
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Immunol. , 24 March 2022
Sec. T Cell Biology
Volume 13 - 2022 | https://doi.org/10.3389/fimmu.2022.867443
This article is part of the Research Topic Molecular Switches of the Immune System: The E Protein/Id Axis in Hematopoietic Development and Function View all 11 articles
 Diogo F. T. Veiga1,2†
Diogo F. T. Veiga1,2† Mathieu Tremblay1†
Mathieu Tremblay1† Bastien Gerby1,3
Bastien Gerby1,3 Sabine Herblot1,4André Haman1Patrick Gendron1
Sabine Herblot1,4André Haman1Patrick Gendron1 Sébastien Lemieux1,5
Sébastien Lemieux1,5 Juan Carlos Zúñiga-Pflücker6
Juan Carlos Zúñiga-Pflücker6 Josée Hébert1,7,8,9Joseph Paul Cohen10,11
Josée Hébert1,7,8,9Joseph Paul Cohen10,11 Trang Hoang1*
Trang Hoang1*Early T-cell development is precisely controlled by E proteins, that indistinguishably include HEB/TCF12 and E2A/TCF3 transcription factors, together with NOTCH1 and pre-T cell receptor (TCR) signalling. Importantly, perturbations of early T-cell regulatory networks are implicated in leukemogenesis. NOTCH1 gain of function mutations invariably lead to T-cell acute lymphoblastic leukemia (T-ALL), whereas inhibition of E proteins accelerates leukemogenesis. Thus, NOTCH1, pre-TCR, E2A and HEB functions are intertwined, but how these pathways contribute individually or synergistically to leukemogenesis remain to be documented. To directly address these questions, we leveraged Cd3e-deficient mice in which pre-TCR signaling and progression through β-selection is abrogated to dissect and decouple the roles of pre-TCR, NOTCH1, E2A and HEB in SCL/TAL1-induced T-ALL, via the use of Notch1 gain of function transgenic (Notch1ICtg) and Tcf12+/- or Tcf3+/- heterozygote mice. As a result, we now provide evidence that both HEB and E2A restrain cell proliferation at the β-selection checkpoint while the clonal expansion of SCL-LMO1-induced pre-leukemic stem cells in T-ALL is uniquely dependent on Tcf12 gene dosage. At the molecular level, HEB protein levels are decreased via proteasomal degradation at the leukemic stage, pointing to a reversible loss of function mechanism. Moreover, in SCL-LMO1-induced T-ALL, loss of one Tcf12 allele is sufficient to bypass pre-TCR signaling which is required for Notch1 gain of function mutations and for progression to T-ALL. In contrast, Tcf12 monoallelic deletion does not accelerate Notch1IC-induced T-ALL, indicating that Tcf12 and Notch1 operate in the same pathway. Finally, we identify a tumor suppressor gene set downstream of HEB, exhibiting significantly lower expression levels in pediatric T-ALL compared to B-ALL and brain cancer samples, the three most frequent pediatric cancers. In summary, our results indicate a tumor suppressor function of HEB/TCF12 in T-ALL to mitigate cell proliferation controlled by NOTCH1 in pre-leukemic stem cells and prevent NOTCH1-driven progression to T-ALL.
Thymocyte reprogramming into self-renewing cells is a mandatory event in T-cell leukemogenesis, induced by aberrantly expressed oncogenic transcription factors (1–6). This initiating event sets a pre-leukemic state, while progression to overt leukemia requires additional collaborating events within pathways that control cell fate in the thymus, to evolve through layers of selective pressure (7–10).
The first acquisition of full T-lineage identity is marked by successful rearrangement of the T cell receptor (Tcr) β locus catalyzed by recombination-activating gene 1 (RAG1) and RAG2 at the CD4/CD8 double negative DN2-DN3 transitional stages (Figure 1A). The β-selection checkpoint is controlled by the pre-TCR resulting from the pairing of the successfully rearranged TCR β chain with the invariant pre-Tα chain and the CD3 signaling complex to trigger a burst of cell proliferation and survival, leading to differentiation of DN thymocytes to the CD4+CD8+ double-positive (DP) stage. Both the pre-TCR and NOTCH1 have obligatory functions at this first checkpoint (11–13). Gain of function mutations of NOTCH1 are found in more than 55% of childhood T-ALL (14), leading to the well-accepted notion that NOTCH1 is a major oncogenic event in T-ALL (15–17). The acquisition of Notch1 mutations in T-ALL absolutely requires pre-TCR/CD3 signaling (4) and involves recombination activating enzymes (RAG1/2) (18). Additionally, the NOTCH1 pathway can also be hyperactive as a consequence of loss of function mutations of FBXW7, the E3 ligase that degrades MYC (19), an essential downstream target of NOTCH1 (20, 21). Nonetheless, the NOTCH-MYC-FBXW7 triad appears to be genetically unaltered in ~1/3 T-ALL cases, raising the question whether additional genes or pathways may contribute to T-ALL progression.
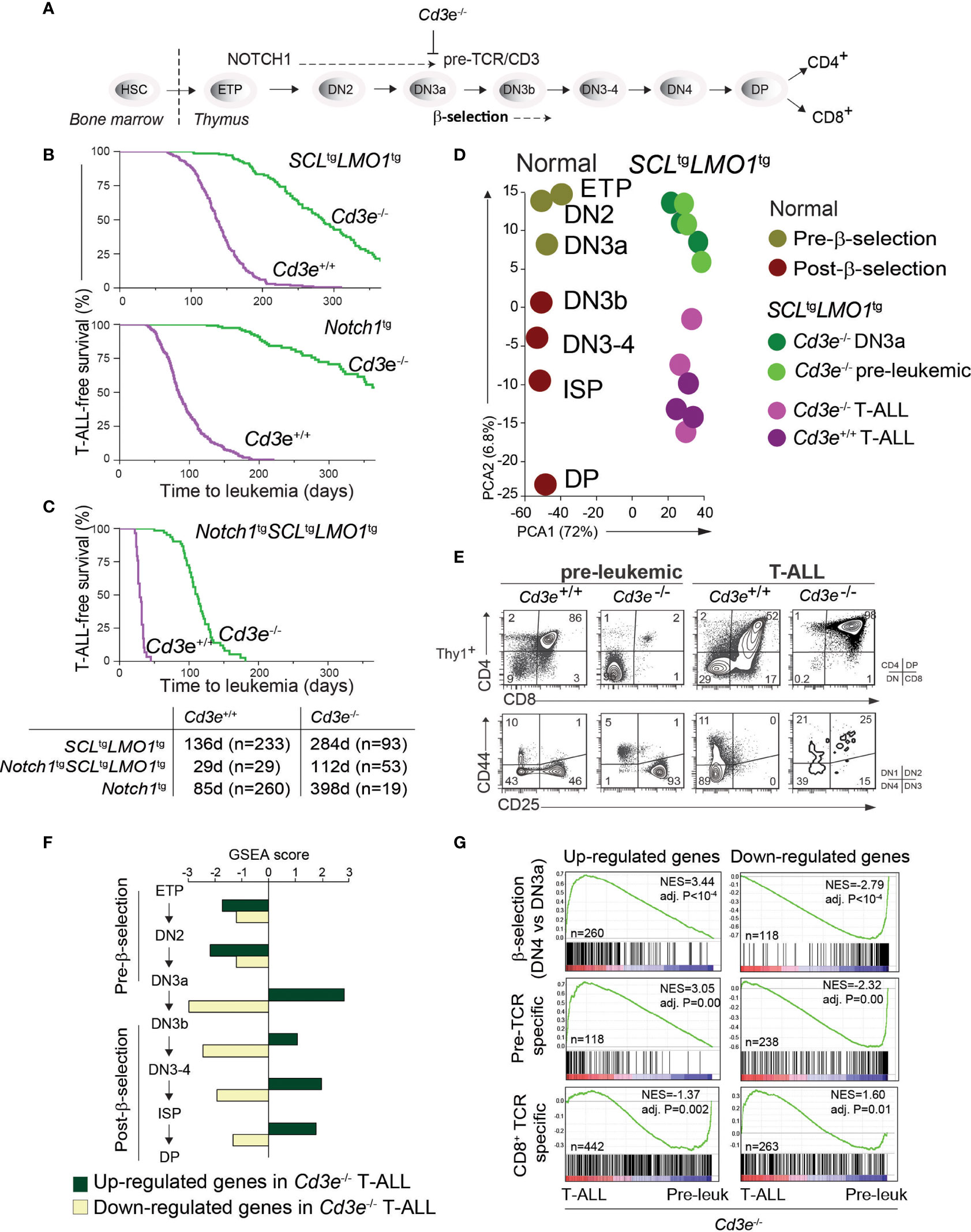
Figure 1 Pre-TCR signaling is functionally important for T-ALL progression. (A) Pre-TCR signalling and thymocytes development. (B, C) Kaplan-Meyer survival curves comparing disease development in the pre-TCR proficient (Cd3e+/+) and deficient (Cd3e-/-) backgrounds in three models of T-ALL. (D) Principal component analysis of the transcriptomes of normal thymocyte subsets compared to SCL-LMO1-induced pre-leukemic and leukemic (T-ALL) thymocytes. (E) FACS phenotypes of SCLtgLMO1tg thymocytes from Cd3e+/+ and Cd3e-/- backgrounds at pre-leukemic and leukemic stages. (F) Gene set enrichment analysis (GSEA) correlates disease progression to stages of thymocyte differentiation. Up-regulated and down-regulated gene sets were computed at each stage of normal thymocyte development from ETP to DP using microarray data from the Immgen project (http://www.immgen.org/), and enrichment was tested during disease progression (pre-leukemia to leukemia). Dark green bars denote enrichment of the up-regulated signature, and yellow bars denote enrichment of down-regulated signatures. (G) GSEA analysis of β-selection, pre-TCR specific and CD8+ TCR specific gene signatures during disease progression. Left panels show the enrichment tests for up-regulated gene signatures, and right panels show enrichment of genes decreased by β-selection, pre-TCR and CD8+ TCR.
Comprehensive high throughput sequencing have unravelled the genomic landscape of T-ALL in children (22–24) and adults (25), uncovering a low mutation burden in leukemias compared to solid tumors (26, 27). These studies identified recurring mutations within genes and pathways that control cell fate in thymocytes, confirming the dominant presence of NOTCH1 as a driver mutation. Unlike NOTCH1, oncogenic transcription factors in T-ALL are not mutated but aberrantly expressed in the T lineage driven by chromosomal translocations. These oncogenic transcription factors belong to two families, the basic helix-loop-helix family (SCL/TAL1, TAL2, LYL1) and associated partners (LMO1, LMO2), as well as homeodomain proteins (TLX1, TLX3, HOXA) (reviewed in (28, 29). Transgenic mice in which oncogene expression is driven in the thymus develop T-ALL with variable latency, indicating the necessary acquisition of collaborating events. This is illustrated by the loss of Bcl11b function, through transcription repression by the TLX1 oncogene or through mono-allelic deletion in mouse models (30) and in 9% of human T-ALL (31).
Similar to BCL11B (32), both E2A (E12 and E47 (33)), and HEB (HEBCan and HEBAlt (34)) are essential for the commitment of progenitor thymocytes to the T-cell lineage (35, 36) by governing a gene expression program that is critical for T-cell development [(37), reviewed in (38)] and includes T-cell specific genes such as Ptcra and Cd4 (39, 40), as well as cell cycle genes such as Cdkn1a (37, 41). Moreover, E2A is antiproliferative in thymocytes (42–44) and E2A-deficient mice develop T-cell lymphomas (42, 45), indicating that E2A has tumor suppressor functions, much like BCL11B. Nonetheless and unlike BCL11B, neither E2A/TCF3 nor HEB/TCF12 was found mutated or affected by copy number variations in human T-ALL (10, 25), raising the possibility of non-genetic inactivation of E2A or HEB that has so far escaped genomic studies. E protein activity can be inhibited by direct heterodimerization with Id proteins, members of the HLH family that lack DNA binding domains (37) or by the SCL and LYL1 oncoproteins (39, 40, 46, 47). Nonetheless, inhibition of E protein by SCL is insufficient for T-cell leukemogenesis which requires transcription activation of a stemness gene expression program by the SCL-LMO1 complex (2) or LMO2 (48, 49). Finally, O’Neil et al. have previously shown a genetic collaboration between Tcf12 or Tcf3-deficiency and SCL/TAL1 in accelerating T-ALL onset (47). Because Heb deficiency would cause reduced pre-TCR expression (50) and decreased cell proliferation (51), it remains to be documented how this would accelerate T-ALL onset. In summary, while it is well recognized that E2A can be a tumor suppressor in mouse models, it is not clear whether E2A or HEB is inactivated in human T-ALL and how inactivation may occur, given the essential and dosage-dependent role of E2A and HEB in the T lineage (50–52).
Given the intricate interaction between NOTCH1, pre-TCR signaling and E proteins, we elected to use Cd3e-/- mice as a powerful genetic model to dissect and decouple the roles of NOTCH1, pre-TCR and HEB in leukemia progression, specifically in DN3 thymocytes, previously shown to be the cell of origin of SCL-LMO1 (2, 4) and LMO2 (48) -induced T-ALL. Thus, by abrogating β-selection and analyzing Notch1 gain of function and Tcf12 loss of function individually, our results unravel a strong selective pressure for down regulation of HEB protein levels driven either by NOTCH1 and/or by pre-TCR signaling as a requirement for progression from the pre-leukemic state to overt T-ALL.
All animals were kept on a C57BL6/J strain background and maintained in pathogen-free conditions according to institutional animal care and use guidelines. Lck-NotchIC9 (Notch1tg) (53), SIL-SCL (A (5)3SCL; SCLtg) (54), Lck-LMO1 (LMO1tg) (55), Cd3e-/- (56), E2a/Tcf3+/- (57) and Heb/Tcf12+/- (52) were described previously. Kaplan-Meier survival and statistical analysis was performed using GraphPad Prism 9.0 software (GraphPad Software, Inc.). T-ALL susceptibility was computed from areas under the curve (AUC) of Kaplan–Meier survival curves using Prism (1-AUC). Generation of human xenograft T-ALL blasts (14H025 and 14H148) were described previously (58). Human blast and primary murine thymocytes were cultured in MEM Alpha culture medium (Thermo Fisher) supplemented with 10% FBS, 10mM HEPES, 1mM sodium pyruvate, 55 μM β-mercaptoethanol, 2 mM glutamax, 5 ng/mL human FLT-3 Ligand, 5 ng/mL murine IL-7 and 20 ng/mL murine SCF. The DN T-cell line AD10.1 and Jurkat were cultured as previously described (50). KOPT-K1 and P12-ICHIKAWA cell lines were obtained from the DSMZ collection, Germany and maintained in RPMI-1640 culture media supplemented with 10% Fetal Bovine Serum (FBS).
Single-cell suspensions were prepared from thymi or thymoma of mice. Flow cytometry analysis and cell sorting were done as described previously (39) using antibodies against Thy1.2, CD4, CD8, CD25 and CD44, using propidium iodide to exclude dead cells. Multiparametric flow cytometry analysis was performed on a FORTESSA flow cytometer, and cell sorting was performed on FACSAria (BD Biosciences, San Jose, CA). Cell cycle analysis using DAPI staining was performed using ModFit (Verity Software Software, USA).
For Cdnk1a gene expression analysis, DN thymocytes from wt and Heb/Tcf12-/- newborn mice were sorted by flow cytometry and cDNAs were prepared as described previously (39). Southern blots of the amplicons were revealed by hybridization using an internal 32P-labeled oligonucleotide fragment (primer sequences are listed in Supplementary Table l). The ribosomal Rps16 expression was used as a control for cDNA quality and quantity.
For Notch1 sequencing, cDNA was prepared from total RNAs as described previously (39). Amplification of Notch1 exons 26, 27, and 34 from leukemias cDNA were Sanger sequenced in both directions. Quantitative gene expression analysis was performed on StepOne system (Life Technologies) using specific primers and Advanced qPCR mix (Wisent). Primer sequences used for specific mRNA amplification are listed in Supplemental Table S1.
Cells were lysed in RIPA buffer containing a cocktail of protease inhibitors. Protein extracts were resolved on bis-acrylamide gel, transferred on PVDF membranes and hybridized with anti-HEB and anti-E2A (Santa-Cruz Biotechnology Inc., CA) and anti-tubulin-β (Sigma) and anti-ERK (Cell Signaling) as a loading control.
Chromatin immunoprecipitation were performed on either Cd3e-/- primary thymocytes or AD10.1 extracts as previously described (50). Quantitative PCR was performed on StepOne system (Life Technologies) using specific primers using Advanced qPCR mix (Wisent). Oligonucleotide sequences used for promoter amplification are shown in Supplementary Table 1.
Total RNAs were prepared from freshly isolated thymocytes from Cd3e-/- (control DN3 thymocytes), Cd3-/-SCLtgLMO1tg T-ALL and pre-leukemic (3-week-old), and Cd3e+/+SCLtgLMO1tg leukemic mice using the RNeasy extraction kit (Qiagen, Mississauga, ON). cDNA synthesis, labeling and hybridization onto Affymetrix Mouse Genome 430A 2.0 arrays were performed at the Ottawa Health Research Institute (Ottawa, ON) as described (2). Raw data were normalized using the RMA procedure implemented in the Affy package from Bioconductor (59).
We obtained raw microarray data for normal thymocyte populations generated by the Immgen project from GEO (accession number GSE15907). Data were normalized using the RMA procedure implemented in the Affy package from Bioconductor (59). We derived “transition” signatures for each differentiation step (i.e. ETP to DN2, DN2 to DN3a, etc.), which contained genes whose expression levels present with at least a 2-fold change (up or down-regulated) in the transition (gene signatures provided in Supplementary File 1). In Figure 1F, gene set enrichment analysis (60) was applied to detect transition signatures that are enriched in the transcriptome of Cd3e-/-SCLtgLMO1tg and Cd3e+/+SCLtgLMO1tg leukemic cells.
Pre-TCR specific signatures (Figure 1G, Supplementary File 1) included genes that increased or decreased at least 2-fold during the DN3a-DN3b transition, and are not regulated by the αβTCR in peripheral CD8+ T cells stimulated by antigen (naive versus activated CD8+ T cells). Conversely, TCR-specific signatures (Figure 1G, Supplementary File 1) included genes exclusively regulated by the αβTCR in activated CD8+ T cells.
We collected genome-wide chromatin occupancy data for 7 transcription factors implicated in pre-TCR signalling (11 ChIP-seq experiments in total, Figure 2A) from Wang et al. (61), Miyazaki et al. (37), and the HemoChIP project (62). ChIP-seq data obtained for E2A (DN3 and DN4) (37) and NOTCH1 (G4A2 and T6E murine cell lines) (61) were processed according to the following steps: (i) sequence reads were mapped to the mouse genome mm9 using Bowtie with default parameters (maximum 2 mismatches); and (ii) peak coordinates were determined by the MACS tool, using the cutoff P < 10–9. Peak coordinates for the HemoChIP dataset mapped to the mouse genome mm9 were downloaded from http://hscl.cimr.cam.ac.uk/ChIP-Seq_Compendium/ChIP- Seq_Compendium2.html. Last, all peaks were associated to their closest transcription start sites in the mouse genome using PeakAnalyzer v.1.4 tool (63). Lists of targets bound by transcription factors included all genes containing at least one binding site for the regulator (Supplementary File 1). We tested enrichment of targets using the Fisher’s exact test (Figure 2A).
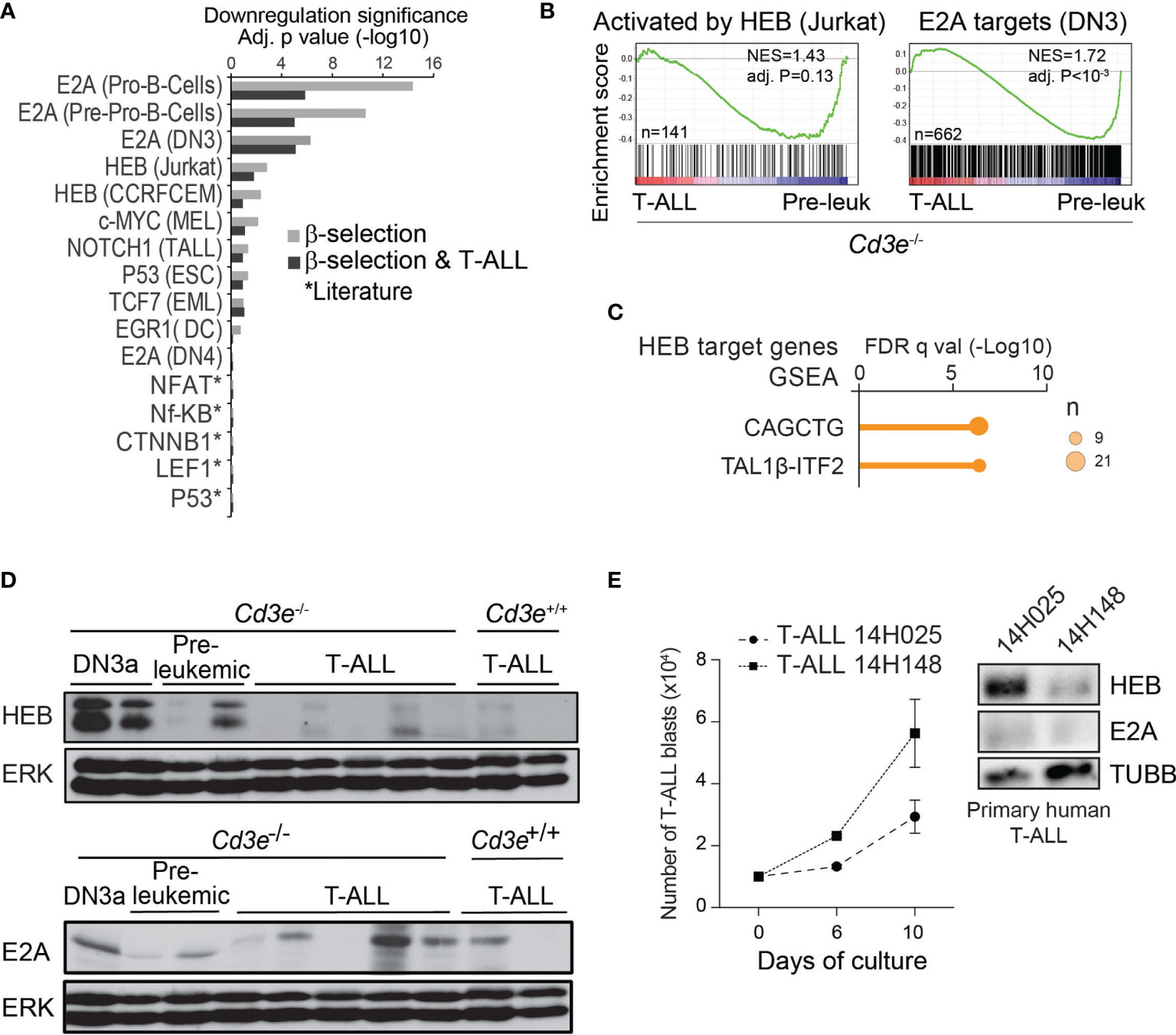
Figure 2 T-ALL progression associated with an inhibition of E proteins targets and downregulation of HEB protein. (A) Regulator analysis identified transcription factors associated to transcriptional repression during T-ALL progression and β-selection. Regulators are rank-ordered according to their enrichment scores. The bars display the downregulation significance (log10 adjusted P, Fisher’s exact test). Targets were extracted from ChIP-seq datasets (cell types in parenthesis). (B) GSEA analysis of genes activated by HEB and E2A targets during progression from pre-leukemic to leukemic state in Cd3e-deficient SCLtgLMO1tg T-ALL. (C) Binding site motif enrichment (MSig) within the proximal promoter regions of HEB target genes down-regulated between Cd3e-/- leukemic and pre-leukemic cells. (D) HEB and E2A protein levels during T-ALL progression measured by Western blotting. (E) Growth in vitro for primary T‐ALL samples (left panel). Western blot analysis of theindicated T-ALL samples (right panel).
DNA was extracted from Cd3e-/-SCLtgLMO1tg leukemias (n=4) and control Cd3e-/- thymocytes (n=2), followed by targeted exome enrichment was performed using the mouse Nimblegen SeqCap EZ kit from Roche. Sequencing was performed in the Illumina HiSeq2000 at the IRIC Genomics platform. Low quality bases (quality below 20) in paired-end reads were trimmed off using the Trimmomatic tool (64), duplicate reads were removed using Picard (http://broadinstitute.github.io/picard), and alignment to the mouse genome (mm10) was performed with bwa (65). The resulting depth of coverage was 20X for at least 85% of the captured exome across all samples. Exome variants in T-ALL samples were predicted using Strelka (66), using Cd3e-/- thymocytes as matching controls, and annotation was performed using ANNOVAR (67). SIFT scores to determine deleterious variants were computed using the Variant Effect Predictor tool (68).
RNA extracted from Cd3e-/-SCLtgLMO1tg leukemias (n=4) and control Cd3e-/- thymocytes (n=2) was prepared using the TruSeq RNA kit (Illumina) and sequenced in the Illumina HiSeq2000 in the IRIC Genomics platform. Low quality bases (quality below 20) in paired-end reads were trimmed off using the Trimmomatic, and processed reads were aligned to the mouse genome mm10 using Tophat2/Bowtie2 v2.0.7 (69). Gene levels were quantified (FPKM values) based on the UCSC reference genes annotation using cuffdiff v.2.1.1.
RNA-seq data from pediatric tumors including a minimum of 313 T-ALL, 720 B-ALL and 350 Brain tumor samples were accessible for data analysis via the St. Jude PeCan data portal (https://pecan.stjude.cloud) (70) using Protein paint to capture RNA expression from the above ALL datasets (71).
The first critical event in leukemogenesis is the reprogramming of DN3 thymocytes into pre-leukemic stem cells (pre-LSCs) by the SCL-LMO1 oncogenes (2). These two oncogenes target the DN3 population (4, 72) but are not sufficient per se for progression to T-ALL. While the initiating reprogramming event is pre-TCR independent (2), progression to T-ALL requires both pre-TCR and NOTCH1 signaling (4), thus emulating the requirement for cooperative signaling between the two important pathways for normal thymocyte differentiation (11). To define the precise contribution of each pathway to disease progression, we took a genetic approach to quantitatively estimate T-ALL progression using disease penetrance and the time to leukemia onset as endpoints to measure leukemogenesis. T-ALL induced by SCLtgLMO1tg or Notch1tg separately is affected by the absence of pre-TCR/CD3 signaling (Figure 1B) as previously reported (4, 72, 73). In contrast, SCL-LMO1 together with the hyperactive Notch1 allele (ICN1, hereafter Notch1tg), induce T-ALL with full penetrance in the presence or in the absence of Cd3e (Figure 1C). Nonetheless, in Cd3e-proficient mice, pre-TCR/CD3 signaling accelerates the disease to 29 days, compared to 112 days in Cd3e-deficient mice. These results indicate that Notch1tg drives the penetrance of T-ALL while the pre-TCR determines the time to leukemia. Of note, T-ALL induced by the three oncogenes together in the absence of Cd3e reproduce the disease induced by the two transcription factor oncogenes SCL and LMO1 in a Cd3e-proficient background (Figure S1). Therefore, in the context of T-ALL induced by the SCL and LMO1 oncogenes, the Notch1 transgene controls disease penetrance while pre-TCR signaling accelerates disease onset.
While pre-TCR signaling has been known to be important for leukemogenesis (74, 75) and more specifically for SCL-LMO1-induced T-ALL (4, 76), the contribution of the pre-TCR and downstream molecular effectors remain to be uncovered. T-ALL that still develops in the absence of CD3 or RAG, completely lacked the typical Notch1 gain of function mutations (4), providing us with a unique genetic tool to dissect the contribution of these pathways to T-ALL.
We therefore conducted a transcriptomic analysis that capitalized on our identification of DN3a as the cell of origin of T-ALL, and in Cd3e-/- mice in which pre-TCR signaling is abrogated, causing thymocyte differentiation blockade at the DN3a stage. This allows for a stringent comparison between the pre-leukemic and the leukemic state to define the molecular signature of progression. During the pre-leukemic stage, Cd3e-/-SCLtgLMO1tg thymocytes are blocked at the DN3a stage, as expected from the absence of CD3.
We next compared the transcriptomes of pre-leukemic (n=3) and of SCLtgLMO1tg leukemic cells (n=6) (Figure 1D). We applied principal component analysis (PCA) to compare the transcriptomes of normal thymocyte populations (obtained from the Immgen project) with pre-leukemic and leukemic samples. The first component reflected the distinct Affymetrix chips used for profiling whereas the second PCA component organized the transcriptomes according to their differentiation trajectories, from ETP to DP cells. Overall, PCA showed that pre-leukemic samples were comparable to the Cd3ϵ-/- DN3a thymocytes. Strikingly, progression to T-ALL correlates with the expression profiles of thymocytes that have undergone β-selection, despite the absence of pre-TCR signaling in Cd3ϵ-/- mice. Consistent with PCA, we observed that leukemic cells acquired a post-β-selection phenotype to become DN3b-DP cells (98% of the thymic mass), compared to 2% at the pre-leukemic stage, despite the complete lack of normal pre-TCR function in Cd3e-/- mice (Figure 1E).
We also correlated disease progression with gene signatures of thymocyte differentiation using gene set enrichment analysis (GSEA). We observed that only signatures associated to post-β-selection thymocytes (DN3b to DN4 cells) were positively correlated with T-ALL progression (Figure 1F). The strongest correlation was associated with the DN3a-DN3b transition, exactly at the stage where the pre-TCR/CD3 triggers a burst of cell proliferation following a productive TCRβ rearrangement and the formation of a functional pre-TCR/CD3 complex.
CD3 signaling is important for pre-TCR function and β-selection, but also for TCR signaling. To distinguish the contribution of these two pathways to T-ALL progression, we applied GSEA to analyse gene signatures of antigen-independent (pre-TCR) and antigen-dependent (TCR) T-cell stimulation in T-ALL progression. Pre-TCR-induced genes (DN3b-DN4) correlated positively with leukemia progression (Figure 1G, adj. P < 10-4). In contrast, the TCR gene signature did not correlate positively with T-ALL progression induced by SCL-LMO1 (Figure 1G).
In summary, our results indicate that leukemic cells display gene signatures of post-β-selection thymocytes, suggesting that a pre-TCR/CD3-like proliferation has occurred even in the absence of a functional pre-TCR (Cd3e-/- background). Moreover, progression to T-ALL overlaps specifically with pre-TCR-driven gene signature. This CD3-independent activation of the pre-TCR molecular signature indicates a strong selective pressure during leukemic progression for pathways that normally control the β-selection checkpoint.
Several transcription factors have been implicated in pre-TCR signaling and/or β-selection (reviewed in (77)). To determine their potential contribution to T-ALL progression, we first performed a systematic regulator analysis based on published ChIP-seq datasets (62).
This analysis predicted that targets of E proteins, E2A and HEB, are down-regulated during β-selection and T-ALL progression (Figure 2A, Fisher’s exact test). In addition, from data obtained with shRNA knock-down of HEB (78),we identified a list of 389 genes activated by HEB in Jurkat cells, (i.e. fold-change > 1.5, t-test P < 0.05). GSEA indicated that these HEB targets (Figure 2B, left panel) as well as E2A-bound genes (Figure 2B, right panel) are down-regulated when comparing Cd3e-/- leukemic and pre-leukemic cells. Last, well known E-Box motifs CAGCTG and TAL1β-ITF2 (79) were found to be enriched in HEB target genes (Figure 2C).
Overall, these analyses indicate that inhibition of E protein activity may be important both at the β-selection checkpoint and during T-ALL progression.
Mice lacking E2a/Tcf3 develop lymphomas, suggesting that E2A is a tumor suppressor (42, 45). Nonetheless, TCF3 mRNA is highly expressed in human T-ALL (9, 80) and the TCF3 gene is neither deleted nor mutated, raising the question how E2A acts as a tumor suppressor. Previous work showed that pre-TCR signaling inhibits E2A activity via upregulation of Id3 (81). However, the very low levels of ID3 in most human T-ALL samples (Figure S2A) and in murine SCLtgLMO1tg T-ALL (Figure S2B) do not support a role for ID3 in sequestering HEB or E2A in T-ALL.
Next, we investigated Heb expression at the mRNA and protein levels in T-ALL progression. Heb/Tcf12 mRNA levels were equally high in control, pre-leukemic and leukemic cells (Figure S2C). In contrast, we found by western blotting that HEB protein was almost absent in murine leukemic cells, contrasting with high expression levels in normal DN3a thymocytes and variable levels in pre-leukemic thymocytes (Figure 2D, upper panel). E2A protein levels also decreased with progression to T-ALL, albeit to a lesser extent (Figure 2D, lower panel) while mRNA levels remained elevated (Figure S2C). Moreover, we observed that HEB levels steadily increased in Jurkat cells treated with the proteasome inhibitor MG132 (Figure S2D), indicating that HEB levels are regulated by proteasomal degradation in leukemic cells. We next inspected E protein levels in two primary T-ALL patient samples. HEB protein levels were undetected in the sample with higher proliferation in culture, whereas E2A was undetectable in both samples (Figure 2E). Taken together, these results indicate a strong selective pressure for HEB protein down-regulation during progression to T-ALL.
To directly address the role of Heb or E2a, we analyzed thymocyte numbers in E2a/Tcf3+/- or Heb/Tcf12+/- mice in the context of the SCLtgLMO1tg mice or their wild type (wt) littermates. In adult wt mice, removal of one Heb allele or one E2a allele did not significantly affect thymocyte numbers at the DN3 to DP stages (Figure 3A), although Heb monoallelic deletion resulted in modest but significantly increased cell numbers within populations undergoing β-selection, i.e. DN3b and DN3-4 (Figure S3), concurring with the view that Heb enforces a proliferation checkpoint at this stage (44, 82). We and others previously showed that the SCL-LMO1 or LMO2 oncogenes expand the DN3 populations due to increased self-renewal capacity (2, 4). Interestingly, Heb haploinsufficiency further increased the expansion of the DN3 and DN4 populations induced by the SCL-LMO1 oncogenes (Figure 3A). To directly address the antiproliferative role of HEB, we compared S/G2/M phase progression in Heb/Tcf12+/- and Heb/Tcf12+/+ DN3 thymocytes from Cd3e-/- mice. In absence of pre-TCR signaling, while loss of one allele of Heb increased the proportion of proliferating cells, we found that the SCL-LMO1 oncogenes decreased the proportion of cycling DN3 thymocytes, consistent with a role for SCL in quiescence control (83). In this context, removing one Heb allele re-established proliferating DN3 thymocytes to normal proportions (Figure 3B). Therefore, in the absence of pre-TCR signaling, the SCL and LMO1 oncogenes revealed Heb haplo-insufficiency in cell cycle control, indicating and that HEB anti-proliferative function is required to control oncogenic stress at the β-selection checkpoint. In addition to the previously reported role for Id3-mediated inhibition of E proteins during β-selection in steady state (84), our data indicate a distinctive requirement for HEB in stress response.
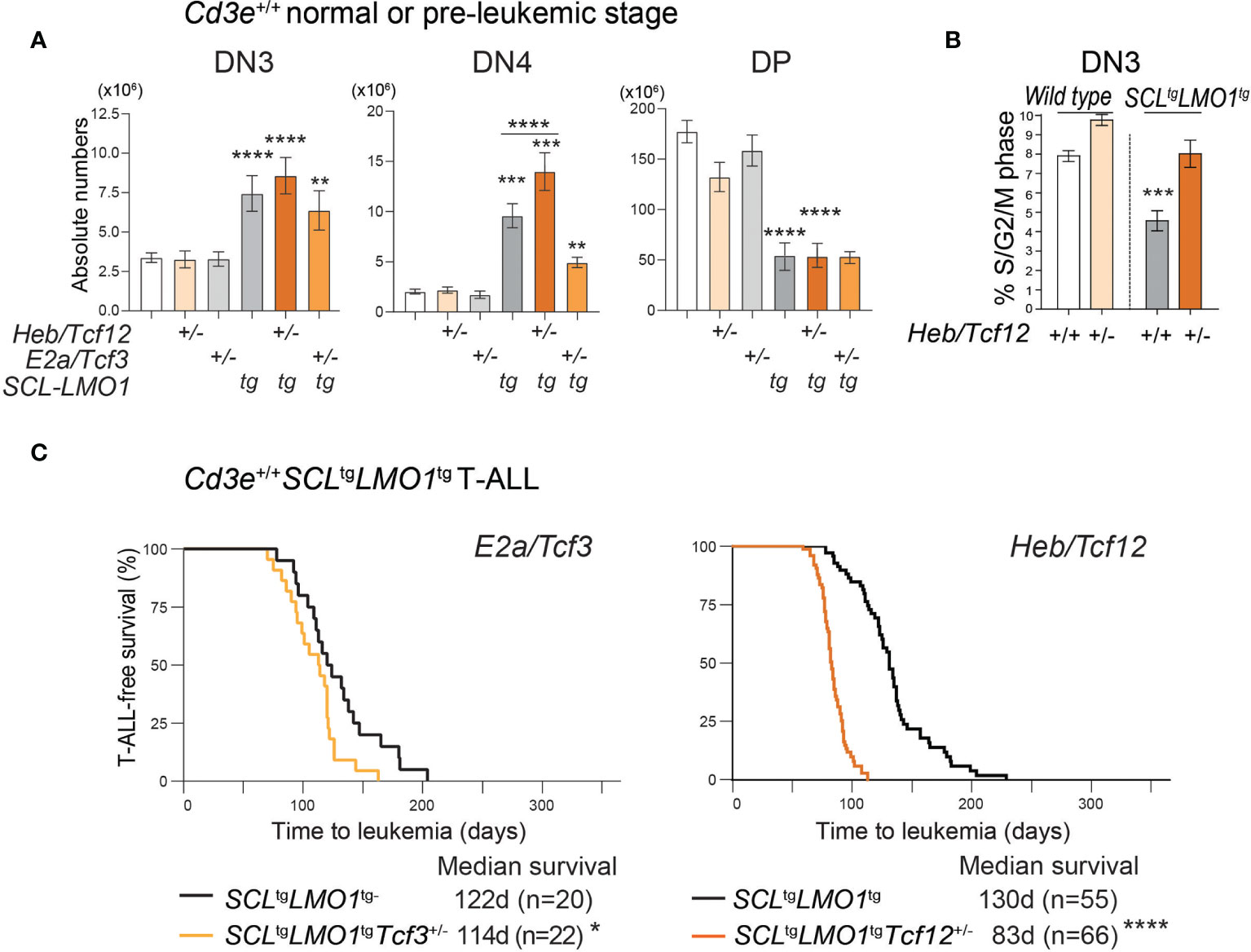
Figure 3 HEB restricts cell proliferation at the b‐selection checkpoint and acts as a tumor suppressor in T-ALL. (A) Effect of loss of one allele of Heb/Tcf12 or E2a/Tcf3 on the absolute numbers of thymocytes within the indicated subsets in a wt or SCLtgLMO1tg background. Shown are the average ± SD of at least 9 mice per group (5-6 weeks). Where not specified P value is compared to wild type control: **p = 0.0039, ***p < 0.002 and ****p < 0.0001. (B) Monoallelic Heb/Tcf12 deletion increased S/G2/M phase in Cd3e-deficientpre-leukemic DN3 thymocytes. Cell cycle analysis of Cd3e-deficient thymocytes in Heb/ Tcf12+/+ andHeb/Tcf12+/- backgrounds. *p value <0.01). (C) Kaplan-Meyer survival curves comparing E2a/Tcf3+/+ with E2a/Tcf3+/- backgrounds (left panel) as well as Heb/Tcf12+/+ and Heb/Tcf12+/- backgrounds in Cd3e-proficient SCLtgLMO1tg leukemias. In both panels, the +/+ genotypes are wild type littermates of +/- mice. N represents the numbers of mice and the median survival in days was computed from the survival curves. *p = 0.039 and ****p < 0.0001.
The E proteins and Id axis has a well-established tumor-suppressor function (42, 45). We therefore addressed the question whether both HEB and E2A have a tumor suppressor function in the context of SCL-LMO1-induced T-ALL (Figure 3C). In Cd3e-proficient mice, loss of one E2a allele caused a modest decrease in latency from 122 days in littermate controls to 114 days (Figure 3C, left panel). In contrast, deletion of one Heb allele accelerated the time of onset to 83 days compared to 130 days in littermate controls (Figure 3C, right panel). Together, our results indicate a tumor suppressor function for Heb which acts in a gene-dosage dependent manner in T-ALL induced by SCL and LMO1.
In Cd3e-deficient mice expressing the SCL and LMO1 oncogenes, inactivation of a single Heb allele bypassed pre-TCR signalling to increase the proportion and numbers of DN4 cells during the pre-leukemic stage (Figures 4A, B and Figure S4). In addition, monoallelic Heb deletion allowed a minor population of SCL-LMO1 expressing thymocytes to progress to the DP stage (1.4%, Figures 4A, B). Last, progression to the leukemic stage is associated with a transition to a post-β-selection phenotype in Cd3e-deficient SCLtgLMO1tg mice, which consistently increased in Heb/Tcf12+/- T-ALL (Figures 4C, D).
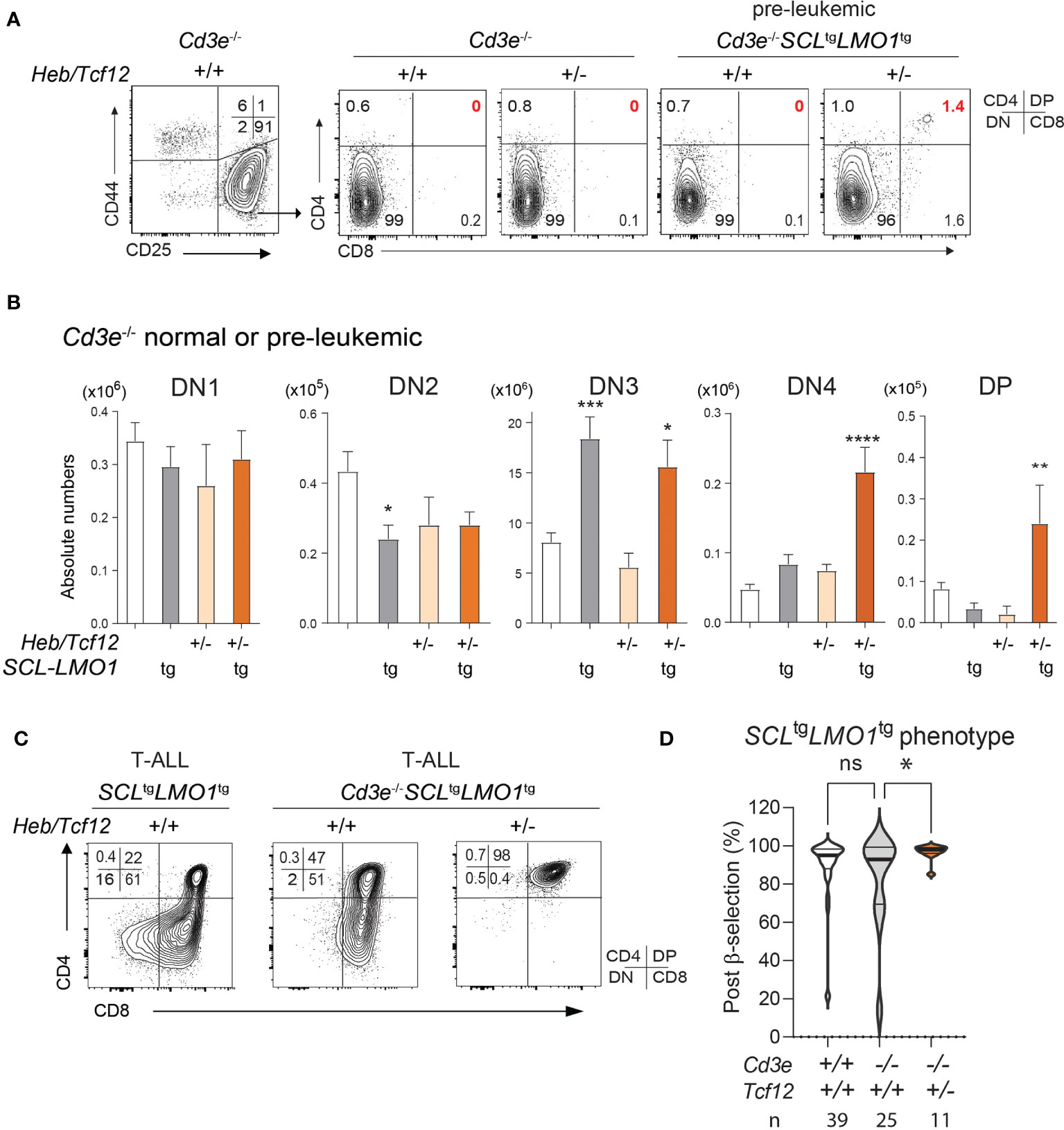
Figure 4 The SCL and LMO1 oncogenes favor the DN-DP transition at the leukemic stage despite the absence of pre-TCR/CD3 signaling: synergy with decreased Heb gene dosage. (A) FACS phenotypes of normal Cd3e-/- or pre-leukemic Cd3e-/-SCLtgLMO1tg thymocytes from Heb/Tcf12+/+ and Heb/Tcf12+/- backgrounds. Flow cytometry profiles for the CD4− CD8− DN populations are shown in Figure S4. (B) Absolute numbers of thymocytes within the indicated subsets in normal Cd3e-/- or pre-leukemic Cd3e-/-SCLtgLMO1tg in Heb/Tcf12+/+ and Heb/Tcf12+/- backgrounds. Shown are the average ± SD of at least 5 mice per group, taken at 4 weeks. Note the significant increase in the post-β selection DN4 and DP populations in SCLtgLMO1tgHeb/Tcf12+/- mice. (C) FACS phenotypes of Cd3e-proficient or deficient SCLtgLMO1tg leukemias from Heb/Tcf12+/+ and Heb/Tcf12+/- backgrounds. (D) T-ALL with a post-β-selection phenotype. Shown are the percentages of cells from each T-ALL with post-β-selection surface phenotypes (DN3-4, DN4, ISP, SP). n represents the numbers of mice analysed. * adj p=0.02. pvalue < 0.05; **p value < 0.005; ***p value < 0.0005; ****p value < 0.0001, ns, Non significant.
We next assessed the impact of Heb gene dosage on disease penetrance and time to leukemia. Strikingly, decreased Heb (Heb/Tcf12+/-) compensates for the absence of pre-TCR signaling in Cd3e-/- mice and allowed SCLtgLMO1tg T-ALL to become fully penetrant, in addition to accelerating disease onset by 107 days (Figures 5A, B). Hence, in Cd3e-/-SCLtgLMO1tg mice in which disease penetrance was 80%, loss of one Heb allele recapitulated the effect of the Notch1 oncogene on restoring full disease penetrance as shown in Figure 1C. Since Notch1tg-induced T-ALL is also dependent on pre-TCR function (Figures 1C and 5C, D), we next addressed the importance of Heb in T-ALL induced by Notch1tg in pre-TCR proficient and pre-TCR deficient mice. In contrast to SCL and LMO1, decreased Heb gene dosage did not affect Notch1tg-induced T-ALL in Cd3e-/- or in Cd3e+/+ mice (Figures 5C–F), indicating that Heb and Notch1 operate in the same genetic pathway in T-ALL, as suggested during normal differentiation (36). In summary, Heb and Notch1 inversely control the penetrance of T-ALL induced by the SCL and LMO1 oncogenes whereas the pre-TCR determines the DN-DP transition during the pre-leukemic stage and the time of disease onset.
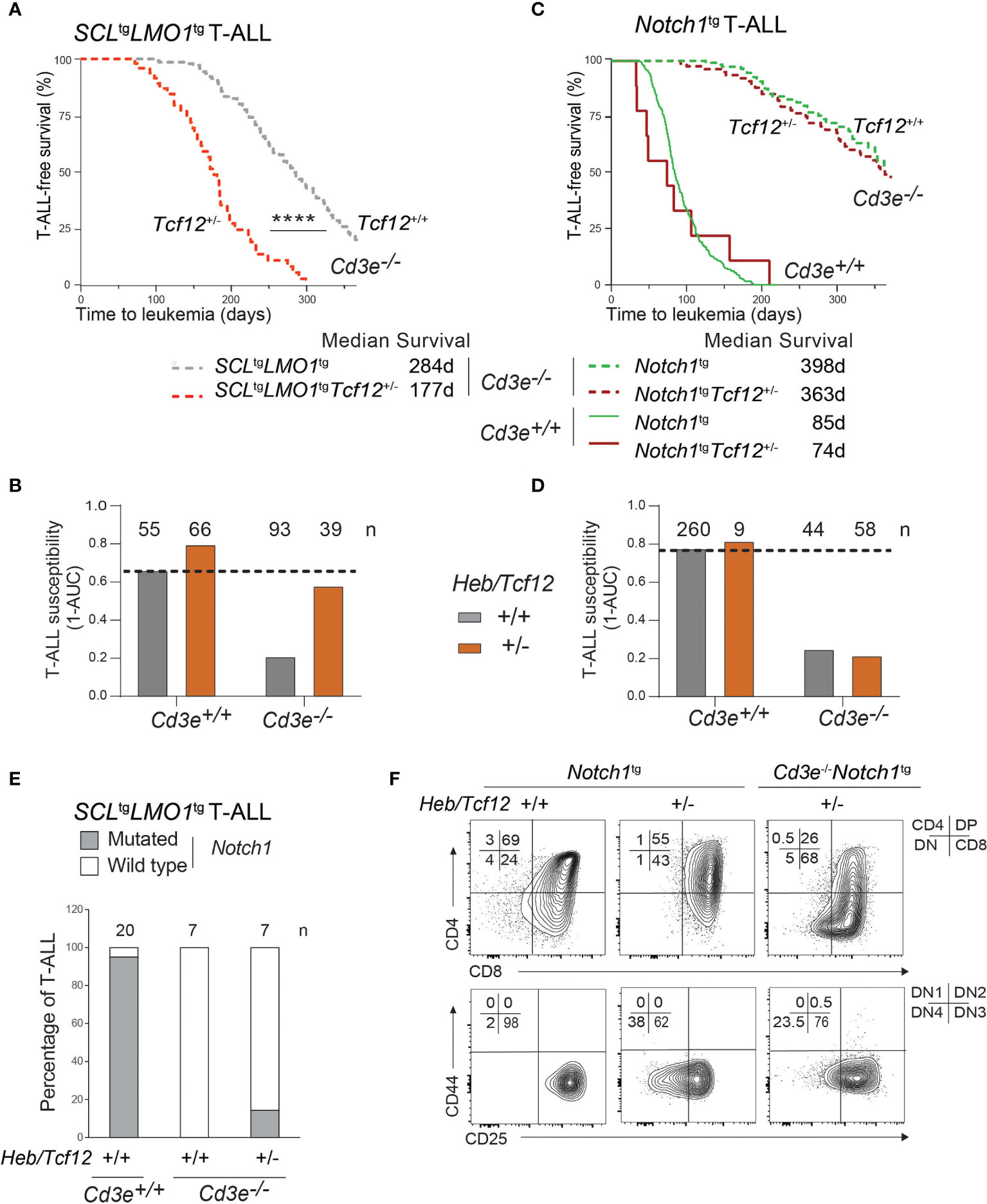
Figure 5 The SCL and LMO1 oncogenes but not the Notch1/IC9 oncogene reveal Heb/Tcf12 as a haplo-insufficient tumor suppressor in T-ALL. (A) Kaplan-Meyer survival curves comparing Cd3e-deficient SCLtgLMO1tg leukemias in Heb/Tcf12+/+ and Heb/Tcf12+/- backgrounds. Shown is the median survival in days. (B) T-ALL susceptibility of SCLtgLMO1tg mice in Cd3e-proficient (Cd3e+/+, Figure 3C) or deficient (Cd3e-/-, Figure 5A) backgrounds was calculated from the area under the curve (AUC) of the above Kaplan-Meyer graph over 365 days. Shown on top are the numbers of mice per group. (C, D) Kaplan-Meyer survival curves (C) and T-ALL susceptibility (D) comparing Notch1/IC9-induced leukemias in Heb/Tcf12+/+ and Heb/Tcf12+/- backgrounds that are Cd3e-deficient or Cd3e-proficient. (E) Presence of Notch1 activating mutations in Cd3e-proficient but not Cd3e-deficient SCLtgLMO1tg T-ALL. Shown are the percentage of T-ALL samples with an activating mutation in the Notch1 locus in Heb/Tcf12+/+ and Heb/Tcf12+/- backgrounds. Shown on top are the numbers of leukemias sequenced in each group. Only one Heb/Tcf12+/-Cd3e-deficient SCLtgLMO1tg T-ALL harbours a mutation affecting the PEST domain of Notch1. (F) Leukemic phenotypes of Cd3e-proficient or Cd3e-deficient Notch1tg leukemias from Heb/Tcf12+/+ and Heb/Tcf12+/- backgrounds **** pvalue<0.0001.
Since the Notch1 oncogene is sufficient to bypass pre-TCR signaling to confer full leukemic penetrance in Cd3e-/-SCLtgLMO1tg mice (Figure 1B), we addressed the question whether the increased penetrance caused by monoallelic Heb deletion shown in Figure 5A could be due to the acquisition of Notch1 gain of function mutations. We analyzed a cohort of mice with Heb/Tcf12+/- and Heb/Tcf12+/+ T-ALL for the presence of Notch1 mutations (Figures 5E and S5). As expected, 19 of 20 SCLtgLMO1tg T-ALL in Cd3e+/+Heb/Tcf12+/+ mice exhibit Notch1 gain of function mutations (4, 85) which affect the PEST domain whereas Cd3e-deficient SCLtgLMO1tgHeb/Tcf12+/+ T-ALL completely lacked Notch1 mutation as reported (4). In comparison, only 1 of 7 Heb/Tcf12+/- T-ALLs acquired Notch1 mutations (Figure 5E and Figure S5). Therefore, the increased aggressiveness of Heb/Tcf12+/- T-ALL is unlikely due to Notch1 gain of function mutations.
These results establish Heb as a tumor suppressor that normally enforces a proliferative checkpoint during β-selection to suppress oncogene-induced T-ALL in a gene dosage-dependent manner. Unlike classical tumor suppressors, the human HEB gene is not affected at the genomic level in T-ALL (10, 25). Here, we show that HEB is regulated at the protein level, pointing to a distinctive loss of function mechanism.
Since HEB/TCF12 is not deleted nor inactivated by deleterious mutations in human T-ALL, we addressed the possibility that down-regulation or genetic inactivation of HEB targets with tumor suppressor function could also be a mechanism associated to and/or selected for during disease progression. We performed exome sequencing of Cd3e-/-SCLtgLMO1tg leukemias (n=4) and control Cd3e-/thymocytes to identify genetic alterations involved in leukemia development in the absence of pre-TCR signaling. The software Strelka (66) was applied to discover somatic SNVs and short indels using the paired tumor-control configuration. After filtering out known polymorphisms reported in the SNP database v.138, we obtained 85 non-synonymous SNVs predicted to be deleterious by the Variant Effect Predictor tool (68). In addition, Strelka predicted 19 stop gains, 10 stop loss, 2 frameshift deletions and 1 frameshift insertion (Supplementary File 2).
We investigated the gene set affected by the above mutations (102 genes in total, Figure 6A) using the MSig database enrichment tool (http://software.broadinstitute.org/gsea/msigdb/annotate.jsp). We found a strong enrichment (adjusted P = 1.2x10-4) for genes containing the E-box motif CAGGTG in their proximal promoters, suggesting they could be regulated by E proteins E2A and HEB (Figure 6B and Supplementary File 2). The AP4 E-box motif CAGCTG was also found to be enriched in promoters of mutated genes (35 genes, adjusted P = 1.2x10-4).
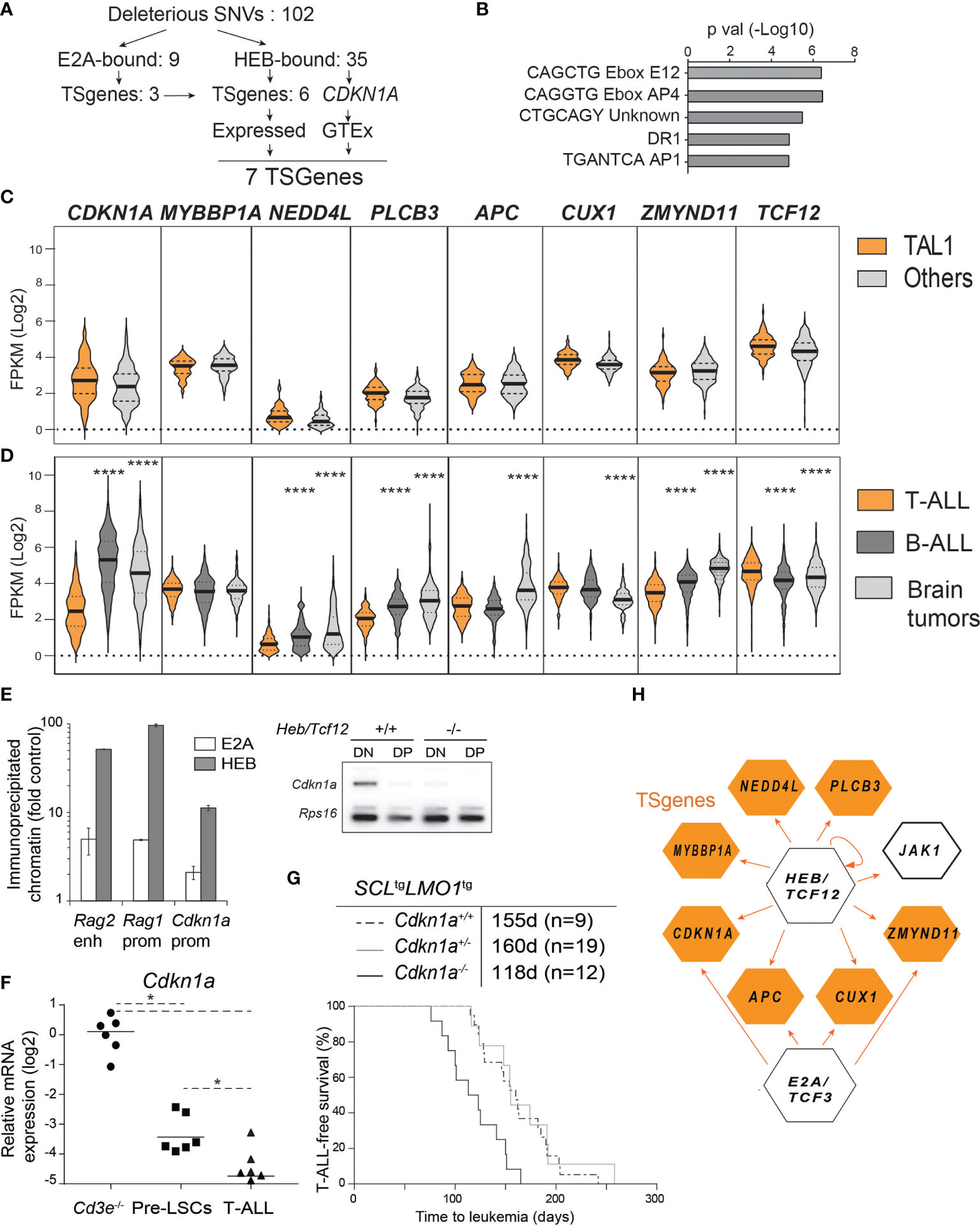
Figure 6 HEB controls a tumor suppressor network in T-ALL. (A) Strategy of tumor suppressor identification amongst HEB target genes that are mutated in Cd3e-/-SCLtgLMO1tg leukemic cells. (B) Enrichment in E boxes within the proximal promoter regions of HEB target genes. (C, D) Expression levels of the indicated TSGenes in TAL1+ vs other molecular subgroups in pediatric T-ALL dataset from Liu et al. (C) and in T-ALL, B-ALL and brain tumors from the Pediatric Cancer Genome Project cohort (D). (E) Chromatin immunoprecipitation of Cdkn1a promoter with HEB and E2A in DN thymocytes. The Rag1 promoter and Rag2 enhancer are included as positive controls (left panel) and RT-PCR analysis of Cdkn1a expression in DN and DP thymocytes in wt and Heb/Tcf12 knockout mice. Amplified bands were revealed by hybridization with an internal 32P-labeled oligonucleotide fragment (right panel). (F) Quantitative RT-PCR analysis of Cdkn1a expression in Cd3e-/- DN3 cells, pre-LSCs and leukemic cells. (G) Kaplan-Meyer survival curves of SCLtgLMO1tg T-ALL with partial and complete loss of Cdkn1a. (H) A tumor suppressor network downstream of HEB or E2A validated by chromatin immunoprecipitation. HEB is autoregulatory but not E2A. *p value < 0.01; ****p value < 0.0001
To confirm binding, we performed ChIP in the AD10 DN cell line and found by PCR that HEB occupies the promoter regions of 29 of the mutated genes within 2 kb of the transcriptional start site, in addition to Cdkn1a, validated here as a HEB target (Figure S6A). Binding enrichment was at least five-fold over a negative control and for 3 genes, the enrichment was almost as high as that observed with Ptcra which was amplified as a positive control (Figure S6A). In contrast, Csf3r which was amplified as a negative control did not show any enrichment.
We next addressed whether HEB-bound genes with loss of function mutations in murine T-ALL might be tumor suppressors in the human disease. We included Cdkn1a in this analysis because Cdkn1a is a well-documented HEB and E2A target gene (41, 44, 86) and because of strong genetic evidence for Cdkn1a tumor suppressor function in mice harboring one additional Cdkn1a allele (87). Out of 102 genes with deleterious mutations (for a total of 103 including Cdkn1a), 35 are HEB-bound genes (34.3%) according to previously published HEB ChIP-seq data with two human TAL1/SCL T-cell lines (78) and ChIP-PCR validation from this study (Figure S6A). Of note, 7 of these 35 HEB-bound genes (20%) are known tumor suppressors (https://bioinfo.uth.edu/TSGene, Figure 6A). We checked gene expression in blood and brain samples from normal tissues in the GTEx portal (https://www.gtexportal.org) and all seven genes were expressed in normal cells and therefore retained for further analysis. When we applied the same filtering strategy to E2A instead of HEB, only 10 of 103 genes are E2A-bound (9.7%) and 4 of the 10 genes are annotated as TSGenes (Figures 6A, E, H). All four genes are present in the seven HEB-bound TSGenes.
These 7 HEB-targets and TS genes were not found to be recurrently mutated in T-ALL (10). Of note, T-ALL can be classified into distinct molecular subgroups based on chromosomal translocations and gene signatures (9, 23, 88). Since our transgenic model is representative of the SCL/TAL1 molecular subgroup, we next addressed the question whether these seven HEB target TSGenes would be expressed at lower levels in the TAL1 subgroup compared to the other T-ALL subgroups, using the dataset published by Liu et al. (10). Gene expression levels were remarkably comparable between TAL1 and non-TAL1 subgroups and were consistently low compared to TCF12 (Figure 6B). We then searched the Pediatric Cancer Genome Project (https://www.stjude.cloud) (89) that covers more diversified cancer types, in order to compare expression levels for these seven genes in T-ALL (10), B-ALL (90) and brain tumors (Figure 6D). Strikingly, CDKN1A expression is five-and four-fold lower in T-ALL compared to B-ALL and brain tumors, respectively (Figure 6D). In addition, four other TSGenes are expressed at two- to eight-fold lower levels in T-ALL compared to brain tumors and/or B-ALL samples: NEDD4L, PLCB3, APC and ZMYND11 (Figure 6D). These expression patterns contrast sharply with those of LCK which is highest in T-ALL as expected (Figure S6C), RUNX1 which is equally expressed in T- and B-ALL but not in brain tumors, or JAK1, encoding a non-receptor tyrosine kinase, which is higher in B-ALL compared to the other two groups, and finally NAXE, encoding a metabolic enzyme which is expressed in all three types of pediatric cancers with modest but significantly lower levels in B-ALL (Figure S6B). Overall, low expression levels of HEB target TSGenes in primary human T-ALL samples concur with decreased HEB function in T-ALL compared to B-ALL and brain tumors, the three most frequent pediatric cancers, and a tumor suppressor function for HEB in pediatric T-ALL. Of note, these potential TSGenes are rarely mutated or deleted in T-ALL. Rather, these TSGenes are expressed at much lower levels in T-ALL compared to the other pediatric tumors.
Cdkn1a, a typical target of E2A and Id (41, 86, 91, 92) is part of the gene set that is downregulated at the β-selection checkpoint (Figure S6C) (93). p21Cdkn1a mediates G1 arrest by inhibiting CDK1 and CDK2 and loss of CDKN1A is a predictor of poor outcome in renal cell carcinoma (94). In agreement with our results (Figurse 6C, D), CDKN1A was found to be very low in human T-ALL (95). We therefore addressed the functional implication of Cdkn1a in this mouse model of SCL/TAL1 human T-ALL. We first confirmed that both E2A and HEB occupy the Cdkn1a promoter in primary DN thymocytes (Figure 6E, left panel). Moreover, Cdkn1a expression was nearly abrogated in Heb/Tcf12-deficient DN thymocytes (Figure 6E, right panel), indicating that HEB is a major transcriptional regulator of Cdkn1a at this developmental stage in the thymus. Treatment of thymocytes with phorbol 12-myristate 13-acetate (PMA) leads to activation of protein kinase C (PKC) which phosphorylates RAF and activates the ERK-MAPK pathway, and thus can be used to emulate pre-TCR/TCR signalling (96). We observed that Cdkn1a levels were significantly down-regulated in DN3 cells incubated with PMA for 6 hours (Figure S6D), consistent with the view that pre-TCR signals down-regulate p21 through the ERK-MAPK pathway (84, 96). In mouse leukemias, Cdkn1a expression was decreased in pre-LSCs compared to wild-type DN3a thymocytes and was further reduced in leukemic cells, in both Cd3e+/+ and Cd3e-/- leukemias (Figure 6F). Finally, we observed that T-ALL onset was accelerated by 37 days in Cdkn1a-deficient mice (Figure 6G), compared to the 53 day acceleration found in Heb/Tcf12+/- mice, confirming a tumor-suppressor function for Cdkn1a (Figure 6G). Of note, Cdkn1a was haplosufficient in this genetic assay (Figure 6G), indicating that a reduction threshold must be attained for leukemic progression to occur. These results concur with the stepwise decrease in Cdkn1a in pre-LSCs and in leukemic blasts at time of overt leukemia, indicating that p21 is a threshold-dependent tumor suppressor in T-ALL. In summary, our genetic approach indicates that in the absence of a hyperactive NOTCH1 and of pre-TCR signaling, progression to acute leukemia in SCL-LMO1-induced T-ALL involves the downregulation of a tumor suppressor network implicating HEB and p21CDKN1A (Figure 6H).
In the present study, we provide genetic evidence for differing requirements of NOTCH1, pre-TCR signaling and HEB in driving progression to T-ALL. By quantifying two disease endpoints, T-ALL penetrance and disease latency, i.e. the median time to overt leukemia, we show that in the context of the SCL and LMO1 oncogenes, the hyperactive Notch1 oncogene on its own determines disease penetrance even in the absence of pre-TCR signaling. Despite the apparent sufficiency of the three oncogenes, SCL, LMO1 and Notch1, an active pre-TCR accelerates the time of onset to 29 days, which is the time required for a single leukemic stem cell to induce T-ALL upon transplantation as shown previously (4). Therefore, SCL and LMO1 acting in synergy with two essential signaling pathways in thymocyte development are sufficient to transform a normal DN3a thymocyte into a fully transformed leukemia initiating cell. Moreover, our data indicate that Notch1 and Heb operate in the same genetic pathway, and mono-allelic deletion of Heb can recapitulate the capacity of the Notch1 oncogene to cause a fully penetrant disease in the absence of pre-TCR signaling.
While pre-TCR signaling has been shown to modulate the aggressiveness of T-ALL in several mouse models, the pre-TCR is dispensable for T-ALL induced by E47- or Trp53-deficiency (97, 98). Hence, the importance of the pre-TCR in T cell transformation induced by oncogenic events remained to be clarified. We propose that the importance of pre-TCR signaling depends on the primary oncogenic transcription factor. Previous transcriptome analyses of pediatric T-ALL identified five molecular subgroups and showed that the TCR signaling pathway is significantly enriched in the TAL1 group while other KEGG pathways related genes were more generally distributed within the other molecular subgroups (9). We now provide evidence that β-selection but not antigen-specific TCR signals are collaborating events. Indeed, the pre-TCR drives cell proliferation, which is required not only for clonal expansion but also for DN-DP differentiation (99) and thymocyte survival (100). A pre-T cell receptor lacking the TCR beta variable domain causes an expansion of the DP population that precedes overt T cell leukemia, suggesting that abnormal pre-TCR function can be oncogenic (101). In addition, the pre-TCR signal is important for Notch1/ICN1, Notch3- and TEL-JAK2-induced leukemias (74, 102, 103), indicating that developmental processes required for normal thymocyte development can be implicated in the pathogenesis of T-ALL (75). In the present study, even though T-ALL can develop in the absence of Cd3e and of Notch1 mutations, our transcriptomic comparison of preleukemic cells with fully transformed leukemic cells indicate a reactivation of β-selection during the progression to T-ALL. The signal strength providing progression to CD4+CD8+ DP T-ALL can either originate from a hyperactive Notch1 allele or the deletion of a single Heb allele. Our data provide compelling evidence for the importance of signal strength of NOTCH1 or HEB in driving the DN to DP transition associated with leukemogenesis.
The pre-TCR complex induces Id expression and consequently inhibition of E protein activity suggesting that the pre-TCR functions upstream of E proteins (81). Nonetheless, E2a-deficient mice develop T-cell lymphomas (42, 45), associated with Notch1 mutations within the PEST domain (104), suggesting that E2a is not downstream of Notch1. Rather, the latter observations suggest that Notch1 and E2a operate in parallel pathways. Moreover, Heb may function parallel to or downstream of the pre-TCR (51). Since NOTCH1 and pre-TCR functions are cooperative during β-selection (11), and both are believed to affect E protein function (81, 105, 106), pre-TCR signaling becomes a confounding factor in assessing the contribution of NOTCH1 and of E proteins in leukemogenesis. Using Cd3e-/- mice in which pre-TCR function is abrogated, we now show that oncogenic Notch1 controls disease penetrance in SCL-LMO1-induced T-ALL while the pre-TCR signal governs the time of leukemia onset. Last, our data indicate that Heb but not E2a operate in the same pathway as Notch1 since monoallelic deletion of Heb does not affect Notch1-induced T-ALL but accelerates the disease induced by SCL-LMO1 in Cd3e-sufficient mice. Finally, loss of one Heb allele in Cd3e-deficient mice compensates in part the Notch1 oncogene to restore full penetrance to T-ALL induced by SCL and LMO1.
We and others previously showed that the SCL transcription factor heterodimerizes with E2A or HEB, thereby inhibiting target gene expression such as Cd4 and Ptcra and thymocyte differentiation (39, 40, 50). Nonetheless, E47 deficiency promotes the aberrant development of Rag1 null thymocytes and appearance of DP cells (84), similar to the appearance of a minor population of DP cells reported here in Cd3e-deficient thymocytes lacking one Heb allele.
E proteins are negatively regulated by ID proteins, another class of bHLH protein. At the β-selection checkpoint, the activation of RAS extracellular-signal-related kinase (ERK)–MAP kinase pathway upregulates ID proteins that bind to E proteins preventing their transcriptional activity (84). E2A inhibits proliferation and differentiation at the β-selection checkpoint in the absence of pre-TCR expression (84), suggesting that inhibition of E2A via Id3 would enforce the two distinctive functional outputs of pre-TCR signaling in favoring both cell proliferation and differentiation. However, Id2 and Id3 suppress lymphomagenesis (107), suggesting a necessary balance of E protein/ID levels during thymocyte differentiation. Partial redundancy may explain that both E2a and Heb are haplosufficient for thymocyte differentiation during steady-state conditions. Nonetheless, the oncogenic stress induced by SCL and LMO1 in thymocytes reveal Heb haploinsufficiency at the DN to DP transition, more specifically at the β-selection checkpoint revealing at the same time that E2a cannot compensate for Heb in stress-response, despite their quasi-redundancy during steady-state conditions.
Our results also bring out the importance of monitoring protein levels in primary tumors, since inactivation of tumor suppressors via protein downregulation could represent a distinct mechanism driving T-ALL.
Master regulators of hematopoietic lineages have been implicated in tumor suppressor function, as reported for C/EBPα (108, 109) and SPI1/PU.1 in acute myeloblastic leukemia (AML) (110) or PAX5 loss of function in B-ALL (90, 111). While E2A/TCF3 is annotated as a tumor suppressor gene (TSGene 2.0) with direct experimental and clinical evidence, HEB/TCF12 tumor suppressor function has been overlooked, despite the critical role of HEB in the T lineage (reviewed in (4, 112)). Due to a non-redundant function to enforce T lineage (36) and at multiple stages of T cell differentiation, we propose that the essentiality of HEB precludes the possibility of identifying loss of function mutations in hematopoietic malignancies. Our study reveals the possibility of temporary loss of function via downregulation of HEB protein levels. Moreover, our analysis of the PCGP cohort identifies decreased gene expression levels in T-ALL compared to other pediatric cancers for at least five TSGenes that are HEB targets, regardless of the T-ALL molecular subgroup (9). In addition to well documented genetic alterations of tumor suppressor genes, our study indicates that reduced expression levels of multiple tumor suppressor genes could also contribute to the process of tumorigenesis, as illustrated here for CDKN1A.
Cdkn1a is a well known E2A and HEB target (44) and tumor suppressor gene (113). More recently, the Cdkn1aSUPER mouse is shown to be more resistant to transformation and exhibit a strong cancer protection phenotype, establishing a direct gene dosage-dependent tumor suppressor function for CDKN1A (87). Our observations indicate that Cdkn1a is down regulated by the SCL-LMO1 oncogenes during the pre-leukemic stage and further down regulated during progression to T-ALL. In Cd3e-deficient T-ALL, our data point to the down-regulation of Cdkn1a as part of the selective pressure to activate molecular mechanisms underlying the β-selection process and drive progression to T-ALL. Supporting this hypothesis, CDKN1A gene expression levels in the PCGP cohort is on average six- to seven-fold lower in pediatric T-ALL compared to B-ALL, AML or brain tumors. In addition to mRNA downregulation, p21Cdkn1a can also be phosphorylated and degraded via ubiquitin-dependent and ubiquitin-independent proteolysis (113). Much like HEB, the CDKN1A gene is not subject to copy number variations nor deleterious mutations in hematopoietic malignancies. Rather, CDKN1A levels can be downregulated at the mRNA level, as illustrated here, or at the level of protein stability.
HEB and E2A exert an anti-proliferative function in thymocytes prior to Tcrb rearrangement, required for the formation of a functional pre-TCR (44), consistent with a tumor suppressor function reported here for HEB. In addition to a well-timed restriction in cell proliferation, it remains possible that tumor suppression involves additional molecular functions secured either by HEB or HEB downstream targets, as reported for TP53 (114). In addition to CDKN1A, HEB and E2A co-occupy three TSG loci, APC, a haplo-insufficient tumor suppressor, ZMYND11 a chromatin reader and tumor suppressor in breast cancer (115) and CUX1, a homeodomain transcription regulator and haploinsufficient tumor suppressor (116). HEB is also found to occupy three other TSGenes, MYBPP1A, potentially involved in nucleolar stress, NEDD4L, an E3 ligase with tumor suppressor function (117) and PLCB3, encoding phospholipase C beta 3 (phosphatidylinositol-specific), involved in G-protein-linked receptor-mediated signal transduction (Figure 6H). These additional targets may account for the more prominent role of HEB as a tumor suppressor in response to the oncogenic stress induced by SCL and LMO1, exactly at the β-selection checkpoint controlled by HEB. Beyond the SCL/TAL1 molecular group, five HEB target-TSGenes are down regulated in T-ALL compared to other pediatric tumors. In T-ALL, we propose that HEB controls a network of tumor suppressor genes to mitigate the oncogenic stress occurring at the β-selection checkpoint. Unlike classical tumor suppressors, these genes are not inactivated by genomic deletion or deleterious mutations but are down-regulated at the mRNA or at the protein levels. This mechanism cannot be detected by assessing DNA copy number variation or whole-genome sequencing, as exemplified by the total absence of HEB copy number loss or point mutations in T-ALL. In this context, future large-scale proteomics studies in primary tumors will be able to address whether downregulation of HEB protein is a recurrent event in human T-ALL.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: http://www.ncbi.nlm.nih.gov/geo, accession ID: GSE198506. RNA-seq data from pediatric tumors used for analysis in this study were obtained from the St. Jude Cloud (https://www.stjude.cloud).
The studies involving human participants were reviewed and approved by Comité d’éthique de la recherche clinique, Université de Montréal. The patients/participants provided their written informed consent to participate in this study.
DV, MT, BG and TH conceived and designed the work. DV, MT, BG, SH and AH conducted the studies and/or contributed to the acquisition of data. DV, MT, BG, SH, PG, SL, JZ-P, JH and JPC contributed to the interpretation of the data and/or data analysis. JH, BG and AH contributed to experiments with primary human leukemic cells. DV, MT, and TH wrote the manuscript. All authors contributed to the article and approved the submitted version.
This work was supported by grants from the Canadian Cancer Society (TH), the Canadian Institute for Health Research (TH), the FRSQ Cancer Network (JH), the Leukemia Lymphoma Society of Canada (TH), a post-doctoral fellowship from LLSC (SH) and fellowships from the Cole Foundation (DV and MT). The infrastructure was supported in part by an FRSQ group grant and the Canadian Foundation for Innovation.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We thank Pearl Campbell from the Ottawa Heath Research Institute for assistance in cDNA arrays hybridization; Véronique Litalien, Karine Lachapelle and Koryne Léveillé for the mouse colony; Danièle Gagné, Annie Gosselin, and Angélique Bellemare-Pelletier for assistance in flow cytometry and cell sorting; Magali Humbert, Geneviève Lavoie and Philippe Roux for help in the western blots; and Julie Gervais and Manon Valiquette for the InVivo Biology platform.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2022.867443/full#supplementary-material
1. Hoang T, Lambert JA, Martin R. SCL/TAL1 in Hematopoiesis and Cellular Reprogramming. Curr Top Dev Biol (2016) 118:163–204. doi: 10.1016/bs.ctdb.2016.01.004
2. Gerby B, Tremblay CS, Tremblay M, Rojas-Sutterlin S, Herblot S, Hebert J, et al. SCL, LMO1 and Notch1 Reprogram Thymocytes Into Self-Renewing Cells. PLoS Genet (2014) 10(12):e1004768. doi: 10.1371/journal.pgen.1004768
3. McCormack MP, Shields BJ, Jackson JT, Nasa C, Shi W, Slater NJ, et al. Requirement for Lyl1 in a Model of Lmo2-Driven Early T-Cell Precursor ALL. Blood (2013) 122(12):2093–103. doi: 10.1182/blood-2012-09-458570
4. Tremblay M, Tremblay CS, Herblot S, Aplan PD, Hebert J, Perreault C, et al. Modeling T-Cell Acute Lymphoblastic Leukemia Induced by the SCL and LMO1 Oncogenes. Genes Dev (2010) 24(11):1093–105. doi: 10.1101/gad.1897910
5. Cleveland SM, Smith S, Tripathi R, Mathias EM, Goodings C, Elliott N, et al. Lmo2 Induces Hematopoietic Stem Cell-Like Features in T-Cell Progenitor Cells Prior to Leukemia. Stem Cells (2013) 31(5):882–94. doi: 10.1002/stem.1345
6. Fregona V, Bayet M, Gerby B. Oncogene-Induced Reprogramming in Acute Lymphoblastic Leukemia: Towards Targeted Therapy of Leukemia-Initiating Cells. Cancers (2021) 13(21):5511. doi: 10.3390/cancers13215511
7. Greaves M, Maley CC. Clonal Evolution in Cancer. Nature (2012) 481(7381):306–13. doi: 10.1038/nature10762
8. Oshima K, Khiabanian H, da Silva-Almeida AC, Tzoneva G, Abate F, Ambesi-Impiombato A, et al. Mutational Landscape, Clonal Evolution Patterns, and Role of RAS Mutations in Relapsed Acute Lymphoblastic Leukemia. Proc Natl Acad Sci (2016) 113(40):11306–11. doi: 10.1073/pnas.1608420113
9. Ferrando AA, Neuberg DS, Staunton J, Loh ML, Huard C, Raimondi SC, et al. Gene Expression Signatures Define Novel Oncogenic Pathways in T Cell Acute Lymphoblastic Leukemia. Cancer Cell (2002) 1(1):75–87. doi: 10.1016/S1535-6108(02)00018-1
10. Liu Y, Easton J, Shao Y, Maciaszek J, Wang Z, Wilkinson MR, et al. The Genomic Landscape of Pediatric and Young Adult T-Lineage Acute Lymphoblastic Leukemia. Nat Genet (2017) 49(8):1211–8. doi: 10.1038/ng.3909
11. Ciofani M, Schmitt TM, Ciofani A, Michie AM, Cuburu N, Aublin A, et al. Obligatory Role for Cooperative Signaling by Pre-TCR and Notch During Thymocyte Differentiation. J Immunol (2004) 172(9):5230–9. doi: 10.4049/jimmunol.172.9.5230
12. Maillard I, Tu L, Sambandam A, Yashiro-Ohtani Y, Millholland J, Keeshan K, et al. The Requirement for Notch Signaling at the Beta-Selection Checkpoint In Vivo is Absolute and Independent of the Pre-T Cell Receptor. J Exp Med (2006) 203(10):2239–45. doi: 10.1084/jem.20061020
13. Ciofani M, Zúñiga-Pflücker JC. Notch Promotes Survival of Pre–T Cells at the β-Selection Checkpoint by Regulating Cellular Metabolism. Nat Immunol (2005) 6(9):881–8. doi: 10.1038/ni1234
14. Weng AP, Ferrando AA, Lee W, Morris J, Silverman LB, Sanchez-Irizarry C, et al. Activating Mutations of NOTCH1 in Human T Cell Acute Lymphoblastic Leukemia. Science (2004) 306(5694):269–71. doi: 10.1126/science.1102160
15. Roy M, Pear WS, Aster JC. The Multifaceted Role of Notch in Cancer. Curr Opin Genet Dev (2007) 17(1):52–9. doi: 10.1016/j.gde.2006.12.001
16. Kushwah R, Guezguez B, Lee JB, Hopkins CI, Bhatia M. Pleiotropic Roles of Notch Signaling in Normal, Malignant, and Developmental Hematopoiesis in the Human. EMBO Rep (2014) 15(11):1128–38. doi: 10.15252/embr.201438842
17. Tatarek J, Cullion K, Ashworth T, Gerstein R, Aster JC, Kelliher MA. Notch1 Inhibition Targets the Leukemia-Initiating Cells in a Tal1/Lmo2 Mouse Model of T-ALL. Blood (2011) 118(6):1579–90. doi: 10.1182/blood-2010-08-300343
18. Ashworth TD, Pear WS, Chiang MY, Blacklow SC, Mastio J, Xu L, et al. Deletion-Based Mechanisms of Notch1 Activation in T-ALL: Key Roles for RAG Recombinase and a Conserved Internal Translational Start Site in Notch1. Blood (2010) 116(25):5455–64. doi: 10.1182/blood-2010-05-286328
19. King B, Trimarchi T, Reavie L, Xu L, Mullenders J, Ntziachristos P, et al. The Ubiquitin Ligase FBXW7 Modulates Leukemia-Initiating Cell Activity by Regulating MYC Stability. Cell (2013) 153(7):1552–66. doi: 10.1016/j.cell.2013.05.041
20. Weng AP, Millholland JM, Yashiro-Ohtani Y, Arcangeli ML, Lau A, Wai C, et al. C-Myc is an Important Direct Target of Notch1 in T-Cell Acute Lymphoblastic Leukemia/Lymphoma. Genes Dev (2006) 20(15):2096–109. doi: 10.1101/gad.1450406
21. Yashiro-Ohtani Y, Wang H, Zang C, Arnett KL, Bailis W, Ho Y, et al. Long-Range Enhancer Activity Determines Myc Sensitivity to Notch Inhibitors in T Cell Leukemia. Proc Natl Acad Sci USA (2014) 111(46):E4946–53. doi: 10.1073/pnas.1407079111
22. De Keersmaecker K, Atak ZK, Li N, Vicente C, Patchett S, Girardi T, et al. Exome Sequencing Identifies Mutation in CNOT3 and Ribosomal Genes RPL5 and RPL10 in T-Cell Acute Lymphoblastic Leukemia. Nat Genet (2013) 45(2):186–90. doi: 10.1038/ng.2508
23. Homminga I, Pieters R, Langerak AW, de Rooi JJ, Stubbs A, Verstegen M, et al. Integrated Transcript and Genome Analyses Reveal NKX2-1 and MEF2C as Potential Oncogenes in T Cell Acute Lymphoblastic Leukemia. Cancer Cell (2011) 19(4):484–97. doi: 10.1016/j.ccr.2011.02.008
24. Cauwelier B, Cave H, Gervais C, Lessard M, Barin C, Perot C, et al. Clinical, Cytogenetic and Molecular Characteristics of 14 T-ALL Patients Carrying the TCRbeta-HOXA Rearrangement: A Study of the Groupe Francophone De Cytogenetique Hematologique. Leukemia (2007) 21(1):121–8. doi: 10.1038/sj.leu.2404410
25. Tate JG, Bamford S, Jubb HC, Sondka Z, Beare DM, Bindal N, et al. COSMIC: The Catalogue Of Somatic Mutations In Cancer. Nucleic Acids Res (2018) 47(D1):D941–D7. doi: 10.1093/nar/gky1015
26. Kandoth C, McLellan MD, Vandin F, Ye K, Niu B, Lu C, et al. Mutational Landscape and Significance Across 12 Major Cancer Types. Nature (2013) 502(7471):333–9. doi: 10.1038/nature12634
27. Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LA, Kinzler KW. Cancer Genome Landscapes. Science (2013) 339(6127):1546–58. doi: 10.1126/science.1235122
28. Look AT. Oncogenic Transcription Factors in the Human Acute Leukemias. Science (1997) 278(5340):1059–64. doi: 10.1126/science.278.5340.1059
29. Van Vlierberghe P, Ferrando A. The Molecular Basis of T Cell Acute Lymphoblastic Leukemia. J Clin Invest (2012) 122(10):3398–406. doi: 10.1172/JCI61269
30. De Keersmaecker K, Real PJ, Gatta GD, Palomero T, Sulis ML, Tosello V, et al. The TLX1 Oncogene Drives Aneuploidy in T Cell Transformation. Nat Med (2010) 16(11):1321–7. doi: 10.1038/nm.2246
31. Gutierrez A, Kentsis A, Sanda T, Holmfeldt L, Chen SC, Zhang J, et al. The BCL11B Tumor Suppressor is Mutated Across the Major Molecular Subtypes of T-Cell Acute Lymphoblastic Leukemia. Blood (2011) 118(15):4169–73. doi: 10.1182/blood-2010-11-318873
32. Li L, Leid M, Rothenberg EV. An Early T Cell Lineage Commitment Checkpoint Dependent on the Transcription Factor Bcl11b. Science (2010) 329(5987):89–93. doi: 10.1126/science.1188989
33. Murre C, McCaw PS, Baltimore D. A New DNA Binding and Dimerization Motif in Immunoglobulin Enhancer Binding, Daughterless, MyoD, and Myc Proteins. Cell (1989) 56(5):777–83. doi: 10.1016/0092-8674(89)90682-X
34. Wang D, Claus CL, Vaccarelli G, Braunstein M, Schmitt TM, Zúñiga-Pflücker JC, et al. The Basic Helix-Loop-Helix Transcription Factor HEBAlt is Expressed in Pro-T Cells and Enhances the Generation of T Cell Precursors. J Immunol (2006) 177(1):109–19. doi: 10.4049/jimmunol.177.1.109
35. Barndt RJ, Dai M, Zhuang Y. Functions of E2A-HEB Heterodimers in T-Cell Development Revealed by a Dominant Negative Mutation of HEB. Mol Cell Biol (2000) 20(18):6677–85. doi: 10.1128/MCB.20.18.6677-6685.2000
36. Braunstein M, Anderson MK. HEB-Deficient T-Cell Precursors Lose T-Cell Potential and Adopt an Alternative Pathway of Differentiation. Mol Cell Biol (2011) 31(5):971–82. doi: 10.1128/MCB.01034-10
37. Miyazaki M, Rivera RR, Miyazaki K, Lin YC, Agata Y, Murre C. The Opposing Roles of the Transcription Factor E2A and its Antagonist Id3 That Orchestrate and Enforce the Naive Fate of T Cells. Nat Immunol (2011) 12(10):992–1001. doi: 10.1038/ni.2086
38. Belle I, Zhuang Y. E Proteins in Lymphocyte Development and Lymphoid Diseases. Curr Top Dev Biol (2014) 110:153–87. doi: 10.1016/B978-0-12-405943-6.00004-X
39. Herblot S, Steff AM, Hugo P, Aplan PD, Hoang T. SCL and LMO1 Alter Thymocyte Differentiation: Inhibition of E2A-HEB Function and Pre-T Alpha Chain Expression. Nat Immunol (2000) 1(2):138–44. doi: 10.1038/77819
40. Chervinsky DS, Zhao XF, Lam DH, Ellsworth M, Gross KW, Aplan PD. Disordered T-Cell Development and T-Cell Malignancies in SCL LMO1 Double-Transgenic Mice: Parallels With E2A-Deficient Mice. Mol Cell Biol (1999) 19(7):5025–35. doi: 10.1128/MCB.19.7.5025
41. Herblot S, Aplan PD, Hoang T. Gradient of E2A Activity in B-Cell Development. Mol Cell Biol (2002) 22(3):886–900. doi: 10.1128/MCB.22.3.886-900.2002
42. Bain G, Engel I, Robanus Maandag EC, te Riele HP, Voland JR, Sharp LL, et al. E2A Deficiency Leads to Abnormalities in Alphabeta T-Cell Development and to Rapid Development of T-Cell Lymphomas. Mol Cell Biol (1997) 17(8):4782–91. doi: 10.1128/MCB.17.8.4782
43. Park ST, Nolan GP, Sun XH. Growth Inhibition and Apoptosis Due to Restoration of E2A Activity in T Cell Acute Lymphoblastic Leukemia Cells. J Exp Med (1999) 189(3):501–8. doi: 10.1084/jem.189.3.501
44. Wojciechowski J, Lai A, Kondo M, Zhuang Y. E2A and HEB are Required to Block Thymocyte Proliferation Prior to Pre-TCR Expression. J Immunol (2007) 178(9):5717–26. doi: 10.4049/jimmunol.178.9.5717
45. Yan W, Young AZ, Soares VC, Kelley R, Benezra R, Zhuang Y. High Incidence of T-Cell Tumors in E2A-Null Mice and E2A/Id1 Double-Knockout Mice. Mol Cell Biol (1997) 17(12):7317–27. doi: 10.1128/MCB.17.12.7317
46. Hsu HL, Wadman I, Baer R. Formation of In Vivo Complexes Between the TAL1 and E2A Polypeptides of Leukemic T Cells. Proc Natl Acad Sci USA (1994) 91(8):3181–5. doi: 10.1073/pnas.91.8.3181
47. O'Neil J, Shank J, Cusson N, Murre C, Kelliher M. TAL1/SCL Induces Leukemia by Inhibiting the Transcriptional Activity of E47/HEB. Cancer Cell (2004) 5(6):587–96. doi: 10.1016/j.ccr.2004.05.023
48. McCormack MP, Young LF, Vasudevan S, de Graaf CA, Codrington R, Rabbitts TH, et al. The Lmo2 Oncogene Initiates Leukemia in Mice by Inducing Thymocyte Self-Renewal. Science (2010) 327(5967):879–83. doi: 10.1126/science.1182378
49. Smith S, Tripathi R, Goodings C, Cleveland S, Mathias E, Hardaway JA, et al. LIM Domain Only-2 (LMO2) Induces T-Cell Leukemia by Two Distinct Pathways. PLoS One (2014) 9(1):e85883. doi: 10.1371/journal.pone.0085883
50. Tremblay M, Herblot S, Lecuyer E, Hoang T. Regulation of pT Alpha Gene Expression by a Dosage of E2A, HEB, and SCL. J Biol Chem (2003) 278(15):12680–7. doi: 10.1074/jbc.M209870200
51. Barndt R, Dai MF, Zhuang Y. A Novel Role for HEB Downstream or Parallel to the Pre-TCR Signaling Pathway During Alpha Beta Thymopoiesis. J Immunol (1999) 163(6):3331–43.
52. Zhuang Y, Cheng P, Weintraub H. B-Lymphocyte Development is Regulated by the Combined Dosage of Three Basic Helix-Loop-Helix Genes, E2A, E2-2, and HEB. Mol Cell Biol (1996) 16(6):2898–905. doi: 10.1128/MCB.16.6.2898
53. Robey E, Chang D, Itano A, Cado D, Alexander H, Lans D, et al. An Activated Form of Notch Influences the Choice Between CD4 and CD8 T Cell Lineages. Cell (1996) 87(3):483–92. doi: 10.1016/S0092-8674(00)81368-9
54. Aplan PD, Jones CA, Chervinsky DS, Zhao X, Ellsworth M, Wu C, et al. An Scl Gene Product Lacking the Transactivation Domain Induces Bony Abnormalities and Cooperates With LMO1 to Generate T-Cell Malignancies in Transgenic Mice. EMBO J (1997) 16(9):2408–19. doi: 10.1093/emboj/16.9.2408
55. McGuire EA, Rintoul CE, Sclar GM, Korsmeyer SJ. Thymic Overexpression of Ttg-1 in Transgenic Mice Results in T-Cell Acute Lymphoblastic Leukemia/Lymphoma. Mol Cell Biol (1992) 12(9):4186–96. doi: 10.1128/mcb.12.9.4186-4196.1992
56. Malissen M, Gillet A, Ardouin L, Bouvier G, Trucy J, Ferrier P, et al. Altered T Cell Development in Mice With a Targeted Mutation of the CD3-Epsilon Gene. EMBO J (1995) 14(19):4641–53. doi: 10.1002/j.1460-2075.1995.tb00146.x
57. Zhuang Y, Kim CG, Bartelmez S, Cheng P, Groudine M, Weintraub H. Helix-Loop-Helix Transcription Factors E12 and E47 are Not Essential for Skeletal or Cardiac Myogenesis, Erythropoiesis, Chondrogenesis, or Neurogenesis. Proc Natl Acad Sci USA (1992) 89(24):12132–6. doi: 10.1073/pnas.89.24.12132
58. Gerby B, Veiga D, Krosl J, Ouellette J, Fares I, Tremblay M, et al. High-Throughput Screening in Niche-Based Assay Identifies Compounds to Target Pre-Leukemic Stem Cells. J Clin Invest (2016) 126(12):4569–84. doi: 10.1172/JCI86489
59. Gautier L, Cope L, Bolstad BM, Irizarry RA. Affy–Analysis of Affymetrix GeneChip Data at the Probe Level. Bioinformatics (2004) 20(3):307–15. doi: 10.1093/bioinformatics/btg405
60. Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, et al. Gene Set Enrichment Analysis: A Knowledge-Based Approach for Interpreting Genome-Wide Expression Profiles. Proc Natl Acad Sci USA (2005) 102(43):15545–50. doi: 10.1073/pnas.0506580102
61. Wang H, Zou J, Zhao B, Johannsen E, Ashworth T, Wong H, et al. Genome-Wide Analysis Reveals Conserved and Divergent Features of Notch1/RBPJ Binding in Human and Murine T-Lymphoblastic Leukemia Cells. Proc Natl Acad Sci USA (2011) 108(36):14908–13. doi: 10.1073/pnas.1109023108
62. Hannah R, Joshi A, Wilson NK, Kinston S, Gottgens B. A Compendium of Genome-Wide Hematopoietic Transcription Factor Maps Supports the Identification of Gene Regulatory Control Mechanisms. Exp Hematol (2011) 39(5):531–41. doi: 10.1016/j.exphem.2011.02.009
63. Salmon-Divon M, Dvinge H, Tammoja K, Bertone P. PeakAnalyzer: Genome-Wide Annotation of Chromatin Binding and Modification Loci. BMC Bioinform (2010) 11:415. doi: 10.1186/1471-2105-11-415
64. Bolger AM, Lohse M, Usadel B. Trimmomatic: A Flexible Trimmer for Illumina Sequence Data. Bioinformatics (2014) 30(15):2114–20. doi: 10.1093/bioinformatics/btu170
65. Li H, Durbin R. Fast and Accurate Short Read Alignment With Burrows-Wheeler Transform. Bioinformatics (2009) 25(14):1754–60. doi: 10.1093/bioinformatics/btp324
66. Saunders CT, Wong WS, Swamy S, Becq J, Murray LJ, Cheetham RK. Strelka: Accurate Somatic Small-Variant Calling From Sequenced Tumor-Normal Sample Pairs. Bioinformatics (2012) 28(14):1811–7. doi: 10.1093/bioinformatics/bts271
67. Wang K, Li M, Hakonarson H. ANNOVAR: Functional Annotation of Genetic Variants From High-Throughput Sequencing Data. Nucleic Acids Res (2010) 38(16):e164. doi: 10.1093/nar/gkq603
68. McLaren W, Pritchard B, Rios D, Chen Y, Flicek P, Cunningham F. Deriving the Consequences of Genomic Variants With the Ensembl API and SNP Effect Predictor. Bioinformatics (2010) 26(16):2069–70. doi: 10.1093/bioinformatics/btq330
69. Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, Salzberg SL. TopHat2: Accurate Alignment of Transcriptomes in the Presence of Insertions, Deletions and Gene Fusions. Genome Biol (2013) 14(4):R36. doi: 10.1186/gb-2013-14-4-r36
70. McLeod C, Gout AM, Zhou X, Thrasher A, Rahbarinia D, Brady SW, et al. St. Jude Cloud: A Pediatric Cancer Genomic Data-Sharing Ecosystem. Cancer Discov (2021) 11(5):1082–99. doi: 10.1158/2159-8290.CD-20-1230
71. Zhou X, Edmonson MN, Wilkinson MR, Patel A, Wu G, Liu Y, et al. Exploring Genomic Alteration in Pediatric Cancer Using ProteinPaint. Nat Genet (2016) 48(1):4–6. doi: 10.1038/ng.3466
72. Chervinsky DS, Lam DH, Melman MP, Gross KW, Aplan PD. Scid Thymocytes With TCRbeta Gene Rearrangements are Targets for the Oncogenic Effect of SCL and LMO1 Transgenes. Cancer Res (2001) 61(17):6382–7.
73. Campese AF, Garbe AI, Zhang F, Grassi F, Screpanti I, von Boehmer H. Notch1-Dependent Lymphomagenesis is Assisted by But Does Not Essentially Require Pre-TCR Signaling. Blood (2006) 108(1):305–10. doi: 10.1182/blood-2006-01-0143
74. Bellavia D, Campese AF, Checquolo S, Balestri A, Biondi A, Cazzaniga G, et al. Combined Expression of Ptalpha and Notch3 in T Cell Leukemia Identifies the Requirement of preTCR for Leukemogenesis. Proc Natl Acad Sci USA (2002) 99(6):3788–93. doi: 10.1073/pnas.062050599
75. Aifantis I, Raetz E, Buonamici S. Molecular Pathogenesis of T-Cell Leukaemia and Lymphoma. Nat Rev Immunol (2008) 8(5):380–90. doi: 10.1038/nri2304
76. Fasseu M, Aplan PD, Chopin M, Boissel N, Bories JC, Soulier J, et al. P16ink4a Tumor Suppressor Gene Expression and CD3epsilon Deficiency But Not Pre-TCR Deficiency Inhibit TAL1-Linked T-Lineage Leukemogenesis. Blood (2007) 110(7):2610–9. doi: 10.1182/blood-2007-01-066209
77. Bailis W, Pear WS. “The Molecular Basis of T Cell Development and How Epigenetic/Transcriptional Deregulation Leads to T-ALL”. In: Bonifer C, Cockerill NP, editors. Transcriptional and Epigenetic Mechanisms Regulating Normal and Aberrant Blood Cell Development, vol. p . Berlin, Heidelberg: Springer Berlin Heidelberg (2014). p. 267–93.
78. Sanda T, Lawton LN, Barrasa MI, Fan ZP, Kohlhammer H, Gutierrez A, et al. Core Transcriptional Regulatory Circuit Controlled by the TAL1 Complex in Human T Cell Acute Lymphoblastic Leukemia. Cancer Cell (2012) 22(2):209–21. doi: 10.1016/j.ccr.2012.06.007
79. Wadman IA, Osada H, Grutz GG, Agulnick AD, Westphal H, Forster A, et al. The LIM-Only Protein Lmo2 is a Bridging Molecule Assembling an Erythroid, DNA-Binding Complex Which Includes the TAL1, E47, GATA-1 and Ldb1/NLI Proteins. EMBO J (1997) 16(11):3145–57. doi: 10.1093/emboj/16.11.3145
80. Simon C, Chagraoui J, Krosl J, Gendron P, Wilhelm B, Lemieux S, et al. A Key Role for EZH2 and Associated Genes in Mouse and Human Adult T-Cell Acute Leukemia. Genes Dev (2012) 26(7):651–6. doi: 10.1101/gad.186411.111
81. Xi H, Schwartz R, Engel I, Murre C, Kersh GJ. Interplay Between RORgammat, Egr3, and E Proteins Controls Proliferation in Response to Pre-TCR Signals. Immunity (2006) 24(6):813–26. doi: 10.1016/j.immuni.2006.03.023
82. Engel I, Murre C. E2A Proteins Enforce a Proliferation Checkpoint in Developing Thymocytes. EMBO J (2004) 23(1):202–11. doi: 10.1038/sj.emboj.7600017
83. Lacombe J, Herblot S, Rojas-Sutterlin S, Haman A, Barakat S, Iscove NN, et al. Scl Regulates the Quiescence and the Long-Term Competence of Hematopoietic Stem Cells. Blood (2010) 115(4):792–803. doi: 10.1182/blood-2009-01-201384
84. Engel I, Johns C, Bain G, Rivera RR, Murre C. Early Thymocyte Development is Regulated by Modulation of E2A Protein Activity. J Exp Med (2001) 194(6):733–45. doi: 10.1084/jem.194.6.733
85. Lin YW, Nichols RA, Letterio JJ, Aplan PD. Notch1 Mutations are Important for Leukemic Transformation in Murine Models of Precursor-T Leukemia/Lymphoma. Blood (2006) 107(6):2540–3. doi: 10.1182/blood-2005-07-3013
86. Schwartz R, Engel I, Fallahi-Sichani M, Petrie HT, Murre C. Gene Expression Patterns Define Novel Roles for E47 in Cell Cycle Progression, Cytokine-Mediated Signaling, and T Lineage Development. Proc Natl Acad Sci USA (2006) 103(26):9976–81. doi: 10.1073/pnas.0603728103
87. Torgovnick A, Heger JM, Liaki V, Isensee J, Schmitt A, Knittel G, et al. The Cdkn1a(SUPER) Mouse as a Tool to Study P53-Mediated Tumor Suppression. Cell Rep (2018) 25(4):1027–39.e6. doi: 10.1016/j.celrep.2018.09.079
88. Soulier J, Clappier E, Cayuela JM, Regnault A, Garcia-Peydro M, Dombret H, et al. HOXA Genes are Included in Genetic and Biologic Networks Defining Human Acute T-Cell Leukemia (T-ALL). Blood (2005) 106(1):274–86. doi: 10.1182/blood-2004-10-3900
89. Downing JR, Wilson RK, Zhang J, Mardis ER, Pui CH, Ding L, et al. The Pediatric Cancer Genome Project. Nat Genet (2012) 44(6):619–22. doi: 10.1038/ng.2287
90. Gu Z, Churchman ML, Roberts KG, Moore I, Zhou X, Nakitandwe J, et al. PAX5-Driven Subtypes of B-Progenitor Acute Lymphoblastic Leukemia. Nat Genet (2019) 51(2):296–307. doi: 10.1038/s41588-018-0315-5
91. Funato N, Ohtani K, Ohyama K, Kuroda T, Nakamura M. Common Regulation of Growth Arrest and Differentiation of Osteoblasts by Helix-Loop-Helix Factors. Mol Cell Biol (2001) 21(21):7416–28. doi: 10.1128/MCB.21.21.7416-7428.2001
92. Prabhu S, Ignatova A, Park ST, Sun XH. Regulation of the Expression of Cyclin-Dependent Kinase Inhibitor P21 by E2A and Id Proteins. Mol Cell Biol (1997) 17(10):5888–96. doi: 10.1128/MCB.17.10.5888
93. Mingueneau M, Kreslavsky T, Gray D, Heng T, Cruse R, Ericson J, et al. The Transcriptional Landscape of Alphabeta T Cell Differentiation. Nat Immunol (2013) 14(6):619–32. doi: 10.1038/ni.2590
94. Ohashi R, Angori S, Batavia AA, Rupp NJ, Ajioka Y, Schraml P, et al. Loss of CDKN1A mRNA and Protein Expression Are Independent Predictors of Poor Outcome in Chromophobe Renal Cell Carcinoma Patients. Cancers (2020) 12(2):465. doi: 10.3390/cancers12020465
95. Davies C, Hogarth LA, Dietrich PA, Bachmann PS, Mackenzie KL, Hall AG, et al. P53-Independent Epigenetic Repression of the P21waf1 Gene in T-Cell Acute Lymphoblastic Leukemia. J Biol Chem (2011) 286(43):37639–50. doi: 10.1074/jbc.M111.272336
96. Bain G, Cravatt CB, Loomans C, Alberola-Ila J, Hedrick SM, Murre C. Regulation of the Helix-Loop-Helix Proteins, E2A and Id3, by the Ras-ERK MAPK Cascade. Nat Immunol (2001) 2(2):165–71. doi: 10.1038/84273
97. Engel I, Murre C. Disruption of Pre-TCR Expression Accelerates Lymphomagenesis in E2A-Deficient Mice. Proc Natl Acad Sci USA (2002) 99(17):11322–7. doi: 10.1073/pnas.162373999
98. Liao MJ, Zhang XX, Hill R, Gao J, Qumsiyeh MB, Nichols W, et al. No Requirement for V(D)J Recombination in P53-Deficient Thymic Lymphoma. Mol Cell Biol (1998) 18(6):3495–501. doi: 10.1128/MCB.18.6.3495
99. Kreslavsky T, Gleimer M, Miyazaki M, Choi Y, Gagnon E, Murre C, et al. β-Selection-Induced Proliferation Is Required for αβ T Cell Differentiation. Immunity (2012) 37(5):840–53. doi: 10.1016/j.immuni.2012.08.020
100. Wong GW, Knowles GC, Mak TW, Ferrando AA, Zuniga-Pflucker JC. HES1 Opposes a PTEN-Dependent Check on Survival, Differentiation, and Proliferation of TCRbeta-Selected Mouse Thymocytes. Blood (2012) 120(7):1439–48. doi: 10.1182/blood-2011-12-395319
101. Jacobs H, Ossendorp F, de Vries E, Ungewiss K, von Boehmer H, Borst J, et al. Oncogenic Potential of a Pre-T Cell Receptor Lacking the TCR Beta Variable Domain. Oncogene (1996) 12(10):2089–99.
102. dos Santos NR, Rickman DS, de Reynies A, Cormier F, Williame M, Blanchard C, et al. Pre-TCR Expression Cooperates With TEL-JAK2 to Transform Immature Thymocytes and Induce T-Cell Leukemia. Blood (2007) 109(9):3972–81. doi: 10.1182/blood-2006-09-048801
103. Allman D, Karnell FG, Punt JA, Bakkour S, Xu L, Myung P, et al. Separation of Notch1 Promoted Lineage Commitment and Expansion/Transformation in Developing T Cells. J Exp Med (2001) 194(1):99–106. doi: 10.1084/jem.194.1.99
104. Reschly EJ, Spaulding C, Vilimas T, Graham WV, Brumbaugh RL, Aifantis I, et al. Notch1 Promotes Survival of E2A-Deficient T Cell Lymphomas Through Pre-T Cell Receptor-Dependent and -Independent Mechanisms. Blood (2006) 107(10):4115–21. doi: 10.1182/blood-2005-09-3551
105. Ikawa T, Kawamoto H, Goldrath AW, Murre C. E Proteins and Notch Signaling Cooperate to Promote T Cell Lineage Specification and Commitment. J Exp Med (2006) 203(5):1329–42. doi: 10.1084/jem.20060268
106. Yashiro-Ohtani Y, He Y, Ohtani T, Jones ME, Shestova O, Xu L, et al. Pre-TCR Signaling Inactivates Notch1 Transcription by Antagonizing E2A. Genes Dev (2009) 23(14):1665–76. doi: 10.1101/gad.1793709
107. Miyazaki M, Miyazaki K, Chen S, Chandra V, Wagatsuma K, Agata Y, et al. The E-Id Protein Axis Modulates the Activities of the PI3K-AKT-Mtorc1-Hif1a and C-Myc/p19Arf Pathways to Suppress Innate Variant TFH Cell Development, Thymocyte Expansion, and Lymphomagenesis. Genes Dev (2015) 29(4):409–25. doi: 10.1101/gad.255331.114
108. Girard N, Tremblay M, Humbert M, Grondin B, Haman A, Labrecque J, et al. RARalpha-PLZF Oncogene Inhibits C/EBPalpha Function in Myeloid Cells. Proc Natl Acad Sci USA (2013) 110(33):13522–7. doi: 10.1073/pnas.1310067110
109. Pabst T, Mueller BU, Zhang P, Radomska HS, Narravula S, Schnittger S, et al. Dominant-Negative Mutations of CEBPA, Encoding CCAAT/enhancer Binding Protein-Alpha (C/EBPalpha), in Acute Myeloid Leukemia. Nat Genet (2001) 27(3):263–70. doi: 10.1038/85820
110. Mueller BU, Pabst T, Osato M, Asou N, Johansen LM, Minden MD, et al. Heterozygous PU.1 Mutations are Associated With Acute Myeloid Leukemia. Blood (2002) 100(3):998–1007. doi: 10.1182/blood-2002-12-3903
111. Familiades J, Bousquet M, Lafage-Pochitaloff M, Bene MC, Beldjord K, De Vos J, et al. PAX5 Mutations Occur Frequently in Adult B-Cell Progenitor Acute Lymphoblastic Leukemia and PAX5 Haploinsufficiency is Associated With BCR-ABL1 and TCF3-PBX1 Fusion Genes: A GRAALL Study. Leukemia (2009) 23(11):1989–98. doi: 10.1038/leu.2009.135
112. Braunstein M, Anderson MK. HEB in the Spotlight: Transcriptional Regulation of T-Cell Specification, Commitment, and Developmental Plasticity. Clin Dev Immunol (2012) 2012:678705. doi: 10.1155/2012/678705
113. Abbas T, Dutta A. P21 in Cancer: Intricate Networks and Multiple Activities. Nat Rev Cancer (2009) 9(6):400–14. doi: 10.1038/nrc2657
114. Li T, Kon N, Jiang L, Tan M, Ludwig T, Zhao Y, et al. Tumor Suppression in the Absence of P53-Mediated Cell-Cycle Arrest, Apoptosis, and Senescence. Cell (2012) 149(6):1269–83. doi: 10.1016/j.cell.2012.04.026
115. Wen H, Li Y, Xi Y, Jiang S, Stratton S, Peng D, et al. ZMYND11 Links Histone H3.3K36me3 to Transcription Elongation and Tumour Suppression. Nature (2014) 508(7495):263–8. doi: 10.1038/nature13045
116. Ramdzan ZM, Nepveu A. CUX1, a Haploinsufficient Tumour Suppressor Gene Overexpressed in Advanced Cancers. Nat Rev Cancer (2014) 14(10):673–82. doi: 10.1038/nrc3805
Keywords: SCL/TAL1, LMO1, HEB/TCF12, E2A/TCF3, NOTCH1, T-cell acute lymphoblastic leukemia, tumor suppressor genes
Citation: Veiga DFT, Tremblay M, Gerby B, Herblot S, Haman A, Gendron P, Lemieux S, Zúñiga-Pflücker JC, Hébert J, Cohen JP and Hoang T (2022) Monoallelic Heb/Tcf12 Deletion Reduces the Requirement for NOTCH1 Hyperactivation in T-Cell Acute Lymphoblastic Leukemia. Front. Immunol. 13:867443. doi: 10.3389/fimmu.2022.867443
Received: 01 February 2022; Accepted: 28 February 2022;
Published: 24 March 2022.
Edited by:
Avinash Bhandoola, National Institutes of Health (NIH), United StatesReviewed by:
Ivan Maillard, University of Pennsylvania, United StatesCopyright © 2022 Veiga, Tremblay, Gerby, Herblot, Haman, Gendron, Lemieux, Zúñiga-Pflücker, Hébert, Cohen and Hoang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Trang Hoang, dHJhbmcuaG9hbmdAdW1vbnRyZWFsLmNh
†These authors have contributed equally to this work and share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.