 Nian Chen
Nian Chen Cheng Peng*
Cheng Peng* Dan Li
Dan Li
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Immunol. , 29 April 2022
Sec. Inflammation
Volume 13 - 2022 | https://doi.org/10.3389/fimmu.2022.869307
This article is part of the Research Topic Epigenetics of the Immune Component of Inflammation View all 43 articles
Glioblastoma (GBM) is the most common malignant brain tumor in adults, and immunotherapies and genetic therapies for GBM have evolved dramatically over the past decade, but GBM therapy is still facing a dilemma due to the high recurrence rate. The inflammatory microenvironment is a general signature of tumors that accelerates epigenetic changes in GBM and helps tumors avoid immunological surveillance. GBM tumor cells and glioma-associated microglia/macrophages are the primary contributors to the inflammatory condition, meanwhile the modification of epigenetic events including DNA methylation, non-coding RNAs, and histone methylation and deacetylases involved in this pathological process of GBM, finally result in exacerbating the proliferation, invasion, and migration of GBM. On the other hand, histone deacetylase inhibitors, DNA methyltransferases inhibitors, and RNA interference could reverse the inflammatory landscapes and inhibit GBM growth and invasion. Here, we systematically review the inflammatory-associated epigenetic changes and regulations in the microenvironment of GBM, aiming to provide a comprehensive epigenetic profile underlying the recognition of inflammation in GBM.
Glioblastoma (GBM) is the most aggressive and common malignant brain tumor arising from neural precursor cells (1). It is defined as grade IV glioma by WHO and divided into two distinct subgroups, namely IDH wild-type (primary GBM, approximately 90%) and IDH mutant-type (secondary GBM, 10%) (2). Both types of GBM shows similar clinical symptoms that include headache, nausea, dizziness, speech difficulties, and cerebral edema (3), and is characterized by poor survival and remarkably high tumor heterogeneity, which invades surrounding brain tissue and quickly develops resistance to therapy (4). Primary GBM is more common and manifests as de novo development with no identifiable precursor lesion, whereas secondary GBM is characterized by precursor diffusion or progression with anaplastic astrocytoma, anaplastic oligoastrocytoma, and anaplastic oligodendroglioma (3, 5). Additionally, EGFR mutations are frequently enriched in primary GBM while p53 mutations are more common in secondary GBM (6, 7). Although the differences between the two subtypes have been found, improvement of therapies for GBM still needs to continue due to unknown risk factors (8). Notably, epigenetic variations have displayed vital roles in the development of tumor progression in recent years (9), such as DNA methylation, histone modification, and other epigenetic modifiers that could modulate the oncogene expression of GBM (10, 11). Thus, epigenetic changes may be a novel perspective for understanding the physiology and pathogenesis of GBM.
The tumor microenvironment (TME) is a host for supporting the growth and invasion of tumors, promoting neoplastic transformation, protecting the tumor from host immunity, and providing niches for dormant metastases to thrive (12), and is created by interactions between malignant and non-transformed cells (13). Besides high tumor heterogeneity, a unique TME is one of vital reasons for dismal results of GBM treatment (5, 14–16). GBM tumor cells have the ability to transform the immune response into chronic inflammation (17), which helps tumor relapse by nurturing GBM stem cells (GSCs), resulting in epithelial-mesenchymal transition and multidrug resistance (18). And then, angiogenesis and inflammatory cytokines induced by GBM expansion increase the abnormal blood brain barrier (BBB) or blood tumor barrier (BTB), which further inhibits the entrance of functional immune cells in the brain (19). Apart from tumor cells, glioma-associated microglia/macrophages (GAMs) are the main types of infiltrating immune cells, and account for approximately 30% of the GBM cell population (20). Notably, GAMs acquire two specific phenotypes including the classical pro-inflammatory state M1 and alternative anti-inflammatory state M2. The latter is found to be the primary phenotype in GBM (21) and can be reprogrammed by GBM tumor cells (22), generating a complex and dynamic network of inflammatory cytokines, chemokines, and matrix remodeling (13). As a result, the autocrine and reciprocal paracrine of GBM tumor cells and GAMs promote an inflammatory TME in favor of tumor promotion (23). Moreover, inflammation cooperates with the Warburg effect in GBM to exacerbate the inflammation response in the TME through producing more inflammatory cytokines, lactate, and immunosuppressive and angiogenetic factors, leading to a more suitable and beneficial condition for GBM progression (24–32). Therefore, targeting the inflammatory TME might be a promising therapeutic strategy for GBM.
Epigenetic modifications are involved in inflammatory procession in GBM via regulating the inflammatory signaling pathways and production of inflammatory cytokines (24, 33–35). Due to the sustained exploration of epigenetic modification in the last decade, various epigenetic changes such as DNA methylation, histone modification, mRNA and non-coding RNA (ncRNA)-mediated targeting regulation, and nucleosome and chromosome remodeling were determined in cancer progression (36). Among these, DNA methylation and nucleosome-mediated inflammatory gene expressions, aberrant histone methylation or acetylation-mediated glioma-associated macrophages/microglia (GAMs) polarization, and ncRNA-mediated high expression of inflammatory signaling pathways contribute to the transcription and growth of GBM tumor cells (37–40). These epigenetic modifications induce inflammatory cytokine release that accelerates the chronic inflammatory response in the TME of GBM, and reversing or inhibiting these changes are beneficial for longer survival in GBM patients, suggesting that epigenetic regulations of inflammation are important to GBM (10, 11). Therefore, we systemically reviewed the associated epigenetic changes and regulation in the inflammatory TME of GBM procession, aiming to comprehensively recognize epigenetic modifications of the inflammatory microenvironment in GBM.
Aberrant activation of inflammatory responses is a significant trait of GBM (41, 42), which endows GBM tumor cells with an immune evasion ability, thus causing immune tolerance of GBM to any therapies (38, 43). GBM tumor cells and GAMs are the main contributors to the inflammatory microenvironment (44, 45), exaggerating tumor invasion and relapse (46, 47). Moreover, GBM tumor cells also collaborate with the extracellular matrix, astrocytes, pericytes, and endothelial cells to secrete multiple inflammatory cytokines and chemokines that foster GAM infiltration and polarization (48–50), which forms a vicious cycle that accelerates the inflammatory response in the microenvironment and further aggravates GBM.
GBM tumor cells exhibit a strong inflammatory signature that persistently produces pro-inflammatory cytokines at chronic low levels, thus generating a chronic inflammatory state in the TME (51). Represented by IL-1 and IL-6, primary mediators of GBM tumor cells in the inflammatory TME can orchestrate immunological activation and inflammatory signaling cascades such as hypoxia-inducible factor-1α (HIF-1α), Wnt-1, nuclear factor-kappa B (NF-κB), and STAT3 in GBM (52–55) (Figure 1). The members of the IL-1 subfamily including IL-1β and IL-33 drive tumor promotion and immune suppression (56). Activations of the NLRP3 inflammasome and CD133 in GBM tumor cells could induce the production of IL-1β and its downstream chemokines CCL3, CXCL3, and CXCL5 (57), which facilitate the migration, proliferation, self-renewal, and invasion of GBM (58–60). IL-1β activates both RAS and Wnt-1 which mediate the elevation of HIF-1α, thus inducing a HIF-1α/IL-1β autocrine loop in GBM tumor cells (61). Meanwhile, IL-1β also activates the p38 mitogen-activated protein kinase (MAPK)-activated protein kinase 2 (MK2)- human antigen R (HuR), toll-like receptor 4 (TLR-4), and other inflammation-associated signaling pathways that significantly increase the levels of IL-6 and IL-8 in GBM tumor cells, eventually developing an inflammatory TME in favor of GBM invasion and growth (61–63). Additionally, IL-33 induced by GBM tumor cells is another important inflammatory mediator that accelerates GBM proliferation, migration, and invasion (47, 64, 65). Interestingly, IL-33 could transform non-stem cells to GBM stem cells (66), activate disintegrin and metalloproteinase with thrombospondin motifs 5 (ADAMTS5) and EGFR by promoting the accumulation of tenascin-C (TNC), which in turn exacerbates GBM tumor cell proliferation (47, 64, 65, 67). Both subfamilies of IL-1 accelerate GBM relapse, however, blocking IL-1β and IL-33 can reduce tumor progression-associated compensation mechanisms including the Warburg effect (47, 68). Furthermore, emerging data suggest that GBM tumor cell-induced IL-6 displays a pro-tumorigenic role, and correlates with poor prognosis and GAM infiltration in GBM (67, 69, 70). IL-6 stimulates the invasion and growth of GBM tumor cells (71) by binding to heterogeneous membrane receptor complexes (formed by IL-6r and glycoprotein 130) and initiating typical IL-6 signal transduction (53), such as STAT3. Therefore, pro-inflammatory cytokines such as IL-1 members and IL-6 secreted from GBM tumor cells aggravate tumor-promoting inflammation and GBM growth.

Figure 1 Inflammation-related changes in GBM tumor cells. GBM tumor cells produce abundant IL-1 and IL-6 through activating various pathways including STAT3, RAS, NFκB, NLRP3, and HIF-1α pathways, thus creating a chronic inflammatory environment, which benefits GBM growth.
GAMs are the domain members of immune cells in GBM when GSCs have a strong evasion ability (38). It is noteworthy that pro-inflammatory and angiogenic pathways including TNF, interferon (IFN), NF-κB, and hypoxia pathways in GAMs accelerate RAS-driven-GBM tumor cell proliferation via upregulation of IL-1β, VEGFA, CCL8, Arg1, CD274, and PD-L1 (72). High levels of inflammatory genes including IL-6, IL-8, IL-1β, IL13RA1, IL13RA2, IL10RB, CXCR4, OSMR, CCR1, MDK, LIF, FAS, CCL2, CCL20, CXCL10, CXCL11, and CXCL14 as well as GAM makers such as CD14, CD163, TLRs, and CHI3L1 co-occur and are positively associated with poor survival in mesenchymal GBM (73). This suggests that GAM-associated inflammation is corrected with GBM progression. GAMs consist of two types of cells, GBM-associated macrophages and microglia. The former are preferentially recruited to the perivascular areas in early GBM, and GBM-associated microglia are localized to peritumoral regions (74, 75). Both types have polarized phenotypes with different functions: anti-tumorigenic and pro-tumorigenic phenotypes, which are defined as M1 or M2-like GAMs, respectively (50). Initially, stimulators like lipopolysaccharide (LPS), interferon (IFN)-γ, TNF, CD80, CD86, and IL-12A/B induce M1-like polarization by activating the TLR4-NF-κB and STAT1 signaling pathways (73, 76), whereas activations of IL-4, IL-6, IL10, CD163, MSR1, MRC1, CD209, CLEC10A, CLEC7A, and CXCR4 induce M2-like polarization through the NF-κB/STAT3 pathway (73). In general, most GAMs display the M2 phenotype due to activation of STAT3 and NF-κB, secretions of immunosuppressive cytokines (e.g., IL-6, IL-10, IL-8, TGF-β1, MCP-1 and PGE-2), and colony stimulating factor (CSF) secretions of the TME in GBM (77–81). M2 GAMs further produce more Arg-1, TGF-β, IL-10, IL-6, and other abundant immunosuppressive cytokines, thus foster immunosuppression and pro-angiogenesis which contribute to growth and invasion of GBM after GBM tumor cells adapt to the pro-inflammatory TME (46). Additionally, other pathways including mTOR also increase activities of STAT3 or NF-κB in M2 GAMs that decrease immune reactivity and functional immune cell infiltration (80). Ultimately, these effects mediate GBM tumor immune evasion and growth (Figure 2), hence, reshaped GAMs have potential as a therapy mode in GBM.
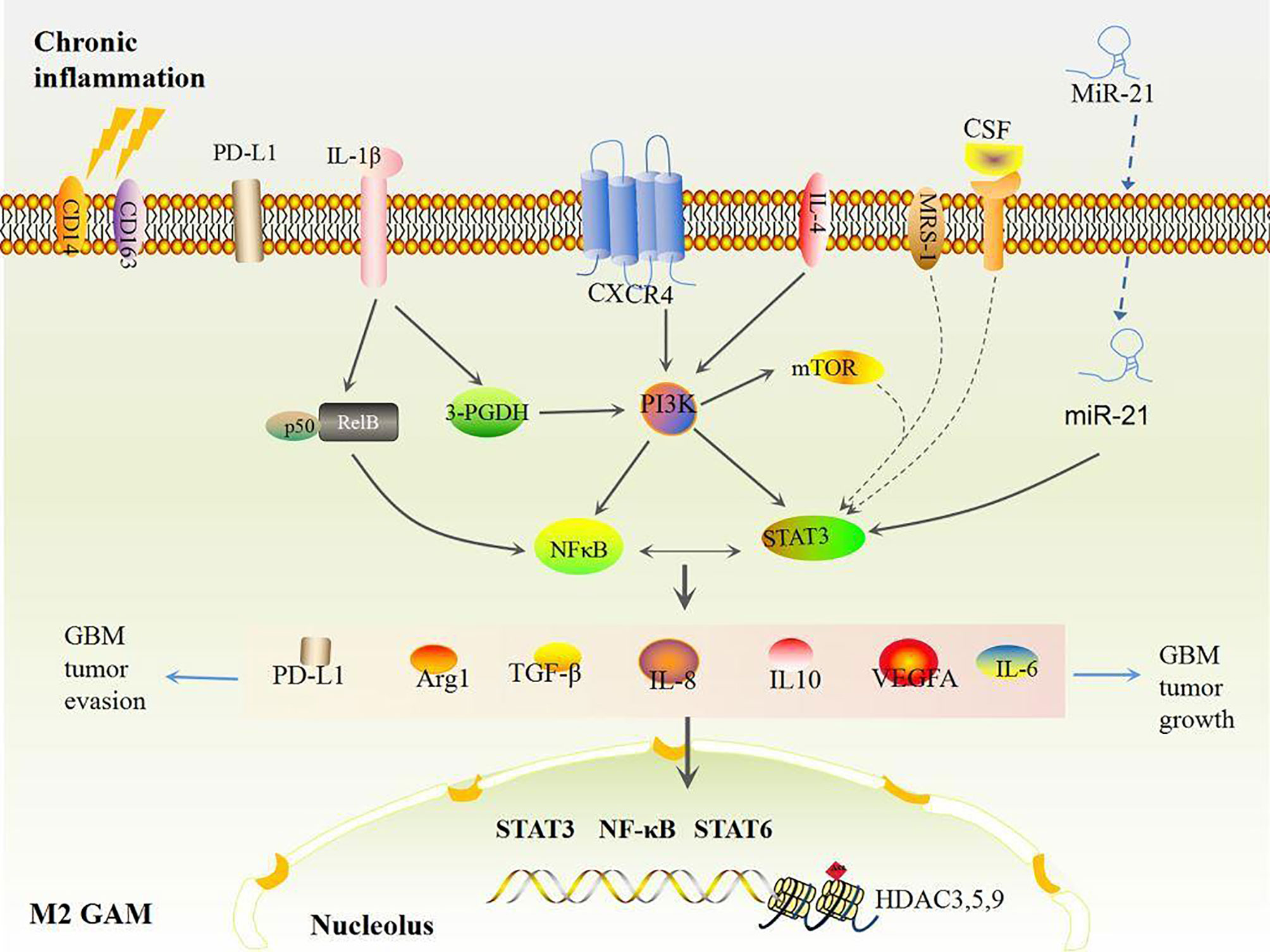
Figure 2 Chronic inflammation induced M2 GAMs. Under chronic inflammation, most GAMs are polarized to the M2 type by STAT3 and NFκB activation, which produces high levels of VEGF and immunosuppressive factors including IL-6, IL-8, IL-10, Arg1, TGF-β, and PD-L1, eventually inducing more M2 polarizations and accelerating GBM growth and evasion in turn.
Cellular communication in GBM also participates in chronic inflammatory procession (Figure 3). At the primary state, the crosstalk between stromal cells and tumor cells that induces inflammatory cytokines like IL-2 can recruit neutrophils, mast cells, T cells, and B cells to the TME of GBM (44). In contrast, GBM tumor cells protect themselves by inducing IFN-γ, IL-10, IL-8, CCL2, IL-6, and TGFβ, which impair the anti-tumor ability of immune cells and reprogram immune cells to inflammatory phenotypes that produce inflammatory mediators, anagenetic factors, and PD-1/PD-L1, resulting in GBM tumor cell evasion and invasion (44, 82, 83). And then, GBM tumor cell-induced IL-6 activates STAT3 in astrocytes, which subsequently drives IL-6 to stimulate GBM tumor cells and further promotes STAT3 signals and enhances downstream events (84). Certainly, STAT3-dependent signal-mediated GBM tumor cells produce more IL-6 thus increasing the level of PD-L1 in immunosuppressive peripheral myeloid cells and facilitating tumor growth (85), which indicates that reciprocal activation of IL-6 and STAT3 continuously fosters chronic inflammation to exacerbate GBM growth and evasion (84). Interestingly, pro-inflammatory cytokines induce tumor granule neuron precursors differentiating into astrocytes (74), and then astrocyte-mediated IL-4 stimulates GAMs to produce insulin-like growth factor 1, thereby further promoting tumor progression (86). GBM tumor cells also foster pericytes and endothelial cells to produce more IL-10 and TGFβ by activating the IL-6/STAT3 signaling pathway, thus inciting tumor growth (87–89). Surrounding anti-inflammatory cytokines, including IL-8 and IL-10, shift M1 GAMs toward M2 GAMs (78), which would express Arg-1 (46) and contribute to abnormal angiogenic activity in GBM (46).
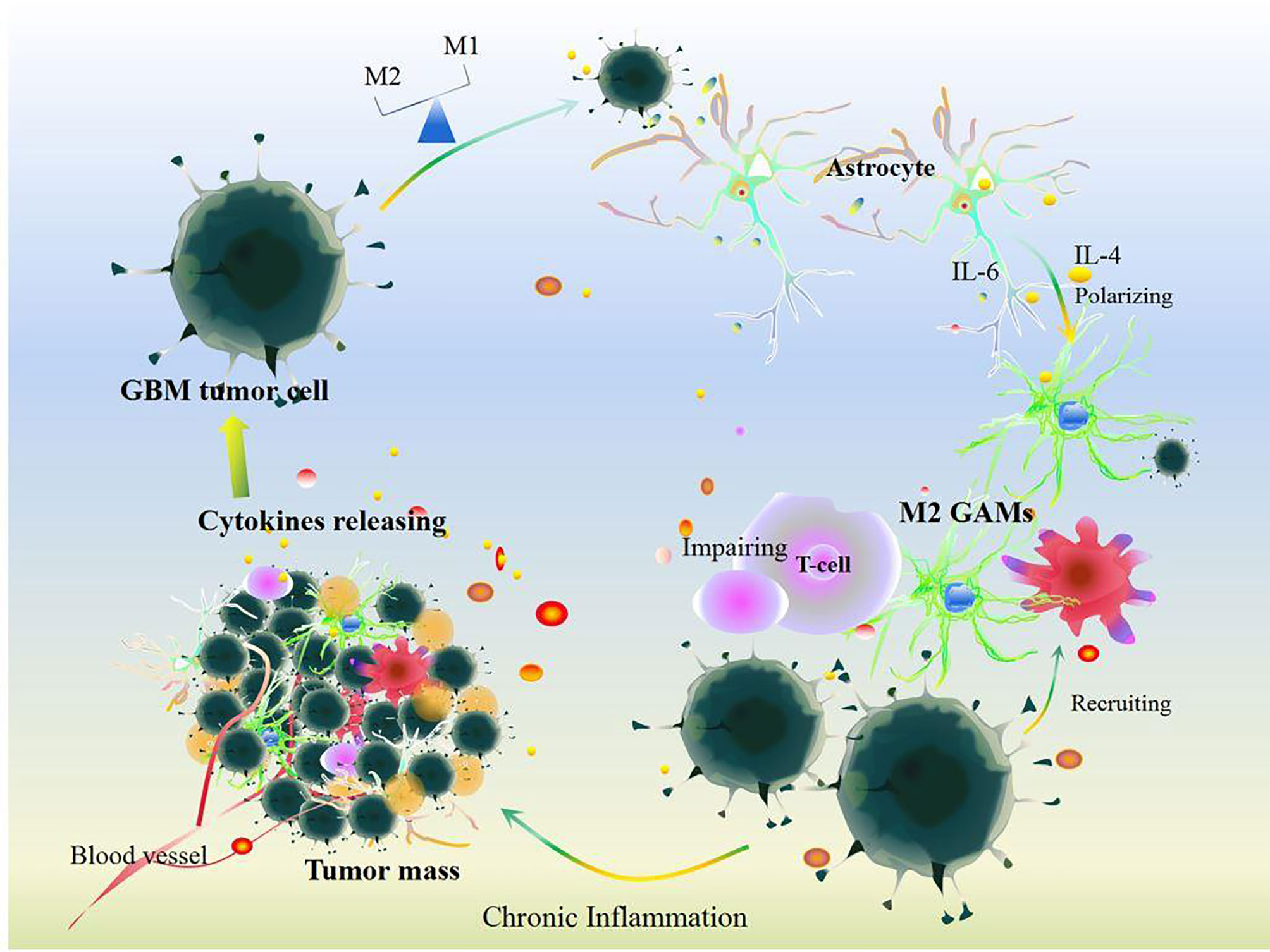
Figure 3 Cellular communication in GBM. Under chronic inflammation, cytokines released by cells including somatic cells and other immune cells can recruit GAM infiltration after the survey and chemotherapy and immunotherapy treatments in GBM. Additionally, GBM tumor cells induce GAM polarization resulting in disrupting the balance of M1 and M2 GAMs. Meanwhile, GBM tumor cells communication with astrocytes and other somatic cells could impair T cell function and release plenty of cytokines including IL-6, IL-4, IL-10, IL-8, IL-1, TGF-β, Arg-1, and VEGF. All these changes aggravate GBM tumor growth.
Furthermore, crosstalk between GBM tumor cells and GAMs is indispensable to the growth of GBM. Firstly, GBM tumor cell-induced IL-1β drives the HIF-1α-IL-1β autocrine loop to maintain a persistently elevated IL-1β level (61) that activates RelB/p50 complexes, thus attracting or polarizing GAMs (90). GBM tumor cells promote M2 GAM polarization by activation of STAT3, NF-κB, Wnt-3a, and mechanistic target of rapamycin (mTOR) (91), resulting in upregulation of IL-10 levels (91). And then, M2 GAMs also release IL-1β that activates protein kinase c (PKC)-delta and mediates phosphorylation of the glycolytic enzyme glycerol-3-phosphate dehydrogenase (3-PGDH), which subsequently activates phosphatidylinositol-3-kinase (PI3K) and promotes a feed-forward inflammatory loop in GBM tumor cells (68, 90). Moreover, GBM tumor cells together with GAMs generate a mass of succinate and lactate through the PI3K/HIF-1α pathway, thus aggravating local inflammation and GBM metastasis (27–31). Specially, lactate accelerates glycolysis, angiogenesis, and chronic inflammation by promoting IL-1β, IL-8, VEGF, and NF-κB signals, which ensures sufficient oxygen and nutrient supply for tumor cell proliferation (32). Collectively, GBM tumor cell communication with GAMs, immune cells, astrocytes, pericytes, and endothelial cells accelerates the development of a chronic inflammatory TME in GBM.
Epigenetic changes occur simultaneously with oncogenic events in cancer progression. In GBM, epigenetic modification including DNA methylation, histone modification, and non-coding RNA regulation could modulate the inflammatory genes-oncogenes loop and associated immunosuppression (92–95), providing a therapeutic strategy and comprehensively recognized pathogenesis of GBM by targeting the inflammatory TME (Table 1).
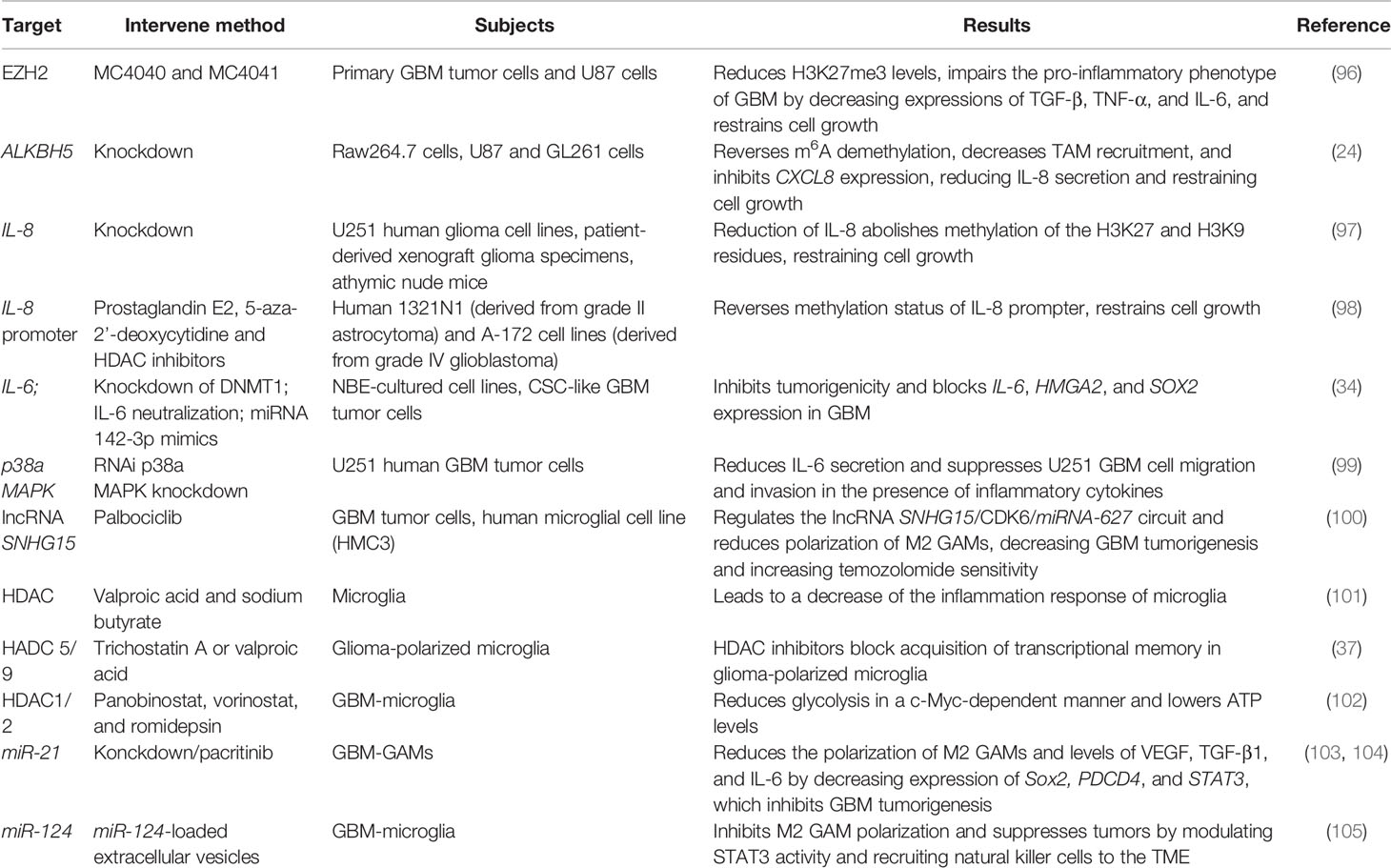
Table 1 Epigenetic control inflammatory response of GBM.
DNA methylation is an epigenetic modification of DNA that is important for the normal regulation of transcription, embryonic development, genomic imprinting, genome stability, and chromatin structure, and is controlled by DNA methyltransferases, methyl-CpG binding proteins, and other chromatin-remodeling factors, thereby controlling gene expression (36). DNA methylation loss of oncogenes can activate inflammatory oncogene expression and extensively promote GBM growth (106, 107). GBM patients with an unmethylated O6-methylguanine-DNA methyltransferase (MGMT) promoter significantly show an elevated expression of inflammatory genes such as IL-6, CCL2, CCXCL2, HLA-A, and Serum amyloid A1 (108, 109), similar to methylation of MGMT, positively associated with IL-6-mediated primary GBM procession (110). This implies that inflammation is associated with high expression of MGMT. Additionally, N6-methyladenine (N6-mA) is highly enriched in histone 3 lysine 9 trimethyl (H3K9me3) in GBM tumor cells, which is induced by ALKBH and exacerbates GSCs’ self-renewal and proliferation (111, 112). Specially, the demethylase activity of ALKBH5 has been demonstrated as an indispensable role in regulating the conditions of hypoxia and inflammatory TME in GBM and is required for NLRP3 inflammasome-related CXCL8 and NEAT1 gene activation (24, 113, 114). Silencing MGMT or ALKBH5 of GBM tumor cells could inhibit GAM infiltration and repress NEAT1 and CXCL8 expression, eventually suppressing GBM tumor cell growth and lengthening the survival of GBM patients (24, 113, 115). This suggests that regulation of the DNA methylation state of inflammation-associated genes is beneficial for GBM treatment.
Histone modification is covalent post-translational modification to histone proteins including methylation, phosphorylation, acetylation, ubiquitylation, and sumoylation, and regulates gene expression by altering chromatin structure or recruiting histone modifiers (116). Histone methylation and acetylation are found frequently in GBM. On one hand, enhancer of zeste homologue 2 (EZH2) and lysine 27 on histone H3 (H3K27) methyltransferase (H3K27me) highly mutate in GBM tumor cells, and lead to a poor outcome of GBM (96). EZH2 increases H3K27me3 in the promoter of PTEN thus silencing PTEN and activating the threonine protein kinase B (AKT)/mTOR pathway, promoting GBM tumor cell proliferation and metastasis (117). Interestingly, EZH2 inhibitors (MC4040 and MC4041) not only reverse the pro-inflammatory phenotype of GBM U-87 and GL-1 cells by downregulating the levels of H3K27me3, but also inhibit epithelial-mesenchymal transition and migration of primary GBM tumor cells by reducing the level of VEGF and VEGFR1, resulting in inhibiting GBM cell proliferation and invasion of GBM (96). On the other hand, histone deacetylases (HDACs) are also positively associated with poor clinical features of GBM (118, 119). HDAC activity is greatly increased in GBM by activating STAT6, IFR-3, IFR-4, NFκB, TLR, and IFN (120), and is regarded as the main effector of epigenetic alterations in M2 GAMs (121, 122). During the GBM process, hyperactivity of HDAC3, 5, and 9 is expressed in IL-4-induced M2 GAMs (101, 123) that increase the levels of TGFβ and IL-10, resulting in exacerbating the immunosuppressive capacity of GBM tumor cells (124). It is worth noting that HDAC inhibitors can thwart M2-type polarization of GAM and retard GBM tumor growth (37, 125) by restricting activation of histone marks or targeting the STAT6 signaling pathway in a HDAC3-dependent manner (123, 124, 126). Unfortunately, EZH or HDAC suppression also induces M1 GAMs secreting IL-1β and IL-6, and activates STAT3 signals in GBM tumor cells, leading to an aggravated inflammatory response in the TME of GBM (124, 127). Thus, other histone modifications and more effective histone-regulating methods of inflammation in GBM need further exploration.
Non-coding RNAs (ncRNAs) related to epigenetic regulation consist of two main groups: long ncRNAs (lncRNAs) and short ncRNAs including miRNA, siRNA, intronic RNA, repetitive RNA, piRNAs, snoRNAs, and lincRNAs, which generally act as cis-acting silencers, but also as trans-acting regulators of site-specific modification and imprinted gene-silencing (128, 129). Among these, miRNA has been frequently explored in GBM progression and invasion (130). A variety of miRNAs, including miR-21, miR-26, miR-221/222, miR-210, miR-155, and miR-10b, promote GBM cell proliferation, apoptosis, and growth by targeting PTEN expression and active Akt and HIF3α. Recently, a growing number of studies revealed the relationship between miRNAs and inflammation (100, 105, 131–133). The MiR142-3p promoter is methylated which decreases miR142-3p gene expression and increases the level of IL-6, thus promoting GBM invasion and migration in a DNA methyltransferase (DNMT) 1-dependent manner (34). In addition, inhibition of miR-93 in GBM tumor cells fosters an inflammatory microenvironment via increasing the levels of IL-6, IL-8, IL-1β, granulocyte-colony stimulating factor, leukemia inhibitory factor, COX2, and CXCL5 (131). MiR-155 induced by inflammatory cytokines, such as IL-1β and TNFα, mediates mesenchymal transition of GBM tumor cells and GBM growth (134, 135). Nevertheless, inhibition of miR-155 restrains the proliferation, migration, and invasion of GBM tumor cell growth by activation of STAT3 (132, 133). Interestingly, miR-124-loaded extracellular vesicles suppress GBM tumors by activating STAT3 activity and recruiting natural killer cells to the TME (100, 105). Downregulation of miRNA-125b or inhibition of mitogen-activated protein kinase (MAPK) mRNA could restrain the p38/MAPK pathway and reduce IL-6 secretion, which suppresses GBM tumor cell migration and invasion (99, 130, 136). MiR142-3p mimics could block IL-6, HMGA2, and SOX2 expression in GBM tumor cells that inhibit tumorigenicity (34). Besides, upregulation of miR-93 can significantly suppress proliferation, migration, and angiogenesis of GBM tumor cells by alleviating the inflammatory environment (131). MiR-21 in exosomes or extracellular vesicles induced by GBM tumor cells can be incepted by GAMs and upregulate IL-6, TGF-β, and Arg-1 in M2 GAMs (103, 104), resulting in downregulating the immune response and accelerating growth and invasion of GBM tumor cells (103). However, inhibition of miR-21 significantly reduces the polarization of M2 GAMs and decreases levels of VEGF, TGF-β1, and IL-6 by decreasing activities of Sox2, PDCD4, and STAT3, ultimately inhibiting GBM tumor cell growth (104). Collectively, RNA interference is important to GBM treatment through multiple regulations of inflammation-related signals.
The inflammatory TME as the primary contributor to the pathogenesis of GBM, which consists of various functional cells including GBM tumor cells, GAMs, and other non-tumor cells, and the autocrine and reciprocal paracrine of these above cells form a chronic low-grade inflammation state that benefits proliferation, invasion, migration, and evasion of GBM. While, epigenetic modification is a new perspective for understanding the pathogenesis of GBM. Here, we find that epigenetic regulators including DNMT, EHZ, HDAC, and miRNAs are involved in the inflammation of GBM, reminding us that epigenetic control of the inflammatory TME may be a rewarding therapy for GBM treatment. However, some problems still need to be further explored in the future. Firstly, the different epigenetic changes between primary and secondary GBM need to be explained, which is meaningful to identify and diagnose the two subtypes. Secondly, DNA methylation and histone modification including phosphorylation, ubiquitylation, and sumoylation of the inflammatory TME in GBM need elaborating. And then, the relationships of the inflammatory TME between other epigenetic modifications, such as chromosome structure, nucleosome transcription, long ncRNAs, and other short ncRNAs need comprehensively uncovering. Finally, the interactive influences of different epigenetic modifications in GBM need to be explained. Overall, we provide insight into the epigenetic regulation of inflammation in GBM and reveal the relationship between inflammation and GBM progression, suggesting some future directions of GBM underlying epigenetic modification.
NC drafted the manuscript and drew the figures. CP and DL conceived and revised the review. All authors contributed to the article and approved the submitted version.
Financial support by the National Natural Science Foundation of China (82104477 and U19A2010) and multidisciplinary interdisciplinary innovation team for multidimensional evaluation of southwestern characteristic Chinese medicine resources (NO. ZYYCXTD-D-202209), and special support from the China Postdoctoral Science Foundation (2019M663456 and 2019TQ0044), Xinglin Scholar Research Promotion Project of Chengdu University of TCM (BSH2019008) are gratefully acknowledged.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Vigneswaran K, Boyd NH, Oh SY, Lallani S, Boucher A, Neill SG, et al. YAP/TAZ Transcriptional Coactivators Create Therapeutic Vulnerability to Verteporfin in EGFR-Mutant Glioblastoma. Clin Cancer Res (2021) 27(5):1553–69. doi: 10.1158/1078-0432.CCR-20-0018
2. Louis DN, Perry A, Reifenberger G, von Deimling A, Figarella-Branger D, Cavenee WK, et al. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: A Summary. Acta Neuropathol (2016) 131(6):803–20. doi: 10.1007/s00401-016-1545-1
3. DeCordova S, Shastri A, Tsolaki AG, Yasmin H, Klein L, Singh SK, et al. Molecular Heterogeneity and Immunosuppressive Microenvironment in Glioblastoma. Front Immunol (2020) 11:1402. doi: 10.3389/fimmu.2020.01402
4. Johnson KC, Houseman EA, King JE, von Herrmann KM, Fadul CE, Christensen BC. 5-Hydroxymethylcytosine Localizes to Enhancer Elements and Is Associated With Survival in Glioblastoma Patients. Nat Commun (2016) 7:13177. doi: 10.1038/ncomms13177
5. Tan AC, Ashley DM, Lopez GY, Malinzak M, Friedman HS, Khasraw M. Management of Glioblastoma: State of the Art and Future Directions. CA Cancer J Clin (2020) 70(4):299–312. doi: 10.3322/caac.21613
6. Aldape K, Zadeh G, Mansouri S, Reifenberger G, von Deimling A. Glioblastoma: Pathology, Molecular Mechanisms and Markers. Acta Neuropathol (2015) 129(6):829–48. doi: 10.1007/s00401-015-1432-1
7. Wang Q, Hu B, Hu X, Kim H, Squatrito M, Scarpace L, et al. Tumor Evolution of Glioma-Intrinsic Gene Expression Subtypes Associates With Immunological Changes in the Microenvironment. Cancer Cell (2017) 32(1):42–56.e6. doi: 10.1016/j.ccell.2017.06.003
8. Le Rhun E, Preusser M, Roth P, Reardon DA, van den Bent M, Wen P, et al. Molecular Targeted Therapy of Glioblastoma. Cancer Treat Rev (2019) 80:101896. doi: 10.1016/j.ctrv.2019.101896
9. Bennett RL, Licht JD. Targeting Epigenetics in Cancer. Annu Rev Pharmacol Toxicol (2018) 58:187–207. doi: 10.1146/annurev-pharmtox-010716-105106
10. Hegi ME, Diserens AC, Gorlia T, Hamou MF, de Tribolet N, Weller M, et al. MGMT Gene Silencing and Benefit From Temozolomide in Glioblastoma. N Engl J Med (2005) 352(10):997–1003. doi: 10.1056/NEJMoa043331
11. Touat M, Idbaih A, Sanson M, Ligon KL. Glioblastoma Targeted Therapy: Updated Approaches From Recent Biological Insights. Ann Oncol (2017) 28(7):1457–72. doi: 10.1093/annonc/mdx106
12. Swartz MA, Iida N, Roberts EW, Sangaletti S, Wong MH, Yull FE, et al. Tumor Microenvironment Complexity: Emerging Roles in Cancer Therapy. Cancer Res (2012) 72(10):2473–80. doi: 10.1158/0008-5472.CAN-12-0122
13. Balkwill FR, Capasso M, Hagemann T. The Tumor Microenvironment at a Glance. J Cell Sci (2012) 12523):5591–6. doi: 10.1242/jcs.116392
14. Desland FA, Hormigo A. The CNS and the Brain Tumor Microenvironment: Implications for Glioblastoma Immunotherapy. Int J Mol Sci (2020) 21(19):7358. doi: 10.3390/ijms21197358
15. Wang J, Toregrosa-Allen S, Elzey BD, Utturkar S, Lanman NA, Bernal-Crespo V, et al. Multispecific Targeting of Glioblastoma With Tumor Microenvironment-Responsive Multifunctional Engineered NK Cells. Proc Natl Acad Sci USA (2021) 118(45):e210757118. doi: 10.1073/pnas.2107507118
16. Foray C, Valtorta S, Barca C, Winkeler A, Roll W, Muther M, et al. Imaging Temozolomide-Induced Changes in the Myeloid Glioma Microenvironment. Theranostics (2021) 11(5):2020–33. doi: 10.7150/thno.47269
17. Litak J, Mazurek M, Grochowski C, Kamieniak P, Rolinski J. PD-L1/PD-1 Axis in Glioblastoma Multiforme. Int J Mol Sci (2019) 20(21):5347. doi: 10.3390/ijms20215347
18. Erin N, Grahovac J, Brozovic A, Efferth T. Tumor Microenvironment and Epithelial Mesenchymal Transition as Targets to Overcome Tumor Multidrug Resistance. Drug Resist Updat (2020) 53:100715. doi: 10.1016/j.drup.2020.100715
19. Arvanitis CD, Ferraro GB, Jain RK. The Blood-Brain Barrier and Blood-Tumour Barrier in Brain Tumours and Metastases. Nat Rev Cancer (2020) 20(1):26–41. doi: 10.1038/s41568-019-0205-x
20. Morisse MC, Jouannet S, Dominguez-Villar M, Sanson M, Idbaih A. Interactions Between Tumor-Associated Macrophages and Tumor Cells in Glioblastoma: Unraveling Promising Targeted Therapies. Expert Rev Neurother (2018) 18(9):729–37. doi: 10.1080/14737175.2018.1510321
21. Yelton CJ, Ray SK. Histone Deacetylase Enzymes and Selective Histone Deacetylase Inhibitors for Antitumor Effects and Enhancement of Antitumor Immunity in Glioblastoma. Neuroimmunol Neuroinflamm (2018) 5:46. doi: 10.20517/2347-8659.2018.58
22. Dasgupta Y, Golovine K, Nieborowska-Skorska M, Luo L, Matlawska-Wasowska K, Mullighan CG, et al. Drugging DNA Repair to Target T-ALL Cells. Leuk Lymphoma (2018) 59(7):1746–9. doi: 10.1080/10428194.2017.1397662
23. Najafi M, Goradel NH, Farhood B, Salehi E, Solhjoo S, Toolee H, et al. Tumor Microenvironment: Interactions and Therapy. J Cell Physiol (2019) 234(5):5700–21. doi: 10.1002/jcp.27425
24. Dong F, Qin X, Wang B, Li Q, Hu J, Cheng X, et al. ALKBH5 Facilitates Hypoxia-Induced Paraspeckle Assembly and IL8 Secretion to Generate an Immunosuppressive Tumor Microenvironment. Cancer Res (2021) 81(23):5876–88. doi: 10.1158/0008-5472.CAN-21-1456
25. Kesarwani P, Kant S, Prabhu A, Chinnaiyan P. The Interplay Between Metabolic Remodeling and Immune Regulation in Glioblastoma. Neuro Oncol (2017) 19(10):1308–15. doi: 10.1093/neuonc/nox079
26. Velpula KK, Bhasin A, Asuthkar S, Tsung AJ. Combined Targeting of PDK1 and EGFR Triggers Regression of Glioblastoma by Reversing the Warburg Effect. Cancer Res (2013) 73(24):7277–89. doi: 10.1158/0008-5472.CAN-13-1868
27. Wu JY, Huang TW, Hsieh YT, Wang YF, Yen CC, Lee GL, et al. Cancer-Derived Succinate Promotes Macrophage Polarization and Cancer Metastasis via Succinate Receptor. Mol Cell (2020) 77(2):213–27.e5. doi: 10.1016/j.molcel.2019.10.023
28. Kes MMG, Van den Bossche J, Griffioen AW, Huijbers EJM. Oncometabolites Lactate and Succinate Drive Pro-Angiogenic Macrophage Response in Tumors. Biochim Biophys Acta Rev Cancer (2020) 1874(2):188427. doi: 10.1016/j.bbcan.2020.188427
29. Zhu L, Zhao Q, Yang T, Ding W, Zhao Y. Cellular Metabolism and Macrophage Functional Polarization. Int Rev Immunol (2015) 34(1):82–100. doi: 10.3109/08830185.2014.969421
30. Ivashkiv LB. The Hypoxia-Lactate Axis Tempers Inflammation. Nat Rev Immunol (2020) 20(2):85–6. doi: 10.1038/s41577-019-0259-8
31. Doherty JR, Cleveland JL. Targeting Lactate Metabolism for Cancer Therapeutics. J Clin Invest (2013) 123(9):3685–92. doi: 10.1172/JCI69741
32. Hirschhaeuser F, Sattler UG, Mueller-Klieser W. Lactate: A Metabolic Key Player in Cancer. Cancer Res (2011) 71(22):6921–5. doi: 10.1158/0008-5472.CAN-11-1457
33. Bayarsaihan D. Epigenetic Mechanisms in Inflammation. J Dent Res (2011) 90(1):9–17. doi: 10.1177/0022034510378683
34. Chiou GY, Chien CS, Wang ML, Chen MT, Yang YP, Yu YL, et al. Epigenetic Regulation of the Mir142-3p/Interleukin-6 Circuit in Glioblastoma. Mol Cell (2013) 52(5):693–706. doi: 10.1016/j.molcel.2013.11.009
35. Dong Z, Cui H. Epigenetic Modulation of Metabolism in Glioblastoma. Semin Cancer Biol (2019) 57:45–51. doi: 10.1016/j.semcancer.2018.09.002
36. Dawson MA, Kouzarides T. Cancer Epigenetics: From Mechanism to Therapy. Cell (2012) 150(1):12–27. doi: 10.1016/j.cell.2012.06.013
37. Maleszewska M, Steranka A, Smiech M, Kaza B, Pilanc P, Dabrowski M, et al. Sequential Changes in Histone Modifications Shape Transcriptional Responses Underlying Microglia Polarization by Glioma. Glia (2021) 69(1):109–23. doi: 10.1002/glia.23887
38. Gangoso E, Southgate B, Bradley L, Rus S, Galvez-Cancino F, McGivern N, et al. Glioblastomas Acquire Myeloid-Affiliated Transcriptional Programs via Epigenetic Immunoediting to Elicit Immune Evasion. Cell (2021) 184(9):2454–70.e26. doi: 10.1016/j.cell.2021.03.023
39. Abels ER, Maas SLN, Nieland L, Wei Z, Cheah PS, Tai E, et al. Glioblastoma-Associated Microglia Reprogramming Is Mediated by Functional Transfer of Extracellular miR-21. Cell Rep (2019) 28(12):3105–19.e7. doi: 10.1016/j.celrep.2019.08.036
40. Sheikh T, Sen E. P53 Affects Epigenetic Signature on SOCS1 Promoter in Response to TLR4 Inhibition. Cytokine (2021) 140:155418. doi: 10.1016/j.cyto.2020.155418
41. Ham SW, Jeon HY, Jin X, Kim EJ, Kim JK, Shin YJ, et al. TP53 Gain-of-Function Mutation Promotes Inflammation in Glioblastoma. Cell Death Differ (2019) 26(3):409–25. doi: 10.1038/s41418-018-0126-3
42. Gromeier M, Brown MC, Zhang G, Lin X, Chen Y, Wei Z, et al. Very Low Mutation Burden Is a Feature of Inflamed Recurrent Glioblastomas Responsive to Cancer Immunotherapy. Nat Commun (2021) 12(1):352. doi: 10.1038/s41467-020-20469-6
43. Murray PJ. Macrophage Polarization. Annu Rev Physiol (2017) 79:541–66. doi: 10.1146/annurev-physiol-022516-034339
44. Broekman ML, Maas SLN, Abels ER, Mempel TR, Krichevsky AM, Breakefield XO. Multidimensional Communication in the Microenvirons of Glioblastoma. Nat Rev Neurol (2018) 14(8):482–95. doi: 10.1038/s41582-018-0025-8
45. Tomaszewski W, Sanchez-Perez L, Gajewski TF, Sampson JH. Brain Tumor Microenvironment and Host State: Implications for Immunotherapy. Clin Cancer Res (2019) 25(14):4202–10. doi: 10.1158/1078-0432.CCR-18-1627
46. Cui X, Morales RT, Qian W, Wang H, Gagner JP, Dolgalev I, et al. Hacking Macrophage-Associated Immunosuppression for Regulating Glioblastoma Angiogenesis. Biomaterials (2018) 161:164–78. doi: 10.1016/j.biomaterials.2018.01.053
47. De Boeck A, Ahn BY, D’Mello C, Lun X, Menon SV, Alshehri MM, et al. Glioma-Derived IL-33 Orchestrates an Inflammatory Brain Tumor Microenvironment That Accelerates Glioma Progression. Nat Commun (2020) 11(1):4997. doi: 10.1038/s41467-020-18569-4
48. Bowman RL, Klemm F, Akkari L, Pyonteck SM, Sevenich L, Quail DF, et al. Macrophage Ontogeny Underlies Differences in Tumor-Specific Education in Brain Malignancies. Cell Rep (2016) 17(9):2445–59. doi: 10.1016/j.celrep.2016.10.052
49. Huang YH, Airas L, Schwab N, Wiendl H. Janus Head: The Dual Role of HLA-G in CNS Immunity. Cell Mol Life Sci (2011) 68(3):407–16. doi: 10.1007/s00018-010-0582-5
50. Sampson JH, Gunn MD, Fecci PE, Ashley DM. Brain Immunology and Immunotherapy in Brain Tumours. Nat Rev Cancer (2020) 20(1):12–25. doi: 10.1038/s41568-019-0224-7
51. Baker KJ, Houston A, Brint E. IL-1 Family Members in Cancer; Two Sides to Every Story. Front Immunol (2019) 10:1197. doi: 10.3389/fimmu.2019.01197
52. Katrib A, Jeong HH, Fransen NL, Henzel KS, Miller JA. An Inflammatory Landscape for Preoperative Neurologic Deficits in Glioblastoma. Front Genet (2019) 10:488. doi: 10.3389/fgene.2019.00488
53. Yeung YT, McDonald KL, Grewal T, Munoz L. Interleukins in Glioblastoma Pathophysiology: Implications for Therapy. Br J Pharmacol (2013) 168(3):591–606. doi: 10.1111/bph.12008
54. Hubner M, Effinger D, Wu T, Strauss G, Pogoda K, Kreth FW, et al. The IL-1 Antagonist Anakinra Attenuates Glioblastoma Aggressiveness by Dampening Tumor-Associated Inflammation. Cancers (Basel) (2020) 12(2):433. doi: 10.3390/cancers12020433
55. Deng J, Fleming JB. Inflammation and Myeloid Cells in Cancer Progression and Metastasis. Front Cell Dev Biol (2021) 9:759691. doi: 10.3389/fcell.2021.759691
56. Mantovani A, Dinarello CA, Molgora M, Garlanda C. Interleukin-1 and Related Cytokines in the Regulation of Inflammation and Immunity. Immunity (2019) 50(4):778–95. doi: 10.1016/j.immuni.2019.03.012
57. Lee SY, Kim JK, Jeon HY, Ham SW, Kim H. CD133 Regulates IL-1beta Signaling and Neutrophil Recruitment in Glioblastoma. Mol Cells (2017) 40(7):515–22. doi: 10.14348/mocells.2017.0089
58. Fathima Hurmath K, Ramaswamy P, Nandakumar DN. IL-1beta Microenvironment Promotes Proliferation, Migration, and Invasion of Human Glioma Cells. Cell Biol Int (2014) 38(12):1415–22. doi: 10.1002/cbin.10353
59. Tezcan G, Garanina EE, Alsaadi M, Gilazieva ZE, Martinova EV, Markelova MI, et al. Therapeutic Potential of Pharmacological Targeting NLRP3 Inflammasome Complex in Cancer. Front Immunol (2020) 11:607881. doi: 10.3389/fimmu.2020.607881
60. Ding Q, Shen L, Nie X, Lu B, Pan X, Su Z, et al. MiR-223-3p Overexpression Inhibits Cell Proliferation and Migration by Regulating Inflammation-Associated Cytokines in Glioblastomas. Pathol Res Pract (2018) 214(9):1330–9. doi: 10.1016/j.prp.2018.05.012
61. Sharma V, Dixit D, Koul N, Mehta VS, Sen E. Ras Regulates Interleukin-1beta-Induced HIF-1alpha Transcriptional Activity in Glioblastoma. J Mol Med (Berl) (2011) 89(2):123–36. doi: 10.1007/s00109-010-0683-5
62. Gurgis FM, Yeung YT, Tang MX, Heng B, Buckland M, Ammit AJ, et al. The P38-MK2-HuR Pathway Potentiates EGFRvIII-IL-1beta-Driven IL-6 Secretion in Glioblastoma Cells. Oncogene (2015) 34(22):2934–42. doi: 10.1038/onc.2014.225
63. Gupta P, Ghosh S, Nagarajan A, Mehta VS, Sen E. Beta-Defensin-3 Negatively Regulates TLR4-HMGB1 Axis Mediated HLA-G Expression in IL-1beta Treated Glioma Cells. Cell Signal (2013) 25(3):682–9. doi: 10.1016/j.cellsig.2012.12.001
64. Zhang JF, Tao T, Wang K, Zhang GX, Yan Y, Lin HR, et al. IL-33/ST2 Axis Promotes Glioblastoma Cell Invasion by Accumulating Tenascin-C. Sci Rep (2019) 9(1):20276. doi: 10.1038/s41598-019-56696-1
65. Akcora-Yildiz D, Yukselten Y, Sunguroglu M, Ugur HC, Sunguroglu A. IL-33 Induces ADAMTS5 Expression and Cell Migration in Glioblastoma Multiforme. Med Oncol (2022) 39(2):22. doi: 10.1007/s12032-021-01590-y
66. Angel I, Pilo Kerman O, Rousso-Noori L, Friedmann-Morvinski D. Tenascin C Promotes Cancer Cell Plasticity in Mesenchymal Glioblastoma. Oncogene (2020) 39(46):6990–7004. doi: 10.1038/s41388-020-01506-6
67. Fang KM, Yang CS, Lin TC, Chan TC, Tzeng SF. Induced Interleukin-33 Expression Enhances the Tumorigenic Activity of Rat Glioma Cells. Neuro Oncol (2014) 16(4):552–66. doi: 10.1093/neuonc/not234
68. Lu J, Xu Z, Duan H, Ji H, Zhen Z, Li B, et al. Tumor-Associated Macrophage Interleukin-Beta Promotes Glycerol-3-Phosphate Dehydrogenase Activation, Glycolysis and Tumorigenesis in Glioma Cells. Cancer Sci (2020) 111(6):1979–90. doi: 10.1111/cas.14408
69. Gramatzki D, Frei K, Cathomas G, Moch H, Weller M, Mertz KD. Interleukin-33 in Human Gliomas: Expression and Prognostic Significance. Oncol Lett (2016) 12(1):445–52. doi: 10.3892/ol.2016.4626
70. Xue H, Yuan G, Guo X, Liu Q, Zhang J, Gao X, et al. A Novel Tumor-Promoting Mechanism of IL6 and the Therapeutic Efficacy of Tocilizumab: Hypoxia-Induced IL6 Is a Potent Autophagy Initiator in Glioblastoma via the P-STAT3-MIR155-3p-CREBRF Pathway. Autophagy (2016) 12(7):1129–52. doi: 10.1080/15548627.2016.1178446
71. Jiang Y, Han S, Cheng W, Wang Z, Wu A. NFAT1-Regulated IL6 Signalling Contributes to Aggressive Phenotypes of Glioma. Cell Commun Signal (2017) 15(1):54. doi: 10.1186/s12964-017-0210-1
72. Rao R, Han R, Ogurek S, Xue C, Wu LM, Zhang L, et al. Glioblastoma Genetic Drivers Dictate the Function of Tumor-Associated Macrophages/Microglia and Responses to CSF1R Inhibition. Neuro Oncol (2021) 24(4):584–97. doi: 10.1093/neuonc/noab228
73. Zanotto-Filho A, Goncalves RM, Klafke K, de Souza PO, Dillenburg FC, Carro L, et al. Inflammatory Landscape of Human Brain Tumors Reveals an NFkappaB Dependent Cytokine Pathway Associated With Mesenchymal Glioblastoma. Cancer Lett (2017) 390:176–87. doi: 10.1016/j.canlet.2016.12.015
74. Chen Z, Feng X, Herting CJ, Garcia VA, Nie K, Pong WW, et al. Cellular and Molecular Identity of Tumor-Associated Macrophages in Glioblastoma. Cancer Res (2017) 77(9):2266–78. doi: 10.1158/0008-5472.CAN-16-2310
75. Darmanis S, Sloan SA, Croote D, Mignardi M, Chernikova S, Samghababi P, et al. Single-Cell RNA-Seq Analysis of Infiltrating Neoplastic Cells at the Migrating Front of Human Glioblastoma. Cell Rep (2017) 21(5):1399–410. doi: 10.1016/j.celrep.2017.10.030
76. Chicoine MR, Zahner M, Won EK, Kalra RR, Kitamura T, Perry A, et al. The In Vivo Antitumoral Effects of Lipopolysaccharide Against Glioblastoma Multiforme Are Mediated in Part by Toll-Like Receptor 4. Neurosurgery (2007) 60(2):372–80. doi: 10.1227/01.NEU.0000249280.61761.2E
77. Zhu C, Kros JM, van der Weiden M, Zheng P, Cheng C, Mustafa DA. Expression Site of P2RY12 in Residential Microglial Cells in Astrocytomas Correlates With M1 and M2 Marker Expression and Tumor Grade. Acta Neuropathol Commun (2017) 5(1):4. doi: 10.1186/s40478-016-0405-5
78. Yang L, Zhang Y. Tumor-Associated Macrophages: From Basic Research to Clinical Application. J Hematol Oncol (2017) 10(1):58. doi: 10.1186/s13045-017-0430-2
79. Pyonteck SM, Akkari L, Schuhmacher AJ, Bowman RL, Sevenich L, Quail DF, et al. CSF-1R Inhibition Alters Macrophage Polarization and Blocks Glioma Progression. Nat Med (2013) 19(10):1264–72. doi: 10.1038/nm.3337
80. Dumas AA, Pomella N, Rosser G, Guglielmi L, Vinel C, Millner TO, et al. Microglia Promote Glioblastoma via mTOR-Mediated Immunosuppression of the Tumour Microenvironment. EMBO J (2020) 39(15):e103790. doi: 10.15252/embj.2019103790
81. Zhai K, Huang Z, Huang Q, Tao W, Fang X, Zhang A, et al. Pharmacological Inhibition of BACE1 Suppresses Glioblastoma Growth by Stimulating Macrophage Phagocytosis of Tumor Cells. Nat Cancer (2021) 2(11):1136–51. doi: 10.1038/s43018-021-00267-9
82. Qian J, Wang C, Wang B, Yang J, Wang Y, Luo F, et al. The IFN-Gamma/PD-L1 Axis Between T Cells and Tumor Microenvironment: Hints for Glioma Anti-PD-1/PD-L1 Therapy. J Neuroinflamm (2018) 15(1):290. doi: 10.1186/s12974-018-1330-2
83. Piperi C, Papavassiliou KA, Papavassiliou AG. Pivotal Role of STAT3 in Shaping Glioblastoma Immune Microenvironment. Cells (2019) 8(11):1398. doi: 10.3390/cells8111398
84. Chen Z, Yuan SJ, Li K, Zhang Q, Li TF, An HC, et al. Doxorubicin-Polyglycerol-Nanodiamond Conjugates Disrupt STAT3/IL-6-Mediated Reciprocal Activation Loop Between Glioblastoma Cells and Astrocytes. J Control Release (2020) 320:469–83. doi: 10.1016/j.jconrel.2020.01.044
85. Lamano JB, Lamano JB, Li YD, DiDomenico JD, Choy W, Veliceasa D, et al. Glioblastoma-Derived IL6 Induces Immunosuppressive Peripheral Myeloid Cell PD-L1 and Promotes Tumor Growth. Clin Cancer Res (2019) 25(12):3643–57. doi: 10.1158/1078-0432.CCR-18-2402
86. Yao M, Ventura PB, Jiang Y, Rodriguez FJ, Wang L, Perry JSA, et al. Astrocytic Trans-Differentiation Completes a Multicellular Paracrine Feedback Loop Required for Medulloblastoma Tumor Growth. Cell (2020) 180(3):502–20.e19. doi: 10.1016/j.cell.2019.12.024
87. Sena IFG, Paiva AE, Prazeres P, Azevedo PO, Lousado L, Bhutia SK, et al. Glioblastoma-Activated Pericytes Support Tumor Growth via Immunosuppression. Cancer Med (2018) 7(4):1232–9. doi: 10.1002/cam4.1375
88. Couto M, Coelho-Santos V, Santos L, Fontes-Ribeiro C, Silva AP, Gomes CMF. The Interplay Between Glioblastoma and Microglia Cells Leads to Endothelial Cell Monolayer Dysfunction via the Interleukin-6-Induced JAK2/STAT3 Pathway. J Cell Physiol (2019) 234(11):19750–60. doi: 10.1002/jcp.28575
89. Tong L, Li J, Li Q, Wang X, Medikonda R, Zhao T, et al. ACT001 Reduces the Expression of PD-L1 by Inhibiting the Phosphorylation of STAT3 in Glioblastoma. Theranostics (2020) 10(13):5943–56. doi: 10.7150/thno.41498
90. Waters MR, Gupta AS, Mockenhaupt K, Brown LN, Biswas DD, Kordula T. RelB Acts as a Molecular Switch Driving Chronic Inflammation in Glioblastoma Multiforme. Oncogenesis (2019) 8(6):37. doi: 10.1038/s41389-019-0146-y
91. Matias D, Dubois LG, Pontes B, Rosario L, Ferrer VP, Balca-Silva J, et al. GBM-Derived Wnt3a Induces M2-Like Phenotype in Microglial Cells Through Wnt/beta-Catenin Signaling. Mol Neurobiol (2019) 56(2):1517–30. doi: 10.1007/s12035-018-1150-5
92. Sica A, Mantovani A. Macrophage Plasticity and Polarization: In Vivo Veritas. J Clin Invest (2012) 122(3):787–95. doi: 10.1172/JCI59643
93. Dou Z, Ghosh K, Vizioli MG, Zhu J, Sen P, Wangensteen KJ, et al. Cytoplasmic Chromatin Triggers Inflammation in Senescence and Cancer. Nature (2017) 550(7676):402–6. doi: 10.1038/nature24050
94. Burgess R, Jenkins R, Zhang Z. Epigenetic Changes in Gliomas. Cancer Biol Ther (2008) 7(9):1326–34. doi: 10.4161/cbt.7.9.6992
95. Liu Z, Ren Y, Meng L, Li L, Beatson R, Deng J, et al. Epigenetic Signaling of Cancer Stem Cells During Inflammation. Front Cell Dev Biol (2021) 9:772211. doi: 10.3389/fcell.2021.772211
96. Stazi G, Taglieri L, Nicolai A, Romanelli A, Fioravanti R, Morrone S, et al. Dissecting the Role of Novel EZH2 Inhibitors in Primary Glioblastoma Cell Cultures: Effects on Proliferation, Epithelial-Mesenchymal Transition, Migration, and on the Pro-Inflammatory Phenotype. Clin Epigenetics (2019) 11(1):173. doi: 10.1186/s13148-019-0763-5
97. Hasan T, Caragher SP, Shireman JM, Park CH, Atashi F, Baisiwala S, et al. Interleukin-8/CXCR2 Signaling Regulates Therapy-Induced Plasticity and Enhances Tumorigenicity in Glioblastoma. Cell Death Dis (2019) 10(4):292. doi: 10.1038/s41419-019-1387-6
98. Venza I, Visalli M, Fortunato C, Ruggeri M, Ratone S, Caffo M, et al. PGE2 Induces Interleukin-8 Derepression in Human Astrocytoma Through Coordinated DNA Demethylation and Histone Hyperacetylation. Epigenetics (2012) 7(11):1315–30. doi: 10.4161/epi.22446
99. Yeung YT, Bryce NS, Adams S, Braidy N, Konayagi M, McDonald KL, et al. P38 MAPK Inhibitors Attenuate Pro-Inflammatory Cytokine Production and the Invasiveness of Human U251 Glioblastoma Cells. J Neurooncol (2012) 109(1):35–44. doi: 10.1007/s11060-012-0875-7
100. Li Z, Zhang J, Zheng H, Li C, Xiong J, Wang W, et al. Modulating lncRNA SNHG15/CDK6/miR-627 Circuit by Palbociclib, Overcomes Temozolomide Resistance and Reduces M2-Polarization of Glioma Associated Microglia in Glioblastoma Multiforme. J Exp Clin Cancer Res (2019) 38(1):380. doi: 10.1186/s13046-019-1371-0
101. Kaminska B, Mota M, Pizzi M. Signal Transduction and Epigenetic Mechanisms in the Control of Microglia Activation During Neuroinflammation. Biochim Biophys Acta (2016) 1862(3):339–51. doi: 10.1016/j.bbadis.2015.10.026
102. Nguyen TTT, Zhang Y, Shang E, Shu C, Torrini C, Zhao J, et al. HDAC Inhibitors Elicit Metabolic Reprogramming by Targeting Super-Enhancers in Glioblastoma Models. J Clin Invest (2020) 130(7):3699–716. doi: 10.1172/JCI129049
103. van der Vos KE, Abels ER, Zhang X, Lai C, Carrizosa E, Oakley D, et al. Directly Visualized Glioblastoma-Derived Extracellular Vesicles Transfer RNA to Microglia/Macrophages in the Brain. Neuro Oncol (2016) 18(1):58–69. doi: 10.1093/neuonc/nov244
104. Chuang HY, Su YK, Liu HW, Chen CH, Chiu SC, Cho DY, et al. Preclinical Evidence of STAT3 Inhibitor Pacritinib Overcoming Temozolomide ResistanceVia Downregulating miR-21-Enriched Exosomes From M2 Glioblastoma-Associated Macrophages. J Clin Med (2019) 8(7):959. doi: 10.3390/jcm8070959
105. Hong S, You JY, Paek K, Park J, Kang SJ, Han EH, et al. Inhibition of Tumor Progression and M2 Microglial Polarization by Extracellular Vesicle-Mediated microRNA-124 in a 3D Microfluidic Glioblastoma Microenvironment. Theranostics (2021) 11(19):9687–704. doi: 10.7150/thno.60851
106. Wenger A, Ferreyra Vega S, Kling T, Bontell TO, Jakola AS, Caren H. Intratumor DNA Methylation Heterogeneity in Glioblastoma: Implications for DNA Methylation-Based Classification. Neuro Oncol (2019) 21(5):616–27. doi: 10.1093/neuonc/noz011
107. Zhou D, Alver BM, Li S, Hlady RA, Thompson JJ, Schroeder MA, et al. Distinctive Epigenomes Characterize Glioma Stem Cells and Their Response to Differentiation Cues. Genome Biol (2018) 19(1):43. doi: 10.1186/s13059-018-1420-6
108. Wang QW, Liu HJ, Zhao Z, Zhang Y, Wang Z, Jiang T, et al. Prognostic Correlation of Autophagy-Related Gene Expression-Based Risk Signature in Patients With Glioblastoma. Onco Targets Ther (2020) 13:95–107. doi: 10.2147/OTT.S238332
109. Zhang H, Xu Y, Deng G, Yuan F, Tan Y, Gao L, et al. SAA1 Knockdown Promotes the Apoptosis of Glioblastoma Cells via Downregulation of AKT Signaling. J Cancer (2021) 12(9):2756–67. doi: 10.7150/jca.48419
110. Piperi C, Themistocleous MS, Papavassiliou GA, Farmaki E, Levidou G, Korkolopoulou P, et al. High Incidence of MGMT and RARbeta Promoter Methylation in Primary Glioblastomas: Association With Histopathological Characteristics, Inflammatory Mediators and Clinical Outcome. Mol Med (2010) 16(1-2):1–9. doi: 10.2119/molmed.2009.00140
111. Xie Q, Wu TP, Gimple RC, Li Z, Prager BC, Wu Q, et al. N(6)-Methyladenine DNA Modification in Glioblastoma. Cell (2018) 175(5):1228–43.e20. doi: 10.1016/j.cell.2018.10.006
112. Zhang S, Zhao BS, Zhou A, Lin K, Zheng S, Lu Z, et al. M(6)A Demethylase ALKBH5 Maintains Tumorigenicity of Glioblastoma Stem-Like Cells by Sustaining FOXM1 Expression and Cell Proliferation Program. Cancer Cell (2017) 31(4):591–606.e6. doi: 10.1016/j.ccell.2017.02.013
113. Wang L, Xia JW, Ke ZP, Zhang BH. Blockade of NEAT1 Represses Inflammation Response and Lipid Uptake via Modulating miR-342-3p in Human Macrophages THP-1 Cells. J Cell Physiol (2019) 234(4):5319–26. doi: 10.1002/jcp.27340
114. Zhang P, Cao L, Zhou R, Yang X, Wu M. The lncRNA Neat1 Promotes Activation of Inflammasomes in Macrophages. Nat Commun (2019) 10(1):1495. doi: 10.1038/s41467-019-09482-6
115. Butler M, Pongor L, Su YT, Xi L, Raffeld M, Quezado M, et al. MGMT Status as a Clinical Biomarker in Glioblastoma. Trends Cancer (2020) 6(5):380–91. doi: 10.1016/j.trecan.2020.02.010
116. Lawrence M, Daujat S, Schneider R. Lateral Thinking: How Histone Modifications Regulate Gene Expression. Trends Genet (2016) 32(1):42–56. doi: 10.1016/j.tig.2015.10.007
117. Yang R, Wang M, Zhang G, Bao Y, Wu Y, Li X, et al. E2F7-EZH2 Axis Regulates PTEN/AKT/mTOR Signalling and Glioblastoma Progression. Br J Cancer (2020) 123(9):1445–55. doi: 10.1038/s41416-020-01032-y
118. Cohen AL, Piccolo SR, Cheng L, Soldi R, Han B, Johnson WE, et al. Genomic Pathway Analysis Reveals That EZH2 and HDAC4 Represent Mutually Exclusive Epigenetic Pathways Across Human Cancers. BMC Med Genomics (2013) 6:35. doi: 10.1186/1755-8794-6-35
119. Yang WB, Hsu CC, Hsu TI, Liou JP, Chang KY, Chen PY, et al. Increased Activation of HDAC1/2/6 and Sp1 Underlies Therapeutic Resistance and Tumor Growth in Glioblastoma. Neuro Oncol (2020) 22(10):1439–51. doi: 10.1093/neuonc/noaa103
120. Larionova I, Kazakova E, Patysheva M, Kzhyshkowska J. Transcriptional, Epigenetic and Metabolic Programming of Tumor-Associated Macrophages. Cancers (Basel) (2020) 12(6):1411. doi: 10.3390/cancers12061411
121. Daskalaki MG, Tsatsanis C, Kampranis SC. Histone Methylation and Acetylation in Macrophages as a Mechanism for Regulation of Inflammatory Responses. J Cell Physiol (2018) 233(9):6495–507. doi: 10.1002/jcp.26497
122. Cenciarini M, Valentino M, Belia S, Sforna L, Rosa P, Ronchetti S, et al. Dexamethasone in Glioblastoma Multiforme Therapy: Mechanisms and Controversies. Front Mol Neurosci (2019) 12:65. doi: 10.3389/fnmol.2019.00065
123. Czimmerer Z, Daniel B, Horvath A, Ruckerl D, Nagy G, Kiss M, et al. The Transcription Factor STAT6 Mediates Direct Repression of Inflammatory Enhancers and Limits Activation of Alternatively Polarized Macrophages. Immunity (2018) 48(1):75–90.e6. doi: 10.1016/j.immuni.2017.12.010
124. Yin Y, Qiu S, Li X, Huang B, Xu Y, Peng Y. EZH2 Suppression in Glioblastoma Shifts Microglia Toward M1 Phenotype in Tumor Microenvironment. J Neuroinflamm (2017) 14(1):220. doi: 10.1186/s12974-017-0993-4
125. He L, Jhong JH, Chen Q, Huang KY, Strittmatter K, Kreuzer J, et al. Global Characterization of Macrophage Polarization Mechanisms and Identification of M2-Type Polarization Inhibitors. Cell Rep (2021) 37(5):109955. doi: 10.1016/j.celrep.2021.109955
126. Ghiboub M, Zhao J, Li Yim AYF, Schilderink R, Verseijden C, van Hamersveld PHP, et al. HDAC3 Mediates the Inflammatory Response and LPS Tolerance in Human Monocytes and Macrophages. Front Immunol (2020) 11:550769. doi: 10.3389/fimmu.2020.550769
127. Buendia Duque M, Pinheiro KV, Thomaz A, da Silva CA, Freire NH, Brunetto AT, et al. Combined Inhibition of HDAC and EGFR Reduces Viability and Proliferation and Enhances STAT3 mRNA Expression in Glioblastoma Cells. J Mol Neurosci (2019) 68(1):49–57. doi: 10.1007/s12031-019-01280-5
128. Esteller M. Non-Coding RNAs in Human Disease. Nat Rev Genet (2011) 12(12):861–74. doi: 10.1038/nrg3074
129. Matsui M, Corey DR. Non-Coding RNAs as Drug Targets. Nat Rev Drug Discov (2017) 16(3):167–79. doi: 10.1038/nrd.2016.117
130. DeOcesano-Pereira C, Machado RAC, Chudzinski-Tavassi AM, Sogayar MC. Emerging Roles and Potential Applications of Non-Coding RNAs in Glioblastoma. Int J Mol Sci (2020) 21(7):2611. doi: 10.3390/ijms21072611
131. Hubner M, Moellhoff N, Effinger D, Hinske CL, Hirschberger S, Wu T, et al. MicroRNA-93 Acts as an “Anti-Inflammatory Tumor Suppressor” in Glioblastoma. Neurooncol Adv (2020) 2(1):vdaa047. doi: 10.1093/noajnl/vdaa047
132. Wu X, Wang Y, Yu T, Nie E, Hu Q, Wu W, et al. Blocking MIR155HG/miR-155 Axis Inhibits Mesenchymal Transition in Glioma. Neuro Oncol (2017) 19(9):1195–205. doi: 10.1093/neuonc/nox017
133. Cui B, Chen L, Zhang S, Mraz M, Fecteau JF, Yu J, et al. MicroRNA-155 Influences B-Cell Receptor Signaling and Associates With Aggressive Disease in Chronic Lymphocytic Leukemia. Blood (2014) 124(4):546–54. doi: 10.1182/blood-2014-03-559690
134. Tarassishin L, Lee SC. Interferon Regulatory Factor 3 Alters Glioma Inflammatory and Invasive Properties. J Neurooncol (2013) 113(2):185–94. doi: 10.1007/s11060-013-1109-3
135. Wu W, Yu T, Wu Y, Tian W, Zhang J, Wang Y. The Mir155hg/miR-185/ANXA2 Loop Contributes to Glioblastoma Growth and Progression. J Exp Clin Cancer Res (2019) 38(1):133. doi: 10.1186/s13046-019-1132-0
Keywords: glioblastoma, inflammation, microenvironment, epigenetic regulation, GBM tumor cells, glioma-associated microglia/macrophages
Citation: Chen N, Peng C and Li D (2022) Epigenetic Underpinnings of Inflammation: A Key to Unlock the Tumor Microenvironment in Glioblastoma. Front. Immunol. 13:869307. doi: 10.3389/fimmu.2022.869307
Received: 04 February 2022; Accepted: 28 March 2022;
Published: 29 April 2022.
Edited by:
Guan-Jun Yang, Ningbo University, ChinaReviewed by:
Shengpeng Wang, University of Macau, Macau, SAR ChinaCopyright © 2022 Chen, Peng and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Cheng Peng, cGVuZ2NoZW5nX2NkQDEyNi5jb20=; Dan Li, bGlkYW5AY2R1dGNtLmVkdS5jbg==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.