 Anne-Marie Aubin1,2Félix Lombard-Vadnais1,3Roxanne Collin1,2,4Holly A. Aliesky5,6Sandra M. McLachlan5,6
Anne-Marie Aubin1,2Félix Lombard-Vadnais1,3Roxanne Collin1,2,4Holly A. Aliesky5,6Sandra M. McLachlan5,6 Sylvie Lesage1,2*
Sylvie Lesage1,2*- 1Immunology-Oncology Division, Maisonneuve-Rosemont Hospital Research Center, Montreal, QC, Canada
- 2Département de Microbiologie, Infectiologie et Immunologie, Université de Montréal, Montréal, QC, Canada
- 3Department of Microbiology and Immunology, McGill University, Montreal, QC, Canada
- 4CellCarta, Montreal, QC, Canada
- 5Thyroid Autoimmune Disease Unit, Cedars-Sinai Research Institute, Los Angeles, CA, United States
- 6Department of Medicine, David Geffen School of Medicine at University of California Los Angeles (UCLA), Los Angeles, CA, United States
Autoimmune diabetes arises spontaneously in Non-Obese Diabetic (NOD) mice, and the pathophysiology of this disease shares many similarities with human type 1 diabetes. Since its generation in 1980, the NOD mouse, derived from the Cataract Shinogi strain, has represented the gold standard of spontaneous disease models, allowing to investigate autoimmune diabetes disease progression and susceptibility traits, as well as to test a wide array of potential treatments and therapies. Beyond autoimmune diabetes, NOD mice also exhibit polyautoimmunity, presenting with a low incidence of autoimmune thyroiditis and Sjögren’s syndrome. Genetic manipulation of the NOD strain has led to the generation of new mouse models facilitating the study of these and other autoimmune pathologies. For instance, following deletion of specific genes or via insertion of resistance alleles at genetic loci, NOD mice can become fully resistant to autoimmune diabetes; yet the newly generated diabetes-resistant NOD strains often show a high incidence of other autoimmune diseases. This suggests that the NOD genetic background is highly autoimmune-prone and that genetic manipulations can shift the autoimmune response from the pancreas to other organs. Overall, multiple NOD variant strains have become invaluable tools for understanding the pathophysiology of and for dissecting the genetic susceptibility of organ-specific autoimmune diseases. An interesting commonality to all autoimmune diseases developing in variant strains of the NOD mice is the presence of autoantibodies. This review will present the NOD mouse as a model for studying autoimmune diseases beyond autoimmune diabetes.
Highlights
1. The Non-Obese Diabetic (NOD) mouse as a model of multiple autoimmune diseases
2. Congenic and transgenic NOD mice represent relevant models of human pathologies
3. Spontaneous occurrence of autoimmune thyroiditis, neuropathy and biliary diseases
4. The NOD mouse can be used to study polyautoimmune phenotypes
Introduction to the Non-Obese Diabetic Mouse Strain
Since its first description by Makino et al. in 1980 (1), the Non-Obese Diabetic (NOD) mouse strain represents the only mouse model that spontaneously develops autoimmune diabetes (2–4). The NOD strain is originally derived from Cataract Shinogi (CTS) mice, an inbred subline of the outbred ICR mouse strain, which develop cataracts (1, 2). In an effort to generate a mouse model for insulin-dependent diabetes, CTS mice with either low or high fasting glucose levels were further interbred. Eventually, mice from the ‘normoglycemic’ colony presented with diabetic symptoms, namely polyuria and glycosuria. These mice were selected for breeding, establishing the original NOD mouse colony (1, 5). Importantly, the autoimmune diabetes pathology in NOD mice shares several characteristics with human type 1 diabetes (T1D) (1, 3, 6, 7). For instance, the major histocompatibility class (MHC) locus is a defining autoimmune diabetes susceptibility factor in both mice and humans, with a common amino acid substitution in an MHC class II gene (4, 8). Studying the NOD mouse has considerably improved our understanding of this autoimmune disease, facilitating the identification of genetic variants contributing to disease susceptibility, of various immune cells causing pancreatic β-cell destruction, and of environmental contributors to disease susceptibility (6, 7, 9, 10). For further information on the use of NOD mice in dissecting the pathophysiology of autoimmune diabetes, the readers are referred to the following reviews on the topic (2–4).
This review will instead focus on the other organ-specific autoimmune diseases that spontaneously develop in NOD mice as well as in genetically manipulated NOD mice. Specifically, several NOD congenic mice and NOD genetic knockout mice are protected from autoimmune diabetes. In these diabetes-resistant mice, other autoimmune diseases spontaneously arise, such as autoimmune thyroiditis, autoimmune polyneuropathies, and autoimmune biliary disease. The use of the NOD mouse and its variants to study polyautoimmune syndromes will also be discussed. While autoantigen-specific T cell responses are a critical part of the pathology in autoimmune diabetes (6, 11), this review will more broadly discuss the presence of immune cells in the target tissues as well as the presence of autoantibodies in variants of the NOD mouse model, for each autoimmune pathology.
Autoimmune Thyroid Disease
Autoimmune thyroid disease (AITD) includes Hashimoto’s thyroiditis, Graves’ disease (autoimmune hyperthyroidism), neonatal Graves’ disease, and postpartum thyroiditis (12). All forms of AITD are characterized by the presence of immune infiltrates (in variable amounts) in the thyroid gland and particularly by the presence of IgG class autoantibodies directed towards specific thyroid autoantigens, namely thyroglobulin, thyroid peroxidase (TPO), and the thyrotropin receptor (TSHR) (13). Of note, while some of these autoantibodies are present in the serum of many individuals with normal thyroid function, the presence of TPO is significantly associated with thyroid disease (14). Interestingly, the prevalence of AITD is more frequent in people living with T1D (PWT1D) than in the general population (15–18). Based on the study of Hwang et al., the prevalence of thyroglobulin and TPO thyroid autoantibodies in PWT1D is around 30% (19), whereas the prevalence in the general population is approximately 10% (14).
The NOD Mouse as a Model of Autoimmune Thyroiditis
As in PWT1D, NOD mice can develop spontaneous autoimmune thyroiditis (SAT). In NOD mice, the cumulative incidence at one year ranges from ~5% to ~15% (20, 21). In both humans and mice, an iodine-rich diet accelerates the development of the disease (21, 22). The iodide excess is toxic for thyroid cells by a mechanism involving oxidative stress (23). This parallel between humans and mice highlights the relevance of the NOD mouse model for understanding autoimmune thyroiditis pathology (24). However, there are limits associated with the use of the NOD mice for the study of SAT. For one, NOD mice have a high incidence of autoimmune diabetes, especially in females where it reaches 70 to 90% by 30 weeks (25). This presents a challenge when attempting to isolate the immunological factors that specifically drive SAT independently of the immune response to autoimmune diabetes. In addition, the incidence of SAT is low in NOD mice in absence of an iodine-rich diet (20), such that very large cohorts of mice must be used to characterize the progression of the pathophysiology. Currently, an autoimmune diabetes-resistant genetic derivative of the NOD mouse model, the NOD.H2h4 congenic mouse, is more commonly used to study SAT.
NOD.H2h4 congenic mice were originally generated to determine the impact of the MHC class II locus on diabetes and insulitis development (26, 27). Specifically, the NOD.H2h4 bears the thyroiditis-prone H2h4 MHC locus from the B10.A(4R) mouse strain, composed of H-2Kk and H-2Db for MHC class I, and I-Ak for MHC class II (26) (Figure 1). In contrast to the low ~5% to ~15% incidence of SAT in NOD mice, 50%-70% of NOD.H2h4 congenic mice develop SAT (21, 26, 28). Moreover, subjecting the mice to an iodine-rich diet enhances the severity and the incidence of thyroid lesions in both NOD and NOD.H2h4 congenic mice, which can reach an incidence of nearly 100% in both strains (21, 22, 24, 28–30).
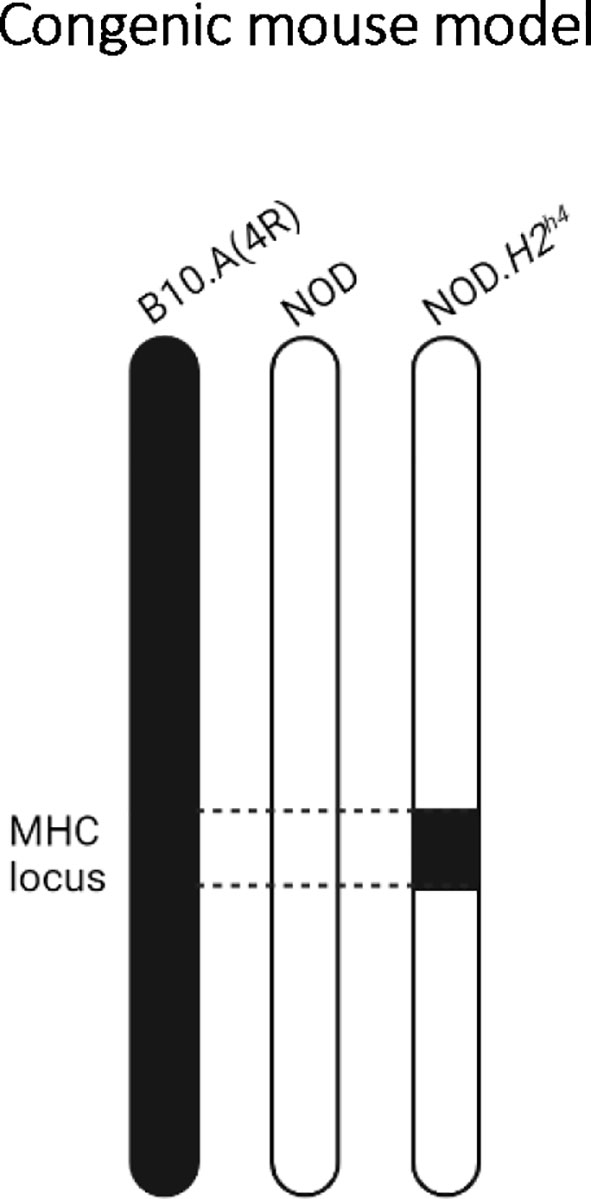
Figure 1 The NOD.H2h4 congenic mouse model. Representation of the mouse chromosome 17 from the parental B10.A(4R) (left), parental NOD (middle), and congenic NOD.H2h4 (right) strains. By backcrossing NOD mice to B10.A(4R), the thyroiditis-prone H2h4 MHC locus from chromosome 17 of the parental B10.A(4R) mouse has been selected at each backcross generation to replace in the NOD-derived H2g7 MHC locus, resulting in the NOD.H2h4 congenic mouse.
While NOD.H2h4 mice develop SAT, this strain is completely protected from diabetes onset (21, 26, 28). This suggests that the progression to diabetes is not necessary for SAT development, and that the break of tolerance towards thyroid autoantigens is favored by the H2h4 MHC haplotype, while the H2g7 MHC haplotype is necessary for diabetes onset. This observation also revealed that the organ-specific autoimmune susceptibility determined by the NOD genetic background can be shifted to other organs by modification of different genetic loci. In other words, different MHC loci in NOD mice can predispose to different organ-specific autoimmune diseases. Studies in families with T1D and AITD also revealed a strong genetic link to the MHC class II locus (17, 31, 32). Specifically, the MHC class II haplotype DR3-DQB1*0201 is a risk haplotype shared by both T1D and AITD (17, 31), while HLA-DR3 is specifically linked to T1D susceptibility (31, 32). Therefore, in both mice and humans, the MHC locus shifts the autoimmune response towards given target organs.
To assess the contribution of the MHC locus in thyroiditis development in mice, a comparative study was done using the NOD.H2h4 mouse and the NOD.H2k mouse (28). The primary difference between the NOD.H2h4 and NOD.H2k mice is the presence of I-E MHC class II molecule in the H2k locus (26, 28). After exposure to an iodine-rich diet, the extent of the autoimmune thyroiditis and the levels of thyroglobulin and TPO autoantibodies were higher in the NOD.H2h4 mice than in the NOD.H2k mice, in which TPO antibodies were essentially absent (28). This suggests that variants in the MHC locus between these two mouse strains influence the thyroid autoantibody profile (28). Consequently, the NOD.H2h4 mouse, which develops both thyroglobulin and TPO autoantibodies, is arguably the most representative mouse model of human AITD pathology (28).
Immune Cells Infiltration Within the Thyroid Gland
One of the key characteristics shared between AITD in humans and SAT in the NOD.H2h4 mouse is the infiltration of immune cells within the thyroid gland. The recruitment and migration of lymphocytes in this gland is supported by adhesion molecules expressed on endothelial cells (33). Of interest, whereas NOD mice express high levels of ICAM-1 on thyrocytes, CBA/J, A/J, BALB/c, and C57 mice show little to no expression of ICAM-1 (33). The high expression of ICAM-1 on thyrocytes driven by the NOD genetic background (and thus also present in NOD.H2h4 mice), is a genetic risk factor to SAT. ICAM-1 promotes the recruitment of immune cells into the thyroid, which then target specific thyroid autoantigens (33).
In the NOD.H2h4 mouse model, as in people living with AITD, the thyroid immune cell infiltrate is predominantly composed of CD4+ and CD8+ T cells, B cells, macrophages, natural killer cells, and dendritic cells (34). Still, in humans, information regarding the kinetics of the infiltration within the thyroid is limited. To better understand the kinetics of thyroid cell infiltration, Bonita et al. took advantage of the NOD.H2h4 mouse model (34). They show that the immune cell infiltration in the NOD.H2h4 thyroid begins with CD4+ T cells, followed by CD8+ T cells and macrophages, and finally by B cells (34).
CD4+ and CD8+ T Cells
T cells are part of the adaptive arm of the immune response and self-reactive T cells are necessary and sufficient for onset and progression of many autoimmune diseases (35–38). Elimination of CD4+ and CD8+ T cells completely prevents thyroiditis development by suppressing thyroid infiltration and thyroid autoantibody production in the NOD mouse, even on iodine-supplemented diet (24). This suggests that T cells are necessary for SAT (24). In addition to promoting thyroid autoantibody production by B cells (30, 34), CD4+ T cells are also required for the maintenance of inflammation in the thyroid gland (30). IFN-γ, secreted by CD4+ T cells, damages thyrocytes (30, 34) and induces the expression of MHC class II and adhesion molecules on thyrocytes, ultimately resulting in the recruitment of other immune cells, such as CD8+ T cells, macrophages, B cells, and plasma cells (30, 34). CD8+ T cells also contribute to disease progression by secreting cytokines, namely IFN-γ and TNFα (34), and by mediating perforin/granzyme-dependent lysis of thyrocytes, resulting in severe damage to the thyroid gland (34).
iNKT Cells
Invariant Natural Killer T (iNKT) cells have first been identified as an unusual T cell population expressing both T cell receptors (TCR) and the NK markers (NK1.1, NKG2D, and Ly49) (39–41). iNKT cells recognize antigens by the non-polymorphic MHC class I-like molecule CD1d (39–41). These cells exhibit a wide array of immunological functions such as the production of chemokines and cytokines, cytolytic activity, and activation and recruitment of other cell types (39, 41). Of interest, an indirect pathogenic role of iNKT cells has been suggested in autoimmune thyroiditis (42). Sharma et al. generated two iNKT cell lines derived from NOD.H2h4 splenocytes (42). After stimulation with thyroglobulin, these iNKT cell lines produce cytokines such as IFN-γ, TNF-α, IL-2, IL-4, and IL-10 (42). The adoptive transfer of thyroglobulin-stimulated iNKT cell lines enhanced autoimmune thyroiditis in NOD.H2h4 mice fed with an iodine-rich diet (42), suggesting that iNKT cells enhanced autoimmune thyroiditis in NOD.H2h4 mice. In addition, it was reported that the spleen of NOD.H2h4 mice contains more iNKT cells than BALB/c mice (43), suggesting a link between iNKT cell abundance and SAT susceptibility. With the availability of CD1d-tetramers allowing to quantify iNKT cells more precisely, we revisited this concept. In contrast to the previous report (43), we observed a higher percentage and number of iNKT cells in the spleen of BALB/c mice when compared to NOD.H2h4 mice (Figure 2). Further studies are required to understand the true implication of these cells in the development of autoimmune thyroiditis.
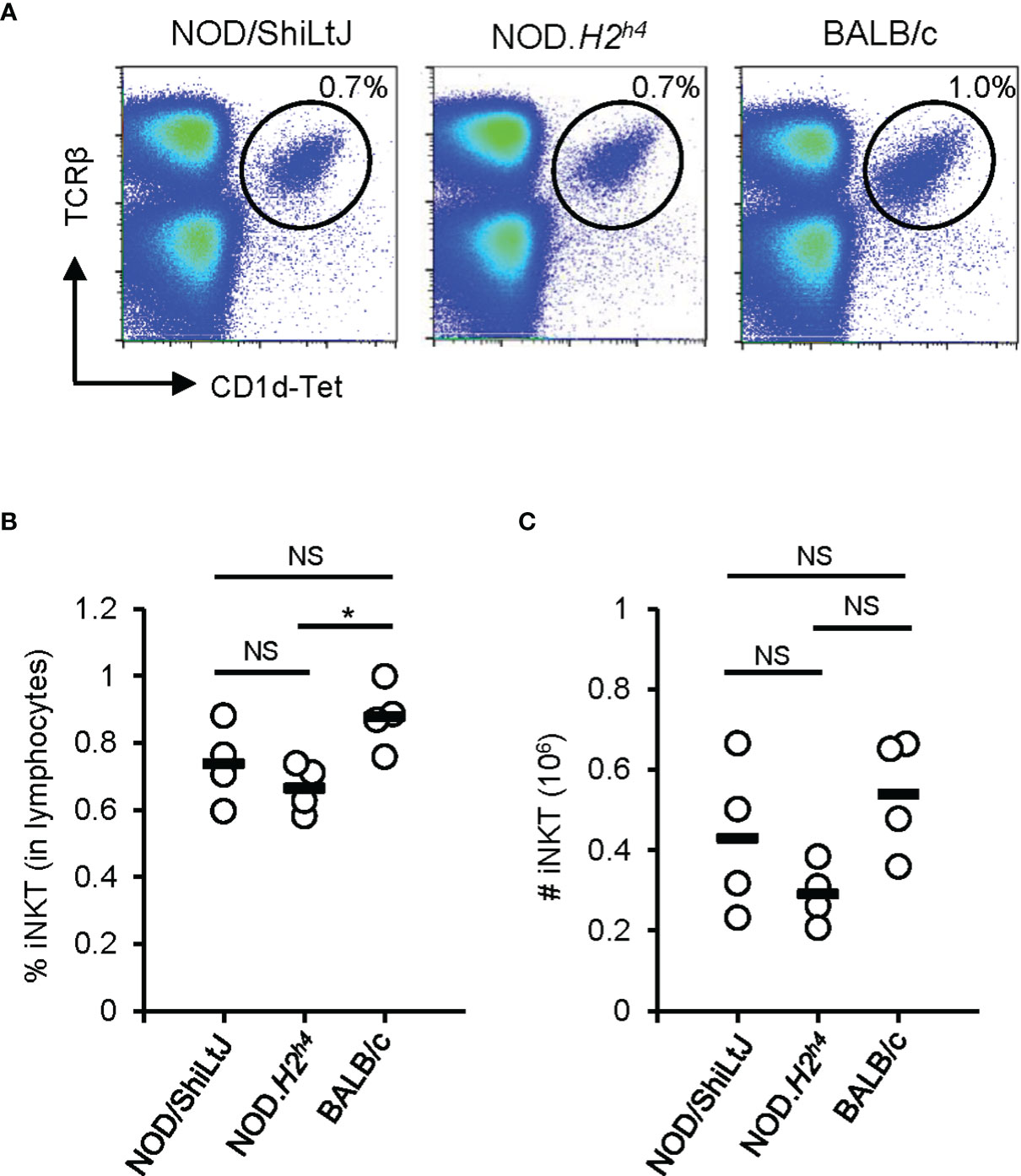
Figure 2 iNKT cell abundance in the spleen. Spleen of NOD/ShiLtJ, NOD.H2h4, and BALB/c mice were stained with antibodies to TCRβ and with CD1d tetramer (NIH Tetramer Core Facility). Data was acquired on BD FACSCanto II flow cytometer and analyzed with FlowJo. (A) Representative flow cytometry profiles of iNKT (TCRβ+CD1d-Tet+) cells in the spleen of NOD/ShiLtJ, NOD.H2h4, and BALB/c mice. (B) Compilation of the percentage of iNKT cells in the spleen of NOD/ShiLtJ, NOD.H2h4, and BALB/c mice (n = 4). (C) Compilation of absolute number of iNKT cells in the spleen of NOD/ShiLtJ, NOD.H2h4, and BALB/c mice (n = 4). One-way ANOVA tests were performed for statistical analysis. Non-significant, NS; P-value > 0.05, and *; P-value = 0.03.
Regulatory T Cells (Treg) and T-Helper (Th) Cells
Tregs are immunomodulatory cells that prevent autoimmune responses and thus could be used as a therapeutic in autoimmune diseases (44). Accordingly, a depletion of CD25+ Tregs before subjecting the mice to an iodine-rich diet increases the severity of thyroiditis in NOD.H2h4 mice (45), suggesting an important role for Tregs in the control of autoimmune thyroiditis.
Apart from Tregs, other Th subsets differentiated from naïve CD4+ T cells include Th1, Th2 and Th17, which are primarily distinguished based on the expression of specific transcription factors and their cytokine profile (46). Th1, Th2 and Th17 respectively express T-BET, GATA-3 and RORγt and secrete IFN-γ, IL-4 and IL-17 as their prototypical cytokine (47). In NOD.H2h4 mice, the presence of IFN-γ in the thyroid before the onset of lesions suggests that Th1 cytokines may play an important role in the initiation of autoimmune thyroiditis (30, 34). In addition, Th2 cytokines, such as IL-4 and IL-13, are maximal after thyroid lesions develop suggesting that these cytokines are involved in the late chronic phase of the disease, maintaining the thyroid inflammatory response (30). Moreover, NOD.H2h4-IFN-γ-/-, NOD.H2h4-IFN-γR-/-, and NOD.H2h4-IL-17-/- are resistant to the development of thyroiditis (48, 49), suggesting that both Th1 and Th17 profiles contribute to the pathology (50).
Of interest, there is an interplay between Tregs and Th cells in immune responses (51). This holds true in susceptibility to thyroiditis. Indeed, while both NOD.H2h4.IL-17-/- and NOD.H2h4 IFN-γR-/- mice are resistant to thyroiditis, depletion of CD25+ Tregs induces thyroiditis in NOD.H2h4.IL-17-/- mice but not in NOD.H2h4 IFN-γR-/- mice (50). This suggests that Tregs may more effectively control Th1-driven thyroiditis than Th17-driven pathology. Altogether, these observations point to a key role for Th cells in the development and progression of thyroiditis. Knowing that Th subsets facilitate the humoral response (46), they may effectively contribute to autoantibody production in thyroiditis.
B Cells
By producing antibodies, B cells can provide immune protection against infections (52). However, B cells can also have pathogenic roles in autoimmune diseases by producing autoantibodies, by promoting immune complexes deposition, antibody dependent cell cytotoxicity (ADCC), and as antigen-presenting cells (APCs) (53). Indeed, B cells are important players in SAT in the NOD.H2h4 mouse. This is exemplified in the NOD.H2h4-μ-/- mouse, devoid of B cells, as well as in NOD.H2h4 mice treated with anti-IgM or anti-CD20 antibodies, to deplete B cells (54–56). In these models, B cell depletion results in a decrease in the severity of thyroid lesions, as well as undetectable levels of thyroid autoantibodies (54–56). Further characterization of B cells in NOD.H2h4 mice revealed that expression of costimulatory molecules, such as CD80 and CD86, is increased on B cells following SAT onset (56). In addition, these B cells produce proinflammatory cytokines such as TNF-α and IL-6 (56). By providing costimulatory signals and secreting proinflammatory cytokines, it was suggested that B cells act as APCs, promoting the activation and expansion of autoreactive T cells (54–56). This model proposes a central role for B cells in autoimmune thyroiditis via their involvement in the activation of pathogenic T cells and their production of autoantibodies (Figure 3). Incidentally, B cells are essential for the development of Graves’ disease in which hyperthyroidism is directly caused by thyroid stimulating antibodies that target the TSHR (13).
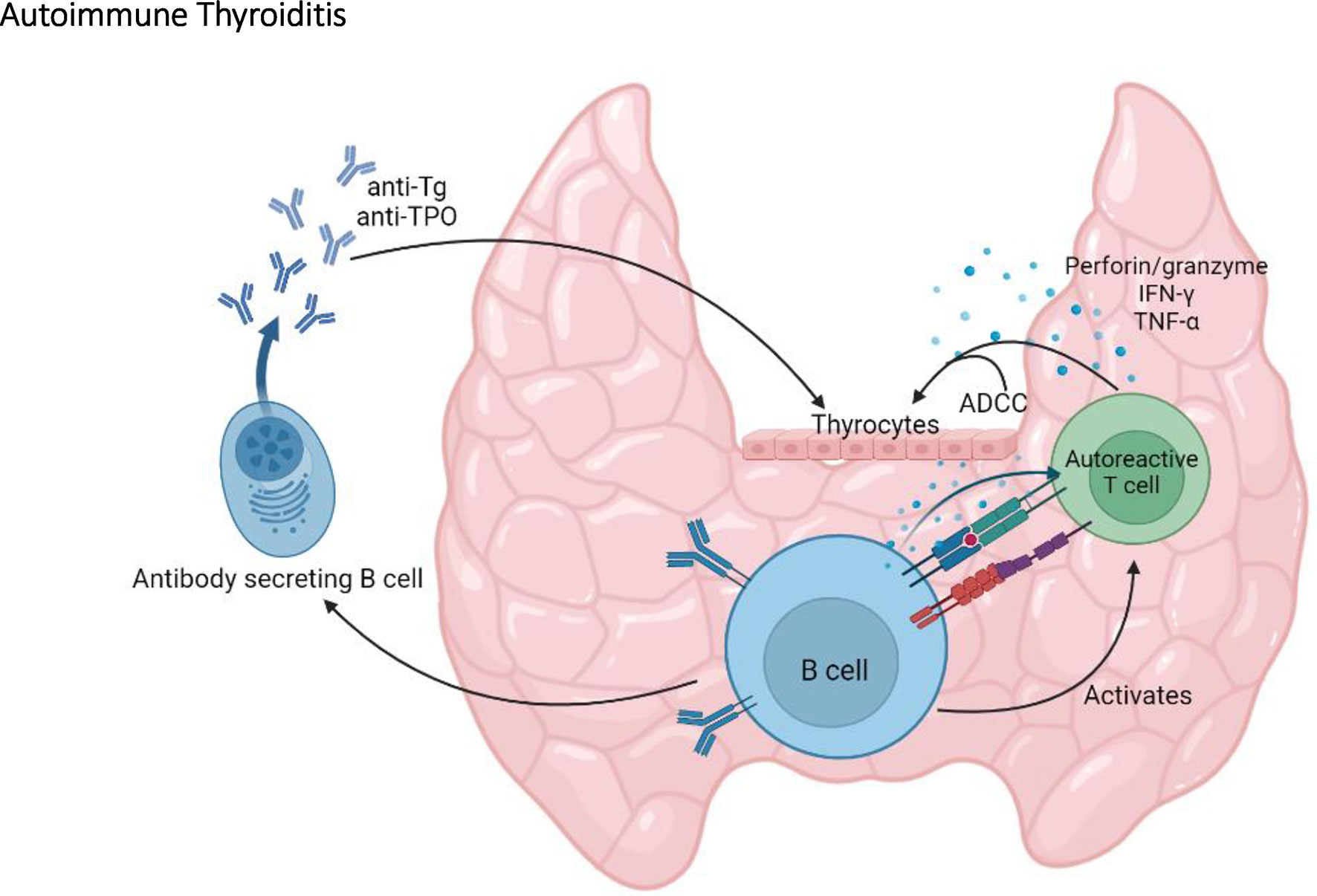
Figure 3 The central role of B cells in autoimmune thyroiditis. By producing anti-thyroglobulin (Tg) and anti-TPO autoantibodies, by activating autoreactive T cells, and by promoting antibody dependent cell cytotoxicity (ADCC), B cells play an important role in the onset and the progression of autoimmune thyroiditis in the NOD mouse.
Production of Thyroid Autoantibodies
In addition to immune infiltration, the breakdown of tolerance towards thyroid autoantigens is shared between autoimmune thyroiditis in humans and NOD.H2h4 mice, as shown by the presence of autoantibodies. In both species, the major thyroid autoantigens are thyroglobulin (13), the predominant component of the thyroid gland, and TPO; both thyroglobulin and TPO are involved in the process of thyroid hormones synthesis (57). The break of tolerance towards these two thyroid autoantigens can be explained by their immunogenicity (13). For example, the abundance and size of the thyroglobulin and TPO proteins promote the generation of a large pool of peptides, which can be presented on MHC to T cells (57). In mice, thyroglobulin autoantibodies appear first followed by TPO autoantibodies (28, 29), suggesting that thyroglobulin is one of the first targeted autoantigens (57). The NOD.H2h4 mouse, when exposed to iodine-supplemented diet, develops thyroglobulin-antibodies of subclasses IgG1 and IgG2b (30). IgG2b thyroglobulin antibodies correlate with thyroid lesions and could therefore represent a biomarker for predicting thyroiditis (29). Of interest, treating NOD.H2h4 mice with blocking antibodies to PD-1 and to CTLA-4 markedly enhances thyroiditis and autoantibodies to thyroglobulin and TPO (58).
In humans, antibody levels to thyroglobulin and TPO are twice as high in women than in men, with reported values of 15.2 U/ml in women vs 7.6 U/ml in men for thyroglobulin and 17 U/ml in women compared to 8.7 U/ml in men for TPO (14). However, in NOD.H2h4 mice, the levels of thyroglobulin antibody levels are higher in males than females (22), whereas TPO antibody levels are higher in females than males (22). Thus, the presence of TPO antibodies in NOD.H2h4 mice more closely resembles the situation in humans than the presence of thyroglobulin antibodies (22). Of note, autoantibodies to thyroglobulin or to TPO in NOD.H2h4 mice are species specific, and do not cross-react with human thyroglobulin or human TPO (30, 57). Importantly, most humans with autoantibodies to thyroglobulin and TPO are euthyroid. Hypothyroidism is only manifest after extensive thyroid lymphocytic infiltration and thyroid tissue damage depletes the substantial thyroid hormone reserves and overwhelms the capacity of TSH to restore thyroid function (14, 59). Consequently, like NOD.H2h4 mice, most patients with autoantibodies to thyroglobulin and/or TPO have subclinical disease (14). It should be emphasized that autoantibodies to thyroglobulin and particularly to TPO are markers of thyroid lymphocytic infiltration and are a risk factor for the development of hypothyroidism (60, 61). Of note, as for autoimmune diabetes, the presence of autoantibodies directed towards thyroid antigens reflects an ongoing humoral response. Yet, the direct pathogenic potential of these autoantibodies has not been clearly demonstrated, except for thyroid stimulating antibodies that target the TSHR in Graves’ disease (13). To that effect, transgenic expression of the human TSHR A-subunit at low levels in the thymus enables hTSHR/NOD.H2h4 females, exposed to iodine-supplemented diet, to develop stimulating antibodies to the TSHR, the hallmark of Graves’ disease (62). These TSHR antibodies stimulate cAMP production by human-TSHR-expressing cells in a bioassay. However hTSHR/NOD.H2h4 mice do not develop hyperthyroidism, because the antibodies target human TSHR and do not cross react with the mouse TSHR (63).
Overall, the NOD.H2h4 mouse has presented itself as an invaluable mouse model for the study of AITD and manifestations of this disease, such as immune cell infiltration and autoantibody production; these traits are similar to those observed in people living with AITD. The NOD.H2h4 mouse strain therefore represents an excellent animal model for the dissection of the mechanisms leading to AITD (30) and for the investigation of potential therapies against autoimmune thyroiditis (21, 22, 28, 45, 64). Moreover, manipulation of this mouse model has revealed that thyroiditis results from complex immune responses, where T cells are necessary for disease progression. Still, the humoral arm of the immune response plays a clear role in this pathology, as the presence of autoantibodies precedes disease diagnosis and eliminating B cells dampens the pathology. There is also evidence to support a role for B cells in antigen presentation to T cells. All of these traits are reminiscent of autoimmune diabetes progression in NOD mice, suggesting a parallel between the organ-specific immune mechanisms leading to these two pathologies.
Neuropathies
In the general population, the prevalence of neuropathy, also called peripheral neuropathy, is around 2% and increases with age up to 8% in people older than 55 years old (65). Peripheral neuropathy is characterized by damage to the axon or myelin of a neuron (66). In contrast, polyneuropathy (PNP) describes a pathology where several nerves of the peripheral nervous system are damaged, such as sensory, motor, and/or autonomic nerves (66). PNPs, with a prevalence of ~5% to 8% (67), are the most common type of peripheral nervous system disorder and are caused by various factors, such as chronic alcoholism, chemotherapeutic drugs, genetic factors, and vitamin deficiency or overdose (66, 67). In Europe and North America, diabetes remains the most common cause of PNP, with diabetic patients representing from 30% to 66% of all PNP cases (65–67). Notably, more people are affected by diabetic neuropathy (DN) than all other types of PNP, including Charcot-Marie-Tooth, Guillain-Barré syndrome, and chronic inflammatory demyelinating polyneuropathy (65–67). Indeed, DN affects from 200 to 600 individuals per 100 000 people each year, whereas the prevalence is less than 15 in 100 000 individuals for all other PNPs combined (66).
DN is a painful disease defined by loss of sensory function and sensation of numbness, prickling, or burning in the distal lower extremities (66, 68). In people living with diabetes, the exact cause of these neuropathic symptoms is unknown, but some hypotheses involve metabolic, neurovascular or autoimmune pathways (69–71). The more common hypothesis suggests that chronic elevation of glucose level in the blood of people living with diabetes leads to redox imbalance and ultimately to oxidative stress (71, 72). This oxidative stress leads to glycation and oxidation of proteins, as well as dyslipidemia characterized by low levels of high-density lipoprotein cholesterol and high levels of total cholesterol, triglycerides, and low-density lipoprotein cholesterol. Dyslipidemia reduces blood flow and nerve perfusion, possibly resulting in neuropathic symptoms (71).
It is estimated that around 50% of people living with diabetes will develop DN (68). Concomitant with the increase in diabetes prevalence, the prevalence of DN is also increasing (73) but remains similar between PWT1D (11–50%) and people living with type 2 diabetes (PWT2D) (8–51%) (68). The incidence of DN is higher in PWT2D (6,100 per 100,000 person-years) than in PWT1D (2,800 per 100,000 person-years) (68). This difference between prevalence and incidence occurring in PWT1D and PWT2D could be due to several factors like differences in the age of diabetes development (68).
The NOD Mouse as a Model of Autoimmune Neuropathy
As for PWT1D, NOD mice are also prone to develop autoimmune damage to the nervous system. Indeed, autoimmune reactions occurring in NOD mice can shift from the pancreatic islets towards nervous tissues after inhibition or disruption of costimulatory pathways, cytokines, or transcription factors that are important in the maintenance of immune tolerance. Here we will discuss some genetically modified NOD mice that develop autoimmune neuropathy and therefore represent a tool for the study of this disease.
Disruption of Immune Tolerance Leading to Autoimmune Neuropathy
T cell activation requires three different signals: signal 1; TCR signaling via recognition of peptides presented by MHC, signal 2; costimulatory molecules, and signal 3; cytokines (74, 75). As mentioned, T cells are necessary for autoimmune diabetes progression in NOD mice. In trying to understand how T cells contribute to autoimmune diabetes, various NOD mouse models where genetically engineered to specifically target signal 1, 2 or 3. Altering either signal 1, 2 or 3 in NOD mice appears to shift the pancreatic β cell-specific autoimmune response towards the nervous system (Figure 4).
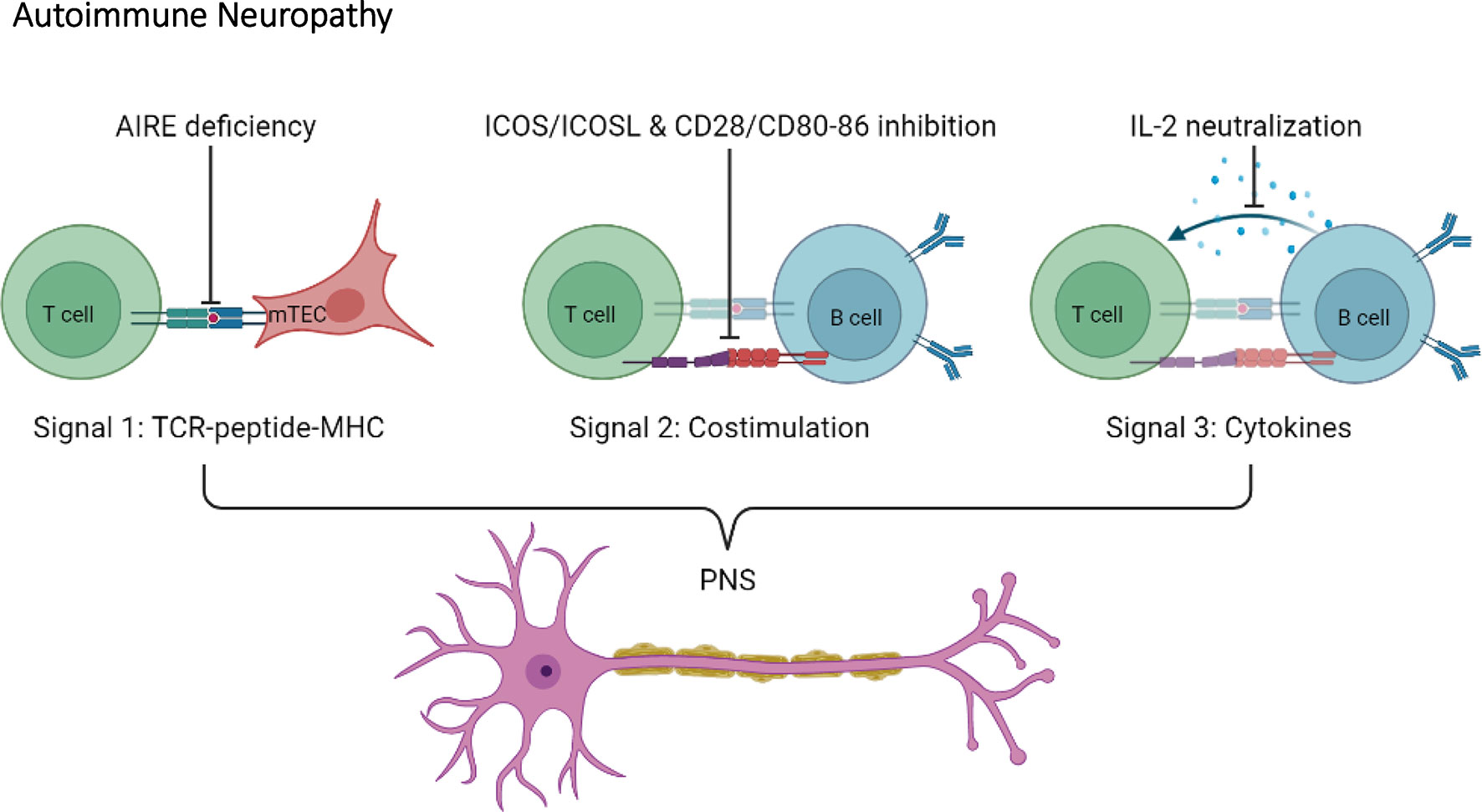
Figure 4 Disruption of T cell activation signals induce autoimmune neuropathy in the NOD mouse. Autoimmune neuropathy can be induced in the NOD mouse after modulation of one of the three T cell activation signals. More specifically, mutations in AIRE transcription factor expression in medullary thymic epithelial cells (mTECs) indirectly impact signal 1. In addition, disruptions in co-stimulation signals, such as ICOS/ICOSL and CD28/CD80-86 (signal 2), or neutralization of IL-2 (signal 3) induce autoimmune neuropathy in the NOD mouse.
AIRE Transcription Factor: An Indirect Impact on Signal 1
The transcription factor AIRE promotes the ectopic expression of tissue-restricted antigens in the thymus (76, 77). The presentation of these self-antigens allows for the negative selection of self-reactive thymocytes and favors the generation of Tregs that mediate peripheral tolerance (78–80). The NOD.AireGW/+ mouse has a dominant G228W mutation in the gene coding for AIRE causing a partial loss of function, such that expression levels of tissue restricted antigens is reduced by 10% relative to NOD mice (81, 82). Of interest, the NOD.AireGW/+ mouse shows a decrease in the thymic expression of myelin protein 0, one of the major autoantigens of the peripheral nervous system, representing more than 50% of the peripheral myelin protein content (83, 84). In addition to revealing that myelin protein 0 expression in the thymus is regulated by AIRE, it also explains the loss of tolerance to this protein in the NOD.AireGW/+ mouse (82, 83). Indeed, the partial loss of AIRE function in NOD.AireGW/+ mice promotes the escape of myelin protein 0 self-reactive T cells into the periphery, which target nervous system elements but also pancreatic tissue (82). This results in the development of autoimmune peripheral neuropathy, similar to human chronic inflammatory demyelinating polyneuropathy, as well as autoimmune diabetes (82, 83). Therefore, a slight shift in the abundance of self-antigen expression in the thymus of NOD.AireGW/+ mice indirectly impacts signal 1, by not providing sufficient self-antigen presentation to the developing thymocytes. This, in turn, allows for the escape of self-reactive T cells, some of which target the nervous system, causing peripheral neuropathy.
Costimulatory Pathways: Signal 2
Costimulatory molecules are expressed at the surface of immune cells and enhance the intracellular signal provided by signal 1. CD28 is the prototypical costimulatory molecule for naïve T cell stimulation (85, 86). It is constitutively expressed on T cells and binds to the CD80 and CD86 receptors expressed on APCs (87). ICOS, a member of the CD28 family, is expressed on activated T cells and binds ICOSL on APCs (88). The interaction of ICOS with ICOSL and/or of CD28 with CD80 and CD86 triggers a co-stimulatory signaling cascade, which facilitates T cell activation (87). Of relevance, these costimulatory pathways are involved in autoimmunity (88, 89).
To define the involvement of ICOS and CD28 costimulatory pathways in T1D, genetic deletion of ICOS, ICOSL or CD86 was performed in NOD mice. Interestingly, NOD.ICOS-/-, NOD.ICOSL-/-, and NOD.CD86-/- mice are all protected from diabetes, suggesting an important role for these costimulatory pathways in autoimmune diabetes (90, 91). However, autoimmune neuropathies spontaneously developed in all of these strains (90, 91). Specifically, the NOD.ICOS-/- and NOD.ICOSL-/- mice show neuromuscular autoimmunity characterized by hind leg paralysis and immune infiltration of T cells, macrophages and granulocytes in the peripheral and central nervous system (PNS, CNS), including peripheral nerves, sensory ganglia, muscles, brain, and spinal cord (91). The reason for the autoimmunity deviation from the pancreas to nervous tissue in the NOD.ICOS-/- and NOD.ICOSL-/- mice remains unknown (91). Analogously, the NOD.CD86-/- mouse develops a spontaneous autoimmune peripheral polyneuropathy (SAPP) (90). And, as for NOD.AireGW/+ mice, NOD.CD86-/- mice display a break of tolerance towards myelin protein 0, the dominant autoantigen in the peripheral nervous system (83, 84). The reasons for the shift in target organ for the autoimmune response may be explained, in part, by the fact that CD86 genetic deletion leads to overexpression of CD80 on myeloid dendritic cells infiltrating the peripheral nerves (90, 92). The overexpression of CD80 on these APCs leads to activation of myelin-specific T cells, myelin sheet destruction and SAPP development (90). In addition, disruption of the CD28 costimulatory pathway leads to a reduction in Treg number, which could ultimately enhance susceptibility to SAPP (93). Exploiting genetically modified NOD mice will help dissect how disruptions in signal 2 facilitate a shift in the autoimmune response towards a different target organ. This is especially important when considering therapeutic approaches that target these pathways, to avoid treatment of T1D that would instead lead to the development of neuropathy.
Although autoimmune diabetes and neuropathy are characterized by different manifestations, the genetic factors promoting these two diseases on the NOD genetic background partially overlap. The H2g7 MHC haplotype of NOD mice not only plays an important role in autoimmune diabetes but is also necessary for the development of autoimmune neuropathy in the NOD.CD86-/- mouse (94). This suggests that the H2g7 haplotype promotes self-reactivity against various organs. The genetic susceptibility overlap can also be attributed to non-MHC loci. For example, diabetes resistance loci were introduced in NOD.CD86-/- mice to generate NOD.CD86-/–Idd3/5 and NOD.CD86-/–Idd3/10/18 congenic mice. These congenic mice are completely protected from both autoimmune diabetes and neuropathy (94). Thus, genetically modified NOD mice allow to study mechanisms as well as genetic factors promoting the development of autoimmune neuropathy.
Cytokines: Signal 3
T cell activation is modulated by the presence of cytokines, which represent the third signal for T cells activation (75). Unbalanced cytokine production is deleterious and may lead to the development of autoimmunity (95–97). A key cytokine in modulating T cell function is IL-2; it facilitates the proliferation of T cells and is involved in immune tolerance by allowing the homeostatic maintenance of Tregs (98–100). Similar to NOD mice with targeted disruption of costimulatory molecules, autoimmune peripheral neuropathy has been described in NOD mice deficient in IL-2 (98). While intraperitoneal injection of anti-IL-2 monoclonal antibodies in NOD mice accelerates diabetes onset, it also induces the development of autoimmune peripheral neuropathy in more than 50% of the treated mice (98). This neuropathy is characterized by ataxia and paralysis of the limbs due to demyelination of the peripheral nerves (98). Anti-IL-2 treatment in NOD mice enhances autoimmunity by reducing Treg number, their activation, and their suppressive function (98). Of interest, IL-2 is one of the key candidate genes in the Idd3 susceptibility locus (101, 102). Idd3, and thus IL-2 variants, may generally predispose NOD mice to autoimmune diseases by altering the function and development of Treg cells (98).
Altogether, genetic manipulations leading to alterations in T cell signal 1, 2 or 3 in NOD mice can shift the immune response from pancreatic β cells towards the nervous system. This break in T cell tolerance allows for infiltration of autoreactive T cells in the peripheral nerves, which ultimately leads to the production of autoantibodies targeting myelin protein 0 by self-reactive B cells (84). Of note, autoantibodies targeting myelin protein 0 have also been found in serum from individuals diagnosed with Guillain-Barré syndrome and chronic inflammatory demyelinating polyneuropathy (103, 104).
Production of Autoantibodies Targeting Nervous System Antigens
Pancreatic islets are surrounded by cells of the autonomous nervous system (105). In addition, pancreatic β-cells and neuronal cells share some autoantigens such as GAD, ICA515, and the neuronal type III intermediate filament protein, peripherin (105, 106). These autoantigens of the pancreatic nervous system are targeted by islet-infiltrating autoreactive T cells as well as autoantibodies (105). The production of autoantibodies against pancreatic nervous system antigens occurs in the early phase of diabetes and could explain certain neurological pathologies occurring in the prediabetic stage in humans and mice (105). In addition, B cell producing peripherin autoantibodies have been isolated directly from the pancreatic islets of NOD mice (106). Altogether, these observations point to a potential cross-reactive autoimmune response to both pancreatic β cells and neuronal cells, resulting in the production of autoantibodies as a reflection of an ongoing humoral immune response, which likely contributes to the pathology.
To specifically study the impact of peripherin-specific B cells in diabetes and neuritis, a BCR-transgenic mouse model (NOD-PerIg) was generated (106). In this mouse, B cells express the H and L chain Ig transgene from the peripherin-specific hybridoma clone H280, isolated from the pancreas of NOD mice (107). Compared to non-transgenic NOD mice, NOD-PerIg mice develop early onset diabetes, with an expansion of diabetogenic T cells, revealing an important association between the pancreas and the nervous system (107). Genetic manipulation of B cell responses in the NOD mouse has identified a clear link between autoimmune diabetes and neuropathy. This link between autoimmune diabetes and neuropathy has also been observed in non-NOD mouse models of autoimmune diabetes (108).
In sum, as for thyroiditis, manipulating the NOD mouse has informed us on cellular processes and genetic pathways linking autoimmune diabetes to peripheral neuropathies. As multiple immune characteristics are shared between autoimmune neuropathy in NOD mice and humans, the genetically modified NOD mice described above continue to be useful to improve our knowledge on autoimmune neuropathy, as well as the connection between the pancreas and the nervous system.
Autoimmune Biliary Diseases
Primary biliary cirrhosis (PBC), primary sclerosing cholangitis (PSC), and IgG4-associated cholangitis (IAC) represent the three main forms of autoimmune biliary diseases (ABD) (109, 110). All ABD share specific symptoms such as bile duct obliteration and cholestasis, characterized by a strong reduction of bile flow (111–114). Here we will focus on the most common form of ABD which is PBC, with an overall prevalence of ~19 to ~40 cases per 100 000 individuals depending on the geographic location (110). PBC is a chronic autoimmune cholestatic liver disease most frequently observed in middle-aged women (115), and is characterized by lymphocytic infiltration of the liver portal tracts, destruction of the epithelial cells of the intrahepatic bile duct, and serologic hallmarks of antimitochondrial autoantibodies (AMA) (116). Notably, 90-95% of people living with PBC (PWPBC) will develop AMA; these autoantibodies long precede clinical symptoms of PBC, often for many years, and yet represent one of the three criteria for the definitive diagnosis of PBC (117).
The NOD Mouse as a Model of ABD
In an attempt to understand the contribution of genetic loci linked to autoimmune diabetes susceptibility in NOD mice, the congenic NOD.c3c4 mouse carrying resistance alleles on chromosomes 3 (Idd3, Idd10, Idd17, Idd18) and 4 (Idd9.1, Idd9.2, Idd9.3), was generated (118, 119). The NOD.c3c4 mouse does not show signs of autoimmune diabetes (119), but about half of the female and a quarter of the male mice spontaneously develop a fatal form of ABD (118). Similar to human PBC, NOD.c3c4 mice exhibit lymphocyte infiltration in the liver, production of autoantibodies, biliary obstruction, and finally liver failure leading to death (118–120). Of interest, the NOD.c3c4 strain was the first mouse model of human PBC (119).
T Cell Infiltration in the Liver
In NOD.c3c4 mice, abundant T cell infiltration can be observed in the liver, with CD4+ and CD8+ T cells primarily located in the biliary epithelium (119). CD4+ T cells in the liver produce pro-inflammatory cytokines such as IFN-γ and IL-2 (119). Importantly, antibody-mediated depletion of T cells leads to a significant reduction in disease onset in NOD.c3c4 mice (119). Moreover, transfer of CD4+ T cells from a NOD.c3c4 mouse to a lymphopenic NOD.c3c4-scid mouse is sufficient to induce ABD development (119). Altogether, these observations demonstrate that T cells are necessary and sufficient for ABD in NOD.c3.c4 mice.
The role of T cells in ABD development has also been investigated in a new congenic mouse model of PBC, the NOD.ABD mouse, derived from the NOD.c3c4 mouse (120). This congenic subline, with shorter resistance loci on chromosomes 3 and 4 than those in NOD.c3c4 mouse, develops ABD as well as autoimmune diabetes (120). This suggests that these two autoimmune diseases are not mutually exclusive in the NOD.ABD congenic mouse model. Of interest, the development of both T1D and PBC has also been reported in humans (121). The NOD.ABD mouse model develops a similar form of ABD as the NOD.c3c4 mouse characterized by common bile duct (CBD) dilation, immune cell infiltration, and biliary epithelial proliferation resulting in cyst formation (120). Of note, NOD.ABD mice show an accumulation of central and effector memory CD8+ T cells in the liver, which effectively produce IFN-γ and TNF-α (120). Additionally, the transfer of NOD.ABD CD8+ T cells alone or with CD4+CD25- T cells into NOD.c3c4-scid mice promotes ABD development in these recipients, suggesting an important role of autoreactive CD8+ T cells in ABD (120). Overall, studies in NOD.ABD and NOD.c3c4 congenic mice highlight an important pathogenic role of T cells in ABD development.
Production of Autoantibodies in ABD
As mentioned above, the presence of autoantibodies, particularly of AMA, is a strong serologic hallmark of disease, with 90-95% of PWPBC presenting with these autoantibodies (117, 122). In NOD.ABD mice, AMA were shown to bind the E2 subunit of the pyruvate dehydrogenase complex (PDC-E2), part of the mitochondrial 2-oxoacid dehydrogenase complexes (120). While PDC-E2 is a ubiquitous autoantigen expressed in all nucleated cells in the body, in PWPBC, only bile duct epithelial cells are targeted (119). The reason for the specific targeting of bile duct epithelial cells is unclear; it suggests that other components are at play, and that the presence of AMA may be secondary to tissue destruction. Of interest, anti-PDC-E2 antibodies are present in both NOD.c3c4 and NOD.ABD mouse models of PBC (119, 120). However, the proportion of NOD.ABD mice presenting with these autoantibodies is rather low, and, in contrast to PWPBC, anti-PDC-E2 antibody-positive mice increases with disease severity and age (120). Still, anti-PDC-E2 antibodies appear before detectable liver immune cells infiltration in both PWPBC and NOD congenic mice (119, 122).
In addition to anti-PDC-E2 antibodies, antinuclear antibodies (ANAs) and anti-Smith antibodies (anti-Sm) are also observed in the sera from PWPBC but at a lower incidence (48% of PWPBC develop ANAs vs 24% for anti-Sm) (123). Notably, the presence or absence of ANAs and anti-Sm varies among the different NOD mice congenic for chromosomes 3 and/or 4. In contrast to NOD and NOD.ABD mice which do not develop ANAs and anti-Sm autoantibodies, these autoantibodies are found in the serum of NOD.c3c4 and other congenic lines (118, 120). Further investigation of congenic sublines suggests that the Idd9.3 locus is sufficient for ANAs and anti-Sm autoantibody production (120, 124). Within the Idd9.3 locus, there is a candidate gene encoding for CD137 (4-1BB), an inducible costimulatory molecule on T cells (124, 125). A three amino acid difference in CD137 between NOD and B10 mice results in a lower CD137 costimulatory signal in NOD mice (124, 126). This may explain why NOD mice carrying non-NOD alleles at this locus show an increased production of autoantibodies, via the enhanced CD137-mediated costimulation between T cells and B cells (124, 126).
Overall, the NOD.c3c4 congenic mouse is a relevant model of PBC; it shares significant characteristics with PBC including key aspects of the humoral autoantibody response (127, 128) (Figure 5). In addition, NOD.ABD congenic subline allows to investigate the relationship between ABD and autoimmune diabetes. These NOD congenic mice further allow the identification of relevant and possibly clinically targetable molecular pathways for the development of new treatments.
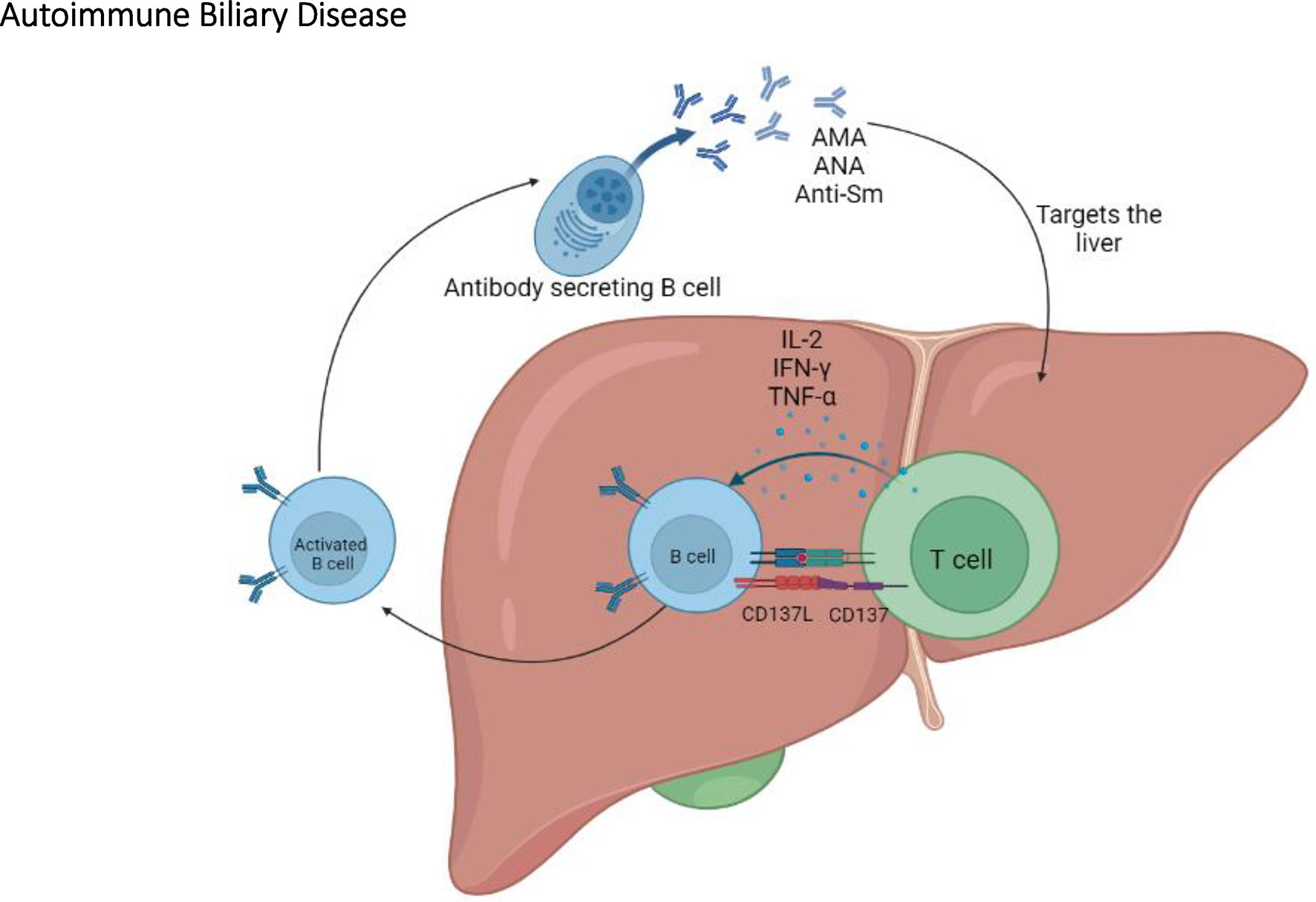
Figure 5 The immunopathogenesis of autoimmune biliary disease. In the NOD congenic mice, T cells play an important role in ABD by activating B cells which will eventually produce AMA, ANA, and anti-Sm autoantibodies.
Polyautoimmunity in NOD Mice
The term polyautoimmunity is used to describe the presence of more than one autoimmune disease in the same individual (129–131). For instance, a given NOD mouse can simultaneously present with multiple autoimmune diseases, such as autoimmune diabetes and thyroiditis (20, 132). The polyautoimmunity does not need to include autoimmune diabetes. In fact, NOD.CCR7-/- mice are protected from diabetes, but develop multiple autoimmune phenotypes, including immune infiltration in the thyroid, sciatic nerve, lung, stomach, intestine, uterus, and testis, among others (133). Notably, the thyroid pathology in these mice most closely resembles the primary hypothyroidism observed in humans (133). In addition, autoimmune diabetes-resistant NOD.H2h4 and NOD.H2h4-IFN-γ-/-CD28-/- mice spontaneously develop thyroiditis and Sjögren’s syndrome (SS) (134–136). While these findings further highlight the remarkable autoimmune-prone background of the NOD mouse, we will mostly focus our discussion to polyautoimmune phenotypes that include autoimmune diabetes.
In addition to autoimmune diabetes and thyroiditis (20, 132), NOD mice can present with both autoimmune diabetes and SS (137–143). T1D and SS can also co-occur in humans, with up to 55% of PWT1D exhibit symptoms of SS, such as keratoconjunctivitis sicca (dry eyes) and xerostomia (dry mouth) (144). SS is a chronic autoimmune exocrinopathy disorder characterized by lymphocyte infiltration and progressive damage to the exocrine glands, mainly the lacrimal and salivary glands (145, 146). These damages lead to decreased tears and saliva secretion, which ultimately result in keratoconjunctivitis sicca and xerostomia (145, 146). SS is notably defined by important B cell alterations of the humoral immunity which result in a polyclonal B cell activation and antibodies production (135, 147). In fact, one of the main hallmarks of SS is the presence of lymphocyte infiltration in the exocrine glands which formed organized lymphoid structures called ectopic follicles (148). In these ectopic follicles, all subsets of B cells are present, including antibody-secreting B cells which produce pathogenic antibodies that are useful for SS diagnosis (135, 148). These autoantibodies, which target non-organ-specific antigens, are Rheumatoid factor, anti-double stranded DNA, ANA, anti-Ro, and anti-La (147–149). Of note, the presence of anti-Ro and anti-La is a criterion for SS diagnosis (149). A study in NOD.H2h4 mice reveals that anti-Ro and anti-La appear before the development of ectopic follicles in the salivary gland whereas antibodies to double stranded DNA only develop after the appearance of ectopic follicles (148). These observations are consistent with anti-Ro and anti-La being the hallmark of SS and particularly as markers identifying patients in the active stage of the disease. SS is most prevalent in women aged between 30 to 60 years old, with a female to male ratio from 20:1 to 9:1 (137, 146, 150). In NOD mice, as in humans, the development of SS seems to be influenced by sex hormones because SS in NOD mice is significantly higher in females than males (137).
Another example of polyautoimmune traits present in humans is T1D and multiple sclerosis (MS) (151). MS is an autoimmune inflammatory disease of the central nervous system characterized by autoimmune responses against the protective myelin sheaths around nerve fibers, leading to severe and progressive neurological impairment (152). PWT1D have a 3 to 20 times higher risk of developing MS compared to the general population (151, 153–156). In addition, these two autoimmune diseases share some genetic and environmental susceptibility factors (151). Exposure to vitamin D seems to protect against the onset of MS and T1D (157). Interestingly, immune responses against pancreatic islet have been observed in people with MS, and, conversely, PWT1D show immune responses against central nervous system antigens (158). In addition to T1D and MS polyautoimmunity, T1D can also be observed in association with other autoimmune diseases such as AITD and DN in the same individual (15–18, 68).
The polyautoimmunity observed in NOD mice allows investigation of the mechanisms underlying this complex trait. Indeed, polyautoimmunity in NOD mice can be exacerbated by genetic manipulation and/or modulation of immune functions (Figure 6). For instance, targeting PD-1, AIRE, IL-2 or performing thymectomy in NOD mice promotes polyautoimmunity, as discussed below.
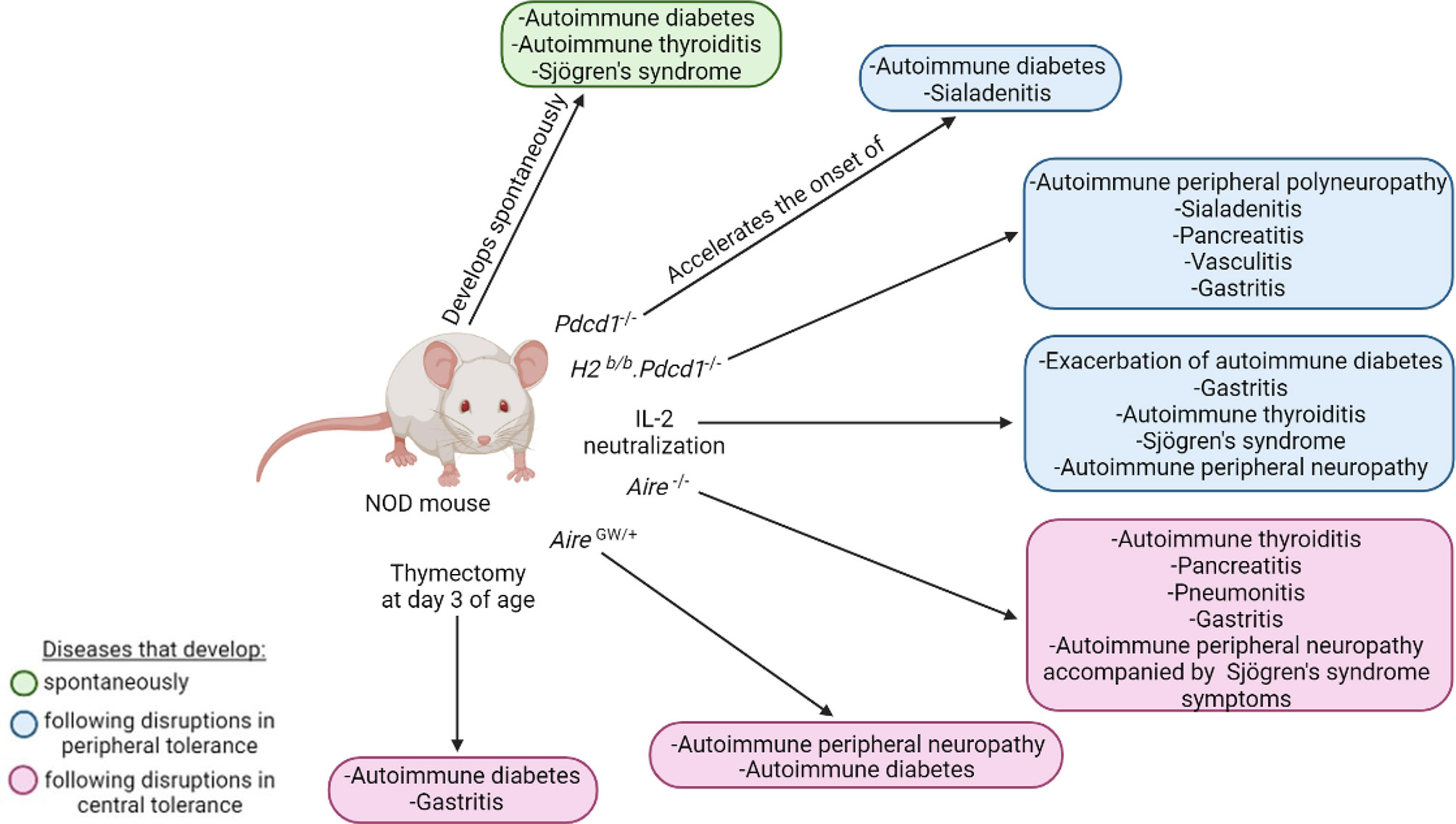
Figure 6 Polyautoimmunity in the NOD mouse. The NOD mouse can spontaneously develop autoimmune diabetes, autoimmune thyroiditis, and Sjögren’s syndrome (green). Disruptions in peripheral (blue) and central tolerance (pink) exacerbate or induce a wide spectrum of other organ-specific autoimmune diseases.
PD-1 Driving Polyautoimmunity
PD-1, which is coded by the Pdcd1 gene, is an immunoreceptor involved in the regulation of peripheral tolerance by inducing and maintaining T cell clonal anergy and homeostatic control of B cells and myeloid cells (159–163). The interaction of PD-1 with its ligands, PD-L1 and PD-L2, suppresses immune responses like autoimmunity and sustained inflammation (163, 164). As such, PD-1 deficiency on the NOD genetic background accelerates the onset and incidence of autoimmune diabetes, with an onset at 5 weeks instead of 12-17 weeks in NOD mice, and an incidence reaching 100% by 10 weeks (164). Sialadenitis is also accelerated and more severe, with significantly greater pathological scores at 6 weeks of age in NOD.Pdcd1-/- relative to NOD mice (164). Sialadenitis is an inflammation of the salivary glands caused by an increase in the activation and effector functions of autoreactive T cells (165). The NOD.Pdcd1-/- mouse, which presents a rapid onset of both autoimmune diabetes and sialadenitis, can thus be used to study polyautoimmunity. The early onset of autoimmune diabetes in the NOD.Pdcd1-/- mice is in part due to early and severe insulitis, resulting in rapid destruction of pancreatic β-cells (164). As for NOD mice, insulitis and autoimmune diabetes in NOD.Pdcd1-/- mice is dependent on the H2g7 MHC locus. Indeed, as for NOD-H2b/b mice (26), NOD-H2b/b.Pdcd1-/- mice are completely protected from insulitis and autoimmune diabetes (160, 166). This indicates that the H2g7 haplotype is absolutely required for autoimmune diabetes development, even in NOD.Pdcd1-/- mice (26, 160). Rather than developing autoimmune diabetes, the NOD-H2b/b mice develop SS (141, 167–169), whereas the NOD-H2b/b.Pdcd1-/- female mice are polyautoimmune; they develop spontaneous peripheral polyneuropathy, sialadenitis, pancreatitis, vasculitis, and gastritis (160, 166). This polyautoimmunity is likely due to a break in T cell tolerance as a consequence of a disruption of the PD-1 pathway (160). To identify the genetic factors that drive this polyautoimmune phenotype, Jiang et al. performed a genetic linkage analysis between NOD-H2b/b.Pdcd1-/- and C57BL/6.Pdcd1-/- mice (166). They identified 14 non-MHC quantitative trait loci linked to these autoimmune traits (166). These studies highlight the relevance of using genetically manipulated NOD mice to study polyautoimmunity to identify additional genetic variants linked to autoimmune diseases (160, 166).
AIRE Transcription Factor as a Prototypical Factor Causing Polyautoimmunity
As mentioned above, the AIRE transcription factor has an important role in maintaining self-tolerance and preventing autoimmunity (78, 170–173). The polyautoimmune syndrome resulting from AIRE mutations is a rare autosomal recessive disease called autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy (APECED) or autoimmune polyendocrine syndrome type-1 (174–176). In addition to developing chronic mucocutaneous candidiasis, hypoparathyroidism, and primary adrenal insufficiency, people living with APECED also develop several organ-specific autoimmune manifestations including T1D, autoimmune thyroiditis, gastritis, and hepatitis (170, 177, 178). As in people living with APECED, AIRE deficiency in mice from various genetic backgrounds, including NOD.Aire-/- mice, have circulating autoantibodies targeting multiple organs and lymphocytic infiltration in various tissues, representing a good model for APECED studies (178, 179). Of note, NOD.Aire-/- mice are protected from autoimmune diabetes but exhibit thyroiditis, pancreatitis, pneumonitis, gastritis, and autoimmune peripheral neuropathy accompanied by the development of some SS symptoms (146, 167, 180). As mentioned above, the NOD.AireGW/+ mouse is also polyautoimmune in that it develops autoimmune peripheral neuropathy and autoimmune diabetes (82, 83). Thus, both the NOD.Aire-/- and NOD.AireGW/+ mice are relevant models to study polyautoimmunity.
To study the impact of the humoral response in polyautoimmunity, Gavanescu et al. compared NOD.Aire-/- and NOD.Aire-/-μMT mice, and showed that the lack of B cells in AIRE-deficient mice strongly reduces autoimmune manifestations such as organ inflammation (179). In addition, depleting B cells in a variant of the NOD.Aire-/- model significantly reduced inflammation and destruction of the pancreas (179). These results suggest that B cells contribute to APECED pathology and that anti-B cell therapies could help alleviate symptoms in people living with APECED (179).
IL-2 and Polyautoimmunity
Polyautoimmunity is also observed in NOD mice treated with IL-2 neutralizing antibodies (98, 166). These mice show an exacerbation of autoimmune diabetes and develop a wide spectrum of organ-specific autoimmune diseases such as gastritis, thyroiditis, SS, and peripheral neuropathy (98). This is likely due to the fact that IL-2 neutralizing antibodies broadly reduce Treg number, as well as their suppressive functions (98–100, 181).
Thymectomized NOD Mice Develop Polyautoimmunity
As for PD-1 deficiency, AIRE mutations, and IL-2 neutralization, thymectomy (Tx) performed at three days of age (d3-Tx) in NOD mice leads to the development of polyautoimmunity (182). While d3-Tx in NOD mice does not impact autoimmune diabetes onset, it concomitantly results in autoimmune gastritis development (182). Autoimmune gastritis is a CD4+ T cell-mediated disease mainly characterized by lymphocytic infiltration in the gastric mucosa and the production of autoantibodies against the parietal cell H+/K+ ATPase proton pump (183–185). The BALB/c mouse is particularly susceptible to autoimmune gastritis (183). D3-Tx BALB/c mice develop autoimmune gastritis that closely resembles human disease and for which the pathologic score is higher than in d3-Tx NOD mice (183). Interestingly, susceptibility loci linked to autoimmune gastritis, namely Gasa1 and 2, are located on mouse chromosome 4 (184, 185) and overlap with the Idd11 and Idd9 loci, respectively (183–185). This suggests a strong genetic association between autoimmune gastritis and diabetes (186). Notably, the prevalence of autoimmune gastritis in PWT1D is 3-to-5-fold higher than in the general population (187). The polyautoimmunity developing in Tx mice is thus relevant to autoimmune gastritis and diabetes.
Overall, disturbances in various components affecting T cell tolerance exacerbates polyautoimmunity in NOD mice, providing clues to the development of polyautoimmunity and potentially revealing therapeutic targets to alleviate the severity of the pathologies.
The NOD Mouse as a Relevant Tool Beyond Spontaneous Autoimmunity
Unarguably, the NOD mouse model is a useful tool to study autoimmune diabetes. By genetic manipulation, derivatives of the NOD mouse model represent relevant spontaneous models for multiple human autoimmune pathologies. However, one cannot ignore other highly relevant uses of the NOD mouse model. For one, intravenous injection of pertussis toxin in NOD mice induces the development of experimental autoimmune encephalitis (158). This new induced model exhibits phases of remission, and closely mimics clinical and histopathological properties of MS; it may help to determine the genetic and environmental factors that promote the progression of MS (158). In addition, following injection of heat-killed bacillus Calmette-Guérin, NOD mice develop a non-organ specific autoimmune rheumatic disease similar to SLE (188–190), creating yet another relevant induced model to study the progression of a human pathology.
Apart from autoimmune diseases, the NOD strain has been used for studying human cells. Indeed, due to a polymorphism in CD172a, the NOD strain allows for better engraftment of human hematopoietic cells than other mice (191, 192). The strong interaction between the CD172a protein on the NOD macrophages and CD47 on human cells leads to a negative regulation of macrophage phagocytosis (193). Engraftment of human cells is typically performed in NOD.SCID, NOD.Rag-/-, NOD.SCID.IL2Rγ-/- or NOD.Rag-/-.IL2Rγ-/- mice, deficient in various components of the adaptive immune system, to further facilitate xenogeneic engraftment (194–199). Additional NOD mouse models are constantly being created to enhance human cell engraftment or to study specific diseases (200–206). For instance, the Human Immune System (HIS)-DRAGA (HLA-A2.HLA-DR4.Rag1-/-.IL-2Rγc-/-.NOD) mouse, grafted with human epithelial cells expressing the human angiotensin-converting enzyme 2 (hACE2) receptor in their lungs, was generated for COVID-19 research (207). Following immune reconstitution with human HLA-matched hematopoietic stem cells and intranasal infection with SARS-CoV-2, the HIS-DRAGA mouse exhibits T cell infiltration in the lungs and develops the different forms of severity of COVID-19 disease, as seen in the human population (207). The HIS-DRAGA mouse strain provides an important model for studying SARS-CoV-2 infection, as well as the immune responses generated against this virus, and can be used to test potential therapeutics and vaccines (207).
Conclusion
The NOD mouse remains one of the best models to study T1D. It is useful to study autoimmune susceptibility as well as genetic and cellular factors contributing to breakdowns of immune tolerance. Genetic manipulation of the NOD mouse has generated excellent models for studying spontaneous organ-specific autoimmune diseases other than diabetes such as thyroiditis, neuropathies, ABD and even polyautoimmunity. The manifestation of these autoimmune diseases in the NOD variant strains share many characteristics with human diseases, particularly immune cell infiltration in the targeted organ and a strong humoral response involving the generation of autoantibodies. Although pancreatic β cells have been shown to be particularly fragile in NOD mice (208), the exact reasons why the NOD mouse develops autoimmune diabetes whereas genetically modified NOD mice spontaneously develop autoimmune responses to other target organs remain unknown. In addition to organ-specific autoimmune diseases, disturbances in peripheral or central tolerance in NOD mice lead to polyautoimmunity, providing key information on the importance of these immune tolerance mechanisms for maintaining health. All in all, the NOD mouse, along with the several NOD congenic mice and NOD genetic knockout mice that have been generated over the years, represent indispensable tools in research that may be exploited for applications much broader than the study of type 1 diabetes. With their close parallel to various human autoimmune pathologies, these models should be exploited to increase our understanding of these specific pathologies as well as to design and test novel therapeutics.
Author Contributions
A-MA wrote the first draft of the manuscript and prepared most of the figures. FL-V contributed to the first draft of the manuscript and prepared some figures. RC analyzed the data for Figure 2. HA generated the data for Figure 2 and revised the final version of the manuscript. SM supervised HA and revised the final version of the manuscript. SL supervised A-MA and FL-V and revised the final version of the manuscript. All authors contributed to the article and approved the submitted version.
Funding
A-MA holds scholarships from the Canadian Institutes of Health Research and the Fondation de l’Hôpital Maisonneuve-Rosemont. FL-V holds scholarships from the Fondation de l’Hôpital Maisonneuve-Rosemont, the Cole Foundation, and the Fonds de Recherche Quebec Santé. Funding for studies by SMM: Supported by NIH DK 54684 (SMM) and DK 19289 (Basil Rapoport). SL is a Research Scholars Emeritus awardee from the Fonds de Recherche Quebec Santé.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Makino S, Kunimoto K, Muraoka Y, Mizushima Y, Katagiri K, Tochino Y. Breeding of a Non-Obese, Diabetic Strain of Mice. Jikken Dobutsu (1980) 29(1):1–13. doi: 10.1538/expanim1978.29.1_1
2. Mullen Y. Development of the Nonobese Diabetic Mouse and Contribution of Animal Models for Understanding Type 1 Diabetes. Pancreas (2017) 46(4):455–66. doi: 10.1097/MPA.0000000000000828
3. Anderson MS, Bluestone JA. The NOD Mouse: A Model of Immune Dysregulation. Annu Rev Immunol (2005) 23:447–85. doi: 10.1146/annurev.immunol.23.021704.115643
4. Chen YG, Mathews CE, Driver JP. The Role of NOD Mice in Type 1 Diabetes Research: Lessons From the Past and Recommendations for the Future. Front Endocrinol (Lausanne) (2018) 9:51. doi: 10.3389/fendo.2018.00051
5. Ikegami H, Makino S, Harada M, Eisenbarth GS, Hattori M. The Cataract Shionogi Mouse, a Sister Strain of the Non-Obese Diabetic Mouse: Similar Class II But Different Class I Gene Products. Diabetologia (1988) 31(4):254–8. doi: 10.1007/BF00290594
6. Pearson JA, Wong FS, Wen L. The Importance of the Non Obese Diabetic (NOD) Mouse Model in Autoimmune Diabetes. J Autoimmun (2016) 66:76–88. doi: 10.1016/j.jaut.2015.08.019
7. Lehuen A, Diana J, Zaccone P, Cooke A. Immune Cell Crosstalk in Type 1 Diabetes. Nat Rev Immunol (2010) 10(7):501–13. doi: 10.1038/nri2787
8. Polychronakos C, Li Q. Understanding Type 1 Diabetes Through Genetics: Advances and Prospects. Nat Rev Genet (2011) 12(11):781–92. doi: 10.1038/nrg3069
9. Drescher KM, Kono K, Bopegamage S, Carson SD, Tracy S. Coxsackievirus B3 Infection and Type 1 Diabetes Development in NOD Mice: Insulitis Determines Susceptibility of Pancreatic Islets to Virus Infection. Virology (2004) 329(2):381–94. doi: 10.1016/j.virol.2004.06.049
10. Hillhouse EE, Collin R, Chabot-Roy G, Guyon MJ, Tessier N, Boulay M, et al. Nearby Construction Impedes the Progression to Overt Autoimmune Diabetes in NOD Mice. J Diabetes Res (2013) 2013:620313. doi: 10.1155/2013/620313
11. Babad J, Geliebter A, DiLorenzo TP. T-Cell Autoantigens in the Non-Obese Diabetic Mouse Model of Autoimmune Diabetes. Immunol (2010) 131(4):459–65. doi: 10.1111/j.1365-2567.2010.03362.x
12. Braverman LE, Utiger RD. Werner & Ingbar’s The Thyroid A Fundamental and Clinical Text. 9th ed. Philadelphia: Lippincott Williams & Wilkins (2005).
13. McLachlan SM, Rapoport B. Breaking Tolerance to Thyroid Antigens: Changing Concepts in Thyroid Autoimmunity. Endocr Rev (2014) 35(1):59–105. doi: 10.1210/er.2013-1055
14. Hollowell JG, Staehling NW, Flanders WD, Hannon WH, Gunter EW, Spencer CA, et al. T(4), and Thyroid Antibodies in the United States Population (1988 to 1994): National Health and Nutrition Examination Survey (NHANES III). J Clin Endocrinol Metab (2002) 87(2):489–99. doi: 10.1210/jcem.87.2.8182
15. Nishi M. Diabetes Mellitus and Thyroid Diseases. Diabetol Int (2018) 9(2):108–12. doi: 10.1007/s13340-018-0352-4
16. Duntas LH, Orgiazzi J, Brabant G. The Interface Between Thyroid and Diabetes Mellitus. Clin Endocrinol (Oxf) (2011) 75(1):1–9. doi: 10.1111/j.1365-2265.2011.04029.x
17. Villano MJ, Huber AK, Greenberg DA, Golden BK, Concepcion E, Tomer Y. Autoimmune Thyroiditis and Diabetes: Dissecting the Joint Genetic Susceptibility in a Large Cohort of Multiplex Families. J Clin Endocrinol Metab (2009) 94(4):1458–66. doi: 10.1210/jc.2008-2193
18. Taniyama M, Kasuga A, Nagayama C, Ito K. Occurrence of Type 1 Diabetes in Graves’ Disease Patients Who Are Positive for Antiglutamic Acid Decarboxylase Antibodies: An 8-Year Followup Study. J Thyroid Res (2010) 2011:306487. doi: 10.4061/2011/306487
19. Hwang GB, Yoon JS, Park KJ, Lee HS, Hwang JS. Prevalence of Autoimmune Thyroiditis in Patients With Type 1 Diabetes: A Long-Term Follow-Up Study. Ann Pediatr Endocrinol Metab (2018) 23(1):33–7. doi: 10.6065/apem.2018.23.1.33
20. Damotte D, Colomb E, Cailleau C, Brousse N, Charreire J, Carnaud C. Analysis of Susceptibility of NOD Mice to Spontaneous and Experimentally Induced Thyroiditis. Eur J Immunol (1997) 27(11):2854–62. doi: 10.1002/eji.1830271117
21. Kolypetri P, Noel NA, Carayanniotis KA, Carayanniotis G. Iodine Content of Thyroglobulin in Nod.H2h4 Mice Developing Iodine-Accelerated Autoimmune Thyroiditis. Hormones (Athens) (2010) 9(2):151–60. doi: 10.14310/horm.2002.1265
22. McLachlan SM, Aliesky HA, Rapoport B. To Reflect Human Autoimmune Thyroiditis, Thyroid Peroxidase (Not Thyroglobulin) Antibodies Should be Measured in Female (Not Sex-Independent) NOD.H2(h4) Mice. Clin Exp Immunol (2019) 196(1):52–8. doi: 10.1111/cei.13249
23. Vitale M, Di Matola T, D’Ascoli F, Salzano S, Bogazzi F, Fenzi G, et al. Iodide Excess Induces Apoptosis in Thyroid Cells Through a P53-Independent Mechanism Involving Oxidative Stress. Endocrinology (2000) 141(2):598–605. doi: 10.1210/endo.141.2.7291
24. Hutchings PR, Verma S, Phillips JM, Harach SZ, Howlett S, Cooke A. Both CD4(+) T Cells and CD8(+) T Cells Are Required for Iodine Accelerated Thyroiditis in NOD Mice. Cell Immunol (1999) 192(2):113–21. doi: 10.1006/cimm.1998.1446
25. Chen D, Thayer TC, Wen L, Wong FS. Mouse Models of Autoimmune Diabetes: The Nonobese Diabetic (NOD) Mouse. Methods Mol Biol (2020) 2128:87–92. doi: 10.1007/978-1-0716-0385-7_6
26. Podolin PL, Pressey A, DeLarato NH, Fischer PA, Peterson LB, Wicker LS. I-E+ Nonobese Diabetic Mice Develop Insulitis and Diabetes. J Exp Med (1993) 178(3):793–803. doi: 10.1084/jem.178.3.793
27. Weatherall D, Sarvetnick N, Shizuru JA. Genetic Control of Diabetes Mellitus. Diabetologia (1992) 35(Suppl 2):S1–7. doi: 10.1007/BF00586273
28. Pelletier AN, Aliesky HA, Banuelos B, Chabot-Roy G, Rapoport B, Lesage S, et al. Evidence That MHC I-E Dampens Thyroid Autoantibodies and Prevents Spreading to a Second Thyroid Autoantigen in I-A(k) NOD Mice. Genes Immun (2015) 16(4):268–74. doi: 10.1038/gene.2015.7
29. Rasooly L, Burek CL, Rose NR. Iodine-Induced Autoimmune Thyroiditis in NOD-H-2h4 Mice. Clin Immunol Immunopathol (1996) 81(3):287–92. doi: 10.1006/clin.1996.0191
30. Braley-Mullen H, Sharp GC, Medling B, Tang H. Spontaneous Autoimmune Thyroiditis in NOD.H-2h4 mice. J Autoimmun (1999) 12(3):157–65. doi: 10.1006/jaut.1999.0272
31. Golden B, Levin L, Ban Y, Concepcion E, Greenberg DA, Tomer Y. Genetic Analysis of Families With Autoimmune Diabetes and Thyroiditis: Evidence for Common and Unique Genes. J Clin Endocrinol Metab (2005) 90(8):4904–11. doi: 10.1210/jc.2004-2236
32. Levin L, Ban Y, Concepcion E, Davies TF, Greenberg DA, Tomer Y. Analysis of HLA Genes in Families With Autoimmune Diabetes and Thyroiditis. Hum Immunol (2004) 65(6):640–7. doi: 10.1016/j.humimm.2004.02.026
33. Bonita RE, Rose NR, Rasooly L, Caturegli P, Burek CL. Adhesion Molecules as Susceptibility Factors in Spontaneous Autoimmune Thyroiditis in the NOD-H2h4 Mouse. Exp Mol Pathol (2002) 73(3):155–63. doi: 10.1006/exmp.2002.2470
34. Bonita RE, Rose NR, Rasooly L, Caturegli P, Burek CL. Kinetics of Mononuclear Cell Infiltration and Cytokine Expression in Iodine-Induced Thyroiditis in the NOD-H2h4 Mouse. Exp Mol Pathol (2003) 74(1):1–12. doi: 10.1016/S0014-4800(03)80002-3
35. Deng Q, Luo Y, Chang C, Wu H, Ding Y, Xiao R. The Emerging Epigenetic Role of CD8+T Cells in Autoimmune Diseases: A Systematic Review. Front Immunol (2019) 10:856. doi: 10.3389/fimmu.2019.00856
36. Bonilla FA, Oettgen HC. Adaptive Immunity. J Allergy Clin Immunol (2010) 125(2 Suppl 2):S33–40. doi: 10.1016/j.jaci.2009.09.017
37. Walter U, Santamaria P. CD8+ T Cells in Autoimmunity. Curr Opin Immunol (2005) 17(6):624–31. doi: 10.1016/j.coi.2005.09.014
38. Pawlak M, Ho AW, Kuchroo VK. Cytokines and Transcription Factors in the Differentiation of CD4(+) T Helper Cell Subsets and Induction of Tissue Inflammation and Autoimmunity. Curr Opin Immunol (2020) 67:57–67. doi: 10.1016/j.coi.2020.09.001
39. Matsuda JL, Mallevaey T, Scott-Browne J, Gapin L. CD1d-Restricted iNKT Cells, the ‘Swiss-Army Knife’ of the Immune System. Curr Opin Immunol (2008) 20(3):358–68. doi: 10.1016/j.coi.2008.03.018
40. Bendelac A, Lantz O, Quimby ME, Yewdell JW, Bennink JR, Brutkiewicz RR. CD1 Recognition by Mouse NK1+ T Lymphocytes. Science (1995) 268(5212):863–5. doi: 10.1126/science.7538697
41. Bendelac A, Savage PB, Teyton L. The Biology of NKT Cells. Annu Rev Immunol (2007) 25:297–336. doi: 10.1146/annurev.immunol.25.022106.141711
42. Sharma RB, Fan X, Caturegli P, Rose NR, Burek CL. Invariant NKT Cell Lines Derived From the NOD·H2 Mouse Enhance Autoimmune Thyroiditis. J Thyroid Res (2011) 2011:895923. doi: 10.4061/2011/895923
43. Burek CL, Sharma RB, Rose NR. NKT Cell Regulation of Autoimmune Thyroiditis. Autoimmunity (2003) 36(6-7):405–8. doi: 10.1080/08916930310001603064
44. Göschl L, Scheinecker C, Bonelli M. Treg Cells in Autoimmunity: From Identification to Treg-Based Therapies. Semin Immunopathol (2019) 41(3):301–14. doi: 10.1007/s00281-019-00741-8
45. Nagayama Y, Horie I, Saitoh O, Nakahara M, Abiru N. CD4+CD25+ Naturally Occurring Regulatory T Cells and Not Lymphopenia Play a Role in the Pathogenesis of Iodide-Induced Autoimmune Thyroiditis in NOD-H2h4 Mice. J Autoimmun (2007) 29(2-3):195–202. doi: 10.1016/j.jaut.2007.07.008
46. Zhu J, Paul WE. Heterogeneity and Plasticity of T Helper Cells. Cell Res (2010) 20(1):4–12. doi: 10.1038/cr.2009.138
47. Zhu J, Paul WE. Peripheral CD4+ T-Cell Differentiation Regulated by Networks of Cytokines and Transcription Factors. Immunol Rev (2010) 238(1):247–62. doi: 10.1111/j.1600-065X.2010.00951.x
48. Yu S, Sharp GC, Braley-Mullen H. Dual Roles for IFN-Gamma, But Not for IL-4, in Spontaneous Autoimmune Thyroiditis in NOD.H-2h4 Mice. J Immunol (2002) 169(7):3999–4007. doi: 10.4049/jimmunol.169.7.3999
49. Horie I, Abiru N, Nagayama Y, Kuriya G, Saitoh O, Ichikawa T, et al. T Helper Type 17 Immune Response Plays an Indispensable Role for Development of Iodine-Induced Autoimmune Thyroiditis in Nonobese Diabetic-H2h4 Mice. Endocrinology (2009) 150(11):5135–42. doi: 10.1210/en.2009-0434
50. Horie I, Abiru N, Sakamoto H, Iwakura Y, Nagayama Y. Induction of Autoimmune Thyroiditis by Depletion of CD4+CD25+ Regulatory T Cells in Thyroiditis-Resistant IL-17, But Not Interferon-Gamma Receptor, Knockout Nonobese Diabetic-H2h4 Mice. Endocrinology (2011) 152(11):4448–54. doi: 10.1210/en.2011-1356
51. Shevyrev D, Tereshchenko V. Treg Heterogeneity, Function, and Homeostasis. Front Immunol (2019) 10:3100. doi: 10.3389/fimmu.2019.03100
52. Bachmann MF, Kopf M. The Role of B Cells in Acute and Chronic Infections. Curr Opin Immunol (1999) 11(3):332–9. doi: 10.1016/S0952-7915(99)80053-3
53. Martin F, Chan AC. Pathogenic Roles of B Cells in Human Autoimmunity; Insights From the Clinic. Immunity (2004) 20(5):517–27. doi: 10.1016/S1074-7613(04)00112-8
54. Braley-Mullen H, Yu S. Early Requirement for B Cells for Development of Spontaneous Autoimmune Thyroiditis in NOD.H-2h4 Mice. J Immunol (2000) 165(12):7262–9. doi: 10.4049/jimmunol.165.12.7262
55. Yu S, Dunn R, Kehry MR, Braley-Mullen H. B Cell Depletion Inhibits Spontaneous Autoimmune Thyroiditis in NOD.H-2h4 Mice. J Immunol (2008) 180(11):7706–13. doi: 10.4049/jimmunol.180.11.7706
56. Hong SH, Braley-Mullen H. Follicular B Cells in Thyroids of Mice With Spontaneous Autoimmune Thyroiditis Contribute to Disease Pathogenesis and Are Targets of Anti-CD20 Antibody Therapy. J Immunol (2014) 192(3):897–905. doi: 10.4049/jimmunol.1301628
57. Chen CR, Hamidi S, Braley-Mullen H, Nagayama Y, Bresee C, Aliesky HA, et al. Antibodies to Thyroid Peroxidase Arise Spontaneously With Age in NOD.H-2h4 Mice and Appear After Thyroglobulin Antibodies. Endocrinology (2010) 151(9):4583–93. doi: 10.1210/en.2010-0321
58. Ippolito S, Di Dalmazi G, Pani F, Sabini E, Caturegli P. Distinct Cytokine Signatures in Thyroiditis Induced by PD-1 or CTLA-4 Blockade: Insights From a New Mouse Model. Thyroid (2021) 31(12):1839–49. doi: 10.1089/thy.2021.0165
59. Caturegli P, De Remigis A, Rose NR. Hashimoto Thyroiditis: Clinical and Diagnostic Criteria. Autoimmun Rev (2014) 13(4-5):391–7. doi: 10.1016/j.autrev.2014.01.007
60. Tunbridge WM, Evered DC, Hall R, Appleton D, Brewis M, Clark F, et al. The Spectrum of Thyroid Disease in a Community: The Whickham Survey. Clin Endocrinol (Oxf) (1977) 7(6):481–93. doi: 10.1111/j.1365-2265.1977.tb01340.x
61. Vanderpump MP, Tunbridge WM, French JM, Appleton D, Bates D, Clark F, et al. The Incidence of Thyroid Disorders in the Community: A Twenty-Year Follow-Up of the Whickham Survey. Clin Endocrinol (Oxf) (1995) 43(1):55–68. doi: 10.1111/j.1365-2265.1995.tb01894.x
62. McLachlan SM, Aliesky HA, Banuelos B, Lesage S, Collin R, Rapoport B. High-Level Intrathymic Thyrotrophin Receptor Expression in Thyroiditis-Prone Mice Protects Against the Spontaneous Generation of Pathogenic Thyrotrophin Receptor Autoantibodies. Clin Exp Immunol (2017) 188(2):243–53. doi: 10.1111/cei.12928
63. Rapoport B, Aliesky HA, Banuelos B, Chen CR, McLachlan SM. A Unique Mouse Strain That Develops Spontaneous, Iodine-Accelerated, Pathogenic Antibodies to the Human Thyrotrophin Receptor. J Immunol (2015) 194(9):4154–61. doi: 10.4049/jimmunol.1500126
64. Sun H, Ye Z, Li N, Jin F, Yan J, Wu K. Effect of Emodin on T Cell Subsets in NOD Mice With NaI−induced Experimental Autoimmune Thyroiditis. Mol Med Rep (2018) 18(5):4303–12. doi: 10.3892/mmr.2018.9434
65. Watson JC, Dyck PJ. Peripheral Neuropathy: A Practical Approach to Diagnosis and Symptom Management. Mayo Clin Proc (2015) 90(7):940–51. doi: 10.1016/j.mayocp.2015.05.004
66. Ramdharry G. Peripheral Nerve Disease. Handb Clin Neurol (2018) 159:403–15. doi: 10.1016/B978-0-444-63916-5.00026-4
67. Sommer C, Geber C, Young P, Forst R, Birklein F, Schoser B. Polyneuropathies. Dtsch Arztebl Int (2018) 115(6):83–90. doi: 10.3238/arztebl.2018.0083
68. Feldman EL, Callaghan BC, Pop-Busui R, Zochodne DW, Wright DE, Bennett DL, et al. Diabetic Neuropathy. Nat Rev Dis Primers (2019) 5(1):42. doi: 10.1038/s41572-019-0092-1
69. Winer S, Tsui H, Lau A, Song A, Li X, Cheung RK, et al. Autoimmune Islet Destruction in Spontaneous Type 1 Diabetes Is Not Beta-Cell Exclusive. Nat Med (2003) 9(2):198–205. doi: 10.1038/nm818
70. Janahi NM, Santos D, Blyth C, Bakhiet M, Ellis M. Diabetic Peripheral Neuropathy, Is It an Autoimmune Disease? Immunol Lett (2015) 168(1):73–9. doi: 10.1016/j.imlet.2015.09.009
71. Singh R, Kishore L, Kaur N. Diabetic Peripheral Neuropathy: Current Perspective and Future Directions. Pharmacol Res (2014) 80:21–35. doi: 10.1016/j.phrs.2013.12.005
72. King GL, Loeken MR. Hyperglycemia-Induced Oxidative Stress in Diabetic Complications. Histochem Cell Biol (2004) 122(4):333–8. doi: 10.1007/s00418-004-0678-9
73. Peltier A, Goutman SA, Callaghan BC. Painful Diabetic Neuropathy. Bmj (2014) 348:g1799. doi: 10.1136/bmj.g1799
74. Smith-Garvin JE, Koretzky GA, Jordan MS. T Cell Activation. Annu Rev Immunol (2009) 27:591–619. doi: 10.1146/annurev.immunol.021908.132706
75. Curtsinger JM, Mescher MF. Inflammatory Cytokines as a Third Signal for T Cell Activation. Curr Opin Immunol (2010) 22(3):333–40. doi: 10.1016/j.coi.2010.02.013
76. Anderson MS, Venanzi ES, Klein L, Chen Z, Berzins SP, Turley SJ, et al. Projection of an Immunological Self Shadow Within the Thymus by the Aire Protein. Science (2002) 298(5597):1395–401. doi: 10.1126/science.1075958
77. Anderson MS, Venanzi ES, Chen Z, Berzins SP, Benoist C, Mathis D. The Cellular Mechanism of Aire Control of T Cell Tolerance. Immunity (2005) 23(2):227–39. doi: 10.1016/j.immuni.2005.07.005
78. Yang S, Fujikado N, Kolodin D, Benoist C, Mathis D. Regulatory T Cells Generated Early in Life Play a Distinct Role in Maintaining Self-Tolerance. Science (2015) 348(6234):589–94. doi: 10.1126/science.aaa7017
79. Liston A, Lesage S, Wilson J, Peltonen L, Goodnow CC. Aire Regulates Negative Selection of Organ-Specific T Cells. Nat Immunol (2003) 4(4):350–4. doi: 10.1038/ni906
80. Liston A, Gray DH, Lesage S, Fletcher AL, Wilson J, Webster KE, et al. Gene Dosage–Limiting Role of Aire in Thymic Expression, Clonal Deletion, and Organ-Specific Autoimmunity. J Exp Med (2004) 200(8):1015–26. doi: 10.1084/jem.20040581
81. Su MA, Giang K, Zumer K, Jiang H, Oven I, Rinn JL, et al. Mechanisms of an Autoimmunity Syndrome in Mice Caused by a Dominant Mutation in Aire. J Clin Invest (2008) 118(5):1712–26. doi: 10.1172/JCI34523
82. Zeng XL, Nagavalli A, Smith CJ, Howard JF, Su MA. Divergent Effects of T Cell Costimulation and Inflammatory Cytokine Production on Autoimmune Peripheral Neuropathy Provoked by Aire Deficiency. J Immunol (2013) 190(8):3895–904. doi: 10.4049/jimmunol.1203001
83. Su MA, Davini D, Cheng P, Giang K, Fan U, DeVoss JJ, et al. Defective Autoimmune Regulator-Dependent Central Tolerance to Myelin Protein Zero Is Linked to Autoimmune Peripheral Neuropathy. J Immunol (2012) 188(10):4906–12. doi: 10.4049/jimmunol.1200493
84. Louvet C, Kabre BG, Davini DW, Martinier N, Su MA, DeVoss JJ, et al. A Novel Myelin P0-Specific T Cell Receptor Transgenic Mouse Develops a Fulminant Autoimmune Peripheral Neuropathy. J Exp Med (2009) 206(3):507–14. doi: 10.1084/jem.20082113
85. Xia F, Qian CR, Xun Z, Hamon Y, Sartre AM, Formisano A, et al. TCR and CD28 Concomitant Stimulation Elicits a Distinctive Calcium Response in Naive T Cells. Front Immunol (2018) 9:2864. doi: 10.3389/fimmu.2018.02864
86. Acuto O, Michel F. CD28-Mediated Co-Stimulation: A Quantitative Support for TCR Signalling. Nat Rev Immunol (2003) 3(12):939–51. doi: 10.1038/nri1248
87. Chen L, Flies DB. Molecular Mechanisms of T Cell Co-Stimulation and Co-Inhibition. Nat Rev Immunol (2013) 13(4):227–42. doi: 10.1038/nri3405
88. Dong C, Juedes AE, Temann UA, Shresta S, Allison JP, Ruddle NH, et al. ICOS Co-Stimulatory Receptor Is Essential for T-Cell Activation and Function. Nature (2001) 409(6816):97–101. doi: 10.1038/35051100
89. Edner NM, Carlesso G, Rush JS, Walker LSK. Targeting Co-Stimulatory Molecules in Autoimmune Disease. Nat Rev Drug Discov (2020) 19(12):860–83. doi: 10.1038/s41573-020-0081-9
90. Salomon B, Rhee L, Bour-Jordan H, Hsin H, Montag A, Soliven B, et al. Development of Spontaneous Autoimmune Peripheral Polyneuropathy in B7-2-Deficient NOD Mice. J Exp Med (2001) 194(5):677–84. doi: 10.1084/jem.194.5.677
91. Prevot N, Briet C, Lassmann H, Tardivel I, Roy E, Morin J, et al. Abrogation of ICOS/ICOS Ligand Costimulation in NOD Mice Results in Autoimmune Deviation Toward the Neuromuscular System. Eur J Immunol (2010) 40(8):2267–76. doi: 10.1002/eji.201040416
92. Lesage S, Goodnow CC. Organ-Specific Autoimmune Disease: A Deficiency of Tolerogenic Stimulation. J Exp Med (2001) 194(5):F31–6. doi: 10.1084/jem.194.5.F31
93. Vogel I, Kasran A, Cremer J, Kim YJ, Boon L, Van Gool SW, et al. CD28/CTLA-4/B7 Costimulatory Pathway Blockade Affects Regulatory T-Cell Function in Autoimmunity. Eur J Immunol (2015) 45(6):1832–41. doi: 10.1002/eji.201445190
94. Bour-Jordan H, Thompson HL, Giampaolo JR, Davini D, Rosenthal W, Bluestone JA. Distinct Genetic Control of Autoimmune Neuropathy and Diabetes in the Non-Obese Diabetic Background. J Autoimmun (2013) 45:58–67. doi: 10.1016/j.jaut.2013.06.005
95. Kunz M, Ibrahim SM. Cytokines and Cytokine Profiles in Human Autoimmune Diseases and Animal Models of Autoimmunity. Mediators Inflamm (2009) 2009:979258. doi: 10.1155/2009/979258
96. Yadav D, Sarvetnick N. Cytokines and Autoimmunity: Redundancy Defines Their Complex Nature. Curr Opin Immunol (2003) 15(6):697–703. doi: 10.1016/j.coi.2003.09.006
97. Hill N, Sarvetnick N. Cytokines: Promoters and Dampeners of Autoimmunity. Curr Opin Immunol (2002) 14(6):791–7. doi: 10.1016/S0952-7915(02)00403-X
98. Setoguchi R, Hori S, Takahashi T, Sakaguchi S. Homeostatic Maintenance of Natural Foxp3(+) CD25(+) CD4(+) Regulatory T Cells by Interleukin (IL)-2 and Induction of Autoimmune Disease by IL-2 Neutralization. J Exp Med (2005) 201(5):723–35. doi: 10.1084/jem.20041982
99. Malek TR. The Biology of Interleukin-2. Annu Rev Immunol (2008) 26:453–79. doi: 10.1146/annurev.immunol.26.021607.090357
100. Abbas AK, Trotta E D, Marson A, Bluestone JA. Revisiting IL-2: Biology and Therapeutic Prospects. Sci Immunol (2018) 3(25):1–8. doi: 10.1126/sciimmunol.aat1482
101. Yamanouchi J, Rainbow D, Serra P, Howlett S, Hunter K, Garner VE, et al. Interleukin-2 Gene Variation Impairs Regulatory T Cell Function and Causes Autoimmunity. Nat Genet (2007) 39(3):329–37. doi: 10.1038/ng1958
102. Mangada J, Pearson T, Brehm MA, Wicker LS, Peterson LB, Shultz LD, et al. Idd Loci Synergize to Prolong Islet Allograft Survival Induced by Costimulation Blockade in NOD Mice. Diabetes (2009) 58(1):165–73. doi: 10.2337/db08-0275
103. Allen D, Giannopoulos K, Gray I, Gregson N, Makowska A, Pritchard J, et al. Antibodies to Peripheral Nerve Myelin Proteins in Chronic Inflammatory Demyelinating Polyradiculoneuropathy. J Peripher Nerv Syst (2005) 10(2):174–80. doi: 10.1111/j.1085-9489.2005.0010207.x
104. Khalili-Shirazi A, Atkinson P, Gregson N, Hughes RA. Antibody Responses to P0 and P2 Myelin Proteins in Guillain-Barré Syndrome and Chronic Idiopathic Demyelinating Polyradiculoneuropathy. J Neuroimmunol (1993) 46(1-2):245–51. doi: 10.1016/0165-5728(93)90255-W
105. Carrillo J, Puertas MC, Alba A, Ampudia RM, Pastor X, Planas R, et al. Islet-Infiltrating B-Cells in Nonobese Diabetic Mice Predominantly Target Nervous System Elements. Diabetes (2005) 54(1):69–77. doi: 10.2337/diabetes.54.1.69
106. Racine JJ, Chapman HD, Doty R, Cairns BM, Hines TJ, Tadenev ALD, et al. T Cells From NOD-PerIg Mice Target Both Pancreatic and Neuronal Tissue. J Immunol (2020) 205(8):2026–38. doi: 10.4049/jimmunol.2000114
107. Leeth CM, Racine J, Chapman HD, Arpa B, Carrillo J, Carrascal J, et al. B-Lymphocytes Expressing an Ig Specificity Recognizing the Pancreatic ß-Cell Autoantigen Peripherin Are Potent Contributors to Type 1 Diabetes Development in NOD Mice. Diabetes (2016) 65(7):1977–87. doi: 10.2337/db15-1606
108. Al-Awar A, Kupai K, Veszelka KD, Szűcs G, Attieh Z, Murlasits Z, et al. Experimental Diabetes Mellitus in Different Animal Models. J Diabetes Res (2016) 2016:1–12. doi: 10.1155/2016/9051426
109. Imam MH, Talwalkar JA, Lindor KD. Clinical Management of Autoimmune Biliary Diseases. J Autoimmun (2013) 46:88–96. doi: 10.1016/j.jaut.2013.06.014
110. Lee CW, Ronnekleiv-Kelly S. Autoimmune Diseases of the Biliary Tract: A Review. Surg Clin North Am (2019) 99(2):185–201. doi: 10.1016/j.suc.2018.11.003
111. Yeh MJ, Kim SY, Jhaveri KS, Behr SC, Seo N, Yeh BM. Imaging of Autoimmune Biliary Disease. Abdom Radiol (NY) (2017) 42(1):3–18. doi: 10.1007/s00261-016-0903-8
112. Bowlus CL, Gershwin ME. The Diagnosis of Primary Biliary Cirrhosis. Autoimmun Rev (2014) 13(4-5):441–4. doi: 10.1016/j.autrev.2014.01.041
113. Silveira MG, Lindor KD. Primary Sclerosing Cholangitis. Can J Gastroenterol (2008) 22(8):689–98. doi: 10.1155/2008/824168
114. Hubers LM, Maillette de Buy Wenniger LJ, Doorenspleet ME, Klarenbeek PL, Verheij J, Rauws EA, et al. IgG4-Associated Cholangitis: A Comprehensive Review. Clin Rev Allergy Immunol (2015) 48(2-3):198–206. doi: 10.1007/s12016-014-8430-2
115. Sherlock S, Scheuer PJ. The Presentation and Diagnosis of 100 Patients With Primary Biliary Cirrhosis. N Engl J Med (1973) 289(13):674–8. doi: 10.1056/NEJM197309272891306
116. Lindor KD, Gershwin ME, Poupon R, Kaplan M, Bergasa NV, Heathcote EJ. Primary Biliary Cirrhosis. Hepatology (2009) 50(1):291–308. doi: 10.1002/hep.22906
117. Kaplan MM, Gershwin ME. Primary Biliary Cirrhosis. N Engl J Med (2005) 353(12):1261–73. doi: 10.1056/NEJMra043898
118. Koarada S, Wu Y, Fertig N, Sass DA, Nalesnik M, Todd JA, et al. Genetic Control of Autoimmunity: Protection From Diabetes, But Spontaneous Autoimmune Biliary Disease in a Nonobese Diabetic Congenic Strain. J Immunol (2004) 173(4):2315–23. doi: 10.4049/jimmunol.173.4.2315
119. Irie J, Wu Y, Wicker LS, Rainbow D, Nalesnik MA, Hirsch R, et al. NOD.c3c4 Congenic Mice Develop Autoimmune Biliary Disease That Serologically and Pathogenetically Models Human Primary Biliary Cirrhosis. J Exp Med (2006) 203(5):1209–19. doi: 10.1084/jem.20051911
120. Yang GX, Wu Y, Tsukamoto H, Leung PS, Lian ZX, Rainbow DB, et al. CD8 T Cells Mediate Direct Biliary Ductule Damage in Nonobese Diabetic Autoimmune Biliary Disease. J Immunol (2011) 186(2):1259–67. doi: 10.4049/jimmunol.1001597
121. Teufel A, Weinmann A, Kahaly GJ, Centner C, Piendl A, Wörns M, et al. Concurrent Autoimmune Diseases in Patients With Autoimmune Hepatitis. J Clin Gastroenterol (2010) 44(3):208–13. doi: 10.1097/MCG.0b013e3181c74e0d
122. Hu S, Zhao F, Wang Q, Chen WX. The Accuracy of the Anti-Mitochondrial Antibody and the M2 Subtype Test for Diagnosis of Primary Biliary Cirrhosis: A Meta-Analysis. Clin Chem Lab Med (2014) 52(11):1533–42. doi: 10.1515/cclm-2013-0926
123. Chou MJ, Lee SL, Chen TY, Tsay GJ. Specificity of Antinuclear Antibodies in Primary Biliary Cirrhosis. Ann Rheum Dis (1995) 54(2):148–51. doi: 10.1136/ard.54.2.148
124. Irie J, Wu Y, Sass DA, Ridgway WM. Genetic Control of Anti-Sm Autoantibody Production in NOD Congenic Mice Narrowed to the Idd9.3 region. Immunogenetics (2006) 58(1):9–14. doi: 10.1007/s00251-005-0066-1
125. Goodwin RG, Din WS, Davis-Smith T, Anderson DM, Gimpel SD, Sato TA, et al. Molecular Cloning of a Ligand for the Inducible T Cell Gene 4-1BB: A Member of an Emerging Family of Cytokines With Homology to Tumor Necrosis Factor. Eur J Immunol (1993) 23(10):2631–41. doi: 10.1002/eji.1830231037
126. Cannons JL, Chamberlain G, Howson J, Smink LJ, Todd JA, Peterson LB, et al. Genetic and Functional Association of the Immune Signaling Molecule 4-1BB (CD137/TNFRSF9) With Type 1 Diabetes. J Autoimmun (2005) 25(1):13–20. doi: 10.1016/j.jaut.2005.04.007
127. Wakabayashi K, Lian ZX, Moritoki Y, Lan RY, Tsuneyama K, Chuang YH, et al. IL-2 Receptor Alpha(-/-) Mice and the Development of Primary Biliary Cirrhosis. Hepatology (2006) 44(5):1240–9. doi: 10.1002/hep.21385
128. Oertelt S, Lian ZX, Cheng CM, Chuang YH, Padgett KA, He XS, et al. Anti-Mitochondrial Antibodies and Primary Biliary Cirrhosis in TGF-Beta Receptor II Dominant-Negative Mice. J Immunol (2006) 177(3):1655–60. doi: 10.4049/jimmunol.177.3.1655
129. Anaya JM. The Diagnosis and Clinical Significance of Polyautoimmunity. Autoimmun Rev (2014) 13(4-5):423–6. doi: 10.1016/j.autrev.2014.01.049
130. Rojas-Villarraga A, Amaya-Amaya J, Rodriguez-Rodriguez A, Mantilla RD, Anaya JM. Introducing Polyautoimmunity: Secondary Autoimmune Diseases No Longer Exist. Autoimmune Dis (2012) 2012:254319. doi: 10.1155/2012/254319
131. Rojas M, Ramírez-Santana C, Acosta-Ampudia Y, Monsalve DM, Rodriguez-Jimenez M, Zapata E, et al. New Insights Into the Taxonomy of Autoimmune Diseases Based on Polyautoimmunity. J Autoimmun (2022) 126:102780. doi: 10.1016/j.jaut.2021.102780
132. Bernard NF, Ertug F, Margolese H. High Incidence of Thyroiditis and Anti-Thyroid Autoantibodies in NOD Mice. Diabetes (1992) 41(1):40–6. doi: 10.2337/diab.41.1.40
133. Martin AP, Marinkovic T, Canasto-Chibuque C, Latif R, Unkeless JC, Davies TF, et al. CCR7 Deficiency in NOD Mice Leads to Thyroiditis and Primary Hypothyroidism. J Immunol (2009) 183(5):3073–80. doi: 10.4049/jimmunol.0900275
134. Braley-Mullen H, Yu S. NOD.H-2h4 Mice: An Important and Underutilized Animal Model of Autoimmune Thyroiditis and Sjogren’s Syndrome. Adv Immunol (2015) 126:1–43. doi: 10.1016/bs.ai.2014.11.001
135. Sáez Moya M, Gutiérrez-Cózar R, Puñet-Ortiz J, Rodríguez de la Concepción ML, Blanco J, Carrillo J, et al. Autoimmune B Cell Repertoire in a Mouse Model of Sjögren’s Syndrome. Front Immunol (2021) 12:666545. doi: 10.3389/fimmu.2021.666545
136. Kayes TD, Weisman GA, Camden JM, Woods LT, Bredehoeft C, Downey EF, et al. New Murine Model of Early Onset Autoimmune Thyroid Disease/Hypothyroidism and Autoimmune Exocrinopathy of the Salivary Gland. J Immunol (2016) 197(6):2119–30. doi: 10.4049/jimmunol.1600133
137. Scuron MD, Fay B, Oliver J, Smith P. Spontaneous Model of Sjögren’s Syndrome in NOD Mice. Curr Protoc Pharmacol (2019) 86(1):e65. doi: 10.1002/cpph.65
138. Robinson CP, Yamachika S, Alford CE, Cooper C, Pichardo EL, Shah N, et al. Elevated Levels of Cysteine Protease Activity in Saliva and Salivary Glands of the Nonobese Diabetic (NOD) Mouse Model for Sjögren Syndrome. Proc Natl Acad Sci USA (1997) 94(11):5767–71. doi: 10.1073/pnas.94.11.5767
139. Burt RA, Watkins L, Tan IK, Wang N, Quirk F, Mackin L, et al. An NZW-Derived Interval on Chromosome 7 Moderates Sialadenitis, But Not Insulitis in Congenic Nonobese Diabetic Mice. J Immunol (2010) 184(2):859–68. doi: 10.4049/jimmunol.0903149
140. Ciecko AE, Foda B, Barr JY, Ramanathan S, Atkinson MA, Serreze DV, et al. Interleukin-27 Is Essential for Type 1 Diabetes Development and Sjögren Syndrome-Like Inflammation. Cell Rep (2019) 29(10):3073–86.e5. doi: 10.1016/j.celrep.2019.11.010
141. Cha S, Peck AB, Humphreys-Beher MG. Progress in Understanding Autoimmune Exocrinopathy Using the Non-Obese Diabetic Mouse: An Update. Crit Rev Oral Biol Med (2002) 13(1):5–16. doi: 10.1177/154411130201300103
142. Robinson CP, Yamamoto H, Peck AB, Humphreys-Beher MG. Genetically Programmed Development of Salivary Gland Abnormalities in the NOD (Nonobese Diabetic)-Scid Mouse in the Absence of Detectable Lymphocytic Infiltration: A Potential Trigger for Sialoadenitis of NOD Mice. Clin Immunol Immunopathol (1996) 79(1):50–9. doi: 10.1006/clin.1996.0050
143. Humphreys-Beher MG, Hu Y, Nakagawa Y, Wang PL, Purushotham KR. Utilization of the Non-Obese Diabetic (NOD) Mouse as an Animal Model for the Study of Secondary Sjögren’s Syndrome. Adv Exp Med Biol (1994) 350:631–6. doi: 10.1007/978-1-4615-2417-5_105
144. Binder A, Maddison PJ, Skinner P, Kurtz A, Isenberg DA. Sjögren’s Syndrome: Association With Type-1 Diabetes Mellitus. Br J Rheumatol (1989) 28(6):518–20. doi: 10.1093/rheumatology/28.6.518
145. Park YS, Gauna AE, Cha S. Mouse Models of Primary Sjogren’s Syndrome. Curr Pharm Des (2015) 21(18):2350–64. doi: 10.2174/1381612821666150316120024
146. Chen FY, Lee A, Ge S, Nathan S, Knox SM, McNamara NA. Aire-Deficient Mice Provide a Model of Corneal and Lacrimal Gland Neuropathy in Sjögren’s Syndrome. PloS One (2017) 12(9):e0184916. doi: 10.1371/journal.pone.0184916
147. Mayo Clinic laboratories. SS-A and SS-B Antibodies, IgG, Serum. Available at: https://neurology.testcatalog.org/show/SSAB.
148. Karnell JL, Mahmoud TI, Herbst R, Ettinger R. Discerning the Kinetics of Autoimmune Manifestations in a Model of Sjögren’s Syndrome. Mol Immunol (2014) 62(2):277–82. doi: 10.1016/j.molimm.2014.05.006
149. Peri Y, Agmon-Levin N, Theodor E, Shoenfeld Y. Sjögren’s Syndrome, the Old and the New. Best Pract Res Clin Rheumatol (2012) 26(1):105–17. doi: 10.1016/j.berh.2012.01.012
150. Kassan SS, Moutsopoulos HM. Clinical Manifestations and Early Diagnosis of Sjögren Syndrome. Arch Intern Med (2004) 164(12):1275–84. doi: 10.1001/archinte.164.12.1275
151. Bechtold S, Blaschek A, Raile K, Dost A, Freiberg C, Askenas M, et al. Higher Relative Risk for Multiple Sclerosis in a Pediatric and Adolescent Diabetic Population: Analysis From DPV Database. Diabetes Care (2014) 37(1):96–101. doi: 10.2337/dc13-1414
152. Comi C, Cappellano G, Chiocchetti A, Orilieri E, Buttini S, Ghezzi L, et al. The Impact of Osteopontin Gene Variations on Multiple Sclerosis Development and Progression. Clin Dev Immunol (2012) 2012:212893. doi: 10.1155/2012/212893
153. Nielsen NM, Westergaard T, Frisch M, Rostgaard K, Wohlfahrt J, Koch-Henriksen N, et al. Type 1 Diabetes and Multiple Sclerosis: A Danish Population-Based Cohort Study. Arch Neurol (2006) 63(7):1001–4. doi: 10.1001/archneur.63.7.1001
154. Marrosu MG, Motzo C, Murru R, Lampis R, Costa G, Zavattari P, et al. The Co-Inheritance of Type 1 Diabetes and Multiple Sclerosis in Sardinia Cannot be Explained by Genotype Variation in the HLA Region Alone. Hum Mol Genet (2004) 13(23):2919–24. doi: 10.1093/hmg/ddh319
155. Pitzalis M, Zavattari P, Murru R, Deidda E, Zoledziewska M, Murru D, et al. Genetic Loci Linked to Type 1 Diabetes and Multiple Sclerosis Families in Sardinia. BMC Med Genet (2008) 9:3. doi: 10.1186/1471-2350-9-3
156. Dorman JS, Steenkiste AR, Burke JP, Songini M. Type 1 Diabetes and Multiple Sclerosis: Together at Last. Diabetes Care (2003) 26(11):3192–3. doi: 10.2337/diacare.26.11.3192
157. Handel AE, Handunnetthi L, Ebers GC, Ramagopalan SV. Type 1 Diabetes Mellitus and Multiple Sclerosis: Common Etiological Features. Nat Rev Endocrinol (2009) 5(12):655–64. doi: 10.1038/nrendo.2009.216
158. Winer S, Astsaturov I, Cheung R, Gunaratnam L, Kubiak V, Cortez MA, et al. Type I Diabetes and Multiple Sclerosis Patients Target Islet Plus Central Nervous System Autoantigens; Nonimmunized Nonobese Diabetic Mice can Develop Autoimmune Encephalitis. J Immunol (2001) 166(4):2831–41. doi: 10.4049/jimmunol.166.4.2831
159. Ansari MJ, Salama AD, Chitnis T, Smith RN, Yagita H, Akiba H, et al. The Programmed Death-1 (PD-1) Pathway Regulates Autoimmune Diabetes in Nonobese Diabetic (NOD) Mice. J Exp Med (2003) 198(1):63–9. doi: 10.1084/jem.20022125
160. Yoshida T, Jiang F, Honjo T, Okazaki T. PD-1 Deficiency Reveals Various Tissue-Specific Autoimmunity by H-2b and Dose-Dependent Requirement of H-2g7 for Diabetes in NOD Mice. Proc Natl Acad Sci USA (2008) 105(9):3533–8. doi: 10.1073/pnas.0710951105
161. Nishimura H, Minato N, Nakano T, Honjo T. Immunological Studies on PD-1 Deficient Mice: Implication of PD-1 as a Negative Regulator for B Cell Responses. Int Immunol (1998) 10(10):1563–72. doi: 10.1093/intimm/10.10.1563
162. Riella LV, Paterson AM, Sharpe AH, Chandraker A. Role of the PD-1 Pathway in the Immune Response. Am J Transplant (2012) 12(10):2575–87. doi: 10.1111/j.1600-6143.2012.04224.x
163. Francisco LM, Sage PT, Sharpe AH. The PD-1 Pathway in Tolerance and Autoimmunity. Immunol Rev (2010) 236:219–42. doi: 10.1111/j.1600-065X.2010.00923.x
164. Wang J, Yoshida T, Nakaki F, Hiai H, Okazaki T, Honjo T. Establishment of NOD-Pdcd1-/- Mice as an Efficient Animal Model of Type I Diabetes. Proc Natl Acad Sci USA (2005) 102(33):11823–8. doi: 10.1073/pnas.0505497102
165. Sumida T, Tsuboi H, Iizuka M, Hirota T, Asashima H, Matsumoto I. The Role of M3 Muscarinic Acetylcholine Receptor Reactive T Cells in Sjögren’s Syndrome: A Critical Review. J Autoimmun (2014) 51:44–50. doi: 10.1016/j.jaut.2013.12.012
166. Jiang F, Yoshida T, Nakaki F, Terawaki S, Chikuma S, Kato Y, et al. Identification of QTLs That Modify Peripheral Neuropathy in NOD.H2b-Pdcd1-/- Mice. Int Immunol (2009) 21(5):499–509. doi: 10.1093/intimm/dxp020
167. Masli S, Dartt DA. Mouse Models of Sjögren’s Syndrome With Ocular Surface Disease. Int J Mol Sci (2020) 21(23):1–19. doi: 10.3390/ijms21239112
168. Robinson CP, Yamachika S, Bounous DI, Brayer J, Jonsson R, Holmdahl R, et al. A Novel NOD-Derived Murine Model of Primary Sjögren’s Syndrome. Arthritis Rheumatol (1998) 41(1):150–6. doi: 10.1002/1529-0131(199801)41:1<150::AID-ART18>3.0.CO;2-T
169. Kiripolsky J, Shen L, Liang Y, Li A, Suresh L, Lian Y, et al. Systemic Manifestations of Primary Sjögren’s Syndrome in the NOD.B10Sn-H2(b)/J Mouse Model. Clin Immunol (2017) 183:225–32. doi: 10.1016/j.clim.2017.04.009
170. Orlova EM, Sozaeva LS, Kareva MA, Oftedal BE, Wolff ASB, Breivik L, et al. Expanding the Phenotypic and Genotypic Landscape of Autoimmune Polyendocrine Syndrome Type 1. J Clin Endocrinol Metab (2017) 102(9):3546–56. doi: 10.1210/jc.2017-00139
171. Mathis D, Benoist C. Aire. Annu Rev Immunol (2009) 27:287–312. doi: 10.1146/annurev.immunol.25.022106.141532
172. Su MA, Anderson MS. Aire: An Update. Curr Opin Immunol (2004) 16(6):746–52. doi: 10.1016/j.coi.2004.09.009
173. Mathis D, Benoist C. A Decade of AIRE. Nat Rev Immunol (2007) 7(8):645–50. doi: 10.1038/nri2136
174. Finnish-German APECED Consortium. An Autoimmune Disease, APECED, Caused by Mutations in a Novel Gene Featuring Two PHD-Type Zinc-Finger Domains. Nat Genet (1997) 17(4):399–403. doi: 10.1038/ng1297-399
175. Nagamine K, Peterson P, Scott HS, Kudoh J, Minoshima S, Heino M, et al. Positional Cloning of the APECED Gene. Nat Genet (1997) 17(4):393–8. doi: 10.1038/ng1297-393
176. Björses P, Aaltonen J, Horelli-Kuitunen N, Yaspo ML, Peltonen L. Gene Defect Behind APECED: A New Clue to Autoimmunity. Hum Mol Genet (1998) 7(10):1547–53. doi: 10.1093/hmg/7.10.1547
177. Bruserud Ø, Oftedal BE, Landegren N, Erichsen MM, Bratland E, Lima K, et al. A Longitudinal Follow-Up of Autoimmune Polyendocrine Syndrome Type 1. J Clin Endocrinol Metab (2016) 101(8):2975–83. doi: 10.1210/jc.2016-1821
178. Ramsey C, Winqvist O, Puhakka L, Halonen M, Moro A, Kämpe O, et al. Aire Deficient Mice Develop Multiple Features of APECED Phenotype and Show Altered Immune Response. Hum Mol Genet (2002) 11(4):397–409. doi: 10.1093/hmg/11.4.397
179. Gavanescu I, Benoist C, Mathis D. B Cells Are Required for Aire-Deficient Mice to Develop Multi-Organ Autoinflammation: A Therapeutic Approach for APECED Patients. Proc Natl Acad Sci USA (2008) 105(35):13009–14. doi: 10.1073/pnas.0806874105
180. Jiang W, Anderson MS, Bronson R, Mathis D, Benoist C. Modifier Loci Condition Autoimmunity Provoked by Aire Deficiency. J Exp Med (2005) 202(6):805–15. doi: 10.1084/jem.20050693
181. Papiernik M, de Moraes ML, Pontoux C, Vasseur F, Pénit C. Regulatory CD4 T Cells: Expression of IL-2R Alpha Chain, Resistance to Clonal Deletion and IL-2 Dependency. Int Immunol (1998) 10(4):371–8. doi: 10.1093/intimm/10.4.371
182. Gagnerault MC, Lanvin O, Pasquier V, Garcia C, Damotte D, Lucas B, et al. Autoimmunity During Thymectomy-Induced Lymphopenia: Role of Thymus Ablation and Initial Effector T Cell Activation Timing in Nonobese Diabetic Mice. J Immunol (2009) 183(8):4913–20. doi: 10.4049/jimmunol.0901954
183. Ang DK, Brodnicki TC, Jordan MA, Wilson WE, Silveira P, Gliddon BL, et al. Two Genetic Loci Independently Confer Susceptibility to Autoimmune Gastritis. Int Immunol (2007) 19(9):1135–44. doi: 10.1093/intimm/dxm087
184. Silveira PA, Baxter AG, Cain WE, van Driel IR. A Major Linkage Region on Distal Chromosome 4 Confers Susceptibility to Mouse Autoimmune Gastritis. J Immunol (1999) 162(9):5106–11.
185. Silveira PA, Wilson WE, Esteban LM, Jordan MA, Hawke CG, van Driel IR, et al. Identification of the Gasa3 and Gasa4 Autoimmune Gastritis Susceptibility Genes Using Congenic Mice and Partitioned, Segregative and Interaction Analyses. Immunogenetics (2001) 53(9):741–50. doi: 10.1007/s00251-001-0391-y
186. Baxter AG, Jordan MA, Silveira PA, Wilson WE, Van Driel IR. Genetic Control of Susceptibility to Autoimmune Gastritis. Int Rev Immunol (2005) 24(1-2):55–62. doi: 10.1080/08830180590884404
187. De Block CE, De Leeuw IH, Van Gaal LF. Autoimmune Gastritis in Type 1 Diabetes: A Clinically Oriented Review. J Clin Endocrinol Metab (2008) 93(2):363–71. doi: 10.1210/jc.2007-2134
188. Baxter AG, Horsfall AC, Healey D, Ozegbe P, Day S, Williams DG, et al. Mycobacteria Precipitate an SLE-Like Syndrome in Diabetes-Prone NOD Mice. Immunology (1994) 83(2):227–31.
189. Baxter AG, Healey D, Cooke A. Mycobacteria Precipitate Autoimmune Rheumatic Disease in NOD Mice via an Adjuvant-Like Activity. Scand J Immunol (1994) 39(6):602–6. doi: 10.1111/j.1365-3083.1994.tb03419.x
190. Horsfall AC, Howson R, Silveira P, Williams DG, Baxter AG. Characterization and Specificity of B-Cell Responses in Lupus Induced by Mycobacterium Bovis in NOD/Lt Mice. Immunology (1998) 95(1):8–17. doi: 10.1046/j.1365-2567.1998.00563.x
191. Takenaka K, Prasolava TK, Wang JC, Mortin-Toth SM, Khalouei S, Gan OI, et al. Polymorphism in Sirpa Modulates Engraftment of Human Hematopoietic Stem Cells. Nat Immunol (2007) 8(12):1313–23. doi: 10.1038/ni1527
192. Yamauchi T, Takenaka K, Urata S, Shima T, Kikushige Y, Tokuyama T, et al. Polymorphic Sirpa Is the Genetic Determinant for NOD-Based Mouse Lines to Achieve Efficient Human Cell Engraftment. Blood (2013) 121(8):1316–25. doi: 10.1182/blood-2012-06-440354
193. Sim J, Sockolosky JT, Sangalang E, Izquierdo S, Pedersen D, Harriman W, et al. Discovery of High Affinity, Pan-Allelic, and Pan-Mammalian Reactive Antibodies Against the Myeloid Checkpoint Receptor Sirpα. MAbs (2019) 11(6):1036–52. doi: 10.1080/19420862.2019.1624123
194. Hudson WA, Li Q, Le C, Kersey JH. Xenotransplantation of Human Lymphoid Malignancies Is Optimized in Mice With Multiple Immunologic Defects. Leukemia (1998) 12(12):2029–33. doi: 10.1038/sj.leu.2401236
195. Shultz LD, Schweitzer PA, Christianson SW, Gott B, Schweitzer IB, Tennent B, et al. Multiple Defects in Innate and Adaptive Immunologic Function in NOD/LtSz-Scid Mice. J Immunol (1995) 154(1):180–91.
196. Shultz LD, Lang PA, Christianson SW, Gott B, Lyons B, Umeda S, et al. NOD/LtSz-Rag1null Mice: An Immunodeficient and Radioresistant Model for Engraftment of Human Hematolymphoid Cells, HIV Infection, and Adoptive Transfer of NOD Mouse Diabetogenic T Cells. J Immunol (2000) 164(5):2496–507. doi: 10.4049/jimmunol.164.5.2496
197. Ito M, Hiramatsu H, Kobayashi K, Suzue K, Kawahata M, Hioki K, et al. NOD/SCID/gamma(c)(null) Mouse: An Excellent Recipient Mouse Model for Engraftment of Human Cells. Blood (2002) 100(9):3175–82. doi: 10.1182/blood-2001-12-0207
198. Shultz LD, Lyons BL, Burzenski LM, Gott B, Chen X, Chaleff S, et al. Human Lymphoid and Myeloid Cell Development in NOD/LtSz-Scid IL2R Gamma Null Mice Engrafted With Mobilized Human Hemopoietic Stem Cells. J Immunol (2005) 174(10):6477–89. doi: 10.4049/jimmunol.174.10.6477
199. Pearson T, Shultz LD, Miller D, King M, Laning J, Fodor W, et al. Non-Obese Diabetic-Recombination Activating Gene-1 (NOD-Rag1 Null) Interleukin (IL)-2 Receptor Common Gamma Chain (IL2r Gamma Null) Null Mice: A Radioresistant Model for Human Lymphohaematopoietic Engraftment. Clin Exp Immunol (2008) 154(2):270–84. doi: 10.1111/j.1365-2249.2008.03753.x
200. Ehx G, Somja J, Warnatz HJ, Ritacco C, Hannon M, Delens L, et al. Xenogeneic Graft-Versus-Host Disease in Humanized NSG and NSG-HLA-A2/HHD Mice. Front Immunol (2018) 9:1943. doi: 10.3389/fimmu.2018.01943
201. King M, Pearson T, Shultz LD, Leif J, Bottino R, Trucco M, et al. A New Hu-PBL Model for the Study of Human Islet Alloreactivity Based on NOD-Scid Mice Bearing a Targeted Mutation in the IL-2 Receptor Gamma Chain Gene. Clin Immunol (2008) 126(3):303–14. doi: 10.1016/j.clim.2007.11.001
202. Sonntag K, Eckert F, Welker C, Müller H, Müller F, Zips D, et al. Chronic Graft-Versus-Host-Disease in CD34(+)-Humanized NSG Mice Is Associated With Human Susceptibility HLA Haplotypes for Autoimmune Disease. J Autoimmun (2015) 62:55–66. doi: 10.1016/j.jaut.2015.06.006
203. Wunderlich M, Chou FS, Sexton C, Presicce P, Chougnet CA, Aliberti J, et al. Improved Multilineage Human Hematopoietic Reconstitution and Function in NSGS Mice. PloS One (2018) 13(12):e0209034. doi: 10.1371/journal.pone.0209034
204. Wang M, Yao LC, Cheng M, Cai D, Martinek J, Pan CX, et al. Humanized Mice in Studying Efficacy and Mechanisms of PD-1-Targeted Cancer Immunotherapy. FASEB J (2018) 32(3):1537–49. doi: 10.1096/fj.201700740R
205. Krevvata M, Shan X, Zhou C, Dos Santos C, Habineza Ndikuyeze G, Secreto A, et al. Cytokines Increase Engraftment of Human Acute Myeloid Leukemia Cells in Immunocompromised Mice But Not Engraftment of Human Myelodysplastic Syndrome Cells. Haematologica (2018) 103(6):959–71. doi: 10.3324/haematol.2017.183202
206. Chen Q, Khoury M, Chen J. Expression of Human Cytokines Dramatically Improves Reconstitution of Specific Human-Blood Lineage Cells in Humanized Mice. Proc Natl Acad Sci USA (2009) 106(51):21783–8. doi: 10.1073/pnas.0912274106
207. Brumeanu TD, Vir P, Karim AF, Kar S, Benetiene D, Lok M, et al. A Human-Immune-System (HIS) Humanized Mouse Model (DRAGA: HLA-A2. HLA-DR4. Rag1 KO.IL-2rγc KO. NOD) for COVID-19. Hum Vaccin Immunother (2022) 29:1–16. doi: 10.1080/21645515.2022.2048622
Keywords: NOD mice, polyautoimmunity, thyroiditis, neuropathy, biliary disease
Citation: Aubin A-M, Lombard-Vadnais F, Collin R, Aliesky HA, McLachlan SM and Lesage S (2022) The NOD Mouse Beyond Autoimmune Diabetes. Front. Immunol. 13:874769. doi: 10.3389/fimmu.2022.874769
Received: 12 February 2022; Accepted: 21 March 2022;
Published: 29 April 2022.
Edited by:
David Serreze, Jackson Laboratory, United StatesReviewed by:
Pau Serra Devecchi, Institut de Recerca Biomèdica August Pi i Sunyer (IDIBAPS), SpainGeorgia Fousteri, San Raffaele Hospital (IRCCS), Italy
Copyright © 2022 Aubin, Lombard-Vadnais, Collin, Aliesky, McLachlan and Lesage. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sylvie Lesage, c3lsdmllLmxlc2FnZUB1bW9udHJlYWwuY2E=; c3lsdmllLmxlc2FnZUBnbWFpbC5jb20=