 Ilse Gille
Ilse Gille Frans H. J. Claas1,3
Frans H. J. Claas1,3 Sebastiaan Heidt
Sebastiaan Heidt- 1Department of Immunology, Leiden University Medical Center, Leiden, Netherlands
- 2Department of Hematology, Leiden University Medical Center, Leiden, Netherlands
- 3Eurotransplant Reference Laboratory, Leiden University Medical Center, Leiden, Netherlands
Solid organ transplantation is the treatment of choice for various end-stage diseases, but requires the continuous need for immunosuppression to prevent allograft rejection. This comes with serious side effects including increased infection rates and development of malignancies. Thus, there is a clinical need to promote transplantation tolerance to prevent organ rejection with minimal or no immunosuppressive treatment. Polyclonal regulatory T-cells (Tregs) are a potential tool to induce transplantation tolerance, but lack specificity and therefore require administration of high doses. Redirecting Tregs towards mismatched donor HLA molecules by modifying these cells with chimeric antigen receptors (CAR) would render Tregs far more effective at preventing allograft rejection. Several studies on HLA-A2 specific CAR Tregs have demonstrated that these cells are highly antigen-specific and show a superior homing capacity to HLA-A2+ allografts compared to polyclonal Tregs. HLA-A2 CAR Tregs have been shown to prolong survival of HLA-A2+ allografts in several pre-clinical humanized mouse models. Although promising, concerns about safety and stability need to be addressed. In this review the current research, obstacles of CAR Treg therapy, and its potential future in solid organ transplantation will be discussed.
Introduction
Organ transplantation remains the best treatment option for patients with end-stage organ failure. Despite its successes, transplantation still faces many obstacles such as the availability of donor organs, side effects of immunosuppressive drugs and immunological rejection. Ideally, donor and patient are fully matched for human leucocyte antigens (HLA), however this is in most instances not feasible due to the extensive polymorphism of HLA. To prevent detrimental immune reactivity towards mismatched HLA between donor and patient, administering immunosuppressive drugs is necessary. Currently, several drugs are used for induction therapy, maintenance therapy, and anti-rejection therapy. Immunosuppressive induction therapy is intense immunosuppression given in the first days after transplantation, of which basiliximab is mostly used. After these critical few days, the patient is left with maintenance immunosuppression to continuously dampen the immune system. Most standard care consists of three immunosuppressive drugs, namely calcineurin inhibitors (e.g. tacrolimus or cyclosporine), antimetabolites (e.g. mycophenolate mofetil (MMF) or azathioprine) and corticosteroids (e.g. methylprednisolone or prednisone) (1). Ideally, lower doses of the maintenance therapy can be administered over time to these patients (2, 3). If rejection occurs, patients are treated with high doses of steroids or lymphocyte depleting antibodies. Unfortunately, immunosuppressive drugs come with many side effects such as cardiovascular complications (4), and chronic immune suppression gives rise to higher infection rates (5) and elevated risk on the development of malignancies (6).
Graft rejection can arise through several immunological pathways. In transplantation, T-cells can be activated by antigen-presenting cells (APCs) through both the direct, indirect and semi-direct pathway. The direct pathway involves activation of T-cells by intact donor HLA molecules on donor APCs. Graft cells that express mismatched donor HLA molecules can therefore be recognized and killed by these T-cells. The indirect pathway is characterized by the activation of T-cells by peptides derived from donor cells (mostly donor HLA) presented by the recipient’s self HLA class II molecules expressed on recipient APCs, akin a normal immune response to a pathogenic peptide presented in self HLA. The semi-direct pathway comprises T-cells that recognize intact HLA on the recipients’ own APCs in a similar manner as to the direct pathway, albeit these allogeneic HLA molecules are acquired from donor cells and expressed on the cell surface of recipient APCs. Direct alloreactivity mainly occurs directly after transplantation and disappears as donor APCs are eliminated in time. In contrast, indirect alloreactivity becomes more prominent late after transplantation. Additionally, alloreactive B-cells are activated by recognizing epitopes on intact HLA molecules. They can further be activated by follicular T helper (Tfh) cells, which results in class switching and affinity maturation, and differentiation to become either long-lived plasma cells or memory B-cells. Indirect allorecognition of donor-derived peptides in self HLA class II on B-cells by Tfh cells is required to achieve this. The ensuing plasma cells secrete HLA antibodies which can bind to mismatched HLA molecules on the graft, resulting in so-called antibody-mediated rejection (AMR). The main route of damage is through activation of the complement system. Additionally, natural killer (NK) cells interact with antibodies by their Fc receptors and induce antibody-dependent cellular cytotoxicity (ADCC). Moreover, NK-cells contribute to allograft rejection by directly targeting donor cells via the detection of missing self HLA class I molecules.
The holy grail of transplantation medicine is to achieve transplantation tolerance, defined as the absence of a detrimental immune response towards the allograft in the absence of immunosuppressive drugs while maintaining protective immunity towards pathogens and malignant cells. Interestingly, it has been shown that spontaneous transplantation tolerance can occur in liver transplantation recipients (7), and to a lesser extent in kidney transplant recipients (8, 9). By studying the immune profile of these tolerant patients, as well as on basis of several studies in animal models (10–14), the main mechanisms of the development and/or maintenance of transplantation tolerance have been identified. Additionally, from animal studies, several potential ways of actively achieving tolerance have been identified, such as the induction of donor chimerism from the same source as the organ donor (15, 16). Additionally, the use of autologous Mesenchymal Stromal Cells (MSC) has been shown to allow for tacrolimus withdrawal in a clinical phase II study (17). It is generally believed that in order to achieve tolerance, the balance of regulatory T-cells (Tregs) and conventional T-cells needs to be skewed towards increased Tregs numbers. Accordingly, Treg immunotherapy is currently being investigated as a clinically feasible way to prevent graft rejection and to ultimately induce allograft tolerance. The first clinical experience of polyclonal Treg therapy in the setting of kidney transplantation was positive (15, 18). Currently, a phase II clinical trial called the TWO study is enrolling kidney transplant patients who will receive expanded polyclonal Tregs in order to achieve minimization of immunosuppressive drugs (EudraCT: 2017-001421-41) (18).
Extensive research has shown that the main type of Tregs is a subset of CD4+ T-cells that plays an important role in the suppression of immune activation and maintaining tolerance to self (19). This T-cell subset is characterized by the constitutive expression of the transcription factor FoxP3 (20, 21). Moreover, Tregs are generally identified as CD4+CD25highCD127lowFoxP3+ cells. Tregs can be divided into peripherally induced Tregs (pTregs) and thymus-derived (tTregs); while tTregs undergo selection in the thymus, pTregs develop from conventional CD4+ T-cells that are converted into pTregs in peripheral organs. Due to this difference in development, tTregs have an autoreactive T-cell receptor (TCR), whereas pTregs do not.
Previously, polyclonal Tregs have been investigated as cellular therapy to prevent the development of Graft-versus-Host-Disease (GVHD) (22, 23). The first report on treatment of GVHD with ex vivo expanded Tregs demonstrated relief of symptoms and the possibility to reduce immunosuppressive drugs in a case of chronic GVHD, and temporarily alleviated clinical symptoms in a patient with acute GVHD (22). Years later, umbilical cord blood-derived Tregs were expanded and administered to 11 patients suffering from GVHD after having received stem cell transplantation for treatment of hematological malignancies. Incidence of grade II-IV acute GVHD was five times lower in patients with receiving Tregs, and chronic GVHD after one year was absent in the Treg treatment group compared to 14% in the control group (23). More recently, the multicenter ONE study focused on regulatory cell-based therapy to determine the safety in the setting of kidney transplantation. The ONE study describes six phase I/IIa trials with kidney transplant patients that received regulatory cellular therapy, including dendritic cells or macrophages, but also polyclonal and donor-antigen reactive Tregs (15, 18). Patients received standard immunosuppression therapy with tapered steroids, MMF and tacrolimus. Induction therapy with basiliximab was not administered due to the potential influence of the anti-CD25 agent on the Treg population. Treatment with human polyclonal Tregs was demonstrated to be safe and even allowing for standard immunosuppression therapy to be reduced in a subset of patients (15). Interestingly, in 8 out of 11 patients receiving polyclonal Tregs (ONEnTreg13), stable tacrolimus monotherapy was achieved (24). In another safety and feasibility trial, autologous polyclonal Tregs were administered to kidney transplant recipients who showed subclinical inflammation on 6-month surveillance biopsies. In this study, it was demonstrated that polyclonal Tregs labeled with deuterated glucose persisted and remained phenotypically stable in kidney transplant patients that were receiving immunosuppression (25). These data together suggest the potential safe use of polyclonally expanded Tregs as a therapeutic means to at least reduce the use of immunosuppressive drugs.
Whereas cellular therapy using polyclonal Tregs is promising, high numbers of cells are required to obtain the desired effects. Importantly, in vitro studies have shown that allospecific Tregs outperform polyclonal Tregs in suppressive capacity (26–33), resulting in lower Treg cell numbers required. Allospecific Tregs can theoretically be generated through several methods including expansion through direct antigen presentation by donor APCs, indirect antigen presentation with patient APCs pulsed with peptide from donor or tetramers made up of patient HLA class II, as recently reviewed by Hu et al. (34). As these methods are rather cumbersome and difficult to standardize, alternative means to achieve donor-specificity are warranted. Already some time ago, it has been shown that T-cell specificity could be redirected by genetic modification to express either a transgenic TCR (35) or chimeric antigen receptor (CAR) (36). In the field of adoptive cell therapy, genetically modified antigen-specific T-cells are an attractive option, since fewer T-cells are needed compared to polyclonal cell populations to induce an effective response. In addition, because of their exquisite antigen-specificity, fewer off-target effects are to be expected. Furthermore, CAR T-cells have the additional advantage of recognizing cell surface antigen in an HLA unrestricted manner.
CAR T-cells and CAR-Treg cells
CAR T-cells are genetically modified T-cells that express CAR molecules on their cell surface and have been originally developed in the setting of hematological malignancies. CAR T-cells recognize antigens via a single-chain variable fragments (scFv) domain which is a fusion protein of the variable regions of the heavy and light chain of a specific immunoglobulin (Ig). This extracellular CAR domain is coupled to a spacer to ensure flexibility, a transmembrane domain, and the intracellular signaling domain CD3ζ, allowing for T-cell activation. By using the antigen recognition domain of an antibody coupled to the signaling domain of a T-cell receptor, T-cell responses with high specificity can be generated. However, the first-generation CARs demonstrated that the CD3ζ chain activation domain was not sufficient to sustain T-cell function in primary T-cells (37). Therefore, second-generation CAR T-cells were developed which additionally comprise an intracellular costimulatory domain, of which 4-1BB (i.e. CD137) and CD28 are most often used (Figure 1A). This modification improved the activation, proliferation and prolonged survival of the CAR T-cells (38, 39). While third-generation CAR T-cells were designed with two co-stimulatory domains with the aim to achieve superior killing, addition of these domains did not show to be beneficial over a single co-stimulatory domain (40, 41). Fourth-generation CARs were subsequently generated, which are also referred to as TRUCKS (T-cell redirected for antigen-unrestricted cytokine-initiated killing). These CARs were engineered to release additional transgene cytokines upon CAR ligation or express additional costimulatory ligands (42). To date, second-generation CARs are most often used in clinical settings.
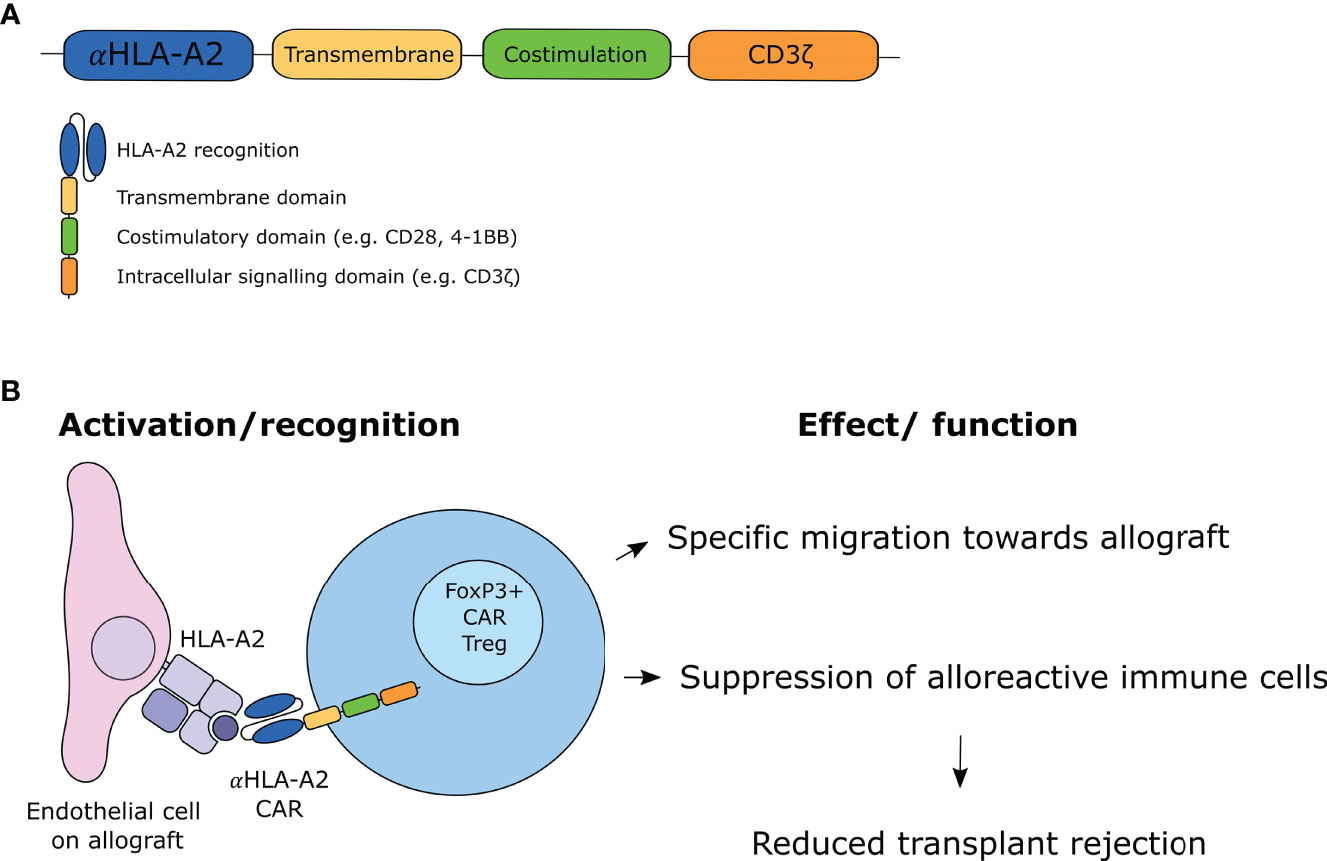
Figure 1 CAR Treg design and working mechanism. (A) Design of second-generation CAR molecule. (B) Proposed mechanism of action of CAR Tregs.
The first CAR T-cell therapies that were authorized by the Food and Drug Administration (FDA) were tisagenlecleucel and axicabtagene ciloleucel, which both are CD19 CAR T-cell therapies for the treatment of B-cell acute lymphoblastic leukemia and B-cell lymphoma, respectively (43, 44). Since then, the interest in CAR T-cell therapy targeting multiple tumor types has increased. More recently, the development of CAR T-cells for treatment of autoimmune diseases, allergy, and induction of transplantation tolerance has been under investigation. In contrast to the oncology field, antigen-specific regulation rather than activation is desired here. Elinav and colleagues were one of the first to redirect Tregs in an experimental setting by designing a CAR to treat colitis in mice (45). They showed specificity of CAR Tregs against the model antigen 2,4,6-trinitrophenol (TNP). These TNP CAR Tregs suppressed the activity of effector T (Teff) cells in an MHC and CD28-independent manner. Furthermore, they demonstrated that TNP CAR Tregs preferentially migrated to TNP-sensitized colon and prevented the development of colitis in wild-type mice. Since then, Tregs have been engineered with CAR technology as potential therapy for several other autoimmune diseases and allergy-related diseases, such as experimental autoimmune encephalomyelitis (EAE) (46, 47), type I diabetes (48), asthma (49), hemophilia A (50, 51), vitiligo (52), and arthritis (53). Additionally, several papers describe the protection of CAR Tregs from the development of GVHD after stem cell transplantation in murine models (54–56). Imura et al. generated CD19 CAR Tregs that were able to suppress IgG antibody production and differentiation of B-cells (55). While CD19 CAR Treg therapy reduced the risk of development of GVHD in immunodeficient mice that were reconstituted with human PBMCs, the nature of the CAR construct still makes this an antigen non-specific therapy. Martin et al. engineered CD4+ T-cells to suppress GVHD in an antigen-specific manner by redirecting them towards the mismatched recipient HLA-A*02:01 alloantigen and to express FoxP3 (56). Furthermore, they showed that these genetically modified cells were superior in suppressing GVHD compared to polyclonal Tregs by means of reducing inflammation and inhibiting pro-inflammatory cytokine production. Along these lines, several research groups are working on the development of CAR Tregs to limit allograft rejection after solid organ transplantation (Figure 1B). In the case of solid organ transplantation, the antigens that can be targeted by CARs are the mismatched HLA molecules that are expressed solely on the allograft. As proof of concept, several CAR Treg products have been developed with a CAR directed against HLA-A2. In addition to CD4+ Tregs, CD8+ Tregs have been identified and mediate a suppressive function in transplantation setting, demonstrated as delayed skin graft rejection in humanized mice (57). CD8+ CAR Tregs have been described to exhibit suppressive activity in pre-clinical models (58). It has been suggested that CD8+ CAR Tregs could have a synergistic role with CD4+ Tregs in suppressive function to induce tolerance.
CAR Treg Design
CAR design is critically important for potent and antigen-specific CAR-T, as well as CAR Treg therapy. The exact CAR Treg design depends on the experimental setting. For research purposes, many viral vectors encode for a gene marker, such as myc, nerve growth factor receptor (NGFR), green fluorescent protein (GFP) or luciferase to purify and identify the transduced cells during experiments. By doing so, cells can easily be identified and data on purity, as well as persistence, can be obtained. When applied in the clinic, these markers are removed, allowing the frequency of CAR T-cells to be determined solely by qPCR based on the unique sequence of the introduced CAR. Additionally, an important factor in the development of CAR transduced cells is obtaining high transduction efficiency, not only because higher yields of antigen-specific cells will be obtained, but also to reduce production time. Currently, therapies with engineered CAR T-cells using both lentiviral or retroviral transduction are accepted and regarded as being safe (59).
Another important aspect of the CAR design is the choice in the costimulatory domain. The costimulatory domains currently used are the same as those used in conventional CAR T-cells. While conventional T cells and Tregs often express the same costimulatory receptors, the intracellular ‘wiring’ is different, resulting in different effector functions. In CAR T-cells, these domains are incorporated into the constructs to improve the activation, proliferation and prolonged. In Tregs, CD28 signaling is essential for development and homeostasis, whereas 4-1BB signaling is reported to enhance Treg proliferation, however the use of costimulatory domains in CAR Tregs has not been studied extensively. So far, most CAR Tregs were designed with either 4-1BB or CD28 as the costimulatory domain. Recently, Dawson investigated second-generation CAR Tregs with 10 different costimulatory domains (e.g. CD28, CD28 Y173F, ICOS, CTLA-4, CTLA-4 Y165G, PD-1, 4-1BB, GITR, OX40, TNFR2) and showed that CD28 costimulatory signaling was superior when tested for function and gene expression profile (54). The use of 4-1BB or TNFR2 caused a decrease in Treg stability and function. In line with these findings, Boroughs et al. and Imura et al. showed a reduced suppressive function of 4-1BB-CAR Tregs compared to CD28-CAR Tregs (55, 60). These findings suggest that 4-1BB is not desired as costimulatory domain for CAR Tregs, in contrast to CAR T-cells. Further research into the different costimulatory domains is needed to reveal which costimulatory domain is the most optimal for CAR Tregs.
Interestingly, Boardman and colleagues demonstrated that the CAR intracellular signaling domain is necessary to elicit a strong regulatory response by comparing an HLA-A2 specific CAR lacking CD28-CD3ζ (ΔCAR) domains with an HLA-A2 CAR that comprised the CD28-CD3ζ signaling domain (27). They showed that CAR activation including CD28-CD3ζ signaling protected HLA-A2+ skin allografts more effectively than merely TCR-mediated allorecognition demonstrated by polyclonal or ΔCAR Tregs. This is in line with the findings of first-generation CAR T-cells being outperformed by second-generation CAR T-cells, due to the costimulatory domain that increased their effectiveness and prolonged survival (38, 39).
Alloantigen-Specificity of CAR Tregs
The most obvious target for antigen-specific Tregs in the setting of solid organ transplantation is mismatched HLA expressed by the allograft. Most CARs investigated in the field of transplantation are based on HLA-A2 specific monoclonal antibodies (mAbs), due to the high prevalence of HLA-A2 in the general population (61). These CARs thus express an scFv constructed of a heavy and light chain of an HLA-A2 mAb and are therefore specific for an epitope present on HLA-A2. For example, MacDonald and colleagues generated an scFv from the variable regions of the anti-HLA-A2 heavy and light chains of the BB7.2 mouse hybridoma for the design of their HLA-A2 specific CAR (62). They and others showed that the scFv domain of the CAR retains their HLA-A2 specificity (62, 63). Generally, sequences from mouse mAbs are more likely to be immunogenic for humans and may induce antibodies that could neutralize the CAR Tregs. Therefore, Dawson and colleagues humanized the heavy and light chains of the BB7.2 mouse mAb to ultimately generate 18 different humanized CARs (64). They reported that both murine and humanized CAR Tregs are suppressive and migrate to HLA-A2+ grafts in vivo, however humanized CAR Tregs showed reduced binding to other HLA alleles. Hence, it could be advantageous to use a fully human HLA-specific mAb as a source of the scFv domain. Along this line, Boardman et al. used a human HLA-A2 specific scFv sequence that was derived from a patient that was sensitized by blood transfusion (27, 65). The epitope recognized by the scFv were the residues 142 to 145 (TTKH) which corresponds to a well-described HLA eplet (66), that besides HLA-A2 is present on the alleles HLA-A68 and HLA-A69 (65). HLA-A2 specificity of the scFv was confirmed by cytokine production and cytotoxicity of CAR Teff cells after stimulation with HLA-A2 positive and HLA-A2 negative cells (27). In addition, Muller et al. used the variable domain sequences of a well-characterized human B-cell derived hybridoma (clone SN607D8) (67) to produce an HLA-A2 specific scFv (68). Importantly, they found that the original anti-HLA-A2 sequences of the SN607D8 hybridoma could not successfully be expressed in the scFv. Only after engrafting the complementarity-determining region (CDR) regions of the heavy and light chain into a scaffold of the anti-HER2 antibody Herceptin, HLA-A2 CAR surface expression could be verified with the same specificity. This highlights some of the hurdles that have to be overcome when genetically modifying T-cells with the aim to redirect specificity.
Additionally, of particular concern is the conservation of the specificity of the CAR molecule, which can be addressed by several assays. Dawson et al. showed that the relative binding of the CAR molecules to HLA-specific beads in FlowPRA revealed a strong correlation with the MFI of tetramer binding (64). Additionally, CAR Treg specificity can be assessed with a panel of APCs expressing different single HLA alleles by measuring activation marker expression (64). Furthermore, tetramer staining (26, 62, 63), proliferation or cytokine secretion upon coculture with antigen-positive cells (26, 27, 63, 68) can be used. To move from the experimental setting to the clinical reality, the possibility to target the most prevalent HLA antigens should be explored, in order to serve the vast majority of transplant recipients. Fortunately, a large number of human HLA-specific mAbs have been developed with a wide range of specificities (66, 69–71). Evidently, it is crucial that the epitope recognized by the mAbs used for CAR generation is well-defined, and that patients who express this particular epitope in their HLA phenotype are excluded.
Migration of CAR Tregs Towards Allografts
Migration of Tregs to the allograft is required to induce local suppression of alloimmunity (72). Using an in vitro model, HLA-A2-specific CAR Treg transmigratory capacity through an endothelial monolayer towards HLA-A2+ target cells was compared to polyclonal Tregs. CAR Tregs demonstrated not only preferential but also faster migration than their polyclonal counterparts, thereby improving the protective function (27). Furthermore, several studies were performed in immunodeficient mice to test whether CAR Tregs preferentially migrate to allografts. In a humanized mouse model, NOD SCID gamma (NSG) mice received adjacent skin transplants from both NSG or NSG-HLA-A*02:01 transgenic mice and subsequently received peripheral blood mononuclear cells (PBMCs) and luciferase labeled HLA-A2 CAR Tregs or nonspecific CAR Tregs. Bioluminescence imaging demonstrated preferential migration of HLA-A2 CAR Tregs towards the HLA-A2+ skin allograft (64), indicating that HLA-specific CAR Tregs can migrate towards the location where regulation is required. Muller et al. demonstrated similar effects where HLA-A2-CAR Tregs migrate to the HLA-A2+ transgenic islets in a model of induced diabetes in immunodeficient mice (68). In addition, Noyan et al. studied a different HLA-A2 CAR, in a model where immunodeficient NOD rag gamma (NRG) mice were transplanted with human HLA-A2+ skin grafts (26). After engraftment, the mice received allogeneic PBMCs either in combination with HLA-A2 CAR Tregs, with polyclonal Tregs, or no Tregs. Here, immune infiltrates containing FoxP3+CD4+ cells were present in tolerated skin grafts 40 days after infusion of either CAR Tregs or polyclonal Tregs (26). In immunocompetent mice, HLA-A2 specific CAR Tregs were demonstrated to migrate in a significantly higher proportion to a transgenic HLA-A2+ skin graft compared to HLA-A2- skin graft (63). Of note, the group of Lombardi suggested that merely preferential migration towards allografts contributes to the protective function of CAR Tregs and is improved by TCR-dependent and CAR-dependent activation (27).
Suppression of Alloimmunity by CAR Tregs
HLA-A2 CAR Tregs have been shown to dose-dependently suppress T-cell responses in a system where HLA-A2+ transgenic B-cells pulsed with the OVA 323-339 peptide were cocultured with OTII CD4+ T-cells in the absence or presence of HLA-A2 or nonspecific CAR Tregs (63). Similarly, HLA-A2 CAR Tregs inhibited allospecific Teff cell proliferation in mixed lymphocyte assays against HLA-A2-positive stimulator cells (26, 62). MacDonald and colleagues were the first to show that HLA-A2 CAR Tregs prevented xenogeneic GVHD induced by HLA-A2 expressing T-cells in immunodeficient mice (62). Consequently, the survival of the mice that received CAR Tregs was improved compared to the groups that received non-specific CAR Tregs or polyclonal Tregs. Similarly, in a human skin xenograft transplant model in immunodeficient mice, HLA-A2 CAR Tregs protected the HLA-A2+ allografts more potently than polyclonal Tregs (27). HLA-A2 CAR Tregs have been shown to reduce gene expression of inflammatory cytokines in HLA-A2+ skin grafts of NSG mice, leading to increased survival in a humanized mouse skin transplantation model (64). At the same time, CAR Tregs were demonstrated to produce the anti-inflammatory interleukin 10 (IL-10) in the presence of alloantigens in vitro, suggesting that they contribute to a graft-specific immunosuppressive environment (27). Interestingly, the constitutive co-expression of IL-10 in CAR Tregs has been shown to be advantageous to the suppressive capacity (73).
Importantly, CAR Tregs do not seem to adequately inhibit the memory T-cell compartment. Several studies have shown that alloreactive memory T-cells and skin allograft rejection were not inhibited (63, 74). They demonstrated that HLA-A2 CAR Tregs did not suppress the formation of interferon gamma (IFN-γ) producing memory T-cells (63). This can pose a potential problem, as T-cells with alloreactive potential make up 1-10% of the peripheral T-cells, including memory T-cells (32, 75–79). In line with these findings, it was shown that mice that were sensitized upon transplantation did not benefit from subsequent CAR Treg therapy, since graft survival was not improved compared to non-specific or no CAR Treg therapy (63). This was different from de novo DSA responses, since antigen-specific CAR Tregs administered prior to sensitization decreased anti-HLA-A2 IgG DSA in vivo when mice were transplanted with HLA-A2+ skin grafts (63). The lack of CAR Treg efficacy in sensitized patients could limit the potential application of CAR Treg therapy in the clinical setting, since around 20% of patients on the Eurotransplant waiting list are sensitized, and around 5% is highly sensitized (80).
Persistence of CAR Tregs
In order to constitute an effective and long-lasting therapy, CAR T-cells need to survive long enough to carry out their function (81). In the case of Tregs, they need to survive in the patient’s body for a sufficient time period to suppress allograft-specific immune activation after transplantation. Lifelong effective suppression by CAR Tregs would be necessary to make immunosuppressive drugs redundant. Several studies using animal models have looked into the persistence of CAR Tregs after administration. In one study, in vitro CAR Tregs were present in blood, spleen and lymph nodes up to 7 days after infusion in immunocompetent mice (63). In comparison, Noyan and colleagues demonstrated longer persistence of HLA-A2 CAR Tregs shown as infiltration in skin allografts of immune reconstituted humanized NRG mice after 40 days (26). Of note, the experiment had to be stopped at that point due to the development of GVHD in these mice. Comparably, Dawson et al. showed persistence of specific CAR Tregs for at least 21 days in the HLA-A2+ graft of NSG mice where they persisted longer than polyclonal Tregs (64). It is clear that at least in the murine setting the effect of CAR Tregs is not long-lasting and that grafts were eventually rejected. Importantly, immunodeficient mice lack human cytokines and chemokines necessary for survival of infused human T-cells. Thus, the persistence of human T-cells is not representative of the persistence in humans in such models. Whereas the mouse models suggest that CAR Treg therapy represents a form of antigen-specific immunosuppression that needs to be administered with regular intervals, this notion can only be substantiated in clinical trials.
It is well known that cytokines are critical for Treg proliferation and survival, and Treg development is dependent on IL-2, transforming growth factor-β (TGFβ) and co-stimulatory molecules. Generally, Tregs are anergic, meaning that they need exogenous IL-2 in order to proliferate (82, 83). CARs can stimulate cells independent of IL-2 in the short term, but the absence of exogenous IL-2 eventually leads to decreased cell viability in the long term (62). Therefore, IL-2 is required for the long-term survival of CAR Tregs. IL-2 therapy can stimulate the proliferation of donor-specific Tregs and thereby contribute to allograft survival (84). Similarly, the administration of low-dose IL-2 is investigated after infusion of polyclonal Tregs in patients with diabetes mellitus type 1 (NCT02772679). It has been described that IL-2 promotes Treg expansion by enhancing antiapoptotic Bcl-2 expression (85). Furthermore, IL-33 is critical for the regulation of Tregs in tissues, including the kidney and IL-7 is necessary for circulation of nTregs between secondary lymphoid organs, as wells as survival and proliferation. Fourth generation CAR T-cells, have been generated to additionally express cytokines such as IL-18 (86), IL-12 (87) and IL-15 (88) to improve CAR T-cell survival or anti-tumor function. This principle has not yet been described in the context of CAR Tregs, however IL-2 co-expression might be beneficial for CAR Treg persistence. It is clear that cytokines play a pivotal role in the development of CAR Tregs and also in maintenance of these cells. This stresses that mouse models, lacking cytokines necessary for CAR Treg maintenance lack the power to describe the persistence of CAR Tregs. In addition, antigen exposure can lead to higher survival rates of the antigen-specific CAR Tregs compared to conditions without antigen exposure This finding is to be expected as it is known that CAR T-cells persistence is related to antigen exposure or indeed antigen loss in tumor field (89). This demonstrates the importance of antigen exposure for the survival of CAR Tregs.
Safety of CAR Tregs
Tonic Signaling in CAR Tregs
Conventional CAR T-cells are known to be susceptible to tonic signaling. Normally, T-cells will only become activated after stimulation of the TCR. As described earlier, CAR Tregs can be stimulated both through their TCR and CAR. In the case of tonic signaling, the T-cell will become constitutively activated in an antigen-independent manner (90). This can also drive T-cell exhaustion, resulting in reduced T-cell persistence and impaired activity (91). Tonic signaling has not been described extensively for CAR Tregs, but it can be assumed that this may occur similar to conventional CAR T-cells. Importantly, this might lead to systemic immune suppression by CAR Tregs. Alternatively, Tregs may become exhausted. Exhaustion of Tregs is more challenging to assess since these cells express typical exhaustion markers characteristic for exhausted Tconvs, such as CTLA4 and PD-1. Interestingly, it has been shown that those cells with a high density of CAR molecules on the cell surface show CAR clustering, resulting in tonic signaling (92). A way to reduce tonic signaling would be to exclude Tregs with high expression levels of the CAR molecules as suggested by MacDonald (62). Furthermore, the usage of 4-1BB as a costimulatory domain potentially reduces T-cell exhaustion induced by tonic signaling whereas CD28 costimulation augments this (91), thereby affecting the persistence of CAR T-cells. This is in line with findings from Frigault et al., who showed that ligand-independent signaling was dependent on CD28 and the intracellular domain CD3ζ of the CAR construct (92). The occurrence and role of tonic signaling should be included in further research on CAR Tregs.
It is important to note that Tregs that are transduced with CAR molecules still express the endogenous TCR. Studies have shown that CAR Tregs can equally be activated through the TCR as the CAR (26, 27, 62). MacDonald and colleagues demonstrated that stimulation of the endogenous TCR does not negatively affect the suppressive function of CAR Tregs (62). Boardman et al. even argue that CAR Tregs confer more protection when they are concurrently activated in a TCR-dependent and CAR-dependent manner (27). They suggest that direct allorecognition contributes to this effect, thereby limiting this additive effect to the early phase after transplantation. However, genome editing can be used to insert the CAR at the T-cell receptor α constant (TRAC) locus. By disrupting the TRAC locus, endogenous TCRs are not expressed anymore and in addition, this will lead to uniform CAR expression and enhanced potency in T-cells (93).
Phenotypic Stability of CAR Tregs
Tregs are generally characterized by constitutive FoxP3 expression and cell surface expression of CD25 and cytotoxic T lymphocyte antigen 4 (CTLA4). Human CD4+ Tregs can be divided into three phenotypical and functional populations based on CD45RA, CD25 and FoxP3 expression, underlining the heterogeneity of Treg cells. Naïve/resting Tregs are CD45RA-positive and have a low FoxP3 expression, whereas memory/activated Treg do not express CD45RA and have a high FoxP3 expression, while both populations have suppressive properties (94). Finally, there is a population that is not suppressive but produces inflammatory cytokines and is defined by absence of CD45RA and low FoxP3 expression. Plasticity of Tregs refers to the ability of Tregs to express lineage-specific molecules or transcription factors and thereby change migratory or functional capabilities but remaining the identity of FoxP3 Tregs. Treg cell plasticity is probably regulated by several signals and can drive Tregs towards an effector phenotype after expressing similar transcription factors used by Teff cells. Treg instability is therefore of major concern in CAR Treg therapy. One of the concerns is that Treg instability will lead to an acquired cytotoxic ability of these T-cells and/or lead to the outgrowth of conventional CAR T-cells with allogeneic specificity. FoxP3+ Treg are predominantly stable, however, it has been suggested that a minority may become unstable under inflammatory circumstances, thereby modifying their functional properties. Especially naïve thymic-derived Tregs are perceived as very stable due to their epigenetic program (95, 96) and are therefore proposed as the preferred source for Treg engineering. CD4+CD25highFoxP3+CD45RAhigh Tregs can be isolated which offers the possibility of obtaining a stable and pure population of Tregs which can be used for clinical application (97).
To ensure safety, Treg phenotype should not be affected by the transduction or any alterations performed on these cells. Several studies demonstrated that expression levels of key regulatory T-cell markers and/or homing receptors, for example, FoxP3, CTLA-A4, CD39, CD45RA, and CCR7 remain unchanged in CAR Tregs (26, 27, 62, 68). Other studies have established that Tregs preserved their in vitro capacity to suppress via the endogenous TCR (62) or CAR (63). In addition, Treg stability can be monitored by analyzing the Treg-specific demethylated region (TSDR) (98). HLA-A2 CAR Tregs were shown to have preserved a stable demethylation pattern of the TSDR, indicating that these cells remained stable Tregs (26, 62). Importantly, cytotoxicity towards HLA-A2+ epithelial cells could not be detected by HLA-A2 CAR Tregs (27). MacDonald et al. demonstrated a very low proportion of induced cell death in HLA-A2-positive cells when co-cultured with high ratios of CAR Tregs, but similar experiments with the presence of HLA-A2-positive PBMCs show that cytolytic activity was negligible (62). Another study in the setting of murine allogeneic islet transplantation indicated that despite the accumulation of CAR Tregs in allografts, no islet destruction could be observed, which suggested that CAR Tregs are not cytotoxic to allogeneic islets (68). Since these studies have described Treg stability directly after generation and infusion, long-term stability still remains to be investigated in order to establish safety.
Suicide Gene
The safe use of CAR Tregs may be increased with the use of safety switches. Suicide genes can be activated and lead to permissive selective cell death after exposure to an activating molecule. Suicide genes that are expressed on the cell surface and can be activated by mAbs, include RQR8 (99) and truncated epidermal growth factor receptor (tEGFR) (100, 101). RQR8 combines epitopes from CD34 and CD20 antigens and can therefore be recognized by both mAbs as well as by the therapeutic antibody rituximab. The truncated form of EGFR retains its specificity and its epitope can be recognized by pharmaceutical drug anti-EGFR monoclonal antibody, cetuximab. Other suicide genes can be activated by small molecules, for example, inducible caspase 9 (iCasp9) (102, 103) and the herpes simplex virus thymidine kinase (HSV1-tk) (104). iCasp is a modified human caspase 9 gene fused to a human FK506 binding protein (FKBP) which becomes activated after dimerization. Dimerizing small molecules thus leads to activated iCAsp9 and consequently the elimination of the iCasp9-transduced T-cells. iCasp9-expressing T-cells have been used in a clinical trial of five patients that developed GVHD after allogeneic stem-cell transplantation (105). After a single application of the dimerizing drug over 90% of modified cells were eliminated. Furthermore, clinical trials with CAR T-cells equipped with the iCasp9 suicide switch are ongoing to target GD2-positive tumors (NCT01822652, NCT02992210). Additional to the possibility of selective elimination of genetically modified cells, these safety switches enable selection and cell tracking (99, 101). Of note, FKBP, which is part of iCasp, is the target molecule for tacrolimus, which is standard maintenance therapy. This potentially complicates the use for this suicide gene in CAR Tregs when patients receive standard maintenance immunosuppressives, but may not be a problem in a setting where tacrolimus is weaned.
Future Perspectives
Thus far, HLA-A2 specific CAR constructs have been studied in detail in mostly pre-clinical models. Besides HLA-A2 specificity, it would be interesting to study CAR Tregs with different HLA specificities to determine if similar safety, efficiency and effectiveness can be obtained as with HLA-A2 CAR Tregs. Presumably, these characteristics are not deviating from data presented for HLA-A2 CAR Tregs. Several established scFv are described to cross-react with other HLA alleles due to epitope sharing, meaning that in theory they can already be applied to target these specificities. By designing CARs with different specificities, a broad range of the described HLA alleles can be covered. An attractive aspect would be to see if CAR Tregs with different specificities could be combined and whether this would enhance their effect, possibly requiring lower cell doses. From a practical point of view, generating a limited number of CAR specificities that could potentially cover most HLA mismatched transplant situations would be desired. Obviously, this set of CARs will have to be different for different geographical locations. To get an estimate for the Eurotransplant population, we looked into cadaveric transplants in the Eurotransplant Kidney Allocation System (ETKAS) from 2017-2019, and determined the frequency of specific mismatches at HLA-A on the antigen level (Table 1). As can be seen in cumulative percentages, 12 HLA antigen mismatches at the split level make up at least 90% of mismatches at this locus, indicating that when looking at HLA-A in isolation, around 12 CAR Treg specificities would be required to serve 90% of the Eurotransplant population. As CAR Tregs in the setting of transplantation use HLA-specific antibodies as antigen receptor, not HLA antigens, but HLA epitopes are targeted (106). Since epitopes are shared between different HLA antigens, this will reduce the number of required CAR-specificities, as they can cover multiple mismatched HLA molecules. In addition, in a transplantation setting including multiple HLA mismatches, the selection of only specific mismatches to target with CAR Tregs would further reduce the number required to cover the majority of donor-patient mismatches.
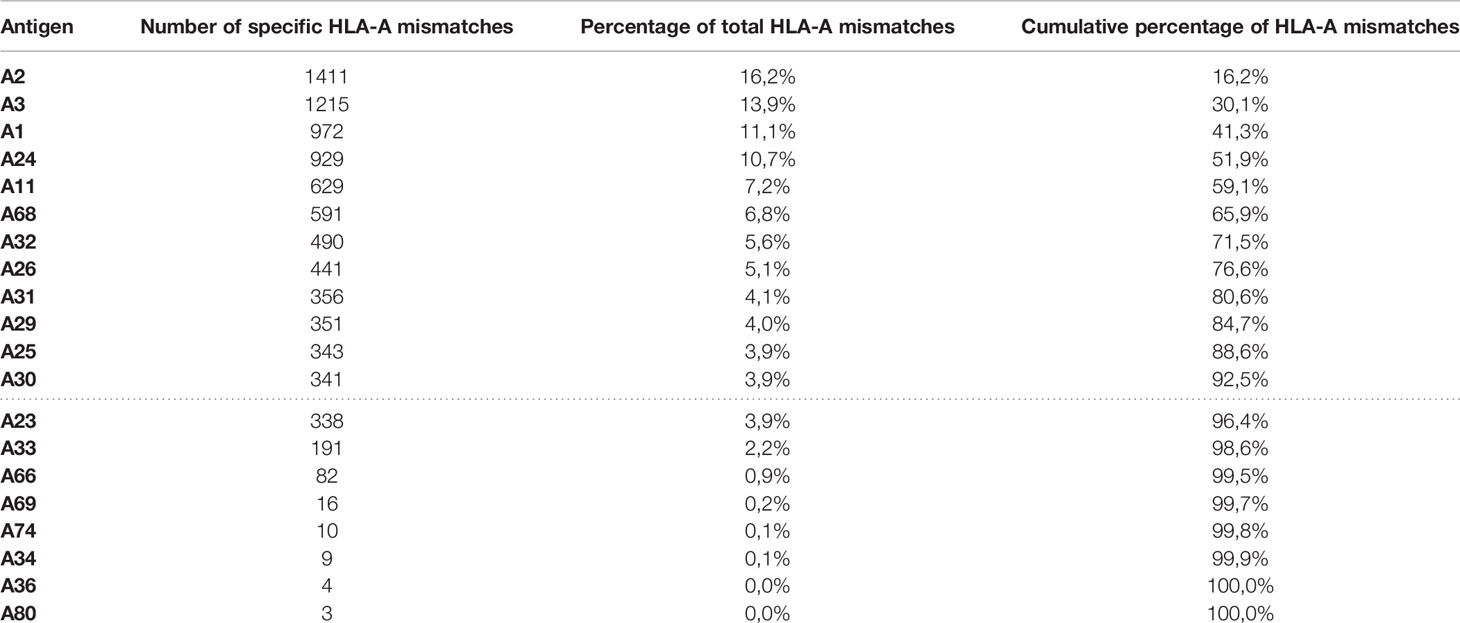
Table 1 Frequency of specific HLA-A antigen mismatches out of 8722 total HLA-A mismatches in ETKAS allocation 2017-2019.
An optimal CAR Treg dosing regimen is pivotal when moving towards clinical trials. The number of CAR Tregs needed to induce a clinically relevant suppressive effect remains to be established. As previously discussed, CAR Tregs will proliferate when they are stimulated by the presence of the antigen, which prolongs their survival. Infusion of low numbers of CAR Tregs might lead to overstimulation of the therapeutic cells which potentially leads to exhaustion. Furthermore, optimal timing for the infusion of CAR Tregs remains to be explored, especially regarding the suboptimal regulation of memory responses. In addition, repeated infusions of CAR Tregs could be investigated to prevent transplant rejection. These challenges are all affected by stability of the CAR Tregs over time and it should be stressed that this is an important aspect of investigation in further clinical trials.
Recently, the first phase I/IIa multicentre study with CAR Tregs started with the recruitment of kidney transplant recipients to test the safety and tolerability of HLA-A2 CAR Tregs (NCT04817774). Living donor kidney transplant patients receiving an HLA-A2+ allograft will receive autologous Tregs that have been expanded ex vivo and transduced with HLA-A2 CARs. Three single ascending dose cohorts of these CAR Tregs and an additional expansion cohort will be investigated. These data will give more insight on further implementation of CAR Tregs in solid organ transplantation. Application of CAR Tregs can be a potent immunotherapy that has the potential to (partially) replace immunosuppression with current immunotherapy strategies. If successful, this might bring the field one step closer to achieving the holy grail of transplantation; graft-specific tolerance. Importantly, the success of such therapy appears to be dependent on the immunological history of the patient. Once sensitized, CAR Treg therapy is no longer successful, at least in preclinical models. Besides this, there are many questions that still need to be resolved; what is the best dosing regimen, what is the required frequency of CAR Treg infusion, what immunosuppressive protocol will give the best result in conjunction with CAR Treg therapy? The potential assays to monitor CAR Treg persistence, longevity, and suppressive capacity should be identified. Finally, stability of the Treg phenotype is pivotal for CAR Treg therapy to become a clinical reality. The development of clinical trials will provide more insight into these questions and the potential utility of CAR Tregs.
Author Contributions
IG, FC, MHMH and SH participated in manuscript writing and editing, GWH performed data analysis. All authors contributed to the article and approved the submitted version.
Funding
IG was supported by research funding from the Dutch Kidney Foundation project code 20OI148.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Halloran PF. Immunosuppressive Drugs for Kidney Transplantation. N Engl J Med (2004) 351(26):2715–29. doi: 10.1056/NEJMra033540
2. Ekberg H, Tedesco-Silva H, Demirbas A, Vítko Š, Nashan B, Gürkan A, et al. Reduced Exposure to Calcineurin Inhibitors in Renal Transplantation. N Engl J Med (2007) 357(25):2562–75. doi: 10.1056/nejmoa067411
3. Ekberg H, Bernasconi C, Tedesco-Silva H, Vítko S, Hugo C, Demirbas A, et al. Calcineurin Inhibitor Minimization in the Symphony Study: Observational Results 3 Years After Transplantation. Am J Transplant (2009) 9(8):1876–85. doi: 10.1111/j.1600-6143.2009.02726.x
4. Miller LW. Cardiovascular Toxicities of Immunosuppressive Agents. Am J Transplant (2002) 2(9):807–18. doi: 10.1034/j.1600-6143.2002.20902.x
5. Dharnidharka VR, Stablein DM, Harmon WE. Post-Transplant Infections Now Exceed Acute Rejection as Cause for Hospitalization: A Report of the Naprtcs1. Am J Transplant (2004) 4(3):384–9. doi: 10.1111/j.1600-6143.2004.00350.x
6. Kasiske BL, Snyder JJ, Gilbertson DT, Wang C. Cancer After Kidney Transplantation in the United States. Am J Transplant (2004) 4(6):905–13. doi: 10.1111/j.1600-6143.2004.00450.x
7. Levitsky J, Feng S. Tolerance in Clinical Liver Transplantation. Hum Immunol (2018) 79(5):283–7. doi: 10.1016/j.humimm.2017.10.007
8. Sagoo P, Perucha E, Sawitzki B, Tomiuk S, Stephens DA, Miqueu P, et al. Development of a Cross-Platform Biomarker Signature to Detect Renal Transplant Tolerance in Humans. J Clin Invest (2010) 120(6):1848–61. doi: 10.1172/jci39922
9. Newell KA, Asare A, Kirk AD, Gisler TD, Bourcier K, Suthanthiran M, et al. Identification of a B Cell Signature Associated With Renal Transplant Tolerance in Humans. J Clin Invest (2010) 120(6):1836–47. doi: 10.1172/jci39933
10. Nadig SN, Więckiewicz J, Wu DC, Warnecke G, Zhang W, Luo S, et al. In Vivo Prevention of Transplant Arteriosclerosis by Ex Vivo–Expanded Human Regulatory T Cells. Nat Med (2010) 16(7):809–13. doi: 10.1038/nm.2154
11. Martínez-Llordella M, Puig-Pey I, Orlando G, Ramoni M, Tisone G, Rimola A, et al. Multiparameter Immune Profiling of Operational Tolerance in Liver Transplantation. Am J Transplant (2007) 7(2):309–19. doi: 10.1111/j.1600-6143.2006.01621.x
12. Ramos HC, Reyes J, Abu-Elmagd K, Zeevi A, Reinsmoen N, Tzakis A, et al. Weaning of Immunosuppression in Long-Term Liver Transplant Recipients. Transplantation (1995) 59(2):212–7. doi: 10.1097/00007890-199501270-00010
13. Mazariegos GV, Reyes J, Marino IR, Demetris AJ, Flynn B, Irish W, et al. Weaning of Immunosuppression in Liver Transplant Recipients12. Transplantation (1997) 63(2):243–9. doi: 10.1097/00007890-199701270-00012
14. Li Y, Koshiba T, Yoshizawa A, Yonekawa Y, Masuda K, Ito A, et al. Analyses of Peripheral Blood Mononuclear Cells in Operational Tolerance After Pediatric Living Donor Liver Transplantation. Am J Transplant (2004) 4(12):2118–25. doi: 10.1111/j.1600-6143.2004.00611.x
15. Sawitzki B, Harden PN, Reinke P, Moreau A, Hutchinson JA, Game DS, et al. Regulatory Cell Therapy in Kidney Transplantation (the One Study): A Harmonised Design and Analysis of Seven Non-Randomised, Single-Arm, Phase 1/2a Trials. Lancet (2020) 395(10237):1627–39. doi: 10.1016/S0140-6736(20)30167-7
16. Kawai T, Cosimi AB, Spitzer TR, Tolkoff-Rubin N, Suthanthiran M, Saidman SL, et al. Hla-Mismatched Renal Transplantation Without Maintenance Immunosuppression. N Engl J Med (2008) 358(4):353–61. doi: 10.1056/nejmoa071074
17. Reinders MEJ, Groeneweg KE, Hendriks SH, Bank JR, Dreyer GJ, Vries APJ, et al. Autologous Bone Marrow-Derived Mesenchymal Stromal Cell Therapy With Early Tacrolimus Withdrawal: The Randomized Prospective, Single-Center, Open-Label Triton Study. Am J Transplant (2021) 21(9):3055–65. doi: 10.1111/ajt.16528
18. Harden PN, Game DS, Sawitzki B, van der Net JB, Hester J, Bushell A, et al. Feasibility, Long-Term Safety, and Immune Monitoring of Regulatory T Cell Therapy in Living Donor Kidney Transplant Recipients. Am J Transplant (2021) 21(4):1603–11. doi: 10.1111/ajt.16395
19. Sakaguchi S, Yamaguchi T, Nomura T, Ono M. Regulatory T Cells and Immune Tolerance. Cell (2008) 133(5):775–87. doi: 10.1016/j.cell.2008.05.009
20. Wan YY, Flavell RA. Regulatory T-Cell Functions Are Subverted and Converted Owing to Attenuated Foxp3 Expression. Nature (2007) 445(7129):766–70. doi: 10.1038/nature05479
21. Hori S, Nomura T, Sakaguchi S. Control of Regulatory T Cell Development by the Transcription Factor Foxp3. Science (2003) 299(5609):1057–61. doi: 10.1126/science.1079490
22. Trzonkowski P, Bieniaszewska M, Juscinska J, Dobyszuk A, Krzystyniak A, Marek N, et al. First-In-Man Clinical Results of the Treatment of Patients With Graft Versus Host Disease With Human Ex Vivo Expanded CD4+Cd25+Cd127- T Regulatory Cells. Clin Immunol (2009) 133(1):22–6. doi: 10.1016/j.clim.2009.06.001
23. Brunstein CG, Miller JS, McKenna DH, Hippen KL, DeFor TE, Sumstad D, et al. Umbilical Cord Blood-Derived T Regulatory Cells to Prevent Gvhd: Kinetics, Toxicity Profile, and Clinical Effect. Blood (2016) 127(8):1044–51. doi: 10.1182/blood-2015-06-653667
24. Roemhild A, Otto NM, Moll G, Abou-El-Enein M, Kaiser D, Bold G, et al. Regulatory T Cells for Minimising Immune Suppression in Kidney Transplantation: Phase I/IIa Clinical Trial. BMJ (2020) 371:m3734. doi: 10.1136/bmj.m3734
25. Chandran S, Tang Q, Sarwal M, Laszik ZG, Putnam AL, Lee K, et al. Polyclonal Regulatory T Cell Therapy for Control of Inflammation in Kidney Transplants. Am J Transplant (2017) 17(11):2945–54. doi: 10.1111/ajt.14415
26. Noyan F, Zimmermann K, Hardtke-Wolenski M, Knoefel A, Schulde E, Geffers R, et al. Prevention of Allograft Rejection by Use of Regulatory T Cells With an Mhc-Specific Chimeric Antigen Receptor. Am J Transplant (2017) 17(4):917–30. doi: 10.1111/ajt.14175
27. Boardman DA, Philippeos C, Fruhwirth GO, Ibrahim MAA, Hannen RF, Cooper D, et al. Expression of a Chimeric Antigen Receptor Specific for Donor HLA Class I Enhances the Potency of Human Regulatory T Cells in Preventing Human Skin Transplant Rejection. Am J Transplant (2017) 17(4):931–43. doi: 10.1111/ajt.14185
28. Brennan TV, Tang Q, Liu F-C, Hoang V, Bi M, Bluestone JA, et al. Requirements for Prolongation of Allograft Survival With Regulatory T Cell Infusion in Lymphosufficient Hosts. J Surg Res (2011) 169(1):e69–75. doi: 10.1016/j.jss.2011.03.021
29. Golshayan D, Jiang S, Tsang J, Garin MI, Mottet C, Lechler RI. In Vitro–Expanded Donor Alloantigen–Specific Cd4+Cd25+ Regulatory T Cells Promote Experimental Transplantation Tolerance. Blood (2007) 109(2):827–35. doi: 10.1182/blood-2006-05-025460
30. Joffre O, Santolaria T, Calise D, Saati TA, Hudrisier D, Romagnoli P, et al. Prevention of Acute and Chronic Allograft Rejection With CD4+CD25+Foxp3+ Regulatory T Lymphocytes. Nat Med (2008) 14(1):88–92. doi: 10.1038/nm1688
31. Sagoo P, Ali N, Garg G, Nestle FO, Lechler RI, Lombardi G. Human Regulatory T Cells With Alloantigen Specificity Are More Potent Inhibitors of Alloimmune Skin Graft Damage Than Polyclonal Regulatory T Cells. Sci Trans Med (2011) 3(83):83ra42–2. doi: 10.1126/scitranslmed.3002076
32. Suchin EJ, Langmuir PB, Palmer E, Sayegh MH, Wells AD, Turka LA. Quantifying the Frequency of Alloreactive T Cells In Vivo: New Answers to an Old Question. J Immunol (2001) 166(2):973–81. doi: 10.4049/jimmunol.166.2.973
33. Tsang JY-S, Tanriver Y, Jiang S, Xue S-A, Ratnasothy K, Chen D, et al. Conferring Indirect Allospecificity on Cd4+Cd25+ Tregs by Tcr Gene Transfer Favors Transplantation Tolerance in Mice. J Clin Invest (2008) 118(11):3619–28. doi: 10.1172/jci33185
34. Hu M, Rogers NM, Li J, Zhang GY, Wang YM, Shaw K, et al. Antigen Specific Regulatory T Cells in Kidney Transplantation and Other Tolerance Settings. Front Immunol (2021) 12:717594. doi: 10.3389/fimmu.2021.717594
35. Dembić Z, Haas W, Weiss S, McCubrey J, Kiefer H, Von Boehmer H, et al. Transfer of Specificity by Murine A and B T-Cell Receptor Genes. Nature (1986) 320(6059):232–8. doi: 10.1038/320232a0
36. Eshhar Z, Waks T, Gross G, Schindler DG. Specific Activation and Targeting of Cytotoxic Lymphocytes Through Chimeric Single Chains Consisting of Antibody-Binding Domains and the Gamma or Zeta Subunits of the Immunoglobulin and T-Cell Receptors. Proc Natl Acad Sci (1993) 90(2):720–4. doi: 10.1073/pnas.90.2.720
37. Brocker T, Karjalainen K. Signals Through T Cell Receptor-Zeta Chain Alone Are Insufficient to Prime Resting T Lymphocytes. J Exp Med (1995) 181(5):1653–9. doi: 10.1084/jem.181.5.1653
38. Krause A, Guo H-F, Latouche J-B, Tan C, Cheung N-KV, Sadelain M. Antigen-Dependent Cd28 Signaling Selectively Enhances Survival and Proliferation in Genetically Modified Activated Human Primary T Lymphocytes. J Exp Med (1998) 188(4):619–26. doi: 10.1084/jem.188.4.619
39. Porter DL, Levine BL, Kalos M, Bagg A, June CH. Chimeric Antigen Receptor–Modified T Cells in Chronic Lymphoid Leukemia. N Engl J Med (2011) 365(8):725–33. doi: 10.1056/nejmoa1103849
40. Abate-Daga D, Lagisetty KH, Tran E, Zheng Z, Gattinoni L, Yu Z, et al. A Novel Chimeric Antigen Receptor Against Prostate Stem Cell Antigen Mediates Tumor Destruction in a Humanized Mouse Model of Pancreatic Cancer. Hum Gene Ther (2014) 25(12):1003–12. doi: 10.1089/hum.2013.209
41. Hombach AA, Rappl G, Abken H. Arming Cytokine-Induced Killer Cells With Chimeric Antigen Receptors: CD28 Outperforms Combined CD28–Ox40 “Super-Stimulation”. Mol Ther (2013) 21(12):2268–77. doi: 10.1038/mt.2013.192
42. Chmielewski M, Hombach AA, Abken H. Of Cars and Trucks: Chimeric Antigen Receptor (Car) T Cells Engineered With an Inducible Cytokine to Modulate the Tumor Stroma. Immunol Rev (2014) 257(1):83–90. doi: 10.1111/imr.12125
43. Maude SL, Teachey DT, Porter DL, Grupp SA. CD19-Targeted Chimeric Antigen Receptor T-Cell Therapy for Acute Lymphoblastic Leukemia. Blood (2015) 125(26):4017–23. doi: 10.1182/blood-2014-12-580068
44. Neelapu SS, Locke FL, Bartlett NL, Lekakis LJ, Miklos DB, Jacobson CA, et al. Axicabtagene Ciloleucel Car T-Cell Therapy in Refractory Large B-Cell Lymphoma. N Engl J Med (2017) 377(26):2531–44. doi: 10.1056/NEJMoa1707447
45. Elinav E, Waks T, Eshhar Z. Redirection of Regulatory T Cells With Predetermined Specificity for the Treatment of Experimental Colitis in Mice. Gastroenterology (2008) 134(7):2014–24. doi: 10.1053/j.gastro.2008.02.060
46. De Paula Pohl A, Schmidt A, Zhang AH, Maldonado T, Konigs C, Scott DW. Engineered Regulatory T Cells Expressing Myelin-Specific Chimeric Antigen Receptors Suppress Eae Progression. Cell Immunol (2020) 358:104222. doi: 10.1016/j.cellimm.2020.104222
47. Fransson M, Piras E, Burman J, Nilsson B, Essand M, Lu B, et al. Car/Foxp3-Engineered T Regulatory Cells Target the CNS and Suppress Eae Upon Intranasal Delivery. J Neuroinflamm (2012) 9(1):112. doi: 10.1186/1742-2094-9-112
48. Tenspolde M, Zimmermann K, Weber LC, Hapke M, Lieber M, Dywicki J, et al. Regulatory T Cells Engineered With a Novel Insulin-Specific Chimeric Antigen Receptor as a Candidate Immunotherapy for Type 1 Diabetes. J Autoimmun (2019) 103:102289. doi: 10.1016/j.jaut.2019.05.017
49. Skuljec J, Chmielewski M, Happle C, Habener A, Busse M, Abken H, et al. Chimeric Antigen Receptor-Redirected Regulatory T Cells Suppress Experimental Allergic Airway Inflammation, a Model of Asthma. Front Immunol (2017) 8:1125. doi: 10.3389/fimmu.2017.01125
50. Rana J, Perry DJ, Kumar SRP, Muñoz-Melero M, Saboungi R, Brusko TM, et al. Car- and Truc-Redirected Regulatory T Cells Differ in Capacity to Control Adaptive Immunity to Fviii. Mol Ther (2021) 29(9):2660–76. doi: 10.1016/j.ymthe.2021.04.034
51. Pohl AD, Venkatesha SH, Zhang AH, Scott DW. Suppression of Fviii-Specific Memory B Cells by Chimeric Bar Receptor-Engineered Natural Regulatory T Cells. Front Immunol (2020) 11:693. doi: 10.3389/fimmu.2020.00693
52. Mukhatayev Z, Dellacecca ER, Cosgrove C, Shivde R, Jaishankar D, Pontarolo-Maag K, et al. Antigen Specificity Enhances Disease Control by Tregs in Vitiligo. Front Immunol (2020) 11:581433. doi: 10.3389/fimmu.2020.581433
53. Whittington KB, Prislovsky A, Beaty J, Albritton L, Radic M, Rosloniec EF. CD8+ T Cells Expressing an HLA-DR1 Chimeric Antigen Receptor Target Autoimmune CD4+ T Cells in an Antigen-Specific Manner and Inhibit the Development of Autoimmune Arthritis. J Immunol (2022) 208(1):16–26. doi: 10.4049/jimmunol.2100643
54. Dawson NAJ, Rosado-Sanchez I, Novakovsky GE, Fung VCW, Huang Q, McIver E, et al. Functional Effects of Chimeric Antigen Receptor Co-Receptor Signaling Domains in Human Regulatory T Cells. Sci Transl Med (2020) 12(557):eaaz3866. doi: 10.1126/scitranslmed.aaz3866
55. Imura Y, Ando M, Kondo T, Ito M, Yoshimura A. Cd19-Targeted Car Regulatory T Cells Suppress B Cell Pathology Without Gvhd. JCI Insight (2020) 5(14):e136185. doi: 10.1172/jci.insight.136185
56. Martin A, Daris M, Johnston JA, Cui J. Hla-A*02:01-Directed Chimeric Antigen Receptor/Forkhead Box P3-Engineered Cd4+ T Cells Adopt a Regulatory Phenotype and Suppress Established Graft-Versus-Host Disease. Cytotherapy (2021) 23(2):131–6. doi: 10.1016/j.jcyt.2020.10.002
57. Bezie S, Meistermann D, Boucault L, Kilens S, Zoppi J, Autrusseau E, et al. Ex Vivo Expanded Human Non-Cytotoxic Cd8(+)Cd45rc(Low/-) Tregs Efficiently Delay Skin Graft Rejection and Gvhd in Humanized Mice. Front Immunol (2017) 8:2014:2014. doi: 10.3389/fimmu.2017.02014
58. Bézie S, Charreau B, Vimond N, Lasselin J, Gérard N, Nerrière-Daguin V, et al. Human Cd8+ Tregs Expressing a Mhc-Specific Car Display Enhanced Suppression of Human Skin Rejection and Gvhd in Nsg Mice. Blood Adv (2019) 3(22):3522–38. doi: 10.1182/bloodadvances.2019000411
59. Marcucci KT, Jadlowsky JK, Hwang W-T, Suhoski-Davis M, Gonzalez VE, Kulikovskaya I, et al. Retroviral and Lentiviral Safety Analysis of Gene-Modified T Cell Products and Infused Hiv and Oncology Patients. Mol Ther (2018) 26(1):269–79. doi: 10.1016/j.ymthe.2017.10.012
60. Boroughs AC, Larson RC, Choi BD, Bouffard AA, Riley LS, Schiferle E, et al. Chimeric Antigen Receptor Costimulation Domains Modulate Human Regulatory T Cell Function. JCI Insight (2019) 4(8):e126194. doi: 10.1172/jci.insight.126194
61. Burt C, Cryer C, Fuggle S, Little AM, Dyer P. Hla-A, -B, -Dr Allele Group Frequencies in 7007 Kidney Transplant List Patients in 27 Uk Centres. Int J Immunogenet (2013) 40(3):209–15. doi: 10.1111/iji.12000
62. MacDonald KG, Hoeppli RE, Huang Q, Gillies J, Luciani DS, Orban PC, et al. Alloantigen-Specific Regulatory T Cells Generated With a Chimeric Antigen Receptor. J Clin Invest (2016) 126(4):1413–24. doi: 10.1172/Jci82771
63. Sicard A, Lamarche C, Speck M, Wong M, Rosado-Sanchez I, Blois M, et al. Donor-Specific Chimeric Antigen Receptor Tregs Limit Rejection in Naive But Not Sensitized Allograft Recipients. Am J Transplant (2020) 20(6):1562–73. doi: 10.1111/ajt.15787
64. Dawson NAJ, Lamarche C, Hoeppli RE, Bergqvist P, Fung VCW, McIver E, et al. Systematic Testing and Specificity Mapping of Alloantigen-Specific Chimeric Antigen Receptors in Regulatory T Cells. JCI Insight (2019) 4(6):e123672. doi: 10.1172/jci.insight.123672
65. Watkins NA, Brown C, Hurd C, Navarrete C, Ouwehand WH. The Isolation and Characterisation of Human Monoclonal HLA-A2 Antibodies From an Immune V Gene Phage Display Library. Tissue Antigens (2000) 55(3):219–28. doi: 10.1034/j.1399-0039.2000.550305.x
66. Mulder A, Kardol M, Regan J, Buelow R, Claas F. Reactivity of Twenty-Two Cytotoxic Human Monoclonal Hla Antibodies Towards Soluble Hla Class I in an Enzyme-Linked Immunosorbent Assay (Pra-Stat). Hum Immunol (1997) 56(1-2):106–13. doi: 10.1016/s0198-8859(97)00146-8
67. Mulder A, Eijsink C, Kardol MJ, Franke-Van Dijk MEI, van der Burg SH, Kester M, et al. Identification, Isolation, and Culture of HLA-A2-Specific B Lymphocytes Using Mhc Class I Tetramers. J Immunol (2003) 171(12):6599–603. doi: 10.4049/jimmunol.171.12.6599
68. Muller YD, Ferreira LMR, Ronin E, Ho P, Nguyen V, Faleo G, et al. Precision Engineering of an Anti-Hla-A2 Chimeric Antigen Receptor in Regulatory T Cells for Transplant Immune Tolerance. Front Immunol (2021) 12:686439. doi: 10.3389/fimmu.2021.686439
69. Kramer CSM, Franke-Van Dijk MEI, Bakker KH, Uyar-Mercankaya M, Karahan GE, Roelen DL, et al. Generation and Reactivity Analysis of Human Recombinant Monoclonal Antibodies Directed Against Epitopes on Hla-Dr. Am J Transplant (2020) 20(12):3341–53. doi: 10.1111/ajt.15950
70. Bezstarosti S, Bakker KH, Kramer CSM, de Fijter JW, Reinders MEJ, Mulder A, et al. A Comprehensive Evaluation of the Antibody-Verified Status of Eplets Listed in the HLA Epitope Registry. Front Immunol (2022) 12:800946. doi: 10.3389/fimmu.2021.800946
71. Mulder A, Kardol MJ, Arn JS, Eijsink C, Franke MEI, Schreuder GMT, et al. Human Monoclonal HLA Antibodies Reveal Interspecies Crossreactive Swine Mhc Class I Epitopes Relevant for Xenotransplantation. Mol Immunol (2010) 47(4):809–15. doi: 10.1016/j.molimm.2009.10.004
72. Zhang N, Schroppel B, Lal G, Jakubzick C, Mao X, Chen D, et al. Regulatory T Cells Sequentially Migrate From Inflamed Tissues to Draining Lymph Nodes to Suppress the Alloimmune Response. Immunity (2009) 30(3):458–69. doi: 10.1016/j.immuni.2008.12.022
73. Mohseni YR, Saleem A, Tung SL, Dudreuilh C, Lang C, Peng Q, et al. Chimeric Antigen Receptor-Modified Human Regulatory T Cells That Constitutively Express Il-10 Maintain Their Phenotype and Are Potently Suppressive. Eur J Immunol (2021) 51(10):2522–30. doi: 10.1002/eji.202048934
74. Yang J, Brook MO, Carvalho-Gaspar M, Zhang J, Ramon HE, Sayegh MH, et al. Allograft Rejection Mediated by Memory T Cells Is Resistant to Regulation. Proc Natl Acad Sci (2007) 104(50):19954–9. doi: 10.1073/pnas.0704397104
75. Macedo C, Orkis EA, Popescu I, Elinoff BD, Zeevi A, Shapiro R, et al. Contribution of Naïve and Memory T-Cell Populations to the Human Alloimmune Response. Am J Transplant (2009) 9(9):2057–66. doi: 10.1111/j.1600-6143.2009.02742.x
76. Felix NJ, Allen PM. Specificity of T-Cell Alloreactivity. Nat Rev Immunol (2007) 7(12):942–53. doi: 10.1038/nri2200
77. Golshayan D, Wyss JC, Buckland M, Hernandez-Fuentes M, Lechler RI. Differential Role of Naïve and Memory CD4+ T-Cell Subsets in Primary Alloresponses. Am J Transplant (2010) 10(8):1749–59. doi: 10.1111/j.1600-6143.2010.03180.x
78. Karahan GE, Claas FHJ, Heidt S. Heterologous Immunity of Virus-Specific T Cells Leading to Alloreactivity: Possible Implications for Solid Organ Transplantation. Viruses (2021) 13(12):2359. doi: 10.3390/v13122359
79. Amir AL, D’Orsogna LJA, Roelen DL, van Loenen MM, Hagedoorn RS, de Boer R, et al. Allo-Hla Reactivity of Virus-Specific Memory T Cells Is Common. Blood (2010) 115(15):3146–57. doi: 10.1182/blood-2009-07-234906
80. Heidt S, Claas FHJ. Transplantation in Highly Sensitized Patients: Challenges and Recommendations. Expert Rev Clin Immunol (2018) 14(8):673–9. doi: 10.1080/1744666x.2018.1498335
81. Guedan S, Posey AD, Shaw C, Wing A, Da T, Patel PR, et al. Enhancing Car T Cell Persistence Through Icos and 4-1bb Costimulation. JCI Insight (2018) 3(1):e96976. doi: 10.1172/jci.insight.96976
82. Levings MK, Sangregorio R, Roncarolo M-G. Human Cd25+Cd4+ T Regulatory Cells Suppress Naive and Memory T Cell Proliferation and Can Be Expanded in Vitro Without Loss of Function. J Exp Med (2001) 193(11):1295–302. doi: 10.1084/jem.193.11.1295
83. Fontenot JD, Rasmussen JP, Gavin MA, Rudensky AY. A Function for Interleukin 2 in Foxp3-Expressing Regulatory T Cells. Nat Immunol (2005) 6(11):1142–51. doi: 10.1038/ni1263
84. Ratnasothy K, Jacob J, Tung S, Boardman D, Lechler RI, Sanchez-Fueyo A, et al. Il-2 Therapy Preferentially Expands Adoptively Transferred Donor-Specific Tregs Improving Skin Allograft Survival. Am J Transplant (2019) 19(7):2092–100. doi: 10.1111/ajt.15306
85. Whitehouse G, Gray E, Mastoridis S, Merritt E, Kodela E, Yang JHM, et al. Il-2 Therapy Restores Regulatory T-Cell Dysfunction Induced by Calcineurin Inhibitors. Proc Natl Acad Sci (2017) 114(27):7083–8. doi: 10.1073/pnas.1620835114
86. Hu B, Ren J, Luo Y, Keith B, Young RM, Scholler J, et al. Augmentation of Antitumor Immunity by Human and Mouse Car T Cells Secreting Il-18. Cell Rep (2017) 20(13):3025–33. doi: 10.1016/j.celrep.2017.09.002
87. Pegram HJ, Lee JC, Hayman EG, Imperato GH, Tedder TF, Sadelain M, et al. Tumor-Targeted T Cells Modified to Secrete Il-12 Eradicate Systemic Tumors Without Need for Prior Conditioning. Blood (2012) 119(18):4133–41. doi: 10.1182/blood-2011-12-400044
88. Hoyos V, Savoldo B, Quintarelli C, Mahendravada A, Zhang M, Vera J, et al. Engineering CD19-Specific T Lymphocytes With Interleukin-15 and a Suicide Gene to Enhance Their Anti-Lymphoma/Leukemia Effects and Safety. Leukemia (2010) 24(6):1160–70. doi: 10.1038/leu.2010.75
89. Shah NN, Fry TJ. Mechanisms of Resistance to Car T Cell Therapy. Nat Rev Clin Oncol (2019) 16(6):372–85. doi: 10.1038/s41571-019-0184-6
90. Ajina A, Maher J. Strategies to Address Chimeric Antigen Receptor Tonic Signaling. Mol Cancer Ther (2018) 17(9):1795–815. doi: 10.1158/1535-7163.mct-17-1097
91. Long AH, Haso WM, Shern JF, Wanhainen KM, Murgai M, Ingaramo M, et al. 4-1bb Costimulation Ameliorates T Cell Exhaustion Induced by Tonic Signaling of Chimeric Antigen Receptors. Nat Med (2015) 21(6):581–90. doi: 10.1038/nm.3838
92. Frigault MJ, Lee J, Basil MC, Carpenito C, Motohashi S, Scholler J, et al. Identification of Chimeric Antigen Receptors That Mediate Constitutive or Inducible Proliferation of T Cells. Cancer Immunol Res (2015) 3(4):356–67. doi: 10.1158/2326-6066.CIR-14-0186
93. Eyquem J, Mansilla-Soto J, Giavridis T, van der Stegen SJC, Hamieh M, Cunanan KM, et al. Targeting a Car to the Trac Locus With Crispr/Cas9 Enhances Tumour Rejection. Nature (2017) 543(7643):113–7. doi: 10.1038/nature21405
94. Miyara M, Yoshioka Y, Kitoh A, Shima T, Wing K, Niwa A, et al. Functional Delineation and Differentiation Dynamics of Human CD4+ T Cells Expressing the Foxp3 Transcription Factor. Immunity (2009) 30(6):899–911. doi: 10.1016/j.immuni.2009.03.019
95. Schmidl C, Delacher M, Huehn J, Feuerer M. Epigenetic Mechanisms Regulating T-Cell Responses. J Allergy Clin Immunol (2018) 142(3):728–43. doi: 10.1016/j.jaci.2018.07.014
96. Arroyo Hornero R, Betts GJ, Sawitzki B, Vogt K, Harden PN, Wood KJ. Cd45ra Distinguishes Cd4+Cd25+Cd127–/Low Tsdr Demethylated Regulatory T Cell Subpopulations With Differential Stability and Susceptibility to Tacrolimus-Mediated Inhibition of Suppression. Transplantation (2017) 101(2):302–9. doi: 10.1097/tp.0000000000001278
97. Fritsche E, Volk H-D, Reinke P, Abou-El-Enein M. Toward an Optimized Process for Clinical Manufacturing of Car-Treg Cell Therapy. Trends Biotechnol (2020) 38(10):1099–112. doi: 10.1016/j.tibtech.2019.12.009
98. Floess S, Freyer J, Siewert C, Baron U, Olek S, Polansky J, et al. Epigenetic Control of the Foxp3 Locus in Regulatory T Cells. PloS Biol (2007) 5(2):e38. doi: 10.1371/journal.pbio.0050038
99. Philip B, Kokalaki E, Mekkaoui L, Thomas S, Straathof K, Flutter B, et al. A Highly Compact Epitope-Based Marker/Suicide Gene for Easier and Safer T-Cell Therapy. Blood (2014) 124(8):1277–87. doi: 10.1182/blood-2014-01-545020
100. Paszkiewicz PJ, Fräßle SP, Srivastava S, Sommermeyer D, Hudecek M, Drexler I, et al. Targeted Antibody-Mediated Depletion of Murine Cd19 Car T Cells Permanently Reverses B Cell Aplasia. J Clin Invest (2016) 126(11):4262–72. doi: 10.1172/jci84813
101. Wang X, Chang W-C, Wong CW, Colcher D, Sherman M, Ostberg JR, et al. A Transgene-Encoded Cell Surface Polypeptide for Selection, In Vivo Tracking, and Ablation of Engineered Cells. Blood (2011) 118(5):1255–63. doi: 10.1182/blood-2011-02-337360
102. Zhou X, Dotti G, Krance RA, Martinez CA, Naik S, Kamble RT, et al. Inducible Caspase-9 Suicide Gene Controls Adverse Effects From Alloreplete T Cells After Haploidentical Stem Cell Transplantation. Blood (2015) 125(26):4103–13. doi: 10.1182/blood-2015-02-628354
103. Straathof KC, Pulè MA, Yotnda P, Dotti G, Vanin EF, Brenner MK, et al. An Inducible Caspase 9 Safety Switch for T-Cell Therapy. Blood (2005) 105(11):4247–54. doi: 10.1182/blood-2004-11-4564
104. Caruso M, Panis Y, Gagandeep S, Houssin D, Salzmann JL, Klatzmann D. Regression of Established Macroscopic Liver Metastases After in Situ Transduction of a Suicide Gene. Proc Natl Acad Sci (1993) 90(15):7024–8. doi: 10.1073/pnas.90.15.7024
105. Di Stasi A, Tey S-K, Dotti G, Fujita Y, Kennedy-Nasser A, Martinez C, et al. Inducible Apoptosis as a Safety Switch for Adoptive Cell Therapy. N Engl J Med (2011) 365(18):1673–83. doi: 10.1056/nejmoa1106152
106. Bezstarosti S, Kramer CSM, Claas FHJ, De Fijter JW, Reinders MEJ, Heidt S. Implementation of Molecular Matching in Transplantation Requires Further Characterization of Both Immunogenicity and Antigenicity of Individual Hla Epitopes. Hum Immunol (2022) 83(3):256–63. doi: 10.1016/j.humimm.2021.12.002
Keywords: CAR (chimeric antigen receptor), Tregs (regulatory T cells), transplantation, antigen specificity, cellular therapy, tolerance
Citation: Gille I, Claas FHJ, Haasnoot GW, Heemskerk MHM and Heidt S (2022) Chimeric Antigen Receptor (CAR) Regulatory T-Cells in Solid Organ Transplantation. Front. Immunol. 13:874157. doi: 10.3389/fimmu.2022.874157
Received: 11 February 2022; Accepted: 01 April 2022;
Published: 26 May 2022.
Edited by:
Effie Wang Petersdorf, Fred Hutchinson Cancer Research Center, United StatesReviewed by:
Thomas Wekerle, Medical University of Vienna, AustriaSuman Mitra, INSERM UMR1277 Hétérogénéité, Plasticité et Résistance aux Thérapies Anticancéreuses (CANTHER), France
Copyright © 2022 Gille, Claas, Haasnoot, Heemskerk and Heidt. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sebastiaan Heidt, Uy5IZWlkdEBsdW1jLm5s