 Long Chen1†
Long Chen1† Fukun Chen2†Huatao Niu3†
Fukun Chen2†Huatao Niu3† Jindan Li1Yongzhu Pu1Conghui Yang1Yue Wang1Rong Huang1Ke Li4*
Jindan Li1Yongzhu Pu1Conghui Yang1Yue Wang1Rong Huang1Ke Li4* Yujie Lei5*
Yujie Lei5* Yunchao Huang5*
Yunchao Huang5*- 1Department of PET/CT Center, Yunnan Cancer Hospital, The Third Affiliated Hospital of Kunming Medical University, Cancer Center of Yunnan Province, Kunming, China
- 2Department of Nuclear Medicine, Yunnan Cancer Hospital, The Third Affiliated Hospital of Kunming Medical University, Cancer Center of Yunnan Province, Kunming, China
- 3Department of Neurosurgery, Yunnan Cancer Hospital, The Third Affiliated Hospital of Kunming Medical University, Cancer Center of Yunnan Province, Kunming, China
- 4Department of Cancer Biotherapy Center, Yunnan Cancer Hospital, The Third Affiliated Hospital of Kunming Medical University, Cancer Center of Yunnan Province, Kunming, China
- 5Department of Thoracic Surgery I, Key Laboratory of Lung Cancer of Yunnan Province, Yunnan Cancer Hospital, The Third Affiliated Hospital of Kunming Medical University, Cancer Center of Yunnan Province, Kunming, China
Different from surgery, chemical therapy, radio-therapy and target therapy, Chimeric antigen receptor-modified T (CAR-T) cells, a novel adoptive immunotherapy strategy, have been used successfully against both hematological tumors and solid tumors. Although several problems have reduced engineered CAR-T cell therapeutic outcomes in clinical trials for the treatment of thoracic malignancies, including the lack of specific antigens, an immunosuppressive tumor microenvironment, a low level of CAR-T cell infiltration into tumor tissues, off-target toxicity, and other safety issues, CAR-T cell treatment is still full of bright future. In this review, we outline the basic structure and characteristics of CAR-T cells among different period, summarize the common tumor-associated antigens in clinical trials of CAR-T cell therapy for thoracic malignancies, and point out the current challenges and new strategies, aiming to provide new ideas and approaches for preclinical experiments and clinical trials of CAR-T cell therapy for thoracic malignancies.
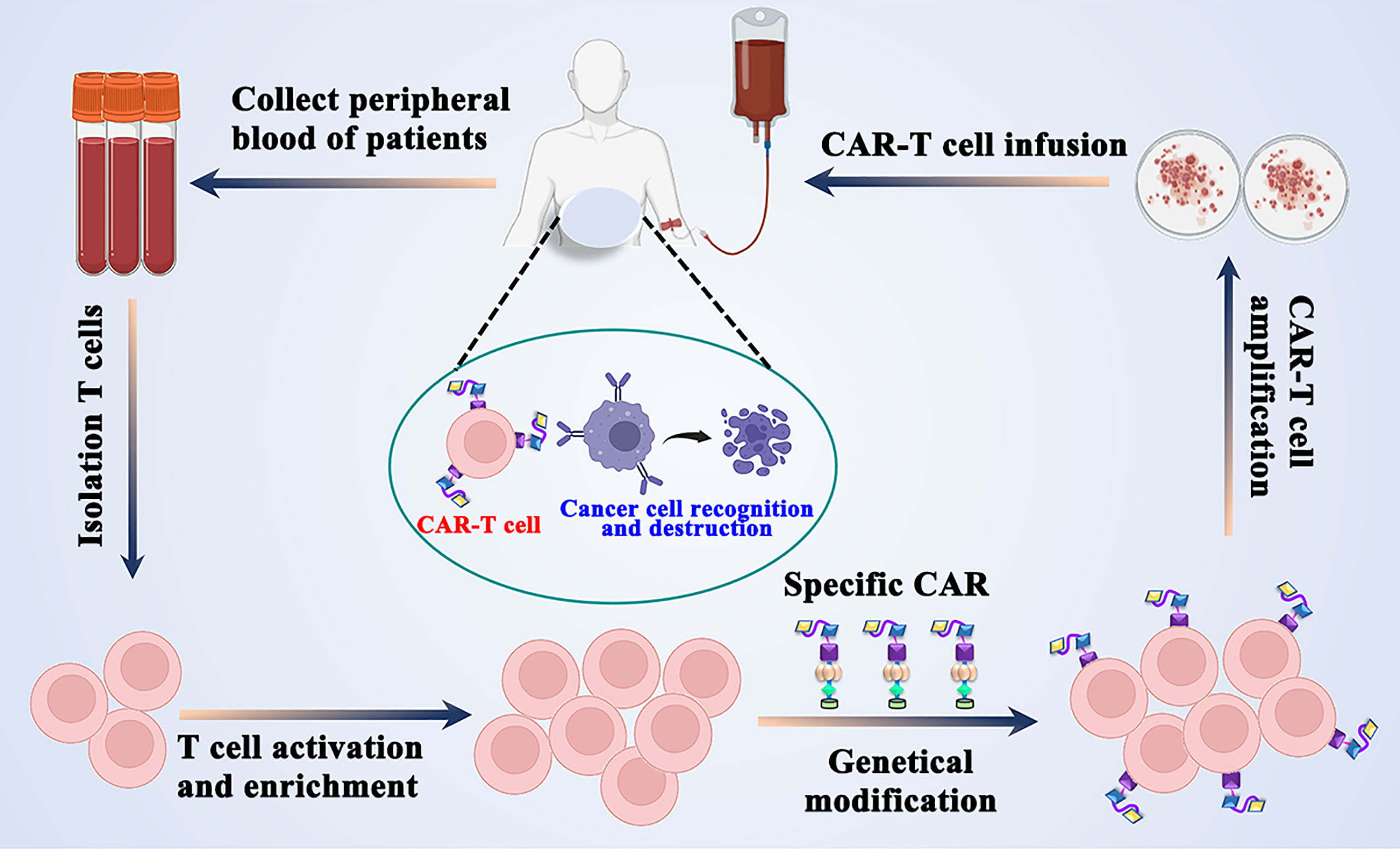
Graphical Abstract A concise workflow of CAR-T T cell therapy in clinical practice.
Introduction
With the continuous improvement of living standards, the incidence and mortality of tumors are rapidly increasing worldwide (1). Among them, thoracic malignancies are common thoracic surgical diseases with high morbidity and mortality, mainly including lung cancer, breast cancer, esophageal cancer, pleural mesothelioma, and thymic cancer (2). According to estimates from the Global Cancer Statistics 2020, there were an estimated 5,103,160 new cases of thoracic cancers and 3,051,494 cancer deaths (3), accounting for 26.45% and 30.64% of new cancers and deaths worldwide, respectively. Thus, thoracic cancer is the leading cause of cancer-related death and a significant obstacle to enhancing life expectancy worldwide. In recent decades, despite advancements in our knowledge of tumor progression and treatment strategies (e.g., radical surgery, chemotherapy, and radiotherapy) that contribute to prolonged survival times of patients with thoracic cancers, the prognosis of thoracic cancers has not improved due to tumor mutation and heterogeneity (4, 5). Moreover, many thoracic cancers are diagnosed at an advanced stage that often miss the optimal treatment time and are prone to recurrence after surgery (6–8). Thus, it is imperative to seek novel methods to stop tumor progression and prolong the survival time of patients with thoracic malignancies.
In the past decade, numerous studies have used immunotherapy with checkpoint inhibitors, especially monoclonal antibody-targeted drugs, for the treatment of malignancies (such as solid tumors and hematological malignancies), but its application in preclinical and clinical studies still has some limitations (9). Moreover, cytotoxic T cells have been reported to act as important immune mediators in controlling tumor progression (10). Additionally, beneficial effects have been reported in patients with melanoma, lung cancer, and breast cancer when treated with adoptive T cell therapy and genetically engineered T cells (11), which indicated that T cells have the potential to eliminate malignant tumors under appropriate conditions. In some cases, thoracic cancers are already being inhibited by T cell therapy, such as esophageal cancer (12), lung cancer (13), and breast cancer (14). Of note, chimeric antigen receptor (CAR)-T cells, which act as modified T cell therapy, have attracted growing interest in malignant tumors in recent years (15) and are also considered safe and reliable immunotherapies in malignant tumors (16). Currently, CAR-T cell immunotherapy has been highly successful in hematologic malignancies, with overall remission rates of more than 80% (17). For example, CAR-T cells targeting CD19 have a long-term remission effect on drug-resistant B cell malignancies, with a cure rate of approximately 85% in patients with relapsed and refractory acute B-lymphocytic leukemia and non-Hodgkin lymphoma (18, 19). Currently, five types of CAR-T cells targeting CD19 have been approved by the US Food and Drug Administration (FDA) for the treatment of hematologic malignancies (20), opening up new directions for tumor immunotherapy and antitumor treatment. Simultaneously, a range of solid tumor CAR-T cell target tumor-associated antigens (TAAs) have been identified and are in early clinical trials (21, 22). Moreover, several studies have focused on CAR-T cell immunotherapy for the treatment of thoracic cancers and have made good progress in clinical trials (22, 23). The above findings suggest that CAR-T cell immunotherapy may be a novel strategy for the treatment of thoracic tumors.
In this review, we summarize the recent research advances in CAR-T cell immunotherapy for thoracic malignancies, including the structure and generation of CAR-T cells and clinical applications. Moreover, we focus on the main challenges and future prospects of CAR-T cell immunotherapy against thoracic cancers, aiming to provide new ideas for the clinical trial design and treatment of thoracic malignancy immunotherapy.
The Structure And Generation Of Car−T Cells
The Structure of CAR−T Cells
CAR-T cells are produced by isolating the patient’s T cells out of the body and re-forcing them into the body and bind to on cancer cells specifically (24). CARs are mainly composed of an extracellular antigen recognition domain, a hinge and transmembrane domain, and an intracellular signal transduction domain (Figure 1). The single-chain variable fragment (scFv) of the target antigen-antibody, consisted by the heavy chain variable regions and the light chain variable regions is specific to the TAA. The hinge and transmembrane structural domains serve to connect the extracellular and intracellular structural domains therefore leads to the CAR-T cell activation (25). Meanwhile, the length or flexibility of the transmembrane structural domain can also affect the function of CAR (26). The intracellular signal transduction structural domain mainly consists of the stimulatory factor CD3ζ chain and is often combined with other costimulatory molecules, activating T cell function (27).
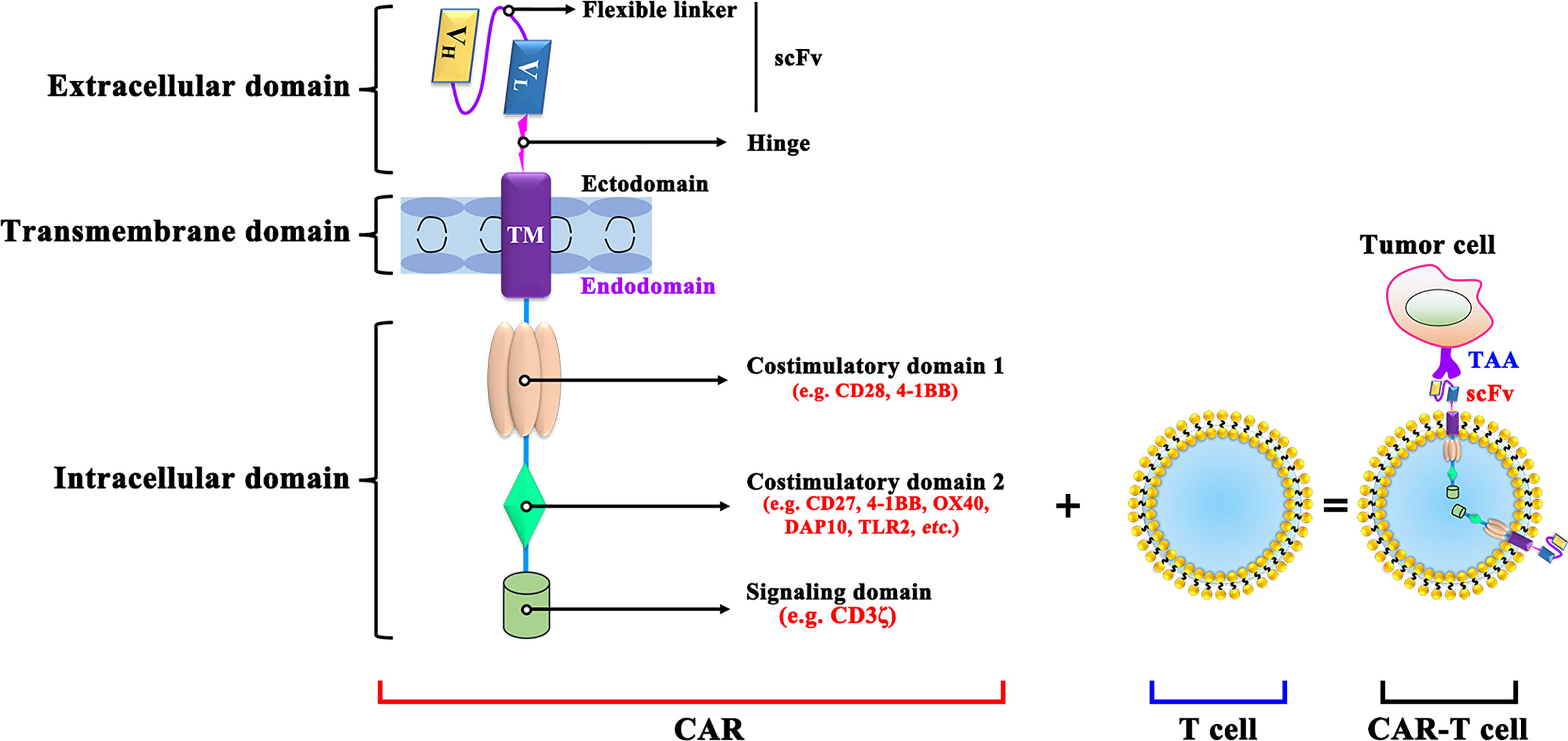
Figure 1 The structure of CAR-T cell. VL, Light chain variable; VH, Heavy chain variable; ICOS, inducible costimulatory; ScFv, Single-chain variable fragment; DAP10, DNAX-activating protein 10; TLR2, Toll-like receptor-2; TAA, tumor-associated antigen; scFv: single-chain fragment variable.
Generation of CAR−T Cells
CAR-T cells are currently classified into five generations based on their intracellular signaling structural domains, with the main differences between CAR-T cell generations being specific costimulatory molecules (Figure 2). The first generation of CAR-T cells is so concise that it included only CD3ζ as an intracellular signaling (28). For lacking costimulatory molecules, the first-generation CAR-T cells cannot provide prolonged triggering of T cell activation and therefore have limited antitumor effect. The second-generation CAR, with costimulatory molecules and inducible costimulatory were added to enhance T cell proliferation (29). Based on the fact that CD28-CAR-T cells are more potent in killing cancer cells, and 4-1BB-CAR-T cells exhibit lower depletion rates and longer-lasting killing effects on cancer cells (30), third-generation CARs added both CD28 and OX-40/4-1BB (31). As cytokine secretion in third-generation CAR-T cells are upregulated and greatly inhibited cancer cell proliferation is enhanced (32, 33), fourth-generation CARs, also known as T cells redirected for universal cytokine-mediated killing (TRUCKs) (34), adds cytokine-encoding genes to enhance cancer killing effect by secreting inflammatory cytokines. Promisingly, the fifth-generation CARs, replacing OX-40/CD27 by IL-2 receptor β, has shown potential effect via activating the Janus kinases and signal transducers and activators of transcription-3/5 pathway in tumors (35, 36). However, both the safety and efficacy of the 5th-generation are need to be investigated and the possibly damaged transduction efficiency of CAR-T cells also should be taken care (37).
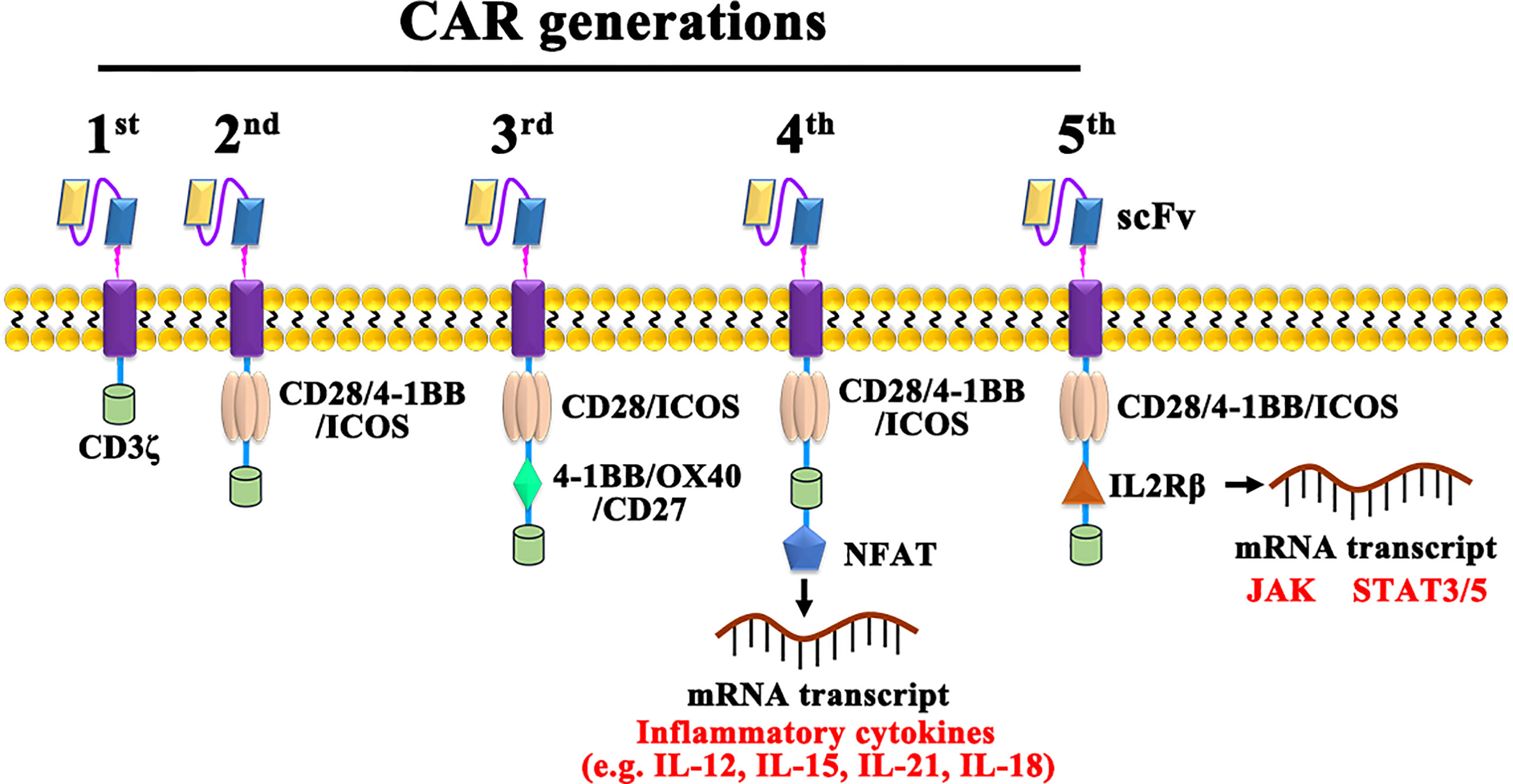
Figure 2 The construction of 1st, 2nd, 3rd, 4th, and 5th generation CARs. NFAT, Nuclear factor of activated T cells; JAK, Janus kinase; STAT, Signal transducer and activator of transcription.
Target Antigens For Car-T Cell Therapy In Clinical Trials For Thoracic Malignancies
In recent decades, the difficulty of CAR-T cell immunotherapy in thoracic malignancies has been mainly due to the lack of ideal targets. The ideal TAA for CAR-T cell immunotherapy is exclusively expressed on all or most tumor cells but not expressed or expressed at very low levels on normal tissues (38), which can enable CAR-T cells to trigger cancer-specific immune responses, thus sparing healthy tissues (39). However, it is difficult to obtain the ideal TAA for CAR-T cell immunotherapy in thoracic malignancies as CD19 in hematologic malignancies (40, 41). Based on previous studies, we summarize a series of TAAs that could be used as antigenic targets for CAR-T cells in patients with thoracic tumors in Figure 3 and Table 1.
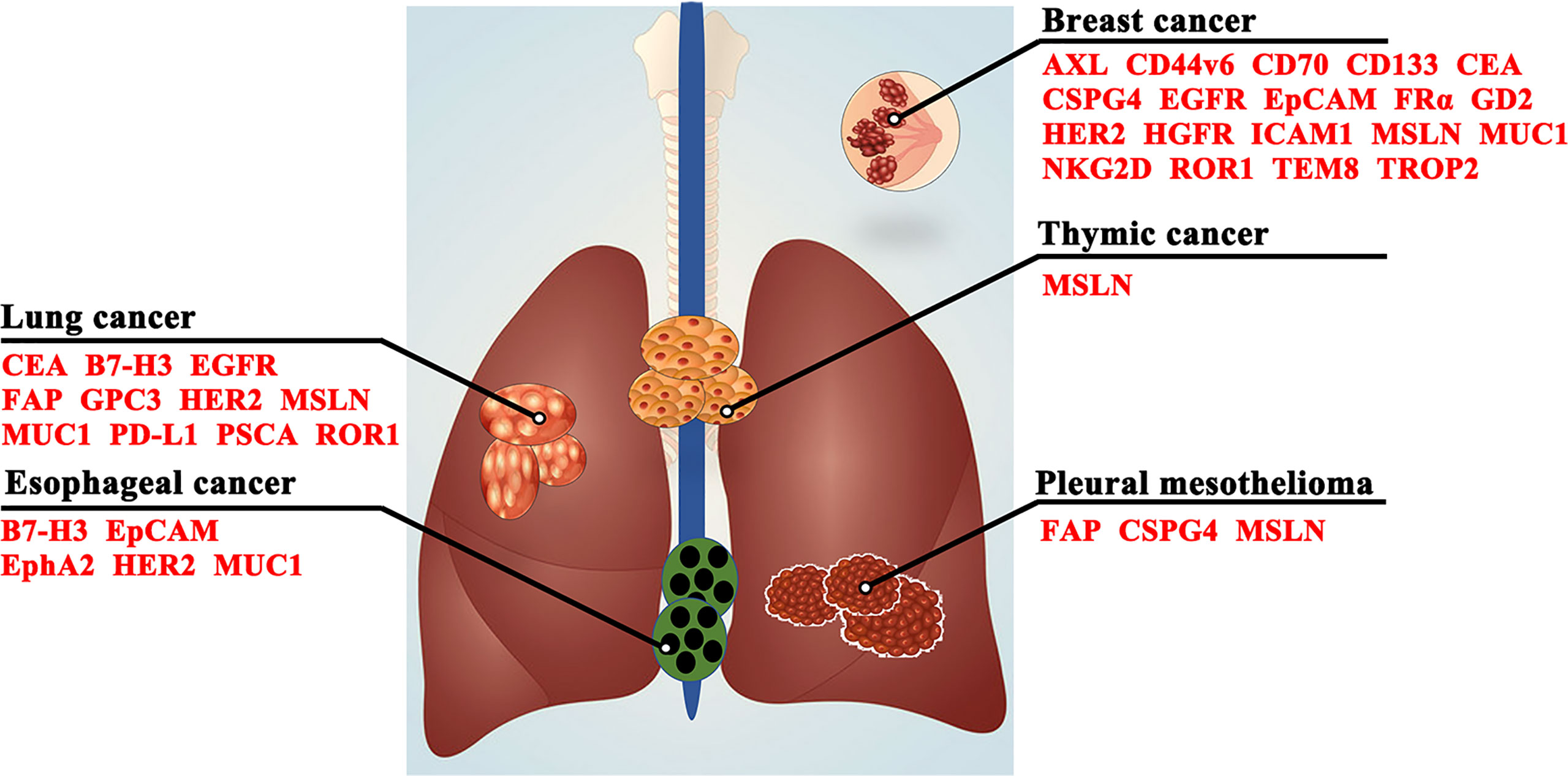
Figure 3 Target antigens for CAR-T cell therapy in thoracic malignancies. CD44v6, CD44 containing variant exon v6; CEA, carcinoembryonic antigen; CSPG4, chondroitin sulfate proteoglycan 4; EGFR, epidermal growth factor receptor; EpCAM, epithelial cell adhesion molecule; EphA2, erythropoietin-producing hepatocellular carcinoma A2; FAP, fibroblast activating protein; FRα, folate receptor α; GD2, glycolipid disialoganglioside; GPC3, glypican-3; HER2, human epidermal growth factor receptor 2; HGFR, hepatocyte growth factor receptor; ICAM1, intercellular adhesion molecule-1; MSLN, mesothelin; MUC1, mucin 1; NKG2D, natural killer group 2, member D; PD-L1, programmed death-ligand 1; PSCA, prostate stem cell antigen; ROR1, receptor tyrosine kinase-like orphan receptor 1; TEM8, tumor endothelial marker 8; TROP2, trophoblast cell surface protein 2.
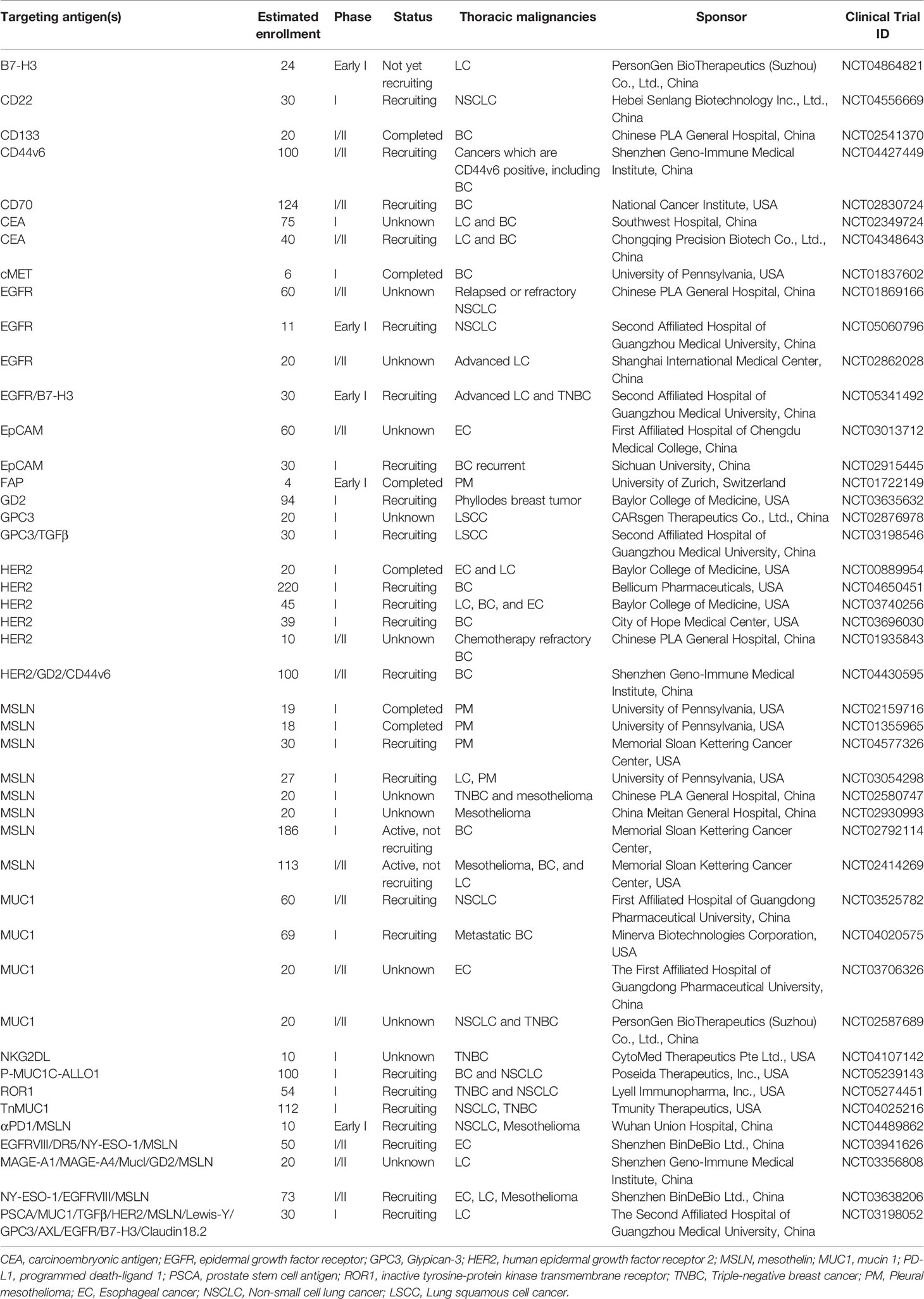
Table 1 Ongoing clinical trials of CAR-T cell therapy for thoracic cancer in ClinicalTrials.gov.
B7-H3
B7-H3 (CD276), which is a member of the B7 immunoglobulin superfamily and is highly expressed in many malignant tumors, serves as a molecular target for cancer immunotherapy (42). Numerous studies have demonstrated that B7-H3 facilitates the development and progression of tumors by promoting the malignant biological behavior of cancer cells (43, 44), such as cell proliferation, migration, invasion, apoptosis, and metabolism. Moreover, overexpression of B7-H3 inhibited the activation of T cells and effectively suppressed the proliferation and cytotoxic functions of activated T cells. For example, inhibition of B7-H3 promoted the viability of cytotoxic T lymphocytes (CTLs) and natural killer (NK) cells and reduced the number of tumor-associated macrophages and tumor load (45). Of note, B7-H3 was overexpressed in tissues of patients with thoracic malignancies (46–48), and antibody immunotherapy targeting B7-H3 did not lead to toxicity to vital organs (49). Scribner et al. (50) reported that the antibody–drug MGC018 targeting B7-H3 possessed antitumor activity in patient-derived xenograft models of breast cancer and lung cancer. The above studies indicated that B7-H3 may be an ideal TAA for cancer cell immunotherapy. Recently, several clinical studies showed that B7-H3-targeted CAR-T cells exhibited effective antitumor activity in hematologic tumors (e.g., acute myeloid leukemia) (51) and solid tumors (e.g., brain tumors, ovarian cancer, prostate cancer, melanoma) (52–54). Meanwhile, several clinical trials have been designed to test the safety, tolerability, and feasibility of B7-H3-targeted CAR-T cells against thoracic tumors, including NCT05341492, NCT04864821, and NCT03198052. Overall, B7-H3-targeted CAR-T cells may be a novel curative approach for B7-H3-positive patients with thoracic tumors.
CEA (Carcinoembryonic Antigen)
CEA is a glycoprotein that belongs to the immunoglobulin superfamily, and its expression is positively correlated with tumor incidence (55). Meanwhile, analysis of the TCGA database revealed that CEA was highly expressed in thoracic tumors (e.g., lung, breast, and esophageal), and patients with high CEA expression showed a worse prognosis. Previous studies have also proven that CEA serves as an ideal target for the treatment of gastrointestinal tumors (56, 57). Preclinical data have confirmed that the serum concentrations of CEA in patients with advanced non-small-cell lung cancer (NSCLC) were correlated with the occurrence of brain metastases (58), and high CEA expression was associated with clinicopathological characteristics in lung cancer patients, including lymph node metastasis and vascular infiltration (59). Recent studies confirmed that CEA-targeted CAR-T cells inhibited tumor growth and enhanced the overall survival time of tumor-bearing mice (60, 61). Importantly, CEA-specific CAR-T cells exhibited an antitumor effect in patients with CEA-positive solid tumors and did not cause cytokine release syndrome (62).
EGFR (Epidermal Growth Factor Receptor)
EGFR, which is highly expressed on the membrane surface of many solid tumor cells and are involved in nearly all aspects of malignant cancer, belongs to the ErbB family of growth factor receptor tyrosine kinases (63, 64). Previous studies have shown that EGFR expression is upregulated in the tissues of patients with thoracic malignancies (65–67), indicating that it can be an effective biomarker for the diagnosis and treatment of thoracic tumors (68). The results of an EGFR-positive relapsed/refractory (R/R) NSCLC clinical trial (NCT01869166) showed that none of the patients experienced significant toxic side effects after anti-EGFR CAR-T cell therapy, two patients achieved partial remission, and five patients had stable disease for 2-8 months. Xia et al. (69) reported that third-generation EGFR-targeted CAR-T cells exerted potent and specific suppression of triple-negative breast cancer (TNBC) cell growth in vitro and in vivo by activating the Fas/FADD/Caspase pathway. The above studies suggested that EGFR-targeted CAR-T cell therapy could be utilized in the treatment of patients with EGFR-positive thoracic malignancies in the future, although additional clinical studies are needed to confirm these results.
Epcam (Epithelial Cell Adhesion Molecule)
EpCAM is a transmembrane glycoprotein also known as CD326. Previous studies have demonstrated that overexpression of EpCAM is associated with poor prognosis in patients with esophageal squamous cell carcinoma (70), lung cancer (71), and breast cancer (72), and it can be used as a marker for circulating tumor cells involved in cancer cell metastasis (73). Meanwhile, EpCAM plays a key role in tumorigenesis and metastasis (74). Hiraga et al. (75) showed that high expression of EpCAM was closely associated with bone metastasis in breast cancer. Importantly, EpCAM is an excellent target for various therapeutic approaches, including immunotherapy, because it is uniformly expressed on the surface of tumor cells (76, 77). As expected, a clinical trial also confirmed that EpCAM-targeted CAR-T cells are safe and effective in the treatment of EpCAM-positive gastric cancer (78). Taken together, EpCAM may be a promising target for CAR-T cell therapy in thoracic malignancies.
FAP (Fibroblast Activating Protein)
FAP is a marker expressed on cancer-associated fibroblasts in human solid tumors (79). Previous studies have found that overexpression of FAP facilitates cancer cell proliferation, invasion, and angiogenesis (80) and serves as a novel target for various cancer therapies (81). In addition, FAP has been reported to be an excellent target for immunotherapy in glioblastoma (82). Wang et al. (83) also demonstrated that FAP-targeted CAR-T cells inhibited the growth of lung transplantation tumors by removing FAP-positive stromal cells without severe toxicity. Another study by Schuberth et al. (84) performed a phase I clinical trial and demonstrated that FAP-targeted redirected CD8+ T cells hampered FAP-positive tumor growth and prolonged the survival of mice with malignant pleural mesothelioma. Therefore, FAP-targeted CAR-T cell therapy may be an effective approach for thoracic malignancy treatment in the clinic, but its safety and efficacy need further evaluation.
HER2 (Human Epidermal Growth Factor Receptor 2)
HER2 is a transmembrane glycoprotein that has become more widely studied as a target for tumor therapy in recent years. Previous studies have confirmed that HER2 is highly expressed in thoracic malignancies (85) and facilitates the proliferation, invasion, and angiogenesis of cancer cells (86). Of note, HER2 serves as a promising biomarker for the diagnosis and treatment of solid tumors (87, 88), which has attracted many scholars to focus on HER2 as a novel target for cancer immunotherapy. For example, HER2-targeted CAR-T cells inhibited xenograft growth in esophageal cancer mouse models and reduced proinflammatory cytokine secretion (89). Another study demonstrated that third-generation HER2-targeted CAR-T cells exhibited an antitumor effect on HER2-positive and trastuzumab-resistant breast cancer in vivo (90). The above studies suggested that HER2 may be clinically effective as a target for CAR-T cell immunotherapy for the treatment of thoracic malignancies.
Mesothelin (MSLN)
MSLN is a cell adhesion glycoprotein and its overexpression was positively correlated with high tumor aggressiveness and poor prognosis in patients with thoracic malignancies (91–94). Importantly, MSLN has been reported to be a more desirable TAA for CAR-T cell therapy in solid tumors (95). For example, MSLN-CAR-T cells could specifically kill various MSLN-positive solid tumor cell lines and release cytokines in vitro and also decreased the growth of MSLN-positive solid tumors (e.g., ovarian, breast, colorectal cancer) in vivo (96). Ye et al. (97) showed that second-generation anti-MSLN CAR-T cells possessed a significantly higher ability to kill NSCLC cells than T cells and reduced the growth of tumors in a xenograft mouse model. Another study reported that exosomes derived from MSLN-targeted CAR-T cells notably inhibited the growth of MSLN-positive triple-negative breast cancer without obvious side effects (98). Moreover, MSLN-CAR-T cells displayed stronger antitumor activity in NSCLC due to enhanced migration and infiltration into tumor tissues induced by the chemokine receptors CCR2b and CCR4 (99). Similarly, an oncolytic adenovirus targeting TGFβ contributed to enhancing the antitumor effect of MSLN-targeted CAR-T cells on breast cancer (100). Using anti-MSLN CAR-T cells for malignant mesothelioma, Castelletti et al. (101) described antitumor activity with a high safety profile in a clinical trial. Collectively, modified CAR-T cells targeting MSLN could be a promising therapeutic approach for MSLN-expressing thoracic malignancies.
MUC1 (Mucin 1)
MUC1 is a transmembrane protein that facilitates cancer cell adhesion and metastasis (102). Previous studies have confirmed that MUC1 is aberrantly overexpressed in thoracic malignancies, including lung cancer (103), breast cancer (104), and esophageal cancer (105), and serves as an oncogene in the tumorigenesis of various human adenocarcinomas. Of note, MUC1 has been reported as a reliable target for immunotherapy of solid malignancies (106). Wei et al. (107) showed that CAR-T cells targeting prostate stem cell antigen (PSCA) and MUC1 significantly eliminated tumor cells that were positive for both PSCA and MUC1 in NSCLC. Another study reported that MUC1-targeted CAR-T cells reduced the proliferation capability of esophageal cancer cells by activating the JAK/STAT pathway and inhibited tumor growth in transplantation models and patient-derived tumor xenograft (PDX) models of esophageal cancer in vivo (108). In addition, 6 clinical trials are currently evaluating the safety and efficacy of anti-MUC1 CAR-T cell therapy in thoracic malignancies (NCT03179007, NCT02587689, NCT03198052, NCT03706326, NCT03525782, and NCT05239143).
Programmed Death-Ligand 1 (PD-L1)
Targeting the programmed death-1 (PD-1)/PD-L1 signaling pathway has made substantial progress in the immunotherapy of thoracic malignancies in recent years (109). Numerous studies have confirmed that PD-L1 serves as an important immune checkpoint that is upregulated in various malignant tumors, including thoracic tumors (110, 111). Previous studies have demonstrated that PD-L1 can inhibit T cell proliferation and activation by binding to PD-1 on T cells, ultimately leading to immune escape of tumor cells (112, 113). Meanwhile, the treatment of malignancies with PD-L1 antibody has shown safe and exciting results in preclinical studies and clinical trials (114). Of note, preclinical studies demonstrated that PD-L1-targeted CAR-T cells possessed potent cytotoxic effects against NSCLC (115) and breast cancer (116). Qin et al. (117) reported that CAR-T cells targeting PD-L1 significantly inhibited the growth of multiple types of solid tumors in PDX mouse models. Another study proved that PD-L1-targeted CAR-T cells exhibited antigen-specific activation, cytokine production, and cytotoxic activity against PD-L1high NSCLC cells and xenograft tumors, and the addition of a subtherapeutic dose of local radiotherapy improved the efficacy of PD-L1-CAR-T cells against PD-L1low NSCLC cells and xenograft tumors (115). Moreover, inactivation of the PD-1/PD-L1 pathway enhanced the toxicity of CAR-T cells against tumor cells (118). Currently, several clinical trials are investigating the safety and efficacy of PD-L1-targeted CAR-T cells in thoracic malignancies (NCT03060343, NCT04556669, NCT04684459). However, a pilot study of anti-PD-L1 CAR-T cell immunotherapy for advanced lung cancer in a phase I trial was terminated due to serious adverse events (NCT03330834). Therefore, further evaluation of the potential applications of anti-PD-L1 CAR-T cell therapy in clinical trials is needed.
ROR1 (Receptor Tyrosine Kinase-Like Orphan Receptor 1)
ROR1, a tyrosine kinase-like orphan receptor, is upregulated in both lung cancer and breast cancer but has very low expression in normal tissues (119). Zheng et al. (120) demonstrated that ROR1 was an independent prognostic biomarker for overall survival. Importantly, the antitumor activity of anti-ROR1 CAR-T cells was equivalent to that of CD19 CAR-T cells in human mantle cell lymphoma (121). In both breast and lung cancer, ROR1-targeted CAR-T cells significantly restricted tumor growth and prolonged tumor survival (122). A recent study demonstrated that treatment with anti-ROR1 CAR-T cells could effectively kill NSCLC and TNBC cells in a three-dimensional tumor model (123). Thus, targeting ROR1 may be an effective strategy to improve CAR-T cell efficacy for the clinical treatment of thoracic malignancies.
Others
Currently, there are many other candidate TAAs for CAR-T cell immunotherapy in thoracic malignancies, including chondroitin sulfate proteoglycan 4 (CSPG4), CD44v6, CD80/CD86, CD56-and Delta-like ligand 3 (DLL-3), erythropoietin-producing hepatocellular carcinoma A2 (EphA2), folate receptor alpha (FRα), glycolipid disialoganglioside (GD2), glypican-3 (GPC3), Lewis-Y antigen, L1 cell adhesion molecule (L1CAM), lung-specific X (LUNX), IL13Rα2, melanoma-associated antigen (MAGE)-A1, MAGE-A4, and PSCA (124–129), which have not yet been validated in clinical trials.
Current Challenges and Strategies of CAR−T Cell Therapy in Thoracic Malignancies
CAR-T cell immunotherapy in solid tumors, especially in thoracic malignancies, still faces many obstacles compared to various types of malignant hematological tumors. The following aspects need to be taken into consideration for CAR-T cell immunotherapy in thoracic malignancies (Figure 4) (1): on-target/off-tumor toxicity (2); tumor antigen escape (3); neurological toxicity (4); immunosuppressive microenvironment (5); CAR-T cell trafficking and tumor infiltration. In summary, overcoming these challenges is the current hot field of CAR-T cell therapy in thoracic malignancies.
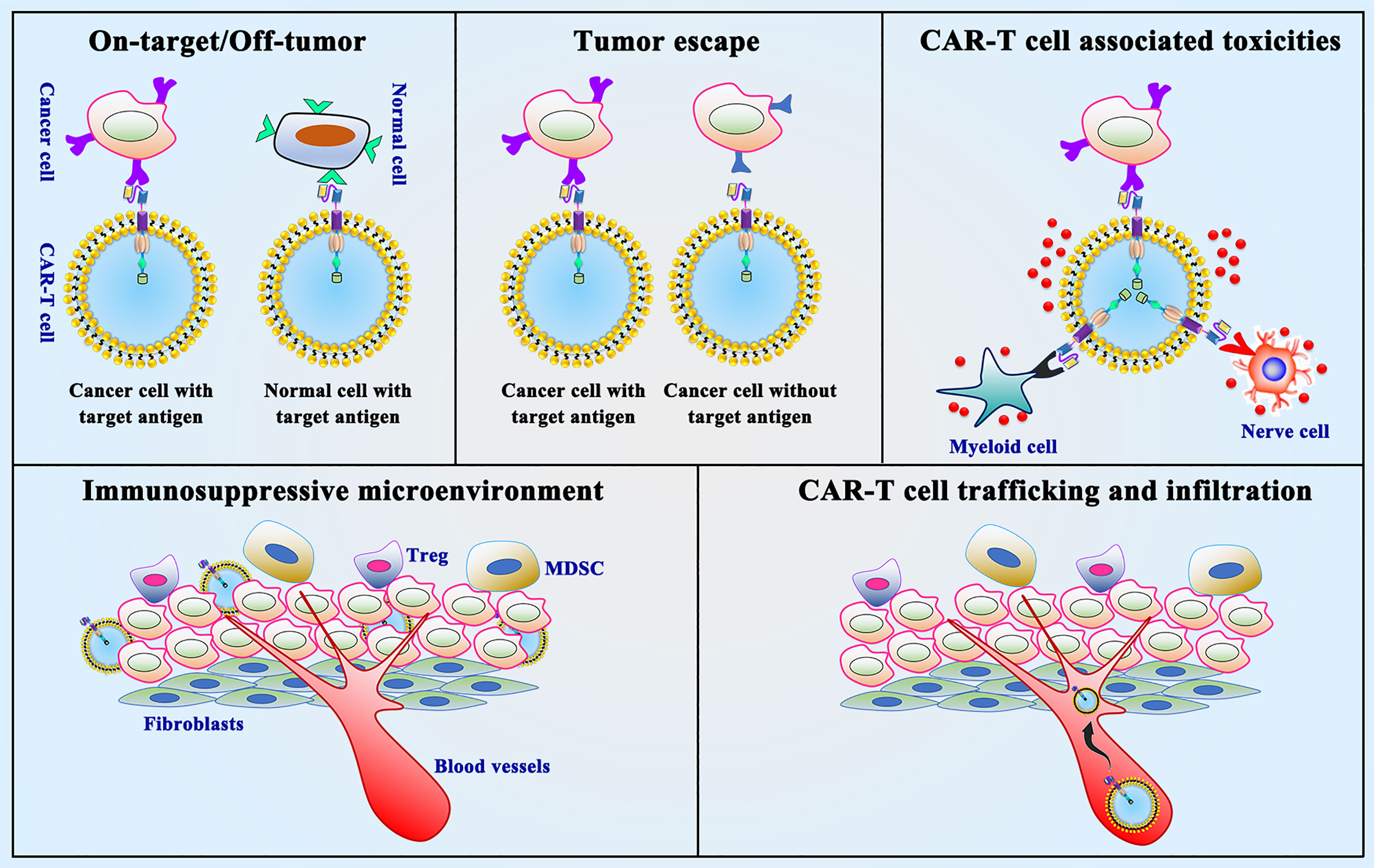
Figure 4 Limitations and challenges of CAR-T cell therapy. On-target/off-tumor toxicity, tumor antigen escape, neurological toxicity, immunosuppressive microenvironment and CAR-T cell trafficking and tumor infiltration are the presented limitations and challenges in CAR-T cell therapy.
On−Target/Off−Tumor Toxicity
The most critical problem with CAR-T cell therapy for solid tumors is the lack of an ideal TAA. The degree of on-target/off-tumor toxicity is the key component to the success of these candidate TAAs for CAR-T cells (130). ERBB2 expression is relatively low in the normal lung tissues, however, Morgan et al. (131) reported that injection with anti-ERBB2 CAR-T cells resulted in a colon cancer patient developed respiratory distress 15 minutes later and eventually died after 5 days. Meanwhile, the off-tumor toxicity of CAR-T cells may cause normal organ dysfunction (132). Screening and discovery of novel tumor antigens (133), dual CAR systems (134) and suicide genes (135) possibly can avoid these risks. Recently, many novel tumor antigens [e.g., intercellular adhesion molecule-1 (ICAM1) (136), NKG2D (137), VEGFR2 (138), MUC4 (139), and cluster of differentiation (CD)70 (140)] were reported to be effective targets for CAR-T cell therapy of solid tumors. Wang et al. (141) showed that chlorotoxin as the targeting domain of CAR-T cells exhibited anti-glioblastoma (GBM) activity and resulted in tumor regression in orthotopic xenograft GBM tumor models with the potential to reduce antigen escape during CAR-T cell therapy. Moreover, a new technology, namely single-cell RNA sequencing, may provide a more accurate target antigen expression profile for TAA selection, which can better predict the efficacy and toxicity of novel CAR-T cell therapy in tumors (142). Choi et al. (143) demonstrated an elegant approach to overcome EGFRvIII antigen loss, with EGFRvIII-targeting CAR-T cells that secrete a bispecific T cell engager (BiTE) against wild-type EGFR, and CAR-T-BiTE cells did not result in toxicity against human skin grafts in vivo compared with EGFR-specific CAR-T cells. Furthermore, designing CAR-T cells targeting multiple targets in combination may also be an effective strategy to enhance tumor eradication (144). For example, Roybal et al. (145) found that anti-GFP and anti-CD19 dual-specific CAR-T cells significantly inhibited K562 cell proliferation and xenograft tumor growth. Meanwhile, preclinical studies showed that GD2/B7-H3 (146) or ROR1/B7-H3 (147) SynNotch CAR-T cells killed tumor cells with high specificity and efficacy and without toxicity to normal cells expressing the target antigen.
Neurological Toxicity
Neurotoxicity is characterized by various neurological symptoms, including headache, aphasia, delirium, and even cerebral hemorrhage, seizures, and death (148). During this process the systemic inflammatory response associated with CRS may contribute to the risk of complications of neurotoxicity (149, 150). The activation of endothelial cells possibly facilitates the occurrence to neurotoxicity (151), which has been verified by autopsy showing that the disrupted endothelial dysfunction and blood–brain barrier disruption (152). Importantly, neurotoxicity can be largely reversible and completely resolved after treatment with tocilizumab and dexamethasone, whereas neurotoxicity recovery was slower after treatment with tocilizumab for neurotoxicity patients with endothelial cell activation (153).
Cytokine Release Syndrome (CRS)
CRS is induced by T cell activation and commonly presented with fever, chills, muscle pain, generalized weakness, and systemic organ failure (154). Activated CAR-T cells is the leading cause of CRS and possibly result in a significant increase in the secretion of proinflammatory factors by immune cells (155). To avoid this disadvantage, a controlled gene “device”, such as herpes simplex virus thymidine kinase (HSV-TK), human inducible caspase 9 (iCasp9), mutant human thymidylate kinase (mTMPK), and human CD20, for CAR-T cells was applied and has shown to be effective in reducing proinflammatory cytokine secretion and clearing CAR-T cells from the body in time for acute toxicity (156–159). Apart from that, dasatinib can also act as a CAR-T cell “switch” to control the biological function of CAR-T cells upon entry into the body and protect mice from CRS (160). Moreover, optimizing CAR gene transfection can regulate the in vivo lifespan and kinetics of CAR-T cells (161) and using nanoparticles can reduce and avoid CRS (162). Overall, avoiding CRS damage after CAR-T cell immunotherapy will be a key issue in the treatment of thoracic malignancies in the future.
Immunosuppressive Microenvironment
Immunosuppressive TME is characterized by hypoxia, oxidative stress, and tumor-derived cytokine suppression, which is greatly restricted the CAR-T cell therapy (22). Suppressive immune cells, including regulatory T cells, myeloid-derived suppressor cells, and tumor-associated macrophages, can be activated by a variety of immunosuppressive factors released by tumor cells (163). Of note, preclinical studies have extensively shown that the TME is hostile to T cells (164, 165). All these studies suggest that altering the immunosuppressive effects on the TME possibly enhance the anticancer effects of CAR-T cells. Some groups have demonstrated that PD-1-blocking scFv secreting CAR-T cells significantly prolonged the survival time of tumor-bearing (166) and CAR-T cells overexpressing the PD-1 dominant negative receptor could act as a “decoy receptor” to bind and block PD-L1/2 inhibitory signals (167). In addition, IL-7/IL-5 exhibited antitumor activity by promoting CAR-T cell proliferation ability, reducing CAR-T cell apoptosis, and reforming the immunosuppressive TME (168). Therefore, CAR-T cells coexpressing immune-related factors may be an effective solution for the clinical treatment of thoracic malignancies.
CAR-T Cell Trafficking and Tumor Infiltration
In the treatment of hematologic malignancies, CAR-T cells can effectively exert their antitumor effects by direct contact with tumor cells. However, the ability of CAR-T cells to infiltrate solid tumors is restricted when treating thoracic malignancies due to physical barriers (e.g., tumor-associated fibroblasts (CAFs) and dense extracellular matrix (ECM)) in the tumor tissue (15), which results in reduced antitumor effects. In addition, the immunosuppressive TME also limits the penetration and movement of CAR-T cells within solid tumors (169). Thus, improving the ability of CAR-T cells to specifically degrade ECM in stroma-rich solid tumors without compromising their cytotoxicity (170) might be an effective strategy to alleviate the above limitations. For example, Caruana et al. (171) reported that engineered CAR-T cells expressed heparinase, which degrades heparan sulfate proteoglycans, the main components of the ECM, and thus promoted T cell infiltration into the tumor and antitumor activity. Wang et al. (83) showed that FAP-targeted CAR-T cells possessed an antitumor effect on solid tumors by reducing tumor fibroblasts and enhancing host immunity without severe toxicity in xenograft models. Recent studies have confirmed that engineered CAR-T cells expressing chemokine receptors (e.g., CXCR1, CXCR2, CXCR4) contribute to enhancing CAR-T cell trafficking and tumor infiltration (172, 173) as well as improving antitumor activity. Overall, further studies are needed to develop new delivery strategies to improve the penetration of CAR-T cells in tumor tissues, which will enhance the efficacy of CAR-T cells in thoracic malignancies.
Tumor Antigen Escape
Currently, other factors affecting the antitumor effect of CAR-T cells on malignancies may be related to antigen escape. For example, anti-CD19 CAR-T cell therapy caused the loss of CD19 target antigen in R/R B cell acute lymphoblastic leukemia (B-ALL) patients (174). In addition, target antigen escape is a major cause of R/R cancer and a key factor in the failure or stronger side effects of expanding the use of CAR-T cells toward solid cancers with multiple surface antigens (175). The construction of CAR-T cells containing dual targets may be an effective strategy to address this problem. For example, the therapeutic effect of CAR-T cells with dual targets of CD19 and CD22 in a phase I clinical trial (NCT03330691) for the treatment of R/R B-ALL was better than that of single-target CD19 or CD22, which could avoid the problem of target antigen escape that occurs with single targets. Moreover, anti-CD19/BAFF-R CAR-T cell therapy showed prolonged in vivo persistence and exhibited antigen-specific cytokine release, degranulation, and cytotoxicity against both CD19-/- and BAFF-R-/- variant human ALL cells in vitro (176). Another study showed that CAR-T cells targeting BAFF-R could overcome CD19 antigen loss in B cell malignancies (177). These findings are important for developing approaches to overcome the risk of tumor antigen escape in CAR-T cell immunotherapy for thoracic tumors.
Opportunities To Improve Car-T Cell Safety And Efficacy
Previous studies have shown that uncontrolled CAR-T cell proliferation in patients with malignancies treated with CAR-T cells can cause severe toxicity (178, 179). Currently, numerous studies have developed many methods to improve the safety and efficacy of CAR-T cell therapy in solid tumors, as described below.
Removal of Residual CAR-T Cells
The integration of “suicide genes” into T cells served as an inducible safety switch that allowed transduced CAR-T cells to kill themselves in the case of adverse events (180). Preliminary studies have shown that different suicide genes, such as HSV-TK, iCasp9, mTMPK, and human CD20, can be expressed in donor T cells (158, 159) and have shown promising safe suicidal effects in early-phase clinical trials of CAR-T cell therapy. Functionally, activation of HSV-TK, iCasp9 and CD20 eventually resulted in effective T cell destruction; however, iCasp9 and CD20 induced immediate cell death, HSV-TK-expressing T cells required 3 d of exposure to ganciclovir, and mTMPK-transduced cells in all T cell killing rates reflected a poorer response (181). Klopp et al. (182) showed that depletion of T cells via iCasp9 increased the safety of adoptive T cell therapy against chronic hepatitis B. Another study showed that the HSV-TK suicide gene could enhance the safety of anti-CD44v6 CAR-T cell therapy in lung cancer (128). To date, only two suicide genes (HSV-TK and iCasp9) have demonstrated an excellent safety profile in clinical trials (NCT00423124; ChiCTR-OOC-16007779).
ON/OFF-Switch for CAR
Currently, engineered CAR-T cells, as autonomous “living drugs” for cancer treatment, lack precise control and may cause toxicity, suggesting that assembling ON/OFF switches for CARs with small molecules may address the above limitations (183, 184). For example, Wu et al. (157) designed ON-switch CARs that enable small-molecule (e.g., AP21967) control over T cell therapeutic functions while still retaining antigen specificity. Similarly, another study established a new CAR structure with an integrated ON-switch system that controls the function of CAR-T cells, and CAR-T cells with integrated controllable transients exhibited antitumor activity under multiple cytotoxic cycles using small molecule drugs without severe toxicity (156). Jan et al. (185) constructed the ON-switch CAR (lenalidomide ON-switch split CAR) and the OFF-switch CAR (lenalidomide OFF-switch degradable CAR). Importantly, treatment with lenalidomide only restricts the short-term toxicity of CAR-T cell immunotherapy but does not affect the long-term antitumor effects of CAR-T cells. Moreover, Frankel et al. (186) proposed that bifunctional molecules could act as a bridge between cytotoxic T cells that can effectively kill cancer cells on one side and T cells that target CD3 molecules and associated antigens on the surface of tumor cells on the other side, thus activating T cells with a double switch and effectively destroying the target cells.
Improving Trafficking
Currently, the application of CAR-T cells for solid tumors can be performed by devices placed surgically (e.g., central nervous system tumors), by intra-arterial delivery, or by direct intratumoral injection. For example, Brown et al. (187) reported that inhibition of tumor growth and upregulation of immune cytokine levels by intracranial infusion of CAR-T cells targeting IL13Rα2 was not associated with toxic effects. Tchou et al. (188) showed that intratumoral injection of anti-cMET CAR-T cells halted tumor growth in patients with metastatic breast cancer and evoked an inflammatory response within tumors, and none of the patients had study drug-related adverse effects greater than grade 1. In addition, prompting CAR-T cells to express chemokine receptors may also be an effective strategy to accelerate CAR-T cell trafficking to tumors. For example, CAR-T cells targeting GD2 could facilitate CAR-T cell migration by expressing CCR2b. Similarly, CCR2b enhanced the migration of CAR-T cells targeting MSLN in vitro and in a mouse xenograft model of NSCLC (99). Perera et al. (189) demonstrated that CCR4 can serve as a novel target antigen for the treatment of T cell malignancies by CAR-T cells. However, there is controversy about the optimal chemokine receptor used to improve CAR-T cell trafficking (190). Furthermore, many chemokines are used as target antigens for CAR-T cells in solid tumor treatment (172, 191–193).
Improving CAR-T Cell Manufacturing
Autologous CAR-T cells are patient-derived personalized products that can achieve long-term antitumor activity but still have many drawbacks, such as treatment delays (2 to 4 weeks), complex manufacturing procedures, and increased costs (194). Importantly, the development of universal CAR-T cells could simplify the manufacturing process and expand production, facilitating immediate delivery of immunotherapy at a lower cost (195). For example, Choi et al. (196) created universal EGFRvIII CAR-T cells using the CRISPR–Cas9 system and showed significant antitumor activity in preclinical glioma models and prolonged survival in mice bearing intracranial tumors. In addition, phase I clinical trials of universal CAR-T cells targeting MSLN (NCT03545815) and NKG2D (NCT03692429) are underway to seek safe and effective therapeutic methods.
Future Perspectives For Car−T Cell Therapy In Thoracic Malignancies
The success of CAR-T cell therapy in hematologic malignancies has inspired the thought dealing with thoracic malignancies and has entered a phase of rapid development (36). Future studies on CAR-T cells may include but not limited in (1): searching for more specific target antigens (2); reforming the CAR structure to enhance the efficacy, specificity, and survival time of CAR-T cells (3); decreasing the toxicity of CAR-T cells (4); constructing CAR-T cells that target the TME of thoracic malignancies (5); exploring combination therapies; and (6) establishing natural ligand–receptor-based CAR-T cells. Importantly, these modified CARs are being studied in animal models and clinical trials in an attempt to mitigate tumor antigen heterogeneity and may eventually form the next generation of CAR-T cells (197). In conclusion, the above efforts will provide safer and more effective clinical applications of CAR-T cell immunotherapy for thoracic malignancies.
Conclusion
We summarized the structure, history of CAR-T cells, the common and uncommon TAAs used in CAR-T cell therapy against thoracic malignancies, as well as pointed out current challenges and possible effective strategies. Thoracic malignancies, including lung cancer, breast cancer, mesenchymal malignancies, esophageal cancer account for nearly one third of new cancers and deaths worldwide. Thus, thoracic cancer is the leading cause of cancer-related death and a significant obstacle to enhancing life expectancy worldwide. Different from chemotherapy, radiotherapy and target therapy, CAR-T cell immunotherapy against thoracic malignancies represents a brand treatment choice. Although there is some limitations, the beneficial results of preliminary trials have provided a prospective future for their application in the subsequent clinical treatment of thoracic malignancies. On-target/off-tumor, tumor antigen escape, CAR-T cell associated toxicities, immunosuppressive microenvironment, CAR-T cell trafficking and infiltration are the major disadvantages. However, via screening specific target antigens, improving trafficking and improving CAR-T cell manufacturing, CAR-T cell therapy may improve its current status in the near future. CAR-T cells have obtained great success in the field of hematological tumors, stimulating many researchers to study the application of CAR-T cells of thoracic malignancies. Luckily, both experimental and clinical trials of CAR-T cells for thoracic malignancies are underway, which will greatly promote the application of CAR-T cell treatment clinically.
Data Availability Statement
The original contributions presented in the study are included in the article/supplementary material. Further inquiries can be directed to the corresponding authors.
Author Contributions
LC, FC and HN designed the study and wrote this manuscript. JL, YP, CY, and YW compiled and analyzed the literature. KL, YL and YH proposed the study, revised, and re-organized the manuscript. All authors read and approved the final manuscript
Funding
This study was supported by XingDianYingCai Support Plan, the National Natural Science Foundation of China (No. 81960496), Academician Zhan Qimin Workstation of Yunnan Province, Yunnan Fundamental Research Projects (202101AT070050, 202001AY070001−247), Yunnan health training project of high-level talents (H-2018006, H-2018055), and the 2017 Medical Oncology Academic leader of Yunnan province (D-2017001).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Turner RR, Steed L, Quirk H, Greasley RU, Saxton JM, Taylor SJ, et al. Interventions for Promoting Habitual Exercise in People Living With and Beyond Cancer. Cochrane Database Syst Rev (2018) 9:Cd010192. doi: 10.1002/14651858.CD010192.pub3
2. Li Y, Sui Y, Chi M, Zhang J, Guo L. Study on the Effect of MRI in the Diagnosis of Benign and Malignant Thoracic Tumors. Dis Markers (2021) 2021:3265561. doi: 10.1155/2021/3265561
3. Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, et al. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J Clin (2021) 71:209–49. doi: 10.3322/caac.21660
4. Ozaki Y, Muto S, Takagi H, Watanabe M, Inoue T, Fukuhara M, et al. Tumor Mutation Burden and Immunological, Genomic, and Clinicopathological Factors as Biomarkers for Checkpoint Inhibitor Treatment of Patients With non-Small-Cell Lung Cancer. Cancer Immunol Immunother (2020) 69:127–34. doi: 10.1007/s00262-019-02446-1
5. Parikh K, Huether R, White K, Hoskinson D, Beaubier N, Dong H, et al. Tumor Mutational Burden From Tumor-Only Sequencing Compared With Germline Subtraction From Paired Tumor and Normal Specimens. JAMA Netw Open (2020) 3:e200202. doi: 10.1001/jamanetworkopen.2020.0202
6. Wang Y, Ye D, Kang M, Zhu L, Pan S, Wang F. Risk Factors and Patterns of Abdominal Lymph Node Recurrence After Radical Surgery for Locally Advanced Thoracic Esophageal Squamous Cell Cancer. Cancer Manag Res (2020) 12:3959–69. doi: 10.2147/cmar.s249810
7. Zhang S, Ikramuddin S, Beckwith HC, Sheka AC, Wirth KM, Blaes AH. The Impact of Bariatric Surgery on Breast Cancer Recurrence: Case Series and Review of Literature. Obes Surg (2020) 30:780–5. doi: 10.1007/s11695-019-04099-6
8. Jones GS, Baldwin DR. Recent Advances in the Management of Lung Cancer. Clin Med (Lond) (2018) 18:s41–6. doi: 10.7861/clinmedicine.18-2-s41
9. Emens LA, Ascierto PA, Darcy PK, Demaria S, Eggermont AMM, Redmond WL, et al. Cancer Immunotherapy: Opportunities and Challenges in the Rapidly Evolving Clinical Landscape. Eur J Cancer (2017) 81:116–29. doi: 10.1016/j.ejca.2017.01.035
10. Del Vecchio F, Martinez-Rodriguez V, Schukking M, Cocks A, Broseghini E, Fabbri M. Professional Killers: The Role of Extracellular Vesicles in the Reciprocal Interactions Between Natural Killer, CD8+ Cytotoxic T-Cells and Tumour Cells. J Extracell Vesicles (2021) 10:e12075. doi: 10.1002/jev2.12075
11. Guedan S, Ruella M, June CH. Emerging Cellular Therapies for Cancer. Annu Rev Immunol (2019) 37:145–71. doi: 10.1146/annurev-immunol-042718-041407
12. Zhu YG, Xiao BF, Zhang JT, Cui XR, Lu ZM, Wu N. Genetically Modified T Cells for Esophageal Cancer Therapy: A Promising Clinical Application. Front Oncol (2021) 11:763806. doi: 10.3389/fonc.2021.763806
13. Ichiki Y, Shigematsu Y, Baba T, Shiota H, Fukuyama T, Nagata Y, et al. Development of Adoptive Immunotherapy With KK-LC-1-Specific TCR-Transduced γδt Cells Against Lung Cancer Cells. Cancer Sci (2020) 111:4021–30. doi: 10.1111/cas.14612
14. Ge Y, Böhm HH, Rathinasamy A, Xydia M, Hu X, Pincha M, et al. Tumor-Specific Regulatory T Cells From the Bone Marrow Orchestrate Antitumor Immunity in Breast Cancer. Cancer Immunol Res (2019) 7:1998–2012. doi: 10.1158/2326-6066.cir-18-0763
15. Sterner RC, Sterner RM. CAR-T Cell Therapy: Current Limitations and Potential Strategies. Blood Cancer J (2021) 11:69. doi: 10.1038/s41408-021-00459-7
16. Hong M, Clubb JD, Chen YY. Engineering CAR-T Cells for Next-Generation Cancer Therapy. Cancer Cell (2020) 38:473–88. doi: 10.1016/j.ccell.2020.07.005
17. King AC, Orozco JS. Axicabtagene Ciloleucel: The First FDA-Approved CAR T-Cell Therapy for Relapsed/Refractory Large B-Cell Lymphoma. J Adv Pract Oncol (2019) 10:878–82. doi: 10.6004/jadpro.2019.10.8.9
18. Anagnostou T, Riaz IB, Hashmi SK, Murad MH, Kenderian SS. Anti-CD19 Chimeric Antigen Receptor T-Cell Therapy in Acute Lymphocytic Leukaemia: A Systematic Review and Meta-Analysis. Lancet Haematol (2020) 7:e816–26. doi: 10.1016/s2352-3026(20)30277-5
19. Makita S, Imaizumi K, Kurosawa S, Tobinai K. Chimeric Antigen Receptor T-Cell Therapy for B-Cell non-Hodgkin Lymphoma: Opportunities and Challenges. Drugs Context (2019) 8:212567. doi: 10.7573/dic.212567
20. Vishwasrao P, Li G, Boucher JC, Smith DL, Hui SK. Emerging CAR T Cell Strategies for the Treatment of AML. Cancers (Basel) (2022) 14:1241. doi: 10.3390/cancers14051241
21. Ma S, Li X, Wang X, Cheng L, Li Z, Zhang C, et al. Current Progress in CAR-T Cell Therapy for Solid Tumors. Int J Biol Sci (2019) 15:2548–60. doi: 10.7150/ijbs.34213
22. Martinez M, Moon EK. CAR T Cells for Solid Tumors: New Strategies for Finding, Infiltrating, and Surviving in the Tumor Microenvironment. Front Immunol (2019) 10:128. doi: 10.3389/fimmu.2019.00128
23. Ghosn M, Cheema W, Zhu A, Livschitz J, Maybody M, Boas FE, et al. Image-Guided Interventional Radiological Delivery of Chimeric Antigen Receptor (CAR) T Cells for Pleural Malignancies in a Phase I/II Clinical Trial. Lung Cancer (2022) 165:1–9. doi: 10.1016/j.lungcan.2022.01.003
24. Srivastava S, Riddell SR. Engineering CAR-T Cells: Design Concepts. Trends Immunol (2015) 36:494–502. doi: 10.1016/j.it.2015.06.004
25. Maus MV, Grupp SA, Porter DL, June CH. Antibody-Modified T Cells: CARs Take the Front Seat for Hematologic Malignancies. Blood (2014) 123:2625–35. doi: 10.1182/blood-2013-11-492231
26. Guest RD, Hawkins RE, Kirillova N, Cheadle EJ, Arnold J, O'Neill A, et al. The Role of Extracellular Spacer Regions in the Optimal Design of Chimeric Immune Receptors: Evaluation of Four Different Scfvs and Antigens. J Immunother (2005) 28:203–11. doi: 10.1097/01.cji.0000161397.96582.59
27. Milone MC, Fish JD, Carpenito C, Carroll RG, Binder GK, Teachey D, et al. Chimeric Receptors Containing CD137 Signal Transduction Domains Mediate Enhanced Survival of T Cells and Increased Antileukemic Efficacy In Vivo. Mol Ther (2009) 17:1453–64. doi: 10.1038/mt.2009.83
28. Gross G, Waks T, Eshhar Z. Expression of Immunoglobulin-T-Cell Receptor Chimeric Molecules as Functional Receptors With Antibody-Type Specificity. Proc Natl Acad Sci U.S.A. (1989) 86:10024–8. doi: 10.1073/pnas.86.24.10024
29. Kawalekar OU, O'Connor RS, Fraietta JA, Guo L, McGettigan SE, Posey AD Jr., et al. Distinct Signaling of Coreceptors Regulates Specific Metabolism Pathways and Impacts Memory Development in CAR T Cells. Immunity (2016) 44:380–90. doi: 10.1016/j.immuni.2016.01.021
30. Cherkassky L, Morello A, Villena-Vargas J, Feng Y, Dimitrov DS, Jones DR, et al. Human CAR T Cells With Cell-Intrinsic PD-1 Checkpoint Blockade Resist Tumor-Mediated Inhibition. J Clin Invest (2016) 126:3130–44. doi: 10.1172/jci83092
31. D'Aloia MM, Zizzari IG, Sacchetti B, Pierelli L, Alimandi M. CAR-T Cells: The Long and Winding Road to Solid Tumors. Cell Death Dis (2018) 9:282. doi: 10.1038/s41419-018-0278-6
32. Enblad G, Karlsson H, Gammelgård G, Wenthe J, Lövgren T, Amini RM, et al. A Phase I/IIa Trial Using CD19-Targeted Third-Generation CAR T Cells for Lymphoma and Leukemia. Clin Cancer Res (2018) 24:6185–94. doi: 10.1158/1078-0432.ccr-18-0426
33. Zhao W, Jia L, Zhang M, Huang X, Qian P, Tang Q, et al. The Killing Effect of Novel Bi-Specific Trop2/PD-L1 CAR-T Cell Targeted Gastric Cancer. Am J Cancer Res (2019) 9:1846–56.
34. Chmielewski M, Abken H. TRUCKs: The Fourth Generation of CARs. Expert Opin Biol Ther (2015) 15:1145–54. doi: 10.1517/14712598.2015.1046430
35. Kim DW, Cho JY. Recent Advances in Allogeneic CAR-T Cells. Biomolecules (2020) 10:263. doi: 10.3390/biom10020263
36. Qu J, Mei Q, Chen L, Zhou J. Chimeric Antigen Receptor (CAR)-T-Cell Therapy in non-Small-Cell Lung Cancer (NSCLC): Current Status and Future Perspectives. Cancer Immunol Immunother (2021) 70:619–31. doi: 10.1007/s00262-020-02735-0
37. Abreu TR, Fonseca NA, Gonçalves N, Moreira JN. Current Challenges and Emerging Opportunities of CAR-T Cell Therapies. J Control Release (2020) 319:246–61. doi: 10.1016/j.jconrel.2019.12.047
38. Hinrichs CS, Restifo NP. Reassessing Target Antigens for Adoptive T-Cell Therapy. Nat Biotechnol (2013) 31:999–1008. doi: 10.1038/nbt.2725
39. Mirzaei HR, Rodriguez A, Shepphird J, Brown CE, Badie B. Chimeric Antigen Receptors T Cell Therapy in Solid Tumor: Challenges and Clinical Applications. Front Immunol (2017) 8:1850. doi: 10.3389/fimmu.2017.01850
40. Wei J, Han X, Bo J, Han W. Target Selection for CAR-T Therapy. J Hematol Oncol (2019) 12:62. doi: 10.1186/s13045-019-0758-x
41. Liu D. CAR-T "the Living Drugs", Immune Checkpoint Inhibitors, and Precision Medicine: A New Era of Cancer Therapy. J Hematol Oncol (2019) 12:113. doi: 10.1186/s13045-019-0819-1
42. Yang S, Wei W, Zhao Q. B7-H3, a Checkpoint Molecule, as a Target for Cancer Immunotherapy. Int J Biol Sci (2020) 16:1767–73. doi: 10.7150/ijbs.41105
43. Huang Y, Zhang HL, Li ZL, Du T, Chen YH, Wang Y, et al. FUT8-Mediated Aberrant N-Glycosylation of B7H3 Suppresses the Immune Response in Triple-Negative Breast Cancer. Nat Commun (2021) 12:2672. doi: 10.1038/s41467-021-22618-x
44. Liu S, Liang J, Liu Z, Zhang C, Wang Y, Watson AH, et al. The Role of CD276 in Cancers. Front Oncol (2021) 11:654684. doi: 10.3389/fonc.2021.654684
45. Lee YH, Martin-Orozco N, Zheng P, Li J, Zhang P, Tan H, et al. Inhibition of the B7-H3 Immune Checkpoint Limits Tumor Growth by Enhancing Cytotoxic Lymphocyte Function. Cell Res (2017) 27:1034–45. doi: 10.1038/cr.2017.90
46. Yu TT, Zhang T, Su F, Li YL, Shan L, Hou XM, et al. ELK1 Promotes Epithelial-Mesenchymal Transition and the Progression of Lung Adenocarcinoma by Upregulating B7-H3. Oxid Med Cell Longev (2021) 2021:2805576. doi: 10.1155/2021/2805576
47. Kim NI, Park MH, Kweon SS, Lee JS. B7-H3 and B7-H4 Expression in Breast Cancer and Their Association With Clinicopathological Variables and T Cell Infiltration. Pathobiology (2020) 87:179–92. doi: 10.1159/000505756
48. Chen L, Chen J, Xu B, Wang Q, Zhou W, Zhang G, et al. B7-H3 Expression Associates With Tumor Invasion and Patient's Poor Survival in Human Esophageal Cancer. Am J Transl Res (2015) 7:2646–60.
49. Modak S, Zanzonico P, Grkovski M, Slotkin EK, Carrasquillo JA, Lyashchenko SK, et al. B7H3-Directed Intraperitoneal Radioimmunotherapy With Radioiodinated Omburtamab for Desmoplastic Small Round Cell Tumor and Other Peritoneal Tumors: Results of a Phase I Study. J Clin Oncol (2020) 38:4283–91. doi: 10.1200/jco.20.01974
50. Scribner JA, Brown JG, Son T, Chiechi M, Li P, Sharma S, et al. Preclinical Development of MGC018, a Duocarmycin-Based Antibody-Drug Conjugate Targeting B7-H3 for Solid Cancer. Mol Cancer Ther (2020) 19:2235–44. doi: 10.1158/1535-7163.mct-20-0116
51. Lichtman EI, Du H, Shou P, Song F, Suzuki K, Ahn S, et al. Preclinical Evaluation of B7-H3-Specific Chimeric Antigen Receptor T Cells for the Treatment of Acute Myeloid Leukemia. Clin Cancer Res (2021) 27:3141–53. doi: 10.1158/1078-0432.ccr-20-2540
52. Majzner RG, Theruvath JL, Nellan A, Heitzeneder S, Cui Y, Mount CW, et al. CAR T Cells Targeting B7-H3, a Pan-Cancer Antigen, Demonstrate Potent Preclinical Activity Against Pediatric Solid Tumors and Brain Tumors. Clin Cancer Res (2019) 25:2560–74. doi: 10.1158/1078-0432.ccr-18-0432
53. Zhang Z, Jiang C, Liu Z, Yang M, Tang X, Wang Y, et al. B7-H3-Targeted CAR-T Cells Exhibit Potent Antitumor Effects on Hematologic and Solid Tumors. Mol Ther Oncolytics (2020) 17:180–9. doi: 10.1016/j.omto.2020.03.019
54. Zhang Y, He L, Sadagopan A, Ma T, Dotti G, Wang Y, et al. Targeting Radiation-Resistant Prostate Cancer Stem Cells by B7-H3 CAR T Cells. Mol Cancer Ther (2021) 20:577–88. doi: 10.1158/1535-7163.mct-20-0446
55. Parkhurst MR, Yang JC, Langan RC, Dudley ME, Nathan DA, Feldman SA, et al. T Cells Targeting Carcinoembryonic Antigen can Mediate Regression of Metastatic Colorectal Cancer But Induce Severe Transient Colitis. Mol Ther (2011) 19:620–6. doi: 10.1038/mt.2010.272
56. Shimada H, Noie T, Ohashi M, Oba K, Takahashi Y. Clinical Significance of Serum Tumor Markers for Gastric Cancer: A Systematic Review of Literature by the Task Force of the Japanese Gastric Cancer Association. Gastric Cancer (2014) 17:26–33. doi: 10.1007/s10120-013-0259-5
57. Sousa AR, Oliveira MJ, Sarmento B. Impact of CEA-Targeting Nanoparticles for Drug Delivery in Colorectal Cancer. J Pharmacol Exp Ther (2019) 370:657–70. doi: 10.1124/jpet.118.254441
58. Grunnet M, Sorensen JB. Carcinoembryonic Antigen (CEA) as Tumor Marker in Lung Cancer. Lung Cancer (2012) 76:138–43. doi: 10.1016/j.lungcan.2011.11.012
59. Matsuguma H, Nakahara R, Igarashi S, Ishikawa Y, Suzuki H, Miyazawa N, et al. Pathologic Stage I non-Small Cell Lung Cancer With High Levels of Preoperative Serum Carcinoembryonic Antigen: Clinicopathologic Characteristics and Prognosis. J Thorac Cardiovasc Surg (2008) 135:44–9. doi: 10.1016/j.jtcvs.2007.09.032
60. Fan J, Das JK, Xiong X, Chen H, Song J. Development of CAR-T Cell Persistence in Adoptive Immunotherapy of Solid Tumors. Front Oncol (2020) 10:574860. doi: 10.3389/fonc.2020.574860
61. Cha SE, Kujawski M, Yazaki PJ, Brown C, Shively JE. Tumor Regression and Immunity in Combination Therapy With Anti-CEA Chimeric Antigen Receptor T Cells and Anti-CEA-IL2 Immunocytokine. Oncoimmunology (2021) 10:1899469. doi: 10.1080/2162402x.2021.1899469
62. Wang L, Ma N, Okamoto S, Amaishi Y, Sato E, Seo N, et al. Efficient Tumor Regression by Adoptively Transferred CEA-Specific CAR-T Cells Associated With Symptoms of Mild Cytokine Release Syndrome. Oncoimmunology (2016) 5:e1211218. doi: 10.1080/2162402x.2016.1211218
63. Roskoski R Jr. Small Molecule Inhibitors Targeting the EGFR/ErbB Family of Protein-Tyrosine Kinases in Human Cancers. Pharmacol Res (2019) 139:395–411. doi: 10.1016/j.phrs.2018.11.014
64. Santos EDS, Nogueira KAB, Fernandes LCC, Martins JRP, Reis AVF, Neto JBV, et al. EGFR Targeting for Cancer Therapy: Pharmacology and Immunoconjugates With Drugs and Nanoparticles. Int J Pharm (2021) 592:120082. doi: 10.1016/j.ijpharm.2020.120082
65. Scharpenseel H, Hanssen A, Loges S, Mohme M, Bernreuther C, Peine S, et al. EGFR and HER3 Expression in Circulating Tumor Cells and Tumor Tissue From non-Small Cell Lung Cancer Patients. Sci Rep (2019) 9:7406. doi: 10.1038/s41598-019-43678-6
66. Halpern AL, Kohtz PD, White AM, Houk AK, Rehring JF, Hanson L, et al. Secretory Phospholipase A2 IIa Mediates Expression of Growth Factor Receptors in Esophageal Adenocarcinoma. Dig Dis Sci (2021) 66:784–95. doi: 10.1007/s10620-020-06241-2
67. Weinberg F, Peckys DB, de Jonge N. EGFR Expression in HER2-Driven Breast Cancer Cells. Int J Mol Sci (2020) 21:9008. doi: 10.3390/ijms21239008
68. Friedlaender A, Subbiah V, Russo A, Banna GL, Malapelle U, Rolfo C, et al. EGFR and HER2 Exon 20 Insertions in Solid Tumours: From Biology to Treatment. Nat Rev Clin Oncol (2022) 19:51–69. doi: 10.1038/s41571-021-00558-1
69. Xia L, Zheng ZZ, Liu JY, Chen YJ, Ding JC, Xia NS, et al. EGFR-Targeted CAR-T Cells are Potent and Specific in Suppressing Triple-Negative Breast Cancer Both In Vitro and In Vivo. Clin Transl Immunol (2020) 9:e01135. doi: 10.1002/cti2.1135
70. Matsuda T, Takeuchi H, Matsuda S, Hiraiwa K, Miyasho T, Okamoto M, et al. EpCAM, a Potential Therapeutic Target for Esophageal Squamous Cell Carcinoma. Ann Surg Oncol (2014) 21(Suppl 3):S356–64. doi: 10.1245/s10434-014-3579-8
71. Alibolandi M, Ramezani M, Abnous K, Sadeghi F, Atyabi F, Asouri M, et al. In Vitro and In Vivo Evaluation of Therapy Targeting Epithelial-Cell Adhesion-Molecule Aptamers for non-Small Cell Lung Cancer. J Control Release (2015) 209:88–100. doi: 10.1016/j.jconrel.2015.04.026
72. Zhang Y, Xie X, Yeganeh PN, Lee DJ, Valle-Garcia D, Meza-Sosa KF, et al. Immunotherapy for Breast Cancer Using EpCAM Aptamer Tumor-Targeted Gene Knockdown. Proc Natl Acad Sci U.S.A. (2021) 118:e2022830118. doi: 10.1073/pnas.2022830118
73. Cetin D, Okan M, Bat E, Kulah H. A Comparative Study on EpCAM Antibody Immobilization on Gold Surfaces and Microfluidic Channels for the Detection of Circulating Tumor Cells. Colloids Surf B Biointerfaces (2020) 188:110808. doi: 10.1016/j.colsurfb.2020.110808
74. Ma X, Kang X, He L, Zhou J, Zhou J, Sturm MB, et al. Identification of Tumor Specific Peptide as EpCAM Ligand and Its Potential Diagnostic and Therapeutic Clinical Application. Mol Pharm (2019) 16:2199–213. doi: 10.1021/acs.molpharmaceut.9b00185
75. Hiraga T, Ito S, Nakamura H. EpCAM Expression in Breast Cancer Cells is Associated With Enhanced Bone Metastasis Formation. Int J Cancer (2016) 138:1698–708. doi: 10.1002/ijc.29921
76. Schneck H, Gierke B, Uppenkamp F, Behrens B, Niederacher D, Stoecklein NH, et al. EpCAM-Independent Enrichment of Circulating Tumor Cells in Metastatic Breast Cancer. PloS One (2015) 10:e0144535. doi: 10.1371/journal.pone.0144535
77. Gu Q, Chen X, Zhou L, Liu X. Exosome EpCAM Promotes the Metastasis of Glioma by Targeting the CD44 Signaling Molecule on the Surface of Glioma Cells. Adv Clin Exp Med (2020) 29:1277–82. doi: 10.17219/acem/126051
78. Knödler M, Körfer J, Kunzmann V, Trojan J, Daum S, Schenk M, et al. Randomised Phase II Trial to Investigate Catumaxomab (Anti-EpCAM × anti-CD3) for Treatment of Peritoneal Carcinomatosis in Patients With Gastric Cancer. Br J Cancer (2018) 119:296–302. doi: 10.1038/s41416-018-0150-6
79. Kakarla S, Chow KK, Mata M, Shaffer DR, Song XT, Wu MF, et al. Antitumor Effects of Chimeric Receptor Engineered Human T Cells Directed to Tumor Stroma. Mol Ther (2013) 21:1611–20. doi: 10.1038/mt.2013.110
80. Busek P, Mateu R, Zubal M, Kotackova L, Sedo A. Targeting Fibroblast Activation Protein in Cancer - Prospects and Caveats. Front Biosci (Landmark Ed) (2018) 23:1933–68. doi: 10.2741/4682
81. Hamson EJ, Keane FM, Tholen S, Schilling O, Gorrell MD. Understanding Fibroblast Activation Protein (FAP): Substrates, Activities, Expression and Targeting for Cancer Therapy. Proteomics Clin Appl (2014) 8:454–63. doi: 10.1002/prca.201300095
82. Ebert LM, Yu W, Gargett T, Toubia J, Kollis PM, Tea MN, et al. Endothelial, Pericyte and Tumor Cell Expression in Glioblastoma Identifies Fibroblast Activation Protein (FAP) as an Excellent Target for Immunotherapy. Clin Transl Immunol (2020) 9:e1191. doi: 10.1002/cti2.1191
83. Wang LC, Lo A, Scholler J, Sun J, Majumdar RS, Kapoor V, et al. Targeting Fibroblast Activation Protein in Tumor Stroma With Chimeric Antigen Receptor T Cells can Inhibit Tumor Growth and Augment Host Immunity Without Severe Toxicity. Cancer Immunol Res (2014) 2:154–66. doi: 10.1158/2326-6066.cir-13-0027
84. Schuberth PC, Hagedorn C, Jensen SM, Gulati P, van den Broek M, Mischo A, et al. Treatment of Malignant Pleural Mesothelioma by Fibroblast Activation Protein-Specific Re-Directed T Cells. J Transl Med (2013) 11:187. doi: 10.1186/1479-5876-11-187
85. Dhritlahre RK, Saneja A. Recent Advances in HER2-Targeted Delivery for Cancer Therapy. Drug Discovery Today (2021) 26:1319–29. doi: 10.1016/j.drudis.2020.12.014
86. Smith TA. Towards Detecting the HER-2 Receptor and Metabolic Changes Induced by HER-2-Targeted Therapies Using Medical Imaging. Br J Radiol (2010) 83:638–44. doi: 10.1259/bjr/31053812
87. Perrier A, Gligorov J, Lefèvre G, Boissan M. The Extracellular Domain of Her2 in Serum as a Biomarker of Breast Cancer. Lab Invest (2018) 98:696–707. doi: 10.1038/s41374-018-0033-8
88. De Cuyper A, Van Den Eynde M, Machiels JP. HER2 as a Predictive Biomarker and Treatment Target in Colorectal Cancer. Clin Colorectal Cancer (2020) 19:65–72. doi: 10.1016/j.clcc.2020.02.007
89. Yu F, Wang X, Shi H, Jiang M, Xu J, Sun M, et al. Development of Chimeric Antigen Receptor-Modified T Cells for the Treatment of Esophageal Cancer. Tumori (2021) 107:341–52. doi: 10.1177/0300891620960223
90. Li H, Yuan W, Bin S, Wu G, Li P, Liu M, et al. Overcome Trastuzumab Resistance of Breast Cancer Using Anti-HER2 Chimeric Antigen Receptor T Cells and PD1 Blockade. Am J Cancer Res (2020) 10:688–703.
91. Coelho R, Ricardo S, Amaral AL, Huang YL, Nunes M, Neves JP, et al. Regulation of Invasion and Peritoneal Dissemination of Ovarian Cancer by Mesothelin Manipulation. Oncogenesis (2020) 9:61. doi: 10.1038/s41389-020-00246-2
92. Levý M, Boublíková L, Büchler T, Šimša J. Treatment of Malignant Peritoneal Mesothelioma. Klin Onkol (2019) 32:333–7. doi: 10.14735/amko2019333
93. Forest F, Patoir A, Dal Col P, Sulaiman A, Camy F, Laville D, et al. Nuclear Grading, BAP1, Mesothelin and PD-L1 Expression in Malignant Pleural Mesothelioma: Prognostic Implications. Pathology (2018) 50:635–41. doi: 10.1016/j.pathol.2018.05.002
94. Lv J, Li P. Mesothelin as a Biomarker for Targeted Therapy. biomark Res (2019) 7:18. doi: 10.1186/s40364-019-0169-8
95. Klampatsa A, Dimou V, Albelda SM. Mesothelin-Targeted CAR-T Cell Therapy for Solid Tumors. Expert Opin Biol Ther (2021) 21:473–86. doi: 10.1080/14712598.2021.1843628
96. Zhang Q, Liu G, Liu J, Yang M, Fu J, Liu G, et al. The Antitumor Capacity of Mesothelin-CAR-T Cells in Targeting Solid Tumors in Mice. Mol Ther Oncolytics (2021) 20:556–68. doi: 10.1016/j.omto.2021.02.013
97. Ye L, Lou Y, Lu L, Fan X. Mesothelin-Targeted Second Generation CAR-T Cells Inhibit Growth of Mesothelin-Expressing Tumors In Vivo. Exp Ther Med (2019) 17:739–47. doi: 10.3892/etm.2018.7015
98. Yang P, Cao X, Cai H, Feng P, Chen X, Zhu Y, et al. The Exosomes Derived From CAR-T Cell Efficiently Target Mesothelin and Reduce Triple-Negative Breast Cancer Growth. Cell Immunol (2021) 360:104262. doi: 10.1016/j.cellimm.2020.104262
99. Wang Y, Wang J, Yang X, Yang J, Lu P, Zhao L, et al. Chemokine Receptor CCR2b Enhanced Anti-Tumor Function of Chimeric Antigen Receptor T Cells Targeting Mesothelin in a Non-Small-Cell Lung Carcinoma Model. Front Immunol (2021) 12:628906. doi: 10.3389/fimmu.2021.628906
100. Li Y, Xiao F, Zhang A, Zhang D, Nie W, Xu T, et al. Oncolytic Adenovirus Targeting TGF-β Enhances Anti-Tumor Responses of Mesothelin-Targeted Chimeric Antigen Receptor T Cell Therapy Against Breast Cancer. Cell Immunol (2020) 348:104041. doi: 10.1016/j.cellimm.2020.104041
101. Castelletti L, Yeo D, van Zandwijk N, Rasko JEJ. Anti-Mesothelin CAR T Cell Therapy for Malignant Mesothelioma. biomark Res (2021) 9:11. doi: 10.1186/s40364-021-00264-1
102. Chen W, Zhang Z, Zhang S, Zhu P, Ko JK, Yung KK. MUC1: Structure, Function, and Clinic Application in Epithelial Cancers. Int J Mol Sci (2021) 22:6567. doi: 10.3390/ijms22126567
103. Xu T, Li D, Wang H, Zheng T, Wang G, Xin Y. MUC1 Downregulation Inhibits non-Small Cell Lung Cancer Progression in Human Cell Lines. Exp Ther Med (2017) 14:4443–7. doi: 10.3892/etm.2017.5062
104. Jing X, Liang H, Hao C, Yang X, Cui X. Overexpression of MUC1 Predicts Poor Prognosis in Patients With Breast Cancer. Oncol Rep (2019) 41:801–10. doi: 10.3892/or.2018.6887
105. Sun ZG, Yu L, Gao W, Wang Z, Zhu LM. Clinical and Prognostic Significance of MUC1 Expression in Patients With Esophageal Squamous Cell Carcinoma After Radical Resection. Saudi J Gastroenterol (2018) 24:165–70. doi: 10.4103/sjg.SJG_420_17
106. Pourjafar M, Samadi P, Saidijam M. MUC1 Antibody-Based Therapeutics: The Promise of Cancer Immunotherapy. Immunotherapy (2020) 12:1269–86. doi: 10.2217/imt-2020-0019
107. Wei X, Lai Y, Li J, Qin L, Xu Y, Zhao R, et al. PSCA and MUC1 in non-Small-Cell Lung Cancer as Targets of Chimeric Antigen Receptor T Cells. Oncoimmunology (2017) 6:e1284722. doi: 10.1080/2162402x.2017.1284722
108. Zhang H, Zhao H, He X, Xi F, Liu J. JAK-STAT Domain Enhanced MUC1-CAR-T Cells Induced Esophageal Cancer Elimination. Cancer Manag Res (2020) 12:9813–24. doi: 10.2147/cmar.s264358
109. Sui H, Ma N, Wang Y, Li H, Liu X, Su Y, et al. Anti-PD-1/PD-L1 Therapy for Non-Small-Cell Lung Cancer: Toward Personalized Medicine and Combination Strategies. J Immunol Res (2018) 2018:6984948. doi: 10.1155/2018/6984948
110. Kitagawa S, Hakozaki T, Kitadai R, Hosomi Y. Switching Administration of Anti-PD-1 and Anti-PD-L1 Antibodies as Immune Checkpoint Inhibitor Rechallenge in Individuals With Advanced non-Small Cell Lung Cancer: Case Series and Literature Review. Thorac Cancer (2020) 11:1927–33. doi: 10.1111/1759-7714.13483
111. Doroshow DB, Bhalla S, Beasley MB, Sholl LM, Kerr KM, Gnjatic S, et al. PD-L1 as a Biomarker of Response to Immune-Checkpoint Inhibitors. Nat Rev Clin Oncol (2021) 18:345–62. doi: 10.1038/s41571-021-00473-5
112. Lotfinejad P, Kazemi T, Mokhtarzadeh A, Shanehbandi D, Jadidi Niaragh F, Safaei S, et al. PD-1/PD-L1 Axis Importance and Tumor Microenvironment Immune Cells. Life Sci (2020) 259:118297. doi: 10.1016/j.lfs.2020.118297
113. Tran TH, Phuong Tran TT. Targeting the PD-1/PD-L1 Axis for Cancer Treatment: A Review on Nanotechnology. R Soc Open Sci (2022) 9:211991. doi: 10.1098/rsos.211991
114. Takamori S, Toyokawa G, Takada K, Shoji F, Okamoto T, Maehara Y. Combination Therapy of Radiotherapy and Anti-PD-1/PD-L1 Treatment in Non-Small-Cell Lung Cancer: A Mini-Review. Clin Lung Cancer (2018) 19:12–6. doi: 10.1016/j.cllc.2017.06.015
115. Liu M, Wang X, Li W, Yu X, Flores-Villanueva P, Xu-Monette ZY, et al. Targeting PD-L1 in non-Small Cell Lung Cancer Using CAR T Cells. Oncogenesis (2020) 9:72. doi: 10.1038/s41389-020-00257-z
116. Li D, English H, Hong J, Liang T, Merlino G, Day CP, et al. A Novel PD-L1-Targeted Shark V(NAR) Single-Domain-Based CAR-T Cell Strategy for Treating Breast Cancer and Liver Cancer. Mol Ther Oncolytics (2022) 24:849–63. doi: 10.1016/j.omto.2022.02.015
117. Qin L, Zhao R, Chen D, Wei X, Wu Q, Long Y, et al. Chimeric Antigen Receptor T Cells Targeting PD-L1 Suppress Tumor Growth. biomark Res (2020) 8:19. doi: 10.1186/s40364-020-00198-0
118. Shen C, Zhang Z, Tian Y, Li F, Zhou L, Jiang W, et al. Sulforaphane Enhances the Antitumor Response of Chimeric Antigen Receptor T Cells by Regulating PD-1/PD-L1 Pathway. BMC Med (2021) 19:283. doi: 10.1186/s12916-021-02161-8
119. Balakrishnan A, Goodpaster T, Randolph-Habecker J, Hoffstrom BG, Jalikis FG, Koch LK, et al. Analysis of ROR1 Protein Expression in Human Cancer and Normal Tissues. Clin Cancer Res (2017) 23:3061–71. doi: 10.1158/1078-0432.ccr-16-2083
120. Zheng YZ, Ma R, Zhou JK, Guo CL, Wang YS, Li ZG, et al. ROR1 is a Novel Prognostic Biomarker in Patients With Lung Adenocarcinoma. Sci Rep (2016) 6:36447. doi: 10.1038/srep36447
121. Hudecek M, Lupo-Stanghellini MT, Kosasih PL, Sommermeyer D, Jensen MC, Rader C, et al. Receptor Affinity and Extracellular Domain Modifications Affect Tumor Recognition by ROR1-Specific Chimeric Antigen Receptor T Cells. Clin Cancer Res (2013) 19:3153–64. doi: 10.1158/1078-0432.ccr-13-0330
122. Srivastava S, Furlan SN, Jaeger-Ruckstuhl CA, Sarvothama M, Berger C, Smythe KS, et al. Immunogenic Chemotherapy Enhances Recruitment of CAR-T Cells to Lung Tumors and Improves Antitumor Efficacy When Combined With Checkpoint Blockade. Cancer Cell (2021) 39:193–208.e10. doi: 10.1016/j.ccell.2020.11.005
123. Wallstabe L, Göttlich C, Nelke LC, Kühnemundt J, Schwarz T, Nerreter T, et al. ROR1-CAR T Cells are Effective Against Lung and Breast Cancer in Advanced Microphysiologic 3D Tumor Models. JCI Insight (2019) 4:e126345. doi: 10.1172/jci.insight.126345
124. Li N, Liu S, Sun M, Chen W, Xu X, Zeng Z, et al. Chimeric Antigen Receptor-Modified T Cells Redirected to EphA2 for the Immunotherapy of Non-Small Cell Lung Cancer. Transl Oncol (2018) 11:11–7. doi: 10.1016/j.tranon.2017.10.009
125. Chu W, Zhou Y, Tang Q, Wang M, Ji Y, Yan J, et al. Bi-Specific Ligand-Controlled Chimeric Antigen Receptor T-Cell Therapy for non-Small Cell Lung Cancer. Biosci Trends (2018) 12:298–308. doi: 10.5582/bst.2018.01048
126. Shimizu Y, Suzuki T, Yoshikawa T, Endo I, Nakatsura T. Next-Generation Cancer Immunotherapy Targeting Glypican-3. Front Oncol (2019) 9:248. doi: 10.3389/fonc.2019.00248
127. Harrer DC, Dörrie J, Schaft N. CSPG4 as Target for CAR-T-Cell Therapy of Various Tumor Entities-Merits and Challenges. Int J Mol Sci (2019) 20:5942. doi: 10.3390/ijms20235942
128. Porcellini S, Asperti C, Corna S, Cicoria E, Valtolina V, Stornaiuolo A, et al. CAR T Cells Redirected to CD44v6 Control Tumor Growth in Lung and Ovary Adenocarcinoma Bearing Mice. Front Immunol (2020) 11:99. doi: 10.3389/fimmu.2020.00099
129. Reppel L, Tsahouridis O, Akulian J, Davis IJ, Lee H, Fucà G, et al. Targeting Disialoganglioside GD2 With Chimeric Antigen Receptor-Redirected T Cells in Lung Cancer. J Immunother Cancer (2022) 10:e003897. doi: 10.1136/jitc-2021-003897
130. Zeltsman M, Dozier J, McGee E, Ngai D, Adusumilli PS. CAR T-Cell Therapy for Lung Cancer and Malignant Pleural Mesothelioma. Transl Res (2017) 187:1–10. doi: 10.1016/j.trsl.2017.04.004
131. Morgan RA, Yang JC, Kitano M, Dudley ME, Laurencot CM, Rosenberg SA. Case Report of a Serious Adverse Event Following the Administration of T Cells Transduced With a Chimeric Antigen Receptor Recognizing ERBB2. Mol Ther (2010) 18:843–51. doi: 10.1038/mt.2010.24
132. Bonifant CL, Jackson HJ, Brentjens RJ, Curran KJ. Toxicity and Management in CAR T-Cell Therapy. Mol Ther Oncolytics (2016) 3:16011. doi: 10.1038/mto.2016.11
133. Raj D, Nikolaidi M, Garces I, Lorizio D, Castro NM, Caiafa SG, et al. CEACAM7 Is an Effective Target for CAR T-Cell Therapy of Pancreatic Ductal Adenocarcinoma. Clin Cancer Res (2021) 27:1538–52. doi: 10.1158/1078-0432.ccr-19-2163
134. Caruso HG, Hurton LV, Najjar A, Rushworth D, Ang S, Olivares S, et al. Tuning Sensitivity of CAR to EGFR Density Limits Recognition of Normal Tissue While Maintaining Potent Antitumor Activity. Cancer Res (2015) 75:3505–18. doi: 10.1158/0008-5472.can-15-0139
135. Jones BS, Lamb LS, Goldman F, Di Stasi A. Improving the Safety of Cell Therapy Products by Suicide Gene Transfer. Front Pharmacol (2014) 5:254. doi: 10.3389/fphar.2014.00254
136. Wei H, Wang Z, Kuang Y, Wu Z, Zhao S, Zhang Z, et al. Intercellular Adhesion Molecule-1 as Target for CAR-T-Cell Therapy of Triple-Negative Breast Cancer. Front Immunol (2020) 11:573823. doi: 10.3389/fimmu.2020.573823
137. Zhang Y, Li X, Zhang J, Mao L. Novel Cellular Immunotherapy Using NKG2D CAR-T for the Treatment of Cervical Cancer. BioMed Pharmacother (2020) 131:110562. doi: 10.1016/j.biopha.2020.110562
138. Englisch A, Altvater B, Kailayangiri S, Hartmann W, Rossig C. VEGFR2 as a Target for CAR T Cell Therapy of Ewing Sarcoma. Pediatr Blood Cancer (2020) 67:e28313. doi: 10.1002/pbc.28313
139. Gautam SK, Kumar S, Dam V, Ghersi D, Jain M, Batra SK. MUCIN-4 (MUC4) is a Novel Tumor Antigen in Pancreatic Cancer Immunotherapy. Semin Immunol (2020) 47:101391. doi: 10.1016/j.smim.2020.101391
140. Jin L, Ge H, Long Y, Yang C, Chang YE, Mu L, et al. CD70, a Novel Target of CAR T-Cell Therapy for Gliomas. Neuro Oncol (2018) 20:55–65. doi: 10.1093/neuonc/nox116
141. Wang D, Starr R, Chang WC, Aguilar B, Alizadeh D, Wright SL, et al. Chlorotoxin-Directed CAR T Cells for Specific and Effective Targeting of Glioblastoma. Sci Transl Med (2020) 12:eaaw2672. doi: 10.1126/scitranslmed.aaw2672
142. Watanabe K, Kuramitsu S, Posey AD Jr., June CH. Expanding the Therapeutic Window for CAR T Cell Therapy in Solid Tumors: The Knowns and Unknowns of CAR T Cell Biology. Front Immunol (2018) 9:2486. doi: 10.3389/fimmu.2018.02486
143. Choi BD, Yu X, Castano AP, Bouffard AA, Schmidts A, Larson RC, et al. CAR-T Cells Secreting BiTEs Circumvent Antigen Escape Without Detectable Toxicity. Nat Biotechnol (2019) 37:1049–58. doi: 10.1038/s41587-019-0192-1
144. June CH, O'Connor RS, Kawalekar OU, Ghassemi S, Milone MC. CAR T Cell Immunotherapy for Human Cancer. Science (2018) 359:1361–5. doi: 10.1126/science.aar6711
145. Roybal KT, Rupp LJ, Morsut L, Walker WJ, McNally KA, Park JS, et al. Precision Tumor Recognition by T Cells With Combinatorial Antigen-Sensing Circuits. Cell (2016) 164:770–9. doi: 10.1016/j.cell.2016.01.011
146. Moghimi B, Muthugounder S, Jambon S, Tibbetts R, Hung L, Bassiri H, et al. Preclinical Assessment of the Efficacy and Specificity of GD2-B7H3 SynNotch CAR-T in Metastatic Neuroblastoma. Nat Commun (2021) 12:511. doi: 10.1038/s41467-020-20785-x
147. Srivastava S, Salter AI, Liggitt D, Yechan-Gunja S, Sarvothama M, Cooper K, et al. Logic-Gated ROR1 Chimeric Antigen Receptor Expression Rescues T Cell-Mediated Toxicity to Normal Tissues and Enables Selective Tumor Targeting. Cancer Cell (2019) 35:489–503.e8. doi: 10.1016/j.ccell.2019.02.003
148. Rubin DB, Danish HH, Ali AB, Li K, LaRose S, Monk AD, et al. Neurological Toxicities Associated With Chimeric Antigen Receptor T-Cell Therapy. Brain (2019) 142:1334–48. doi: 10.1093/brain/awz053
149. Norelli M, Camisa B, Barbiera G, Falcone L, Purevdorj A, Genua M, et al. Monocyte-Derived IL-1 and IL-6 are Differentially Required for Cytokine-Release Syndrome and Neurotoxicity Due to CAR T Cells. Nat Med (2018) 24:739–48. doi: 10.1038/s41591-018-0036-4
150. Freyer CW, Porter DL. Cytokine Release Syndrome and Neurotoxicity Following CAR T-Cell Therapy for Hematologic Malignancies. J Allergy Clin Immunol (2020) 146:940–8. doi: 10.1016/j.jaci.2020.07.025
151. Gust J, Taraseviciute A, Turtle CJ. Neurotoxicity Associated With CD19-Targeted CAR-T Cell Therapies. CNS Drugs (2018) 32:1091–101. doi: 10.1007/s40263-018-0582-9
152. Torre M, Solomon IH, Sutherland CL, Nikiforow S, DeAngelo DJ, Stone RM, et al. Neuropathology of a Case With Fatal CAR T-Cell-Associated Cerebral Edema. J Neuropathol Exp Neurol (2018) 77:877–82. doi: 10.1093/jnen/nly064
153. Gust J, Hay KA, Hanafi LA, Li D, Myerson D, Gonzalez-Cuyar LF, et al. Endothelial Activation and Blood-Brain Barrier Disruption in Neurotoxicity After Adoptive Immunotherapy With CD19 CAR-T Cells. Cancer Discovery (2017) 7:1404–19. doi: 10.1158/2159-8290.cd-17-0698
154. Neelapu SS, Tummala S, Kebriaei P, Wierda W, Gutierrez C, Locke FL, et al. Chimeric Antigen Receptor T-Cell Therapy - Assessment and Management of Toxicities. Nat Rev Clin Oncol (2018) 15:47–62. doi: 10.1038/nrclinonc.2017.148
155. Hay KA, Hanafi LA, Li D, Gust J, Liles WC, Wurfel MM, et al. Kinetics and Biomarkers of Severe Cytokine Release Syndrome After CD19 Chimeric Antigen Receptor-Modified T-Cell Therapy. Blood (2017) 130:2295–306. doi: 10.1182/blood-2017-06-793141
156. Juillerat A, Marechal A, Filhol JM, Valton J, Duclert A, Poirot L, et al. Design of Chimeric Antigen Receptors With Integrated Controllable Transient Functions. Sci Rep (2016) 6:18950. doi: 10.1038/srep18950
157. Wu CY, Roybal KT, Puchner EM, Onuffer J, Lim WA. Remote Control of Therapeutic T Cells Through a Small Molecule-Gated Chimeric Receptor. Science (2015) 350:aab4077. doi: 10.1126/science.aab4077
158. Philip B, Kokalaki E, Mekkaoui L, Thomas S, Straathof K, Flutter B, et al. A Highly Compact Epitope-Based Marker/Suicide Gene for Easier and Safer T-Cell Therapy. Blood (2014) 124:1277–87. doi: 10.1182/blood-2014-01-545020
159. Di Stasi A, Tey SK, Dotti G, Fujita Y, Kennedy-Nasser A, Martinez C, et al. Inducible Apoptosis as a Safety Switch for Adoptive Cell Therapy. N Engl J Med (2011) 365:1673–83. doi: 10.1056/NEJMoa1106152
160. Mestermann K, Giavridis T, Weber J, Rydzek J, Frenz S, Nerreter T, et al. The Tyrosine Kinase Inhibitor Dasatinib Acts as a Pharmacologic on/Off Switch for CAR T Cells. Sci Transl Med (2019) 11:eaau5907. doi: 10.1126/scitranslmed.aau5907
161. Schmidt P, Raftery MJ, Pecher G. Engineering NK Cells for CAR Therapy-Recent Advances in Gene Transfer Methodology. Front Immunol (2020) 11:611163. doi: 10.3389/fimmu.2020.611163
162. Nguyen NT, Huang K, Zeng H, Jing J, Wang R, Fang S, et al. Nano-Optogenetic Engineering of CAR T Cells for Precision Immunotherapy With Enhanced Safety. Nat Nanotechnol (2021) 16:1424–34. doi: 10.1038/s41565-021-00982-5
163. Qi FL, Wang MF, Li BZ, Lu ZF, Nie GJ, Li SP. Reversal of the Immunosuppressive Tumor Microenvironment by Nanoparticle-Based Activation of Immune-Associated Cells. Acta Pharmacol Sin (2020) 41:895–901. doi: 10.1038/s41401-020-0423-5
164. Renner K, Singer K, Koehl GE, Geissler EK, Peter K, Siska PJ, et al. Metabolic Hallmarks of Tumor and Immune Cells in the Tumor Microenvironment. Front Immunol (2017) 8:248. doi: 10.3389/fimmu.2017.00248
165. Oliver AJ, Lau PKH, Unsworth AS, Loi S, Darcy PK, Kershaw MH, et al. Tissue-Dependent Tumor Microenvironments and Their Impact on Immunotherapy Responses. Front Immunol (2018) 9:70. doi: 10.3389/fimmu.2018.00070
166. Rafiq S, Yeku OO, Jackson HJ, Purdon TJ, van Leeuwen DG, Drakes DJ, et al. Targeted Delivery of a PD-1-Blocking scFv by CAR-T Cells Enhances Anti-Tumor Efficacy In Vivo. Nat Biotechnol (2018) 36:847–56. doi: 10.1038/nbt.4195
167. Chen N, Morello A, Tano Z, Adusumilli PS. CAR T-Cell Intrinsic PD-1 Checkpoint Blockade: A Two-in-One Approach for Solid Tumor Immunotherapy. Oncoimmunology (2017) 6:e1273302. doi: 10.1080/2162402x.2016.1273302
168. Zhou J, Jin L, Wang F, Zhang Y, Liu B, Zhao T. Chimeric Antigen Receptor T (CAR-T) Cells Expanded With IL-7/IL-15 Mediate Superior Antitumor Effects. Protein Cell (2019) 10:764–9. doi: 10.1007/s13238-019-0643-y
169. Maggs L, Cattaneo G, Dal AE, Moghaddam AS, Ferrone S. CAR T Cell-Based Immunotherapy for the Treatment of Glioblastoma. Front Neurosci (2021) 15:662064. doi: 10.3389/fnins.2021.662064
170. Zhang E, Gu J, Xu H. Prospects for Chimeric Antigen Receptor-Modified T Cell Therapy for Solid Tumors. Mol Cancer (2018) 17:7. doi: 10.1186/s12943-018-0759-3
171. Caruana I, Savoldo B, Hoyos V, Weber G, Liu H, Kim ES, et al. Heparanase Promotes Tumor Infiltration and Antitumor Activity of CAR-Redirected T Lymphocytes. Nat Med (2015) 21:524–9. doi: 10.1038/nm.3833
172. Jin L, Tao H, Karachi A, Long Y, Hou AY, Na M, et al. CXCR1- or CXCR2-Modified CAR T Cells Co-Opt IL-8 for Maximal Antitumor Efficacy in Solid Tumors. Nat Commun (2019) 10:4016. doi: 10.1038/s41467-019-11869-4
173. Sun R, Luo H, Su J, Di S, Zhou M, Shi B, et al. Olaparib Suppresses MDSC Recruitment via SDF1α/CXCR4 Axis to Improve the Anti-Tumor Efficacy of CAR-T Cells on Breast Cancer in Mice. Mol Ther (2021) 29:60–74. doi: 10.1016/j.ymthe.2020.09.034
174. Fry TJ, Shah NN, Orentas RJ, Stetler-Stevenson M, Yuan CM, Ramakrishna S, et al. CD22-Targeted CAR T Cells Induce Remission in B-ALL That is Naive or Resistant to CD19-Targeted CAR Immunotherapy. Nat Med (2018) 24:20–8. doi: 10.1038/nm.4441
175. Kailayangiri S, Altvater B, Wiebel M, Jamitzky S, Rossig C. Overcoming Heterogeneity of Antigen Expression for Effective CAR T Cell Targeting of Cancers. Cancers (Basel) (2020) 12:1075. doi: 10.3390/cancers12051075
176. Wang X, Dong Z, Awuah D, Chang WC, Cheng WA, Vyas V, et al. CD19/BAFF-R Dual-Targeted CAR T Cells for the Treatment of Mixed Antigen-Negative Variants of Acute Lymphoblastic Leukemia. Leukemia (2022) 36:1015–24. doi: 10.1038/s41375-021-01477-x
177. Qin H, Dong Z, Wang X, Cheng WA, Wen F, Xue W, et al. CAR T Cells Targeting BAFF-R can Overcome CD19 Antigen Loss in B Cell Malignancies. Sci Transl Med (2019) 11:eaaw9414. doi: 10.1126/scitranslmed.aaw9414
178. Grigor EJM, Fergusson D, Kekre N, Montroy J, Atkins H, Seftel MD, et al. Risks and Benefits of Chimeric Antigen Receptor T-Cell (CAR-T) Therapy in Cancer: A Systematic Review and Meta-Analysis. Transfus Med Rev (2019) 33:98–110. doi: 10.1016/j.tmrv.2019.01.005
179. Cao JX, Wang H, Gao WJ, You J, Wu LH, Wang ZX. The Incidence of Cytokine Release Syndrome and Neurotoxicity of CD19 Chimeric Antigen Receptor-T Cell Therapy in the Patient With Acute Lymphoblastic Leukemia and Lymphoma. Cytotherapy (2020) 22:214–26. doi: 10.1016/j.jcyt.2020.01.015
180. Gargett T, Brown MP. The Inducible Caspase-9 Suicide Gene System as a "Safety Switch" to Limit on-Target, Off-Tumor Toxicities of Chimeric Antigen Receptor T Cells. Front Pharmacol (2014) 5:235. doi: 10.3389/fphar.2014.00235
181. Marin V, Cribioli E, Philip B, Tettamanti S, Pizzitola I, Biondi A, et al. Comparison of Different Suicide-Gene Strategies for the Safety Improvement of Genetically Manipulated T Cells. Hum Gene Ther Methods (2012) 23:376–86. doi: 10.1089/hgtb.2012.050
182. Klopp A, Schreiber S, Kosinska AD, Pulé M, Protzer U, Wisskirchen K. Depletion of T Cells via Inducible Caspase 9 Increases Safety of Adoptive T-Cell Therapy Against Chronic Hepatitis B. Front Immunol (2021) 12:734246. doi: 10.3389/fimmu.2021.734246
183. Miao L, Zhang J, Huang B, Zhang Z, Wang S, Tang F, et al. Special Chimeric Antigen Receptor (CAR) Modifications of T Cells: A Review. Front Oncol (2022) 12:832765. doi: 10.3389/fonc.2022.832765
184. Zajc CU, Dobersberger M, Schaffner I, Mlynek G, Pühringer D, Salzer B, et al. A Conformation-Specific ON-Switch for Controlling CAR T Cells With an Orally Available Drug. Proc Natl Acad Sci U.S.A. (2020) 117:14926–35. doi: 10.1073/pnas.1911154117
185. Jan M, Scarfò I, Larson RC, Walker A, Schmidts A, Guirguis AA, et al. Reversible ON- and OFF-Switch Chimeric Antigen Receptors Controlled by Lenalidomide. Sci Transl Med (2021) 13:eabb6295. doi: 10.1126/scitranslmed.abb6295
186. Frankel SR, Baeuerle PA. Targeting T Cells to Tumor Cells Using Bispecific Antibodies. Curr Opin Chem Biol (2013) 17:385–92. doi: 10.1016/j.cbpa.2013.03.029
187. Brown CE, Alizadeh D, Starr R, Weng L, Wagner JR, Naranjo A, et al. Regression of Glioblastoma After Chimeric Antigen Receptor T-Cell Therapy. N Engl J Med (2016) 375:2561–9. doi: 10.1056/NEJMoa1610497
188. Tchou J, Zhao Y, Levine BL, Zhang PJ, Davis MM, Melenhorst JJ, et al. Safety and Efficacy of Intratumoral Injections of Chimeric Antigen Receptor (CAR) T Cells in Metastatic Breast Cancer. Cancer Immunol Res (2017) 5:1152–61. doi: 10.1158/2326-6066.cir-17-0189
189. Perera LP, Zhang M, Nakagawa M, Petrus MN, Maeda M, Kadin ME, et al. Chimeric Antigen Receptor Modified T Cells That Target Chemokine Receptor CCR4 as a Therapeutic Modality for T-Cell Malignancies. Am J Hematol (2017) 92:892–901. doi: 10.1002/ajh.24794
190. Mikucki ME, Fisher DT, Matsuzaki J, Skitzki JJ, Gaulin NB, Muhitch JB, et al. Non-Redundant Requirement for CXCR3 Signalling During Tumoricidal T-Cell Trafficking Across Tumour Vascular Checkpoints. Nat Commun (2015) 6:7458. doi: 10.1038/ncomms8458
191. Tian Y, Wen C, Zhang Z, Liu Y, Li F, Zhao Q, et al. CXCL9-Modified CAR T Cells Improve Immune Cell Infiltration and Antitumor Efficacy. Cancer Immunol Immunother (2022). doi: 10.1007/s00262-022-03193-6
192. Li G, Guo J, Zheng Y, Ding W, Han Z, Qin L, et al. CXCR5 Guides Migration and Tumor Eradication of Anti-EGFR Chimeric Antigen Receptor T Cells. Mol Ther Oncolytics (2021) 22:507–17. doi: 10.1016/j.omto.2021.07.003
193. Liu G, Rui W, Zheng H, Huang D, Yu F, Zhang Y, et al. CXCR2-Modified CAR-T Cells Have Enhanced Trafficking Ability That Improves Treatment of Hepatocellular Carcinoma. Eur J Immunol (2020) 50:712–24. doi: 10.1002/eji.201948457
194. Edeline J, Houot R, Marabelle A, Alcantara M. CAR-T Cells and BiTEs in Solid Tumors: Challenges and Perspectives. J Hematol Oncol (2021) 14:65. doi: 10.1186/s13045-021-01067-5
195. Depil S, Duchateau P, Grupp SA, Mufti G, Poirot L. 'Off-the-Shelf' Allogeneic CAR T Cells: Development and Challenges. Nat Rev Drug Discov (2020) 19:185–99. doi: 10.1038/s41573-019-0051-2
196. Choi BD, Yu X, Castano AP, Darr H, Henderson DB, Bouffard AA, et al. CRISPR-Cas9 Disruption of PD-1 Enhances Activity of Universal EGFRvIII CAR T Cells in a Preclinical Model of Human Glioblastoma. J Immunother Cancer (2019) 7:304. doi: 10.1186/s40425-019-0806-7
Keywords: thoracic malignancies, chimeric antigen receptor-modified T cells, immunotherapy, targeting specific antigens, solid tumor
Citation: Chen L, Chen F, Niu H, Li J, Pu Y, Yang C, Wang Y, Huang R, Li K, Lei Y and Huang Y (2022) Chimeric Antigen Receptor (CAR)-T Cell Immunotherapy Against Thoracic Malignancies: Challenges and Opportunities. Front. Immunol. 13:871661. doi: 10.3389/fimmu.2022.871661
Received: 08 February 2022; Accepted: 14 June 2022;
Published: 14 July 2022.
Edited by:
Sergei Kusmartsev, University of Florida, United StatesReviewed by:
William Donelan, University of Florida, United StatesLeonardo M. R. Ferreira, Medical University of South Carolina, United States
Copyright © 2022 Chen, Chen, Niu, Li, Pu, Yang, Wang, Huang, Li, Lei and Huang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yunchao Huang, aHVhbmd5dW5jaDIwMTdAMTI2LmNvbQ==; Yujie Lei, NTgzMjcxMDI1QHFxLmNvbQ==; Ke Li, bGlrZWxpa2VsaWtlQDEyNi5jb20=
†These authors have contributed equally to this work