 Cillian Gartlan1*
Cillian Gartlan1* Tom Tipton1
Tom Tipton1 Francisco J. Salguero2
Francisco J. Salguero2 Quentin Sattentau3
Quentin Sattentau3 Andrew Gorringe2
Andrew Gorringe2 Miles W. Carroll1*
Miles W. Carroll1*- 1Wellcome Centre for Human Genetics and Pandemic Sciences Institute, Nuffield Department of Medicine, University of Oxford, Oxford, United Kingdom
- 2Research and Evaluation, UK Health Security Agency, Porton Down, Salisbury, United Kingdom
- 3The Sir William Dunn School of Pathology, University of Oxford, Oxford, United Kingdom
Vaccine-associated enhanced disease (VAED) is a difficult phenomenon to define and can be confused with vaccine failure. Using studies on respiratory syncytial virus (RSV) vaccination and dengue virus infection, we highlight known and theoretical mechanisms of VAED, including antibody-dependent enhancement (ADE), antibody-enhanced disease (AED) and Th2-mediated pathology. We also critically review the literature surrounding this phenomenon in pathogenic human coronaviruses, including MERS-CoV, SARS-CoV-1 and SARS-CoV-2. Poor quality histopathological data and a lack of consistency in defining severe pathology and VAED in preclinical studies of MERS-CoV and SARS-CoV-1 vaccines in particular make it difficult to interrogate potential cases of VAED. Fortuitously, there have been only few reports of mild VAED in SARS-CoV-2 vaccination in preclinical models and no observations in their clinical use. We describe the problem areas and discuss methods to improve the characterisation of VAED in the future.
Introduction
Vaccine-associated enhanced disease (VAED) is a rarely-observed phenomenon whereby vaccination promotes immune responses that exacerbate the disease caused by subsequent infection with the associated pathogen. VAED has been observed in humans in three vaccine trials, for vaccines against dengue virus, respiratory syncytial virus (RSV), and measles (1–3). Of these vaccines, only the dengue vaccine has been approved, under particular conditions to avoid inducing VAED (4). In this review, we outline the mechanisms of VAED (Figure 1), how it is characterised, critically evaluate the evidence surrounding VAED with a focus on pathogenic coronaviruses, and propose a framework which can be used to investigate VAED. While VAED is usually associated with antibody responses, there are also potential roles for T cells and complement, each of which will be discussed. To note; VAED is sometimes also referred to as vaccine-associated enhanced respiratory disease (VAERD). It can be difficult to distinguish between VAED and vaccine failure, however the Coalition for Epidemic Preparedness Innovations (CEPI) have recently developed a case definition for VAED (5). The guidelines put forward by this group of experts can be used to investigate potential cases of VAED in future clinical trials and may also help to inform such investigations in preclinical models.
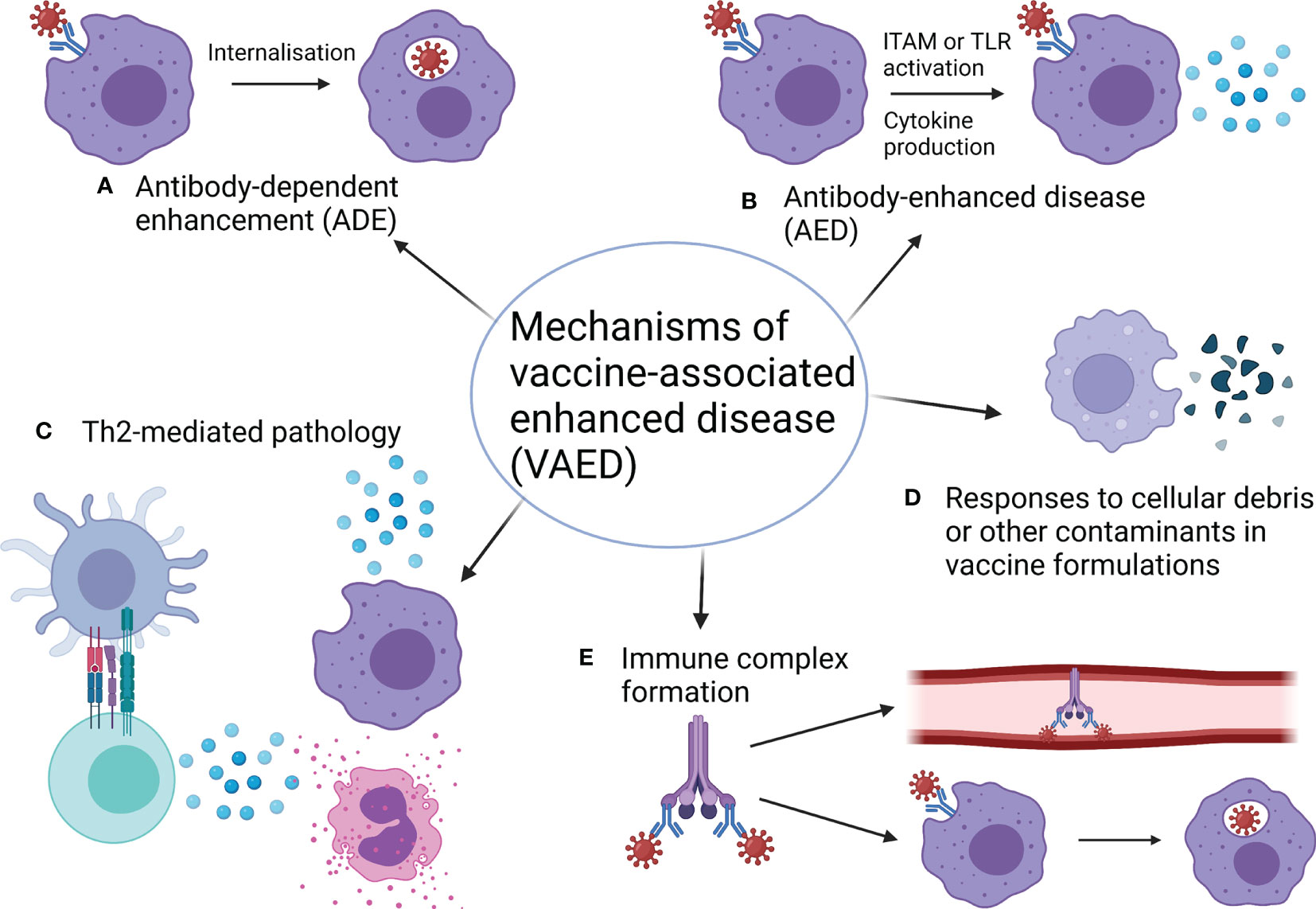
Figure 1 Mechanisms of vaccine-associated enhanced disease. (A) Antibody-dependent enhancement (ADE) occurs when antibodies increase the ability of a virus to infect cells (also see Figure 2). (B) Antibody-enhanced disease (AED) occurs when antibodies exacerbate inflammation, resulting in pathology (also see Figure 2). (C) Th2-skewed responses can be pathogenic for some infections and so vaccines that induce Th2 responses in this case can cause pathology. Usually Th2 pathology is associated with eosinophil infiltration. (D) Components of vaccine formulations such as bovine serum albumin (BSA) and cellular debris can mediate pathogenic cellular responses to these components when encountered again as contaminants in the challenge material. While these components are normally removed during vaccine preparation, some preclinical studies have not included appropriate washing and centrifugation steps to facilitate this. (E) Immune complexes between viral proteins, antibodies and/or complement can lead to a build-up of deposits in blood vessels and organs or facilitate enhanced uptake of virus through myeloid cells, causing ADE. Both of these outcomes can enhance pathology. Made with BioRender.com.
Antibody-dependent enhancement (ADE) is arguably the most well-understood mechanistic explanation of VAED, although in many cases it may not be the major contributor to VAED. ADE describes how antibodies enhance uptake of a virus into cells that can facilitate productive infection and viral dissemination (Figure 2). ADE has been best characterised in dengue virus reinfections and has been a major challenge in dengue virus vaccine development (6). ADE can be caused by binding of non-neutralising antibodies or sub-neutralising concentrations of antibodies. The former is an issue of antibody quality, while the latter is an issue of quantity. Both of these mechanisms result in virus uptake by phagocytic cells through Fc gamma receptors (FcγRs) on myeloid cells - including monocytes, macrophages and dendritic cells (DCs) - without neutralising the virus to protect against further infection. For viruses that can proliferate and propagate through infection of phagocytes, such as dengue virus, these mechanisms worsen disease outcomes. However, even in cases where viruses can mediate ADE, many antibodies provide a background of protection and only a minority of antibodies will contribute to ADE. Where antibody titre is the primary correlate, there is thought to be a range at which antibody concentrations are pathogenic rather than neutralising, as shown with dengue infection (7). At very low levels, there may not be enough antibody binding to facilitate ADE and at very high levels, antibodies may be able to neutralise viruses by preventing viral proteins binding to their receptor targets.
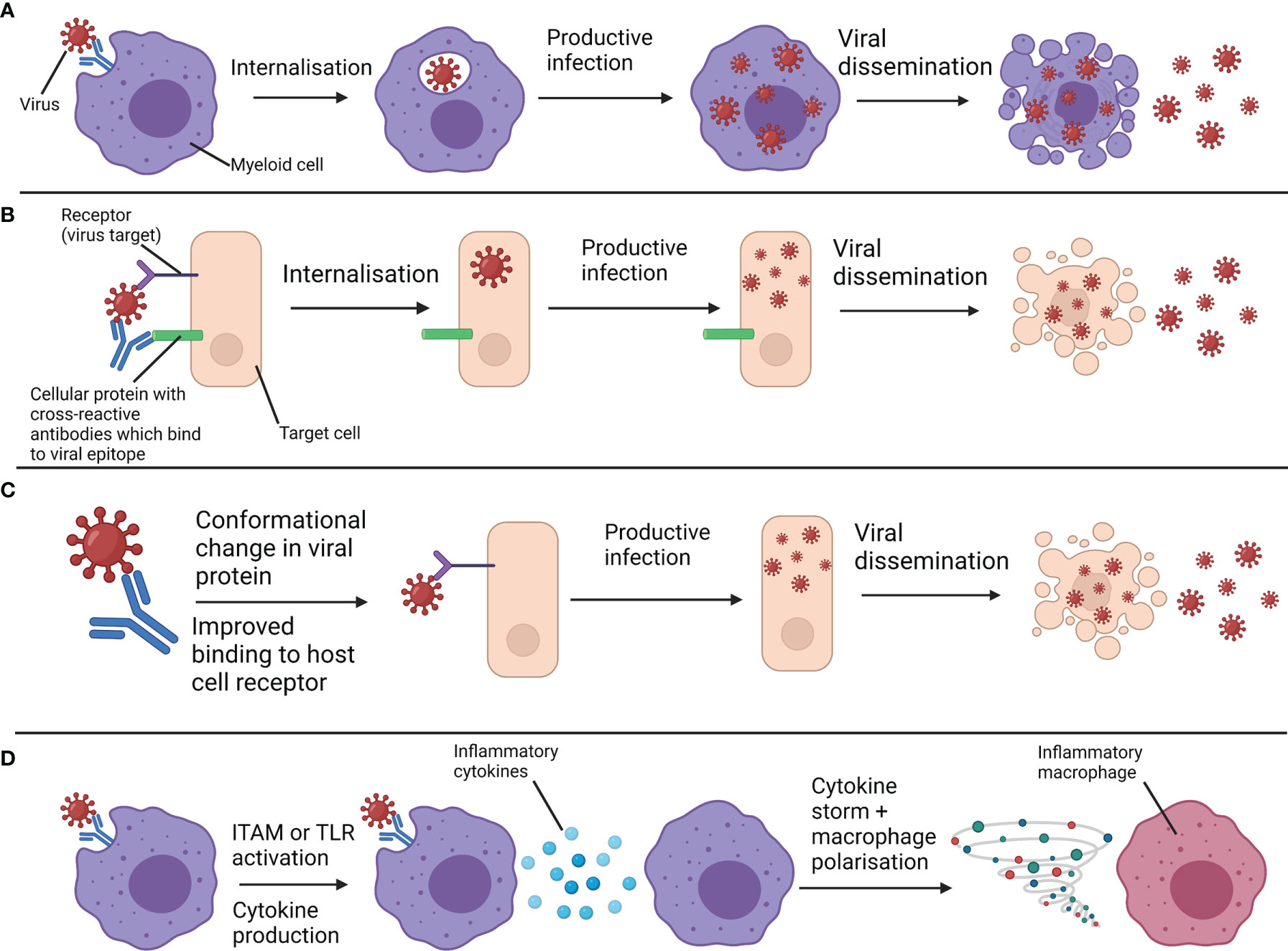
Figure 2 Overview of antibody-dependent enhancement (ADE) and antibody-enhanced disease (AED). (A) Non-neutralising antibodies or sub-neutralising antibody concentrations bind to viruses and interact with Fc receptors on myeloid cells. This facilitates the internalisation of viruses. Viruses that can productively infect myeloid cells can proliferate and spread following their uptake, enhancing infection. This is a form of ADE. (B) Cross-reactive antibodies bind to both virus and host cell components, bringing viruses in close contact with their receptor. Receptor-mediated uptake and enhanced infection follows. This is another form of ADE. (C) Antibodies against a particular epitope drive a conformational change in a viral protein which enhances infection through improved binding to the host cell receptor. This is another form of ADE. (D) Antibodies bound to virus interact with Fc receptors on myeloid cells and either activate immunoreceptor tyrosine-based activation motifs (ITAMs) associated with these receptors, or facilitate viral uptake and subsequent activation of endosomal toll-like receptors (TLRs). Through either of these mechanisms, inflammatory cytokines and chemokines are produced, exacerbating inflammation to a pathogenic extent and polarising myeloid cells towards more inflammatory phenotypes. These are forms of AED. Productive infection of myeloid cells is not required for this mechanism. Created with BioRender.com.
While ADE concerns enhancement of viral entry into susceptible cells, another term, antibody-enhanced disease (AED), concerns enhancement of immunopathology (see Figure 2). Sometimes, AED is also referred to as “ADE of disease” or “enhanced respiratory disease”. In this review we will use the terms “ADE” and “AED” separately. AED occurs when antibodies bound to a virus also bind to FcγRs of myeloid cells and increase inflammation to such an extent as to cause immunopathology. The increase in inflammatory cytokine production can be due to activation of immunoreceptor tyrosine-based activation motifs (ITAMs) through FcγRs (8) or activation of endosomal toll-like receptors (TLRs) by pathogen-associated molecular patterns (PAMPs) on a pathogen following antibody-mediated uptake. It is important to note that while FcγR binding or activation can be pathogenic in an ADE or AED context respectively, FcγRs also play a significant role in antiviral immunity, sometimes more than potent neutralising activity (9). The level of fucosylation of IgG1 antibodies is known to impact their ability to bind to and activate the activatory receptor FcγRIIIa, with lower fucosylation associated with stronger binding (10). Immune complexes can also contribute to ADE or AED. Immune complexes are formed by antibody-antigen aggregates and sometimes also involve binding of complement binding proteins. These complexes can be either protective or pathogenic, as they can stimulate antigen presentation and protective immune responses or be deposited in blood vessels and organs and trigger pathogenic inflammation (11). Immune complexes can also bind FcγRs to facilitate ADE (12).
At the onset of the COVID-19 pandemic there were significant safety concerns with the development of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) vaccines (13), particularly given prior reports of VAED in preclinical models of SARS-CoV-1 and Middle East respiratory syndrome (MERS) vaccination. These studies will be a focus of this review. Animal models are essential for investigating VAED. However, progression of infection and severity of disease can vary widely between species and so different animal models have particular advantages in terms of immunological and pathological insights, although none completely mimic the human case (14–16). Beyond physiology, the molecular mechanisms that characterise coronavirus disease 2019 (COVID-19) progression can also vary between species. For instance, the gene encoding ACE2 is an interferon-stimulated gene (ISG) in human cells but not in murine tissue (17). This means that interferon induction in response to SARS-CoV-2 will likely have quite different pathological consequences in humans compared to mice. Fc receptor functions also vary widely between species, with humans exhibiting features absent in other species (18). This has implications for the study of the antibody-mediated elements of VAED in animal models. Overall, non-human primate (NHP) models are likely to provide the most relevant pathological insights to the human case, given similarities in physiology and immune mechanisms, although both rhesus and cynomolgus macaques both present only mild to moderate forms of COVID-19 (16). As many VAED concerns have been associated with formalin-inactivated vaccines, we will also discuss recent studies examining formalin-inactivated SARS-CoV-2 based vaccines in preclinical models, which were designed with the goal of stimulating VAED (19, 20). In order to investigate VAED in animal models, in vivo pathology studies are essential as disease progression and its consequences are difficult to determine from serological and other in vitro data alone.
Background
Vaccines against dengue virus and RSV have demonstrated VAED in humans. While the mechanism of VAED is well-characterised as being mediated by ADE in dengue virus, the mechanisms behind VAED in RSV are less clear (Table 1).

Table 1 An overview of ADE, AED and VAED concerns relevant to dengue virus and RSV.
Antibody-Dependent Enhancement
Dengue virus provides a clear example of VAED through ADE, enabled by its tropism for monocytes and macrophages in particular (6). As a consequence of ADE, secondary infections with a different serotype of dengue virus are much more pathogenic than primary infections (37). This is because many cross-reactive antibodies in the secondary infection lack neutralising activity but can still bind to the virus and assist its entry into FcγR-expressing phagocytes. Ramadhany et al. found that engineering of the Fc region on antibodies, which changes the antibody subclass, affects binding to FcγRs and subsequently the extent of ADE observed (38). The mutations that increased or decreased ADE activity sometimes depended on the cell type being infected, as different cell types express different levels of FcγR subtypes and other mutations decreased ADE in more than one cell type (38).
A monocyte-independent mechanism of ADE has also been proposed in dengue, although it remains to be seen if this occurs with other viruses. This mechanism requires molecular mimicry between virions and host cell components (self-antigens), such as the prM protein in dengue virions and heat shock protein 60 (HSP60) (21). However, the mechanism has not been completely elucidated and may involve conformational changes in virion proteins that mediate cell entry or simply bringing virions in close proximity with target cells so that they can interact with their receptor. As with other forms of ADE, its significance depends on the concentration of enhancing antibodies vs the concentration of neutralising antibodies. A bispecific antibody targeting host cells and dengue virus has provided evidence for this FcγR-independent form of ADE in dengue in vitro (22). Recent findings suggest that as antibody levels increase in dengue infection, the severity of ADE declines, as more neutralising antibodies are produced than non-neutralising antibodies (23). This suggests that only low levels of cross-reactive antibodies will cause ADE. While antibodies in dengue can contribute to ADE, antibodies are also important in preventing dengue replication, at least at the appropriate concentrations (24). An intermediate concentration range of cross-reactive antibodies is thought to contribute to ADE because higher titres contribute to neutralisation, while titres below this range are insufficient to cause ADE (7).
From dengue virus studies, it is apparent that several conditions can contribute to the development of ADE. Firstly, there are several serotypes of dengue circulating, each with antigenic variability. Secondly, binding of antibodies to these variable antigens can facilitate productive infection of myeloid cells through FcγRs Thirdly, subsequent infections with serotypes that differ from the first serotype encountered will provoke memory B cell responses which produce cross-reactive antibodies with non-neutralising or poorly-neutralising activity, facilitating productive infection of myeloid cells. Finally, when antibody levels wane after infection or vaccination, sub-neutralising concentrations of antibodies can enhance disease rather than prevent it (1, 7). Another factor that may contribute to ADE in dengue virus infection is the level of fucosylation of anti-dengue virus antibodies (25). Bournazos et al. found that dengue infection induced IgG1 afucosylation, which was associated with worsened disease outcomes upon secondary infection. Interestingly, the authors found no association between neutralising activity or antibody titres and severity of infection (25), contradicting the aforementioned idea that particular concentrations of non-neutralising antibodies are the primary contributors to disease severity. Afucosylation of IgG1 increases its binding affinity to FcγRIIIa (10). Therefore, afucosylated anti-dengue virus antibodies in secondary infection may improve the ability of the virus to invade monocytes through ADE. Bournazos et al. did not observe afucosylation of antibodies in patients infected with West Nile virus or Zika virus, which are other flaviviruses (25). While West Nile virus and Zika virus pathogenesis is exacerbated by monocytes, ADE is not thought to play a major role in the pathogenesis of either of these viruses (39–41).
Efficacy trials of a three-dose tetravalent dengue vaccine (CYD-TDV) demonstrated that serostatus upon vaccination determined whether the vaccine was protective or caused enhanced disease (1). Those who were seropositive for dengue prior to vaccination demonstrated lower levels of hospitalisation compared to controls, while those who were seronegative prior to vaccination demonstrated higher levels of hospitalisation when compared to controls. This phenomenon was not dependent on the serotype that vaccinated individuals were infected with, but was most prevalent in those infected by serotype 2. CYD-TDV administration was also associated with higher risks of severe thrombocytopenia (1). However, the vaccine has been shown to be safe and effective in seropositive individuals and has been approved for use in seropositive individuals aged 9 or above in many countries and the European Union (4).
ADE has also been observed following infection with the filoviruses Ebola virus (EBOV) and Marburg virus (MARV) in vitro and in vivo (42–45). In line with these findings, ADE has also been found to occur in humans regardless of serum antibody affinity, class or specificity and is instead dependent on antibody concentration, with sub-neutralising concentrations of antibodies enhancing infection (46). This study also found that antibodies with high affinity for FcγRs contribute more to ADE, although antibodies with low affinity for FcγRs can also make a contribution, consistent with the findings by Ramadhany et al. in relation to dengue virus (38). As with dengue, neutralising antibodies are important in protection against Ebola virus disease (EVD) (47), so in vaccination it is important to induce effective antibody titres to ensure protection and not disease enhancement. However, in rhesus macaques, it has been found that passive immunotherapy, involving transfer of convalescent plasma from EBOV-immune macaques to naïve macaques, can lead to enhanced infection, causing viral titres at death to rise over 100-fold above controls (48). Monoclonal antibodies have also been found to enhance viral replication in EBOV infection, unless used at high concentrations, when they become protective (46, 49–52). In humans, convalescent plasma was not associated with improved survival in treatment of Ebola infection (53).
Antibody-Enhanced Disease and Th2 Pathology
RSV and measles are paramyxoviruses that have been linked with VAED and this has been reviewed elsewhere (26, 27, 54). Both viruses cause moderate symptoms in most infected people but are more likely to become severe or fatal in young children. In 1969, Kim et al. published a study on a formalin-inactivated alum-adjuvanted vaccine for RSV, which became the first study that demonstrated VAED in humans (2). Kim et al. reported that 80% of the vaccinated cohort that became infected required hospitalisation, compared to 5% of the infected control group and two young children in the vaccinated group died as a result (2). As with dengue, VAED was observed in previously seronegative individuals and not in those who were seropositive before vaccination (26). While much research has gone into what went wrong, there is still no licenced vaccine for RSV. Fears of VAED have delayed RSV vaccine development, so understanding the phenomenon is paramount to ensure rapid development of safe and effective vaccines (13). VAED in the inactivated RSV vaccine is thought to have been mediated by immune complex deposition in the lungs and pathogenic Th2 responses, which caused eosinophil infiltration of the lungs (26).
As well as the formalin-inactivated vaccine, vaccinia virus expressing the G protein (which RSV uses to attach to host cells) has been widely used to replicate the eosinophil infiltration seen in VAED pathology (26, 28, 29). While the goal of this research was to dissect a mechanism of VAED, it is unknown whether VAED in the vaccinia virus vaccines and the formalin-inactivated vaccine is mediated by the same mechanism, as pointed out by Acosta et al. (26). These authors have also pointed out that enhanced disease in cotton rat (55) and bovine (56) models does not involve eosinophils and that the role of eosinophils in mediating VAED has been questioned (30). Knudson et al. demonstrated using a formalin-inactivated alum-adjuvanted vaccine that it is Th2-biased CD4+ T cells that mediate VAED and not eosinophils or antibody levels (30). The role of CD4+ T cells and their production of IL-4 and IL-10 had also been highlighted many years earlier (57, 58), but only more recently could the eosinophilia that followed be ruled out as the cause of pathology (30). Consistent with the Th2 immune phenotype, the authors also noted a lack of CD8+ T cell induction by a formalin-inactivated RSV vaccine. Therefore, eosinophil infiltration may be linked to Th2 pathology in some cases without being the mediating factor of the enhanced disease. The role of eosinophils in VAED more generally may vary depending on the pathogen in question, the animal model and the characteristics of the vaccine preparation.
While immune complexes and weakly neutralising antibodies have also been suggested to contribute to VAED in RSV (31, 32), they do not have as convincing a mechanistic explanation as Th2-biased CD4+ T cells (30). As for what mediates the Th2 bias, there are several potential explanations (26). It is possible that the use of aluminium hydroxide (alum) as an adjuvant in many RSV vaccines could explain Th2 polarisation, as alum is a known Th2-skewing adjuvant (33).The Th2-skewing effect of carbonyl groups created by formalin inactivation itself could also cause the pathology seen in the formalin-inactivated RSV vaccine (34). Alternatively, a particular peptide in the G protein may cause the Th2 bias that leads to pathology and the associated eosinophilia (29). Formalin inactivation alone cannot explain Th2 polarisation, as studies using isolated G or F glycoproteins without formalin in BALB/c mice and cotton rats respectively also demonstrated this effect (59, 60). Both of these studies used formulations containing alum. Immune complexes could also possibly contribute to Th2 polarisation through ligation of FcγRs on macrophages (35). While one study examined weakly neutralising antibodies as potential inducers of Th2 responses, the TLR agonists used in this study may offer an alternative interpretation of the results (32). Addition of TLR agonists to a formalin-inactivated RSV vaccine led to greater affinity maturation and enabled the vaccine to induce protective instead of disease-enhancing responses, however the authors note that T cell polarisation resulting from the addition of the TLR agonists may be another explanation for prevention of VAED (32). Alternatively, a defined level of antibody neutralisation may have been able to overpower the pathogenic effect of Th2 responses. It is also possible that weak neutralisation activity only allows antibodies to carry out the more pathogenic side of their activity, forming immune complexes through complement activation, which have been shown to promote VAED upon subsequent RSV infection (31), possibly due to Th2-skewing (35). Affinity maturation induced by previous natural infection may explain why seropositive vaccine recipients did not show enhanced disease (2), as their antibodies likely had greater neutralising activity.
The balance of correlates of protection vs the correlates of enhanced disease in RSV vaccination are poorly defined (61). A study in the cotton rat model demonstrates that the use of a Th1-biasing adjuvant with the F protein is not enough, and that high doses of antigen are also required to provide protection against pathology (60). This highlights the significance of having both appropriate T cell bias and dosage to minimise pathology and induce protective responses, although these correlates of protection have been difficult to quantify. In this study, despite low virus titres and induction of neutralising antibody levels previously found to be protective, pathology was observed in the low-dose group that received a Th1-biasing adjuvant (60). More recently, live-attenuated and vector platforms administered intranasally have appeared to provide protection without any VAED in children (36) and several vaccine platforms targeting pre-fusion F protein are currently in phase III clinical trials. Viral vector platforms induce Th1-biased immune responses (62), which may explain the lack of VAED.
VAED has also been observed in children who received a formalin-inactivated measles vaccine (3) and is thought to have a similar mechanism to VAED in RSV (54). In measles, the symptomatic disease mediated by the vaccine was characterised as ‘atypical measles’ and as in RSV, could be abrogated by neutralising antibodies induced by infection with live virus (54). This is the earliest known case of VAED in humans, although the RSV case was the first to be correctly characterised as VAED. Immune complex formation by non-neutralising antibodies and the subsequent deposition of these complexes are also thought to contribute to VAED in atypical measles (63).
If a virus cannot infect and replicate within myeloid cells (through ADE), AED can still occur. For AED, the infection of myeloid cells is replaced with induction of other antibody-mediated activities that exacerbate immunopathology. In the context of vaccination, antigenic variability in some cases of ADE/AED, such as that seen in dengue, could be replaced by the use of an antigen in the vaccine preparation that generates non-neutralising or sub-neutralising antibody responses. This could arise through use of an unintentionally modified and/or conformationally incorrect antigen. For instance, formalin inactivation can alter the structure of antigens through addition of cross-links, or the presence of reactive carbonyls may alter their Th bias, as discussed previously in the context of RSV (34). Alternatively, the use of inappropriate cell lines to produce vaccine antigens can lead to weakly neutralising antibody responses due to differences in post-translational modifications when compared to the case of human infection, which has been shown in influenza vaccination (64). While this has not resulted in VAED, this mechanism could theoretically cause VAED in other viral infections.
Vaccine-Associated Enhanced Disease in Pathogenic Coronaviruses
ADE in coronaviruses infections has been best-characterised in feline infectious peritonitis (FIP) (65), which affects cats and not humans. Like dengue virus, FIP virus (FIPV) productively infects macrophages (66). Unlike dengue virus, re-infection with the same serotype of FIPV can lead to ADE (67), possibly caused by sub-neutralising antibody concentrations. However, unlike dengue virus, the more severe pathogenesis of FIP arises when the spike protein of FIPV mutates within the host to allow infection of macrophages (68). While some FIPV vaccine efforts have demonstrated efficacy, others have caused VAED (69). In human coronaviruses, there is conflicting evidence concerning ADE and AED in particular (Table 2) (100). We will review the evidence for and against, focusing on potential mechanisms and in vivo pathology. Some authors have made claims of ADE in human coronavirus infections after providing evidence for their antibody-mediated entry of myeloid cells. However, without productive infection within these cells, it is unlikely that any pathological consequences in vivo caused by ADE can occur. Antibody-mediated entry of viruses into myeloid cells is therefore not sufficient evidence for ADE, as viruses are often eliminated by macrophages through this mechanism, known as antibody-dependent cellular phagocytosis (ADCP). In order to avoid AED, vaccines must generate responses against appropriate targets which provide effective neutralisation, such as the viral protein used to interact with cellular receptors. In coronaviruses, the receptor-binding domain (RBD) of the spike is the most appropriate target, as blocking it prevents binding to its cellular target. Some of the VAED in preclinical vaccines against coronaviruses may have resulted from responses generated against other structural proteins or modified spike protein such that the antibodies generated against it are sub-neutralising. Alternatively, VAED may sometimes be explained through inappropriate skewing of immune responses towards inflammatory phenotypes that exacerbate pathology or presence of contaminants in vaccine preparations.

Table 2 An overview of ADE, AED and VAED concerns in pathogenic coronaviruses.
Preclinical vaccine studies with other coronaviruses raised concerns for the development of SARS-CoV-2 vaccines, owing to reports of vaccine-enhanced disease (VAED). While Feline infectious peritonitis virus, MERS-CoV and SARS-CoV-1 have shown evidence for ADE, AED and/or VAED in animal models, this has never been demonstrated in humans. SARS-CoV-2 preclinical studies did not highlight VAED as a concern.
In MERS, the mechanism of antibody-mediated entry into myeloid cells is thought to involve a conformational change in the spike protein upon antibody binding to the RBD, but not to other sites (70). This uptake is mediated by the same pathway as that induced by its cellular target, DPP4. Similar to the in vivo ADE dengue virus findings mentioned previously (7), it was found that the extent of viral entry was dependent on intermediate concentrations of antibodies against MERS-CoV in vitro (70). However, as only low levels of productive infection of macrophages and dendritic cells have been reported (71, 72), anti-MERS-CoV antibody concentrations are not linked to pathogenesis to the same extent as anti-dengue virus antibody concentrations (7). When anti-MERS-CoV antibodies are seen to enhance pathology, it is through AED. MERS-CoV has been found to induce inflammatory cytokines and chemokines in infected macrophages (71), which contribute to the cytokine storm associated with the immunopathology of severe MERS-CoV infection (101). MERS-CoV reinfection in rabbits has been shown to cause severe immunopathology, partly owing to the lack of neutralising antibodies, which were only produced following this second exposure, as well as the disease-enhancing activity of non-neutralising antibodies (73). The authors suggested AED via complement activation rather than ADE, as no increase in viral load was seen during the reinfection, demonstrating in vivo that the extent of productive infection of macrophages in MERS is not involved in pathogenesis. Importantly, the authors found neutralising antibodies to be protective (73), thus highlighting the importance of including appropriate antigens in vaccines to induce neutralising rather than non-neutralising antibodies which may be pathogenic.
There have also been suggestions that immunisation against MERS can cause Th2 immunopathology in mice (74). The authors used transgenic mice with the human MERS virus receptor DPP4. Immunohistochemical (IHC) staining was used to visualise eosinophils but published images showed very high background, and as the authors did not show haematoxylin and eosin (H+E) stained images, it is difficult to interpret whether the MBP-positive cells are eosinophils as they are claimed to be. Immunisation of mice with a parainfluenza viral vector was found to be effective and did not cause pathology, while a UV-inactivated MERS-CoV vaccine was protective but also associated with immunopathology (75).
Successful MERS vaccination has been observed in rhesus macaques with the use of the ChAdOx viral vector platform (76), which is also used in a widely-administered and effective human SARS-CoV-2 vaccine (102). Rather than enhancing disease, as might be expected from the aforementioned in vitro studies and in vivo murine studies, this MERS vaccine protected the macaques from respiratory pathology across six different MERS-CoV strains (76). This demonstrates the importance of carrying out in vivo histopathological analysis in appropriate models when studying VAED. This vaccine has been assessed in phase 1 trials with promising safety and immunogenicity data (103) but any concerns of VAED can only be monitored if larger trials are conducted where vaccinees are exposed to MERS-CoV infection. Safe and effective vaccination against MERS virus using a modified vaccinia Ankara (MVA) viral vector has also been observed in mice (104, 105) and in dromedary camels, the reservoir host of the virus (106). The MVA vector has also been assessed in phase 1 trials (107).
With regards to SARS-CoV-1, a closer relative of SARS-CoV-2, antibodies have been reported to enhance in vitro infection of myeloid cells (77, 78) but crucially these cases were non-productive or ‘abortive’ infections, meaning the viral particles were unable to replicate within these infected cells and disseminate further. Instead of antibody-macrophage interactions enhancing disease, macrophages in SARS-CoV-1 infection appear to be essential for antibody-mediated viral clearance, as demonstrated in mouse models (79). In this study, the importance of ADCP by macrophages was highlighted by the fact that neutralising activity was not essential for viral clearance and species-matched Fc receptors were required for clearance (79). However, in SARS-CoV-1 vaccination, the story is more complex.
Liu et al. demonstrated that vaccination of Chinese macaques with an MVA vector encoding the SARS-CoV-1 spike glycoprotein induced high levels of neutralising antibodies that reduced viral loads following challenge. However, histopathological examination revealed that these animals had greater lung damage following challenge compared to controls (80). Through adoptive transfer of vaccine-induced neutralising antibodies to unvaccinated macaques, Liu et al. showed that anti-SARS-CoV-1 spike antibodies conferred more severe lung damage compared to controls upon challenge. The features of diffuse alveolar damage (DAD) described include the presence hyaline membranes in Figure 1. However, it is not clear if hyaline membranes are present in the images in this figure and the pathology may be incorrectly interpreted. Similarly, in Figure 2, the pathology that the authors allude to in the figure legend is difficult to observe and the scoring system used is unclear. The enhanced pathology correlated with infiltration of inflammatory macrophages and rising serum IL-8 levels is much clearer, as areas of interest are magnified (80). The presence of anti-spike antibodies skewed lung macrophages towards inflammatory phenotypes and away from wound-healing phenotypes found in higher numbers in controls (80). Consistent with these findings, sera from deceased SARS-CoV-1 patients demonstrated higher levels of neutralising antibodies and of inflammatory macrophages when compared to SARS-CoV-1 survivors and the antibodies from deceased patients could skew macrophages towards inflammatory phenotypes. The skewing effect was partially attributable to engagement of FcγRs (80). However, these skewing effects were noted in isolated monocytes from the infected macaques, and so this in vitro analysis lacks the context of other factors in vivo that may have contributed to AED in this case, such as formation of immune complexes following complement deposition.
In another study testing candidate MVA vector vaccines expressing SARS-CoV-1 spike protein or nucleocapsid, carried out in ferrets, neither vaccine was protective and the MVA-spike vaccine was reported to be associated with hepatitis (81). Less severe pathology was noted in the MVA-nucleocapsid vaccine and the MVA-spike vaccine that was associated with hepatitis also induced neutralising antibodies (81). However, for findings in ferrets it must be noted that hepatitis is often a background finding in experimental settings for this animal model, possibly caused by prior chronic viral infection with hepatitis E (82).
A live-attenuated mucosal vaccine against SARS-CoV-1, based on an attenuated parainfluenza virus vector expressing the spike protein, has shown efficacy in preventing viral shedding following SARS-CoV-1 challenge in African green monkeys (83). The vaccine was administered via both a nasal and intratracheal inoculation and no evidence of VAED was observed, although the study did not closely examine pathology. Another intranasal viral vector SARS-CoV-1 vaccine, using a recombinant adeno-associated virus platform, has also been shown to be protective in mice and was associated with less pulmonary pathology (84).
Formalin-inactivated SARS-CoV-1 vaccines have also demonstrated both protection (85) and pathology (86) in vaccinated macaques following challenge. Alongside possible VAED from a formalin-inactivated vaccine, Wang et al. found that human antibodies against a particular epitope, S597-603, on the spike protein could enhance infection of Vero E6 cells (derived from kidney cells extracted from African green monkeys), which lack FcγRs (86). Combining these findings with the pathology they observed in macaques, Wang et al. suggested possible ADE owing to the previously described mechanism of dual-specific antibody binding due to molecular mimicry between virion and host cell components (21) (Figure 2B), implying that the host cell component in question is present both in macaques and Vero E6 cells. While this phenomenon has been observed with dengue virus, it could also apply to SARS-CoV-1 and other coronaviruses. When a peptide (S597-625) containing this epitope was used to vaccinate macaques, it was found to cause more severe lung histopathology compared to other peptides and it was also not protective (86). A monoclonal antibody against this epitope, at a dose of 1.8 mg/kg, caused some pathology in the lungs following challenge, as well as macrophage infiltration and higher numbers of cells infected with SARS-CoV-1 (86). Antibodies against this epitope are likely non-neutralising because this region is not within the RBD. However, in this paper, the gross pathology is difficult to interpret, given the poor quality of the images showing abundant post-mortem artefacts. The H&E staining is also difficult to interpret as the authors focus on small areas with high magnification. Ideally, Wang et al. would have used virus detection and a robust histopathology scoring system or digital image analysis to interpret their findings. The study lacks an objective quantitative analysis for pathology observed by IHC, which would be useful and less prone to bias. A SARS-CoV-1 reinfection study of macaques could determine whether vaccine-induced antibodies are to blame for enhanced pathology following challenge or if neutralising antibodies resulting from natural infection also cause pathology in the same way. This could lay to rest whether the mechanism of enhanced pathologies seen in SARS-CoV-1 preclinical studies is due to poor quality vaccine preparations/design or whether it is the result of a viral AED mechanism. Further studies are required to determine the balance between neutralising and non-neutralising (or potentially enhancing) antibodies in re-infected vs vaccinated challenge models and how these antibody ratios relate to observed pathology.
While mouse models are not ideal for studying the pathology of coronaviruses, some murine studies have demonstrated signs of protection or enhanced pathology with SARS-CoV-1 vaccines. A SARS-CoV-1 vaccine that was double-inactivated using both formalin and ultraviolet (UV) light inactivation demonstrated signs of enhanced pathology associated with eosinophilic infiltration and provided poor protection against heterologous challenge in aged mice (87). Heterologous challenge involving infection with a lethal zoonotic virus (icHC/SZ/61/03-S) led to worsened pathology in young vaccinated mice compared to young unvaccinated mice. While immune infiltrates were noted, the observed pathology did not correlate with weight loss or mortality by day 4 post challenge. The authors hypothesise that anti-nucleocapsid responses contribute to the observed pathology. An alum-adjuvanted version of this vaccine protected young mice from both homologous and heterologous challenge but demonstrated eosinophil-associated pathology following heterologous challenge (87). The eosinophil-associated pathology in response to a UV-inactivated vaccine in mice can be overcome through the use of TLR agonists, which also maintain the protection observed in this vaccine (88).
Another study found pulmonary eosinophilic infiltrates in a variety of vaccines in Balb/c mice, however these mice were also ultimately protected against challenge (89). Tseng et al. demonstrated that Th2 immunopathology is induced by a variety of vaccines and hypothesised that non-neutralising antibody responses against nucleocapsid protein might explain much of the observed immunopathology, as shown previously (90, 91). This is because although the recombinant spike protein vaccine (SV) induced immunopathology, it was to a lesser extent than that observed in the other vaccines evaluated (89). Tseng et al. also point to a paper examining a spike-expressing vector which did not show immunopathology in mice and a nucleocapsid-expressing vector that did (91). The other vaccines included evaluated by Tseng et al. included a virus-like particle (VLP) vaccine, a double- inactivated vaccine (DIV), which was inactivated with both formalin and UV radiation and a whole virus vaccine inactivated with propiolactone and formulated with alum (BPV). The histopathology and IHC staining demonstrate eosinophilic infiltrates and pulmonary pathology in Balb/c mice given the SV, DIV or BPV vaccines two days following challenge. However, Tseng et al. do not quantify the pathology. Furthermore, the authors have not included an image of a bronchus, bronchiole and a large blood vessel for the H+E stain control, making it difficult to compare the histopathology between the control and challenged mice. In contrast to the study by Tseng et al. in which a spike protein vaccine demonstrated signs of immunopathology (albeit less than the other vaccines evaluated) (89), another subunit vaccine based on the RBD of the spike has demonstrated protection and no vaccine-associated pathology following challenge in mice (92).
A potential reason for observed ADE and AED in formalin-inactivated vaccines is that formaldehyde treatment may alter the structure of the spike protein through cross-linking such that the antibodies generated have reduced neutralising activity against wild-type SARS-CoV-1. The potential for this mechanism was demonstrated with a formalin-inactivated virus studied by our group, which then generated suboptimal vaccine responses, which we hypothesised was due to reduced exposure of the spike RBD following formaldehyde treatment (19). While this has not yet been shown to be true of the SARS-CoV-1 spike, a similar mechanism may be at play given the homology between the viruses. The impact of formalin inactivation will be described further in the SARS-CoV-2 section. Other forms of neutralisation may not alter protein conformations to the same extent. For instance, a SARS-CoV-1 vaccine containing beta-propiolactone-inactivated SARS-CoV-1 has demonstrated no AED-associated pathology in rhesus macaques, despite low levels of neutralising antibodies (93).
VAED in formalin-inactivated vaccines may also be caused by contamination of the vaccine preparation, with culture medium or cellular debris for example. Shaw and colleagues found that a formalin-inactivated RSV vaccine caused respiratory disease in the cotton rat model, which is widely used for investigation of RSV vaccine VAED following challenge, even when the RSV components were removed from the vaccine (94). Thus, it was found that the pathology was mediated by T cell responses to non-viral antigens, which included vaccine contaminants like bovine serum albumin (BSA). We hypothesised that we did not observe enhanced pathology in our NHP VAED study of a formalin-inactivated SARS-CoV-2 because of successful removal of such contaminants (19).
As previously mentioned, animal models have their obvious limitations and so the most convincing evidence for a lack of VAED in coronavirus vaccination would come from SARS-CoV-1 and MERS-CoV vaccine efficacy clinical trials. Phase 1 clinical trials found these vaccines to be safe and immunogenic and there were no reports of enhanced disease in vaccinated individuals (95–97), although vaccine effectiveness could only be assessed in phase III efficacy trials, which did not go ahead due to low infection rates as the epidemics were controlled. In an analysis of FcγRIIa polymorphisms on SARS-CoV-1 infection outcome, it was found that patients with a high-affinity FcγRIIa allele (H131) were more likely to survive and had reduced severity of disease (108). The low-affinity allele (R131) on the other hand was prevalent in higher proportions in patients who were hospitalised or who had died following infection. If AED contributed to pathology in natural infection with SARS-CoV-1, one would expect the opposite findings because in AED, engagement of activatory Fc receptors, such as FcγRIIa, is pathogenic. The findings instead indicate that FcγRIIa in SARS-CoV-1 may instead aid viral clearance through ADCP.
Vaccine-Associated Enhanced Disease in the Context of SARS-CoV-2
Considering the aforementioned suggestions of AED in other coronaviruses discussed above, there has been much discussion around the potential dangers of AED in SARS-CoV-2 (109–112). As of yet there is no evidence of FcγR-mediated ADE in SARS-CoV-2 infection and unlike MERS, which demonstrated some evidence of productive infection of macrophages, SARS-CoV-2 does not productively infect macrophages (113, 114). While reinfection data in animal models and humans is lacking for other coronaviruses, infection of macaques with SARS-CoV-2 has been found to reduce disease severity upon reinfection, with reduced viral loads and high neutralising antibody titres noted (115, 116). There have also been no indications that the severity of reinfections with different variants can be exacerbated by antibody responses, as occurs with infections with different serotypes of dengue virus. However, a different mechanism of ADE has been proposed for SARS-CoV-2, involving antibodies against particular epitopes in the N-terminal domain (NTD) of the S1 component of the spike protein (98). These ‘enhancing antibodies’ have been shown to open the RBD, increasing its affinity for ACE2 and thereby enhancing infectivity. Each individual enhancing antibody is thought to bind to a particular site of the NTD on two different spike proteins at once to facilitate the opening of the RBD. Interestingly, these ‘enhancing antibodies’ have been found in both uninfected and infected individuals, with severe COVID-19 patients having a higher enhancing:neutralising antibody ratio (98). The authors also found that neutralising anti-RBD monoclonal antibodies could reduce the fold-change in ACE-2 binding conferred by enhancing antibodies, particularly at higher concentrations. However, this enhancing activity was not completely eliminated, up to a neutralising antibody concentration of 1ug/ml. It is important to note that this work was carried out in vitro and specifically in an ACE2-transfected HEK293T cell line (98), which lacks the SARS-CoV-2 co-receptor TMPRSS2, so does not mimic the prominent viral entry mechanism seen in natural infection (117). A recent study that used in vitro enhancing antibodies in vivo in mice and macaques found no evidence of enhancement of disease progression and instead found these antibodies to be protective across dose levels (99). The study found three of the 46 monkeys infused with enhancing bodies to have higher inflammation scores than controls and one monkey which had higher inflammatory cytokine levels in the bronchoalveolar lavage. This indicates that while enhancing antibodies may enhance infection in vitro, they may also sometimes cause an increase in lung inflammation, perhaps through an FcγR- and ITAM-mediated AED mechanism. In a vaccine context, these enhancing antibodies are not likely to be produced in large enough quantities to overcome the protection conferred by neutralising antibodies and T cells, which is consistent with the protection rather than enhancement observed in vaccine studies. Some other potential concerns of AED in COVID-19 come from studies of antibody fucosylation. The spike-specific and the RBD-specific antibodies of COVID-19 patients with more severe disease have been found to have lower fucosylation of their Fc domains (118), which is associated with increased binding to the activatory Fc receptor FcγRIIIa, hypothesised to cause immunopathology. However, in vitro, antibodies from COVID-19 patient convalescent plasma have been shown not to contribute to aberrant cytokine production by macrophages upon binding to FcγRIIa or FcγRIIIa (119). There is also no convincing evidence of AED contributing to pathology in vivo in SARS-CoV-2 vaccine studies, in contrast to the conflicting findings and misinterpretations of VAED in preclinical vaccine studies for SARS-CoV-1 and MERS-CoV vaccines.
As previously mentioned, we have demonstrated that formaldehyde treatment of the SARS-CoV-2 spike leads to cross-linking in this protein (19). We proposed that formaldehyde fixation secured the conformation of trimers in the spike protein such that approximately half of the trimers were in a ‘RBD down’ conformation, while the other half of the trimers were in the ‘RBD up’ conformation. The ‘RBD up’ conformation is also known as the prefusion conformation. By fixing the conformation with formaldehyde, the trimers are no longer free to sample both conformations, meaning the neutralising epitopes of the RBD will likely have lower immunogenicity due to their exposure being reduced in half of the trimers (19). As a consequence, antibody titres developed against SARS-CoV-2 in a formaldehyde-fixed vaccine may be sub-neutralising when challenged with live virus. While this mechanism can lead to AED in theory (Figure 3), we did not observe signs of VAED in rhesus macaques and only transient signs of pathology in ferrets, despite using a formaldehyde-inactivated vaccine formulated with Alhydrogel, designed to elicit VAED (19). Similarly, a study using a formaldehyde-inactivated vaccine in a Syrian hamster model found protective effects and no enhanced pathology after viral challenge (20). This is despite the use of a regimen designed to enhance disease and the Syrian hamster is a more severe disease model than the ferret or macaque (14, 15). Some protective effects were noted even if the vaccine was administered just before or shortly after infection, when antibody levels are more likely to be found at sub-neutralising concentrations and are less likely to have high affinities conferred by affinity maturation (20).
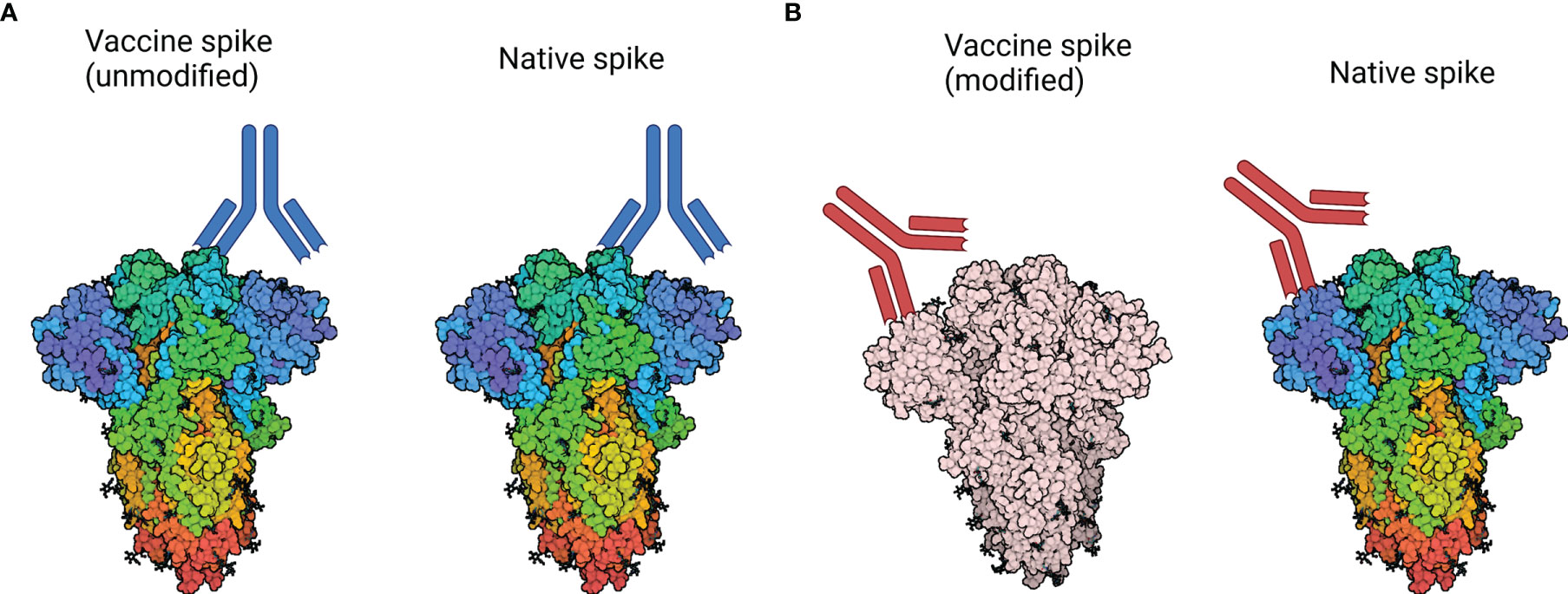
Figure 3 How modification of the spike protein could impact neutralising activity. (A) Native spike: Antibodies are generated against a neutralising epitope on the vaccine-derived spike protein, which closely represents the neutralising epitope found on circulating virus. As a result, neutralising activity against the vaccine-derived spike corresponds to neutralisation of circulating virus. (B) Modified spike: Antibody responses may be generated against a modified epitope, which no longer represents a neutralising epitope on circulating virus. As a result, antibodies produced in response to native spike may be sub-neutralising or non-neutralising and may contribute to VAED. Spike protein structure accessed through Protein Data Bank, PBD entry 6VXX (120). Created with BioRender.com.
The spike protein epitope responsible for observed AED in a formaldehyde-inactivated SARS-CoV-1 preclinical vaccine study, mentioned previously (86), is also present in SARS-CoV-2. The amino acid sequence is LYQDVNC and is found at S597-603 in the SARS-CoV-1 spike protein and S611-617 in the SARS-CoV-2 spike protein. While formaldehyde inactivation may explain VAED to an extent, the enhancing effect of high doses of a monoclonal antibody against this peptide in SARS-CoV-1 (86) warrants investigation for similar phenomena in SARS-CoV-2. However, VAED has not been commonly observed in animal models and there are no reports in human trials of SARS-CoV-2 vaccines to date. While subunit vaccines containing the spike protein are immunogenic, subunit vaccines that use only the RBD of the spike are thought to require adjuvants in order to stimulate protective immunity (121, 122).
Beta-propiolactone inactivation of SARS-CoV-2 has been reported to cause viral aggregation at high concentrations, which can lead to a loss of antigenic potential, owing either to this aggregation or chemical modification of viral amino acids (123). However, the concentration (1:1000) at which this loss of antigenic potential was seen, was found to be much greater than the concentration of beta-propiolactone that is sufficient for inactivation. Beta-propiolactone-inactivated SARS-CoV-2 vaccines induce protective neutralising antibody responses without AED in macaques (124). Another preclinical study in macaques found such a vaccine to be safe and effective, with no AED despite low neutralising antibody titres in low-dose groups (125). A vaccine based on beta-propiolactone inactivation has been shown to be safe and effective (126) and is now in widespread use.
While an MVA vaccine for SARS-CoV-1 demonstrated possible AED in macaques (80), an MVA vaccine for SARS-CoV-2 protected against immunopathology following challenge and induces potent antibody and CD8 T cell responses in macaques (127). The MVA vaccine used in the latter study, MVA/S, expressed a membrane-anchored full-length spike protein and contained two mutations that ensure the spike is always in a prefusion (RBD up) conformation, meaning the RBD would always be completely exposed. This likely contributed to its potent immunogenicity. It is possible that this MVA/S vaccine approach generated antibodies with stronger neutralising capabilities and this could partially explain why VAED was observed in the SARS-CoV-1 MVA vaccine and not in the SARS-CoV-2 vaccine.
A DNA vaccine for SARS-CoV-2 has been shown to induce protective immune responses in rhesus macaques (128). Pathological analysis following challenge demonstrated protection in the majority of the animals in the one- and two-dose groups. However, one of the animals in the one-dose group had a much higher lung histopathology score compared to the others in the group and the controls. This animal did not generate an antibody response to the vaccine, as the antibody levels pre-challenge were comparable to those in the unvaccinated cohort (128). Therefore, it may be that the pathology associated with this animal is linked to a lack of protection rather than VAED, which would also explain the lack of eosinophilic infiltrates usually associated with VAED. However, this case warrants further investigation.
Discussion and Conclusions
A variety of techniques can be deployed to investigate VAED and elucidate potential mechanisms of VAED. These include traditional histopathological staining, such as H+E staining, as well as immunohistochemistry (IHC), in situ hybridisation (e.g. RNAscope), qPCR as well as spatial and systemic immunological analyses. There are several potential biomarkers for VAED, although the relevance of these biomarkers will vary widely depending on disease kinetics, whether the pathogen can productively infect monocytes and what immune responses are protective vs pathogenic in a particular case. These potential biomarkers following challenge could include eosinophil infiltration, weak antibody neutralisation, inflammatory monocyte infiltration, Th2-associated cytokines and immune complexes. While some of these biomarkers can be measured in vitro, in vivo evidence is required in order to demonstrate VAED. While there is no specific immunological assay for AED, assays can be developed to investigate ADE. The importance of background protection by antibodies means that an assay that measures the ratio of neutralising antibodies to enhancing antibodies, such as that developed for dengue virus, is particularly useful (23). However, observing enhanced uptake and spread of virus in vitro largely excludes the contributions of Fc-mediated immune functions that could contribute to elimination of infection in vivo, or could exacerbate pathology through induction of excessive inflammation (9). As described by Bournazos et al, concerns of ADE in Human Immunodeficiency Virus (HIV) and other viruses such as influenza and Ebola virus stemmed from in vitro studies but even non-neutralising antibodies with strong FcγR interactions have been shown not to mediate pathology in vivo and have instead been shown to be protective (9).
The use of animal models and the understanding of what aspects of these are similar or differ to humans is essential for the study of VAED (14, 15). As histopathological analysis is essential for evaluating VAED concerns, it is important that pathology is quantified so that it can be objectively assessed and compared between studies. The use of a histopathological scoring system, such as one we have described previously (16), could help to minimise overinterpretation of pathological findings and lead to the creation of standard thresholds for pathology as it relates to enhanced disease. As we have demonstrated, the timing of the post-challenge cull can influence the pathology observed (19). Including groups for short-term pathology vs long-term pathology could be useful for determining how severe clinical consequences are likely to be at particular timepoints if the pathology was replicated in humans. In order to do this, it is important to dissect the importance of transient pathology that is later resolved so that decisions can be made based on the acceptable level of transient pathology if long-term protection against severe disease is achieved. In many cases of VAED observed at a particular timepoint, protection against severe disease has also been noted. It will also be useful to compare vaccinated post-challenge models of pathology with challenge and re-challenge models. The lack of re-challenge studies in SARS-CoV-1 and MERS-CoV makes it more difficult to determine the mechanism and consequences of potential VAED. For instance, if similar pathology is seen in both possible VAED and in re-infection models, and this pathology is greater than that seen in unvaccinated animals, the immune system is likely to be enhancing infection. However, if vaccinated animals have transient pathology upon challenge, but re-infected animals have more severe pathology that is long-lasting, the vaccine is having a protective effect and preventing disease enhancement seen in natural reinfection. With some viruses, such as dengue virus, pathology is often more pronounced during re-infection than during initial infection (37), so reinfection models would help to answer the question of whether or not vaccines that enhance disease do so in a similar way to the natural course of the immune response. If not, then a component of the vaccine preparation may be to blame.
T cell contributions to VAED are generally thought to occur through Th2 cells mediating inflammation and pulmonary eosinophil infiltration. Th1 responses to vaccinations are preferable for dealing with viral infections without inducing pathology. In mouse studies, it is known that C57BL/6 mice are predisposed towards Th1-biased immune responses, while BALB/c mice are predisposed towards Th2-biased responses. Therefore, in cases of possible VAED in murine models, authors should highlight how their choice of model may have impacted any observed pathology.
For SARS-CoV-2 and other pathogenic coronaviruses, there is no vaccine preparation that is used as a control for inducing VAED. We previously attempted to create such a control for SARS-CoV-2 with a formalin-inactivated vaccine that included alhydrogel, with the goal of inducing non-neutralising antibodies and skewing T cells towards Th2 phenotypes (19). Others have also attempted to induce VAED by administering a formalin-inactivated vaccine shortly before or after challenge, when levels of circulating antibodies are likely to be quite low (20). Other ways to create a model of VAED may be to use monoclonal or polyclonal recombinant non-neutralising antibodies, with species-matched Fc regions, which could give insights into ADE and AED specifically. Recombinant antibodies against particular ‘enhancing epitopes’ such as those potentially found in SARS-CoV-1 (86) could also be used to create such a model. Ideally, a model of VAED should also take differences between homologous and heterologous challenge into account. For some viruses, such as dengue virus, differences between serotypes mean that antibodies that were neutralising against one serotype can be non-neutralising against another, mediating enhanced disease. In the context of SARS-CoV-2, this could be evaluated for different circulating variants, although the differences between virus variants are more subtle than those between virus serotypes. For in vitro assays examining antibody binding to viral antigens, it is important to keep the viral antigen in its native state, particularly as specific epitopes elicit neutralising antibodies whereas others may be able to confer disease enhancement. Direct coating of antigens onto ELISA plates for example, can alter the structure of antigens, as can formalin inactivation (19, 129). In contrast, capture ELISA platforms can ensure that an antigen is not modified and so responses to all epitopes can be assessed.
Earlier in the SARS-CoV-2 pandemic, another fear around VAED concerned cross-reactivity between other coronaviruses and SARS-CoV-2. One hypothesis suggested that COVID-19 may be more severe in adults than in children because adults had been exposed to a wider array of coronaviruses throughout their lifetime and as a result were producing cross-reactive but non-neutralising antibodies against SARS-CoV-2, which could cause ADE (130). Similarly, the authors also suggested imprinting as a potential reason as to why IgG responses appeared to develop much faster in many patients than they do during the course of other viral infections (130). Imprinting (also known as original antigenic sin or the Hoskin’s effect) occurs on first exposure to an immunodominant antigen on a virus, through infection or vaccination, and prevents the development of robust immune responses to other antigenic determinants on the same virus or its variants upon subsequent exposures/vaccinations. This phenomenon has been very well-characterised in antibody responses against influenza virus in particular, where it has been shown to be both protective and pathogenic, depending on how distantly related an encountered virus is from that which has been ‘imprinted’ (131). Several hypotheses have been proposed to explain its mechanism (131). Imprinting in T cell responses is however a much more controversial area. In dengue virus, it is unknown whether cross-reactive T cells are more protective or pathogenic and this may depend on the HLA alleles of individuals (132). Imprinting and cross-reactive pathogenic T cell responses were first proposed for lymphocytic choriomeningitis virus (133). The idea of imprinting and T cells has come under scrutiny, as some argue against its existence from a mechanistic standpoint, stating that T cell receptors (TCRs) with poor affinity for a variant will not be able to outcompete TCRs with a higher affinity (134).
SARS-CoV-2 vaccines have not been associated with significant VAED in preclinical studies or clinical use. It is unknown why this is the case, considering VAED was associated with other types of human coronavirus in preclinical studies. We suggest that some of the reasons we have highlighted, from formaldehyde fixation & cellular debris contaminants in formulations of experimental vaccines to over-interpretation of pathology, may explain much of the VAED in the literature. It may be that vaccines against SARS-CoV-2, even in preclinical studies, were better at avoiding VAED because of improvements in vaccine preparation. Experiments that demonstrated VAED for SARS-CoV-1 preclinical vaccines should be repeated so that these concerns can be re-evaluated.
There is potential for future variants/serotypes of viruses, including SARS-CoV-2, to provoke sub-neutralising antibodies in individuals who have encountered similar (but poorly cross-reactive) epitopes. This was the case for the SARS-CoV-2 variant known as Omicron, which demonstrates a 41-fold drop in neutralising antibody titres in patients who have received two doses of the mRNA vaccine BNT162b2 (135). Despite this drop in neutralisation, no enhancement of disease has been reported. Infection with other variants of SARS-CoV-2 have also been shown to impact antibody binding to SARS-CoV-2 and its variants post-vaccination through imprinting, but no disease enhancement has been reported in these cases either (136). Seasonal coronaviruses also appear to provide a level of back-boosting or cross-protection in some individuals (137, 138). Cross-reactivity has been observed between SARS-CoV-1 and SARS-CoV-2, which results in improved vaccine-induced immune responses by provoking the generation of broadly-neutralising antibodies against a wide variety of coronaviruses (139). However, imprinting may also have negative consequences for future vaccines based on spike proteins from SARS-CoV-2 variants, due to back-boosting of conserved but non-neutralising epitopes (140). Waning antibody levels, which are a cause of ADE in dengue virus infection, have been observed 6 months following vaccination (141, 142), however a level of protection is still being observed to date in vaccinated people and there have been no documented cases of ADE owing to this or any other cause in SARS-CoV-2. Memory T cells induced in response to vaccination have been shown to have highly heterogenous antigen-specific responses, which are thought to contribute to long-term protection against severe disease (143). Even with robust antibody escape as seen in Omicron, T cell responses are likely to be sustained (144).
Overall, genetic vaccine platforms (mRNA and viral vectors) are in theory be less likely to induce AED than inactivated vaccines or natural infection. This is because genetic platforms ensure responses are generated against unmodified neutralising epitopes, encoded by the platform, while inactivated whole-virus vaccines have a wider variety of epitopes for the immune system to generate responses against. Some of these epitopes will be non-neutralising and could therefore contribute to AED. Formalin-inactivated vaccines also pose the risk of altering the structure of antigens, as we have shown for the SARS-CoV-2 spike, however we demonstrated that this structural change does not lead to VAED in NHPs (19). The concern of formalin inactivation stimulating Th2 pathology through carbonyl group formation, which was demonstrated in mouse models of RSV (34), was also not observed in our study. Genetic vaccine platforms also have the advantage of using host-generated glycosylation when the protein of interest is synthesised, which mimics the case in natural infection. Protein-based vaccines which use non-human cell lines to produce the protein of interest could generate viral proteins with glycosylation patterns that differ from that produced during natural infection. Grant et al. found that glycans shield approximately 40% of the SARS-CoV-2 spike protein surface, with implications for human leucocyte antigen (HLA) complex binding and antigen-specific immune responses as a result (145). Many influenza vaccine antigens are produced in fertilised chicken eggs, and a glycosylation site in H3N2 influenza has been found to alter antibody binding such that weak neutralising responses to this site were elicited by both ferrets and humans (64). As differential glycosylation patterns between vaccine antigens and wild-type antigens can induce weakly neutralising antibodies in response to particular epitopes, this is another theoretical concern for VAED, provided the pathogen in question can facilitate ADE or AED.
VAED is a concern that should be carefully evaluated for new vaccines. While the term VAED is usually associated with viral infections, the phenomenon also has potential to occur with other microbial infections, which show potential for ADE and/or AED (12, 146). While VAED is a rare phenomenon, it should be studied in greater detail in preclinical models considering its clinical consequences, its potential to stall vaccine development and its ability to undermine public confidence in safe & effective licenced vaccines. We have outlined the potential mechanisms of VAED and described improved methods that can be employed so that potential problems can be identified at a pre-clinical stage and potentially false VAED signals, resulting in delays in vaccine development, can be avoided.
Author Contributions
CG wrote the manuscript and designed the tables and figures. TT, FJS, QS, AG, and MC provided guidance and revised the manuscript. FJS, contributed to the discussion of histopathology. All authors contributed to the article and approved the submitted version.
Funding
TT and MC were funded by the US Food and Drug Administration 75F40120C00085: Characterization of severe coronavirus infection in humans and model systems for MCM development and evaluation. TT and MC were also funded by a grant from the Coalition for Epidemic Preparedness Innovations (CEPI): Applying systems immunology to NHP COVID-19 vaccine challenge study data sets to define Correlates of Protection to SARS-CoV-2.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Sridhar S, Luedtke A, Langevin E, Zhu M, Bonaparte M, Machabert T, et al. Effect of Dengue Serostatus on Dengue Vaccine Safety and Efficacy. N Engl J Med (2018) 379(4):327–40. doi: 10.1056/NEJMoa1800820
2. Kim HWHA, Canchola JG, Brandt CD, Pyles G, Chanock RM, Jensen K, et al. Respiratory Syncytial Virus Disease in Infants Despite Prior Administration of Antigenic Inactivated Vaccine. Am J Epidemiol (1969) 89(4):422–34. doi: 10.1093/oxfordjournals.aje.a120955
3. Fulginiti VA, Eller JJ, Downie AW, Kempe CH. Altered Reactivity to Measles Virus: Atypical Measles in Children Previously Immunized With Inactivated Measles Virus Vaccines. JAMA (1967) 202(12):1075–80. doi: 10.1001/jama.1967.03130250057008
4. Coronel D, García-Rivera EJ, Rivera DM, Arredondo-García JL, Dietze R, Perroud AP, et al. Immune Response Persistence and Safety of a Booster Dose of the Tetravalent Dengue Vaccine in Adolescents and Adults Who Previously Completed the 3-Dose Schedule 4-5 Years Earlier in Latin America: A Randomized Placebo-Controlled Trial. Pediatr Infect Dis J (2020) 39(10):961–8. doi: 10.1097/INF.0000000000002830
5. Munoz FM, Cramer JP, Dekker CL, Dudley MZ, Graham BS, Gurwith M, et al. Vaccine-Associated Enhanced Disease: Case Definition and Guidelines for Data Collection, Analysis, and Presentation of Immunization Safety Data. Vaccine (2021) 39(22):3053–66. doi: 10.1016/j.vaccine.2021.01.055
6. Shukla R, Ramasamy V, Shanmugam RK, Ahuja R, Khanna N. Antibody-Dependent Enhancement: A Challenge for Developing a Safe Dengue Vaccine. Front Cell Infection Microbiol (2020) 10:1–12. doi: 10.3389/fcimb.2020.572681
7. Katzelnick LC, Gresh L, Halloran ME, Mercado JC, Kuan G, Gordon A, et al. Antibody-Dependent Enhancement of Severe Dengue Disease in Humans. Science (2017) 358(6365):929–32. doi: 10.1126/science.aan6836
8. ben Mkaddem S, Benhamou M, Monteiro RC. Understanding Fc Receptor Involvement in Inflammatory Diseases: From Mechanisms to New Therapeutic Tools. Front Immunol (2019) 10:811. doi: 10.3389/fimmu.2019.00811
9. Bournazos S, Gupta A, Ravetch JV. The Role of IgG Fc Receptors in Antibody-Dependent Enhancement. Nat Rev Immunol (2020) 20(10):633–43. doi: 10.1038/s41577-020-00410-0
10. Li T, DiLillo DJ, Bournazos S, Giddens JP, Ravetch JV, Wang LX. Modulating IgG Effector Function by Fc Glycan Engineering. Proc Natl Acad Sci USA (2017) 114(13):3485–90. doi: 10.1073/pnas.1702173114
11. Wang XY, Wang B, Wen YM. From Therapeutic Antibodies to Immune Complex Vaccines. NPJ Vaccines (2019) 4(1):1–8. doi: 10.1038/s41541-018-0095-z
12. Halstead SB, Mahalingam S, Marovich MA, Ubol S, Mosser DM. Intrinsic Antibody-Dependent Enhancement of Microbial Infection in Macrophages: Disease Regulation by Immune Complexes. Lancet Infect Dis (2010) 10(10):712–22. doi: 10.1016/S1473-3099(10)70166-3
13. Graham BBS. Rapid COVID-19 Vaccine Development. Sci (2020) 368(6494):945–6. doi: 10.1126/science.abb8923
14. Muñoz-Fontela C, Widerspick L, Albrecht RA, Beer M, Carroll MW, de Wit E, et al. Advances and Gaps in SARS-CoV-2 Infection Models. PloS Pathog (2022) 18(1):e1010161. doi: 10.1371/journal.ppat.1010161
15. Muñoz-fontela C, Dowling WE, Funnell SGP, Gsell P, Carroll MW, Cavaleri M, et al. Animal Models for COVID-19. Nature (2020) 586:509–15. doi: 10.1038/s41586-020-2787-6
16. Salguero FJ, White AD, Slack GS, Fotheringham SA, Bewley KR, Gooch KE, et al. Comparison of Rhesus and Cynomolgus Macaques as an Infection Model for COVID-19. Nat Commun (2021) 12(1):1–14. doi: 10.1038/s41467-021-21389-9
17. Ziegler CGK, Allon SJ, Nyquist SK, Mbano IM, Miao VN, Tzouanas CN, et al. SARS-CoV-2 Receptor ACE2 Is an Interferon-Stimulated Gene in Human Airway Epithelial Cells and Is Detected in Specific Cell Subsets Across Tissues. Cell (2020) 181(5):1016–35.e19. doi: 10.1016/j.cell.2020.04.035
18. Bournazos S. IgG Fc Receptors: Evolutionary Considerations - Fc Mediated Activity of Antibodies: Structural and Functional Diversity. In: Ravetch JV, Nimmerjahn F, editors. Fc Mediated Activity of Antibodies. Cham: Springer International Publishing (2019). p. 1–11. doi: 10.1007/82_2019_149
19. Bewley KR, Gooch K, Thomas KM, Longet S, Wiblin N, Hunter L, et al. Immunological and Pathological Outcomes of SARS-CoV-2 Challenge Following Formalin-Inactivated Vaccine in Ferrets and Rhesus Macaques. Sci Adv (2021) 7:7996. doi: 10.1126/sciadv.abg7996
20. Li C, Chen Y-X, Liu F-F, Lee AC-Y, Zhao Y, Ye Z-H, et al. Absence of Vaccine-Enhanced Disease With Unexpected Positive Protection Against Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2) by Inactivated Vaccine Given Within 3 Days of Virus Challenge in Syrian Hamster Model. Clin Infect Dis (2021) 73(3):e719–34. doi: 10.1093/cid/ciab083
21. Huang K-J, Yang Y-C, Lin Y-S, Huang J-H, Liu H-S, Yeh T-M, et al. The Dual-Specific Binding of Dengue Virus and Target Cells for the Antibody-Dependent Enhancement of Dengue Virus Infection. J Immunol (2006) 176(5):2825–32. doi: 10.4049/jimmunol.176.5.2825
22. Mady BJ, Erbe DV, Kurane I, Fanger MW, Ennis FA. Antibody-Dependent Enhancement of Dengue Virus Infection Mediated by Bispecific Antibodies Against Cell Surface Molecules Other Than Fc Gamma Receptors. J Immunol (1991) 147(9):3139–44.
23. Yamanaka A, Imad HA, Phumratanaprapin W, Phadungsombat J, Konishi E, Shioda T. Antibody-Dependent Enhancement Representing In Vitro Infective Progeny Virus Titer Correlates With the Viremia Level in Dengue Patients. Sci Rep (2021) 11(1):1–11. doi: 10.1038/s41598-021-91793-0
24. Clapham HE, Quyen TH, Kien DTH, Dorigatti I, Simmons CP, Ferguson NM. Modelling Virus and Antibody Dynamics During Dengue Virus Infection Suggests a Role for Antibody in Virus Clearance. PloS Comput Biol (2016) 12(5):e1004951. doi: 10.1371/journal.pcbi.1004951
25. Bournazos S, Vo HTM, Duong V, Auerswald H, Ly S, Sakuntabhai A, et al. Antibody Fucosylation Predicts Disease Severity in Secondary Dengue Infection. Science (2021) 372(6546):1102–5. doi: 10.1126/science.abc7303
26. Acosta PL, Caballero MT, Polack FP. Brief History and Characterization of Enhanced Respiratory Syncytial Virus Disease. Clin Vaccine Immunol (2016) 23(3):189–95. doi: 10.1128/CVI.00609-15
27. Ruckwardt TJ, Morabito KM, Graham BS. Immunological Lessons From Respiratory Syncytial Virus Vaccine Development. Immun (2019) 51(3):429–42. doi: 10.1016/j.immuni.2019.08.007
28. Tang YW, Graham BS. Anti-IL-4 Treatment at Immunization Modulates Cytokine Expression, Reduces Illness, and Increases Cytotoxic T Lymphocyte Activity in Mice Challenged With Respiratory Syncytial Virus. J Clin Invest (1994) 94(5):1953–8. doi: 10.1172/JCI117546
29. Tebbey PW, Hagen M, Hancock GE. Atypical Pulmonary Eosinophilia Is Mediated by a Specific Amino Acid Sequence of the Attachment (G) Protein of Respiratory Syncytial Virus. J Exp Med (1998) 188(10):1967–72. doi: 10.1084/jem.188.10.1967
30. Knudson CJ, Hartwig SM, Meyerholz DK, Varga SM. RSV Vaccine-Enhanced Disease Is Orchestrated by the Combined Actions of Distinct CD4 T Cell Subsets. PloS Pathog (2015) 11(3):e1004757. doi: 10.1371/journal.ppat.1004757
31. Polack FP, Teng MN, Collins PL, Prince GA, Exner M, Regele H, et al. A Role for Immune Complexes in Enhanced Respiratory Syncytial Virus Disease. J Exp Med (2002) 196(6):859–65. doi: 10.1084/jem.20020781
32. Delgado MF, Coviello S, Monsalvo AC, Melendi GA, Hernandez JZ, Batalle JP, et al. Lack of Antibody Affinity Maturation Due to Poor Toll-Like Receptor Stimulation Leads to Enhanced Respiratory Syncytial Virus Disease. Nat Med (2009) 15(1):34–41. doi: 10.1038/nm.1894
33. Brewer JM, Conacher M, Hunter CA, Mohrs M, Brombacher F, Alexander J. Aluminium Hydroxide Adjuvant Initiates Strong Antigen-Specific Th2 Responses in the Absence of IL-4- or IL-13-Mediated Signaling. J Immunol (Baltimore Md: 1950) (1999) 163(12):6448–54.
34. Moghaddam A, Olszewska W, Wang B, Tregoning JS, Helson R, Sattentau QJ, et al. A Potential Molecular Mechanism for Hypersensitivity Caused by Formalin-Inactivated Vaccines. Nat Med (2006) 12(8):905–7. doi: 10.1038/nm1456
35. Anderson CF, Mosser DM. Cutting Edge: Biasing Immune Responses by Directing Antigen to Macrophage Fcγ Receptors. J Immunol (2002) 168(8):3697–701. doi: 10.4049/jimmunol.168.8.3697
36. Graham BS. Vaccines Against Respiratory Syncytial Virus: The Time has Finally Come. Vaccine (2016) 34(30):3535–41. doi: 10.1016/j.vaccine.2016.04.083
37. st. John AL, Rathore APS. Adaptive Immune Responses to Primary and Secondary Dengue Virus Infections. Nat Rev Immunol (2019) 19(4):218–30. doi: 10.1038/s41577-019-0123-x
38. Ramadhany R, Hirai I, Sasaki T, Ono KI, Ramasoota P, Ikuta K, et al. Antibody With an Engineered Fc Region as a Therapeutic Agent Against Dengue Virus Infection. Antiviral Res (2015) 124:61–8. doi: 10.1016/j.antiviral.2015.10.012
39. Mehlhop E, Ansarah-Sobrinho C, Johnson S, Engle M, Fremont DH, Pierson TCC, et al. Complement Protein C1q Inhibits Antibody-Dependent Enhancement of Flavivirus Infection in an IgG Subclass-Specific Manner. Cell Host Microbe (2007) 2(6):417–26. doi: 10.1016/j.chom.2007.09.015
40. Getts DR, Terry RL, Getts MT, Müller M, Rana S, Shrestha B, et al. Ly6c+ “Inflammatory Monocytes” Are Microglial Precursors Recruited in a Pathogenic Manner in West Nile Virus Encephalitis. J Exp Med (2008) 205(10):2319–37. doi: 10.1084/jem.20080421
41. Ayala-Nunez NV, Follain G, Delalande F, Hirschler A, Partiot E, Hale GL, et al. Zika Virus Enhances Monocyte Adhesion and Transmigration Favoring Viral Dissemination to Neural Cells. Nat Commun (2019) 10(1):1–16. doi: 10.1038/s41467-019-12408-x
42. Takada A, Watanabe S, Okazaki K, Kida H, Kawaoka Y. Infectivity-Enhancing Antibodies to Ebola Virus Glycoprotein. J Virol (2001) 75(5):2324–30. doi: 10.1128/JVI.75.5.2324-2330.2001
43. Takada A, Feldmann H, Ksiazek TG, Kawaoka Y. Antibody-Dependent Enhancement of Ebola Virus Infection. J Virol (2003) 77(13):7539–44. doi: 10.1128/JVI.77.13.7539-7544.2003
44. Takada A, Ebihara H, Feldmann H, Geisbert TW, Kawaoka Y. Epitopes Required for Antibody-Dependent Enhancement of Ebola Virus Infection. J Infect Dis (2007) 196 Suppl:S347–56. doi: 10.1086/520581
45. Nakayama E, Tomabechi D, Matsuno K, Kishida N, Yoshida R, Feldmann H, et al. Antibody-Dependent Enhancement of Marburg Virus Infection. J Infect Dis (2011) 204 Suppl(Suppl 3):S978–85. doi: 10.1093/infdis/jir334
46. Kuzmina NA, Younan P, Gilchuk P, Santos RI, Andrew I, Ilinykh PA, et al. Antibody-Dependent Enhancement of Ebola Virus Infection by Human Antibodies Isolated from Survivors. Cell Rep (2019) 24(7):1802–15. doi: 10.1016/j.celrep.2018.07.035
47. Hargreaves A, Brady C, Mellors J, Tipton T, Carroll MW, Longet S. Filovirus Neutralising Antibodies: Mechanisms of Action and Therapeutic Application. Pathogens (2021) 10:1201. doi: 10.3390/pathogens10091201
48. Jahrling PB, Geisbert JB, Swearengen JR, Larsen T, Geisbert TW. Ebola Hemorrhagic Fever: Evaluation of Passive Immunotherapy in Nonhuman Primates. J Infect Dis (2007) 196 Suppl:S400–3. doi: 10.1086/520587
49. Oswald WB, Geisbert TW, Davis KJ, Geisbert JB, Sullivan NJ, Jahrling PB, et al. Neutralizing Antibody Fails to Impact the Course of Ebola Virus Infection in Monkeys. PloS Pathog (2007) 3(1):e9. doi: 10.1371/journal.ppat.0030009
50. Qiu X, Audet J, Wong G, Pillet S, Bello A, Cabral T, et al. Successful Treatment of Ebola Virus-Infected Cynomolgus Macaques With Monoclonal Antibodies. Sci Trans Med (2012) 4(138):138ra81. doi: 10.1126/scitranslmed.3003876
51. Qiu X, Wong G, Audet J, Bello A, Fernando L, Alimonti JB, et al. Reversion of Advanced Ebola Virus Disease in Nonhuman Primates With ZMapp. Nature (2014) 514(7520):47–53. doi: 10.1038/nature13777
52. Qiu X, Audet J, Lv M, He S, Wong G, Wei H, et al. Two-mAb Cocktail Protects Macaques Against the Makona Variant of Ebola Virus. Sci Trans Med (2016) 8(329):329ra33–329ra33. doi: 10.1126/scitranslmed.aad9875
53. van Griensven J, Edwards T, de Lamballerie X, Semple MG, Gallian P, Baize S, et al. Evaluation of Convalescent Plasma for Ebola Virus Disease in Guinea. N Engl J Med (2016) 374(1):33–42. doi: 10.1056/NEJMoa1511812
54. Polack FP. Atypical Measles and Enhanced Respiratory Syncytial Virus Disease (ERD) Made Simple. Pediatr Res (2007) 62(1):111–5. doi: 10.1203/PDR.0b013e3180686ce0
55. Prince GA, Curtis SJ, Yim KC, Porter DD. Vaccine-Enhanced Respiratory Syncytial Virus Disease in Cotton Rats Following Immunization With Lot 100 or a Newly Prepared Reference Vaccine. J Gen Virol (2001) 82(12):2881–8. doi: 10.1099/0022-1317-82-12-2881
56. Gershwin LJ, Schelegle ES, Gunther RA, Anderson ML, Woolums AR, Larochelle DR, et al. A Bovine Model of Vaccine Enhanced Respiratory Syncytial Virus Pathophysiology. Vaccine (1998) 16(11):1225–36. doi: 10.1016/S0264-410X(98)80123-0
57. Connors M, Kulkarni AB, Firestone CY, Holmes KL, Morse HC 3rd, Sotnikov AV, et al. Pulmonary Histopathology Induced by Respiratory Syncytial Virus (RSV) Challenge of Formalin-Inactivated RSV-Immunized BALB/c Mice Is Abrogated by Depletion of CD4+ T Cells. J Virol (1992) 66(12):7444–51. doi: 10.1128/jvi.66.12.7444-7451.1992
58. Connors M, Giese NA, Kulkarni AB, Firestone CY, Morse HC, Murphy BR. Enhanced Pulmonary Histopathology Induced by Respiratory Syncytial Virus (RSV) Challenge of Formalin-Inactivated RSV-Immunized BALB/c Mice Is Abrogated by Depletion of Interleukin-4 (IL-4) and IL-10. J Virol (1994) 68(8):5321–5. doi: 10.1128/jvi.68.8.5321-5325.1994
59. Hancock GE, Speelman DJ, Heers K, Bortell E, Smith J, Cosco C. Generation of Atypical Pulmonary Inflammatory Responses in BALB/c Mice After Immunization With the Native Attachment (G) Glycoprotein of Respiratory Syncytial Virus. J Virol (1996) 70(11):7783–91. doi: 10.1128/jvi.70.11.7783-7791.1996
60. Murphy BR, Sotnikov AV, Lawrence LA, Banks SM, Prince GA. Enhanced Pulmonary Histopathology Is Observed in Cotton Rats Immunized With Formalin-Inactivated Respiratory Syncytial Virus (RSV) or Purified F Glycoprotein and Challenged With RSV 3-6 Months After Immunization. Vaccine (1990) 8(5):497–502. doi: 10.1016/0264-410X(90)90253-I
61. Jorquera PA, Anderson L, Tripp RA. Understanding Respiratory Syncytial Virus (RSV) Vaccine Development and Aspects of Disease Pathogenesis. Expert Rev Vaccines (2016) 15(2):173–87. doi: 10.1586/14760584.2016.1115353
62. Jeyanathan M, Afkhami S, Smaill F, Miller MS, Lichty BD, Xing Z. Immunological Considerations for COVID-19 Vaccine Strategies. Nat Rev Immunol (2020) 20(10):615–32. doi: 10.1038/s41577-020-00434-6
63. Polack FP, Hoffman SJ, Crujeiras G, Griffin DE. A Role for Nonprotective Complement-Fixing Antibodies With Low Avidity for Measles Virus in Atypical Measles. Nat Med (2003) 9(9):1209–13. doi: 10.1038/nm918
64. Zost SJ, Parkhouse K, Gumina ME, Kim K, Perez SD, Wilson PC, et al. Contemporary H3N2 Influenza Viruses Have a Glycosylation Site That Alters Binding of Antibodies Elicited by Egg-Adapted Vaccine Strains. Proc Natl Acad Sci USA (2017) 114(47):12578–83. doi: 10.1073/pnas.1712377114
65. Hohdatsu T, Yamada M, Tominaga R, Makino K, Kida K, Koyama H. Antibody-Dependent Enhancement of Feline Infectious Peritonitis Virus Infection in Feline Alveolar Macrophages and Human Monocyte Cell Line U937 by Serum of Cats Experimentally or Naturally Infected With Feline Coronavirus. J Veterinary Med Sci (1998) 60(1):49–55. doi: 10.1292/jvms.60.49
66. Rottier PJM, Nakamura K, Schellen P, Volders H, Haijema BJ. Acquisition of Macrophage Tropism During the Pathogenesis of Feline Infectious Peritonitis Is Determined by Mutations in the Feline Coronavirus Spike Protein. J Virol (2005) 79(22):14122–30. doi: 10.1128/JVI.79.22.14122-14130.2005
67. Takano T, Kawakami C, Yamada S, Satoh R, Hohdatsu T. Antibody-Dependent Enhancement Occurs Upon Re-Infection With the Identical Serotype Virus in Feline Infectious Peritonitis Virus Infection. J Veterinary Med Sci (2008) 70(12):1315–21. doi: 10.1292/jvms.70.1315
68. Emmler L, Felten S, Matiasek K, Balzer HJ, Pantchev N, Leutenegger C, et al. Feline Coronavirus With and Without Spike Gene Mutations Detected by Real-Time RT-PCRs in Cats With Feline Infectious Peritonitis. J Feline Med Surg (2020) 22(8):791–9. doi: 10.1177/1098612X19886671
69. Pedersen NC. An Update on Feline Infectious Peritonitis: Virology and Immunopathogenesis. Veterinary J (2014) 201(2):123–32. doi: 10.1016/j.tvjl.2014.04.017
70. Wan Y, Shang J, Sun S, Tai W, Chen J, Geng Q, et al. Molecular Mechanism for Antibody-Dependent Enhancement of Coronavirus Entry. J Virol (2020) 94(5):1–15. doi: 10.1128/JVI.02015-19
71. Zhou J, Chu H, Li C, Wong BH, Cheng Z, Poon VK, et al. Active Replication of Middle East Respiratory Syndrome Coronavirus and Aberrant Induction of Inflammatory Cytokines and Chemokines in Human Macrophages: Implications for Pathogenesis. J Infect Dis (2014) 43:1331–42. doi: 10.1093/infdis/jit504
72. Tynell J, Westenius V, Rönkkö E, Munster VJ, Melén K, Österlund P, et al. Middle East Respiratory Syndrome Coronavirus Shows Poor Replication But Significant Induction of Antiviral Responses in Human Monocyte-Derived Macrophages and Dendritic Cells. J Gen Virol (2016) 97(2):344–55. doi: 10.1099/jgv.0.000351
73. Houser KV, Broadbent AJ, Gretebeck L, Vogel L, Lamirande EW, Sutton T, et al. Enhanced Inflammation in New Zealand White Rabbits When MERS-CoV Reinfection Occurs in the Absence of Neutralizing Antibody. PloS Pathogens (2017) 13(8):1–25. doi: 10.1371/journal.ppat.1006565
74. Agrawal AS, Tao X, Algaissi A, Garron T, Narayanan K, Peng BH, et al. Immunization With Inactivated Middle East Respiratory Syndrome Coronavirus Vaccine Leads to Lung Immunopathology on Challenge With Live Virus. Hum Vaccines Immunother (2016) 12(9):2351–6. doi: 10.1080/21645515.2016.1177688
75. Li K, Li Z, Wohlford-Lenane C, Meyerholz DK, Channappanavar R, An D, et al. Single-Dose, Intranasal Immunization With Recombinant Parainfluenza Virus 5 Expressing Middle East Respiratory Syndrome Coronavirus (MERS-CoV) Spike Protein Protects Mice From Fatal MERS-CoV Infection. mBio (2020) 11(2):e00554–20. doi: 10.1128/mBio.00554-20
76. van Doremalen N, Haddock E, Feldmann F, Meade-White K, Bushmaker T, Fischer RJ, et al. A Single Dose of ChAdOx1 MERS Provides Protective Immunity in Rhesus Macaques. Sci Adv (2020) 6(24):eaba8399. doi: 10.1126/sciadv.aba8399
77. Jaume M, Yip MS, Cheung CY, Leung HL, Li PH, Kien F, et al. Anti-Severe Acute Respiratory Syndrome Coronavirus Spike Antibodies Trigger Infection of Human Immune Cells via a pH- and Cysteine Protease-Independent Fc R Pathway. J Virol (2011) 85(20):10582–97. doi: 10.1128/JVI.00671-11
78. Yip MS, Leung NHL, Cheung CY, Li PH, Lee HHY, Daëron M, et al. Antibody-Dependent Infection of Human Macrophages by Severe Acute Respiratory Syndrome Coronavirus. Virol J (2014) 11(1):1–11. doi: 10.1186/1743-422X-11-82
79. Yasui F, Kohara M, Kitabatake M, Nishiwaki T, Fujii H, Tateno C, et al. Phagocytic Cells Contribute to the Antibody-Mediated Elimination of Pulmonary-Infected SARS Coronavirus. Virology (2014) 454–455(1):157–68. doi: 10.1016/j.virol.2014.02.005
80. Liu L, Wei Q, Lin Q, Fang J, Wang H, Kwok H, et al. Anti-Spike IgG Causes Severe Acute Lung Injury by Skewing Macrophage Responses During Acute SARS-CoV Infection. JCI Insight (2019) 4(4):e123158. doi: 10.1172/jci.insight.123158
81. Czub M, Weingartl H, Czub S, He R, Cao J. Evaluation of Modified Vaccinia Virus Ankara Based Recombinant SARS Vaccine in Ferrets. Vaccine (2005) 23(17–18):2273–9. doi: 10.1016/j.vaccine.2005.01.033
82. Li T-C, Yang T, Yoshizaki S, Ami Y, Suzaki Y, Ishii K, et al. Ferret Hepatitis E Virus Infection Induces Acute Hepatitis and Persistent Infection in Ferrets. Veterinary Microbiol (2016) 183:30–6. doi: 10.1016/j.vetmic.2015.11.014
83. Bukreyev A, Lamirande EW, Buchholz UJ, Vogel LN, Elkins WR, St Claire M, et al. Mucosal Immunisation of African Green Monkeys (Cercopithecus Aethiops) With an Attenuated Parainfluenza Virus Expressing the SARS Coronavirus Spike Protein for the Prevention of SARS. Lancet (2004) 363(9427):2122–7. doi: 10.1016/S0140-6736(04)16501-X
84. Du L, Zhao G, Lin Y, Sui H, Chan C, Ma S, et al. Intranasal Vaccination of Recombinant Adeno-Associated Virus Encoding Receptor-Binding Domain of Severe Acute Respiratory Syndrome Coronavirus (SARS-CoV) Spike Protein Induces Strong Mucosal Immune Responses and Provides Long-Term Protection Against SARS-. J Immunol (2008) 180(2):948–56. doi: 10.4049/jimmunol.180.2.948
85. Zhou J, Wang W, Zhong Q, Hou W, Yang Z, Xiao SY, et al. Immunogenicity, Safety, and Protective Efficacy of an Inactivated SARS-Associated Coronavirus Vaccine in Rhesus Monkeys. Vaccine (2005) 23(24):3202–9. doi: 10.1016/j.vaccine.2004.11.075
86. Wang Q, Zhang L, Kuwahara K, Li L, Liu Z, Li T, et al. Immunodominant SARS Coronavirus Epitopes in Humans Elicited Both Enhancing and Neutralizing Effects on Infection in Non-Human Primates. ACS Infect Dis (2016) 2(5):361–76. doi: 10.1021/acsinfecdis.6b00006
87. Bolles M, Deming D, Long K, Agnihothram S, Whitmore A, Ferris M, et al. A Double-Inactivated Severe Acute Respiratory Syndrome Coronavirus Vaccine Provides Incomplete Protection in Mice and Induces Increased Eosinophilic Proinflammatory Pulmonary Response Upon Challenge. J Virol (2011) 85(23):12201–15. doi: 10.1128/JVI.06048-11
88. Iwata-Yoshikawa N, Uda A, Suzuki T, Tsunetsugu-Yokota Y, Sato Y, Morikawa S, et al. Effects of Toll-Like Receptor Stimulation on Eosinophilic Infiltration in Lungs of BALB/c Mice Immunized With UV-Inactivated Severe Acute Respiratory Syndrome-Related Coronavirus Vaccine. J Virol (2014) 88(15):8597–614. doi: 10.1128/JVI.00983-14
89. Tseng C-T, Sbrana E, Iwata-Yoshikawa N, Newman PC, Garron T, Atmar RL, et al. Immunization With SARS Coronavirus Vaccines Leads to Pulmonary Immunopathology on Challenge With the SARS Virus. PloS One (2012) 7(4):e35421. doi: 10.1371/journal.pone.0035421
90. Yasui F, Kai C, Kitabatake M, Inoue S, Yoneda M, Yokochi S, et al. Prior Immunization With Severe Acute Respiratory Syndrome (SARS)-Associated Coronavirus (SARS-CoV) Nucleocapsid Protein Causes Severe Pneumonia in Mice Infected With SARS-CoV. J Immunol (2008) 181(9):6337–48. doi: 10.4049/jimmunol.181.9.6337
91. Deming D, Sheahan T, Heise M, Yount B, Davis N, Sims A, et al. Vaccine Efficacy in Senescent Mice Challenged With Recombinant SARS-CoV Bearing Epidemic and Zoonotic Spike Variants. PloS Med (2006) 3(12):e525. doi: 10.1371/journal.pmed.0030525
92. Du L, Zhao G, He Y, Guo Y, Zheng BJ, Jiang S, et al. Receptor-Binding Domain of SARS-CoV Spike Protein Induces Long-Term Protective Immunity in an Animal Model. Vaccine (2007) 25(15):2832–8. doi: 10.1016/j.vaccine.2006.10.031
93. Luo F, Liao FL, Wang H, Tang HB, Yang ZQ, Hou W. Evaluation of Antibody-Dependent Enhancement of SARS-CoV Infection in Rhesus Macaques Immunized With an Inactivated SARS-CoV Vaccine. Virol Sin (2018) 33(2):201–4. doi: 10.1007/s12250-018-0009-2
94. Shaw CA, Galarneau JR, Bowenkamp KE, Swanson KA, Palmer GA, Palladino G, et al. The Role of Non-Viral Antigens in the Cotton Rat Model of Respiratory Syncytial Virus Vaccine-Enhanced Disease. Vaccine (2013) 31(2):306–12. doi: 10.1016/j.vaccine.2012.11.006
95. Lin JT, Zhang JS, Su N, Xu JG, Wang N, Chen JT, et al. Safety and Immunogenicity From a Phase I Trial of Inactivated Severe Acute Respiratory Syndrome Coronavirus Vaccine. Antiviral Ther (2007) 12(7):1107–13.
96. Martin JE, Louder MK, Holman LSA, Gordon IJ, Enama ME, Larkin BD, et al. A SARS DNA Vaccine Induces Neutralizing Antibody and Cellular Immune Responses in Healthy Adults in a Phase I Clinical Trial. Vaccine (2008) 26(50):6338–43. doi: 10.1016/j.vaccine.2008.09.026
97. Folegatti PM, Bittaye M, Flaxman A, Lopez FR, Bellamy D, Kupke A, et al. Safety and Immunogenicity of a Candidate Middle East Respiratory Syndrome Coronavirus Viral-Vectored Vaccine: A Dose-Escalation, Open-Label, Non-Randomised, Uncontrolled, Phase 1 Trial. Lancet Infect Dis (2020) 20(7):816–26. doi: 10.1016/S1473-3099(20)30160-2
98. Liu Y, Soh WT, Kishikawa J, Hirose M, Nakayama EE, Li S, et al. An Infectivity-Enhancing Site on the SARS-CoV-2 Spike Protein Targeted by Antibodies. Cell (2021) 184(13):3452–66.e18. doi: 10.1016/j.cell.2021.05.032
99. Li D, Edwards RJ, Manne K, Martinez DR, Schäfer A, Alam SM, et al. In Vitro and In Vivo Functions of SARS-CoV-2 Infection-Enhancing and Neutralizing Antibodies. Cell (2021) 184(16):4203–19.e32. doi: 10.1016/j.cell.2021.06.021
100. Huang AT, Garcia-Carreras B, Hitchings MDT, Yang B, Katzelnick LC, Rattigan SM, et al. A Systematic Review of Antibody Mediated Immunity to Coronaviruses: Kinetics, Correlates of Protection, and Association With Severity. Nat Commun (2020) 11(1):1–16. doi: 10.1038/s41467-020-18450-4
101. Channappanavar R, Perlman S. Pathogenic Human Coronavirus Infections: Causes and Consequences of Cytokine Storm and Immunopathology. Semin Immunopathol (2017) 39(5):529–39. doi: 10.1007/s00281-017-0629-x
102. Voysey M, Clemens SAC, Madhi SA, Weckx LY, Folegatti PM, Aley PK, et al. Safety and Efficacy of the ChAdOx1 Ncov-19 Vaccine (AZD1222) Against SARS-CoV-2: An Interim Analysis of Four Randomised Controlled Trials in Brazil, South Africa, and the UK. Lancet (London England) (2021) 397(10269):99–111. doi: 10.1016/S0140-6736(20)32661-1
103. Bosaeed M, Balkhy HH, Almaziad S, Aljami HA, Alhatmi H, Alanazi H, et al. Safety and Immunogenicity of ChAdOx1 MERS Vaccine Candidate in Healthy Middle Eastern Adults (MERS002): An Open-Label, Non-Randomised, Dose-Escalation, Phase 1b Trial. Lancet Microbe (2022) 3(1):e11–20. doi: 10.1016/S2666-5247(21)00193-2
104. Song F, Fux R, Provacia LB, Volz A, Eickmann M, Becker S, et al. Middle East Respiratory Syndrome Coronavirus Spike Protein Delivered by Modified Vaccinia Virus Ankara Efficiently Induces Virus-Neutralizing Antibodies. J Virol (2013) 87(21):11950–4. doi: 10.1128/JVI.01672-13
105. Volz A, Kupke A, Song F, Jany S, Fux R, Shams-Eldin H, et al. Protective Efficacy of Recombinant Modified Vaccinia Virus Ankara Delivering Middle East Respiratory Syndrome Coronavirus Spike Glycoprotein. J Virol (2015) 89(16):8651–6. doi: 10.1128/JVI.00614-15
106. Haagmans BL, van den Brand JMA, Raj VS, Volz A, Wohlsein P, Smits SL, et al. An Orthopoxvirus-Based Vaccine Reduces Virus Excretion After MERS-CoV Infection in Dromedary Camels. Sci (New York NY) (2016) 351(6268):77–81. doi: 10.1126/science.aad1283
107. Koch T, Dahlke C, Fathi A, Kupke A, Krähling V, Okba NMA, et al. Safety and Immunogenicity of a Modified Vaccinia Virus Ankara Vector Vaccine Candidate for Middle East Respiratory Syndrome: An Open-Label, Phase 1 Trial. Lancet Infect Dis (2020) 20(7):827–38. doi: 10.1016/S1473-3099(20)30248-6
108. Yuan FF, Tanner J, Chan PKS, Biffin S, Dyer WB, Geczy AF, et al. Influence of Fcγ RIIA and MBL Polymorphisms on Severe Acute Respiratory Syndrome. Tissue Antigens (2005) 66(4):291–6. doi: 10.1111/j.1399-0039.2005.00476.x
109. Arvin AM, Fink K, Schmid MA, Cathcart A, Spreafico R, Havenar-Daughton C, et al. A Perspective on Potential Antibody-Dependent Enhancement of SARS-CoV-2. Nat (2020) 584(7821):353–63. doi: 10.1038/s41586-020-2538-8
110. Iwasaki A, Yang Y. The Potential Danger of Suboptimal Antibody Responses in COVID-19. Nat Rev Immunol (2020) 20(6):339–41. doi: 10.1038/s41577-020-0321-6
111. Lee WS, Wheatley AK, Kent SJ, DeKosky BJ. Antibody-Dependent Enhancement and SARS-CoV-2 Vaccines and Therapies. Nat Microbiol (2020) 5(10):1185–91. doi: 10.1038/s41564-020-00789-5
112. Ricke DO. Two Different Antibody-Dependent Enhancement (ADE) Risks for SARS-CoV-2 Antibodies. Front Immunol (2021) 12:1–9. doi: 10.3389/fimmu.2021.640093
113. Hui KPY, Cheung MC, Perera RAPM, Ng KC, Bui CHT, Ho JCW, et al. Tropism, Replication Competence, and Innate Immune Responses of the Coronavirus SARS-CoV-2 in Human Respiratory Tract and Conjunctiva: An Analysis in Ex-Vivo and In-Vitro Cultures. Lancet Respir Med (2020) 8(7):687–95. doi: 10.1016/S2213-2600(20)30193-4
114. Niles MA, Gogesch P, Kronhart S, Ortega Iannazzo S, Kochs G, Waibler Z, et al. Macrophages and Dendritic Cells Are Not the Major Source of Pro-Inflammatory Cytokines Upon SARS-CoV-2 Infection. Front Immunol (2021) 12:647824. doi: 10.3389/fimmu.2021.647824
115. Chandrashekar A, Liu J, Martino AJ, McMahan K, Mercad NB, Peter L, et al. SARS-CoV-2 Infection Protects Against Rechallenge in Rhesus Macaques. Science (2020) 369(6505):812–7. doi: 10.1126/science.abc4776
116. Deng W, Bao L, Liu J, Xiao C, Liu J, Xue J, et al. Primary Exposure to SARS-CoV-2 Protects Against Reinfection in Rhesus Macaques. Sci (New York NY) (2020) 369(6505):818–23. doi: 10.1126/science.abc5343
117. Jackson CB, Farzan M, Chen B, Choe H. Mechanisms of SARS-CoV-2 Entry Into Cells. Nat Rev Mol Cell Biol (2022) 23(1):3–20. doi: 10.1038/s41580-021-00418-x
118. Larsen MD, de Graaf EL, Sonneveld ME, Plomp HR, Nouta J, Hoepel W, et al. Afucosylated IgG Characterizes Enveloped Viral Responses and Correlates With COVID-19 Severity. Science (2021) 371(6532):eabc8378. doi: 10.1126/science.abc8378
119. Maemura T, Kuroda M, Armbrust T, Yamayoshi S, Halfmann PJ, Kawaoka Y. Antibody-Dependent Enhancement of SARS-CoV-2 Infection Is Mediated by the IgG Receptors Fcγriia and Fcγriiia But Does Not Contribute to Aberrant Cytokine Production by Macrophages. mBio (2021) 12(5):e0198721. doi: 10.1128/mBio.01987-21
120. Walls AC, Park Y-J, Tortorici MA, Wall A, McGuire AT, Veesler D. Structure, Function, and Antigenicity of the SARS-CoV-2 Spike Glycoprotein. Cell (2020) 181(2):281–92.e6. doi: 10.1016/j.cell.2020.02.058
121. Tan H, Juno JA, Lee WS, Barber-axthelm I, Kelly HG, Wragg KM, et al. Immunogenicity of Prime-Boost Protein Subunit Vaccine Strategies Against SARS-CoV-2 in Mice and Macaques. Nat Commun (2021) 12(1):4–13. doi: 10.1038/s41467-021-21665-8
122. Arunachalam PS, Walls AC, Golden N, Atyeo C, Fischinger S, Li C, et al. Adjuvanting a Subunit COVID-19 Vaccine to Induce Protective Immunity. Nat (2021) 594(7862):253–8. doi: 10.1038/s41586-021-03530-2
123. Gupta D, Parthasarathy H, Sah V, Tandel D, Vedagiri D, Reddy S, et al. Inactivation of SARS-CoV-2 by β-Propiolactone Causes Aggregation of Viral Particles and Loss of Antigenic Potential. Virus Res (2021) 305:198555. doi: 10.1016/j.virusres.2021.198555
124. Wang H, Zhang Y, Huang B, Deng W, Quan Y, Wang W, et al. Development of an Inactivated Vaccine Candidate, BBIBP-CorV, With Potent Protection Against SARS-CoV-2. Cell (2020) 182(3):713–721.e9. doi: 10.1016/j.cell.2020.06.008
125. Gao Q, Bao L, Mao H, Wang L, Xu K, Yang M, et al. Development of an Inactivated Vaccine Candidate for SARS-CoV-2. Science (2020) 369(6499):77–81. doi: 10.1126/science.abc1932
126. Tanriover MD, Doğanay HL, Akova M, Güner HR, Azap A, Akhan S, et al. Efficacy and Safety of an Inactivated Whole-Virion SARS-CoV-2 Vaccine (CoronaVac): Interim Results of a Double-Blind, Randomised, Placebo-Controlled, Phase 3 Trial in Turkey. Lancet (London England) (2021) 398(10296):213–22. doi: 10.1016/S0140-6736(21)01429-X
127. Routhu NK, Cheedarla N, Gangadhara S, Bollimpelli VS, Boddapati AK, Shiferaw A, et al. A Modified Vaccinia Ankara Vector-Based Vaccine Protects Macaques From SARS-CoV-2 Infection, Immune Pathology, and Dysfunction in the Lungs. Immunity (2021) 54(3):542–56. doi: 10.1016/j.immuni.2021.02.001
128. Gooch KE, Smith TRF, Salguero FJ, Fotheringham SA, Watson RJ, Dennis MJ, et al. One or Two Dose Regimen of the SARS-CoV-2 Synthetic DNA Vaccine INO-4800 Protects Against Respiratory Tract Disease Burden in Nonhuman Primate Challenge Model. Vaccine (2021) 39(34):4885–94. doi: 10.1016/j.vaccine.2021.06.057
129. Schiffner T, de Val N, Russell RA, de Taeye SW, de la Peña AT, Ozorowski G, et al. Chemical Cross-Linking Stabilizes Native-Like HIV-1 Envelope Glycoprotein Trimer Antigens. J Virol (2016) 90(2):813–28. doi: 10.1128/JVI.01942-15
130. Fierz W, Walz B. Antibody Dependent Enhancement Due to Original Antigenic Sin and the Development of SARS. Front Immunol (2020) 11:1–5. doi: 10.3389/fimmu.2020.01120
131. Zhang A, Stacey HD, Mullarkey CE, Miller MS. Original Antigenic Sin: How First Exposure Shapes Lifelong Anti–Influenza Virus Immune Responses. J Immunol (2019) 202(2):335–40. doi: 10.4049/jimmunol.1801149
132. Screaton G, Mongkolsapaya J, Yacoub S, Roberts C. New Insights Into the Immunopathology and Control of Dengue Virus Infection. Nat Rev Immunol (2015) 15(12):745–59. doi: 10.1038/nri3916
133. Klenerman P, Zinkernagel RM. Original Antigenic Sin Impairs Cytotoxic T Lymphocyte Responses to Viruses Bearing Variant Epitopes. Nature (1998) 394(6692):482–5. doi: 10.1038/28860
134. Zehn D, Turner MJ, Lefrançois L, Bevan MJ. Lack of Original Antigenic Sin in Recall CD8 + T Cell Responses. J Immunol (2010) 184(11):6320–6. doi: 10.4049/jimmunol.1000149
135. Cele S, Jackson L, Khoury DS, Khan K, Moyo-Gwete T, Tegally H, et al. Omicron Extensively But Incompletely Escapes Pfizer BNT162b2 Neutralization. Nature (2022) 602(7898):654–56. doi: 10.1038/s41586-021-04387-1
136. Reynolds CJ, Gibbons JM, Pade C, Lin K-M, Sandoval DM, Pieper F, et al. Heterologous Infection and Vaccination Shapes Immunity Against SARS-CoV-2 Variants. Science (2021) 375(6577):183–92. doi: 10.1126/science.abm0811
137. Aydillo T, Rombauts A, Stadlbauer D, Aslam S, Abelenda-Alonso G, Escalera A, et al. Immunological Imprinting of the Antibody Response in COVID-19 Patients. Nat Commun (2021) 12(1):3781. doi: 10.1038/s41467-021-23977-1
138. Swadling L, Diniz MO, Schmidt NM, Amin OE, Chandran A, Shaw E, et al. Pre-Existing Polymerase-Specific T Cells Expand in Abortive Seronegative SARS-CoV-2. Nature (2021) 601(7891):110–17. doi: 10.1038/s41586-021-04186-8
139. Tan C-W, Chia W-N, Young BE, Zhu F, Lim B-L, Sia W-R, et al. Pan-Sarbecovirus Neutralizing Antibodies in BNT162b2-Immunized SARS-CoV-1 Survivors. N Engl J Med (2021) 385(15):1401–6. doi: 10.1056/NEJMoa2108453
140. Wheatley AK, Fox A, Tan H-X, Juno JA, Davenport MP, Subbarao K, et al. Immune Imprinting and SARS-CoV-2 Vaccine Design. Trends Immunol (2021) 42(11):956–9. doi: 10.1016/j.it.2021.09.001
141. Naaber P, Tserel L, Kangro K, Sepp E, Jürjenson V, Adamson A, et al. Dynamics of Antibody Response to BNT162b2 Vaccine After Six Months: A Longitudinal Prospective Study. Lancet Regional Health Europe (2021) 10:100208. doi: 10.1016/j.lanepe.2021.100208
142. Thomas SJ, Moreira ED, Kitchin N, Absalon J, Gurtman A, Lockhart S, et al. Safety and Efficacy of the BNT162b2 mRNA Covid-19 Vaccine Through 6 Months. N Engl J Med (2021) 385(19):1761–73. doi: 10.1056/NEJMoa2110345
143. Bertoletti A, le Bert N, Qui M, Tan AT. SARS-CoV-2-Specific T Cells in Infection and Vaccination. Cell Mol Immunol (2021) 18(10):2307–12. doi: 10.1038/s41423-021-00743-3
144. Keeton R, Tincho MB, Ngomti A, Baguma R, Benede N, Suzuki A, et al. T Cell Responses to SARS-CoV-2 Spike Cross-Recognize Omicron. Nature (2022) 603(7901):488–492. doi: 10.1038/s41586-022-04460-3
145. Grant OC, Montgomery D, Ito K, Woods RJ. Analysis of the SARS-CoV-2 Spike Protein Glycan Shield Reveals Implications for Immune Recognition. Sci Rep (2020) 10(1):1–11. doi: 10.1038/s41598-020-71748-7
Keywords: vaccine-associated enhanced disease, COVID-19, SARS-CoV-2, coronavirus, vaccine, enhancement, safety
Citation: Gartlan C, Tipton T, Salguero FJ, Sattentau Q, Gorringe A and Carroll MW (2022) Vaccine-Associated Enhanced Disease and Pathogenic Human Coronaviruses. Front. Immunol. 13:882972. doi: 10.3389/fimmu.2022.882972
Received: 24 February 2022; Accepted: 14 March 2022;
Published: 04 April 2022.
Edited by:
Xuguang (Sean) Li, Health Canada, CanadaReviewed by:
Gary Wong, Institut Pasteur of Shanghai (CAS), ChinaRoger Y. Tam, Health Canada, Canada
Aaron Farnsworth, Health Canada, Canada
Takuo Mizukami, National Institute of Infectious Diseases (NIID), Japan
Copyright © 2022 Gartlan, Tipton, Salguero, Sattentau, Gorringe and Carroll. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Cillian Gartlan, Y2dhcnRsYW5Ad2VsbC5veC5hYy51aw==; Miles W. Carroll, TWlsZXMuY2Fycm9sbEBuZG0ub3guYWMudWs=