 Gina López-Cantillo1
Gina López-Cantillo1 Bernardo Armando Camacho
Bernardo Armando Camacho Cesar Ramírez-Segura
Cesar Ramírez-Segura- 1Laboratorio de Investigación en Ingeniería Celular y Molecular, Instituto Distrital de Ciencia Biotecnología e Innovación en Salud (IDCBIS), Bogotá, Colombia
- 2Grupo de Inmunobiología y Biología Celular, Facultad de Ciencias, Pontificia Universidad Javeriana, Bogotá, Colombia
- 3Instituto Distrital de Ciencia Biotecnología e Innovación en Salud (IDCBIS), Bogotá, Colombia
Adoptive cell therapy with T cells reprogrammed to express chimeric antigen receptors (CAR-T cells) has been highly successful in patients with hematological neoplasms. However, its therapeutic benefits have been limited in solid tumor cases. Even those patients who respond to this immunotherapy remain at risk of relapse due to the short-term persistence or non-expansion of CAR-T cells; moreover, the hostile tumor microenvironment (TME) leads to the dysfunction of these cells after reinfusion. Some research has shown that, in adoptive T-cell therapies, the presence of less differentiated T-cell subsets within the infusion product is associated with better clinical outcomes. Naive and memory T cells persist longer and exhibit greater antitumor activity than effector T cells. Therefore, new methods are being studied to overcome the limitations of this therapy to generate CAR-T cells with these ideal phenotypes. In this paper, we review the characteristics of T-cell subsets and their implications in the clinical outcomes of adoptive therapy with CAR-T cells. In addition, we describe some strategies developed to overcome the reduced persistence of CAR T-cells and alternatives to improve this therapy by increasing the expansion ability and longevity of modified T cells. These methods include cell culture optimization, incorporating homeostatic cytokines during the expansion phase of manufacturing, modulation of CAR-T cell metabolism, manipulating signaling pathways involved in T-cell differentiation, and strategies related to CAR construct designs.
Introduction
On August 30, 2017, the US Food and Drug Administration (FDA) approved the use of tisagenlecleucel (CTL-019; Kymriah®, Novartis, Basel, Switzerland), a CD19-directed chimeric antigen receptor (CAR) T cell, for adoptive cell therapy (ACT) to treat patients with relapsed/refractory B-cell acute lymphoblastic leukemia (r/r B-ALL) (1–3). The complete remission rate (CRR) was 63% [95% confidence interval (CI), 50%–75%], and all patients in complete remission (CR) attained minimal residual disease (MRD) less than 0.01% after a median follow-up of 4.8 months (2). In October 2017, the FDA granted regular approval for axicabtagene ciloleucel (Yescarta), another CAR-T cell directed against CD19, to treat adult patients with relapsed/refractory large B-cell lymphoma after two or more lines of systemic therapy (4). Efficacy was assessed in terms of CRR and duration of response in 101 adult patients with relapsed/refractory large B-cell lymphoma (median of 3 prior systemic regimens) who underwent treatment in a single-arm trial (5). In 2020, brexucabtagene autoleucel was approved for the treatment of relapsed/refractory diffuse large B-cell and mantle cell lymphomas (6, 7). Likewise, lisocabtagene maraleucel was approved in February 2021 as therapy for refractory large B-cell lymphoma (7, 8). Recently, the FDA has approved a CAR-T cell immunotherapy for multiple myeloma; the first-in-class B-cell maturation antigen (BCMA)-targeted CAR-T cell therapy received the agency’s approval on March 26, 2021, to treat adults with relapsed/refractory multiple myeloma (9, 10).
The clinical response to CAR-T cell therapy has been associated with the in vivo expansion and long-term persistence of functional CAR-T cells (11). Mounting evidence suggests that successful outcomes in patients treated with CAR-T cells depend on the cells’ ability to expand and persist after infusion. One of the major issues of using CAR-T cells for the treatment of solid tumors is the low persistence of the cells infused within the tumor mass (12). Long-term persistence and robust in vivo expansion of CAR-T cells infused during ACT are associated with sustained clinical remission and survival of recipient patients (11, 13–15). In 2019, Hay et al. reported a study on adult patients with B-ALL infused with CD19-directed CAR-T cells. Although the MRD was negative in 85% of individuals and the CRR was high, 49% of patients relapsed after CAR-T cell infusion. Before or at relapse, the CAR transgene copies were low or undetectable in peripheral blood (16).
Different factors have been associated with long-lasting remission after the adoptive transfer of CAR-T cells. One of the critical aspects that determine the efficacy of CAR-T cell therapy is the in vivo persistence of the cells infused. Some studies have shown that the persistence of CAR-T cells is correlated with the phenotype of the T cells infused and that prolonged detection of CAR-T cells is associated with superior responses even in patients with high-grade diseases (17). In search of determinants of therapeutic response to CD19-directed CAR-T cells in patients with chronic lymphocytic leukemia (CLL), a study evaluated the genomic and phenotypic features of the cells infused. The authors found that CAR-T cells from patients in CR exhibited upregulation of genes related to a memory cell phenotype, whereas CAR-T cells from patients with no clinical response showed upregulation of genes associated with an effector or exhausted cell phenotype (18). In 2014, Maude et al. reported a pilot clinical trial of 25 patients with r/r B-ALL treated with CD19-directed CAR-T cells. Of these patients, 90% achieved CR on day 28 and 6 months after infusion; the rate of relapse-free survival was 80%. Furthermore, they found an association between the persistence of CAR-T cells in peripheral blood and B-cell aplasia in patients who had a response (13). The recovery of CD19+ lymphocytes from peripheral blood within the first semester following infusion of CAR-T cells indicated the disappearance of these CAR-T cells or the loss of their function (19).
The quantification of CAR-T cells is usually performed by flow cytometry, to detect the surface expression of CAR, or by quantitative polymerase chain reaction, to detect the CAR gene, but not its expression. However, CAR detection does not imply clinical response as cells can be not functionally active (20). An indirect parameter of persistent CD19-directed CAR-T cells after adoptive transfer is B-cell aplasia (20). Several studies have shown an association between the length of cancer remission and B-cell aplasia. B-cell aplasia is an indicator of functionally active CAR-T cells that deplete CD19+ B cells and is associated with a sustained therapy response (11, 13).
This review focuses on the different approaches used by researchers worldwide to achieve the persistence of CAR-T cells and improve their immunophenotype for better treatment response.
CAR-T Cell Differentiation Stage and Treatment Response
The differentiation stage of T cells affects their proliferative and survival abilities. The proliferation and the survival of adoptively transferred T cells strongly correlate with their antitumor activity (21–23). The immunophenotype of cells used to start the manufacture of CAR-T cells relates to the treatment outcomes. For instance, long-term remission is related to the enrichment of CD27+/CD45RO−/CD8+ T cells with memory-like features (18, 24).
The antitumor activity of adoptively transferred T cells depends on their expansion and long-term activity. Clinical results have shown that less differentiated memory T cells are required for the sustained in vivo persistence of adoptively transferred CAR-T cells, while naive (TN), central memory (TCM), and stem-like memory (TSCM) lymphocytes are related to a good response due to their ability to proliferate and live longer (25). Effector T-lymphocyte (TE) subsets exhibit low self-renewal ability, reduced homing to tumor niches, and lower survival than memory lymphocyte (TM) subpopulations (26–28). Preclinical models have been used to examine the longevity and functional features of CAR-T cells derived from memory and naive T-cell subsets. The results have demonstrated that CAR-T cells produced from the CD4+ and CD8+ TN and TCM subsets have greater antitumor potency and proliferation than those derived from effector memory T lymphocytes (TEM) (29). These data indicate that naive and memory T cells are important in CAR-T cell therapy because they display sustained proliferation and higher persistence in vivo (30, 31).
The diversity of the T-cell subpopulations results from the microenvironment stimulus and the cell–antigen interaction. Both CD4+ and CD8+ CAR-T cells can participate in killing malignant cells (32). Indeed, the combination of the most potent CD4+ and CD8+ CAR-expressing T-cell subsets has synergistic antitumor effects in vivo (29). For example, in ACT with GD2-directed CAR-T cells for the treatment of neuroblastoma, the number of CD4+ T cells and TCM cells (CD45RO+/CD62L+) within the infused product showed high concordance with the length of persistence of CAR-T cells (17). Therefore, understanding the generation and maintenance of the different T-cell subsets is critical for proposing strategies that improve the clinical outcomes of CAR-T cell therapy (31, 33).
T-Cell Subsets
T cells can be subdivided into several subsets identified according to the combination of molecules expressed on the cell surface (34). These phenotypic differences are related to the migratory and functional characteristics of each T-cell subpopulation (35, 36).
Naive T Cells
Immature T lymphocytes are characterized by the high expression of the transmembrane phosphatase CD45RA isoform (33, 36). Cells are considered naive until they interact with their cognate antigen. This interaction activates TN lymphocytes to proliferate and differentiate into TM and/or TE lymphocytes (27). Other surface markers expressed by TN lymphocytes are CCR7 and CD62L (L-selectin), which guide the homing of T cells to the secondary lymphoid organs; CD27 and CD28, which provide co-stimulatory signals; and CXCR4 and IL7Rα (CD127) (24, 36). The TN lymphocyte subset lacks the expression of CD45RO, CD95, CD11a, CD122, CD31, and KLRG1 (Figure 1 and Supplementary Table S1) (28, 34, 37) and is also characterized by high proliferative ability (36).
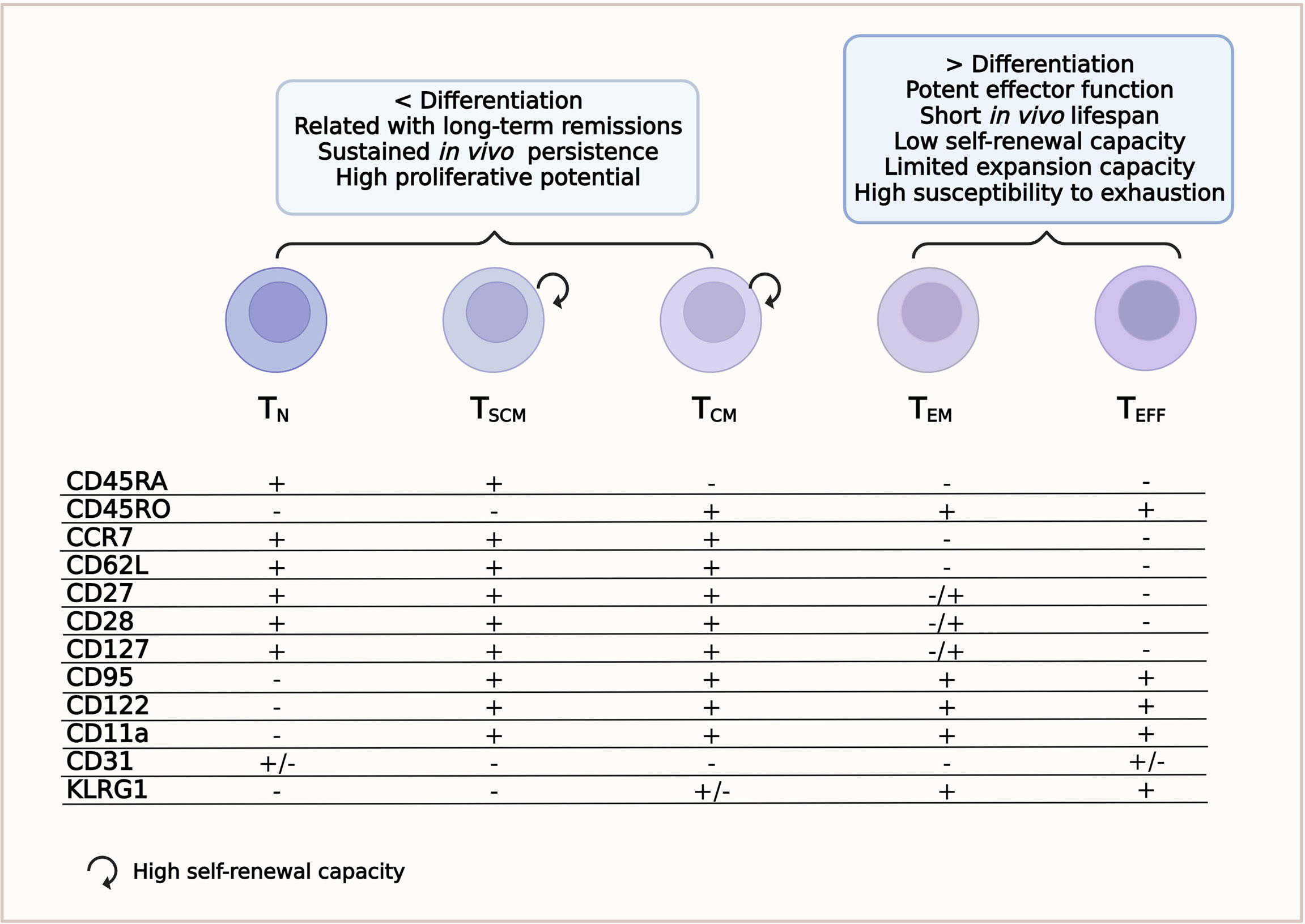
Figure 1 T-cell subsets. Phenotype and relevant features for a successful chimeric antigen receptor T-cell (CAR-T cell) therapy.
Stem Cell Memory T Cells
This subset is the least differentiated of the memory T-cell subpopulations. TSCM cells have been recently identified as CD4+ or CD8+ T cells with a TN/TM-like phenotype. TSCM cells express CD45RA, CCR7, CD95, CXCR3, CD11a, IL-2Rβ receptor (CD122), and CD127 (34, 36, 38) and have high levels of the transcriptional regulator T cell factor 1 (TCF-1), but do not express CD45RO (Figure 1 and Supplementary Table S1) (37). They have a self-renewal ability similar to that of stem cells and can reconstitute all populations of TM and TE cells (24, 39). Compared to other TM subpopulations, the TSCM subset develops a faster response upon antigenic stimulation and can persist for a long time (27, 40). Because of these characteristics, the TSCM subset has received significant attention in the ACT field (31). Despite their features implying several rounds of division [low expressions of T-cell receptor excision circles (TRECs) and Ki-67high], they maintain their naive-like phenotype (36).
Memory T Cells with Naive Phenotypes
In 2016, Pulko et al. identified a CD8+ T-cell subpopulation that produced effector cytokines after stimulation through the T-cell receptor (TCR). The phenotypes of these cells were CXCR3high, CD49d+, INF-γ+, CD45RA+, CCR7+, CD95low, and CD28int. Although these cells shared several features with TN cells, they had a restricted TCR Vβ repertoire, suggesting antigen-driven stimulation and expansion, and differed transcriptionally from TM and TE cells (36, 39, 41).
Central Memory T Cells
Central memory T cells circulate through secondary lymphoid organs and are characterized by a long life span. Although TCM have less cytotoxic capacity than TE, they can proliferate rapidly and provide an early batch of cytokines after being stimulated by the specific antigen (27, 42). They secrete tumor necrosis factor alpha (TNF-α), although more efficiently interleukin 2 (IL-2), and express CD45RO, CD62L, CD28, CD27, CCR7, CD127, CD11a, IL-18Rα, CXCR4, and CXCR3, but not CD45RA (Figure 1 and Supplementary Table S1) (24, 34, 37, 43). The loss of CCR7 and CD62L accompanies the transition from TCM to the TEM phenotype and, therefore, the cells can no longer migrate to lymphoid tissues (27).
Effector Memory T Cells
Effector memory T cells mainly circulate to non-lymphoid tissues and typically express CD45RO, CD122, CD95, KLRG1, LFA-1, IL-18Rα, chemokine receptors, and tissue homing receptors, but are negative for CD62L, CCR7, and CD31 (Figure 1 and Table Supplementary S1) (34, 37, 43). The TEM cell subset also secretes TNF-α, but has a greater capacity to release interferon gamma (IFN-γ) and are more cytotoxic than TCM lymphocytes (24, 31).
Tissue-Resident Memory T Cells
These cells are very similar to TEM cells, but differ by the expression of CD103 and CD69 (33, 34). These cells do not express CD62L, CD25, CD38, and HLA-DR (34). Moreover, they remain in non-lymphoid tissues and can self-renew in situ and respond to secondary infections (38). Their ability to infiltrate solid tumors is well known and makes them potentially useful for developing CAR-T cells to treat these types of neoplasias (27). The surface marker expression of tissue-resident memory T cells (TRM) differs among tissues. On the skin, TRM cells express cutaneous lymphocyte-associated antigen (CLA), CCR4, and CCR6, and about 50% of them are CCR5+/CXCR3+; in the gut, TRM cells express CD69, CCR6, CCR9, and CD49d; and in the lung, they express CD49a, PSGL-1, CCR5, CXCR3, and CCR6 (34, 44).
Terminal Effector T Cells
This cell subset comprises fully differentiated T cells ready for rapid responses and potent effector functions. However, they have a short life span and a very low self-renewal ability (27). Phenotypically, terminal effector T cells (TEFF) are positive for CD95, CD122, KLGR1 and several homing receptors to migrate to sites of inflammation, such as CCR5 and LFA-1; furthermore, they re-express CD45RA (34, 45). These cells do not express CD45RO, CCR7, CD62L, IL7Rα (CD127), CD27, or CD28 (Figure 1 and Supplementary Table S1) (45). They also have limited expansion ability and rapidly die or become exhausted (33, 38).
Exhausted T Cells
The persisting antigenic stimulation of antigen-specific CD8+ T cells throughout the responses to chronic infections or cancer leads to a gradual loss of the effector functions, with T cells becoming dysfunctional (33, 34, 38). The exhaustion features comprise the sustained expression of inhibitory receptors, altered metabolism fitness, low proliferative capacity, and a reduced secretion of effector cytokines (46). There are two main subsets of exhausted T cells (TEX): precursor of exhausted T cells (TPEX) and terminally exhausted T cells (34).
TPEX have only recently been identified. This small cell subpopulation exhibits memory and exhaustion features, such as the expression of TCF-1, CD62L, ID3, and PD-1 and reduced cytokine secretion. These cells mediate the response to immune checkpoint inhibitors and can self-renew and differentiate into terminally exhausted T cells (TTEX) (33, 46).
TTEX cells typically co-express PD-1, LAG-3, TIM-3, CD160, and TIGIT (34, 38). However, they do not respond to immune checkpoint blockade and their proliferation potential is impaired (47).
T-cell exhaustion is a dynamic process from progenitor to terminally exhausted cells, characterized by different stages, each with distinct features. Understanding this process is necessary to designing more precise immunotherapy strategies. These approaches would help block the differentiation toward exhaustion and reverse certain stages of exhausted T cells (48).
CD4+ T-Cell Subsets
CD4+ T cells can differentiate into several subpopulations, namely, T helper (Th) 1, Th2, Th9, Th17, Th22, follicular helper T cells (TFH), and regulatory T cells (Treg). Cell differentiation depends on the cytokines present in the environment and the strength of TCR signaling (Figure 2) (49). Each cell subset has particular characteristics and releases a cytokine cocktail that defines its functions. These roles can either be anti- or pro-inflammatory, related to protection, survival, or immune homeostasis (34). Several preclinical trial data have demonstrated that, in ACT with CAR-T cells, CD4+ T cells showed a direct antitumor activity comparable to cytotoxic CD8+ CAR-T cells (50–52). Moreover, the function of CD8+ CAR-T cells was characterized by exhaustion and apoptosis in the presence of antigen-specific TCR stimulation, whereas CD4+ CAR-T cells retained equivalent cytotoxicity despite TCR stimulation (50). CD4+ CAR-T cells helped augment the proliferation of CD8+ T cells, but they did not ameliorate the potency of CD8+ CAR-T cell effectors (51). Interestingly, in a murine model using CD28-based second-generation CAR-T cells, Th1 and Th2 cells released cytokines that led to different types of cytotoxicity (53).
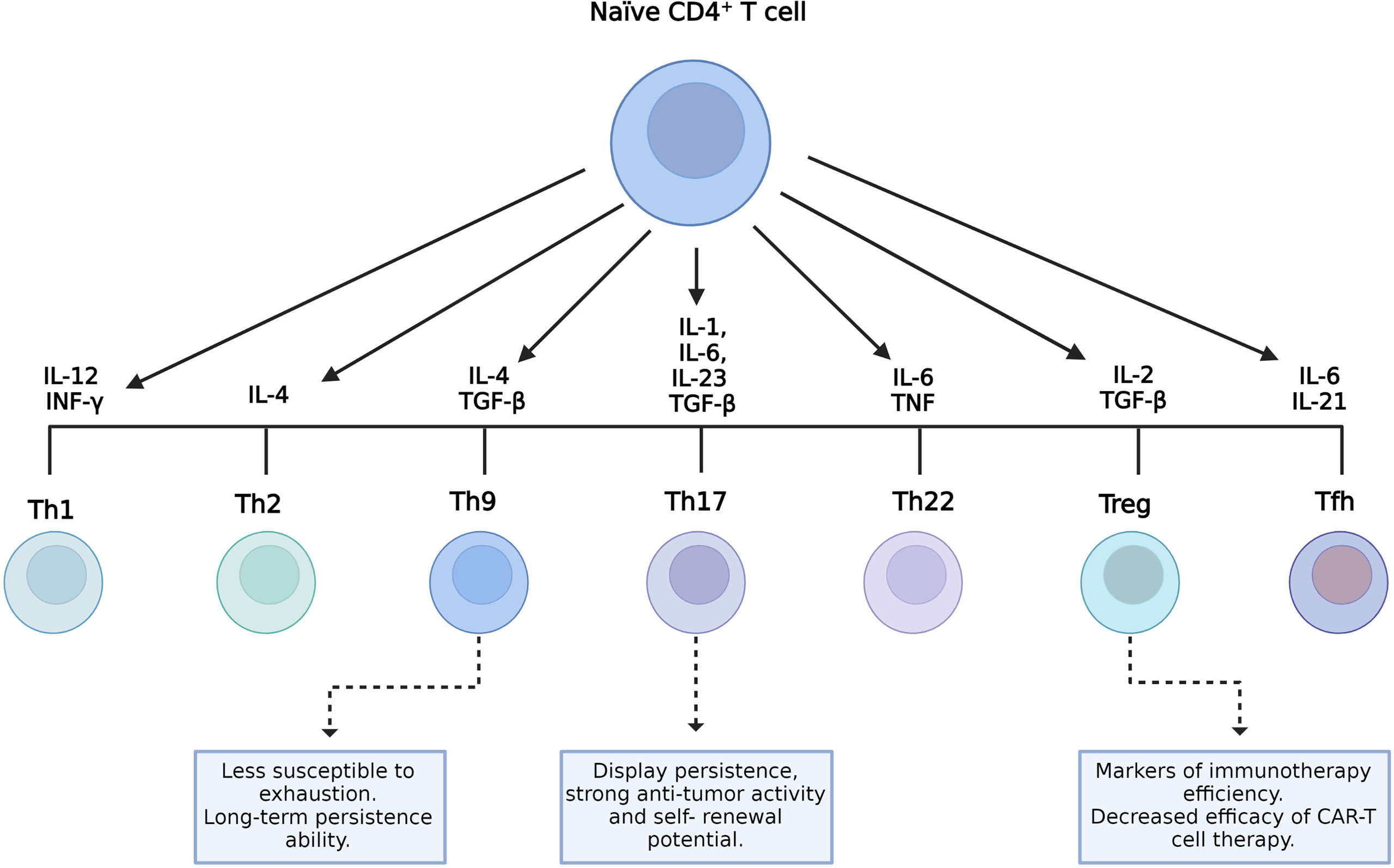
Figure 2 CD4+ T-cell subsets and their role on the final manufactured chimeric antigen receptor T-cell (CAR-T cell) product. Different environment cytokine combinations are required to generate different CD4+ T-cell subsets. Characteristics of T helper 9 (Th9) and Th17 cells with the potential to improve the efficacy of CAR-T cells and the role of regulatory T cells (Tregs) as indicators of therapeutic efficacy are shown.
In addition, tumor-specific Th9 cells are less susceptible to exhaustion, cause complete lysis of tumor cells, and persist longer given their unique hyperproliferative feature (Figure 2) (54). In patients undergoing ACT, resistance can rise due to the outgrowth of antigen-loss-variant (ALV) cancer cells (55). In a recent study, adoptively transferred tumor-specific Th9 cells have been shown to be able to eradicate established ALV-containing tumors (56). Human autologous mesothelin-specific CAR Th9 cells, but not regular or high doses of Th1+Tc1 CAR-T cells, were able to eradicate human ovarian cancer (OvCa) patient-derived xenograft (PDX) in humanized NSG (NOD scid gamma) mice (55). Interestingly, a recent study has shown that the intrinsic activation of CD4+ T cells potentiates the antitumor effects of Th9 cells upon adoptive transfer in mice and that human Th9 cell differentiation can be enhanced through STING activation (57). In addition, other studies found that STING agonists synergize with CAR-T cells, enhancing their ability to control tumor growth (58), and administration of the STING ligand cyclic guanosine monophosphate–adenosine monophosphate (cGAMP) improved the antitumor responses in models of melanoma and colon cancer (59, 60).
On the other hand, Th17 cells are characterized by their plasticity since they can transdifferentiate into other effector subsets, including Th1-like Th17 cells that express the transcription factor T-box-expressed-in-T-cells (T-bet), IL-17, and IFN-γ (61–63). The role of this cell subset in tumor immunity remains partly elucidated. According to some studies, Th17 cells can either promote or eliminate tumors depending on the context of the tumor (64, 65). Interestingly, several reports have shown that the antitumor activity of Th17 cells is related to their ability to recruit and activate cytotoxic T lymphocytes, natural killer (NK) cells, dendritic cells (DCs), and neutrophils into the tumor and also to the plasticity of Th17 cells to differentiate toward the Th1 phenotype that eliminates tumors via the secretion of IFN-γ (62, 66, 67). IL-17, one of the Th1-like Th17-related cytokines, is associated with a pro-tumorigenic role by controlling tumor angiogenesis, increasing cell proliferation, and preventing cell apoptosis (62). Although IL-17 shows some antitumor effects, it is IFN-γ rather than IL-17 from Th1-like Th17 cells that appears essential to an efficient antitumor response (68–70). Indeed, the combination of STING agonists with Th/Tc17 CAR-T cells increased the trafficking, persistence, and tumor control in a murine model of breast cancer (58). Furthermore, Guedan et al. reported the enhanced antitumor activity and increased persistence of CAR-T cells in a preclinical model of ACT using CAR Th17 cells engineered with an inducible T-cell co-stimulator (ICOS) domain (71). In recent years, it has become clear that Th17 cells display persistence, self-renewal potential, and the ability to drive potent antitumor responses (Figure 2) (65).
Treg cells exert an immunosuppressive function and play a key role in maintaining immune homeostasis. They prevent unwanted immune reactions, such as autoimmunity and allergies. Usually, they express CD95+ and CD127low (34). However, their presence in tumors is related to disease progression as they inhibit the antitumor immune response (Figure 2) (72).
The Treg/TE cell ratio is a key marker of the efficacy of immunotherapy (71). Infiltrating CD4+ Treg cells in solid tumors decrease the efficacy of CAR-T cell therapy (73). Deletion of the Lck-binding region within the CD28 endodomain, which is linked to IL-2 production, reverses Treg cell-induced tumor infiltration and enhances the antitumor activity of CAR-T cells (73).
Given the diversity of CD4+ T-cell subpopulations and the cytokines they secrete, it is essential to characterize them in the final product of the CAR-T cell manufacturing process. This way, the conditions necessary to enrich less differentiated T cells in the final CAR-T cell product can be defined to improve their antitumor efficacy in vivo (74).
Strategies to Improve the Persistence of CAR-T Cells
CAR Architecture
Since the introduction of ACT with CAR-T cells, the clinical outcomes have been hindered by the poor persistence of the engineered T cells; therefore, several CAR-T cell generations have been developed to improve cell persistence and functionality (75). The first generation of CAR-T cells had the simplest architecture, i.e., an extracellular single-chain variable fragment (scFv) specific for a cancer marker; hinge and transmembrane regions usually derived from the CH2–CH3 region of IgG1, IgG4, or CD8; and the cytoplasmic CD3ζ signaling domain (Figure 3A) (76, 77). Signaling through the CD3ζ domain did not suffice to prime resting T cells, and the first CAR-T cell generation could achieve neither sustained response nor cytokine release due to this limited signaling ability (78–80).
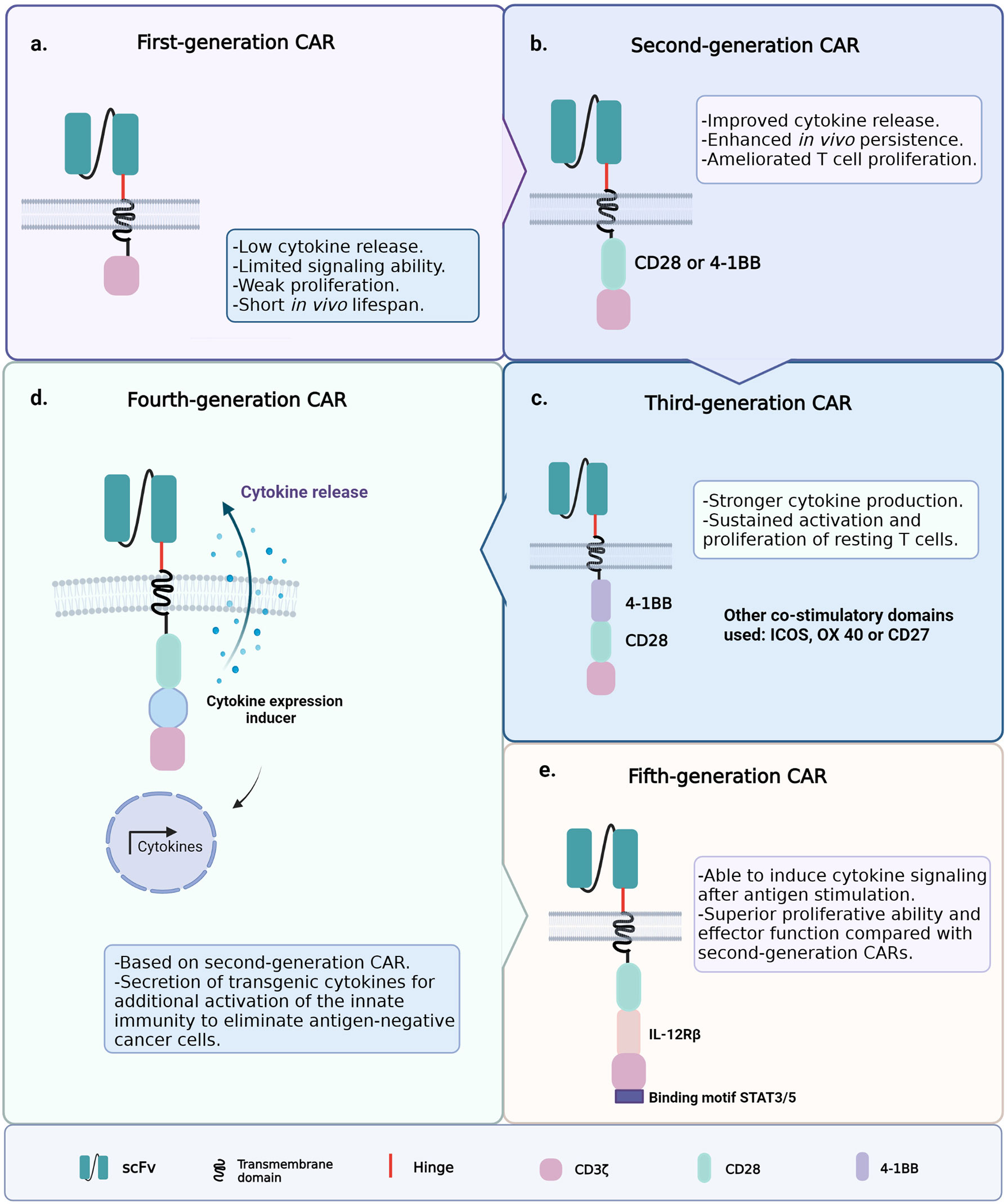
Figure 3 Generations of chimeric antigen receptor T-cell (CAR-T cell) construct designs. (A) First-generation chimeric antigen receptors (CARs) were composed of a single-chain variable fragment (scFv) specific for a cancer marker, hinge and transmembrane domains, and the cytoplasmic CD3ζ signaling domain. (B) Second-generation CARs included the coupling of a co-stimulatory signaling domain. (C) Third-generation CARs incorporated a second co-stimulatory signaling domain. (D) Fourth-generation CARs were based on the structure of the second-generation CARs, plus an inducible gene expression cassette encoding a transgenic cytokine. (E) Fifth-generation CARs contain an IL-2 receptor β-chain domain and a binding site for STAT3.
To improve T-cell signaling, the second generation of CAR-T cells included the coupling of a co-stimulatory signaling domain (e.g., CD28 or 4-1BB) that improves activation, enhances survival, and promotes the efficient expansion of the modified T cells (Figure 3B) (81, 82). Other common T-cell co-stimulatory molecules such as ICOS, CD27, and OX40 have been studied (83, 84). The currently approved FDA therapies Kymriah and Yescarta belong to this second-generation CAR-T cells. The experience gained from the application of second-generation CAR-T cells highlighted the relevance of the co-stimulatory molecule on the function and fate of the engineered cells within the TME (85). The addition of the 4-1BB (CD137) domain to CAR constructs promoted the induction of CD8+ T cells with increased respiratory capacity and heightened mitochondrial biogenesis, two characteristics of the least differentiated memory T cells (24, 25, 86). On the other hand, the inclusion of the CD28 co-stimulatory domain induced the expansion of TEM lymphocytes with a gene signature of glycolytic metabolism (87, 88). Consistent with the above, chimeric antigen receptors (CARs) containing CD28ζ or 4-1BBζ are more likely to activate genes associated with the TE or TM phenotype, respectively (89). This is why CD28ζ-containing CAR-T cells persist about 30 days, while those with 4-1BBζ are found even 4 years after the ACT in some patients (88).
Third-generation CARs incorporated a second co-stimulatory signaling domain to achieve greater functional potency (Figure 3C) (90, 91). For example, the addition of the CD28 and OX40 domains to a CD3ζ chain leads to the sustained activation, proliferation, and effector function of resting T cells through the NFκB signaling pathway (85). Furthermore, ICOS-dependent signaling in CAR-T cells has been shown to result in an enhanced cell survival following the ACT. This evidence highlights the importance of testing novel CAR-T cell constructs to counter solid tumors and non-lymphoid hematologic malignancies; approaches to enhance CAR-T cell persistence remain an unmet medical need to date (71, 92). The design of fourth-generation CARs, known as T cells redirected for universal cytokine killing (TRUCKs), was based on the structure of the second-generation CARs. They contain an inducible gene expression cassette coding for a transgenic cytokine, such as IL-12 (IL-8, IL-9, IL-15, and IL-18 are still under investigation), to be delivered into the targeted tissue (Figure 3D) (91, 93). The accumulation of IL-12 can effectively recruit innate immune cells to the TME and attack antigen-negative cancer cells that CAR-T cells cannot recognize (94, 95). Recently, fifth-generation CARs have been studied and engineered based on the second-generation CARs. They contain an IL-2 receptor β-chain domain and include a binding site for STAT3 (Figure 3E) (96). This CAR construct induces a robust cytokine secretion through the activation of the JAK/STAT signaling pathway in the targeted tumor after antigen stimulation (91).
It appears that each part of the CAR construct can influence the persistence and phenotype of CAR-T cells. For instance, the CD3ζ chain generally employed as a signaling domain on CAR constructs contains three immunoreceptor tyrosine-based activation motifs (ITAMs) (97). An ITAM domain consists of two consecutive YxxL/I motifs separated by a defined number of amino acids (YxxL/I-X6−8-YxxL/I) (98). TCR binding to the peptide–MHC complex leads to the activation of the Src family kinase Lck, which phosphorylates two tyrosine residues in each of the CD3ζ ITAMs (97, 99). CD3ζ ITAMs have different roles in the regulation of T-cell activation; for example, mutations of CD3ζ ITAM1 and ITAM2 significantly impaired signal transduction and induced cell death. However, mutation of CD3ζ ITAM3 did not induce cell death, but rather increased IL-2 secretion and MAPK phosphorylation (99, 100). CD28ζ-based CAR-T cells that only contain the ITAM1 domain resulted in higher percentages of TSCM and TCM and a lower fraction of TEFF cells and yielded long-lasting and complete tumor remission in an in vivo animal model (Figure 4) (101). The CD28 cytoplasmic domain contains a YMNM motif that gets phosphorylated upon binding to CD80/CD86 ligands, which can bind to Grb2/Gads through the asparagine residue (102). Besides, the CD28 proline-rich regions can interact with Itk, Tec, Lck, Grb2/Vav, and filamin A (FLNA) (99, 103). CAR-T cells bearing a mutated CD28 domain (CD28-YMFM) promote a long-lasting antitumor control (Figure 4). Besides, CD28-YMFM CAR-T cells exhibit reduced differentiation and exhaustion and increased skewing toward the Th17 profile (104).
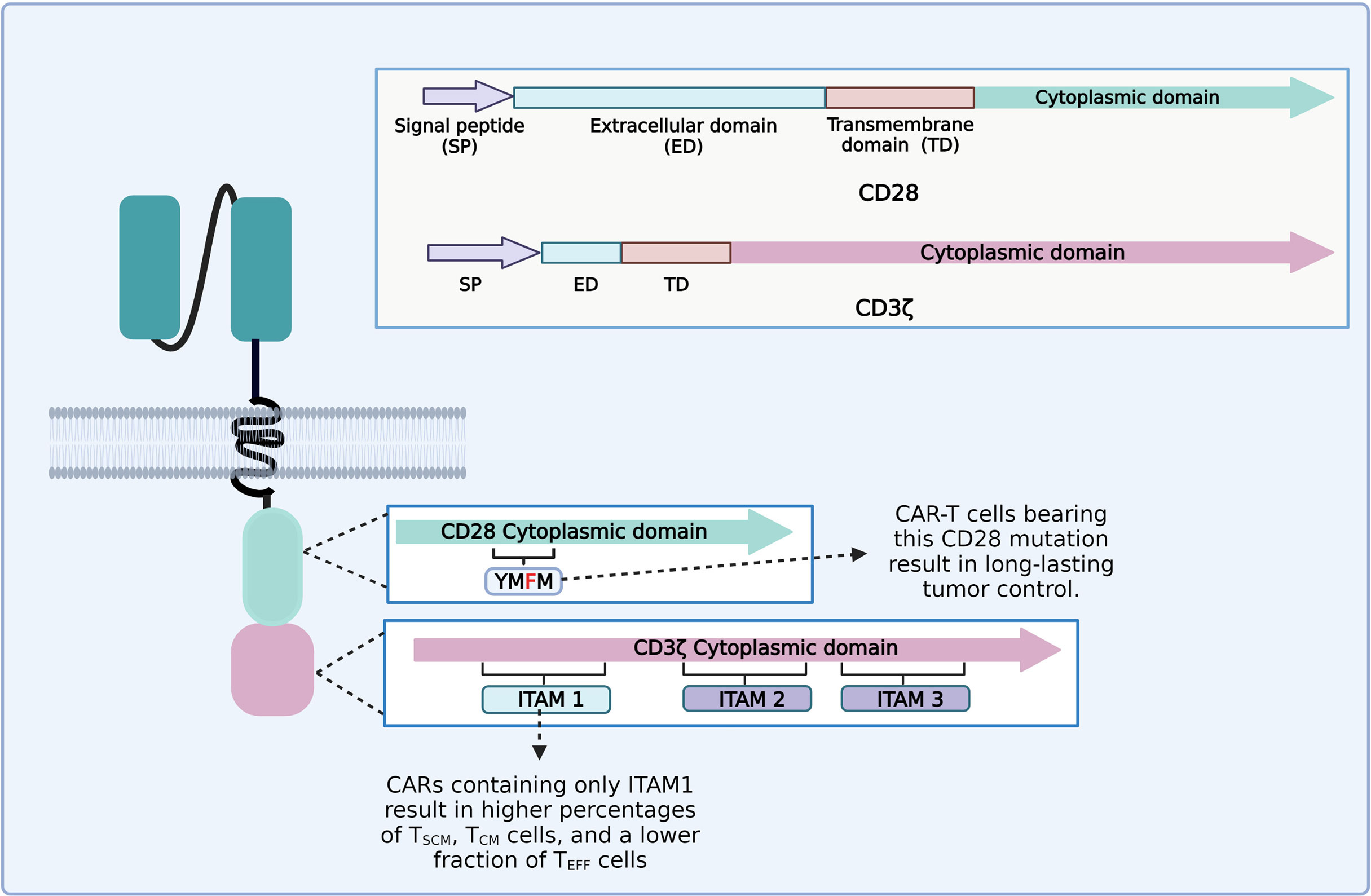
Figure 4 Diagram of the modifications on chimeric antigen receptor (CAR) cytoplasmic domains leading to enhanced tumor control and enrichment of less differentiated T-cell subsets related to better clinical outcomes.
Several studies have shown that the extracellular spacer module (the hinge) significantly impacts the performance of CAR-T cell. Tumor membrane-proximal epitopes are best accessible to CARs with long spacers, while CARs with short spacers exhibit the highest activity against distal epitopes (105–107). These facts support the hypothesis that an optimal distance between T cells and target cells is required for CAR-T cells to be able to trigger an effective immune response. Therefore, a spacer is not required when the epitope is far from the T cells and, conversely, when the tumor epitope is very close to the membrane; the lack of a spacer region might result in a length inadequate for optimal T-cell activity (105). Recent advances have revealed that the TCR acts as a mechanoreceptor. The difference in the dimensions of the ligated peptide MHC (pMHC)/TCR complexes (~15 nm) and the surrounding molecules provides tensive forces that transiently bend the membrane around TCR microclusters. Conformational changes induced by the initial TCR signaling may thus act as a molecular spring to provide defined forces on the engaged TCRs, and such forces might be an amplifying source for T-cell activation (108). Thus, the intercellular space length between T cells and antigen-presenting cells (APCs) that leads to the best activation is equivalent to ~4 Ig-like domains (~15 nm) (108, 109). However, high-affinity TCR ligands can effectively induce TCR triggering even at large interspatial distances between T cells and APCs (109). There is evidence that the CD22-specific chimeric TCR signal strength and Ag sensitivity can be modulated by selecting target epitopes according to their distances from the cell membrane, allowing discrimination between targets with disparate Ag density (107).
Modified CARs have been developed to reduce the binding to soluble Fc gamma receptors (FcγRs); for instance, the CD19R(EQ) contains IgG4-Fc spacers carrying two mutations within the CH2 region (L235E and N297Q), and the CD19Rch2Δ incorporates a CH2 deletion. These CAR-T cells exhibit improved persistence and more potent CD19-specific anti-lymphoma efficacy in NSG mice (110). A novel class of CAR spacers with similar attributes to IgG spacers, but without unspecific off-target binding derived from the sialic acid-binding immunoglobulin-type lectins (Siglecs), was used to build a CAR directed against a membrane-proximal (TSPAN8) epitope of a pancreatic ductal adenocarcinoma model. In vivo settings using these CAR-T cells led to the generation of advantageous TCM cells that released low levels of inflammatory cytokines and retained excellent tumor-killing functions (111).
Another important strategy consists in modulating the affinity to the scFv. A novel CD19 CAR (CAT), with a lower affinity than FMC63, showed higher in vitro proliferation and cytotoxicity and greater in vivo proliferative and antitumor activity compared with FMC63 CAR-T cells (112). In this sense, there is a minimal TCR affinity needed for T-cell activation and, additionally, a plateau to achieve maximal T-cell activity, revealing a TCR affinity threshold (113). Indeed, at low-density peptide ligands, the response is expected to be dominated by the low TCR-to-peptide interaction capable of using serial triggering to achieve the threshold required for T-cell activation. In contrast, high-affinity TCR-to-peptide interactions cannot achieve the activation threshold (114).
An additional strategy for optimizing the transferred construct includes the expression of non-coding RNAs. For instance, recent studies have shown that CAR-T cells engineered to express and deliver the RN7SL1 promote the expansion and effector memory differentiation of CAR-T cells characterized by high persistence and less exhaustion (115–118).
New concepts derived from the synthetic biology field for developing novel approaches in cell therapy are becoming appealing, such as the design of engineered cells harboring synthetic gene circuits able to biologically sense and compute signals derived from intracellular or extracellular biomarkers (119). These biological devices could ultimately be integrated into increasingly complex systems (119). The possibility of engineering T cells with synthetic systems responding to multiple inputs would benefit ACT with CAR-T cells and will probably open the door to the next generation of smarter self-decision-making CAR-T cells (120). For example, a generation of CAR-T cells that are only effective locally might also increase the choice of tumor targetable antigens. In this sense, incorporating the oxygen-sensitive domain (HIF-1a) could generate a CAR construct with gene expression induced by a low oxygen concentration, a characteristic of the TME (120).
Enhance Expansion/Persistence by Vaccination
A multicenter phase I/II study of donor CD19-directed CAR-transduced Epstein–Barr virus (EBV)-specific cytotoxic T lymphocytes (CTLs) in pediatric patients with acute ALL showed that the use of donor EBV-specific CTLs to manufacture CD19CAR could enhance the CAR-T cell expansion/persistence after vaccination with EBV-specific peptides (121). A different approach to enhancing the CAR-T cell function against solid tumors is by directly boosting donor cells with a vaccine that interacts with the chimeric receptor in vivo. The idea is to attach a small target molecule (peptide or protein–ligand) of a CAR into the membrane of the APCs of lymph nodes using the amphiphile ligand (amph-ligand). In this way, the target epitope is displayed on the APC surface together with a native cytokine receptor. The amph-ligand strategy has safely expanded CAR-T cells in vivo, increased their functionality, and enhanced their antitumor activity in multiple models of solid tumors (122). A new approach uses a nanoparticulate RNA vaccine designed for wide delivery of the CAR antigen into lymphoid compartments throughout the body. This vaccine stimulates the presentation of the natively folded target of the CAR-T cells on resident dendritic cells to promote the cognate and selective expansion of CAR-T cells. This strategy improves the engraftment of CAR-T cells and the regression of large tumors in mouse models (123).
The upgrading of the previously described approaches requires more studies to evaluate the usefulness of combined CAR designs, including different generations of CARs and advanced vaccination strategies. In addition, to further improve these strategies, the CAR expression could be placed under the control of the TRAC locus, which has been shown to avert the tonic CAR signaling and delay the effector T-cell differentiation and exhaustion (122, 124).
CAR-T Cell Therapy Combined With Oncolytic Viruses
Several preclinical studies have shown that oncolytic viruses (OVs) can synergize with CAR-T cells to overcome the multiple challenges that CAR-T cell therapy encounters in solid tumors by increasing CAR-T cell trafficking within the tumor and enhancing antitumor activity, as well as eliminating antigen-negative cancer cells (125, 126). In 2014, Nishio et al. armed an oncolytic adenovirus (Ad5Δ24) with the chemokines RANTES (regulated upon activation, normal T cells expressed and secreted) and IL-15 to enhance the trafficking and survival of a third generation of anti-GD2 CAR-T cells on a neuroblastoma xenograft model. They observed that intratumoral administration of this OV led to the improved persistence and migration of the infused CAR-T cells (127). Furthermore, the use of OV-IL15C, an oncolytic virus expressing the IL15/IL15Ra complex, enhanced the persistence of EGFR-CAR T cells that elicited strong antitumor responses in glioblastoma and improved this therapy in an immunocompetent mouse model (128). Another example of this strategy is T-SIGn, an oncolytic viral vector encoding IFNα, MIP1α, and CD80 that acts in synergy with anti-EGFR CAR-T cells. Tumor lysis induced by T-SIGn releases neoantigens and upregulates the antigen processing and presentation machinery to promote epitope spreading. Therefore, the virus reprograms the immunosuppressive TME into a pro-inflammatory one to attract and activate CAR-T cells and innate antigen-presenting cells, amplifying the antitumor response. This strategy was intravenously administered to eliminate pulmonary metastases in a murine model (129).
Despite the promising results obtained in murine models, this approach must be evaluated. Currently, only one ongoing clinical trial (NCT03740256) is being conducted in patients with human epidermal growth factor receptor 2 (HER2)-positive cancer. This study aimed to evaluate the safety and efficacy of anti-HER2 CAR-T cells combined with intratumoral injection of CAdVEC (an oncolytic adenovirus designed to enhance the antitumor immune response). This trial is on recruitment status and there are no results yet (130).
Cell Culture Optimization
The culture medium used for the expansion phase of CAR-T cells impacts the cell performance in vivo (33). Components of the culture medium can influence not only the gene delivery but also the differentiation, proliferation, and potency of CAR-T cells (131, 132).
Usually, the culture medium is supplemented with serum of animal or human origin. Fetal bovine serum (FBS) is often used in research in a broad range of cell cultures as a source of nutrients and growth factors. However, it has several issues: firstly, FBS does not simulate the human microenvironment, which limits its translational application (131); secondly, it involves the risk of transmitting bovine spongiform encephalopathy and some viral infections; thirdly, it can promote the development of unwanted immunological reactions; and, finally, variations between the brands and batches of sera can affect the reproducibility of the experiments (131, 133, 134). Human serum (HS) does not contain any xenogeneic components and supplements the medium with trophogens and additional stimuli that favor cell growth and survival. However, the serum can inhibit cell growth (at high concentrations), is expensive, and there is a marked variability between different batches (131). Medvec et al. observed that expanding T cells in a medium without human serum improved their functionality and persistence (135).
Since many of the metabolites and growth factors required for cell proliferation originate in cells such as erythrocytes, platelets, and endothelial cells, some researchers have studied whether extracts derived from these cells, obtained from whole blood fractions, can support the differentiation and proliferation of T cells (131, 133). For example, blood platelets contain strong mitogens such as growth factors, chemokines, and cytokines (136). Recent studies have provided support that the human platelet lysate (HPL) allows the expansion of CAR-T cells and increases the percentage of TCM cells in the final product compared to those obtained in media supplemented with FBS or HS (136). This observation suggests that HPL-supplemented media for culturing CAR-T cell improves the cell functionality in vivo and enriches the TCM cell subset associated with increased cell persistence in patients following ACT (136). The impact of HPL-exposed CAR-T cells was evaluated in vivo in a mouse xenograft model. The cell proliferation and antitumor effects were more significant compared to those of CAR-T cells cultured in media supplemented with FBS or HS (137).
Ghassemi et al. found that, among other alternatives, the serum used for CAR-T cell expansion culture can be substituted by Physiologix™ (Phx), an extract of growth factors obtained from whole blood. Compared to CAR-T cells expanded with HS-supplemented media, CAR-T cells cultured in Phx-containing media displayed increased transduction efficiency, as evidenced by their in vitro cytotoxic activity and superior in vivo cell survival ability in neuroblastoma models. Additional metabolomic analyses of the composition of Phx showed a modest enrichment in carnosine, a dipeptide composed of the isomers β-alanine and L-histidine. Carnosine is a critical factor that improves the CAR transduction efficiency in activated T cells; it can also decrease the media acidification and induce a glycolytic-to-oxidative metabolic change, a characteristic related to better antitumor effects (131). Smith et al. demonstrated that xeno-free CTS™ Immune Cell Serum Replacement allows the efficient expansion of gene-modified T cells with similar yields to those generated when FBS or HS was used as a supplement (138). Moreover, as an alternative to the expensive serum-free specific culture media, acellular Wharton’s jelly can be utilized as a supportive substance; furthermore, it increases the memory properties of T cells (139).
Other approaches that have been studied include reducing the length of the cell expansion phase and the effect of RetroNectin on T cell culture. The protocols for T-cell engineering routinely expand T cells ex vivo for 9–14 days. However, Ghassemi et al., in 2018, reported that CAR-T cells targeting CD19 (CART19) expanded for 3–5 days proliferated more and showed greater cytotoxic ability in vitro, as well as in a murine xenograft model of ALL, showing that the antileukemic activity inversely correlated with the ex vivo culture time. In addition, these cells persisted longer and showed more robust antitumor activity in a murine model (140). A recently published work has shown that quickly generated (24-h expansion) non-activated CAR-T cells exhibited higher in vivo antileukemic activity per cell than the activated CAR-T cells produced using the standard protocol. The former protocol for the rapid manufacturing of CAR-T cells may reduce the production costs and broaden their applicability. However, immunosuppressive factors in the TME may hinder the ability to generate functional CAR-T cells using this approach (141). The facts previously described must be considered relevant in the microenvironment of solid tumors.
On the other hand, RetroNectin (a recombinant human fibronectin fragment containing the VLA-4 and VLA-5 binding domains) is generally used to enhance the transduction efficiency given its ability to co-localize viral vectors and cells of interest, such as hematopoietic progenitor cells and T lymphocytes (142, 143). When RetroNectin was used in conjunction with anti-CD3 monoclonal antibodies (mAbs), it also enhanced T-cell expansion while preserving the CD45RAhigh CCR7high phenotype, characteristic of TN and TCM cells (144–147). RetroNectin influences the CD4+/CD8+ composition of T-cell products by inhibiting the apoptosis of CD8+ T cells and shifting the cell composition toward a cytolytic phenotype over in vitro culture (144) and during their in vivo persistence (148).
Cytokines Used to Yield Undifferentiated CAR-T Cells
Modulation of the interleukin cocktails can affect the memory functions of T cells and are used as an alternative approach to increase the efficacy of CAR-T cell therapy (144, 149–152). Cytokines are biologically active peptides that act by binding to their specific receptors located on cell surfaces (153). Interleukins are a subgroup of cytokines that allow communication between cells of the immune system; they determine processes such as the activation, differentiation, maintenance, function, and proliferation of immune cells (154). The interleukins chosen for the manufacturing process influences the T-cell proliferation and differentiation in CAR-T cell cultures (144). The most used and studied interleukin is IL-2, which has an essential role in the manufacturing process of CAR-T cells since it stimulates cell proliferation and maintains cell viability during the expansion phase (153). However, the stimulation of T cells with IL-2 favors the differentiation of short-life-span and exhausted cells because IL-2 induces a switch to glycolysis, a feature of TE cells (33, 155).
The substitution of IL-2 with other γ-chain cytokines such as IL-7, IL-15, and IL-21 plays a crucial role in the functionality, homeostasis, differentiation, and expansion of T cells and allows obtaining more significant proportions of less differentiated lymphocytes (156). There is evidence that the culture of CAR-T cells in the presence of IL-15 reduces the activity of mTORC1 and conserves the stem cell memory phenotype that has higher antitumor activity and proliferative ability (33, 155). Some studies have shown that, during the expansion phase of CD28-based CD19 CAR-T cells, a mixture of IL-7 + IL-15 increased the number and proportion of a T-cell subpopulation with TSCM- and TCM-like phenotypes. Moreover, these CAR-T cells showed higher expansion and effector function abilities and more significant migration to secondary lymphoid organs, leading to longer cell persistence and antitumor activity in vivo (149, 157, 158).
Moreover, IL-21 is another member of the γ-chain cytokine family that has shown favorable effects on the T-cell expansion process. Loschinski et al. found that exposure of T cells to IL-21 reduced the glycolytic activity and increased the fatty acid oxidation (FAO), a pathway essential for TCM generation (156). Furthermore, some studies have reported that a mixture of IL-21 + IL-4 + IL-7 added to the culture media maintained the memory phenotype and reduced the expressions of inhibitory receptors including PD-1 and TIM3 in CAR-T cells (159). The combination of IL-12 + IL-7 or IL-21 in the ex vivo cell expansion process yielded CD8+ T cells with enhanced persistence in a NOD/SCID/γc−/− mouse model (150). IL-12 is a non-γ-chain cytokine important in regulating T-cell differentiation and memory generation (160).
Metabolic Reprogramming of CAR-T Cells
The metabolic requirements of T cells depend on their degree of activation, differentiation, and functionality. For example, cells in a quiescent state, such as TN cells, rely on a catabolic metabolism of low-energy consumption, which uses the oxidation of fatty acids, amino acids, and glucose as energy sources, mainly through oxidative phosphorylation pathways (161). However, upon cell activation, the metabolism becomes highly glycolytic to generate the intermediate biomolecules required for cell proliferation. Specific metabolic and epigenetic changes must occur in cells in order to proliferate and differentiate. As can be seen, metabolism is intimately linked to cell activation, proliferation, migration, and function, and therefore to the very fate of T cells (85, 161, 162).
As for the metabolic conditioning of CAR-T cells, the CAR design itself can define their metabolism and functionality. In this sense, it is known that the CD28 co-stimulating domain increases the glycolytic metabolism and differentiation of T cells toward the TEM cell subpopulations. In contrast, the 4-1BB co-stimulating domain increases oxidative metabolism and mitochondrial biogenesis, promoting differentiation to the TCM phenotype, characterized by improved cell proliferation and persistence (88, 161). Hence, it can be concluded that promoting a low metabolic activity over the CAR-T cell manufacturing process could favor the production of T cells with a less differentiated phenotype, which would have greater longevity and antitumor potential. Conversely, a high metabolic activity could favor an effector lymphocyte enrichment; therefore, following infusion into patients, these cells would be quickly depleted, resulting in reduced antitumor activity (162).
Strategies to Target the T-Cell Metabolism
While studying the metabolome and proteome of activated CD4+ TN cells, Geiger et al., in 2016, discovered that increasing the levels of L-arginine in the culture medium induced a metabolic shift from glycolysis to oxidative phosphorylation. This change promoted T-cell differentiation toward a TCM-like phenotype characterized by the expressions of CCR7 and CD62L (163).
Another approach is inhibiting the glycolytic metabolism using drugs or expanding cells in a medium with low glucose concentrations; for example, in 2013, Sukumar et al. used 2-deoxy-D-glucose, a hexokinase 2 inhibitor, to limit the glycolytic metabolism of CD8+ T lymphocytes. This strategy increased the development of TM cells. Additionally, the researchers found that inhibition of the glycolytic pathway was associated with the expression of transcription factors that drive cell differentiation into the memory phenotype (164). The PI3K/AKT/mTor pathway is essential to regulate T-cell differentiation and memory generation. Its activation promotes the expression of the GLUT1 gene, a glucose transporter, which promotes glycolytic metabolism by increasing glucose intake from the medium. Consequently, it facilitates the differentiation of T cells into an effector subset (162). The effect of this pathway inhibition on CAR-T cells has been studied. In 2018, Perkins et al. expanded BCMA-directed CAR-T cells together with a PI3K inhibitor to investigate its activity in vivo. The adoptive transfer of these CAR-T cells resulted in complete long-term tumor regression in animal models of Burkitt lymphoma and multiple myeloma. The animals were even immune to a re-challenge with tumor cells. In addition, the phenotypic analysis of these BCMA-directed CAR-T cells showed a high frequency of CD8+CD62L+ T cells. These results suggest that PI3K inhibition during ex vivo cell expansion generates a product with better antitumor efficacy in vivo (165). Similarly, in 2019, Zhang et al. demonstrated that the culture of epithelial cell adhesion molecule (EpCAM)-directed CAR-T cells with the AKT inhibitor MK2206 did not affect cell proliferation or viability. However, the AKT inhibitor prevented the terminal differentiation of CAR-T cells. These cells exhibited higher expansion and antitumor efficacy in an animal model of colon cancer. Also noteworthy was the finding that AKT inhibition increased the CAR rate expression (166).
Moreover, there is evidence that crosstalk between the Wnt pathway and IL-12 signaling inhibits the T-bet and mTOR pathways and impairs memory programming, which can be recovered in part by rapamycin (167). Furthermore, mTOR inhibition in activated TN cells using a high concentration of rapamycin or TWS119 (an activator of the Wnt-β/catenin pathway) induced the generation of a TSCM phenotype. The inhibition of the pathway induced a switch in the metabolism of T cells characterized by an increase in FAO. The TSCM subpopulation exhibited superior functional characteristics and a more remarkable repopulation ability after adoptive transfer (168).
In 2016, the study of Bengsch et al. revealed that the peroxisome proliferator-activated receptor-gamma co-activator 1-α (PGC-1α) is a central regulator of oxidative phosphorylation. Moreover, when PGC-1α was overexpressed in TEX cells, it corrected the dysregulated mitochondrial function, improving metabolic fitness and effector function (169). More recently, Dumauthioz et al. enhanced the mitochondrial biogenesis in CD8+ T cells by overexpressing PGC-1α and observed that this strategy improved the antitumor effect by promoting the generation of CD8+ memory T cells (170).
Furthermore, some reports have demonstrated that PD-1 blocking in CAR-T cells improved tumor control and overall cell survival. Moreover, the PD-1 inhibition in T cells led to a metabolic switch from glycolysis toward an increased FAO; these cells exhibited enhanced survival and similarities to memory T cells (171).
Conclusion
Cancer-specific chimeric antigen receptor (CAR) T cells have emerged as one of the most promising immunotherapies to target various types of cancer. However, many barriers must still be overcome to generate highly successful clinical outcomes. One of these unmet needs relates to the persistence of CAR-T cells, and multiple strategies focused on CAR’s architecture, cellular metabolism, T-cell phenotype, vaccination boost, and cell culture optimization are under development to improve it (Figure 5).
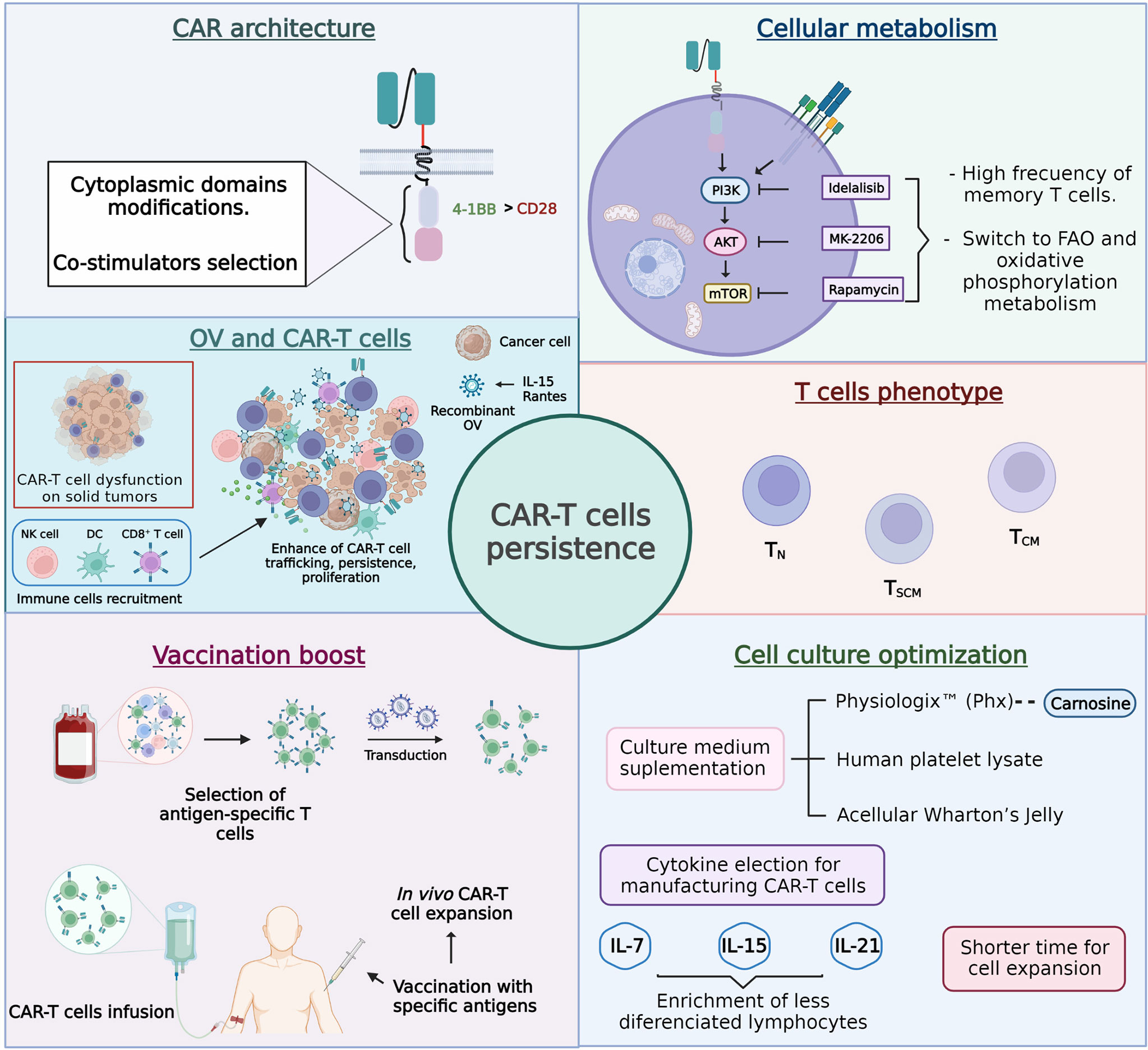
Figure 5 Approaches designed to improve chimeric antigen receptor T cell (CAR-T cell) persistence.
In agreement with the described facts, a plausible strategy to extend the in vivo persistence of CAR-T cells could require combinatory approaches, mainly when the therapy is directed against solid tumors because their hostile microenvironment induces the dysfunction of T cells, making the cell persistence requirements much higher. In this sense, understanding the underlying knowledge supporting each approach under study is extremely relevant. This way, rational combinatory strategies pursuing synergistic antitumor effects in vivo could be better chosen. Finally, it is essential to keep in mind that most of the reviewed approaches come from NSG mice research. Although this is the acknowledged model to assess cellular therapeutic efficacy, some factors cannot be accurately predicted, such as the interactions of CAR-T cells with the TME that could directly affect the persistence of CAR-T cells.
In this review, we summarized some of the approaches developed to circumvent the CAR-T cell short persistence barrier and offered ideas to tackle this hurdle to those researchers who have begun to work on CAR-T cell production.
Author Contributions
GLC and CRS wrote and edited the manuscript. All authors reviewed, made an intellectual contribution, and approved the submitted version.
Funding
GLC received financial support from the project entitled “Investigación Orientada a la Implementación de Buenas Prácticas para la Aplicación Clínica de Terapias Celulares. Modelo: tph en Bogotá” (Research Oriented to the Implementation of Good Practices for the Clinical Application of Cellular Therapies. Model Tph in Bogotá) (code BPIN2016000100035; agreement 0182 of 2018), signed with the District Health Financial Fund. CU was financed by resources of the “Colombia Científica” program of the Ministry of Science and Technology allocated to the Pontificia Universidad Javeriana. “Ecosistema Científico” call (contract no. FP44842-221-2018) and DreemBio. BAC was funded by the IDCBIS institute. CRS was financed with resources transferred from the District Health Financial Fund (FFDS, by its abbreviation in Spanish) to IDCBIS, according to Resolution 515 of April 12, 2021, issued by the District Health Secretariat.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
The authors thank the Science, Technology, and Innovation Funding (code BPIN2016000100035), from the Royalties General System (SGR, by its abbreviation in Spanish) of the National Planning Department of Colombia, and Dr. Martha Mesa for the English language review and correction. Figures were created with BioRender (BioRender.com), whose license we own.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2022.878209/full#supplementary-material
References
1. Liu Y, Chen X, Han W, Zhang Y. Tisagenlecleucel, an Approved Anti-CD19 Chimeric Antigen Receptor T-Cell Therapy for the Treatment of Leukemia. Drugs Today (2017) 53:597. doi: 10.1358/dot.2017.53.11.2725754
2. O’Leary MC, Lu X, Huang Y, Lin X, Mahmood I, Przepiorka D, et al. FDA Approval Summary: Tisagenlecleucel for Treatment of Patients With Relapsed or Refractory B-Cell Precursor Acute Lymphoblastic Leukemia. Clin Cancer Res (2019) 25:1142–6. doi: 10.1158/1078-0432.CCR-18-2035
3. Maude SL, Laetsch TW, Buechner J, Rives S, Boyer M, Bittencourt H, et al. Tisagenlecleucel in Children and Young Adults With B-Cell Lymphoblastic Leukemia. N Engl J Med (2018) 378(5):439–48. doi: 10.1056/NEJMoa1709866
4. Neelapu SS, Locke FL, Bartlett NL, Lekakis LJ, Miklos DB, Jacobson CA, et al. Axicabtagene Ciloleucel CAR T-Cell Therapy in Refractory Large B-Cell Lymphoma. N Engl J Med (2017) 377:2531–44. doi: 10.1056/NEJMoa1707447
5. Bouchkouj N, Kasamon YL, de Claro RA, George B, Lin X, Lee S, et al. FDA Approval Summary: Axicabtagene Ciloleucel for Relapsed or Refractory Large B-Cell Lymphoma. Clin Cancer Res (2019) 25:1702–8. doi: 10.1158/1078-0432.CCR-18-2743
6. Reagan PM, Friedberg JW. Axicabtagene Ciloleucel and Brexucabtagene Autoleucel in Relapsed and Refractory Diffuse Large B-Cell and Mantle Cell Lymphomas. Future Oncol (2021) 17:1269–83. doi: 10.2217/fon-2020-0291
7. Mullard A. FDA Approves Fourth CAR-T Cell Therapy. Nat Rev Drug Discov (2021) 20:166. doi: 10.1038/d41573-021-00031-9
8. Abramson JS, Palomba ML, Gordon LI, Lunning MA, Wang M, Arnason J, et al. Lisocabtagene Maraleucel for Patients With Relapsed or Refractory Large B-Cell Lymphomas (TRANSCEND NHL 001): A Multicentre Seamless Design Study. Lancet (2020) 396:839–52. doi: 10.1016/S0140-6736(20)31366-0
9. First CAR-T Therapy to Target BCMA Gets FDA Nod. Nat Biotechnol (2021) 39:531–1. doi: 10.1038/s41587-021-00929-0
10. Munshi NC, Anderson LD, Shah N, Madduri D, Berdeja J, Lonial S, et al. Idecabtagene Vicleucel in Relapsed and Refractory Multiple Myeloma. N Engl J Med (2021) 384:705–16. doi: 10.1056/NEJMoa2024850
11. Porter DL, Hwang W-T, Frey NV, Lacey SF, Shaw PA, Loren AW, et al. Chimeric Antigen Receptor T Cells Persist and Induce Sustained Remissions in Relapsed Refractory Chronic Lymphocytic Leukemia. Sci Transl Med (2015) 7:303ra139. doi: 10.1126/scitranslmed.aac5415
12. Fan J, Das JK, Xiong X, Chen H, Song J. Development of CAR-T Cell Persistence in Adoptive Immunotherapy of Solid Tumors. Front Oncol (2021) 10:574860. doi: 10.3389/fonc.2020.574860
13. Maude SL, Frey N, Shaw PA, Aplenc R, Barrett DM, Bunin NJ, et al. Chimeric Antigen Receptor T Cells for Sustained Remissions in Leukemia. N Engl J Med (2014) 371:1507–17. doi: 10.1056/NEJMoa1407222
14. Kershaw MH, Westwood JA, Parker LL, Wang G, Eshhar Z, Mavroukakis SA, et al. A Phase I Study on Adoptive Immunotherapy Using Gene-Modified T Cells for Ovarian Cancer. Clin Cancer Res (2006) 12:6106–15. doi: 10.1158/1078-0432.CCR-06-1183
15. Hsieh EM, Scherer LD, Rouce RH. Replacing CAR-T Cell Resistance With Persistence by Changing a Single Residue. J Clin Invest (2020) 130:2806–8. doi: 10.1172/JCI136872
16. Hay KA, Gauthier J, Hirayama AV, Voutsinas JM, Wu Q, Li D, et al. Factors Associated With Durable EFS in Adult B-Cell ALL Patients Achieving MRD-Negative CR After CD19 CAR T-Cell Therapy. Blood (2019) 133:1652–63. doi: 10.1182/blood-2018-11-883710
17. Louis CU, Savoldo B, Dotti G, Pule M, Yvon E, Myers GD, et al. Antitumor Activity and Long-Term Fate of Chimeric Antigen Receptor–Positive T Cells in Patients With Neuroblastoma. Blood (2011) 118:6050–6. doi: 10.1182/blood-2011-05-354449
18. Fraietta JA, Lacey SF, Orlando EJ, Pruteanu-Malinici I, Gohil M, Lundh S, et al. Determinants of Response and Resistance to CD19 Chimeric Antigen Receptor (CAR) T Cell Therapy of Chronic Lymphocytic Leukemia. Nat Med (2018) 24:563–71. doi: 10.1038/s41591-018-0010-1
19. Si Lim SJ, Grupp SA, DiNofia AM. Tisagenlecleucel for Treatment of Children and Young Adults With Relapsed/Refractory B-Cell Acute Lymphoblastic Leukemia. Pediatr Blood Cancer (2021) 68:1–10. doi: 10.1002/pbc.29123
20. Gupta A, Gill S. CAR-T Cell Persistence in the Treatment of Leukemia and Lymphoma. Leukemia Lymphoma (2021) 62(11):1–13. doi: 10.1080/10428194.2021.1913146
21. Gattinoni L. Acquisition of Full Effector Function In Vitro Paradoxically Impairs the In Vivo Antitumor Efficacy of Adoptively Transferred CD8+ T Cells. J Clin Invest (2005) 115:1616–26. doi: 10.1172/JCI24480
22. Hinrichs CS, Borman ZA, Cassard L, Gattinoni L, Spolski R, Yu Z, et al. Adoptively Transferred Effector Cells Derived From Naive Rather Than Central Memory CD8+ T Cells Mediate Superior Antitumor Immunity. Proc Natl Acad Sci (2009) 106:17469–74. doi: 10.1073/pnas.0907448106
23. Gattinoni L, Lugli E, Ji Y, Pos Z, Paulos CM, Quigley MF, et al. A Human Memory T Cell Subset With Stem Cell–Like Properties. Nat Med (2011) 17:1290–7. doi: 10.1038/nm.2446
24. Gattinoni L, Klebanoff CA, Restifo NP. Paths to Stemness: Building the Ultimate Antitumour T Cell. Nat Rev Cancer (2012) 12:671–84. doi: 10.1038/nrc3322
25. Turtle CJ, Hanafi L-A, Berger C, Gooley TA, Cherian S, Hudecek M, et al. CD19 CAR–T Cells of Defined CD4+:CD8+ Composition in Adult B Cell ALL Patients. J Clin Invest (2016) 126:2123–38. doi: 10.1172/JCI85309
26. Kaartinen T, Luostarinen A, Maliniemi P, Keto J, Arvas M, Belt H, et al. Low Interleukin-2 Concentration Favors Generation of Early Memory T Cells Over Effector Phenotypes During Chimeric Antigen Receptor T-Cell Expansion. Cytotherapy (2017) 19:689–702. doi: 10.1016/j.jcyt.2017.03.067
27. McLellan AD, Ali Hosseini Rad SM. Chimeric Antigen Receptor T Cell Persistence and Memory Cell Formation. Immunol Cell Biol (2019) 97:664–74. doi: 10.1111/imcb.12254
28. Blaeschke F, Stenger D, Kaeuferle T, Willier S, Lotfi R, Kaiser AD, et al. Induction of a Central Memory and Stem Cell Memory Phenotype in Functionally Active CD4+ and CD8+ CAR T Cells Produced in an Automated Good Manufacturing Practice System for the Treatment of CD19+ Acute Lymphoblastic Leukemia. Cancer Immunol Immunother (2018) 67:1053–66. doi: 10.1007/s00262-018-2155-7
29. Sommermeyer D, Hudecek M, Kosasih PL, Gogishvili T, Maloney DG, Turtle CJ, et al, Sommermeyer D, et al. Chimeric Antigen Receptor-Modified T Cells Derived From Defined CD8+ and CD4+ Subsets Confer Superior Antitumor Reactivity. Vivo Leukemia (2016) 30:492–500. doi: 10.1038/leu.2015.247
30. Riddell SR, Sommermeyer D, Berger C, Liu L (Steven), Balakrishnan A, Salter A, et al. Adoptive Therapy With Chimeric Antigen Receptor–Modified T Cells of Defined Subset Composition. Cancer J (2014) 20:141–4. doi: 10.1097/PPO.0000000000000036
31. Busch DH, Fräßle SP, Sommermeyer D, Buchholz VR, Riddell SR. Role of Memory T Cell Subsets for Adoptive Immunotherapy. Semin Immunol (2016) 28:28–34. doi: 10.1016/j.smim.2016.02.001
32. Liadi I, Singh H, Romain G, Rey-Villamizar N, Merouane A, Adolacion JRT, et al. Individual Motile CD4+ T Cells Can Participate in Efficient Multikilling Through Conjugation to Multiple Tumor Cells. Cancer Immunol Res (2015) 3:473–82. doi: 10.1158/2326-6066.CIR-14-0195
33. Chan JD, Lai J, Slaney CY, Kallies A, Beavis PA, Darcy PK. Cellular Networks Controlling T Cell Persistence in Adoptive Cell Therapy. Nat Rev Immunol (2021) 21(12):769–84. doi: 10.1038/s41577-021-00539-6
34. Brummelman J, Pilipow K, Lugli E. The Single-Cell Phenotypic Identity of Human CD8+ and CD4+ T Cells. Int Rev Cell Mol Biol (2018) 341:63–124. doi: 10.1016/bs.ircmb.2018.05.007
35. Henning AN, Roychoudhuri R, Restifo NP. Epigenetic Control of CD8+ T Cell Differentiation. Nat Rev Immunol (2018) 18:340–56. doi: 10.1038/nri.2017.146
36. van den Broek T, Borghans JAM, van Wijk F. The Full Spectrum of Human Naive T Cells. Nat Rev Immunol (2018) 18:363–73. doi: 10.1038/s41577-018-0001-y
37. Tantalo DG, Oliver AJ, von Scheidt B, Harrison AJ, Mueller SN, Kershaw MH, et al. Understanding T Cell Phenotype for the Design of Effective Chimeric Antigen Receptor T Cell Therapies. J Immunother Cancer (2021) 9:e002555. doi: 10.1136/jitc-2021-002555
38. Ando M, Ito M, Srirat T, Kondo T, Yoshimura A. Memory T Cell, Exhaustion, and Tumor Immunity. Immunol Med (2020) 43:1–9. doi: 10.1080/25785826.2019.1698261
39. Frumento G, Verma K, Croft W, White A, Zuo J, Nagy Z, et al. Homeostatic Cytokines Drive Epigenetic Reprogramming of Activated T Cells Into a “Naive-Memory” Phenotype. iScience (2020) 23:100989. doi: 10.1016/j.isci.2020.100989
40. Li M, Yao D, Zeng X, Kasakovski D, Zhang Y, Chen S, et al. Age Related Human T Cell Subset Evolution and Senescence. Immun Ageing (2019) 16:24. doi: 10.1186/s12979-019-0165-8
41. Pulko V, Davies JS, Martinez C, Lanteri MC, Busch MP, Diamond MS, et al. Human Memory T Cells With a Naive Phenotype Accumulate With Aging and Respond to Persistent Viruses. Nat Immunol (2016) 17:966–75. doi: 10.1038/ni.3483
42. Samji T, Khanna KM. Understanding Memory CD8 + T Cells. Immunol Lett (2017) 185:32–9. doi: 10.1016/j.imlet.2017.02.012
43. Martin MD, Badovinac VP. Defining Memory CD8 T Cell. Front Immunol (2018) 9:2692. doi: 10.3389/fimmu.2018.02692
44. Kumar BV, Ma W, Miron M, Granot T, Guyer RS, Carpenter DJ, et al. Human Tissue-Resident Memory T Cells Are Defined by Core Transcriptional and Functional Signatures in Lymphoid and Mucosal Sites. Cell Rep (2017) 20:2921–34. doi: 10.1016/j.celrep.2017.08.078
45. Henson SM, Riddell NE, Akbar AN. Properties of End-Stage Human T Cells Defined by CD45RA Re-Expression. Curr Opin Immunol (2012) 24:476–81. doi: 10.1016/j.coi.2012.04.001
46. Hashimoto M, Kamphorst AO, Im SJ, Kissick HT, Pillai RN, Ramalingam SS, et al. CD8 T Cell Exhaustion in Chronic Infection and Cancer: Opportunities for Interventions. Annu Rev Med (2018) 69:301–18. doi: 10.1146/annurev-med-012017-043208
47. Guo Y, Xie Y-Q, Gao M, Zhao Y, Franco F, Wenes M, et al. Metabolic Reprogramming of Terminally Exhausted CD8+ T Cells by IL-10 Enhances Anti-Tumor Immunity. Nat Immunol (2021) 22:746–56. doi: 10.1038/s41590-021-00940-2
48. Jiang W, He Y, He W, Wu G, Zhou X, Sheng Q, et al. Exhausted Cd8+T Cells in the Tumor Immune Microenvironment: New Pathways to Therapy. Front Immunol (2021) 11:622509. doi: 10.3389/fimmu.2020.622509
49. Golubovskaya V, Wu L. Different Subsets of T Cells, Memory, Effector Functions, and CAR-T Immunotherapy. Cancers (2016) 8:36. doi: 10.3390/cancers8030036
50. Yang Y, Kohler ME, Chien CD, Sauter CT, Jacoby E, Yan C, et al. TCR Engagement Negatively Affects CD8 But Not CD4 CAR T Cell Expansion and Leukemic Clearance. Sci Transl Med (2017) 9:eaag1209. doi: 10.1126/scitranslmed.aag1209
51. Wang D, Aguilar B, Starr R, Alizadeh D, Brito A, Sarkissian A, et al. Glioblastoma-Targeted CD4+ CAR T Cells Mediate Superior Antitumor Activity. JCI Insight (2018) 3:e99048. doi: 10.1172/jci.insight.99048
52. Csaplár M, Szöllősi J, Gottschalk S, Vereb G, Szöőr Á. Cytolytic Activity of CAR T Cells and Maintenance of Their CD4+ Subset Is Critical for Optimal Antitumor Activity in Preclinical Solid Tumor Models. Cancers (2021) 13:4301. doi: 10.3390/cancers13174301
53. Cheadle EJ, Sheard V, Rothwell DG, Bridgeman JS, Ashton G, Hanson V, et al. Differential Role of Th1 and Th2 Cytokines in Autotoxicity Driven by CD19-Specific Second-Generation Chimeric Antigen Receptor T Cells in a Mouse Model. J. Immunol (2014) 192:3654–65. doi: 10.4049/jimmunol.1302148
54. Lu Y, Wang Q, Xue G, Bi E, Ma X, Wang A, et al. Th9 Cells Represent a Unique Subset of CD4+ T Cells Endowed With the Ability to Eradicate Advanced Tumors. Cancer Cell (2018) 33:1048–60.e7. doi: 10.1016/j.ccell.2018.05.004
55. Xue G, Zheng N, Fang J, Jin G, Li X, Dotti G, et al. Adoptive Cell Therapy With Tumor-Specific Th9 Cells Induces Viral Mimicry to Eliminate Antigen-Loss-Variant Tumor Cells. Cancer Cell (2021) ;39(12):1610–22.e9. doi: 10.1016/j.ccell.2021.09.011
56. Sek K, Chan CW, Beavis PA, Darcy PK. Adoptive Transfer of Tumor-Specific Th9 Cells Eradicates Heterogeneous Antigen-Expressing Tumor Cells. Cancer Cell (2021) 39(12):1564–66. doi: 10.1016/j.ccell.2021.10.013
57. Benoit-Lizon I, Jacquin E, Rivera Vargas T, Richard C, Roussey A, Dal Zuffo L, et al. CD4 T Cell-Intrinsic STING Signaling Controls the Differentiation and Effector Functions of T H 1 and T H 9 Cells. J Immunother Cancer (2022) 10:e003459. doi: 10.1136/jitc-2021-003459
58. Xu N, Palmer DC, Robeson AC, Shou P, Bommiasamy H, Laurie SJ, et al. STING Agonist Promotes CAR T Cell Trafficking and Persistence in Breast Cancer. J Exp Med (2021) 218:e20200844. doi: 10.1084/jem.20200844
59. Demaria O, De Gassart A, Coso S, Gestermann N, Di Domizio J, Flatz L, et al. STING Activation of Tumor Endothelial Cells Initiates Spontaneous and Therapeutic Antitumor Immunity. Proc Natl Acad Sci USA (2015) 112:15408–13. doi: 10.1073/pnas.1512832112
60. Ramanjulu JM, Pesiridis GS, Yang J, Concha N, Singhaus R, Zhang SY, et al. Design of Amidobenzimidazole STING Receptor Agonists With Systemic Activity. Nature (2018) 564(7736):439–43. doi: 10.1038/s41586-018-0705-y
61. Kamali AN, Noorbakhsh SM, Hamedifar H, Jadidi-Niaragh F, Yazdani R, Bautista JM, et al. A Role for Th1-Like Th17 Cells in the Pathogenesis of Inflammatory and Autoimmune Disorders. Mol Immunol (2019) 105:107–15. doi: 10.1016/j.molimm.2018.11.015
62. Asadzadeh Z, Mohammadi H, Safarzadeh E, Hemmatzadeh M, Mahdian-shakib A, Jadidi-Niaragh F, et al. The Paradox of Th17 Cell Functions in Tumor Immunity. Cell Immunol (2017) 322:15–25. doi: 10.1016/j.cellimm.2017.10.015
63. Wacleche V, Landay A, Routy J-P, Ancuta P. The Th17 Lineage: From Barrier Surfaces Homeostasis to Autoimmunity, Cancer, and HIV-1 Pathogenesis. Viruses (2017) 9:303. doi: 10.3390/v9100303
64. Vitiello GA, Miller G. Targeting the Interleukin-17 Immune Axis for Cancer Immunotherapy. J Exp Med (2020) 217:e20190456. doi: 10.1084/jem.20190456
65. Majchrzak K, Nelson MH, Bailey SR, Bowers JS, Yu X-Z, Rubinstein MP, et al. Exploiting IL-17-Producing CD4+ and CD8+ T Cells to Improve Cancer Immunotherapy in the Clinic. Cancer Immunol Immunother (2016) 65:247–59. doi: 10.1007/s00262-016-1797-6
66. Najafi S, Mirshafiey A. The Role of T Helper 17 and Regulatory T Cells in Tumor Microenvironment. Immunopharmacol Immunotoxicol (2019) 41:16–24. doi: 10.1080/08923973.2019.1566925
67. Martin-Orozco N, Muranski P, Chung Y, Yang XO, Yamazaki T, Lu S, et al. T Helper 17 Cells Promote Cytotoxic T Cell Activation in Tumor Immunity. Immunity (2009) 31:787–98. doi: 10.1016/j.immuni.2009.09.014
68. Muranski P, Borman ZA, Kerkar SP, Klebanoff CA, Ji Y, Sanchez-Perez L, et al. Th17 Cells Are Long Lived and Retain a Stem Cell-Like Molecular Signature. Immunity (2011) 35:972–85. doi: 10.1016/j.immuni.2011.09.019
69. Marques HS, de Brito BB, da Silva FAF, Santos MLC, de Souza JCB, Correia TML, et al. Relationship Between Th17 Immune Response and Cancer. WJCO (2021) 12:845–67. doi: 10.5306/wjco.v12.i10.845
70. Alizadeh D, Katsanis E, Larmonier N. The Multifaceted Role of Th17 Lymphocytes and Their Associated Cytokines in Cancer. Clin Dev Immunol (2013) 2013:1–11. doi: 10.1155/2013/957878
71. Guedan S, Chen X, Madar A, Carpenito C, McGettigan SE, Frigault MJ, et al. ICOS-Based Chimeric Antigen Receptors Program Bipolar TH17/TH1 Cells. Blood (2014) 124:1070–80. doi: 10.1182/blood-2013-10-535245
72. Wolf D, Sopper S, Pircher A, Gastl G, Wolf AM. Treg(s) in Cancer: Friends or Foe?: THE AMBIGUOUS ROLE OF TREG IN CANCER. J Cell Physiol (2015) 230:2598–605. doi: 10.1002/jcp.25016
73. Wang X, Popplewell LL, Wagner JR, Naranjo A, Blanchard MS, Mott MR, et al. Phase 1 Studies of Central Memory–Derived CD19 CAR T–cell Therapy Following Autologous HSCT in Patients With B-Cell NHL. Blood (2016) 127:2980–90. doi: 10.1182/blood-2015-12-686725
74. Tay RE, Richardson EK, Toh HC. Revisiting the Role of CD4+ T Cells in Cancer Immunotherapy—New Insights Into Old Paradigms. Cancer Gene Ther (2021) 28:5–17. doi: 10.1038/s41417-020-0183-x
75. Milone MC, Fish JD, Carpenito C, Carroll RG, Binder GK, Teachey D, et al. Chimeric Receptors Containing CD137 Signal Transduction Domains Mediate Enhanced Survival of T Cells and Increased Antileukemic Efficacy In Vivo. Mol Ther (2009) 17:1453–64. doi: 10.1038/mt.2009.83
76. Eshhar Z, Waks T, Gross G, Schindler DG. Specific Activation and Targeting of Cytotoxic Lymphocytes Through Chimeric Single Chains Consisting of Antibody-Binding Domains and the Gamma or Zeta Subunits of the Immunoglobulin and T-Cell Receptors. Proc Natl Acad Sci (1993) 90:720–4. doi: 10.1073/pnas.90.2.720
77. Gross G, Waks T, Eshhar Z. Expression of Immunoglobulin-T-Cell Receptor Chimeric Molecules as Functional Receptors With Antibody-Type Specificity. Proc Natl Acad Sci (1989) 86:10024–8. doi: 10.1073/pnas.86.24.10024
78. Brocker T, Karjalainen K. Signals Through T Cell Receptor-Zeta Chain Alone are Insufficient to Prime Resting T Lymphocytes. J Exp Med (1995) 181:1653–9. doi: 10.1084/jem.181.5.1653
79. Subklewe M, von Bergwelt-Baildon M, Humpe A. Chimeric Antigen Receptor T Cells: A Race to Revolutionize Cancer Therapy. Transfus Med Hemother (2019) 46:15–24. doi: 10.1159/000496870
80. Brocker T. Chimeric Fv-Zeta or Fv-Epsilon Receptors are Not Sufficient to Induce Activation or Cytokine Production in Peripheral T Cells. Blood (2000) 96:1999–2001. doi: 10.1182/blood.V96.5.1999.h8001999_1999_2001
81. Krause A, Guo H-F, Latouche J-B , Tan C, Cheung N-KV, Sadelain M. Antigen-Dependent CD28 Signaling Selectively Enhances Survival and Proliferation in Genetically Modified Activated Human Primary T Lymphocytes. J Exp Med (1998) 188:619–26. doi: 10.1084/jem.188.4.619
82. Porter DL, Levine BL, Kalos M, Bagg A, June CH. Chimeric Antigen Receptor–Modified T Cells in Chronic Lymphoid Leukemia. N Engl J Med (2011) 365:725–33. doi: 10.1056/NEJMoa1103849
83. Andrea AE, Chiron A, Bessoles S, Hacein-Bey-Abina S. Engineering Next-Generation CAR-T Cells for Better Toxicity Management. IJMS (2020) 21:8620. doi: 10.3390/ijms21228620
84. Larson RC, Maus MV. Recent Advances and Discoveries in the Mechanisms and Functions of CAR T Cells. Nat Rev Cancer (2021) 21:145–61. doi: 10.1038/s41568-020-00323-z
85. Mangal JL, Handlos JL, Esrafili A, Inamdar S, Mcmillian S, Wankhede M, et al. Engineering Metabolism of Chimeric Antigen Receptor (CAR) Cells for Developing Efficient Immunotherapies. Cancers (2021) 13:1123. doi: 10.3390/cancers13051123
86. Guest RD, Kirillova N, Mowbray S, Gornall H, Rothwell DG, Cheadle EJ, et al. Definition and Application of Good Manufacturing Process-Compliant Production of CEA-Specific Chimeric Antigen Receptor Expressing T-Cells for Phase I/II Clinical Trial. Cancer Immunol Immunother (2014) 63:133–45. doi: 10.1007/s00262-013-1492-9
87. Weinkove R, George P, Dasyam N, McLellan AD. Selecting Costimulatory Domains for Chimeric Antigen Receptors: Functional and Clinical Considerations. Clin Transl Immunol (2019) 8:e1049. doi: 10.1002/cti2.1049
88. Kawalekar OU, O’Connor RS, Fraietta JA, Guo L, McGettigan SE, Posey AD, et al. Distinct Signaling of Coreceptors Regulates Specific Metabolism Pathways and Impacts Memory Development in CAR T Cells. Immunity (2016) 44:380–90. doi: 10.1016/j.immuni.2016.01.021
89. Salter AI, Ivey RG, Kennedy JJ, Voillet V, Rajan A, Alderman EJ, et al. Phosphoproteomic Analysis of Chimeric Antigen Receptor Signaling Reveals Kinetic and Quantitative Differences That Affect Cell Function. Sci Signal (2018) 11:eaat6753. doi: 10.1126/scisignal.aat6753
90. Guedan S, Posey AD, Shaw C, Wing A, Da T, Patel PR, et al. Enhancing CAR T Cell Persistence Through ICOS and 4-1BB Costimulation. JCI Insight (2018) 3:e96976. doi: 10.1172/jci.insight.96976
91. Lin W-Y, Wang H-H, Chen Y-W, Lin C-F, Fan H-C, Lee Y-Y, et al. Gene Modified CAR-T Cellular Therapy for Hematologic Malignancies. IJMS (2020) 21:8655. doi: 10.3390/ijms21228655
92. Talebi M, Nozad Charoudeh H, Movassagpour Akbari AA, Baradaran B, Kazemi T. Effect of Cellular-Based Artificial Antigen Presenting Cells Expressing ICOSL, in T-Cell Subtypes Differentiation and Activation. Adv Pharm Bull (2020) 11:537–42. doi: 10.34172/apb.2021.062
93. Chmielewski M, Abken H. TRUCKS, the Fourth-Generation CAR T Cells: Current Developments and Clinical Translation. Adv Cell Gene Ther (2020) 3:1–9. doi: 10.1002/acg2.84
94. Chmielewski M, Abken H. TRUCKs: The Fourth Generation of CARs. Expert Opin Biol Ther (2015) 15:1145–54. doi: 10.1517/14712598.2015.1046430
95. Bell M, Gottschalk S. Engineered Cytokine Signaling to Improve CAR T Cell Effector Function. Front Immunol (2021) 12:684642. doi: 10.3389/fimmu.2021.684642
96. Tokarew N, Ogonek J, Endres S, von Bergwelt-Baildon M, Kobold S. Teaching an Old Dog New Tricks: Next-Generation CAR T Cells. Br J Cancer (2019) 120:26–37. doi: 10.1038/s41416-018-0325-1
97. Malissen B, Bongrand P. Early T Cell Activation: Integrating Biochemical, Structural, and Biophysical Cues. Annu Rev Immunol (2015) 33:539–61. doi: 10.1146/annurev-immunol-032414-112158
98. Abram CL, Lowell CA. The Expanding Role for ITAM-Based Signaling Pathways in Immune Cells. Sci STKE (2007) 2007:1–6. doi: 10.1126/stke.3772007re2
99. Meng X, Jing R, Qian L, Zhou C, Sun J. Engineering Cytoplasmic Signaling of CD28ζ CARs for Improved Therapeutic Functions. Front Immunol (2020) 11:1046. doi: 10.3389/fimmu.2020.01046
100. Chae W-J, Lee H-K, Han J-H, Kim S-WV, Bothwell ALM, Morio T, et al. Qualitatively Differential Regulation of T Cell Activation and Apoptosis by T Cell Receptor ζ Chain ITAMs and Their Tyrosine Residues. Int Immunol (2004) 16:1225–36. doi: 10.1093/intimm/dxh120
101. Feucht J, Sun J, Eyquem J, Ho Y-J, Zhao Z, Leibold J, et al. Calibration of CAR Activation Potential Directs Alternative T Cell Fates and Therapeutic Potency. Nat Med (2019) 25:82–8. doi: 10.1038/s41591-018-0290-5
102. Harada Y, Ohgai D, Watanabe R, Okano K, Koiwai O, Tanabe K, et al. A Single Amino Acid Alteration in Cytoplasmic Domain Determines IL-2 Promoter Activation by Ligation of CD28 But Not Inducible Costimulator (ICOS). J Exp Med (2003) 197:257–62. doi: 10.1084/jem.20021305
103. Riha P, Rudd CE. CD28 Co-Signaling in the Adaptive Immune Response. Self/Nonself (2010) 1:231–40. doi: 10.4161/self.1.3.12968
104. Guedan S, Madar A, Casado-Medrano V, Shaw C, Wing A, Liu F, et al. Single Residue in CD28-Costimulated CAR-T Cells Limits Long-Term Persistence and Antitumor Durability. J Clin Invest (2020) 130:3087–97. doi: 10.1172/JCI133215
105. Guest RD, Hawkins RE, Kirillova N, Cheadle EJ, Arnold J, O’Neill A, et al. The Role of Extracellular Spacer Regions in the Optimal Design of Chimeric Immune Receptors: Evaluation of Four Different Scfvs and Antigens. J Immunother (2005) 28:203–11. doi: 10.1097/01.cji.0000161397.96582.59
106. Hudecek M, Lupo-Stanghellini M-T, Kosasih PL, Sommermeyer D, Jensen MC, Rader C, et al. Receptor Affinity and Extracellular Domain Modifications Affect Tumor Recognition by ROR1-Specific Chimeric Antigen Receptor T Cells. Clin Cancer Res (2013) 19:3153–64. doi: 10.1158/1078-0432.CCR-13-0330
107. James SE, Greenberg PD, Jensen MC, Lin Y, Wang J, Till BG, et al. Antigen Sensitivity of CD22-Specific Chimeric TCR Is Modulated by Target Epitope Distance From the Cell Membrane. J Immunol (2008) 180:7028–38. doi: 10.4049/jimmunol.180.10.7028
108. Al-Aghbar MA, Jainarayanan AK, Dustin ML, Roffler SR. The Interplay Between Membrane Topology and Mechanical Forces in Regulating T Cell Receptor Activity. Commun Biol (2022) 5:40. doi: 10.1038/s42003-021-02995-1
109. Chen B-M, Al-Aghbar MA, Lee C-H, Chang T-C, Su Y-C, Li Y-C, et al. The Affinity of Elongated Membrane-Tethered Ligands Determines Potency of T Cell Receptor Triggering. Front Immunol (2017) 8:793. doi: 10.3389/fimmu.2017.00793
110. Jonnalagadda M, Mardiros A, Urak R, Wang X, Hoffman LJ, Bernanke A, et al. Chimeric Antigen Receptors With Mutated IgG4 Fc Spacer Avoid Fc Receptor Binding and Improve T Cell Persistence and Antitumor Efficacy. Mol Ther (2015) 23:757–68. doi: 10.1038/mt.2014.208
111. Schäfer D, Henze J, Pfeifer R, Schleicher A, Brauner J, Mockel-Tenbrinck N, et al. A Novel Siglec-4 Derived Spacer Improves the Functionality of CAR T Cells Against Membrane-Proximal Epitopes. Front Immunol (2020) 11:1704. doi: 10.3389/fimmu.2020.01704
112. Ghorashian S, Kramer AM, Onuoha S, Wright G, Bartram J, Richardson R, et al. Enhanced CAR T Cell Expansion and Prolonged Persistence in Pediatric Patients With ALL Treated With a Low-Affinity CD19 CAR. Nat Med (2019) 25:1408–14. doi: 10.1038/s41591-019-0549-5
113. Schmid DA, Irving MB, Posevitz V, Hebeisen M, Posevitz-Fejfar A, Sarria J-CF, et al. Evidence for a TCR Affinity Threshold Delimiting Maximal CD8 T Cell Function. J Immunol (2010) 184:4936–46. doi: 10.4049/jimmunol.1000173
114. Thomas S, Xue S-A, Bangham CRM, Jakobsen BK, Morris EC, Stauss HJ. Human T Cells Expressing Affinity-Matured TCR Display Accelerated Responses But Fail to Recognize Low Density of MHC-Peptide Antigen. Blood (2011) 118:319–29. doi: 10.1182/blood-2010-12-326736
115. Johnson LR, Lee DY, Eacret JS, Ye D, June CH, Minn AJ. The Immunostimulatory RNA RN7SL1 Enables CAR-T Cells to Enhance Autonomous and Endogenous Immune Function. Cell (2021) 184(19):4981–95.e14. doi: 10.1016/j.cell.2021.08.004
116. Immunostimulatory RNA Expression Enhances CAR T-Cell Function In Vivo. Cancer Discov (2021) 11:OF6–6. doi: 10.1158/2159-8290.CD-RW2021-130
117. Nutt WS, Srivastava S. Special Delivery! CAR-T Cells Transport RN7SL1 to the Tumor Microenvironment. Trends Mol Med (2021) 27:1019–21. doi: 10.1016/j.molmed.2021.09.002
118. Wang M, Zhang C, Jiang X. CAR-T: A Potential Gene Carrier Targeting Solid Tumor Immune Microenvironment. Sig Transduct Target Ther (2021) 6:393. doi: 10.1038/s41392-021-00812-z
119. Cubillos-Ruiz A, Guo T, Sokolovska A, Miller PF, Collins JJ, Lu TK, et al. Engineering Living Therapeutics With Synthetic Biology. Nat Rev Drug Discov (2021) 20:941–60. doi: 10.1038/s41573-021-00285-3
120. Juillerat A, Marechal A, Filhol JM, Valogne Y, Valton J, Duclert A, et al. An Oxygen Sensitive Self-Decision Making Engineered CAR T-Cell. Sci Rep (2017) 7:39833. doi: 10.1038/srep39833
121. Rossig C, Pule M, Altvater B, Saiagh S, Wright G, Ghorashian S, et al. Vaccination to Improve the Persistence of CD19CAR Gene-Modified T Cells in Relapsed Pediatric Acute Lymphoblastic Leukemia. Leukemia (2017) 31:1087–95. doi: 10.1038/leu.2017.39
122. Ma L, Dichwalkar T, Chang JYH, Cossette B, Garafola D, Zhang AQ, et al. Enhanced CAR–T Cell Activity Against Solid Tumors by Vaccine Boosting Through the Chimeric Receptor. Science (2019) 365:162–8. doi: 10.1126/science.aav8692
123. Reinhard K, Rengstl B, Oehm P, Michel K, Billmeier A, Hayduk N, et al. An RNA Vaccine Drives Expansion and Efficacy of Claudin-CAR-T Cells Against Solid Tumors. Science (2020) 367:446–53. doi: 10.1126/science.aay5967
124. Eyquem J, Mansilla-Soto J, Giavridis T, van derStegen SJC, Hamieh M, Cunanan KM, et al. Targeting a CAR to the TRAC Locus With CRISPR/Cas9 Enhances Tumour Rejection. Nature (2017) 543:113–7. doi: 10.1038/nature21405
125. Bommareddy PK, Shettigar M, Kaufman HL. Integrating Oncolytic Viruses in Combination Cancer Immunotherapy. Nat Rev Immunol (2018) 18:498–513. doi: 10.1038/s41577-018-0014-6
126. Guedan S, Alemany R. CAR-T Cells and Oncolytic Viruses: Joining Forces to Overcome the Solid Tumor Challenge. Front Immunol (2018) 9:2460. doi: 10.3389/fimmu.2018.02460
127. Nishio N, Diaconu I, Liu H, Cerullo V, Caruana I, Hoyos V, et al. Armed Oncolytic Virus Enhances Immune Functions of Chimeric Antigen Receptor–Modified T Cells in Solid Tumors. Cancer Res (2014) 74:5195–205. doi: 10.1158/0008-5472.CAN-14-0697
128. Ma R, Lu T, Li Z, Teng K-Y, Mansour AG, Yu M, et al. An Oncolytic Virus Expressing Il15/Il15rα Combined With Off-The-Shelf EGFR-CAR NK Cells Targets Glioblastoma. Cancer Res (2021) 81:3635–48. doi: 10.1158/0008-5472.CAN-21-0035
129. Sonzogni O, Zak DE, Sasso MS, Lear R, Muntzer A, Zonca M, et al. T-SIGn Tumor Reengineering Therapy and CAR T Cells Synergize in Combination Therapy to Clear Human Lung Tumor Xenografts and Lung Metastases in NSG Mice. OncoImmunology (2022) 11:2029070. doi: 10.1080/2162402X.2022.2029070
130. Binary Oncolytic Adenovirus in Combination With HER2-Specific Autologous CAR VST, Advanced HER2 Positive Solid Tumors - Tabular View. Available at: https://clinicaltrials.gov/ct2/show/record/NCT03740256.
131. Ghassemi S, Martinez-Becerra FJ, Master AM, Richman SA, Heo D, Leferovich J, et al. Enhancing Chimeric Antigen Receptor T Cell Anti-Tumor Function Through Advanced Media Design. Mol Ther - Methods Clin Dev (2020) 18:595–606. doi: 10.1016/j.omtm.2020.07.008
132. Barro L, Burnouf P-A, Chou M-L, Nebie O, Wu Y-W, Chen M-S, et al. Human Platelet Lysates for Human Cell Propagation. Platelets (2021) 32:152–62. doi: 10.1080/09537104.2020.1849602
133. Bieback K, Fernandez-Muñoz B, Pati S, Schäfer R. Gaps in the Knowledge of Human Platelet Lysate as a Cell Culture Supplement for Cell Therapy: A Joint Publication From the AABB and the International Society for Cell & Gene Therapy. Cytotherapy (2019) 21:911–24. doi: 10.1016/j.jcyt.2019.06.006
134. Subbiahanadar Chelladurai K, Selvan Christyraj JD, Rajagopalan K, Yesudhason BV, Venkatachalam S, Mohan M, et al. Alternative to FBS in Animal Cell Culture - An Overview and Future Perspective. Heliyon (2021) 7:e07686. doi: 10.1016/j.heliyon.2021.e07686
135. Medvec AR, Ecker C, Kong H, Winters EA, Glover J, Varela-Rohena A, et al. Improved Expansion and In Vivo Function of Patient T Cells by a Serum-Free Medium. Mol Ther - Methods Clin Dev (2018) 8:65–74. doi: 10.1016/j.omtm.2017.11.001
136. Canestrari E, Steidinger HR, McSwain B, Charlebois SJ, Dann CT. Human Platelet Lysate Media Supplement Supports Lentiviral Transduction and Expansion of Human T Lymphocytes While Maintaining Memory Phenotype. J Immunol Res (2019) 2019:1–11. doi: 10.1155/2019/3616120
137. Torres Chavez A, McKenna MK, Canestrari E, Dann CT, Ramos CA, Lulla P, et al. Expanding CAR T Cells in Human Platelet Lysate Renders T Cells With In Vivo Longevity. J Immunother Cancer (2019) 7:330. doi: 10.1186/s40425-019-0804-9
138. Smith C, Økern G, Rehan S, Beagley L, Lee SK, Aarvak T, et al. Ex Vivo Expansion of Human T Cells for Adoptive Immunotherapy Using the Novel Xeno-Free CTS Immune Cell Serum Replacement. Clin Trans Immunol (2015) 4:e31. doi: 10.1038/cti.2014.31
139. Talebi M, Nozad Charoudeh H, Movassaghpour Akbari AA, Baradaran B, Kazemi T. Acellular Wharton’s Jelly, Potentials in T-Cell Subtypes Differentiation, Activation and Proliferation. Adv Pharm Bull (2020) 10:617–22. doi: 10.34172/apb.2020.074
140. Ghassemi S, Nunez-Cruz S, O’Connor RS, Fraietta JA, Patel PR, Scholler J, et al. Reducing Ex Vivo Culture Improves the Antileukemic Activity of Chimeric Antigen Receptor (CAR) T Cells. Cancer Immunol Res (2018) 6:1100–9. doi: 10.1158/2326-6066.CIR-17-0405
141. Ghassemi S, Durgin JS, Nunez-Cruz S, Patel J, Leferovich J, Pinzone M, et al. Rapid Manufacturing of Non-Activated Potent CAR T Cells. Nat BioMed Eng (2022) 6:118–28. doi: 10.1038/s41551-021-00842-6
142. Lee H-J, Lee Y-S, Kim H-S, Kim Y-K, Kim J-H, Jeon S-H, et al. Retronectin Enhances Lentivirus-Mediated Gene Delivery Into Hematopoietic Progenitor Cells. Biologicals (2009) 37:203–9. doi: 10.1016/j.biologicals.2009.01.008
143. Dodo K, Chono H, Saito N, Tanaka Y, Tahara K, Nukaya I, et al. An Efficient Large-Scale Retroviral Transduction Method Involving Preloading the Vector Into a RetroNectin-Coated Bag With Low-Temperature Shaking. PloS One (2014) 9:e86275. doi: 10.1371/journal.pone.0086275
144. Gargett T, Brown MP. Different Cytokine and Stimulation Conditions Influence the Expansion and Immune Phenotype of Third-Generation Chimeric Antigen Receptor T Cells Specific for Tumor Antigen GD2. Cytotherapy (2015) 17:487–95. doi: 10.1016/j.jcyt.2014.12.002
145. Yu SS, Nukaya I, Enoki T, Chatani E, Kato A, Goto Y, et al. In Vivo Persistence of Genetically Modified T Cells Generated Ex Vivo Using the Fibronectin CH296 Stimulation Method. Cancer Gene Ther (2008) 15:508–16. doi: 10.1038/cgt.2008.21
146. Stock S, Hoffmann J-M, Schubert M-L, Wang L, Wang S, Gong W, et al. Influence of Retronectin-Mediated T-Cell Activation on Expansion and Phenotype of CD19-Specific Chimeric Antigen Receptor T Cells. Hum Gene Ther (2018) 29:1167–82. doi: 10.1089/hum.2017.237
147. Gong W, Wang L, Stock S, Ni M, Schubert M-L, Neuber B, et al. Evaluation of Production Protocols for the Generation of NY-ESO-1-Specific T Cells. Cells (2021) 10:152. doi: 10.3390/cells10010152
148. Hosoi H, Ikeda H, Imai N, Amaike C, Wang L, Orito Y, et al. Stimulation Through Very Late Antigen-4 and -5 Improves the Multifunctionality and Memory Formation of CD8 + T Cells: Immunomodulation. Eur J Immunol (2014) 44:1747–58. doi: 10.1002/eji.201343969
149. Xu Y, Zhang M, Ramos CA, Durett A, Liu E, Dakhova O, et al. Closely Related T-Memory Stem Cells Correlate With In Vivo Expansion of CAR.CD19-T Cells and are Preserved by IL-7 and IL-15. Blood (2014) 123:3750–9. doi: 10.1182/blood-2014-01-552174
150. Yang S, Ji Y, Gattinoni L, Zhang L, Yu Z, Restifo NP, et al. Modulating the Differentiation Status of Ex Vivo-Cultured Anti-Tumor T Cells Using Cytokine Cocktails. Cancer Immunol Immunother (2013) 62:727–36. doi: 10.1007/s00262-012-1378-2
151. Lamers CHJ, van Steenbergen-Langeveld S, van Brakel M, Groot-van Ruijven CM, van Elzakker PMML, van Krimpen B, et al. T Cell Receptor-Engineered T Cells to Treat Solid Tumors: T Cell Processing Toward Optimal T Cell Fitness. Hum Gene Ther Methods (2014) 25:345–57. doi: 10.1089/hgtb.2014.051
152. Klebanoff CA, Finkelstein SE, Surman DR, Lichtman MK, Gattinoni L, Theoret MR, et al. IL-15 Enhances the In Vivo Antitumor Activity of Tumor-Reactive CD8+ T Cells. Proc Natl Acad Sci USA (2004) 101:1969–74. doi: 10.1073/pnas.0307298101
153. Gershovich PM, Karabelskii AV, Ulitin AB, Ivanov RA. The Role of Checkpoint Inhibitors and Cytokines in Adoptive Cell-Based Cancer Immunotherapy With Genetically Modified T Cells. Biochem Moscow (2019) 84:695–710. doi: 10.1134/S0006297919070022
154. Anestakis D, Petanidis S, Kalyvas S, Nday C, Tsave O, Kioseoglou E, et al. Mechanisms and αpplications of ιnterleukins in Cancer Immunotherapy. IJMS (2015) 16:1691–710. doi: 10.3390/ijms16011691
155. Alizadeh D, Wong RA, Yang X, Wang D, Pecoraro JR, Kuo C-F, et al. IL15 Enhances CAR-T Cell Antitumor Activity by Reducing Mtorc1 Activity and Preserving Their Stem Cell Memory Phenotype. Cancer Immunol Res (2019) 7:759–72. doi: 10.1158/2326-6066.CIR-18-0466
156. Loschinski R, Böttcher M, Stoll A, Bruns H, Mackensen A, Mougiakakos D. IL-21 Modulates Memory and Exhaustion Phenotype of T-Cells in a Fatty Acid Oxidation-Dependent Manner. Oncotarget (2018) 9:13125–38. doi: 10.18632/oncotarget.24442
157. Ghassemi S, Bedoya F, Nunez-Cruz S, June C, Melenhorst J, Milone M. Shortened T Cell Culture With IL-7 and IL-15 Provides the Most Potent Chimeric Antigen Receptor (CAR)-Modified T Cells for Adoptive Immunotherapy. Mol Ther (2016) 24:S79. doi: 10.1016/S1525-0016(16)33012-X
158. Cieri N, Camisa B, Cocchiarella F, Forcato M, Oliveira G, Provasi E, et al. IL-7 and IL-15 Instruct the Generation of Human Memory Stem T Cells From Naive Precursors. Blood (2013) 121:573–84. doi: 10.1182/blood-2012-05-431718
159. Ptáčková P, Musil J, Štach M, Lesný P, Němečková Š, Král V, et al. A New Approach to CAR T-Cell Gene Engineering and Cultivation Using Piggybac Transposon in the Presence of IL-4, IL-7 and IL-21. Cytotherapy (2018) 20:507–20. doi: 10.1016/j.jcyt.2017.10.001
160. Lee J-B, Lee K-A, Chang J. Phenotypic Changes Induced by IL-12 Priming Regulate Effector and Memory CD8 T Cell Differentiation. Int Immunol (2007) 19:1039–48. doi: 10.1093/intimm/dxm072
161. Jenkins Y, Zabkiewicz J, Ottmann O, Jones N. Tinkering Under the Hood: Metabolic Optimisation of CAR-T Cell Therapy. Antibodies (2021) 10:17. doi: 10.3390/antib10020017
162. Rostamian H, Fallah-Mehrjardi K, Khakpoor-Koosheh M, Pawelek JM, Hadjati J, Brown CE, et al. A Metabolic Switch to Memory CAR T Cells: Implications for Cancer Treatment. Cancer Lett (2021) 500:107–18. doi: 10.1016/j.canlet.2020.12.004
163. Geiger R, Rieckmann JC, Wolf T, Basso C, Feng Y, Fuhrer T, et al. L-Arginine Modulates T Cell Metabolism and Enhances Survival and Anti-Tumor Activity. Cell (2016) 167(3):829–42.e13. doi: 10.1016/j.cell.2016.09.031
164. Sukumar M, Liu J, Ji Y, Subramanian M, Crompton JG, Yu Z, et al. Inhibiting Glycolytic Metabolism Enhances CD8+ T Cell Memory and Antitumor Function. J Clin Invest (2013) 123:4479–88. doi: 10.1172/JCI69589
165. Perkins MR, Grande S, Hamel A, Horton HM, Garrett TE, Miller SM, et al. Manufacturing an Enhanced CAR T Cell Product By Inhibition of the PI3K/Akt Pathway During T Cell Expansion Results in Improved In Vivo Efficacy of Anti-BCMA CAR T Cells. Blood (2015) 126:1893–3. doi: 10.1182/blood.V126.23.1893.1893
166. Zhang Q, Ding J, Sun S, Liu H, Lu M, Wei X, et al. Akt Inhibition at the Initial Stage of CAR-T Preparation Enhances the CAR-Positive Expression Rate, Memory Phenotype and In Vivo Efficacy. Am J Cancer Res (2019) Nov 1;9(11):2379–96.
167. Xiao Z, Sun Z, Smyth K, Li L. Wnt Signaling Inhibits CTL Memory Programming. Mol Immunol (2013) 56:423–33. doi: 10.1016/j.molimm.2013.06.008
168. Scholz G, Jandus C, Zhang L, Grandclément C, Lopez-Mejia IC, Soneson C, et al. Modulation of mTOR Signalling Triggers the Formation of Stem Cell-Like Memory T Cells. EBioMedicine (2016) 4:50–61. doi: 10.1016/j.ebiom.2016.01.019
169. Bengsch B, Johnson AL, Kurachi M, Odorizzi PM, Pauken KE, Attanasio J, et al. Bioenergetic Insufficiencies Due to Metabolic Alterations Regulated by the Inhibitory Receptor PD-1 Are an Early Driver of CD8 + T Cell Exhaustion. Immunity (2016) 45:358–73. doi: 10.1016/j.immuni.2016.07.008
170. Dumauthioz N, Tschumi B, Wenes M, Marti B, Wang H, Franco F, et al. Enforced PGC-1α Expression Promotes CD8 T Cell Fitness, Memory Formation and Antitumor Immunity. Cell Mol Immunol (2021) 18:1761–71. doi: 10.1038/s41423-020-0365-3
Keywords: CAR-T cells, persistence, metabolism, differentiation status, culture optimization
Citation: López-Cantillo G, Urueña C, Camacho BA and Ramírez-Segura C (2022) CAR-T Cell Performance: How to Improve Their Persistence? Front. Immunol. 13:878209. doi: 10.3389/fimmu.2022.878209
Received: 17 February 2022; Accepted: 25 March 2022;
Published: 28 April 2022.
Edited by:
Song Zhang, Nankai University, ChinaReviewed by:
Yang Xu, Southern University of Science and Technology, ChinaNuo Xu, AstraZeneca, United States
Copyright © 2022 López-Cantillo, Urueña, Camacho and Ramírez-Segura. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Cesar Ramírez-Segura, Y3JhbWlyZXpAaWRjYmlzLm9yZy5jbw==