 Yunzhou Pu
Yunzhou Pu Qing Ji
Qing Ji
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 03 May 2022
Sec. Cancer Immunity and Immunotherapy
Volume 13 - 2022 | https://doi.org/10.3389/fimmu.2022.874589
Anti-programmed cell death 1 (PD-1) or anti-PD-ligand (L) 1 drugs, as classic immune checkpoint inhibitors, are considered promising treatment strategies for tumors. In clinical practice, some cancer patients experience drug resistance and disease progression in the process of anti-PD-1/PD-L1 immunotherapy. Tumor-associated macrophages (TAMs) play key roles in regulating PD-1/PD-L1 immunosuppression by inhibiting the recruitment and function of T cells through cytokines, superficial immune checkpoint ligands, and exosomes. There are several therapies available to recover the anticancer efficacy of PD-1/PD-L1 inhibitors by targeting TAMs, including the inhibition of TAM differentiation and re-education of TAM activation. In this review, we will summarize the roles and mechanisms of TAMs in PD-1/PD-L1 blocker resistance. Furthermore, we will discuss the therapies that were designed to deplete TAMs, re-educate TAMs, and intervene with chemokines secreted by TAMs and exosomes from M1 macrophages, providing more potential options to improve the efficacy of PD-1/PD-L1 inhibitors.
Cancer is a worldwide health problem, with an increasing number of confirmed cases and a high mortality (1). Immune escape is one of the most important characteristics of cancers. Tumors reduce immunogenicity as they divide and proliferate, leading to immune escape. Immune checkpoint inhibitors (ICIs) are a new method for tumor immune escape that yields survival benefits for tumor patients. The first ICI that was developed targeted the protein cytotoxic T-lymphocyte antigen 4 (CTLA-4)-ipilimumab (2), which increased survival by 3.7 months in patients with advanced melanoma and boosted the field of cancer treatment. ICIs bind to CTLA-4 or PD-1 and its ligand PD-L1, the key targets related to T-cell activation and exhaustion, and then eliminate immune suppression by tumors.
In the tumor microenvironment (TME), PD-L1 is expressed on the surface of tumors and binds to PD-1 on T cells to resist the killing effect of T cells, ultimately causing tumor immune escape. The application of anti-PD-1/PD-L1 monoclonal antibodies (mAbs) to block the PD-1/PD-L1 signaling pathway has shown excellent antitumor efficacy in a variety of solid tumors (3). However, clinical studies have demonstrated that some patients do not respond to the therapy, and some patients even exhibit tumor recurrence after a period of remission (4). Drug resistance is a crucial factor that determines the efficiency of anti-PD-1/PD-L1 ICIs. Therefore, a deeper understanding of the regulation of the PD-1/PD-L1 axis is essential for the improvement of antitumor immunotherapy.
The mechanisms of resistance to PD-1/PD-L1 blockade mainly include dysfunction or activation disorder of T cells, depletion or reduced infiltration of T cells, and changes in PD-L1 expression (5). The infiltration of T cells in the TME is the precondition of antitumor immunity, while the infiltration of immunosuppressive cells is the premise of tumor immune escape. Tumor-associated macrophages (TAMs) are immunosuppressive cells that induce drug resistance to PD-1/PD-L1 therapy. As one of the most abundant cell types in solid tumors, TAMs contribute to T-cell dysfunction and exhaustion through the secretion of cytokines and metabolic products (6–8) and increase PD-L1 expression in tumor cells and other immunosuppressive cells (9–11). In diagnosed cancers, high macrophage infiltration is often closely related to the occurrence of drug resistance to PD-1/PD-L1 immune suppressants (12–14). Therefore, TAMs have been suggested as important targets to reverse the resistance to anti-PD-1/PD-L1 therapy. In this review, we highlight the recent findings of the suppressive effects of TAMs on PD-1/PD-L1 checkpoint inhibitors. To facilitate precision medicine and expand the target population, we further discuss combination therapies that may improve the efficacy of ICIs targeting PD-1/PD-L1.
Macrophages have powerful functions in identifying, phagocytosing, and removing bacteria and foreign bodies in the immune system. In the process of tumorigenesis, macrophages evolve, resulting in the properties of TAMs that promote tumor growth (15). Mounting evidence suggests that secretions or exosomes from tumor cells shift the transcriptional program of TAMs from the M1-like phenotype to the M2-like phenotype (16–20). In a variety of cancers, the infiltration of M2 TAMs is significantly related to poor prognosis, tumor progression, and other adverse clinical outcomes (14, 21–24). Moreover, in the process of anti-PD-1/PD-L1 immunotherapy, M2 TAMs can also suppress immunotherapy efficacy by inhibiting T-cell activity and enhancing the expression of PD-L1 in the TME. Specifically, M2 TAMs inhibit the function of PD-1/PD-L1 blockers by secreting anti-inflammatory cytokines and exosomes, increasing superficial immune checkpoint ligands (Figure 1).
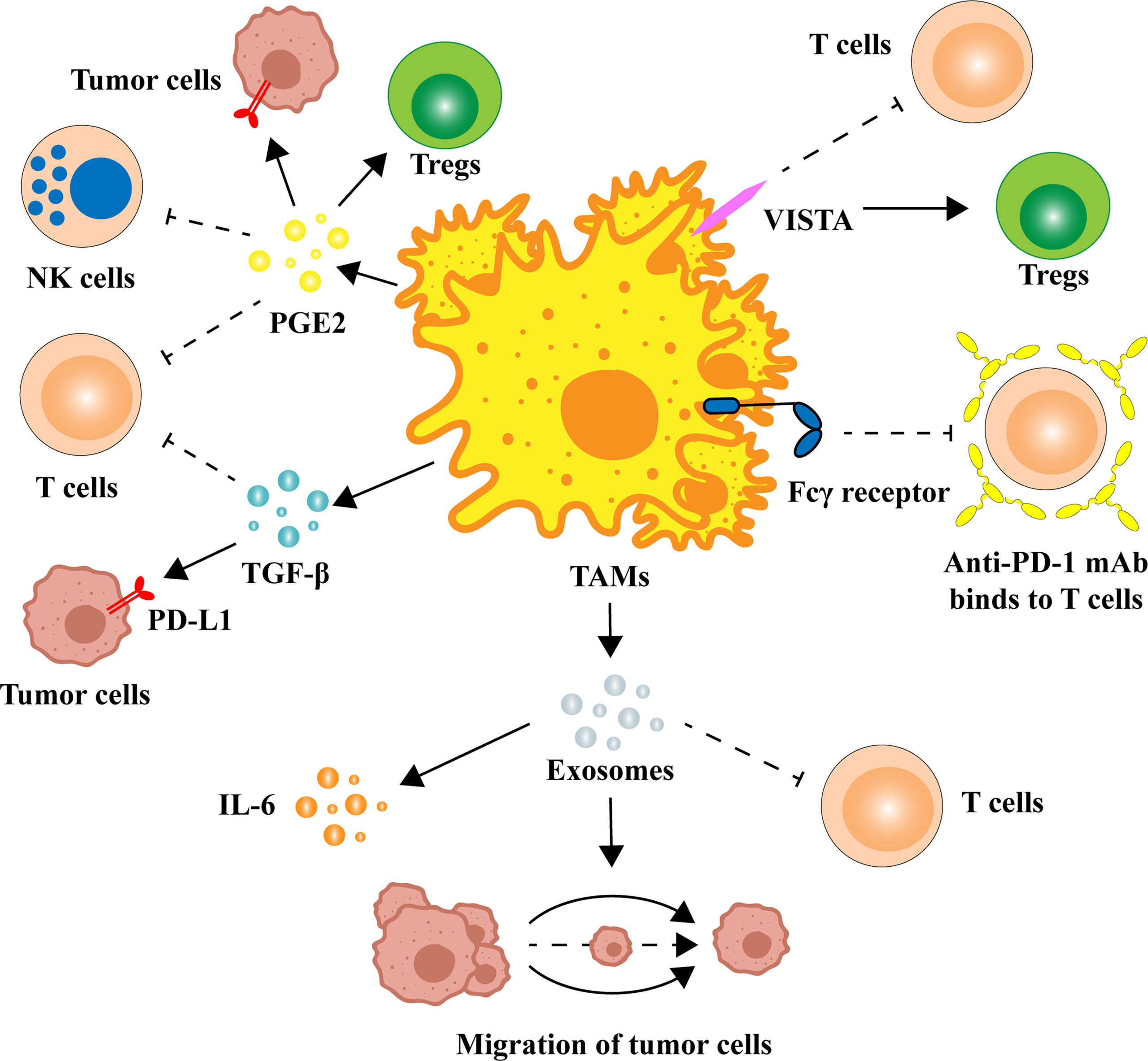
Figure 1 Multiple ways in which TAMs regulate the expression and function of PD-1/PD-L1. TAMs can release a variety of cytokines to alter the TME, such as TGF-β and PGE2. There are also homologous immune checkpoint ligands on the surface of TAMs that can block anti-PD-1/PD-L1 immune efficacy. M2 TAM-derived exosomes are also potentially associated with PD-1/PD-L1 inhibitors. IL, interleukin; mAb, monoclonal antibody; NK cell, natural killer cell; PD-1, programmed cell death 1; PD-L1, programmed cell death ligand-1; PGE2, prostaglandin E2; TAM, tumor-associated macrophage; TGF-β, transforming growth factor-β; Treg, regulatory T cell; VISTA, V-domain Ig-containing suppressor of T-cell activation.
TAMs are capable of secreting several cytokines, which mediate tumor-promoting activity and immunosuppression in the TME. Among them, transforming growth factor-β (TGF-β) and prostaglandin E2 (PGE2) are noteworthy due to their strong immunosuppressive effect and high correlation with TAM (25–29).
TGF-β has been shown to affect anti-PD-1/PD-L1 immunotherapy by inhibiting the activation of T-cells and the expression of PD-L1 (30). TGF-β derived from TAMs inhibits T-cell activity by decreasing the expression levels of IFN-γ and Granzyme B (indicating cytotoxic activity) in T cells through phosphorylation of the Smad2/3 protein and the inhibition of mitochondrial respiration (31, 32). The expression of TGF-β was associated with the infiltration of T cells. Tumors with higher expression of TGF-β presented lower infiltration of CD8+ T cells. TGF-β can also modulate the function of PD-1/PD-L1 by regulating PD-L1 expression. In solid tumors such as breast cancer, TGF-β can induce the upregulation of PD-L1 in tumor cells and tumor-associated angiogenesis, which may be associated with the accumulation of succinate in tumor cells (11). Increased TGF-β levels in the TME not only promoted T-cell exclusion and accumulation of regulatory T cells (Tregs) (33) but also blocked the acquisition of the Th1 effector phenotype (34). In addition, since TGF-β in the TME originates from a variety of cells (35), immunosuppression induced by crosstalk between these cells should also be noted.
PGE2 can inhibit T-cell activation and function by increasing the expression of PD-L1 (36–38). As a downstream of cyclooxygenase 2 (COX-2), the level of PGE2 in the TME is regulated by the expression of COX-2 and microsomal PGE2 synthase 1 (mPGES1) (39). In bladder cancer, TAMs can increase the expression of PD-L1 in tumor-infiltrating myeloid cells through the COX-2/mPGES1/PGE2 pathway, which leads to the exclusion of CD8+ T cells (7). Similarly, PGE2 upregulates PD-L1 expression in ovarian cancer cells by activating the PI3K-AKT-mTOR pathway (40). Moreover, PGE2 can induce the expression of Forkhead Box P3 (Foxp3) to stimulate the differentiation of immunosuppressive Tregs from naïve T cells (41). Of particular note is that therapies targeting PGE2 with NSAIDs or COX-2 inhibitors fail in clinical trials due to global prostaglandin inhibition, which in turn could cause serious side effects (42). Therefore, precision therapy targeting macrophages should be proposed, which may be the next step in reversing drug resistance to PD-1/PD-L1 therapy.
In addition to the expression of PD-L1 on the surface of TAMs, there are also homologous immune checkpoint ligands that can block anti-PD-1/PD-L1 immune efficacy. V-domain Ig-containing suppressor of T-cell activation (VISTA), an immune checkpoint ligand expressed by TAMs (43–46), is an immunosuppressive molecule that reduces T-cell proliferation and cytokine production while sustaining Treg function (46). The expression of VISTA is not only positively correlated with the expression of PD-L1 on the surface of tumor cells but also correlated with the patient’s poor prognosis, pathological grade, and lymph node status (47, 48). In fact, a recent study demonstrated a strong correlation between VISTA expression and tumor infiltration by myeloid cells and PD-1+ inflammatory cells (49). Targeting VISTA antibodies can regulate innate immunity and adaptive immunity by promoting T-cell infiltration, thereby slowing tumor growth in mouse cancer models (50). However, VISTA is also highly expressed in hematopoietic and microglial cells (51, 52), and more studies on the systemic responses to VISTA-targeted therapies are needed. We look forward to its future application in combination with anti-PD-1/PD-L1 mAbs.
Moreover, TAMs can prevent the interaction of anti-immune checkpoint mAbs with the targets through the Fcγ receptor present on cell surfaces (53). Indeed, studies have demonstrated that after administration, anti-PD-1 mAb binds to tumor-infiltrating T cells at an early stage but is subsequently captured by TAMs due to the presence of Fcγ receptors, which ultimately leads to drug failure (53, 54). More importantly, activation of Fcγ receptors by an anti-PD-1 mAb results in depletion of activated CD8+ T cells in vitro and in vivo, reducing the therapeutic effect (55). Therefore, the design of FC-null anti-PD-1 mAbs (55) or specific competitive inhibitors is one of the future strategies necessary to block Fcγ receptor-mediated resistance and increase T-cell infiltration.
Exosomes in the TME have been reported as a medium of communication between cells for the occurrence and invasion of tumors (56–58). Exosomes are able to promote the migration of cancer cells through the PI3K-AKT signaling pathway activated by apolipoprotein E (59). The effect of M2 TAM-derived exosomes on drug resistance has also been reported (60), and it has been verified that microRNAs in exosomes are key regulators of resistance to gemcitabine (60). Analogously, miRNAs of M2 TAM-derived exosomes have also been implicated in the regulation of anti-PD-1/PD-L1 immunotherapy. MicroRNA-21 (MiR-21) expression is relatively high in glioma and associated with low infiltration of CD8+ T cells. Inhibiting miR-21 in exosomes not only improves the proliferation and cytotoxic activity of CD8+ T cells but also reduces the level of TGF-β1, which prevents immune escape of glioma cells (61). In addition, another study showed that the combination of miR-21 deletion and anti-PD-1 treatment demonstrates better antitumor activity than either drug alone (62). Moreover, in vivo, miR-155-5p in exosomes secreted by M2 TAMs can promote the expression of interleukin-6 (IL-6) in tumor cells, thereby inhibiting the T-cell immune response (63).
As mentioned above, TAMs have a multichannel inhibitory effect on anti-PD-1/PD-L1 immunotherapy. Therefore, targeting TAMs is of great significance to improve the efficacy of anti-PD-1/PD-L1 immunotherapy. Currently, strategies are designed to deplete TAMs, re-educate TAMs, and intervene with chemokines secreted by TAMs. The combination of the exosomes secreted by M1 macrophages and other nanoimmunotherapy strategies provides more potential options to reduce the occurrence of PD-1/PD-L1 immunosuppression (Figure 2). In addition, we summarize the current clinical trials on different targets of TAMs in combination with anti-PD-1/PD-L1 mAbs (Table 1).
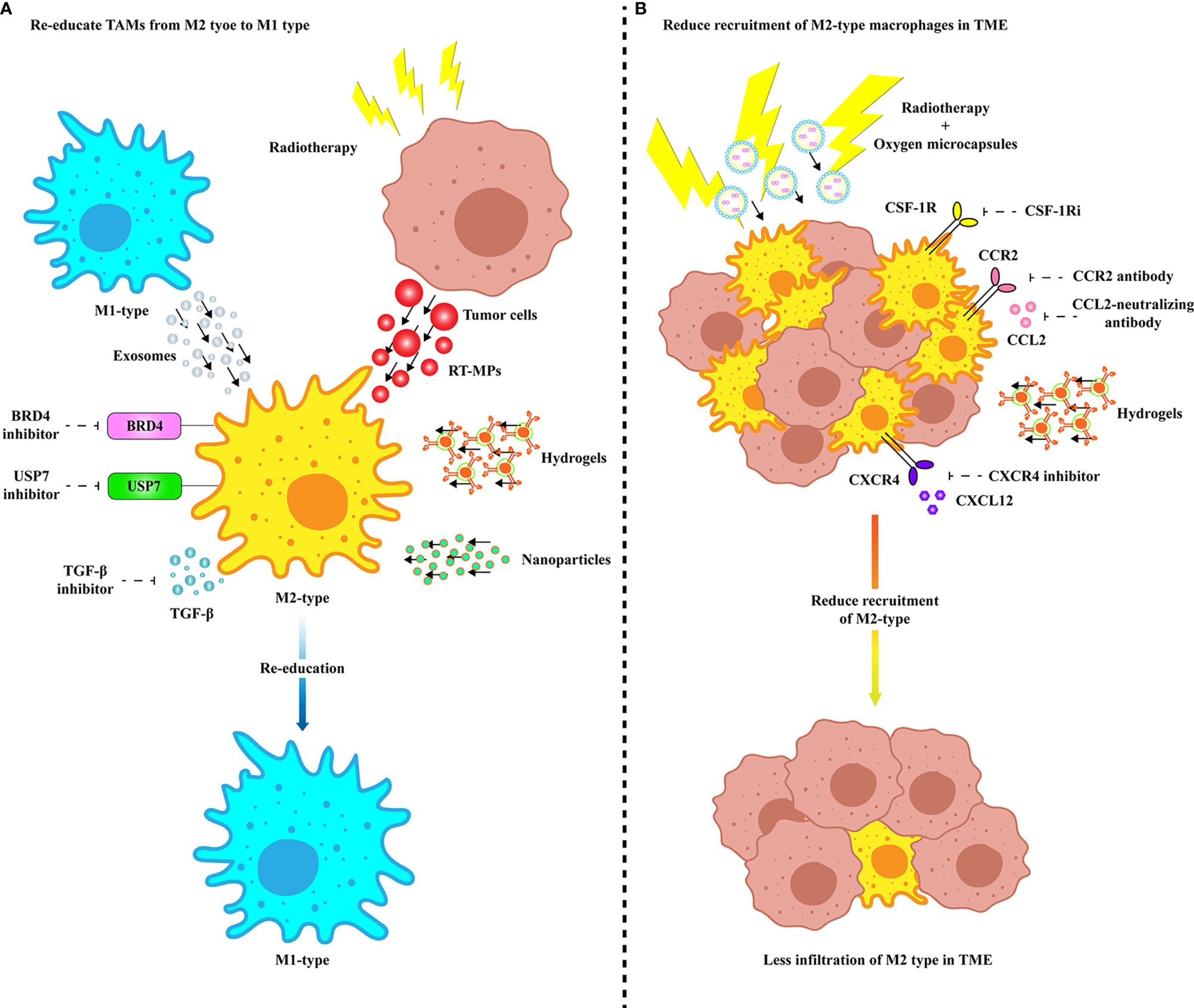
Figure 2 Treatments targeting TAMs to improve anti-PD-1/PD-L1 efficacy. (A) Multiple modes of re-education from M2 TAMs to M1 phenotype. Specific inhibition of membrane proteins of M2 TAMs and TGF-β secretion can enable TAMs to obtain tumoricidal phenotype. M2 TAMs are repolarized to M1 type by internalizing vesicles from M1-type macrophages and radiated tumor cells. Precise targeting of TAMs in TME improves the efficacy of anti-PD-1/PD-L1 therapy through drug delivery platforms such as nanovesicles and hydrogels. (B) Various ways to inhibit TAM recruitment and infiltration in TME. Infiltration of TAMs in TME can be inhibited by specific inhibition of receptors expressed on the surface of TAM cell membrane or by delivery of drugs via hydrogel. Radiotherapy combined with ameliorating the hypoxia microenvironment can effectively eliminate the infiltration of TAMs in TME. BRD4, Bromodomain-containing protein 4; CCL, C-C motif chemokine ligand; CCR, C-C motif chemokine receptor; COX-2, Cyclooxygenase 2; CSF-1R, Colony-stimulating factor 1 receptor; CSF-1Ri, CSF-1R inhibitor; CXCL, CXC-motif chemokine ligand; CXCR, C-X-C chemokine receptor; RT-MP, Microparticles released by radiated tumor cell; TGF-β, Transforming growth factor-β; USP7, Ubiquitin-specific protease 7.
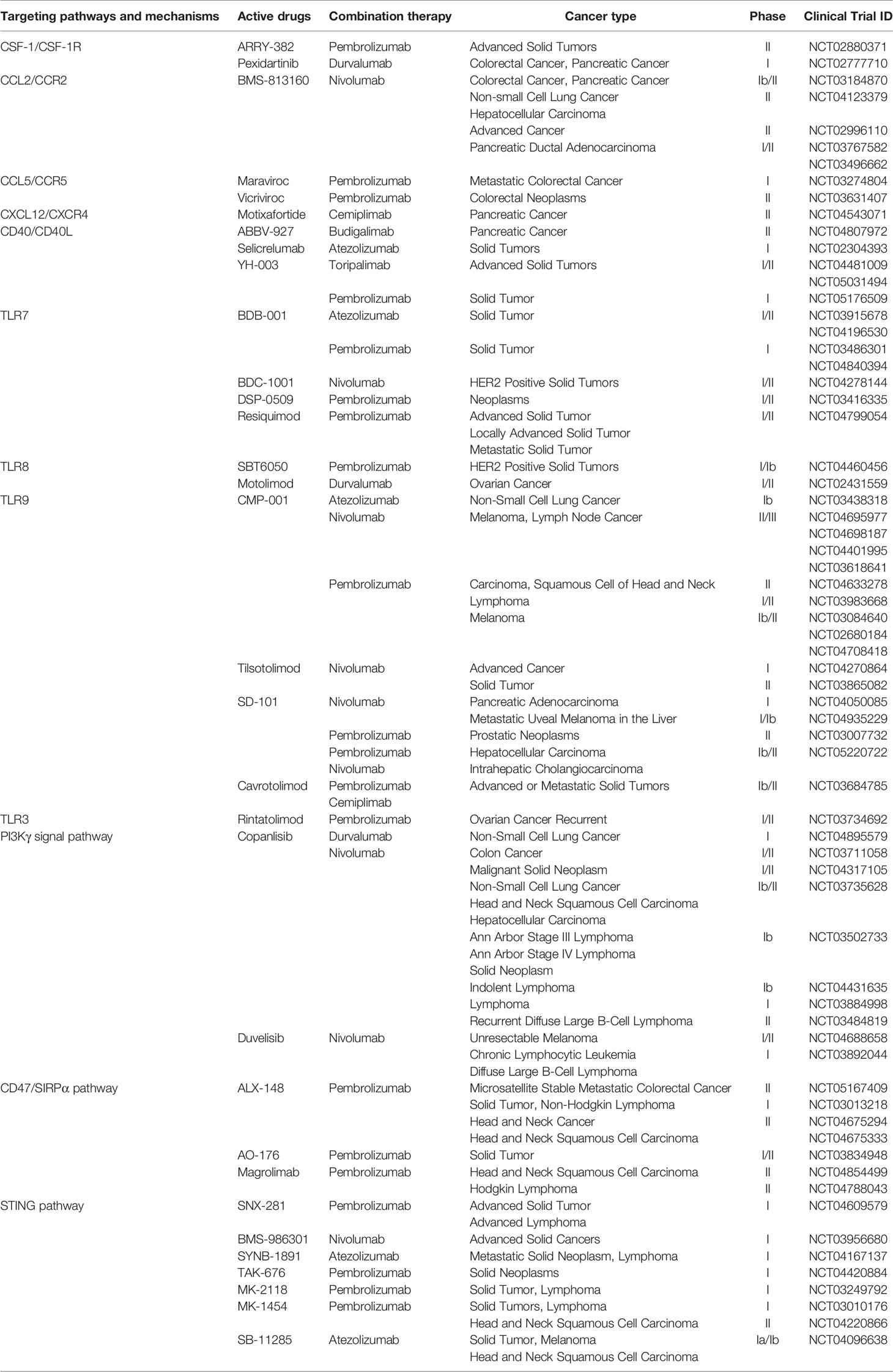
Table 1 Characteristics of clinical trials and drugs on TAM-targeted therapy stratified by targeting mechanisms.
As the infiltration of TAMs limits clinically relevant immune responses (64–66), depleting TAMs seems to be an attractive strategy. Colony-stimulating factor 1 receptor (CSF-1R) blockers are the main method by which to deplete M2 TAMs (67, 68). CSF-1 binds to its receptor, and the latter then undergoes autophosphorylation, which plays an important role in the proliferation, differentiation, and maintenance of macrophages (69). Therefore, a CSF-1R inhibitor (CSF-1Ri) may improve the efficacy of anti-PD-1/PD-L1 immunity. PLX3397 (pexidartinib), a CSF-1R kinase inhibitor, can increase the infiltration and antitumor function of CD8+ T cells in tumors when combined with anti-PD-1 therapy (70, 71). At the same time, it can also effectively reduce the appearance of tumor neovascularization and ascites (72). BLZ945, another CSF-1Ri, combined with a PD-1/PD-L1 blocking antibody is also effective in controlling tumor growth (13, 73). Inherent antitumor drugs can also target CSF-1R to provide a new method for combined immunotherapy. Erlotinib, a first-generation small-molecule inhibitor targeting the epidermal growth factor receptor (EGFR) tyrosine kinase, is suitable for the first-line treatment of EGFR mutation-positive non-small cell lung cancer (NSCLC). Combination therapy consisting of its derivative TD-92 and anti-PD-1 contributes to reduced tumor growth and increased survival in vivo (74).
Recently, several findings have suggested that re-education of M2 TAMs rather than depletion may represent a more effective strategy. Previous studies have reported that the protumor M2 phenotype can be re-educated to the tumoricidal M1 phenotype, thereby inhibiting the supporting role of TAMs in tumors (75). BRD4, a bromodomain and extraterminal (BET) family protein, can enhance the expression of CCL2 by activating the NF-κB signaling pathway, which, in turn, causes the recruitment of macrophages in tumors (76). The BRD4 inhibitor AZD5153 can re-educate TAMs from M2 to M1 and promote the secretion of proinflammatory cytokines, thereby activating cytotoxic T lymphocytes (CTLs) in vitro (77). More importantly, AZD5153 was proven to render ovarian cancer sensitive to anti-PD-L1 treatment through a 3-D microfluidic model (77). SF2523, another BRD4 inhibitor that can block the polarization of TAMs, restores the activity of CD8+ T cells and then stimulates the antitumor immune response (78). Furthermore, the BET inhibitor JQ1 can significantly reduce PD-L1 expression on tumor cells and TAMs and limit tumor progression in a cytotoxic T-cell-dependent manner (79).
Ubiquitin-specific protease 7 (USP7), a deubiquitinating enzyme, is considered a promising therapeutic target because of its regulatory role in DNA damage and epigenetic inheritance (80). USP7 has been identified as a highly expressed M2 TAM gene, and specific inhibition of USP7 can reprogram M2 TAMs into M1 through the P38 MAPK pathway (81). In addition, targeting USP7 promoted the infiltration and cytotoxicity of CD8+ T cells in the TME and decreased PD-L1 expression in tumor cells (81, 82).
In addition to targeting M2 TAMs themselves, the immunosuppressive effects of M2 TAMs can also be reversed by inhibiting cytokines secreted by M2 TAMs.
As one of the most important immunosuppressive cytokines secreted by M2 TAMs, TGF-β changes the TME by limiting the infiltration of T cells to inhibit antitumor immunity (8). Combination therapy with a TGF-β inhibitor (1D11 and galunisertib) and anti-PD-1/PD-L1 resulted in the upregulation of immune response genes, restoring the cytotoxic activity of T cells and the antitumor activity of anti-PD-L1 (32, 83). In addition to the above effects, combinatorial treatment consisting of tranilast, a TGF-β inhibitor, with Doxil nanomedicine has been shown to improve M1 macrophage content in the tumor tissue, which results in the increased efficacy of anti-PD-1 (84). Phase II research is ongoing to investigate the combined effect of TGF-β inhibitor (vactosertib) with anti-PD-L1 (durvalumab) (NCT04064190), while another phase II study is assessing the efficacy and safety of NIS793 with and without spartalizumab (NCT04390763) in untreated metastatic pancreatic ductal adenocarcinoma (mPDAC). In addition, M7824 is a bifunctional fusion protein comprising a mAb against PD-L1 fused to the extracellular domain of TGF-β receptor 2 (85). Compared with targeting TGF-β alone, M7824 has been proven to reduce tumor burden and improve overall survival (OS) (85). A phase II trial was performed to determine the efficacy of M7824 plus topotecan or temozolomide in recurrent SCLC (NCT03554473).
CCL2 is a chemokine that attracts a number of CCR2-high-expressing monocytes to the tumor site. The role of C-C motif chemokine receptor 2 (CCR2) seems to influence TAM recruitment at the tumor site. In a recent study, it was proven that CCR2 is involved in the recruitment and initiation of tumor-promoting inflammation (86). Many preclinical studies showed high efficacy of CCL2/CCR2 antagonists; for instance, targeting CCR2 with a small-molecule inhibitor not only reduced recruitment of M2-type macrophages but also induced tumor infiltration of activated CD8+ T cells (87). Many other preclinical studies on different tumor types showed that either depletion of CCR2 or disruption of the CCL2-CCR2 interaction has an impact on the inhibition of TAM recruitment and tumor regression or inhibition of metastasis (88–91). A phase I/II trial of combination immunotherapy with nivolumab and a CCR2/CCR5 dual antagonist (BMS-813160) is in progress to evaluate whether this therapy is safe in patients with locally advanced pancreatic cancer (LAPC) (NCT03767582).
CXC-motif chemokine ligand 12 (CXCL12) is another chemokine that regulates the migration of monocytes (92). Elevated C-X-C chemokine receptor 4 (CXCR4) is correlated with the tumorigenesis of NSCLC (93). CXCL12 secretion could be induced in response to radiation therapy and cause the accumulation of TAMs in the tumor (94). BL-8040 (motixafortide), one of the CXCR4 antagonists, plus the anti-PD-1 pembrolizumab in the COMBAT trial contributes to the improvement found in pancreatic ductal adenocarcinoma (PDAC) patients (95). After using a new CXCR4 inhibitor peptide R, the expression of CD73, CD38, and IL-10 in non-small cell lung cancer is reduced, which can rescue the cytotoxic activity of T cells and prevent TAM polarization (96). Another CXCR4 antagonist, Pep R, demonstrated efficacy in combination with nivolumab in melanoma. In addition, there are already 2 observational studies in progress to study whether Pep R can reverse anti-PD1 resistance (NCT03891485 and NCT03628859).
Many people have recognized the potential of TGF-β, and there are currently many clinical trials combining TGF-β targeting with PD-1/PD-L1 treatment (97). However, most studies have stalled due to serious adverse events or the observation of minimal clinical benefit (98), which may be associated with higher global drug levels. As CXCR4 is widely expressed in hematopoietic cells and a variety of stem cells (99), therapies targeting CXCR4 face a similar dilemma. In contrast, therapies that block the CCL2–CCr2 axis appear to be safer and are expected to elucidate the mechanism of action in different cancer species. In addition, for the current targeted drugs yielding a poor systemic response, the combination of cell-targeted drug delivery systems is an ideal choice to counteract their limitations.
Compared with traditional delivery systems, nanoparticles that can specifically deliver drugs to TAMs and modulate their polarized states may be an effective method in cancer immunotherapy. For example, tumor cell-derived microparticles containing the chemotherapeutic drug methotrexate (TMP-MTX), nanoparticles delivering shikonin and PD-L1 knockdown siRNA (SK/siR-NPs), and Gadofullerene (GF-Ala) nanoparticles can all reprogram M2 TAMs to an M1-like phenotype and increase the infiltration of CTLs, thereby effectively inhibiting tumor growth (100–102). CMPB90-1, a new natural polysaccharide from Cordyceps, converts immunosuppressive TAMs by binding to toll-like receptor 2 (TLR2), polarizes TAMs to the M1 phenotype, and has antitumor effects and a better safety profile (103). These studies may provide a promising strategy for the development of high-efficiency, low-toxicity immunotherapy based on nanotechnology.
Due to the unpredictable toxicity and poor scalability of nanocarriers in the human body (104), other advanced drug carriers have gradually attracted attention, such as hydrogels (105), exosomes (106), and enucleated cells (107). A Melittin-Rada32 hybrid peptide hydrogel loaded with doxorubicin (DOX) was designed to reshape the tumor immune microenvironment in the treatment of melanoma, which specifically consumes M2 TAMs and increases activated CTL infiltration (108). Dai et al. designed a hydrogel scaffold loaded with KN93, a Ca2+/calmodulin-dependent protein kinase II (CAMKII) inhibitor, which can reprogram TAMs into the M1 phenotype (109). After this hydrogel treatment, CTL infiltration in the TME increased, and the expression of macrophage PD-L1 increased, suggesting that it has good prospects for anti-PD-1 treatment (109).
Studies have reported that exosomes derived from macrophages have immunomodulatory effects (110). Exosomes secreted by M1 macrophages are reported to inhibit the development of gastric cancer and activate T-cell-dependent immune responses (111). Endogenous macrophage exosomes have been shown to have absolute advantages over their safety (112). Therefore, M1 macrophage-derived exosomes (M1-exos) can be used to deliver various anticancer drugs for tumor therapy. Mannose-modified macrophage-derived microparticles (Man-MPs) loaded with metformin have been developed to efficiently target M2-like TAMs to repolarize them into the M1-like phenotype (113). More importantly, the collagen-degrading capacity of Man-MPs contributes to the infiltration of CD8+ T cells into tumor interiors and enhances tumor accumulation and penetration of anti-PD-1 (113). Macrophage-derived exosomes loaded with PTX and Dox were developed to treat triple-negative breast cancer (TNBC) in vivo (114). With the development of nanotechnology, exosome-mimetic nanovesicles derived from M1 macrophages (M1NVs) were designed to repolarize M2 TAMs to M1 macrophages (115). Moreover, injection of a combination of M1NVs and anti-PD-L1 further reduced the tumor size compared with the injection of either M1NVs or aPD-L1 alone (115).
As a conventional means of tumor treatment, radiotherapy leads to increased expression of PD-L1 in tumor cells (116), which is one of the markers of anti-PD-1/PD-L1 mAb therapy (117). Interestingly, in a model of malignant pleural effusions, microparticles released by radiated tumor cells (RT-MPs) can precisely locate M2-TAMs in the TME and convert the latter into M1-TAMs by activating the JAK-STAT and MAPK pathways (118). It is noteworthy that the combination of RT-MPs and anti-PD-1 exhibits good biocompatibility and memory immune response, which TMP-MTX cannot match (118). Since hypoxia has been proven to be an important factor affecting the clinical outcome after radiotherapy (119), strategies to interfere with hypoxia have been developed to optimize radiotherapy (120). Oxygen microcapsules are designed to rapidly increase the oxygen concentration in the TME, resulting in reduced infiltration of TAMs while enhancing the efficacy of radiotherapy (121). Of particular note is the microcapsules’ ability to repolarize TAMs into the M1 phenotype, which, in turn, activates the T-cell-mediated antitumor immune response (121). Obviously, this nanotherapy can further enhance the efficacy of radiotherapy combined with anti-PD-1/PD-L1 mAbs.
The application of ICIs in clinical practice has completely changed the therapeutic strategies for cancer patients by improving the prognosis and reducing the impact on their quality of life (QoL) compared with standard approaches (117, 122–129). However, the high cost of anti-PD-1 and PD-L1 agents (130, 131) highlights the need to select patients who will benefit most from the treatment early, supporting the research on predictive biomarkers of response and strategies to overcome resistance and optimize the efficacy of these drugs.
Targeting macrophages to treat cancer is a young but rapidly developing area of research and therapy. Despite great interest, the optimum therapeutic approach has not yet been identified because TAMs represent a heterogeneous population, and their role in tumors varies depending on many environmental conditions. The other difficulty arises from the TME, which is a very dynamic tissue and contains various infiltrating immune cells and external factors that influence tumor progression, macrophage polarization, and therapeutic response. Some macrophage-targeting therapeutics are effective as monotherapies. However, more evidence exists that targeting TAMs could improve the efficacy of conventional therapies and immune therapeutics. Currently, two main approaches that target TAMs with apparent opposite effects have been developed. One approach is to deplete macrophages, and the other is to re-educate them to kill cancer cells. Depending on the macrophage infiltration status and the chosen therapy as a combination treatment, various approaches will be chosen. For example, through their Fcγ receptors, macrophages were shown to take up therapeutic antibodies such as anti-PD-L1, limiting the efficacy of such therapeutic modalities in animal models. In fact, in several recent studies, it was shown that depleting macrophages with the use of CCL2/CCR2 antagonists improves the efficacy of PD-L1-targeting antibodies and possibly other ICIs (132, 133).
In addition, some important issues should be resolved before TAM antagonists are used to overcome resistance to immunotherapy. More convincing clinical studies are needed to confirm the correlation of macrophage infiltration or phenotype with the outcomes of patients under anti-PD-1/PD-L1 therapy. It is essential to identify subpopulations that have the potential to benefit from different therapies targeting macrophages. Despite these difficulties, there is still great potential to harness macrophage biology to improve the efficiency of anti-PD-1/PD-L1 ICIs in oncology.
YP and QJ conceived the structure of manuscript and revised the manuscript. YP created the figures. QJ reviewed the manuscript. All authors contributed to the article and approved the submitted version.
This work was supported by the National Science Foundation of China (82074225 to QJ).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
AKT: Protein kinase B; BET: Bromodomain and extraterminal; BRD4: Bromodomain-containing protein 4; CAMKII: Ca2+/calmodulin-dependent protein kinase II; CCL: C-C motif chemokine ligand; CCR: C-C motif chemokine receptor; COX-2: Cyclooxygenase 2; CSF-1R: Colony-stimulating factor 1 receptor; CSF-1Ri: CSF-1R inhibitor; CTL: Cytotoxic T lymphocyte; CTLA-4: Cytotoxic T lymphocyte antigen 4; CXCL: CXC-motif chemokine ligand; CXCR: C-X-C chemokine receptor; DC: Dendritic cell; EGFR: Epidermal growth factor receptor; Foxp3: Forkhead Box P3; GF-Ala: Gadofullerene; ICI: Immune checkpoint inhibitor; IL: Interleukin; LAPC: Locally advanced pancreatic cancer; M1-exo: M1 macrophage-derived exosome; M1NV: Nanovesicle derived from M1 macrophages; mAb: Monoclonal antibody; Man-MP: Mannose-modified macrophage-derived microparticle; miR: MicroRNA; mPDAC: Metastatic pancreatic ductal adenocarcinoma; mPGES1: Microsomal PGE2 synthase1; NF-κB: Nuclear factor kappa-B; NK cell: Natural killer cell; NSCLC: Non-small cell lung cancer; OS: Overall survival; PDAC: Pancreatic ductal adenocarcinoma; PD-1: Programmed cell death 1; PD-L1: Programmed cell death ligand-1; PGE2: Prostaglandin E2; PI3K: Phosphatidylinositol 3-kinase; QoL: Quality of life; RT-MP: Microparticles released by radiated tumor cell; SK: Shikonin; SK/siR-NP: Versatile nanoparticle codelivering SK and PD-L1 knockdown siRNA; TAM: Tumor-associated macrophage; TGF-β: Transforming growth factor-β; TLR: Toll-like receptor; TME: Tumor microenvironment; TMP-MTX: Tumor cell-derived microparticles containing the chemotherapeutic drug methotrexate; TNBC: Triple-negative breast cancer; Treg: Regulatory T cell; USP7: Ubiquitin-specific protease 7; VISTA: V-domain Ig-containing suppressor of T-cell activation.
AKT: Protein kinase B
BET: Bromodomain and extraterminal
BRD4: Bromodomain-containing protein 4
CAMKII: Ca2+/calmodulin-dependent protein kinase II
CCL: C-C motif chemokine ligand
CCR: C-C motif chemokine receptor
COX-2: Cyclooxygenase 2
CSF-1R: Colony-stimulating factor 1 receptor
CSF-1Ri: CSF-1R inhibitor
CTL: Cytotoxic T lymphocyte
CTLA-4: Cytotoxic T lymphocyte antigen 4
CXCL: CXC-motif chemokine ligand
CXCR: C-X-C chemokine receptor
DC: Dendritic cell
EGFR: Epidermal growth factor receptor
Foxp3: Forkhead Box P3
GF-Ala: Gadofullerene
ICI: Immune checkpoint inhibitor
IL: Interleukin
LAPC: Locally advanced pancreatic cancer
M1-exo: M1 macrophage-derived exosome
M1NV: Nanovesicle derived from M1 macrophages
mAb: Monoclonal antibody
Man-MP: Mannose-modified macrophage-derived microparticle
miR: MicroRNA
mPDAC: Metastatic pancreatic ductal adenocarcinoma
mPGES1: Microsomal PGE2 synthase1
NF-κB: Nuclear factor kappa-B
NK cell: Natural killer cell
NSCLC: Non-small cell lung cancer
OS: Overall survival
PDAC: Pancreatic ductal adenocarcinoma
PD-1: Programmed cell death 1
PD-L1: Programmed cell death ligand-1
PGE2: Prostaglandin E2
PI3K: Phosphatidylinositol 3-kinase
QoL: Quality of life
RT-MP: Microparticles released by radiated tumor cell
SK: Shikonin
SK/siR-NP: Versatile nanoparticle codelivering SK and PD-L1 knockdown siRNA
TAM: Tumor-associated macrophage
TGF-β: Transforming growth factor-β
TLR: Toll-like receptor
TME: Tumor microenvironment
TMP-MTX: Tumor cell-derived microparticles containing the chemotherapeutic drug methotrexate
TNBC: Triple-negative breast cancer
Treg: Regulatory T cell
USP7: Ubiquitin-specific protease 7
VISTA: V-domain Ig-containing suppressor of T-cell activation
1. Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, et al. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J Clin (2021) 71:209–49. doi: 10.3322/caac.21660
3. Gordon SR, Maute RL, Dulken BW, Hutter G, George BM, McCracken MN, et al. PD-1 Expression by Tumour-Associated Macrophages Inhibits Phagocytosis and Tumour Immunity. Nature (2017) 545:495–9. doi: 10.1038/nature22396
4. Beaver JA, Hazarika M, Mulkey F, Mushti S, Chen H, He K, et al. Patients With Melanoma Treated With an Anti-PD-1 Antibody Beyond RECIST Progression: A US Food and Drug Administration Pooled Analysis. Lancet Oncol (2018) 19:229–39. doi: 10.1016/S1470-2045(17)30846-X
5. Ren D, Hua Y, Yu B, Ye X, He Z, Li C, et al. Predictive Biomarkers and Mechanisms Underlying Resistance to PD1/PD-L1 Blockade Cancer Immunotherapy. Mol Cancer (2020) 19:19. doi: 10.1186/s12943-020-1144-6
6. Dong L, Chen C, Zhang Y, Guo P, Wang Z, Li J, et al. The Loss of RNA N(6)-Adenosine Methyltransferase Mettl14 in Tumor-Associated Macrophages Promotes CD8(+) T Cell Dysfunction and Tumor Growth. Cancer Cell (2021) 39:945–57. doi: 10.1016/j.ccell.2021.04.016
7. Prima V, Kaliberova LN, Kaliberov S, Curiel DT, Kusmartsev S. COX2/mPGES1/PGE2 Pathway Regulates PD-L1 Expression in Tumor-Associated Macrophages and Myeloid-Derived Suppressor Cells. Proc Natl Acad Sci USA (2017) 114:1117–22. doi: 10.1073/pnas.1612920114
8. Mariathasan S, Turley SJ, Nickles D, Castiglioni A, Yuen K, Wang Y, et al. TGFbeta Attenuates Tumour Response to PD-L1 Blockade by Contributing to Exclusion of T Cells. Nature (2018) 554:544–8. doi: 10.1038/nature25501
9. Zhang X, Zeng Y, Qu Q, Zhu J, Liu Z, Ning W, et al. PD-L1 Induced by IFN-Gamma From Tumor-Associated Macrophages via the JAK/STAT3 and PI3K/AKT Signaling Pathways Promoted Progression of Lung Cancer. Int J Clin Oncol (2017) 22:1026–33. doi: 10.1007/s10147-017-1161-7
10. Lin C, He H, Liu H, Li R, Chen Y, Qi Y, et al. Tumour-Associated Macrophages-Derived CXCL8 Determines Immune Evasion Through Autonomous PD-L1 Expression in Gastric Cancer. Gut (2019) 68:1764–73. doi: 10.1136/gutjnl-2018-316324
11. Gomez V, Eykyn TR, Mustapha R, Flores-Borja F, Male V, Barber PR, et al. Breast Cancer-Associated Macrophages Promote Tumorigenesis by Suppressing Succinate Dehydrogenase in Tumor Cells. Sci Signal (2020) 13:eaax4585. doi: 10.1126/scisignal.aax4585
12. Zhu Z, Zhang H, Chen B, Liu X, Zhang S, Zong Z, et al. PD-L1-Mediated Immunosuppression in Glioblastoma Is Associated With the Infiltration and M2-Polarization of Tumor-Associated Macrophages. Front Immunol (2020) 11:588552. doi: 10.3389/fimmu.2020.588552
13. Cui X, Ma C, Vasudevaraja V, Serrano J, Tong J, Peng Y, et al. Dissecting the Immunosuppressive Tumor Microenvironments in Glioblastoma-On-a-Chip for Optimized PD-1 Immunotherapy. Elife (2020) 9:e52253. doi: 10.7554/eLife.52253
14. Kim YJ, Won CH, Lee MW, Choi JH, Chang SE, Lee WJ. Correlation Between Tumor-Associated Macrophage and Immune Checkpoint Molecule Expression and Its Prognostic Significance in Cutaneous Melanoma. J Clin Med (2020) 9(8):2500. doi: 10.3390/jcm9082500
15. Ostuni R, Kratochvill F, Murray PJ, Natoli G. Macrophages and Cancer: From Mechanisms to Therapeutic Implications. Trends Immunol (2015) 36:229–39. doi: 10.1016/j.it.2015.02.004
16. Chen X, Ying X, Wang X, Wu X, Zhu Q, Wang X. Exosomes Derived From Hypoxic Epithelial Ovarian Cancer Deliver microRNA-940 to Induce Macrophage M2 Polarization. Oncol Rep (2017) 38:522–8. doi: 10.3892/or.2017.5697
17. Ying X, Wu Q, Wu X, Zhu Q, Wang X, Jiang L, et al. Epithelial Ovarian Cancer-Secreted Exosomal miR-222-3p Induces Polarization of Tumor-Associated Macrophages. Oncotarget (2016) 7:43076–87. doi: 10.18632/oncotarget.9246
18. Yin C, Han Q, Xu D, Zheng B, Zhao X, Zhang J. SALL4-Mediated Upregulation of Exosomal miR-146a-5p Drives T-Cell Exhaustion by M2 Tumor-Associated Macrophages in HCC. Oncoimmunology (2019) 8:1601479. doi: 10.1080/2162402X.2019.1601479
19. Lin Y, Xu J, Lan H. Tumor-Associated Macrophages in Tumor Metastasis: Biological Roles and Clinical Therapeutic Applications. J Hematol Oncol (2019) 12:76. doi: 10.1186/s13045-019-0760-3
20. Sarode P, Zheng X, Giotopoulou GA, Weigert A, Kuenne C, Gunther S, et al. Reprogramming of Tumor-Associated Macrophages by Targeting Beta-Catenin/FOSL2/ARID5A Signaling: A Potential Treatment of Lung Cancer. Sci Adv (2020) 6:z6105. doi: 10.1126/sciadv.aaz6105
21. Sumitomo R, Hirai T, Fujita M, Murakami H, Otake Y, Huang CL. M2 Tumor-Associated Macrophages Promote Tumor Progression in Non-Small-Cell Lung Cancer. Exp Ther Med (2019) 18:4490–8. doi: 10.3892/etm.2019.8068
22. Sumitomo R, Hirai T, Fujita M, Murakami H, Otake Y, Huang CL. PD-L1 Expression on Tumor-Infiltrating Immune Cells is Highly Associated With M2 TAM and Aggressive Malignant Potential in Patients With Resected Non-Small Cell Lung Cancer. Lung Cancer (2019) 136:136–44. doi: 10.1016/j.lungcan.2019.08.023
23. Marchesi F, Cirillo M, Bianchi A, Gately M, Olimpieri OM, Cerchiara E, et al. High Density of CD68+/CD163+ Tumour-Associated Macrophages (M2-TAM) at Diagnosis is Significantly Correlated to Unfavorable Prognostic Factors and to Poor Clinical Outcomes in Patients With Diffuse Large B-Cell Lymphoma. Hematol Oncol (2015) 33:110–2. doi: 10.1002/hon.2142
24. Zhang M, He Y, Sun X, Li Q, Wang W, Zhao A, et al. High M1/M2 Ratio of Tumor-Associated Macrophages is Associated With Extended Survival in Ovarian Cancer Patients. J Ovarian Res (2014) 7:19. doi: 10.1186/1757-2215-7-19
25. Steitz AM, Steffes A, Finkernagel F, Unger A, Sommerfeld L, Jansen JM, et al. Tumor-Associated Macrophages Promote Ovarian Cancer Cell Migration by Secreting Transforming Growth Factor Beta Induced (TGFBI) and Tenascin C. Cell Death Dis (2020) 11:249. doi: 10.1038/s41419-020-2438-8
26. Liu C, Zhang W, Wang J, Si T, Xing W. Tumor-Associated Macrophage-Derived Transforming Growth Factor-Beta Promotes Colorectal Cancer Progression Through HIF1-TRIB3 Signaling. Cancer Sci (2021) 112:4198–207. doi: 10.1111/cas.15101
27. Santoni M, Romagnoli E, Saladino T, Foghini L, Guarino S, Capponi M, et al. Triple Negative Breast Cancer: Key Role of Tumor-Associated Macrophages in Regulating the Activity of Anti-PD-1/PD-L1 Agents. Biochim Biophys Acta Rev Cancer (2018) 1869:78–84. doi: 10.1016/j.bbcan.2017.10.007
28. Eisengart CA, Mestre JR, Naama HA, Mackrell PJ, Rivadeneira DE, Murphy EM, et al. Prostaglandins Regulate Melanoma-Induced Cytokine Production in Macrophages. Cell Immunol (2000) 204:143–9. doi: 10.1006/cimm.2000.1686
29. Wei J, Zhang J, Wang D, Cen B, Lang JD, DuBois RN. The COX-2-PGE2 Pathway Promotes Tumor Evasion in Colorectal Adenomas. Cancer Prev Res (Phila) (2022). doi: 10.1158/1940-6207.CAPR-21-0572
30. Batlle E, Massague J. Transforming Growth Factor-Beta Signaling in Immunity and Cancer. Immunity (2019) 50:924–40. doi: 10.1016/j.immuni.2019.03.024
31. Dimeloe S, Gubser P, Loeliger J, Frick C, Develioglu L, Fischer M, et al. Tumor-Derived TGF-Beta Inhibits Mitochondrial Respiration to Suppress IFN-Gamma Production by Human CD4(+) T Cells. Sci Signal (2019) 12:eaav3334. doi: 10.1126/scisignal.aav3334
32. Li L, Yang L, Wang L, Wang F, Zhang Z, Li J, et al. Impaired T Cell Function in Malignant Pleural Effusion is Caused by TGF-Beta Derived Predominantly From Macrophages. Int J Cancer (2016) 139:2261–9. doi: 10.1002/ijc.30289
33. Konkel JE, Zhang D, Zanvit P, Chia C, Zangarle-Murray T, Jin W, et al. Transforming Growth Factor-Beta Signaling in Regulatory T Cells Controls T Helper-17 Cells and Tissue-Specific Immune Responses. Immunity (2017) 46:660–74. doi: 10.1016/j.immuni.2017.03.015
34. Tauriello D, Palomo-Ponce S, Stork D, Berenguer-Llergo A, Badia-Ramentol J, Iglesias M, et al. TGFbeta Drives Immune Evasion in Genetically Reconstituted Colon Cancer Metastasis. Nature (2018) 554:538–43. doi: 10.1038/nature25492
36. Sha W, Olesch C, Hanaka H, Radmark O, Weigert A, Brune B. Necrosis in DU145 Prostate Cancer Spheroids Induces COX-2/mPGES-1-Derived PGE2 to Promote Tumor Growth and to Inhibit T Cell Activation. Int J Cancer (2013) 133:1578–88. doi: 10.1002/ijc.28181
37. Tanaka K, Koga Y, Taniguchi K, Kamikaseda K, Nihashi Y, Nomoto K. T-Cell Recruitment From the Thymus to the Spleen in Tumor-Bearing Mice. II. Functional Characteristics of Recruited T-Cells. J Natl Cancer Inst (1986) 77:733–8. doi: 10.1093/jnci/77.3.733
38. Mulligan JK, Rosenzweig SA, Young MR. Tumor Secretion of VEGF Induces Endothelial Cells to Suppress T Cell Functions Through the Production of PGE2. J Immunother (2010) 33:126–35. doi: 10.1097/CJI.0b013e3181b91c9c
39. Weigert A, Strack E, Snodgrass RG, Brune B. mPGES-1 and ALOX5/-15 in Tumor-Associated Macrophages. Cancer Metastasis Rev (2018) 37:317–34. doi: 10.1007/s10555-018-9731-3
40. Komura N, Mabuchi S, Shimura K, Yokoi E, Kozasa K, Kuroda H, et al. The Role of Myeloid-Derived Suppressor Cells in Increasing Cancer Stem-Like Cells and Promoting PD-L1 Expression in Epithelial Ovarian Cancer. Cancer Immunol Immunother (2020) 69:2477–99. doi: 10.1007/s00262-020-02628-2
41. Nakanishi M, Rosenberg DW. Multifaceted Roles of PGE2 in Inflammation and Cancer. Semin Immunopathol (2013) 35:123–37. doi: 10.1007/s00281-012-0342-8
42. Solomon SD, McMurray JJ, Pfeffer MA, Wittes J, Fowler R, Finn P, et al. Cardiovascular Risk Associated With Celecoxib in a Clinical Trial for Colorectal Adenoma Prevention. N Engl J Med (2005) 352:1071–80. doi: 10.1056/NEJMoa050405
43. Gao J, Ward JF, Pettaway CA, Shi LZ, Subudhi SK, Vence LM, et al. VISTA is an Inhibitory Immune Checkpoint That is Increased After Ipilimumab Therapy in Patients With Prostate Cancer. Nat Med (2017) 23:551–5. doi: 10.1038/nm.4308
44. Kryczek I, Zou L, Rodriguez P, Zhu G, Wei S, Mottram P, et al. B7-H4 Expression Identifies a Novel Suppressive Macrophage Population in Human Ovarian Carcinoma. J Exp Med (2006) 203:871–81. doi: 10.1084/jem.20050930
45. Ni L, Dong C. New Checkpoints in Cancer Immunotherapy. Immunol Rev (2017) 276:52–65. doi: 10.1111/imr.12524
46. Nowak EC, Lines JL, Varn FS, Deng J, Sarde A, Mabaera R, et al. Immunoregulatory Functions of VISTA. Immunol Rev (2017) 276:66–79. doi: 10.1111/imr.12525
47. Xie X, Zhang J, Shi Z, Liu W, Hu X, Qie C, et al. The Expression Pattern and Clinical Significance of the Immune Checkpoint Regulator VISTA in Human Breast Cancer. Front Immunol (2020) 11:563044. doi: 10.3389/fimmu.2020.563044
48. Schoop H, Bregenzer A, Halske C, Behrens HM, Kruger S, Egberts JH, et al. Therapy Resistance in Neoadjuvantly Treated Gastric Cancer and Cancer of the Gastroesophageal Junction is Associated With an Increased Expression of Immune Checkpoint Inhibitors-Comparison Against a Therapy Naive Cohort. Transl Oncol (2020) 13:165–76. doi: 10.1016/j.tranon.2019.11.004
49. Kuklinski LF, Yan S, Li Z, Fisher JL, Cheng C, Noelle RJ, et al. VISTA Expression on Tumor-Infiltrating Inflammatory Cells in Primary Cutaneous Melanoma Correlates With Poor Disease-Specific Survival. Cancer Immunol Immunother (2018) 67:1113–21. doi: 10.1007/s00262-018-2169-1
50. Le Mercier I, Chen W, Lines JL, Day M, Li J, Sergent P, et al. VISTA Regulates the Development of Protective Antitumor Immunity. Cancer Res (2014) 74:1933–44. doi: 10.1158/0008-5472.CAN-13-1506
51. Wang L, Rubinstein R, Lines JL, Wasiuk A, Ahonen C, Guo Y, et al. VISTA, a Novel Mouse Ig Superfamily Ligand That Negatively Regulates T Cell Responses. J Exp Med (2011) 208:577–92. doi: 10.1084/jem.20100619
52. Borggrewe M, Grit C, Den Dunnen W, Burm SM, Bajramovic JJ, Noelle RJ, et al. VISTA Expression by Microglia Decreases During Inflammation and is Differentially Regulated in CNS Diseases. Glia (2018) 66:2645–58. doi: 10.1002/glia.23517
53. Arlauckas SP, Garris CS, Kohler RH, Kitaoka M, Cuccarese MF, Yang KS, et al. In Vivo Imaging Reveals a Tumor-Associated Macrophage-Mediated Resistance Pathway in Anti-PD-1 Therapy. Sci Transl Med (2017) 9(389):eaal3604. doi: 10.1126/scitranslmed.aal3604
54. Lo RG, Moro M, Sommariva M, Cancila V, Boeri M, Centonze G, et al. Antibody-Fc/FcR Interaction on Macrophages as a Mechanism for Hyperprogressive Disease in Non-Small Cell Lung Cancer Subsequent to PD-1/PD-L1 Blockade. Clin Cancer Res (2019) 25:989–99. doi: 10.1158/1078-0432.CCR-18-1390
55. Moreno-Vicente J, Willoughby JE, Taylor MC, Booth SG, English VL, Williams EL, et al. Fc-Null Anti-PD-1 Monoclonal Antibodies Deliver Optimal Checkpoint Blockade in Diverse Immune Environments. J Immunother Cancer (2022) 10:e003735. doi: 10.1136/jitc-2021-003735
56. Milane L, Singh A, Mattheolabakis G, Suresh M, Amiji MM. Exosome Mediated Communication Within the Tumor Microenvironment. J Control Release (2015) 219:278–94. doi: 10.1016/j.jconrel.2015.06.029
57. Wu Q, Zhou L, Lv D, Zhu X, Tang H. Exosome-Mediated Communication in the Tumor Microenvironment Contributes to Hepatocellular Carcinoma Development and Progression. J Hematol Oncol (2019) 12:53. doi: 10.1186/s13045-019-0739-0
58. Whiteside TL. Exosome and Mesenchymal Stem Cell Cross-Talk in the Tumor Microenvironment. Semin Immunol (2018) 35:69–79. doi: 10.1016/j.smim.2017.12.003
59. Zheng P, Luo Q, Wang W, Li J, Wang T, Wang P, et al. Tumor-Associated Macrophages-Derived Exosomes Promote the Migration of Gastric Cancer Cells by Transfer of Functional Apolipoprotein E. Cell Death Dis (2018) 9:434. doi: 10.1038/s41419-018-0465-5
60. Binenbaum Y, Fridman E, Yaari Z, Milman N, Schroeder A, Ben DG, et al. Transfer of miRNA in Macrophage-Derived Exosomes Induces Drug Resistance in Pancreatic Adenocarcinoma. Cancer Res (2018) 78:5287–99. doi: 10.1158/0008-5472.CAN-18-0124
61. Yang F, Wang T, Du P, Fan H, Dong X, Guo H. M2 Bone Marrow-Derived Macrophage-Derived Exosomes Shuffle microRNA-21 to Accelerate Immune Escape of Glioma by Modulating PEG3. Cancer Cell Int (2020) 20:93. doi: 10.1186/s12935-020-1163-9
62. Xi J, Huang Q, Wang L, Ma X, Deng Q, Kumar M, et al. miR-21 Depletion in Macrophages Promotes Tumoricidal Polarization and Enhances PD-1 Immunotherapy. Oncogene (2018) 37:3151–65. doi: 10.1038/s41388-018-0178-3
63. Ma YS, Wu TM, Ling CC, Yu F, Zhang J, Cao PS, et al. M2 Macrophage-Derived Exosomal microRNA-155-5p Promotes the Immune Escape of Colon Cancer by Downregulating ZC3H12B. Mol Ther Oncolytics (2021) 20:484–98. doi: 10.1016/j.omto.2021.02.005
64. Hensler M, Kasikova L, Fiser K, Rakova J, Skapa P, Laco J, et al. M2-Like Macrophages Dictate Clinically Relevant Immunosuppression in Metastatic Ovarian Cancer. J Immunother Cancer (2020) 8:e000979. doi: 10.1136/jitc-2020-000979
65. Chen L, Cao MF, Xiao JF, Ma QH, Zhang H, Cai RL, et al. Stromal PD-1(+) Tumor-Associated Macrophages Predict Poor Prognosis in Lung Adenocarcinoma. Hum Pathol (2020) 97:68–79. doi: 10.1016/j.humpath.2019.12.007
66. Cao L, Che X, Qiu X, Li Z, Yang B, Wang S, et al. M2 Macrophage Infiltration Into Tumor Islets Leads to Poor Prognosis in Non-Small-Cell Lung Cancer. Cancer Manag Res (2019) 11:6125–38. doi: 10.2147/CMAR.S199832
67. Gomez-Roca CA, Italiano A, Le Tourneau C, Cassier PA, Toulmonde M, D'Angelo SP, et al. Phase I Study of Emactuzumab Single Agent or in Combination With Paclitaxel in Patients With Advanced/Metastatic Solid Tumors Reveals Depletion of Immunosuppressive M2-Like Macrophages. Ann Oncol (2019) 30:1381–92. doi: 10.1093/annonc/mdz163
68. Ries CH, Cannarile MA, Hoves S, Benz J, Wartha K, Runza V, et al. Targeting Tumor-Associated Macrophages With Anti-CSF-1R Antibody Reveals a Strategy for Cancer Therapy. Cancer Cell (2014) 25:846–59. doi: 10.1016/j.ccr.2014.05.016
69. Sherr CJ, Ashmun RA, Downing JR, Ohtsuka M, Quan SG, Golde DW, et al. Inhibition of Colony-Stimulating Factor-1 Activity by Monoclonal Antibodies to the Human CSF-1 Receptor. Blood (1989) 73:1786–93. doi: 10.1182/blood.V73.7.1786.1786
70. Peranzoni E, Lemoine J, Vimeux L, Feuillet V, Barrin S, Kantari-Mimoun C, et al. Macrophages Impede CD8 T Cells From Reaching Tumor Cells and Limit the Efficacy of Anti-PD-1 Treatment. Proc Natl Acad Sci USA (2018) 115:E4041–50. doi: 10.1073/pnas.1720948115
71. Shi G, Yang Q, Zhang Y, Jiang Q, Lin Y, Yang S, et al. Modulating the Tumor Microenvironment via Oncolytic Viruses and CSF-1r Inhibition Synergistically Enhances Anti-PD-1 Immunotherapy. Mol Ther (2019) 27:244–60. doi: 10.1016/j.ymthe.2018.11.010
72. Dammeijer F, Lievense LA, Kaijen-Lambers ME, van Nimwegen M, Bezemer K, Hegmans JP, et al. Depletion of Tumor-Associated Macrophages With a CSF-1r Kinase Inhibitor Enhances Antitumor Immunity and Survival Induced by DC Immunotherapy. Cancer Immunol Res (2017) 5:535–46. doi: 10.1158/2326-6066.CIR-16-0309
73. Mao Y, Eissler N, Blanc KL, Johnsen JI, Kogner P, Kiessling R. Targeting Suppressive Myeloid Cells Potentiates Checkpoint Inhibitors to Control Spontaneous Neuroblastoma. Clin Cancer Res (2016) 22:3849–59. doi: 10.1158/1078-0432.CCR-15-1912
74. Shih CT, Shiau CW, Chen YL, Chen LJ, Chao TI, Wang CY, et al. TD-92, a Novel Erlotinib Derivative, Depletes Tumor-Associated Macrophages in Non-Small Cell Lung Cancer via Down-Regulation of CSF-1R and Enhances the Anti-Tumor Effects of Anti-PD-1. Cancer Lett (2021) 498:142–51. doi: 10.1016/j.canlet.2020.10.043
75. Beatty GL, Chiorean EG, Fishman MP, Saboury B, Teitelbaum UR, Sun W, et al. CD40 Agonists Alter Tumor Stroma and Show Efficacy Against Pancreatic Carcinoma in Mice and Humans. Science (2011) 331:1612–6. doi: 10.1126/science.1198443
76. Mu J, Sun P, Ma Z, Sun P. BRD4 Promotes Tumor Progression and NF-Kappab/CCL2-Dependent Tumor-Associated Macrophage Recruitment in GIST. Cell Death Dis (2019) 10:935. doi: 10.1038/s41419-019-2170-4
77. Li X, Fu Y, Yang B, Guo E, Wu Y, Huang J, et al. BRD4 Inhibition by AZD5153 Promotes Antitumor Immunity via Depolarizing M2 Macrophages. Front Immunol (2020) 11:89. doi: 10.3389/fimmu.2020.00089
78. Joshi S, Singh AR, Liu KX, Pham TV, Zulcic M, Skola D, et al. SF2523: Dual PI3K/BRD4 Inhibitor Blocks Tumor Immunosuppression and Promotes Adaptive Immune Responses in Cancer. Mol Cancer Ther (2019) 18:1036–44. doi: 10.1158/1535-7163.MCT-18-1206
79. Zhu H, Bengsch F, Svoronos N, Rutkowski MR, Bitler BG, Allegrezza MJ, et al. BET Bromodomain Inhibition Promotes Anti-Tumor Immunity by Suppressing PD-L1 Expression. Cell Rep (2016) 16:2829–37. doi: 10.1016/j.celrep.2016.08.032
80. Wang Z, Kang W, You Y, Pang J, Ren H, Suo Z, et al. USP7: Novel Drug Target in Cancer Therapy. Front Pharmacol (2019) 10:427. doi: 10.3389/fphar.2019.00427
81. Dai X, Lu L, Deng S, Meng J, Wan C, Huang J, et al. USP7 Targeting Modulates Anti-Tumor Immune Response by Reprogramming Tumor-Associated Macrophages in Lung Cancer. Theranostics (2020) 10:9332–47. doi: 10.7150/thno.47137
82. Wang Z, Kang W, Li O, Qi F, Wang J, You Y, et al. Abrogation of USP7 is an Alternative Strategy to Downregulate PD-L1 and Sensitize Gastric Cancer Cells to T Cells Killing. Acta Pharm Sin B (2021) 11:694–707. doi: 10.1016/j.apsb.2020.11.005
83. Lim YW, Coles GL, Sandhu SK, Johnson DS, Adler AS, Stone EL. Single-Cell Transcriptomics Reveals the Effect of PD-L1/TGF-Beta Blockade on the Tumor Microenvironment. BMC Biol (2021) 19:107. doi: 10.1186/s12915-021-01034-z
84. Panagi M, Voutouri C, Mpekris F, Papageorgis P, Martin MR, Martin JD, et al. TGF-Beta Inhibition Combined With Cytotoxic Nanomedicine Normalizes Triple Negative Breast Cancer Microenvironment Towards Anti-Tumor Immunity. Theranostics (2020) 10:1910–22. doi: 10.7150/thno.36936
85. Knudson KM, Hicks KC, Luo X, Chen JQ, Schlom J, Gameiro SR. M7824, a Novel Bifunctional Anti-PD-L1/TGFbeta Trap Fusion Protein, Promotes Anti-Tumor Efficacy as Monotherapy and in Combination With Vaccine. Oncoimmunology (2018) 7:e1426519. doi: 10.1080/2162402X.2018.1426519
86. Jala VR, Bodduluri SR, Ghosh S, Chheda Z, Singh R, Smith ME, et al. Absence of CCR2 Reduces Spontaneous Intestinal Tumorigenesis in the Apc(Min) (/+) Mouse Model. Int J Cancer (2021) 148:2594–607. doi: 10.1002/ijc.33477
87. Mittal P, Wang L, Akimova T, Leach CA, Clemente JC, Sender MR, et al. The CCR2/MCP-1 Chemokine Pathway and Lung Adenocarcinoma. Cancers (Basel) (2020) 12(12):3723. doi: 10.3390/cancers12123723
88. Yumimoto K, Akiyoshi S, Ueo H, Sagara Y, Onoyama I, Ueo H, et al. F-Box Protein FBXW7 Inhibits Cancer Metastasis in a Non-Cell-Autonomous Manner. J Clin Invest (2015) 125:621–35. doi: 10.1172/JCI78782
89. Arakaki R, Yamasaki T, Kanno T, Shibasaki N, Sakamoto H, Utsunomiya N, et al. CCL2 as a Potential Therapeutic Target for Clear Cell Renal Cell Carcinoma. Cancer Med (2016) 5:2920–33. doi: 10.1002/cam4.886
90. Li X, Yao W, Yuan Y, Chen P, Li B, Li J, et al. Targeting of Tumour-Infiltrating Macrophages via CCL2/CCR2 Signalling as a Therapeutic Strategy Against Hepatocellular Carcinoma. Gut (2017) 66:157–67. doi: 10.1136/gutjnl-2015-310514
91. Loberg RD, Ying C, Craig M, Day LL, Sargent E, Neeley C, et al. Targeting CCL2 With Systemic Delivery of Neutralizing Antibodies Induces Prostate Cancer Tumor Regression In Vivo. Cancer Res (2007) 67:9417–24. doi: 10.1158/0008-5472.CAN-07-1286
92. Beider K, Bitner H, Leiba M, Gutwein O, Koren-Michowitz M, Ostrovsky O, et al. Multiple Myeloma Cells Recruit Tumor-Supportive Macrophages Through the CXCR4/CXCL12 Axis and Promote Their Polarization Toward the M2 Phenotype. Oncotarget (2014) 5:11283–96. doi: 10.18632/oncotarget.2207
93. Yusen W, Xia W, Shengjun Y, Shaohui Z, Hongzhen Z. The Expression and Significance of Tumor Associated Macrophages and CXCR4 in Non-Small Cell Lung Cancer. J BUON (2018) 23:398–402.
94. Argyle D, Kitamura T. Targeting Macrophage-Recruiting Chemokines as a Novel Therapeutic Strategy to Prevent the Progression of Solid Tumors. Front Immunol (2018) 9:2629. doi: 10.3389/fimmu.2018.02629
95. Bockorny B, Semenisty V, Macarulla T, Borazanci E, Wolpin BM, Stemmer SM, et al. BL-8040, a CXCR4 Antagonist, in Combination With Pembrolizumab and Chemotherapy for Pancreatic Cancer: The COMBAT Trial. Nat Med (2020) 26:878–85. doi: 10.1038/s41591-020-0880-x
96. Fortunato O, Belisario DC, Compagno M, Giovinazzo F, Bracci C, Pastorino U, et al. CXCR4 Inhibition Counteracts Immunosuppressive Properties of Metastatic NSCLC Stem Cells. Front Immunol (2020) 11:2168. doi: 10.3389/fimmu.2020.02168
97. Gulley JL, Schlom J, Barcellos-Hoff MH, Wang XJ, Seoane J, Audhuy F, et al. Dual Inhibition of TGF-Beta and PD-L1: A Novel Approach to Cancer Treatment. Mol Oncol (2021) 00:1–18. doi: 10.1002/1878-0261.13146
98. van den Bulk J, de Miranda N, Ten DP. Therapeutic Targeting of TGF-Beta in Cancer: Hacking a Master Switch of Immune Suppression. Clin Sci (Lond) (2021) 135:35–52. doi: 10.1042/CS20201236
99. Teicher BA, Fricker SP. CXCL12 (SDF-1)/CXCR4 Pathway in Cancer. Clin Cancer Res (2010) 16:2927–31. doi: 10.1158/1078-0432.CCR-09-2329
100. Cavalcante RS, Ishikawa U, Silva ES, Silva-Junior AA, Araujo AA, Cruz LJ, et al. STAT3/NF-kappaB Signalling Disruption in M2 Tumour-Associated Macrophages is a Major Target of PLGA Nanocarriers/PD-L1 Antibody Immunomodulatory Therapy in Breast Cancer. Br J Pharmacol (2021) 178:2284–304. doi: 10.1111/bph.15373
101. Li J, Zhao M, Sun M, Wu S, Zhang H, Dai Y, et al. Multifunctional Nanoparticles Boost Cancer Immunotherapy Based on Modulating the Immunosuppressive Tumor Microenvironment. ACS Appl Mater Interfaces (2020) 12:50734–47. doi: 10.1021/acsami.0c14909
102. Li L, Zhen M, Wang H, Sun Z, Jia W, Zhao Z, et al. Functional Gadofullerene Nanoparticles Trigger Robust Cancer Immunotherapy Based on Rebuilding an Immunosuppressive Tumor Microenvironment. Nano Lett (2020) 20:4487–96. doi: 10.1021/acs.nanolett.0c01287
103. Bi S, Huang W, Chen S, Huang C, Li C, Guo Z, et al. Cordyceps Militaris Polysaccharide Converts Immunosuppressive Macrophages Into M1-Like Phenotype and Activates T Lymphocytes by Inhibiting the PD-L1/PD-1 Axis Between TAMs and T Lymphocytes. Int J Biol Macromol (2020) 150:261–80. doi: 10.1016/j.ijbiomac.2020.02.050
104. Xiao Q, Li X, Li Y, Wu Z, Xu C, Chen Z, et al. Biological Drug and Drug Delivery-Mediated Immunotherapy. Acta Pharm Sin B (2021) 11:941–60. doi: 10.1016/j.apsb.2020.12.018
105. Oliva N, Conde J, Wang K, Artzi N. Designing Hydrogels for On-Demand Therapy. Acc Chem Res (2017) 50:669–79. doi: 10.1021/acs.accounts.6b00536
106. Srivastava A, Rathore S, Munshi A, Ramesh R. Organically Derived Exosomes as Carriers of Anticancer Drugs and Imaging Agents for Cancer Treatment. Semin Cancer Biol (2022). doi: 10.1016/j.semcancer.2022.02.020
107. Wang H, Alarcon CN, Liu B, Watson F, Searles S, Lee CK, et al. Genetically Engineered and Enucleated Human Mesenchymal Stromal Cells for the Targeted Delivery of Therapeutics to Diseased Tissue. Nat BioMed Eng (2021). doi: 10.1038/s41551-021-00815-9
108. Jin H, Wan C, Zou Z, Zhao G, Zhang L, Geng Y, et al. Tumor Ablation and Therapeutic Immunity Induction by an Injectable Peptide Hydrogel. ACS Nano (2018) 12:3295–310. doi: 10.1021/acsnano.7b08148
109. Dai X, Meng J, Deng S, Zhang L, Wan C, Lu L, et al. Targeting CAMKII to Reprogram Tumor-Associated Macrophages and Inhibit Tumor Cells for Cancer Immunotherapy With an Injectable Hybrid Peptide Hydrogel. Theranostics (2020) 10:3049–63. doi: 10.7150/thno.42385
110. Bhatnagar S, Shinagawa K, Castellino FJ, Schorey JS. Exosomes Released From Macrophages Infected With Intracellular Pathogens Stimulate a Proinflammatory Response In Vitro and In Vivo. Blood (2007) 110:3234–44. doi: 10.1182/blood-2007-03-079152
111. Li Z, Suo B, Long G, Gao Y, Song J, Zhang M, et al. Exosomal miRNA-16-5p Derived From M1 Macrophages Enhances T Cell-Dependent Immune Response by Regulating PD-L1 in Gastric Cancer. Front Cell Dev Biol (2020) 8:572689. doi: 10.3389/fcell.2020.572689
112. Gao ZS, Zhang CJ, Xia N, Tian H, Li DY, Lin JQ, et al. Berberine-Loaded M2 Macrophage-Derived Exosomes for Spinal Cord Injury Therapy. Acta Biomater (2021) 126:211–23. doi: 10.1016/j.actbio.2021.03.018
113. Wei Z, Zhang X, Yong T, Bie N, Zhan G, Li X, et al. Boosting Anti-PD-1 Therapy With Metformin-Loaded Macrophage-Derived Microparticles. Nat Commun (2021) 12:440. doi: 10.1038/s41467-020-20723-x
114. Haney MJ, Zhao Y, Jin YS, Li SM, Bago JR, Klyachko NL, et al. Macrophage-Derived Extracellular Vesicles as Drug Delivery Systems for Triple Negative Breast Cancer (TNBC) Therapy. J Neuroimmune Pharmacol (2020) 15:487–500. doi: 10.1007/s11481-019-09884-9
115. Choo YW, Kang M, Kim HY, Han J, Kang S, Lee JR, et al. M1 Macrophage-Derived Nanovesicles Potentiate the Anticancer Efficacy of Immune Checkpoint Inhibitors. ACS Nano (2018) 12:8977–93. doi: 10.1021/acsnano.8b02446
116. Sato H, Okonogi N, Nakano T. Rationale of Combination of Anti-PD-1/PD-L1 Antibody Therapy and Radiotherapy for Cancer Treatment. Int J Clin Oncol (2020) 25:801–9. doi: 10.1007/s10147-020-01666-1
117. Garon EB, Rizvi NA, Hui R, Leighl N, Balmanoukian AS, Eder JP, et al. Pembrolizumab for the Treatment of Non-Small-Cell Lung Cancer. N Engl J Med (2015) 372:2018–28. doi: 10.1056/NEJMoa1501824
118. Wan C, Sun Y, Tian Y, Lu L, Dai X, Meng J, et al. Irradiated Tumor Cell-Derived Microparticles Mediate Tumor Eradication via Cell Killing and Immune Reprogramming. Sci Adv (2020) 6:y9789. doi: 10.1126/sciadv.aay9789
119. Horsman MR, Mortensen LS, Petersen JB, Busk M, Overgaard J. Imaging Hypoxia to Improve Radiotherapy Outcome. Nat Rev Clin Oncol (2012) 9:674–87. doi: 10.1038/nrclinonc.2012.171
120. Telarovic I, Wenger RH, Pruschy M. Interfering With Tumor Hypoxia for Radiotherapy Optimization. J Exp Clin Cancer Res (2021) 40:197. doi: 10.1186/s13046-021-02000-x
121. Dai X, Ruan J, Guo Y, Sun Z, Liu J, Bao X, et al. Enhanced Radiotherapy Efficacy and Induced Anti-Tumor Immunity in HCC by Improving Hypoxia Microenvironment Using Oxygen Microcapsules. Chem Eng J (2021) 422:130109. doi: 10.1016/j.cej.2021.130109
122. Hodi FS, O'Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, et al. Improved Survival With Ipilimumab in Patients With Metastatic Melanoma. N Engl J Med (2010) 363:711–23. doi: 10.1056/NEJMoa1003466
123. Weber JS, D'Angelo SP, Minor D, Hodi FS, Gutzmer R, Neyns B, et al. Nivolumab Versus Chemotherapy in Patients With Advanced Melanoma Who Progressed After Anti-CTLA-4 Treatment (CheckMate 037): A Randomised, Controlled, Open-Label, Phase 3 Trial. Lancet Oncol (2015) 16:375–84. doi: 10.1016/S1470-2045(15)70076-8
124. Robert C, Long GV, Brady B, Dutriaux C, Maio M, Mortier L, et al. Nivolumab in Previously Untreated Melanoma Without BRAF Mutation. N Engl J Med (2015) 372:320–30. doi: 10.1056/NEJMoa1412082
125. Robert C, Schachter J, Long GV, Arance A, Grob JJ, Mortier L, et al. Pembrolizumab Versus Ipilimumab in Advanced Melanoma. N Engl J Med (2015) 372:2521–32. doi: 10.1056/NEJMoa1503093
126. Larkin J, Chiarion-Sileni V, Gonzalez R, Grob JJ, Cowey CL, Lao CD, et al. Combined Nivolumab and Ipilimumab or Monotherapy in Untreated Melanoma. N Engl J Med (2015) 373:23–34. doi: 10.1056/NEJMoa1504030
127. Brahmer J, Reckamp KL, Baas P, Crino L, Eberhardt WE, Poddubskaya E, et al. Nivolumab Versus Docetaxel in Advanced Squamous-Cell Non-Small-Cell Lung Cancer. N Engl J Med (2015) 373:123–35. doi: 10.1056/NEJMoa1504627
128. Borghaei H, Paz-Ares L, Horn L, Spigel DR, Steins M, Ready NE, et al. Nivolumab Versus Docetaxel in Advanced Nonsquamous Non-Small-Cell Lung Cancer. N Engl J Med (2015) 373:1627–39. doi: 10.1056/NEJMoa1507643
129. Motzer RJ, Escudier B, McDermott DF, George S, Hammers HJ, Srinivas S, et al. Nivolumab Versus Everolimus in Advanced Renal-Cell Carcinoma. N Engl J Med (2015) 373:1803–13. doi: 10.1056/NEJMoa1510665
130. Tartari F, Santoni M, Burattini L, Mazzanti P, Onofri A, Berardi R. Economic Sustainability of Anti-PD-1 Agents Nivolumab and Pembrolizumab in Cancer Patients: Recent Insights and Future Challenges. Cancer Treat Rev (2016) 48:20–4. doi: 10.1016/j.ctrv.2016.06.002
131. Montironi R, Santoni M, Tartari F, Lopez-Beltran A, Cheng L, Berardi R, et al. Testing PD-1/PD-L1 Expression in Cancer Therapy: Pathologic Insights and Economic Sustainability. Arch Pathol Lab Med (2016) 140:501–2. doi: 10.5858/arpa.2015-0529-LE
132. Flores-Toro JA, Luo D, Gopinath A, Sarkisian MR, Campbell JJ, Charo IF, et al. CCR2 Inhibition Reduces Tumor Myeloid Cells and Unmasks a Checkpoint Inhibitor Effect to Slow Progression of Resistant Murine Gliomas. Proc Natl Acad Sci USA (2020) 117:1129–38. doi: 10.1073/pnas.1910856117
Keywords: immune checkpoint inhibitor (ICI), PD-1/PD-L1 axis, immunosuppression, tumor-associated macrophages (TAMs), immune microenvironment
Citation: Pu Y and Ji Q (2022) Tumor-Associated Macrophages Regulate PD-1/PD-L1 Immunosuppression. Front. Immunol. 13:874589. doi: 10.3389/fimmu.2022.874589
Received: 12 February 2022; Accepted: 06 April 2022;
Published: 03 May 2022.
Edited by:
Honglin Jin, Huazhong University of Science and Technology, ChinaReviewed by:
Xiaomeng Dai, Zhejiang University, ChinaCopyright © 2022 Pu and Ji. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Qing Ji, dHR0OTkxMThAaG90bWFpbC5jb20=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.