 Emiko Kranz
Emiko Kranz Charles J. Kuhlmann
Charles J. Kuhlmann Joshua Chan3
Joshua Chan3 Patrick Y. Kim
Patrick Y. Kim Masakazu Kamata
Masakazu Kamata- 1Division of Hematology-Oncology, David Geffen School of Medicine at University of California, Los Angeles (UCLA), Los Angeles, CA, United States
- 2Department of Microbiology, School of Medicine, University of Alabama at Birmingham, Birmingham, AL, United States
- 3Department of Microbiology, Immunology, and Molecular Genetics, David Geffen School of Medicine at University of California, Los Angeles (UCLA), Los Angeles, CA, United States
Chimeric-antigen receptor (CAR) T-cell immunotherapy employs autologous-T cells modified with an antigen-specific CAR. Current CAR-T manufacturing processes tend to yield products dominated by effector T cells and relatively small proportions of long-lived memory T cells. Those few cells are a so-called stem cell memory T (TSCM) subset, which express naïve T-cell markers and are capable of self-renewal and oligopotent differentiation into effector phenotypes. Increasing the proportion of this subset may lead to more effective therapies by improving CAR-T persistence; however, there is currently no standardized protocol for the effective generation of CAR-TSCM cells. Here we present a simplified protocol enabling efficient derivation of gene-modified TSCM cells: Stimulation of naïve CD8+ T cells with only soluble anti-CD3 antibody and culture with IL-7 and IL-15 was sufficient for derivation of CD8+ T cells harboring TSCM phenotypes and oligopotent capabilities. These in-vitro expanded TSCM cells were engineered with CARs targeting the HIV-1 envelope protein as well as the CD19 molecule and demonstrated effector activity both in vitro and in a xenograft mouse model. This simple protocol for the derivation of CAR-TSCM cells may facilitate improved adoptive immunotherapy.
Introduction
Gene-engineered T cells with desired antigen-specific receptors such as chimeric-antigen receptors (CARs), aim to confer directed and enhanced cytotoxic T lymphocyte responses (1–4). CARs contain an antigen-binding domain specific for targets and an internal-signaling domain derived from CD3ζ chain as well as 41BB, CD28, or other co-stimulatory molecules (5, 6). When a CAR encounters its target ligand, it signals the cell in a T cell receptor-like, but human-leukocyte antigen (HLA)-independent manner, thus allowing this approach to be used in any patient. In human clinical trials for B-cell malignancies, CAR-modified autologous T cells targeting CD19 have shown robust effector responses (5–10). For HIV-1 infection, CD4ζ CAR has been widely and longitudinally tested in patients. Treatment was safe and well-tolerated for over a decade, but antiviral effects were limited due to low levels of gene-modified cell persistence (11–14).
The novel memory T cell subset known as stem cell-memory T (TSCM) harbors self-renewing and oligopotency capabilities. The TSCM subset has been identified in mice (15), non-human primates (16, 17), and humans (18, 19). In humans, approximately 2-4% of peripheral blood T cells consist of TSCM cells (20). The TSCM cells are phenotypically defined as naïve T (TN) cells by the expression of TN cell markers, such as CD45RA, CD62L and CCR7, but are distinguishable from TN cells by two memory T cell markers: CD95 and CD122. Thus, they exhibit a gene profile between TN and central-memory T (TCM) cells. Of note, the TSCM cells ― unlike other memory T cells ― can be expanded ex vivo while maintaining their stemness, allowing enrichment of the gene-modified population prior to transplant.
Increasing evidence indicates that the TSCM cells exhibit a lesser extent of proliferation and effector activities compared to other memory T cells. Upon antigen stimulation, the TSCM cells have the ability to differentiate into TCM (which are thought to be a premature memory subset that differentiates into the effector subset upon antigen re-encounter), followed by effector-memory T (TEM) cells (which are considered to be committed progenitor cells that undergo terminal differentiation after a limited number of cell divisions) (18, 21–24). The frequency and activity of TSCM cells from clinical samples also supports their prolonged precursor potential in humans (25, 26). Importantly, individual gene-modified TSCM cell clones did not show the emergence of clonal dominance for over a decade after infusion, indicating that gene engineering of TSCM cells does not bring in them a tumorigenic change. These evidence well support that TSCM cells would be an ideal host cell for CAR engineering.
We here developed a simple condition for the derivation and expansion of gene-modified-T cells harboring TSCM-surface phenotype and validated its applicability for gene engineering of T cells using both anti-HIV-1 and anti-CD19 CARs. Our results further demonstrate that these cells can effectively differentiate to functional-T cells conferring CAR-dependent effector activity against target cells in vitro as well as in a xenograft NSG mouse model.
Methods
Cells
Peripheral blood mononuclear cells (PBMCs) from healthy human donors were obtained from the CFAR Virology Core at UCLA without personal identifying information. To minimize a potential induction of T-cell stimulation by events such as crosslinking of cell-surface molecules, TN-cell population was negatively selected in one step using an EasySep™ Human Naive CD8+ T Cell Enrichment Kit (StemCell Technologies, Inc., Vancouver, Canada), which consistently provides >95% purity of CD8+ TN cells (Supplementary Figure 1). Cells were maintained in Iscove’s Modified Dulbecco’s Medium (IMDM) supplemented with 1% GlutaMAX supplement and Antibiotic-Antimycotic (Life Technologies, Grand Island, NY), 20% FCS (SH30070.03E; GE Healthcare Life Sciences, South Logan, UT), and 0.1 mM 2-mercaptoethanol (Sigma-Aldrich St. Louis, MO) (T-cell medium) as reported previously (27). Prior to lentiviral vector transduction, CD8+ TN cells were incubated with various concentrations of anti-CD3 antibody (Hit3a; BioLegend, San Diego, CA) with or without 2 µg/mL of anti-CD28 antibodies (CD28.2; BioLegend) for 2 days in T-cell medium supplemented with 5 ng/mL IL-7 and IL-15 (R&D systems, Minneapolis, MN) as summarized in Figure 1A. All cells were incubated at 37°C in 5% CO2.
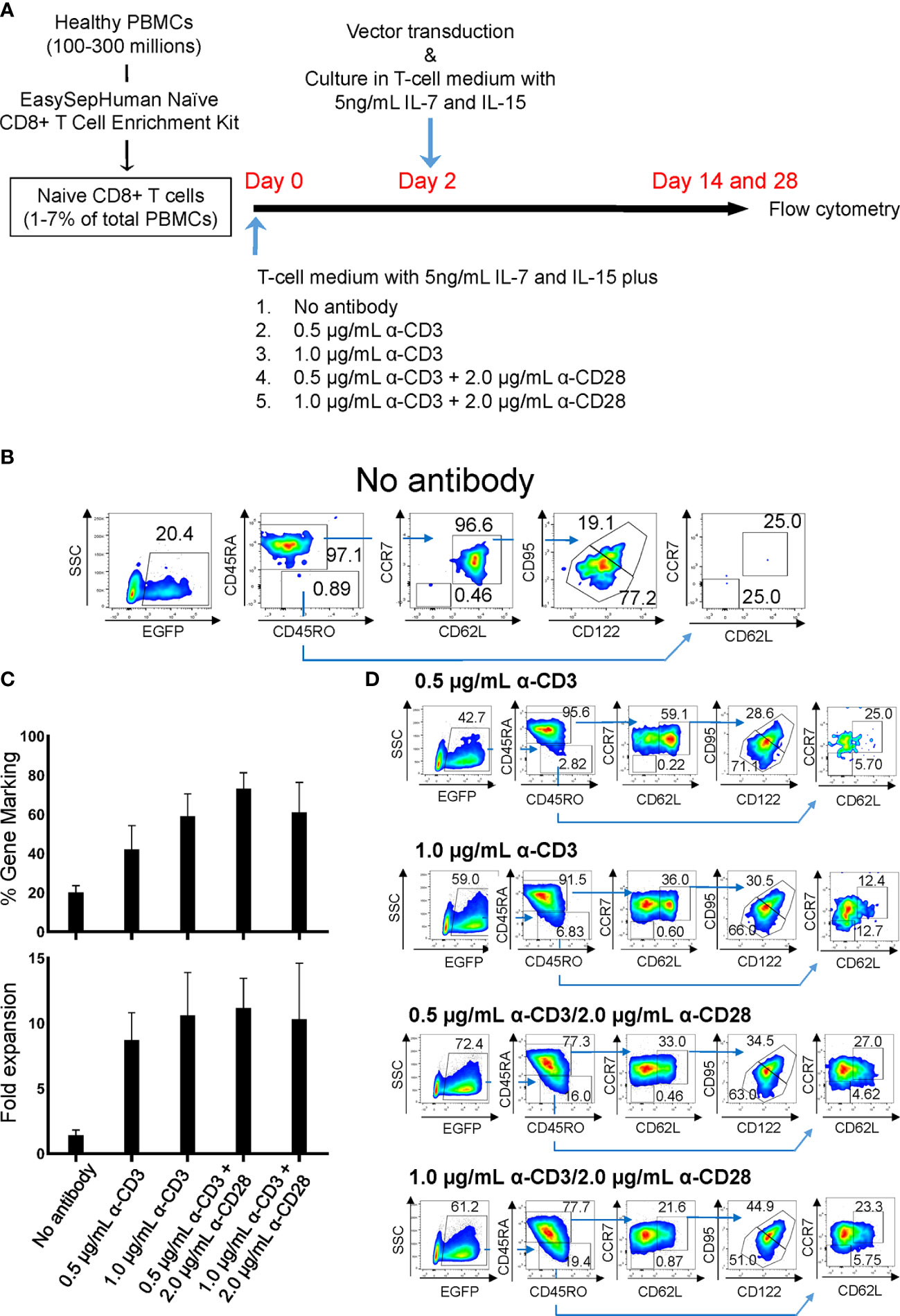
Figure 1 Derivation of gene-marked CD8+ T cells harboring TSCM-surface phenotype under different stimulation conditions. Freshly isolated human PBMCs were separated for CD8+ TN cells using an EasySep™ Human Naïve CD8+ T Cell Enrichment kit. Cells were stimulated for 2 days with the condition as in (A), followed by transduction with lentiviral vector encoding EGFP as a transduction marker. Cells were cultured for an additional 12 days in the presence of 5 ng/mL of IL-7 and IL-15, and cell-surface marker profiles were analyzed by flow cytometry. (A) Summary for derivation of TSCM cells procedure. (B) Flow cytometry analyses of CD8+ TN cells genetically marked by EGFP with no antibody stimulation. (C) % positivity of EGFP-marked cells (Top bars) and fold changes in cell number following 14 days of culture (Bottom bars). (D) Flow cytometry analyses of EGFP-engineered CD8+ TN cells. All experiments were repeated at least three times. Error bars in (C) show the standard deviation of a data set. One representative experiment is shown for (B, D).
Cytotoxic assay
Triple-CD4ζ (27) was used an anti-HIV CAR. For anti-HIV CAR cytotoxicity assays, Jurkat cells encoded without (ΔKS, non-target cells) or with HIV-1HXBC2 envelope protein (HXBC2, target cells), where the expression can be induced by removal of doxycycline from culture medium (28), were used. As an additional target cell for in vivo cytotoxicity assay, TF228.1.16 cells ― which are BJAB cells constitutively expressing HIV-1BH10 envelope protein (29) ― were used. ΔKS, HXBC2, and TF228.1.16 cells were genetically labeled with TagBFP (30), mCherry (31), and mStrawberry (31), respectively. In an assay for anti-CD19 CAR (FMC63-IgG4ζ (32)), we used BCBL-1 (33) (CD19-, non-target cells) and Ramos cells (CD19+, target cells). BCBL-1 and Ramos cells were genetically labeled with TagBFP and mStrawberry, respectively. All cells were obtained from the NIH-AIDS Reagent Program and cultured as recommended.
For in vitro cytotoxicity assay, 5 x 104 CAR- or EGFP-modified CD8+ T cells as a negative control were plated using 100µL of T-cell medium in a 96-well round bottom plate. The same numbers of genetically-labeled ΔKS and HXBC2 cells for CD4ζ or BCBL-1 and Ramos cells for FMC63-IgG4ζ were added to each well and co-incubated for 4 or 16 hours [Effector: Target (E:T) ratio = 1:1]. Antigen specificity of both CARs was also validated with HXBC2 cells culturing with doxycycline to suppress HIV-1HXBC2 envelope protein expression for CD4ζ CAR which cells cannot be a target for CD4ζ CAR, as well as human CD19 overexpressing HXBC2 cells culturing with doxycycline for FMC63-IgG4ζ CAR (data not shown). Total numbers of each cell were determined by MACSQuant (Milteny Biotech, Germany) and relative cytotoxicity of target cells relative to non-target cells was determined by the following formula: Relative cytotoxicity (%) =100 x (1 – target cell number/non-target cell number).
Viruses
Lentiviral vectors were generated and transduced as described elsewhere (27, 34, 35). p24Gag ELISA assays were performed by the CFAR Virology Core at UCLA. Lentiviral vector information used in this research will be provided upon request.
Flow cytometry
The following antibodies were used in flow cytometry: BV711-CD4 (OKT4), BV605-CD8 (RPA-T8), APC/Cy7-CD45RO (UCHL1), AlexaFlour700-CD45RA (H100), PerCP/Cy5.5-CD62L (DREG-56), BV785-CCR7 (G043H7) (all from BioLegend), eFluor650NC-CD3 (OKT3; eBioscience, San Diego, CA), APC-CD95 (LT95; Thermo Fisher, Pittsburgh, PA), and BV421-CD122 (Mik-β3; BD Biosciences, San Jose, CA). Cells were acquired using FACSDiva on BD LSRFortessa. Data for each cell with different cell surface phenotypes were analyzed using FlowJo software (BD Biosciences) as summarized in Supplementary Figure 2. Absolute cell counts were determined using MACSQuant analyzer. Cell sorting was performed by the CFAR Flow Cytometry Core Facility at UCLA.
In vivo tumor-killing assay
Animal research was conducted under UCLA’s Chancellor’s Animal Research Committee. Two million of TF228.1.16 or HXBC2 cells in 50 μL of PBS were mixed with 50 μL of Matrigel (BD Biosciences) and subcutaneously engrafted to the left or right hind limbs of NOD.CB17-Prkdcscid/J (NOD-SCID) mice (n=4) (Jackson Laboratory, Bar Harbor, Maine), respectively. On day 14, either Triple-CD4ζ- or FMC63-IgG4ζ–modified T cells (5x105) were infused via the retro-orbital vein. Biofluorescence images and the weight of xenograft tumors were obtained 42 days post-engraftment.
Statistical analyses
Results are expressed as mean ± standard deviations (SDs). Errors depict SD. Comparisons between two groups were performed using an unpaired two-tailed t-test with Welch’s correction. A p-value less than 0.05 was considered statistically significant.
Results
Stimulation of CD8+ TN cells with soluble anti-CD3 antibody enables efficient gene-modification by lentiviral vector while minimizing T-cell differentiation
Two independent protocols for the derivation of TSCM have been described by Gattinoni et al. (18) and Cieri et al. (19). The major differences between those two protocols are that the former used an inhibitor of glycogen synthase kinase 3β (GSK-3β), TWS119 (15, 36), and the latter used a low concentration of two cytokines, IL-7 and IL-15 with no chemical inhibitor. Our preliminary experiments for a side-by-side comparison of those two protocols showed that there was minimal benefit of the use of TWS119 for the derivation of TSCM cells due to a poor cell expansion by the former protocol. Whereas, the latter protocol was able to yield a nearly 5-fold higher number of cells harboring surface phenotypes corresponding to TSCM (0.41 x 106 vs 1.88 x 106, Supplementary Table 1). However, a considerable number of TEM cells were derivated under both conditions, presumably due to the potent stimulation condition of using anti-CD3/CD28 antibody coated beads. We thus assessed alternative stimulation conditions for their ability to derive large numbers of TSCM while minimizing T-cell differentiation and allowing for gene modification via lentiviral vectors. Negatively selected CD8+ TN cells were incubated with different amounts of soluble anti-CD3 antibody with or without anti-CD28 antibody. The cells were then transduced with a lentiviral vector encoding EGFP as a marker gene and cultured for an additional 12 days with IL-7 and IL-15 followed with Cieri’s protocol (Figure 1A). In the no antibody condition, approximately 20% of CD8+ TN cells were successfully gene-modified, indicating that a small portion of CD8+ T cells are transducable without active stimulation and that culturing in the presence of IL-7 and IL-15 allows maintenance of the CD8+ TN cell phenotype for at least 14 days (Figure 1B). However, the no antibody condition resulted in extremely poor expansion of the cells (<2-fold, Figure 1C). The inclusion of increasing concentrations of anti-CD3 antibody and/or the presence of anti-CD28 antibody increased the levels of gene marking and T-cell expansion (Figure 1C), but also increased the proportion of differentiated cells as defined by the loss of CD45RA or CD62L expression (37–39) (Figures 1D, 2A). The differences shown above became more obvious after an additional 14 days of culture (total 28 days of culture) (Figure 2B, Day 28), indicating that cells harboring TSCM-surface phenotype slowly but continuously expanded in the presence of IL-7 and IL-15. The average numbers from each population in EGFP-positive cells after a total of 28 days of culture in Figure 2B were summarized in Supplementary Table 2: a higher recovery of the EGFP-marked TSCM cells was observed with 0.5 µg (7.35 x 106) or 1.0 µg/mL anti-CD3 (7.84 x 106) stimulation rather than the condition with 0.5 µg/mL anti-CD3 + 2.0 µg of anti-CD28 antibody (5.61 x 106), which generated the highest gene marking as in Figure 1C. Importantly, EGFP-marked TSCM cells were expanded nearly 2-fold following a total of 28 days of culture, whereas the number of TSCM cells in other populations decreased over the course of the culture (Supplementary Figure 3). These results indicate that stimulation of CD8+ TN cells with only soluble anti-CD3 antibody (0.5-1.0 µg/mL) and prolonged-expansion (28 days) in the presence of IL-7 and IL-15 allowed for the most efficient derivation of gene-marked CD8+ T cells harboring TSCM-surface phenotype with a minimum level of T-cell differentiation.
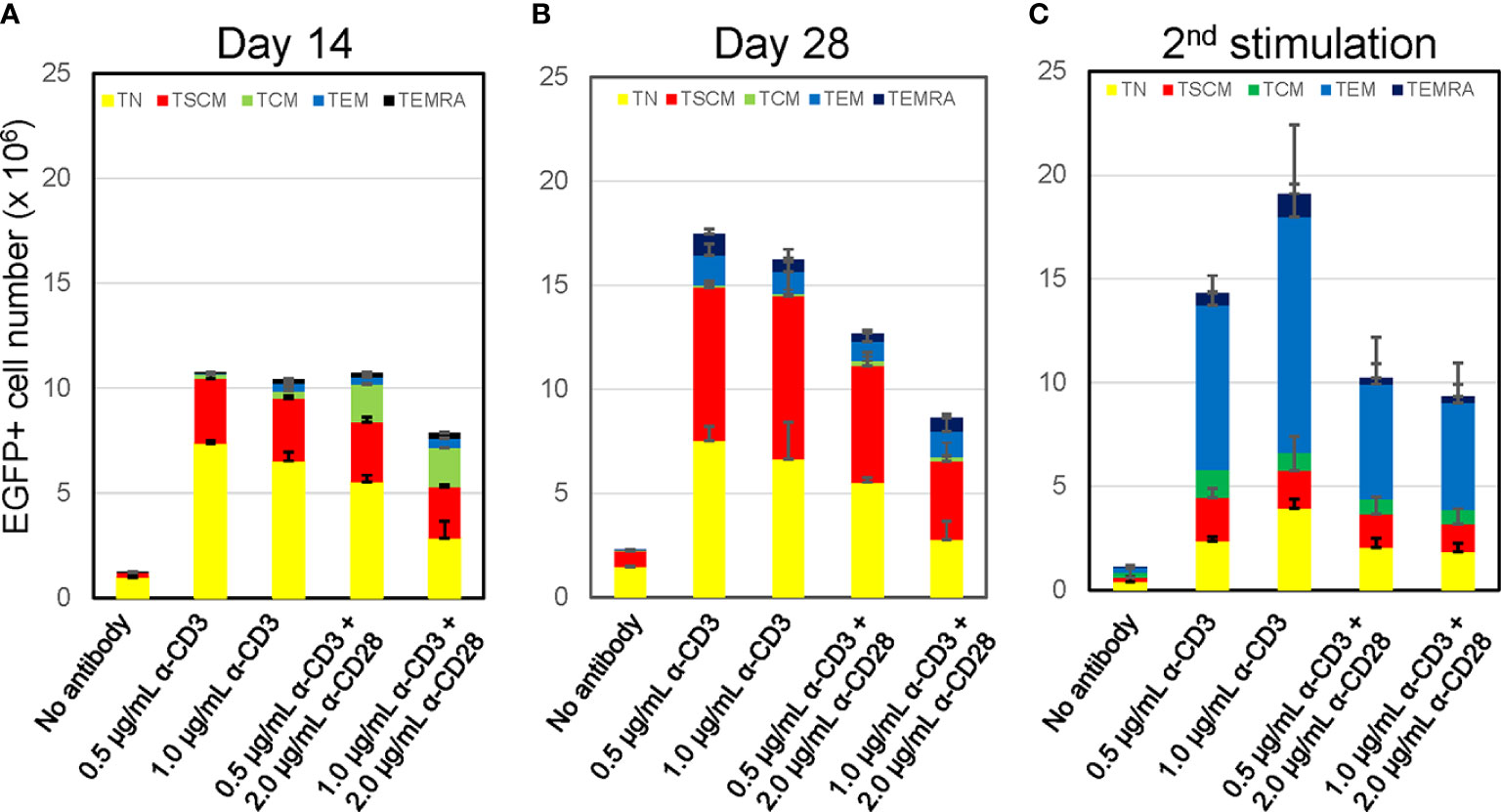
Figure 2 Prolonged culture increases % population of CD8+ T cells harboring TSCM-surface phenotype with maintaining oligopotency Freshly isolated CD8+ TN cells were transduced with lentiviral vector encoding EGFP following stimulation with various antibody conditions shown in Figure 1A. The cells were cultured for an additional 12 days (A, Day14) or 26 days (B, Day 28) in the presence of 5 ng/mL of IL-7 and IL-15. A half million of cells in A was co-stimulated by 0.5 μg/mL anti-CD3 and 2.0 μg/mL anti-CD28 antibodies and further cultures for 14 days (C, 2nd stimulation). Cells were staining for CD45RA, CD45RO, CCR7, CD62L, CD95 and CD122, and surface marker of EGFP-marked cells were analyzed by flow cytometry. Each cell number of EGFP-marked cells with TN, TSCM, TCM, TEM, and TEMRA phenotypes was plotted. Experiments were repeated three times. Error bars show the standard deviation of a data set.
A key feature of the TSCM subset is its oligopotency allowing differentiation into more terminal subsets upon antigen stimulation (18, 19). We confirmed the differentiation ability of these in-vitro generated TSCM cells via anti-CD3/CD28 co-stimulation. The status of T-cell differentiation was evaluated by surface phenotypes at 14-days after the 2nd stimulation (Figure 2C, 2nd stimulation). The number and prevalence of CD8+ T cells harboring the TSCM and TN phenotype was substantially reduced after the 2nd stimulation. Concomitantly, the proportion of differentiated cells ─ especially the TEM population ─ increased under this condition. Thus, these in-vitro generated TSCM cells retain their oligopotency.
Derivation of CAR-modified CD8+ T cells with TSCM-surface phenotype
As above, we were able to validate the oligopotency of the in-vitro generated gene-engineered cells with TSCM-surface phenotypes. We next applied this method for the generation of anti-HIV-1 CAR-T cells. We utilized the Triple-CD4ζ CAR that targets HIV-1 gp120 on the infected cell surface (27) with co-expression of two anti-HIV-1 shRNAs: sh1005 which suppresses surface expression of the key HIV-1 co-receptor CCR5 (40) and sh516 which prevents HIV-1 infection to both CD4+ T and CD8+ T cells mediated via CD4ζ expression (34). Thus, the Triple-CD4ζ CAR can exert potent anti-HIV-1-effector activity while protecting its transduced cells from HIV-1 infection. We previously demonstrated its potent anti-HIV-1 effects in vivo using an HIV-1 challenged-humanized mouse model (41, 41). Although the condition established above allowed successful modification of CD8+ TN cells with the Triple-CD4ζ CAR while maintaining a population with the TSCM-surface phenotype, the proportion of transduced cells was relatively lower compared to that of control EGFP vector (42.9% vs 8.8%, Supplementary Figure 4). This is potentially due to a larger size of the transgene (42). Nevertheless, these gene-marked cells were successfully differentiated to cells harboring surface phenotypes of TCM and TEM cells upon anti-CD3/CD28 stimulation (Figure 3A).
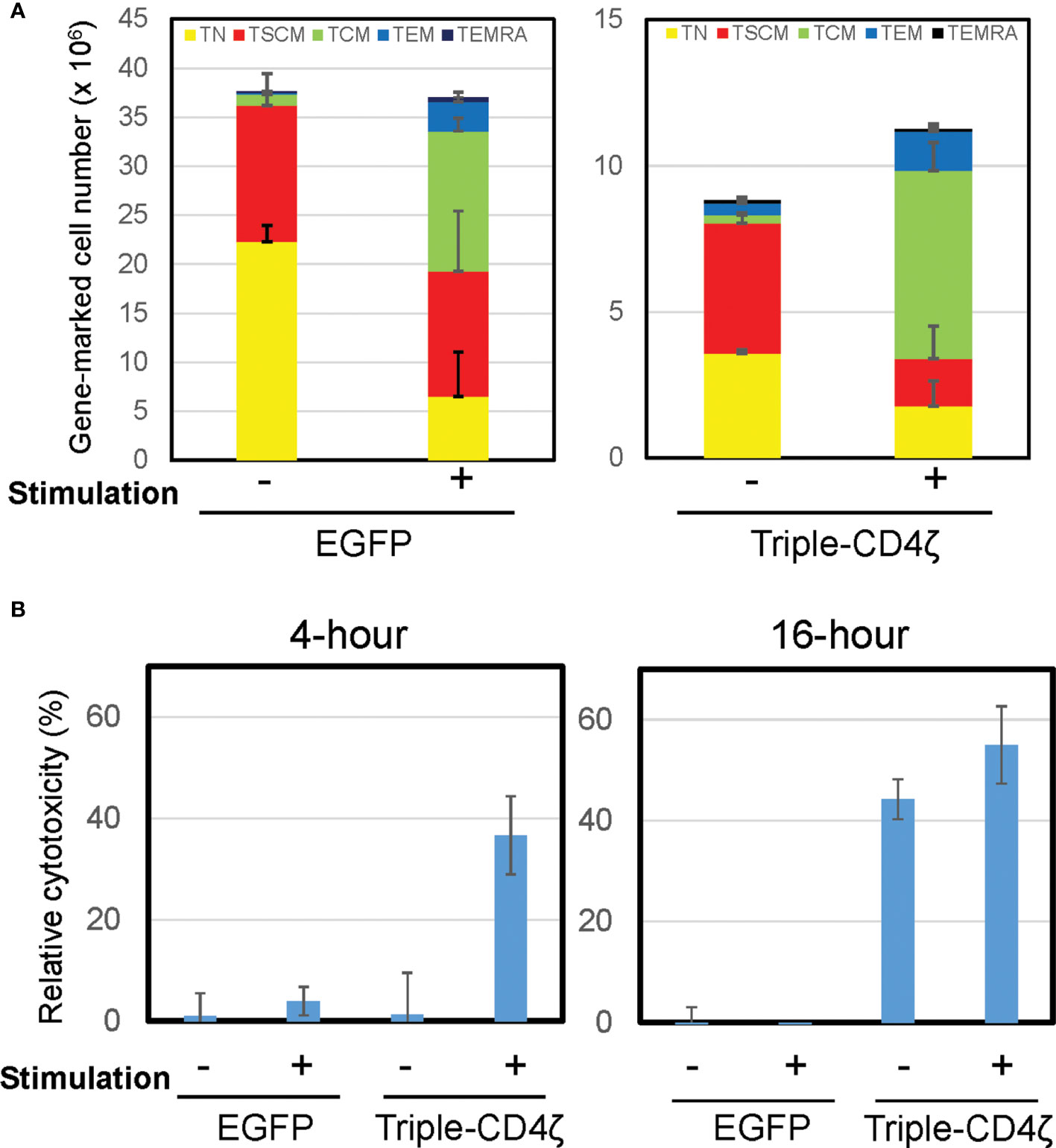
Figure 3 Induced differentiation of Triple-CD4ζ modified CD8+ T cells harboring TSCM-surface phenotype with anti-CD3 and CD28 co-stimulation. (A) Triple-CD4ζ or EGFP-modified CD8+ T cells were co-stimulated with 0.5 µg/mL of anti-CD3 and 2.0 µg/mL of CD28 antibodies for 2 days (2nd stimulation) at day14 post-1st stimulation. The cells were further cultured for an additional 12 days in the presence of 5 ng/mL of IL-7 and IL-15. Cell-surface profiles of gene-marked CD8+ T cells were analyzed by flow cytometry. Each cell number of EGFP- or Triple-CD4ζ-marked cells with TN, TSCM, TCM, TEM, and TEMRA phenotypes was plotted with or without 2nd stimulation. Experiments were repeated three times. Error bars show the standard deviation of a data set. (B) The cells were plated at 5 x 104 cells/100 μL in a 96-well plate. The same number of TagBFP-labeled Jurkat cells (ΔKS, non-target control) and mCherry-labeled Jurkat cells constitutively expressing HIV-1HXBC2 envelope protein (HXBC2, target cells) were added to the wells and incubated for 4 or 16 hours. Total numbers of each population were determined by MACSQuant, and relative cytotoxicity of target cells relative to non-target cells was determined. -: unstimulated, +: 2nd stimulated. Experiments were repeated three times. Error bars show the standard deviation of a data set. Cytotoxicity assays were performed in biological triplicate.
We next assessed the effector function of Triple-CD4ζ-modified CD8+ T cells by a flow-cytometry based cytotoxicity assay. The cells modified with Triple-CD4ζ cells were incubated with their target Jurkat cells expressing HIV-1HXBC2 envelope protein (HXBC2) upon doxycycline removal from culture medium (28). To validate the level of non-specific target cell killing, control Jurkat cells which do not encode HIV-1-envelope protein (ΔKS, non-target cell) were included (28). The former is labeled by mCherry and the latter by Tag-BFP, thus both cells can be distinguished by flow cytometry. The numbers of remaining mCherry+ and TagBFP+ cells following incubation with Triple-CD4ζ-modified CD8+ T cells correspond to levels of anti-HIV-1 CAR-dependent and independent cytotoxicity, respectively. Following a co-culture with the above three cells for 4 and 16 hours, the total numbers of mCherry+ and TagBFP+ cells were analyzed by MACSQuant, and relative cytotoxicity against target cells compared to that against non-target cells was determined (Figure 3B). The pre-differentiated cells shown in Figure 3A were used as a positive control for the assay. There were minimal levels of change in the cell surface phenotypes of Triple-CD4ζ following 4-hour and 16-hour incubations with the target cells (Supplementary Figure 5). A 4-hour incubation was sufficient for the induction of cytotoxic activity by the pre-differentiated cells transduced with Triple-CD4ζ, whereas a 16-hour incubation was required to achieve a similar level of cytotoxicity without pre-differentiation, indicating that longer incubation was required to exert an effector activity for undifferentiated Triple-CD4ζ-modified CD8+ T cells.
Additionally, we generated anti-CD19 (FMC63-IgG4ζ) modified CD8+ T cells using the same conditions as above (Supplementary Figure 6). The level of gene marking by FMC63-IgG4ζ was similarly lower than control vector as seen with Triple-CD4ζ. FMC63-IgG4ζ-modified cells also maintained high levels of expression for TSCM cell markers, and a large portion of the cells harbored TN- and TSCM-surface phenotypes. Similar to Triple-CD4ζ modified-CD8+ T cells, FMC63-IgG4ζ-modified cells also exerted a potent antigen dependent cytotoxicity following 16, but not 4, hours of incubation.
Triple-CD4ζ CAR-modified CD8+ T cells generated with sole anti-CD3 antibody stimulation exert antigen-specific effector functions in a xenograft NOD-SCID mouse model
As above, CD8+ CAR-T cells carrying the TSCM-surface phenotype were generated in vitro with an antigen-specific effector activity. We next tested their effector activity in vivo using a xenograft NOD-SCID mouse model. NOD-SCID mice were engrafted with two different lymphoma cell lines: TF228.1.16 ― a derivative of BJAB (29) or HXBC2 ― a derivative of Jurkat (28). These lines express envelope protein from either the HIV-1BH10 or HIV-1HXBC2 strains, respectively. In vitro, the TF228.1.16 cells were killed by Triple-CD4ζ-modified CD8+ T cells like the HXBC2 cells, but relatively weakly by FMC63-IgG4ζ-modified CD8+ T cells. Although TF228.1.16 is CD19+ and can be a target for FMC63-IgG4ζ-modified CD8+ T cells in vitro (Supplementary Figure 7), our FMC63-IgG4ζ construct contains an IgG hinge-CH2-CH3 domain, which makes ineffective this CAR in vivo due to an Fcγ receptor 1 mediated CAR-T cell elimination (43–46). Thus, the FMC63-IgG4ζ serves as negative control. Those two tumor cells were genetically marked by mStrawberry (TF228.1.16) or mCherry (HXBC2), and subcutaneously engrafted into the left and right hind limbs of NOD-SCID mice, respectively. We confirmed that the expression levels of HIV-1 envelope proteins in tumor tissues developed in xenograft mice were similar to those in cells maintained in vitro by western blotting using anti-GP120 (2G12) and anti-GP41 (2F5) antibodies (data not shown).
At 14 days post-engraftment, we infused CD8+ T cells engineered with either Triple-CD4ζ CAR or FMC63-IgG4ζ CAR derived as above. We analyzed these CAR-dependent effector activities by bioimaging using a Maestro 2 multispectral imaging system (Figure 4). As expected, we observed poor in vivo effector activity of the cells modified by FMC63-IgG4ζ CAR. On the other hand, the cells modified by Triple-CD4ζ CAR showed stronger anti-tumor effector activity against both tumors, with tumor burdens decreasing approximately 1.9-fold for TF228.1.16 tumors (blue bars, Figure 4B) and 5.5-fold for HXBC2 tumors (orange bars, Figure 4B). Compared to HXBC2 tumor, the effector activity on TF228.1.16 tumor was weaker with no statistical significance; this may be due to the faster growth rate of TF228.1.16 cells. These results provide additional evidence that our simplified protocol allows for the derivation of CAR-modified CD8+ T cells harboring TSCM-surface phenotype while maintaining antigen-specific effector activity in vivo.
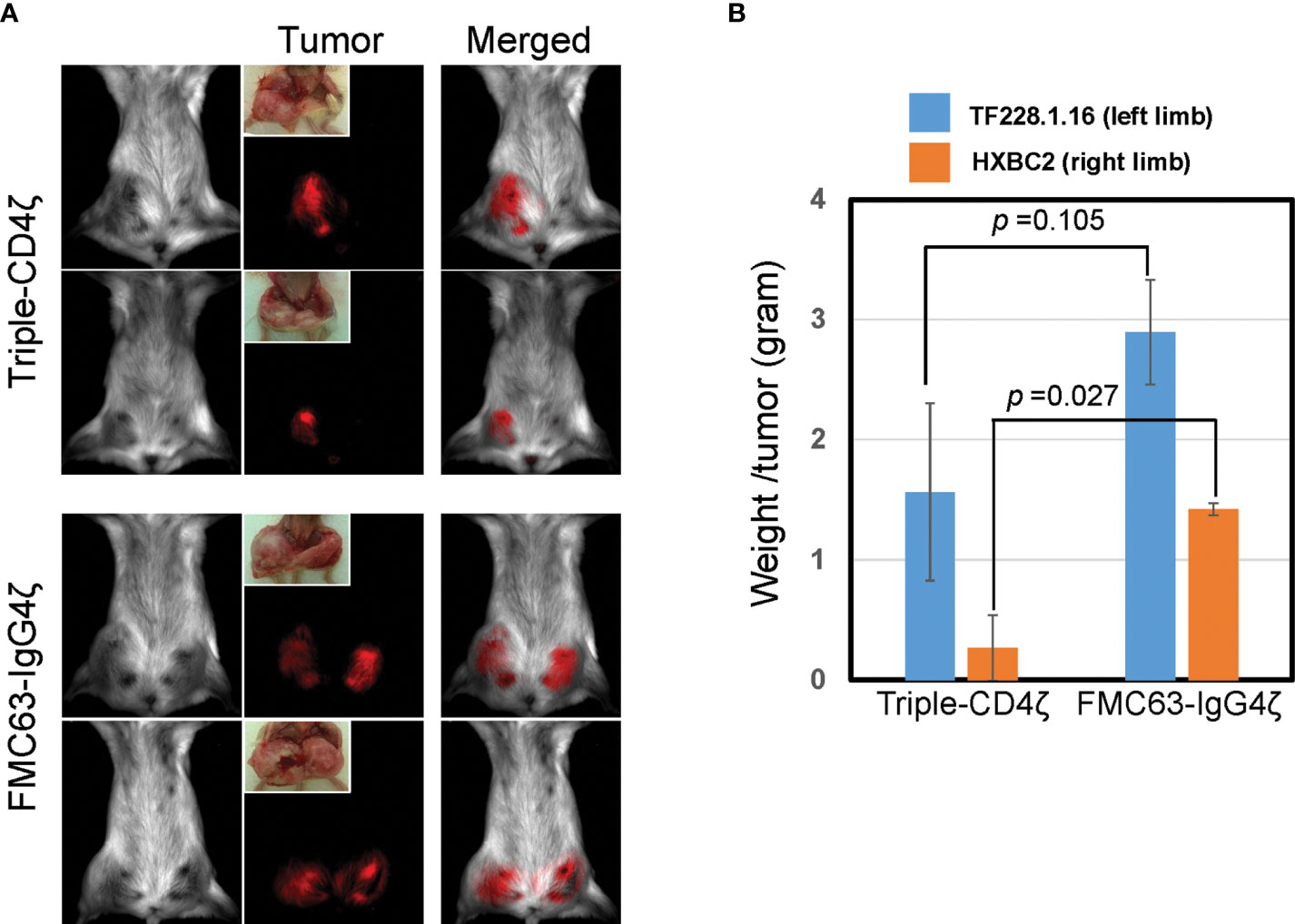
Figure 4 Triple-CD4ζ CAR-modified CD8+ T cells harboring TSCM-surface phenotype eliminate tumor cells expressing HIV-1 envelope proteins in a xenograft mouse model. Two million mStrawberry-labeled CD19+ TF228.1.16 cells expressing envelope protein from HIV-1BH10 or mCherry-labeled Jurkat cells expressing envelope protein from HIV-1HXBC2 (HXBC2), were mixed with Matrigel at a 1:1 ratio and subcutaneously engrafted to the left or right hind limbs of NOD-SCID mice from ventral side, respectively (n = 4). Freshly isolated CD8+ TN cells were stimulated for 2 days with 0.5 µg/mL of anti-CD3 antibody in T-cell medium, followed by transduction with a lentiviral vector encoding either Triple-CD4ζ or FMC63-IgG4ζ. Following 26 days of culture in the presence of 5 ng/mL of IL-7 and IL-15, cells corresponding to 5 x 105 CAR-modified cells were intravenously injected via the retro-orbital vein on day 14 post-engraftment of TF228.1.16 and HXBC2. Biofluorescence images (A) and the weight of xenograft tumors (B) were obtained on day 42 post-engraftment (on day 28 post-transplant of CAR cells). (B) Blue or orange bars: average weight of xenograft tumors from TF228.1.16 (left limb, blue bars) or HXBC2 (right limb, orange bars) on day 42 post-engraftment. Experiments were repeated three times with similar results. Two representative animals from each group were shown.
Discussion
Though there is currently no standardized protocol for CAR-TSCM cell manufacturing, the use of CAR-TSCM products represents a promising approach for improving the outcome of CAR-T based therapies. Here, we refined the protocol for the derivation of CAR-modified CD8+ T cells harboring TSCM-surface phenotype and TSCM-like oligopotency.
The first key to generating CAR-TSCM-like cells is the purification of CD8+ TN cells as a starting population. Flow-cytometry based cell sorting is commonly used to isolate this population, but has 2 major drawbacks: decreased cell viability (47, 48), and the potential for sorting antibodies to induce unwanted activation via binding to TN cell surface markers. By this reasoning, negative selection may be more effective for isolating TN populations because these processes leave the cells ‘untouched’ and also without any antibodies remaining bound to the cells in the final product. By using an EasySep™ Human Naïve CD8+ T Cell Enrichment Kit which enables negative selection of CD8+ TN cells, we consistently obtained CD8+ TN cells with >95% purity (Supplementary Figure 1).
Next, we evaluated the utility of different stimulation conditions for generating gene-modified TSCM-like CD8+ T cells and found that stimulation with only soluble anti-CD3 antibody in the presence of IL-7 and IL-15 was the most efficient. Since this condition does not require the use of novel agents, it is readily translatable for clinical applications. Cieri et al. also tested a stimulation condition with only anti-CD3 antibody for expansion of cells with a CD8+/CD45RA+/CD62L+/CD95- phenotype, but this attempt was unsuccessful (19). There were several differences compared to our protocol; they used a different anti-CD3 antibody clone ― OKT3 ― at >16-times lower concentration (30 ng/mL) in a plate-bound format. With this condition, they observed a poor expansion of CD8+ TN cells compared to the cells co-stimulated with anti-CD3/CD28 antibody coated beads. Surprisingly, most of the cells lost expression of CD45RA and CD62L after 16 days of culture with their experimental conditions (19). We did not observe this by the use of soluble form of anti-CD3 antibody, suggesting that the precoated anti-CD3 antibodies probably induced a stronger stimulation via TCR crosslinking, resulting in T-cell differentiation.
Whereas both CD4+ and CD8+ T cells are needed for an efficient CAR-T cell therapy (49–51); the former mainly serve as CAR-dependent helper cells and the latter as CAR-dependent effector cells (52). We have already adopted the derivation of CAR-modified CD4+ TSCM cells by the use of EasySep Human Naive CD4+ T cell Isolation kit, which enables negative selection of CD4+ TN cells (Stemcell technologies, #19555). Unlike CD8+ TN cells, CD4+ TN cells required co-stimulation by soluble forms of anti-CD3 (1.0 μg/mL) and anti-CD28 (0.1 μg/mL) antibodies for an efficient expansion of cells with TSCM-surface phenotype (data not shown). The level of gene marking as well as oligopotential activity of CAR-engineered CD4+ TSCM cells were similarly reproduced as those confirmed in CAR-engineered CD8+ TSCM cells. We are currently validating the effector activity of CAR-engineered CD4+ T cells as well as seeking to derivate CAR-engineered TSCM cells from negatively selected naive Pan-T cells using EasySep Human Naive Pan T cell Isolation kit (Stemcell technology, #17961) without pre-separating CD4+ or CD8+ TN cells. We believe that these approaches will enable more efficient and reproducible procedures for the manufacturing of highly effective CAR-T cells.
CAR-modified CD8+ T cells harboring TSCM-surface phenotype required longer incubation to exert CAR-dependent effector activity compared to the pre-differentiated cells (16 hours vs 4 hours, Figures 3B and Supplementary Figure 6B). The pre-differentiated cells consisted of more differentiated cells, which have potent cytotoxicity than cells harboring TSCM cell phenotype. We also confirmed that 16 hours of incubation was not sufficient to fully induce differentiation of the cells with TSCM-surface phenotype to cells with differentiated phenotypes, such as TCM or TEM (Supplementary Figure 5). We thus expected the cells derived with our protocol to have less effector activity than those with differentiated phenotypes, and to require longer incubation before acquiring cytotoxicity. These possibilities are currently under investigation.
In recent years, immunotherapy utilizing CAR-engineered T cells has become a highly promising approach, especially for the treatment of blood cancers. TSCM cells have the capacity for both self-renewal and oligopotent differentiation into effector cells upon encounter with antigens; thus, TSCM cells re-directed against their targets could be more effective than mature T cells employed in current clinical trials. Due to the low frequency of TSCM cells in peripheral blood, establishing techniques for efficient expansion with high levels of gene modification will be important for translation to clinical purposes. Our protocol described here is applicable for a prompt implementation of TSCM cell-based immunotherapies against not only cancer but also infectious diseases such as AIDS.
Data availability statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Ethics statement
The animal study was reviewed and approved by Guido Eibl, UCLA.
Author contributions
EK, CJK, wrote the paper. EK, JC, and PYK performed the experiments. ISY, interpreted data. MK designed the research, performed the experiments, analyzed data, interpreted data, and wrote the paper. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the California HIV/AIDS Research Grants Program ID13-LA-563 (M.K.), NIH grants R01AI110200 (MK), and RO1 CA232015 (MK). The following reagents were obtained through the NIH AIDS Reagent Program, Division of AIDS, NIAID, NIH: BCBL-1 (Drs. Michael McGrath and Don Ganem), Jurkat-ΔKS and Jurkat-HXBC2 (4) (Dr. Joseph Sodroski), and TF228.1.16 (Drs. Zdenka Jonak and Steve Trulli). Maestro 2 multispectral imaging system was performed at the CNSI Advanced Light Microscopy/Spectroscopy Shared Resource Facility at UCLA, supported by NIH-NCRR shared resources grant (CJX1-443835-WS-29646) and NSF Major Research Instrumentation grant (CHE-0722519).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2022.877682/full#supplementary-material
Abbreviations
TSCM, stem cell memory T; CAR, chimeric antigen receptor; HIV-1, human immunodeficiency virus type 1.
References
1. Wilkie S, van Schalkwyk MC, Hobbs S, Davies DM, van der Stegen SJ, Pereira AC, et al. Dual targeting of ErbB2 and MUC1 in breast cancer using chimeric antigen receptors engineered to provide complementary signaling. J Clin Immunol (2012) 32:1059–70. doi: 10.1007/s10875-012-9689-9
2. June CH. Adoptive T cell therapy for cancer in the clinic. J Clin Invest (2007) 117:1466–76. doi: 10.1172/JCI32446
3. Gattinoni L, Powell DJ Jr., Rosenberg SA, Restifo NP. Adoptive immunotherapy for cancer: building on success. Nat Rev Immunol (2006) 6:383–93. doi: 10.1038/nri1842
4. Gill S, Maus MV, Porter DL. Chimeric antigen receptor T cell therapy: 25years in the making. Blood Rev (2016) 30:157–67. doi: 10.1016/j.blre.2015.10.003
5. Lindner SE, Johnson SM, Brown CE, Wang LD. Chimeric antigen receptor signaling: Functional consequences and design implications. Sci Adv (2020) 6:eaaz3223. doi: 10.1126/sciadv.aaz3223
6. Cappell KM, Kochenderfer JN. A comparison of chimeric antigen receptors containing CD28 versus 4-1BB costimulatory domains. Nat Rev Clin Oncol (2021) 18:715–27. doi: 10.1038/s41571-021-00530-z
7. Porter DL, Hwang WT, Frey NV, Lacey SF, Shaw PA, Loren AW, et al. Chimeric antigen receptor T cells persist and induce sustained remissions in relapsed refractory chronic lymphocytic leukemia. Sci Transl Med (2015) 7:303ra139. doi: 10.1126/scitranslmed.aac5415
8. Maude SL, Frey N, Shaw PA, Aplenc R, Barrett DM, Bunin NJ, et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N Engl J Med (2014) 371:1507–17. doi: 10.1056/NEJMoa1407222
9. Kochenderfer JN, Dudley ME, Feldman SA, Wilson WH, Spaner DE, Maric I, et al. B-cell depletion and remissions of malignancy along with cytokine-associated toxicity in a clinical trial of anti-CD19 chimeric-antigen-receptor-transduced T cells. Blood (2012) 119:2709–20. doi: 10.1182/blood-2011-10-384388
10. Kochenderfer JN, Dudley ME, Kassim SH, Somerville RP, Carpenter RO, Stetler-Stevenson M, et al. Chemotherapy-refractory diffuse large b-cell lymphoma and indolent b-cell malignancies can be effectively treated with autologous T cells expressing an anti-CD19 chimeric antigen receptor. J Clin Oncol (2015) 33:540–9. doi: 10.1200/JCO.2014.56.2025
11. Mitsuyasu R, Anton P, Deeks S, Scadden D, Connick E, Downs M, et al. Prolonged survival and tissue trafficking following adoptive transfer of CD4zeta gene-modified autologous CD4(+) and CD8(+) T cells in human immunodeficiency virus-infected subjects. Blood (2000) 96:785–93. doi: 10.1182/blood.V96.3.785.015k10_785_793
12. Deeks S, Wagner B, Anton P, Mitsuyasu R, Scadden D, Huang C, et al. A phase II randomized study of HIV-specific T-cell gene therapy in subjects with undetectable plasma viremia on combination antiretroviral therapy. Mol Ther (2002) 5:788–97. doi: 10.1006/mthe.2002.0611
13. Mitsuyasu R, Zack J, Macpherson J, Symonds G. Phase I/II clinical trials using gene-modified adult hematopoietic stem cells for HIV: Lessons learnt. Stem Cells Int (2011). vol. 2011, 393698–705. doi: 10.4061/2011/393698
14. Scholler J, Brady T, Binder-Scholl G, Hwang W, Plesa G, Hege K, et al. Decade-long safety and function of retroviral-modified chimeric antigen receptor T cells. Sci Trans Med (2012) 4:132ra53. doi: 10.1126/scitranslmed.3003761
15. Gattinoni L, Zhong XS, Palmer DC, Ji Y, Hinrichs CS, Yu Z, et al. Wnt signaling arrests effector T cell differentiation and generates CD8+ memory stem cells. Nat Med (2009) 15:808–13. doi: 10.1038/nm.1982
16. Lugli E, Dominguez M, Gattinoni L, Chattopadhyay P, Bolton D, Song K, et al. Superior T memory stem cell persistence supports long-lived T cell memory. J Clin Invest (2013) 123:594–9. doi: 10.1172/JCI66327
17. Lugli E, Gattinoni L, Roberto A, Mavilio D, Price D, Restifo N, et al. Identification, isolation and in vitro expansion of human and nonhuman primate T stem cell memory cells. Nat Protocol (2013) 8:33–42. doi: 10.1038/nprot.2012.143
18. Gattinoni L, Lugli E, Ji Y, Pos Z, Paulos C, Quigley M, et al. A human memory T cell subset with stem cell-like properties. Nat Med (2011) 17:1290–7. doi: 10.1038/nm.2446
19. Cieri N, Camisa B, Cocchiarella F, Forcato M, Oliveira G, Provasi E, et al. IL-7 and IL-15 instruct the generation of human memory stem T cells from naive precursors. Blood (2013) 121:573–84. doi: 10.1182/blood-2012-05-431718
20. Li Y, Wu D, Yang X, Zhou S. Immunotherapeutic potential of T memory stem cells. Front Oncol (2021) 11:723888. doi: 10.3389/fonc.2021.723888
21. Costa Del Amo P, Lahoz-Beneytez J, Boelen L, Ahmed R, Miners KL, Zhang Y, et al. Human TSCM cell dynamics in vivo are compatible with long-lived immunological memory and stemness. PloS Biol (2018) 16:e2005523. doi: 10.1371/journal.pbio.2005523
22. Gattinoni L, Restifo N. Moving T memory stem cells to the clinic. Blood (2013) 121:567–8. doi: 10.1182/blood-2012-11-468660
23. Gattinoni L, Speiser DE, Lichterfeld M, Bonini C. T Memory stem cells in health and disease. Nat Med (2017) 23:18–27. doi: 10.1038/nm.4241
24. Henning AN, Klebanoff CA, Restifo NP. Silencing stemness in T cell differentiation. Science (2018) 359:163–4. doi: 10.1126/science.aar5541
25. Biasco L, Scala S, Basso Ricci L, Dionisio F, Baricordi C, Calabria A, et al. In vivo tracking of T cells in humans unveils decade-long survival and activity of genetically modified T memory stem cells. Sci Transl Med (2015) 7:273ra13. doi: 10.1126/scitranslmed.3010314
26. Biasco L, Izotova N, Rivat C, Ghorashian S, Richardson R, Guvenel A, et al. Clonal expansion of T memory stem cells determines early anti-leukemic responses and long-term CAR T cell persistence in patients. Nat Cancer (2021) 2:629–42. doi: 10.1038/s43018-021-00207-7
27. Kamata M, Kim P, Ng H, Ringpis G, Kranz E, Chan J, et al. Ectopic expression of anti-HIV-1 shRNAs protects CD8+ T cells modified with CD4ζ CAR from HIV-1 infection and alleviates impairment of cell proliferation. Biochem Biophys Res Commun (2015) 463:216–21. doi: 10.1016/j.bbrc.2015.05.026
28. Cao J, Park IW, Cooper A, Sodroski J. Molecular determinants of acute single-cell lysis by human immunodeficiency virus type 1. J Virol (1996) 70:1340–54. doi: 10.1128/jvi.70.3.1340-1354.1996
29. Jonak ZL, Clark RK, Matour D, Trulli S, Craig R, Henri E, et al. A human lymphoid recombinant cell line with functional human immunodeficiency virus type 1 envelope. AIDS Res Hum Retroviruses (1993) 9:23–32. doi: 10.1089/aid.1993.9.23
30. Subach OM, Gundorov IS, Yoshimura M, Subach FV, Zhang J, Gruenwald D, et al. Conversion of red fluorescent protein into a bright blue probe. Chem Biol (2008) 15:1116–24. doi: 10.1016/j.chembiol.2008.08.006
31. Shaner N, Campbell R, Steinbach P, Giepmans B, Palmer A, Tsien R. Improved monomeric red, orange and yellow fluorescent proteins derived from discosoma sp. red fluorescent protein. Nat Biotechnol (2004) 22:1524–5. doi: 10.1038/nbt1037
32. Kochenderfer JN, Feldman SA, Zhao Y, Xu H, Black MA, Morgan RA, et al. Construction and preclinical evaluation of an anti-CD19 chimeric antigen receptor. J Immunother (2009) 32:689–702. doi: 10.1097/CJI.0b013e3181ac6138
33. Renne R, Zhong W, Herndier B, McGrath M, Abbey N, Kedes D, et al. Lytic growth of kaposi's sarcoma-associated herpesvirus (human herpesvirus 8) in culture. Nat Med (1996) 2:342–6. doi: 10.1038/nm0396-342
34. Ringpis G, Shimizu S, Arokium H, Camba-Colón J, Carroll M, Cortado R, et al. Engineering HIV-1-resistant T-cells from short-hairpin RNA-expressing hematopoietic stem/progenitor cells in humanized BLT mice. PloS One (2012) 7:e53492. doi: 10.1371/journal.pone.0053492
35. Kamata M, Nagaoka Y, Chen I. Reassessing the role of APOBEC3G in human immunodeficiency virus type 1 infection of quiescent CD4+ T-cells. PloS Pathog (2009) 5:1000342. doi: 10.1371/journal.ppat.1000342
36. Schroeder JH, Bell LS, Janas ML, Turner M. Pharmacological inhibition of glycogen synthase kinase 3 regulates T cell development in vitro. PloS One (2013) 8:e58501. doi: 10.1371/journal.pone.0058501
37. Brinkman CC, Rouhani SJ, Srinivasan N, Engelhard VH. Peripheral tissue homing receptors enable T cell entry into lymph nodes and affect the anatomical distribution of memory cells. J Immunol (2013) 191:2412–25. doi: 10.4049/jimmunol.1300651
38. Butcher EC, Picker LJ. Lymphocyte homing and homeostasis. Science (1996) 272:60–6. doi: 10.1126/science.272.5258.60
39. Gunn MD, Kyuwa S, Tam C, Kakiuchi T, Matsuzawa A, Williams LT, et al. Mice lacking expression of secondary lymphoid organ chemokine have defects in lymphocyte homing and dendritic cell localization. J Exp Med (1999) 189:451–60. doi: 10.1084/jem.189.3.451
40. An D, Donahue R, Kamata M, Poon B, Metzger M, Mao S, et al. Stable reduction of CCR5 by RNAi through hematopoietic stem cell transplant in non-human primates. Proc Natl Acad Sci U S A. (2007) 104:13110–5. doi: 10.1073/pnas.0705474104
41. Zhen A, Kamata M, Rezek V, Rick J, Levin B, Kasparian S, et al. HIV-Specific immunity derived from chimeric antigen receptor-engineered stem cells. Mol Ther (2015) 23:1358–67. doi: 10.1038/mt.2015.102
42. N. al Yacoub M, Haritonova N. And j. foerster, optimized production and concentration of lentiviral vectors containing large inserts. J Gene Med (2007) 9:579–84. doi: 10.1002/jgm.1052
43. Hudecek M, Sommermeyer D, Kosasih PL, Silva-Benedict A, Liu L, Rader C, et al. The nonsignaling extracellular spacer domain of chimeric antigen receptors is decisive for in vivo antitumor activity. Cancer Immunol Res (2015) 3:125–35. doi: 10.1158/2326-6066.CIR-14-0127
44. Jonnalagadda M, Mardiros A, Urak R, Wang X, Hoffman LJ, Bernanke A, et al. Chimeric antigen receptors with mutated IgG4 fc spacer avoid fc receptor binding and improve T cell persistence and antitumor efficacy. Mol Ther (2015) 23:757–68. doi: 10.1038/mt.2014.208
45. Almasbak H, Walseng E, Kristian A, Myhre MR, Suso EM, Munthe LA, et al. Inclusion of an IgG1-fc spacer abrogates efficacy of CD19 CAR T cells in a xenograft mouse model. Gene Ther (2015) 22:391–403. doi: 10.1038/gt.2015.4
46. Hombach A, Hombach AA, Abken H. Adoptive immunotherapy with genetically engineered T cells: modification of the IgG1 fc 'spacer' domain in the extracellular moiety of chimeric antigen receptors avoids 'off-target' activation and unintended initiation of an innate immune response. Gene Ther (2010) 17:1206–13. doi: 10.1038/gt.2010.91
47. M. al-Rubeai AN, Chalder S. And M.H. Goldman, a flow cytometric study of hydrodynamic damage to mammalian cells. J Biotechnol (1993) 31:161–77. doi: 10.1016/0168-1656(93)90158-J
48. Tomlinson MJ, Tomlinson S, Yang XB, Kirkham J. Cell separation: Terminology and practical considerations. J Tissue Eng (2013) 4:2041731412472690. doi: 10.1177/2041731412472690
49. Turtle CJ, Hanafi LA, Berger C, Gooley TA, Cherian S, Hudecek M, et al. CD19 CAR-T cells of defined CD4+:CD8+ composition in adult b cell ALL patients. J Clin Invest (2016) 126:2123–38. doi: 10.1172/JCI85309
50. Shah NN, Highfill SL, Shalabi H, Yates B, Jin J, Wolters PL, et al. CD4/CD8 T-cell selection affects chimeric antigen receptor (CAR) T-cell potency and toxicity: Updated results from a phase I anti-CD22 CAR T-cell trial. J Clin Oncol (2020) 38:1938–50. doi: 10.1200/JCO.19.03279
51. Zhang H, Zhao P, Huang H. Engineering better chimeric antigen receptor T cells. Exp Hematol Oncol (2020) 9:34. doi: 10.1186/s40164-020-00190-2
Keywords: TSCM, CAR, gene therapy, adoptive immunotherapy, HIV-1
Citation: Kranz E, Kuhlmann CJ, Chan J, Kim PY, Chen ISY and Kamata M (2022) Efficient derivation of chimeric-antigen receptor-modified TSCM cells. Front. Immunol. 13:877682. doi: 10.3389/fimmu.2022.877682
Received: 17 February 2022; Accepted: 07 July 2022;
Published: 28 July 2022.
Edited by:
Shen-Ying Zhang, The Rockefeller University, United StatesReviewed by:
Kyung-Ho Roh, University of Alabama in Huntsville, United StatesKailin Xu, Xuzhou Medical University, China
Copyright © 2022 Kranz, Kuhlmann, Chan, Kim, Chen and Kamata. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Masakazu Kamata, bWFzYTNrQHVhYi5lZHU=
†These authors have contributed equally to this work