussard JA, et al. Translational Implications of Th17-skewed Inflammation Due to Genetic Deficiency of a Cadherin Stress Sensor. J Clin Invest (2022) 132(3):e144363. doi: 10.1172/JCI144363
61. Zimmer SE, Takeichi T, Conway DE, Kubo A, Suga Y, Akiyama M, et al. Differential Pathomechanisms of Desmoglein 1 Transmembrane Domain Mutations in Skin Disease. J Invest Dermatol (2022) 142(2):323–32.e8. doi: 10.1016/j.jid.2021.07.154
62. Norgett EE, Hatsell SJ, Carvajal-Huerta L, Cabezas JC, Common J, Purkis PE, et al. Recessive Mutation in Desmoplakin Disrupts Desmoplakin-Intermediate Filament Interactions and Causes Dilated Cardiomyopathy, Woolly Hair and Keratoderma. Hum Mol Genet (2000) 9(18):2761–6. doi: 10.1093/hmg/9.18.2761
63. Jonkman MF, Pasmooij AM, Pasmans SG, van den Berg MP, Ter Horst HJ, Timmer A, et al. Loss of Desmoplakin Tail Causes Lethal Acantholytic Epidermolysis Bullosa. Am J Hum Genet (2005) 77(4):653–60. doi: 10.1086/496901
64. Waschke J. The Desmosome and Pemphigus. Histochem Cell Biol (2008) 130(1):21–54. doi: 10.1007/s00418-008-0420-0
65. Bhuiyan ZA, Jongbloed JD, van der Smagt J, Lombardi PM, Wiesfeld AC, Nelen M, et al. Desmoglein-2 and Desmocollin-2 Mutations in Dutch Arrhythmogenic Right Ventricular Dysplasia/Cardiomypathy Patients: Results From a Multicenter Study. Circ Cardiovasc Genet (2009) 2(5):418–27. doi: 10.1161/CIRCGENETICS.108.839829
66. Yeruva S, Waschke J. Structure and Regulation of Desmosomes in Intercalated Discs: Lessons From Epithelia. J Anat (2022). doi: 10.1111/joa.13634
67. Kato M, Shimizu A, Yokoyama Y, Kaira K, Shimomura Y, Ishida-Yamamoto A, et al. An Autosomal Recessive Mutation of DSG4 Causes Monilethrix Through the ER Stress Response. J Invest Dermatol (2015) 135(5):1253–60. doi: 10.1038/jid.2015.12
68. Ullah A, Raza SI, Ali RH, Naveed AK, Jan A, Rizvi SD, et al. A Novel Deletion Mutation in the DSG4 Gene Underlies Autosomal Recessive Hypotrichosis With Variable Phenotype in Two Unrelated Consanguineous Families. Clin Exp Dermatol (2015) 40(1):78–84. doi: 10.1111/ced.12457
69. Kljuic A, Bazzi H, Sundberg JP, Martinez-Mir A, O’Shaughnessy R, Mahoney MG, et al. Desmoglein 4 in Hair Follicle Differentiation and Epidermal Adhesion: Evidence From Inherited Hypotrichosis and Acquired Pemphigus Vulgaris. Cell (2003) 113(2):249–60. doi: 10.1016/S0092-8674(03)00273-3
70. Ayub M, Basit S, Jelani M, Ur Rehman F, Iqbal M, Yasinzai M, et al. A Homozygous Nonsense Mutation in the Human Desmocollin-3 (DSC3) Gene Underlies Hereditary Hypotrichosis and Recurrent Skin Vesicles. Am J Hum Genet (2009) 85(4):515–20. doi: 10.1016/j.ajhg.2009.08.015
71. Kountikov EI, Poe JC, Maclver NJ, Rathmell JC, Tedder TF. A Spontaneous Deletion Within the Desmoglein 3 Extracellular Domain of Mice Results in Hypomorphic Protein Expression, Immunodeficiency, and a Wasting Disease Phenotype. Am J Pathol (2015) 185(3):617–30. doi: 10.1016/j.ajpath.2014.10.025
72. Hegazy M, Perl AL, Svoboda SA, Green KJ. Desmosomal Cadherins in Health and Disease. Annu Rev Pathol (2022) 17:47–72. doi: 10.1146/annurev-pathol-042320-092912
73. Amagai M, Matsuyoshi N, Wang ZH, Andl C, Stanley JR. Toxin in Bullous Impetigo and Staphylococcal Scalded-Skin Syndrome Targets Desmoglein 1. Nat Med (2000) 6(11):1275–7. doi: 10.1038/81385
74. Wang H, Li ZY, Liu Y, Persson J, Beyer I, Möller T, et al. Desmoglein 2 is a Receptor for Adenovirus Serotypes 3, 7, 11 and 14. Nat Med (2011) 17(1):96–104. doi: 10.1038/nm.2270
75. Kamekura R, Kolegraff KN, Nava P, Hilgarth RS, Feng M, Parkos CA, et al. Loss of the Desmosomal Cadherin Desmoglein-2 Suppresses Colon Cancer Cell Proliferation Through EGFR Signaling. Oncogene (2014) 33(36):4531–6. doi: 10.1038/onc.2013.442
76. Chen YJ, Lee LY, Chao YK, Chang JT, Lu YC, Li HF, et al. DSG3 Facilitates Cancer Cell Growth and Invasion Through the DSG3-plakoglobin-TCF/LEF-Myc/cyclin D1/MMP Signaling Pathway. PloS One (2013) 8(5):e64088. doi: 10.1371/journal.pone.0064088
77. Spindler V, Meir M, Vigh B, Flemming S, Hütz K, Germer CT, et al. Loss of Desmoglein 2 Contributes to the Pathogenesis of Crohn’s Disease. Inflammation Bowel Dis (2015) 21(10):2349–59. doi: 10.1097/MIB.0000000000000486
78. Meir M, Burkard N, Ungewiß H, Diefenbacher M, Flemming S, Kannapin F, et al. Neurotrophic Factor GDNF Regulates Intestinal Barrier Function in Inflammatory Bowel Disease. J Clin Invest (2019) 129(7):2824–40. doi: 10.1172/JCI120261
79. Gross A, Pack LAP, Schacht GM, Kant S, Ungewiss H, Meir M, et al. Desmoglein 2, But Not Desmocollin 2, Protects Intestinal Epithelia From Injury. Mucosal Immunol (2018) 11(6):1630–9. doi: 10.1038/s41385-018-0062-z
80. Flemming S, Luissint AC, Kusters DHM, Raya-Sandino A, Fan S, Zhou DW, et al. Desmocollin-2 Promotes Intestinal Mucosal Repair by Controlling Integrin-Dependent Cell Adhesion and Migration. Mol Biol Cell (2020) 31(6):407–18. doi: 10.1091/mbc.E19-12-0692
81. Raya-Sandino A, Luissint AC, Kusters DHM, Narayanan V, Flemming S, Garcia-Hernandez V, et al. Regulation of Intestinal Epithelial Intercellular Adhesion and Barrier Function by Desmosomal Cadherin Desmocollin-2. Mol Biol Cell (2021) 32(8):753–68. doi: 10.1091/mbc.E20-12-0775
82. Gross A, Zhou B, Bewersdorf L, Schwarz N, Schacht GM, Boor P, et al. Desmoplakin Maintains Transcellular Keratin Scaffolding and Protects From Intestinal Injury. Cell Mol Gastroenterol Hepatol (2021) 13(4):1181–200. doi: 10.1016/j.jcmgh.2021.12.009
83. Schlegel N, Boerner K, Waschke J. Targeting Desmosomal Adhesion and Signalling for Intestinal Barrier Stabilization in Inflammatory Bowel diseases-Lessons From Experimental Models and Patients. Acta Physiol (Oxf) (2021) 231(1):e13492. doi: 10.1111/apha.13492
84. Schlegel N, Meir M, Heupel WM, Holthöfer B, Leube RE, Waschke J. Desmoglein 2-Mediated Adhesion is Required for Intestinal Epithelial Barrier Integrity. Am J Physiol Gastrointest Liver Physiol (2010) 298(5):G774–83. doi: 10.1152/ajpgi.00239.2009
85. Burkard N, Meir M, Kannapin F, Otto C, Petzke M, Germer C-T, et al. Desmoglein2 Regulates Claudin2 Expression by Sequestering Pi-3-Kinase in Intestinal Epithelial Cells. Front Immunol (2021) 12. doi: 10.3389/fimmu.2021.756321
86. Amagai M, Tsunoda K, Zillikens D, Nagai T, Nishikawa T. The Clinical Phenotype of Pemphigus is Defined by the Anti-Desmoglein Autoantibody Profile. J Am Acad Dermatol (1999) 40(2 Pt 1):167–70. doi: 10.1016/S0190-9622(99)70183-0
87. Belloni-Fortina A, Faggion D, Pigozzi B, Peserico A, Bordignon M, Baldo V, et al. Detection of Autoantibodies Against Recombinant Desmoglein 1 and 3 Molecules in Patients With Pemphigus Vulgaris: Correlation With Disease Extent at the Time of Diagnosis and During Follow-Up. Clin Dev Immunol (2009) 2009:187864. doi: 10.1155/2009/187864
88. Cheng SW, Kobayashi M, Kinoshita-Kuroda K, Tanikawa A, Amagai M, Nishikawa T. Monitoring Disease Activity in Pemphigus With Enzyme-Linked Immunosorbent Assay Using Recombinant Desmogleins 1 and 3. Br J Dermatol (2002) 147(2):261–5. doi: 10.1046/j.1365-2133.2002.04838.x
89. Kamiya K, Aoyama Y, Shirafuji Y, Hamada T, Morizane S, Fujii K, et al. A Higher Correlation of the Antibody Activities Against the Calcium-Dependent Epitopes of Desmoglein 3 Quantified by Ethylenediaminetetraacetic Acid-Treated Enzyme-Linked Immunosorbent Assay With Clinical Disease Activities of Pemphigus Vulgaris. J Dermatol Sci (2013) 70(3):190–5. doi: 10.1016/j.jdermsci.2013.02.011
90. Tsunoda K, Ota T, Aoki M, Yamada T, Nagai T, Nakagawa T, et al. Induction of Pemphigus Phenotype by a Mouse Monoclonal Antibody Against the Amino-Terminal Adhesive Interface of Desmoglein 3. J Immunol (2003) 170(4):2170–8. doi: 10.4049/jimmunol.170.4.2170
91. Payne AS, Ishii K, Kacir S, Lin C, Li H, Hanakawa Y, et al. Genetic and Functional Characterization of Human Pemphigus Vulgaris Monoclonal Autoantibodies Isolated by Phage Display. J Clin Invest (2005) 115(4):888–99. doi: 10.1172/JCI24185
92. Di Zenzo G, Di Lullo G, Corti D, Calabresi V, Sinistro A, Vanzetta F, et al. Pemphigus Autoantibodies Generated Through Somatic Mutations Target the Desmoglein-3 Cis-Interface. J Clin Invest (2012) 122(10):3781–90. doi: 10.1172/JCI64413
93. Ishii K, Lin C, Siegel DL, Stanley JR. Isolation of Pathogenic Monoclonal Anti-Desmoglein 1 Human Antibodies by Phage Display of Pemphigus Foliaceus Autoantibodies. J Invest Dermatol (2008) 128(4):939–48. doi: 10.1038/sj.jid.5701132
94. Amagai M, Karpati S, Prussick R, Klaus-Kovtun V, Stanley JR. Autoantibodies Against the Amino-Terminal Cadherin-Like Binding Domain of Pemphigus Vulgaris Antigen are Pathogenic. J Clin Invest (1992) 90(3):919–26. doi: 10.1172/JCI115968
95. Sekiguchi M, Futei Y, Fujii Y, Iwasaki T, Nishikawa T, Amagai M. Dominant Autoimmune Epitopes Recognized by Pemphigus Antibodies Map to the N-terminal Adhesive Region of Desmogleins. J Immunol (2001) 167(9):5439–48. doi: 10.4049/jimmunol.167.9.5439
96. Shimizu H, Masunaga T, Ishiko A, Kikuchi A, Hashimoto T, Nishikawa T. Pemphigus Vulgaris and Pemphigus Foliaceus Sera Show an Inversely Graded Binding Pattern to Extracellular Regions of Desmosomes in Different Layers of Human Epidermis. J Invest Dermatol (1995) 105(2):153–9. doi: 10.1111/1523-1747.ep12316695
97. Amagai M, Koch PJ, Nishikawa T, Stanley JR. Pemphigus Vulgaris Antigen (Desmoglein 3) is Localized in the Lower Epidermis, the Site of Blister Formation in Patients. J Invest Dermatol (1996) 106(2):351–5. doi: 10.1111/1523-1747.ep12343081
98. Amagai M. [Desmoglein, the Target Molecule in Autoimmunity and Infection]. Nihon Rinsho Meneki Gakkai Kaishi (2006) 29(5):325–33. doi: 10.2177/jsci.29.325
99. Ahmed AR, Carrozzo M, Caux F, Cirillo N, Dmochowski M, Alonso AE, et al. Monopathogenic vs Multipathogenic Explanations of Pemphigus Pathophysiology. Exp Dermatol (2016) 25(11):839–46. doi: 10.1111/exd.13106
100. Sinha AA, Sajda T. The Evolving Story of Autoantibodies in Pemphigus Vulgaris: Development of the “Super Compensation Hypothesis”. Front Med (Lausanne) (2018) 5:218. doi: 10.3389/fmed.2018.00218
101. Vu TN, Lee TX, Ndoye A, Shultz LD, Pittelkow MR, Dahl MV, et al. The Pathophysiological Significance of Nondesmoglein Targets of Pemphigus Autoimmunity. Development of Antibodies Against Keratinocyte Cholinergic Receptors in Patients With Pemphigus Vulgaris and Pemphigus Foliaceus. Arch Dermatol (1998) 134(8):971–80. doi: 10.1001/archderm.134.8.971
102. Marchenko S, Chernyavsky AI, Arredondo J, Gindi V, Grando SA. Antimitochondrial Autoantibodies in Pemphigus Vulgaris: A Missing Link in Disease Pathophysiology. J Biol Chem (2010) 285(6):3695–704. doi: 10.1074/jbc.M109.081570
103. Dmochowski M, Hashimoto T, Garrod DR, Nishikawa T. Desmocollins I and II are Recognized by Certain Sera From Patients With Various Types of Pemphigus, Particularly Brazilian Pemphigus Foliaceus. J Invest Dermatol (1993) 100(4):380–4. doi: 10.1111/1523-1747.ep12471934
104. Mao X, Nagler AR, Farber SA, Choi EJ, Jackson LH, Leiferman KM, et al. Autoimmunity to Desmocollin 3 in Pemphigus Vulgaris. Am J Pathol (2010) 177(6):2724–30. doi: 10.2353/ajpath.2010.100483
105. Lambert J, Bracke S, van Roy F, Pas HH, Bonné S, De Schepper S. Serum Plakophilin-3 Autoreactivity in Paraneoplastic Pemphigus. Br J Dermatol (2010) 163(3):630–2. doi: 10.1111/j.1365-2133.2010.09845.x
106. Evangelista F, Dasher DA, Diaz LA, Prisayanh PS, Li N. E-Cadherin is an Additional Immunological Target for Pemphigus Autoantibodies. J Invest Dermatol (2008) 128(7):1710–8. doi: 10.1038/sj.jid.5701260
107. Aoyama Y, Kitajima Y. Pemphigus vulgaris-IgG Causes a Rapid Depletion of Desmoglein 3 (Dsg3) From the Triton X-100 Soluble Pools, Leading to the Formation of Dsg3-depleted Desmosomes in a Human Squamous Carcinoma Cell Line, DJM-1 Cells. J Invest Dermatol (1999) 112(1):67–71. doi: 10.1046/j.1523-1747.1999.00463.x
108. Shu E, Yamamoto Y, Aoyama Y, Kitajima Y. Intraperitoneal Injection of Pemphigus vulgaris-IgG Into Mouse Depletes Epidermal Keratinocytes of Desmoglein 3 Associated With Generation of Acantholysis. Arch Dermatol Res (2007) 299(3):165–7. doi: 10.1007/s00403-007-0754-9
109. Yamamoto Y, Aoyama Y, Shu E, Tsunoda K, Amagai M, Kitajima Y. Anti-Desmoglein 3 (Dsg3) Monoclonal Antibodies Deplete Desmosomes of Dsg3 and Differ in Their Dsg3-depleting Activities Related to Pathogenicity. J Biol Chem (2007) 282(24):17866–76. doi: 10.1074/jbc.M607963200
110. Spindler V, Endlich A, Hartlieb E, Vielmuth F, Schmidt E, Waschke J. The Extent of Desmoglein 3 Depletion in Pemphigus Vulgaris is Dependent on Ca(2+)-induced Differentiation: A Role in Suprabasal Epidermal Skin Splitting? Am J Pathol (2011) 179(4):1905–16. doi: 10.1016/j.ajpath.2011.06.043
111. Calkins CC, Setzer SV, Jennings JM, Summers S, Tsunoda K, Amagai M, et al. Desmoglein Endocytosis and Desmosome Disassembly are Coordinated Responses to Pemphigus Autoantibodies. J Biol Chem (2006) 281(11):7623–34. doi: 10.1074/jbc.M512447200
112. Jolly PS, Berkowitz P, Bektas M, Lee HE, Chua M, Diaz LA, et al. p38MAPK Signaling and Desmoglein-3 Internalization are Linked Events in Pemphigus Acantholysis. J Biol Chem (2010) 285(12):8936–41. doi: 10.1074/jbc.M109.087999
113. Amagai M, Hashimoto T, Shimizu N, Nishikawa T. Absorption of Pathogenic Autoantibodies by the Extracellular Domain of Pemphigus Vulgaris Antigen (Dsg3) Produced by Baculovirus. J Clin Invest (1994) 94(1):59–67. doi: 10.1172/JCI117349
114. Hofrichter M, Dworschak J, Emtenani S, Langenhan J, Weiß F, Komorowski L, et al. Immunoadsorption of Desmoglein-3-Specific Igg Abolishes the Blister-Inducing Capacity of Pemphigus Vulgaris IgG in Neonatal Mice. Front Immunol (2018) 9. doi: 10.3389/fimmu.2018.01935
115. van der Wier G, Pas HH, Jonkman MF. Experimental Human Cell and Tissue Models of Pemphigus. Dermatol Res Pract (2010) 2010:143871. doi: 10.1155/2010/143871
116. Boukamp P, Petrussevska RT, Breitkreutz D, Hornung J, Markham A, Fusenig NE. Normal Keratinization in a Spontaneously Immortalized Aneuploid Human Keratinocyte Cell Line. J Cell Biol (1988) 106(3):761–71. doi: 10.1083/jcb.106.3.761
117. Reijnders CM, van Lier A, Roffel S, Kramer D, Scheper RJ, Gibbs S. Development of a Full-Thickness Human Skin Equivalent In Vitro Model Derived From TERT-Immortalized Keratinocytes and Fibroblasts. Tissue Eng Part A (2015) 21(17-18):2448–59. doi: 10.1089/ten.tea.2015.0139
118. Beckert B, Panico F, Pollmann R, Eming R, Banning A, Tikkanen R. Immortalized Human Htert/KER-CT Keratinocytes a Model System for Research on Desmosomal Adhesion and Pathogenesis of Pemphigus Vulgaris. Int J Mol Sci (2019) 20(13):3113. doi: 10.3390/ijms20133113
119. Ishii K, Harada R, Matsuo I, Shirakata Y, Hashimoto K, Amagai M. In Vitro Keratinocyte Dissociation Assay for Evaluation of the Pathogenicity of Anti-Desmoglein 3 IgG Autoantibodies in Pemphigus Vulgaris. J Invest Dermatol (2005) 124(5):939–46. doi: 10.1111/j.0022-202X.2005.23714.x
120. Ishii K, Amagai M. In Vitro Pathogenicity Assay for Anti-Desmoglein Autoantibodies in Pemphigus. Methods Mol Biol (2013) 961:219–25. doi: 10.1007/978-1-62703-227-8_13
121. Saleh AM, El-Samanoudy SI, Rashed LA, Saleh MA. Evaluation of the Pathogenicity of Anti-Desmoglein Antibodies of Pemphigus Vulgaris Patients Using Human Organ Culture Assay. Arch Dermatol Res (2020) 312(4):289–94. doi: 10.1007/s00403-019-01988-9
122. Iwata H, Bieber K, Hirose M, Ludwig RJ. Animal Models to Investigate Pathomechanisms and Evaluate Novel Treatments for Autoimmune Bullous Dermatoses. Curr Pharm Des (2015) 21(18):2422–39. doi: 10.2174/1381612821666150316122502
123. Anhalt GJ, Labib RS, Voorhees JJ, Beals TF, Diaz LA. Induction of Pemphigus in Neonatal Mice by Passive Transfer of IgG From Patients With the Disease. N Engl J Med (1982) 306(20):1189–96. doi: 10.1056/NEJM198205203062001
124. Takahashi Y, Patel HP, Labib RS, Diaz LA, Anhalt GJ. Experimentally Induced Pemphigus Vulgaris in Neonatal BALB/c Mice: A Time-Course Study of Clinical, Immunologic, Ultrastructural, and Cytochemical Changes. J Invest Dermatol (1985) 84(1):41–6. doi: 10.1111/1523-1747.ep12274679
125. Kugelmann D, Radeva MY, Spindler V, Waschke J. Desmoglein 1 Deficiency Causes Lethal Skin Blistering. J Invest Dermatol (2019) 139(7):1596–9.e2. doi: 10.1016/j.jid.2019.01.002
126. Ohyama M, Amagai M, Tsunoda K, Ota T, Koyasu S, Hata J, et al. Immunologic and Histopathologic Characterization of an Active Disease Mouse Model for Pemphigus Vulgaris. J Invest Dermatol (2002) 118(1):199–204. doi: 10.1046/j.0022-202x.2001.01643.x
127. Amagai M, Stanley JR. Desmoglein as a Target in Skin Disease and Beyond. J Invest Dermatol (2012) 132(3 Pt 2):776–84. doi: 10.1038/jid.2011.390
128. Aoki-Ota M, Tsunoda K, Ota T, Iwasaki T, Koyasu S, Amagai M, et al. A Mouse Model of Pemphigus Vulgaris by Adoptive Transfer of Naive Splenocytes From Desmoglein 3 Knockout Mice. Br J Dermatol (2004) 151(2):346–54. doi: 10.1111/j.1365-2133.2004.06056.x
129. Shimizu A, Ishiko A, Ota T, Tsunoda K, Koyasu S, Amagai M, et al. Ultrastructural Changes in Mice Actively Producing Antibodies to Desmoglein 3 Parallel Those in Patients With Pemphigus Vulgaris. Arch Dermatol Res (2002) 294(7):318–23. doi: 10.1007/s00403-002-0341-z
130. Schiltz JR, Michel B. Production of Epidermal Acantholysis in Normal Human Skin In Vitro by the IgG Fraction From Pemphigus Serum. J Invest Dermatol (1976) 67(2):254–60. doi: 10.1111/1523-1747.ep12513454
131. Waschke J, Spindler V, Bruggeman P, Zillikens D, Schmidt G, Drenckhahn D. Inhibition of Rho A Activity Causes Pemphigus Skin Blistering. J Cell Biol (2006) 175(5):721–7. doi: 10.1083/jcb.200605125
132. Spindler V, Drenckhahn D, Zillikens D, Waschke J. Pemphigus IgG Causes Skin Splitting in the Presence of Both Desmoglein 1 and Desmoglein 3. Am J Pathol (2007) 171(3):906–16. doi: 10.2353/ajpath.2007.070028
133. Egu DT, Schmitt T, Sigmund AM, Waschke J. Electron Microscopy Reveals That Phospholipase C and Ca(2+) Signaling Regulate Keratin Filament Uncoupling From Desmosomes in Pemphigus. Ann Anat (2022) 241:151904. doi: 10.1016/j.aanat.2022.151904
134. Egu DT, Sigmund AM, Schmidt E, Spindler V, Walter E, Waschke J. A New Ex Vivo Human Oral Mucosa Model Reveals That p38MAPK Inhibition is Not Effective to Prevent Autoantibody-Induced Mucosal Blistering in Pemphigus. Br J Dermatol (2019) 182(4):987–94. doi: 10.1111/bjd.18237
135. Egu DT, Walter E, Spindler V, Waschke J. Inhibition of p38MAPK Signalling Prevents Epidermal Blistering and Alterations of Desmosome Structure Induced by Pemphigus Autoantibodies in Human Epidermis. Br J Dermatol (2017) 177(6):1612–8. doi: 10.1111/bjd.15721
136. Schmitt T, Egu DT, Walter E, Sigmund AM, Eichkorn R, Yazdi A, et al. Ca(2+) Signalling is Critical for Autoantibody-Induced Blistering of Human Epidermis in Pemphigus. Br J Dermatol (2021) 185(3):595–604. doi: 10.22541/au.160027523.38160657
137. Wilgram GF, Caulfield JB, Lever WF. An Electron Microscopic Study of Acantholysis in Pemphigus Vulgaris. J Invest Dermatol (1961) 36:373–82. doi: 10.1038/jid.1961.58
138. Sokol E, Kramer D, Diercks GF, Kuipers J, Jonkman MF, Pas HH, et al. Large-Scale Electron Microscopy Maps of Patient Skin and Mucosa Provide Insight Into Pathogenesis of Blistering Diseases. J Invest Dermatol (2015) 135(7):1763–70. doi: 10.1038/jid.2015.109
139. Beggs RR, Rao TC, Dean WF, Kowalczyk AP, Mattheyses AL. Desmosomes Undergo Dynamic Architectural Changes During Assembly and Maturation. Tissue Barriers (2022), 2017225. doi: 10.1080/21688370.2021.2017225
140. Spindler V, Rotzer V, Dehner C, Kempf B, Gliem M, Radeva M, et al. Peptide-Mediated Desmoglein 3 Crosslinking Prevents Pemphigus Vulgaris Autoantibody-Induced Skin Blistering. J Clin Invest (2013) 123(2):800–11. doi: 10.1172/JCI60139
141. Spindler V, Eming R, Schmidt E, Amagai M, Grando S, Jonkman MF, et al. Mechanisms Causing Loss of Keratinocyte Cohesion in Pemphigus. J Invest Dermatol (2018) 138(1):32–7. doi: 10.1016/j.jid.2017.06.022
142. Hu CH, Michel B, Schiltz JR. Epidermal Acantholysis Induced In Vitro by Pemphigus Autoantibody. An Ultrastructural Study. Am J Pathol (1978) 90(2):345–62.
143. Ding X, Diaz LA, Fairley JA, Giudice GJ, Liu Z. The Anti-Desmoglein 1 Autoantibodies in Pemphigus Vulgaris Sera are Pathogenic. J Invest Dermatol (1999) 112(5):739–43. doi: 10.1046/j.1523-1747.1999.00585.x
144. Chan PT, Ohyama B, Nishifuji K, Yoshida K, Ishii K, Hashimoto T, et al. Immune Response Towards the Amino-Terminus of Desmoglein 1 Prevails Across Different Activity Stages in Nonendemic Pemphigus Foliaceus. Br J Dermatol (2010) 162(6):1242–50. doi: 10.1111/j.1365-2133.2010.09696.x
145. Ohyama B, Nishifuji K, Chan PT, Kawaguchi A, Yamashita T, Ishii N, et al. Epitope Spreading is Rarely Found in Pemphigus Vulgaris by Large-Scale Longitudinal Study Using Desmoglein 2-Based Swapped Molecules. J Invest Dermatol (2012) 132(4):1158–68. doi: 10.1038/jid.2011.448
146. Vielmuth F, Waschke J, Spindler V. Loss of Desmoglein Binding Is Not Sufficient for Keratinocyte Dissociation in Pemphigus. J Invest Dermatol (2015) 135(12):3068–77. doi: 10.1038/jid.2015.324
147. Esaki C, Seishima M, Yamada T, Osada K, Kitajima Y. Pharmacologic Evidence for Involvement of Phospholipase C in Pemphigus IgG-induced Inositol 1,4,5-Trisphosphate Generation, Intracellular Calcium Increase, and Plasminogen Activator Secretion in DJM-1 Cells, a Squamous Cell Carcinoma Line. J Invest Dermatol (1995) 105(3):329–33. doi: 10.1111/1523-1747.ep12319948
148. Caldelari R, de Bruin A, Baumann D, Suter MM, Bierkamp C, Balmer V, et al. A Central Role for the Armadillo Protein Plakoglobin in the Autoimmune Disease Pemphigus Vulgaris. J Cell Biol (2001) 153(4):823–34. doi: 10.1083/jcb.153.4.823
149. Schulze K, Galichet A, Sayar BS, Scothern A, Howald D, Zymann H, et al. An Adult Passive Transfer Mouse Model to Study Desmoglein 3 Signaling in Pemphigus Vulgaris. J Invest Dermatol (2012) 132(2):346–55. doi: 10.1038/jid.2011.299
150. Luyet C, Schulze K, Sayar BS, Howald D, Müller EJ, Galichet A. Preclinical Studies Identify non-Apoptotic Low-Level Caspase-3 as Therapeutic Target in Pemphigus Vulgaris. PloS One (2015) 10(3):e0119809. doi: 10.1371/journal.pone.0119809
151. Berkowitz P, Hu P, Liu Z, Diaz LA, Enghild JJ, Chua MP, et al. Desmosome Signaling. Inhibition of p38MAPK Prevents Pemphigus Vulgaris IgG-induced Cytoskeleton Reorganization. J Biol Chem (2005) 280(25):23778–84. doi: 10.1074/jbc.M501365200
152. Berkowitz P, Hu P, Warren S, Liu Z, Diaz LA, Rubenstein DS. p38MAPK Inhibition Prevents Disease in Pemphigus Vulgaris Mice. Proc Natl Acad Sci USA (2006) 103(34):12855–60. doi: 10.1073/pnas.0602973103
153. Berkowitz P, Diaz LA, Hall RP, Rubenstein DS. Induction of p38MAPK and HSP27 Phosphorylation in Pemphigus Patient Skin. J Invest Dermatol (2008) 128(3):738–40. doi: 10.1038/sj.jid.5701080
154. Kawasaki H, Tsunoda K, Hata T, Ishii K, Yamada T, Amagai M. Synergistic Pathogenic Effects of Combined Mouse Monoclonal Anti-Desmoglein 3 IgG Antibodies on Pemphigus Vulgaris Blister Formation. J Invest Dermatol (2006) 126(12):2621–30. doi: 10.1038/sj.jid.5700450
155. Sajda T, Sinha AA. Autoantibody Signaling in Pemphigus Vulgaris: Development of an Integrated Model. Front Immunol (2018) 9:692. doi: 10.3389/fimmu.2018.00692
156. Osada K, Seishima M, Kitajima Y. Pemphigus IgG Activates and Translocates Protein Kinase C From the Cytosol to the Particulate/Cytoskeleton Fractions in Human Keratinocytes. J Invest Dermatol (1997) 108(4):482–7. doi: 10.1111/1523-1747.ep12289726
157. Sánchez-Carpintero I, España A, Pelacho B, López Moratalla N, Rubenstein DS, Diaz LA, et al. In Vivo Blockade of Pemphigus Vulgaris Acantholysis by Inhibition of Intracellular Signal Transduction Cascades. Br J Dermatol (2004) 151(3):565–70. doi: 10.1111/j.1365-2133.2004.06147.x
158. Frusić-Zlotkin M, Raichenberg D, Wang X, David M, Michel B, Milner Y. Apoptotic Mechanism in Pemphigus Autoimmunoglobulins-Induced Acantholysis–Possible Involvement of the EGF Receptor. Autoimmunity (2006) 39(7):563–75. doi: 10.1080/08916930600971836
159. Getsios S, Simpson CL, Kojima S, Harmon R, Sheu LJ, Dusek RL, et al. Desmoglein 1-Dependent Suppression of EGFR Signaling Promotes Epidermal Differentiation and Morphogenesis. J Cell Biol (2009) 185(7):1243–58. doi: 10.1083/jcb.200809044
160. España A, Mòdol T, Gil MP, López-Zabalza MJ. Neural Nitric Oxide Synthase Participates in Pemphigus Vulgaris Acantholysis Through Upregulation of Rous Sarcoma, Mammalian Target of Rapamycin and Focal Adhesion Kinase. Exp Dermatol (2013) 22(2):125–30. doi: 10.1111/exd.12088
161. Radeva MY, Walter E, Stach RA, Yazdi AS, Schlegel N, Sarig O, et al. St18 Enhances PV-Igg-Induced Loss of Keratinocyte Cohesion in Parallel to Increased Erk Activation. Front Immunol (2019) 10:770. doi: 10.3389/fimmu.2019.00770
162. Walter E, Vielmuth F, Wanuske MT, Seifert M, Pollmann R, Eming R, et al. Role of Dsg1- and Dsg3-Mediated Signaling in Pemphigus Autoantibody-Induced Loss of Keratinocyte Cohesion. Front Immunol (2019) 10:1128. doi: 10.3389/fimmu.2019.01128
163. Spindler V, Dehner C, Hübner S, Waschke J. Plakoglobin But Not Desmoplakin Regulates Keratinocyte Cohesion Via Modulation of p38MAPK Signaling. J Invest Dermatol (2014) 134(6):1655–64. doi: 10.1038/jid.2014.21
164. Jiang Y, Chen C, Li Z, Guo W, Gegner JA, Lin S, et al. Characterization of the Structure and Function of a New Mitogen-Activated Protein Kinase (P38beta). J Biol Chem (1996) 271(30):17920–6. doi: 10.1074/jbc.271.30.17920
165. Jiang Y, Gram H, Zhao M, New L, Gu J, Feng L, et al. Characterization of the Structure and Function of the Fourth Member of p38 Group Mitogen-Activated Protein Kinases, P38delta. J Biol Chem (1997) 272(48):30122–8. doi: 10.1074/jbc.272.48.30122
166. Cuenda A, Rousseau S. P38 MAP-kinases Pathway Regulation, Function and Role in Human Diseases. Biochim Biophys Acta (2007) 1773(8):1358–75. doi: 10.1016/j.bbamcr.2007.03.010
167. Cargnello M, Roux PP. Activation and Function of the MAPKs and Their Substrates, the MAPK-activated Protein Kinases. Microbiol Mol Biol Rev (2011) 75(1):50–83. doi: 10.1128/MMBR.00031-10
168. Kawasaki Y, Aoyama Y, Tsunoda K, Amagai M, Kitajima Y. Pathogenic Monoclonal Antibody Against Desmoglein 3 Augments Desmoglein 3 and P38 MAPK Phosphorylation in Human Squamous Carcinoma Cell Line. Autoimmunity (2006) 39(7):587–90. doi: 10.1080/08916930600971943
169. Rötzer V, Hartlieb E, Vielmuth F, Gliem M, Spindler V, Waschke J. E-Cadherin and Src Associate With Extradesmosomal Dsg3 and Modulate Desmosome Assembly and Adhesion. Cell Mol Life Sci (2015) 72(24):4885–97. doi: 10.1007/s00018-015-1977-0
170. Stahley SN, Warren MF, Feldman RJ, Swerlick RA, Mattheyses AL, Kowalczyk AP. Super-Resolution Microscopy Reveals Altered Desmosomal Protein Organization in Tissue From Patients With Pemphigus Vulgaris. J Invest Dermatol (2016) 136(1):59–66. doi: 10.1038/JID.2015.353
171. Saito M, Stahley SN, Caughman CY, Mao X, Tucker DK, Payne AS, et al. Signaling Dependent and Independent Mechanisms in Pemphigus Vulgaris Blister Formation. PloS One (2012) 7(12):e50696. doi: 10.1371/journal.pone.0050696
172. Mao X, Sano Y, Park JM, Payne AS. P38 MAPK Activation is Downstream of the Loss of Intercellular Adhesion in Pemphigus Vulgaris. J Biol Chem (2011) 286(2):1283–91. doi: 10.1074/jbc.M110.172874
173. Mao X, Li H, Sano Y, Gaestel M, Mo Park J, Payne AS. MAPKAP Kinase 2 (MK2)-Dependent and -Independent Models of Blister Formation in Pemphigus Vulgaris. J Invest Dermatol (2014) 134(1):68–76. doi: 10.1038/jid.2013.224
174. Berkowitz P, Chua M, Liu Z, Diaz LA, Rubenstein DS. Autoantibodies in the Autoimmune Disease Pemphigus Foliaceus Induce Blistering Via p38 Mitogen-Activated Protein Kinase-Dependent Signaling in the Skin. Am J Pathol (2008) 173(6):1628–36. doi: 10.2353/ajpath.2008.080391
175. Yoshida K, Ishii K, Shimizu A, Yokouchi M, Amagai M, Shiraishi K, et al. Non-Pathogenic Pemphigus Foliaceus (PF) IgG Acts Synergistically With a Directly Pathogenic PF Igg to Increase Blistering by p38MAPK-dependent Desmoglein 1 Clustering. J Dermatol Sci (2017) 85(3):197–207. doi: 10.1016/j.jdermsci.2016.12.010
176. Kim C, Sano Y, Todorova K, Carlson BA, Arpa L, Celada A, et al. The Kinase p38 Alpha Serves Cell Type-Specific Inflammatory Functions in Skin Injury and Coordinates Pro- and Anti-Inflammatory Gene Expression. Nat Immunol (2008) 9(9):1019–27. doi: 10.1038/ni.1640
177. Kotlyarov A, Yannoni Y, Fritz S, Laass K, Telliez JB, Pitman D, et al. Distinct Cellular Functions of MK2. Mol Cell Biol (2002) 22(13):4827–35. doi: 10.1128/MCB.22.13.4827-4835.2002
178. Ellebrecht CT, Maseda D, Payne AS. Pemphigus and Pemphigoid: From Disease Mechanisms to Druggable Pathways. J Invest Dermatol (2022) 142(3 Pt B):907–14. doi: 10.1016/j.jid.2021.04.040
179. Harmon RM, Simpson CL, Johnson JL, Koetsier JL, Dubash AD, Najor NA, et al. Desmoglein-1/Erbin Interaction Suppresses ERK Activation to Support Epidermal Differentiation. J Clin Invest (2013) 123(4):1556–70. doi: 10.1172/JCI65220
180. Cirillo N, Lanza A, Prime SS. Induction of Hyper-Adhesion Attenuates Autoimmune-Induced Keratinocyte Cell-Cell Detachment and Processing of Adhesion Molecules Via Mechanisms That Involve PKC. Exp Cell Res (2010) 316(4):580–92. doi: 10.1016/j.yexcr.2009.10.005
181. Egu DT, Kugelmann D, Waschke J. Role of PKC and ERK Signaling in Epidermal Blistering and Desmosome Regulation in Pemphigus. Front Immunol (2019) 10:2883. doi: 10.3389/fimmu.2019.02883
182. Tsang SM, Brown L, Lin K, Liu L, Piper K, O’Toole EA, et al. Non-Junctional Human Desmoglein 3 Acts as an Upstream Regulator of Src in E-cadherin Adhesion, a Pathway Possibly Involved in the Pathogenesis of Pemphigus Vulgaris. J Pathol (2012) 227(1):81–93. doi: 10.1002/path.3982
183. Chernyavsky AI, Arredondo J, Kitajima Y, Sato-Nagai M, Grando SA. Desmoglein Versus non-Desmoglein Signaling in Pemphigus Acantholysis: Characterization of Novel Signaling Pathways Downstream of Pemphigus Vulgaris Antigens. J Biol Chem (2007) 282(18):13804–12. doi: 10.1074/jbc.M611365200
184. Cirillo N, AlShwaimi E, McCullough M, Prime SS. Pemphigus Vulgaris Autoimmune Globulin Induces Src-dependent Tyrosine-Phosphorylation of Plakophilin 3 and its Detachment From Desmoglein 3. Autoimmunity (2014) 47(2):134–40. doi: 10.3109/08916934.2013.866100
185. Ivars M, España A, Alzuguren P, Pelacho B, Lasarte JJ, López-Zabalza MJ. The Involvement of ADAM10 in Acantholysis in Mucocutaneous Pemphigus Vulgaris Depends on the Autoantibody Profile of Each Patient. Br J Dermatol (2020) 182(5):1194–204. doi: 10.1111/bjd.18382
186. Seishima M, Esaki C, Osada K, Mori S, Hashimoto T, Kitajima Y. Pemphigus IgG, But Not Bullous Pemphigoid IgG, Causes a Transient Increase in Intracellular Calcium and Inositol 1,4,5-Triphosphate in DJM-1 Cells, a Squamous Cell Carcinoma Line. J Invest Dermatol (1995) 104(1):33–7. doi: 10.1111/1523-1747.ep12613469
187. Nishizuka Y. The Role of Protein Kinase C in Cell Surface Signal Transduction and Tumour Promotion. Nature (1984) 308(5961):693–8. doi: 10.1038/308693a0
188. Kitajima Y. 150(Th) Anniversary Series: Desmosomes and Autoimmune Disease, Perspective of Dynamic Desmosome Remodeling and its Impairments in Pemphigus. Cell Commun Adhes (2014) 21(6):269–80. doi: 10.3109/15419061.2014.943397
189. Kitajima Y. Mechanisms of Desmosome Assembly and Disassembly. Clin Exp Dermatol (2002) 27(8):684–90. doi: 10.1046/j.1365-2230.2002.01116.x
190. Delva E, Jennings JM, Calkins CC, Kottke MD, Faundez V, Kowalczyk AP. Pemphigus Vulgaris IgG-induced Desmoglein-3 Endocytosis and Desmosomal Disassembly are Mediated by a Clathrin- and Dynamin-Independent Mechanism. J Biol Chem (2008) 283(26):18303–13. doi: 10.1074/jbc.M710046200
191. Jennings JM, Tucker DK, Kottke MD, Saito M, Delva E, Hanakawa Y, et al. Desmosome Disassembly in Response to Pemphigus Vulgaris IgG Occurs in Distinct Phases and can be Reversed by Expression of Exogenous Dsg3. J Invest Dermatol (2011) 131(3):706–18. doi: 10.1038/jid.2010.389
192. Stahley SN, Saito M, Faundez V, Koval M, Mattheyses AL, Kowalczyk AP. Desmosome Assembly and Disassembly are Membrane Raft-Dependent. PloS One (2014) 9(1):e87809. doi: 10.1371/journal.pone.0087809
193. Choi YJ, Laclef C, Yang N, Andreu-Cervera A, Lewis J, Mao X, et al. RPGRIP1L is Required for Stabilizing Epidermal Keratinocyte Adhesion Through Regulating Desmoglein Endocytosis. PloS Genet (2019) 15(1):e1007914. doi: 10.1371/journal.pgen.1007914
194. Fülle JB, Huppert H, Liebl D, Liu J, Alves de Almeida R, Yanes B, et al. Desmosome Dualism - Most of the Junction is Stable, But a Plakophilin Moiety is Persistently Dynamic. J Cell Sci (2021) 134(21):jcs258906. doi: 10.1242/jcs.258906
195. Shimizu A, Ishiko A, Ota T, Tsunoda K, Amagai M, Nishikawa T. Igg Binds to Desmoglein 3 in Desmosomes and Causes a Desmosomal Split Without Keratin Retraction in a Pemphigus Mouse Model. J Invest Dermatol (2004) 122(5):1145–53. doi: 10.1111/j.0022-202X.2004.22426.x
196. Wang W, Amagai M, Ishiko A. Desmosome Splitting is a Primary Ultrastructural Change in the Acantholysis of Pemphigus. J Dermatol Sci (2009) 54(1):59–61. doi: 10.1016/j.jdermsci.2008.10.010
197. van der Wier G, Jonkman MF, Pas HH, Diercks GF. Ultrastructure of Acantholysis in Pemphigus Foliaceus Re-Examined From the Current Perspective. Br J Dermatol (2012) 167(6):1265–71. doi: 10.1111/j.1365-2133.2012.11173.x
198. Stahley SN, Warren MF, Feldman RJ, Swerlick RA, Mattheyses AL, Kowalczyk AP. Super-Resolution Microscopy Reveals Altered Desmosomal Protein Organization in Tissue From Patients With Pemphigus Vulgaris. J Cell Sci (2016) 136(1):59–66. doi: 10.1038/JID.2015.353
199. Hobbs RP, Green KJ. Desmoplakin Regulates Desmosome Hyperadhesion. J Invest Dermatol (2012) 132(2):482–5. doi: 10.1038/jid.2011.318
200. Tucker DK, Stahley SN, Kowalczyk AP. Plakophilin-1 Protects Keratinocytes From Pemphigus Vulgaris IgG by Forming Calcium-Independent Desmosomes. J Invest Dermatol (2014) 134(4):1033–43. doi: 10.1038/jid.2013.401
201. Bartle EI, Rao TC, Beggs RR, Dean WF, Urner TM, Kowalczyk AP, et al. Protein Exchange is Reduced in Calcium-Independent Epithelial Junctions. J Cell Biol (2020) 219(6):e201906153. doi: 10.1083/jcb.201906153
202. Dehner C, Rötzer V, Waschke J, Spindler V. A Desmoplakin Point Mutation With Enhanced Keratin Association Ameliorates Pemphigus Vulgaris Autoantibody-Mediated Loss of Cell Cohesion. Am J Pathol (2014) 184(9):2528–36. doi: 10.1016/j.ajpath.2014.05.016
Keywords: pemphigus, signaling, desmosomes, ultrastructure, ex vivo skin model, electron microscope
Citation: Egu DT, Schmitt T and Waschke J (2022) Mechanisms Causing Acantholysis in Pemphigus-Lessons from Human Skin. Front. Immunol. 13:884067. doi: 10.3389/fimmu.2022.884067
Received: 25 February 2022; Accepted: 12 April 2022;
Published: 20 May 2022.
Edited by:
Enno Schmidt, University of Lübeck, GermanyReviewed by:
Yasuo Kitajima, Kizawa Memorial Hospital, JapanJong Hoon Kim, Yonsei University, South Korea
Copyright © 2022 Egu, Schmitt and Waschke. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jens Waschke, SmVucy5XYXNjaGtlQG1lZC51bmktbXVlbmNoZW4uZGU=