
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
HYPOTHESIS AND THEORY article
Front. Immunol., 12 May 2022
Sec. Viral Immunology
Volume 13 - 2022 | https://doi.org/10.3389/fimmu.2022.880961
This article is part of the Research TopicDefinition of the Immune Parameters Related to COVID-19 SeverityView all 45 articles
 Karim M. Aloul1
Karim M. Aloul1 Josefine Eilsø Nielsen1,2
Josefine Eilsø Nielsen1,2 Erwin B. Defensor3
Erwin B. Defensor3 Jennifer S. Lin1
Jennifer S. Lin1 John A. Fortkort1Mehrdad Shamloo3
John A. Fortkort1Mehrdad Shamloo3 Jeffrey D. Cirillo4
Jeffrey D. Cirillo4 Adrian F. Gombart5,6
Adrian F. Gombart5,6 Annelise E. Barron1*
Annelise E. Barron1*COVID-19 is characterized by hyperactivation by inflammatory cytokines and recruitment of macrophages, neutrophils, and other immune cells, all hallmarks of a strong inflammatory response that can lead to severe complications and multi-organ damage. Mortality in COVID-19 patients is associated with a high prevalence of neutrophil extracellular trap (NET) formation and microthrombosis that are exacerbated by hyperglycemia, diabetes, and old age. SARS-CoV-2 infection in humans and non-human primates have revealed long-term neurological consequences of COVID-19, possibly concomitant with the formation of Lewy bodies in the brain and invasion of the nervous system via the olfactory bulb. In this paper, we review the relevance of the human cathelicidin LL-37 in SARS-CoV-2 infections. LL-37 is an immunomodulatory, host defense peptide with direct anti-SARS-CoV-2 activity, and pleiotropic effects on the inflammatory response, neovascularization, Lewy body formation, and pancreatic islet cell function. The bioactive form of vitamin D and a number of other compounds induce LL-37 expression and one might predict its upregulation, could reduce the prevalence of severe COVID-19. We hypothesize upregulation of LL-37 will act therapeutically, facilitating efficient NET clearance by macrophages, speeding endothelial repair after inflammatory tissue damage, preventing α-synuclein aggregation, and supporting blood-glucose level stabilization by facilitating insulin release and islet β-cell neogenesis. In addition, it has been postulated that LL-37 can directly bind the S1 domain of SARS-CoV-2, mask angiotensin converting enzyme 2 (ACE2) receptors, and limit SARS-CoV-2 infection. Purposeful upregulation of LL-37 could also serve as a preventative and therapeutic strategy for SARS-CoV-2 infections.
The virus SARS-CoV-2 has caused more than 6 million deaths worldwide since its arrival in December of 2019 (1). SARS-CoV-2 has many features that make it highly infectious including its glycoproteins, rapid entry through furin cleavage and TMPRSS2 (2–4), and suppression of host translation through Nsp1 (5–7). In response to the COVID-19 pandemic, the scientific community rallied to create new therapeutics and evaluate the effectiveness of any strategies that had been previously developed.
In humans, the cathelicidin antimicrobial peptide (CAMP) gene encodes the pro-protein hCAP-18. Proteinase 3-mediated extracellular cleavage processes hCAP18 into the active 37 amino acid peptide, LL-37 (8). LL-37 is an amphipathic alpha-helical peptide that carries a positive charge of +6 at physiological pH (structure and chemical sequence are displayed in Figure 1). Many different cell types including barrier epithelial cells, macrophages, and neutrophils express this pro-protein and peptide throughout the body (10). The vitamin D pathway primarily regulates the CAMP gene (11–14). Recognition of bacterial or viral pathogens by Toll-like receptors (TLRs) activates cells to metabolize 25-hydroxyvitamin D [25(OH)D] to the active 1α,25-dihydroxyvitamin D [1,25(OH)2D]. This active form of vitamin D, binds to the vitamin D receptor (VDR), thus inducing the CAMP gene and numerous other vitamin D target genes involved in the immune response (14). Monocytes, macrophages and lung epithelial cells upregulate LL-37 expression via TLR-mediated activation of the vitamin D pathway (14–16).

Figure 1 (A) The structure of (13)C,(15)N-labeled LL-37 determined by three-dimensional triple-resonance NMR spectroscopy for LL-37 in complex with micelles. (9) LL-37’s alpha-helical secondary structure is evident. (B) The 37 amino acid sequence of LL-37. At physiological pH LL-37 has a resulting net charge of +6.
LL-37 inhibits the propagation of SARS-CoV-2 through a direct mechanism. In silico docking studies have shown that LL-37 binds directly with the angiotensin converting enzyme-2 (ACE-2) binding domain that is critical to SARS-CoV-2 entry to host cells (17–19). These results were corroborated by in vitro and in vivo experimentation that found LL-37 not only blocks the receptor-binding domain (RBD) of SARS-CoV-2, but also cloaks the ACE-2 receptor preventing pseudovirion infection in cell culture and after intranasal application of LL-37 in mice (20). LL-37 also combats other viruses, such as influenza, rhinovirus, and respiratory syncytial virus, by causing disruption of viral membranes (21–24).
LL-37 functions as an antiviral and antibacterial peptide by inhibiting early steps in the viral replication cycle and perforating cytoplasmic bacterial membranes (25–27). In addition to penetrating bacterial membranes, LL-37 prevents biofilm formation and enhances bacterial phagocytosis (28, 29). LL-37 also kills Candida albicans, most effectively as a cleavage fragment RK-31 (30). Covid patients in the ICU for more than a few days commonly suffer from co-infections, and often with resistant species of bacteria and fungi, worsening prognosis (31–33).
In addition to its direct antimicrobial function, LL-37 modulates immune response and influences inflammation, cell proliferation and migration, wound healing, angiogenesis and the release of cytokines and histamine (34). Furthermore, recent studies indicate LL-37 plays an important role in neutrophil NETosis, which in turn can affect the formation and clearance of microthrombi (35–37). In effective NETosis, neutrophils respond to inflammatory stimuli by migrating to the infected tissue and decondensing their nuclear and mitochondrial DNA lined with granule proteins that incapacitate pathogens (NETs), followed by plasma rupture and NET release. These released NETs can trap the pathogens, which are subsequently cleared by DNase 1 and macrophages (38, 39). In COVID-19 patients, we see over accumulated NETs and that healthy neutrophils are more likely to engage in NETosis when prompted with SARS-CoV-2 patient serum (40). We hypothesis that LL-37 is important in both regulating the activation and clearance of NETs. These multiple functions are potentially highly relevant to SARS-CoV-2 infection and ameliorating COVID-19 symptoms and suggest LL-37 could function as a powerful therapeutic agent.
Studies from around the world investigating the correlation between health markers and COVID-19 severity have found statistically significant differences in serum 25-hydroxyvitamin D [25(OH)D] levels between patients having more and less severe COVID-19 outcomes, but others claim no correlation between serum 25-hydroxyvitamin D [25(OH)D] levels and infection or death rates (41–46) and many others discuss the therapeutic effects of vitamin D3 supplementation, referencing upregulation of cathelicidin gene expression and its antiviral capacity as a key component of the therapeutic and prophylactic power of vitamin D3 (44, 45, 47).
The effects of LL-37 are not limited to its ability to inhibit viral replication and infection. In this paper, we offer hypotheses rooted in previously published work to discuss the potential therapeutic and prophylactic uses of LL-37 as an effective tool in ameliorating COVID-19 pathology and reducing severe effects of COVID-19 infections. In addition, we describe strategies to regulate its expression with small molecules to achieve these goals.
The lethality of SARS-CoV-2 is often attributed to its ability to induce thrombosis. Autopsies of COVID-19 patients reveal thrombosis in many of the narrow vessels as the primary cause of death. In one study involving autopsies of 10 COVID-19 patients, thrombosis and microangiopathy in the small vessels and capillaries led to an associated haemorrhage that significantly contributed to death. The lungs were found to have entangled neutrophils entrapped in fibrin and platelets forming thrombi in alveolar capillaries (38). Another study involving autopsies of 11 COVID-19 patients found thrombosis in the pulmonary arteries of all patients. This thrombosis was associated with heart attacks in eight of the patients and bronchopneumonia in six of the patients (48). A meta-analysis of 341 autopsies of COVID-19 patients bolstered the finding of thrombi in microvessels of the lungs, and additionally noted alveolar damage resulting in hyaline membrane formation (49). A study of blood from COVID-19 patients found that NET clearance was diminished and that these NETs instigate inflammation through interactions with anti-NET antibodies and macrophages. They found complications of NETs to include induction of alveolar cell apoptosis, mucus plugs, and capilaritis (50). These thrombi and observed alveolar damage develop from a complex interplay of inflammation and dysregulation of homeostasis.
One potential factor in this interplay is vasoconstriction in the lungs and other parts of the body due to the infection of pericytes by SARS-CoV-2. The binding domain affinity of SARS-CoV-2 for ACE-2 prevents the conversion of angiotensin II to angiotensin-(1-7), instigating constrictive behavior in pericytes. This effect has been observed in the brain of a Syrian golden hamster, which has an ACE2 sequence similar to human ACE2, and is hypothesized to occur in the pericytes of the heart and the kidney (51). Dysregulation of pericytes by the S protein of SARS-Cov-2 was also observed in pericytes sourced from human myocardial tissue (52). Constrictive behavior in pericytes present in the lungs may contribute to pulmonary thrombosis in COVID-19 patients by the mechanism discussed above, or through another interaction with inflammation pathways. Pericytes are associated with hypertension in vessels of the lungs of humans and rat models and increase in proliferation preceding hypertension after interaction with the inflammatory cytokine interleukin-6 (IL-6) (53). This cytokine is correlated with more severe COVID-19 cases (54).
NETs are nuclear and mitochondrial DNA strands expelled from neutrophils through a process called NETosis. These strands form the backbone of weblike complexes studded with various peptides and proteins such as histones, lactoferrin, myeloperoxidase, neutrophil elastase, High Mobility Group Box 1, and LL-37 (39). The resulting NET complexes can bind and destroy infected cells, viruses, bacteria, and other pathogens around the neutrophils by exposing them to these peptides and proteins (39).
Beyond protecting against infectious agents, NETs can also have negative impacts on physiology by promoting tissue damage and thrombogenesis. These effects have previously been observed in vitro in neutrophils responding to endothelial damage in Escherichia coli infections (55). Toll-like receptor 4 (TLR4), which is present on the surface of platelets and activated by lipopolysaccharides (LPS) secreted during bacterial infection, encourages binding to neutrophils and promotes NETosis events in pulmonary capillaries (55). NETs can also adhere to platelets and form thrombi, and the histone proteins in NETs are sufficient to nucleate thrombi that matches those of deep vein thrombosis based on extracellular DNA presence and histone concentration in thrombi, as shown in a baboon animal model (36). The SARS-CoV-2 virus directly induces NETosis in healthy neutrophils isolated from patient serum, which demonstrates that COVID-19 patients face increased neutrophil NETosis rates as a direct result of the infection (40). This formation of thrombus scaffolds associated with increased NET production is compounded with increased thrombotic risk due to activation of the complement cascade/coagulation system by SARS-CoV-2 (56). Neutrophils from COVID-19 patients have been found to carry tissue factor (TF), an integral membrane protein involved in blood coagulation, and express elevated TF that is found within NETs (57).
The combination of vasoconstriction, nucleation of thrombi by SARS-CoV-2 induced NETs, and activation of the cascade/coagulation system is important to the pathology of COVID-19. Investigations into the histopathology of COVID-19 patients found vascular occlusions caused by NETs occurring in lung, kidney and liver tissue, and these occlusions disrupted circulation and induced endothelial damage (58). In lung specimens from patients with influenza and SARS-CoV-2 infection, researchers found neutrophil recruitment, NETosis, and subsequent immunothrombosis to be typical for severe COVID-19, but less prominent in influenza pneumonia (59). In Germany, a study of COVID-19 patients and healthy controls noted a significantly increase in NET markers in the plasma of COVID-19 patients (59, 60). Epithelial damage due to neutrophils activated by SARS-CoV-2 caused apoptosis of the A549 epithelial cell line, and this apoptosis was larger in magnitude than what was observed in neutrophils that were not activated by SARS-CoV-2. The neutrophils activated by SARS-CoV-2 released more NETs and were more cytotoxic than cells not activated by SARS-CoV-2 (61). NETs can also contribute to cytokine storm, a potentially fatal overstimulation of inflammation responses, through the stimulation of amongst others IL-6 (62). The foregoing factors—thrombosis, contribution to cytokine storm, and the impact of the granule proteins released by neutrophil congregations on endothelial cells— explain why increased levels of neutrophils are such a strong clinical indicator of severe COVID-19 outcomes (63). Consistent with this, in one recent analysis of a potentially fatal large vessel thrombus in a 28-year-old woman with COVID-19 infection, investigators ruled out all other instigators of thrombosis except infection, and they found the thrombus to have high neutrophil counts and a predominance of platelets (64).
The impact of age and other comorbidities on NETosis may explain the observed association with COVID-19. Researchers have identified chronic low-grade sterile inflammation, i.e. not caused by a pathogen, in elderly populations. High baseline serum concentrations of C reactive protein (CRP), IL-6, IL-8 and other cytokines characterize this inflammation (65). SARS-CoV-2 infection also appears to increase the production of IL-6 and IL-8, since higher levels of these cytokines are associated with more severe disease pathophysiology in COVID-19 patients (54). Amplification of these cytokines has also been observed in epithelial cells infected by previous coronaviruses, specifically SARS-CoV (66, 67). IL-8 chemoattracts neutrophils and T cells (68). IL-6 helps differentiate CD4+ T-cells, triggers the release of platelets from bone marrow, and reduces serological iron in response to lesions (69). Elevated IL-6 and IL-8 levels in the proximity of infected epithelial cells may lead to an increased concentration of neutrophils and platelets, and may thus result in thrombi instigated by the NET-nucleated thrombi interactions discussed above.
Based on the foregoing, we argue that NETs are highly relevant to COVID-19 pathology. Since neutrophil NETs are not observed to accumulate in extracellular regions in healthy tissues, physiological processes must exist for their removal after they have performed their immunological function. Although these processes are still not fully understood, some aspects of NET removal, and the cells that recycle them, have been elucidated. Current literature implicates deoxyribonuclease 1 (DNase 1), an enzyme that allows for the cleavage and modification of DNA, as an essential tool in NET removal. DNase helps with the degradation of the cellular and mitochondrial DNA that comprises the backbones of NETs. Curiously, while DNase can remove extracellular DNA, it failed to remove the majority of NET components adhered to the glycoproteins and glycosaminoglycans covering the endothelium (glycocalyx) which caused liver damage in a mice MRSA sepsis model (70). Moreover, simple upregulation of DNAse is insufficient to achieve NET removal, since DNAse by itself does not completely degrade NETs (71). Indeed, upregulating DNAse 1 could be detrimental, since NET fragments can boost certain bacterial coinfections such as Haemophilus influenzae (72, 73). Further elucidation of the mechanisms by which NET removal is achieved is critical to the development of procedures for effectively reducing NETosis in COVID-19 patients, and underscore the need for a greater understanding of the processes by which NET proteins that attach to the endothelium can reduce host tissue damage.
After NET processing, NET removal is achieved by macrophages through phagocytosis. Macrophages in the M2 state process the NETs and induce a pro-inflammatory response by releasing tumor necrosis factor-alpha (TNF-α), interferon gamma (IFN-γ), chemokine ligand 8 (CXCL8), chemokine ligand 10 (CXCL10/IP-10), chemokine ligand 12 (CXCL12/SDF-1) and clear extracellular DNA (74). Macrophages in the M1 state spew their own external DNA in a PAD-4 dependent manner during the initial interaction with NETosis, then utilize caspase activated DNase 1 to process the extDNA, and thereby clear extracellular DNA (extDNA) in roughly 24 hours. During the late phase this happens through a non-inflammatory mechanism (74). PAD4 catalyzes the conversion of protein-bound arginine into citrulline, resulting in a loss of positive charge thereby affecting chromatin structure. This distinction between macrophage states is particularly important for COVID-19. Indeed, it has been argued that macrophage activation and dysfunction is the key driver of COVID-19, and macrophage chemokine signatures indicate they are in the M1 state (74, 75). The issue of whether extracellular DNA from M1 macrophages can nucleate thrombi is important for determining major instigators of thrombosis in COVID-19 patients. The foregoing two-step process may represent an incomplete picture of NET clearance.
Although clearance of NETs requires LL-37, the specific mechanisms involved are unclear. Removal of all the proteins attached to the DNA and histone backbone of the NETs abrogates phagocytosis by macrophages. Incubating these “naked” NETs with LL-37 restored in vitro phagocytosis of NETs, thus showing the importance of LL-37 in this process (35).
The existing literature reveals potentially relevant roles of LL-37 in NET clearance. LL-37 facilitates the in-vitro endocytosis of extDNA by dendritic cells through a process of DNA aggregation and condensation (76). LL-37 participates in the binding (through electrostatic forces), condensation, and uptake of extDNA in in vitro studies involving bacterial lysis and mammalian cell responses (77–79). The highly positive net charge of LL-37 (+6 at physiologically relevant ranges of pH) promotes interaction with DNA. Here, it is notable that DNA and LL-37 are both helical molecules, therefore, in some ways; they are structurally similar to each other. Their opposite charges allow them to interact via attractive electrostatic forces, a property allowing DNA condensation with LL-37 and promoting efficient phagocytosis by macrophages (70). We hypothesize that LL-37 aids clearance of NETs released by neutrophils and extDNA of M1 macrophages in COVID-19 patients, by binding to extDNA and condensing it into denser assemblies that activated macrophages can more effectively phagocytose. This may explain how administration of LL-37 helped clear histone-DNA complexes released from NETs in a murine sepsis model (80). Macrophages themselves can release LL-37 to facilitate this process further.
LL-37 also interacts with macrophages in other ways. It may serve as a signaling molecule for macrophages in the clearance of NETs. LL-37 regulates autophagy by macrophages in-vitro by activating transcription of autophagy-related genes (81). LL-37 also neutralizes LPS-mediated activation of macrophages and drastically reduces their production of TNF-α and IL-6 (82, 83). In some pathologies such as sepsis, LL-37 modulates many of the same cytokines seen in COVID-19 infections. LL-37 immunomodulation reduces levels of IL-6, TNF-α, IL-1β, and macrophage pyroptosis in sepsis-induced mice (84). In addition, LL-37 inhibits IL-6 production in macrophages treated with IFN-γ by inhibiting the p65 NF-κB signaling pathway (85). IL-6 is of particular concern because it has been identified as one of the most prominent cytokines in severe COVID-19 infections (54). Further investigation of LL-37 as a therapeutic to minimize macrophage dysfunction and the cytokine production associated with Macrophage Activation Syndrome (MAS) is warranted (86).
In addition to regulating macrophage activation, preincubation of influenza A virus (IAV) with LL-37 was shown to reduce expression of inflammatory cytokine IL-8 by neutrophils; IL-8 elevation is associated with more severe COVID-19 (54, 87). We hypothesize LL-37 is critical to the amelioration of COVID-19 and its sequelae through the condensation of neutrophilic DNA released in NETs, prevention of cytokine storm, and the signaling of macrophages to clear NETs as shown in Figure 2.
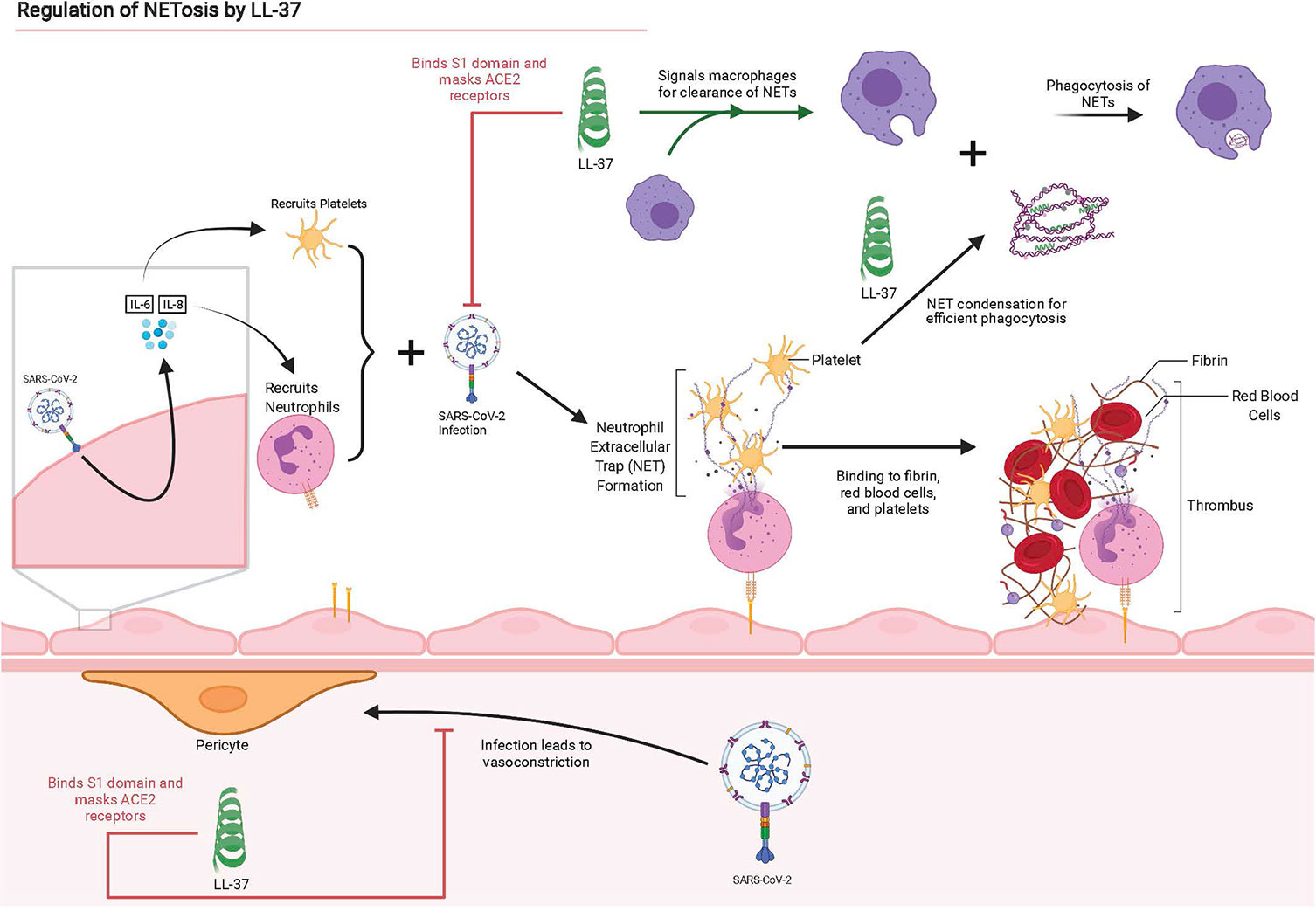
Figure 2 Graphical summary of interactions between SARS-CoV-2 and lung epithelium, instigating NET production and the disruption or antagonistic behavior of LL-37 for these interactions. Adapted from “The Propagation of Immunothrombosis by Leukocytes and Platelets”, by BioRender.com (2021). Retrieved from https://app.biorender.com/biorender-templates.
The power of citrullination is relevant to this discussion. Air pollution increases the citrullination of proteins and the formation of autoantibodies to these citrullinated proteins (88). This correlates with increased inhibition of NET clearance and defective NETs by a few mechanisms. The enzyme PAD4 catalyzes citrullination and decondensation of chromatin-releasing DNA and histones, which are expelled as NETs (89). As noted above, PAD4 facilitates extDNA release in macrophages (74). In addition, PAD4 can citrullinate LL-37, alter its antiviral activity, and increase the immune response by epithelial cells (90). In addition to air pollution, PAD4 is overexpressed in neutrophils from diabetic patients, making such neutrophils more likely to undergo NETosis and lose antiviral activity due to LL-37 citrullination. This modification also reduces the wound healing impact of LL-37 in instigating neovascularization and angiogenesis (91).
Another important consideration is that LL-37 helps induce and stabilize NETs. LL-37 is critical in the formation of NETs in vitro, which makes it a potential target for inhibiting the expulsion of NETs (37). It has also been found to increase NET production in response to IAV pre-incubated with LL-37, although research into its effects when introduced post-infection are undetermined (87). In one study, phorbol myristate acetate (PMA) was used to incite NET release in cultures of neutrophils. In some cultures, NET production doubled when treated with 10 μM of LL-37. In studies performed by Neumann et al. using parallel assays with a random LL-37 fragment library, a positive correlation was observed between NET induction and the hydrophobicity of LL-37 fragments, thus suggesting that hydrophobicity plays a role in the induction of NETs (37). Of course, one should critically analyze studies utilizing PMA for NET induction because differences in inflammation signatures from pathogens versus PMA induction exist.
LL-37 is essential to NET survival and persistence once deployed, because LL-37 inclusion within NET structures protects neutrophil DNA from cleavage by bacterial nucleases, including Staphylococcus aureus and Streptococcus pneumoniae (92). In this study Neumann et al., again, used parallel assays with a random LL-37 fragment library and found that cationicity was a critical factor to the function of LL-37 in protecting neutrophil DNA from cleavage by bacterial nucleases (92). While it is unclear whether LL-37 prevents degradation by human nucleases, it has been found that LL-37, and cationic antimicrobial peptides in general, help to stabilize neutrophil-derived DNA and NETs against bacterial nuclease degradation.
Another concern of LL-37 is a possible contribution to extracellular DNA concentrations. Previous studies have found that LL-37 induces macrophages toward M1 differentiation which we have already seen produce extracellular DNA similar to that seen in NETs since both are released via PAD4 dependent mechanisms (74, 93). If this extracellular DNA nucleates thrombi such as NETs or contributes to non-productive inflammation, then experiments to determine the ability of LL-37 to aid in phagocytosis of extracellular DNA as NETs should be weighed against the ability of LL-37 to stimulate extracellular DNA release from macrophages. The ability of LL-37 to reduce macrophage IL-6 production should also be weighed against its impact on IL-6 production in epithelial cells. Previous studies raise the potential concern that introduction of LL-37 to bronchial epithelial cells may increase expression of IL-6 (94). The study was performed in epithelial cells that were not introduced to pathogens and research into whether LL-37 increases IL-6 production in epithelial cells that have been infected should be done. In some applications, this concern should be taken into consideration or could be managed with drugs that interrupt the NF-κB signaling pathway that induces increased expression of IL-6 (94).
Diabetes and hyperglycemia are significant comorbidities of COVID-19. Diabetic patients in a report from China were shown to have higher inflammatory serum markers and D-Dimer levels, which are linked to higher mortality in COVID-19 (95, 96). When comparing diabetic COVID-19 patients with non-diabetic patients the mortality risks increase with 1.9 odds ratio (OR), and risk of severe COVID-19 with 2.75 OR (97). A meta-analysis of 16 observational studies found that the OR of mortality among hyperglycemic patients relative to non-hyperglycemic patients was 3.45 and 2.08 for severe COVID-19 (98). The marked increases in COVID-19 severity and mortality associated with diabetes and hyperglycemia make them important areas of investigation to reducing the impacts of COVID-19.
Diabetes influences the host response to viruses in many ways. Notably, neutrophils from both type 1 and type 2 diabetic patients are primed to undergo NETosis, possibly due to an upregulation of PAD4, and wound healing is significantly delayed due to their presence (91). In addition to being primed to deploy NETs, neutrophils in diabetic mice were less likely to undergo apoptosis and clearance by macrophages. This leads to elevated levels of inflammatory cytokines such as TNF-α (99). These findings are consistent with clinical data, which shows patients with Type 2 Diabetes (T2D) have higher neutrophil-to-lymphocyte ratios and more severe COVID-19 outcomes than their non-diabetic counterparts (100). T2D has also been associated with higher calcium levels throughout the body (101, 102). This free calcium can play an important function in the regulation of NETosis events. Neutrophils isolated from human blood and stimulated by LPS and IL-8 show an increase in intracellular calcium. Moreover, treatment of neutrophils isolated from human blood with calcium ionophores promotes NET release (103–105). Hyperglycemia, which is related to diabetes (particularly T2D), is another major instigator of NETosis.
Hyperglycemia leads to activation of the polyol pathway, which enhances the formation of advanced glycation products, promotes the formation of reactive oxygen species that contribute to inflammation, and effectively reduces neutrophil opsonophagocytosis (106). Infection of β-cell islets by SARS-CoV-2 increases MAPK signaling, promotes β-cell apoptosis and exacerbates hyperglycemia (107–109). Hyperglycemia also “leads to greater MAP kinase signaling, NF-κB activity, and production of cytokines such as IL-6” (106). This general engagement of inflammatory cytokines is also associated with an increase in macrovascular complications (106). Another mechanism explaining the increase in macrovascular complications involves hyperglycemia and insulin resistance, which results in reduced intracellular Mg2+ (110). Magnesium deficiency increases the production of cytokines such as IL-1β, IL-6, TNF-α, vascular cell adhesion molecule-1, and plasminogen activator inhibitor-1 (111). IL-6 and TNF-α instigate neutrophil recruitment and activation (112). These results are consistent with the finding that hyperglycemia increases the release of NETs and circulating markers of NETosis (113). Lower serum magnesium is also associated with increased thrombotic risk and slowed fibrinolysis, which may contribute to, or stem from, NET-platelet thrombi (114). Hyperglycemia, and its associated hypertension, also, has a major impact on pericytes, causing vessels to weaken and form aneurysms in the case of diabetic retinopathy. This phenomenon may aid in explaining the increased severity of COVID-19 in diabetic patients (115).
Hyperglycemia causes significant disruptions in the renin–angiotensin–aldosterone system (RAAS). SARS-CoV-2 also disrupts the RAAS by increasing bradykinin levels through the downregulation of ACE, which clears bradykinin (116). As bradykinin accumulates, it increases the permeability of the local vasculature facilitating the recruitment of neutrophils, which instigates inflammation responses by releasing cytokines and perpetuating the cytokine storm (117–119). This explanation is consistent with the observation that polymorphonuclear leukocyte (PMN) infiltration in pulmonary capillaries and neutrophilic mucositis is observed in lung autopsies obtained from COVID-19 patients (120). This increase in vasopermeability and recruitment of neutrophils leads us to neutrophil extracellular traps (NETs), the next and arguably most important part of this complex inflammatory response interplay.
In an in vivo murine model of Type 1 Diabetes (T1D), cathelicidin-related antimicrobial peptide (CRAMP) is expressed in insulin producing β-cell islets. CAMP/LL-37 served as a stimulator of pancreatic β-cell Ca2+ release and promoted the subsequent release of insulin or glucagon. CAMP/LL-37 treatment also stimulated β-cell neogenesis and enhanced the upregulation of potentially beneficial gut microbes in murine models (121). In addition, CRAMP/LL-37 modulated the inflammatory profile of pancreatic macrophages near β-cell islets in a dose-dependent manner (122). Reductions in the expression of cytokines such as TNF-α and IL-12 have been observed in macrophages from diabetic mice treated with LL-37 (122). Thus, LL-37 may be useful in addressing MAS that appears to be an essential driver of COVID-19 pathology (86) and of recently emerging cases of T1D induced by COVID-19 (123). These findings suggest that LL-37 upregulation may be instrumental in controlling blood sugar levels and to the healthy survival and growth of insulin producing cells. It may also be important in addressing the complications of COVID-19 associated with diabetes and hyperglycemia.
In patients with T2D, NETs were found to contain LL-37, but its antibacterial abilities were found to be abrogated (124). Previous studies have shown that citrullination of LL-37 by PAD4 causes it to lose its antibacterial capacity, ability to promote clearance of extracellular DNA by dendritic cells, and even increases inflammatory responses in cells by abrogating some of LL-37’s immunomodulatory effects (125–128). We were unable to find literature investigating the impact of citrullination on LL-37’s antiviral capacity and recommend investigation into the field. It is known that T2D patients often overexpress PAD4, produce more NETs than non-diabetic reduced NET clearance, and mice models of T2D have increased difficulty in clearing NETs leading to decreased wound healing abilities (91). Patients treated with clarithromycin experience increased LL-37 load on NETs which enhanced wound healing and antibacterial and antiviral activity (124). We hypothesize that upregulated LL-37 can help control blood sugar levels, aid in combating diabetes, and act in a positive feedback loop to prevent citrullination and preserve its antimicrobial, immunomodulatory, and NET clearance activity (Figure 3).
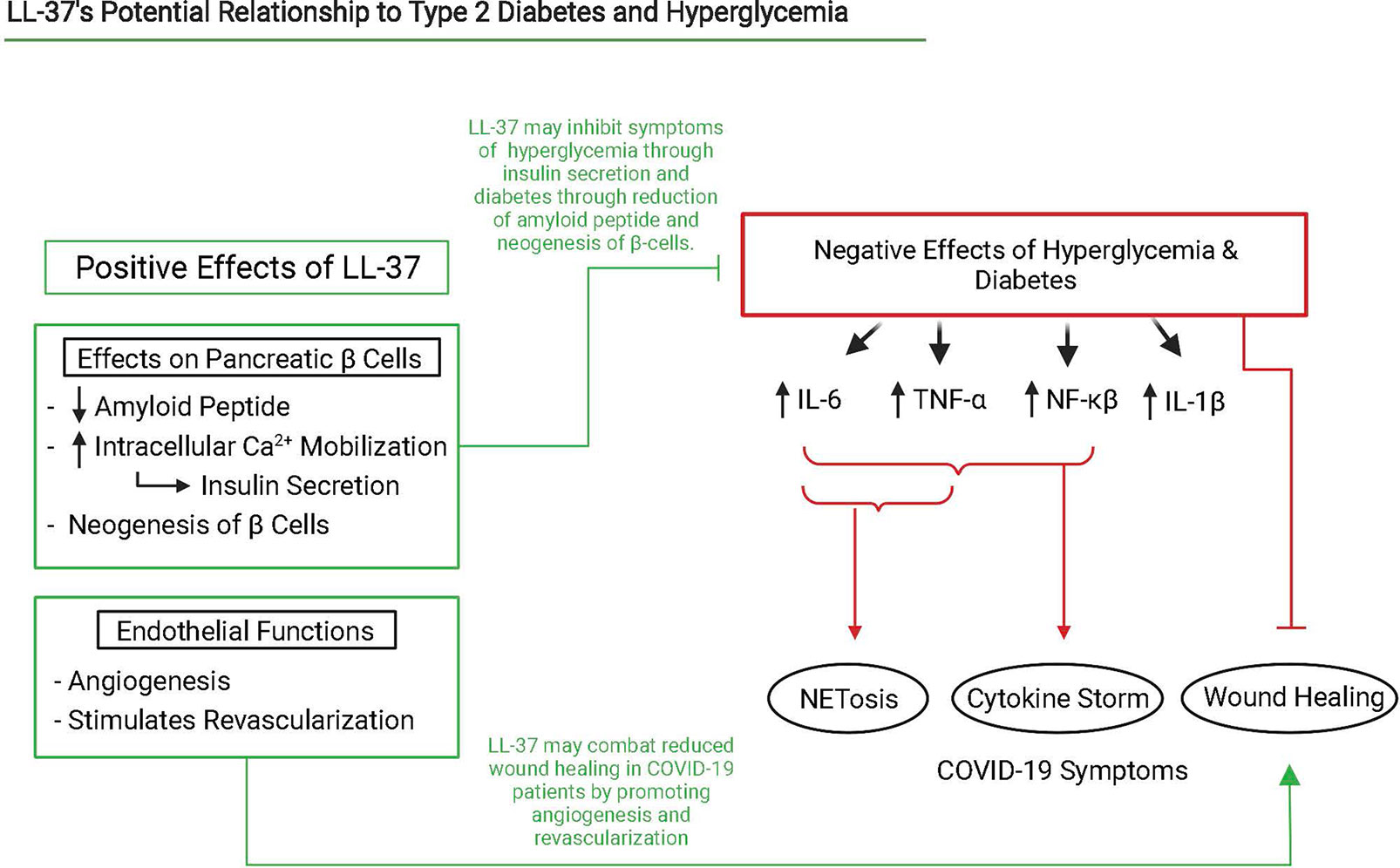
Figure 3 Interaction scheme of LL-37, NETosis, hyperglycemia, and diabetes that lead to severe COVID-19 symptoms. Relevant interactions are cited below. Created with BioRender.com.
In murine models, LL-37 improves wound healing in diabetic mice through its antimicrobial properties, while also directly activating endothelial cells in carrying out angiogenesis and neovascularization in repairing wounds (82). We hypothesize this is a function for addressing endothelial damage resulting from neutrophils and NETs responding to SARS-CoV-2 infections. LL-37 also prevents islet amyloid polypeptide (IAPP) self-assembly and subsequent β-cell damage in vitro (129).
Observed neurological consequences of COVID-19 include chronic fatigue, confusion, dizziness, seizures, visual deficits, encephalopathy, encephalitis, loss of smell and taste, Guillan-Barre Syndrome, and more (130, 131). In the brain, cerebrovascular consequences of COVID-19 such as ischemic stroke and intracerebral hemorrhage can also lead to neurological complication and even death (132). In one case, a COVID-19 patient had acute necrotizing encephalopathy marked with the presence of SARS-CoV-2 RNA, astrocytic activation markers, neuronal injury markers, and more in the cerebral spinal fluid, ultimately leading to them in a coma (133).
There are many potential mechanisms by which COVID-19 can induce these neurological symptoms. In addition to thrombotic risk, NETs also pose potential harm to the central nervous system. It was found that virally activated neutrophils and hypothesized specifically that their NETs and reactive oxygen species (ROS) cause demyelination of the central nervous system in mice infected with a neurotropic coronavirus (134). SARS-CoV-2 activated neutrophils, which have been shown to produce more NETs than control neutrophils, may produce NETs and ROS that contribute to the cognitive dysfunction referred to as “brain fog” that COVID-19 patients experience (40). Knowledge of the ability of NETs to serve as scaffolds for thrombi make them a possible instigator of ischemic stroke seen as well (36, 58, 135).
Neurological complications may also result from infection of astrocytes in the brain by SARS-CoV-2. SARS-CoV-2 has been found to enter the CNS of rhesus monkeys through the olfactory route (136). Once in the CNS, they can trigger inflammatory sequences and dysfunction of surrounding cells. At least one study preprint found astrocytes are disproportionately infected by SARS-CoV-2 and express cell stress signals such as ARCN1 (137). We also saw increased astrocytic activation markers in the autopsy of the patient with acute necrotizing encephalopathy (133). Another preprint study analyzing infection by SARS-CoV-2 in macaques found the formation of Lewy bodies (138). These plaques, which are predominantly composed of alpha-synuclein, are associated with the pathology of Parkinson’s disease and have been hypothesized to derive from a viral etiology including influenza A, norovirus and others (139–141). In a preprint, data from autopsies corroborated the presence of SARS-CoV-2 in all lobes of the brains of COVID-19 patients although there was a lack of inflammation (142).
A high frequency of anti-neuronal autoantibodies in COVID-19 patients could also contribute to cognitive dysfunction (143). The infection of pericytes inducing their dysfunction, and possibly, their function in effectively regulating the blood-brain barrier (BBB) raises concern. Pericytes prevent vessel degeneration and BBB disruption, and act as phagocytes by performing pinocytosis (115). One in vivo preprint study of SARS-CoV-2 infection of pericytes revealed vasoconstriction in the BBB (51).
Since the macaque model of COVID-19 has shown elevated levels in the brain of alpha-synuclein plaques, the role of alpha-synuclein production (if any) in common brain-fog and the attendant possible long-term impacts of COVID-19 is of interest (138). Similarly, since the brain has some of the highest levels of LL-37 expression (144) and LL-37 has previously been shown to suppress alpha-synuclein amyloid formation in cell culture, the impact of cathelicidin induction in addressing these sequelae deserves further investigation (145). We hypothesize enhanced LL-37 expression may address some consequences of COVID-19 by inhibiting alpha-synuclein aggregation and oligomer-induced cell damage and preventing infection of astrocytes as shown in Figure 4. Alpha synuclein plaques are associated with progression of Parkinson’s disease and LL-37 may also serve as a mechanism to reduce progression of the disease in COVID-19 patients. Vitamin D3, an up regulator of LL-37, has been hinted as helpful in addressing Parkinson’s in COVID-19 patients due to its super-promoter activity of Nrf2-KEAP, which promotes protective antioxidant and Ca2+ production and may work in conjunction with the potentially protective effect of LL-37 (146).
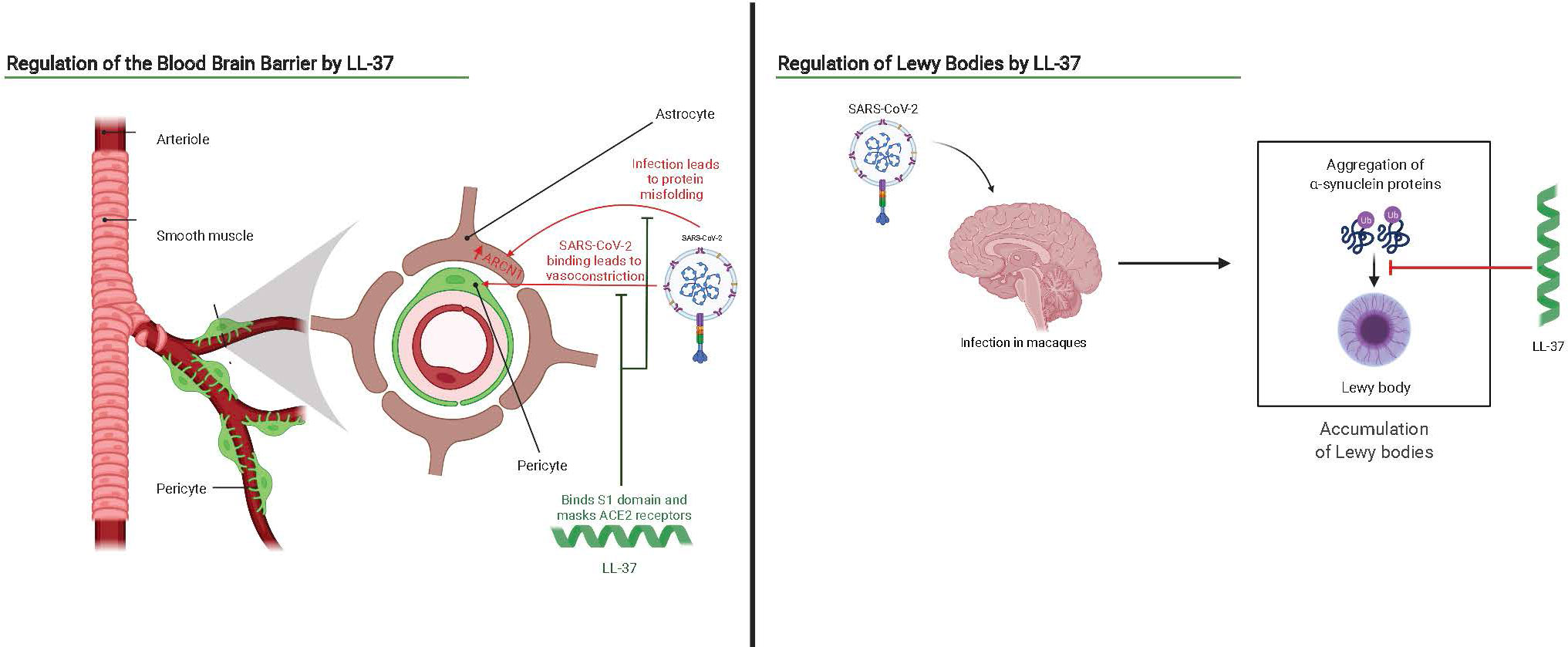
Figure 4 Interaction scheme of SARS-CoV-2 and the brain overlaid with potential antagonistic or therapeutic benefits of LL-37. Adapted from “Brain Vascular System” and “Progression of Parkinson’s Disease by the Substantia Niagra”, by BioRender.com (2021). Retrieved from https://app.biorender.com/biorender-templates.
LL-37 may also provide a role in preventing pericyte dysregulation and astrocyte dysfunction through binding the S1 domain of SARS-CoV-2 and cloaking ACE-2 receptors (20). CRAMP has been shown to have antimicrobial activity in astrocytes of mice models responding to bacterial supernatants, demonstrating the co-location of cathelidin and astrocytes in response to pathogens (147). LL-37 may also prepare astrocytes for infection by gearing them into a pro-inflammatory state with upregulated IL-1β and IL-6, but this inflammatory state must be reduced upon infection to prevent chronic inflammation and disease (144). Research into the impacts of LL-37 in various cell lines of the brain post infection should also be conducted as a dual effect of LL-37 as pro-inflammatory in early infection but anti-inflammatory in cells that have encountered a pathogen has been previously noted (148).
1,25(OH)2D3 upregulation of LL-37 is a highly conserved pathway in humans and one of the major inducers of LL-37 in the body (149, 150). In addition to vitamin D, some short-chain fatty acids are able to induce the expression of the CAMP gene, such as butyrate (151). However, butyrate is undesirable as a therapeutic compound, due to its noxious odor. Phenylbutyrate is a highly effective substitute, since it also induces CAMP gene expression but does not have a disagreeable odor (152). The combination of 1,25(OH)2D3 and phenylbutyrate is synergistic in its ability to induce CAMP gene expression in humans, providing enhanced expression of LL-37 and antibacterial activity, relative to what is achieved with just one of the two compounds (153). In the United States and most European countries, phenylbutyrate is an approved drug for the treatment of Urea-Cycle Disorders in both adult and pediatric patients (154). It was shown that phenylbutyrate induction of the CAMP gene required the vitamin D receptor (155). The synergistic induction of the CAMP gene by the combination of vitamin D3 and phenylbutyrate holds potential as a novel adjunct therapy for bacterial infections, particularly tuberculosis. Based on a dosage study done in humans, the ideal dose for induction of cathelicidin to treat lung infection is 5000 IU vitamin D3 taken daily, plus 500 mg phenylbutyrate per dose taken twice per day (156–159). Phenylbutyrate also showed promise in a preclinical animal trial using rabbits for the treatment of enteropathogenic E. coli-induced diarrhea (160). Certain phenylbutyrate analogs also induce CAMP gene expression. These include, for example, α-methylhydrocinnamate (ST7) (152).
Other compounds also induce the expression of the CAMP gene, although many of these inducers operate via mechanisms that are not fully understood and are independent of the VDR. For instance, the compound curcumin induces CAMP gene expression by a VDR-independent mechanism (161). Additionally, compounds from the family of stilbenoids (in particular resveratrol or pterostilbene) also induce CAMP gene expression by a VDR-independent mechanism, which is also synergistic with 1,25(OH)2D3 (162). Resveratrol induces cathelicidin expression by a novel mechanism involving sphingosine-1-phosphate pathway signaling (163, 164). Genistein, a soy-derived isoflavanoid, also induces cathelicidin expression by a sphingosine-1-phosphate stimulation mechanism (165).
Additional inducers of CAMP gene expression continue to emerge. In 2016, a novel family of compounds called aroylated phenylenediamines was developed to potently induce expression of the CAMP gene. This family includes the compound Entinostat, which has proven efficacious in treating shigellosis and cholera in rabbit models (166–168). Polysaccharide extracts from Vaccaria segetalis seeds (VSP) upregulated CRAMP expression in treated mice and LL-37 expression in A498 cells (169).
In this respect, it is notable that CAMP gene expression is observed in subjects upon exposure to certain external stimuli. For example, exercise induces cathelicidin expression in mice, even after very short (10-minute) sessions (170). Indeed, LL-37 is expressed in sweat, and is localized to both the eccrine gland and sweat ductal epithelial cells (171). Cathelicidin was also strongly induced by exposure to UVB ultraviolet light (172).
Careful consideration of the drugs used to treat COVID-19 reveals relationships to LL-37. Clarithromycin, which is identified above as a regulator of LL-37 concentration on the surface of NETs, is currently under investigation in clinical trials. Metformin, an AMPK activator, has been found to facilitate clearance of NETs (173), decrease the production of pro-inflammatory cytokines and nuclear factors (such as NF-κB) when bound to certain Toll-like-receptors (such as TLR4) (174), and increase insulin sensitivity to reduce hyperglycemia by downregulating NF-κB and TLR4 (175). LL-37 shows similar characteristics as it stimulates the P2X7 receptor which stimulates autophagy in macrophages, in combination with phenylbutyrate and AMPK signaling (176). LL-37 also reduces the expression of TLR4 in murine dendritic cells (177) and lessens TLR4 activation by LPS in J774 macrophages (178). No studies analyzing LL-37’s impact on virally stimulated macrophages were found in our search. TLR4 has been hypothesized to regulate the severity of COVID-19 by binding to SARS-CoV-2 and upregulating cell-surface expression of ACE-2 (179). LL-37 has also been found to modulate blood glucose level effectors and participate in NET clearance as discussed above. These studies suggest a possible clinical benefit in upregulating LL-37.
The therapeutic benefits of inducing LL-37 (Table 1) warrant investigation, not only as a tool for combating SARS-CoV-2 infection but also for applications to the ongoing discovery of longitudinal symptoms and known consequences of infection. The unexplored role of LL-37 in NET formation and clearance may prove to be of critical importance in preventing and ameliorating COVID-19-associated microthrombosis. Furthermore, the ability of LL-37 to instigate bronchial revascularization and angiogenesis has the potential to be critical for recovery. The ability of LL-37 to encourage insulin release and its role in the proper function of NETs in diabetic and hyperglycemic patients could provide another tool in the fight against SARS-CoV-2, since it addresses a major comorbidity of severe COVID-19. Lastly, LL-37 may disrupt some of the consequences of infection, such as alpha-synuclein plaque deposition in the brain, and IAPP plaque deposition in the pancreas associated with the development of insulin insensitivity. The immunomodulatory powers of LL-37 expression have been referenced by recent studies on the effects of vitamin D3 supplementation in modulating COVID-19 disease progression (44, 180). The vitamin D pathway directly induces LL-37 in humans, contributes to a myriad of other benefits to the immune system, and appears to reduce mortality rates by 80% in COVID-19 patients in the UK (181, 182). This relationship is relevant in the lungs, the tissue most impacted by the disease. Lung epithelial cells can change inactive vitamin D3 to its active form, which subsequently produces active LL-37 peptide locally (15, 183). Further investigations of the vitamin D3/LL-37 axis in relation to SARS-CoV-2 may be crucial to the creation of a widely accessible therapeutic strategy to combat infection and disease caused by this rapidly evolving virus.
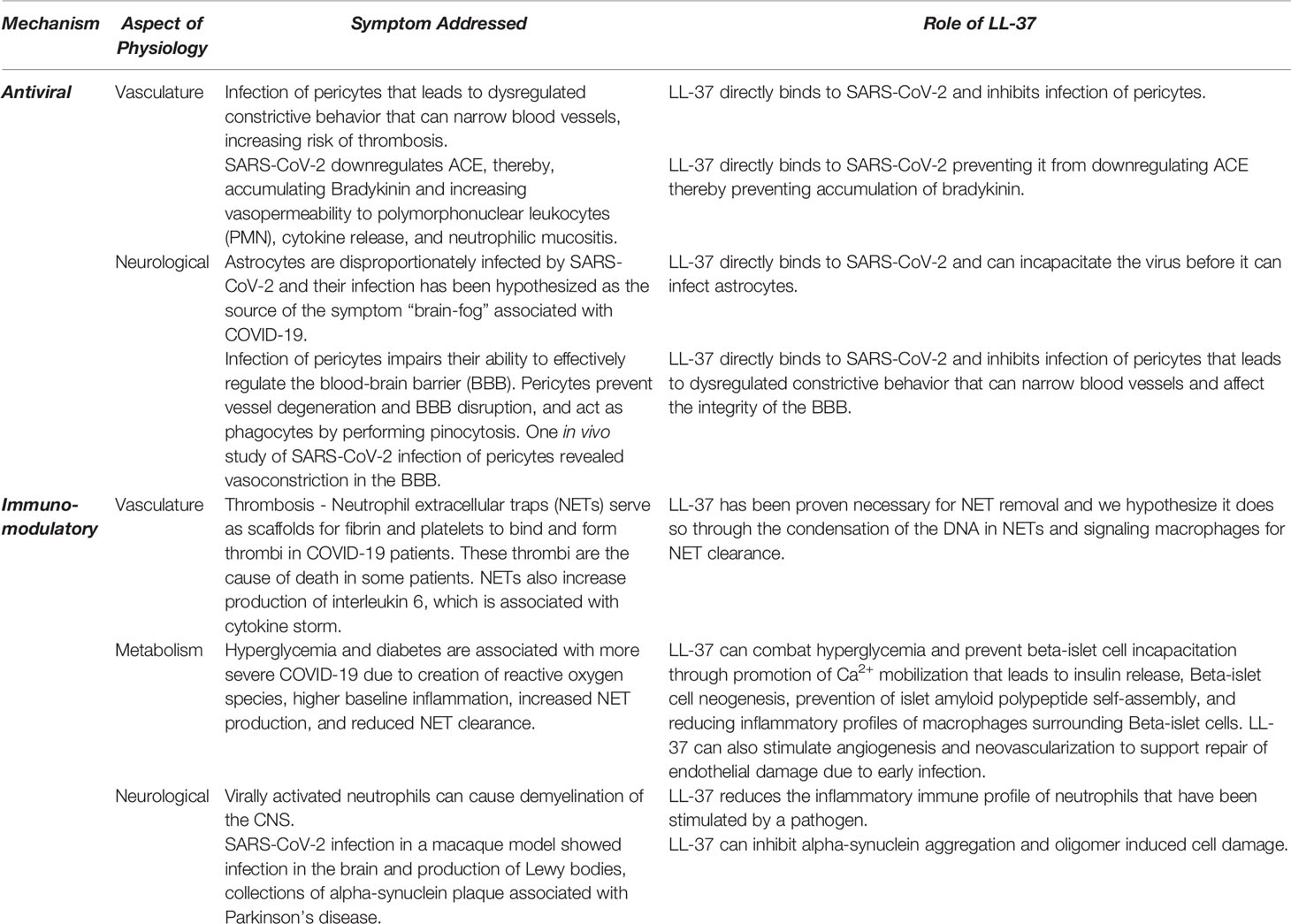
Table 1 List of potential therapeutic effects of LL-37 against COVID-19 separated based on mechanism of action and system targeted.
The original contributions presented in the study are included in the article/supplementary material. Further inquiries can be directed to the corresponding author.
KMA and JAF drafted the manuscript. JEN, EBD, JSL, MS, JDC, AFG, and AEB reviewed, discussed, and performed critical editing. All authors read and approved the final manuscript.
We would like to thank the NIH for funding this work with a Pioneer Award to Annelise Barron, grant # 1DP1 OD029517-01. AB also acknowledges funding from Stanford University’s Discovery Innovation Fund; from the Cisco University Research Program Fund and the Silicon Valley Community Foundation, from Stephen Pearse, and from Dr. James J. Truchard and the Truchard Foundation. JEN was funded by grant NNF21OC0068675 from the Novo Nordisk Foundation and the Stanford Bio-X Program. JC is supported in part by funds provided by the Texas A&M University System.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. JHU CSSE. (2022). Available at: https://www.arcgis.com/apps/dashboards/bda7594740fd40299423467b48e9ecf6 (Accessed 26 April 2022).
2. Hoffmann M, Kleine-Weber H, Schroeder S, Kr̈ger N, Herrler T, Erichsen S, et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell (2020) 181:271. doi: 10.1016/j.cell.2020.02.052
3. Sungnak W, Huang N, Bécavin C, Berg M, Queen R, Litvinukova M, et al. SARS-CoV-2 Entry Factors Are Highly Expressed in Nasal Epithelial Cells Together With Innate Immune Genes. Nat Med (2020) 26:681–7. doi: 10.1038/s41591-020-0868-6
4. Peacock TP, Goldhill DH, Zhou J, Baillon L, Frise R, Swann OC, et al. The Furin Cleavage Site in the SARS-CoV-2 Spike Protein Is Required for Transmission in Ferrets. Nat Microbiol (2021) 6:899–909. doi: 10.1038/s41564-021-00908-w
5. Scudellari M. How the Coronavirus Infects Cells — And Why Delta Is So Dangerous. Nature (2021) 595:640–4. doi: 10.1038/d41586-021-02039-y
6. Finkel Y, Gluck A, Nachshon A, Winkler R, Fisher T, Rozman B, et al. SARS-CoV-2 Uses a Multipronged Strategy to Impede Host Protein Synthesis. Nature (2021) 594:240–5. doi: 10.1038/s41586-021-03610-3
7. Huang C, Lokugamage KG, Rozovics JM, Narayanan K, Semler BL, Makino S. SARS Coronavirus Nsp1 Protein Induces Template-Dependent Endonucleolytic Cleavage of mRNAs: Viral mRNAs Are Resistant to Nsp1-Induced RNA Cleavage. PloS Pathog (2011) 7:e1002433. doi: 10.1371/journal.ppat.1002433
8. Sørensen OE, Follin P, Johnsen AH, Calafat J, Tjabringa GS, Hiemstra PS, et al. Human Cathelicidin, hCAP-18, Is Processed to the Antimicrobial Peptide LL-37 by Extracellular Cleavage With Proteinase 3. Blood (2001) 97:3951–9. doi: 10.1182/blood.v97.12.3951
9. Wang G. Structures of Human Host Defense Cathelicidin LL-37 and Its Smallest Antimicrobial Peptide KR-12 in Lipid Micelles *. J Biol Chem (2008) 283:32637–43. doi: 10.1074/jbc.M805533200
10. Lehrer RI, Ganz T. Cathelicidins: A Family of Endogenous Antimicrobial Peptides. Curr Opin Hematol (2002) 9:18–22. doi: 10.1097/00062752-200201000-00004
11. Wang T-T, Nestel FP, Bourdeau V, Nagai Y, Wang Q, Liao J, et al. Cutting Edge: 1,25-Dihydroxyvitamin D3 Is a Direct Inducer of Antimicrobial Peptide Gene Expression. J Immunol (2004) 173:2909–12. doi: 10.4049/jimmunol.173.5.2909
12. Gombart AF, Borregaard N, Koeffler HP. Human Cathelicidin Antimicrobial Peptide (CAMP) Gene Is a Direct Target of the Vitamin D Receptor and Is Strongly Up-Regulated in Myeloid Cells by 1,25-Dihydroxyvitamin D3. FASEB J (2005) 19:1067–77. doi: 10.1096/fj.04-3284com
13. Weber G, Heilborn JD, Chamorro Jimenez CI, Hammarsjo A, Törmä H, Stahle M. Vitamin D Induces the Antimicrobial Protein Hcap18 in Human Skin. J Invest Dermatol (2005) 124:1080–2. doi: 10.1111/j.0022-202X.2005.23687.x
14. Liu PT, Stenger S, Li H, Wenzel L, Tan BH, Krutzik SR, et al. Toll-Like Receptor Triggering of a Vitamin D-Mediated Human Antimicrobial Response. Science (2006) 311:1770–3. doi: 10.1126/science.1123933
15. Hansdottir S, Monick MM, Hinde SL, Lovan N, Look DC, Hunninghake GW. Respiratory Epithelial Cells Convert Inactive Vitamin D to Its Active Form. J Immunol (2008) 181:7090–9. doi: 10.4049/jimmunol.181.10.7090
16. Adams JS, Ren S, Liu PT, Chun RF, Lagishetty V, Gombart AF, et al. Vitamin D-Directed Rheostatic Regulation of Monocyte Antibacterial Responses. J Immunol (2009) 182:4289–95. doi: 10.4049/jimmunol.0803736
17. Li D, Chen P, Shi T, Mehmood A, Qiu J. HD5 and LL-37 Inhibit SARS-CoV and SARS- CoV-2 Binding to Human ACE2 by Molecular Simulation. Interdiscip Sci (2021) 13:766–77. doi: 10.1007/s12539-021-00462-3
18. Roth A, L̈tke S, Meinberger D, Hermes G, Sengle G, Koch M, et al. LL-37 Fights SARS-CoV-2: The Vitamin D-Inducible Peptide LL-37 Inhibits Binding of SARS-CoV-2 Spike Protein to Its Cellular Receptor Angiotensin Converting Enzyme 2 In Vitro. bioRxiv (2020). doi: 10.1101/2020.12.02.408153
19. Lokhande KB, Banerjee T, Swamy KV, Ghosh P, Deshpande M. An in Silico Scientific Basis for LL-37 as a Therapeutic for Covid-19. Proteins (2022) 90(5):1029-43. doi: 10.1002/prot.26198
20. Wang C, Wang S, Li D, Chen P, Han S, Zhao G, et al. Human Cathelicidin Inhibits SARS-CoV-2 Infection: Killing Two Birds With One Stone. ACS Infect Dis (2021) 7:1545–54. doi: 10.1021/acsinfecdis.1c00096
21. Tripathi S, Tecle T, Verma A, Crouch E, White M, Hartshorn KL. The Human Cathelicidin LL-37 Inhibits Influenza A Viruses Through a Mechanism Distinct From That of Surfactant Protein D or Defensins. J Gen Virol (2013) 94:40–9. doi: 10.1099/vir.0.045013-0
22. Currie SM, Findlay EG, McHugh BJ, Mackellar A, Man T, Macmillan D, et al. The Human Cathelicidin LL-37 has Antiviral Activity Against Respiratory Syncytial Virus. PloS One (2013) 8:e73659. doi: 10.1371/journal.pone.0073659
23. Currie SM, Gwyer Findlay E, McFarlane AJ, Fitch PM, Böttcher B, Colegrave N, et al. Cathelicidins Have Direct Antiviral Activity Against Respiratory Syncytial Virus In Vitro and Protective Function In Vivo in Mice and Humans. J Immunol (2016) 196:2699–710. doi: 10.4049/jimmunol.1502478
24. Sousa FH, Casanova V, Findlay F, Stevens C, Svoboda P, Pohl J, et al. Cathelicidins Display Conserved Direct Antiviral Activity Towards Rhinovirus. Peptides (2017) 95:76–83. doi: 10.1016/j.peptides.2017.07.013
25. Sancho-Vaello E, Gil-Carton D, François P, Bonetti E-J, Kreir M, Pothula KR, et al. The Structure of the Antimicrobial Human Cathelicidin LL-37 Shows Oligomerization and Channel Formation in the Presence of Membrane Mimics. Sci Rep (2020) 10:17356. doi: 10.1038/s41598-020-74401-5
26. Steinstraesser L, Tippler B, Mertens J, Lamme E, Homann H-H, Lehnhardt M, et al. Inhibition of Early Steps in the Lentiviral Replication Cycle by Cathelicidin Host Defense Peptides. Retrovirology (2005) 2:2. doi: 10.1186/1742-4690-2-2
27. Bergman P, Walter-Jallow L, Broliden K, Agerberth B, Söderlund J. The Antimicrobial Peptide LL-37 Inhibits HIV-1 Replication. Curr HIV Res (2007) 5:410–5. doi: 10.2174/157016207781023947
28. Wan M, van der Does AM, Tang X, Lindbom L, Agerberth B, Haeggström JZ. Antimicrobial Peptide LL-37 Promotes Bacterial Phagocytosis by Human Macrophages. J Leukocyte Biol (2014) 95:971–81. doi: 10.1189/jlb.0513304
29. Overhage J, Campisano A, Bains M, Torfs ECW, Rehm BHA, Hancock REW. Human Host Defense Peptide LL-37 Prevents Bacterial Biofilm Formation. Infect Immun (2008) 76:4176–82. doi: 10.1128/IAI.00318-08
30. López-García B, Lee PHA, Yamasaki K, Gallo RL. Anti-Fungal Activity of Cathelicidins and Their Potential Role in Candida Albicans Skin Infection. J Invest Dermatol (2005) 125:108–15. doi: 10.1111/j.0022-202X.2005.23713.x
31. Pasero D, Cossu AP, Terragni P. Multi-Drug Resistance Bacterial Infections in Critically Ill Patients Admitted With COVID-19. Microorganisms (2021) 9:1773. doi: 10.3390/microorganisms9081773
32. Ezeokoli OT, Gcilitshana O, Pohl CH. Risk Factors for Fungal Co-Infections in Critically Ill COVID-19 Patients, With a Focus on Immunosuppressants. J Fungi (2021) 7:545. doi: 10.3390/jof7070545
33. Baskaran V, Lawrence H, Lansbury LE, Webb K, Safavi S, Zainuddin NI, et al. Co-Infection in Critically Ill Patients With COVID-19: An Observational Cohort Study From England. J Med Microbiol (2021)70:1350. doi: 10.1099/jmm.0.001350
34. Bals R, Wilson JM. Cathelicidins - A Family of Multifunctional Antimicrobial Peptides. Cell Mol Life Sci (2003) 60:711–20. doi: 10.1007/s00018-003-2186-9
35. Lazzaretto B, Fadeel B. Intra- and Extracellular Degradation of Neutrophil Extracellular Traps by Macrophages and Dendritic Cells. J Immunol (2019) 203(8):2276–90. doi: 10.4049/jimmunol.1800159
36. Fuchs TA, Brill A, Duerschmied D, Schatzberg D, Monestier M, Myers DD, et al. Extracellular DNA Traps Promote Thrombosis. Proc Natl Acad Sci (2010) 107:15880–5. doi: 10.1073/pnas.1005743107
37. Neumann A, Berends ETM, Nerlich A, Molhoek EM, Gallo RL, Meerloo T, et al. The Antimicrobial Peptide LL-37 Facilitates the Formation of Neutrophil Extracellular Traps. Biochem J (2014) 464:3–11. doi: 10.1042/BJ20140778
38. Fox SE, Akmatbekov A, Harbert JL, Li G, Quincy Brown J, Vander Heide RS. Pulmonary and Cardiac Pathology in African American Patients With COVID-19: An Autopsy Series From New Orleans. Lancet Respir Med (2020) 8:681–6. doi: 10.1016/S2213-2600(20)30243-5
39. Brinkmann V, Reichard U, Goosmann C, Fauler B, Uhlemann Y, Weiss DS, et al. Neutrophil Extracellular Traps Kill Bacteria. Science (2004) 303:1532–5. doi: 10.1126/science.1092385
40. Arcanjo A, Logullo J, Menezes CCB, de Souza Carvalho Giangiarulo TC, dos Reis MC, de Castro GMM, et al. The Emerging Role of Neutrophil Extracellular Traps in Severe Acute Respiratory Syndrome Coronavirus 2 (COVID-19). Sci Rep (2020) 10:19630. doi: 10.1038/s41598-020-76781-0
41. Alpcan A, Tursun S, Kandur Y. Vitamin D Levels in Children With COVID-19: A Report From Turkey. Epidemiol Infect (2021) 149:e180, 1–4. doi: 10.1017/S0950268821001825
42. Nimavat N, Singh S, Singh P, Singh SK, Sinha N. Vitamin D Deficiency and COVID-19: A Case-Control Study at a Tertiary Care Hospital in India. Ann Med Surg (2021) 68:102661. doi: 10.1016/j.amsu.2021.102661
43. Chen J, Mei K, Xie L, Yuan P, Ma J, Yu P, et al. Low Vitamin D Levels do Not Aggravate COVID-19 Risk or Death, and Vitamin D Supplementation Does Not Improve Outcomes in Hospitalized Patients With COVID-19: A Meta-Analysis and GRADE Assessment of Cohort Studies and RCTs. Nutr J (2021) 20:89. doi: 10.1186/s12937-021-00744-y
44. Fakhoury HMA, Kvietys PR, Shakir I, Shams H, Grant WB, Alkattan K. Lung-Centric Inflammation of COVID-19: Potential Modulation by Vitamin D. Nutrients (2021) 13:2216. doi: 10.3390/nu13072216
45. Crane-Godreau MA, Clem KJ, Payne P, Fiering S. Vitamin D Deficiency and Air Pollution Exacerbate COVID-19 Through Suppression of Antiviral Peptide Ll37. Front Public Health (2020) 8:232. doi: 10.3389/fpubh.2020.00232
46. di Filippo L, Allora A, Doga M, Formenti AM, Locatelli M, Rovere Querini P, et al. Vitamin D Levels Are Associated With Blood Glucose and BMI in COVID-19 Patients, Predicting Disease Severity. J Clin Endocrinol Metab (2022) 107:e348–60. doi: 10.1210/clinem/dgab599
47. Ghosh SK, Weinberg A. Ramping Up Antimicrobial Peptides Against Severe Acute Respiratory Syndrome Coronavirus-2. Front Mol Biosci (2021) 8:620806. doi: 10.3389/fmolb.2021.620806
48. Lax SF, Skok K, Zechner P, Kessler HH, Kaufmann N, Koelblinger C, et al. Pulmonary Arterial Thrombosis in COVID-19 With Fatal Outcome. Ann Intern Med (2020) 173:350–61. doi: 10.7326/M20-2566
49. Maiese A, Manetti AC, La Russa R, Di Paolo M, Turillazzi E, Frati P, et al. Autopsy Findings in COVID-19-Related Deaths: A Literature Review. Forensic Sci Med Pathol (2021) 17:279–96. doi: 10.1007/s12024-020-00310-8
50. Torres-Ruiz J, Absalón-Aguilar A, Nuñez-Aguirre M, Pérez-Fragoso A, Carrillo-Vázquez DA, Maravillas-Montero JL, et al. Neutrophil Extracellular Traps Contribute to COVID-19 Hyperinflammation and Humoral Autoimmunity. Cells (2021) 10:2545. doi: 10.3390/cells10102545
51. Hirunpattarasilp C, James G, Freitas F, Sethi H, Kittler JT, Huo J, et al. SARS-CoV-2 Binding to ACE2 Triggers Pericyte-Mediated Angiotensin-Evoked Cerebral Capillary Constriction. [Preprint]. Neuroscience (2021) 1-23. doi: 10.1101/2021.04.01.438122
52. Avolio E, Carrabba M, Milligan R, Kavanagh Williamson M, Beltrami AP, Gupta K, et al. The SARS-CoV-2 Spike Protein Disrupts Human Cardiac Pericytes Function Through CD147 Receptor-Mediated Signalling: A Potential non-Infective Mechanism of COVID-19 Microvascular Disease. Clin Sci (2021) 135:2667–89. doi: 10.1042/CS20210735
53. Ricard N, Tu L, Le Hiress M, Huertas A, Phan C, Thuillet R, et al. Increased Pericyte Coverage Mediated by Endothelial-Derived Fibroblast Growth Factor-2 and Interleukin-6 Is a Source of Smooth Muscle–Like Cells in Pulmonary Hypertension. Circulation (2014) 129:1586–97. doi: 10.1161/CIRCULATIONAHA.113.007469
54. Li Q, Xie Y, Cui Z, Tang S, Yuan B, Huang H, et al. Analysis of Peripheral Blood IL-6 and Leukocyte Characteristics in 364 COVID-19 Patients of Wuhan. Front Immunol (2020) 11:559716. doi: 10.3389/fimmu.2020.559716
55. Clark SR, Ma AC, Tavener SA, McDonald B, Goodarzi Z, Kelly MM, et al. Platelet TLR4 Activates Neutrophil Extracellular Traps to Ensnare Bacteria in Septic Blood. Nat Med (2007) 13:463–9. doi: 10.1038/nm1565
56. Magro CM, Mulvey J, Kubiak J, Mikhail S, Suster D, Crowson AN, et al. Severe COVID-19: A Multifaceted Viral Vasculopathy Syndrome. Ann Diagn Pathol (2021) 50:151645. doi: 10.1016/j.anndiagpath.2020.151645
57. Skendros P, Mitsios A, Chrysanthopoulou A, Mastellos DC, Metallidis S, Rafailidis P, et al. Complement and Tissue Factor–Enriched Neutrophil Extracellular Traps are Key Drivers in COVID-19 Immunothrombosis. J Clin Invest (2020) 130:6151–7. doi: 10.1172/JCI141374
58. Leppkes M, Knopf J, Naschberger E, Lindemann A, Singh J, Herrmann I, et al. Vascular Occlusion by Neutrophil Extracellular Traps in COVID-19. EBioMedicine (2020) 58:102925. doi: 10.1016/j.ebiom.2020.102925
59. Nicolai L, Leunig A, Brambs S, Kaiser R, Joppich M, Hoffknecht M-L, et al. Vascular Neutrophilic Inflammation and Immunothrombosis Distinguish Severe COVID-19 From Influenza Pneumonia. J Thromb Haemostasis (2021) 19:574–81. doi: 10.1111/jth.15179
60. de Buhr N, Parplys AC, Schroeder M, Henneck T, Schaumburg B, Stanelle-Bertram S, et al. Impaired Degradation of Neutrophil Extracellular Traps: A Possible Severity Factor of Elderly Male COVID-19 Patients. JIN (2022), 1–16. doi: 10.1159/000521594
61. Veras FP, Pontelli MC, Silva CM, Toller-Kawahisa JE, de Lima M, Nascimento DC, et al. SARS-CoV-2–Triggered Neutrophil Extracellular Traps Mediate COVID-19 pathologySARS-CoV-2 Directly Triggers ACE- Dependent NETs. J Exp Med (2020) 217:e20201129. doi: 10.1084/jem.20201129
62. Janiuk K, Jabłońska E, Garley M. Significance of NETs Formation in COVID-19. Cells (2021) 10:151. doi: 10.3390/cells10010151
63. Zuo Y, Yalavarthi S, Shi H, Gockman K, Zuo M, Madison JA, et al. Neutrophil Extracellular Traps in COVID-19. JCI Insight (2020) 5:138999. doi: 10.1172/jci.insight.138999
64. Boeckh-Behrens T, Golkowski D, Ikenberg B, Schlegel J, Protzer U, Schulz C, et al. COVID-19-Associated Large Vessel Stroke in a 28- Year-Old Patient. Clin Neuroradiol (2021) 31:511–4. doi: 10.1007/s00062-020-00992-1
65. Akbar AN, Gilroy DW. Aging Immunity May Exacerbate COVID-19. Science (2020) 369:256–7. doi: 10.1126/science.abb0762
66. Del Valle DM, Kim-Schulze S, Huang H-H, Beckmann ND, Nirenberg S, Wang B, et al. An Inflammatory Cytokine Signature Predicts COVID-19 Severity and Survival. Nat Med (2020) 26:1636–43. doi: 10.1038/s41591-020-1051-9
67. Yoshikawa T, Hill T, Li K, Peters CJ, Tseng C-TK. Severe Acute Respiratory Syndrome (SARS) Coronavirus-Induced Lung Epithelial Cytokines Exacerbate SARS Pathogenesis by Modulating Intrinsic Functions of Monocyte-Derived Macrophages and Dendritic Cells. J Virol (2009) 83:3039–48. doi: 10.1128/JVI.01792-08
68. Yuki K, Fujiogi M, Koutsogiannaki S. COVID-19 Pathophysiology: A Review. Clin Immunol (2020) 215:108427. doi: 10.1016/j.clim.2020.108427
69. Tanaka T, Narazaki M, Kishimoto T. IL-6 in Inflammation, Immunity, and Disease. Cold Spring Harb Perspect Biol (2014) 6:a016295. doi: 10.1101/cshperspect.a016295
70. Kolaczkowska E, Jenne CN, Surewaard BGJ, Thanabalasuriar A, Lee W-Y, Sanz M-J, et al. Molecular Mechanisms of NET Formation and Degradation Revealed by Intravital Imaging in the Liver Vasculature. Nat Commun (2015) 6:6673. doi: 10.1038/ncomms7673
71. Farrera C, Fadeel B. Macrophage Clearance of Neutrophil Extracellular Traps is a Silent Process. J Immunol (2013) 191:2647–56. doi: 10.4049/jimmunol.1300436
72. de Buhr N, Von Köckritz-Blickwede M. The Balance of Neutrophil Extracellular Trap Formation and Nuclease Degradation: An Unknown Role of Bacterial Coinfections in COVID-19 Patients? mBio (2021) 12:e03304–20. doi: 10.1128/mBio.03304-20
73. Sreenath K, Batra P, Vinayaraj EV, Bhatia R, SaiKiran K, Singh V, et al. Coinfections With Other Respiratory Pathogens Among Patients With COVID-19. Microbiol Spectr (2021) 9:e00163–21. doi: 10.1128/Spectrum.00163-21
74. Nakazawa D, Shida H, Kusunoki Y, Miyoshi A, Nishio S, Tomaru U, et al. The Responses of Macrophages in Interaction With Neutrophils That Undergo NETosis. J Autoimmun (2016) 67:19–28. doi: 10.1016/j.jaut.2015.08.018
75. Marik PE, Iglesias J, Varon J, Kory P. A Scoping Review of the Pathophysiology of COVID-19. Int J Immunopathol Pharmacol (2021) 35:20587384211048024. doi: 10.1177/20587384211048026
76. Lande R, Gregorio J, Facchinetti V, Chatterjee B, Wang Y-H, Homey B, et al. Plasmacytoid Dendritic Cells Sense Self-DNA Coupled With Antimicrobial Peptide. Nature (2007) 449:564–9. doi: 10.1038/nature06116
77. Sandgren S, Wittrup A, Cheng F, Jönsson M, Eklund E, Busch S, et al. The Human Antimicrobial Peptide LL-37 Transfers Extracellular DNA Plasmid to the Nuclear Compartment of Mammalian Cells via Lipid Rafts and Proteoglycan-Dependent Endocytosis. J Biol Chem (2004) 279:17951–6. doi: 10.1074/jbc.M311440200
78. Duan Z, Fang Y, Sun Y, Luan N, Chen X, Chen M, et al. Antimicrobial Peptide LL-37 Forms Complex With Bacterial DNA to Facilitate Blood Translocation of Bacterial DNA and Aggravate Ulcerative Colitis. Sci Bull (2018) 63:1364–75. doi: 10.1016/j.scib.2018.09.014
79. Hurtado P, Peh CA. LL-37 Promotes Rapid Sensing of CpG Oligodeoxynucleotides by B Lymphocytes and Plasmacytoid Dendritic Cells. J Immunol (2010) 184:1425–35. doi: 10.4049/jimmunol.0902305
80. Hosoda H, Nakamura K, Hu Z, Tamura H, Reich J, Kuwahara-Arai K, et al. Antimicrobial Cathelicidin Peptide LL-37 Induces NET Formation and Suppresses the Inflammatory Response in a Mouse Septic Model. Mol Med Rep (2017) 16:5618–26. doi: 10.3892/mmr.2017.7267
81. Yuk J-M, Shin D-M, Lee H-M, Yang C-S, Jin HS, Kim K-K, et al. Vitamin D3 Induces Autophagy in Human Monocytes/Macrophages via Cathelicidin. Cell Host Microbe (2009) 6:231–43. doi: 10.1016/j.chom.2009.08.004
82. Ramos R, Silva JP, Rodrigues AC, Costa R, Guardão L, Schmitt F, et al. Wound Healing Activity of the Human Antimicrobial Peptide LL37. Peptides (2011) 32:1469–76. doi: 10.1016/j.peptides.2011.06.005
83. Wu W, Wang S, Liu Q, Wang X, Shan T, Wang Y. Cathelicidin-WA Attenuates LPS-Induced Inflammation and Redox Imbalance Through Activation of AMPK Signaling. Free Radical Biol Med (2018) 129:338–53. doi: 10.1016/j.freeradbiomed.2018.09.045
84. Hu Z, Murakami T, Suzuki K, Tamura H, Reich J, Kuwahara-Arai K, et al. Antimicrobial Cathelicidin Peptide LL-37 Inhibits the Pyroptosis of Macrophages and Improves the Survival of Polybacterial Septic Mice. Int Immunol (2016) 28:245–53. doi: 10.1093/intimm/dxv113
85. Nijnik A, Pistolic J, Wyatt A, Tam S, Hancock REW. Human Cathelicidin Peptide LL-37 Modulates the Effects of IFN-γ on APCs. J Immunol (2009) 183:5788–98. doi: 10.4049/jimmunol.0901491
86. Otsuka R, Seino K. Macrophage Activation Syndrome and COVID-19. Inflamm Regen (2020) 40:19. doi: 10.1186/s41232-020-00131-w
87. Tripathi S, Verma A, Kim E-J, White MR, Hartshorn KL. LL-37 Modulates Human Neutrophil Responses to Influenza A Virus. J Leukocyte Biol (2014) 96:931. doi: 10.1189/jlb.4A1113-604RR
88. Mohamed BM, Boyle NT, Schinwald A, Murer B, Ward R, Mahfoud OK, et al. Induction of Protein Citrullination and Auto-Antibodies Production in Murine Exposed to Nickel Nanomaterials. Sci Rep (2018) 8:679. doi: 10.1038/s41598-017-19068-1
89. Thiam HR, Wong SL, Qiu R, Kittisopikul M, Vahabikashi A, Goldman AE, et al. NETosis Proceeds by Cytoskeleton and Endomembrane Disassembly and PAD4-Mediated Chromatin Decondensation and Nuclear Envelope Rupture. Proc Natl Acad Sci (2020) 117:7326–37. doi: 10.1073/pnas.1909546117
90. Casanova V, Sousa FH, Shakamuri P, Svoboda P, Buch C, D’Acremont M, et al. Citrullination Alters the Antiviral and Immunomodulatory Activities of the Human Cathelicidin LL-37 During Rhinovirus Infection. Front Immunol (2020) 11:85. doi: 10.3389/fimmu.2020.00085
91. Wong SL, Demers M, Martinod K, Gallant M, Wang Y, Goldfine AB, et al. Diabetes Primes Neutrophils to Undergo NETosis, Which Impairs Wound Healing. Nat Med (2015) 21:815–9. doi: 10.1038/nm.3887
92. Neumann A, Völlger L, Berends ETM, Molhoek EM, Stapels DAC, Midon M, et al. Novel Role of the Antimicrobial Peptide LL-37 in the Protection of Neutrophil Extracellular Traps Against Degradation by Bacterial Nucleases. J Innate Immun (2014) 6:860–8. doi: 10.1159/000363699
93. van der Does AM, Beekhuizen H, Ravensbergen B, Vos T, Ottenhoff THM, van Dissel JT, et al. LL-37 Directs Macrophage Differentiation Toward Macrophages With a Proinflammatory Signature. J Immunol (2010) 185:1442–9. doi: 10.4049/jimmunol.1000376
94. Pistolic J, Cosseau C, Li Y, Yu J, Filewod NCJ, Gellatly S, et al. Host Defence Peptide LL-37 Induces IL-6 Expression in Human Bronchial Epithelial Cells by Activation of the NF-κb Signaling Pathway. J Innate Immun (2009) 1:254–67. doi: 10.1159/000171533
95. Guo W, Li M, Dong Y, Zhou H, Zhang Z, Tian C, et al. Diabetes is a Risk Factor for the Progression and Prognosis of COVID-19. Diabetes Metab Res Rev (2020) 82:e3319. doi: 10.1002/dmrr.3319
96. Tang N, Li D, Wang X, Sun Z. Abnormal Coagulation Parameters are Associated With Poor Prognosis in Patients With Novel Coronavirus Pneumonia. J Thromb Haemost (2020) 18:844–7. doi: 10.1111/jth.14768
97. Kumar A, Arora A, Sharma P, Anikhindi SA, Bansal N, Singla V, et al. Is Diabetes Mellitus Associated With Mortality and Severity of COVID-19? A Meta-Analysis. Diabetes Metab Syndr (2020) 14:535–45. doi: 10.1016/j.dsx.2020.04.044
98. Yang Y, Cai Z, Zhang J. Hyperglycemia at Admission is a Strong Predictor of Mortality and Severe/Critical Complications in COVID-19 Patients: A Meta-Analysis. Biosci Rep (2021) 41:BSR20203584. doi: 10.1042/BSR20203584
99. Hanses F, Park S, Rich J, Lee JC. Reduced Neutrophil Apoptosis in Diabetic Mice During Staphylococcal Infection Leads to Prolonged Tnfα Production and Reduced Neutrophil Clearance. PloS One (2011) 6:e23633. doi: 10.1371/journal.pone.0023633
100. Liu G, Zhang S, Hu H, Liu T, Huang J. The Role of Neutrophil-Lymphocyte Ratio and Lymphocyte–Monocyte Ratio in the Prognosis of Type 2 Diabetics With COVID-19. Scott Med J (2020) 65:154–60. doi: 10.1177/0036933020953516
101. Levy J, Stern Z, Gutman A, Naparstek Y, Gavin JR, Avioli LV. Plasma Calcium and Phosphate Levels in an Adult Noninsulin-Dependent Diabetic Population. Calcif Tissue Int (1986) 39:316–8. doi: 10.1007/BF02555197
102. Sorva A, Tilvis RS. Low Serum Ionized to Total Calcium Ratio: Association With Geriatric Diabetes Mellitus and With Other Cardiovascular Risk Factors? GER (1990) 36:212–6. doi: 10.1159/000213202
103. Wang Y, Li M, Stadler S, Correll S, Li P, Wang D, et al. Histone Hypercitrullination Mediates Chromatin Decondensation and Neutrophil Extracellular Trap Formation. J Cell Biol (2009) 184:205–13. doi: 10.1083/jcb.200806072
104. de Bont CM, Koopman WJH, Boelens WC, Pruijn GJM. Stimulus-Dependent Chromatin Dynamics, Citrullination, Calcium Signalling and ROS Production During NET Formation. Biochim Biophys Acta (BBA) - Mol Cell Res (2018) 1865:1621–9. doi: 10.1016/j.bbamcr.2018.08.014
105. Gupta AK, Giaglis S, Hasler P, Hahn S. Efficient Neutrophil Extracellular Trap Induction Requires Mobilization of Both Intracellular and Extracellular Calcium Pools and Is Modulated by Cyclosporine a. PloS One (2014) 9:e97088. doi: 10.1371/journal.pone.0097088
106. Graves DT, Kayal RA. Diabetic Complications and Dysregulated Innate Immunity. Front Biosci (2008) 13:1227–39. doi: 10.2741/2757
107. Clark AL, Mirmira RG. SARS-CoV-2 Infection of Islet β Cells: Evidence and Implications. CR Med (2021) 2:1-3. doi: 10.1016/j.xcrm.2021.100380
108. Wu C-T, Lidsky PV, Xiao Y, Lee IT, Cheng R, Nakayama T, et al. SARS-CoV-2 Infects Human Pancreatic β Cells and Elicits β Cell Impairment. Cell Metab (2021) 33:1565–1576.e5. doi: 10.1016/j.cmet.2021.05.013
109. Tang X, Uhl S, Zhang T, Xue D, Li B, Vandana JJ, et al. SARS-CoV-2 Infection Induces Beta Cell Transdifferentiation. Cell Metab (2021) 33:1577–91.e7. doi: 10.1016/j.cmet.2021.05.015
110. DiNicolantonio JJ, O’Keefe JH, Wilson W. Subclinical Magnesium Deficiency: A Principal Driver of Cardiovascular Disease and a Public Health Crisis. Open Heart (2018) 5:e000668. doi: 10.1136/openhrt-2017-000668
111. Kostov K. Effects of Magnesium Deficiency on Mechanisms of Insulin Resistance in Type 2 Diabetes: Focusing on the Processes of Insulin Secretion and Signaling. Int J Mol Sci (2019) 20:E1351. doi: 10.3390/ijms20061351
112. Keshari RS, Jyoti A, Dubey M, Kothari N, Kohli M, Bogra J, et al. Cytokines Induced Neutrophil Extracellular Traps Formation: Implication for the Inflammatory Disease Condition. PloS One (2012) 7:e48111. doi: 10.1371/journal.pone.0048111
113. Menegazzo L, Ciciliot S, Poncina N, Mazzucato M, Persano M, Bonora B, et al. NETosis Is Induced by High Glucose and Associated With Type 2 Diabetes. Acta Diabetol (2015) 52:497–503. doi: 10.1007/s00592-014-0676-x
114. Zheltova AA, Kharitonova MV, Iezhitsa IN, Spasov AA. Magnesium Deficiency and Oxidative Stress: An Update. Biomed (Taipei) (2016) 6:20. doi: 10.7603/s40681-016-0020-6
115. Bergers G, Song S. The Role of Pericytes in Blood-Vessel Formation and Maintenance. Neuro Oncol (2005) 7:452–64. doi: 10.1215/S1152851705000232
116. Garvin MR, Alvarez C, Miller JI, Prates ET, Walker AM, Amos BK, et al. A Mechanistic Model and Therapeutic Interventions for COVID-19 Involving a RAS-Mediated Bradykinin Storm. eLife (2020) 9:e59177. doi: 10.7554/eLife.59177
117. Stuardo M, Gonzalez CB, Nualart F, Boric M, Corthorn J, Bhoola KD, et al. Stimulated Human Neutrophils Form Biologically Active Kinin Peptides From High and Low Molecular Weight Kininogens. J Leukoc Biol (2004) 75:631–40. doi: 10.1189/jlb.1103546
118. Araújo RC, Kettritz R, Fichtner I, Paiva AC, Pesquero JB, Bader M. Altered Neutrophil Homeostasis in Kinin B1 Receptor-Deficient Mice. Biol Chem (2001) 382:91–5. doi: 10.1515/BC.2001.014
119. Hofman Z, de Maat S, Hack CE, Maas C. Bradykinin: Inflammatory Product of the Coagulation System. Clin Rev Allergy Immunol (2016) 51:152–61. doi: 10.1007/s12016-016-8540-0
120. Yao XH, Li TY, He ZC, Ping YF, Liu HW, Yu SC, et al. A Pathological Report of Three COVID-19 Cases by Minimal Invasive Autopsies. Zhonghua Bing Li Xue Za Zhi (2020) 49:411–7. doi: 10.3760/cma.j.cn112151-20200312-00193
121. Pound LD, Patrick C, Eberhard CE, Mottawea W, Wang G-S, Abujamel T, et al. Cathelicidin Antimicrobial Peptide: A Novel Regulator of Islet Function, Islet Regeneration, and Selected Gut Bacteria. Diabetes (2015) 64:4135–47. doi: 10.2337/db15-0788
122. Sun J, Furio L, Mecheri R, van der Does AM, Lundeberg E, Saveanu L, et al. Pancreatic β-Cells Limit Autoimmune Diabetes via an Immunoregulatory Antimicrobial Peptide Expressed Under the Influence of the Gut Microbiota. Immunity (2015) 43:304–17. doi: 10.1016/j.immuni.2015.07.013
123. Marchand L, Pecquet M, Luyton C. Type 1 Diabetes Onset Triggered by COVID-19. Acta Diabetol (2020) 57:1265–6. doi: 10.1007/s00592-020-01570-0
124. Arampatzioglou A, Papazoglou D, Konstantinidis T, Chrysanthopoulou A, Mitsios A, Angelidou I, et al. Clarithromycin Enhances the Antibacterial Activity and Wound Healing Capacity in Type 2 Diabetes Mellitus by Increasing LL-37 Load on Neutrophil Extracellular Traps. Front Immunol (2018) 9:2064. doi: 10.3389/fimmu.2018.02064
125. Wong A, Bryzek D, Dobosz E, Scavenius C, Svoboda P, Rapala-Kozik M, et al. A Novel Biological Role for Peptidyl-Arginine Deiminases: Citrullination of Cathelicidin LL-37 Controls the Immunostimulatory Potential of Cell-Free DNA. J Immunol (2018) 200:2327–40. doi: 10.4049/jimmunol.1701391
126. Al-Adwani S, Wallin C, Balhuizen MD, Veldhuizen EJA, Coorens M, Landreh M, et al. Studies on Citrullinated LL-37: Detection in Human Airways, Antibacterial Effects and Biophysical Properties. Sci Rep (2020) 10:2376. doi: 10.1038/s41598-020-59071-7
127. Kilsgård O, Andersson P, Malmsten M, Nordin SL, Linge HM, Eliasson M, et al. Peptidylarginine Deiminases Present in the Airways During Tobacco Smoking and Inflammation Can Citrullinate the Host Defense Peptide LL-37, Resulting in Altered Activities. Am J Respir Cell Mol Biol (2012) 46:240–8. doi: 10.1165/rcmb.2010-0500oc
128. Koziel J, Bryzek D, Sroka A, Maresz K, Glowczyk I, Bielecka E, et al. Citrullination Alters Immunomodulatory Function of LL-37 Essential for Prevention of Endotoxin-Induced Sepsis. J Immunol (Baltimore Md 1950) (2014) 192:5363. doi: 10.4049/jimmunol.1303062
129. Armiento V, Hille K, Naltsas D, Lin JS, Barron AE, Kapurniotu A. The Human Host- Defense Peptide Cathelicidin LL-37 is a Nanomolar Inhibitor of Amyloid Self-Assembly of Islet Amyloid Polypeptide (IAPP). Angewandte Chemie Int Edition (2020) 59:12837–41. doi: 10.1002/anie.202000148
130. Sarubbo F, El Haji K, Vidal-Balle A, Bargay Lleonart J. Neurological Consequences of COVID-19 and Brain Related Pathogenic Mechanisms: A New Challenge for Neuroscience. Brain Behav Immun - Health (2022) 19:100399. doi: 10.1016/j.bbih.2021.100399
131. Izquierdo-Domínguez A, Rojas-Lechuga MJ, Chiesa-Estomba C, Calvo-Henríquez C, Ninchritz-Becerra E, Soriano-Reixach M, et al. Smell and Taste Dysfunction in COVID-19 Is Associated With Younger Age in Ambulatory Settings: A Multicenter Cross-Sectional Study. J Investig Allergol Clin Immunol (2020) 30:346–57. doi: 10.18176/jiaci.0595
132. Paterson RW, Brown RL, Benjamin L, Nortley R, Wiethoff S, Bharucha T, et al. The Emerging Spectrum of COVID-19 Neurology: Clinical, Radiological and Laboratory Findings. Brain (2020) 143:3104–20. doi: 10.1093/brain/awaa240
133. Virhammar J, Kumlien E, Fällmar D, Frithiof R, Jackmann S, Sköld MK, et al. Acute Necrotizing Encephalopathy With SARS-CoV-2 RNA Confirmed in Cerebrospinal Fluid. Neurology (2020) 95:445–9. doi: 10.1212/WNL.0000000000010250
134. Cheng Y, Skinner DD, Lane TE. Innate Immune Responses and Viral-Induced Neurologic Disease. J Clin Med (2018) 8:E3. doi: 10.3390/jcm8010003
135. Laridan E, Denorme F, Desender L, François O, Andersson T, Deckmyn H, et al. Neutrophil Extracellular Traps in Ischemic Stroke Thrombi. Ann Neurol (2017) 82:223–32. doi: 10.1002/ana.24993
136. Jiao L, Yang Y, Yu W, Zhao Y, Long H, Gao J, et al. The Olfactory Route is a Potential Way for SARS-CoV-2 to Invade the Central Nervous System of Rhesus Monkeys. Sig Transduct Target Ther (2021) 6:1–11. doi: 10.1038/s41392-021-00591-7
137. Andrews MG, Mukhtar T, Eze UC, Simoneau CR, Perez Y, Mostajo-Radji MA, et al. Tropism of SARS-CoV-2 for Developing Human Cortical Astrocytes. bioRxiv (2021). doi: 10.1101/2021.01.17.427024
138. Philippens IHCHM, Böszörményi KP, Wubben JA, Fagrouch ZC, van Driel N, Mayenburg AQ, et al. SARS-CoV-2 Causes Brain Inflammation and Induces Lewy Body Formation in Macaques. bioRxiv (2021). doi: 10.1101/2021.02.23.432474
139. Labrie V, Brundin P. Alpha-Synuclein to the Rescue: Immune Cell Recruitment by Alpha- Synuclein During Gastrointestinal Infection. J Innate Immun (2017) 9:437–40. doi: 10.1159/000479653
140. Takahashi M, Yamada T. Viral Etiology for Parkinson’s Disease–A Possible Role of Influenza A Virus Infection. Jpn J Infect Dis (1999) 52:89–98. doi: 10.7883/yoken.52.89
141. Estrada E. Cascading From SARS-CoV-2 to Parkinson’s Disease Through Protein-Protein Interactions. Viruses (2021) 13:897. doi: 10.3390/v13050897
142. Chertow D, Stein S, Ramelli S, Grazioli A, Chung J-Y, Singh M, et al. SARS-CoV-2 Infection and Persistence Throughout the Human Body and Brain. Res Sq (2022). doi: 10.21203/rs.3.rs-1139035/v1
143. Franke C, Ferse C, Kreye J, Reincke SM, Sanchez-Sendin E, Rocco A, et al. High Frequency of Cerebrospinal Fluid Autoantibodies in COVID-19 Patients With Neurological Symptoms. Brain Behav Immun (2021) 93:415–9. doi: 10.1016/j.bbi.2020.12.022
144. Lee M, Shi X, Barron AE, McGeer E, McGeer PL. Human Antimicrobial Peptide LL-37 Induces Glial-Mediated Neuroinflammation. Biochem Pharmacol (2015) 94:130–41. doi: 10.1016/j.bcp.2015.02.003
145. Santos J, Gracia P, Navarro S, Peña-Díaz S, Pujols J, Cremades N, et al. α-Helical Peptidic Scaffolds to Target α-Synuclein Toxic Species With Nanomolar Affinity. Nat Commun (2021) 12:3752. doi: 10.1038/s41467-021-24039-2
146. Hribar CA, Cobbold PH, Church FC. Potential Role of Vitamin D in the Elderly to Resist COVID-19 and to Slow Progression of Parkinson’s Disease. Brain Sci (2020) 10:E284. doi: 10.3390/brainsci10050284
147. Brandenburg L-O, Varoga D, Nicolaeva N, Leib SL, Wilms H, Podschun R, et al. Role of Glial Cells in the Functional Expression of LL-37/Rat Cathelin-Related Antimicrobial Peptide in Meningitis. J Neuropathol Exp Neurol (2008) 67:1041–54. doi: 10.1097/NEN.0b013e31818b4801
148. Yang B, Good D, Mosaiab T, Liu W, Ni G, Kaur J, et al. Significance of LL-37 on Immunomodulation and Disease Outcome. BioMed Res Int (2020) 2020:e8349712. doi: 10.1155/2020/8349712
149. Hertting O, Holm Å, L̈thje P, Brauner H, Dyrdak R, Jonasson AF, et al. Vitamin D Induction of the Human Antimicrobial Peptide Cathelicidin in the Urinary Bladder. PloS One (2010) 5:e15580. doi: 10.1371/journal.pone.0015580
150. Gombart AF, Saito T, Koeffler HP. Exaptation of an Ancient Alu Short Interspersed Element Provides a Highly Conserved Vitamin D-Mediated Innate Immune Response in Humans and Primates. BMC Genomics (2009) 10:321. doi: 10.1186/1471-2164-10-321
151. Schauber J, Svanholm C, Termén S, Iffland K, Menzel T, Scheppach W, et al. Expression of the Cathelicidin LL-37 Is Modulated by Short Chain Fatty Acids in Colonocytes: Relevance of Signalling Pathways. Gut (2003) 52:735–41. doi: 10.1136/gut.52.5.735
152. Steinmann J, Halldórsson S, Agerberth B, Gudmundsson GH. Phenylbutyrate Induces Antimicrobial Peptide Expression. Antimicrob Agents Chemother (2009) 53:5127–33. doi: 10.1128/AAC.00818-09
153. van der Does AM, Kenne E, Koppelaar E, Agerberth B, Lindbom L. Vitamin D3 and Phenylbutyrate Promote Development of a Human Dendritic Cell Subset Displaying Enhanced Antimicrobial Properties. J Leukoc Biol (2014) 95:883–91. doi: 10.1189/jlb.1013549
154. Peña-Quintana L, Llarena M, Reyes-Suárez D, Aldámiz-Echevarria L. Profile of Sodium Phenylbutyrate Granules for the Treatment of Urea-Cycle Disorders: Patient Perspectives. Patient Prefer Adherence (2017) 11:1489–96. doi: 10.2147/PPA.S136754
155. Kulkarni NN, Yi Z, Huehnken C, Agerberth B, Gudmundsson GH. Phenylbutyrate Induces Cathelicidin Expression via the Vitamin D Receptor: Linkage to Inflammatory and Growth Factor Cytokines Pathways. Mol Immunol (2015) 63:530–9. doi: 10.1016/j.molimm.2014.10.007
156. van der Does AM, Bergman P, Agerberth B, Lindbom L. Induction of the Human Cathelicidin LL-37 as a Novel Treatment Against Bacterial Infections. J Leukoc Biol (2012) 92:735–42. doi: 10.1189/jlb.0412178
157. Mily A, Rekha RS, Kamal SMM, Akhtar E, Sarker P, Rahim Z, et al. Oral Intake of Phenylbutyrate With or Without Vitamin D3upregulates the Cathelicidin LL-37 in Human Macrophages: A Dose Finding Study for Treatment of Tuberculosis. BMC Pulmon Med (2013) 13:23. doi: 10.1186/1471-2466-13-23
158. Mily A, Rekha RS, Kamal SMM, Arifuzzaman ASM, Rahim Z, Khan L, et al. Significant Effects of Oral Phenylbutyrate and Vitamin D3 Adjunctive Therapy in Pulmonary Tuberculosis: A Randomized Controlled Trial. PloS One (2015) 10:e0138340. doi: 10.1371/journal.pone.0138340
159. Rekha RS, Mily A, Sultana T, Haq A, Ahmed S, Mostafa Kamal SM, et al. Immune Responses in the Treatment of Drug-Sensitive Pulmonary Tuberculosis With Phenylbutyrate and Vitamin D3 as Host Directed Therapy. BMC Infect Dis (2018) 18:303. doi: 10.1186/s12879-018-3203-9
160. Al-Mamun A, Mily A, Sarker P, Tiash S, Navarro A, Akter M, et al. Treatment With Phenylbutyrate in a Pre-Clinical Trial Reduces Diarrhea Due to Enteropathogenic Escherichia Coli: Link to Cathelicidin Induction. Microbes Infect (2013) 15:939–50. doi: 10.1016/j.micinf.2013.08.007
161. Guo C, Rosoha E, Lowry MB, Borregaard N, Gombart AF. Curcumin Induces Human Cathelicidin Antimicrobial Peptide Gene Expression Through a Vitamin D Receptor- Independent Pathway. J Nutr Biochem (2013) 24:754–9. doi: 10.1016/j.jnutbio.2012.04.002
162. Guo C, Sinnott B, Niu B, Lowry MB, Fantacone ML, Gombart AF. Synergistic Induction of Human Cathelicidin Antimicrobial Peptide Gene Expression by Vitamin D and Stilbenoids. Mol Nutr Food Res (2014) 58:528–36. doi: 10.1002/mnfr.201300266
163. Park K, Elias PM, Hupe M, Borkowski AW, Gallo RL, Shin K-O, et al. Resveratrol Stimulates Sphingosine-1-Phosphate Signaling of Cathelicidin Production. J Invest Dermatol (2013) 133:1942–9. doi: 10.1038/jid.2013.133
164. Jeong SK, Kim YI, Shin K-O, Kim B-W, Lee SH, Jeon JE, et al. Sphingosine Kinase 1 Activation Enhances Epidermal Innate Immunity Through Sphingosine-1-Phosphate Stimulation of Cathelicidin Production. J Dermatol Sci (2015) 79:229–34. doi: 10.1016/j.jdermsci.2015.06.007
165. Park K, Kim Y-I, Shin K-O, Seo HS, Kim JY, Mann T, et al. The Dietary Ingredient, Genistein, Stimulates Cathelicidin Antimicrobial Peptide Expression Through a Novel S1P-Dependent Mechanism. J Nutr Biochem (2014) 25:734–40. doi: 10.1016/j.jnutbio.2014.03.005
166. Ottosson H, Nylén F, Sarker P, Miraglia E, Bergman P, Gudmundsson GH, et al. Potent Inducers of Endogenous Antimicrobial Peptides for Host Directed Therapy of Infections. Sci Rep (2016) 6:36692. doi: 10.1038/srep36692
167. Miraglia E, Nylén F, Johansson K, Arnér E, Cebula M, Farmand S, et al. Entinostat Up-Regulates the CAMP Gene Encoding LL-37 via Activation of STAT3 and HIF-1α Transcription Factors. Sci Rep (2016) 6:33274. doi: 10.1038/srep33274
168. Sarker P, Banik A, Stromberg R, Gudmundsson GH, Raqib R, Agerberth B. Treatment With Entinostat Heals Experimental Cholera by Affecting Physical and Chemical Barrier Functions of Intestinal Epithelia. Antimicrob Agents Chemother (2017) 61:e02570–16. doi: 10.1128/AAC.02570-16
169. Mao X, Yao R, Guo H, Bao L, Bao Y, Xu Y, et al. Polysaccharides Extract From Vaccaria Segetalis Seeds Inhibits Kidney Infection by Regulating Cathelicidin Expression. J Ethnopharmacol (2021) 267:113505. doi: 10.1016/j.jep.2020.113505
170. Lee J-K, Luchian T, Park Y. Effect of Regular Exercise on Inflammation Induced by Drug- Resistant Staphylococcus Aureus 3089 in ICR Mice. Sci Rep (2015) 5:16364. doi: 10.1038/srep16364
171. Murakami M, Ohtake T, Dorschner RA, Gallo RL, Schittek B, Garbe C. Cathelicidin Anti- Microbial Peptide Expression in Sweat, an Innate Defense System for the Skin. J Invest Dermatol (2002) 119:1090–5. doi: 10.1046/j.1523-1747.2002.19507.x
172. Mallbris L, Edström DW, Sundblad L, Granath F, Stahle M. UVB Upregulates the Antimicrobial Protein Hcap18 mRNA in Human Skin. J Invest Dermatol (2005) 125:1072–4. doi: 10.1111/j.0022-202X.2005.23872.x
173. Grégoire M, Uhel F, Lesouhaitier M, Gacouin A, Guirriec M, Mourcin F, et al. Impaired Efferocytosis and Neutrophil Extracellular Trap Clearance by Macrophages in ARDS. Eur Respir J (2018) 52:1702590. doi: 10.1183/13993003.02590-2017
174. Zhao X, Zmijewski JW, Lorne E, Liu G, Park Y-J, Tsuruta Y, et al. Activation of AMPK Attenuates Neutrophil Proinflammatory Activity and Decreases the Severity of Acute Lung Injury. Am J Physiol Lung Cell Mol Physiol (2008) 295:L497–504. doi: 10.1152/ajplung.90210.2008
175. Peixoto LG, Teixeira RR, Vilela DD, Barbosa LN, Caixeta DC, Deconte SR, et al. Metformin Attenuates the TLR4 Inflammatory Pathway in Skeletal Muscle of Diabetic Rats. Acta Diabetol (2017) 54:943–51. doi: 10.1007/s00592-017-1027-5
176. Rekha RS, Rao Muvva SJ, Wan M, Raqib R, Bergman P, Brighenti S, et al. Phenylbutyrate Induces LL-37-Dependent Autophagy and Intracellular Killing of Mycobacterium Tuberculosis in Human Macrophages. Autophagy (2015) 11:1688–99. doi: 10.1080/15548627.2015.1075110
177. Nardo AD, Braff MH, Taylor KR, Na C, Granstein RD, McInturff JE, et al. Cathelicidin Antimicrobial Peptides Block Dendritic Cell TLR4 Activation and Allergic Contact Sensitization. J Immunol (2007) 178:1829–34. doi: 10.4049/jimmunol.178.3.1829
178. Hu Z, Murakami T, Suzuki K, Tamura H, Kuwahara-Arai K, Iba T, et al. Antimicrobial Cathelicidin Peptide LL-37 Inhibits the LPS/ATP-Induced Pyroptosis of Macrophages by Dual Mechanism. PloS One (2014) 9:e85765. doi: 10.1371/journal.pone.0085765
179. Aboudounya MM, Heads RJ. COVID-19 and Toll-Like Receptor 4 (TLR4): SARS-CoV-2 May Bind and Activate TLR4 to Increase ACE2 Expression, Facilitating Entry and Causing Hyperinflammation. Mediators Inflamm (2021) 2021:8874339. doi: 10.1155/2021/8874339
180. White JH. Emerging Roles of Vitamin D-Induced Antimicrobial Peptides in Antiviral Innate Immunity. Nutrients (2022) 14:284. doi: 10.3390/nu14020284
181. Ling SF, Broad E, Murphy R, Pappachan JM, Pardesi-Newton S, Kong M-F, et al. High- Dose Cholecalciferol Booster Therapy Is Associated With a Reduced Risk of Mortality in Patients With COVID-19: A Cross-Sectional Multi-Centre Observational Study. Nutrients (2020) 12:E3799. doi: 10.3390/nu12123799
182. Chiodini I, Gatti D, Soranna D, Merlotti D, Mingiano C, Fassio A, et al. Vitamin D Status and SARS-CoV-2 Infection and COVID-19 Clinical Outcomes. Front Public Health (2021) 9:736665. doi: 10.3389/fpubh.2021.736665
Keywords: NET clearance, LL-37, COVID-19, SARS-CoV-2, alpha synuclein, diabetes, neutrophil extracellular trap (NET), cathelicidin
Citation: Aloul KM, Nielsen JE, Defensor EB, Lin JS, Fortkort JA, Shamloo M, Cirillo JD, Gombart AF and Barron AE (2022) Upregulating Human Cathelicidin Antimicrobial Peptide LL-37 Expression May Prevent Severe COVID-19 Inflammatory Responses and Reduce Microthrombosis. Front. Immunol. 13:880961. doi: 10.3389/fimmu.2022.880961
Received: 22 February 2022; Accepted: 11 April 2022;
Published: 12 May 2022.
Edited by:
Milos Jesenak, Comenius University, SlovakiaReviewed by:
Noha Mousaad Elemam, University of Sharjah, United Arab EmiratesCopyright © 2022 Aloul, Nielsen, Defensor, Lin, Fortkort, Shamloo, Cirillo, Gombart and Barron. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Annelise E. Barron, YWViYXJyb25Ac3RhbmZvcmQuZWR1
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.