 Nahir Garabatos
Nahir Garabatos Pere Santamaria
Pere Santamaria
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 27 April 2022
Sec. Mucosal Immunity
Volume 13 - 2022 | https://doi.org/10.3389/fimmu.2022.873607
This article is part of the Research Topic Mucosal Microbiota Specific T and B cells: From Homeostasis to Diseases View all 6 articles
The gut microbiota plays a major role in the developmental biology and homeostasis of cells belonging to the adaptive and innate arms of the immune system. Alterations in its composition, which are known to be regulated by both genetic and environmental factors, can either promote or suppress the pathogenic processes underlying the development of various autoimmune diseases, including inflammatory bowel disease, multiple sclerosis, systemic lupus erythematosus, type 1 diabetes and rheumatoid arthritis, to just name a few. Cross-recognition of gut microbial antigens by autoreactive T cells as well as gut microbe-driven alterations in the activation and homeostasis of effector and regulatory T cells have been implicated in this process. Here, we summarize our current understanding of the positive and negative associations between alterations in the composition of the gut microbiota and the development of various autoimmune disorders, with a special emphasis on antigenic mimicry.
Our natural anatomic barriers, including the skin and mucous membranes, are colonized by billions of microorganisms that live in a symbiotic relationship with the host. The gut microbiota, for example, is composed of different species of commensal bacteria, fungi, viruses and archaeas. During natural evolution, the host and the commensal microorganisms that colonize it have co-evolved to develop complex relationships that impact numerous host biological processes, including immune system homeostasis. Multiple gut microbial species, dietary compounds and/or microbial metabolites contribute to these processes. Gut dysbiosis or disruption of the gut barrier function can trigger a loss of tolerance to gut microbial antigens, eliciting immune responses that can potentially promote not only local inflammation, but also distal autoimmune phenomena. Here, we review our current understanding of the positive and negative associations between alterations in the composition of the gut microbiota and autoimmunity, including known examples of antigenic mimicry. Altogether, this information paints a complex landscape that exposes knowledge gaps and research opportunities.
The crosstalk between the immune system and the gut microbiome begins in the immediate postnatal period. The host immune system matures during the first few years of life in a dynamic relationship with the gut microbiome, leading to a state of equilibrium at around 3 years of age (1, 2). The largest microbial colonization of the gut occurs during and immediately after birth (3), and is impacted by factors such as the delivery mode (4) and breast feeding (5, 6). It has been recently shown that a weaning reaction to the microbiota, leading to the generation of RORgamma(+) Treg cells via bacterial and dietary metabolites, including short-chain fatty acids (SCFAs) and retinoic acid, is required for resistance to immunopathologies in the adult, such as colitis and allergic inflammation (7). Related to this, intestinal secretion of the antimicrobial peptide cathelicidin upon exposure to commensal bacteria has been reported to shape a protective neonatal gut microbiota against pancreatic autoimmunity (8).
Maintenance of immune tolerance against the gut microbiota is regulated by complex processes that are orchestrated in the gut-associated lymphoid tissue (GALT). GALT-associated innate immune cells can distinguish between potentially pathogenic microbial components and their harmless commensal counterparts by recognizing pathogen-associated pattern recognition receptors (PRRs), which ultimately lead to the activation of antigen-specific CD4+ and CD8+ T cell effectors. In the healthy steady state, the GALT-associated B cells produce gut microbial antigen-specific IgAs to suppress mucosal penetration by commensals, hence the induction of potentially harmful local immune responses by effector T cells (9). This process is supported by both dendritic cells and T-follicular helper (TFH) cells. In addition, Peyer´s patch-associated Th17 cells promote Ig class switching and production of soluble IgA via IL-21 (10). Local induced FoxP3+ Treg cells (iTregs) also contribute to the maintenance of normal immune homeostasis, by both suppressing pathogenic effector T cell responses and promoting IgA production (11). Interestingly, there is evidence suggesting that most of the TFH cells in the Peyer’s patches arise from pre-existing Treg cells and Th17-type cells (12, 13).
Commensal bacteria and metabolites play an active role in the development and regulation of adaptive immune responses in the gut (Figure 1). In mice, segmented filamentous bacteria (SFB) living in the small intestine help promote the induction of protective, pathogen-specific Th17 responses (14, 15) by triggering the intestinal production of serum amyloid A protein (SAA) and reactive oxygen species (ROS) (16). In humans, the Bifidobacterium B. adolescentis might play a similar role (17). Other commensals promote the activation of Th17-suppressing Treg cell responses by eliciting the production of intestinal thymic stromal lymphopoietin (TSLP) (18). Gut bacteria can also modulate Th1 responses to promote gut microbial tolerance. In mice, for example, gut microbes can suppress local Th1 cell responses via CX3CR1+ mononuclear phagocytes to favor a local tolerant state (19). In contrast, when Klebsiella, which is normally found in the oral cavity, ectopically colonizes the gut, it activates CD11b–CD103+ dendritic cells (DCs), promoting the activation of pro-inflammatory Th1 cell responses (20).
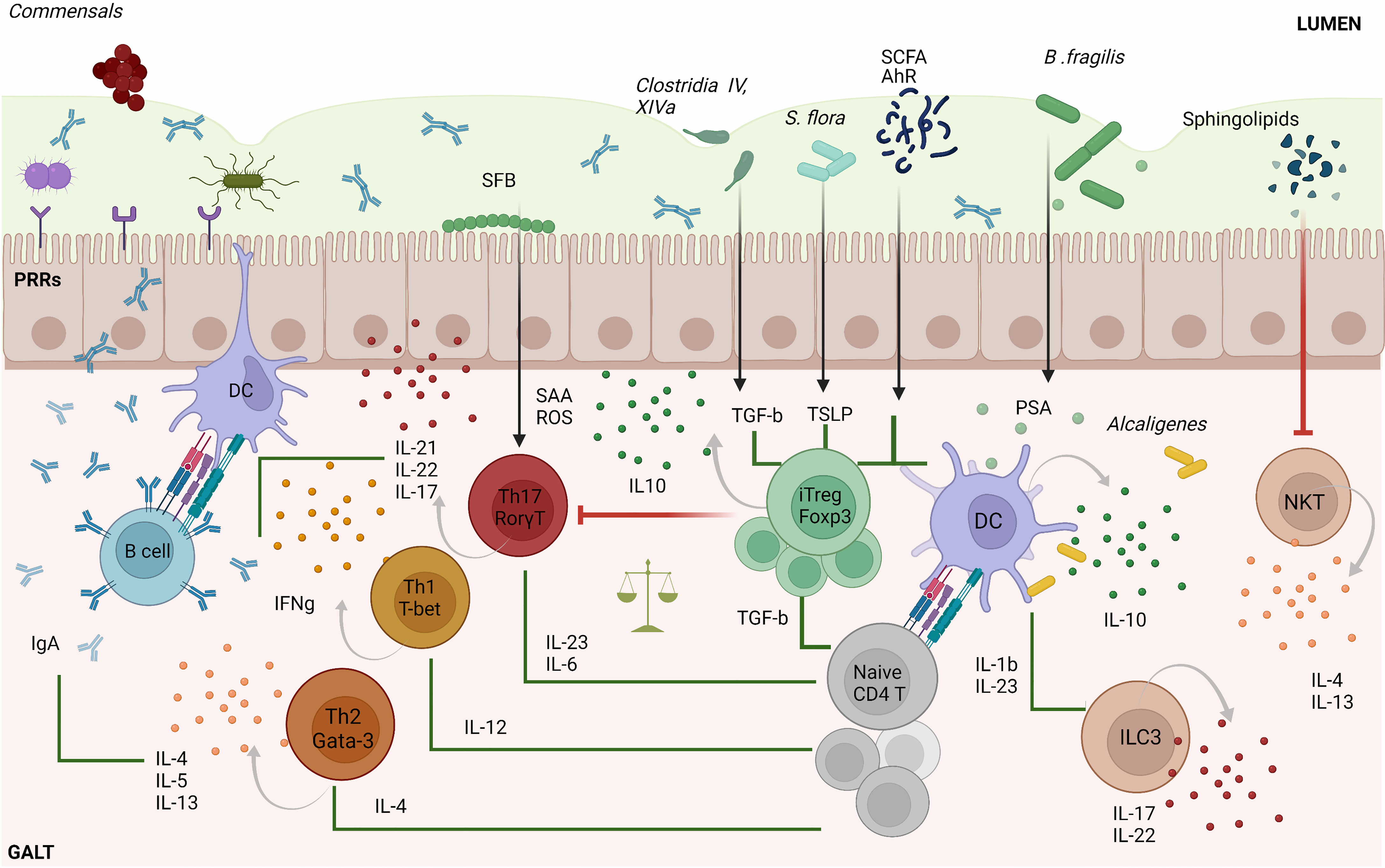
Figure 1 Microbiota–T cell crosstalk in the maintenance of gut homeostasis. Commensal bacteria can trigger pattern recognition receptors (PPRs) on enterocytes and/or activate antigen specific CD4+T cell responses via dendritic cells (DC). Naïve CD4+ T cells can differentiate into four major cell types: Th1, Th2, Th17 and Tregs. The differentiation of each Th type requires specific transcription factors and cytokine sets, as shown in the figure. Th1 cells play an important role in eliminating intracellular pathogens while Th2 control parasitic infections and extracellular pathogens trough the induction of antibody responses. The primary role of Th17 cells is to control infection, but also contributes to intestinal homeostasis by inducing protective IgA responses. SFB commensal bacteria promote gut Th17 cell responses by triggering the intestinal production of SAA and ROS. iTreg cells play a key role in controlling Th cell responses and in maintaining gut immune homeostasis. Several commensal bacteria such as Clostridia spp., dietary compounds (SCFA) and AhR ligands participate in the maintenance of tolerance by inducing gut Treg cell responses or by imprinting tolerogenic features on DCs, as is the case for Alcaligenes spp. Other immune cell types such as invariant natural killer T-cells (iNKT) are suppressed and controlled by bacterial sphingolipids preventing intestinal pro-inflammatory responses. In addition, type 3 innate lymphoid cells (ILC3) promote protective Th17 responses via IL-22 and IL-17. The types of bacteria implicated in particular T cell differentiation pathways as well as metabolites are indicated in the figure. SFB, segmented filamentous bacteria; AhR, Aryl hydrocarbon receptor; TGF-β, transforming growth factor-beta; SCFA, short-chain fatty acids; PSA, polysaccharide A; SAA, serum amyloid A protein; ROS, reactive oxygen species; GALT, gut-associated lymphoid tissue; TSLP, thymic stromal lymphopoietin; iTreg, induced regulatory T cell.
Other gut bacteria contribute to this process by inducing local Treg cell responses. Clostridium clusters IV and XIVa stimulate the secretion of transforming growth factor (TGF)-β by intestinal epithelial cells, promoting the differentiation and expansion of Treg cells in the colonic lamina propria (21). Likewise, F. prausnitzii induces the formation of T-regulatory type 1 (TR1)-like cells via TLR4-mediated activation of DCs (22). Moreover, R. hominis has been associated with activation of FoxP3+ Treg cells in the lamina propria (23). Commensal microbial metabolites also contribute to promoting local Treg cell responses. Eubacterium spp. produce SCFAs, mainly butyrate, that contribute to local immune homeostasis via several mechanisms (24–29). Microbial polysaccharides (PS) have also been associated with this process (30). In mice, for example, B. fragilis promotes the formation of tolerogenic CD103+ DCs and IL-10-producing FoxP3+ Treg cells via PSA-TLR2 signaling (31, 32).
Maintenance of gut microbial tolerance also involves the induction and regulation of other types of T cell responses. Recently, 11 bacterial strains were identified in healthy humans that induce protective IFNγ-producing CD8+ T cell responses in the intestine (33). In mice, invariant natural killer T-cells (iNKT), which bridge the innate and adaptive immune systems, have also been shown to be regulated by the host microbiota. B. fragilis sphingolipids, for example, modulate host colonic iNKT cell homeostasis, promoting gut barrier integrity (34).
Although most gut microbes reside in the lumen or on the gut epithelium, some commensal bacteria such as Alcaligenes spp., Achromobacter spp., Bordetella spp. and Ochrobactrum spp, exist in lymphoid follicles, Peyer’s patches and mesenteric lymph nodes of both healthy mice and humans (35). These bacteria trigger local interleukin-10 (IL-10) and IL-22 production from DCs and Type 3 Innate Lymphoid cells (ILC3), respectively. Whereas IL-10 suppresses the development of pro-inflammatory Th17 responses against commensals, IL-22 signaling favors bacterial colonization of lymphoid tissues.
Thus, the microbiota and the host immune system co-exist in a unique symbiotic relationship, where the host fosters gut colonization by microbes that are beneficial to the host, and/or help it suppress immune responses against these commensals.
Dysregulation of tolerance to gut microbes can lead to the development of intestinal inflammatory processes, such as Crohn’s disease (CD) and ulcerative colitis (UC). Changes in the lifestyle of individuals living in industrialized societies during the last century have transformed how humans are exposed to environmental microbes. Excessive use of antibiotics, increase in hygiene, changes in childbirth mode and maternal breast-feeding patterns and poor nutritional habits have conspired with normal genetic determinants to increase the incidence of various immune-mediated diseases, betraying the beneficial role that these genetic determinants have on the host in the absence of these behavioral/societal changes (Figure 2). This putative association between decreased exposure to microbes and the rapid rise in the incidence and prevalence of chronic inflammatory disorders in industrialized societies has been conceptualized in the “hygiene hypothesis” (36). Various lines of experimental evidence in rodents support this hypothesis. Nonobese diabetic (NOD) mice as well as Biobreeding (BB) rats housed in conventional, non-specific pathogen-free (SPF) conditions develop a significantly decreased incidence of type 1 diabetes (T1D). In addition, infection of these rodent strains with various pathogens suppresses their autoimmune disease proclivity (37–39). Similar effects have been observed in systemic lupus erythematosus (SLE)-prone (NZB x NZW) F1 mice, where infection with P. berghei suppressed the development of lupus nephritis and prolonged survival (40), and in murine models of allergy, where microbial pathogen exposure suppresses disease. Although the precise mechanisms underlying these associations remain unclear, it has been suggested that excessive “hygiene” somehow interferes with adequate development of Treg cells.
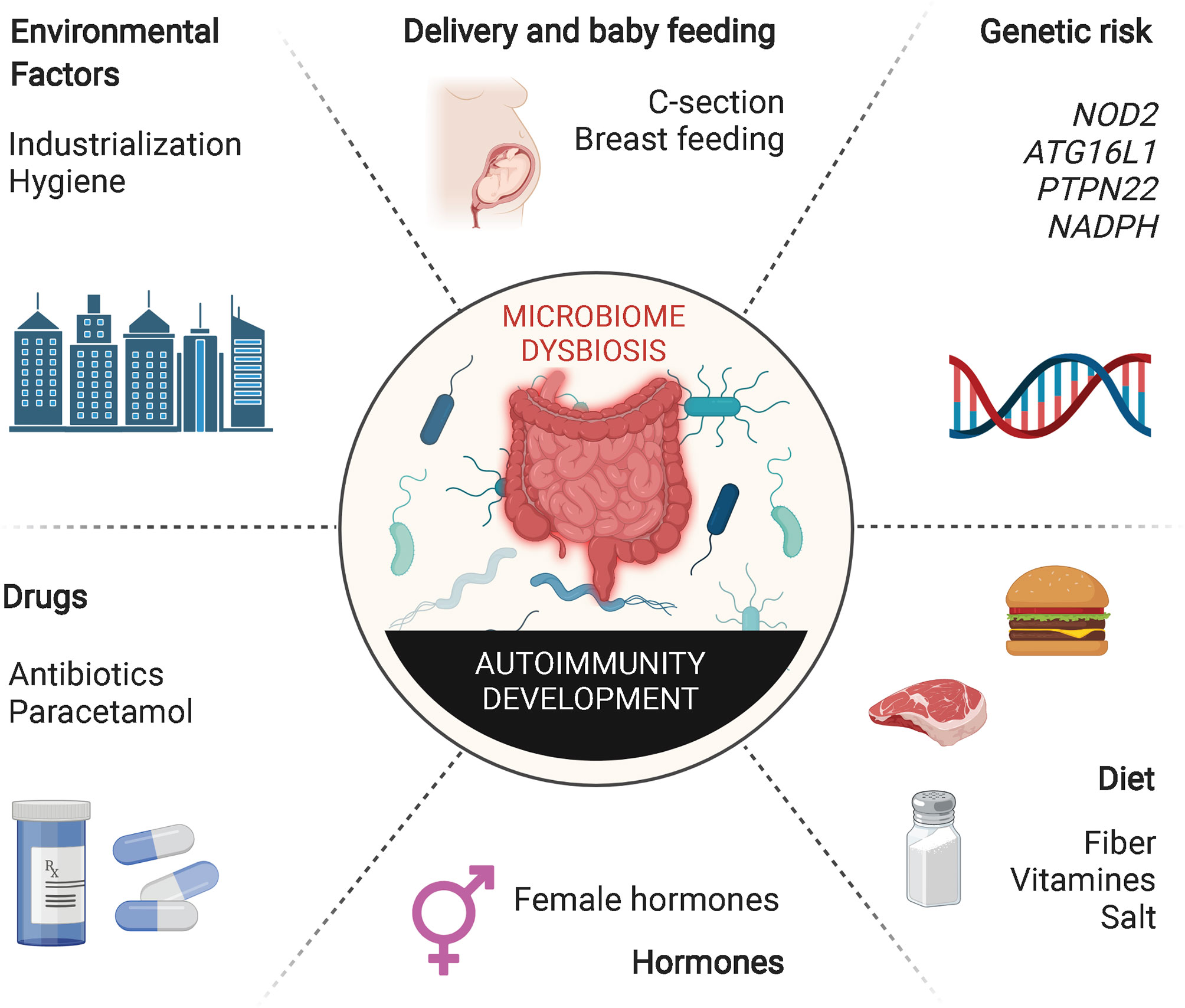
Figure 2 Extrinsic and intrinsic factors inducing gut dysbiosis. Host genetic susceptibility and hormones as well as various host-extrinsic factors such as intake of specific drugs, unhealthy diets, inappropriate microbial exposure, childbirth delivery or breast feeding may induce alterations in the composition of the gut microbiota. Decreased richness and perturbations in taxonomic commensal and metabolite composition have been extensively associated with the development of multiple autoimmune inflammatory disorders.
Thus, antibiotics can have a profound impact on microbiome diversity and, as a result, on the host´s susceptibility to allergic and/or autoimmune diseases. In mice, antibiotics can increase the susceptibility of murine models to these pathological processes. In humans, the effects of antibiotic exposure on these disorders vary as a function of the timing of administration. Whereas excessive use of antibiotics during childhood may increase the susceptibility of children to atopic diseases, antibiotic use during adulthood may help suppress certain autoimmune disease processes (41). For example, excessive oral antibiotic use by mothers or newborns has been associated with increased susceptibility to T1D and asthma during childhood. In contrast, antibiotic-mediated resolution of A. actinomycetemcomitans infections (e.g. in periodontitis) in adults have been associated with suppression of rheumatoid arthritis (RA) (42), presumably due to the ability of the pore-forming toxin of this facultative anaerobe to promote protein citrullination, a major target of RA-associated autoantibodies.
Genetics and sex are additional key variables. Allelic variation at genes such as NOD2, encoding the intracellular PRR Nucleotide Binding Oligomerization Domain Containing 2, is strongly associated with susceptibility or resistance to inflammatory bowel disease (IBD). Microbial colonization of NOD male mice results in increased levels of serum testosterone and protection against T1D development (43, 44).
Despite all these important observations linking alterations in the gut microbiota with different autoimmune and allergic responses, the precise underlying mechanisms are not fully understood. Nevertheless, there is evidence suggesting that this is a multifactorial process, involving microbial-induced polarization of gut-associated T cells toward pathogenic subsets, bystander activation of autoreactive T cells, activation of T cells co-expressing dual (autoreactive and gut microbial antigen-specific) TCRs, and antigenic mimicry (Figure 3).
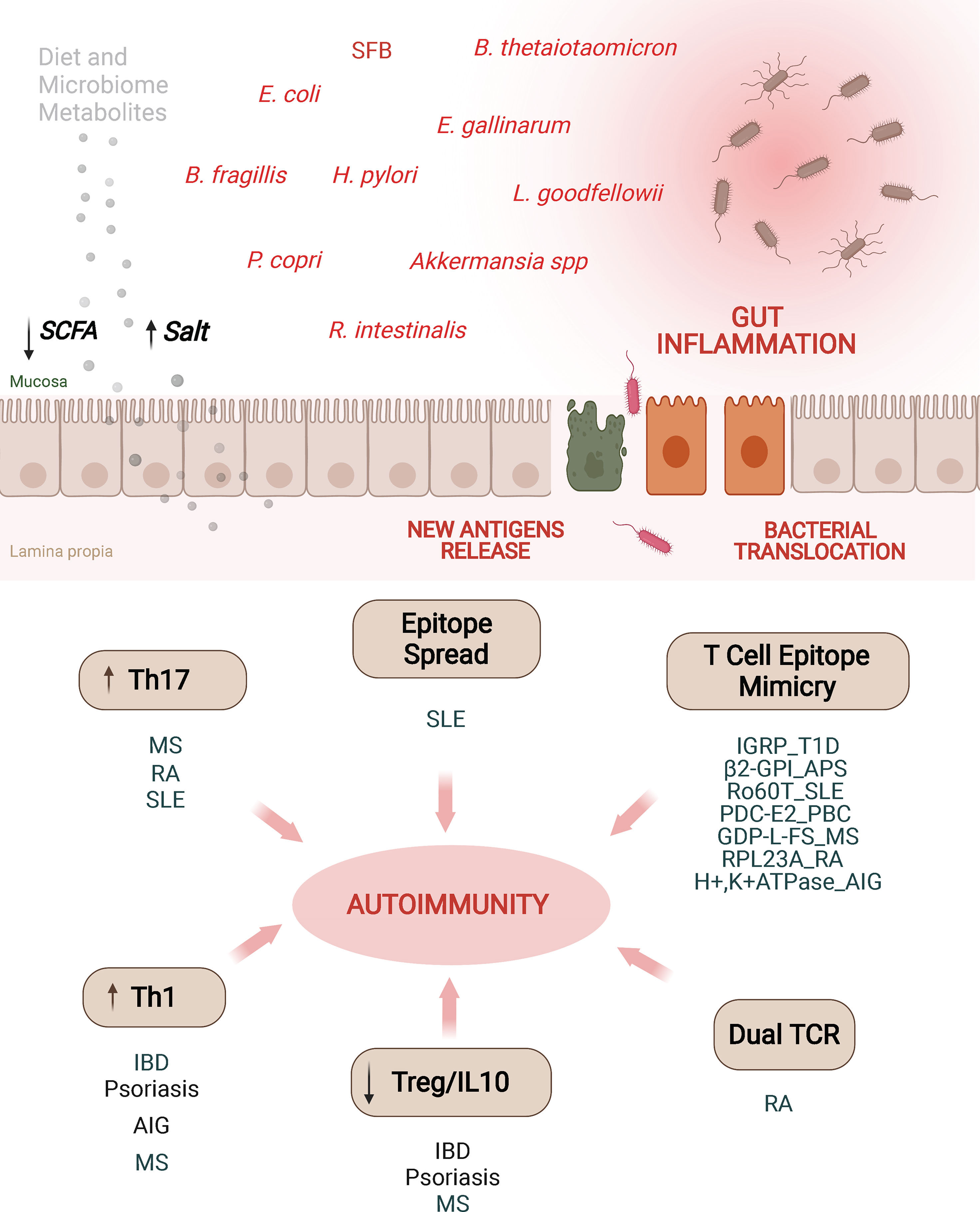
Figure 3 Alterations in the microbiota may promote autoimmunity through different mechanisms. Alterations of intestinal permeability caused by diet, bacterial metabolites, dysbiosis or pathobionts might increase exposure of gut microbial antigens to the gut associated lymphoid tissue. These adverse events have been associated with various autoimmune disorders through different mechanisms. Induction of Th17/Th1 cell responses, impaired or low levels of IL10-secreting Treg cell types, epitope spreading, dual TCR recognition or antigenic mimicry are some of the mechanisms. T1D, type 1 diabetes; AIG, autoimmune gastritis; IBD, inflammatory bowel disease; RA, rheumatoid arthritis; SLE, systemic lupus erythematosus; PBC, primary biliary cholangitis; MS, multiple sclerosis; SCFA, short chain fatty acids; MS, multiple sclerosis; β2-GPI, β2-glycoprotein I; APS, anti-phospholipid syndrome; PDC-E2, pyruvate dehydrogenase complex; GDP-L-FS, guanosine diphosphate-L-fucose synthase; RPL23A, arthritis-related autoantigen 60S ribosomal protein L23a; IGRP, islet-specific glucose-6-phosphatase catalytic subunit-related protein.
Gut dysbiosis or infections with pathobionts may disrupt gut immune homeostasis by polarizing T cell responses. Apoptosis during microbial infection drives autoreactive Th17 cell responses (45). Furthermore, there is evidence that commensals and gut pathogens can trigger differential cytokine expression patterns in gut T-helper cell subsets. Thus, whereas murine SFB promote the formation of IL-10-expressing (non-inflammatory) Th17 cells in the steady state, C. rodentium infection induces the formation of interferon-γ+ (pro-inflammatory) Th17 cells instead (46). In mice, another bacterium, A. muciniphila, promotes TFH cell formation under physiological conditions, but Th17 cell formation in the context of inflammation (47). In humans, A. muciniphila, which is enriched in the microbiota of patients with Multiple Sclerosis (MS), can skew differentiation of T cells into the Th1 cell subset in vitro (48, 49).
Antigenic cross-reactivity or molecular mimicry is another mechanism by which certain gut microbial antigens might be able to trigger T cell responses against autoantigens. Persistent colonization of the host with bacteria expressing cross-reactive epitopes in a host carrying high-risk Human Leukocyte Antigen (HLA) genes might trigger the sustained activation of cross-reactive autoreactive T cells in the gut, particularly if there is a loss in gut barrier integrity. Various autoantigen orthologues and non-orthologous mimotopes of autoantigens encoded in the microbiota have been implicated in the activation of autoreactive T cell responses in various autoimmune disorders (Table 1). Ro60-specific CD4+ T cell hybridomas (targeting the Sjögren’s syndrome Antigen A (SSA)), have been shown to cross-react, in HLA-DR3 transgenic mice, with an orthologous antigen expressed by Capnocytphaga ochracea (52). Likewise, DRB1*04:01-restricted T cells targeting β2-glycoprotein I (β2GPI) epitopes in patients with anti-phospholipid syndrome (APS) have been shown to cross-react with a bacterial peptide from R. intestinalis, inducing pro-inflammatory Th1 cell responses in vitro (51). Another example of molecular mimicry involves the rheumatoid arthritis (RA)-relevant autoantigens N-acetylglucosamine-6-sulfatase (GNS) and filamin A (FLNA). Multiple gut microbial peptide epitopes are structural mimics of these synovial proteins (50). Likewise, the murine diabetogenic IGRP206-214 epitope is a structural and agonistic mimic of a highly homologous epitope from the Bacteroides integrase (53). There is also evidence suggesting an association between CD4+ T-cell cross-reactivity against an E. coli antigen and the pyruvate dehydrogenase complex (PDC), a major autoantigenic target in human Primary Biliary Chollangitis (PBC) (54). Likewise, human autoimmune gastritis has been associated with T cell cross-reactivity against the H. pilory H+, K+–ATPase (55). Furthermore, there is evidence supporting an association between central nervous system (CNS) autoimmunity and cross-reactivity between gut microbial antigens and autoantigenic targets in MS, such as myelin basic protein (MBP) and guanosine diphosphate-L-fucose synthase protein (56–58).
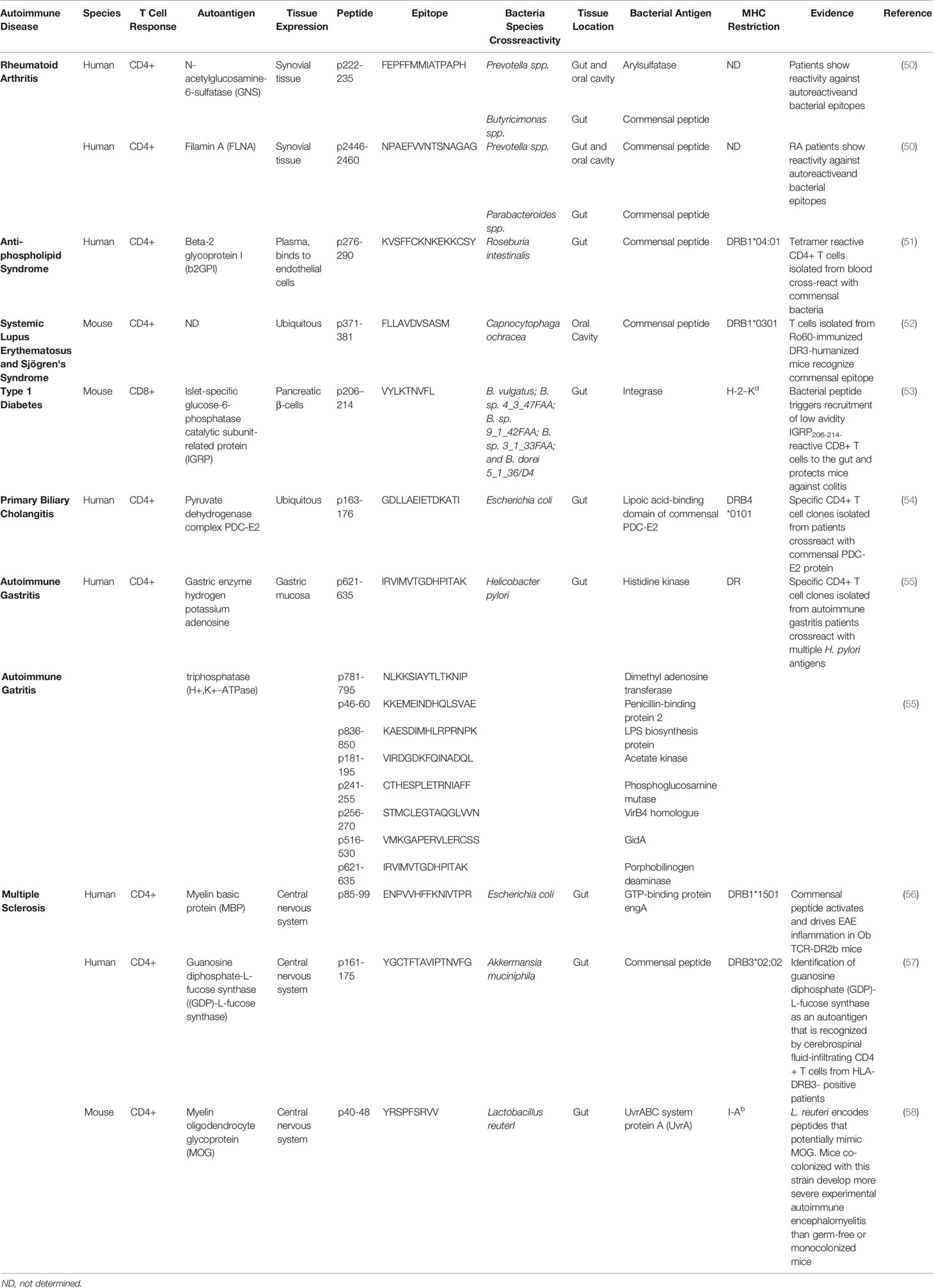
Table 1 Antigenic cross-reactivity between autoimmune disease relevant autoantigens and gut/oral microbial T-cell antigens.
Certain pathobionts can promote autoimmune responses through mechanisms other than molecular mimicry. For example, E. gallinarum, a pathobiont associated with SLE autoimmunity and autoantibody responses against various SLE and APS-relevant autoantigens, such as RNA, double-stranded DNA and β2GPI, has been detected in gut-distal organs of patients, suggesting a role for bacterial translocation in this process (59). As noted above, the pore-forming toxin of the oral pathobiont A. actinomycetemcomitans can citrullinate proteins, leading to neoantigen formation and production of RA-associated autoantibodies (60). Other pathogens can activate autoreactive responses in a non-antigen specific way, by creating an inflammatory environment that promotes bystander lymphocyte activation. For example, in a mouse model of arthritis, SFB antigens induced autoimmune lung inflammation by triggering the formation of autoreactive Th17 cells from naïve T cell precursors co-expressing SFB antigen specific TCRs (61). Notwithstanding these associations, the precise mechanisms and the potential role of these processes in human autoimmune diseases remain unclear.
Dysbiosis and disruption of the integrity or barrier function of the intestinal epithelium have been associated with various autoimmune diseases. Below, we discuss such associations with a focus on potential mechanisms, including antigenic mimicry (Table 2 and Figure 4).
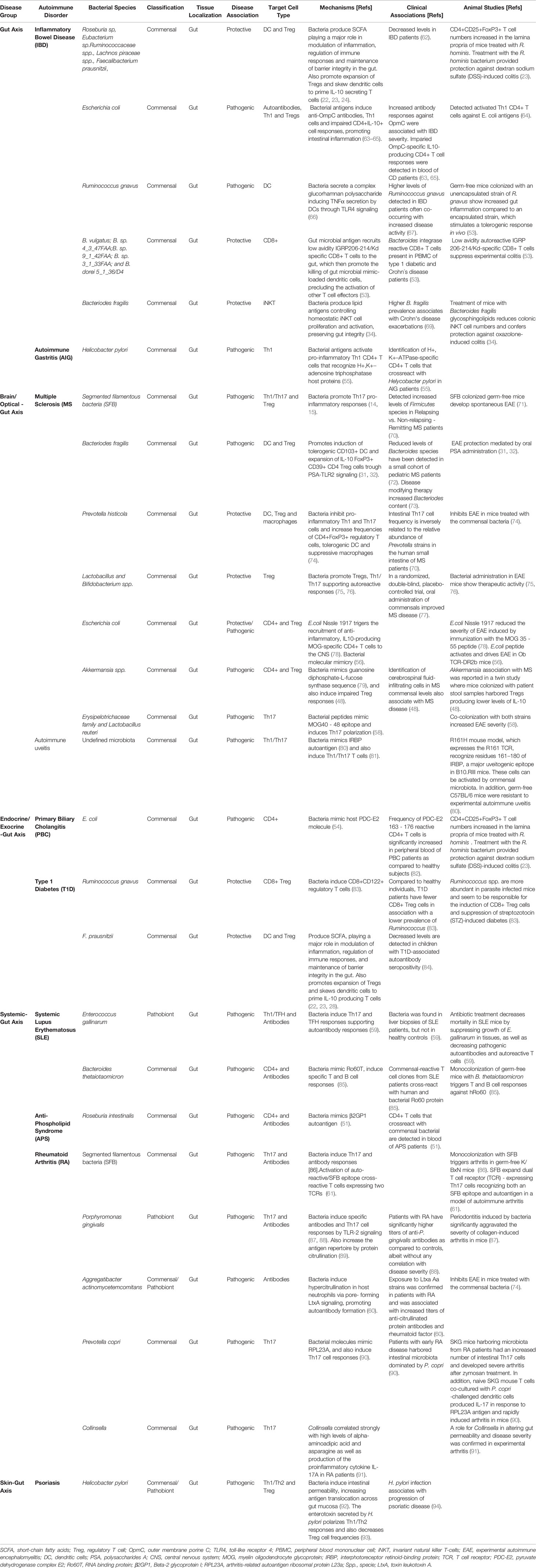
Table 2 Gut and oral bacterial species associated with autoimmune disorders.
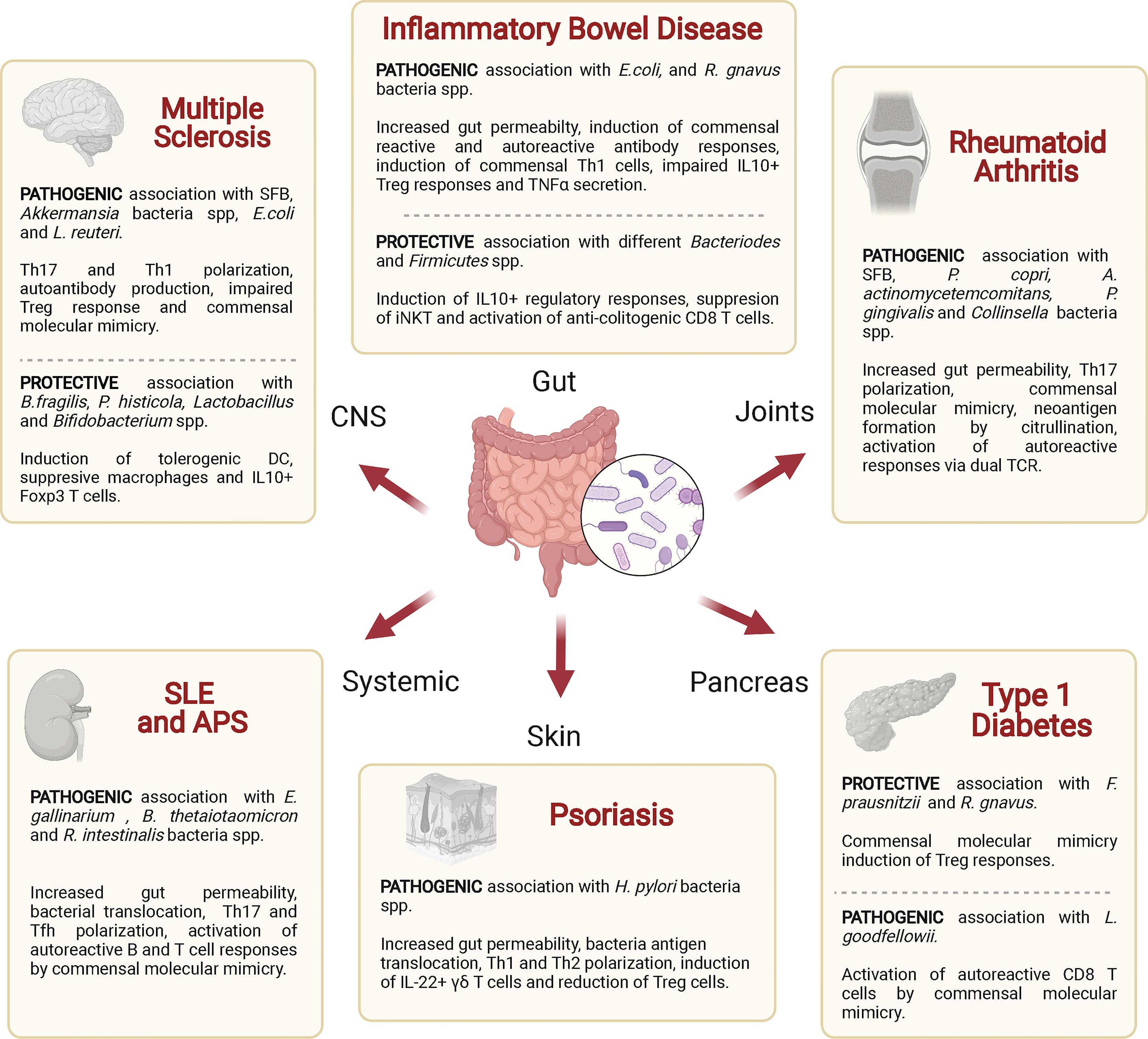
Figure 4 Associations of various autoimmune diseases with commensal bacteria. Specific commensal bacteria may enhance or reduce the host’s susceptibility to specific autoimmune diseases by altering intestinal permeability, polarizing effector or regulatory T cell responses and/or by triggering autoreactive T cell responses via antigen mimicry. SFB, segmented filamentous bacteria; DC, dendritic cell; iNKT, natural killer T-cells; Treg, regulatory T cell; TCR, T cell receptor.
CD is a form of IBD that can affect any part of the gastrointestinal tract, but predominantly targets the terminal ileum and colon. Ulcerative colitis (UC) is another form of IBD which only targets the colon. Despite the fact that the incidence and prevalence of IBD are increasing worldwide and are appearing earlier in life, the etiology and pathogenesis of CD and UC remain ill-defined (95). Although there is an important underlying genetic component (96), disease development requires an environmental trigger (97). Many studies have provided evidence for the loss of gut microbial tolerance in human IBD (98), including the development of B and T cell responses against gut microbial antigens and autoantigens (99–106).
Whereas CD has been generally associated with increased Th17- and Th1-type responses, UC is primarily associated with Th17- and Th2-type responses (107). In CD patients, Th1-associated transcription factors such as STAT4 and T-bet, and cytokine receptors such as IL-12Rβ2 are highly expressed in the lamina propria of the inflamed gut (108). Likewise, the development of CD-like ileitis in SAMP1/YitFc and TnfΔARE mice has been associated with Th1-driven inflammation (109, 110). Indeed, Th1-type cells appear to be necessary for gut inflammation since immune cells from Ifng–/–, Tbx21−/−, and Stat4−/− donors cannot transfer intestinal inflammation into immunocompromised hosts (111–113).
There is also strong evidence for the contribution of Th17-type responses to human IBD. Mucosal biopsies from both CD and UC patients contain increased levels of Th17 cell-derived cytokines (114, 115). In addition, human IBD is associated with genetic polymorphisms at loci encoding Th17 pathway components (e.g., IL6ST, JAK2, STAT3, RORC, IL23R, CCR6) (116). In agreement with these observations, Stat3–/– and Rorc–/– mice are resistant to experimental colitis (117, 118). However, since Th17-type cells are known to contribute to the maintenance of normal gut microbial homeostasis, it seems likely that the pathogenic Th17-like cells that contribute to IBD are a different subset (119, 120). Th17-type cells, like other Th cell subsets, are plastic, and the associations between Th17-type cells and both IBD and other autoimmune disorders appear to be mediated by Th17-like cells co-expressing IFNγ and IL-17A (121, 122). In mice, pathogenic Th17-type cells express high levels of the IL-23 receptor (123, 124), and the IL23R gene is strongly associated with human IBD (116). Interestingly, a subset of intestinal human memory CCR6+CXCR3+ T cells co-expressing Th17 and Th1 markers from CD patients express the Multidrug Resistant Mutation MDR1, a plasma membrane drug efflux pump (125) that is encoded in a gene strongly associated with IBD (ABCB1) (126).
Although the role of Th2-type cells in the pathogenesis of IBD remains unclear, studies in both humans and mice support their involvement. For example, biopsies of UC patients contain increased levels of IL-4 (127) and sera from both UC patients and mice with oxazolone-induced colitis contain elevated levels of IgE, an IL-4-regulated immunoglobulin isotype (128, 129). Likewise, development of ileitis in SAMP1/YitFc mice, and colitis in the TNBS-induced model are associated with Th2-type responses (130, 131).
The onset of IBD has been linked to both microbial dysbiosis and disruption of gut epithelial permeability (132). Decreased abundance on Firmicutes bacteria belonging to the Ruminococcaceae spp., Lachnospiraceae spp, F. prausnitzii and Roseburia spp. families is a signature of microbial dysbiosis in IBD (62, 133). Since these bacteria are butyrate producers, this association may be driven by altered (reduced) induction of iTreg cells in the gut (134, 135). In agreement with this, polymorphisms in genes coding for the immunoregulatory cytokine IL-10 (IL10) or subunits of the IL-10 receptor (IL10RA, IL10RB) are strongly associated with human IBD, particularly with early onset forms of colitis (136). Increased prevalence of pro-inflammatory commensals is yet another mechanism through which dysbiosis may contribute to the pathogenesis of IBD. For example, gut inflammation in patients and murine models has been associated with increased prevalence of R. gnavus (67, 68), which triggers the production of inflammatory cytokines (e.g. TNFα) by DCs via polysaccharide signaling (66). The role of other commensals is less clear. For example, although B. fragilis metabolites promote barrier integrity, the prevalence of this bacteria has been associated with disease exacerbation in CD patients (69), suggesting context-dependent effects.
Increased antibody responses against various gut microbial antigens have been described in IBD. For example, CD patients have increased serum titers of antibodies against the E. coli membrane porin C (OmpC), yeast S. cerevisiae mannose epitopes (ASCA) and bacterial flagellins (CBir) (105, 106), and both the presence and titers of these antibodies are associated with disease severity (63). CD4+ T cell responses against some of these gut microbial antigens have also been associated with IBD. For example, activated OmpC-specific CD4+ T cells are colitogenic in mice (64), and OmpC-specific CD4+ T-cells have been detected in the peripheral blood of IBD patients (65). There is also evidence for a role of flagellin-specific CD4+ T cells; increased frequencies of flagellin-specific CD4+ T cells with an activated, gut homing phenotype were detected in CD and UC patients versus controls. Furthermore, CD, albeit not UC, is associated with increased serum levels of anti-flagellin IgG and IgA antibodies (137).
Many studies have provided evidence for the contribution of an autoimmune component in the maintenance of chronic intestinal inflammation. Most UC, and to a lesser extent CD patients develop peri-nuclear anti-neutrophil cytoplasmic antibodies (pANCA) (99, 138). These antibodies cross-react with the OmpC protein, suggesting a possible role for B cell autoreactivity and gut microbial antigenic cross-reactivity in the pathogenesis of IBD (139). Likewise, IBD has been associated with autoantibody responses against Glycoprotein 2 (GP2) (100, 101), a receptor for bacterial adhesin FimH that is upregulated in the gut epithelium of patients (100). Of note, high levels of anti-GP2 IgA antibodies have been described in pediatric IBD patients (101). An increased prevalence of autoantibodies against FAM84A, a neuronal sensory protein expressed in the gastrointestinal tract, has also been associated with IBD (102).
The contribution of autoreactive T-cell responses to IBD is much less clear. Some studies have reported the ability of commensal bacteria to activate colitogenic T-cells or autoreactive T cells. A Citrobacter infection in ovalbumin specific TCR transgenic mice triggered the apoptotic cell death of infected colonic epithelial cells, promoting intestinal inflammation via the activation of autoreactive Th17 CD4+ T cells (45). Importantly, there is also evidence for protective autoreactive T cell responses in IBD. Specifically, a Bacteroides integrase epitope was shown to induce the recruitment of a highly prevalent low avidity IGRP206-214 specific CD8+ T cell subset to the gut, affording the mice protection against experimental colitis (53).
The above observations suggest that the relative contribution of autoimmune vs. non-autoimmune phenomena to UC and CD is different. Thus, whereas autoreactivity against colonic epithelial cells may play a role in UC, immune reactivity against the intestinal flora is primarily a feature of CD. Multiple environmental factors, genetic determinants as well as the specific contribution of commensal bacteria to dysbiosis could bias the inflammatory response and the disease phenotype in each of these two inflammatory bowel diseases.
MS is a CNS-specific autoimmune disease that is largely driven by Th17-type cells and is characterized by CNS inflammation, demyelination, and progressive neurodegeneration (140). Most patients suffer a relapsing-remitting form of disease (RR-MS). Although the etiology of MS is complex and incompletely understood, both genetic and environmental factors clearly play a role (141, 142).
Several studies have provided evidence for associations between dysbiosis and MS, such as increases in the prevalence of Methanobrevibacter (Archaea) and Akkermansia (143) or firmicutes (70), as well as a reduction in the prevalence of Butyricimonas (143).
In experimental autoimmune encephalomyelitis (EAE), a mouse model of MS, induction of autoreactive B cell responses against myelin oligodendrocyte glycoprotein (MOG) requires the presence of the microbiota (144). In addition, induction of EAE in germ-free mice was associated with reduced levels of IL17 and IFNγ in both the intestine and spinal cord as well as increased levels of Treg cells. Interestingly, colonization of these germ-free mice with SFB restored EAE susceptibility, implicating the microbiota on the development of encephalitogenic Th17 responses (71). More recently, two different bacteria from the Erysipelotrichaceae family and L. reuteri have been associated with the severity of EAE via effects on Th17 cells and MOG molecular mimicry, respectively (58). Another study reported defective production of IL-10 by Treg cells from mice colonized with fecal samples from MS patients, suggesting that MS patients harbor a specific repertoire of commensals that favor CNS autoimmunity (48).
Cross-reactive T cell responses against gut microbial antigens have also been described in MS. In a recent study, cerebrospinal fluid (CSF)-infiltrating T cells specific for GDP-L-fucose synthase cross-reacted with Akkermansia antigens (79). Furthermore, GDP-L specific clones recognized the myelin basic protein (MBP) epitope MBP83-99, suggesting that T cell cross-reactivity between gut microbial and CNS autoantigens could act as a trigger of CNS inflammation.
Other studies reported protective effects of certain commensal bacteria against CNS autoimmunity. Administration of B. fragilis PSA has been shown to protect mice against CNS autoimmune inflammation by promoting the expansion of Foxp3+ Tregs expressing CD39 (31) and by inducing tolerogenic DCs (32). Likewise, administration of P. histicola resulted in reduced frequencies pro-inflammatory Th1 and Th17 cells and increased frequencies of FoxP3+ Treg cells, tolerogenic DCs and suppressive macrophages (74). Other species, such as Bifidobacterium and Lactobacillus have been shown to protect mice against EAE by promoting Treg cell responses and reducing Th1- and Th17-type responses (75, 76). The E. coli Nissle 1917 strain was also shown to suppress CNS inflammation by promoting the formation of IL-10-producing autoreactive Treg cells (78).
Similar observations have been reported in humans. A study in a small cohort of pediatric MS patients reported a reduced prevalence of Bacteroides (72). In addition, increases in the Bacteroides content of the gut microbiota of RRMS patients with commensal modifying therapies was associated with disease-protective effects (73). Likewise, a reduced prevalence of Prevotella strains has been associated with increased frequencies of Th17 cells and disease activity in MS patients (70), suggesting a potential protective role for these bacteria against CNS inflammation. In addition, gut microbiota from MS patients imprinted defective IL-10 responses in fecal transplanted host mice, promoting the development of spontaneous EAE (48). In a recent human clinical trial, oral delivery of Lactobacillus and Bifidobacterium spp ameliorated MS symptoms (77).
SLE is a systemic (multi-organ) autoimmune disease characterized by development of autoantibody responses against nucleic acids, histones and ribonucleoproteins, leading to the formation and deposition of pathogenic immune complexes in various organs, including the kidney. Th17 polarization and higher frequencies of TFH cells have been described in the peripheral blood of SLE patients (145), consistent with the extensive autoantibody response underlying disease pathogenesis. The etiology of SLE, as is also the case for most other autoimmune diseases, remains unclear. There is an important genetic component that, although necessary, is insufficient for disease development (146). Environmental cues, such as infectious agents, are suspected to play a role as triggers of disease development in individuals at risk.
Recent evidence points to the microbiota as another potential contributing factor to the development of SLE (147, 148). Disruption of the barrier function of the gut, leading to translocation of commensal bacteria and pathobionts is one of the possible mechanisms underlying this association. For example, E. gallinarum was detected in the liver of SLE patients (as well as in the liver of patients with autoimmune hepatitis) but not in the liver of healthy controls (59). In a murine model of SLE, antibiotic treatment reduced mortality and decreased the production of pathogenic autoantibodies and autoreactive T cells, in part by suppressing the growth of E. gallinarum in tissues (59).
The composition of the early gut microbiota in mice also appears to have an impact on the development of anti-nuclear autoantibodies (149). SLE patients develop autoantibodies against the evolutionarily conserved RNA binding protein Ro60 (150, 151). Multiple gut commensals, such as B. thetaiotaomicron, express Ro60 orthologues with high sequence homology to human Ro60. Furthermore, colonization of germ-free mice with B. thetaiotaomicron led to the development of T and B cell reactivity against Ro60, as well as to glomerular immune complex deposition mimicking lupus nephritis (85). The same study reported that bacterial Ro60-specific T cell clones isolated from SLE patients cross-reacted with the human orthologue. Together, these data provided evidence for gut microbial molecular mimicry as a potential contributor to the development of SLE.
T1D is a multifactorial autoimmune disorder characterized by immune-mediated destruction of the pancreatic β-cells, in which numerous genetic elements and putative environmental triggers play a role. Several different alterations of gut microbial health have been associated with T1D in both animal models and humans (152–154), including alterations of intestinal permeability (155, 156), as well as loss of gut microbial diversity before the onset of disease (157). Pro-diabetogenic, oral antibiotic-induced gut dysbiosis in NOD mice has been associated with impaired enteric Th17/Treg responses (158). More recently, R. gnavus has been suggested to protect mice against streptozotocin (STZ)-induced diabetes, as well as to promote the development of anti-diabetogenic CD8+CD122+ Treg cells in T1D patients (83).
Gut microbial molecular mimicry has also been implicated as a possible mechanism of autoreactive T-cell activation in the pathogenesis of T1D. A protein from L. goodfellowii was suggested to function as a structural mimic of the murine diabetogenic IGRP206‐214 epitope, as it could promote the activation of cognate TCR-transgenic CD8+ T-cells in vitro (159). In another study, however, metagenomic sequencing of the gut microbiota failed to verify the presence of L. goodfellowii in the gut microbiota of both mice and patients (53). Most importantly, the latter study identified the Bacteroides integrase, an abundant gut microbial antigen, as a true structural and functional mimic of IGRP206‐214 (53). However, experiments in mono-colonized germ-free mice indicated that this gut microbial epitope promotes the recruitment and activation of anti-colitogenic, low avidity IGRP206-214-specific CD8+ T-cells, rather than the activation of their diabetogenic high-avidity counterparts (53).
Gut microbial metabolites have also been implicated in the immunopathogenesis of T1D. Increased prevalence of Bacteroides species as well as deficiencies in bacteria that produce SCFAs have been described in T1D patients (84, 160). For example, children with T1D-associated autoantibody seropositivity have a reduction in the abundance of the butyrate producer F. prausnitzii (84). Another multicenter study of 783 children showed that the microbiota of healthy children is enriched in SCFA-producers, without obvious associations with any taxa, suggesting that microbial function rather than composition might contribute to T1D development (161). In agreement with these observations, NOD mice fed with diets promoting gut microbial production of acetate and butyrate were almost completely protected from T1D via SCFA-mediated immunomodulation (162).
RA is an organ-specific autoimmune disease that is characterized by chronic inflammation and progressive destruction of the joint tissues by arthritogenic T cells and autoantibodies. Although the pathogenesis of RA remains incompletely defined, both genetic and environmental factors, including alterations in the gut microbiota, have been implicated in its development. As is the case for the other autoimmune diseases discussed above, alterations in intestinal permeability (163) and gut microbial composition (164) have been found to predate the onset of disease in RA. Commensal bacteria such as Collinsella have been associated with increased gut permeability and disease severity in both an experimental model of arthritis and in human RA. In RA patients, for example, pro-arthritogenic IL-17A responses in a subset of RA patients were associated with an increased prevalence of Collinsella (91). In another study, colonization of germ-free mice with SFB bacteria was sufficient to induce arthritogenic Th17 responses (86).
Other studies have suggested a role for molecular mimicry as a trigger of arthritogenic autoimmune responses in both animal models and patients. An early work reported the presence of immunoreactivity against an E. coli epitope, QKRAA, in the synovial fluid of patients as compared to controls (165). A more recent study found that autoreactive CD4+ T cells against the autoantigens Filamin A (FLNA) and N-acetylglucosamine-6-sulfatase (GNS) cross-react with similar sequences found in Prevotella, Butyricimonas and Parabacteroides species (50). Furthermore, increased prevalence of Prevotella species, such as P. copri were detected in patients with new-onset RA (166). In mice, Prevotella has also been proposed to contribute to RA development, in this case by both, activating autoreactive T cells specific for the arthritis-relevant autoantigen Ribosomal Protein L23a (RPL23A), and by inducing pro-inflammatory Th17 responses (90).
The oral microbiota has also been implicated in RA. Periodontitis induced by P. gingivalis, an established oral pathobiont linked to this condition, has been associated with the exacerbation of autoimmune arthritis, presumably by inducing pathogenic Th17 responses via TLR2- and IL-1-signalling (87). Of interest, P. gingivalis has been found to contribute also to the generation of citrullinated proteins (antigenic targets of RA) in the oral cavity of RA patients (89, 167), suggesting a potential link between immune responses against these post-translationally modified oral proteins and downstream joint inflammation (168). This property has also been documented for another RA-associated oral pathobiont, A. actinomycetemcomitans (60).
Psoriasis is a prevalent autoimmune disease characterized by keratinocyte hyperproliferation and skin inflammation, where both genetic and environmental factors also play a role (169). Psoriatic skin lesions are associated with dermal and epidermal infiltration of leukocytes, triggered and maintained by T lymphocytes (170, 171). Most of the T cells that infiltrate the psoriatic dermis are CD4+, whereas those that infiltrate the epidermis are primarily CD8+ (171, 172). Different clinical phenotypes have been associated with the presence of bacterial skin commensals capable of inducing local pro-inflammatory Th17 responses (173). However, multiple studies have also underscored the importance of the gut-skin axis on cutaneous autoimmunity. Gut dysbiosis induced by oral antibiotic treatment in neonatal mice promoted the development of psoriasis by increasing the frequency of cutaneous IL-22 producing γδ+T cells (174). In addition, induction of experimental psoriasis via imiquimod exposure is blunted in germ free or antibiotic treated mice, in association with a reduction in Th17 cells (175, 176). In particular, Helicobacter pylori infection has been associated with psoriasis (94), potentially via both local and systemic effects of the inflammatory response (177, 178), such as increased permeability of the gastric mucosa to food antigens, among others (92). In addition, the H. pylori enterotoxin binds to the T cell receptor and induces the expression of T cell skin homing receptors (179, 180).
Vitiligo is another T cell-dependent autoimmune disorder of the skin characterized by skin depigmentation due to immune mediated killing of melanocytes (181). Recently, in a murine model of vitiligo harboring tyrosinase-reactive T cells, oral ampicillin treatment decreased disease severity, suggesting that the gut microbiota may also play a role in this disease (182).
The specific role that gut microbes, metabolites or gut microbial antigens play in the pathogenesis of autoimmune disease is complex and remain ill-defined. There are clear associations between gut dysbiosis and increased intestinal permeability with several autoimmune phenomena. However, whether these abnormalities contribute to, or are merely a bystander effect of disease progression remains to be addressed. Although experiments in gnotobiotic mice have provided useful information in this regard, it is unclear to what extent the presence of an altered immune system in the germ-free mice that were used in these studies might have affected the study outcome. Bacterial translocation due to gut barrier disruption can lead to increased presentation of gut microbial antigens to the immune system. As a result, activation of autoreactive T and B cells by cross-reactive gut microbial antigens remains a potential mechanism, but the evidence providing direct links between gut microbial antigen cross-reactivity and pathogenic autoimmunity remain largely circumstantial in nature. A more extensive use of reductionist systems of autoimmunity (e.g., TCR-transgenic mice), coupled to mono-colonization of germ-free mice with wild-type and mutant gut microbial species (53) should help address this knowledge gap. The links between the effects of gut microbe-derived metabolites (e.g., SCFA) on the gut-associated lymphoid tissue and autoimmune disease are compelling and intriguing but will need to be integrated into the poorly understood sequence of events underlying the corresponding autoimmune diseases, including their genetic underpinnings.
Notwithstanding these limitations, the studies summarized herein strongly support multifaceted roles for the gut microbiota on autoimmune disease susceptibility or resistance. A precise understanding of each of the many potential mechanisms through which commensal bacteria can promote or protect against autoimmune disorders will help conceptualize novel therapeutic applications in this area.
NG and PS wrote the manuscript. Both authors contributed to the article and approved the submitted version.
PS is scientific founder of Parvus Therapeutics Inc. and has a financial interest in the company.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We thank the members of our group for their contributions. The authors’ work is funded by the Canadian Institutes of Health Research (CIHR), the Praespero Foundation, the Ministerio de Educación y Ciencia of Spain (RTI2018-093694-B-100), RETICS, and Generalitat de Catalunya (SGR and CERCA Programmes) Cartoons in Figures 1–4 were created with www.BioRender.com.
1. Yatsunenko T, Rey FE, Manary MJ, Trehan I, Dominguez-Bello MG, Contreras M, et al. Human Gut Microbiome Viewed Across Age and Geography. Nature (2012) 486:222–7. doi: 10.1038/nature11053
2. Koenig JE, Spor A, Scalfone N, Fricker AD, Stombaugh J, Knight R, et al. Succession of Microbial Consortia in the Developing Infant Gut Microbiome. Proc Natl Acad Sci (2011) 108:4578–85. doi: 10.1073/pnas.1000081107
3. Gomez de Agüero M, Ganal-Vonarburg SC, Fuhrer T, Rupp S, Uchimura Y, Li H, et al. The Maternal Microbiota Drives Early Postnatal Innate Immune Development. Science (2016) 351:1296–302. doi: 10.1126/science.aad2571
4. Dominguez-Bello MG, Costello EK, Contreras M, Magris M, Hidalgo G, Fierer N, et al. Delivery Mode Shapes the Acquisition and Structure of the Initial Microbiota Across Multiple Body Habitats in Newborns. Proc Natl Acad Sci USA (2010) 107:11971–5. doi: 10.1073/pnas.1002601107
5. Caballero-Flores G, Sakamoto K, Zeng MY, Wang Y, Hakim J, Matus-Acuña V, et al. Maternal Immunization Confers Protection to the Offspring Against an Attaching and Effacing Pathogen Through Delivery of IgG in Breast Milk. Cell Host Microbe (2019) 25:313–23.e4. doi: 10.1016/j.chom.2018.12.015
6. Zheng W, Zhao W, Wu M, Song X, Caro F, Sun X, et al. Microbiota-Targeted Maternal Antibodies Protect Neonates From Enteric Infection. Nature (2020) 577:543–8. doi: 10.1038/s41586-019-1898-4
7. Al Nabhani Z, Dulauroy S, Marques R, Cousu C, Al Bounny S, Dejardin F, et al. A Weaning Reaction to Microbiota Is Required for Resistance to Immunopathologies in the Adult. Immunity (2019) 50:1276–88.e5. doi: 10.1016/j.immuni.2019.02.014
8. Liang W, Enee E, Andre-Vallee C, Falcone M, Sun J, Diana J. Intestinal Cathelicidin Antimicrobial Peptide Shapes a Protective Neonatal Gut Microbiota Against Pancreatic Autoimmunity. Gastroenterology (2021) 162(4):1288–302. doi: 10.1053/j.gastro.2021.12.272
9. Macpherson AJ, Uhr T. Induction of Protective IgA by Intestinal Dendritic Cells Carrying Commensal Bacteria. Science (2004) 303:1662–5. doi: 10.1126/science.1091334
10. Cao AT, Yao S, Gong B, Nurieva RI, Elson CO, Cong Y. Interleukin (IL)-21 Promotes Intestinal IgA Response to Microbiota. Mucosal Immunol (2015) 8:1072–82. doi: 10.1038/mi.2014.134
11. Cong Y, Feng T, Fujihashi K, Schoeb TR, Elson CO, dominant A. Coordinated T Regulatory Cell-IgA Response to the Intestinal Microbiota. Proc Natl Acad Sci USA (2009) 106:19256–61. doi: 10.1073/pnas.0812681106
12. Tsuji M, Komatsu N, Kawamoto S, Suzuki K, Kanagawa O, Honjo T, et al. Preferential Generation of Follicular B Helper T Cells From Foxp3+ T Cells in Gut Peyer's Patches. Science (2009) 323:1488–92. doi: 10.1126/science.1169152
13. Hirota K, Turner JE, Villa M, Duarte JH, Demengeot J, Steinmetz OM, et al. Plasticity of Th17 Cells in Peyer's Patches is Responsible for the Induction of T Cell-Dependent IgA Responses. Nat Immunol (2013) 14:372–9. doi: 10.1038/ni.2552
14. Klaasen HL, van der Heijden PJ, Stok W, Poelma FG, Koopman JP, Van den Brink ME, et al. Apathogenic, Intestinal, Segmented, Filamentous Bacteria Stimulate the Mucosal Immune System of Mice. Infect Immun (1993) 61:303–6. doi: 10.1128/iai.61.1.303-306.1993
15. Ivanov II, Atarashi K, Manel N, Brodie EL, Shima T, Karaoz U, et al. Induction of Intestinal Th17 Cells by Segmented Filamentous Bacteria. Cell (2009) 139:485–98. doi: 10.1016/j.cell.2009.09.033
16. Atarashi K, Tanoue T, Ando M, Kamada N, Nagano Y, Narushima S, et al. Th17 Cell Induction by Adhesion of Microbes to Intestinal Epithelial Cells. Cell (2015) 163:367–80. doi: 10.1016/j.cell.2015.08.058
17. Tan TG, Sefik E, Geva-Zatorsky N, Kua L, Naskar D, Teng F, et al. Identifying Species of Symbiont Bacteria From the Human Gut That, Alone, Can Induce Intestinal Th17 Cells in Mice. Proc Natl Acad Sci USA (2016) 113:E8141–50. doi: 10.1073/pnas.1617460113
18. Mosconi I, Geuking MB, Zaiss MM, Massacand JC, Aschwanden C, Kwong Chung CK, et al. Intestinal Bacteria Induce TSLP to Promote Mutualistic T-Cell Responses. Mucosal Immunol (2013) 6:1157–67. doi: 10.1038/mi.2013.12
19. Kim M, Galan C, Hill AA, Wu WJ, Fehlner-Peach H, Song HW, et al. Critical Role for the Microbiota in CX(3)CR1(+) Intestinal Mononuclear Phagocyte Regulation of Intestinal T Cell Responses. Immunity (2018) 49:151–63.e5. doi: 10.1016/j.immuni.2018.05.009
20. Atarashi K, Suda W, Luo C, Kawaguchi T, Motoo I, Narushima S, et al. Ectopic Colonization of Oral Bacteria in the Intestine Drives T(H)1 Cell Induction and Inflammation. Science (2017) 358:359–65. doi: 10.1126/science.aan4526
21. Narushima S, Sugiura Y, Oshima K, Atarashi K, Hattori M, Suematsu M, et al. Characterization of the 17 Strains of Regulatory T Cell-Inducing Human-Derived Clostridia. Gut Microbes (2014) 5:333–9. doi: 10.4161/gmic.28572
22. Alameddine J, Godefroy E, Papargyris L, Sarrabayrouse G, Tabiasco J, Bridonneau C, et al. Faecalibacterium Prausnitzii Skews Human DC to Prime IL10-Producing T Cells Through TLR2/6/JNK Signaling and IL-10, IL-27, CD39, and IDO-1 Induction. Front Immunol (2019) 10:143. doi: 10.3389/fimmu.2019.00143
23. Patterson AM, Mulder IE, Travis AJ, Lan A, Cerf-Bensussan N, Gaboriau-Routhiau V, et al. Human Gut Symbiont Roseburia Hominis Promotes and Regulates Innate Immunity. Front Immunol (2017) 8:1166. doi: 10.3389/fimmu.2017.01166
24. Kaisar MMM, Pelgrom LR, van der Ham AJ, Yazdanbakhsh M, Everts B. Butyrate Conditions Human Dendritic Cells to Prime Type 1 Regulatory T Cells via Both Histone Deacetylase Inhibition and G Protein-Coupled Receptor 109a Signaling. Front Immunol (2017) 8:1429. doi: 10.3389/fimmu.2017.01429
25. Neff CP, Rhodes ME, Arnolds KL, Collins CB, Donnelly J, Nusbacher N, et al. Diverse Intestinal Bacteria Contain Putative Zwitterionic Capsular Polysaccharides With Anti-Inflammatory Properties. Cell Host Microbe (2016) 20:535–47. doi: 10.1016/j.chom.2016.09.002
26. Danne C, Ryzhakov G, Martínez-López M, Ilott NE, Franchini F, Cuskin F, et al. A Large Polysaccharide Produced by Helicobacter Hepaticus Induces an Anti-Inflammatory Gene Signature in Macrophages. Cell Host Microbe (2017) 22:733–745.e5. doi: 10.1016/j.chom.2017.11.002
27. Schulthess J, Pandey S, Capitani M, Rue-Albrecht KC, Arnold I, Franchini F, et al. The Short Chain Fatty Acid Butyrate Imprints an Antimicrobial Program in Macrophages. Immunity (2019) 50:432–445.e7. doi: 10.1016/j.immuni.2018.12.018
28. Mukherjee A, Lordan C, Ross RP, Cotter PD. Gut Microbes From the Phylogenetically Diverse Genus Eubacterium and Their Various Contributions to Gut Health. Gut Microbes (2020) 12:1802866. doi: 10.1080/19490976.2020.1802866
29. Singh N, Gurav A, Sivaprakasam S, Brady E, Padia R, Shi H, et al. Activation of Gpr109a, Receptor for Niacin and the Commensal Metabolite Butyrate, Suppresses Colonic Inflammation and Carcinogenesis. Immunity (2014) 40:128–39. doi: 10.1016/j.immuni.2013.12.007
30. Verma R, Lee C, Jeun EJ, Yi J, Kim KS, Ghosh A, et al. Cell Surface Polysaccharides of Bifidobacterium Bifidum Induce the Generation of Foxp3(+) Regulatory T Cells. Sci Immunol (2018) 3. (28):eaat6975. doi: 10.1126/sciimmunol.aat6975
31. Wang Y, Telesford KM, Ochoa-Repáraz J, Haque-Begum S, Christy M, Kasper EJ, et al. An Intestinal Commensal Symbiosis Factor Controls Neuroinflammation via TLR2-Mediated CD39 Signalling. Nat Commun (2014) 5:4432. doi: 10.1038/ncomms5432
32. Ochoa-Repáraz J, Mielcarz DW, Wang Y, Begum-Haque S, Dasgupta S, Kasper DL, et al. A Polysaccharide From the Human Commensal Bacteroides Fragilis Protects Against CNS Demyelinating Disease. Mucosal Immunol (2010) 3:487–95. doi: 10.1038/mi.2010.29
33. Tanoue T, Morita S, Plichta DR, Skelly AN, Suda W, Sugiura Y, et al. A Defined Commensal Consortium Elicits CD8 T Cells and Anti-Cancer Immunity. Nature (2019) 565:600–5. doi: 10.1038/s41586-019-0878-z
34. An D, Oh SF, Olszak T, Neves JF, Avci FY, Erturk-Hasdemir D, et al. Sphingolipids From a Symbiotic Microbe Regulate Homeostasis of Host Intestinal Natural Killer T Cells. Cell (2014) 156:123–33. doi: 10.1016/j.cell.2013.11.042
35. Fung TC, Bessman NJ, Hepworth MR, Kumar N, Shibata N, Kobuley D, et al. Lymphoid-Tissue-Resident Commensal Bacteria Promote Members of the IL-10 Cytokine Family to Establish Mutualism. Immunity (2016) 44:634–46. doi: 10.1016/j.immuni.2016.02.019
36. Bach JF. The Hygiene Hypothesis in Autoimmunity: The Role of Pathogens and Commensals. Nat Rev Immunol (2018) 18:105–20. doi: 10.1038/nri.2017.111
37. Martins TC, Aguas AP. Mechanisms of Mycobacterium Avium-Induced Resistance Against Insulin-Dependent Diabetes Mellitus (IDDM) in Non-Obese Diabetic (NOD) Mice: Role of Fas and Th1 Cells. Clin Exp Immunol (1999) 115:248–54. doi: 10.1046/j.1365-2249.1999.00781.x
38. Takei I, Asaba Y, Kasatani T, Maruyama T, Watanabe K, Yanagawa T, et al. Suppression of Development of Diabetes in NOD Mice by Lactate Dehydrogenase Virus Infection. J Autoimmun (1992) 5:665–73. doi: 10.1016/0896-8411(92)90184-R
39. Oldstone MB, Ahmed R, Salvato M. Viruses as Therapeutic Agents. II. Viral Reassortants Map Prevention of Insulin-Dependent Diabetes Mellitus to the Small RNA of Lymphocytic Choriomeningitis Virus. J Exp Med (1990) 171:2091–100. doi: 10.1084/jem.171.6.2091
40. Greenwood BM, Herrick EM, Voller A. Suppression of Autoimmune Disease in NZB and (NZB X NZW) F1 Hybrid Mice by Infection With Malaria. Nature (1970) 226:266–7. doi: 10.1038/226266a0
41. Strzępa A, Lobo FM, Majewska-Szczepanik M, Szczepanik M. Antibiotics and Autoimmune and Allergy Diseases: Causative Factor or Treatment? Int Immunopharmacol (2018) 65:328–41. doi: 10.1016/j.intimp.2018.10.021
42. Mukherjee A, Jantsch V, Khan R, Hartung W, Fischer R, Jantsch J, et al. Rheumatoid Arthritis-Associated Autoimmunity Due to Aggregatibacter Actinomycetemcomitans and Its Resolution With Antibiotic Therapy. Front Immunol (2018) 9:2352. doi: 10.3389/fimmu.2018.02352
43. Markle JG, Frank DN, Mortin-Toth S, Robertson CE, Feazel LM, Rolle-Kampczyk U, et al. Sex Differences in the Gut Microbiome Drive Hormone-Dependent Regulation of Autoimmunity. Science (2013) 339:1084–8. doi: 10.1126/science.1233521
44. Yurkovetskiy L, Burrows M, Khan AA, Graham L, Volchkov P, Becker L, et al. Gender Bias in Autoimmunity Is Influenced by Microbiota. Immunity (2013) 39:400–12. doi: 10.1016/j.immuni.2013.08.013
45. Campisi L, Barbet G, Ding Y, Esplugues E, Flavell RA, Blander JM. Apoptosis in Response to Microbial Infection Induces Autoreactive TH17 Cells. Nat Immunol (2016) 17:1084–92. doi: 10.1038/ni.3512
46. Omenetti S, Bussi C, Metidji A, Iseppon A, Lee S, Tolaini M, et al. The Intestine Harbors Functionally Distinct Homeostatic Tissue-Resident and Inflammatory Th17 Cells. Immunity (2019) 51:77–89.e6. doi: 10.1016/j.immuni.2019.05.004
47. Ansaldo E, Slayden LC, Ching KL, Koch MA, Wolf NK, Plichta DR, et al. Akkermansia Muciniphila Induces Intestinal Adaptive Immune Responses During Homeostasis. Science (2019) 364:1179–84. doi: 10.1126/science.aaw7479
48. Berer K, Gerdes LA, Cekanaviciute E, Jia X, Xiao L, Xia Z, et al. Gut Microbiota From Multiple Sclerosis Patients Enables Spontaneous Autoimmune Encephalomyelitis in Mice. Proc Natl Acad Sci USA (2017) 114:10719–24. doi: 10.1073/pnas.1711233114
49. Cekanaviciute E, Yoo BB, Runia TF, Debelius JW, Singh S, Nelson CA, et al. Gut Bacteria From Multiple Sclerosis Patients Modulate Human T Cells and Exacerbate Symptoms in Mouse Models. Proc Natl Acad Sci USA (2017) 114:10713–8. doi: 10.1073/pnas.1711235114
50. Pianta A, Arvikar SL, Strle K, Drouin EE, Wang Q, Costello CE, et al. Two Rheumatoid Arthritis-Specific Autoantigens Correlate Microbial Immunity With Autoimmune Responses in Joints. J Clin Invest (2017) 127:2946–56. doi: 10.1172/JCI93450
51. Ruff WE, Dehner C, Kim WJ, Pagovich O, Aguiar CL, Yu AT, et al. And B Cells Cross-React With Mimotopes Expressed by a Common Human Gut Commensal to Trigger Autoimmunity. Cell Host Microbe (2019) 26:100–113.e8. doi: 10.1016/j.chom.2019.05.003
52. Szymula A, Rosenthal J, Szczerba BM, Bagavant H, Fu SM, Deshmukh US. T Cell Epitope Mimicry Between Sjögren's Syndrome Antigen A (SSA)/Ro60 and Oral, Gut, Skin and Vaginal Bacteria. Clin Immunol (2014) 152:1–9. doi: 10.1016/j.clim.2014.02.004
53. Hebbandi Nanjundappa R, Ronchi F, Wang J, Clemente-Casares X, Yamanouchi J, Sokke Umeshappa C, et al. A Gut Microbial Mimic That Hijacks Diabetogenic Autoreactivity to Suppress Colitis. Cell (2017) pp:655–667.e17. doi: 10.1016/j.cell.2017.09.022
54. Shimoda S, Nakamura M, Ishibashi H, Hayashida K, Niho Y. HLA DRB4 0101-Restricted Immunodominant T Cell Autoepitope of Pyruvate Dehydrogenase Complex in Primary Biliary Cirrhosis: Evidence of Molecular Mimicry in Human Autoimmune Diseases. J Exp Med (1995) 181:1835–45. doi: 10.1084/jem.181.5.1835
55. Amedei A, Bergman MP, Appelmelk BJ, Azzurri A, Benagiano M, Tamburini C, et al. Molecular Mimicry Between Helicobacter Pylori Antigens and H+, K+ –Adenosine Triphosphatase in Human Gastric Autoimmunity. J Exp Med (2003) 198:1147–56. doi: 10.1084/jem.20030530
56. Harkiolaki M, Holmes SL, Svendsen P, Gregersen JW, Jensen LT, McMahon R, et al. T Cell-Mediated Autoimmune Disease Due to Low-Affinity Crossreactivity to Common Microbial Peptides. Immunity (2009) 30:348–57. doi: 10.1016/j.immuni.2009.01.009
57. Planas R, Santos R, Tomas-Ojer P, Cruciani C, Lutterotti A, Faigle W, et al. GDP-L-Fucose Synthase Is a CD4+ T Cell-Specific Autoantigen in DRB3∗02:02 Patients With Multiple Sclerosis, Science Translational Medicine. Am Assoc Adv Sci (2018) 30(3):348–57. doi: 10.1126/scitranslmed.aat4301
58. Miyauchi E, Kim S-W, Suda W, Kawasumi M, Onawa S, Taguchi-Atarashi N, et al. Gut Microorganisms Act Together to Exacerbate Inflammation in Spinal Cords. Nature (2020) 585:102–6. doi: 10.1038/s41586-020-2634-9
59. Vieira SM, Hiltensperger M, Kumar V, Zegarra-Ruiz D, Dehner C, Khan N, et al. Translocation of a Gut Pathobiont Drives Autoimmunity in Mice and Humans. Science (2018) 359:1156–61. doi: 10.1126/science.aar7201
60. Konig MF, Abusleme L, Reinholdt J, Palmer RJ, Teles RP, Sampson K, et al. Aggregatibacter Actinomycetemcomitans-Induced Hypercitrullination Links Periodontal Infection to Autoimmunity in Rheumatoid Arthritis. Sci Transl Med (2016) 8:369ra176. doi: 10.1126/scitranslmed.aaj1921
61. Bradley CP, Teng F, Felix KM, Sano T, Naskar D, Block KE, et al. Segmented Filamentous Bacteria Provoke Lung Autoimmunity by Inducing Gut-Lung Axis Th17 Cells Expressing Dual TCRs. Cell Host Microbe (2017) 22:697–704.e4. doi: 10.1016/j.chom.2017.10.007
62. Vijay A, Valdes AM. Role of the Gut Microbiome in Chronic Diseases: A Narrative Review. Eur J Clin Nutr (2021) 1–13. doi: 10.1038/s41430-021-00991-6
63. Papp M, Altorjay I, Dotan N, Palatka K, Foldi I, Tumpek J, et al. New Serological Markers for Inflammatory Bowel Disease Are Associated With Earlier Age at Onset, Complicated Disease Behavior, Risk for Surgery, and NOD2/CARD15 Genotype in a Hungarian IBD Cohort. Am J Gastroenterol (2008) 103:665–81. doi: 10.1111/j.1572-0241.2007.01652.x
64. Cong Y, Brandwein SL, McCabe RP, Lazenby A, Birkenmeier EH, Sundberg JP, et al. CD4+ T Cells Reactive to Enteric Bacterial Antigens in Spontaneously Colitic C3H/HeJBir Mice: Increased T Helper Cell Type 1 Response and Ability to Transfer Disease. J Exp Med (1998) 187:855–64. doi: 10.1084/jem.187.6.855
65. Uchida AM, Boden EK, James EA, Shows DM, Konecny AJ, Lord JD. Escherichiacoli-Specific CD4+ T Cells Have Public T-Cell Receptors and Low Interleukin 10 Production in Crohn's Disease. Cell Mol Gastroenterol Hepatol (2020) 10:507–26. doi: 10.1016/j.jcmgh.2020.04.013
66. Henke MT, Kenny DJ, Cassilly CD, Vlamakis H, Xavier RJ, Clardy J. Ruminococcus Gnavus, a Member of the Human Gut Microbiome Associated With Crohn's Disease, Produces an Inflammatory Polysaccharide. Proc Natl Acad Sci USA (2019) 116:12672–7. doi: 10.1186/s13073-017-0490-5
67. Hall AB, Yassour M, Sauk J, Garner A, Jiang X, Arthur T, et al. A Novel Ruminococcus Gnavus Clade Enriched in Inflammatory Bowel Disease Patients. Genome Med (2017) 9:103. doi: 10.1186/s13073-017-0490-5
68. Henke MT, Brown EM, Cassilly CD, Vlamakis H, Xavier RJ, Clardy J. Capsular Polysaccharide Correlates With Immune Response to the Human Gut Microbe Ruminococcus Gnavus. Proc Natl Acad Sci USA (2021) 118. (20):e2007595118. doi: 10.1073/pnas.2007595118
69. Becker HEF, Jamin C, Bervoets L, Boleij A, Xu P, Pierik MJ, et al. Higher Prevalence of Bacteroides Fragilis in Crohn’s Disease Exacerbations and Strain-Dependent Increase of Epithelial Resistance. Front Microbiol (2021) 12:598232. doi: 10.3389/fmicb.2021.598232
70. Cosorich I, Dalla-Costa G, Sorini C, Ferrarese R, Messina MJ, Dolpady J, et al. High Frequency of Intestinal TH17 Cells Correlates With Microbiota Alterations and Disease Activity in Multiple Sclerosis. Sci Adv (2017) 3:e1700492. doi: 10.1126/sciadv.1700492
71. Lee YK, Menezes JS, Umesaki Y, Mazmanian SK. Proinflammatory T-Cell Responses to Gut Microbiota Promote Experimental Autoimmune Encephalomyelitis. Proc Natl Acad Sci USA (2011) 108 Suppl 1:4615–22.
72. Tremlett H, Fadrosh DW, Faruqi AA, Zhu F, Hart J, Roalstad S, et al. Gut Microbiota in Early Pediatric Multiple Sclerosis: A Case-Control Study. Eur J Neurol (2016) 23:1308–21. doi: 10.1111/ene.13026
73. Katz Sand I, Zhu Y, Ntranos A, Clemente JC, Cekanaviciute E, Brandstadter R, et al. Disease-Modifying Therapies Alter Gut Microbial Composition in MS. Neurol Neuroimmunol Neuroinflamm (2019) 6:e517.
74. Mangalam A, Shahi SK, Luckey D, Karau M, Marietta E, Luo N, et al. Human Gut-Derived Commensal Bacteria Suppress CNS Inflammatory and Demyelinating Disease. Cell Rep (2017) 20:1269–77. doi: 10.1016/j.celrep.2017.07.031
75. Lavasani S, Dzhambazov B, Nouri M, Fåk F, Buske S, Molin G, et al. A Novel Probiotic Mixture Exerts a Therapeutic Effect on Experimental Autoimmune Encephalomyelitis Mediated by IL-10 Producing Regulatory T Cells. PloS One (2010) 5:e9009. doi: 10.1371/journal.pone.0009009
76. Salehipour Z, Haghmorad D, Sankian M, Rastin M, Nosratabadi R, Soltan Dallal MM, et al. Bifidobacterium Animalis in Combination With Human Origin of Lactobacillus Plantarum Ameliorate Neuroinflammation in Experimental Model of Multiple Sclerosis by Altering CD4+ T Cell Subset Balance. BioMed Pharmacother (2017) 95:1535–48. doi: 10.1016/j.biopha.2017.08.117
77. Kouchaki E, Tamtaji OR, Salami M, Bahmani F, Daneshvar Kakhaki R, Akbari E, et al. Clinical and Metabolic Response to Probiotic Supplementation in Patients With Multiple Sclerosis: A Randomized, Double-Blind, Placebo-Controlled Trial. Clin Nutr (2017) 36:1245–9. doi: 10.1016/j.clnu.2016.08.015
78. Secher T, Kassem S, Benamar M, Bernard I, Boury M, Barreau F, et al. Oral Administration of the Probiotic Strain Escherichia Coli Nissle 1917 Reduces Susceptibility to Neuroinflammation and Repairs Experimental Autoimmune Encephalomyelitis-Induced Intestinal Barrier Dysfunction. Front Immunol (2017) 8:1096. doi: 10.3389/fimmu.2017.01096
79. Planas R, Santos R, Tomas-Ojer P, Cruciani C, Lutterotti A, Faigle W, et al. GDP-L-Fucose Synthase Is a CD4(+) T Cell-Specific Autoantigen in DRB3*02:02 Patients With Multiple Sclerosis. Sci Transl Med (2018) 10. (462):eaat4301. doi: 10.1126/scitranslmed.aat4301
80. Horai R, Zárate-Bladés CR, Dillenburg-Pilla P, Chen J, Kielczewski JL, Silver PB, et al. Microbiota-Dependent Activation of an Autoreactive T Cell Receptor Provokes Autoimmunity in an Immunologically Privileged Site. Immunity (2015) 43:343–53. doi: 10.1016/j.immuni.2015.07.014
81. Heissigerova J, Seidler Stangova P, Klimova A, Svozilkova P, Hrncir T, Stepankova R, et al. The Microbiota Determines Susceptibility to Experimental Autoimmune Uveoretinitis. J Immunol Res (2016) 2016:5065703. doi: 10.1155/2016/5065703
82. Ichiki Y, Shimoda S, Hara H, Shigematsu H, Nakamura M, Hayashida K, et al. Analysis of T-Cell Receptor Beta of the T-Cell Clones Reactive to the Human PDC-E2 163-176 Peptide in the Context of HLA-DR53 in Patients With Primary Biliary Cirrhosis. Hepatology (1997) 26:728–33. doi: 10.1053/jhep.1997.v26.pm0009303504
83. Shimokawa C, Kato T, Takeuchi T, Ohshima N, Furuki T, Ohtsu Y, et al. CD8+ Regulatory T Cells Are Critical in Prevention of Autoimmune-Mediated Diabetes. Nat Commun (2020) 11:1922. doi: 10.1038/s41467-020-15857-x
84. de Goffau MC, Luopajärvi K, Knip M, Ilonen J, Ruohtula T, Härkönen T, et al. Fecal Microbiota Composition Differs Between Children With β-Cell Autoimmunity and Those Without. Diabetes (2013) 62:1238–44. doi: 10.2337/db12-0526
85. Greiling TM, Dehner C, Chen X, Hughes K, Iñiguez AJ, Boccitto M, et al. Commensal Orthologs of the Human Autoantigen Ro60 as Triggers of Autoimmunity in Lupus. Sci Transl Med (2018) 10(434):eaan2306. doi: 10.1126/scitranslmed.aan2306
86. Wu HJ, Ivanov II, Darce J, Hattori K, Shima T, Umesaki Y, et al. Gut-Residing Segmented Filamentous Bacteria Drive Autoimmune Arthritis via T Helper 17 Cells. Immunity (2010) 32:815–27. doi: 10.1016/j.immuni.2010.06.001
87. de Aquino SG, Abdollahi-Roodsaz S, Koenders MI, van de Loo FA, Pruijn GJ, Marijnissen RJ, et al. Periodontal Pathogens Directly Promote Autoimmune Experimental Arthritis by Inducing a TLR2- and IL-1-Driven Th17 Response. J Immunol (2014) 192:4103–11. doi: 10.4049/jimmunol.1301970
88. Lee JY, Choi IA, Kim JH, Kim KH, Lee EY, Lee EB, et al. Association Between Anti-Porphyromonas Gingivalis or Anti-α-Enolase Antibody and Severity of Periodontitis or Rheumatoid Arthritis (RA) Disease Activity in RA. BMC Musculoskelet Disord (2015) 16:190. doi: 10.1186/s12891-015-0647-6
89. Wegner N, Wait R, Sroka A, Eick S, Nguyen KA, Lundberg K, et al. Peptidylarginine Deiminase From Porphyromonas Gingivalis Citrullinates Human Fibrinogen and α-Enolase: Implications for Autoimmunity in Rheumatoid Arthritis. Arthritis Rheum (2010) 62:2662–72. doi: 10.1002/art.27552
90. Maeda Y, Kurakawa T, Umemoto E, Motooka D, Ito Y, Gotoh K, et al. Dysbiosis Contributes to Arthritis Development via Activation of Autoreactive T Cells in the Intestine. Arthritis Rheumatol (2016) 68:2646–61. doi: 10.1002/art.39783
91. Chen J, Wright K, Davis JM, Jeraldo P, Marietta EV, Murray J, et al. An Expansion of Rare Lineage Intestinal Microbes Characterizes Rheumatoid Arthritis. Genome Med (2016) 8:43. doi: 10.1186/s13073-016-0299-7
92. Matysiak-Budnik T, Coffin B, Lavergne-Slove A, Sabate JM, Mégraud F, Heyman M. Helicobacter Pylori Increases the Epithelial Permeability to a Food Antigen in Human Gastric Biopsies. Am J Gastroenterol (2004) 99:225–32. doi: 10.1111/j.1572-0241.2004.04080.x
93. Wang SK, Zhu HF, He BS, Zhang ZY, Chen ZT, Wang ZZ, et al. CagA+ H Pylori Infection Is Associated With Polarization of T Helper Cell Immune Responses in Gastric Carcinogenesis. World J Gastroenterol (2007) 13:2923–31. doi: 10.3748/wjg.v13.i21.2923
94. Yu M, Zhang R, Ni P, Chen S, Duan G. Helicobacter Pylori Infection and Psoriasis: A Systematic Review and Meta-Analysis. Med (Kaunas) (2019) 63. (3)193–200. doi: 10.3390/medicina55100645
95. Clevers H. Inflammatory Bowel Disease, Stress, and the Endoplasmic Reticulum. N Engl J Med (2009) 360:726–7. doi: 10.1056/NEJMcibr0809591
96. Khor B, Gardet A, Xavier RJ. Genetics and Pathogenesis of Inflammatory Bowel Disease. Nature (2011) 474:307–17. doi: 10.1038/nature10209
97. Cho JH, Brant SR. Recent Insights Into the Genetics of Inflammatory Bowel Disease. Gastroenterology (2011) 140:1704–12. doi: 10.1053/j.gastro.2011.02.046
98. Duchmann R, Kaiser I, Hermann E, Mayet W, Ewe K, Meyer zum Büschenfelde KH. Tolerance Exists Towards Resident Intestinal Flora But Is Broken in Active Inflammatory Bowel Disease (IBD). Clin Exp Immunol (1995) 102:448–55. doi: 10.1111/j.1365-2249.1995.tb03836.x
99. Olives JP, Breton A, Hugot JP, Oksman F, Johannet C, Ghisolfi J, et al. Antineutrophil Cytoplasmic Antibodies in Children With Inflammatory Bowel Disease: Prevalence and Diagnostic Value. J Pediatr Gastroenterol Nutr (1997) 25:142–8. doi: 10.1097/00005176-199708000-00003
100. Derer S, Brethack AK, Pietsch C, Jendrek ST, Nitzsche T, Bokemeyer A, et al. Inflammatory Bowel Disease-Associated GP2 Autoantibodies Inhibit Mucosal Immune Response to Adherent-Invasive Bacteria. Inflammation Bowel Dis (2020) 26:1856–68. doi: 10.1093/ibd/izaa069
101. Shpoliansky M, Roggenbuck D, Pinsker M, Salamon N, Weiss B, Shouval DS, et al. Antibodies Against Glycoprotein 2 Are Specific Biomarkers for Pediatric Crohn's Disease. Dig Dis Sci (2021) 66:2619–26. doi: 10.1007/s10620-020-06589-5
102. Vermeulen N, de Béeck KO, Vermeire S, Van Steen K, Michiels G, Ballet V, et al. Identification of a Novel Autoantigen in Inflammatory Bowel Disease by Protein Microarray. Inflammation Bowel Dis (2011) 17:1291–300. doi: 10.1002/ibd.21508
103. Hegazy AN, West NR, Stubbington MJT, Wendt E, Suijker KIM, Datsi A, et al. Circulating and Tissue-Resident CD4(+) T Cells With Reactivity to Intestinal Microbiota Are Abundant in Healthy Individuals and Function Is Altered During Inflammation. Gastroenterology (2017) 153:1320–1337.e16. doi: 10.1053/j.gastro.2017.07.047
104. Pirzer U, Schönhaar A, Fleischer B, Hermann E, Meyer zum Büschenfelde KH. Reactivity of Infiltrating T Lymphocytes With Microbial Antigens in Crohn's Disease. Lancet (1991) 338:1238–9. doi: 10.1016/0140-6736(91)92104-A
105. Landers CJ, Cohavy O, Misra R, Yang H, Lin YC, Braun J, et al. Selected Loss of Tolerance Evidenced by Crohn's Disease-Associated Immune Responses to Auto- and Microbial Antigens. Gastroenterology (2002) 123:689–99. doi: 10.1053/gast.2002.35379
106. Targan SR, Landers CJ, Yang H, Lodes MJ, Cong Y, Papadakis KA, et al. Antibodies to CBir1 Flagellin Define a Unique Response That Is Associated Independently With Complicated Crohn's Disease. Gastroenterology (2005) 128:2020–8. doi: 10.1053/j.gastro.2005.03.046
107. Fuss IJ, Neurath M, Boirivant M, Klein JS, de la Motte C, Strong SA, et al. Disparate CD4+ Lamina Propria (LP) Lymphokine Secretion Profiles in Inflammatory Bowel Disease. Crohn's Disease LP Cells Manifest Increased Secretion of IFN-Gamma, Whereas Ulcerative Colitis LP Cells Manifest Increased Secretion of IL-5. J Immunol (1996) 157:1261–70.
108. Matsuoka K, Inoue N, Sato T, Okamoto S, Hisamatsu T, Kishi Y, et al. T-Bet Upregulation and Subsequent Interleukin 12 Stimulation Are Essential for Induction of Th1 Mediated Immunopathology in Crohn's Disease. Gut (2004) 53:1303–8. doi: 10.1136/gut.2003.024190
109. Kosiewicz MM, Nast CC, Krishnan A, Rivera-Nieves J, Moskaluk CA, Matsumoto S, et al. Th1-Type Responses Mediate Spontaneous Ileitis in a Novel Murine Model of Crohn's Disease. J Clin Invest (2001) 107:695–702. doi: 10.1172/JCI10956
110. Kontoyiannis D, Pasparakis M, Pizarro TT, Cominelli F, Kollias G. Impaired On/Off Regulation of TNF Biosynthesis in Mice Lacking TNF AU-Rich Elements: Implications for Joint and Gut-Associated Immunopathologies. Immunity (1999) 10:387–98. doi: 10.1016/S1074-7613(00)80038-2
111. Ito H, Fathman CG. CD45RBhigh CD4+ T Cells From IFN-Gamma Knockout Mice Do Not Induce Wasting Disease. J Autoimmun (1997) 10:455–9. doi: 10.1016/S0896-8411(97)90152-9
112. Neurath MF, Weigmann B, Finotto S, Glickman J, Nieuwenhuis E, Iijima H, et al. The Transcription Factor T-Bet Regulates Mucosal T Cell Activation in Experimental Colitis and Crohn's Disease. J Exp Med (2002) 195:1129–43. doi: 10.1084/jem.20011956
113. Claesson MH, Bregenholt S, Bonhagen K, Thoma S, Möller P, Grusby MJ, et al. Colitis-Inducing Potency of CD4+ T Cells in Immunodeficient, Adoptive Hosts Depends on Their State of Activation, IL-12 Responsiveness, and CD45RB Surface Phenotype. J Immunol (1999) 162:3702–10.
114. Jiang W, Su J, Zhang X, Cheng X, Zhou J, Shi R, et al. Elevated Levels of Th17 Cells and Th17-Related Cytokines Are Associated With Disease Activity in Patients With Inflammatory Bowel Disease. Inflamm Res (2014) 63:943–50. doi: 10.1007/s00011-014-0768-7
115. Olsen T, Rismo R, Cui G, Goll R, Christiansen I, Florholmen J. TH1 and TH17 Interactions in Untreated Inflamed Mucosa of Inflammatory Bowel Disease, and Their Potential to Mediate the Inflammation. Cytokine (2011) 56:633–40. doi: 10.1016/j.cyto.2011.08.036
116. Jostins L, Ripke S, Weersma RK, Duerr RH, McGovern DP, Hui KY, et al. Host-Microbe Interactions Have Shaped the Genetic Architecture of Inflammatory Bowel Disease. Nature (2012) 491:119–24. doi: 10.1038/nature11582
117. Durant L, Watford WT, Ramos HL, Laurence A, Vahedi G, Wei L, et al. Diverse Targets of the Transcription Factor STAT3 Contribute to T Cell Pathogenicity and Homeostasis. Immunity (2010) 32:605–15. doi: 10.1016/j.immuni.2010.05.003
118. Leppkes M, Becker C, Ivanov II, Hirth S, Wirtz S, Neufert C, et al. RORgamma-Expressing Th17 Cells Induce Murine Chronic Intestinal Inflammation via Redundant Effects of IL-17A and IL-17f. Gastroenterology (2009) 136:257–67. doi: 10.1053/j.gastro.2008.10.018
119. Reynolds JM, Martinez GJ, Nallaparaju KC, Chang SH, Wang YH, Dong C. Cutting Edge: Regulation of Intestinal Inflammation and Barrier Function by IL-17c. J Immunol (2012) 189:4226–30. doi: 10.4049/jimmunol.1103014
120. Song X, Dai D, He X, Zhu S, Yao Y, Gao H, et al. Growth Factor FGF2 Cooperates With Interleukin-17 to Repair Intestinal Epithelial Damage. Immunity (2015) 43:488–501. doi: 10.1016/j.immuni.2015.06.024
121. Cosmi L, Maggi L, Santarlasci V, Liotta F, Annunziato F. T Helper Cells Plasticity in Inflammation. Cytom A (2014) 85:36–42. doi: 10.1002/cyto.a.22348
122. Sundrud MS, Trivigno C. Identity Crisis of Th17 Cells: Many Forms, Many Functions, Many Questions. Semin Immunol (2013) 25:263–72. doi: 10.1016/j.smim.2013.10.021
123. Ghoreschi K, Laurence A, Yang XP, Tato CM, McGeachy MJ, Konkel JE, et al. Generation of Pathogenic T(H)17 Cells in the Absence of TGF-β Signalling. Nature (2010) 467:967–71. doi: 10.1038/nature09447
124. Lee Y, Awasthi A, Yosef N, Quintana FJ, Xiao S, Peters A, et al. Induction and Molecular Signature of Pathogenic TH17 Cells. Nat Immunol (2012) 13:991–9. doi: 10.1038/ni.2416
125. Ramesh R, Kozhaya L, McKevitt K, Djuretic IM, Carlson TJ, Quintero MA, et al. Pro-Inflammatory Human Th17 Cells Selectively Express P-Glycoprotein and Are Refractory to Glucocorticoids. J Exp Med (2014) 211:89–104. doi: 10.1084/jem.20130301
126. Annese V, Valvano MR, Palmieri O, Latiano A, Bossa F, Andriulli A. Multidrug Resistance 1 Gene in Inflammatory Bowel Disease: A Meta-Analysis. World J Gastroenterol (2006) 12:3636–44. doi: 10.3748/wjg.v12.i23.3636
127. Mann ER, Bernardo D, Ng SC, Rigby RJ, Al-Hassi HO, Landy J, et al. Human Gut Dendritic Cells Drive Aberrant Gut-Specific T-Cell Responses in Ulcerative Colitis, Characterized by Increased IL-4 Production and Loss of IL-22 and Ifnγ. Inflammation Bowel Dis (2014) 20:2299–307. doi: 10.1097/MIB.0000000000000223
128. Levo Y, Shalit M, Wollner S, Fich A. Serum IgE Levels in Patients With Inflammatory Bowel Disease. Ann Allergy (1986) 56:85–7.
129. Hoving JC, Kirstein F, Nieuwenhuizen NE, Fick LC, Hobeika E, Reth M, et al. B Cells That Produce Immunoglobulin E Mediate Colitis in BALB/c Mice. Gastroenterology (2012) 142:96–108. doi: 10.1053/j.gastro.2011.09.044
130. Bamias G, Martin C, Mishina M, Ross WG, Rivera-Nieves J, Marini M, et al. Proinflammatory Effects of TH2 Cytokines in a Murine Model of Chronic Small Intestinal Inflammation. Gastroenterology (2005) 128:654–66. doi: 10.1053/j.gastro.2004.11.053
131. Fichtner-Feigl S, Strober W, Geissler EK, Schlitt HJ. Cytokines Mediating the Induction of Chronic Colitis and Colitis-Associated Fibrosis. Mucosal Immunol (2008) 1 Suppl 1:S24–7. doi: 10.1038/mi.2008.41
132. Mottawea W, Chiang CK, Mühlbauer M, Starr AE, Butcher J, Abujamel T, et al. Altered Intestinal Microbiota-Host Mitochondria Crosstalk in New Onset Crohn's Disease. Nat Commun (2016) 7:13419. doi: 10.1038/ncomms13419
133. Geirnaert A, Calatayud M, Grootaert C, Laukens D, Devriese S, Smagghe G, et al. Butyrate-Producing Bacteria Supplemented In Vitro to Crohn’s Disease Patient Microbiota Increased Butyrate Production and Enhanced Intestinal Epithelial Barrier Integrity. Sci Rep (2017) 7:11450. doi: 10.1038/s41598-017-11734-8
134. Marchesi JR, Holmes E, Khan F, Kochhar S, Scanlan P, Shanahan F, et al. Rapid and Noninvasive Metabonomic Characterization of Inflammatory Bowel Disease. J Proteome Res (2007) 6:546–51. doi: 10.1021/pr060470d
135. Veltkamp C, Anstaett M, Wahl K, Möller S, Gangl S, Bachmann O, et al. Apoptosis of Regulatory T Lymphocytes Is Increased in Chronic Inflammatory Bowel Disease and Reversed by Anti-Tnfα Treatment. Gut (2011) 60:1345–53. doi: 10.1136/gut.2010.217117
136. Glocker EO, Kotlarz D, Boztug K, Gertz EM, Schäffer AA, Noyan F, et al. Inflammatory Bowel Disease and Mutations Affecting the Interleukin-10 Receptor. N Engl J Med (2009) 361:2033–45. doi: 10.1056/NEJMoa0907206
137. Cook L, Lisko DJ, Wong MQ, Garcia RV, Himmel ME, Seidman EG, et al. Analysis of Flagellin-Specific Adaptive Immunity Reveals Links to Dysbiosis in Patients With Inflammatory Bowel Disease. Cell Mol Gastroenterol Hepatol (2020) 9:485–506. doi: 10.1016/j.jcmgh.2019.11.012
138. Targan SR, Landers CJ, Cobb L, MacDermott RP, Vidrich A. Perinuclear Anti-Neutrophil Cytoplasmic Antibodies Are Spontaneously Produced by Mucosal B Cells of Ulcerative Colitis Patients. J Immunol (1995) 155:3262–7.
139. Cohavy O, Bruckner D, Gordon LK, Misra R, Wei B, Eggena ME, et al. Colonic Bacteria Express an Ulcerative Colitis pANCA-Related Protein Epitope. Infect Immun (2000) 68:1542–8. doi: 10.1128/IAI.68.3.1542-1548.2000
140. Sospedra M, Martin R. Immunology of Multiple Sclerosis. Semin Neurol (2016) 36:115–27. doi: 10.1055/s-0036-1579739
141. Ascherio A. Environmental Factors in Multiple Sclerosis. Expert Rev Neurother (2013) 13:3–9. doi: 10.1586/14737175.2013.865866
142. Oksenberg JR. Decoding Multiple Sclerosis: An Update on Genomics and Future Directions. Expert Rev Neurother (2013) 13:11–9. doi: 10.1586/14737175.2013.865867
143. Jangi S, Gandhi R, Cox LM, Li N, von Glehn F, Yan R, et al. Alterations of the Human Gut Microbiome in Multiple Sclerosis. Nat Commun (2016) 7:12015. doi: 10.1038/ncomms12015
144. Berer K, Mues M, Koutrolos M, Rasbi ZA, Boziki M, Johner C, et al. Commensal Microbiota and Myelin Autoantigen Cooperate to Trigger Autoimmune Demyelination. Nature (2011) 479:538–41. doi: 10.1038/nature10554
145. Suárez-Fueyo A, Bradley SJ, Tsokos GC. T Cells in Systemic Lupus Erythematosus. Curr Opin Immunol (2016) 43:32–8. doi: 10.1016/j.coi.2016.09.001
146. Deng Y, Tsao BP. Updates in Lupus Genetics. Curr Rheumatol Rep (2017) 19:68. doi: 10.1007/s11926-017-0695-z
147. Rosenbaum JT, Silverman GJ. The Microbiome and Systemic Lupus Erythematosus. N Engl J Med (2018) 378:2236–7. doi: 10.1056/NEJMcibr1804368
148. Silverman GJ. The Microbiome in SLE Pathogenesis. Nat Rev Rheumatol (2019) 15:72–4. doi: 10.1038/s41584-018-0152-z
149. Van Praet JT, Donovan E, Vanassche I, Drennan MB, Windels F, Dendooven A, et al. Commensal Microbiota Influence Systemic Autoimmune Responses. EMBO J (2015) 34:466–74. doi: 10.15252/embj.201489966
150. Wolin SL, Reinisch KM. The Ro 60 kDa Autoantigen Comes Into Focus: Interpreting Epitope Mapping Experiments on the Basis of Structure. Autoimmun Rev (2006) 5:367–72. doi: 10.1016/j.autrev.2005.10.004
151. Deshmukh US, Lewis JE, Gaskin F, Kannapell CC, Waters ST, Lou YH, et al. Immune Responses to Ro60 and Its Peptides in Mice. I. The Nature of the Immunogen and Endogenous Autoantigen Determine the Specificities of the Induced Autoantibodies. J Exp Med (1999) 189:531–40. doi: 10.1084/jem.189.3.531
152. Knip M, Siljander H. The Role of the Intestinal Microbiota in Type 1 Diabetes Mellitus. Nat Rev Endocrinol (2016) 12:154–67. doi: 10.1038/nrendo.2015.218
153. Paun A, Yau C, Danska JS. The Influence of the Microbiome on Type 1 Diabetes. J Immunol (2017) 198:590–5. doi: 10.4049/jimmunol.1601519
154. Needell JC, Zipris D. The Role of the Intestinal Microbiome in Type 1 Diabetes Pathogenesis. Curr Diabetes Rep (2016) 16:89. doi: 10.1007/s11892-016-0781-z
155. Li X, Atkinson MA. The Role for Gut Permeability in the Pathogenesis of Type 1 Diabetes–A Solid or Leaky Concept? Pediatr Diabetes (2015) 16:485–92. doi: 10.1111/pedi.12305
156. Maffeis C, Martina A, Corradi M, Quarella S, Nori N, Torriani S, et al. Association Between Intestinal Permeability and Faecal Microbiota Composition in Italian Children With Beta Cell Autoimmunity at Risk for Type 1 Diabetes. Diabetes Metab Res Rev (2016) 32:700–9. doi: 10.1002/dmrr.2790
157. Kostic AD, Gevers D, Siljander H, Vatanen T, Hyötyläinen T, Hämäläinen AM, et al. The Dynamics of the Human Infant Gut Microbiome in Development and in Progression Toward Type 1 Diabetes. Cell Host Microbe (2015) 17:260–73. doi: 10.1016/j.chom.2015.01.001
158. Livanos AE, Greiner TU, Vangay P, Pathmasiri W, Stewart D, McRitchie S, et al. Antibiotic-Mediated Gut Microbiome Perturbation Accelerates Development of Type 1 Diabetes in Mice. Nat Microbiol (2016) 1:16140. doi: 10.1038/nmicrobiol.2016.140
159. Tai N, Peng J, Liu F, Gulden E, Hu Y, Zhang X, et al. Microbial Antigen Mimics Activate Diabetogenic CD8 T Cells in NOD Mice. J Exp Med (2016) 213:2129–46. doi: 10.1084/jem.20160526
160. de Goffau MC, Fuentes S, van den Bogert B, Honkanen H, de Vos WM, Welling GW, et al. Aberrant Gut Microbiota Composition at the Onset of Type 1 Diabetes in Young Children. Diabetologia (2014) 57:1569–77. doi: 10.1007/s00125-014-3274-0
161. Vatanen T, Franzosa EA, Schwager R, Tripathi S, Arthur TD, Vehik K, et al. The Human Gut Microbiome in Early-Onset Type 1 Diabetes From the TEDDY Study. Nature (2018) 562:589–94. doi: 10.1038/s41586-018-0620-2
162. Mariño E, Richards JL, McLeod KH, Stanley D, Yap YA, Knight J, et al. Gut Microbial Metabolites Limit the Frequency of Autoimmune T Cells and Protect Against Type 1 Diabetes. Nat Immunol (2017) 18:552–62. doi: 10.1038/ni.3713
163. Tajik N, Frech M, Schulz O, Schälter F, Lucas S, Azizov V, et al. Targeting Zonulin and Intestinal Epithelial Barrier Function to Prevent Onset of Arthritis. Nat Commun (2020) 11:1995. doi: 10.1038/s41467-020-15831-7
164. Zhang X, Zhang D, Jia H, Feng Q, Wang D, Liang D, et al. The Oral and Gut Microbiomes Are Perturbed in Rheumatoid Arthritis and Partly Normalized After Treatment. Nat Med (2015) 21:895–905. doi: 10.1038/nm.3914
165. Albani S, Keystone EC, Nelson JL, Ollier WE, La Cava A, Montemayor AC, et al. Positive Selection in Autoimmunity: Abnormal Immune Responses to a Bacterial dnaJ Antigenic Determinant in Patients With Early Rheumatoid Arthritis. Nat Med (1995) 1:448–52. doi: 10.1038/nm0595-448
166. Scher JU, Sczesnak A, Longman RS, Segata N, Ubeda C, Bielski C, et al. Expansion of Intestinal Prevotella Copri Correlates With Enhanced Susceptibility to Arthritis. Elife (2013) 2:e01202. doi: 10.7554/eLife.01202
167. Romero V, Fert-Bober J, Nigrovic PA, Darrah E, Haque UJ, Lee DM, et al. Immune-Mediated Pore-Forming Pathways Induce Cellular Hypercitrullination and Generate Citrullinated Autoantigens in Rheumatoid Arthritis. Sci Transl Med (2013) 5:209ra150. doi: 10.1126/scitranslmed.3006869
168. Rantapää-Dahlqvist S, de Jong BA, Berglin E, Hallmans G, Wadell G, Stenlund H, et al. Antibodies Against Cyclic Citrullinated Peptide and IgA Rheumatoid Factor Predict the Development of Rheumatoid Arthritis. Arthritis Rheum (2003) 48:2741–9. doi: 10.1002/art.11223
169. Nestle FO, Kaplan DH, Barker J. Psoriasis. N Engl J Med (2009) 361:496–509. doi: 10.1056/NEJMra0804595
170. Valdimarsson H, Bake BS, Jónsdótdr I, Fry L. Psoriasis: A Disease of Abnormal Keratinocyte Proliferation Induced by T Lymphocytes. Immunol Today (1986) 7:256–9. doi: 10.1016/0167-5699(86)90005-8
171. Conrad C, Boyman O, Tonel G, Tun-Kyi A, Laggner U, de Fougerolles A, et al. Alpha1beta1 Integrin Is Crucial for Accumulation of Epidermal T Cells and the Development of Psoriasis. Nat Med (2007) 13:836–42. doi: 10.1038/nm1605
172. Kryczek I, Bruce AT, Gudjonsson JE, Johnston A, Aphale A, Vatan L, et al. Induction of IL-17+ T Cell Trafficking and Development by IFN-Gamma: Mechanism and Pathological Relevance in Psoriasis. J Immunol (2008) 181:4733–41. doi: 10.4049/jimmunol.181.7.4733
173. Ruiz-Romeu E, Ferran M, de Jesus-Gil C, Garcia P, Sagrista M, Casanova JM, et al. Microbe-Dependent Induction of IL-9 by CLA(+) T Cells in Psoriasis and Relationship With IL-17a. J Invest Dermatol (2018) 138:580–7. doi: 10.1016/j.jid.2017.08.048
174. Zanvit P, Konkel JE, Jiao X, Kasagi S, Zhang D, Wu R, et al. Antibiotics in Neonatal Life Increase Murine Susceptibility to Experimental Psoriasis. Nat Commun (2015) 6:8424. doi: 10.1038/ncomms9424
175. Stehlikova Z, Kostovcikova K, Kverka M, Rossmann P, Dvorak J, Novosadova I, et al. Crucial Role of Microbiota in Experimental Psoriasis Revealed by a Gnotobiotic Mouse Model. Front Microbiol (2019) 10:236. doi: 10.3389/fmicb.2019.00236
176. Zakostelska Z, Malkova J, Klimesova K, Rossmann P, Hornova M, Novosadova I, et al. Intestinal Microbiota Promotes Psoriasis-Like Skin Inflammation by Enhancing Th17 Response. PloS One (2016) 11:e0159539. doi: 10.1371/journal.pone.0159539
177. Testerman TL, Morris J. Beyond the Stomach: An Updated View of Helicobacter Pylori Pathogenesis, Diagnosis, and Treatment. World J Gastroenterol (2014) 20:12781–808. doi: 10.3748/wjg.v20.i36.12781
178. Kao CY, Sheu BS, Sheu SM, Yang HB, Chang WL, Cheng HC, et al. Higher Motility Enhances Bacterial Density and Inflammatory Response in Dyspeptic Patients Infected With Helicobacter Pylori. Helicobacter (2012) 17:411–6. doi: 10.1111/j.1523-5378.2012.00974.x
179. Magen E, Delgado JS. Helicobacter Pylori and Skin Autoimmune Diseases. World J Gastroenterol (2014) 20:1510–6. doi: 10.3748/wjg.v20.i6.1510
180. Radić M. Role of Helicobacter Pylori Infection in Autoimmune Systemic Rheumatic Diseases. World J Gastroenterol (2014) 20:12839–46. doi: 10.3748/wjg.v20.i36.12839
181. Picardo M, Dell'Anna ML, Ezzedine K, Hamzavi I, Harris JE, Parsad D, et al. Vitiligo. Nat Rev Dis Primers (2015) 1:15011. doi: 10.1038/nrdp.2015.11
Keywords: gut microbiota, dysbiosis, autoreactive T-cell responses, autoimmune disease, molecular mimicry, gut microbial metabolites, immunoregulation, gut microbial homeostasis
Citation: Garabatos N and Santamaria P (2022) Gut Microbial Antigenic Mimicry in Autoimmunity. Front. Immunol. 13:873607. doi: 10.3389/fimmu.2022.873607
Received: 11 February 2022; Accepted: 14 March 2022;
Published: 27 April 2022.
Edited by:
Guillaume Sarrabayrouse, Université de Paris, FranceReviewed by:
Julien Diana, Institut National de la Santé et de la Recherche Médicale (INSERM), FranceCopyright © 2022 Garabatos and Santamaria. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Pere Santamaria, cHNhbnRhbWFAdWNhbGdhcnkuY2E=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.