 Melanie Kny
Melanie Kny Jens Fielitz
Jens Fielitz
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 09 May 2022
Sec. Molecular Innate Immunity
Volume 13 - 2022 | https://doi.org/10.3389/fimmu.2022.878755
This article is part of the Research Topic The Role of ER Stress and the UPR in Shaping Immunity: Volume II View all 7 articles
Critically ill patients at the intensive care unit (ICU) often develop a generalized weakness, called ICU-acquired weakness (ICUAW). A major contributor to ICUAW is muscle atrophy, a loss of skeletal muscle mass and function. Skeletal muscle assures almost all of the vital functions of our body. It adapts rapidly in response to physiological as well as pathological stress, such as inactivity, immobilization, and inflammation. In response to a reduced workload or inflammation muscle atrophy develops. Recent work suggests that adaptive or maladaptive processes in the endoplasmic reticulum (ER), also known as sarcoplasmic reticulum, contributes to this process. In muscle cells, the ER is a highly specialized cellular organelle that assures calcium homeostasis and therefore muscle contraction. The ER also assures correct folding of proteins that are secreted or localized to the cell membrane. Protein folding is a highly error prone process and accumulation of misfolded or unfolded proteins can cause ER stress, which is counteracted by the activation of a signaling network known as the unfolded protein response (UPR). Three ER membrane residing molecules, protein kinase R-like endoplasmic reticulum kinase (PERK), inositol requiring protein 1a (IRE1a), and activating transcription factor 6 (ATF6) initiate the UPR. The UPR aims to restore ER homeostasis by reducing overall protein synthesis and increasing gene expression of various ER chaperone proteins. If ER stress persists or cannot be resolved cell death pathways are activated. Although, ER stress-induced UPR pathways are known to be important for regulation of skeletal muscle mass and function as well as for inflammation and immune response its function in ICUAW is still elusive. Given recent advances in the development of ER stress modifying molecules for neurodegenerative diseases and cancer, it is important to know whether or not therapeutic interventions in ER stress pathways have favorable effects and these compounds can be used to prevent or treat ICUAW. In this review, we focus on the role of ER stress-induced UPR in skeletal muscle during critical illness and in response to predisposing risk factors such as immobilization, starvation and inflammation as well as ICUAW treatment to foster research for this devastating clinical problem.
Skeletal muscle is one of the biggest organs in our body and of utmost importance for human health. It assures our vital body functions, such as breathing, protects our inner organs and maintains posture. Skeletal muscle serves as protein reservoir, which during consuming diseases assures survival. As a highly plastic organ skeletal muscle adapts to varying loading conditions, where increased load (e.g., training) leads to muscle growth and reduced load (e.g., immobilization) cause a decrease in muscle mass and muscle function, referred to as muscle atrophy. One of the most severe forms of muscle atrophy occurs in critically ill patients who often experience a diffuse symmetric weakness of all extremities and the diaphragm that is accompanied by muscle wasting, called intensive care unit-acquired weakness (ICUAW). ICUAW prolongs hospital-stay, prolongs mechanical ventilation and increases mortality. Some risk factors for ICUAW such as disease severity, inflammation and sepsis, insulin resistance, feeding status, and long-lasting mechanical ventilation have been identified but the molecular mechanisms are not well defined. However, a disturbed protein homeostasis with an increased protein degradation and a decreased protein synthesis has been implicated. Protein synthesis in myocytes is stringently controlled in the endoplasmic reticulum (ER, also called sarcoplasmic reticulum in myocytes) that also assures contractile calcium handling. The ER coordinates the synthesis, folding and posttranslational modification of membrane residing or secreted proteins. One third of newly synthesized proteins are defective, un- or misfolded. Also, physiological requirements and pathological conditions such as inflammation, viral infection, and hypoxia, may result in accumulation of defective proteins in the ER. Importantly, accumulation of damaged or unfolded proteins threatens ER function and is called ER stress. To resolve ER stress the Unfolded Protein Response (UPR) that aims to restore protein homeostasis by decreasing general protein synthesis, enhancing ER folding capacity, and mediating the degradation of unfolded proteins is initiated. If the UPR cannot resolve ER stress programmed cell death is induced. The UPR was reported to regulate muscle mass, inflammation, insulin resistance and starvation. Although these conditions are also implicated in ICUAW a direct link between ER stress-induced UPR and ICUAW has not been drawn.
Here we review the role of ER stress-induced UPR in skeletal muscle during critical illness and in response to predisposing risk factors. We first describe mechanisms of skeletal muscle atrophy and elute on the clinical syndrome of ICUAW. We then give an overview of protein quality control, ER stress and the different branches of UPR signaling. Recently, several ICUAW risk factors have been identified, and therefore we discuss how starvation, insulin resistance, immobilization, inflammation and sepsis, regeneration as well as physiotherapy and mobilization are interrelated with ER stress-induced UPR in muscle (Figure 1). Finally, we review the effects of UPR modulating compounds and chaperons on the UPR and discuss their potential to prevent or treat ICUAW.
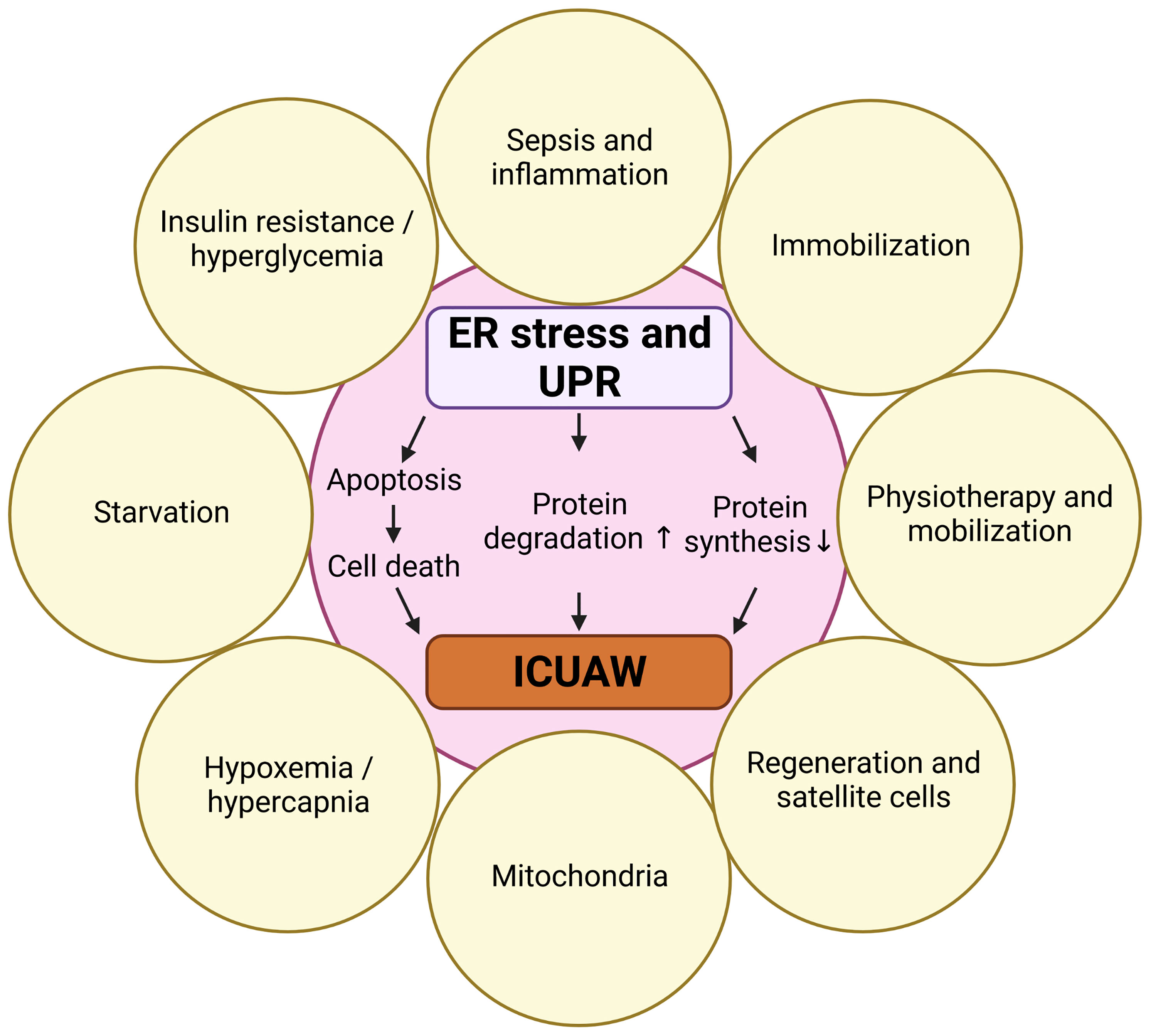
Figure 1 Risk factors for ICUAW that are also involved in ER stress and UPR. Created with BioRender.com.
The skeletal muscle is one of the biggest organs in the human body and contributes approximately 40% of our body weight. It serves as a vital part in the musculoskeletal system and assures a multitude of functions, such as breathing, movement, maintaining body posture, protection of joints and inner organs as well as chewing and swallowing. It also serves as a huge protein reservoir that is used during periods of starvation or in life-threatening situations such as critical illness. Although the main purpose of this is to support survival mechanisms a continued usage of muscle as an energy source will eventually compromise its function. Because muscle contraction and movement are energy demanding skeletal muscle is a tissue that is metabolically active. Muscle uses glucose as a main energy source and any dysfunction in glucose uptake, such as occurring in insulin-resistance, will not only compromise muscle function but also has systemic effects (e.g., hyperglycemia).
Skeletal muscle mass is highly dynamic and is regulated by physiological and pathological stress such as exercise, nutrition, postnatal and adolescent growth, aging, cancer and inflammation. The response of the skeletal muscle to these stresses is an increase or a decrease in muscle mass and function. Deliberately, we here focus on circumstances that will reduce muscle mass. A physiological reduction in muscle mass (i.e., physiological muscle atrophy) mainly occurs due to unloading, such as in athletes with discontinued training, people with little physical activity, those having a sedentary life style or astronauts during space flight with a reduced gravitational load. Pathological muscle atrophy is caused by fasting, loss of innervation, due to neurological diseases (e.g., stroke, spinal cord or nerve injury), and accompanies various diseases, such as cancer (1), end-stage heart disease (2), end-stage renal disease (3), chronic obstructive pulmonary disease [COPD; (4)] or sepsis (5). A reduction in muscle mass and function has severe consequences for affected patients and its prevention is of utmost importance to human health. In general, protein homeostasis (i.e., proteostasis) in muscle is assured by a critical balance of protein synthesis, quality control and degradation. During muscle atrophy contractile proteins are degraded by the ubiquitin proteasome system (UPS), the autophagy-lysosomal pathway (ALP), and proteases such as calpains and caspases (6). Briefly, UPS-mediated protein degradation is a sequential process that leads to the degradation of target proteins by a large multiprotease complex called the proteasome. ALP works via the self-destruction of target containing lysosomes and mediates degradation of bulks of proteins and whole organelles. Calcium-dependent papain-like proteinases (Calpains) are lysosome independent cysteine proteinases and Caspases are Cysteine-Aspartate proteinases involved in programmed cell death. The function of the UPS and the ALP in physiological and pathological muscle atrophy are well understood. Whereas the ALP employs lysosomal proteolytic enzymes to degrade protein aggregates and membrane proteins, the UPS is a cytosolic and nuclear machinery that mediates targeted degradation of cytosolic and nuclear proteins. As part of protein quality control, the UPS is responsible for the degradation of defective proteins, such as incomplete, misfolded, denatured or oxidized proteins which tend to accumulate and form cytotoxic aggregates. Recently we have eluted on the role and regulation of UPS-dependent protein degradation in ICUAW [for review (6)]. Briefly, UPS-dependent protein degradation is key for many cellular processes, such as cell cycle control, development, signal transduction, and many more. The UPS is implicated in many diseases, such as cancer, neurological disorders or inflammation (7). During a process called ubiquitination, the 7.6 kDa polypeptide Ubiquitin (Ub) that is ubiquitously expressed (8, 9), is conjugated to the target protein resulting in mono- or polyubiquitination (8). This process requires the sequential action of three classes of enzymes: E1 (ubiquitin-activating), E2 (ubiquitin-conjugating) and E3 (ubiquitin-protein ligating) enzymes (10). In this cascade, the E1 ubiquitin-activating enzyme activates ubiquitin in an ATP-dependent manner facilitating the formation of a thioester and transferring the so modified ubiquitin to an E2 ubiquitin-conjugating enzyme. The E2 enzyme than interacts with its cognate E3 ubiquitin ligase to transfer the ubiquitin covalently to a target protein via an isopeptide bond. This cascade is repeatedly executed to generate ubiquitin chains of a minimum of four ubiquitin moieties on target proteins. The ubiquitin moieties can be linked at different lysine-residues of the ubiquitin amino acid sequence. For protein degradation lysine 48 (K48) linked poly-ubiquitin chains are most important as they are eventually recognized by the 26S proteasome were the so modified proteins are degraded (11). The specificity of the reaction is assured by E3 ubiquitin ligases that are also the rate limiting step. E3 ubiquitin ligases often display a tissue specific expression and a distinct subcellular distribution (12), which is thought to facilitate binding to their clients. Several E3 ubiquitin ligases have been described to play a role in skeletal muscle atrophy such as Muscle RING (really interesting new gene) finger 1 (MuRF1 encoded by Trim63), atrogin-1/MAFbx (encoded by Fbxo32), Muscle ubiquitin ligase of the SCF Complex in atrophy-1 (MUSA1, encoded by Fbxo30), Neural precursor cell expressed developmentally down-regulated protein 4 (NEDD4), and TNF receptor-associated factor 6 (TRAF6) (13–16). E3 ubiquitin ligases that are involved in muscle atrophy are often upregulated in muscle wasting conditions, and are therefore called atrogenes (13). They also assure substrate specificity for proteins to be degraded. For example, MuRF1 has been shown to associate with, ubiquitinate and mediate UPS-dependent degradation of the contractile proteins myosin heavy chain (MyHC) 2 and 4 (17). MuRF1 has also been shown to mediate the degradation of other structural proteins, such as alpha-actin, troponin I, troponin-T, telethonin, titin, nebulin, the nebulin-related protein NRAP, myosin light chain 2, myotilin and T-cap (18–21), as well as proteins that are involved in muscular energy metabolism such as muscle-type creatine kinase and glucocorticoid modulatory element binding protein-1 (22,23). Recent work suggests that MuRF1 functions as a component of a Cullin-type ubiquitin ligase encompassing Cullin 4A (Cul4A), DDB1, Rbx1 and DCAF8 to mediate ubiquitination and degradation of MyHC (24). Mice deficient for MuRF1/Trim63 and atrogin-1/Fbxo32 are protected against various forms of muscle atrophy (13) and pharmacological inhibition of MuRF1 has been shown to attenuate muscle atrophy (25,26). Previously we showed that MuRF1/Trim63 and atrogin-1/Fbxo32 mRNA and protein expression are increased in the vastus lateralis of critically ill human patients, especially those that were at risk to develop muscle wasting (27). Both E3 ligases were also upregulated in the tibialis anterior and gastrocnemius and plantaris of mouse models of neurogenic, inflammation- and starvation induced muscle atrophy (28–32), where their increased expression was associated with the atrophy phenotype and the loss of MyHC. We and others recently summarized the current knowledge about factors and signaling pathways involved in the regulation of E3 ubiquitin ligases relevant for muscle atrophy (6,33,34). For this review it is relevant to know that proinflammatory cytokines (e.g., Tumor Necrosis Factor (TNF), Interleukin 6 (IL-6), IL-1β, TNF-Related Weak Inducer of Apoptosis (TWEAK), members of the Transforming Growth Factor (TGFs) family) and acute phase proteins [e.g., Serum Amyloid A1 (SAA1)] are well known factors involved in muscle wasting (6,33,34). To cause muscle atrophy, these cytokines bind to their receptors at the myocyte cell surface and activate transcription factors and signaling proteins such as Nuclear Factor-κB (NF-κB), JAK/STAT, SMAD, MAPK, TFEB/TFE3, and IGF/PI3K/AKT to regulate MuRF1/Trim63 and atrogin-1/Fbxo32 expression that in turn mediate muscle atrophy (6,33,34). Importantly, in addition to the immune system the skeletal muscle itself contributes to the production and release of acute phase response proteins and proinflammatory cytokines (28,31,32,35), which may aggravate muscle loss especially during chronic inflammation and sepsis. Although we start to understand the role of protein degradation and its regulation in muscle atrophy only few therapeutic concepts arose from these insights.
Generalized weakness that is accompanied by muscle wasting is a dominant clinical problem for many critically ill patients during treatment at the intensive care unit (ICU) but also after ICU discharge. This syndrome is called intensive care unit (ICU)-acquired weakness (ICUAW), which is defined as clinically detected weakness in critically ill patients where the only plausible etiology is the critical illness itself (36). To rule out alternative causes of weakness, it is requested that the manifestation of ICUAW must follow the commencement of critical illness. About 40% of critically ill patients experience ICUAW. A major contributor to ICUAW is a reduced muscle mass and function. Indeed, during the first 10 days in ICU critically ill patients lose around 20% of muscle mass (37). Due to the nature of muscle function tests that require cooperative patients, in most patients the decreased muscle function is not diagnosed until after the acute disease phase, e.g., when the patients awake from sedation (38). Therapeutic interventions at this stage are tedious and require lots of resources. An early detection of ICUAW and a risk factor-based treatment could be more effective, and such clinical studies have been performed (39–41). Patients with ICUAW show a pronounced disparity between movement and their level of consciousness and cooperativity. Physical examination often reveals diffuse, symmetric weakness of all extremities and the diaphragm. The decreased muscle function is associated with a prolonged ICU- and hospital-stay, and an increased mortality. Additionally, weakness of the diaphragm and the auxiliary respiratory muscles, prolongs mechanical ventilation and hampers ventilator weaning (42–44). Persisting weakness is associated with functional limitations, lower employment rates and a diminished quality of life (45,46). ICUAW patients often encounter limitations even in simple daily activities such as getting out of bed, getting out of a chair and going up and down stairs. Sequelae of ICUAW may persist for years after ICU- or hospital-discharge, which is in contrast to physiological muscle atrophy, which is reversible and responds well to physiotherapy and exercise. The pathophysiology of ICUAW is still elusive. A recent meta-analysis revealed that disease severity (i.e., Acute Physiology and Chronic Health Evaluation II score), sepsis, multiple organ failure, hyperglycemia (i.e., insulin resistance), electrolyte disturbances, parenteral nutrition (i.e., feeding status), female sex, medication (e.g., neuromuscular blocking agents, aminoglycoside antibiotics and norepinephrine) and long-lasting mechanical ventilation are risk factors for ICUAW (47). Because 60 to 100% of patients with sepsis develop ICUAW, this risk factor is of utmost importance. Sepsis, which is defined as a life-threatening organ dysfunction caused by a dysregulated host response to an infection (48,49), is the predominant cause of ICU-admission and the prevailing cause of death in ICU (50). Almost 30% of critically ill patients are either admitted with or become septic during ICU treatment (51, 52). Since systemic inflammation in sepsis is strongly associated with muscle atrophy and ICUAW in nearly all patients, a better understanding of the underlying pathomechanisms is required to prevent or treat the disease.
The reduction in muscle mass of critically ill patients is associated with a decreased myofiber cross-sectional area (MCSA). In general, skeletal muscle is composed of myofibers with different contractile behaviors, so called type I/slow- and type II/fast-twitch myofibers. The contractile behavior of myofibers depends on the myosin heavy chain (MyHC) that they predominantly express and based on these differences fast and slow contracting muscles are differentiated. For example, the soleus muscle predominantly contains type I/slow contracting MyHC-I, encoded by MYH7 and is therefore considered a slow muscle, whereas the tibialis anterior muscle mainly contains type II/fast contracting MyHC-IIA, MyHC-IIB or MyHC-IIx, encoded by MYH2, MYH4 and MYH1, respectively, and is therefore referred to as a fast muscle. Importantly, the degree of slow/type I- and fast/type II-fiber atrophy is different. Specifically, although histological analyses of biopsy specimens from the vastus lateralis muscle of ICU patients revealed that the MCSA of all fiber types is reduced myofiber atrophy is much more pronounced in fast twitch/type II-myofibers as opposed to slow twitch/type I-myofibers. This is associated with a reduction in fast and slow myosin heavy chain (MyHC) protein and mRNA expression (27). The consequences of myofiber atrophy are a decrease in muscle function with reduced specific force and endurance (53). In this review we will focus on the importance of the ICUAW-risk factors inflammation and sepsis, insulin resistance, feeding status and immobilization as well as muscle activation for the disease course.
Like protein degradation also protein synthesis is stringently controlled in myocytes. These control mechanisms occur in the endoplasmic reticulum (ER) that in muscle is also called sarcoplasmic reticulum. The ER is a cytoplasmatic membrane network consisting of tubules, vesicles and cavities that reaches from the nuclear membrane into the cytoplasm. In myocytes, the ER is highly specialized for contractile calcium handling. Importantly, the ER also coordinates the synthesis, folding, posttranslational modification and maturation of proteins, which is particularly relevant for secreted and membrane residing proteins. Soluble proteins that are meant to be processed in the ER harbor an N-terminal signal peptide, which is often cut upon import into the ER (54). The recognition of the signal peptide occurs either during translation (co-translational protein import) inducing a translational arrest until the ribosome has docked to the ER or after translation is completed, which requires cytosolic chaperones (e.g., heat shock protein 70 and 40 family members) that hold the proteins in an import competent unfolded state (55, 56). The oxidizing and calcium containing ER lumen provides an environment that enables protein modification and maturation, which is virtually impossible in the cytoplasm, e.g., disulfide bond formation, glycophosphatidylinositol (GPI)-anchor addition, N-linked glycosylation, protein folding and chaperone functionality (57). Within the process of translation, translocation and processing, nascent proteins are constantly monitored by a complex system of ER chaperones assuring assistance for posttranslational modification as well as folding and quality control. Among these chaperones are heat shock proteins, such as glucose regulated protein 78 (GRP78, also referred to as binding immunoglobulin protein (BiP) or HSPA5), lectins, such as Calnexin or Calreticulin, or thiol-disulfide oxidoreductases such as protein disulfide isomerase (PDI) (58, 59). Despite this well-regulated quality control system, around 30% of newly synthesized proteins are defective, un- or misfolded (60). Moreover, mature proteins can be damaged by environmental or pathophysiological conditions. Also, the protein folding capacity of the ER can be disturbed by a perturbed ER calcium homeostasis, hypoxia, viral infection, increased protein cargo or altered protein glycosylation. These conditions can lead to the accumulation of defective proteins in the ER, which is toxic to the cell and threaten cell viability, and is called ER stress. To counteract and resolve accumulation of damaged or unfolded proteins, ER stress induces signaling pathways, which are altogether named the Unfolded Protein Response (UPR). The UPR causes a decrease in general protein synthesis, enhances the ER folding capacity, and mediates the degradation of unfolded proteins. If the UPR is not sufficient to resolve ER stress, Janus Kinases (JNK) and Caspases 3, 7 and 12 are activated to induce programmed cell death (61, 62).
The UPR is mediated by three ER transmembrane UPR-signal transducers, the inositol-requiring protein 1 (IRE1), the protein kinase R (PKR)-like endoplasmic reticulum kinase (PERK) and the cAMP-dependent transcription factor 6 (ATF6) (63). Roughly, in the absence of ER stress, the ER residing chaperone protein GRP78/BiP is bound to PERK, IRE1 and ATF6, and keeps them in an inactive state. Upon ER stress, GRP78 dissociates from these receptors and binds to misfolded proteins, which activates the UPR signaling proteins (Figure 2) (64).
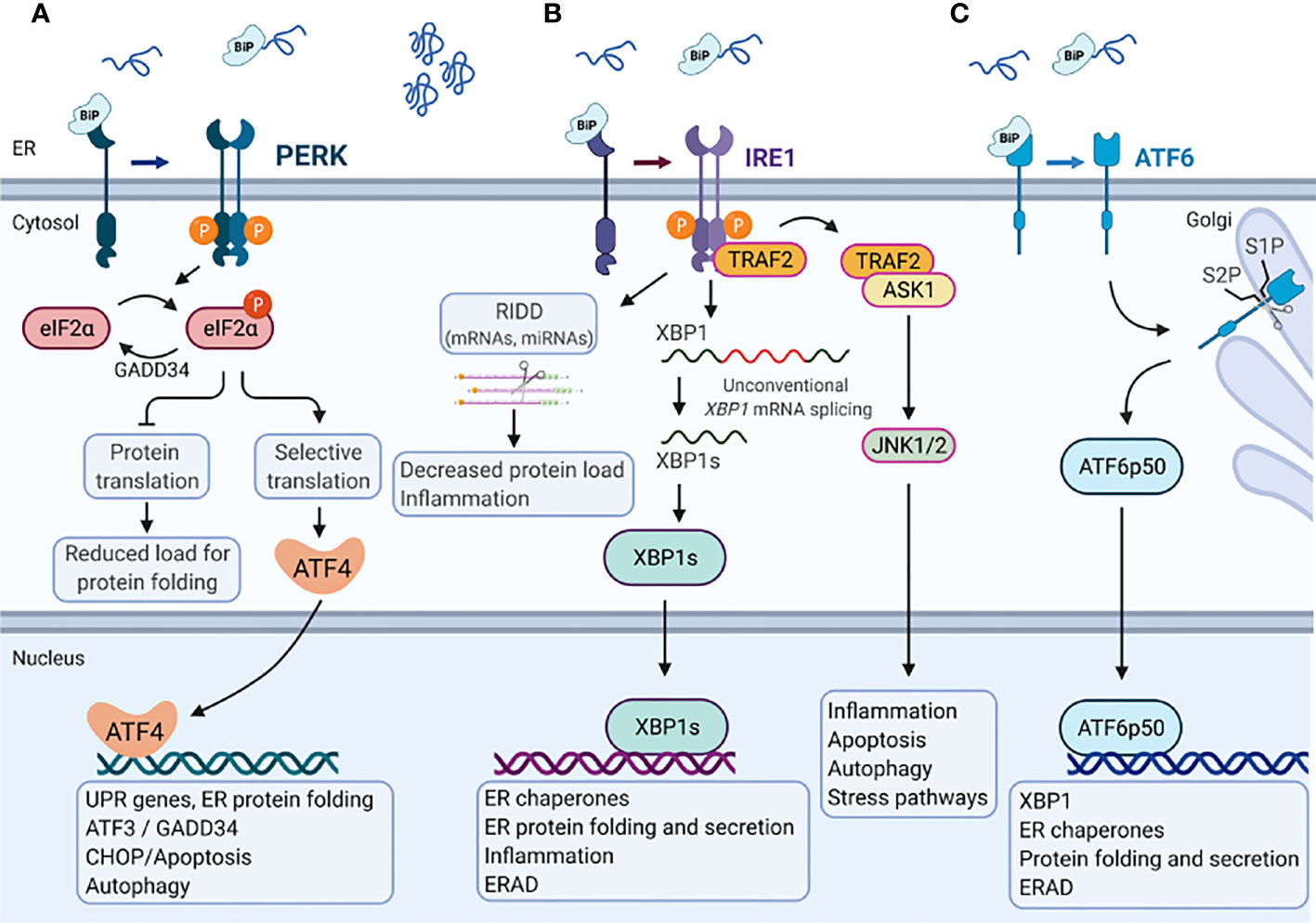
Figure 2 Endoplasmic reticulum stress-mediated Unfolded protein response. In the absence of ER stress, the ER chaperone protein GRP78/BiP is bound to the three major branches of the unfolded protein response (UPR) (A) PERK, (B) IRE1, and (C) ATF6 and keeps them in an inactive state. Upon accumulation of misfolded or unfolded proteins that cause Endoplasmic reticulum (ER) stress, GRP78/BiP dissociates from these receptors and binds to misfolded proteins, which activates the UPR signaling proteins. (A) PERK phosphorylates eukaryotic translation initiation factor 2 subunit-α (eIF2α), decreasing mRNA translation. However, specific mRNAs (e.g., ATF4) mRNA can be translated in the presence of phosphorylated eIF2α. ATF4 activates the transcription of UPR target genes that encode proteins involved in ER protein folding, autophagy and apoptosis (e.g., CCAAT/enhancer- binding protein homologous protein (CHOP) and GADD34). GADD34 targets protein phosphatase 1 (PP1) to dephosphorylate eIF2α which reactivates mRNA translation. (B) IRE1α has endoribonuclease activity and splices XBP1 mRNA to XBP1s, which encodes a transcription factor that activates expression of UPR target genes such as chaperones, ER folding and secretion, inflammation and ERAD. Activation of the IRE1α endoribonuclease activity can also induce the degradation of mRNAs encoding membrane or secreted proteins by regulated IRE1-dependent decay (RIDD), which affects the protein folding load, inflammation and inflammasome signaling. The cytosolic domain of IRE1α is also able to interact with tumor necrosis factor receptor-associated factor 2 (TRAF2), which can activate ASK1 and JNK1/2 to mediate inflammation, apoptosis, autophagy and stress pathways. (C) ATF6 moves from the ER to the Golgi apparatus, where it is cleaved by site-1 protease (S1P) and site-2 protease (S2P). The cleaved product (ATF6p50) migrates to the nucleus and activates the transcription of XBP1 as well as genes that are involved in ER protein folding and secretion, encode ER chaperones, and ERAD components. Created with BioRender.com.
IRE1, a type I transmembrane protein, is conserved from yeast to human with two paralogues in Mammalia, IRE1α and IRE1β. Their cytosolic domain comprises a serine-threonine kinase and an endoribonuclease activity. In response to ER stress GRP78 dissociates from IRE1, which in turn dimer- or oligomerize, become autophosphorylated and activates its own ribonuclease activity. IRE1 activation results in spliceosome-independent splicing of a 26-base intron from X-box-binding protein 1 (XBP1) mRNA (65). Spliced XBP1 (XBP1s) acts as a transcription factor that increases the expression of genes encoding ER chaperones, transcription factors (e.g., XBP1) and components of the secretory pathway as well as proteins involved in ER-associated degradation (ERAD) (66). Activation of IRE1 also induces rapid turnover of mRNAs encoding membrane or secreted proteins in a process called regulated IRE1-dependent decay (RIDD) (67). However, long lasting ER stress causes binding of IRE1α to TNF receptor-associated factor 2 (TRAF2), which in turn recruit’s apoptosis signal-regulating kinase 1 (ASK1) and activates the apoptotic c-Jun amino-terminal kinase (JNK) as well as the p38 pathway to initiate cell death (68, 69).
PERK, a type I transmembrane protein, binds GRP78 in its inactive state and has a cytosolic domain with kinase activity. In response to ER stress, PERK undergoes autophosphorylation and oligomerization. Activated PERK phosphorylates eukaryotic translation initiation factor 2α (eIF2-α) on serine 51, resulting in general inhibition of translation. However, eIF2-α also increases the translation of some mRNAs, such as that of the transcription factor activating transcription factor-4 (ATF4), which in turn induces UPR target genes (70). ATF4 increases the expression of the transcription factors ATF3 and C/EBP homologous protein (CHOP). ATF3 together with ATF4 promote the expression of growth arrest and DNA damage-inducible protein (GADD34), a regulatory subunit of protein phosphatase 1 (PP1), leading to dephosphorylation of eIF2-α which re-activates protein synthesis (62). In contrast, CHOP induces multiple pro-apoptotic molecules and is therefore considered a pro-apoptotic protein (71).
ATF6 is a type II transmembrane protein, that in response to ER stress moves from the ER to the Golgi apparatus where it is cleaved by Site-1 protease and Site-2 protease. This process is called regulated intermembrane proteolysis. The cleaved N-terminal fragment of ATF6 is a basic leucine zipper (bZIP) transcription factor that harbors DNA binding and transcription activation domains (72, 73). Upon cleavage ATF6 translocates to the nucleus where it cooperates with XBP1s to bind to ER stress response elements (ERSE) in the promoter region of genes encoding proteins that counteract ER stress, such as GRP78, GRP94 and Calnexin to induce their expression. The activation and downstream signaling pathways of the different UPR branches have been reviewed in further detail recently (63).
For removal of misfolded proteins from the ER, the quality control system of the ER sorts them for ER-associated protein degradation (ERAD) via the proteasome. The UPR activates the transcription of ERAD genes (74). The process of ERAD follows four consecutive steps: misfolded proteins are 1) recognized by GRP78/BiP and transferred to the ER membrane where they are 2) ubiquitinated and retrotranslocated by E3 ubiquitin ligase containing multiprotein complexes and are 3) extracted from the ER membrane by the p97 complex, and 4) are finally transferred to the 26S proteasome for degradation [reviewed in (75)].
The critical importance of the UPR for cell survival and organ function is supported by genetic studies. For example, homozygous Grp78/BiP knockout mice show pre-implantation lethality by embryonic day 3.5, with greatly reduced proliferation of embryonic cells and increased apoptotic death of the inner cell mass that are the precursors of embryonic stem cells (76). These data indicate that GRP78 is indispensable for embryonic cell growth and pluripotent cell survival. In contrast, heterozygous Grp78/BiP mice, in which the GRP78/BiP levels are halved in adult tissues, are viable and phenotypically normal (76). In addition, although Perk1-knockout mice are morphologically normal at birth, they die postnatally due to β-cell degeneration in the pancreas resulting in diabetes mellitus, reduced exocrine function of the pancreas with attenuated secretion of digestive enzymes, and skeletal defects (77). Similarly, homozygous Eif2aS51A-mutant mice that express a phosphorylation resistant form of eIF2α die within 18 hours after birth due to hypoglycemia associated with defective gluconeogenesis (78). Both Ire1- and Xbp1-deficient mice are embryonically lethal due to defective B cell lymphopoiesis and liver failure, respectively (79). Single Atf6a and Atf6b-knockout mice develop normally, whereas Atf6a and Atf6b-double knockout mice are embryonically lethal (80). In addition, when exposed to ER stress-inducing reagent tunicamycin (an inhibitor of N-glycosylation in the ER) by intraperitoneal injection the Atf6a-knockout mice display liver dysfunction and steatosis (81). In summary, these data show that the UPR is particularly important for proliferative and secretory cells, which facilitated research especially in cells and organs and their pathologies were these aspects are important. In contrast, in skeletal myocytes and muscle associated pathologies these features have not been recognized as that important for a long time. Despite the fact that the skeletal muscle harbors a large network of ER, the function of ER stress and the UPR in the regulation of physiological skeletal muscle function and diseases is not well understood. However, current studies reveal that an activation of the UPR can have beneficial as well as deleterious effects for the skeletal muscle (82). In the following paragraphs, we review the available literature about the role of the UPR in the skeletal muscle focusing on ICUAW and associated pathologies.
Appropriate nutrition is often an issue in the care of critically ill patients especially during the first days in ICU that often lead to missed caloric goals. Adequate feeding and outcome are closely connected with each other. Both underfeeding and overfeeding is often observed in critically ill patients, which can be harmful to critically ill patients suggesting that caloric goals need to be correctly determined and reached (83). Underfeeding and overfeed are related to uncertainties about the energy expenditure (resting metabolic rate, additional requirements due to the illness that is also dynamic with the disease course), the underlying disease itself, unknown body composition, difficulties to administer food, and the route of enteral versus parenteral nutrition. Patients biological age and sex differences may also have an effect on energy expenditure. Uncertainties about the right composition of the administered calories complicate the situation. As we describe later in this review hyperglycemia and insulin resistance are risk factors for ICUAW. It is therefore reasonable to assume that treating these risk factors may improve clinical outcome. This hypothesis led to the clinical concept of permissive underfeeding, which has been shown to reduce blood glucose and insulin levels indicative for an improved insulin sensitivity (266). However, Arabi et al., investigated if permissive underfeeding has an effect on clinical outcome in critically ill patients. They showed that administration of a hypocaloric diet with 70% of calculated caloric requirements (permissive underfeeding) did not affect mortality, ICU stay and other secondary outcomes when compared to standard nutrition (266). The study design required that both groups received the same amount of proteins implicating that not the number of calories administered but that the amount of proteins could have an effect on outcome of critically ill patients. The problematic situation on feeding of critically ill patients has been reviewed elsewhere (267–270). Although experimental models suggest an association between the feeding status and UPR, its involvement in this complex situation is far from being understood.
Importantly, fasting is a strong stimulus for skeletal muscle atrophy (29), which shares common pathways with other causes of muscle wasting (84). The UPS and the ALP are the best described pathways involved in starvation-induced muscle atrophy. Recent work indicates that also the UPR is involved in this pathology (85). Specifically, 24 hours of starvation caused an elevation of mRNA and protein expression of UPR markers such as Atf4, Chop, Grp94, and Gadd34 and an increased splicing of XBP1 in the gastrocnemius muscle of mice. Also, 48 hours of starvation in cows leads to an increased IRE1α phosphorylation and an increased XBP1 splicing in hepatocytes, indicating a connection between ER stress and starvation (86).
Additionally, TNF Receptor Associated Factor 6 (TRAF6) an E3 ubiquitin ligase that belongs to a family of conserved intracellular adaptor proteins has been shown to be involved in UPR. In response to cytokines and microbial products, TRAF6 regulates multiple signaling pathways, such as NF-κB, MAPK, and PI3K/Akt. Skeletal muscle specific deletion of Traf6 attenuates skeletal muscle atrophy caused by starvation, denervation, and cancer cachexia (15, 85). Of note, TRAF6 is essential for the inducible expression of ER stress response-related genes in response to fasting. Specifically, the starvation-induced increase in mRNA and protein expression of the UPR markers ATF4, CHOP, GRP94, and GADD34 in the gastrocnemius muscle was attenuated in muscle specific Traf6 knockout mice (85). However, the mechanism by which TRAF6 and the UPR are connected in skeletal muscle during starvation remains to be uncovered.
The activating transcription factor 4 (ATF4) that is downstream of the PERK arm, was also shown to mediate starvation-induced muscle atrophy. Of note, 24 hours of fasting increased the Atf4 mRNA expression in tibialis anterior of male mice (87). Inhibition of ATF4 expression by electroporation of a small interfering RNA targeting ATF4 or overexpression of a phosphorylation-resistant form of eIF2α (eIF2α-S51A) attenuated myofiber atrophy in tibialis anterior muscle during fasting (87). Overexpression of ATF4 caused a reduction in myofiber size even in the absence of fasting. Importantly, electroporation of a transcriptionally inactive ATF4 construct (ATF4ΔbZIP) did not reduce myofiber size, suggesting that ATF4-dependent gene expression is required for ATF4-mediated atrophic effects (87). These data show that ATF4 mediates starvation-induced muscle atrophy but it can also cause muscle atrophy in the absence of starvation.
Starvation has also been shown to activate the nutrient-sensitive transcription factors transcription factor EB (TFEB) and transcription factor E3 (TFE3). Both TFEB and TFE3 belong to the microphthalmia/transcription factor E (MiT/TFE) family of basic bZIP transcription factors that recognize a unique E-box motif within the proximal promoters of lysosomal and autophagy genes (88, 89), and regulate cellular catabolism and nutrient-dependent lysosomal response (90, 91). Both TFEB (92) and TFE3 (89) are predominantly localized in the cytoplasm but also to the nucleus (93, 94). In the presence of nutrients, mTORC1 (mammalian target of rapamycin complex 1) phosphorylates TFEB and TFE3, which facilitate their binding to 14-3-3 chaperone proteins and mediates their retention in the cytoplasm. Conversely, during starvation reduced mTORC1 activity increases the shuttling of TFEB (89) and TFE3 (89) into the nucleus increasing the expression of TFEB- and TFE3-dependent genes. Importantly, ER stress also induces the nuclear translocation of TFEB and TFE3 and this effect was shown to be dependent on PERK and calcineurin but not mTORC1 (95). We recently described that TFEB (93) and TFE3 (94) are also localized to the nucleus where TFEB is bound to conserved E-box elements in the MuRF1/Trim63 promoter to regulate its expression (93) implicating a role in muscle atrophy. Both TFEB and TFE3 are inhibited by class IIa histone deacetylases (i.e., HDAC4, 5, and 7) and this inhibition is reversed by the Protein kinase D family (i.e., PKD1, 2 and 3). However, if this pathway is important in ER stress induced muscle atrophy has not been investigated.
Insulin resistance accompanied by hyperglycemia is common in critically ill patients and strongly associated with increased morbidity and mortality (96). The underlying mechanism is still elusive but includes a decreased activity at the level of the insulin receptor and the post-receptor level (97). The insulin receptor is a receptor tyrosine kinase and its activation by insulin causes the cytosolic domains to autophosphorylate. The activated insulin receptor phosphorylates tyrosine residues in proteins such as insulin receptor substrate 1 (IRS-1) and IRS-2 that mediate downstream signaling. IRS-1 activates PI3K and Akt, which regulate muscle protein homeostasis (98). In addition, activation of the insulin receptor via the Akt substrate of 160 kDa leads to translocation of the glucose transporter (GLUT)-4 into the plasma membrane, which increases cellular glucose uptake (99). Insulin resistance causes decreased glucose uptake in skeletal muscle. Under physiological conditions myocytes internalize circulating glucose via their main glucose transporter glucose transporter-4 (GLUT4). GLUT4 is contained in storage vesicles that are localized near the sarcolemma, the transverse tubular system and in the trans-Golgi region (100). Normally, these GLUT4 storage vesicles are translocated to the plasma membrane and glucose uptake is increased in skeletal muscle in response to insulin (101). In insulin resistance insulin receptor signaling is perturbed, GLUT4 translocation is attenuated and circulating glucose cannot be taken up. Since glucose is the main source of energy in muscle, less energy-rich phosphates (e.g., ATP) are formed, which reduces muscular energy supply (102). This impairs muscle function, especially strength and endurance. At the same time, insulin has an anabolic effect on muscle by increasing protein synthesis and inhibiting protein breakdown (98, 103), so that insulin resistance leads to disturbed protein homeostasis with increased protein breakdown and reduced protein synthesis, resulting in muscle atrophy and weakness (27, 37). Importantly, insulin resistance has been reported to be a predictor for ICUAW (47). Indeed, previously we reported that GLUT4 was trapped in the perinuclear space of myofibers of critically ill patients whereas it was localized to the sarcolemma of myofibers of control subjects (102). This observation was most pronounced in patients with critical illness myopathy, a severe form of ICUAW further supporting that insulin resistance is involved in ICUAW.
Although the contribution of the UPR to insulin resistance in skeletal muscles of the critically ill is uncertain, published evidence supports this assumption. For example, ER stress has been shown to be associated with insulin resistance, which was accompanied by a suppression of insulin receptor signaling and serine phosphorylation of insulin receptor substrate-1 (IRS-1), in vivo and in vitro (104). Insulin resistance was also described in Xbp1 knockout mice (104). Inhibition of ER stress by the chemical chaperones 4-phenyl butyric acid (4-PBA) or taurine-conjugated ursodeoxycholic acid (TUDCA) restored systemic insulin sensitivity, enhanced the action of insulin on muscle, and normalized hyperglycemia in obese and diabetic mice (105). These data indicate that inhibition of ER stress by the chemical chaperones 4-PBA and TUDCA counteracts insulin resistance.
The adenosine 5’-monophosphate (AMP)-activated protein kinase (AMPK) that senses and regulates cellular energy states increases the expression of GLUT4 (106). Deletion of Ampk2a (encoding AMPK2α) in mice causes a reduction in glucose utilization in skeletal muscle (107). Activation of ER stress by tunicamycin or thapsigargin (an inhibitor of ER-specific calcium ATPase) reduces AMPK activity, insulin signaling and glucose uptake in rat-derived L6 myotubes, mainly due to an increased extracellular signal-regulated kinase (ERK) phosphorylation. Inhibition of ERK restored AMPK phosphorylation and glucose uptake that were deteriorated in response to ER stress (108). These data implicate a cross talk between ER stress and the major energy sensor AMPK in myocytes.
Insulin resistance caused by the saturated fatty acid palmitic acid also caused ER stress as indicated by an increased mRNA and protein expression of the ER stress-markers ATF6, ATF3, GRP78, CHOP, and Tribbles 3 (TRIB3) in C2C12 myotubes. Palmitic acid-induced ER stress, insulin resistance and, inflammation were attenuated by low concentrations of the proteasome inhibitor Bortezomib and these effects were mediated by AMPK. Importantly, knockdown of AMPK abolished the protective effects of Bortezomib (109). These studies indicate that ER stress induced insulin resistance is at least partially mediated by inhibition of AMPK activity.
The pseudokinase Tribbles 3 (TRIB3) that is expressed in various tissues such as liver, adipose tissue, heart, and skeletal muscle (110) has also been shown to impede insulin signaling (111). TRIB3 is increased in skeletal muscle of patients with type 2 diabetes mellitus known to be insulin resistant (110). TRIB3 binds to Akt and inhibits Akt phosphorylation eventually preventing insulin-stimulated glucose uptake in C2C12 myocytes (112). Activation of ER stress by tunicamycin and thapsigargin causes an increase in TRIB3 in C2C12 cells and skeletal muscle, which possibly mediates ER stress-induced insulin resistance. Accordingly, when TRIB3 was overexpressed in tibialis anterior of mice insulin signaling was greatly impaired (113). In addition, deletion of Trib3 in mice prevented high-fat diet induced insulin resistance in skeletal muscle (113). If Tribbles 3 plays a role in insulin resistance in skeletal muscle of the critically ill is not known.
The Protein tyrosine phosphatase 1B (PTP1B) that is a key negative regulator of insulin signaling by dephosphorylation of tyrosine residues of the insulin receptor which inhibits insulin receptor signaling (114). PTP1B that is localized on the ER membrane has also been shown to mediate insulin resistance in response to ER stress (115). Specifically, high-fat diet (for 20-weeks) induced ER stress (e.g., GRP78) caused an increase in PTP1B expression in the gastrocnemius of mice. This response was attenuated by administration of TUDCA for 5-weeks. Likewise, tunicamycin-induced ER stress caused an increase in PTP1B levels in C2C12 myotubes. In addition, tunicamycin-induced phosphorylation of eIF2α and JNK2 were attenuated in Ptpn1 knockout mice, encoding PTP1B. Downregulation of PTP1B or inhibition of ER stress by TUDCA treatment improved glucose uptake in tunicamycin treated C2C12 myotubes (116). These data indicate that ER stress activates PTP1B and that PTP1B is required for activation of ER stress pathways to mediate insulin resistance in skeletal muscle. In addition, it has been reported that mice harboring a germline deletion of Ptpn1 were protected against high-fat diet induced insulin resistance, weight gain, and hepatic steatosis. These mice also exhibited improved glucose uptake and Akt phosphorylation in the gastrocnemius. In addition, high-fat diet-induced increase in GRP78/BiP and phosphorylation of eIF2α as well as JNK2 were significantly blunted in Ptpn1 knockout mice. These data support the hypothesis that ER stress induces the expression of PTP1B which mediates insulin resistance in the skeletal muscle in response to high-fat diet (117). It has also been shown that PTP1B potentiates IRE1-mediated ER stress signaling pathways in mouse embryonic fibroblasts (118).
Hyperglycemia leads to elevated glucosamine levels that are also able to induce insulin resistance and ER stress as indicated by an increased expression of the ER stress markers GRP78/BiP, XBP1 and ATF6 in both human and L6 myotubes (119). Pretreatment of both rat and human myotubes with 4-PBA or TUDCA, attenuated glucosamine-induced ER stress and insulin resistance. Glucosamine reduced the expression of GLUT4, myocyte enhancer factor 2A (MEF2A) and peroxisome proliferator-activated receptor gamma coactivator-1 alpha (PGC-1α) expression, which was prevented by 4-PBA and TUDCA in both rat and human myotubes. Importantly, glucosamine-mediated downregulation of GLUT4, MEF2A and PGC-1α was attenuated by ATF6 silencing. These data strongly indicate that the ATF6 arm of the UPR mediates glucosamine-induced insulin resistance (119).
In summary, these studies indicate that ER stress and UPR pathways are involved in systemic and muscular insulin resistance and muscular glucose uptake. However, if this pathomechanism is involved in ICUAW and if its targeting has therapeutic potential requires further investigation.
Immobilization of critically ill patients will inevitably lead to disuse and unloading of skeletal muscle which are major risk factors for ICUAW (21). Although immobilization allows complex treatments in ICU, the accompanying unloading of the skeletal muscle will lead to skeletal muscle atrophy and a reduction in muscle force. The loss of muscle mass and function delays recovery and attempts to prevent muscle atrophy through physical therapy or other interventions have been insufficient. Despite the fact that skeletal muscle atrophy due to immobilization is a prevalent and severe clinical problem in many different medical situations, the underlying mechanisms are not well understood. Of note, bed rest in humans and immobilization of animals have been shown to cause skeletal muscle atrophy mainly by an increased activity of UPS-mediated protein degradation with enhanced expression of MuRF1 and atrogin1 (27, 29–32, 120). Since skeletal muscle atrophy due to disuse leads to an upregulation of genes encoding sarcoplasmic reticulum (SR) calcium-handling proteins and considering that many of the proteins that are induced with ER stress are also calcium-handling proteins a connection between disuse atrophy and ER stress is likely. This hypothesis was tested in soleus muscle of rats after seven days of unloading. In this model, the expression of the UPR markers GRP78/BiP, calreticulin, CHOP and eIF2α remained unchanged in response to inactivity (121). Similar observations have been made by Baehr et al., were hindlimb unloading did not cause ER stress in soleus of adult (9 month) and old (29 month) male rats (122). However, ER stress was different between soleus and tibialis anterior of the same animals and revealed age-specific effects. The authors also investigated if reloading of the muscle show age-specific effects on ER stress. Indeed, in response to 14 days of reloading, both GRP78/BiP and CHOP were increased in soleus of adult but not old mice. Also, CHOP increased upon reloading in tibialis anterior of old but not adult rats, whereas GRP78/BiP remained unchanged (122). Contrary to the mentioned data, an upregulation of ER stress and UPR pathway genes in response to immobilization was reported by other groups. For example, 10-days of bed rest resulted in a persistent upregulation of the UPR in skeletal muscle of young men, as shown by microarray analysis (123). In addition, the expression of ATF4, a downstream transcription factor of PERK branch, increases in response to 3 days of unilateral hindlimb immobilization in tibialis anterior in mice (124). Furthermore, conditional Atf4-knockout mice lacking ATF4 in muscle are resistant to immobilization-induced muscle atrophy, fasting-induced muscle atrophy, and age-related muscle atrophy (87, 124–126). Conversely, forced expression of ATF4 is sufficient to induce muscle atrophy even in the absence of inactivity (87, 126). Further studies that address the individual UPR pathways are needed to elucidate their specific role of ER stress in disuse-induced skeletal muscle atrophy.
Denervation is another model to investigate mechanisms of immobility-induced muscle atrophy. To achieve this phenotype the sciatic nerve is cut and dissected in rodents (29). It has been shown that denervation leads to an increased phosphorylation of eIF2α and splicing of XBP1 mRNA in the gastrocnemius 3 days and 7 days post denervation indicative for an activation of the PERK and IRE1 arm of the UPR. In denervation, also the expression of Grp78/BiP and Chop was also increased (127). These data show that denervation activates the PERK and IRE1 arm of the UPR in skeletal muscle. It also showed that the proapoptotic transcription factor CHOP, also a target gene of the PERK arm of the UPR, is involved in denervation induced muscle atrophy. Specifically, CHOP deficient mice exhibited an increased skeletal muscle wasting in response to denervation (127). Parveen et al., demonstrated an increased mRNA expression of Grp78, Atf4, and Atf6 and an increased protein content of PERK, ATF4, CHOP, IRE1α, and XBP1s in the gastrocnemius of mice in response to 14 days of denervation (128). They also found that a myofiber-specific ablation of Xbp1 attenuated denervation-induced skeletal muscle atrophy in mice (128). A further study in mice revealed that the PERK target gene Sestrin2 (Sesn2) that is a stress responsive protein and protects cells from cell death by maintaining mitochondrial homeostasis (129) is upregulated in the gastrocnemius two weeks after denervation (130). Knockdown of Sesn2 aggravated denervation-induced skeletal muscle atrophy. These data indicate that the PERK arm of the UPR via upregulation of Sestrin2 attenuates denervation-induced skeletal muscle atrophy (130). However, because the IRE1/XBP1 arm of the UPR has also been shown to increase Sesn2 expression, this effect is not unique for the PERK arm (131). In summary, these data reveal a complex involvement of the different UPR arms in various atrophy models depending on age and muscle types. Further studies are needed to decipher the involvement of the individual UPR branches in specific atrophy conditions. Generally, it seems that dependent on the muscle atrophy trigger, different branches of the UPR are activated. One further example for this hypothesis is the differential expression of the UPR in sarcopenic patients suffering from either chronic obstructive pulmonary disease (COPD) or lung cancer cachexia. While lung cancer cachexia-patients show an increase in many ER stress markers and all three UPR branches, only few ER stress markers and the IRE1α branch of the UPR are increased in COPD patients (132).
Almost all major ER stress related genes have been found to be upregulated in sepsis, as summarized by Khan and colleagues (133). Moreover, the UPR is able to regulate cytokine production at various steps, from stimulation of pattern recognition receptors (PRR), to modulation of inflammatory signaling pathways, and the regulation of cytokine transcription factors (134). These reviews (133, 134) based on a significant number of original articles demonstrating that there is a connection between sepsis, inflammation and the UPR pathway. However, if these UPR pathways are also relevant for ICUAW has not been shown.
In sepsis, expression of the pattern recognition receptors Toll-like receptor (TLR) 2 and TLR4 as well as their downstream pathways are induced in skeletal muscle (31). Specifically, we recently showed that the acute phase protein serum amyloid A1 (SAA1) via TLR2 and TLR4 activates NF-κB p65, that translocates to the nucleus, binds to NF-κB response elements in the promoter region of its target genes and increases their expression (31). Inhibition of NF-κB inhibited sepsis-induced muscle wasting in mice (31). In addition, TLR2 and TLR4 have been shown to engage IRE1α to promote cytosolic splicing and activation of XBP1 to increase the expression of proinflammatory cytokines, such as IL-6, to regulate innate immune responses in macrophages (135). Our data also revealed that SAA1 via TLR2 and TLR4 and the NF-κB pathway increased the expression of IL-6 in myocytes (28, 31). However, if SAA1 binding to TLR2 and TLR4 also affects the IRE1α/XBP1/IL-6 axis in myocytes has not been tested. Moreover, the expression of several TLR as well as their downstream signaling adaptor, myeloid differentiation primary response 88 (MyD88), are increased in response to denervation in skeletal muscle of mice. Importantly, deletion of Myd88 inhibited the activation of XBP1 and attenuated muscle atrophy in response to denervation (128). These data indicate that TLR and TLR signaling are connected to UPR during inflammation.
The skeletal muscle has been shown to be affected by and to react to immunologic conditions such as proinflammatory stimuli (30–32, 136). The involvement of the UPR in the pathogenesis of different myopathies such as Sporadic inclusion body myositis, Myasthenia gravis, Duchenne muscular dystrophy has been reviewed recently (137). In immune cells, ER stress and the UPR are implicated in the regulation of the inflammatory state (138). A couple of studies demonstrated that also in myocytes, proinflammatory stimuli regulate UPR signaling. For example, acute muscle injury by cardiotoxin injections resulted in an inflammatory response and an increased expression of ER stress and UPR genes, including chaperones and genes from all three branches of the UPR. A link between increased cytokines and ER stress in skeletal muscle has also been suggested by the observation that the UPR is disrupted in inflammatory muscle diseases (139).
Interestingly, Interferon gamma (IFN-γ) treatment of myocytes causes a muscular immune response and activates the IRE1α and PERK but not the ATF6 arm of the UPR. Specifically, IFN-γ caused ER stress in mouse myogenic progenitor cell-derived myotubes in vitro. In addition, ER stress was also caused by cardiotoxin injections in tibialis anterior in vivo. At base line myoblasts and differentiated myotubes do not express class I major histocompatibility complex (MHC) I and MHCII (140). However, when myotubes were treated with IFN-γ the expression of MHCI, TLRs and proinflammatory cytokines (i.e., IL-1β, IL-6) was considerably increased. This response was further triggered by inhibition of ER stress through 4-PBA. The ER stress inducing agents tunicamycin and thapsigargin prevented the IFN-γ-induced expression of MHCII and MHCI molecules. Inhibition of IRE1α attenuated the UPR stressor-mediated reduction of these immunobiological molecules (141). These data indicate that the IRE1α arm mediates immunobiological suppression in myocytes during inflammation. Notably, not only does inflammation affect the UPR but this also works the other way around. IRE1α induces JNK signaling, which regulates many inflammatory genes (68). IRE1α signaling also activates the NF-κB signaling pathway that induces multiple inflammatory cytokines, such as IL-6. In addition it has been demonstrated that IRE1α and IKK are linked by TRAF2 to activate NF-κB (142) and this pathway was also activated in inflammatory skeletal muscle atrophy (30, 31).
There is strong evidence that ER stress induces inflammation by activating inflammasomes (143). Inflammasomes are multiprotein complexes that act as scaffolds for caspase-1-mediated maturation and secretion of inactive pro-IL-1β and pro-IL-18 to active IL-1β and IL-18 (144). Both IL-1β and IL-18 are involved in sepsis (145), sepsis-induced muscle failure (30) and sepsis-induced cardiomyopathy (35). Cytoplasmic receptors of the nucleotide-binding domain (NOD)-like receptor (NLR) family are key inflammasome components (144). The NOD-, leucine-rich repeat (LRR)- and pyrin domain-containing protein-3 (NLRP3) is one of the best-characterized NLR proteins able to form an inflammasome complex and all components of the NLRP3 inflammasome are contained in the skeletal muscle (146). Importantly, ER stress has been reported to activate the NLRP3 inflammasome with a subsequent increase in IL-1β secretion. Because this effect was independent of the UPR a contribution of a novel ER stress response signaling pathway was proposed (143). These findings taken together strongly indicate that there is a direct link between ER stress and inflammation in skeletal muscle and given the role of proinflammatory cytokines in muscle atrophy also a connection between ER stress and ICUAW is very likely. This however needs to be proven.
The role of circulating and tissue residing immune cells for ICUAW in general and ER stress and UPR in particular is not well understood. However, novel technical advances may provide new insights into the role of the various different cell types contained in muscle for the pathogenesis of muscle wasting in critically ill patients. For example, using a combined approach of single-cell RNA sequencing and single-cells mass cytometry, ten different mononuclear cell types (i.e., muscle satellite cells, fibro-adipogenic progenitors, macrophages, neutrophils, endothelial cells, B cells, T cells, glial cells, smooth muscle and mesenchymal cells and scleraxis-positiv tenocytes) were mapped in adult mouse muscle (271). Another study used single-cell RNA-sequencing to classify single cells isolated from tibialis anterior muscle into myocytes, endothelial cells, fibroblasts, mesenchymal stem cells, macrophages, neutrophils, T-cells, B-cells, and dendritic cells. These data indicate that several classes of immune cells are contained in muscle. Importantly, early during sepsis, a decrease in the proportion of most non-immune cell populations and an increase in immune cells (i.e., neutrophils and macrophages) was observed in muscle of male and female mice subjected to the cecal ligation and puncture (CLP) and faecal-induced peritonitis (FIP) models of sepsis. These findings persisted for one month after sepsis (272). In summary, these data implicate that immune cells may be involved in sepsis-induced muscle wasting. However, if UPR in any of these muscle residing cells contributes to ICUAW has not been investigated and is far from being understood. Likewise, if ER stress and UPR affect the levels of immune cells in muscle tissue during acute or chronic injury is also unknown. Nevertheless, these novel techniques will allow us to investigate the function of UPR in any of these cells and its importance for ICUAW.
Both hypoxemia and hypercapnia are associated with an increased mortality in critically ill patients, particularly in those patients with acute respiratory distress syndrome (ARDS).
Hypoxemia induces skeletal muscle atrophy in male Sprague-Dawley rats (273) and male C57BL/6J mice (274), and was implicated to play a role in muscle wasting of patients with chronic obstructive pulmonary disease (275). Hypoxemia-induced muscle wasting was shown to increase MuRF1/Trim63 and atrogin-1/Fbxo32 expression possibly mediating muscle wasting (de Theije et al., 2018). However, hypoxia is also a strong activator of the oxygen sensitive transcription factor hypoxia-inducible factor 1 (HIF-1). Once activated, HIF-1 increases the expression of genes involved in angiogenesis such as vascular endothelial growth factor A (VEGFA) and erythropoiesis as well as glucose and energy metabolism [for Review (276)]. Previously it has been shown that hypoxia and UPR are closely connected with each other. In their study Pereira et al., nicely demonstrated in human neuroblastoma cells that UPR induces VEGF mRNA expression and that ATF4 but not XBP-1s mediated this response (277). They also showed that cooperation of the HIF-1 and UPR pathways led to a greater induction of VEGF expression when compared to activation of either pathway alone (277). It is very likely, especially in critically ill septic patients that pneumonia, edema, and use of vasoconstrictive agents such as norepinephrine hinder oxygen delivery causing hypoxia in peripheral tissues such as skeletal muscle. Hypoxia mediated activation of UPR and HIF-1 may in turn accelerate muscle wasting. However, if the cooperation of both pathways contributes to ICUAW warrants further investigation.
An elevation of blood carbon dioxide levels called hypercapnia is often observed in critically ill patients with acute respiratory distress syndrome (ARDS) during protective mechanical ventilation (278) and may contribute to ICUAW. It is also frequently observed in patients with chronic obstructive pulmonary disease (COPD). Hypercapnia has been shown to cause skeletal muscle atrophy in human patients and male C57Bl/6 mice as well as cultivated myotubes (279, 280). Hypercapnia-induced muscle wasting was shown to be mediated by a decreased muscular protein synthesis (280) and an increased protein degradation via the AMP-activated kinase/FOXO3a/MuRF1 pathway (279). Recent evidence suggests that both acute and chronic hypercapnia impair ER function and cause UPR. A current review eluted on the distinct mechanisms by which hypercapnia and UPR pathways are interconnected in the setting of acute and chronic pulmonary diseases (281). Nevertheless, the importance of these pathways or their interaction for ICUAW is not well understood.
Many ICU survivors experience a long-lasting impairment of muscle function that may even be apparent five years after their acute illness (44, 45), which is indicative for a perturbed muscle regeneration. This is surprising given the fact that skeletal muscle has a tremendous capacity for regeneration (147). Regeneration of skeletal muscle is realized by muscle residing stem cells, called satellite cells, that are located between the sarcolemma and the basal membrane of myofibers in a quiescent state (147). In response to myofiber damage, satellite cells get activated, proliferate and differentiate into myoblasts that in turn fuse with injured myofibers (147). Although the majority of satellite cells differentiate into the myogenic lineage, few of them undergo self-renewal, return to a quiescent state and replenish the satellite cell pool. Myogenic differentiation requires a well-controlled expression of myogenic transcription factors (148), myoblast-fusion proteins (149, 150) and contractile proteins (151). This involves the sequential expression of the transcription factors paired box gene 3 (Pax3) and Pax7 followed by the expression of myogenic regulatory factors (MRFs) such as Myf5, MyoD, Myogenin, and MRF4. The role of myogenesis in maintaining skeletal muscle size is well described (152); however, its importance in critical illness in general and ICUAW in particular is unexplored. Previously is has been reported that regenerative muscle capacity is impaired in ICU patients (153) and septic mice (154); although, genes involved in skeletal muscle regeneration are upregulated in muscle of survivors of critical illness (155). The process of myogenesis and the underlying mechanisms are investigated in vitro by using primary satellite cells and myoblasts, mouse-derived C2C12 myoblasts or rat-derived L6 myoblasts, which form multi-nucleated myotubes in low serum conditions. To investigate myogenesis in vivo, intramuscular injections of barium chloride (156) or cardiotoxin, a snake venom toxin that causes myofiber necrosis without effects on satellite cells viability (157), and subsequent time course analyses are used.
Previously it has been reported that the UPR mediates selective apoptosis of differentiation-incompetent myoblasts during skeletal muscle formation (158). In these cells, ER stress activated caspase-12, which in turn activated caspase-9 and caspase-3 to mediate apoptosis. Previously it was shown that exclusively the ATF6 arm of the UPR was increased in those myoblasts that undergo apoptosis in response to induction of myogenic differentiation (159). When ATF6 or caspase-12 were inhibited, myoblast apoptosis was reduced and the formation of multi-nucleated C2C12 myotubes was attenuated. An increase in caspase-12 was also observed in skeletal muscle during embryonic development. These data suggest that the ATF6 arm of the UPR is involved in selective apoptosis of those myoblasts that are susceptible to stress (159). However, caspase-12 deficient mice develop normally (160), indicating that caspase-12–dependent apoptosis is not essential for muscle development. Interestingly, activation of ER stress has been shown to promote myofiber formation (161). When ER stress was activated by tunicamycin or thapsigargin differentiation-associated apoptosis of myoblasts was increased. Nevertheless, the surviving myoblasts were resistant to apoptosis and differentiated well into contracting myotubes. These data suggest that during myogenesis differentiation incompetent myoblasts are specifically targeted by ER stress (161).
The other arms of the UPR are also implicated during myogenesis. A transient activation of the PERK/eIF2α arm of the UPR and its downstream target, CHOP, has been observed in a subset of myoblasts during differentiation (162). Knockdown of CHOP expression lead to an earlier and more robust myogenic differentiation, whereas its overexpression delayed differentiation. Cells that expressed CHOP did not express the myogenic regulatory factors MyoD and Myogenin indicating that CHOP inhibits myogenic differentiation by inhibition of Myod gene expression.
The IRE1 arm of the UPR leads to the generation of XBP1s, a potent transcription factor (163). XBP1 was shown to be a target gene of the transcription factors MyoD and Myogenin (164) that are essential for myogenic differentiation suggesting a connection between XBP1 and myogenesis. XBP1 regulates the expression of genes involved in maintenance of ER function, growth, and DNA damage and repair pathways in myoblasts as well as in myotubes (165). Overexpression of XBP1 inhibited the expression of myogenic differentiation markers, such as Myh7, Myh4 and Mef2c, and led to formation of shorter and less mature C2C12 myotubes during differentiation. This effect was caused by XBP1-mediated induction of Muscle, Intestine and Stomach Expression 1 (Mist1)/Basic Helix-Loop-Helix Family Member A15 (BHLHA15) expression. Specifically, XBP1 binds to the Mist1 promoter in myoblasts, XBP1s and ER stress increase the expression of Mist1 and downregulation of XBP1 reduced the ER stress-induced Mist1 expression (166). Mist1 is a transcription factor that inhibits the activity of MyoD (166). In summary, these data indicate that XBP1 plays a role in myogenic differentiation by induction of Mist1-mediated inhibition of MyoD (165).
The role of satellite cells in general and the UPR in particular in ICUAW is not well defined. Nevertheless, previously it has been shown that the downstream target of the PERK arm of the UPR eIF2α is constitutively phosphorylated in quiescent satellite cells and that this phosphorylation is decreased in activated satellite cells (167). When a phosphorylation resistant mutant of eIF2α was expressed (i.e., eIF2αS51A) or PERK was deleted satellite cell quiescence was lost as indicated by an increased Myf5 and MyoD expression. Satellite cells that express eIF2αS51A are able to undergo terminal differentiation and subsequent fusion with myofibers. However, these satellite cells are incapable to undergo self-renewal. Interestingly, this study also showed that inhibition of eIF2α dephosphorylation using the salubrinal-derivative Sal003 promoted satellite cell self-renewal ex vivo (168). Sal003 inhibits the serine/threonine phosphatase GADD34/PP1, which dephosphorylates phosphorylated eIF2α, and therefore keeps eIF2α in a phosphorylated state (169). In addition, inhibition of eIF2α dephosphorylation by Sal003 retained the myogenic capacity of satellite cells when transplanted in skeletal muscle of a mouse model of Duchenne muscular dystrophy in vivo (168). However, these data are in contrast to recently published work were eIF2αS51A overexpression was shown to cause cell death and diminish the differentiation of cultured myoblasts (162). Differences in the experimental approaches and techniques as well as the cell types used for analyses might account for these discrepancies (162, 168).
In summary, these data show that ER stress and UPR play a key role in satellite cell function and fate, skeletal muscle regeneration and myogenesis. However, it is currently unknown to which extend satellite cell function and regeneration are involved in ICUAW and even more so if ER stress and UPR are involved in this process.
To relive ER stress higher amounts of ATP are needed especially for a proper function of newly synthesized ER chaperone proteins. This suggests a close connection between the ER and mitochondria, which are the main site of ATP production in myocytes. Because muscle is a highly energy consuming tissue the interaction between the ER and mitochondria during ER stress possibly affects its function and adaptation processes. This is supported by the observation that tunicamycin-induced ER stress causes a translocation of mitochondria to the ER. These translocated mitochondria were reported to increase ATP production, which increases the intracellular ATP pool (282). The transport of ATP and signaling molecules between the ER and mitochondria occurs at interfaces between both organelles, which are called mitochondria-associated ER membranes (MAM) (283). This interface is realized by specific proteins localized to the outer mitochondrial membrane and the ER (284). MAM are involved in ER stress, calcium signaling, mitochondrial dynamics, lipid trafficking, initiation of apoptosis, and autophagosome formation [for review (284–286]. Interactions between ER and mitochondria in skeletal muscle and its effects on calcium homeostasis and reactive oxygen species has been reviewed recently (287). MAM have also been shown to influence insulin signaling through different pathways, including those associated with ER stress responses, mitochondrial function, and inflammation. A recent review addressed possible mechanisms underlying MAM-associated insulin resistance (Cheng et al., 2020). In addition, MAM have also been shown to be affected by insulin resistance, obesity, and type 2 diabetes. However, a decrease (Tubbs et al., 2018) as well as an increase (Arruda et al., 2014;Thoudam et al., 2019) of MAM in murine and human skeletal muscle as well as cultivated myocytes has been shown in response to metabolic and ER stress. These conflicting results are possibly due to differences in the techniques used to investigate MAM structure and function in skeletal muscle and myocytes. Likewise, the data on changes of the amount of proteins that mediate tethering of mitochondria to the ER in response to metabolic stress are not uniform. It is therefore difficult to assess the functional impact of MAM modifications for muscle. Also, no data are available for the role of MAM in muscle of critically ill patients, therefore, further analyses are needed to conclude if MAM are involved in the pathogenesis of ICUAW.
To prevent sequelae of ICU treatment, critically ill patients receive physiotherapy and if applicable early mobilization. The goal is to prevent muscle loss and to maintain and support muscle function, as reviewed elsewhere (41, 170). Protocol-based early mobilization has been reported to improve muscle function and reduce insulin resistance in ICU patients, and is recommended in guidelines (171–174). In general ICU patients, early mobilization has been shown to be clinically beneficial (174–176). If additional physiotherapeutic measures, such as neuromuscular electrical stimulation (NMES), have an effect on muscular structure or function is debated. Some reports described that such interventions prevent muscle atrophy and improve glucose metabolism and physical function, whereas others did not find any effect (102, 177–182). Whole-body vibration (WBV) has been shown to be effective for maintenance of muscle mass and strength in elderly individuals. However, its impact on long-term outcome of ICU patients has not been investigated, although it is feasible in critically ill patients (40, 183, 184). We recently investigated if additional muscle activating measures (e.g., NMES or WBV) have an effect on muscle structure and function when applied in combination with daily protocol-based physiotherapy in critically ill patients (SOFA ≥ 9). We observed that muscle integrity was maintained and found an increased myocyte cross sectional area in the intervention group as compared to those patients that received daily protocol-based physiotherapy only. However, early administration of additional muscle activating measures did not improve muscle strength or function in the critically ill (41). Although early mobilization, physiotherapy, muscle activating measures and physical training are quite distinct in terms of the intended goals and the applied work load, it is noticeable that acute and chronic exercise have been associated with ER stress. For example, a single strong bout of exercise and chronic exercise caused an activation of different branches of the UPR. However, whether or not mobilization therapy increases or decreases ER stress and UPR, if this interferes with the effects of other ICUAW risk factors on ER stress and if this has an effect on muscle structure and function is not known. It is possible, but not proven, that muscle activation measures aggravate UPR that is already induced by immobilization, inflammation, and treatment and that this is time, context, dose and frequency dependent.
If applicable critically ill patients receive a structured physiotherapy during ICU treatment (185). The effects of physiotherapy and mobilization on ER stress-induced UPR with regards to muscle structure and function are uncertain. Although not directly comparable to physiotherapy endurance and resistance exercise have been shown to regulate UPR in muscle. The effects of exercise on UPR were often investigated in rodent models that combined exercise with metabolic stress induced by, among others, type 2 diabetes mellitus and or high-fat diet. In these models, type 2 diabetes mellitus induced by streptozotocin injection and high-fat diet caused glucose intolerance, induced ER stress and endoplasmic reticulum-associated degradation (ERAD) in skeletal muscle of male mice. Swimming (1h per day, 5 days per week for 6 weeks) of these mice improved glucose intolerance and alleviated ER stress in skeletal muscle (186). Specifically, type 2 diabetes mellitus significantly increased the phosphorylation of IRE1α and ATF6 expression in skeletal muscle and swimming alleviated this response. In contrast, the PERK branch of the UPR remained unaffected by type 2 diabetes mellitus and exercise. Similar findings have been reported in muscle biopsy specimens from vastus lateralis from young and old untrained men in response to resistance exercise. Acute resistance exercise led to an increase in both XBP1 and XBP1s, indicating that exercise may stimulate the IRE1α and ATF6 arms of the UPR. In contrast, acute resistance exercise did not affect the PERK pathway as no increase in Atf4, Chop, Gadd34, or eif2a mRNA expression and eIF2α phosphorylation were found 24 hours and 48 hours after exercise (187). In contrast to swimming and resistance exercise, it was shown that endurance exercise, when applied in addition to high-fat diet in mice, further increased the expression of several UPR markers when compared to high-fat diet alone. This response was different between the muscle (i.e., soleus vs. tibialis anterior) and organs (i.e., skeletal muscle, liver, pancreas) investigated (188). Importantly, it has been reported that UPR is absent in the heart in response to short-term exercise in adult male Sprague Dawley rats (189) and male and female mice (190). It is also interesting to note that a single bout of running exercise activates the UPR in some (e.g., quadriceps, gastrocnemius) but no other muscles (e.g., erector spinae) (190) indicating a muscle specific regulation and possibly function of the UPR. Muscle specific (soleus vs. tibialis anterior) and age-related regulations of ER stress markers have also been observed in adult (9 month) and old (29 month) male rats following 14 days of hindlimb unloading by tail-suspension and 13 days of reloading (122). In summary, these data imply that the UPR is differentially regulated in a muscle- and tissue-specific manner that depends on the type of exercise applied (e.g., swimming, resistance- or endurance exercise) and its duration (e.g., short- or long-term, single bout) as well as the preexisting metabolic situation and age. An additional level of complexity is added when the different branches of the UPR are considered. Whether or not physiotherapy and early mobilization are able to increase or decrease UPR, if this adds on to preexisting UPR caused by inflammation, immobilization, insulin resistance, and if this has beneficial or detrimental effects on skeletal muscle in critically ill patients needs further investigation. Given the kinetics of UPR it will be interesting to know if there is a time window where critical illness-induced and therapy related ER stress and UPR interfere with each other and aggravate muscle damage, which most certainly will have clinical implications.
These data show that exercise as well as immobilization are able to activate ER stress and UPR and that UPR is involved in the control of muscle mass and function. However, to which degree immobilization- vs. mobilization-mediated UPR activity affects the occurrence of ICUAW and if this knowledge can be used therapeutically is not known.
Given its importance in multiple cellular processes and human diseases such as cancer, neurodegenerative disease (e.g., Alzheimer’s disease, Parkinson’s disease, amyotrophic lateral sclerosis, Huntington’s disease) and metabolic disorders (e.g., type 2 diabetes mellitus) attempts have been undertaken to pharmacologically target the UPR. These efforts have been summarized recently (191). However, UPR modulators have rarely been tested in critically ill patients or ICUAW, which is not surprising as several critical functions of the UPR in muscle are not fully understood and sometimes controversial. As already mentioned, preclinical animal models revealed that targeting specific UPR branches or mediators may have beneficial or detrimental consequences for disease severity or progression which depends on the disease context. Among others, disease-, tissue- and fiber type-specific activation of distinct UPR pathways that often follow a time course complicate decision making for the therapeutic usage of UPR modulators. Many preclinical disease models are based on otherwise healthy young rodents that do often not have contributing risk factors that may affect the disease course, which is in contrast to the critically ill especially on medical ICUs. These studies are mainly performed in male, some in female rodents but usually not in animals of both sexes. In order to evaluate therapeutic effects especially in muscle results from multiple muscles (e.g., slow- and fast-twitch muscles) need to be obtained, which is often not the case. Finally, although there are many valid reasons to choose a preemptive treatment in situations where the onset of a disease and even its cause is very well defined, this is in great contrast to the clinical reality. These limitations need to be considered when decisions are made to translate from preclinical to clinical applications of UPR modulators.
One consequence of UPR activation is an increased expression of chaperone proteins, which will enhance the protein folding capacity of the ER. Therefore, it appears logical to apply chaperones to assist protein folding and to reduce ER stress. The two most commonly used chaperons that are used in vitro and in vivo in animal models as well as in human patients are the chemical chaperones Tauroursodeoxycholic acid (TUDCA) and 4-phenyl butyric acid (4-PBA). On the basis of cell culture and pre-clinical studies described in the previous chapters, TUDCA and 4-PBA represent promising therapeutic options for those myopathies that are associated with ER stress. Both have previously been shown to be effective in inhibiting the ER stress-mediated lipotoxicity of pancreatic beta-cells (192). For example, 4-PBA enhanced glucose metabolism by increasing hexokinase activity, glucose consumption and Glut4 mRNA expression in L6 myotubes (193). 4-PBA also increased GLUT4 expression and enhanced glucose metabolism in C2C12 myotubes (194). In addition, TUDCA has been shown to improve the blunted glucose uptake in response to tunicamycin in C2C12 myotubes (116).
TUDCA was also found to be effective in preclinical disease models. For example, loss-of-function mutations of the ER-resident phosphatidate phosphatase lipin1 (LPIN1) causes severe muscle injury in children. Skeletal muscle specific Lpin1 knockout mice develop a myopathy (e.g., reduced muscle force, myofiber necrosis, centronucleated fibers, fibrosis, immune cell infiltration) that is linked to an increased UPR and ER fragmentation. When these mice were treated with TUDCA, ER stress was alleviated and the myopathic phenotype was rescued [e.g., increased muscle force, reduction in myofiber necrosis; (195)]. In addition, TUDCA treatment of obese (i.e., leptin-deficient ob/ob mice) mice attenuated high-fat diet induced ER stress and insulin resistance. When applied to inducible muscle-specific Opa1 knockout mice, which show ER stress, systemic inflammation, and muscle atrophy TUDCA treatment reduced ER stress, improved the number of mitochondria and prevented muscle wasting. These data suggest that attenuation of ER stress improves muscle function in a disease model that is characterized by mitochondrial dysfunction (196). In addition, the effects of 4-PBA were investigated in mice with an I4895T mutation in the type I ryanodine receptor (RyR1), which display muscle weakness and atrophy that is associated with an increase in ER stress markers (e.g., GRP94, CHOP and BIP) in skeletal muscle. Chronic treatment of these mice with 4-PBA reduced ER stress and improved muscle strength (197). Finally, in a rat model of severe burn injury, 4-PBA treatment prevented burn-induced ER stress (i.e., ER swelling, expression of ER stress markers) and attenuated skeletal muscle injury and wasting (198).
However, work done by others showed that neither TUDCA nor 4-PBA restored palmitate-induced insulin resistance in C2C12 myotubes (199). Interestingly, when applied to 20 obese subjects (8 men, 12 women) for 4 weeks TUDCA (1,750 mg/day) improved muscular and hepatic insulin sensitivity, but this response was independent of its effects on ER stress since muscular ER stress markers remained unchanged (200). Because ER stress markers are highly activated in skeletal muscle of Lewis lung carcinoma (LLC) and ApcMin/+ (intestinal and mammary tumors) mouse models of cancer cachexia it was hypothesized that inhibition of ER stress could prevent cancer associated muscle wasting. However, 4-PBA treatment not only accelerated the reduction in muscle strength and mass in LLC-tumor bearing mice but also in healthy control animals. 4-PBA also increased the proportion of fast-twitch myofibers in soleus muscle of both control and LLC-bearing mice. 4-PBA also induced atrophy of primary myotubes in vitro. These data indicate that ER stress and UPR pathways are essential for maintaining skeletal muscle mass and strength and maybe protective against cancer cachexia (201). Further studies are needed to address the context-dependent and sometimes negative effects that have been observed with TUDCA and 4-PBA.
Targeting ER stress-induced UPR may be beneficial for treatment of myopathies, but it is very important to determine which branch of the UPR is involved in which myopathy because inhibiting all three branches may not be safe as it could lead to unexpected side effects. Consequently, targeting specific UPR branches and their downstream regulators may be a better option. The development of UPR modulators, specifically IRE1α RNase inhibitors, IRE1α kinase inhibitors, PERK inhibitors, eIF2α phosphatase inhibitors, eIF2B activators, and ATF6 activators and inhibitors has been reviewed recently (191). Only few of these compounds have been tested in myocytes or muscle pathologies. We have here listed some examples were UPR modulators have been used (Table 1).
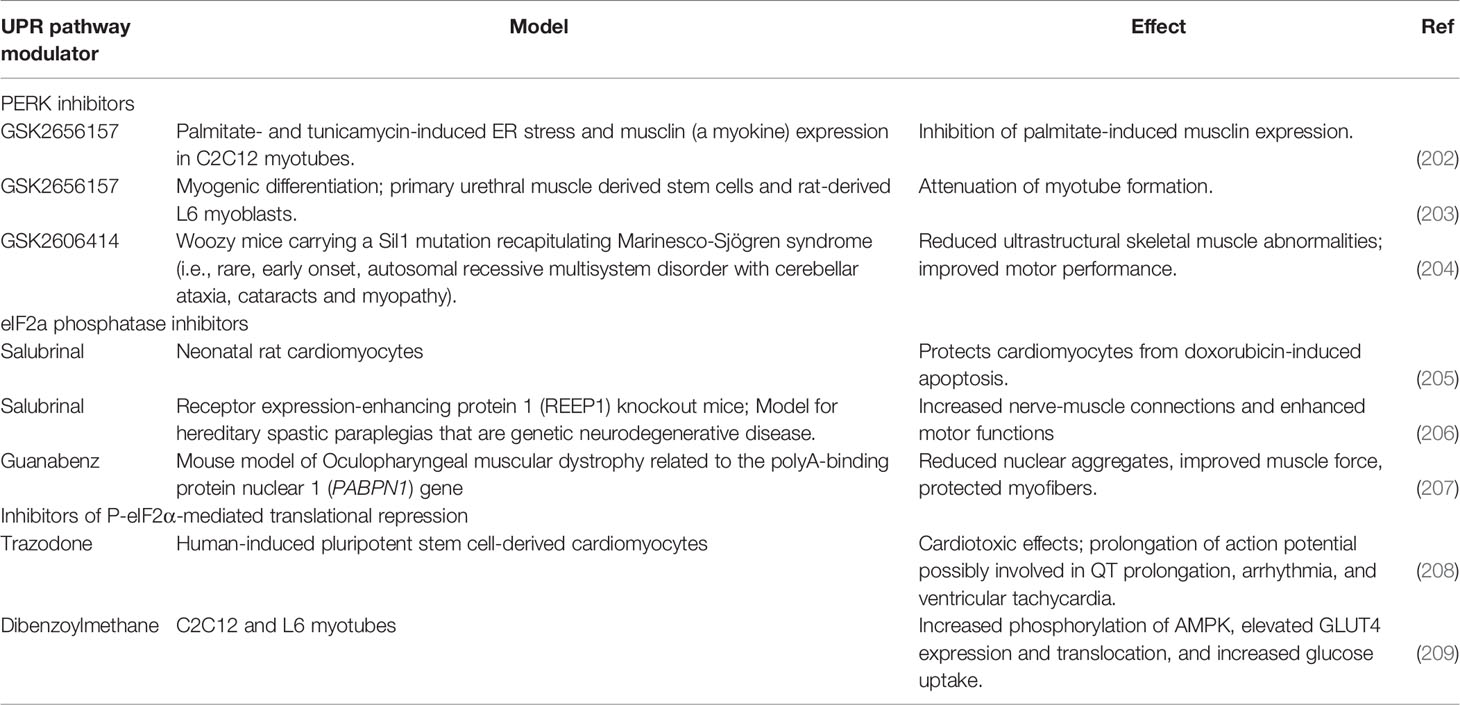
Table 1 Effects of UPR pathway modulators.
The compound GSK2656157, an inhibitor of the PERK branch of the UPR, has been used in C2C12 cells. Specifically, Gu et al., investigated the effects of ER stress on the expression of musclin (encoded by Ostn, osteocrin), which is a muscle-secreted cytokine (myokine) that is associated with insulin resistance in type 2 diabetes mellitus. Treatment of C2C12 cells with palmitate or tunicamycin caused ER stress and increased the expression of Ostn, which was attenuated by 4-PBA. Importantly, inhibition of the PERK branch of the UPR by GSK2656157 reduced palmitate-induced Ostn expression whereas inhibition of the IRE1 or ATF6 branch of the UPR had no effect. These data indicate that the PERK- but not the IRE1 or the ATF6 branch of the UPR is involved in palmitate-induced Ostn gene expression (202). In summary, it is possible to inhibit specific UPR branches and these inhibitors are useful to perform mechanistic studies in myocytes. If these inhibitors are also useful to prevent or treat ICUAW in vivo warrants further investigation.
Novel UPR modulators were found to be contained in natural products (210), which could serve as a source for novel lead compounds. Such natural UPR modulators provide an innovative and novel strategy to prevent or treat ICUAW. We have here listed a few promising natural compounds, their source and the model systems they were investigated in (Table 2). However, for most of these compounds their effects on ER stress have been investigated in heart-failure models in vivo or in cardiomyocytes in vitro. Although favorable effects of these compounds have been shown in human patients and animal models, further analyses are needed to elucidate if they also inhibit ER stress and UPR pathways in skeletal muscle as part of their mode of action and if this has favorable effects for the critically ill.

Table 2 Natural compounds with effects of ER stress and UPR in muscle.
Intensive care unit (ICU)-acquired weakness (ICUAW) is a complex clinical syndrome that complicates treatment of the critically ill and may persist for years in survivors. No effective therapy is in place to prevent or treat this devastating syndrome. Several predisposing risk factors have been identified, which allows mechanistical analyses and may pave the road for therapeutic interventions. A dysbalanced protein homeostasis with increased degradation (via UPS and ALP) and reduced synthesis plays a major role in ICUAW. However, ER stress and the UPR that are involved in many physiological and pathological processes in skeletal muscle are also implicated. Specifically, almost all established ICUAW risk factors (e.g., infection, inflammation, sepsis, multiple organ failure, insulin resistance, immobilization) and treatments (e.g., nutrition, physiotherapy/early mobilization) have also been shown to be directly or indirectly associated with ER stress and the UPR. Nevertheless, due to the complexity of multiple UPR pathways and its numerous upstream activators as well as downstream targets the precise function of the UPR in ICUAW is far from being understood. This situation is complicated by the observation that activation of ER stress-induced UPR may have beneficial or deleterious effects, that its activation differs different muscle types, and that it follows a time-course under some conditions. Although ER stress and the UPR are involved in regulation of skeletal muscle mass, it is unknown how they regulate skeletal muscle atrophy and hypertrophy. If ER stress and the UPR within satellite cells are also important for the long-term residuals of ICUAW needs further investigation. Nevertheless, modulation of ER stress and UPR pathways may be useful to treat ICUAW risk factors as well as ICUAW itself. For that, several activators and inhibitors of ER stress are available. However, before these pharmacological compounds can be used, a deeper understanding of the underlying pathomechanisms is required.
MK drafted and revised the manuscript, drafted the Figures and the Tables. JF edited and revised the manuscript, edited the Figures and the Tables. All authors contributed to the article and approved the submitted version
This study was supported by the Deutsche Forschungsgemeinschaft (FI 965/5-1, FI 965/5-2, FI 965/9-1, FI 965/10-1 (to JF)) and the German Center for Cardiovascular Research, partner site Greifswald [DZHK 81Z5400153 (to JF)].
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We acknowledge support for the Article Processing Charge from the DFG and the Open Access Publication Fund of the University of Greifswald.
1. Paval DR, Patton R, Mcdonald J, Skipworth RJE, Gallagher IJ, Laird BJ, et al. A Systematic Review Examining the Relationship Between Cytokines and Cachexia in Incurable Cancer. J Cachexia Sarcopenia Muscle (2022) 13:824–838. doi: 10.1002/jcsm.12912
2. Beltrami M, Fumagalli C, Milli M. Frailty, Sarcopenia and Cachexia in Heart Failure Patients: Different Clinical Entities of the Same Painting. World J Cardiol (2021) 13:1–10. doi: 10.4330/wjc.v13.i1.1
3. Shu X, Lin T, Wang H, Zhao Y, Jiang T, Peng X, et al. Diagnosis, Prevalence, and Mortality of Sarcopenia in Dialysis Patients: A Systematic Review and Meta-Analysis. J Cachexia Sarcopenia Muscle (2022) 13:145–58. doi: 10.1002/jcsm.12890
4. Sepulveda-Loyola W, Osadnik C, Phu S, Morita AA, Duque G, Probst VS. Diagnosis, Prevalence, and Clinical Impact of Sarcopenia in COPD: A Systematic Review and Meta-Analysis. J Cachexia Sarcopenia Muscle (2020) 11:1164–76. doi: 10.1002/jcsm.12600
5. Schefold JC, Wollersheim T, Grunow JJ, Luedi MM, Z’graggen WJ, Weber-Carstens S. Muscular Weakness and Muscle Wasting in the Critically Ill. J Cachexia Sarcopenia Muscle (2020) 11:1399–412. doi: 10.1002/jcsm.12620
6. Haberecht-Muller S, Kruger E, Fielitz J. Out of Control: The Role of the Ubiquitin Proteasome System in Skeletal Muscle During Inflammation. Biomolecules (2021) 11. doi: 10.3390/biom11091327
7. Wolf DH, Hilt W. The Proteasome: A Proteolytic Nanomachine of Cell Regulation and Waste Disposal. Biochim Biophys Acta (2004) 1695:19–31. doi: 10.1016/j.bbamcr.2004.10.007
8. Ciehanover A, Hod Y, Hershko A. A Heat-Stable Polypeptide Component of an ATP-Dependent Proteolytic System From Reticulocytes. Biochem Biophys Res Commun (1978) 81:1100–5. doi: 10.1016/0006-291X(78)91249-4
9. Wilkinson KD, Urban MK, Haas AL. Ubiquitin is the ATP-Dependent Proteolysis Factor I of Rabbit Reticulocytes. J Biol Chem (1980) 255:7529–32. doi: 10.1016/S0021-9258(19)43857-X
10. Hershko A, Heller H, Elias S, Ciechanover A. Components of Ubiquitin-Protein Ligase System. Resolution, Affinity Purification, and Role in Protein Breakdown. J Biol Chem (1983) 258:8206–14. doi: 10.1016/S0021-9258(20)82050-X
11. Tanaka K. The Proteasome: Overview of Structure and Functions. Proc Jpn Acad Ser B Phys Biol Sci (2009) 85:12–36. doi: 10.2183/pjab.85.12
12. Reymond A, Meroni G, Fantozzi A, Merla G, Cairo S, Luzi L, et al. The Tripartite Motif Family Identifies Cell Compartments. EMBO J (2001) 20:2140–51. doi: 10.1093/emboj/20.9.2140
13. Bodine SC, Latres E, Baumhueter S, Lai VK, Nunez L, Clarke BA, et al. Identification of Ubiquitin Ligases Required for Skeletal Muscle Atrophy. Science (2001) 294:1704–8. doi: 10.1126/science.1065874
14. Koncarevic A, Jackman RW, Kandarian SC. The Ubiquitin-Protein Ligase Nedd4 Targets Notch1 in Skeletal Muscle and Distinguishes the Subset of Atrophies Caused by Reduced Muscle Tension. FASEB J (2007) 21:427–37. doi: 10.1096/fj.06-6665com
15. Paul PK, Gupta SK, Bhatnagar S, Panguluri SK, Darnay BG, Choi Y, et al. Targeted Ablation of TRAF6 Inhibits Skeletal Muscle Wasting in Mice. J Cell Biol (2010) 191:1395–411. doi: 10.1083/jcb.201006098
16. Milan G, Romanello V, Pescatore F, Armani A, Paik JH, Frasson L, et al. Regulation of Autophagy and the Ubiquitin-Proteasome System by the FoxO Transcriptional Network During Muscle Atrophy. Nat Commun (2015) 6:6670. doi: 10.1038/ncomms7670
17. Fielitz J, Kim MS, Shelton JM, Latif S, Spencer JA, Glass DJ, et al. Myosin Accumulation and Striated Muscle Myopathy Result From the Loss of Muscle RING Finger 1 and 3. J Clin Invest (2007) 117:2486–95. doi: 10.1172/JCI32827
18. Kedar V, Mcdonough H, Arya R, Li HH, Rockman HA, Patterson C. Muscle-Specific RING Finger 1 is a Bona Fide Ubiquitin Ligase That Degrades Cardiac Troponin I. Proc Natl Acad Sci USA (2004) 101:18135–40. doi: 10.1073/pnas.0404341102
19. Witt SH, Granzier H, Witt CC, Labeit S. MURF-1 and MURF-2 Target a Specific Subset of Myofibrillar Proteins Redundantly: Towards Understanding MURF-Dependent Muscle Ubiquitination. J Mol Biol (2005) 350:713–22. doi: 10.1016/j.jmb.2005.05.021
20. Polge C, Heng AE, Jarzaguet M, Ventadour S, Claustre A, Combaret L, et al. Muscle Actin is Polyubiquitinylated In Vitro and In Vivo and Targeted for Breakdown by the E3 Ligase Murf1. FASEB J (2011) 25:3790–802. doi: 10.1096/fj.11-180968
21. Polge C, Cabantous S, Deval C, Claustre A, Hauvette A, Bouchenot C, et al. A Muscle-Specific MuRF1-E2 Network Requires Stabilization of MuRF1-E2 Complexes by Telethonin, a Newly Identified Substrate. J Cachexia Sarcopenia Muscle (2018) 9:129–45. doi: 10.1002/jcsm.12249
22. Mcelhinny AS, Kakinuma K, Sorimachi H, Labeit S, Gregorio CC. Muscle-Specific RING Finger-1 Interacts With Titin to Regulate Sarcomeric M-Line and Thick Filament Structure and may Have Nuclear Functions via its Interaction With Glucocorticoid Modulatory Element Binding Protein-1. J Cell Biol (2002) 157:125–36. doi: 10.1083/jcb.200108089
23. Koyama S, Hata S, Witt CC, Ono Y, Lerche S, Ojima K, et al. Muscle RING-Finger Protein-1 (MuRF1) as a Connector of Muscle Energy Metabolism and Protein Synthesis. J Mol Biol (2008) 376:1224–36. doi: 10.1016/j.jmb.2007.11.049
24. Nowak M, Suenkel B, Porras P, Migotti R, Schmidt F, Kny M, et al. DCAF8, a Novel MuRF1 Interaction Partner, Promotes Muscle Atrophy. J Cell Sci (2019) 132. doi: 10.1242/jcs.233395
25. Bowen TS, Adams V, Werner S, Fischer T, Vinke P, Brogger MN, et al. Small-Molecule Inhibition of MuRF1 Attenuates Skeletal Muscle Atrophy and Dysfunction in Cardiac Cachexia. J Cachexia Sarcopenia Muscle (2017) 8:939–53. doi: 10.1002/jcsm.12233
26. Adams V, Gussen V, Zozulya S, Cruz A, Moriscot A, Linke A, et al. Small-Molecule Chemical Knockdown of MuRF1 in Melanoma Bearing Mice Attenuates Tumor Cachexia Associated Myopathy. Cells (2020) 9. doi: 10.3390/cells9102272
27. Wollersheim T, Woehlecke J, Krebs M, Hamati J, Lodka D, Luther-Schroeder A, et al. Dynamics of Myosin Degradation in Intensive Care Unit-Acquired Weakness During Severe Critical Illness. Intensive Care Med (2014) 40:528–38. doi: 10.1007/s00134-014-3224-9
28. Langhans C, Weber-Carstens S, Schmidt F, Hamati J, Kny M, Zhu X, et al. Inflammation-Induced Acute Phase Response in Skeletal Muscle and Critical Illness Myopathy. PloS One (2014) 9:e92048. doi: 10.1371/journal.pone.0092048
29. Schmidt F, Kny M, Zhu X, Wollersheim T, Persicke K, Langhans C, et al. The E3 Ubiquitin Ligase TRIM62 and Inflammation-Induced Skeletal Muscle Atrophy. Crit Care (2014) 18:545. doi: 10.1186/s13054-014-0545-6
30. Huang N, Kny M, Riediger F, Busch K, Schmidt S, Luft FC, et al. Deletion of Nlrp3 Protects From Inflammation-Induced Skeletal Muscle Atrophy. Intensive Care Med Exp (2017) 5:3. doi: 10.1186/s40635-016-0115-0
31. Hahn A, Kny M, Pablo-Tortola C, Todiras M, Willenbrock M, Schmidt S, et al. Serum Amyloid A1 Mediates Myotube Atrophy via Toll-Like Receptors. J Cachexia Sarcopenia Muscle (2020) 11:103–19. doi: 10.1002/jcsm.12491
32. Zanders L, Kny M, Hahn A, Schmidt S, Wundersitz S, Todiras M, et al. Sepsis Induces Interleukin 6, Gp130/JAK2/STAT3, and Muscle Wasting. J Cachexia Sarcopenia Muscle (2022) 13:713–27. doi: 10.1002/jcsm.12867
33. Aweida D, Cohen S. Breakdown of Filamentous Myofibrils by the UPS-Step by Step. Biomolecules (2021) 11. doi: 10.3390/biom11010110
34. Peris-Moreno D, Cussonneau L, Combaret L, Polge C, Taillandier D. Ubiquitin Ligases at the Heart of Skeletal Muscle Atrophy Control. Molecules (2021) 26. doi: 10.3390/molecules26020407
35. Busch K, Kny M, Huang N, Klassert TE, Stock M, Hahn A, et al. Inhibition of the NLRP3/IL-1beta Axis Protects Against Sepsis-Induced Cardiomyopathy. J Cachexia Sarcopenia Muscle (2021) 12:1653–68. doi: 10.1002/jcsm.12763
36. Fan E, Cheek F, Chlan L, Gosselink R, Hart N, Herridge MS, et al. An Official American Thoracic Society Clinical Practice Guideline: The Diagnosis of Intensive Care Unit-Acquired Weakness in Adults. Am J Respir Crit Care Med (2014) 190:1437–46. doi: 10.1164/rccm.201411-2011ST
37. Puthucheary ZA, Rawal J, Mcphail M, Connolly B, Ratnayake G, Chan P, et al. Acute Skeletal Muscle Wasting in Critical Illness. JAMA (2013) 310:1591–600. doi: 10.1001/jama.2013.278481
38. Schweickert WD, Hall J. ICU-Acquired Weakness. Chest (2007) 131:1541–9. doi: 10.1378/chest.06-2065
39. Weber-Carstens S, Koch S, Spuler S, Spies CD, Bubser F, Wernecke KD, et al. Nonexcitable Muscle Membrane Predicts Intensive Care Unit-Acquired Paresis in Mechanically Ventilated, Sedated Patients. Crit Care Med (2009) 37:2632–7. doi: 10.1097/CCM.0b013e3181a92f28
40. Wollersheim T, Haas K, Wolf S, Mai K, Spies C, Steinhagen-Thiessen E, et al. Whole-Body Vibration to Prevent Intensive Care Unit-Acquired Weakness: Safety, Feasibility, and Metabolic Response. Crit Care (2017) 21:9. doi: 10.1186/s13054-016-1576-y
41. Wollersheim T, Grunow JJ, Carbon NM, Haas K, Malleike J, Ramme SF, et al. Muscle Wasting and Function After Muscle Activation and Early Protocol-Based Physiotherapy: An Explorative Trial. J Cachexia Sarcopenia Muscle (2019) 10:734–47. doi: 10.1002/jcsm.12428
42. De Jonghe B, Bastuji-Garin S, Durand MC, Malissin I, Rodrigues P, Cerf C, et al. Respiratory Weakness is Associated With Limb Weakness and Delayed Weaning in Critical Illness. Crit Care Med (2007) 35:2007–15. doi: 10.1097/01.ccm.0000281450.01881.d8
43. Sharshar T, Bastuji-Garin S, Stevens RD, Durand MC, Malissin I, Rodriguez P, et al. Presence and Severity of Intensive Care Unit-Acquired Paresis at Time of Awakening are Associated With Increased Intensive Care Unit and Hospital Mortality. Crit Care Med (2009) 37:3047–53. doi: 10.1097/CCM.0b013e3181b027e9
44. Van Aerde N, Meersseman P, Debaveye Y, Wilmer A, Gunst J, Casaer MP, et al. Five-Year Impact of ICU-Acquired Neuromuscular Complications: A Prospective, Observational Study. Intensive Care Med (2020) 46:1184–93. doi: 10.1007/s00134-020-05927-5
45. Herridge MS, Tansey CM, Matte A, Tomlinson G, Diaz-Granados N, Cooper A, et al. Functional Disability 5 Years After Acute Respiratory Distress Syndrome. N Engl J Med (2011) 364:1293–304. doi: 10.1056/NEJMoa1011802
46. Koch S, Wollersheim T, Bierbrauer J, Haas K, Morgeli R, Deja M, et al. Long-Term Recovery In Critical Illness Myopathy is Complete, Contrary to Polyneuropathy. Muscle Nerve (2014) 50:431–6. doi: 10.1002/mus.24175
47. Yang T, Li Z, Jiang L, Wang Y, Xi X. Risk Factors for Intensive Care Unit-Acquired Weakness: A Systematic Review and Meta-Analysis. Acta Neurol Scand (2018) 138:104–14. doi: 10.1111/ane.12964
48. Berek K, Margreiter J, Willeit J, Berek A, Schmutzhard E, Mutz NJ. Polyneuropathies in Critically Ill Patients: A Prospective Evaluation. Intensive Care Med (1996) 22:849–55. doi: 10.1007/BF02044106
49. De Jonghe B, Cook D, Sharshar T, Lefaucheur JP, Carlet J, Outin H. Acquired Neuromuscular Disorders in Critically Ill Patients: A Systematic Review. Groupe De Reflexion Et D’etude Sur Les Neuromyopathies En Reanimation. Intensive Care Med (1998) 24:1242–50. doi: 10.1007/s001340050757
50. Perner A, Gordon AC, De Backer D, Dimopoulos G, Russell JA, Lipman J, et al. Sepsis: Frontiers in Diagnosis, Resuscitation and Antibiotic Therapy. Intensive Care Med (2016) 42:1958–69. doi: 10.1007/s00134-016-4577-z
51. Sakr Y, Jaschinski U, Wittebole X, Szakmany T, Lipman J, Namendys-Silva SA, et al. Sepsis in Intensive Care Unit Patients: Worldwide Data From the Intensive Care Over Nations Audit. Open Forum Infect Dis (2018) 5:ofy313. doi: 10.1093/ofid/ofy313
52. Vincent JL, Lefrant JY, Kotfis K, Nanchal R, Martin-Loeches I, Wittebole X, et al. Icon, Investigators, sComparison of European ICU Patients in 2012 (ICON) Versus 2002 (SOAP). Intensive Care Med (2018) 44:337–44. doi: 10.1007/s00134-017-5043-2
53. Ochala J, Larsson L. Effects of a Preferential Myosin Loss on Ca2+ Activation of Force Generation in Single Human Skeletal Muscle Fibres. Exp Physiol (2008) 93:486–95. doi: 10.1113/expphysiol.2007.041798
54. Blobel G, Dobberstein B. Transfer of Proteins Across Membranes. I. Presence of Proteolytically Processed and Unprocessed Nascent Immunoglobulin Light Chains on Membrane-Bound Ribosomes of Murine Myeloma. J Cell Biol (1975) 67:835–51. doi: 10.1083/jcb.67.3.835
55. Walter P, Lingappa VR. Mechanism of Protein Translocation Across the Endoplasmic Reticulum Membrane. Annu Rev Cell Biol (1986) 2:499–516. doi: 10.1146/annurev.cb.02.110186.002435
56. Kunze M, Berger J. The Similarity Between N-Terminal Targeting Signals for Protein Import Into Different Organelles and its Evolutionary Relevance. Front Physiol (2015) 6:259. doi: 10.3389/fphys.2015.00259
57. Malhotra JD, Kaufman RJ. The Endoplasmic Reticulum and the Unfolded Protein Response. Semin Cell Dev Biol (2007) 18:716–31. doi: 10.1016/j.semcdb.2007.09.003
58. Ellgaard L, Helenius A. Quality Control in the Endoplasmic Reticulum. Nat Rev Mol Cell Biol (2003) 4:181–91. doi: 10.1038/nrm1052
59. Nishikawa S, Brodsky JL, Nakatsukasa K. Roles of Molecular Chaperones in Endoplasmic Reticulum (ER) Quality Control and ER-Associated Degradation (ERAD). J Biochem (2005) 137:551–5. doi: 10.1093/jb/mvi068
60. Schubert U, Anton LC, Gibbs J, Norbury CC, Yewdell JW, Bennink JR. Rapid Degradation of a Large Fraction of Newly Synthesized Proteins by Proteasomes. Nature (2000) 404:770–4. doi: 10.1038/35008096
61. Schroder M, Kaufman RJ. ER Stress and the Unfolded Protein Response. Mutat Res (2005) 569:29–63. doi: 10.1016/j.mrfmmm.2004.06.056
62. Read A, Schroder M. The Unfolded Protein Response: An Overview. Biol (Basel) (2021) 10. doi: 10.3390/biology10050384
63. Hetz C, Zhang K, Kaufman RJ. Mechanisms, Regulation and Functions of the Unfolded Protein Response. Nat Rev Mol Cell Biol (2020) 21:421–38. doi: 10.1038/s41580-020-0250-z
64. Kohno K. How Transmembrane Proteins Sense Endoplasmic Reticulum Stress. Antioxid Redox Signal (2007) 9:2295–303. doi: 10.1089/ars.2007.1819
65. Sidrauski C, Cox JS, Walter P. tRNA Ligase is Required for Regulated mRNA Splicing in the Unfolded Protein Response. Cell (1996) 87:405–13. doi: 10.1016/S0092-8674(00)81361-6
66. Yoshida H. Unconventional Splicing of XBP-1 mRNA in the Unfolded Protein Response. Antioxid Redox Signal (2007) 9:2323–33. doi: 10.1089/ars.2007.1800
67. Hollien J, Lin JH, Li H, Stevens N, Walter P, Weissman JS. Regulated Ire1-Dependent Decay of Messenger RNAs in Mammalian Cells. J Cell Biol (2009) 186:323–31. doi: 10.1083/jcb.200903014
68. Urano F, Wang X, Bertolotti A, Zhang Y, Chung P, Harding HP, et al. Coupling of Stress in the ER to Activation of JNK Protein Kinases by Transmembrane Protein Kinase Ire1. Science (2000) 287:664–6. doi: 10.1126/science.287.5453.664
69. Nishitoh H, Matsuzawa A, Tobiume K, Saegusa K, Takeda K, Inoue K, et al. ASK1 is Essential for Endoplasmic Reticulum Stress-Induced Neuronal Cell Death Triggered by Expanded Polyglutamine Repeats. Genes Dev (2002) 16:1345–55. doi: 10.1101/gad.992302
70. Harding HP, Zhang Y, Ron D. Protein Translation and Folding are Coupled by an Endoplasmic-Reticulum-Resident Kinase. Nature (1999) 397:271–4. doi: 10.1038/16729
71. Marciniak SJ, Yun CY, Oyadomari S, Novoa I, Zhang Y, Jungreis R, et al. CHOP Induces Death by Promoting Protein Synthesis and Oxidation in the Stressed Endoplasmic Reticulum. Genes Dev (2004) 18:3066–77. doi: 10.1101/gad.1250704
72. Yoshida H, Haze K, Yanagi H, Yura T, Mori K. Identification of the Cis-Acting Endoplasmic Reticulum Stress Response Element Responsible for Transcriptional Induction of Mammalian Glucose-Regulated Proteins. Involvement of Basic Leucine Zipper Transcription Factors. J Biol Chem (1998) 273:33741–9. doi: 10.1074/jbc.273.50.33741
73. Ye J, Rawson RB, Komuro R, Chen X, Dave UP, Prywes R, et al. ER Stress Induces Cleavage of Membrane-Bound ATF6 by the Same Proteases That Process SREBPs. Mol Cell (2000) 6:1355–64. doi: 10.1016/S1097-2765(00)00133-7
74. Travers KJ, Patil CK, Wodicka L, Lockhart DJ, Weissman JS, Walter P. Functional and Genomic Analyses Reveal an Essential Coordination Between the Unfolded Protein Response and ER-Associated Degradation. Cell (2000) 101:249–58. doi: 10.1016/S0092-8674(00)80835-1
75. Qu J, Zou T, Lin Z. The Roles of the Ubiquitin-Proteasome System in the Endoplasmic Reticulum Stress Pathway. Int J Mol Sci (2021) 22. doi: 10.3390/ijms22041526
76. Luo S, Mao C, Lee B, Lee AS. GRP78/BiP is Required for Cell Proliferation and Protecting the Inner Cell Mass From Apoptosis During Early Mouse Embryonic Development. Mol Cell Biol (2006) 26:5688–97. doi: 10.1128/MCB.00779-06
77. Zhang P, Mcgrath B, Li S, Frank A, Zambito F, Reinert J, et al. The PERK Eukaryotic Initiation Factor 2 Alpha Kinase is Required for the Development of the Skeletal System, Postnatal Growth, and the Function and Viability of the Pancreas. Mol Cell Biol (2002) 22:3864–74. doi: 10.1128/MCB.22.11.3864-3874.2002
78. Scheuner D, Song B, Mcewen E, Liu C, Laybutt R, Gillespie P, et al. Translational Control is Required for the Unfolded Protein Response and In Vivo Glucose Homeostasis. Mol Cell (2001) 7:1165–76. doi: 10.1016/S1097-2765(01)00265-9
79. Zhang K, Wong HN, Song B, Miller CN, Scheuner D, Kaufman RJ. The Unfolded Protein Response Sensor IRE1alpha is Required at 2 Distinct Steps in B Cell Lymphopoiesis. J Clin Invest (2005) 115:268–81. doi: 10.1172/JCI200521848
80. Yamamoto K, Sato T, Matsui T, Sato M, Okada T, Yoshida H, et al. Transcriptional Induction of Mammalian ER Quality Control Proteins is Mediated by Single or Combined Action of ATF6alpha and XBP1. Dev Cell (2007) 13:365–76. doi: 10.1016/j.devcel.2007.07.018
81. Yamamoto K, Takahara K, Oyadomari S, Okada T, Sato T, Harada A, et al. Induction of Liver Steatosis and Lipid Droplet Formation in ATF6alpha-Knockout Mice Burdened With Pharmacological Endoplasmic Reticulum Stress. Mol Biol Cell (2010) 21:2975–86. doi: 10.1091/mbc.e09-02-0133
82. Bohnert KR, Mcmillan JD, Kumar A. Emerging Roles of ER Stress and Unfolded Protein Response Pathways in Skeletal Muscle Health and Disease. J Cell Physiol (2018) 233:67–78. doi: 10.1002/jcp.25852
83. Zusman O, Theilla M, Cohen J, Kagan I, Bendavid I, Singer P. Resting Energy Expenditure, Calorie and Protein Consumption in Critically Ill Patients: A Retrospective Cohort Study. Crit Care (2016) 20:367. doi: 10.1186/s13054-016-1538-4
84. Lecker SH, Jagoe RT, Gilbert A, Gomes M, Baracos V, Bailey J, et al. Multiple Types of Skeletal Muscle Atrophy Involve a Common Program of Changes in Gene Expression. FASEB J (2004) 18:39–51. doi: 10.1096/fj.03-0610com
85. Paul PK, Bhatnagar S, Mishra V, Srivastava S, Darnay BG, Choi Y, et al. The E3 Ubiquitin Ligase TRAF6 Intercedes in Starvation-Induced Skeletal Muscle Atrophy Through Multiple Mechanisms. Mol Cell Biol (2012) 32:1248–59. doi: 10.1128/MCB.06351-11
86. Islam MA, Adachi S, Shiiba Y, Takeda KI, Haga S, Yonekura S. Effects of Starvation-Induced Negative Energy Balance on Endoplasmic Reticulum Stress in the Liver of Cows. Anim Biosci (2022) 35:22–28. doi: 10.5713/ab.21.0140
87. Ebert SM, Monteys AM, Fox DK, Bongers KS, Shields BE, Malmberg SE, et al. The Transcription Factor ATF4 Promotes Skeletal Myofiber Atrophy During Fasting. Mol Endocrinol (2010) 24:790–9. doi: 10.1210/me.2009-0345
88. Sardiello M, Palmieri M, Di Ronza A, Medina DL, Valenza M, Gennarino VA, et al. A Gene Network Regulating Lysosomal Biogenesis and Function. Science (2009) 325:473–7. doi: 10.1126/science.1174447
89. Martina JA, Diab HI, Lishu L, Jeong AL, Patange S, Raben N, et al. The Nutrient-Responsive Transcription Factor TFE3 Promotes Autophagy, Lysosomal Biogenesis, and Clearance of Cellular Debris. Sci Signal (2014) 7:ra9. doi: 10.1126/scisignal.2004754
90. Settembre C, Di Malta C, Polito VA, Garcia Arencibia M, Vetrini F, Erdin S, et al. TFEB Links Autophagy to Lysosomal Biogenesis. Science (2011) 332:1429–33. doi: 10.1126/science.1204592
91. Slade L, Pulinilkunnil T. The MiTF/TFE Family of Transcription Factors: Master Regulators of Organelle Signaling, Metabolism, and Stress Adaptation. Mol Cancer Res (2017) 15:1637–43. doi: 10.1158/1541-7786.MCR-17-0320
92. Settembre C, Zoncu R, Medina DL, Vetrini F, Erdin S, Erdin S, et al. A Lysosome-To-Nucleus Signalling Mechanism Senses and Regulates the Lysosome via mTOR and TFEB. EMBO J (2012) 31:1095–108. doi: 10.1038/emboj.2012.32
93. Du Bois P, Pablo Tortola C, Lodka D, Kny M, Schmidt F, Song K, et al. Angiotensin II Induces Skeletal Muscle Atrophy by Activating TFEB-Mediated MuRF1 Expression. Circ Res (2015) 117:424–36. doi: 10.1161/CIRCRESAHA.114.305393
94. Pablo Tortola C, Fielitz B, Li Y, Rudebusch J, Luft FC, Fielitz J. Activation of Tripartite Motif Containing 63 Expression by Transcription Factor EB and Transcription Factor Binding to Immunoglobulin Heavy Chain Enhancer 3 Is Regulated by Protein Kinase D and Class IIa Histone Deacetylases. Front Physiol (2020) 11:550506. doi: 10.3389/fphys.2020.550506
95. Martina JA, Diab HI, Brady OA, Puertollano R. TFEB and TFE3 are Novel Components of the Integrated Stress Response. EMBO J (2016) 35:479–95. doi: 10.15252/embj.201593428
96. Krinsley JS. Association Between Hyperglycemia and Increased Hospital Mortality in a Heterogeneous Population of Critically Ill Patients. Mayo Clin Proc (2003) 78:1471–8. doi: 10.4065/78.12.1471
97. Samuel VT, Shulman GI. Mechanisms for Insulin Resistance: Common Threads and Missing Links. Cell (2012) 148:852–71. doi: 10.1016/j.cell.2012.02.017
98. Goodman CA, Mayhew DL, Hornberger TA. Recent Progress Toward Understanding the Molecular Mechanisms That Regulate Skeletal Muscle Mass. Cell Signal (2011) 23:1896–906. doi: 10.1016/j.cellsig.2011.07.013
99. Bruss MD, Arias EB, Lienhard GE, Cartee GD. Increased Phosphorylation of Akt Substrate of 160 kDa (AS160) in Rat Skeletal Muscle in Response to Insulin or Contractile Activity. Diabetes (2005) 54:41–50. doi: 10.2337/diabetes.54.1.41
100. Slot JW, Geuze HJ, Gigengack S, James DE, Lienhard GE. Translocation of the Glucose Transporter GLUT4 in Cardiac Myocytes of the Rat. Proc Natl Acad Sci U.S.A. (1991) 88:7815–9. doi: 10.1073/pnas.88.17.7815
101. Hayashi T, Hirshman MF, Fujii N, Habinowski SA, Witters LA, Goodyear LJ. Metabolic Stress and Altered Glucose Transport: Activation of AMP-Activated Protein Kinase as a Unifying Coupling Mechanism. Diabetes (2000) 49:527–31. doi: 10.2337/diabetes.49.4.527
102. Weber-Carstens S, Schneider J, Wollersheim T, Assmann A, Bierbrauer J, Marg A, et al. Critical Illness Myopathy and GLUT4: Significance of Insulin and Muscle Contraction. Am J Respir Crit Care Med (2013) 187:387–96. doi: 10.1164/rccm.201209-1649OC
103. Jespersen JG, Nedergaard A, Reitelseder S, Mikkelsen UR, Dideriksen KJ, Agergaard J, et al. Activated Protein Synthesis and Suppressed Protein Breakdown Signaling in Skeletal Muscle of Critically Ill Patients. PloS One (2011) 6:e18090. doi: 10.1371/journal.pone.0018090
104. Ozcan U, Cao Q, Yilmaz E, Lee AH, Iwakoshi NN, Ozdelen E, et al. Endoplasmic Reticulum Stress Links Obesity, Insulin Action, and Type 2 Diabetes. Science (2004) 306:457–61. doi: 10.1126/science.1103160
105. Ozcan U, Yilmaz E, Ozcan L, Furuhashi M, Vaillancourt E, Smith RO, et al. Chemical Chaperones Reduce ER Stress and Restore Glucose Homeostasis in a Mouse Model of Type 2 Diabetes. Science (2006) 313:1137–40. doi: 10.1126/science.1128294
106. Kurth-Kraczek EJ, Hirshman MF, Goodyear LJ, Winder WW. 5’ AMP-Activated Protein Kinase Activation Causes GLUT4 Translocation in Skeletal Muscle. Diabetes (1999) 48:1667–71. doi: 10.2337/diabetes.48.8.1667
107. Viollet B, Andreelli F, Jorgensen SB, Perrin C, Geloen A, Flamez D, et al. The AMP-Activated Protein Kinase Alpha2 Catalytic Subunit Controls Whole-Body Insulin Sensitivity. J Clin Invest (2003) 111:91–8. doi: 10.1172/JCI16567
108. Hwang SL, Jeong YT, Li X, Kim YD, Lu Y, Chang YC, et al. Inhibitory Cross-Talk Between the AMPK and ERK Pathways Mediates Endoplasmic Reticulum Stress-Induced Insulin Resistance in Skeletal Muscle. Br J Pharmacol (2013) 169:69–81. doi: 10.1111/bph.12124
109. Kwak HJ, Choi HE, Jang J, Park SK, Bae YA, Cheon HG. Bortezomib Attenuates Palmitic Acid-Induced ER Stress, Inflammation and Insulin Resistance in Myotubes via AMPK Dependent Mechanism. Cell Signal (2016) 28:788–97. doi: 10.1016/j.cellsig.2016.03.015
110. Eyers PA, Keeshan K, Kannan N. Tribbles in the 21st Century: The Evolving Roles of Tribbles Pseudokinases in Biology and Disease. Trends Cell Biol (2017) 27:284–98. doi: 10.1016/j.tcb.2016.11.002
111. Du K, Herzig S, Kulkarni RN, Montminy M. TRB3: A Tribbles Homolog That Inhibits Akt/PKB Activation by Insulin in Liver. Science (2003) 300:1574–7. doi: 10.1126/science.1079817
112. Koh HJ, Arnolds DE, Fujii N, Tran TT, Rogers MJ, Jessen N, et al. Skeletal Muscle-Selective Knockout of LKB1 Increases Insulin Sensitivity, Improves Glucose Homeostasis, and Decreases Trb3. Mol Cell Biol (2006) 26:8217–27. doi: 10.1128/MCB.00979-06
113. Koh HJ, Toyoda T, Didesch MM, Lee MY, Sleeman MW, Kulkarni RN, et al. Tribbles 3 Mediates Endoplasmic Reticulum Stress-Induced Insulin Resistance in Skeletal Muscle. Nat Commun (2013) 4:1871. doi: 10.1038/ncomms2851
114. Kenner KA, Anyanwu E, Olefsky JM, Kusari J. Protein-Tyrosine Phosphatase 1B is a Negative Regulator of Insulin- and Insulin-Like Growth Factor-I-Stimulated Signaling. J Biol Chem (1996) 271:19810–6. doi: 10.1074/jbc.271.33.19810
115. Panzhinskiy E, Ren J, Nair S. Pharmacological Inhibition of Protein Tyrosine Phosphatase 1b: A Promising Strategy for the Treatment of Obesity and Type 2 Diabetes Mellitus. Curr Med Chem (2013) 20:2609–25. doi: 10.2174/0929867311320210001
116. Panzhinskiy E, Hua Y, Culver B, Ren J, Nair S. Endoplasmic Reticulum Stress Upregulates Protein Tyrosine Phosphatase 1B and Impairs Glucose Uptake in Cultured Myotubes. Diabetologia (2013) 56:598–607. doi: 10.1007/s00125-012-2782-z
117. Panzhinskiy E, Ren J, Nair S. Protein Tyrosine Phosphatase 1B and Insulin Resistance: Role of Endoplasmic Reticulum Stress/Reactive Oxygen Species/Nuclear Factor Kappa B Axis. PloS One (2013) 8:e77228. doi: 10.1371/journal.pone.0077228
118. Gu F, Nguyen DT, Stuible M, Dube N, Tremblay ML, Chevet E. Protein-Tyrosine Phosphatase 1b Potentiates IRE1 Signaling During Endoplasmic Reticulum Stress. J Biol Chem (2004) 279:49689–93. doi: 10.1074/jbc.C400261200
119. Raciti GA, Iadicicco C, Ulianich L, Vind BF, Gaster M, Andreozzi F, et al. Glucosamine-Induced Endoplasmic Reticulum Stress Affects GLUT4 Expression via Activating Transcription Factor 6 in Rat and Human Skeletal Muscle Cells. Diabetologia (2010) 53:955–65. doi: 10.1007/s00125-010-1676-1
120. Bierbrauer J, Koch S, Olbricht C, Hamati J, Lodka D, Schneider J, et al. Early Type II Fiber Atrophy in Intensive Care Unit Patients With Nonexcitable Muscle Membrane. Crit Care Med (2012) 40:647–50. doi: 10.1097/CCM.0b013e31823295e6
121. Hunter RB, Mitchell-Felton H, Essig DA, Kandarian SC. Expression of Endoplasmic Reticulum Stress Proteins During Skeletal Muscle Disuse Atrophy. Am J Physiol Cell Physiol (2001) 281:C1285–1290. doi: 10.1152/ajpcell.2001.281.4.C1285
122. Baehr LM, West DW, Marcotte G, Marshall AG, De Sousa LG, Baar K, et al. Age-Related Deficits in Skeletal Muscle Recovery Following Disuse are Associated With Neuromuscular Junction Instability and ER Stress, Not Impaired Protein Synthesis. Aging (Albany NY) (2016) 8:127–46. doi: 10.18632/aging.100879
123. Alibegovic AC, Sonne MP, Hojbjerre L, Bork-Jensen J, Jacobsen S, Nilsson E, et al. Insulin Resistance Induced by Physical Inactivity is Associated With Multiple Transcriptional Changes in Skeletal Muscle in Young Men. Am J Physiol Endocrinol Metab (2010) 299:E752–763. doi: 10.1152/ajpendo.00590.2009
124. Fox DK, Ebert SM, Bongers KS, Dyle MC, Bullard SA, Dierdorff JM, et al. P53 and ATF4 Mediate Distinct and Additive Pathways to Skeletal Muscle Atrophy During Limb Immobilization. Am J Physiol Endocrinol Metab (2014) 307:E245–261. doi: 10.1152/ajpendo.00010.2014
125. Ebert SM, Dyle MC, Kunkel SD, Bullard SA, Bongers KS, Fox DK, et al. Stress-Induced Skeletal Muscle Gadd45a Expression Reprograms Myonuclei and Causes Muscle Atrophy. J Biol Chem (2012) 287:27290–301. doi: 10.1074/jbc.M112.374777
126. Ebert SM, Dyle MC, Bullard SA, Dierdorff JM, Murry DJ, Fox DK, et al. Identification and Small Molecule Inhibition of an Activating Transcription Factor 4 (ATF4)-Dependent Pathway to Age-Related Skeletal Muscle Weakness and Atrophy. J Biol Chem (2015) 290:25497–511. doi: 10.1074/jbc.M115.681445
127. Yu Z, Wang AM, Adachi H, Katsuno M, Sobue G, Yue Z, et al. Macroautophagy Is Regulated by the UPR-Mediator CHOP and Accentuates the Phenotype of SBMA Mice. PloS Genet (2011) 7:e1002321. doi: 10.1371/journal.pgen.1002321
128. Parveen A, Bohnert KR, Tomaz Da Silva M, Wen Y, Bhat R, Roy A, et al. MyD88-Mediated Signaling Intercedes in Neurogenic Muscle Atrophy Through Multiple Mechanisms. FASEB J (2021) 35:e21821. doi: 10.1096/fj.202100777RR
129. Ding B, Parmigiani A, Divakaruni AS, Archer K, Murphy AN, Budanov AV. Sestrin2 is Induced by Glucose Starvation via the Unfolded Protein Response and Protects Cells From Non-Canonical Necroptotic Cell Death. Sci Rep (2016) 6:22538. doi: 10.1038/srep22538
130. Yang X, Xue P, Yuan M, Xu X, Wang C, Li W, et al. SESN2 Protects Against Denervated Muscle Atrophy Through Unfolded Protein Response and Mitophagy. Cell Death Dis (2021) 12:805. doi: 10.1038/s41419-021-04094-9
131. Saveljeva S, Cleary P, Mnich K, Ayo A, Pakos-Zebrucka K, Patterson JB, et al. Endoplasmic Reticulum Stress-Mediated Induction of SESTRIN 2 Potentiates Cell Survival. Oncotarget (2016) 7:12254–66. doi: 10.18632/oncotarget.7601
132. Barreiro E, Salazar-Degracia A, Sancho-Munoz A, Gea J. Endoplasmic Reticulum Stress and Unfolded Protein Response Profile in Quadriceps of Sarcopenic Patients With Respiratory Diseases. J Cell Physiol (2019) 234:11315–29. doi: 10.1002/jcp.27789
133. Khan MM, Yang WL, Wang P. Endoplasmic Reticulum Stress in Sepsis. Shock (2015) 44:294–304. doi: 10.1097/SHK.0000000000000425
134. Smith JA. Regulation of Cytokine Production by the Unfolded Protein Response; Implications for Infection and Autoimmunity. Front Immunol (2018) 9:422. doi: 10.3389/fimmu.2018.00422
135. Martinon F, Chen X, Lee AH, Glimcher LH. TLR Activation of the Transcription Factor XBP1 Regulates Innate Immune Responses in Macrophages. Nat Immunol (2010) 11:411–8. doi: 10.1038/ni.1857
136. Zhu X, Kny M, Schmidt F, Hahn A, Wollersheim T, Kleber C, et al. Secreted Frizzled-Related Protein 2 and Inflammation-Induced Skeletal Muscle Atrophy. Crit Care Med (2017) 45:e169-e183. doi: 10.1097/CCM.0000000000002056
137. Gallot YS, Bohnert KR. Confounding Roles of ER Stress and the Unfolded Protein Response in Skeletal Muscle Atrophy. Int J Mol Sci (2021) 22. doi: 10.3390/ijms22052567
138. Chipurupalli S, Samavedam U, Robinson N. Crosstalk Between ER Stress, Autophagy and Inflammation. Front Med (Lausanne) (2021) 8:758311. doi: 10.3389/fmed.2021.758311
139. Isaac ST, Tan TC, Polly P. Endoplasmic Reticulum Stress, Calcium Dysregulation and Altered Protein Translation: Intersection of Processes That Contribute to Cancer Cachexia Induced Skeletal Muscle Wasting. Curr Drug Targets (2016) 17:1140–6. doi: 10.2174/1389450116666150416115721
140. Afzali AM, Muntefering T, Wiendl H, Meuth SG, Ruck T. Skeletal Muscle Cells Actively Shape (Auto)Immune Responses. Autoimmun Rev (2018) 17:518–29. doi: 10.1016/j.autrev.2017.12.005
141. Gu R, Huang T, Xiao J, Liao Z, Li J, Lan H, et al. The IRE1alpha Arm of UPR Regulates Muscle Cells Immune Characters by Restraining P38 MAPK Activation. Front Physiol (2019) 10:1198. doi: 10.3389/fphys.2019.01198
142. Hu P, Han Z, Couvillon AD, Kaufman RJ, Exton JH. Autocrine Tumor Necrosis Factor Alpha Links Endoplasmic Reticulum Stress to the Membrane Death Receptor Pathway Through IRE1alpha-Mediated NF-kappaB Activation and Down-Regulation of TRAF2 Expression. Mol Cell Biol (2006) 26:3071–84. doi: 10.1128/MCB.26.8.3071-3084.2006
143. Menu P, Mayor A, Zhou R, Tardivel A, Ichijo H, Mori K, et al. ER Stress Activates the NLRP3 Inflammasome via an UPR-Independent Pathway. Cell Death Dis (2012) 3:e261. doi: 10.1038/cddis.2011.132
144. Schroder K, Tschopp J. The Inflammasomes. Cell (2010) 140:821–32. doi: 10.1016/j.cell.2010.01.040
145. Lamkanfi M, Dixit VM. Inflammasomes and Their Roles in Health and Disease. Annu Rev Cell Dev Biol (2012) 28:137–61. doi: 10.1146/annurev-cellbio-101011-155745
146. Rawat R, Cohen TV, Ampong B, Francia D, Henriques-Pons A, Hoffman EP, et al. Inflammasome Up-Regulation and Activation in Dysferlin-Deficient Skeletal Muscle. Am J Pathol (2010) 176:2891–900. doi: 10.2353/ajpath.2010.090058
147. Relaix F, Zammit PS. Satellite Cells are Essential for Skeletal Muscle Regeneration: The Cell on the Edge Returns Centre Stage. Development (2012) 139:2845–56. doi: 10.1242/dev.069088
148. Bentzinger CF, Wang YX, Rudnicki MA. Building Muscle: Molecular Regulation of Myogenesis. Cold Spring Harb Perspect Biol (2012) 4. doi: 10.1101/cshperspect.a008342
149. Millay DP, O’rourke JR, Sutherland LB, Bezprozvannaya S, Shelton JM, Bassel-Duby R, et al. Myomaker Is a Membrane Activator of Myoblast Fusion and Muscle Formation. Nature (2013) 499:301–5. doi: 10.1038/nature12343
150. Quinn ME, Goh Q, Kurosaka M, Gamage DG, Petrany MJ, Prasad V, et al. Myomerger Induces Fusion of Non-Fusogenic Cells and is Required for Skeletal Muscle Development. Nat Commun (2017) 8:15665. doi: 10.1038/ncomms15665
151. Schiaffino S, Rossi AC, Smerdu V, Leinwand LA, Reggiani C. Developmental Myosins: Expression Patterns and Functional Significance. Skelet Muscle (2015) 5:22. doi: 10.1186/s13395-015-0046-6
152. Charge SB, Rudnicki MA. Cellular and Molecular Regulation of Muscle Regeneration. Physiol Rev (2004) 84:209–38. doi: 10.1152/physrev.00019.2003
153. Dos Santos C, Hussain SN, Mathur S, Picard M, Herridge M, Correa J, et al. Mechanisms of Chronic Muscle Wasting and Dysfunction After an Intensive Care Unit Stay. A Pilot Study. Am J Respir Crit Care Med (2016) 194:821–30. doi: 10.1164/rccm.201512-2344OC
154. Rocheteau P, Chatre L, Briand D, Mebarki M, Jouvion G, Bardon J, et al. Sepsis Induces Long-Term Metabolic and Mitochondrial Muscle Stem Cell Dysfunction Amenable by Mesenchymal Stem Cell Therapy. Nat Commun (2015) 6:10145. doi: 10.1038/ncomms10145
155. Walsh CJ, Batt J, Herridge MS, Mathur S, Bader GD, Hu P, et al. Transcriptomic Analysis Reveals Abnormal Muscle Repair and Remodeling in Survivors of Critical Illness With Sustained Weakness. Sci Rep (2016) 6:29334. doi: 10.1038/srep29334
156. Caldwell CJ, Mattey DL, Weller RO. Role of the Basement Membrane in the Regeneration of Skeletal Muscle. Neuropathol Appl Neurobiol (1990) 16:225–38. doi: 10.1111/j.1365-2990.1990.tb01159.x
157. Couteaux R, Mira JC, D’albis A. Regeneration of Muscles After Cardiotoxin Injury. I. Cytological Aspects. Biol Cell (1988) 62:171–82. doi: 10.1111/j.1768-322X.1988.tb00719.x
158. Olson EN. Interplay Between Proliferation and Differentiation Within the Myogenic Lineage. Dev Biol (1992) 154:261–72. doi: 10.1016/0012-1606(92)90066-P
159. Nakanishi K, Sudo T, Morishima N. Endoplasmic Reticulum Stress Signaling Transmitted by ATF6 Mediates Apoptosis During Muscle Development. J Cell Biol (2005) 169:555–60. doi: 10.1083/jcb.200412024
160. Nakagawa T, Zhu H, Morishima N, Li E, Xu J, Yankner BA, et al. Caspase-12 Mediates Endoplasmic-Reticulum-Specific Apoptosis and Cytotoxicity by Amyloid-Beta. Nature (2000) 403:98–103. doi: 10.1038/47513
161. Nakanishi K, Dohmae N, Morishima N. Endoplasmic Reticulum Stress Increases Myofiber Formation In Vitro. FASEB J (2007) 21:2994–3003. doi: 10.1096/fj.06-6408com
162. Alter J, Bengal E. Stress-Induced C/EBP Homology Protein (CHOP) Represses MyoD Transcription to Delay Myoblast Differentiation. PloS One (2011) 6:e29498. doi: 10.1371/journal.pone.0029498
163. Wang M, Kaufman RJ. The Impact of the Endoplasmic Reticulum Protein-Folding Environment on Cancer Development. Nat Rev Cancer (2014) 14:581–97. doi: 10.1038/nrc3800
164. Blais A, Tsikitis M, Acosta-Alvear D, Sharan R, Kluger Y, Dynlacht BD. An Initial Blueprint for Myogenic Differentiation. Genes Dev (2005) 19:553–69. doi: 10.1101/gad.1281105
165. Acosta-Alvear D, Zhou Y, Blais A, Tsikitis M, Lents NH, Arias C, et al. XBP1 Controls Diverse Cell Type- and Condition-Specific Transcriptional Regulatory Networks. Mol Cell (2007) 27:53–66. doi: 10.1016/j.molcel.2007.06.011
166. Lemercier C, To RQ, Carrasco RA, Konieczny SF. The Basic Helix-Loop-Helix Transcription Factor Mist1 Functions as a Transcriptional Repressor of myoD. EMBO J (1998) 17:1412–22. doi: 10.1093/emboj/17.5.1412
167. Zismanov V, Chichkov V, Colangelo V, Jamet S, Wang S, Syme A, et al. Phosphorylation of Eif2alpha Is a Translational Control Mechanism Regulating Muscle Stem Cell Quiescence and Self-Renewal. Cell Stem Cell (2016) 18:79–90. doi: 10.1016/j.stem.2015.09.020
168. Robert F, Kapp LD, Khan SN, Acker MG, Kolitz S, Kazemi S, et al. Initiation of Protein Synthesis by Hepatitis C Virus is Refractory to Reduced Eif2.GTP.Met-tRNA(I)(Met) Ternary Complex Availability. Mol Biol Cell (2006) 17:4632–44. doi: 10.1091/mbc.e06-06-0478
169. Boyce M, Bryant KF, Jousse C, Long K, Harding HP, Scheuner D, et al. A Selective Inhibitor of Eif2alpha Dephosphorylation Protects Cells From ER Stress. Science (2005) 307:935–9. doi: 10.1126/science.1101902
170. Hodgson CL, Tipping CJ. Physiotherapy Management of Intensive Care Unit-Acquired Weakness. J Physiother (2017) 63:4–10. doi: 10.1016/j.jphys.2016.10.011
171. Schweickert WD, Pohlman MC, Pohlman AS, Nigos C, Pawlik AJ, Esbrook CL, et al. Early Physical and Occupational Therapy in Mechanically Ventilated, Critically Ill Patients: A Randomised Controlled Trial. Lancet (2009) 373:1874–82. doi: 10.1016/S0140-6736(09)60658-9
172. Denehy L, Skinner EH, Edbrooke L, Haines K, Warrillow S, Hawthorne G, et al. Exercise Rehabilitation for Patients With Critical Illness: A Randomized Controlled Trial With 12 Months of Follow-Up. Crit Care (2013) 17:R156. doi: 10.1186/cc12835
173. Bein T, Bischoff M, Bruckner U, Gebhardt K, Henzler D, Hermes C, et al. S2e Guideline: Positioning and Early Mobilisation in Prophylaxis or Therapy of Pulmonary Disorders : Revision 2015: S2e Guideline of the German Society of Anaesthesiology and Intensive Care Medicine (DGAI). Anaesthesist (2015) 64 Suppl 1:1–26. doi: 10.1007/s00101-015-0071-1
174. Schaller SJ, Anstey M, Blobner M, Edrich T, Grabitz SD, Gradwohl-Matis I, et al. Early, Goal-Directed Mobilisation in the Surgical Intensive Care Unit: A Randomised Controlled Trial. Lancet (2016) 388:1377–88. doi: 10.1016/S0140-6736(16)31637-3
175. Morris PE, Griffin L, Berry M, Thompson C, Hite RD, Winkelman C, et al. Receiving Early Mobility During an Intensive Care Unit Admission is a Predictor of Improved Outcomes in Acute Respiratory Failure. Am J Med Sci (2011) 341:373–7. doi: 10.1097/MAJ.0b013e31820ab4f6
176. Morris PE, Berry MJ, Files DC, Thompson JC, Hauser J, Flores L, et al. Standardized Rehabilitation and Hospital Length of Stay Among Patients With Acute Respiratory Failure: A Randomized Clinical Trial. JAMA (2016) 315:2694–702. doi: 10.1001/jama.2016.7201
177. Gerovasili V, Stefanidis K, Vitzilaios K, Karatzanos E, Politis P, Koroneos A, et al. Electrical Muscle Stimulation Preserves the Muscle Mass of Critically Ill Patients: A Randomized Study. Crit Care (2009) 13:R161. doi: 10.1186/cc8123
178. Gerovasili V, Tripodaki E, Karatzanos E, Pitsolis T, Markaki V, Zervakis D, et al. Short-Term Systemic Effect of Electrical Muscle Stimulation in Critically Ill Patients. Chest (2009) 136:1249–56. doi: 10.1378/chest.08-2888
179. Routsi C, Gerovasili V, Vasileiadis I, Karatzanos E, Pitsolis T, Tripodaki E, et al. Electrical Muscle Stimulation Prevents Critical Illness Polyneuromyopathy: A Randomized Parallel Intervention Trial. Crit Care (2010) 14:R74. doi: 10.1186/cc8987
180. Karatzanos E, Gerovasili V, Zervakis D, Tripodaki ES, Apostolou K, Vasileiadis I, et al. Electrical Muscle Stimulation: An Effective Form of Exercise and Early Mobilization to Preserve Muscle Strength in Critically Ill Patients. Crit Care Res Pract (2012) 2012:432752. doi: 10.1155/2012/432752
181. Patsaki I, Gerovasili V, Sidiras G, Karatzanos E, Mitsiou G, Papadopoulos E, et al. Effect of Neuromuscular Stimulation and Individualized Rehabilitation on Muscle Strength in Intensive Care Unit Survivors: A Randomized Trial. J Crit Care (2017) 40:76–82. doi: 10.1016/j.jcrc.2017.03.014
182. Fossat G, Baudin F, Courtes L, Bobet S, Dupont A, Bretagnol A, et al. Effect of In-Bed Leg Cycling and Electrical Stimulation of the Quadriceps on Global Muscle Strength in Critically Ill Adults: A Randomized Clinical Trial. JAMA (2018) 320:368–78. doi: 10.1001/jama.2018.9592
183. Bogaerts A, Delecluse C, Claessens AL, Coudyzer W, Boonen S, Verschueren SM. Impact of Whole-Body Vibration Training Versus Fitness Training on Muscle Strength and Muscle Mass in Older Men: A 1-Year Randomized Controlled Trial. J Gerontol A Biol Sci Med Sci (2007) 62:630–5. doi: 10.1093/gerona/62.6.630
184. Machado A, Garcia-Lopez D, Gonzalez-Gallego J, Garatachea N. Whole-Body Vibration Training Increases Muscle Strength and Mass in Older Women: A Randomized-Controlled Trial. Scand J Med Sci Sports (2010) 20:200–7. doi: 10.1111/j.1600-0838.2009.00919.x
185. Connolly B, O’neill B, Salisbury L, Blackwood B, Enhanced Recovery after Critical Illness Programme G. Physical Rehabilitation Interventions for Adult Patients During Critical Illness: An Overview of Systematic Reviews. Thorax (2016) 71:881–90. doi: 10.1136/thoraxjnl-2015-208273
186. Zhang Z, Cui D, Zhang T, Sun Y, Ding S. Swimming Differentially Affects T2DM-Induced Skeletal Muscle ER Stress and Mitochondrial Dysfunction Related to MAM. Diabetes Metab Syndr Obes (2020) 13:1417–28. doi: 10.2147/DMSO.S243024
187. Ogborn DI, Mckay BR, Crane JD, Parise G, Tarnopolsky MA. The Unfolded Protein Response is Triggered Following a Single, Unaccustomed Resistance-Exercise Bout. Am J Physiol Regul Integr Comp Physiol (2014) 307:R664–669. doi: 10.1152/ajpregu.00511.2013
188. Deldicque L, Cani PD, Delzenne NM, Baar K, Francaux M. Endurance Training in Mice Increases the Unfolded Protein Response Induced by a High-Fat Diet. J Physiol Biochem (2013) 69:215–25. doi: 10.1007/s13105-012-0204-9
189. Murlasits Z, Lee Y, Powers SK. Short-Term Exercise Does Not Increase ER Stress Protein Expression in Cardiac Muscle. Med Sci Sports Exerc (2007) 39:1522–8. doi: 10.1249/mss.0b013e3180cc25c7
190. Wu J, Ruas JL, Estall JL, Rasbach KA, Choi JH, Ye L, et al. The Unfolded Protein Response Mediates Adaptation to Exercise in Skeletal Muscle Through a PGC-1alpha/ATF6alpha Complex. Cell Metab (2011) 13:160–9. doi: 10.1016/j.cmet.2011.01.003
191. Hetz C, Axten JM, Patterson JB. Pharmacological Targeting of the Unfolded Protein Response for Disease Intervention. Nat Chem Biol (2019) 15:764–75. doi: 10.1038/s41589-019-0326-2
192. Choi SE, Lee YJ, Jang HJ, Lee KW, Kim YS, Jun HS, et al. A Chemical Chaperone 4-PBA Ameliorates Palmitate-Induced Inhibition of Glucose-Stimulated Insulin Secretion (GSIS). Arch Biochem Biophys (2008) 475:109–14. doi: 10.1016/j.abb.2008.04.015
193. Da Silva MH, Pecanha FLM, De Oliveira AM, Da-Silva WS. 4-Phenyl Butyric Acid Increases Particulate Hexokinase Activity and Protects Against ROS Injury in L6 Myotubes. Life Sci (2017) 179:98–102. doi: 10.1016/j.lfs.2017.05.004
194. Hu H, Li L, Wang C, He H, Mao K, Ma X, et al. 4-Phenylbutyric Acid Increases GLUT4 Gene Expression Through Suppression of HDAC5 But Not Endoplasmic Reticulum Stress. Cell Physiol Biochem (2014) 33:1899–910. doi: 10.1159/000362967
195. Rashid T, Nemazanyy I, Paolini C, Tatsuta T, Crespin P, De Villeneuve D, et al. Lipin1 Deficiency Causes Sarcoplasmic Reticulum Stress and Chaperone-Responsive Myopathy. EMBO J (2019) 38. doi: 10.15252/embj.201899576
196. Tezze C, Romanello V, Desbats MA, Fadini GP, Albiero M, Favaro G, et al. Age-Associated Loss of OPA1 in Muscle Impacts Muscle Mass, Metabolic Homeostasis, Systemic Inflammation, and Epithelial Senescence. Cell Metab (2017) 251374-1389:e1376. doi: 10.1016/j.cmet.2017.04.021
197. Lee CS, Hanna AD, Wang H, Dagnino-Acosta A, Joshi AD, Knoblauch M, et al. A Chemical Chaperone Improves Muscle Function in Mice With a RyR1 Mutation. Nat Commun (2017) 8:14659. doi: 10.1038/ncomms14659
198. Ma L, Chu W, Chai J, Shen C, Li D, Wang X. ER Stress and Subsequent Activated Calpain Play a Pivotal Role in Skeletal Muscle Wasting After Severe Burn Injury. PloS One (2017) 12:e0186128. doi: 10.1371/journal.pone.0186128
199. Rieusset J, Chauvin MA, Durand A, Bravard A, Laugerette F, Michalski MC, et al. Reduction of Endoplasmic Reticulum Stress Using Chemical Chaperones or Grp78 Overexpression Does Not Protect Muscle Cells From Palmitate-Induced Insulin Resistance. Biochem Biophys Res Commun (2012) 417:439–45. doi: 10.1016/j.bbrc.2011.11.135
200. Kars M, Yang L, Gregor MF, Mohammed BS, Pietka TA, Finck BN, et al. Tauroursodeoxycholic Acid may Improve Liver and Muscle But Not Adipose Tissue Insulin Sensitivity in Obese Men and Women. Diabetes (2010) 59:1899–905. doi: 10.2337/db10-0308
201. Bohnert KR, Gallot YS, Sato S, Xiong G, Hindi SM, Kumar A. Inhibition of ER Stress and Unfolding Protein Response Pathways Causes Skeletal Muscle Wasting During Cancer Cachexia. FASEB J (2016) 30:3053–68. doi: 10.1096/fj.201600250RR
202. Gu N, Guo Q, Mao K, Hu H, Jin S, Zhou Y, et al. Palmitate Increases Musclin Gene Expression Through Activation of PERK Signaling Pathway in C2C12 Myotubes. Biochem Biophys Res Commun (2015) 467:521–6. doi: 10.1016/j.bbrc.2015.10.005
203. Wang B, Zhou J, Banie L, Reed-Maldonado AB, Ning H, Lu Z, et al. Low-Intensity Extracorporeal Shock Wave Therapy Promotes Myogenesis Through PERK/ATF4 Pathway. Neurourol Urodyn (2018) 37:699–707. doi: 10.1002/nau.23380
204. Grande V, Ornaghi F, Comerio L, Restelli E, Masone A, Corbelli A, et al. PERK Inhibition Delays Neurodegeneration and Improves Motor Function in a Mouse Model of Marinesco-Sjogren Syndrome. Hum Mol Genet (2018) 27:2477–89. doi: 10.1093/hmg/ddy152
205. Gong N, Wu JH, Liang ZS, Jiang WH, Wang XW. Role of Salubrinal in Protecting Cardiomyocytes From Doxorubicin-Induced Apoptosis. Genet Mol Res (2015) 14:12377–85. doi: 10.4238/2015.October.16.4
206. Wang B, Yu Y, Wei L, Zhang Y. Inhibition of ER Stress Improves Progressive Motor Deficits in a REEP1-Null Mouse Model of Hereditary Spastic Paraplegia. Biol Open (2020) 9. doi: 10.1242/bio.054296
207. Malerba A, Roth F, Harish P, Dhiab J, Lu-Nguyen N, Cappellari O, et al. Pharmacological Modulation of the ER Stress Response Ameliorates Oculopharyngeal Muscular Dystrophy. Hum Mol Genet (2019) 28:1694–708. doi: 10.1093/hmg/ddz007
208. Lee S, Lee HA, Kim SJ, Kim KS. Cellular Mechanisms for Trazodone-Induced Cardiotoxicity. Hum Exp Toxicol (2016) 35:501–10. doi: 10.1177/0960327115595683
209. Kim N, Kim HM, Lee ES, Lee JO, Lee HJ, Lee SK, et al. Dibenzoylmethane Exerts Metabolic Activity Through Regulation of AMP-Activated Protein Kinase (AMPK)-Mediated Glucose Uptake and Adipogenesis Pathways. PloS One (2015) 10:e0120104. doi: 10.1371/journal.pone.0120104
210. Ren J, Bi Y, Sowers JR, Hetz C, Zhang Y. Endoplasmic Reticulum Stress and Unfolded Protein Response in Cardiovascular Diseases. Nat Rev Cardiol (2021) 18:499–521. doi: 10.1038/s41569-021-00511-w
211. Yin XL, Shen H, Zhang W, Yang Y. Inhibition of Endoplasm Reticulum Stress by Anisodamine Protects Against Myocardial Injury After Cardiac Arrest and Resuscitation in Rats. Am J Chin Med (2011) 39:853–66. doi: 10.1142/S0192415X11009251
212. Yu Y, Zhu C, Hong Y, Chen L, Huang Z, Zhou J, et al. Effectiveness of Anisodamine for the Treatment of Critically Ill Patients With Septic Shock: A Multicentre Randomized Controlled Trial. Crit Care (2021) 25:349. doi: 10.1186/s13054-021-03774-4
213. Wei X, Zhang Z, Han L, Wei Y. Protective Effect of Anisodamine on Reperfusion Injury of Skeletal Muscles in Rabbit. Acta Orthop Scand (1998) 69:633–7. doi: 10.3109/17453679808999270
214. Xu ZQ, Shao BZ, Ke P, Liu JG, Liu GK, Chen XW, et al. Combined Administration of Anisodamine and Neostigmine Rescued Acute Lethal Crush Syndrome Through Alpha7nachr-Dependent JAK2-STAT3 Signaling. Sci Rep (2016) 6:37709. doi: 10.1038/srep37709
215. Shen M, Wang L, Yang G, Gao L, Wang B, Guo X, et al. Baicalin Protects the Cardiomyocytes From ER Stress-Induced Apoptosis: Inhibition of CHOP Through Induction of Endothelial Nitric Oxide Synthase. PloS One (2014) 9:e88389. doi: 10.1371/journal.pone.0088389
216. Shi T, Li T, Jiang X, Jiang X, Zhang Q, Wang Y, et al. Baicalin Protects Mice From Infection With Methicillin-Resistant Staphylococcus Aureus via Alleviating Inflammatory Response. J Leukoc Biol (2020) 108:1829–39. doi: 10.1002/JLB.3AB0820-576RRR
217. Li CG, Yan L, Mai FY, Shi ZJ, Xu LH, Jing YY, et al. Baicalin Inhibits NOD-Like Receptor Family, Pyrin Containing Domain 3 Inflammasome Activation in Murine Macrophages by Augmenting Protein Kinase A Signaling. Front Immunol (2017) 8:1409. doi: 10.3389/fimmu.2017.01409
218. Pan Y, Song D, Zhou W, Lu X, Wang H, Li Z. Baicalin Inhibits C2C12 Myoblast Apoptosis and Prevents Against Skeletal Muscle Injury. Mol Med Rep (2019) 20:709–18. doi: 10.3892/mmr.2019.10298
219. Emanuele E, Bertona M, Pareja-Galeano H, Fiuza-Luces C, Morales JS, Sanchis-Gomar F, et al. Baicalin Supplementation Reduces Serum Biomarkers of Skeletal Muscle Wasting and may Protect Against Lean Body Mass Reduction in Cancer Patients: Results From a Pilot Open-Label Study. Neuro Endocrinol Lett (2016) 37:213–6.
220. Fang P, Yu M, Min W, Wan D, Han S, Shan Y, et al. Effect of Baicalin on GLUT4 Expression and Glucose Uptake in Myotubes of Rats. Life Sci (2018) 196:156–61. doi: 10.1016/j.lfs.2018.01.022
221. Zhao GL, Yu LM, Gao WL, Duan WX, Jiang B, Liu XD, et al. Berberine Protects Rat Heart From Ischemia/Reperfusion Injury via Activating JAK2/STAT3 Signaling and Attenuating Endoplasmic Reticulum Stress. Acta Pharmacol Sin (2016) 37:354–67. doi: 10.1038/aps.2015.136
222. Chen L, Su X, Hu Y. Berberine Down-Regulated Myostatin Expression and Facilitated Metabolism via Smad Pathway in Insulin Resistant Mice. Diabetes Metab Syndr Obes (2020) 13:4561–9. doi: 10.2147/DMSO.S275301
223. Wang H, Liu D, Cao P, Lecker S, Hu Z. Atrogin-1 Affects Muscle Protein Synthesis and Degradation When Energy Metabolism is Impaired by the Antidiabetes Drug Berberine. Diabetes (2010) 59:1879–89. doi: 10.2337/db10-0207
224. Ding K, Chen L, He J, Wang J, Yu C, Wang H. Tetrahydropalmatine Alleviates Hyperlipidemia by Regulating Lipid Peroxidation, Endoplasmic Reticulum Stress, and Inflammasome Activation by Inhibiting the TLR4-NF-kappaB Pathway. Evid Based Complement Alternat Med (2021) 2021:6614985. doi: 10.1155/2021/6614985
225. Lee SJ, Yoo M, Go GY, Hwang J, Lee HG, Kim YK, et al. Tetrahydropalmatine Promotes Myoblast Differentiation Through Activation of P38mapk and MyoD. Biochem Biophys Res Commun (2014) 455:147–52. doi: 10.1016/j.bbrc.2014.10.115
226. Arumugam S, Thandavarayan RA, Arozal W, Sari FR, Giridharan VV, Soetikno V, et al. Quercetin Offers Cardioprotection Against Progression of Experimental Autoimmune Myocarditis by Suppression of Oxidative and Endoplasmic Reticulum Stress via Endothelin-1/MAPK Signalling. Free Radic Res (2012) 46:154–63. doi: 10.3109/10715762.2011.647010
227. Le NH, Kim CS, Park T, Park JH, Sung MK, Lee DG, et al. Quercetin Protects Against Obesity-Induced Skeletal Muscle Inflammation and Atrophy. Mediators Inflammation (2014) 2014:834294. doi: 10.1155/2014/834294
228. Mukai R, Matsui N, Fujikura Y, Matsumoto N, Hou DX, Kanzaki N, et al. Preventive Effect of Dietary Quercetin on Disuse Muscle Atrophy by Targeting Mitochondria in Denervated Mice. J Nutr Biochem (2016) 31:67–76. doi: 10.1016/j.jnutbio.2016.02.001
229. Mukai R, Nakao R, Yamamoto H, Nikawa T, Takeda E, Terao J. Quercetin Prevents Unloading-Derived Disused Muscle Atrophy by Attenuating the Induction of Ubiquitin Ligases in Tail-Suspension Mice. J Nat Prod (2010) 73:1708–10. doi: 10.1021/np100240y
230. Kim Y, Kim CS, Joe Y, Chung HT, Ha TY, Yu R. Quercetin Reduces Tumor Necrosis Factor Alpha-Induced Muscle Atrophy by Upregulation of Heme Oxygenase-1. J Med Food (2018) 21:551–9. doi: 10.1089/jmf.2017.4108
231. Chen C, Yang JS, Lu CC, Chiu YJ, Chen HC, Chung MI, et al. Effect of Quercetin on Dexamethasone-Induced C2C12 Skeletal Muscle Cell Injury. Molecules (2020) 25. doi: 10.3390/molecules25143267
232. Bazzucchi I, Patrizio F, Ceci R, Duranti G, Sgro P, Sabatini S, et al. The Effects of Quercetin Supplementation on Eccentric Exercise-Induced Muscle Damage. Nutrients (2019) 11. doi: 10.3390/nu11010205
233. Guo R, Liu W, Liu B, Zhang B, Li W, Xu Y. SIRT1 Suppresses Cardiomyocyte Apoptosis in Diabetic Cardiomyopathy: An Insight Into Endoplasmic Reticulum Stress Response Mechanism. Int J Cardiol (2015) 191:36–45. doi: 10.1016/j.ijcard.2015.04.245
234. Sun LJ, Sun YN, Chen SJ, Liu S, Jiang GR. Resveratrol Attenuates Skeletal Muscle Atrophy Induced by Chronic Kidney Disease via MuRF1 Signaling Pathway. Biochem Biophys Res Commun (2017) 487:83–9. doi: 10.1016/j.bbrc.2017.04.022
235. Asami Y, Aizawa M, Kinoshita M, Ishikawa J, Sakuma K. Resveratrol Attenuates Denervation-Induced Muscle Atrophy Due to the Blockade of Atrogin-1 and P62 Accumulation. Int J Med Sci (2018) 15:628–37. doi: 10.7150/ijms.22723
236. Wang DT, Yin Y, Yang YJ, Lv PJ, Shi Y, Lu L, et al. Resveratrol Prevents TNF-Alpha-Induced Muscle Atrophy via Regulation of Akt/mTOR/FoxO1 Signaling in C2C12 Myotubes. Int Immunopharmacol (2014) 19:206–13. doi: 10.1016/j.intimp.2014.02.002
237. Manas-Garcia L, Denhard C, Mateu J, Duran X, Gea J, Barreiro E. Beneficial Effects of Resveratrol in Mouse Gastrocnemius: A Hint to Muscle Phenotype and Proteolysis. Cells (2021) 10. doi: 10.3390/cells10092436
238. Yang J, Wang Z, Chen DL. Shikonin Ameliorates Isoproterenol (ISO)-Induced Myocardial Damage Through Suppressing Fibrosis, Inflammation, Apoptosis and ER Stress. BioMed Pharmacother (2017) 93:1343–57. doi: 10.1016/j.biopha.2017.06.086
239. Oberg AI, Yassin K, Csikasz RI, Dehvari N, Shabalina IG, Hutchinson DS, et al. Shikonin Increases Glucose Uptake in Skeletal Muscle Cells and Improves Plasma Glucose Levels in Diabetic Goto-Kakizaki Rats. PloS One (2011) 6:e22510. doi: 10.1371/journal.pone.0022510
240. Li YP, Wang SL, Liu B, Tang L, Kuang RR, Wang XB, et al. Sulforaphane Prevents Rat Cardiomyocytes From Hypoxia/Reoxygenation Injury In Vitro via Activating SIRT1 and Subsequently Inhibiting ER Stress. Acta Pharmacol Sin (2016) 37:344–53. doi: 10.1038/aps.2015.130
241. Son YH, Jang EJ, Kim YW, Lee JH. Sulforaphane Prevents Dexamethasone-Induced Muscle Atrophy via Regulation of the Akt/Foxo1 Axis in C2C12 Myotubes. BioMed Pharmacother (2017) 95:1486–92. doi: 10.1016/j.biopha.2017.09.002
242. Moon JY, Kim DJ, Kim HS. Sulforaphane Ameliorates Serum Starvation-Induced Muscle Atrophy via Activation of the Nrf2 Pathway in Cultured C2C12 Cells. Cell Biol Int (2020) 44:1831–9. doi: 10.1002/cbin.11377
243. Arabi YM, Aldawood AS, Haddad SH, Al-Dorzi HM, Tamim HM, Jones G, et al. Permissive Underfeeding or Standard Enteral Feeding in Critically Ill Adults. N Engl J Med (2015) 372:2398–2408. doi: 10.1056/NEJMoa1502826
244. Elke G, Hartl WH, Kreymann KG, Adolph M, Felbinger TW, Graf T, et al. Clinical Nutrition in Critical Care Medicine - Guideline of the German Society for Nutritional Medicine (DGEM). Clin Nutr ESPEN (2019) 33:220–275. doi: 10.1016/j.clnesp.2019.05.002
245. Van Zanten ARH, De Waele E, Wischmeyer PE. Nutrition Therapy and Critical Illness: Practical Guidance for the ICU, Post-ICU, and Long-Term Convalescence Phases. Crit Care (2019) 23:368. doi: 10.1186/s13054-019-2657-5
246. De Waele E, Jonckheer J, Wischmeyer PE. Indirect Calorimetry in Critical Illness: A New Standard of Care? Curr Opin Crit Care (2021) 27:334–343. doi: 10.1097/MCC.0000000000000844
247. Hill A, Elke G, Weimann A. Nutrition in the Intensive Care Unit-A Narrative Review. Nutrients (2021) 13(8):2851. doi: 10.3390/nu13082851
248. Giordani L, He GJ, Negroni E, Sakai H, Law JYC, Siu MM, et al. High-Dimensional Single-Cell Cartography Reveals Novel Skeletal Muscle-Resident Cell Populations. Mol Cell (2019) 74:609–621.e606. doi: 10.1016/j.molcel.2019.02.026
249. Cho DS, Schmitt RE, Dasgupta A, Ducharme AM, Doles JD. Single-Cell Deconstruction of Post-Sepsis Skeletal Muscle and Adipose Tissue Microenvironments. J Cachexia Sarcopenia Muscle (2020) 11, 1351–1363. doi: 10.1002/jcsm.12596
250. Chaudhary P, Suryakumar G, Prasad R, Singh SN, Ali S, Ilavazhagan G, et al. Chronic Hypobaric Hypoxia Mediated Skeletal Muscle Atrophy: Role of Ubiquitin-Proteasome Pathway and Calpains. Mol Cell Biochem (2012) 364:101–113. doi: 10.1007/s11010-011-1210-x
251. De Theije CC, Schols A, Lamers WH, Ceelen JJM, Van Gorp RH, Hermans JJR, et al. Glucocorticoid Receptor Signaling Impairs Protein Turnover Regulation in Hypoxia-Induced Muscle Atrophy in Male Mice. Endocrinology (2018) 159:519–534. doi: 10.1210/en.2017-00603
252. Langen RC, Gosker HR, Remels AH, Schols AM. Triggers and Mechanisms of Skeletal Muscle Wasting in Chronic Obstructive Pulmonary Disease. Int J Biochem Cell Biol (2013) 45:2245–2256. doi: 10.1016/j.biocel.2013.06.015
253. Schofield CJ, Ratcliffe PJ. Oxygen Sensing by HIF Hydroxylases. Nat Rev Mol Cell Biol (2004) 5:343–354. doi: 10.1038/nrm1366
254. Kwon SJ, Song JJ, Lee YJ. Signal Pathway of Hypoxia-Inducible Factor-1alpha Phosphorylation and Its Interaction With Von Hippel-Lindau Tumor Suppressor Protein During Ischemia in MiaPaCa-2 Pancreatic Cancer Cells. Clin Cancer Res (2005) 11:7607–13. doi: 10.1158/1078-0432.CCR-05-0981
255. Barnes T, Zochios V, Parhar K. Re-examining Permissive Hypercapnia in ARDS: A Narrative Review. Chest (2018) 154:185–195. doi: 10.1016/j.chest.2017.11.010
256. Jaitovich A, Angulo M, Lecuona E, Dada LA, Welch LC, Cheng Y, et al. High CO2 levels cause skeletal muscle atrophy via AMP-activated kinase (AMPK), FoxO3a protein, and muscle-specific Ring finger protein 1 (MuRF1). J Biol Chem (2015) 290:9183–9194. doi: 10.1074/jbc.M114.625715
257. Korponay TC, Balnis J, Vincent CE, Singer DV, Chopra A, Adam AP, et al. High CO2 Downregulates Skeletal Muscle Protein Anabolism via AMP-activated Protein Kinase alpha2-mediated Depressed Ribosomal Biogenesis. Am J Respir Cell Mol Biol (2020) 62:74–86. doi: 10.1165/rcmb.2019-0061OC
258. Kryvenko V, Vadasz I. Mechanisms of Hypercapnia-Induced Endoplasmic Reticulum Dysfunction. Front Physiol (2021) 12:735580. doi: 10.3389/fphys.2021.735580
259. Bravo R, Vicencio JM, Parra V, Troncoso R, Munoz JP, Bui M, et al. Increased ER-Mitochondrial Coupling Promotes Mitochondrial Respiration and Bioenergetics During Early Phases of ER Stress. J Cell Sci (2011) 124:2143–52. doi: 10.1242/jcs.080762
260. Rusinol AE, Cui Z, Chen MH, Vance JE. A Unique Mitochondria-Associated Membrane Fraction From Rat Liver Has a High Capacity for Lipid Synthesis and Contains Pre-Golgi Secretory Proteins Including Nascent Lipoproteins. J Biol Chem (1994) 269:27494–27502.
261. Gordaliza-Alaguero I, Canto C, Zorzano A. Metabolic Implications of Organelle-Mitochondria Communication. EMBO Rep (2019) 20:e47928. doi: 10.15252/embr.201947928
262. Van Vliet AR, Agostinis P. Mitochondria-Associated Membranes and ER Stress. Curr Top Microbiol Immunol (2018) 414:73–102. doi: 10.1007/82_2017_2
263. Gordaliza-Alaguero I, Canto C, Zorzano , A. Metabolic implications of organelle-mitochondria communication. EMBO Rep (2019) 20:e47928. doi: 10.15252/embr.201947928
264. Kumar V, Maity S. ER Stress-Sensor Proteins and ER-Mitochondrial Crosstalk-Signaling Beyond (ER) Stress Response. Biomolecules (2021) 11(2):173. doi: 10.3390/biom11020173
265. Eisner V, Csordas G, Hajnoczky G. Interactions between sarco-endoplasmic reticulum and mitochondria in cardiac and skeletal muscle - pivotal roles in Ca2+ and reactive oxygen species signaling. J Cell Sci. (2013) 126(Pt 14):2965–78. doi: 10.1242/jcs.093609
266. Arabi YM, Aldawood AS, Haddad SH, Al-Dorzi HM, Tamim HM, Jones G, et al. Permissive Underfeeding or Standard Enteral Feeding in Critically Ill Adults. N Engl J Med (2015) 372(25):2398–408. doi: 10.1056/NEJMoa1502826
267. Elke G, Hartl WH, Kreymann KG, Adolph M, Felbinger TW, Graf T, et al. Clinical Nutrition in Critical Care Medicine - Guideline of the German Society for Nutritional Medicine (DGEM). Clin Nutr ESPEN (2019) 33:220–75. doi: 10.1016/j.clnesp.2019.05.002
268. Van Zanten ARH, De Waele E, Wischmeyer PE. Nutrition Therapy and Critical Illness: Practical Guidance for the ICU, Post-ICU, and Long-Term Convalescence Phases. Crit Care (2019) 23(1):368. doi: 10.1186/s13054-019-2657-5
269. De Waele E, Jonckheer J, Wischmeyer PE. Indirect Calorimetry in Critical Illness: A New Standard of Care? Curr Opin Crit Care (2021) 27(4):334–43. doi: 10.1097/MCC.0000000000000844
270. Hill A, Elke G, Weimann A. Nutrition in the Intensive Care Unit-A Narrative Review. Nutrients (2021) 13(8):2851. doi: 10.3390/nu13082851
271. Giordani L, He GJ, Negroni E, Sakai H, Law JYC, Siu MM, et al. High-Dimensional Single-Cell Cartography Reveals Novel Skeletal Muscle-Resident Cell Populations. Mol Cell (2019) 74(3):609–621.e606. doi: 10.1016/j.molcel.2019.02.026
272. Cho DS, Schmitt RE, Dasgupta A, Ducharme AM, Doles JD. Single-Cell Deconstruction of Post-Sepsis Skeletal Muscle and Adipose Tissue Microenvironments. J Cachexia Sarcopenia Muscle (2020) 11(5):1351–63. doi: 10.1002/jcsm.12596
273. Chaudhary P, Suryakumar G, Prasad R, Singh SN, Ali S, Ilavazhagan G. Chronic Hypobaric Hypoxia Mediated Skeletal Muscle Atrophy: Role of Ubiquitin-Proteasome Pathway and Calpains. Mol Cell Biochem (2012) 364(1–2):101–13. doi: 10.1007/s11010-011-1210-x
274. De Theije CC, Schols A, Lamers WH, Ceelen JJM, Van Gorp RH, Hermans JJR, et al. Glucocorticoid Receptor Signaling Impairs Protein Turnover Regulation in Hypoxia-Induced Muscle Atrophy in Male Mice. Endocrinology (2018) 159(1):519–34. doi: 10.1210/en.2017-00603
275. Langen RC, Gosker HR, Remels AH, Schols AM. Triggers and Mechanisms of Skeletal Muscle Wasting in Chronic Obstructive Pulmonary Disease. Int J Biochem Cell Biol (2013) 45(10):2245–56. doi: 10.1016/j.biocel.2013.06.015
276. Schofield CJ, Ratcliffe PJ. Oxygen Sensing by HIF Hydroxylases. Nat Rev Mol Cell Biol (2004) 5(5):343–54. doi: 10.1038/nrm1366
277. Kwon SJ, Song JJ, Lee YJ. Signal Pathway of Hypoxia-Inducible Factor-1alpha Phosphorylation and Its Interaction With von Hippel-Lindau Tumor Suppressor Protein During Ischemia in MiaPaCa-2 Pancreatic Cancer Cells. Clin Cancer Res (2005) 11(21):7607–13. doi: 10.1158/1078-0432.CCR-05-0981
278. Barnes T, Zochios V, Parhar K. Re-examining Permissive Hypercapnia in ARDS: A Narrative Review. Chest (2018) 154(1):185–95. doi: 10.1016/j.chest.2017.11.010
279. Jaitovich A, Angulo M, Lecuona E, Dada LA, Welch LC, Cheng Y, et al. High CO2 Levels Cause Skeletal Muscle Atrophy via AMP-Activated Kinase (AMPK), FoxO3a Protein, and Muscle-Specific Ring Finger Protein 1 (MuRF1). J Biol Chem (2015) 290(14):9183–94. doi: 10.1074/jbc.M114.625715
280. Korponay TC, Balnis J, Vincent CE, Singer DV, Chopra A, Adam AP, et al. High CO2 Downregulates Skeletal Muscle Protein Anabolism via AMP-Activated Protein Kinase alpha2-Mediated Depressed Ribosomal Biogenesis. Am J Respir Cell Mol Biol (2020) 62(1):74–86. doi: 10.1165/rcmb.2019-0061OC
281. Kryvenko V, Vadasz I. Mechanisms of Hypercapnia-Induced Endoplasmic Reticulum Dysfunction. Front Physiol (2021) 12:735580. doi: 10.3389/fphys.2021.735580
282. Bravo R, Vicencio JM, Parra V, Troncoso R, Munoz JP, Bui M, et al. Increased ER-Mitochondrial Coupling Promotes Mitochondrial Respiration and Bioenergetics During Early Phases of ER Stress. J Cell Sci (2011) 124(Pt 13):2143–52. doi: 10.1242/jcs.080762
283. Rusinol AE, Cui Z, Chen MH, Vance JE. A Unique Mitochondria-Associated Membrane Fraction From Rat Liver has a High Capacity for Lipid Synthesis and Contains Pre-Golgi Secretory Proteins Including Nascent Lipoproteins. J Biol Chem (1994) 269(44):27494–502.
284. Gordaliza-Alaguero I, Canto C, Zorzano A. Metabolic Implications of Organelle-Mitochondria Communication. EMBO Rep (2019) 20(9):e47928. doi: 10.15252/embr.201947928
285. Van Vliet AR, Agostinis P. Mitochondria-Associated Membranes and ER Stress. Curr Top Microbiol Immunol (2018) 414:73–102. doi: 10.1007/82_2017_2
286. Kumar V, and Maity S. ER Stress-Sensor Proteins and ER-Mitochondrial Crosstalk-Signaling Beyond (ER) Stress Response. Biomolecules (2021) 11(2):173. doi: 10.3390/biom11020173
Keywords: endoplasmic reticulum stress, inflammation, intensive care unit acquired weakness, unfolded protein response, sepsis
Citation: Kny M and Fielitz J (2022) Hidden Agenda - The Involvement of Endoplasmic Reticulum Stress and Unfolded Protein Response in Inflammation-Induced Muscle Wasting. Front. Immunol. 13:878755. doi: 10.3389/fimmu.2022.878755
Received: 18 February 2022; Accepted: 04 April 2022;
Published: 09 May 2022.
Edited by:
Cláudia Pereira, University of Coimbra, PortugalReviewed by:
Arturo Briva, Universidad de la República, UruguayCopyright © 2022 Kny and Fielitz. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jens Fielitz, amVucy5maWVsaXR6QHVuaS1ncmVpZnN3YWxkLmRl
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.