
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Immunol. , 27 May 2022
Sec. Microbial Immunology
Volume 13 - 2022 | https://doi.org/10.3389/fimmu.2022.872286
 Xiaoman Guo1
Xiaoman Guo1 Xinru Mao1
Xinru Mao1 Di Tian2
Di Tian2 Yixin Liao2
Yixin Liao2 Bintao Su1,3
Bintao Su1,3 Chaoliang Ye1Dongling Shi4
Chaoliang Ye1Dongling Shi4 Tie Fu Liu2*
Tie Fu Liu2* Yun Ling4*
Yun Ling4* Yi Hao1,5*
Yi Hao1,5*Cryptococcus neoformans infection in the central nervous system is a severe infectious disease with poor outcomes and high mortality. It has been estimated that there are 220,000 new cases each year. Over 90% of C. neoformans meningitis cases were diagnosed in AIDS patients with CD4+ T cell count <100 cells/μl; however, the mechanism of cryptococcal meningitis in patients with normal immune functions remains unclear. IL-17 is a pro-inflammatory cytokine and plays an important role in anti-fungal immunity. Here we report that significantly high levels of IL-17 were predominantly detected in the cerebrospinal fluid of patients with either AIDS- or non-AIDS-associated C. neoformans meningitis but not in patients with tuberculous meningitis or non-neurosyphilis. Antifungal therapy minimized the IL-17 level in the cerebrospinal fluid. An in vitro mechanistic study showed that C. neoformans stimulation of healthy peripheral blood mononuclear cells prompted IL-17 production, and CD4+ T cells were the predominant IL-17-producing cells. IL-17 production by C. neoformans stimulation was STAT3 signaling dependent. Inhibition of STAT3 phosphorylation attenuated the C. neoformans-mediated IL-17 expression. Our data highlighted the significance of CD4+ T cells in antifungal immunity and suggested IL-17 as a diagnostic biomarker of C. neoformans infection and STAT3 as a checkpoint for antifungal targeted therapies.
Cryptococcus neoformans (C. neoformans) meningitis (CM) is a systemic and opportunistic fungal infectious disease with morbidity and mortality between 10% and 25% in medically advanced countries (1) and is often diagnosed in immunocompromised patients relevant or irrelevant to HIV/AIDS (2). It contributes about 15% of AIDS-associated opportunistic infection (3). In recent years, more and more cases of non-AIDS-associated C. neoformans meningitis have been reported, and about 220,000 new cases and 181,000 deaths have been estimated each year (3).
Previous studies of anti-fungal immunity in animal models suggested that Th17-type response is important for survival in C. neoformans infection (4). Pulmonary infection with C. neoformans strain H99c increased the pulmonary IL-17 (commonly known as IL-17A) production (5), and the IL-17 level of cerebrospinal fluids (CSF) in HIV-1 infection-associated CM is significantly higher than that in tuberculous meningitis (TBM) (6). IL-17 is a cytokine of CD4+ T helper subset, T helper 17 (Th17) cells, and has originally received attention for its pro-inflammatory function in autoimmunity (7–9). Previous research in a mouse model of C. neoformans infection suggested that a Th17-type response and IL-17 production are important for modulating survival against cryptococcosis (4). Besides this, the genetic defects of the IL-17 signaling pathway contribute to severe mucocutaneous Candida, oropharyngeal Candida, and Aspergillus infections in humans (10–12).
The differentiation of Th17 cells is regulated by a variety of signal pathways, among which the JAK2/STAT3 signal pathway plays a key role. The activated CD4+ T cells produce IL-6 to stimulate the JAK2/STAT3 pathway (13), thereby inducing the expression of the lineage-specific master regulator RORγt to promote the differentiation of Th17 cells (14, 15). Impaired Th17 cell differentiation is a consistent immune defect in STAT3 hyper-IgE syndromes, which may mediate protection against C. neoformans (16).
STAT3 is a potential transcription factor that transduces extracellular signals such as growth factors and cytokines through interaction with polypeptide receptors on the cell surface (17). It is post-translationally activated mainly by tyrosine phosphorylation to form STAT3 dimer, translocate from the cytoplasm into the nucleus, and bind to sequence-specific DNA elements of target genes (18). STAT3 is a consistently expressed protein, but its production is rapidly increased through self-regulation upon activation, as its promoter contains a binding site for its own dimers (19, 20). However, the mechanism of IL-17 induction remains unclear in non-HIV-associated C. neoformans meningitis.
The aim of this study was to investigate the mechanism of IL-17 production and the clinical effect of anti-fungal therapy on it in human C. neoformans meningitis. We found that the CSF of C. neoformans meningitis contains a high level of IL-17, and anti-fungal treatment minimizes the CSF IL-17 level and improves the clinical outcomes. A mechanistical study showed that STAT3 phosphorylation is essential for IL-17 induction. Our findings suggest that IL-17 is an important component in anti-fungal immunity and a potential biomarker for the diagnosis of C. neoformans meningitis.
This study was approved by the ethics committee of Shanghai Public Health Clinical Center, and informed consent was obtained from the participants. The patients were hospitalized at the Department of Infectious Disease in Shanghai Public Health Clinical Center. Healthy individuals were recruited from employee volunteers in Scientific Research Center of Shanghai Public Health Clinical Center. The CSF and blood samples and the clinical data were preserved in accordance with the Declaration of Helsinki and local legislations.
Eleven patients with non-AIDS-associated (HIV-CM) and five patients with AIDS-associated (HIV+CM) C. neoformans meningitis and fifteen patients with TBM (drug-resistant TB) participated in this study. Nine patients with non-neurotic syphilis served as the meningitis-free disease control. The above-mentioned diseases were diagnosed based on the typical clinical presentations of infection, together with neuroimaging characteristics and positive laboratory findings, including abnormal routine CSF biochemical examinations for CM and TBM, and positive specific antibodies or pathogen tests, according to the guidelines from the Centers for Disease Control and Prevention (CDC) and World Health Organization (WHO). The patients with pregnancy and secondary or primary immunodeficiency diseases were excluded. Only adult (>16 years) patients with confirmed infectious diseases were recruited in this study. Although the entire treatment will last around 1 year, in the current study, the patients with non-AIDS CM were under treatment for approximately 90 days. In the meantime, the patients with TBM were under medical treatment for about 100 days. The CSF and blood samples were collected on admission and at about 3 months after the pathogen-specific therapies. The laboratory findings for the participants were summarized in Table 1.
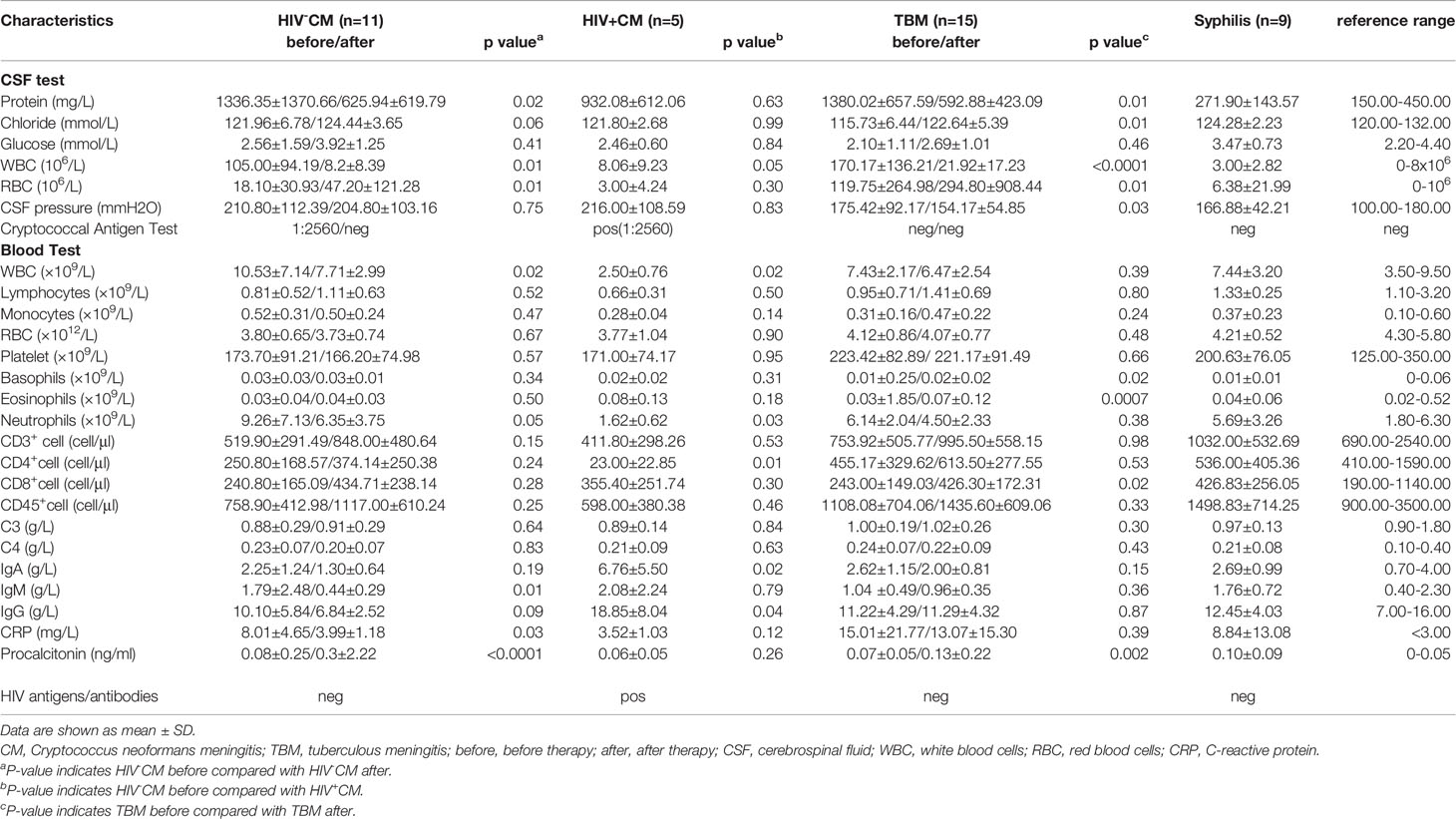
Table 1 Clinical laboratory indexes in patients and their changes after therapies.
Whole blood— anti-coagulated with heparin—from healthy individuals was diluted in 1:2 with RPMI 1640 and was centrifugated on Hypaque-Ficoll gradients (GE, density 1.077 g/ml) at 400g for 30 min. Peripheral blood mononuclear cells (PBMCs) were then collected and washed twice with phosphate-buffered saline (PBS). The cells were cultured in RPMI 1640 (Biological Industries) supplemented with 10% fetal bovine serum (Biological Industries) and 1% penicillin/streptomycin (Beyotime). PBMCs (1 × 106/ml) were stimulated for the indicated times with or without C. neoformans (1 × 106/ml, provided by our clinical laboratory) and bacillus Calmette–Guérin (BCG, 1 × 105/ml, provided by Professor Feifei Wang, Fudan University). In specific experiments, PBMCs were pretreated for 30 min with 2.5 μM of STAT3 inhibitor Stattic (Abcam) before stimulation for phospho-STAT3 analysis by flow cytometry.
The quantitative evaluation of intracellular cytokines was performed using the Human Cytometric Bead Array (CBA) Th1/Th2/Th17 Cytokine Kit (BD Biosciences), according to the manufacturer’s protocol. Equal volumes of assay beads, detection reagent, and the studied sample or standard were added consecutively to each tube and incubated in the dark for 3 h at room temperature. The samples were then washed with 1 ml of wash buffer and centrifuged at 500g for 5 min. After discarding the supernatant, the pellet was resuspended in 300 µl of buffer and analyzed on the same day by flow cytometry. FCAP Array Software, version 3.0, from BD Biosciences was employed to translate the images into data.
PBMCs were incubated in FACS buffer (PBS supplemented with 0.5% bovine serum albumin) for 20 min at 4°C with the following antibodies: anti-CD4 (L200, BD Biosciences), anti-CD8 (RPA-T8, BD Biosciences), anti-CD3 (OKT3, Invitrogen), anti-γδTCR (B1.1, Invitrogen), and anti-CD19 (HIB19, eBioscience). The cells were subsequently washed twice and were resuspended with FACS buffer. The flow cytometry data was acquired on BD LSR Fortessa and analyzed by FlowJo V10. Dead cells were excluded by using Fixable Viability Dye (Invitrogen).
PBMCs (1 × 106/well) were incubated in 24-well plates for 30 min or 3 days with different stimuli. Protein transport inhibitor cocktail (eBioscience) was added for 5 h before the end of the cell culture. After surface staining, the cells were fixed and permeabilized using an intracellular fixation and permeabilization buffer set (eBioscience). Intracellular IL-17 were stained with anti-IL-17 (IL-17A) antibodies (eBio64DEC17, eBioscience) according to the manufacturers’ protocols and analyzed by flow cytometry.
PBMCs were stimulated for 30 min with different stimuli. After surface marker staining, the cells were fixed with 4% paraformaldehyde (Biosharp) for 15 min at room temperature and permeabilized for a minimum of 10 min on ice by slowly adding ice-cold methanol (Sangon Biotech) to a final concentration of 90%. The cells were then stained with rabbit anti-human phospho-STAT3-Tyr705 monoclonal antibody (D3A7, Cell Signaling Technology) or human phospho-STAT1 (Tyr701) Rabbit mAb (58D6, Cell Signaling Technology) for 1 h and were followed by incubation with Alexa Fluor 488-goat anti-rabbit IgG (H+L) (Invitrogen) for 30 min at room temperature. The cells were then washed in PBS twice and were resuspended in 300 μl PBS supplemented with 0.5% bovine serum albumin for the flow cytometry analysis.
The levels of IL-6, IL-10, IL-17, and IFN-γ in the CSF and serum of patients with meningitis, syphilis, and healthy controls, and the levels of IL-17 and IFN-γ in the cell culture supernatants were measured by enzyme-linked immunosorbent assay (ELISA) using ELISA kits of human IL-6 (Absin), human IL-10 (Absin), human IL-17 (Abcam), and human IFN-γ (Abcam), respectively. The OD values at 450 nm were read in duplicates using an automatic microplate reader (BioTek, Synergy2). Serial dilutions of recombinant cytokines were used to generate a standard curve.
PBMCs were lysed on ice for 30 min using RIPA lysis buffer (Beyotime) with protease inhibitors (Beyotime) and phosphatase inhibitors cocktail (Beyotime), and the protein concentration was determined by a bicinchoninic acid assay kit (Biosharp). The proteins (50 µg per lane) were mixed with one-fifth volume of 5× loading buffer, separated by 10% SDS-PAGE, and were then transferred onto polyvinylidene difluoride membranes (Millipore). The membranes were blocked for 1 h with 5% nonfat milk at room temperature and were incubated overnight at 4°C with primary antibodies against STAT3 rabbit mAb (79D7, Cell Signaling Technology), phospho-STAT3-Tyr705 rabbit mAb (D3A7, Cell Signaling Technology), NF-κB p65 rabbit mAb (D14E12, Cell Signaling Technology), anti-NF-kB p65-phospho-S536 antibody (EP2294Y, Abcam), and β-actin rabbit monoclonal antibody (Beyotime). After 3 washes with Tris-buffered saline with 0.1% Tween® 20 detergent, the membranes were incubated for 1 h at room temperature with anti-rabbit horseradish peroxidase-conjugated secondary antibody (GE) and were detected with enhanced chemiluminescence (Beyotime). The results were analyzed using Image Studio software.
The statistical differences between the two groups were analyzed by Student’s t-test using GraphPad Prism Software 8.0 (GraphPad Software, Inc), and the statistical differences among multiple groups were analyzed by Tukey’s post-hoc test. P-value <0.05 was considered a statistically significant difference. Data were presented as mean ± standard deviation or standard error.
The confirmed meningitis patients including 11 CM (7 male and 4 female patients) and 15 TBM (9 male and 6 female patients) had been treated according to the Infectious Diseases Society of America guidelines. The CM patients received a combined anti-fungal therapy of fluconazole, flucytosine, and amphotericin B (21), and the TBM patients were treated with first-line anti-tuberculous drugs of rifampin, isoniazid, ethambutol, and pyrazinamide. The first-line drug-resistant patients were subsequently treated with the second-line drugs moxifloxacin and cycloserine (22). Besides this, all meningitis patients were given a corticosteroid therapy including prednisolone and dexamethasone in the early stage of the disease to relieve acute inflammatory responses and brain edema. The CSF and blood samples were examined before and after therapies. The clinical laboratory characteristics were compared in Table 1. Anti-fungal therapy cleared the CSF pathogen and significantly decreased the levels of protein, white blood cell (WBC), and red blood cell (RBC) counts in the CSF, WBC and neutrophil counts, and IgM, CRP, and procalcitonin in peripheral blood. Similarly, the anti-tuberculous therapy significantly decreased the levels of protein, chloride, WBC, and RBC counts in the CSF and the cell counts of basophils, eosinophils, and CD8+ T cells and procalcitonin in peripheral blood. All laboratory results of meningitis-free syphilis cases were in the normal range and were not re-checked after the anti-syphilis therapy.
To determine the cytokine expression of meningitis, we compared the CSF levels of proinflammatory cytokines IL-6, IL-17, and IFN-γ and anti-inflammatory cytokine IL-10 in patients with non-HIV and HIV-positive CM, TBM, and patients with meningitis-free syphilis. In comparison with the trace CSF levels of all 4 cytokines in non-neurotic syphilis, both non-HIV and HIV-positive CM and TBM significantly increased the CSF IL-6 and IL-10 levels, and TBM considerably increased the CSF IFN-γ and slightly increased the IL-17 levels. In contrast, non-HIV and HIV-positive CM selectively increased the CSF IL-17 level with a minimal induction of IFN-γ (Figures 1A, B). Similar to the findings in the CSF, both non-HIV and HIV-positive CM and TBM significantly induced serum IL-6, IL-10, and IFN-γ. In contrast, non-HIV CM, but not TBM, significantly induced serum IL-17 in comparison with the healthy controls (Figures 1C, D).
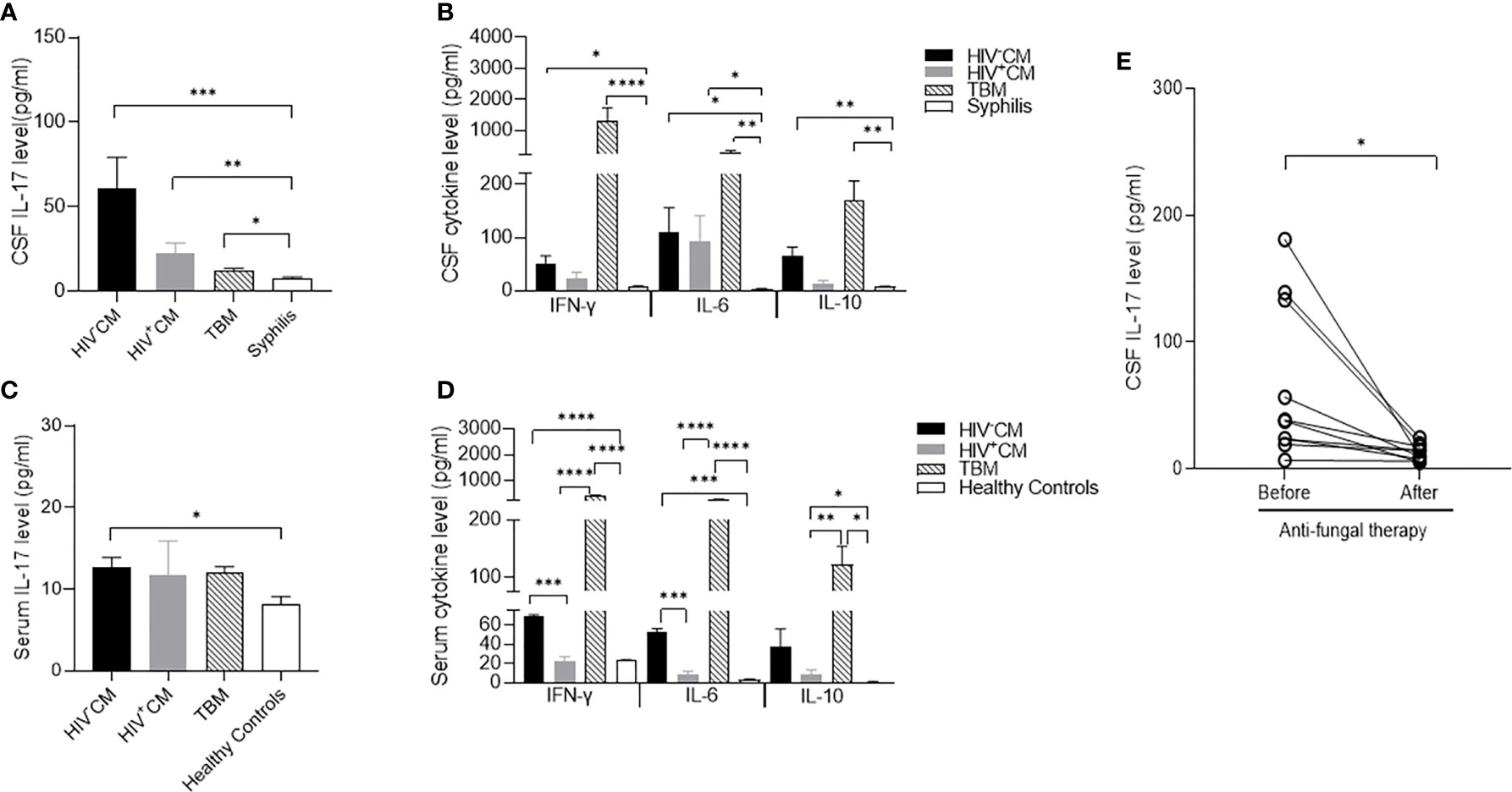
Figure 1 Cytokine levels of cerebrospinal fluid (CSF) and serum in patients with C. neoformans and tuberculosis meningitis. CSF was collected before and at about 3 months after the pathogen-specific therapies, while serum samples were collected on admission. The cytokine levels were measured by ELISA. (A) CSF levels of IL-17 in non-HIV CM (n = 11), HIV-positive CM (n = 5), TBM (n = 15), and syphilis (n = 9) on admission. (B) CSF levels of IFN-γ, IL-10, and IL-6 in non-HIV CM (n = 11), HIV-positive CM (n = 5), TBM (n = 15), and syphilis (n = 9) on admission. Serum levels of IL-17 (C) and IFN-γ, IL-10, and IL-6 (D) in healthy controls (n = 3), non-HIV CM (n = 4), HIV-positive CM (n = 5), and TBM (n = 4) at admission. (E) Changes of CSF levels of IL-17 in non-HIV CM (n = 11) and TBM (n = 15) before and after pathogen-specific therapies. Statistical significance was indicated as *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. Data are shown as mean ± SE of the samples. CM, C. neoformans meningitis; TBM, tuberculous meningitis.
We next followed the changes of CSF cytokine levels in response to pathogen-specific therapies. Consistent with the clinical improvement, the anti-fungal therapy minimized the CSF IL-17 level of non-HIV CM compared to that in meningitis-free syphilis (Figure 1E). The anti-fungal therapy also diminished CSF IL-6, IL-10, and IFN-γ (Supplementary Figure S1A). The anti-tuberculous therapy restored the CSF IFN-γ, IL-6, and IL-10 to basal levels but did not change the CSF IL-17 level (Supplementary Figure S1B).
To model an in vivo infection, we next stimulated healthy PBMCs for 3, 7, or 14 days with C. neoformans or BCG, which, as an efficient vaccine, is well known to best mimic in vivo conditional pathogenic infections and stimulate effective anti-tuberculosis immunity. Here BCG served as a stimulus to induce an anti-bacterial response in vitro. The levels of Th1 (IFN-γ and TNF-α) (Figures 2B, C), Th2 (IL-4, IL-10, and IL-6) (Figures 2D–F), and Th17 (IL-17) (Figure 2A) cytokines were followed by flow cytometric bead array. C. neoformans stimulation significantly induced IL-17 at day 3, which peaked at day 7 and was maintained at a high level at day 14, but it did not significantly change the levels of other cytokines, except IL-6 that was significantly increased at day 14. In contrast, BCG stimulation significantly induced IFN-γ, TNF-α, and IL-6 and IL-10 at day 3 and gradually decreased their levels thereafter; however, BCG did not significantly induce IL-17 production. Thus, in contrast to the fast Th1/Th2 response by BCG stimulation, C. neoformans stimulation induces a postponed Th17 response.
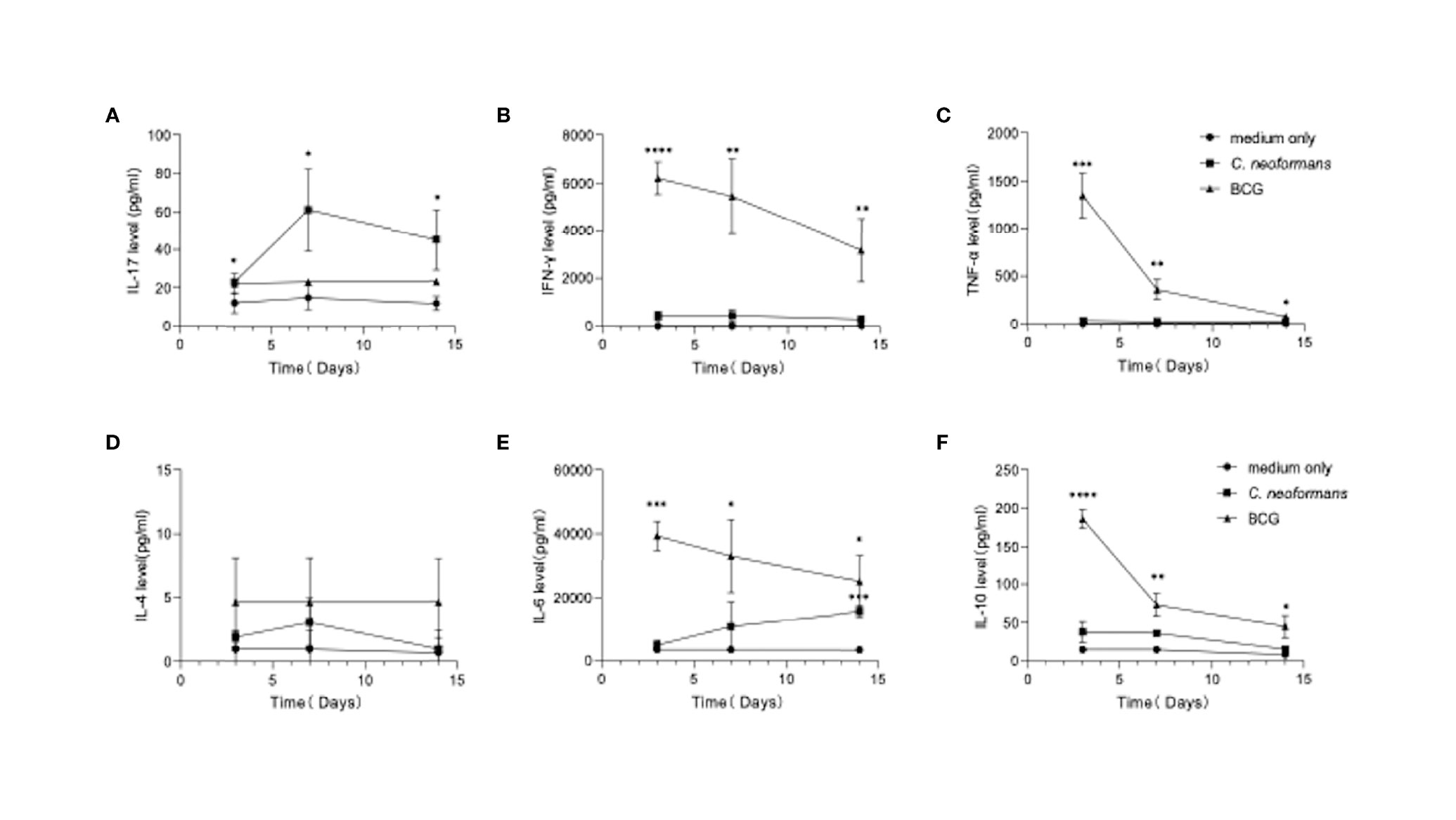
Figure 2 Dynamics of cytokine production by peripheral blood mononuclear cells (PBMCs) in response to C. neoformans or bacillus Calmette–Guérin (BCG) stimulation. Healthy PBMCs were stimulated with C. neoformans or BCG for different times (day 3/7/14). The supernatant cytokine levels were measured by ELISA. The dynamics of IL-17 (A), IFN-γ (B), TNF-α (C), IL-4. (D), IL-6 (E), and IL-10 (F) are shown as mean ± SE of three individual experiments. *0.01<p<0.05, **0.001<p<0.01, ***<0.0001<p<0.001, ****p<0.0001
To determine cytokine-producing cells, we next stimulated healthy PBMCs for 3 days with C. neoformans or BCG and analyzed IL-17-producing cells in αβT (CD4+ or CD8+ T cells) and γδT (CD3+γδTCR+) subpopulations by flow cytometry assay. C. neoformans stimulation significantly increased the IL-17-producing cells in the CD4+ T subset but not in the CD8+ or γδTCR+ subpopulations. In contrast, BCG marginally induced IL-17 positive cells (Figures 3A, B). To confirm the flow cytometry data, the supernatant IL-17 level in C. neoformans or BCG-stimulated cultures were compared by ELISA. As shown in Figure 3C, the IL-17 level was significantly induced by C. neoformans in contrast to the slight increase by BCG stimulation. Consistent with the CSF data, IFN-γ was considerably induced by BCG but minimally induced by C. neoformans stimulation (Supplementary Figure S2).
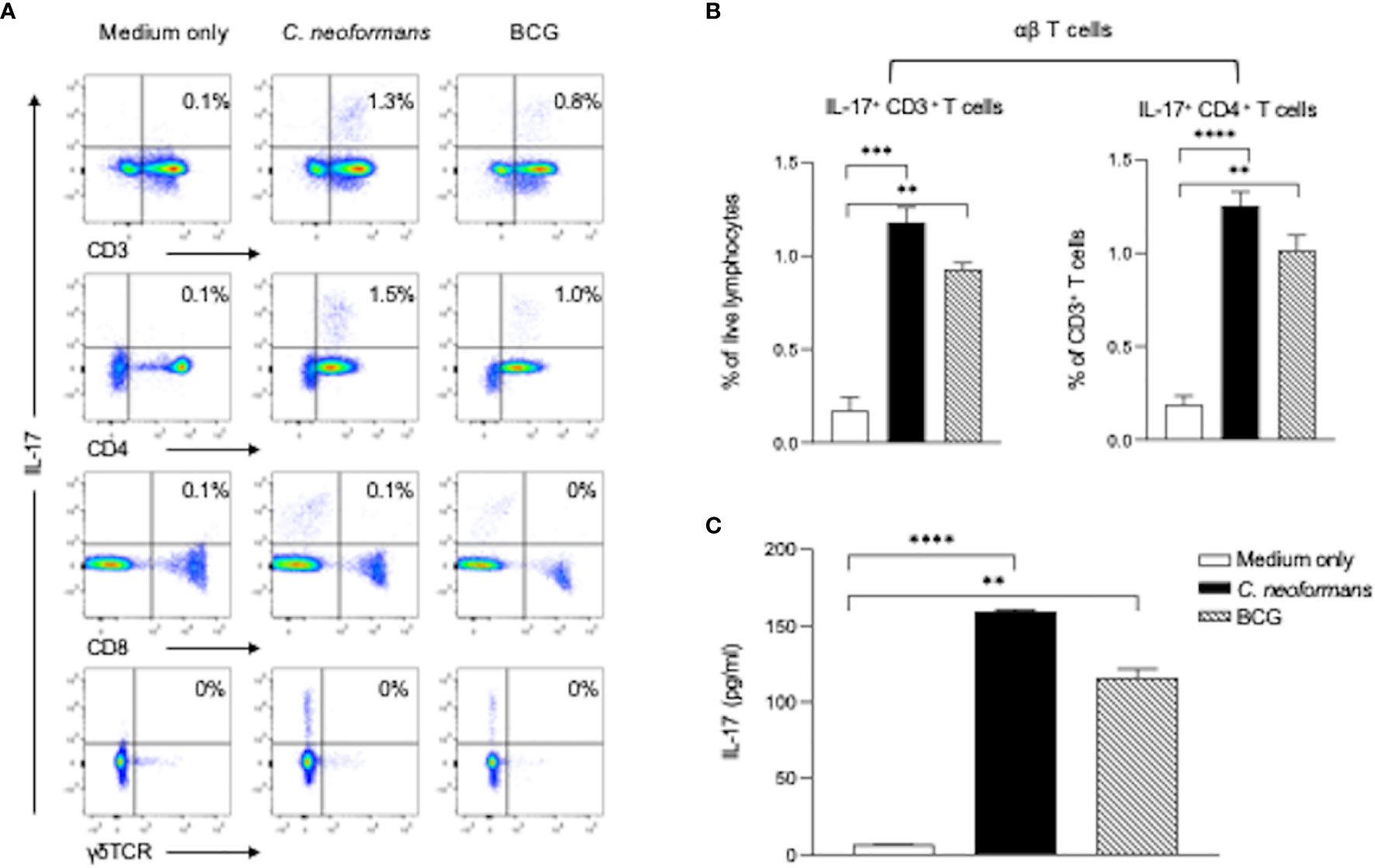
Figure 3 C. neoformans stimulates IL-17 production by CD4+ T cells in vitro. Peripheral blood mononuclear cells were stimulated with C. neoformans or bacillus Calmette–Guérin for 3 days with protein transport inhibitor cocktail for 5 h before the cells were collected. The IL-17-producing cells were analyzed by flow cytometry: representative data of IL-17+ T cells in αβTCR+ T cells (CD3+γδTCR- cells) and γδTCR+ T cells (A) and percentages of IL-17+ CD3+ cells in live lymphocytes and percentages of IL-17+ CD4+ cells in live CD3+ cells (B). The corresponding IL-17 level in the supernatant (C) is shown as mean ± SE of three individual experiments. **p < 0.01, ***p < 0.001, ****p < 0.0001.
To investigate the mechanism of IL-17 production by C. neoformans stimulation, we stimulated healthy PBMCs for 30 min with C. neoformans or BCG and analyzed the changes of the phosphorylation status of STAT3 and its downstream p65 by western blot analysis. Lipopolysaccharide (LPS, 100 ng/ml) stimulation was set as the positive control. As shown in Figure 4A, both LPS and C. neoformans significantly stimulated the phosphorylation of STAT3, in contrast to the negligible phospho-STAT3 by BCG; however, both C. neoformans and BCG were equally effective in stimulating p65 phosphorylation, suggesting that they activate the p65-dependent pathway via different signaling (Figure 4A). To determine if C. neoformans-mediated STAT3 phosphorylation occurs in IL-17-producing cells, we analyzed the phospho-STAT3+ T cell subpopulations by flow cytometry analysis. We found that, among T cells, about 30% of CD4+ T subpopulation, but only about 3% of CD8+ T subpopulation, was phospho-STAT3-positive in LPS or C. neoformans-stimulated cultures, whereas BCG stimulated STAT3 phosphorylation only in about 4% of CD4+ T cells and 0.7% of CD8+ T cells (Figures 4B, C). The densitometry analysis showed that C. neoformans stimulated a significantly higher fluorescence intensity of phospho-STAT3 than BCG did in CD4+ T cells (Figure 4D). To further confirm whether the phosphorylation of STAT3 was specific for C. neoformans stimulation, we analyzed the phospho-STAT1+ T cell by flow cytometry (Supplementary Figure S3). The results showed that about 0.7% of CD4+ T cells were phospho-STAT1-positive in response to C. neoformans stimulation and in contrast to the nearly 3.2% of phospho-STAT1+CD4+ T cells under BCG stimulation (Supplementary Figures S3A, B), which was consistent with the results of the mean fluorescence intensity analysis (Supplementary Figure S3C). Moreover, to verify the relationship of STAT3 phosphorylation and IL-17 production at an early stage, we detected intracellular IL-17 expression in T cells. We found that, similar to STAT3 phosphorylation, IL-17 was produced especially by CD4+ T cells, but not by other cells, in response to C. neoformans (Supplementary Figure S4). Taken together, our data indicate that CD4+IL-17+ cells participate in cryptococcal immune response via STAT3 phosphorylation.
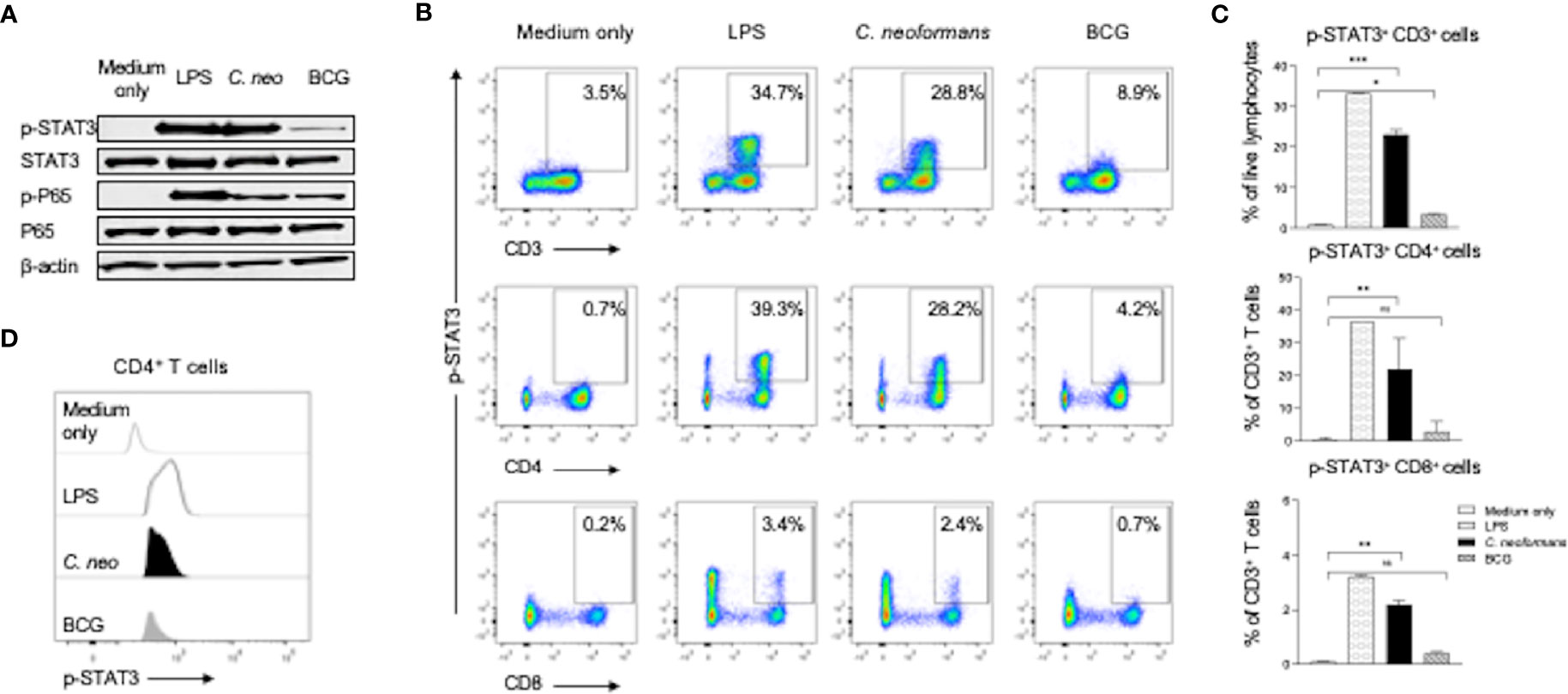
Figure 4 C. neoformans stimulates the phosphorylation of STAT3 in CD4+ T cells. Peripheral blood mononuclear cells were stimulated for 30 min with lipopolysaccharide (100 ng/ml), C. neoformans, or BCG. The phosphorylation status of STAT3 and its downstream P65 at the protein level were analyzed by western blot (A). Phosphorylation of STAT3 in T cell subsets (B, C), and its fluorescence intensity was analyzed by flow cytometry (D). Representative data of three individual experiments are shown. *0.01<p<0.05, **0.001<p<0.01, ***<0.0001<p<0.001.
To determine if the STAT3-dependent pathway is exclusively important for IL-17 production during C. neoformans stimulation, we used a small molecule inhibitor stattic (2.5 μM) to specifically inhibit STAT3 phosphorylation and followed the changes of IL-17+ CD4+ T cells during C. neoformans stimulation of healthy PBMCs. As shown in Figure 5A, C. neoformans stimulation induced p-STAT3 in 30.4% of total T (CD3+) cells, 32.2% of CD4+ T cells, and 2.9% of CD8+ T cells; pretreatment of PBMCs for 30 mins with stattic, p-STAT3+CD3+, p-STAT3+CD4+, and p-STAT3+CD8+ cells resulted in dropping to 1.2, 0.3, and 0.2%, respectively. In accordance with the changes of STAT3 phosphorylation, C. neoformans stimulation yielded 1.2% IL-17+ CD3+ cells in live lymphocytes and 1.6% IL-17+ CD4+T in αβT cells, which were respectively reduced to 0.6 and 0.8% by stattic pretreatment (Figures 5B, C). To confirm the flow cytometry data, C. neoformans stimulation for 3 days produced over 400 pg/ml of supernatant IL-17 that was diminished by stattic pretreatment (Figure 5D). These observations collectively suggested that C. neoformans stimulates IL-17 production by CD4+ T cells via the STAT3-dependent signaling pathway.
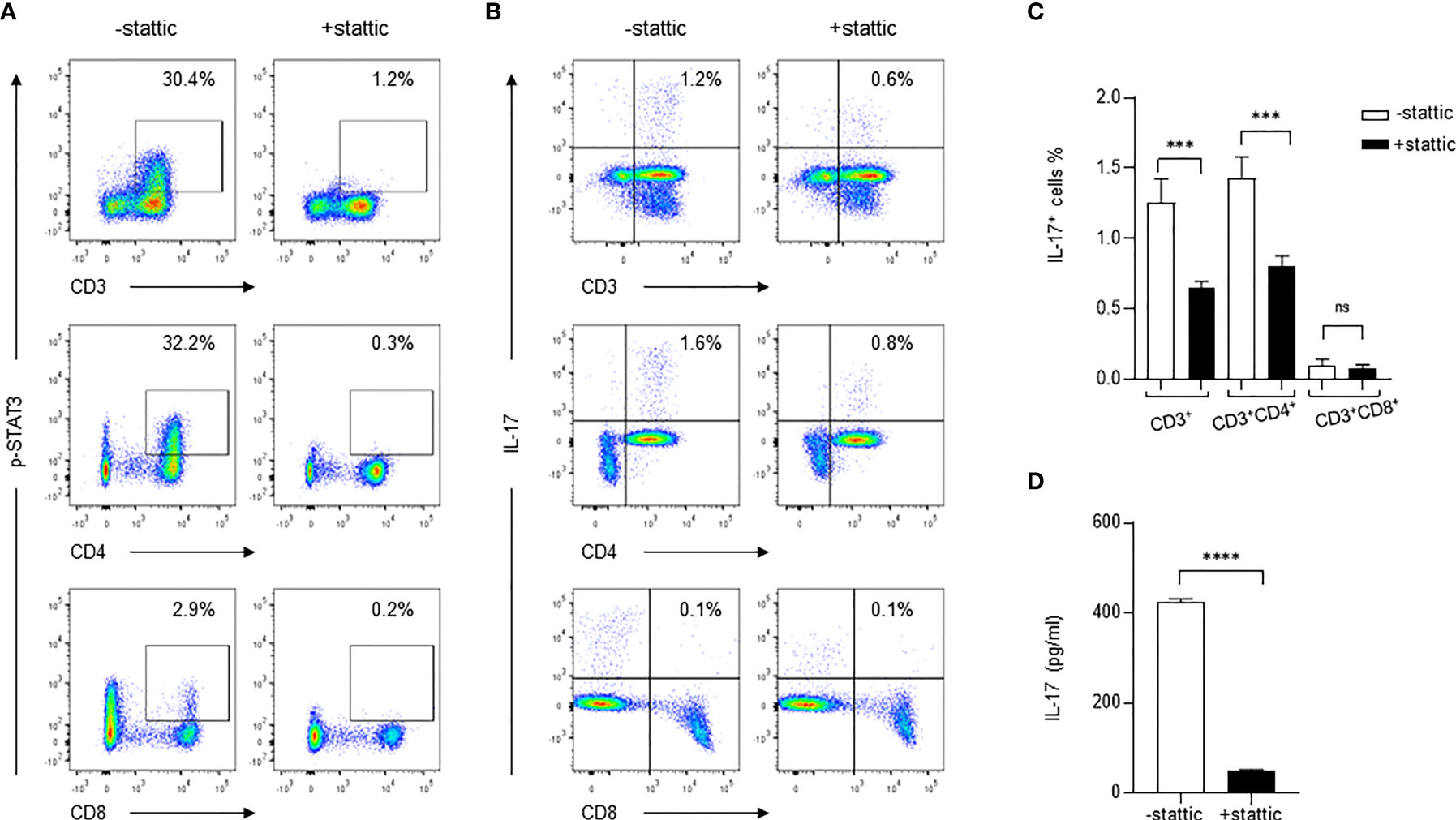
Figure 5 Inhibition of STAT3 phosphorylation diminishes IL-17 production in response to C. neoformans. Peripheral blood mononuclear cells were pretreated for 30 min with STAT3 phosphorylation inhibitor stattic (2.5μM) and were subjected to stimulation for 3 days with C. neoformans. Changes of the phosphorylated STAT3 (A) and IL-17-producing cells (B, C) were analyzed by flow cytometry assay, and the supernatant IL-17 level (mean ± SE) was evaluated by ELISA (D). Representative data of three individual experiments are shown. ***0.0001<p<0.01, ****p < 0.0001; ns, no significance.
The pathogenesis of fungal infections and anti-fungal immunity have recently attracted more and more attentions and effective anti-fungal therapies have become an urgent clinical need. There are more than 300 human pathogenic fungi, and immunodeficiency patients are more susceptible to fungal infections. With the increasing prevalence of HIV infection worldwide, opportunistic fungal infection has been the main driver of HIV mortality. In Africa, C. neoformans meningitis accounts for about 70% (23) of the global deaths of AIDS-associated fungal infections, including candidiasis, Penicillium marneffei infection, pulmonary aspergillus, and so on (24). In recent years, more and more non-HIV-infected sufferers have been reported with CM (25, 26).
IL-17 production has been proposed as an important mechanism of anti-fungal immunity. This inflammatory cytokine IL-17 could be secreted by CD4+, CD8+, γδ T, and NK cells (27, 28). In a mice model of C. neoformans infection, CD4+ T cells were a crucial component of cell-mediated fungal clearance (29–31). In humans, majority of C. neoformans infections were related with CD4+ T cell deficiency due to HIV co-infection, while IL-17-producing CD4+ T (Th17) cells were needed for vaccine-mediated protection against C. neoformans (32–34). Consistent with previous findings, our research suggested that IL-17 was secreted especially by CD4+ T cells, but not by other cells, in response to the stimulation of C. neoformans. Although the CD4+ cell counts in HIV-negative CM patients were lower than the normal reference range but much higher than those of HIV-positive CM patients, all non-AIDS-associated CM patients in the current study had no evidence of immunodeficiency. In addition, previous studies had found that in vitro Th1-type cytokine IFN-γ also plays a significant role to enhance the phagocytic activity of macrophages against C. neoformans (35–37). A mouse model of C. neoformans H99 gamma strain infection produced IFN-γ that protected the invasion of C. neoformans pulmonale (5, 38).
Predominantly increased CSF IL-17 has been reported in patients with HIV+ C. neoformans meningitis (6, 39). Consistent with those observations, our data showed that the CSF IL-17 level was selectively increased in CM patients with or without HIV, but not in TBM patients whose CSF contained a high level of IFN-γ. Interestingly, the CSF IL-17 in non-HIV CM patients was significantly elevated compared with that of HIV-positive CM, which indicated preferential Th17 responses to C. neoformans in immunocompetent individuals and led to speculations of other sources of IL-17-producing cells in case of CD4+ T cell deficiency. We have also found a slightly higher-than-normal serum IL-17 level in non-HIV CM patients, but not in HIV-positive CM. The increased CSF IL-17 or IFN-γ in C. neoformans or tuberculous meningitis was minimized by anti-fungal or anti-tuberculous therapies, respectively. However, the mechanism of IL-17 in the pathogenesis of C. neoformans meningitis in non-AIDS patients is not clear.
It is well known that the pro-inflammatory cytokine production by infections involves cell interactions between immune cells (40–42). To study the immune function of human fungal infection, we used fungus-stimulated healthy PBMC to mimic the in vivo situation and investigated the dynamics of IL-17 production and its signaling pathway. We found that C. neoformans stimulation induced high levels of IL-17 both in the cell and the supernatant, and T cells, especially CD4+ T cells, were the only IL-17 producer; no IL-17+CD8+ T cells or IL-17+γδ T cells were detected.
Protein phosphorylation is one of the post-translational modifications that are particularly important for the regulation of cellular activities (43). NF-κB P65 is a well-known gene transactivator in the innate immune signaling pathway against infection (44). We found that it participates in anti-fungal immunity by showing its phosphorylation activation by cryptococcal stimulation. An earlier study had linked the STAT3 signaling pathway with differentiation of naive CD4+ T cells to Th17 phenotype and IL-17 production (45–47). We demonstrated here that STAT3 signaling plays an important role in the development of IL-17-secreting cells in response to fungal stimulation. In contrast to trace phospho-STAT3 in CD4+ T cells by BCG stimulation, C. neoformans stimulation selectively induced the phosphorylation of STAT3 mainly in CD4+ T cells. No phospho-STAT3-positive cells were detected in CD8+ or γδ T cell subsets. Previous studies suggested that STAT1 is mainly involved in IFN-γ induction. This is aligned with our findings that phospho-STAT1 in T cells is not associated with C. neoformans stimulation (48, 49). However, BCG is a live attenuated tuberculosis vaccine. Although it can produce effective anti-tuberculosis immunity and has well reproduced the immune response of TBM in the current in vitro PBMC stimulation, it has a defect in stimulating T cell immunity due to the deletion of germline DNA fragments called region of difference (RD)-1, -2, -3, and -4, which encode virulent genes in the wild-type tuberculosis strain Mtb H37Rv (50, 51). In particular, RD-1 encodes two secretory proteins, CFP-10 and ESAT-6 (52–54), both of which have T-cell activation and macrophage deactivation functions (55). Thus, the difference of full immune response by infections of BCG and Mtb H37Rv strains should be carefully evaluated in the future.
The STAT3-dependent IL-17 production was further confirmed by the pretreatment of cells with a small molecule compound stattic, a selective STAT3 inhibitor that inhibits the activation, dimerization, and nuclear translocation of STAT3 by interacting with its SH2 domain (56, 57) and represses STAT3 phosphorylation (58). In the current study, the pretreatment of PBMCs significantly repressed STAT3 phosphorylation at Tyr705 by cryptococcal stimulation in CD4+ T cells and further diminished the IL-17-producing CD4+ T cells and secretory IL-17 in the cell culture supernatant. In addition, C. neoformans stimulation selectively triggers STAT3 phosphorylation rather than STAT1 phosphorylation, which further confirmed the STAT3-dependent IL-17 production.
Signal transduction to gene transcription to protein translation is a complex regulatory process. It is common in all signaling pathways that the phosphorylation of upstream signal molecules occurs almost immediately after the activation of the signaling pathway. Thus, as the very upstream of IL-17-producing signaling pathway, STAT3 was rapidly phosphorylated in response to C. neoformans stimulation, whereas IL-17 protein expression was significantly postponed probably due to the down-stream multiple regulatory processes, such as the transcription of target gene and/or daughter genes to finally induce IL-17 gene transcription and translation (59).
In conclusion, our study demonstrated that C. neoformans infection stimulates the development of Th17 cells to produce IL-17 by activating the STAT3-dependent signal pathway, and IL-17 could be a potential biomarker of and STAT3 a checkpoint of targeted therapies for fungal infection.
The raw data supporting the conclusions of this article will be made available by the authors without undue reservation.
The studies involving human participants were reviewed and approved by the Ethical Committee of Shanghai Public Health Clinical Center [approval number (2020) 2020-S144-01]. The patients/participants provided their written informed consent to participate in this study.
YH and YLing conceptualized this study and designed the experiments. XG performed the experiments and data analysis. XM helped in the flow cytometry assays. DT, YL, BS, CY, and DS collected the CSF and blood samples. XG, TL, YLing, and YH wrote the manuscript. All authors contributed to the article and approved the submitted version.
This work was sponsored by the National Natural Science Foundation of China (82072260 and 32070946), the Natural Science Foundation of Shanghai (number 16ZR1429500), the Shanghai Pujiang Program (number 16PJ1408700), the Shanghai “Rising Stars of Medical Talent” Youth Development Program, the Outstanding Youth Medical Talents [SHWJRS (2021)-99], and the Fundamental Research Funds for the Central Universities (HUST: 2015ZHYX007 and 2018KFYYXJJ076).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We would like to thank the patients, the volunteers, and the nurses of Shanghai Public Health Clinical Center for their kind cooperation. We appreciate the technical assistance in flow cytometry from the Medical Subcenter of HUST Analytical and Testing Center.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2022.872286/full#supplementary-material
1. Powderly WG. Cryptococcal Meningitis and AIDS. Clin Infect Diseases: An Off Publ Infect Dis Soc America (1993) 17:837–42. doi: 10.1093/clinids/17.5.837
2. Okongo M, Morgan D, Mayanja B, Ross A, Whitworth J. Causes of Death in a Rural, Population-Based Human Immunodeficiency Virus Type 1 (HIV-1) Natural History Cohort in Uganda. Int J Epidemiol (1998) 27:698–702. doi: 10.1093/ije/27.4.698
3. Rajasingham R, Smith RM, Park BJ, Jarvis JN, Govender NP, Chiller TM, et al. Global Burden of Disease of HIV-Associated Cryptococcal Meningitis: An Updated Analysis. Lancet Infect Dis (2017) 17:873–81. doi: 10.1016/s1473-3099(17)30243-8
4. Müller U, Stenzel W, Köhler G, Werner C, Polte T, Hansen G, et al. IL-13 Induces Disease-Promoting Type 2 Cytokines, Alternatively Activated Macrophages and Allergic Inflammation During Pulmonary Infection of Mice With Cryptococcus Neoformans. J Immunol (Baltimore Md.: 1950) (2007) 179:5367–77. doi: 10.4049/jimmunol.179.8.5367
5. Wormley FL Jr, Perfect JR, Steele C, Cox GM. Protection Against Cryptococcosis by Using a Murine Gamma Interferon-Producing Cryptococcus Neoformans Strain. Infection Immun (2007) 75:1453–62. doi: 10.1128/iai.00274-06
6. Marais S, Meintjes G, Lesosky M, Wilkinson KA, Wilkinson RJ. Interleukin-17 Mediated Differences in the Pathogenesis of HIV-1-Associated Tuberculous and Cryptococcal Meningitis. AIDS (Lond Engl) (2016) 30:395–404. doi: 10.1097/qad.0000000000000904
7. Albanesi C, Cavani A, Girolomoni G. IL-17 is Produced by Nickel-Specific T Lymphocytes and Regulates ICAM-1 Expression and Chemokine Production in Human Keratinocytes: Synergistic or Antagonist Effects With IFN-Gamma and TNF-Alpha. J Immunol (Baltimore Md.: 1950) (1999) 162:494–502.
8. Antonysamy MA, Fanslow WC, Fu F, Li W, Qian S, Troutt AB, et al. Evidence for a Role of IL-17 in Organ Allograft Rejection: IL-17 Promotes the Functional Differentiation of Dendritic Cell Progenitors. J Immunol (Baltimore Md.: 1950) (1999) 162:577–84.
9. Infante-Duarte C, Horton HF, Byrne MC, Kamradt T. Microbial Lipopeptides Induce the Production of IL-17 in Th Cells. J Immunol (Baltimore Md.: 1950) (2000) 165:6107–15. doi: 10.4049/jimmunol.165.11.6107
10. Conti HR, Gaffen SL. IL-17-Mediated Immunity to the Opportunistic Fungal Pathogen Candida Albicans. J Immunol (Baltimore Md.: 1950) (2015) 195:780–8. doi: 10.4049/jimmunol.1500909
11. Drummond RA, Lionakis MS. Organ-Specific Mechanisms Linking Innate and Adaptive Antifungal Immunity. Semin Cell Dev Biol (2019) 89:78–90. doi: 10.1016/j.semcdb.2018.01.008
12. Taylor PR, Roy S, Meszaros EC, Sun Y, Howell SJ, Malemud CJ, et al. JAK/STAT Regulation of Aspergillus Fumigatus Corneal Infections and IL-6/23-Stimulated Neutrophil, IL-17, Elastase, and MMP9 Activity. J leukocyte Biol (2016) 100:213–22. doi: 10.1189/jlb.4A1015-483R
13. Zhu J, Paul WE. Peripheral CD4+ T-Cell Differentiation Regulated by Networks of Cytokines and Transcription Factors. Immunol Rev (2010) 238:247–62. doi: 10.1111/j.1600-065X.2010.00951.x
14. Wu X, Shou Q, Chen C, Cai H, Zhang J, Tang S, et al. An Herbal Formula Attenuates Collagen-Induced Arthritis via Inhibition of JAK2-STAT3 Signaling and Regulation of Th17 Cells in Mice. Oncotarget (2017) 8:44242–54. doi: 10.18632/oncotarget.17797
15. Milner JD, Brenchley JM, Laurence A, Freeman AF, Hill BJ, Elias KM, et al. Impaired T(H)17 Cell Differentiation in Subjects With Autosomal Dominant Hyper-IgE Syndrome. Nature (2008) 452:773–6. doi: 10.1038/nature06764
16. Valdez PA, Vithayathil PJ, Janelsins BM, Shaffer AL, Williamson PR, Datta SK. Prostaglandin E2 Suppresses Antifungal Immunity by Inhibiting Interferon Regulatory Factor 4 Function and Interleukin-17 Expression in T Cells. Immunity (2012) 36:668–79. doi: 10.1016/j.immuni.2012.02.013
17. Levy DE, Darnell JE Jr. Stats: Transcriptional Control and Biological Impact. Nat Rev Mol Cell Biol (2002) 3:651–62. doi: 10.1038/nrm909
18. Akira S, Nishio Y, Inoue M, Wang XJ, Wei S, Matsusaka T, et al. Molecular Cloning of APRF, a Novel IFN-Stimulated Gene Factor 3 P91-Related Transcription Factor Involved in the Gp130-Mediated Signaling Pathway. Cell (1994) 77:63–71. doi: 10.1016/0092-8674(94)90235-6
19. Narimatsu M, Maeda H, Itoh S, Atsumi T, Ohtani T, Nishida K, et al. Tissue-Specific Autoregulation of the Stat3 Gene and its Role in Interleukin-6-Induced Survival Signals in T Cells. Mol Cell Biol (2001) 21:6615–25. doi: 10.1128/mcb.21.19.6615-6625.2001
20. Yang J, Chatterjee-Kishore M, Staugaitis SM, Nguyen H, Schlessinger K, Levy DE, et al. Novel Roles of Unphosphorylated STAT3 in Oncogenesis and Transcriptional Regulation. Cancer Res (2005) 65:939–47.
21. Perfect JR, Dismukes WE, Dromer F, Goldman DL, Graybill JR, Hamill RJ, et al. Clinical Practice Guidelines for the Management of Cryptococcal Disease: 2010 Update by the Infectious Diseases Society of America. Clin Infect diseases: an Off Publ Infect Dis Soc Am (2010) 50:291–322. doi: 10.1086/649858
22. Glimåker M, Johansson B, Grindborg Ö, Bottai M, Lindquist L, Sjölin J. Adult Bacterial Meningitis: Earlier Treatment and Improved Outcome Following Guideline Revision Promoting Prompt Lumbar Puncture. Clin Infect Diseases: An Off Publ Infect Dis Soc Am (2015) 60:1162–9. doi: 10.1093/cid/civ011
23. Wu TC, Chan JW, Ng CK, Tsang DN, Lee MP, Li PC. Clinical Presentations and Outcomes of Penicillium Marneffei Infections: A Series From 1994 to 2004. Hong Kong Med J = Xianggang yi xue za zhi (2008) 14:103–9.
24. Cooper CR Jr, McGinnis MR. Pathology of Penicillium Marneffei. An Emerging Acquired Immunodeficiency Syndrome-Related Pathogen. Arch Pathol Lab Med (1997) 121:798–804.
25. Chen M, Xu Y, Hong N, Yang Y, Lei W, Du L, et al. Epidemiology of Fungal Infections in China. Front Med (2018) 12:58–75. doi: 10.1007/s11684-017-0601-0
26. Yuchong C, Fubin C, Jianghan C, Fenglian W, Nan X, Minghui Y, et al. Cryptococcosis in China (1985-2010): Review of Cases From Chinese Database. Mycopathologia (2012) 173:329–35. doi: 10.1007/s11046-011-9471-1
27. Mills KH. Induction, Function and Regulation of IL-17-Producing T Cells. Eur J Immunol (2008) 38:2636–49. doi: 10.1002/eji.200838535
28. Peck A, Mellins ED. Precarious Balance: Th17 Cells in Host Defense. Infection Immun (2010) 78:32–8. doi: 10.1128/iai.00929-09
29. Mody CH, Lipscomb MF, Street NE, Toews GB. Depletion of CD4+ (L3T4+) Lymphocytes In Vivo Impairs Murine Host Defense to Cryptococcus Neoformans. J Immunol (Baltimore Md.: 1950) (1990) 144:1472–7.
30. Hill JO, Aguirre KM. CD4+ T Cell-Dependent Acquired State of Immunity That Protects the Brain Against Cryptococcus Neoformans. J Immunol (Baltimore Md.: 1950) (1994) 152:2344–50.
31. Buchanan KL, Doyle HA. Requirement for CD4(+) T Lymphocytes in Host Resistance Against Cryptococcus Neoformans in the Central Nervous System of Immunized Mice. Infection Immun (2000) 68:456–62. doi: 10.1128/iai.68.2.456-462.2000
32. Jarvis JN, Harrison TS. HIV-Associated Cryptococcal Meningitis. AIDS (Lond Engl) (2007) 21:2119–29. doi: 10.1097/QAD.0b013e3282a4a64d
33. Tugume L, Rhein J, Hullsiek KH, Mpoza E, Kiggundu R, Ssebambulidde K, et al. HIV-Associated Cryptococcal Meningitis Occurring at Relatively Higher CD4 Counts. J Infect Dis (2019) 219:877–83. doi: 10.1093/infdis/jiy602
34. Hole CR, Wormley FL Jr. Vaccine and Immunotherapeutic Approaches for the Prevention of Cryptococcosis: Lessons Learned From Animal Models. Front Microbiol (2012) 3:291. doi: 10.3389/fmicb.2012.00291
35. Collins HL, Bancroft GJ. Cytokine Enhancement of Complement-Dependent Phagocytosis by Macrophages: Synergy of Tumor Necrosis Factor-Alpha and Granulocyte-Macrophage Colony-Stimulating Factor for Phagocytosis of Cryptococcus Neoformans. Eur J Immunol (1992) 22:1447–54. doi: 10.1002/eji.1830220617
36. Flesch IE, Schwamberger G, Kaufmann SH. Fungicidal Activity of IFN-Gamma-Activated Macrophages. Extracellular Killing of Cryptococcus Neoformans. J Immunol (Baltimore Md.: 1950) (1989) 142:3219–24.
37. Rivera J, Mukherjee J, Weiss LM, Casadevall A. Antibody Efficacy in Murine Pulmonary Cryptococcus Neoformans Infection: A Role for Nitric Oxide. J Immunol (Baltimore Md.: 1950) (2002) 168:3419–27. doi: 10.4049/jimmunol.168.7.3419
38. Wozniak KL, Ravi S, Macias S, Young ML, Olszewski MA, Steele C, et al. Insights Into the Mechanisms of Protective Immunity Against Cryptococcus Neoformans Infection Using a Mouse Model of Pulmonary Cryptococcosis. PLoS One (2009) 4:e6854. doi: 10.1371/journal.pone.0006854
39. Liu Q, Gao Y, Zhang B, Sun F, Yang Q, Liu Y, et al. Cytokine Profiles in Cerebrospinal Fluid of Patients With Meningitis at a Tertiary General Hospital in China. J Microbiology Immunology Infection = Wei mian yu gan ran za zhi (2020) 53:216–24. doi: 10.1016/j.jmii.2018.08.019
40. Tran CN, Lundy SK, White PT, Endres JL, Motyl CD, Gupta R, et al. Molecular Interactions Between T Cells and Fibroblast-Like Synoviocytes: Role of Membrane Tumor Necrosis Factor-Alpha on Cytokine-Activated T Cells. Am J Pathol (2007) 171:1588–98. doi: 10.2353/ajpath.2007.070004
41. Cho ML, Yoon CH, Hwang SY, Park MK, Min SY, Lee SH, et al. Effector Function of Type II Collagen-Stimulated T Cells From Rheumatoid Arthritis Patients: Cross-Talk Between T Cells and Synovial Fibroblasts. Arthritis Rheumatism (2004) 50:776–84. doi: 10.1002/art.20106
42. Miranda-Carús ME, Balsa A, Benito-Miguel M, Pérez de Ayala C, Martín-Mola E. IL-15 and the Initiation of Cell Contact-Dependent Synovial Fibroblast-T Lymphocyte Cross-Talk in Rheumatoid Arthritis: Effect of Methotrexate. J Immunol (Baltimore Md.: 1950) (2004) 173:1463–76. doi: 10.4049/jimmunol.173.2.1463
43. Li X, Wilmanns M, Thornton J, Köhn M. Elucidating Human Phosphatase-Substrate Networks. Sci Signaling (2013) 6:rs10. doi: 10.1126/scisignal.2003203
44. Karin M, Cao Y, Greten FR, Li ZW. NF-kappaB in Cancer: From Innocent Bystander to Major Culprit. Nat Rev Cancer (2002) 2:301–10. doi: 10.1038/nrc780
45. Wang L, Yi T, Kortylewski M, Pardoll DM, Zeng D, Yu H. IL-17 can Promote Tumor Growth Through an IL-6-Stat3 Signaling Pathway. J Exp Med (2009) 206:1457–64. doi: 10.1084/jem.20090207
46. Yang XO, Panopoulos AD, Nurieva R, Chang SH, Wang D, Watowich SS, et al. STAT3 Regulates Cytokine-Mediated Generation of Inflammatory Helper T Cells. J Biol Chem (2007) 282:9358–63. doi: 10.1074/jbc.C600321200
47. Mathur AN, Chang HC, Zisoulis DG, Stritesky GL, Yu Q, O’Malley JT, et al. Stat3 and Stat4 Direct Development of IL-17-Secreting Th Cells. J Immunol (Baltimore Md.: 1950) (2007) 178:4901–7. doi: 10.4049/jimmunol.178.8.4901
48. Haan S, Keller JF, Behrmann I, Heinrich PC, Haan C. Multiple Reasons for an Inefficient STAT1 Response Upon IL-6-Type Cytokine Stimulation. Cell signalling (2005) 17:1542–50. doi: 10.1016/j.cellsig.2005.03.010
49. Ramana CV, Chatterjee-Kishore M, Nguyen H, Stark GR. Complex Roles of Stat1 in Regulating Gene Expression. Oncogene (2000) 19:2619–27. doi: 10.1038/sj.onc.1203525
50. Mahairas GG, Sabo PJ, Hickey MJ, Singh DC, Stover CK. Molecular Analysis of Genetic Differences Between Mycobacterium Bovis BCG and Virulent M. bovis. J Bacteriology (1996) 178:1274–82. doi: 10.1128/jb.178.5.1274-1282.1996
51. Ru H, Liu X, Lin C, Yang J, Chen F, Sun R, et al. The Impact of Genome Region of Difference 4 (RD4) on Mycobacterial Virulence and BCG Efficacy. Front Cell Infection Microbiol (2017) 7:239. doi: 10.3389/fcimb.2017.00239
52. Brodin P, Rosenkrands I, Andersen P, Cole ST, Brosch R. ESAT-6 Proteins: Protective Antigens and Virulence Factors? Trends Microbiol (2004) 12:500–8. doi: 10.1016/j.tim.2004.09.007
53. Berthet FX, Rasmussen PB, Rosenkrands I, Andersen P, Gicquel B. A Mycobacterium Tuberculosis Operon Encoding ESAT-6 and a Novel Low-Molecular-Mass Culture Filtrate Protein (CFP-10). Microbiol (Reading England) (1998) 144(Pt 11):3195–203. doi: 10.1099/00221287-144-11-3195
54. Parsons LM, Brosch R, Cole ST, Somoskövi A, Loder A, Bretzel G, et al. Rapid and Simple Approach for Identification of Mycobacterium Tuberculosis Complex Isolates by PCR-Based Genomic Deletion Analysis. J Clin Microbiol (2002) 40:2339–45. doi: 10.1128/jcm.40.7.2339-2345.2002
55. Ganguly N, Siddiqui I, Sharma P. Role of M. Tuberculosis RD-1 Region Encoded Secretory Proteins in Protective Response and Virulence. Tuberculosis (Edinburgh Scotland) (2008) 88:510–7. doi: 10.1016/j.tube.2008.05.002
56. Wang CM, Hsu CT, Niu HS, Chang CH, Cheng JT, Shieh JM. Lung Damage Induced by Hyperglycemia in Diabetic Rats: The Role of Signal Transducer and Activator of Transcription 3 (STAT3). J Diabetes Its Complications (2016) 30:1426–33. doi: 10.1016/j.jdiacomp.2016.07.005
57. Boengler K, Ungefug E, Heusch G, Schulz R. The STAT3 Inhibitor Stattic Impairs Cardiomyocyte Mitochondrial Function Through Increased Reactive Oxygen Species Formation. Curr Pharm Design (2013) 19:6890–5. doi: 10.2174/138161281939131127115940
58. Zhang Z, Zhou H, Zhou J. Neuritin Inhibits Astrogliosis to Ameliorate Diabetic Cognitive Dysfunction. J Mol Endocrinol (2021) 66:259–72. doi: 10.1530/jme-20-0321
Keywords: IL-17, STAT3, CD4+ T cells, Cryptococcus neoformans meningitis, phosphorylation
Citation: Guo X, Mao X, Tian D, Liao Y, Su B, Ye C, Shi D, Liu TF, Ling Y and Hao Y (2022) Cryptococcus neoformans Infection Induces IL-17 Production by Promoting STAT3 Phosphorylation in CD4+ T Cells. Front. Immunol. 13:872286. doi: 10.3389/fimmu.2022.872286
Received: 09 February 2022; Accepted: 14 April 2022;
Published: 27 May 2022.
Edited by:
Zongxin Ling, Zhejiang University, ChinaReviewed by:
Satoshi Okada, Hiroshima University, JapanCopyright © 2022 Guo, Mao, Tian, Liao, Su, Ye, Shi, Liu, Ling and Hao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Tie Fu Liu, bGl1dGllZnVAc2hwaGMub3JnLmNu; Yun Ling, eXVuLmxpbmdAc2hwaGMub3JnLmNu; Yi Hao, aGFveWlAaHVzdC5lZHUuY24=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.