 Jonathan Florentin1†
Jonathan Florentin1† Scott P. O’Neil1†
Scott P. O’Neil1† Lee L. Ohayon1Afaz Uddin1
Lee L. Ohayon1Afaz Uddin1 Sathish Babu Vasamsetti1Anagha Arunkumar1
Sathish Babu Vasamsetti1Anagha Arunkumar1 Samit Ghosh2Jennifer C. Boatz2Justin Sui2Corrine R. Kliment2
Samit Ghosh2Jennifer C. Boatz2Justin Sui2Corrine R. Kliment2 Stephen Y. Chan1
Stephen Y. Chan1 Partha Dutta1,3*
Partha Dutta1,3*- 1Division of Cardiology, Department of Medicine, Pittsburgh Heart, Lung, Blood, and Vascular Medicine Institute, University of Pittsburgh Medical Center, University of Pittsburgh School of Medicine, Pittsburgh, PA, United States
- 2Department of Medicine, Division of Pulmonary and Critical Care, University of Pittsburgh, Pittsburgh, PA, United States
- 3Department of Immunology, University of Pittsburgh School of Medicine, Pittsburgh, PA, United States
Although it is well known that hypoxia incites unleashed cellular inflammation, the mechanisms of exaggerated cellular inflammation in hypoxic conditions are not known. We observed augmented proliferation of hematopoietic stem and progenitor cells (HSPC), precursors of inflammatory leukocytes, in mice under hypoxia. Consistently, a transcriptomic analysis of human HSPC exposed to hypoxic conditions revealed elevated expression of genes involved in progenitor proliferation and differentiation. Additionally, bone marrow cells in mice expressed high amount of vascular endothelial growth factor (VEGF), and HSPC elevated VEGF receptor 1 (VEGFr1) and its target genes in hypoxic conditions. In line with this, VEGFr1 blockade in vivo and in vitro decreased HSPC proliferation and attenuated inflammation. In silico and ChIP experiments demonstrated that HIF-1α binds to the promoter region of VEGFR1. Correspondingly, HIF1a silencing decreased VEGFr1 expression in HSPC and diminished their proliferation. These results indicate that VEGF signaling in HSPC is an important mediator of their proliferation and differentiation in hypoxia-induced inflammation and represents a potential therapeutic target to prevent aberrant inflammation in hypoxia-associated diseases.
Introduction
Diseases characterized by alveolar hypoxia, such as chronic obstructive pulmonary disease (COPD) and obstructive sleep apnea (OSA), are highly prevalent and contribute to significant morbidity and mortality around the world (1–5). Hypoxia-associated diseases bear high comorbidities including metabolic, cardiovascular, neoplastic and neurologic disease, which a growing body of literature attributes to systemic inflammation (1, 6–8). To date, studies of alveolar hypoxia induced inflammation have focused primarily on the role of resident macrophages, and their interaction with alveolar endothelial cells mediating production of inflammatory cytokines (9–12). Monocyte chemoattractant protein 1 (MCP-1), c-reactive protein, tumor necrosis factor alpha (TNFα), IL-1, IL-6 and IL-8 are increased in hypoxic lung diseases and generally correlate with severity of disease (11, 13, 14). While there is strong evidence that local and systemic inflammation are key components of hypoxic diseases, the mechanisms of exaggerated cellular inflammation in hypoxic conditions have yet to be defined.
Mounting evidence has demonstrated key inflammatory cells involved in the pathogenesis of various diseases including insulin resistance and atherosclerosis (4, 15–17). These cells have been proposed to originate from hematopoietic stem and progenitor cells (HSPC) in the bone marrow and spleen (18–21). Patients with hypoxic lung disease exhibit elevated inflammation and leukocytosis. The leukocytosis observed in chronic inflammation likely represents production of inflammatory leukocytes by their progenitors. However, a direct link between hypoxia induced systemic inflammation and proliferation of HSPC has not been established. Furthermore, the mechanisms of HSPC proliferation and differentiation in hypoxia have not been studied.
Vascular endothelial growth factor (VEGF) has been identified as an important regulator of neovascularization by encouraging endothelial cell proliferation in various diseases such as age-related macular degeneration and neoplastic diseases (22–25). Several recent studies have demonstrated elevated VEGF concentrations in peripheral blood of patients with hypoxic lung disease, which closely correlated with severity of disease (26–31). However, the role of VEGF in HSC proliferation and inflammatory leukocyte production is not known.
We used two independent mouse models of hypoxia to understand the effect of hypoxia in HSPC proliferation: C57BL/6 mice exposed to A) 10% O2 for three weeks, an established mouse model of chronic hypoxia (32–34) and B) cigarette smoke for six months, a COPD mouse model (35, 36). Mice under chronic hypoxic conditions and exposed to cigarette smoke had elevated numbers of neutrophils, monocytes and macrophages in the blood and lungs suggesting augmented hematopoiesis. In line with this, a whole genome RNA sequencing analysis of human HSPC cultured under hypoxic conditions showed increased expression of the genes involved in hematopoiesis. The number and proliferation of HSPC were increased when mice were exposed to hypoxia or cigarette smoke. Concomitantly, we measured increased expression of VEGFr1 in these activated progenitors indicating a possible role of VEGFr1 in hematopoiesis. Indeed, VEGFr1 blockade in vitro and in vivo diminished HSPC proliferation and their differentiation into myeloid cells, resulting in attenuated inflammation in hypoxic conditions. Mechanistically, we observed that HIF-1α binds to the VEGFr1 promoter and increases its expression in HSPC under hypoxic conditions. HIF1a silencing reduced HSPC proliferation and their differentiation into inflammatory leukocytes.
As a whole, the present study describes a role of the VEGFr1 in the activation of hematopoietic progenitors and the production of inflammatory myeloid cells in hypoxia. Additionally, this work proposes the VEGFr1 as a possible therapeutic target to attenuate inflammatory burden in patients with hypoxia.
Methods
Animals
All animal experiments were conducted following NIH and ARRIVE (Animal Research: Reporting of In Vivo Experiments, https://arriveguidelines.org) guidelines under protocols approved by the Institutional Animal Care and Use Committee of the University of Pittsburgh. Ten to twelve-week-old C57BL/6J male mice were exposed to normobaric hypoxic–10% oxygen, which represents 50% of the normal amount of oxygen, or normoxic conditions for 3 weeks. This oxygen concentration resulted in increased right ventricular systolic pressure, Fulton index and pulmonary vascular remodeling (37, 38). Of note, we have observed a very high mortality when we further decreased O2 concentration. After 21 days in hypoxic chamber, mice underwent right ventricular catheterization, followed by tissue and blood collection. The mice were anesthetized with ketamine/xylazine and ventilated through a transtracheal catheter.
Flow Cytometry
C57BL/6 mice were anesthetized, and a small volume of peripheral blood was collected by cardiac puncture, followed by transcardial perfusion with 15mL of ice-cold PBS. One lobe of the lung was harvested, minced and digested with collagenase I, collagenase XI, and hyaluronidase for one hour at 37C. After incubation, a single cell suspension was prepared by passing the digested lung tissue through a 70 μm nylon strainer, followed by washing with 0.5% bovine serum albumin in PBS (FACS buffer). The filtrate was washed with 10 mL of FACS buffer and centrifuged at 4°C for 7 minutes at 350 g. The supernatant was discarded, and the samples were re-suspended and labeled with 600-fold diluted antibody mixture with the following antibodies: anti-CD45.2 (BD Biosciences clone 104, cat#560693), CD11b (BD Biosciences, clone M1/70, cat#557657), CD115 (BD Biosciences, clone T38-320, cat# 565249), Ly6G (BD Biosciences, clone 1A8, cat#564979) and CD64 (BD Biosciences, clone X54-5/7.1, cat#558455). B and T cells were identified as CD45+, CD11b-, and CD19+ or CD3+, respectively. Monocytes were identified as CD45+, CD11b+, CDLy-6G- and CD115+. Neutrophils were considered as CD45+, CD11b+, CD115- and Ly6G+. For bone marrow hematopoietic stem and progenitor cells analyses, cells were stained with biotin conjugated antibodies against lineage markers including B220 (Biolegend, clone RA3-6B2, cat# 103203), CD4 (GK1.5, BD Biosciences, cat# 555345), CD8a (53-6.7, BD Biosciences, cat# 555365), NK1.1 (PK136, Biolegend, cat# 109704), CD11b (M1/70, Biolegend, cat# 101204), CD11c (N418, Biolegend, cat# 117304), Gr-1 (Biolegend, clone RB6-8C5, cat# 108403) and, Ter119 (TER-119, Biolegend, cat# 116204) followed by streptavidin APC/Cy7 conjugation, and antibodies against c- Kit (2B8, Biolegend, cat# 105822), Sca-1 (D7, Biolegend, cat# 108120), IL7Ra (SB/199, BDBiosciences, cat# 565490), CD16/32 (2.4G2, Biolegend, cat# 101318), CD34 (RAM34, BD Biosciences, cat# 553733), CD48 (HM48-1, Biolegend, cat# 103418) and CD150 (TC15-12F12.2, Biolegend, 115918). Hematopoietic stem cells (HSC) were identified as Lin- c-Kit+ Sca-1+ CD48- CD150+, and LSK were deemed to be Lin- c-Kit+ Sca-1+. Granulocyte-macrophage progenitors (GMP) were identified as Lin- c-Kit+ Sca-1- CD16/32+ CD34+. Samples were incubated on ice for 1 hour. Cell numbers per femur were calculated by multiplying total cell counts using a hemocytometer by cell subset frequencies obtained from flow cytometry analysis. Murine Vegfr1 expression was assessed using a Vegfr1 antibody (Thermofisher Scientific, clone 3A6, cat # BSM-52338R). A Fortessa Flow Cytometer (BD) was used to acquire data. Data were analyzed with FlowJo software (Tree Star).
Biochemical Assays
Concentrations of cytokines in plasma and parenchymal tissues were quantified by enzyme-linked immunosorbent assay (ELISA). IL-6 levels were quantified in lung tissue from hypoxic and normoxic conditions using a mouse IL-6 capture ELISA kit following the manufacturer instructions (Invitrogen, Cat # BMS603-2). Comparisons between conditions were performed using Graphpad Prism, and absolute concentration was calculated based on a lyophilized standard included in the kit.
RT PCR
Lungs were harvested and immediately snap frozen in liquid nitrogen until they could be processed. RNA extraction was performed using the PicoPure RNA isolation kit (Applied BioSystems, Cat # KIT0204) without modification to the manufacturer protocol. Total RNA was quantified using a NanoDrop spectrophotometer. Complementary DNA was generated from 100 ng of mRNA per sample using the high capacity cDNA Reverse Transcription kit (Applied BioSystems, Cat # 4368814). Relative gene expression was determined by qPCR with the PowerUp SYBR Green reporter (Applied BioSystems, Cat # A25742) and primers supplied by IDT; gene expression was represented as ΔCt normalized to beta-actin expression.
Whole Mount Imaging
Freshly dissected sternums from C57BL/6 mice exposed to normoxic or hypoxic conditions were fixed in cold 4% paraformaldehyde (PFA, ThermoFisher Scientific, Cat # 28908) in PBS for 25 minutes without agitation. The bones were washed in PBS three times with 15-minute incubations before incubating with common lineage biotinylated antibodies, including anti-TER119 (Biolegend, clone TER-119, cat#116204), CD11b (BD Biosciences, clone M1/70, cat#553309), CD11c (Biolegend, clone N418, cat#117304), CD45R/B220 (Biolegend, clone RA3-6B2, cat#103204), NK1.1 (Biolegend, clone PK136, cat#108704), GR-1 (Biolegend, clone RB6-8C5, cat#108404), CD4 (Biolegend, clone GK1.5, cat#100404), CD8 (Biolegend, clone 53-6.7, cat#100704) and CD127 (Biolegend, clone A7R34, cat#135006). Samples were incubated for 24 hours at 4° C, then washed three times with 1% FBS in PBS with 15-minute incubations, then labeled with streptavidin FITC antibody, followed by overnight incubation at 4°C. The samples were sequentially labeled with anti-VE Cadherin (BD Biosciences, clone 11D4.1, cat# 562242) and anti-CD31 APC antibodies (BD Biosciences, clone MEC 13.3, cat# 551262), followed by rabbit anti-mouse CD150 then rat anti-rabbit Cy3 (Invitrogen) with three wash steps between primary and secondary antibody overnight incubations. Lastly, the samples were washed three times and imaged on a Nikon A1 confocal microscope; Images were analyzed using ImageJ software.
Human Whole Blood data
All human data were collected in accordance with a University of Pittsburgh Independent Review Board approved protocol. Patients with intermittent hypoxia, defined by an apnea hypoxia index (AHI) > 5, were eligible for inclusion in the obstructive sleep apnea group, and matched controls with AHI less than 5 were eligible for inclusion in the control group. Patients with pulmonary function testing (PFT) confirmed chronic obstructive lung disease were eligible for inclusion in the COPD group. Patients with concurrent conditions known to cause leukocytosis were excluded, including known history of CVD, dyslipidemia, diabetes mellitus, allergic, pulmonary or hematological disease, malignancy, recent injury or surgery, recent systemic steroid use, current infection or any systemic medication use (e.g. hypolipidemic or anti-platelet agents). Complete blood cell count laboratory data were collected prior to medical interventions.
HSPC Cell Culture
Hematopoietic stem and progenitor cells were enriched by negative magnetic-activated cell sorting from vertebral, femoral and tibial bone marrow (StemCell Technologies, Cat # 17665). Briefly, we crushed the bones and filtered the cells through a 40μm mesh filter. The cells were then spun down and resuspended in 1mL of FACS buffer. They were then stained with biotin-conjugated lineage antibodies (B220, TER119, Ly6G, CD4, CD8, CD11b, CD11c, and IL7R) at 1:300 dilution for 20 minutes on ice. The cells were spun down and resuspended in 2 ml of buffer (DPBS+2%FBS+1mM EDTA), and 100 ul of biotin selection cocktail was added for 15 minutes at room temperature. Another 50 μL of magnetic nanoparticles was added to the mix for 10 minutes at room temperature. The cells were then placed in a FACS tube in an EasySep magnet (Stem Cell Technologies, Cat# 18000). After 6-7 minutes, the unbound cells were collected. Cells were then cultured in the presence of G-CSF on 96 well round-bottom cell culture plates with 10, 50, or 100 nM concentrations of Sugen (SU5416, MedChem Express), or with siRNA control (siCtl, IDT) or against Vegfr1 (siVegfr1, IDT), for 72 hours prior to hypoxia challenge. Samples were exposed to normobaric hypoxic conditions (10% O2) for 24 hours prior to RNA extraction and flow cytometry analysis.
RNA Sequencing
Publicly available differentially expressed genes from HSPC exposed to hypoxic conditions (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE54663) were assessed for genes of interest including VEFGA and VEGFr1, and heatmap representations of the data were prepared. Enriched pathways were discerned using Ingenuity Pathway Analysis.
ChIP Sequencing
To assess the interaction between HIF-1a and VEGFr1, we first located the promoter region of VEGFr1 using the UCSC genome browser (University of California Santa Cruz). Then, we looked for potential HIF-1-alpha binding sites onto the promoter region of VEGFr1 using Transfac software (GeneXplain). The primers for each HIF-1α binding site were designed using NCBI Primer-BLAST. Murine HSPC were harvested from the bone marrow of C57BL/6 mice. Cells were resuspended in complete SFEM media and plated onto 10 cm dishes. Cells were incubated for 24 hours in normoxia or hypoxia. Cells were then harvested and resuspended in PBS at a concentration 106 cells/mL. Cells were then fixed, and DNA was extracted and immunoprecipitated by a ChIP grade HIF-1α antibody (Rb polyclonal, Novusbio, Cat # NB100-479SS) as previously described (39). Finally, real time PCR was run to quantify the amount of HIF1A bound to VEGFr1 promoter region in each condition.
Adoptive Transfer Experiment
C57BL/6 mice were placed under either normoxic or hypoxic conditions (10% O2) for 10 days. At day 7 of hypoxia, bone marrow HSPC from KIkGR+ (Kikume Green Red) mice were negatively enriched using a magnetic separation method and retro-orbitally injected into either normoxic or hypoxic C57BL/6 mice at a concentration of 20 million cell/mL. Mice were sacrificed, and lungs, bone marrow, spleen and blood were harvested at day 10 of hypoxia. The percentages of progenies derived from the adoptively transferred cells were measured by flow cytometry.
Statistical Analysis
Data were compiled using Prism (GraphPad). Statistics were generated and are presented as the mean ± SEM. Statistical significance between two categories of analyzed samples was calculated using two-tailed Student’s t tests. For multiple category comparisons, one-way ANOVA was used with a post hoc Bonferroni test. Differences with P values <0.05 were considered statistically significant.
Results
Hypoxic Mice Have Elevated Numbers of Inflammatory Leukocytes in the Blood
Various diseases associated with intermittent or chronic hypoxia are characterized by exaggerated systemic inflammation (5, 6, 16, 17, 40–43). Leukocytes, primarily myeloid cells, play a major role in inflammatory diseases including atherosclerosis and diabetes (15, 42). To study alteration in hematopoietic progenitors and their lineage output, we used two established mouse models of hypoxia- mice housed in hypoxic chambers containing 10% oxygen for three weeks and mice exposed to cigarette smoke for six months (35, 36). Mice exposed to cigarette smoke contained significantly heightened numbers of lymphocytes, such as B and T cells, in the bone marrow while myeloid cell content decreased (Supplementary Figures 1A, B). In contrast, splenic lymphocyte numbers were unaltered (Supplementary Figure 1C), and myeloid cells were more numerous in smoke-exposed mice compared to the control group (Supplementary Figure 1C). Additionally, we found that the cytokine expression increased in the lungs of cigarette smoke-exposed mice compared to air-exposed mice (Supplementary Figure 1D). Although these data suggest differentiation of bone marrow progenitors into hematopoietic cells after smoke exposure, we cannot rule out decreased apoptosis of these cells after smoke exposure. Furthermore, the data suggest a preferential egress of myeloid cells from the bone marrow after smoke exposure.
To further delineate the contributions of hypoxia in systemic inflammation, we used a mouse model of chronic hypoxia (10% oxygen). We quantified inflammatory cytokines and leukocytes in mice exposed to hypoxic conditions compared to normoxic controls. We observed increased expression of Il-1b, Il-6, Il-18 and Tnfa in the lungs of hypoxic mice (Figure 1A). Consistent with this heightened expression of the inflammatory cytokine genes, we found elevated concentrations of Il-6, TNF-α and IL-1β in lung parenchyma (Figure 1B). Furthermore, we found that mice in chronic hypoxic conditions had increased numbers of monocytes, neutrophils, and B and T lymphocytes in the blood compared to normoxic control mice (Figure 1C and Supplementary Figures 2A, B). Similarly, the lungs of hypoxic mice harbored augmented numbers of inflammatory cells, including interstitial macrophages (Figure 1D and Supplementary Figure 2C), which are reported to have pathologic roles in pulmonary diseases characterized by hypoxia (44–46). The frequency of bone marrow monocytes and neutrophils increased after hypoxia exposures (Supplementary Figures 2C, D). Additionally, we evaluated leukocyte populations in the spleen of normoxic v. hypoxic mice. We found increased numbers of monocytes and B cells, decreased number of T cells and unchanged numbers of neutrophils (Supplementary Figure 2E). We also measured the ratio of blood/BM leukocyte numbers and noticed a significant increase in this ratio for monocytes and neutrophils suggesting an active egress from the BM to the blood of these cells (Supplementary Figure 2F). Altogether, these data demonstrate leukocytosis in patients and mice with hypoxia.
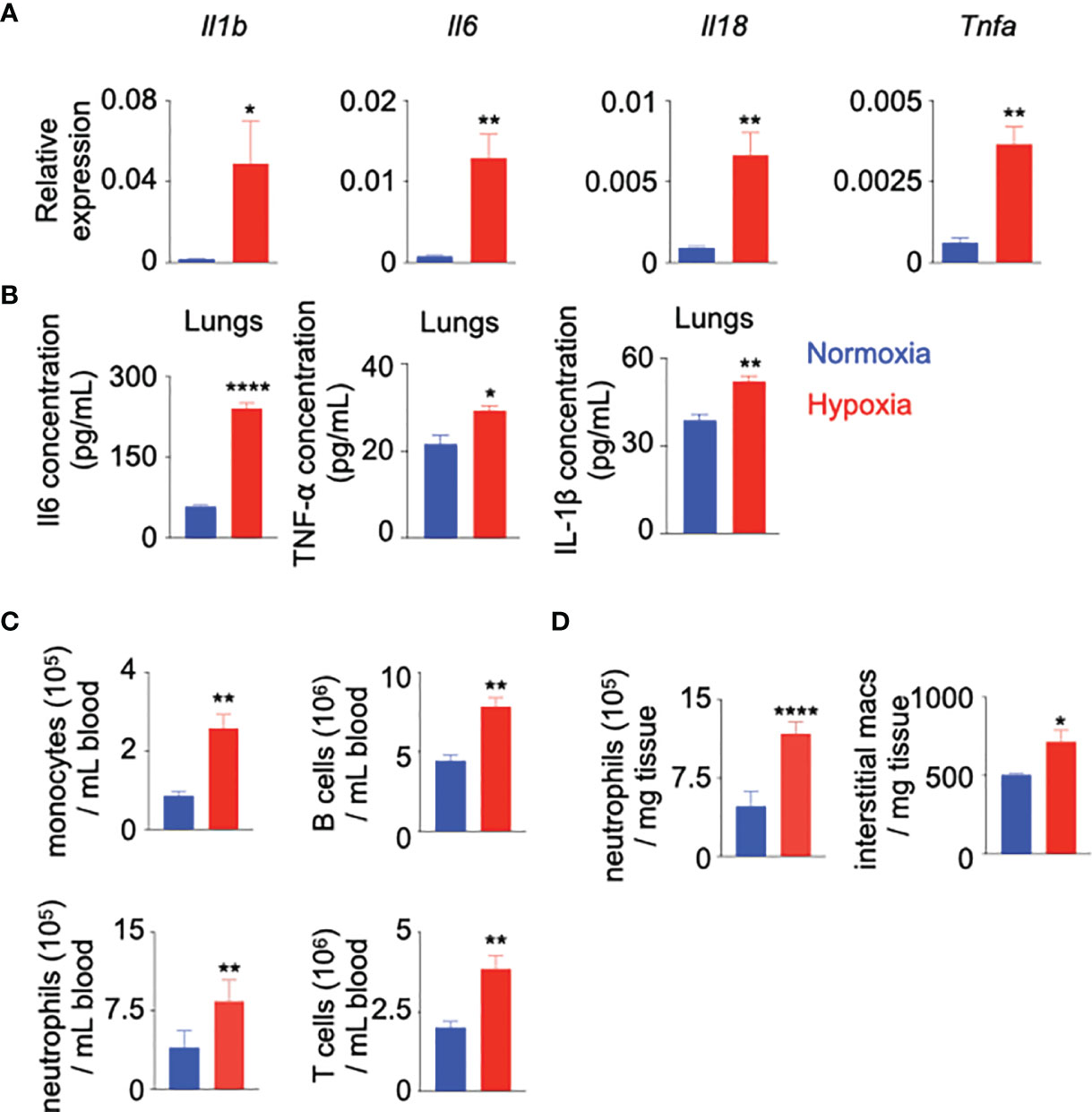
Figure 1 Hypoxia induces inflammation. C57BL/6 mice were placed under either normoxic or hypoxic conditions for 21 days. Lungs, blood and bone marrow were collected. (A) Il1b, Il6, Il18 and Tnfa expression was assessed by RT-qPCR.(B) Il-6, TNF-α and IL1-β protein expression was evaluated in the whole lung and serum by ELISA. (C) The numbers of blood monocytes, neutrophils, B and T cells in hypoxic versus normoxic mice were determined by flow cytometry. (D) The numbers of lung infiltrating neutrophils and interstitial macrophages in these mice were quantified by flow cytometry. n = 5 mice per condition. Data are shown as mean ± s.e.m. *P < 0.05, **P < 0.01, ****P < 0.001.
Hypoxia Increases Proliferation of Hematopoietic Stem and Progenitor Cells
Increased leukocyte numbers in the blood of hypoxic mice may indicate two underlying mechanisms: A) accelerated egress of bone marrow leukocyte into the blood and B) escalated production of leukocytes by hematopoietic stem and progenitor cells (HSPC) in the bone marrow. To determine whether the observed hypoxia-induced leukocytosis represents increased production of mature leukocytes, we assessed the number and proliferation of bone marrow HSPC, which consist of hematopoietic stem cells (HSC), lineage- Sca-1+ c-Kit+ (LSK or HSPC) cells and granulocyte macrophage progenitors (GMP) (Supplementary Figure 3A). Flow cytometry analysis revealed that hypoxia expanded the numbers of HSC, LSK and GMP in the femur and tibia (Figure 2A). Confocal microscopy confirmed a higher density of HSPC in the bone marrow of hypoxic mice compared to the normoxic control (Figure 2B). In line with augmented leukocyte and HSPC numbers, mice in hypoxic conditions had higher proportions of proliferating HSC, LSK and GMP (Figures 2C–E). Studies have shown that chemotherapy and inflammatory cytokines can induce phenotypic shift in Sca-1 surface expression, which may lead to contamination of the phenotypic HSC gate with non-HSC cell types (47–49). To assess if hypoxia alters Sca-1 expression on long term and short term HSC, HSPC, and GMP in the bone marrow, we measured mean fluorescent intensity (MFI) of Sca-1 in these cells by flow cytometry. We did not observe any statistical difference in the expression of Sca-1 in the progenitors between the two groups (Supplementary Figure 3B). Additionally, we isolated HSPC and GMPs from mice kept under either normoxic or hypoxic conditions for three weeks. qPCR for the cell cycle genes revealed that the progenitors expressed higher levels of these genes in hypoxic conditions (Supplementary Figure 4A). HSPC proliferation is prerequisite for their differentiation into leukocytes. To understand if HSPC expansion in hypoxic conditions can result in their higher differentiation, we analyzed a whole genome transcriptome data comparing human CD34+ HSPC cultured under hypoxic vs. normoxic conditions (50). A pathway analysis revealed that the genes involved in differentiation of stem cell, cell cycle and stimulation of cells were enriched in HSPC in hypoxic conditions (Supplementary Figures 4B, 5–7). To discern if hypoxia accelerates HSPC differentiation, we adoptively transferred GFP+ HSPC into mice housed under normoxic and hypoxic conditions, and enumerated progenies by flow cytometry. The bone marrow, blood and lungs of hypoxic mice contained augmented percentages of donor-derived differentiated leukocytes, lymphocytes, monocytes and neutrophils (Figure 2F and Supplementary Figure 8A). This difference of unleashed leukocyte production could not be attributed to higher engraftment of GFP+ HSPC under hypoxia (Supplementary Figure 8B). In summary, these data indicate that hypoxia drives HSPC into the cell cycle and increases their differentiation into inflammatory cells.
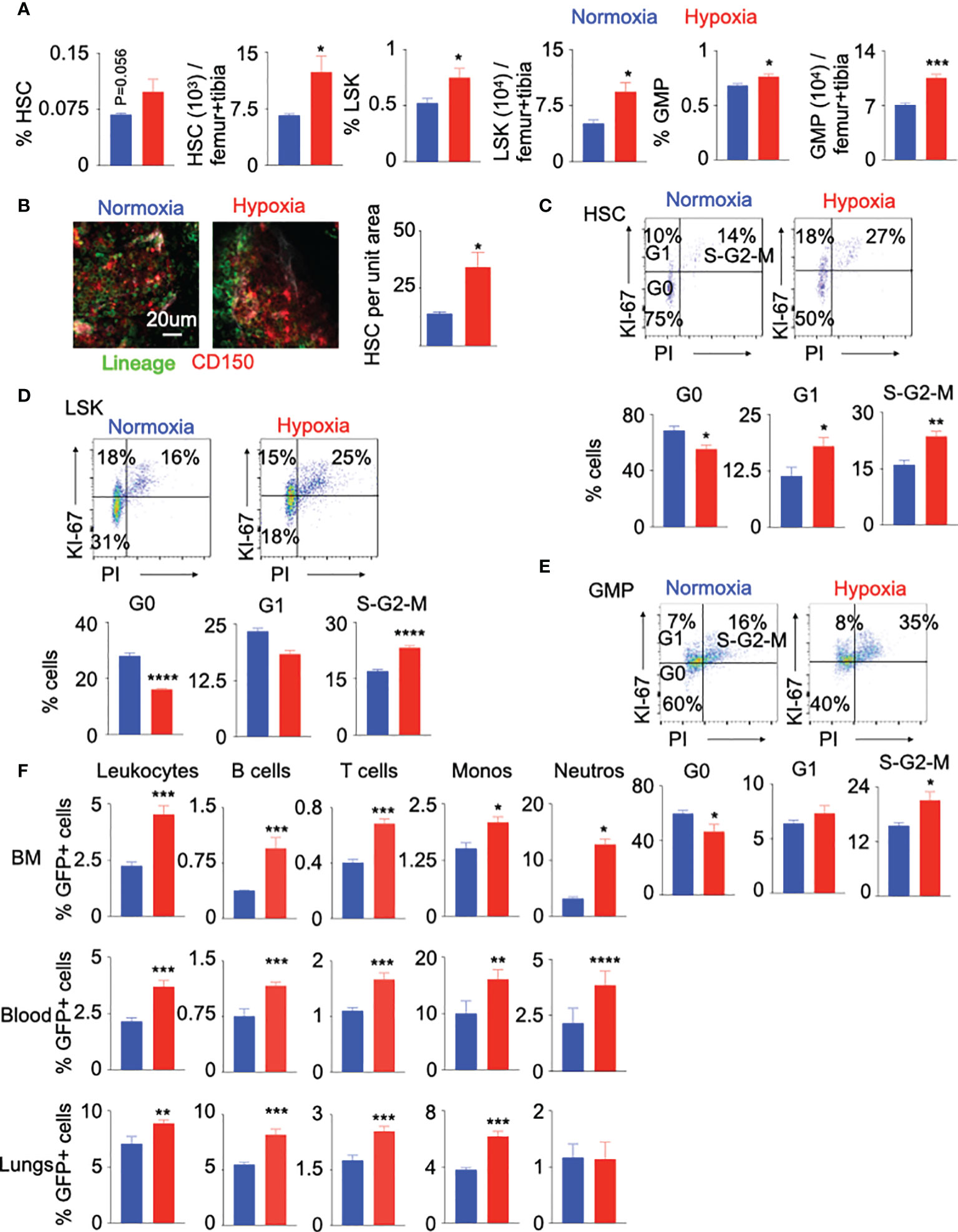
Figure 2 Hypoxia drives HSPC into the cell cycle. C57BL/6 mice were placed under either normoxic or hypoxic conditions for 21 days. Bone marrow was collected. The frequencies and numbers of the hematopoietic progenitors in the bone marrow were determined by flow cytometry (A) and whole mount confocal microscopy (B). The frequencies of HSC (C), LSK (D) and GMP (E) in the cell cycle stages were assessed using intracellular staining of Ki-67 and PI. (F) KikGR-GFP+ (Kikume Green Red) HSPC were adoptively transferred into mice housed in either normoxic or hypoxic conditions for seven days. GFP+ progenies were quantified four days later. n = 5 mice per condition. Data are shown as mean ± s.e.m. *P < 0.05, **P < 0.01, ***P < 0.005, ****P < 0.001.
VEGFr1 Expression Is Increased in Progenitor Cells Following Hypoxia Exposure
The role of VEGF/VEGFr1 in hypoxia-induced angiogenesis is well-documented (29, 51, 52). To investigate the possible role of VEGF-A/VEGFr1 in hypoxia-mediated HSPC proliferation, we first assessed RNA expression of these genes in the whole bone marrow of hypoxic mice. The expression of Vegfa, but not Vegfr1, was increased in bone marrow cells of hypoxic mice compared to their normoxic counterparts (Figure 3A). We observed that LSK and HSC express Vegfr1 in the steady state (Figure 3B). Additionally, the expression of Vegfr1 mRNA and protein in bone marrow GMP increased in hypoxia (Figures 3C, D). In line with these results, we observed that mice exposed to cigarette smoke harbored higher frequency of Vegfr1+ HSPC (Supplementary Figure 9A). VEGFR1 expression in the progenitors also increased (Supplementary Figure 9B). Next, we identified the genes downstream to the VEGFr1 signaling using Ingenuity Pathway Analysis (Supplementary Figure 9C). The expression of these genes was elevated in HSPC cultured under hypoxic conditions (Figure 3E). These data indicate that hypoxia increases VEGFr1 signaling in HSPC. Hypoxia-inducible factor 1α (HIF-1α), a key transcription factor increased in hypoxia, binds to several genes, and increases their expression (53–56). We observed increased Hif1a expression in BM HSPC and GMP of hypoxic mice (Supplementary Figure 9D). Flow cytometry revealed increased numbers of Hif-1α+ HSPC and GMP in hypoxic mice (Supplementary Figure 9E). Additionally, we confirmed decreased expression of prolyl-2 hydroxylase, which accelerates ubiquitination and lysosomal degradation of Hif-1α, in HSPC of hypoxic mice (Supplementary Figure 9F). Corresponding to the fact that Hif-1α increases glycolysis, we observed elevated expression of glycolytic genes in bone marrow HSPC of hypoxic mice (Supplementary Figure 9G). To decipher the mechanisms of increased Vegfr1 expression in hypoxia, we ascertained if the VEGFR1 promoter region has HIF-1α binding sites. Our in silico analysis revealed that HIF-1α has several binding sites on the VEGFr1 promoters in both humans and mice (Figure 3F). Chromatin immunoprecipitation experiments confirmed that HIF-1α binds to the Vegfr1 promoter in mouse HSPC (Figure 3G). HIF-1α inhibition significantly diminished hypoxia-mediated expression of VEGFr1 in human (Figure 3H) and mouse (Figure 3I) HSPC. To delineate the role of HIF-1α in hypoxia-induced HSPC proliferation, we silenced this transcription factor in mouse HSPC and observed significant downregulation of the genes involved in cell cycle progression (Figure 3J) as well as the genes downstream to the VEGFr1 signaling (Supplementary Figure 9H). In aggregate, these data indicate that HIF-1α directly binds to the VEGFr1 promoter in HSPC, increases VEGFr1 expression and augments hematopoietic progenitor proliferation under hypoxic conditions.
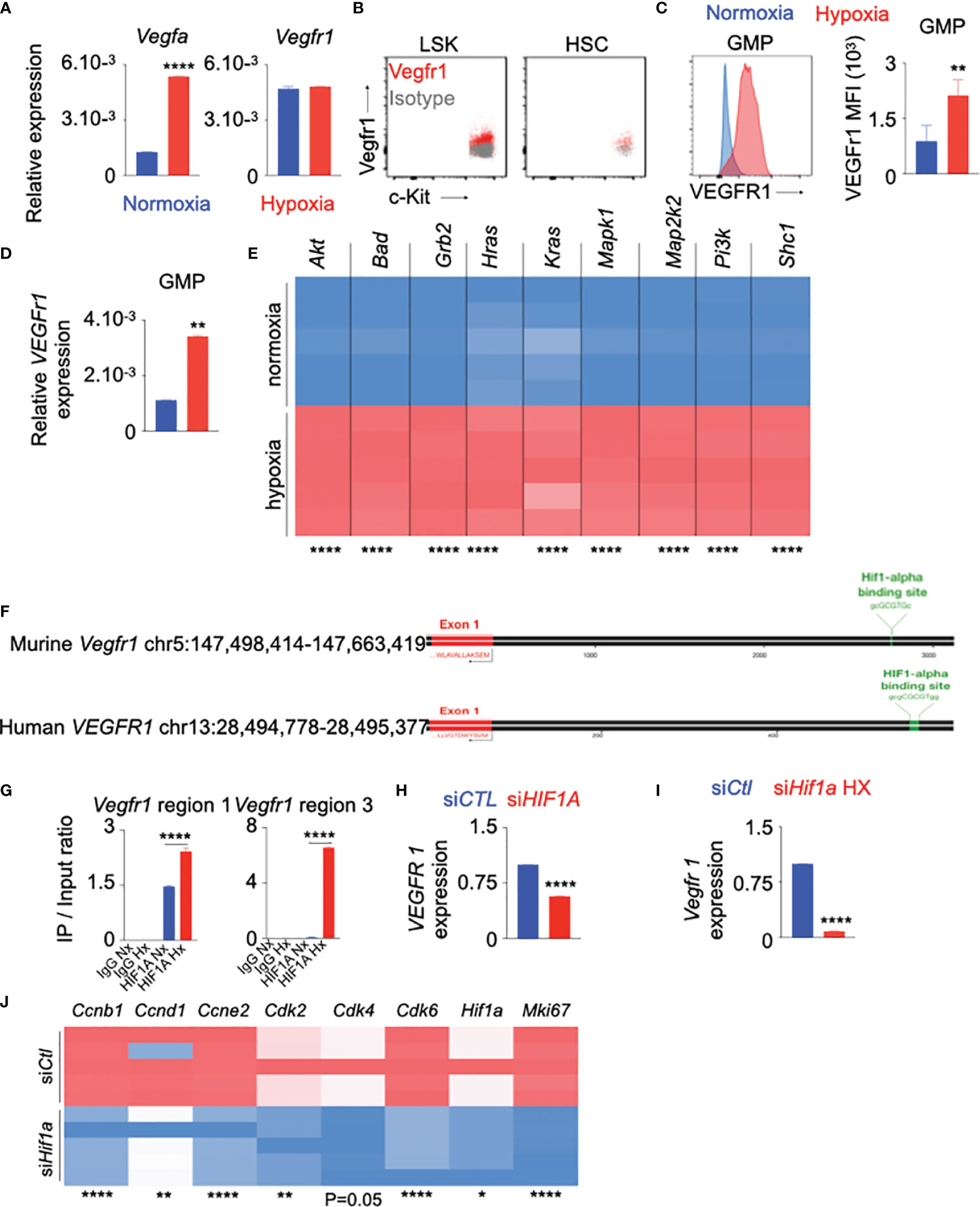
Figure 3 VEGFr1 expression is increased in HSPC in hypoxia. (A–C) C57BL/6 mice were placed under either normoxic or hypoxic conditions for 21 days. Bone marrow was collected. (A) Vegfa and Vegfr 1 expression in the whole bone marrow was assessed by RT-qPCR. B–C) Vegfr 1 expression was measured in bone marrow LSK and HSC in normoxic conditions (B) and GMP in normoxic and hypoxic conditions (C) by flow cytometry. The results are represented as mean fluorescent intensity (MFI). (D) Vegfr 1 gene expression was measured in sorted GMP by RT-qPCR. (E–G) HSPC were sorted from bone marrow of C57BL/6 mice and cultured in complete SFEM media in either normoxic or hypoxic conditions. (E) Heatmap representing expression of the genes downstream to Vegf/Vegfr 1 in HSPC cultured under normoxic and hypoxic conditions. (F) Schematic depicting HIF-1α binding sites on both murine and human VEGFr 1 promoter regions. (G) Confirmation of the binding of HIF-1α to the murine Vegfr 1 promoter region assessed by ChIP qPCR. (H, I) VEGFr 1 expression assessed in human (H) and murine (I) HSPC transfected with either siCTL or siHIF1A in hypoxic conditions. (J) Heatmap showing the expression of the cell cycle genes in HSPC transfected with either siCtl or siHif1a in hypoxic conditions. n = 5 mice per condition (A–D), 5 replicates per condition (E–J). Data are shown as mean ± s.e.m. *P < 0.05, **P < 0.01, ****P < 0.001.
VEGFr Inhibition Decreases HSPC Proliferation
The role of the VEGF signaling is well documented in angiogenesis in different diseases such as cancer and age-related macular degeneration (23–25, 52, 57). However, the contribution of VEGFr in HSPC proliferation is not well studied. To understand the role of VEGFr in hypoxia-mediated HSPC proliferation, we cultured HSPC sorted from mouse bone marrow with SU5416, a VEGFr inhibitor (58, 59), under hypoxic and normoxic conditions. VEGFr inhibition significantly decreased the expression of cell cycle genes responsible for HSPC proliferation (Figure 4A and Supplementary Figure 10A). However, SU5416 inhibits both Vegfr1 and Vegfr2. To ascertain if Vegfrr2 is also important in hypoxia-mediated HSC proliferation, we measured Vegfr2 expression in bone marrow cells of normoxic and hypoxic mice by qPCR. We did not observe any significant difference in Vegfr2 expression in bone marrow cells between these two groups of mice (Supplementary Figure 10B). To evaluate the importance of Vegfr2 in HSC proliferation, we have knocked down Vegfr2 in HSPC isolated from B6 mice using siCtl and siVegfr2 and have measured their proliferation. Vegfr2 does not have any significant effect on progenitor proliferation (Supplementary Figures 10C–F). To further examine the role of VEGFr signaling in proliferation of HSPC, we assessed proliferation of GMP in hypoxic mice injected with the VEGFr inhibitor. Compared to untreated controls, VEGFr inhibitor-injected mice had lower proportions of GMPs in the S-G2-M cell cycles (Figures 4B, C). These mice had increased numbers of quiescent cells in the G0 phase. Interestingly, VEGFr1 inhibition also increased the frequency of GMP in the G1 phase, indicating a G1 to S-G2M arrest in the absence of VEGFr signaling. Additionally, there was a significant reduction in GMP numbers in the bone marrow of hypoxic mice after VEGFr inhibition (Figure 4D).
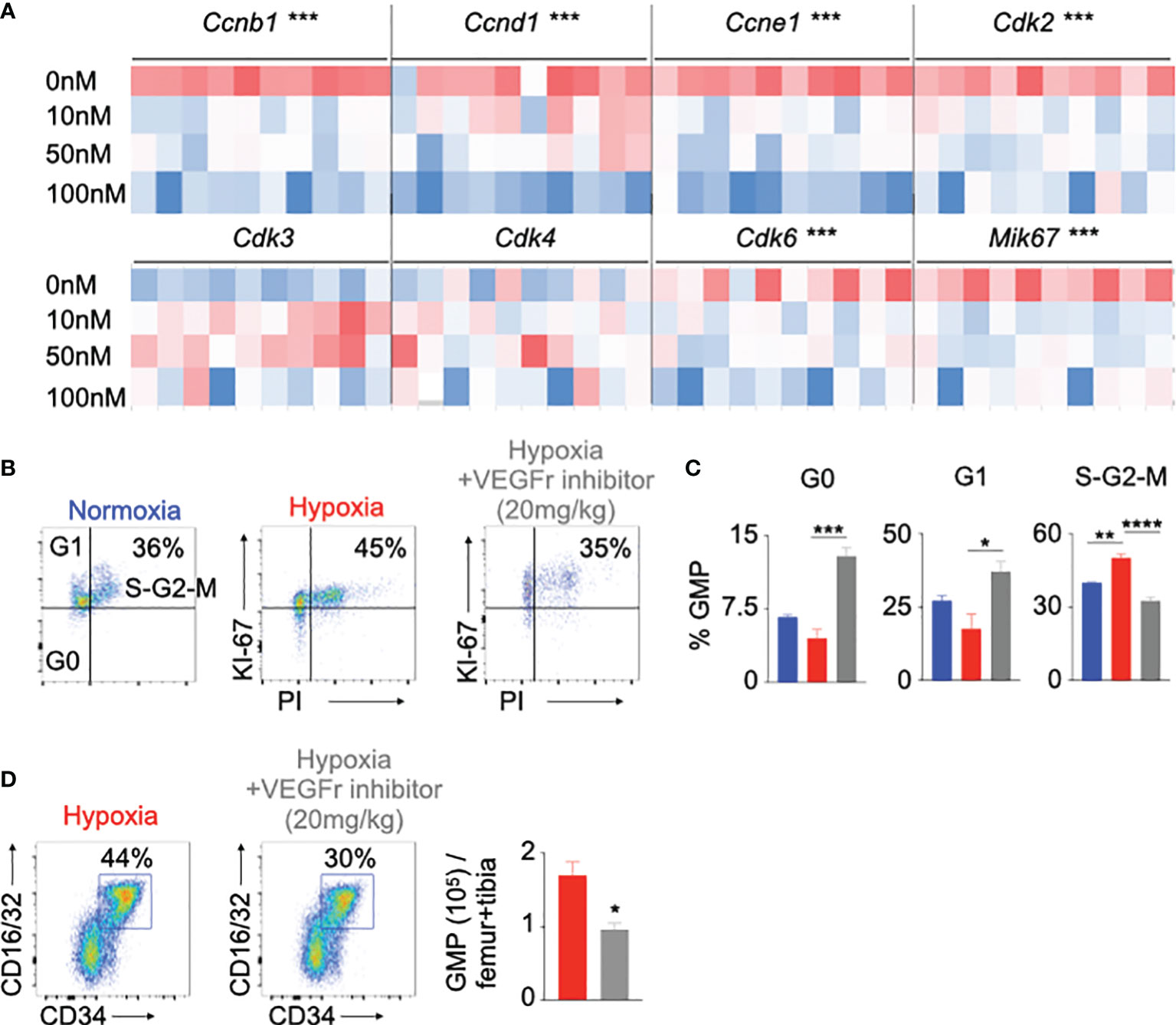
Figure 4 Vegfr inhibition decreased hematopoietic progenitor proliferation in hypoxia. (A) Heatmap depicting expression of the cell cycle genes in mouse HSPC treated with increasing amounts of a Vegfr inhibitor (SU5416, 10-100nM) or vehicle. (B-D) C57BL/6 mice were treated with either SU5416 or vehicle diluent and placed in either normoxia or hypoxia for 21 days. Bone marrow cells were analyzed using flow cytometry to assess GMP proliferation (B, C) and number (D). n = 5 replicates or mice per condition. Data are shown as mean ± s.e.m. *P < 0.05, **P <. 0.01, ***P < 0.001, ****P < 0.001.
VEGFr Inhibition Decreased Inflammation and Inflammatory Cell Numbers
We wanted to evaluate whether the decreased proliferation of HSPC after VEGFr inhibition would have an impact on inflammatory leukocyte generation. To this end, we differentiated HSPC isolated from mouse bone marrow in presence of the VEGFr inhibitor in hypoxic and normoxic conditions. VEGFr inhibition in hypoxic and normoxic HSPC significantly suppressed their differentiation into lymphoid cells, myeloid cells, B cells, monocytes and neutrophils (Figure 5A and Supplementary Figure 11A). Next, we investigated if VEGFr inhibition decreases hypoxia-induced leukocytosis in vivo. Compared to untreated control mice, VEGFr inhibitor-treated mice had decreased percentages and numbers of monocytes and neutrophils in peripheral blood and bone marrow (Supplementary Figures 11B, C and Figures 5B, C). Additionally, we utilized an additional loss of function approach since SU5416 is not specific for VEGFr1. We silenced Vegfr1 in sorted murine HSPC placed in hypoxia for 24 hours. We found that the ability of HSPC to differentiate into mature leukocytes, especially B cells, myeloid cells, monocytes and neutrophils, was reduced in HSPC treated with siVegfr1 compared to siCtl (Supplementary Figure 12A). Additionally, we found decreased expression of the cell cycle check point genes such as Cdk3, Cdk4, and Mki67 after Vegfr1 knock down (Supplementary Figure 12B). Overall, these data indicate the importance of VEGFr1 in the proliferation and differentiation of HSPC into leukocytes under hypoxic conditions.
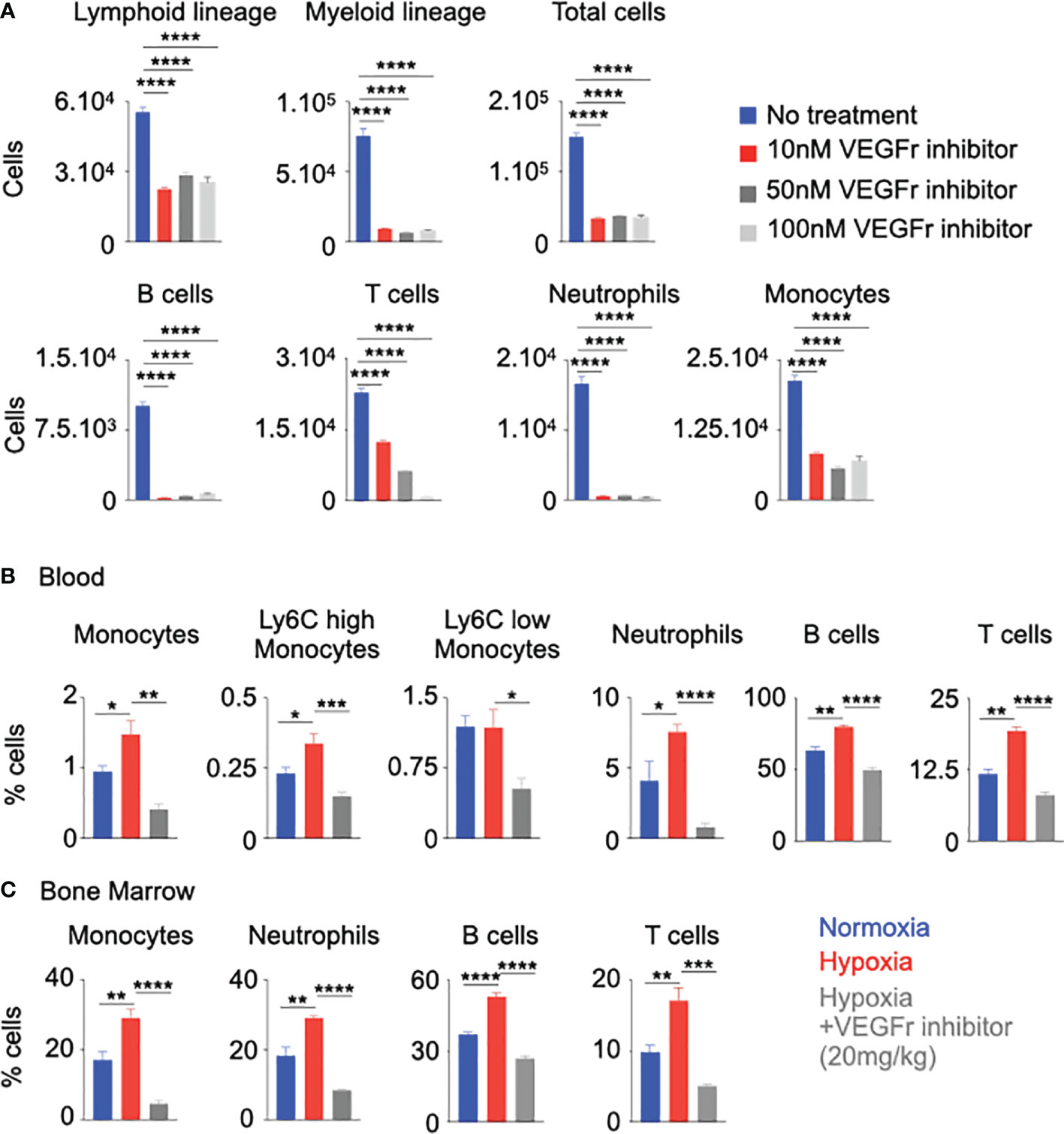
Figure 5 Vegfr inhibition decreased inflammation in hypoxia. (A) Mouse HSPC were treated with increasing amounts of a Vegfr inhibitor (SU5416, 10-100nM) or vehicle diluent (no treatment). Lineage commitment of HSPC was assessed by flow cytometry. B&C) C57BL/6 mice were treated with either a Vegfr inhibitor (SU5416) or vehicle diluent and placed in hypoxia for 21 days. Bone marrow and blood leukocytes were analyzed using flow cytometry. Percentages of blood (B) and bone marrow (C) monocytes and neutrophils were ascertained. n = 5 replicates or mice per condition. Data are shown as mean ± s.e.m. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
Patients With OSA and COPD Recapitulate the Observations Made in Hypoxic Animals
To ascertain systemic inflammation in patients with hypoxic lung diseases, we performed a retrospective chart review of patients with OSA characterized by intermittent hypoxia and COPD marked by chronic hypoxia, and age-matched control patients to enumerate inflammatory cells. The total numbers of neutrophils, lymphocytes, and monocytes were significantly increased in the patients with OSA and COPD compared to the healthy controls (Figure 6 and Supplementary Figure 13A). Additionally, the oxygen saturation levels of OSA and COPD patients included in this study were significantly reduced compared to healthy controls (Supplementary Figure 13B), indicating hypoxia in these patients.
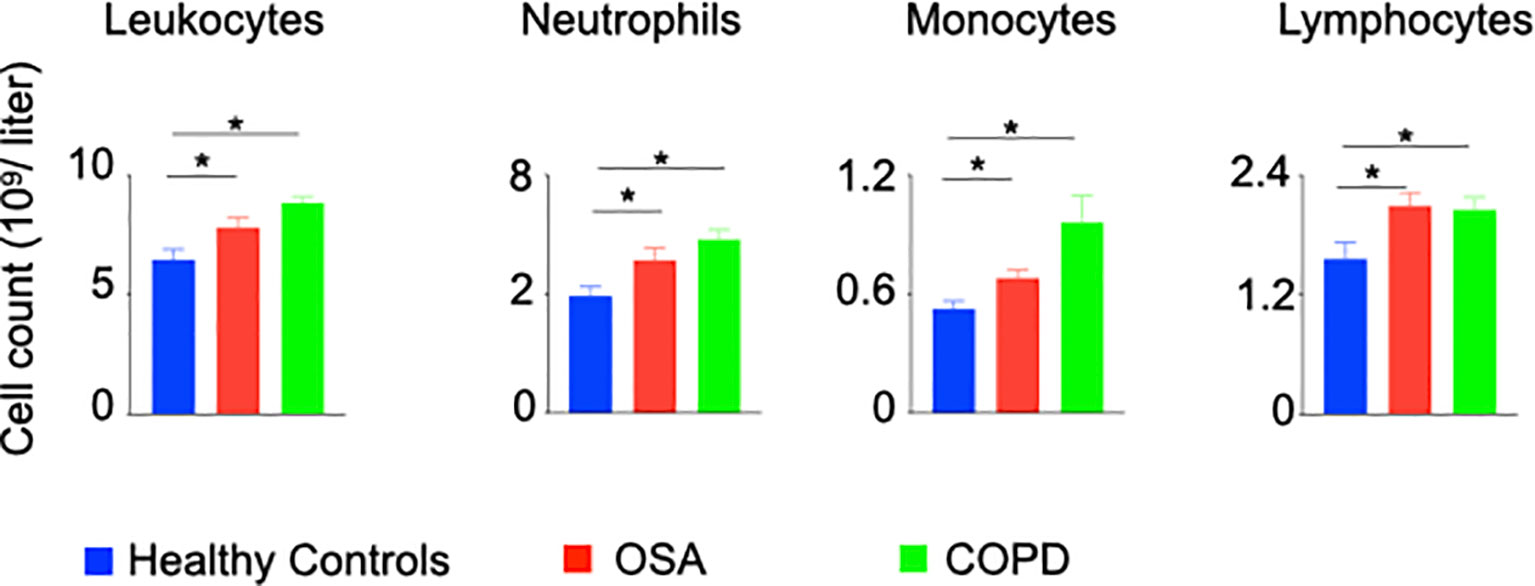
Figure 6 Patients with hypoxia have elevated numbers of inflammatory leukocytes. A restrospective chart review was performed to enumerate inflammatory leukocytes in peripheral blood of patients with OSA and COPD. n = 26 (OSA), 10 (COPD) and 22 (Healthy controls). Data are shown as mean ± s.e.m. *P < 0.05.
Discussion
Patients with hypoxic lung disease, including COPD and OSA, demonstrate chronic inflammation evidenced by elevated inflammatory cytokine levels and inflammatory leukocyte numbers. The degree of systemic inflammation portends clinical outcomes of hypoxic lung disease as well as severity of comorbid conditions (6, 11, 41, 60, 61). There is growing evidence that correction of underlying hypoxia can reduce systemic inflammation highlighting the importance of hypoxia in inflammation (62).
Consistent with serum cytokine levels observed in COPD and OSA, we observed increased levels of IL-1b, IL-6, IL-18, and TNF-a in peripheral blood of mice exposed to hypoxia. Both TNF-a and IL-6 have been shown to be independent risk factors of increased morbidity and mortality in hypoxic lung disease, particularly in COPD (63). IL-6 has been shown to promote proliferation of HSC (64), but interestingly, TNF-α has exhibited a contrasting function regulating hematopoiesis (65). Systemic inflammation has long been postulated to drive the pathogenesis of diseases such as atherosclerosis. The CANTOS trial, assessing monoclonal antibody inhibition of IL-1b, has clearly demonstrated a benefit of attenuating inflammation independent of cholesterol lowering therapies in atherosclerosis (66). Together, these findings illustrate an important role of inflammation in hypoxic lung disease-associated comorbidities.
Elevated numbers of leukocytes in peripheral blood has received recent attention as a marker of inflammation and independent risk factor for diseases such as atherosclerosis (67, 68) and diabetes (69). Lodge et al. illustrated the role of neutrophil-mediated lung damage in patients with COPD (70). Similarly, neutrophilia has been demonstrated as a key feature of obstructive sleep apnea and postulated to contribute to the pathogenesis of OSA (71). The present study demonstrates a mechanism of hypoxia-mediated expansion of inflammatory leukocytes, which likely contribute to the pathogenesis and comorbid conditions of hypoxic lung diseases.
In response to tissue injury, mature leukocytes may be mobilized from sequestered sites or generated de novo from hematopoietic tissues (72, 73). It is reasonable to postulate acute increase in recruitment of mature leukocytes from hematopoietic sites contributes to a relative paucity of leukocytes in the bone marrow and abundance in peripheral circulation. In contrast, under chronic inflammation, expansion of HSPC maintains reservoirs of leukocytes in hematopoietic tissues (74, 75). Additionally, studies have shown that HSPC expand profusely in hypoxic conditions in vitro (76–78). In line with this, we observed a symmetric expansion of leukocytes in the peripheral blood and bone marrow in hypoxic conditions.
VEGFr1 signaling in the proliferation and differentiation of endothelial cells in angiogenesis, particularly in the development of cancer (79, 80), has been well documented. However, the role of VEGF signaling in the proliferation of hematopoietic stem cells and hematopoiesis is understudied. In line with our findings, a few other studies have shown that inhibition of Vegfr1 diminished HSC cell cycling and lineage differentiation after bone marrow suppression (81), and pharmacological stabilization of HIF-1α increases HSC quiescence (82). Our study demonstrates that the proliferation of HSPC and leukocytosis under hypoxic conditions is mediated by VEGFr1 although the data do not rule out the contribution of VEGFr2 in this process. Thus, the current study identifies VEGFr1 as a novel target to dampen inflammation in diseases characterized by hypoxia. Randomized clinical trials will be required to validate the therapeutic efficacy of this target.
HIF-1α is an important regulator of VEGFA expression in local hypoxia, such as tumor microenvironment (83) and on the development and survival of the hematopoietic system (84, 85). HIF-1α also regulates VEGFA transcription, and mobilization of HSPC increases VEGF-A expression (86). Our in silico and molecular experiments demonstrated that HIF-1α directly interacts with the VEGFr1 promoters in human and mouse HSPC under hypoxic conditions contributing to increased expression of this receptor. The importance of this finding was further supported by suppression of HSPC proliferation and leukocyte differentiation after Hif1a silencing in HSPC. However, we acknowledge that we do not know if higher HSPC differentiation is due to the direct effect of hypoxia/HIF-1α or the resulting inflammation. Although our data show that Hif1a silencing decreased HSPC proliferation, HIF-1α also promotes inflammation. Congruently, our and other groups have shown that hypoxia results in systemic inflammation (37, 38, 87). Proinflammatory cytokines can drive HSPC into the cell cycle and increase their proliferation. Thus, exaggerated inflammation may increase lineage output from HSPC in hypoxic mice. Future studies are warranted to understand these mechanisms to reveal potential molecular targets to reduce inflammation in hypoxic diseases.
Data Availability Statement
Publicly available datasets were analyzed in this study. This data can be found here: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE54663.
Ethics Statement
The studies involving human participants were reviewed and approved by University of Pittsburgh Internal Review Board. The patients/participants provided their written informed consent to participate in this study. The animal study was reviewed and approved by IACUC.
Author Contributions
JF and SPO conducted experiments, data analysis, and wrote the manuscript. SBV, LLO and AA conducted experiments. SG and SPO helped with the SpO2 measurement experiment. CRK, SG, JCB and JS helped with the experiments involving the mouse model of COPD. SYC reviewed the manuscript and provided us with hypoxic chambers for culturing HSCPC. PD designed the research study and composed the manuscript.
Conflict of Interest
SYC has served as a consultant for Zogenix, Aerpio, and United Therapeutics. SYC holds research grants from Actelion and Pfizer. SYC has filed patent applications regarding the targeting of metabolism in pulmonary hypertension.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Funding
This work was supported by National Institute of Health grants R00HL121076-03, R01HL14396 7, R01HL142629, R01AG069399, AHA Transformational Project Award (19TPA34910142), AHA Innovative Project Award (19IPLOI34760566) and ALA Innovation Project Award (IA- 629694) (to PD), NIH grants R01 HL124021, HL 122596, HL 138437, and UH2/UH3 TR002073 as well as the American Heart Association Established Investigator Award 18EIA33900027 (to SC), and the VMI Postdoctoral Training Program in Translational Research and Entrepreneurship in Pulmonary and Vascular Biology T32 funded by the National, Heart, Lung and Blood Institute (NHLBI) (to JF).
Acknowledgments
We thank the NIH supported microscopy resources in the Center for Biologic Imaging (NIH grant 1S10OD019973-01). We thank The Center for Organ Recovery & Education (CORE) as well as organ donors and their families for the generous donation of tissues used in this study.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2022.882484/full#supplementary-material
References
1. Maselli DJ, Bhatt SP, Anzueto A, Bowler RP, DeMeo DL, Diaz AA, et al. Clinical Epidemiology of COPD: Insights From 10 Years of the COPDGene Study. Chest (2019) 156(2):228–38. doi: 10.1016/j.chest.2019.04.135
2. Brat K, Plutinsky M, Hejduk K, Svoboda M, Popelkova P, Zatloukal J, et al. Respiratory Parameters Predict Poor Outcome in COPD Patients, Category GOLD 2017 B. Int J Chron Obstruct Pulmon Dis (2018) 13:1037–52. doi: 10.2147/COPD.S147262
3. Abuzaid AS, Al Ashry HS, Elbadawi A, Ld H, Saad M, Elgendy IY, et al. Meta-Analysis of Cardiovascular Outcomes With Continuous Positive Airway Pressure Therapy in Patients With Obstructive Sleep Apnea. Am J Cardiol (2017) 120(4):693–9. doi: 10.1016/j.amjcard.2017.05.042
4. Geovanini GR, Wang R, Weng J, Jenny NS, Shea S, Allison M, et al. Association Between Obstructive Sleep Apnea and Cardiovascular Risk Factors: Variation by Age, Sex, and Race. The Multi-Ethnic Study of Atherosclerosis. Ann Am Thorac Soc (2018) 15(8):970–7. doi: 10.1513/AnnalsATS.201802-121OC
5. Song SO, He K, Narla RR, Kang HG, Ryu HU, Boyko EJ. Metabolic Consequences of Obstructive Sleep Apnea Especially Pertaining to Diabetes Mellitus and Insulin Sensitivity. Diabetes Metab J (2019) 43(2):144–55. doi: 10.4093/dmj.2018.0256
6. Vujic T, Nagorni O, Maric G, Popovic L, Jankovic J. Metabolic Syndrome in Patients With Chronic Obstructive Pulmonary Disease: Frequency and Relationship With Systemic Inflammation. Hippokratia (2016) 20(2):110–4.
7. Chen W, Thomas J, Sadatsafavi M, FitzGerald JM. Risk of Cardiovascular Comorbidity in Patients With Chronic Obstructive Pulmonary Disease: A Systematic Review and Meta-Analysis. Lancet Respir Med (2015) 3(8):631–9. doi: 10.1016/S2213-2600(15)00241-6
8. Cubillos-Zapata C, Hernandez-Jimenez E, Avendano-Ortiz J, Toledano V, Varela-Serrano A, Fernandez-Navarro I, et al. Obstructive Sleep Apnea Monocytes Exhibit High Levels of Vascular Endothelial Growth Factor Secretion, Augmenting Tumor Progression. Mediators Inflamm (2018) 2018:7373921. doi: 10.1155/2018/7373921
9. Chao J, Wood JG, Gonzalez NC. Alveolar Macrophages Initiate the Systemic Microvascular Inflammatory Response to Alveolar Hypoxia. Respir Physiol Neurobiol (2011) 178(3):439–48. doi: 10.1016/j.resp.2011.03.008
10. Chao J, Wood JG, Blanco VG, Gonzalez NC. The Systemic Inflammation of Alveolar Hypoxia Is Initiated by Alveolar Macrophage-Borne Mediator(s). Am J Respir Cell Mol Biol (2009) 41(5):573–82. doi: 10.1165/rcmb.2008-0417OC
11. Kheirandish-Gozal L, Gozal D. Obstructive Sleep Apnea and Inflammation: Proof of Concept Based on Two Illustrative Cytokines. Int J Mol Sci (2019) 20(3):459–77. doi: 10.3390/ijms20030459
12. Gonzalez NC, Wood JG. Alveolar Hypoxia-Induced Systemic Inflammation: What Low PO(2) Does and Does Not Do. Adv Exp Med Biol (2010) 662:27–32. doi: 10.1007/978-1-4419-1241-1_3
13. Lopez-Campos JL, Calero-Acuna C, Lopez-Ramirez C, Abad-Arranz M, Marquez-Martin E, Ortega-Ruiz F, et al. Implications of the Inflammatory Response for the Identification of Biomarkers of Chronic Obstructive Pulmonary Disease. Biomark Med (2016) 10(2):109–22. doi: 10.2217/bmm.15.87
14. MacNee W. Systemic Inflammatory Biomarkers and Co-Morbidities of Chronic Obstructive Pulmonary Disease. Ann Med (2013) 45(3):291–300. doi: 10.3109/07853890.2012.732703
15. de Winther MPJ, Lutgens E. The Link Between Hematopoiesis and Atherosclerosis. N Engl J Med (2019) 380(19):1869–71. doi: 10.1056/NEJMcibr1901397
16. Mo L, Gupta V, Modi R, Munnur K, Cameron JD, Seneviratne S, et al. Severe Obstructive Sleep Apnea Is Associated With Significant Coronary Artery Plaque Burden Independent of Traditional Cardiovascular Risk Factors. Int J Cardiovasc Imaging (2020) 36(2):347–55. doi: 10.1007/s10554-019-01710-w
17. Murphy AM, Thomas A, Crinion SJ, Kent BD, Tambuwala MM, Fabre A, et al. Intermittent Hypoxia in Obstructive Sleep Apnoea Mediates Insulin Resistance Through Adipose Tissue Inflammation. Eur Respir J (2017) 49(4):1601731–41. doi: 10.1183/13993003.01731-2016
18. Baldridge MT, King KY, Boles NC, Weksberg DC, Goodell MA. Quiescent Haematopoietic Stem Cells Are Activated by IFN-Gamma in Response to Chronic Infection. Nature (2010) 465(7299):793–7. doi: 10.1038/nature09135
19. Shi C, Jia T, Mendez-Ferrer S, Hohl TM, Serbina NV, Lipuma L, et al. Bone Marrow Mesenchymal Stem and Progenitor Cells Induce Monocyte Emigration in Response to Circulating Toll-Like Receptor Ligands. Immunity (2011) 34(4):590–601. doi: 10.1016/j.immuni.2011.02.016
20. Heidt T, Courties G, Dutta P, Sager HB, Sebas M, Iwamoto Y, et al. Differential Contribution of Monocytes to Heart Macrophages in Steady-State and After Myocardial Infarction. Circ Res (2014) 115(2):284–95. doi: 10.1161/CIRCRESAHA.115.303567
21. Zigmond E, Varol C, Farache J, Elmaliah E, Satpathy AT, Friedlander G, et al. Ly6C Hi Monocytes in the Inflamed Colon Give Rise to Proinflammatory Effector Cells and Migratory Antigen-Presenting Cells. Immunity (2012) 37(6):1076–90. doi: 10.1016/j.immuni.2012.08.026
22. Valipour A, Schreder M, Wolzt M, Saliba S, Kapiotis S, Eickhoff P, et al. Circulating Vascular Endothelial Growth Factor and Systemic Inflammatory Markers in Patients With Stable and Exacerbated Chronic Obstructive Pulmonary Disease. Clin Sci (Lond) (2008) 115(7):225–32. doi: 10.1042/CS20070382
23. Yang F, Qin Z, Shao C, Liu W, Ma L, Shu Y, et al. Association Between VEGF Gene Polymorphisms and the Susceptibility to Lung Cancer: An Updated Meta-Analysis. BioMed Res Int (2018) 2018:9271215. doi: 10.1155/2018/9271215
24. Zong S, Li H, Shi Q, Liu S, Li W, Hou F. Prognostic Significance of VEGF-C Immunohistochemical Expression in Colorectal Cancer: A Meta-Analysis. Clin Chim Acta (2016) 458:106–14. doi: 10.1016/j.cca.2016.04.037
25. Liu W, Dong Z, Hu R, Wang C. Association of Vascular Endothelial Growth Factor (VEGF) Gene Polymorphisms With Gastric Cancer and Its Development, Prognosis, and Survival. Technol Cancer Res Treat (2018) 17:1533034617753810. doi: 10.1177/1533034617753810
26. Archontogeorgis K, Nena E, Papanas N, Xanthoudaki M, Hatzizisi O, Kyriazis G, et al. Serum Levels of Vascular Endothelial Growth Factor and Insulin-Like Growth Factor Binding Protein-3 in Obstructive Sleep Apnea Patients: Effect of Continuous Positive Airway Pressure Treatment. Open Cardiovasc Med J (2015) 9:133–8. doi: 10.2174/1874192401509010133
27. Baz-Davila R, Espinoza-Jimenez A, Rodriguez-Perez MDC, Zulueta J, Varo N, Montejo A, et al. Role of HIF1A, VEGFA and VEGFR2 SNPs in the Susceptibility and Progression of COPD in a Spanish Population. PloS One (2016) 11(5):e0154998. doi: 10.1371/journal.pone.0154998
28. Hwangbo C, Lee HW, Kang H, Ju H, Wiley DS, Papangeli I, et al. Modulation of Endothelial Bone Morphogenetic Protein Receptor Type 2 Activity by Vascular Endothelial Growth Factor Receptor 3 in Pulmonary Arterial Hypertension. Circulation (2017) 135(23):2288–98. doi: 10.1161/CIRCULATIONAHA.116.025390
29. Ramakrishnan S, Anand V, Roy S. Vascular Endothelial Growth Factor Signaling in Hypoxia and Inflammation. J Neuroimmune Pharmacol (2014) 9(2):142–60. doi: 10.1007/s11481-014-9531-7
30. Fu X, Zhang F. Role of the HIF-1 Signaling Pathway in Chronic Obstructive Pulmonary Disease. Exp Ther Med (2018) 16(6):4553–61. doi: 10.3892/etm.2018.6785
31. Farid Hosseini R, Jabbari Azad F, Yousefzadeh H, Rafatpanah H, Hafizi S, Tehrani H, et al. Serum Levels of Vascular Endothelial Growth Factor in Chronic Obstructive Pulmonary Disease. Med J Islam Repub Iran (2014) 28:85.
32. Gomez-Arroyo J, Saleem SJ, Mizuno S, Syed AA, Bogaard HJ, Abbate A, et al. A Brief Overview of Mouse Models of Pulmonary Arterial Hypertension: Problems and Prospects. Am J Physiol Lung Cell Mol Physiol (2012) 302(10):L977–91. doi: 10.1152/ajplung.00362.2011
33. Vitali SH, Hansmann G, Rose C, Fernandez-Gonzalez A, Scheid A, Mitsialis SA, et al. The Sugen 5416/Hypoxia Mouse Model of Pulmonary Hypertension Revisited: Long-Term Follow-Up. Pulm Circ (2014) 4(4):619–29. doi: 10.1086/678508
34. Stenmark KR, Meyrick B, Galie N, Mooi WJ, McMurtry IF. Animal Models of Pulmonary Arterial Hypertension: The Hope for Etiological Discovery and Pharmacological Cure. Am J Physiol Lung Cell Mol Physiol (2009) 297(6):L1013–L32. doi: 10.1152/ajplung.00217.2009
35. Kliment CR, Nguyen JMK, Kaltreider MJ, Lu Y, Claypool SM, Radder JE, et al. Adenine Nucleotide Translocase Regulates Airway Epithelial Metabolism, Surface Hydration and Ciliary Function. J Cell Sci (2021) 134(4). doi: 10.1242/jcs.257162
36. Hautamaki RD, Kobayashi DK, Senior RM, Shapiro SD. Requirement for Macrophage Elastase for Cigarette Smoke-Induced Emphysema in Mice. Science (1997) 277(5334):2002–4. doi: 10.1126/science.277.5334.2002
37. Florentin J, Coppin E, Vasamsetti SB, Zhao J, Tai YY, Tang Y, et al. Inflammatory Macrophage Expansion in Pulmonary Hypertension Depends Upon Mobilization of Blood-Borne Monocytes. J Immunol (2018) 200(10):3612–25. doi: 10.4049/jimmunol.1701287
38. Florentin J, Zhao J, Tai YY, Vasamsetti SB, O’Neil SP, Kumar R, et al. Interleukin-6 Mediates Neutrophil Mobilization From Bone Marrow in Pulmonary Hypertension. Cell Mol Immunol (2021) 18(2):374–84. doi: 10.1038/s41423-020-00608-1
39. Dahl JA, Collas P. A Rapid Micro Chromatin Immunoprecipitation Assay (ChIP). Nat Protoc (2008) 3(6):1032–45. doi: 10.1038/nprot.2008.68
40. Andrijevic L, Milutinov S, Andrijevic I, Jokic D, Vukoja M. Association Between the Inflammatory Biomarkers and Left Ventricular Systolic Dysfunction in Patients With Exacerbations of Chronic Obstructive Pulmonary Disease. Balkan Med J (2017) 34(3):226–31. doi: 10.4274/balkanmedj.2016.1114
41. Thomsen M, Dahl M, Lange P, Vestbo J, Nordestgaard BG. Inflammatory Biomarkers and Comorbidities in Chronic Obstructive Pulmonary Disease. Am J Respir Crit Care Med (2012) 186(10):982–8. doi: 10.1164/rccm.201206-1113OC
42. Yaggi HK, Mittleman MA, Bravata DM, Concato J, Ware J, Stoney CM, et al. Reducing Cardiovascular Risk Through Treatment of Obstructive Sleep Apnea: 2 Methodological Approaches. Am Heart J (2016) 172:135–43. doi: 10.1016/j.ahj.2015.07.033
43. Yona S, Kim KW, Wolf Y, Mildner A, Varol D, Breker M, et al. Fate Mapping Reveals Origins and Dynamics of Monocytes and Tissue Macrophages Under Homeostasis. Immunity (2013) 38(1):79–91. doi: 10.1016/j.immuni.2012.12.001
44. Pugliese SC, Kumar S, Janssen WJ, Graham BB, Frid MG, Riddle SR, et al. A Time- and Compartment-Specific Activation of Lung Macrophages in Hypoxic Pulmonary Hypertension. J Immunol (2017) 198(12):4802–12. doi: 10.4049/jimmunol.1601692
45. Tuleta I, Stockigt F, Juergens UR, Pizarro C, Schrickel JW, Kristiansen G, et al. Intermittent Hypoxia Contributes to the Lung Damage by Increased Oxidative Stress, Inflammation, and Disbalance in Protease/Antiprotease System. Lung (2016) 194(6):1015–20. doi: 10.1007/s00408-016-9946-4
46. Kojima H, Tokunou T, Takahara Y, Sunagawa K, Hirooka Y, Ichiki T, et al. Hypoxia-Inducible Factor-1 Alpha Deletion in Myeloid Lineage Attenuates Hypoxia-Induced Pulmonary Hypertension. Physiol Rep (2019) 7(7):e14025. doi: 10.14814/phy2.14025
47. Domen J, Weissman IL. Hematopoietic Stem Cells and Other Hematopoietic Cells Show Broad Resistance to Chemotherapeutic Agents In Vivo When Overexpressing Bcl-2. Exp Hematol (2003) 31(7):631–9. doi: 10.1016/S0301-472X(03)00084-5
48. Vazquez SE, Inlay MA, Serwold T. CD201 and CD27 Identify Hematopoietic Stem and Progenitor Cells Across Multiple Murine Strains Independently of Kit and Sca-1. Exp Hematol (2015) 43(7):578–85. doi: 10.1016/j.exphem.2015.04.001
49. Pietras EM, Lakshminarasimhan R, Techner JM, Fong S, Flach J, Binnewies M, et al. Re-Entry Into Quiescence Protects Hematopoietic Stem Cells From the Killing Effect of Chronic Exposure to Type I Interferons. J Exp Med (2014) 211(2):245–62. doi: 10.1084/jem.20131043
50. Wierenga AT, Vellenga E, Schuringa JJ. Convergence of Hypoxia and Tgfβ Pathways on Cell Cycle Regulation in Human Hematopoietic Stem/Progenitor Cells. PloS One (2014) 9(3):e93494. doi: 10.1371/journal.pone.0093494
51. Semenza GL. Regulation of Hypoxia-Induced Angiogenesis: A Chaperone Escorts VEGF to the Dance. J Clin Invest (2001) 108(1):39–40. doi: 10.1172/JCI13374
52. Zhu H, Zhang S. Hypoxia Inducible Factor-1α/Vascular Endothelial Growth Factor Signaling Activation Correlates With Response to Radiotherapy and Its Inhibition Reduces Hypoxia-Induced Angiogenesis in Lung Cancer. J Cell Biochem (2018) 119(9):7707–18. doi: 10.1002/jcb.27120
53. Benita Y, Kikuchi H, Smith AD, Zhang MQ, Chung DC, Xavier RJ. An Integrative Genomics Approach Identifies Hypoxia Inducible Factor-1 (HIF-1)-Target Genes That Form the Core Response to Hypoxia. Nucleic Acids Res (2009) 37(14):4587–602. doi: 10.1093/nar/gkp425
54. Semenza GL. Hypoxia-Inducible Factors: Mediators of Cancer Progression and Targets for Cancer Therapy. Trends Pharmacol Sci (2012) 33(4):207–14. doi: 10.1016/j.tips.2012.01.005
55. Takeda N, Maemura K, Imai Y, Harada T, Kawanami D, Nojiri T, et al. Endothelial PAS Domain Protein 1 Gene Promotes Angiogenesis Through the Transactivation of Both Vascular Endothelial Growth Factor and Its Receptor, Flt-1. Circ Res (2004) 95(2):146–53. doi: 10.1161/01.RES.0000134920.10128.b4
56. Mole DR, Blancher C, Copley RR, Pollard PJ, Gleadle JM, Ragoussis J, et al. Genome-Wide Association of Hypoxia-Inducible Factor (HIF)-1alpha and HIF-2alpha DNA Binding With Expression Profiling of Hypoxia-Inducible Transcripts. J Biol Chem (2009) 284(25):16767–75. doi: 10.1074/jbc.M901790200
57. Gao S, Ma JJ, Lu C. Prognostic Significance of VEGF-C Immunohistochemical Expression in Breast Cancer: A Meta-Analysis. Tumour Biol (2014) 35(2):1523–9. doi: 10.1007/s13277-013-1211-3
58. Robinson GS, Ju M, Shih SC, Xu X, McMahon G, Caldwell RB, et al. Nonvascular Role for VEGF: VEGFR-1, 2 Activity Is Critical for Neural Retinal Development. FASEB J (2001) 15(7):1215–7. doi: 10.1096/fj.00-0598fje
59. Dikov MM, Ohm JE, Ray N, Tchekneva EE, Burlison J, Moghanaki D, et al. Differential Roles of Vascular Endothelial Growth Factor Receptors 1 and 2 in Dendritic Cell Differentiation. J Immunol (2005) 174(1):215–22. doi: 10.4049/jimmunol.174.1.215
60. Celli BR, Locantore N, Yates J, Tal-Singer R, Miller BE, Bakke P, et al. Inflammatory Biomarkers Improve Clinical Prediction of Mortality in Chronic Obstructive Pulmonary Disease. Am J Respir Crit Care Med (2012) 185(10):1065–72. doi: 10.1164/rccm.201110-1792OC
61. Kelly E, Owen CA, Pinto-Plata V, Celli BR. The Role of Systemic Inflammatory Biomarkers to Predict Mortality in Chronic Obstructive Pulmonary Disease. Expert Rev Respir Med (2013) 7(1):57–64. doi: 10.1586/ers.12.82
62. Perrini S, Cignarelli A, Quaranta VN, Falcone VA, Kounaki S, Porro S, et al. Correction of Intermittent Hypoxia Reduces Inflammation in Obese Subjects With Obstructive Sleep Apnea. JCI Insight (2017) 2(17). doi: 10.1172/jci.insight.94379
63. Bradford E, Jacobson S, Varasteh J, Comellas AP, Woodruff P, O’Neal W, et al. The Value of Blood Cytokines and Chemokines in Assessing COPD. Respir Res (2017) 18(1):180. doi: 10.1186/s12931-017-0662-2
64. Tie R, Li H, Cai S, Liang Z, Shan W, Wang B, et al. Interleukin-6 Signaling Regulates Hematopoietic Stem Cell Emergence. Exp Mol Med (2019) 51(10):1–12. doi: 10.1038/s12276-019-0320-5
65. Pronk CJ, Veiby OP, Bryder D, Jacobsen SE. Tumor Necrosis Factor Restricts Hematopoietic Stem Cell Activity in Mice: Involvement of Two Distinct Receptors. J Exp Med (2011) 208(8):1563–70. doi: 10.1084/jem.20110752
66. Ridker PM. Inflammation, Cardiovascular Disease and Cancer: Moving Toward Predictive Medicine. CMAJ (2017) 189(10):E382–E3. doi: 10.1503/cmaj.161033
67. Kim JH, Lim S, Park KS, Jang HC, Choi SH. Total and Differential WBC Counts are Related With Coronary Artery Atherosclerosis and Increase the Risk for Cardiovascular Disease in Koreans. PloS One (2017) 12(7):e0180332. doi: 10.1371/journal.pone.0180332
68. Welsh C, Welsh P, Mark PB, Celis-Morales CA, Lewsey J, Gray SR, et al. Association of Total and Differential Leukocyte Counts With Cardiovascular Disease and Mortality in the UK Biobank. Arterioscler Thromb Vasc Biol (2018) 38(6):1415–23. doi: 10.1161/ATVBAHA.118.310945
69. Koo HK, Kang HK, Song P, Park HK, Lee SS, Jung H. Systemic White Blood Cell Count as a Biomarker Associated With Severity of Chronic Obstructive Lung Disease. Tuberc Respir Dis (Seoul) (2017) 80(3):304–10. doi: 10.4046/trd.2017.80.3.304
70. Lodge KM, Hoenderdos K, Robbins AJ, Chilvers ER, Li W, Condliffe AM. S114 Hypoxia Drives Neutrophil-Mediated Endothelial Damage in Copd. Thorax (2017) 72(Suppl 3):A69. doi: 10.1136/thoraxjnl-2017-210983.120
71. Geovanini GR, Wang R, Weng J, Tracy R, Jenny NS, Goldberger AL, et al. Elevations in Neutrophils With Obstructive Sleep Apnea: The Multi-Ethnic Study of Atherosclerosis (MESA). Int J Cardiol (2018) 257:318–23. doi: 10.1016/j.ijcard.2017.10.121
72. Vasamsetti SB, Florentin J, Coppin E, Stiekema LCA, Zheng KH, Nisar MU, et al. Sympathetic Neuronal Activation Triggers Myeloid Progenitor Proliferation and Differentiation. Immunity (2018) 49(1):93–106.e7. doi: 10.1016/j.immuni.2018.05.004
73. Li B, Cohen A, Hudson TE, Motlagh D, Amrani DL, Duffield JS. Mobilized Human Hematopoietic Stem/Progenitor Cells Promote Kidney Repair After Ischemia/Reperfusion Injury. Circulation (2010) 121(20):2211–20. doi: 10.1161/CIRCULATIONAHA.109.928796
74. Boettcher S, Manz MG. Regulation of Inflammation- and Infection-Driven Hematopoiesis. Trends Immunol (2017) 38(5):345–57. doi: 10.1016/j.it.2017.01.004
75. Fuchs A, Monlish DA, Ghosh S, Chang S-W, Bochicchio GV, Schuettpelz LG, et al. Trauma Induces Emergency Hematopoiesis Through IL-1/MyD88–Dependent Production of G-CSF. J Immunol (2019) 202(10):3020–32. doi: 10.4049/jimmunol.1801456
76. Cipolleschi MG, Dello Sbarba P, Olivotto M. The Role of Hypoxia in the Maintenance of Hematopoietic Stem Cells. Blood (1993) 82(7):2031–7. doi: 10.1182/blood.V82.7.2031.bloodjournal8272031
77. Ivanović Z, Bartolozzi B, Bernabei PA, Cipolleschi MG, Rovida E, Milenković P, et al. Incubation of Murine Bone Marrow Cells in Hypoxia Ensures the Maintenance of Marrow-Repopulating Ability Together With the Expansion of Committed Progenitors. Br J Haematol (2000) 108(2):424–9. doi: 10.1046/j.1365-2141.2000.01842.x
78. Danet GH, Pan Y, Luongo JL, Bonnet DA, Simon MC. Expansion of Human SCID-Repopulating Cells Under Hypoxic Conditions. J Clin Invest (2003) 112(1):126–35. doi: 10.1172/JCI17669
79. Sui H, Zhao J, Zhou L, Wen H, Deng W, Li C, et al. Tanshinone IIA Inhibits Beta-Catenin/VEGF-Mediated Angiogenesis by Targeting TGF-Beta1 in Normoxic and HIF-1alpha in Hypoxic Microenvironments in Human Colorectal Cancer. Cancer Lett (2017) 403:86–97. doi: 10.1016/j.canlet.2017.05.013
80. Bao B, Ali S, Ahmad A, Azmi AS, Li Y, Banerjee S, et al. Hypoxia-Induced Aggressiveness of Pancreatic Cancer Cells Is Due to Increased Expression of VEGF, IL-6 and miR-21, Which Can Be Attenuated by CDF Treatment. PloS One (2012) 7(12):e50165. doi: 10.1371/journal.pone.0050165
81. Hattori K, Heissig B, Wu Y, Dias S, Tejada R, Ferris B, et al. Placental Growth Factor Reconstitutes Hematopoiesis by Recruiting VEGFR1(+) Stem Cells From Bone-Marrow Microenvironment. Nat Med (2002) 8(8):841–9. doi: 10.1038/nm740
82. Forristal CE, Winkler IG, Nowlan B, Barbier V, Walkinshaw G, Levesque JP. Pharmacologic Stabilization of HIF-1α Increases Hematopoietic Stem Cell Quiescence In Vivo and Accelerates Blood Recovery After Severe Irradiation. Blood (2013) 121(5):759–69. doi: 10.1182/blood-2012-02-408419
83. Chen X, Liu J, He B, Li Y, Liu S, Wu B, et al. Vascular Endothelial Growth Factor (VEGF) Regulation by Hypoxia Inducible Factor-1 Alpha (HIF1A) Starts and Peaks During Endometrial Breakdown, Not Repair, in a Mouse Menstrual-Like Model. Hum Reprod (2015) 30(9):2160–70. doi: 10.1093/humrep/dev156
84. Adelman DM, Maltepe E, Simon MC. Multilineage Embryonic Hematopoiesis Requires Hypoxic ARNT Activity. Genes Dev (1999) 13(19):2478–83. doi: 10.1101/gad.13.19.2478
85. Scortegagna M, Morris MA, Oktay Y, Bennett M, Garcia JA. The HIF Family Member EPAS1/HIF-2alpha Is Required for Normal Hematopoiesis in Mice. Blood (2003) 102(5):1634–40. doi: 10.1182/blood-2003-02-0448
86. Lévesque JP, Winkler IG, Hendy J, Williams B, Helwani F, Barbier V, et al. Hematopoietic Progenitor Cell Mobilization Results in Hypoxia With Increased Hypoxia-Inducible Transcription Factor-1 Alpha and Vascular Endothelial Growth Factor A in Bone Marrow. Stem Cells (2007) 25(8):1954–65. doi: 10.1634/stemcells.2006-0688
Keywords: VEGFR1, hematopoietic progenitor, intermittent hypoxia, inflammation, innate immune cell
Citation: Florentin J, O’Neil SP, Ohayon LL, Uddin A, Vasamsetti SB, Arunkumar A, Ghosh S, Boatz JC, Sui J, Kliment CR, Chan SY and Dutta P (2022) VEGF Receptor 1 Promotes Hypoxia-Induced Hematopoietic Progenitor Proliferation and Differentiation. Front. Immunol. 13:882484. doi: 10.3389/fimmu.2022.882484
Received: 23 February 2022; Accepted: 19 April 2022;
Published: 12 May 2022.
Edited by:
Philippe Saas, INSERM U1098 Interactions Hôte-Greffon-Tumeur and Ingénierie Cellulaire et Génique, FranceReviewed by:
Kavita Bisht, The University of Queensland, AustraliaLaurent Delva, INSERM U1231 Lipides, Nutrition, Cancer (LNC), France
Copyright © 2022 Florentin, O’Neil, Ohayon, Uddin, Vasamsetti, Arunkumar, Ghosh, Boatz, Sui, Kliment, Chan and Dutta. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Partha Dutta, ZHV0dGFwYUBwaXR0LmVkdQ==
†These authors have contributed equally to this work